
Submitted:
29 April 2025
Posted:
29 April 2025
You are already at the latest version
Abstract
Developing efficient and environmentally friendly catalysts for sustainable biodiesel production is a promising approach to meet global energy demands and address environmental concerns. In this study, four 2-alkyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium ionic liquids (ASBILs) were synthesized as catalysts for biodiesel production and characterized using FT-IR, NMR, and MS analyses. Their catalytic activities were evaluated in a model esterification reaction between oleic acid and bioethanol. Proton NMR spectroscopy (400 MHz) was employed to determine the conversion of oleic acid to ethyl oleate. A simple methodology for the quantification of ethyl oleate in unpurified reaction mixtures directly in the 1H NMR spectra through the relationship between the areas of a characteristic signal was proposed and tested. The experimental results identified the efficient catalyst amount of 20 mol% (based on oleic acid mass) for optimal reaction parameters: 16.7:1 molar ratio of ethanol to oleic acid and reaction temperature of 102°C. The reactions reached equilibrium within approximately 75 minutes, achieving conversions of 97.5 – 100%. Notably, 100% conversion was obtained using the ASBIL with a C11 alkyl chain, and its catalytic activity showed no significant decline even after ten cycles of reuse.
Keywords:
ionic liquids
; biodiesel
; esterification catalysis
; fatty acids
; bioethanol
; sulfonic acids
; quantitative 1H-NMR
1. Introduction
Due to the growing concerns of the exhaustion of fossil fuel resources and degradation of the environment, renewable and environmentally friendly biofuels are currently explored to achieve energy security, diversifying the energy pool, and mitigating greenhouse gas emissions. Among the various biofuel resources, biodiesel has attracted significant interest as an alternative transport fuel to conventional petroleum diesel. Biodiesel, apart from being environmentally friendly, sustainable, biodegradable, non-flammable, and non-toxic, scores over other biofuels due to a variety of feedstocks used in its production (e.g. vegetable oils: rapeseed in Continental Europe, soybean and canola in North America and palm oil in South East Asia, waste cooking oil, algae oil, animal fats etc.). In addition, the production of biodiesel can help reduce environmental waste, especially the production of second-generation biodiesel, which is made from domestic waste. Furthermore, unlike conventional diesel production, biodiesel production does not produce waste or produces very minimal waste [1,2,3,4,5,6].
The routine procedure for biodiesel production is catalytic esterification of free fatty acids (FFAs) and transesterification of triacylglycerols (TGs). Both of these reactions proceed with short-chain mono-alcohols, such as methanol and ethanol. The transesterification reaction of TGs in vegetable oils with short-chain alcohol in the presence of suitable catalytic systems produces biodiesel, known as a mixture of alkyl esters of fatty acids, e.g., fatty acid methyl ester (FAME) or fatty acid ethyl ester (FAEE), and glycerol (the major by product). The esterification reaction of oil feedstocks with high FFA content with mono-alcohol produces alkyl esters of fatty acids and water as a side product. Catalysts are used to accelerate the reaction rate during the esterification and transesterification processes. Different types of catalysts can be used for biodiesel production, such as heterogeneous catalysts, homogeneous catalysts, and enzymatic catalysts. Numerous interesting patents on catalysts can be used in different reactor configurations to improve the biodiesel production process by producing minimal impurities, eliminating the need for neutralization and water washing of the waste salts. Furthermore, these catalysts may be recycled and reused through a simple process, which reduces production cost and environmental impact [2,6,7,8,9].
Among many new technologies for producing biodiesel, the use of microwaves (MW) has emerged as a highly encouraging prospect. MW irradiation helps reduce the reaction time and results in a higher biodiesel yield than conventional methods because of its distinct thermal and non-thermal effects. It can also improve the catalytic activity, so the reaction time could have been reduced to 10 seconds [10].
One main problem encountered in the study or industrial applications of biodiesel production processes is how to measure the methyl and ethyl ester content [11]. Proton NMR spectroscopy is one of the most suitable techniques for the quantitative analysis of small molecules in their crude forms and in mixtures. The use of the 1H NMR spectra has the advantage of the high natural abundance and magnetic moment of 1H nuclei, which allows the acquisition of 1H NMR spectra with greater signal. Another advantage of NMR is its primary analytical characteristic, because of which it can be applied in quantitative analysis without using any specific reference standard [12,13,14,15,16]. Quantitative NMR spectroscopy has been used to determine fatty acids in oils and fats for its many advantages over other techniques, for example, simplicity, the lack of need for sample pre-treatment such as derivatization or extractions, and especially due to the large amount of information that can be extracted from the NMR spectra. From a single NMR spectrum acquired directly from the sample under investigation without any kind of treatment, which could cause changes to the samples themselves, it is possible to identify and quantify a large number of individual components in a complex mixture through the characteristic NMR signals of each compound. Investigations have demonstrated that the relative fatty acid composition can be obtained just by measuring the areas of each fatty acid’s characteristic signal in the NMR spectra, with no need for a standard [17]. The quantitative inaccuracy of qNMR has been reported to be less than 2.0%, which is an acceptable limit for precise, accurate quantification. Validation processes (e.g., precision, accuracy, linearity, reproducibility, robustness, selectivity, and specificity) have proved that NMR spectroscopy is a good analytical technique for quantitative estimation [18,19,20,21,22]. NMR can provide detailed molecular information once a spectrum is acquired with a sufficiently high signal-to-noise ratio. For most samples generated in the biodiesel industry, sample quantity is not an issue, and NMR can be applied to biodiesel, and careful analysis with NMR can also determine relative amounts of identified components within a mixture such as biodiesel [23]. The detailed experimental conditions for a truly quantitative NMR analysis of biodiesel formation, such as the longitudinal relaxation time (T1), which is the determining factor for quantitative analysis, the linear determination coefficient, and time delay between pulses, were presented in the literature [24]. Proton NMR spectroscopy (200 MHz) was used for quantifying the content of ethyl esters in biodiesel produced by soybean oil ethanolysis. The transesterification values determined in this way were compared with viscosity and total glycerol determinations, and a good correlation was obtained [11]. The 1H NMR methodology elaborated for the quantification of biodiesel in unpurified reaction mixtures, ethyl esters, mono-, di-, triacyclglycerols were detected and quantified in the crude biodiesel using 1H NMR spectroscopic and GC-FID chromatographic methods with conclusion that the quantification methods were efficient for determination of yields of ethyl esters (biodiesel) in mixtures with mono-, di- and triacylglycerols [25]. The ethyl ester conversion calculation methodology is based on a reliable 1H NMR characterization strategy of mono-, di-, and tri-acylglycerols, as well as fatty acid methyl esters. This methodology was established during the last decade by numerous literature contributions [26,27,28,29,30,31,32,33,34,35].
In previous work [36], we synthesized three types of sulfonic acid group functionalized ionic liquids (SAILs) with different numbers of catalytic groups and lipophilicity, and studied their catalytic activities in the esterification of oleic acid and ethanol, by using either conventional or microwave-assisted heating. As the best catalyst showed the SAIL with the lipophilic alkyl chain due to its increased solubility in the reaction mixture and affinity with the reactants, which affects a great deal of its catalytic reactivity. The reaction has been preceded in a homogeneous mixture under reflux, after which the product was separated from the catalyst system by a simple liquid/liquid phase separation at room temperature with excellent yields. With the simple post-process, the catalyst is reusable at least 5 times. Therefore, the catalyst combines the advantages of a homogeneous reaction (high activity, selectivity) with the operational advantages of heterogeneous systems (easy separation, easy recyclability, reusability) and shows very stable activity.
The object of this work is to differentiate the catalytic reactivity of 2-alkyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium ionic liquids (ASBILs) with different alkyl chains and examine reusability performance for biodiesel synthesis. For the purpose of investigating effects of the length of carbon chains on catalytic activity of these ASBILs, four new ASBILs with different alkyl chain (C9, C11, C13 and C17) are prepared by optimized procedure, and together with that of C15 alkyl chain from previous work [36], their catalytic performance is examined in esterification of oleic acid with ethanol. The conversion based on the consumption of carboxylic acids during catalytic esterification reaction experiments is determined by quantitative 1H NMR spectroscopy.
2. Results and Discussion
2.1. Synthesis and Characterization of ASBIL Catalysts 5a - d
C9-, C11-, C13-, and C17-ASBILs 5a - d were synthesized following the procedure described in our previous work [36], as illustrated in Scheme 1. The 2-alkyl-1H-benzo[d]imidazoles 3a - d were obtained through the condensation of o-phenylenediamine with fatty acids 2a - d. Subsequently, 1,3-propanesultone underwent bimolecular nucleophilic substitution (SN2 opening) with benzimidazoles 3a - d, activated by NaH, to yield the 3-(2-alkyl-1-(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium-3-yl)propane-1-sulfonates 4a - d, which were then purified by column chromatography. Finally, treatment with H₂SO₄ yielded the desired catalysts 5a - d.
In the reaction of 1,3-propanesultone with NaH-activated benzimidazoles, the first molecule of 1,3-propanesultone readily bonded to the nitrogen atom in the heterocycle within an hour. However, the attachment of the second 1,3-propanesultone molecule proceeded much more slowly. Consequently, the reaction mixtures were continuously heated at 110 °C for four weeks to ensure completion.
The 2-alkyl-1H-benzo[d]imidazoles 3a - d, intermediates 4a - d, and final catalysts 5a - d were fully characterized by FT-IR, NMR, and MS analyses, as detailed in our previous work [36].
2.2. Catalytic Performance of the Prepared ASBILs
2.2.1. Development and Validation of the NMR Analysis Methodology
To determine the conversion of oleic acid to ethyl oleate during the catalytic esterification reaction, a simple and effective quantitative ¹H NMR analysis method was developed and tested to quantify both ethyl oleate and unreacted oleic acid in the reaction mixture. Figure 1 and Figure 2 illustrate the general ¹H NMR chemical shift assignments for different types of hydrogen atoms in oleic acid and ethyl oleate.
The CH₃ hydrogens of the acyl chains in both oleic acid and ethyl oleate exhibit nearly identical chemical shifts at 0.87 ppm. Meanwhile, the CH₂ hydrogens of the ethanol moiety in ethyl oleate produce a distinct signal at 4.11 ppm. These signals are easily identifiable, well-defined, and do not overlap with other hydrogen signals in the NMR spectra.
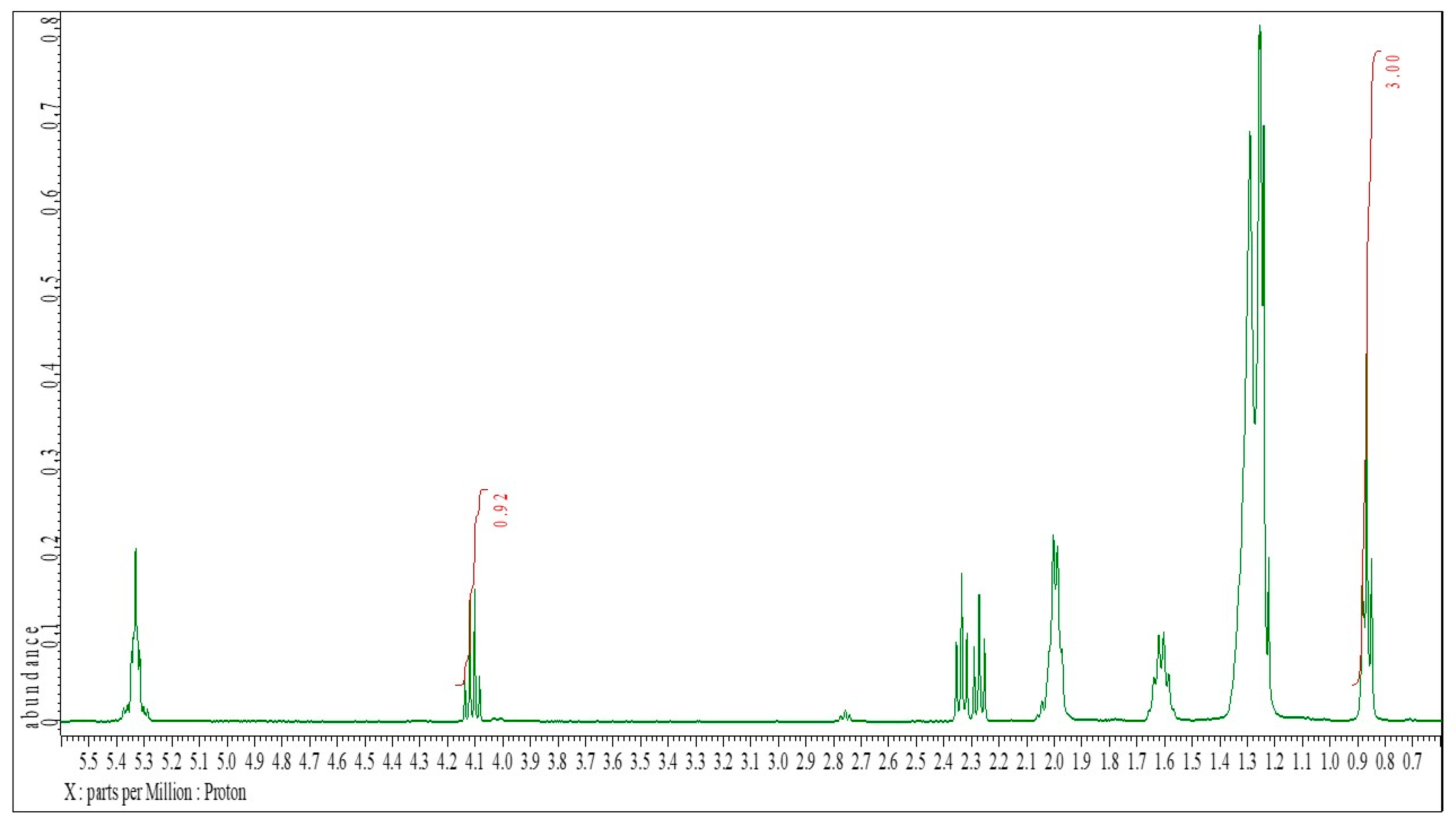
Figure 3 displays a ¹H NMR spectrum of a known mixture containing oleic acid and ethyl oleate (test sample 3). The core principle of this methodology is based on the fact that peak areas in ¹H NMR spectra are proportional to the number of corresponding hydrogen atoms in the sample. Since the signal at 0.87 ppm represents the CH₃ hydrogens of the acyl chains in both oleic acid and ethyl oleate, its integration value corresponds to the total oleic acid content at the start of the reaction and remains unchanged throughout esterification.
The integration value of the peak at 4.11 ppm corresponds to the amount of ethyl oleate formed during esterification, which directly reflects the quantity of oleic acid consumed in the reaction. Therefore, the conversion of oleic acid can be determined by comparing the integration areas of the characteristic ethyl ester signal at 4.11 ppm with the common CH₃ signal of the acyl chain at 0.87 ppm in the ¹H NMR spectrum (Figure 3).
To validate the ¹H NMR analysis methodology, a series of test samples were prepared to simulate reaction mixtures at different stages of esterification and subsequently analysed by ¹H NMR. First, stock solutions of oleic acid and ethyl oleate in deuterated chloroform were prepared by accurately weighing and dissolving oleic acid and ethyl oleate in a precisely measured amount of deuterated chloroform. These stock solutions had precisely known concentrations (0.1078714859 w/w and 0.1161772477 w/w, respectively), calculated from their exact weight values. Next, test samples were prepared by mixing varying amounts of these stock solutions (80+0, 60+20, 40+40, 20+60, and 0+80 μL) with an additional 800 μL of CDCl₃ in NMR tubes. Each dose of stock solution was accurately weighed, allowing for precise calculation of the mole quantities of oleic acid n(OA) and ethyl oleate n(EO) based on the known concentrations of the stock solutions. The simulated conversions were determined from the quantities of oleic acid and ethyl oleate in each sample as follows:
and from the areas of the signals at 0.87 ppm and 4.11 ppm in the 1H NMR spectra as follows:
I(4.11 ppm) represents the ¹H NMR integration value of the CH₂ hydrogen atoms in ethyl oleate (EO-CH₂, δ = 4.11 ppm), while I(0.87 ppm) corresponds to the integration value of the CH₃ hydrogen atoms in the acyl chain (-CH₃, δ = 0.87 ppm), which are common to both ethyl oleate and oleic acid. The normalization factors (2 and 3) used to divide the integration values account for the number of hydrogen atoms in each respective group. As shown in Table 1., the conversions determined by ¹H NMR spectroscopy are in good agreement with the known simulated conversions, confirming the reliability of the developed methodology.
2.2.2. Optimization of Catalyst Dosage and Reaction Conditions
The esterification of oleic acid with ethanol was used as a model reaction to evaluate the catalytic activities of ASBILs. The first step involved determining the optimal reaction conditions. For this purpose, we used the catalyst 2-pentadecyl-1,3-bis(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate (C15-ASBIL), which was synthesized in our previous work [36].
In general, the reaction rate mainly depends on the concentration of the reactants, temperature, and the nature and amount of the catalyst. The optimal reaction temperature was determined based on the temperature of the oil bath (102 °C), at which ethanol in the flask began to boil, and the reaction mixture temperature remained constant despite further heating of the oil bath. A molar ratio of ethanol to oleic acid of 16.7:1 was used, as reported in [36].
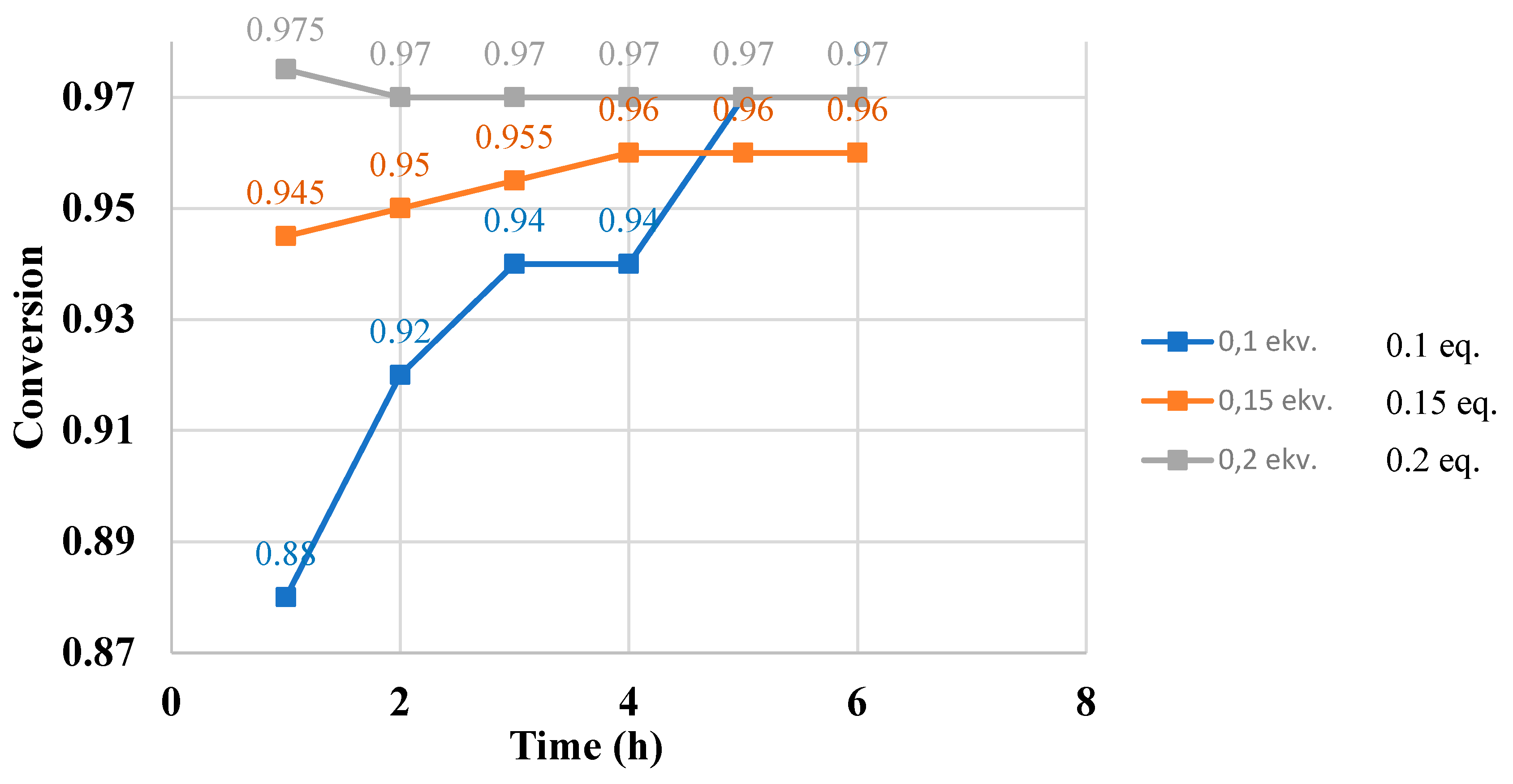
To determine the optimal catalyst dosage, esterification reactions were carried out with various catalyst amounts (10, 15, and 20 mol% based on the mass of oleic acid), and conversions were monitored over six hours at one-hour intervals. For each ¹H NMR sample, 0.2 mL of the reaction mixture was withdrawn, ensuring that approximately 50 mg of oleic acid and ethyl oleate were present in 0.6 mL of deuterated chloroform for quantitative NMR measurements. The total volume of the collected samples did not exceed 10% of the total reaction mixture, ensuring that the sampling process did not significantly influence the reaction progress.
The kinetic plot clearly shows that the reactions approached equilibrium significantly within 5, 4, and 1 hours, respectively, indicating that 20 mol% (based on the mass of oleic acid) of the catalyst is required for efficient catalysis.
The NMR analyses also demonstrated high purity of the ethyl ester product (biodiesel).

Table 2.
Esterification of oleic acid and ethanol with various amounts of C15-ASBIL. Conversions of oleic acid during 6 hours.
Table 2.
Esterification of oleic acid and ethanol with various amounts of C15-ASBIL. Conversions of oleic acid during 6 hours.
| Amount of catalyst C15-ASBIL [mol% based on oleic acid mass] |
Conversion | |||||
|---|---|---|---|---|---|---|
| 1 h | 2 h | 3 h | 4 h | 5 h | 6 h | |
| 10 | 0.88 | 0.92 | 0.94 | 0.94 | 0.97 | 0.97 |
| 15 20 |
0.945 0.975 |
0.95 0.97 |
0.955 0.97 |
0.96 0.97 |
0.96 0.97 |
0.96 0.97 |
Graph 1.
Esterification of oleic acid and ethanol with various amounts of C15- ASBIL. Conversions of oleic acid during 6 hours.
Graph 1.
Esterification of oleic acid and ethanol with various amounts of C15- ASBIL. Conversions of oleic acid during 6 hours.
2.2.3. Catalytic Activity of the Prepared ASBILs
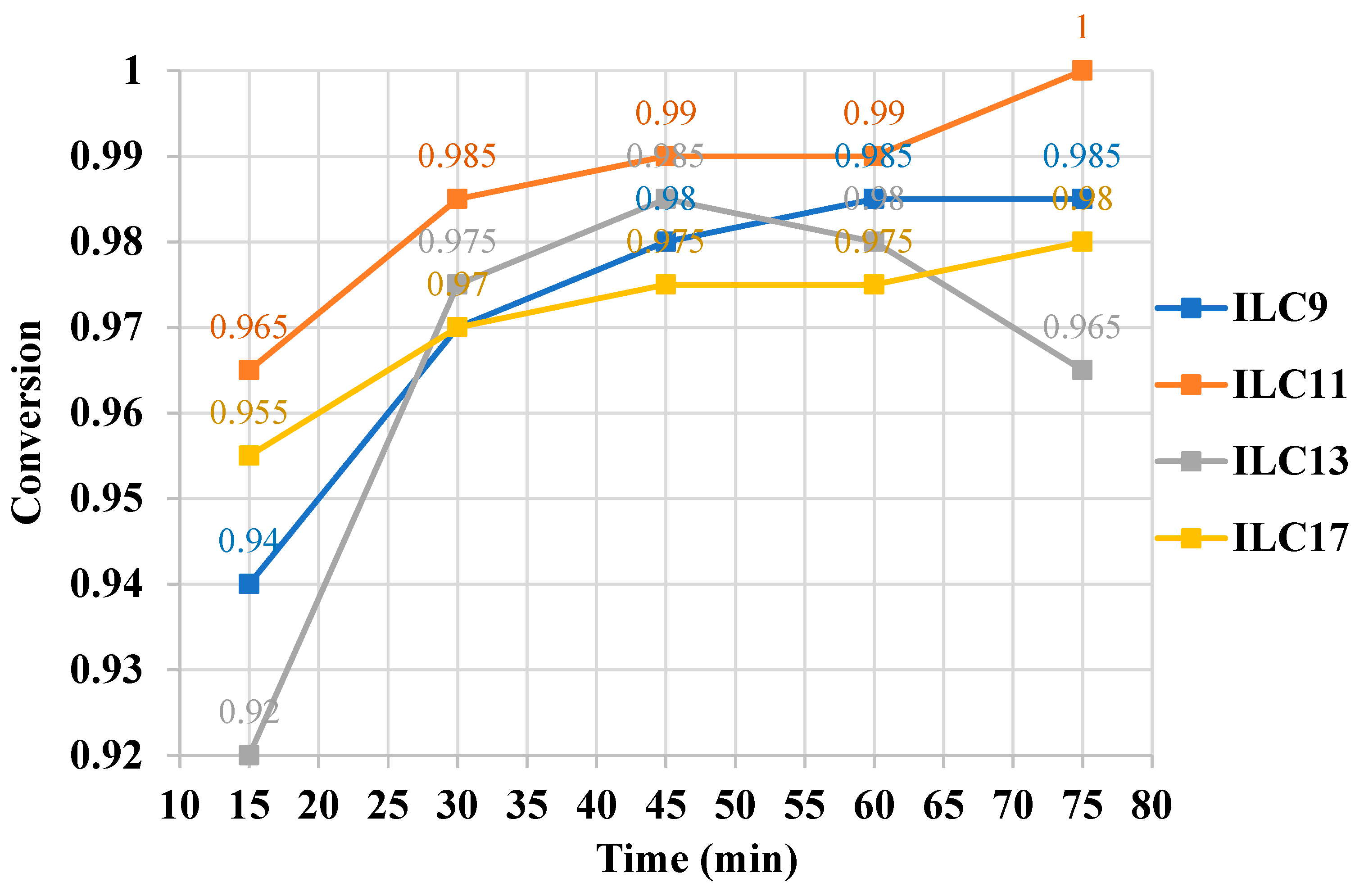
Further, we evaluated the catalytic efficiency of the newly synthesized ASBILs. For this purpose, esterification experiments were conducted under the optimal reaction conditions: a 16.7:1 molar ratio of ethanol to oleic acid, a reaction temperature of 102 °C, and 20 mol% of catalyst (based on the mass of oleic acid), in line with previous experiments. Conversions were monitored in 15-minute intervals. As shown in Table 3 and Graph 2, the reactions reached equilibrium within approximately 45 - 75 minutes.
2.2.4. Catalyst Recyclability
The time required to reach reaction equilibrium, as determined from Graph 2, ranges from 45 to 75 minutes, with the best catalytic efficiency observed for the C11 alkyl chain. Consequently, we selected catalyst 5b (C11-ASBIL) to evaluate its catalytic reusability. Ten cycles of esterification were conducted under the same experimental conditions. After each cycle, the reaction mixture was cooled in an ice bath until the two phases separated, allowing the crude products to be easily removed from the reaction. The catalyst in the lower layer was then recycled and used in the subsequent cycle. As shown in Graph 3, the oleic acid conversions varied between 97.5% and 99%, indicating excellent catalyst reusability.
3. Materials and Methods
3.1. Materials and Reagents
All reagents used in this study were supplied by Sigma–Aldrich, Acros Organics, Penta, and Lachema: 1,3-propansultone: 97.0%, Silica gel: silica gel for chromatography, ultrapure, 46-60 μm, 60A (ACROS ORGANICS); Stearic Acid: 97.0%, Palmitic acid: 98.0%, Myristic Acid: 99.0%, Lauric acid: 99.0%, Capric acid: 99.0% (THERMO SCIENTIFIC); Oleic acid: 70.0%, Activated charcoal (LACH: NER), Ethyl Oleate: 99.5%, o-Phenylenediamine: 99.5%, Dimethylformamide: 99.8%, Sodium Hydride: 60.0% suspension (ALDRICH); Ethanol: 96.0%, Methanol: 99.0%, Diethyl Ether: 99.0%, Acetic Acid Ethyl Ester: 99.7%, Dichloromethane: 99.5%, Hexane: 95.0%, Toluene: 99.0%, Potassium Hydroxide: 85.0%, Sulfuric Acid: 96.0% (PENTA); Hydrochloric acid: 35.0%, Phosphomolybdic acid: pure, (LACHEMA).
Dimethylformamide (DMF) was distilled under argon, hexane was dried with sodium and distilled under argon. Solution of HCl in methanol (approximately 2.7 M) was obtained through bubbling HCl (formed from the reaction of 16 g NaCl and 16 mL conc H2SO4) into 100 mL methanol. Synthetic procedures were carried out under an inert atmosphere until acidic workup.
The catalyst 2-pentadecyl-1,3-bis(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate C15-ASBIL was synthesized in our previous work [36].
3.2. Characterization Methods
The structures of the ionic liquids and their intermediates were analysed by FT-IR, 1H, 13C NMR spectroscopy and mass spectrometry. The supporting spectroscopic data can be found in the Supplementary Materials (Figures S1–S16).
Infrared spectra were measured on a Nicolet 6700 FT-IR spectrometer from Thermo Scientific with the ATR technique using a single reflection horizontal ZnSe crystal. The measurement was carried out at a resolution of 4 cm-1, and the number of scans was 150. The spectra were recorded and processed using OMNIC 7.3 program.
1H and 13C NMR spectra were obtained on a JEOL 400 MHz JNM-ECZ400R/M1 nuclear magnetic resonance spectrometer. Chemical shifts are reported in ppm, and spin-spin coupling constants are presented in Hz.
Mass spectrometry (MS) spectra were performed on a Thermo Scientific Inc. LCQ Fleet Ion Trap instrument using electrospray (ESI) ionization with helium (6.0 Messer, Czech Republic) as a collision gas. Samples dissolved in 5% (v/v) aqueous acetic acid mixed with methanol (1:1, v:v) (concentrations approximately 100 ng mL-1) were introduced to the ion source from a Hamilton syringe using an infusion of 15 μL min-1, source voltage 5.0 kV, tube lens voltage -110.0 V, capillary voltage -35.0 V, capillary temperature 275 °C and N2 as a nebulizing sheath gas 15 p.d.u. In all cases, positive ions corresponding to the molecular ions were observed for the highest peak in the isotopic distribution plot. The isotopic distribution and the whole shape of the spectra of all detected peaks were in agreement with the calculated spectra. The measurement range was 150 to 2000 a.m.u. Flash column chromatography was carried out on silica gel 60. TLC was performed on plates of silica gel 60 F254, detection was executed by spraying with a solution of Phosphomolybdic acid (0.5g) in a mixture of ethanol (10 mL) and water (10 mL) and subsequent heating on the hot plate.
3.3. Synthesis of the Catalysts
ASBIL catalysts 5a - d were synthesized according to the reported procedure [36] with an optimalization as explained below.
- 2-Alkyl-1H-benzo[d]imidazoles 3a - d
A mixture of a fatty acid (0.10 mol) and o-phenylenediamine (16.2 g, 0.15 mol) was placed in a 250-mL round-bottom flask equipped with a magnetic stirrer. The apparatus was evacuated and purged with argon, and this process was repeated twice. The reaction mixture was then heated to 185 °C and stirred vigorously for 2 h. The resulting purple melt was dissolved in ethanol (60 mL) at 80 °C, and the solution was allowed to crystallize overnight. The following day, water (20 mL) was added, and the crude product was collected by filtration and washed with a 1:1 mixture of ethanol and water (140 mL). The product was then dissolved in hot ethanol (60 mL), and a small amount of activated charcoal was added. The hot solution was filtered, and water was added dropwise until slight turbidity formed. Crystallization was carried out at room temperature. The solid was collected by suction filtration, washed with a 1:1 ethanol–water mixture, and dried to afford the pure white crystalline product.
- 2-Nonyl-1H-benzo[d]imidazole 3a
11,11 g of white crystalline product was obtained, yield 45%; mp 123 - 124 °C; 1H NMR (400 MHz, CDCl3): 0.84 (t, J = 6.8 Hz, 3 H), 1.09 – 1.30 (m, 10 H), 1.32 – 1.39 (m, 2 H), 1.82 – 1.90 (m, 2 H), 2.95 (t, J = 7.8 Hz 2 H), 7.18 – 7.22 (m, 2 H), 7.54 (bs, 2 H), 11.58 (bs, 1 H). 13C NMR (101 MHz, CDCl3): 14.04 (s), 22.60 (s), 28.38 (s), 29.22 – 29.40 (m), 31.79 (s), 77.00 (s), 122.02 (s), 155.55 (s); ESI MS m/z [M + H]+ 245 (100 %), calculated for C16H25N2: 245.38; IR (ATR, cm-1): 3180 – 2360 (C-H, benzimidazole), 2926, 2858 (C-H, C9H19), 1636, 1546, 1460 (C=C, C=N, benzimidazole), 752, 728 (C-H, benzimidazole).
- 2-Undecyl-1H-benzo[d]imidazole 3b
17,00 g of white crystalline product was obtained, yield 62%; mp 108 - 109 °C; 1H-NMR (400 MHz, CDCl3): 0.86 (t, J = 6.8 Hz, 3 H), 1.12 – 1.30 (m, 14 H), 1.31 – 1.44 (m, 2 H), 1.80 – 1,88 (m, 2 H), 2.92 (t, J = 7.8 Hz 2 H), 7.18 – 7.22 (m, 2 H), 7.53 (bs, 2 H), 10.59 (bs, 1 H); 13C NMR (101 MHz, CDCl3): 14.20 (s), 22.75 (s), 28.40 (s), 29.40 – 29.68 (m), 31.97 (s), 77.12 (s), 122.18 (s), 155.36 (s); ESI MS m/z [M + H]+ 273 (100 %), calculated for C18H29N2: 273.44; IR (ATR, cm-1): 3169 – 2361 (C-H, benzimidazole); 2919, 2852 (C-H, C11H23); 1626, 1546, 1460 (C=C, C=N, benzimidazole); 754, 719 (C-H, benzimidazole).
- 2-Tridecyl-1H-benzo[d]imidazole 3c
16,20 g of white crystalline product was obtained, yield 54%; mp 106 - 107 °C; 1H NMR (400 MHz, CDCl3): 0.86 (t, J = 6.8 Hz, 3 H), 1.12 – 1.31 (m, 18 H), 1.31 – 1.41(m, 2 H), 1.81 – 1.88 (m, 2 H), 2.92 (t, J = 7.6 Hz 2 H), 7.18 – 7.22 (m, 2 H), 7.53 (bs, 2 H), 10.47 (bs, 1 H); 13C NMR (101 MHz, CDCl3): 14.20 (s), 22.77 (s), 28.39 (s), 29.43 – 29.75 (m), 32.00 (s), 77.12 (s), 122.19 (s), 155.35 (s); ESI MS m/z [M + H]+ 301 (100 %), calculated for C20H33N2: 301.49; IR (ATR, cm-1): 3172 – 2359 (C-H, benzimidazole); 2921, 2847 (C-H, C13H27); 1628, 1548, 1462 (C=C, C=N, benzimidazole); 754, 726 (C-H, benzimidazole).
- 2-Heptadecyl-1H-benzo[d]imidazole 3d
The product still contained a small amount of stearic acid and was therefore further purified as following. It was dissolved in a solution of potassium hydroxide (4 g) in ethanol (100 mL), with a small amount of water was added until slight turbidity appeared. The mixture was then allowed to cool and crystallize. The resulting crystalline product was collected by filtration and washed with warm water until the filtrate gave a neutral pH on indicator paper.
22,57 g of white crystalline product was obtained, yield 63%; mp 91 - 92 °C; 1H NMR (400 MHz, CDCl3): 0.86 (t, J = 6.8 Hz, 3 H), 1.14 – 1.33 (m, 26 H), 1.33 – 1.43(m, 2 H), 1.80 – 1.88 (m, 2 H), 2.91 (t, J = 7.6 Hz 2 H), 7.18 – 7.22 (m, 2 H), 7.53 (bs, 2 H); 13C NMR (101 MHz, CDCl3): 14.23 (s), 22.79 (s), 28.33 (s), 29.43 – 29.80 (m), 32.02 (s), 77.12 (s), 122.24 (s), 155.16 (s). ESI MS m/z [M + H]+ 357 (100 %), calculated for C22H37N2: 357.60; IR (ATR, cm-1): 3175 – 2364 (C-H, benzimidazole); 2918, 2847 (C-H, C17H35); 1631, 1536, 1468 (C=C, C=N, benzimidazole); 751, 717 (C-H, benzimidazole).
- 3-(2-Alkyl-1-(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium-3-yl)propane-1-sulfonates 4a - d
Sodium hydride (60% dispersion in mineral oil, 2.40 g, 60 mmol) was placed in a 250-mL two-neck round-bottom flask fitted with a reflux condenser. The flask was evacuated and refilled with argon three times. The suspension was washed with anhydrous hexane (3 × 10 mL) under argon. Pre-distilled dimethylformamide (DMF, 100 mL) and 2-alkyl-1H-benzo[d]imidazole 4a – d (0.020 mol) were added, and the reaction mixture was stirred vigorously to afford a slightly turbid solution of sodium 2-alkyl-1H-benzo[d]imidazol-1-ide. The mixture was cooled in an ice bath for 15 min before 1,3-propanesultone (7.3 mL, 83 mmol) was added dropwise. The cooling bath was then removed, and the mixture was heated at 110 °C for 4 weeks under an inert atmosphere. After completion, DMF (85–90 mL) was removed under reduced pressure at 110 °C. The remaining residue was washed with diethyl ether (3 × 50 mL). The crude product was suspended in methanol (50 mL), and a solution of HCl in methanol was added dropwise until the mixture gave an acidic pH on indicator paper. The suspension was centrifuged, and the supernatant was evaporated under reduced pressure using a rotary evaporator. The residue was purified by column chromatography on silica gel (110 g), eluting with CH₂Cl₂ (200 mL), CH₂Cl₂–MeOH 9:1 (200 mL), and CH₂Cl₂–MeOH 4:1 (1500 mL), with 100 mL fractions collected.
- 3-(2-Nonyl-1-(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium-3-yl)propane-1-sulfonate 4a
5.94 g of oily product was obtained, yield 61%; 1H NMR (400 MHz, DMSO-d6): 0.82 (t, J = 6.8 Hz, 3 H), 1.14 – 1.34 (m, 10 H), 1.35 – 1.53 (m, 2 H), 1.56 – 1.71 (m, 2 H), 2.02 – 2.16 (m, 4 H), 2.55 (t, J = 6.4 Hz, 4 H), 3.26 (t, J = 8,4 Hz, 2 H), 4.62 (t, J = 7.8 Hz, 4 H), 7.55 – 7.63 (m, 2 H), 8.00 – 8.09 (m, 2 H), 8.15 (bs, 1 H). 13C NMR (101 MHz, DMSO-d6): 22.63 (s), 25.92 (s), 29.22 – 29.30 (m), 34.91 (s), 47.98 (s), 113.82 (s), 126.54 (s), 131,51 (s), 154.13 (s). ESI MS m/z [M + H]+ 489 (8 %), calculated for C22H37N2O6S2: 489.67; IR (ATR, cm-1): 3452 (C-H, benzimidazole); 2927, 2855 (C-H, C9H19); 1642, 1516, 1471 (C=C, C=N, benzimidazole); 1154, 1034 (S=O, -SO3H, -SO3-).
- 3-(2-Undecyl-1-(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium-3-yl)propane-1-sulfonate 4b
5.09 g of oily product was obtained, yield 49%; 1H NMR (400 MHz, DMSO-d6): 0.80 (t, J = 7 Hz, 3 H), 1.12 – 1.31 (m, 14 H), 1.38 – 1.48 (m, 2 H), 1.57 – 1.73 (m, 2 H), 2.02 – 2.18 (m, 4 H), 2.59 (t, J = 6.8 Hz, 4 H), 3.27 (t, J = 7,8 Hz, 2 H), 4.63 (t, J = 7.4 Hz, 4 H), 7.55 – 7.63 (m, 2 H), 8.01 – 8.09 (m, 2 H), 8.27 (bs, 1 H); 13C NMR (101 MHz, DMSO-d6): 22.63 (s), 25.89 (s), 29.25 – 29.59 (m), 34.90 (s), 47.99 (s), 113.83 (s), 126.56 (s), 131.50 (s), 154.14 (s); ESI MS m/z [M + H]+ 517 (6 %) calculated for C24H41N2O6S2: 517.72; IR (ATR, cm-1): 3381 (C-H, benzimidazole); 2927, 2844 (C-H, C11H23); 1648, 1522, 1476 (C=C, C=N, benzimidazole); 1179, 1040 (S=O, -SO3H, -SO3-).
- 3-(2-Tridecyl-1-(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium-3-yl)propane-1-sulfonate 4c
6.75 g of oily product was obtained, yield 92%; 1H NMR (400 MHz, DMSO-d6): 0.80 (t, J = 6.8 Hz, 3 H), 1.11 – 1.33 (m, 18 H), 1.37 – 1.52 (m, 2 H), 1.55 – 1.74 (m, 2 H), 2.04 – 2.15 (m, 4 H), 2.57 (t, J = 6.6 Hz, 4 H), 3.27 (t, J = 8,0 Hz, 2 H), 4.62 (t, J = 7.6 Hz, 4 H), 7.56 – 7.61 (m, 2 H), 8.02 – 8.08 (m, 2 H), 8.23 (bs, 1 H); 13C NMR (101 MHz, DMSO-d6): 22.63 (s), 25.89 (s), 29.25 – 29.61 (m), 34.90 (s), 47.99 (s), 113.83 (s), 126.55 (s), 131.50 (s), 154.14 (s); ESI MS m/z [M + H]+ 545 (3 %) calculated for C26H45N2O6S2: 545.77; IR (ATR, cm-1): 3458 (C-H, benzimidazole); 2924, 2850 (C-H, C13H27); 1653, 1513, 1471 (C=C, C=N, benzimidazole); 1162, 1028 (S=O, -SO3H, -SO3-).
- 3-(2-Heptadecyl-1-(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium-3-yl)propane-1-sulfonate 4d
6.08 g of oily product was obtained, yield 51%; 1H NMR (400 MHz, DMSO-d6): 0.80 (t, J = 6.8 Hz, 3 H), 1.12 – 1.37 (m, 26 H), 1.31 – 1.51 (m, 2 H), 1.54 – 1.73 (m, 2 H), 2.02 – 2.19 (m, 4 H), 2.57 (t, J = 6.6 Hz, 4 H), 3.26 (t, J = 8,0 Hz, 2 H), 4.62 (t, J = 7.6 Hz, 4 H), 7.54 – 7.70 (m, 2 H), 8.00 – 8.10 (m, 2 H), 8.21 (bs, 1 H); 13C NMR (101 MHz, DMSO-d6): 22.63 (s), 25.89 (s), 29.24 – 29.61 (m), 34.90 (s), 48.00 (s), 113.82 (s), 126.55 (s), 131.50 (s), 154.12 (s); ESI MS m/z [M + H]+ 601 (2 %) calculated for C30H53N2O6S2: 601.88; IR (ATR, cm-1): 3429 (C-H, benzimidazole); 2921, 2855 (C-H, C17H35); 1642, 1516, 1468 (C=C, C=N, benzimidazole); 1159, 1037 (S=O, -SO3H, -SO3-).
- 2-Alkyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfates 5a - d
Concentrated sulfuric acid was added to a solution of 3-(2-alkyl-1-(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium-3-yl)propane-1-sulfonate 5a–d in ethanol (90 mL), and the mixture was stirred vigorously until a clear solution was obtained. The solution was then heated in an oil bath at 80 °C for 1 h. After the reaction, the solvent was removed under reduced pressure, and the residue was repeatedly washed with diethyl ether and dried under vacuum. The product was further dried in a desiccator.
- 2-Nonyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate 5a
5a was prepared from 4a (4.89 g, 10 mmol) and sulfuric acid (2.23 mL, 40 mmol) to yield 4.04 g of oily product, yield 69%; ESI MS m/z [M + H]+ 489 (95%) calculated for C22H37N2O6S2: 489.67; IR (ATR, cm-1): 3383 (C-H, benzimidazolium); 2961, 2818 (C-H, C9H19); 1665, 1516, 1471 (C=C, C=N, benzimidazolium); 1159, 1022 (S=O, SO3H, HSO4-).
- 2-Undecyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate 5b
5b was prepared from 4b (3.06 g, 6 mmol) and sulfuric acid (1.32 mL, 24 mmol) to yield 3.68 g of oily product, yield 99%; ESI MS m/z [M + H]+ 517 (48%) calculated for C24H41N2O6S2: 517.72; IR (ATR, cm-1): 3415 (C-H, benzimidazolium); 2941, 2810 (C-H, C11H23); 1705, 1525, 1471 (C=C, C=N, benzimidazolium); 1139, 1014 (S=O, SO3H, HSO4-).
- 2-Tridecyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate 5c
5c was prepared from 4c (5.72 g, 11 mmol) and sulfuric acid (2.34 mL, 42 mmol) to yield 6.21 g of oily product, yield 92%; ESI MS m/z [M + H]+ 545 (100%) calculated for C26H45N2O6S2: 545.77; IR (ATR, cm-1): 3409 (C-H, benzimidazolium); 2929, 2835 (C-H, C13H27); 1673, 1525, 1476 (C=C, C=N, benzimidazolium); 1142, 1020 (S=O, SO3H, HSO4-).
- 2-Heptadecyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate 5d
5d was prepared from 4d (5.23 g, 9 mmol) and sulfuric acid (1.94 mL, 35 mmol) to yield 6.12 g of oily product, yield 100%; ESI MS m/z [M + H]+ 601 (42%), calculated for C30H53N2O6S2: 601.88; IR (ATR, cm-1): 3403 (C-H, benzimidazolium); 2955, 2812 (C-H, C17H35); 1642, 1468, 1411 (C=C, C=N, benzimidazolium); 1148, 1020 (S=O, SO3H, HSO4-).
3.4. Evaluation of Catalytic Performance
The optimum reaction temperature was determined as the oil bath temperature (102 °C) at which the ethanol in the flask began to boil and the temperature of the liquid in the flask ceased to increase, despite a continued rise in the oil bath temperature.
3.4.1. General Procedure for the Esterification of Fatty Acids with Ethanol and Sampling
Oleic acid (2.82 g, 10 mmol), ethanol (10 mL, 167 mmol), and a specified amount of ASBIL catalyst were added to a 25 mL round-bottom flask. The mixture was refluxed with stirring in an oil bath at 102 °C. Samples were taken at specific time intervals.
For sampling, the reaction mixture was first cooled in an ice bath until phase separation occurred. A 0.2 mL aliquot was taken from the upper layer and transferred to a 10 mL flask. The sample was then evaporated to constant weight using a rotary evaporator under reduced pressure. The residue was dissolved in deuterated chloroform (0.6 mL) and transferred to NMR tubes. The conversion of oleic acid was determined by quantitative 1H NMR spectroscopy.
3.4.2. Optimization of Catalyst Quantity and Reaction Time
Esterification reactions were performed using varying amounts (1.0, 1.5, and 2.0 mmol) of the catalyst 2-pentadecyl-1,3-bis(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate (C15-ASBIL). Samples were collected at hourly intervals over a period of 6 h.
3.4.3. Evaluation of Catalytic Performance
Esterification reactions were carried out using 2.0 mmol (the optimum amount) of catalysts 5a – d and 2-pentadecyl-1,3-bis(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium hydrogen sulfate. Samples were taken at 15-minute intervals over a total reaction time of 75 minutes (the optimum reaction time).
3.4.4. Recycling of the Catalyst 5b and Evaluation of Its Stability
Esterification reactions were performed using the optimum amount of catalyst 5b (2.0 mmol) for the optimum reaction time (75 min). Samples were taken, and the catalyst was recycled as follows. The reaction mixture was cooled in an ice bath until phase separation occurred. The upper layer was removed and evaporated to constant weight using a rotary evaporator; 35 mg of the residue was transferred to an NMR tube for analysis. The lower layer, containing the ionic liquid, was also evaporated under reduced pressure. The remaining ionic liquid was washed repeatedly with diethyl ether to remove residual reactants and products (ethanol and ethyl oleate). The purified ionic liquid was then dried under vacuum to constant weight and reused in the next reaction cycle.
3.4.5. Determination of the Conversion by 1H NMR
For 1H NMR analysis, samples were dissolved in 800 µl CDCl3 in 5-mm NMR tubes. The 1H NMR spectra were acquired on a JEOL 400 MHz JNM-ECZ400R/M1 nuclear magnetic resonance spectrometer operating at 9.4 T observing the 1H nuclei at 399.8 MHz. 32 scans of each sample were recorded using the following parameters: temperature 22.1 ◦C,flip angle 45°, pulse length (P1) of 5.75 μs, sweep width (SW) of 5997 Hz, spectral width of 15.0 ppm, transmitter frequency offset of 5 ppm (1998.91 Hz), acquisition time of 2.186 s, and relaxation delay of 40 s. The 1H-NMR spectra were processed by applying an exponential multiplication of the FIDs by a factor of 0.3 Hz prior to Fourier transform with zero-filling to 256 K data points. The relaxation delay for use in the acquisition of quantitative 1H NMR spectra was determined by T1 measurements with the aid of the pulse sequence inversion recovery, with similar parameters as for 1H spectra and changing τ values from 0.1 to 20 s. The 1H NMR chemical shifts are expressed in ppm related to the CHCl3 signal at 7.26 ppm as internal reference.
3.4.6. NMR Sensitivity Test
Testing samples were prepared as follows. Oleic acid (0.1343 g) and deuterated chloroform (1.1107 g) were thoroughly mixed in a 2-mL vial. In a separate 2-mL vial, ethyl oleate (0.1539 g) and deuterated chloroform (1.1708 g) were mixed to form stock solutions of oleic acid and oleic acid ethyl ester with precisely calculated concentrations. Testing samples were then prepared in five NMR tubes by mixing varying amounts (80 + 0, 60 + 20, 40 + 40, 20 + 60, and 0 + 80 μL) of these stock solutions. All doses were weighed using an analytical balance (KERN ABP 200-5DAM). Finally, the contents were diluted with 800 μL of CDCl₃.
4. Conclusions
In summary, four 2-alkyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium ionic liquids (ASBILs) with different alkyl chains were successfully synthesized using an optimized procedure [36]. These ASBILs demonstrated promising potential for highly efficient catalytic biodiesel production, enabling rapid reactions, high conversion rates, easy and clear phase separation, and high-purity biodiesel. They also exhibited excellent cycling stability.
The presence of two sulfonic acid groups ensures strong acid catalysis for the esterification reaction, while the long C9 – C17 alkyl chains provide good affinity with the fatty acid reactant. These properties contribute to the remarkable catalytic activity of ASBILs in the esterification of fatty acids. All ASBILs with C9, C11, C13, C15, and C17 alkyl chains exhibited excellent catalytic activity and stability in the esterification of oleic acid with ethanol.
The optimal catalyst loading was determined to be 20 mol% (based on the mass of oleic acid), allowing the reaction to reach equilibrium within a short time, approximately 45 to 75 minutes. Among the investigated ASBILs, 2-undecyl-1,3-di(3-sulfopropyl)-1H-benzo[d]imidazol-3-ium ionic liquid (C11-ASBIL) exhibited the highest catalytic activity for the conversion of oleic acid into ethyl oleate. Under the optimized conditions - an ethanol-to-oleic acid molar ratio of 16.7:1, a catalyst loading of 20 mol% (based on oleic acid mass), and a reaction temperature of 102°C - the reaction reached equilibrium within 75 minutes, achieving 100% conversion of oleic acid into ethyl oleate. The conversion remained consistently high even after ten reuse cycles, demonstrating exceptional catalytic stability.
Supplementary Materials
The following supporting information can be downloaded at website of this paper posted on Preprints.org, Figure S1: 1H NMR of 4a; Figure S2: 13C NMR of 4a; Figure S3: ESI MS of 4a; Figure S4: 1H NMR of 4b; Figure S5: 13C NMR of 4b; Figure S6: ESI MS of 4b; Figure S7: 1H NMR of 4c; Figure S8: 13C NMR of 4c; Figure S9: ESI MS of 4c; Figure S10: 1H NMR of 4d; Figure S11: 13C NMR of 4d; Figure S12: ESI MS of 4d; Figure S13: FTIR of 4a & 5a; Figure S14: FTIR of 4a & 5b; Figure S15: FTIR of 4c & 5c; Figure S16: FTIR of 4d & 5d.
Author Contributions
Conceptualization and methodology, T.H.N.T.; experiments, J.K. and T.H.N.T.; NMR and MS measurement, P.K; FTIR measurement L.V.; and T.H.N.T.; writing—original draft preparation, T.H.N.T.; writing—review and editing, T.H.N.T., L.V., P.K.; .; supervision, T.H.N.T. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- S. Prasad, K.K. Yadav, S. Kumar, P. Pandita, J.K. Bhutto, M.A. Alreshidi, B. Ravindran, Z.M. Yaseen, S.M. Osman, M.M.S. Cabral-Pinto, Review on biofuel production: Sustainable development scenario, environment, and climate change perspectives − A sustainable approach, Journal of Environmental Chemical Engineering 12 (2024) 111996. [CrossRef]
- K. Srikumar, Y.H. Tan, J. Kansedo, I.S. Tan, N.M. Mubarak, M.L. Ibrahim, P.N.Y. Yek, H.C.Y. Foo, R.R. Karri, M. Khalid, A review on the environmental life cycle assessment of biodiesel production: Selection of catalyst and oil feedstock, Biomass and Bioenergy 185 (2024) 107239. [CrossRef]
- S.M. Farouk, A.M. Tayeb, S.M.S. Abdel-Hamid, R.M. Osman, Recent advances in transesterification for sustainable biodiesel production, challenges, and prospects: a comprehensive review, Environ Sci Pollut Res 31 (2024) 12722–12747. [CrossRef]
- S. Pandey, I. Narayanan, R. Selvaraj, T. Varadavenkatesan, R. Vinayagam, Biodiesel production from microalgae: A comprehensive review on influential factors, transesterification processes, and challenges, Fuel 367 (2024) 131547. [CrossRef]
- J. Julkipli, S. Babel, A.M. Bilyaminu, E.R. Rene, Hydrogen and biodiesel production from food waste: a review, Environ Chem Lett 22 (2024) 585–607. [CrossRef]
- T.M.I. Mahlia, Z.A.H.S. Syazmi, M. Mofijur, A.E.P. Abas, M.R. Bilad, H.C. Ong, A.S. Silitonga, Patent landscape review on biodiesel production: Technology updates, Renewable and Sustainable Energy Reviews 118 (2020) 109526. [CrossRef]
- N. Anil, P.K. Rao, A. Sarkar, J. Kubavat, S. Vadivel, N.R. Manwar, B. Paul, Advancements in sustainable biodiesel production: A comprehensive review of bio-waste derived catalysts, Energy Conversion and Management 318 (2024) 118884. [CrossRef]
- V.G. Nguyen, P. Sharma, M. Dzida, V.H. Bui, H.S. Le, A.S. El-Shafay, H.C. Le, D.T.N. Le, V.D. Tran, A Review on Metal–Organic Framework as a Promising Catalyst for Biodiesel Production, Energy Fuels 38 (2024) 2654–2689. [CrossRef]
- Y. Zhang, S. Sun, A review on biodiesel production using basic ionic liquids as catalysts, Industrial Crops and Products 202 (2023) 117099. [CrossRef]
- R. D., N. Ghosh, S. Lalthazuala Rokhum, G. Halder, Current progress and future outlooks of microwave-irradiated biodiesel production: A holistic review, Chemical Engineering Journal 482 (2024) 149033. [CrossRef]
- P.R. Costa Neto, M.S. Balparda Caro, L.M. Mazzuco, M. da G. Nascimento, Quantification of soybean oil ethanolysis with1 H NMR, J Americ Oil Chem Soc 81 (2004) 1111–1114. [CrossRef]
- S.K. Bharti, R. Roy, Quantitative 1H NMR spectroscopy, TrAC Trends in Analytical Chemistry 35 (2012) 5–26. [CrossRef]
- G.F. Pauli, B.U. Jaki, D.C. Lankin, Quantitative1 H NMR: Development and Potential of a Method for Natural Products Analysis, J. Nat. Prod. 68 (2005) 133–149. [CrossRef]
- G.F. Pauli, T. Gödecke, B.U. Jaki, D.C. Lankin, Quantitative1 H NMR. Development and Potential of an Analytical Method: An Update, J. Nat. Prod. 75 (2012) 834–851. [CrossRef]
- G.F. Pauli, S.-N. Chen, C. Simmler, D.C. Lankin, T. Gödecke, B.U. Jaki, J.B. Friesen, J.B. McAlpine, J.G. Napolitano, Importance of Purity Evaluation and the Potential of Quantitative1 H NMR as a Purity Assay: Miniperspective, J. Med. Chem. 57 (2014) 9220–9231. [CrossRef]
- U. Holzgrabe, Quantitative NMR spectroscopy in pharmaceutical applications, Progress in Nuclear Magnetic Resonance Spectroscopy 57 (2010) 229–240. [CrossRef]
- A. Barison, C.W. Pereira da Silva, F.R. Campos, F. Simonelli, C.A. Lenz, A.G. Ferreira, A simple methodology for the determination of fatty acid composition in edible oils through1 H NMR spectroscopy, Magnetic Reson in Chemistry 48 (2010) 642–650. [CrossRef]
- J.K. Satyarthi, D. Srinivas, P. Ratnasamy, Estimation of Free Fatty Acid Content in Oils, Fats, and Biodiesel by1 H NMR Spectroscopy, Energy Fuels 23 (2009) 2273–2277. [CrossRef]
- V.M. Mello, F.C.C. Oliveira, W.G. Fraga, C.J. do Nascimento, P.A.Z. Suarez, Determination of the content of fatty acid methyl esters (FAME) in biodiesel samples obtained by esterification using1 H-NMR spectroscopy, Magnetic Reson in Chemistry 46 (2008) 1051–1054. [CrossRef]
- G. Knothe, J.A. Kenar, Determination of the fatty acid profile by1 H-NMR spectroscopy, Euro J Lipid Sci & Tech 106 (2004) 88–96. [CrossRef]
- C. Siciliano, E. Belsito, R. De Marco, M.L. Di Gioia, A. Leggio, A. Liguori, Quantitative determination of fatty acid chain composition in pork meat products by high resolution 1H NMR spectroscopy, Food Chemistry 136 (2013) 546–554. [CrossRef]
- P. Siudem, A. Zielińska, K. Paradowska, Application of 1H NMR in the study of fatty acids composition of vegetable oils, Journal of Pharmaceutical and Biomedical Analysis 212 (2022) 114658. [CrossRef]
- M. ter Horst, S. Urbin, R. Burton, C. McMillan, Using proton nuclear magnetic resonance as a rapid response research tool for methyl ester characterization in biodiesel, Lipid Technology 21 (2009) 39–41. [CrossRef]
- G.P. Mambrini, C. Ribeiro, L.A. Colnago, Nuclear magnetic resonance spectroscopic analysis of ethyl ester yield in the transesterification of vegetable oil: an accurate method for a truly quantitative analysis, Magnetic Reson in Chemistry 50 (2012) 1–4. [CrossRef]
- I.G. Rosset, M.C.H. Tavares, E.M. Assaf, A.L.M. Porto, Catalytic ethanolysis of soybean oil with immobilized lipase from Candida antarctica and 1H NMR and GC quantification of the ethyl esters (biodiesel) produced, Applied Catalysis A: General 392 (2011) 136–142. [CrossRef]
- R. Guzatto, D. Defferrari, Q.B. Reiznautt, Í.R. Cadore, D. Samios, Transesterification double step process modification for ethyl ester biodiesel production from vegetable and waste oils, Fuel 92 (2012) 197–203. [CrossRef]
- R.C.M. dos Santos, P.C. Gurgel, N.S. Pereira, R.A. Breves, P.R.R. de Matos, L.P. Silva, M.J.A. Sales, R. de V.V. Lopes, Ethyl esters obtained from pequi and macaúba oils by transesterification with homogeneous acid catalysis, Fuel 259 (2020) 116206. [CrossRef]
- F. Faraguna, M. Racar, Z. Glasovac, A. Jukić, Correlation Method for Conversion Determination of Biodiesel Obtained from Different Alcohols by1 H NMR Spectroscopy, Energy Fuels 31 (2017) 3943–3948. [CrossRef]
- M. dos P.M. de Jesus, L.N. de Melo, J.P.V. da Silva, A.C. Crispim, I.M. Figueiredo, J.H. Bortoluzzi, S.M.P. Meneghetti, Evaluation of Proton Nuclear Magnetic Resonance Spectroscopy for Determining the Yield of Fatty Acid Ethyl Esters Obtained by Transesterification, Energy Fuels 29 (2015) 7343–7349. [CrossRef]
- K.I. Doudin, Quantitative and qualitative analysis of biodiesel by NMR spectroscopic methods, Fuel 284 (2021) 119114. [CrossRef]
- G.G. Shimamoto, L.F. Bianchessi, M. Tubino, Alternative method to quantify biodiesel and vegetable oil in diesel-biodiesel blends through 1 H NMR spectroscopy, Talanta 168 (2017) 121–125. [CrossRef]
- V.M. Mello, F.C.C. Oliveira, W.G. Fraga, C.J. do Nascimento, P.A.Z. Suarez, Determination of the content of fatty acid methyl esters (FAME) in biodiesel samples obtained by esterification using1 H-NMR spectroscopy, Magnetic Reson in Chemistry 46 (2008) 1051–1054. [CrossRef]
- M. Morgenstern, J. Cline, S. Meyer, S. Cataldo, Determination of the Kinetics of Biodiesel Production Using Proton Nuclear Magnetic Resonance Spectroscopy (1 H NMR), Energy Fuels 20 (2006) 1350–1353. [CrossRef]
- L.A. Anderson, A.K. Franz, Real-Time Monitoring of Transesterification by1 H NMR Spectroscopy: Catalyst Comparison and Improved Calculation for Biodiesel Conversion, Energy Fuels 26 (2012) 6404–6410. [CrossRef]
- G.F. Ghesti, J.L. de Macedo, I.S. Resck, J.A. Dias, S.C.L. Dias, FT-Raman Spectroscopy Quantification of Biodiesel in a Progressive Soybean Oil Transesterification Reaction and Its Correlation with1 H NMR Spectroscopy Methods, Energy Fuels 21 (2007) 2475–2480. [CrossRef]
- T.H. Nguyen Thi, J. Koutecká, P. Kaule, L. Vrtoch, V. Šícha, J. Čermák, Design and Synthesis of New Sulfonic Acid Functionalized Ionic Liquids as Catalysts for Esterification of Fatty Acids with Bioethanol, Molecules 28 (2023) 5231. [CrossRef]
Scheme 1.
Synthesis of ASBILs.
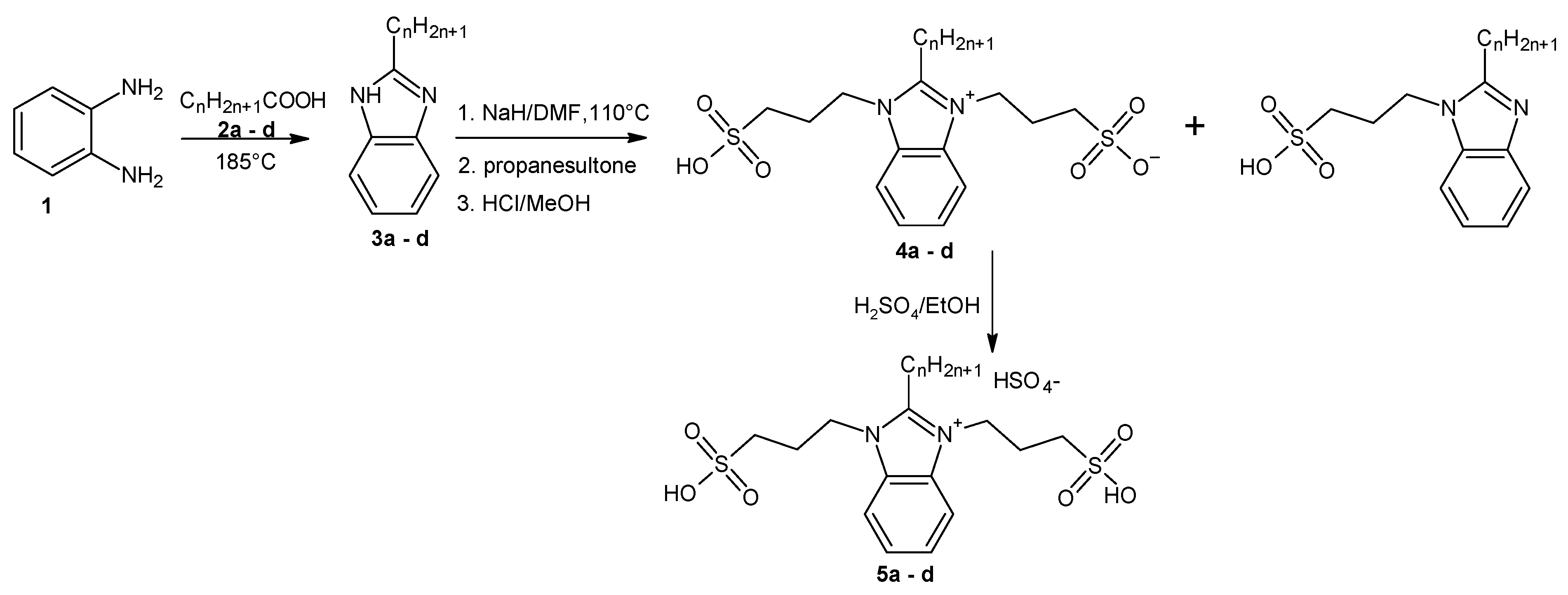
Figure 1.
1H NMR spectrum of oleic acid, the assignments to the individual signals are inscribed.
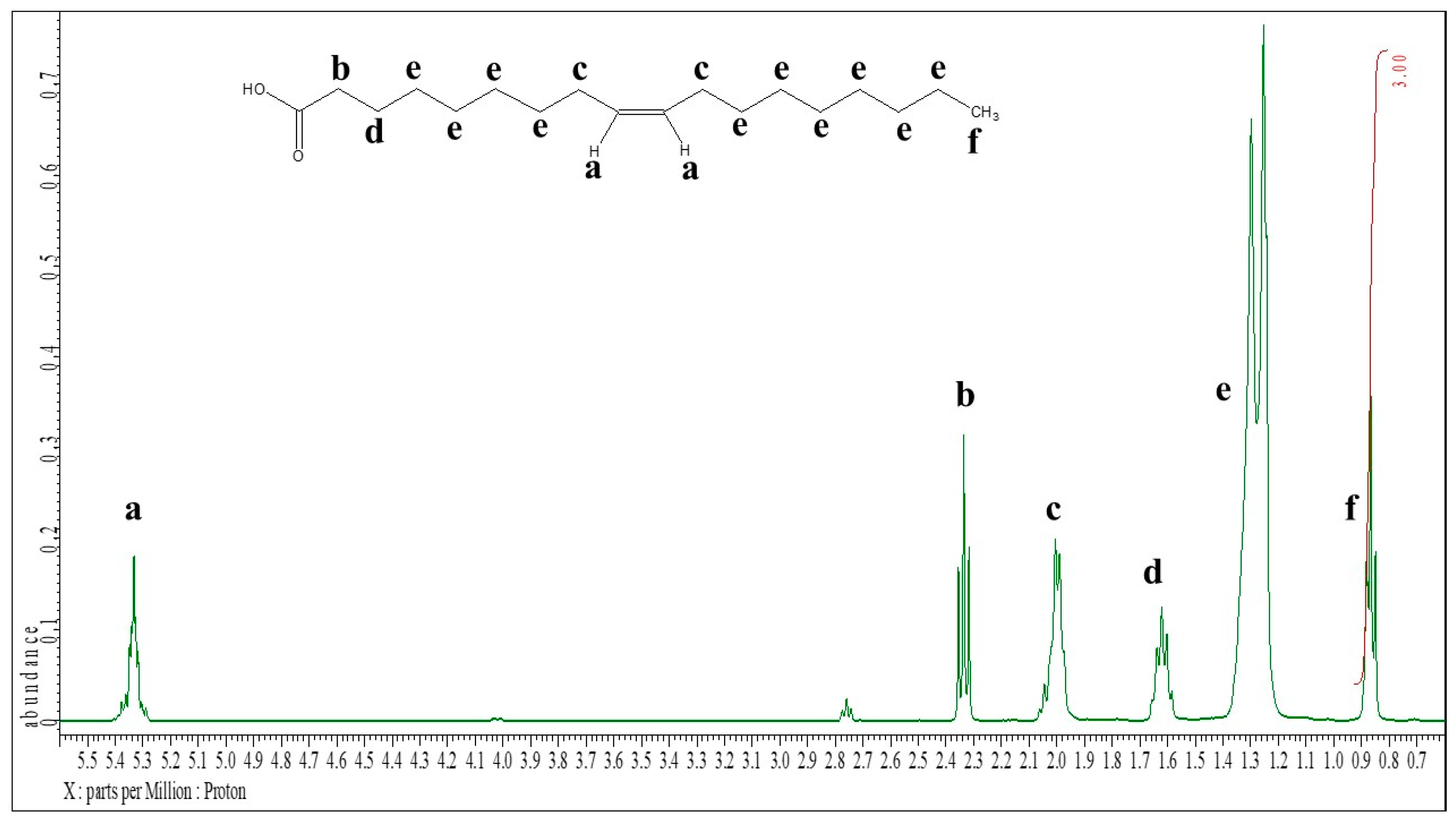
Figure 2.
1H NMR spectrum of ethyl oleate, the assignments to the individual signals are inscribed.
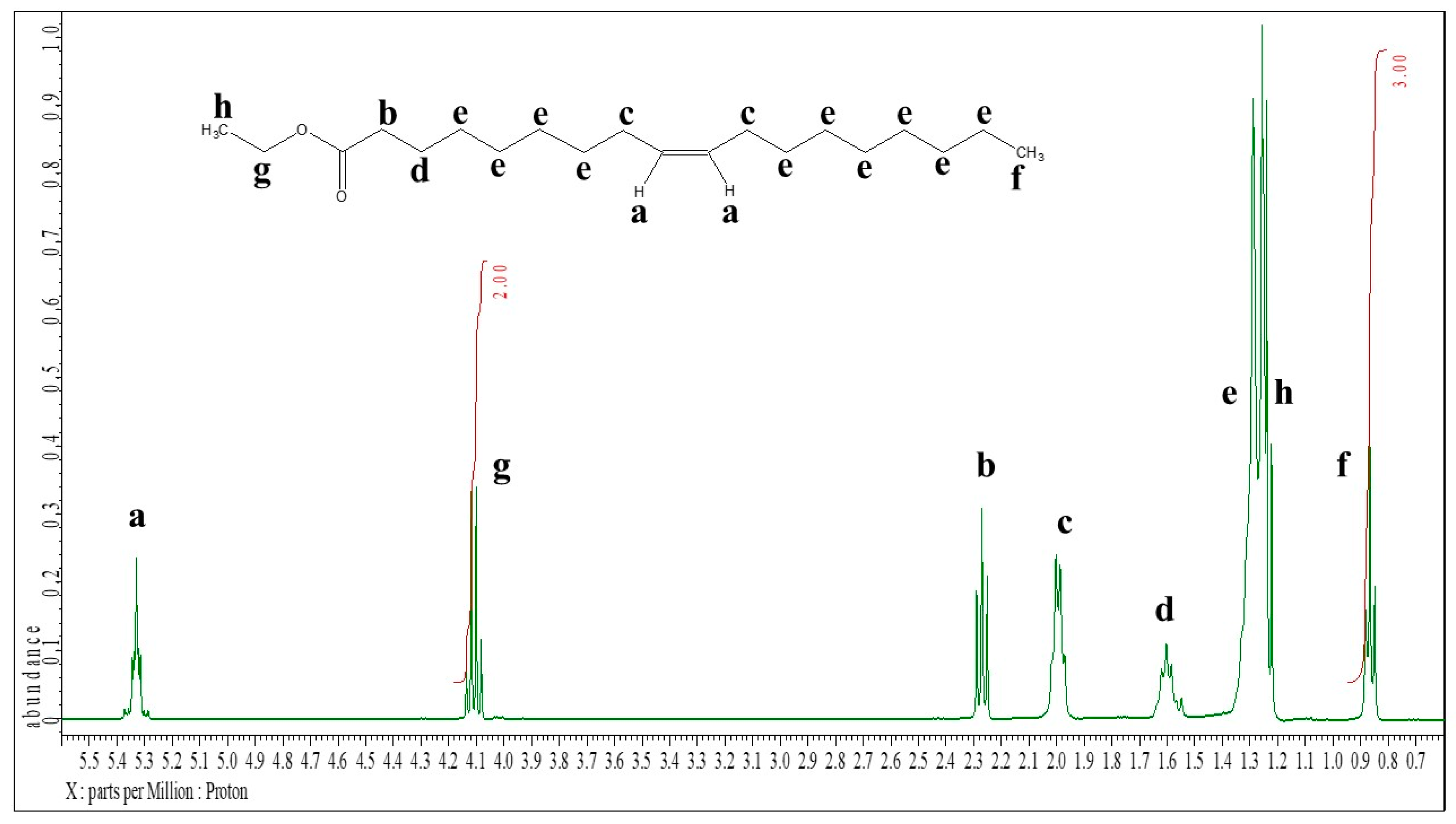
Figure 3.
1H NMR spectrum of a known mixture of oleic acid and ethyl oleate (Sample 3).
Graph 2.
Esterification of oleic acid and ethanol with 0.2 eq of catalysts 5a - d. Conversions of oleic acid during 75 minutes.
Graph 2.
Esterification of oleic acid and ethanol with 0.2 eq of catalysts 5a - d. Conversions of oleic acid during 75 minutes.
Graph 3.
Esterification of oleic acid and ethanol with recycled C11- ASBILs. Conversions of oleic acid after 75 minutes.
Graph 3.
Esterification of oleic acid and ethanol with recycled C11- ASBILs. Conversions of oleic acid after 75 minutes.
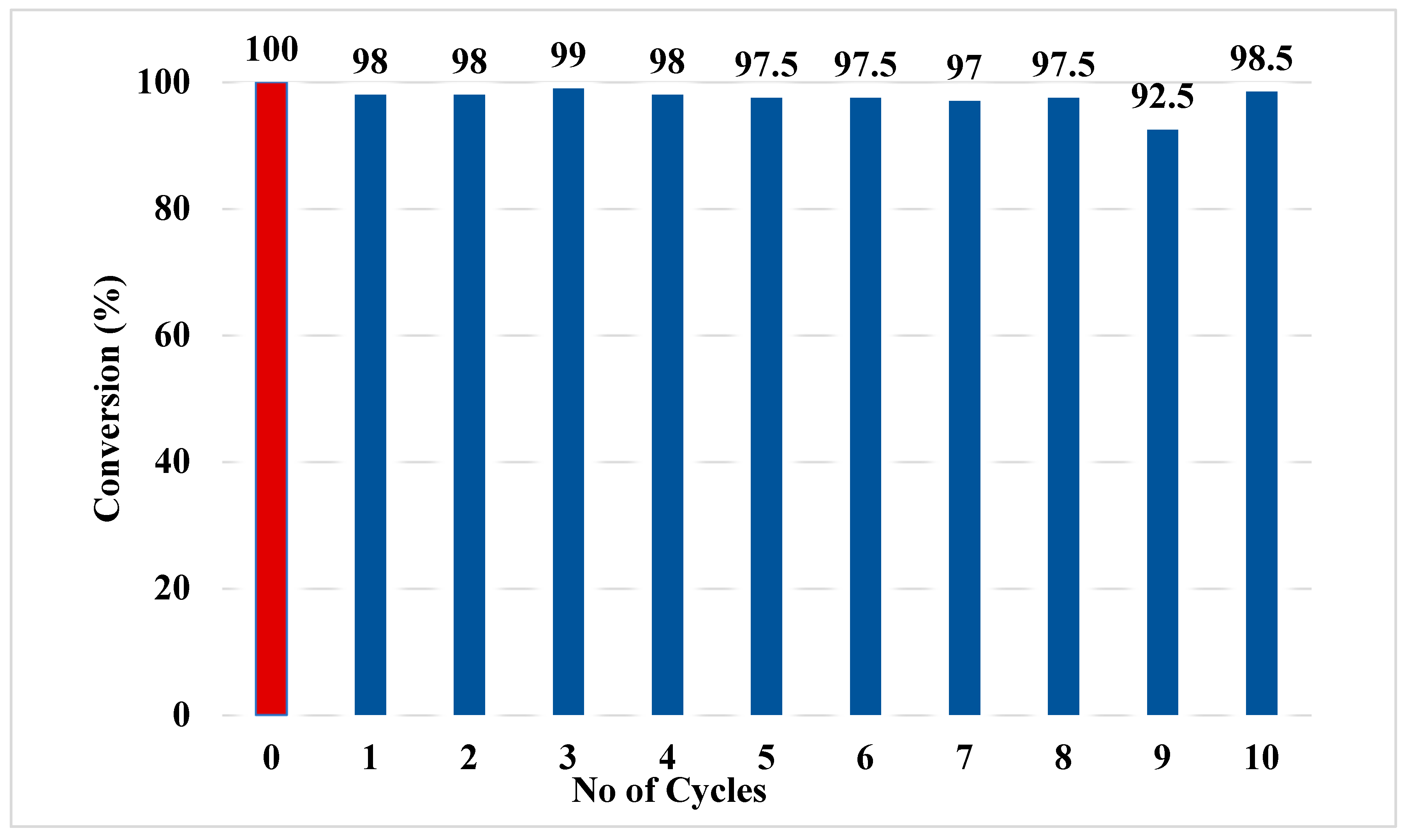
Table 1.
Simulated conversions of test samples and their determined conversions by 1H NMR spectroscopy.
Table 1.
Simulated conversions of test samples and their determined conversions by 1H NMR spectroscopy.
| Sample | n(OA) [mmol] |
n(EO) [mmol] |
inaccuracy of qNMR [%] |
||
|---|---|---|---|---|---|
|
1 2 3 4 5 |
0.03215484516 0.03016903525 0.02016360837 |
0.0000000000 0,008081617983 0,01855782901 |
0.0000000000 0,2112805220 0,4792649825 |
0.000 0,205 0,460 |
0.00 2,97 4.02 |
| 0.009974653801 | 0,03034354703 | 0,7526017135 | 0,740 | 1.67 | |
| 0.00000000000 | 0,03894899193 | 1.0000000000 | 1.000 | 0.00 |
Table 3.
Esterification of oleic acid and ethanol with 2 eq. of catalysts 5a - d. Conversions of oleic acid during 75 minutes.
Table 3.
Esterification of oleic acid and ethanol with 2 eq. of catalysts 5a - d. Conversions of oleic acid during 75 minutes.
| Catalysts | Conversion | Times required to reach reaction equilibrium [min] |
||||
|---|---|---|---|---|---|---|
| 1 5 min | 30 min | 4 5 min | 60 min | 7 5 min | ||
| 5a (C9-ASBIL) | 0.940 0.965 0.920 0.955 |
0.970 0.985 0.975 0.970 |
0.980 0.990 0.985 0.975 |
0.985 0.990 0.980 0.975 |
0.985 1.000 0.965 0.980 |
60 45 45 75 |
|
5b (C11-ASBIL) 5c (C13-ASBIL) 5d (C17-ASBIL) | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



