Submitted:
24 April 2025
Posted:
25 April 2025
You are already at the latest version
Abstract
Inflammation is a key hallmark in cardiomyopathy where misdirected immune activation causes chronic myocardial dysfunction. Among the emerging mechanisms implicated in the dysfunction, the cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) signaling pathway has attracted significant attention. Acting as a critical DNA sensor, the cGAS/STING pathway coordinates inflammatory responses triggered by microbial invasions or endogenous stressors such as autophagic or apoptotic cell death. Despite its pivotal role, the precise molecular mechanisms regulating this pathway and its contribution to aberrant inflammation in cardiomyopathy remain poorly understood and controversial. To address this scientific gap, we first summarized the key findings on the cGAS/STING pathway in different cardiomyopathies using in vivo/in vitro models as well as clinical samples. In the next step, we explored how the cGAS/STING pathway could be modulated by it agonists and antagonists in cardiomyopathy. Finally, leveraging publicly available human single-cell ribonucleic acid sequencing (RNA-seq) dataset and systematic literature review, we identified existing molecular interventions and highlighted potential therapeutic targets to mitigate cGAS/STING-driven inflammation. This integrative approach underscores the therapeutic potential of targeting the cGAS/STING pathway and provides a foundation for developing novel interventions aimed at alleviating inflammatory cardiomyopathy and improving patient outcomes. Future studies will be crucial in validating these findings and translating them into clinics.
Keywords:
Cardiomyopathy
; cGAS/STING pathway
; mitochondria
; DNA damage
1. Introduction
Cardiomyopathy is a type of cardiac disease in which the heart muscle is structurally and functionally altered [1]. Cardiomyopathies can be divided into dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), restrictive cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and unclassified cardiomyopathies [2,3]. Patients with cardiomyopathies may experience a range of symptoms, varying with the type and severity of the condition. Common manifestations include dyspnea, fatigue, chest pain, arrhythmia and even sudden cardiac death [1]. Current treatment for cardiomyopathy mainly focuses on managing the symptoms and preventing complications. To date, no specific drug has been approved for the causal treatment of cardiomyopathies [4]. Therefore, it is crucial to understand the underlying mechanism to identify novel therapeutic targets for the disease.
Many studies have investigated signaling pathways in different cardiomyopathy models and found that several cellular and metabolic pathways, which include β-adrenergic signaling, MAPK/ERK signaling, WNT signaling, Hippo-Yes-associated protein signaling, CaM-kinase signaling, and autophagy signaling, are altered [5,6,7,8]. Recently, there has been extensive research into the cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase/stimulator of interferon genes (cGAS/STING) pathway, highlighting its function in sensing cytosolic DNA and triggering downstream inflammation and cell death [9,10,11,12]. However, the role of the cGAS/STING pathway in cardiomyopathy and its regulatory mechanisms are not yet fully understood. This review seeks to provide comprehensive knowledge about molecular intervention targeting the cGAS/STING pathway in cardiomyopathy.
2. Overview of the cGAS/STING Signaling Pathway
The cGAS/STING pathway is a crucial element of the innate immune response which detects the presence of cytoplasmic DNA and subsequently triggers the expression of inflammatory genes. Cytosolic DNA can originate from both exogenous and endogenous sources [13]. Viruses and bacteria can be sources of exogenous DNA, while endogenous DNA mainly originates from cell apoptosis, auto cellular death, and mitochondrial damage. As shown in Figure 1, The main components of this pathway include cyclic GMP-AMP synthase (cGAS), cyclic GMP-AMP (cGAMP), stimulator of interferon genes (STING), TANK-binding Kinase 1 (TBK1), interferon regulatory factor 3 (IRF3), Type I Interferons (e.g., IFN-β), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). cGAS exists ubiquitously in the cytoplasm as a so-called DNA sensor [14]. Upon binding to double-stranded DNA (dsDNA), it undergoes conformational change and catalyzes the production of cGAMP from ATP and GTP. cGAMP then acts as a second messenger and activates STING. The activation of STING mediates its translocation from the endoplasmic reticulum membrane to the endoplasmic reticulum-Golgi intermediate compartment and Golgi apparatus, phosphorylating TBK1. Phosphorylated TBK1 then phosphorylates and activates IRF3, which serves as a transcription factor. Phosphorylated IRF3 moves into the cell nucleus and triggers the expression of type I interferons [15,16]. Simultaneously, STING activation also triggers NF-κB activation, which subsequently enhances the expression of pro-inflammatory cytokines.
3. Activation of cGAS/STING Pathway in Different Cardiomyopathies
Inflammation is a key factor in the development and progression of a variety of cardiomyopathies [17,18,19,20]. Recent evidence suggests that cGAS/STING-mediated inflammation and apoptosis are involved in myocardial damage and remodeling, which might contribute to the development of cardiomyopathy [21,22,23]. To better demonstrate the essential function of cGAS/STING-signaling in inflammation-mediated cardiomyopathies, we first systematically review the most important studies of the cGAS/STING signaling pathway in various types of cardiomyopathies (see Table 1).
3.1. Dilated Cardiomyopathy
Dilated cardiomyopathy is marked by enlargement and impaired contraction of one or both ventricles. It is a leading cause for heart failure and a primary indication for heart transplantation [24,25,26]. Up to 40% of all dilated cardiomyopathies in humans are of genetic origin, with most mutations affecting genes coding for cytoskeletal or contractile proteins [27,28]. The following systematically reviews some of the most common mutations and the corresponding animal models.
3.1.1. LMNA Cardiomyopathy
Mutations in the LMNA gene, which encodes for the nuclear envelope proteins lamin A and C, represent the second most common genetic cause of DCM [29,30]. Lamin A/C are components of the nuclear lamina, providing structural support to the nucleus and serving as a platform for protein interactions involved in gene regulation, DNA replication, and genome stability [31,32,33,34]. Disruption in lamin A/C can result in abnormal nuclear shape, impaired DNA repair, and altered gene expression [35,36,37,38].
In 2022, Cheedipudi et al. reported on the activation of the DNA damage response pathway in LMNA-deficient mice and found that genetic blockade of cGAS in these mice increased survival, improved cardiac function, reduced cardiac fibrosis and apoptosis39. In mice with LMNA cardiomyopathy, multiple components of the cGAS/STING pathway as well as its downstream targets, including cGAS, TBK1, STING1, pIRF3, and NF-kB, showed marked upregulation in the heart. Genetic knockout of cGAS in these mice significantly reduced the levels of all these components except for STING1 [39]. However, in 2024, En et al. challenged these findings and proposed that cGAS/STING does not contribute to lamin A/C-dependent cardiomyopathy in adult mice. They found that the phenotypes of adult Lmna-deficient mice could not be rescued by either cGAS or STING knockout [40]. They showed that cGAS/STING was not activated in cardiomyocytes and attributed this to the generally low expression levels of cGAS and STING in adult cardiac myocytes. Instead, after enriching the upregulated genes in the hearts of LMNA cardiomyopathy mice from single-nucleus RNA-seq data, they proposed that extracellular matrix signaling is activated instead and acts as a potential inflammatory mediator [40]. More recently, Zuela-Sopilniak et al. conducted a multi-level transcriptomic analysis of LMNA DCM and characterized two subclusters of cardiomyocytes responsible for the DCM pathogenesis [41]. Within these subclusters, despite the low transcriptional expression level of cGAS, STING was found to be activated, possibly due to another cytoplasmic DNA sensor: Gamma-interferon-inducible protein 16 (IFI16). Interestingly, they hypothesized that the activation of cGAS-independent cytosolic pattern recognition pathways and DNA damage response pathways in cardiomyocytes subsequently triggered transcriptomic changes in cardiac fibroblasts, the recruitment of immune cells and activation of extracellular matrix remodeling, which eventually led to cardiac dysfunction [41].
Several reasons could possibly explain these contrary findings. Firstly, the LMNA DCM animal models utilized by these research groups differed. Cheedipudi et al. used Myh6-Cre:LmnaF/F mice in which LMNA was deleted postnatally, while En et al. used inducible Cre recombinase to delete LMNA in cardiomyocytes at 6-8 weeks of age [40]. The activated pathways may vary depending on the timing of LMNA knockout, especially as different pathways undergo changes during cardiac development. It remains uncertain which approach more accurately replicates human LMNA cardiomyopathy. Secondly, the methods used to assess cGAS/STING activity differ between the two studies. Cheedipudi et al. employed immunoblot analysis to assess the expression levels of key proteins involved in the cGAS/STING pathway, while En et al. investigated the activity of key genes from this pathway at the transcriptional level. Evidence showed that mRNA and protein levels often show a weak correlation, likely due to post-transcriptional regulation and post-translational modifications. As a result, transcript abundance may not reliably reflect cGAS/STING signaling activity, which could explain the discrepancies between the findings of En et al. and Zuela-Sopilniak et al. compared to those of Cheedipudi et al. [39,40,41].
3.1.2. LEMD2-Associated Cardiomyopathy
Similar to lamin A/C, the LEM domain-containing protein 2 (LEMD2) is a critical component of the nuclear membrane which maintains nuclear structure [42,43]. Mutations in the LEMD2 gene, specifically the c.T38>G (p.L13>R) mutation in humans, cause dilated cardiomyopathy with arrhythmic features [44,45,46].
In 2022, Caravia et al. generated the first LEMD2-associated cardiomyopathy mouse model to investigate the role of LEMD2 in cardiac development and heart disease. They found that both the DNA damage response pathway and the apoptosis pathway were activated in LEMD2 cardiac-specific knockout mouse hearts. DNA damage was confirmed by immunofluorescence staining on heart sections and isolated cardiomyocytes, which showed upregulation of γ-H2AX [45]. In 2023, Chen et al. demonstrated that in LEMD2-mutated mouse cardiomyocytes or Hela-cells, sustained nuclear envelope rupture led to the leakage of DNA repair factors into the cytoplasm, triggering cell cycle arrest. Nuclear envelop rupture also exposed genomic DNA to the cytosol, activating the cGAS/STING/IFN pathway. As a consequence, the activation of cGAS/STING/IFN signaling drove the production of senescence-associated secretory phenotype factors, leading to cellular senescence [47].
3.1.3. Diabetic Cardiomyopathy
Diabetic cardiomyopathy is a distinct heart condition that arises from diabetes-induced changes in heart structure and function, independent of other cardiovascular risk factors like hypertension or coronary artery disease. It is marked by myocardial hypertrophy, inflammation, fibrosis, and cellular death [48,49]. Patients with diabetic cardiomyopathy will eventually develop heart failure if left untreated [50]. Unlike other forms of cardiomyopathy, diabetic cardiomyopathy is uniquely tied to metabolic disturbances, including enhanced fatty acid utilization and increased oxidative stress, making it a challenging condition to treat and manage [48,51,52].
In 2022, Ma et al. first reported increased mtDNA in the cytosol and activation of the cGAS/STING pathway in an obesity-related DCM model [53]. Transmission electron microscopy revealed significant alterations in mitochondrial structure, and an increased amount of free cytosolic mtDNA was detected by both co-immunolabeling and qRT-PCR. Meanwhile, cGAS and STING, as well as their downstream targets, NF-κB, IRF3, and IL-1β, were activated in the hearts of diabetic mice at both mRNA and protein levels detected by both co-immunolabeling and qRT-PCR. To demonstrate that mtDNA alone can activate cGAS, they transfected purified mtDNA into H9C2 cells and performed western blot and qRT-PCR analyses on those cells. Several components, such as cGAS, STING, IL-1β and IL-18, were upregulated upon mtDNA stimulation. In addition, they were able to show increased STING aggregation to the Golgi apparatus, indicating STING was functionally activated by the mtDNA treatment. Both genetic and pharmacological inhibition of STING rescued cardiac dysfunction, improved myocardial hypertrophy and fibrosis, and reduced inflammation in this mouse model of diabetic cardiomyopathy [53]. Almost at the same time, another research group pointed out that in a mouse model of diabetic DCM, oxidative damage to the mitochondria from lipid toxicity led to the release of mtDNA into the cytosol, subsequently activating the cGAS/STING pathway. In addition, they showed NLRP3 inflammasome-dependent pyroptosis and pro-inflammatory response were also activated as downstream pathways, which eventually caused myocardial hypertrophy in diabetic DCM [54]. Based on these findings, researchers began to find therapies for diabetic DCM. In 2022, Lu et al. found that cardiomyocyte-specific overexpression of Meteorin-like hormone (Metrnl) ameliorated the phenotypes of diabetic cardiomyopathy via activation of the autophagy pathway and inhibition of the cGAS/STING pathway. Mechanistically, Metrnl-induced ULK1 phosphorylation promoted the dephosphorylation and mitochondrial translocation of STING, where it formed a complex with tumor necrosis factor receptor-associated factor 2 (TRAF2). This interaction accelerated the ubiquitination and degradation of STING, ultimately making cardiomyocytes more susceptible to autophagy activation [55]. In 2023, another research group reported that irisin, by activating mitochondrial ubiquitin ligase (MITOL/MARCH5) and suppressing NLRP3 inflammasome through cGAS/STING pathway, rescued the cardiac dysfunction in diabetic cardiomyopathy [56]. In 2024, Chen et al. found that deficiency of Brahma-related gene 1, also known as SMARCA4, resulted in the accumulation of cytoplasmic dsDNA and triggered cGAS/STING activation, exacerbating cardiomyocyte inflammation and apoptosis induced by hyperglycemia and hyperlipidemia in diabetic cardiomyopathy [57]. At the same time, Huang et al. showed that both recombinant IL-37 administration and inducing IL-37 expression could alleviate cardiac dysfunction and myocardial fibrosis in diabetic DCM mice, providing a novel therapeutic target for the disease. Interestingly, they proposed that during the pathogenesis of diabetic DCM, hyperglycemia aggravated mitochondrial damage through SIRT1/AMPK/PGC1α signaling, leading to cell death and the release of extracellular vesicles which contains mtDNA. Fibroblasts then engulf these mtDNA-enriched vesicles, activating TLR9 signaling and the cGAS/STING pathway to initiate pro-fibrotic process and cardiac remodeling. IL-37 exerted its therapeutic effects by inhibiting these events [58].
3.2. Arrhythmogenic Cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM), also known as arrhythmogenic right ventricular cardiomyopathy/dysplasia, comprises a broad category of primary myocardial disorders that manifest as ventricular arrhythmias, heart failure, and sudden cardiac death [59]. The majority of ACM is caused by genetic mutations affecting desmosomal proteins, such as Desmoplakin (DSP), Plakophilin-2 (PKP2), Desmocollin-2 (DSC2), Desmoglein-2 (DSG2), Junction Plakoglobin (JUP), etc. Other genetic factors include mutations in non-desmosomal proteins, such as Phospholamban (PLN), Transmembrane Protein 43 (TMEM43), and Cadherin-2 (CDH2) [60,61,62,63].
Transmembrane protein 43 is a highly conserved membrane protein localizing to the inner membrane of the nucleus and may have an important role in maintaining nuclear envelope structure [64,65]. In 2021, Rouhi et al. found that TMEM43 haploinsufficiency is linked to the activation of the DNA damage response and the TP53 pathway, leading to increased expression of the senescence-associated secretory phenotype and pro-fibrotic factors in cardiomyopathy. cGAS and STING1, part of the DNA damage response pathway, were only activated at a later stage of the disease, which is consistent with the observed late onset of cardiac phenotypes [66].
3.3. Doxorubicin-Induced Cardiomyopathy
Doxorubicin, an anthracycline antibiotic, is commonly used to treat breast cancer, lymphomas, and leukemias [67]. Doxorubicin works by DNA intercalation, generating reactive oxygen species (ROS) and inducing apoptosis in cancer cells [68]. However, a major concern regarding the clinical use of doxorubicin and other anthracyclines is their cardiotoxicity [69,70,71]. Doxorubicin-induced cardiomyopathy (DIC) exhibits morphological and functional characteristics similar to those of dilated cardiomyopathy. Current hypotheses for the pathogenesis of DIC include oxidative stress, mitochondrial dysfunction, DNA damage, calcium overload, and inflammation [72]. Dexrazoxane, which chelates iron and reduces ROS formation, is the only approved medication for DIC [73,74,75]. Recent evidence suggests cGAS/STING-mediated inflammation might be involved in the pathogenesis of DIC [21,22,76].
DIC exists in two forms: acute and chronic. Acute DIC occurs within hours to weeks after administration, while chronic DIC develops months to years later. The pathophysiology of acute DIC mainly involves direct myocardial toxicity, oxidative stress, and inflammatory responses [77]. Chronic DIC, on the other hand, is characterized by chronic oxidative stress, mitochondrial dysfunction, persistent DNA damage, and fibrosis [69]. In 2023, Xiao et al. demonstrated a significantly increased protein level of cGAS/STING in an acute DIC mouse model and showed that STING knockdown prolonged survival and improved cardiac function. Morphological examinations of the heart revealed a reduction in the vacuolation of the myofibrils and the number of myofibrils in mice with STING knockdown. In addition, STING inhibition reduced apoptosis and inflammation in cardiomyocytes. Specifically, a reduced ratio of C-Caspase3 to T-Caspase3, a lower ratio of BAX to BCL2, fewer TUNEL-positive cells, and lower levels of pro-inflammatory cytokines, were seen in STING knockdown mice [76]. In a chronic DIC mouse model, Luo et al. first showed the activation of the cGAS/STING pathway in the heart, and global cGAS or STING knockout prevented DIC. More importantly, they noted that the activation of cGAS/STING was seen in cardiac endothelial cells and macrophages instead of cardiomyocytes and fibroblasts. Endothelial cell-specific STING knockdown rescued DIC and endothelial cell dysfunction. Specifically, cGAS/STING triggered endothelial cell inflammation and regulated mitochondrial dysfunction via CD38-induced nicotinamide adenine dinucleotide depletion in endothelial cells. In addition, the cGAS/STING pathway of cardiac endothelial cells also regulated cellular injury and cardiomyocyte mitochondrial bioenergetics via CD38 ecto-NADase-mediated cardiomyocyte NAD decline [21].
3.4 Sepsis-Induced Cardiomyopathy
Sepsis is a life-threatening syndrome characterized by a systemic inflammatory response to infection [78,79]. The inflammatory response may cause multiple organ dysfunctions if not controlled properly80. Sepsis-induced cardiomyopathy (SIC) develops when cardiac function is impaired during sepsis. Unlike many other types of cardiomyopathies, SIC is usually reversible with the resolution of sepsis and appropriate treatment [81,82].
In 2019, Li et al. first disclosed that STING-IRF3 could trigger lipopolysaccharide (LPS)-induced cardiac dysfunction, inflammation, apoptosis, and pyroptosis by activating NOD-like receptor protein 3 in SIC. STING deficiency could ameliorate these pathological events and improve cardiac function and overall survival. Mechanistically, LPS injection did not alter the expression level of the STING protein but promoted its perinuclear translocation and the nuclear translocation of IRF3. Knockdown of STING inhibited phosphorylation and nuclear translocation of IRF3, thus alleviating the pathological events triggered by IRF3 [83]. In contrast to their findings, Kong et al. showed that the protein expression of STING was indeed increased after LPS injection and its activation was also facilitated. They identified a novel role for Islet cell autoantigen 69 (ICA69) as a positive regulator of STING in the pathogenesis of septic cardiac injury. They discovered significant colocalization of ICA69 and STING in both cardiac tissue and macrophages and found that Ica69 knockdown can mitigate LPS-induced cardiac damage by inhibiting STING-mediated inflammation and ferroptosis [84]. In 2023, Liu et al. observed upregulation of cGAS and STING in the cardiac tissue of LPS-treated mice [85]. In addition, they found that in normal H9C2 cells, silencing of cGAS did not affect the expression levels of cGAS downstream targets, including STING, IRF3, and TBK1. However, in LPS-stimulated H9C2 cells, the expression levels of these proteins were significantly reduced when cGAS was blocked. Furthermore, following cGAS knockdown, inflammation, apoptosis, and excessive ROS production induced by LPS were also alleviated. In summary, the cGAS/STING pathway also plays a significant role in the development and maintenance of cardiomyopathy in SIC [85].
3.5. Other Cardiomyopathies
Carnitine acetyltransferase (CRAT) is a mitochondrial enzyme that catalyzes the conversion of acetyl-CoA to acetylcarnitine, which is then transferred out of the mitochondria [86]. CRAT is important in regulating cellular energy metabolism, and its deficiency leads to mitochondrial dysfunction [87,88]. In a fibroblast-specific CRAT silencing model, mitochondrial DNA was found to be released into the cytosol and activated downstream cGAS/STING/NF-κB signaling [89]. In 2023, Mao et al. generated a cardiomyocyte-specific Crat-deficient mouse model and observed the phenotypes of dilated cardiomyopathy. They also showed that depletion of Crat promoted the release of mitochondrial DNA into the cytoplasm via the mitochondrial permeability transition pore (mPTP) and triggered the cell-intrinsic type I interferon response in myocytes. Multiple cytosolic RNA and DNA sensors, including cGAS, Ddx58, Ifih1, and Aim2, were activated. Knockdown of cGAS reduced interferon-stimulated genes (ISGs) expression, inhibited AIM2 inflammasome activation, and improved cardiac contractile function in CRAT-deficient DCM [90].
Chagas cardiomyopathy is a form of cardiomyopathy resulting from Trypanosoma cruzi infection. The pathological features include myocardial inflammation, fibrosis, and severe arrhythmias [91,92,93]. In Chagas cardiomyopathy, Choudhuri et al. showed that extracellular vesicles from Trypanosoma cruzi-infected cells lead to increased levels of IL-1β, IL-6, and TNF-α in macrophages. In addition, with the use of cGAS inhibitor PF-06928215, a significant reduction in the levels of IL-1β, IL-6c, and TNF-α were detected [94].
Stress cardiomyopathy, also known as Takotsubo cardiomyopathy or broken heart syndrome, occurs when a person experiences acute emotional or physical stress that leads to a sudden reversible weakening of the myocardium. A massive surge of catecholamines, such as adrenaline and norepinephrine, has been suggested as a major mechanism for this disease. Cardiac inflammation, as a consequence of catecholamine surge, has been documented to exacerbate myocardial injury and contribute to disease progression [95,96,97,98]. In 2024, Wang et al. showed in a stress cardiomyopathy mouse model, following acute catecholamine surge, necrotic death was triggered in cardiomyocytes, which then released self-DNA and other damage-associated molecular patterns (DAMPs) [99]. These molecules were recognized by macrophages and activated cytosolic DNA-sensing pathways inside macrophages. STING, as a key component of the DNA-sensing pathway, subsequently triggered inflammatory responses in these macrophages, promoting the release of pro-inflammatory factors, such as TNF, IL-6, and CCL2, eventually leading to myocardial inflammation and injury. They believed self-DNA served as a bridge between myocyte necrosis and macrophage activation. Interestingly, the DNA sensors involved in the process were not limited to cGAS. RNA sequencing revealed several other DNA sensors, including Ddx41, Ddx58, and Zbp1, were also upregulated at the transcriptional level [99].
4. Molecular Intervention and Potential Targets on cGAS/STING Pathway in Cardiomyopathy
4.1. Molecular Intervention on cGAS/STING Pathway in Cardiomyopathy
During the last decades, different animal models have been developed to investigate the pathomechanisms involved in cardiomyopathy, which has contributed substantially to our understanding of the function of the cGAS/STING pathway in cardiomyopathy pathology. Through a literature review of experiments investigating cardiomyopathy, we found a significant activation of the cGAS/STING pathway in the cardiomyopathy group (DCM and HCM) compared to the control group (non-failing hearts, NF). Moreover, inhibition of the cGAS/STING pathway can significantly slow down disease progression. In particular, cGAS and STING inhibitors show therapeutic benefits in various cardiomyopathies. A detailed list of these compounds and their mechanisms of action are chronologically summarized in Table 2.
4.2. Mitochondrial Alteration as a Hotspot for cGAS/STING Pathway Activation
As demonstrated in Figure 1, damaged mitochondria can release endogenous DNA and lead to subsequent activation of the cGAS/STING pathway. By performing bioinformatic analyses using publicly available human single-cell RNA-seq data by Chaffin et al. [100], we found that mitochondrial DNA is significantly reduced in the cardiac muscle cell in the cardiomyopathy group (DCM and HCM) when compared to the control group (non-failing hearts, NF) (see Figure 2). Similarly, a most recent study by Wang et al. showed that mitochondrial function-related pathways (e.g., mitochondrial matrix, mitochondrial inner membrane, mitochondrial respiratory chain complex I, oxidative phosphorylation, and electron transport chain) were found to be significantly downregulated in a stress cardiomyopathy mouse model [99]. These pilot studies shed light on the importance of mitochondrial balance in the pathogenesis of cardiomyopathies. The specific receptors responsible for recognizing mitochondrial DNA in the activation of the cGAS/STING signaling pathway in cardiomyopathy pathology have yet to be identified. Targeting mitochondrial dysfunction presents a promising therapeutic target to preserve mitochondrial balance, prevent excessive cGAS-STING activation, and ultimately avert a severe type I interferon immune response.
4.3. Future Perspectives in Understanding the cGAS/STING Pathway in Cardiomyopathy
Since Chen's group at the University of Texas Southwestern Medical Center discovered the cGAS/STING pathway, cGAS has been identified as a cytosolic DNA sensor that triggers interferon production [9,10]. However, the exact role of the cGAS/STING pathway in cardiomyopathy remains unclear. To better understand this, the following questions need to be answered. (i) Does DNA damage directly promote disease progression or is it just a reflection of late-stage damage to cardiomyocytes and progressive cardiomyocytes loss? (ii) How is the cGAS/STING pathway activated and regulated in myocytes compared to other cell types during cardiomyopathy (e.g., macrophages, fibroblasts, endothelial cells)? what is the specific downstream targets of cGAS during the cytodegeneration of cardiac muscle cells (iii) Considering mtDNA is capable of triggering cGAS/STING pathway during cardiomyopathy [101], it would be intriguing to understand how the mtDNA is released into the cytosol and whether this released mtDNA in the cyotosol could be targeted as a therapeutic approach during cardiac failure?
Last but not least, we deem it mandatory to point out that it is of particular interest to consider the cGAS/STING pathway under the context of aging and senescence during disease progression. The aging and elderly population is particularly susceptible to cardiovascular diseases. Age is an independent risk factor for cardiomyopathy [102]. Elderly patients usually suffer from both neurodegenerative and cardiovascular diseases, and the comorbidity worsens clinically relevant outcomes, including the severity of disability at diagnosis and rate of disability worsening after diagnosis103. Further investigation is warranted to determine if the cGAS/STING pathway serves as the cornerstone in regulating chronic cardiomyopathy.
Author Contributions
W.W. performed the literature review and made the Table under the guidance of J.Z. and H.K. H.K. made the schematic figure with input from J.Z. Y.G. and J.Z. performed the bioinformatics analysis and related figures using publicly available dataset under supervision from H.K.L. A.C.Y. M.K. and H.K.L. contributed additionally to the conceptualization, review/editing and the supervision of the manuscript. All authors contributed to the paper writing and approved the final manuscript.
Funding
Y.G. is supported by the National Natural Science Foundation of China (No. 82171416 and No. 32100766). J.Z. is supported by the National Multiple Sclerosis Society (FG-2407-43793)
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| DCM | Dilated cardiomyopathy |
| HCM | Hypertrophic cardiomyopathy |
| cGAS | cyclic GMP-AMP synthase |
| STING | Stimulator of interferon genes |
| TBK1 | TANK-binding Kinase 1 |
| IRF3 | interferon regulatory factor 3 |
| dsDNA | double-stranded DNA |
References
- Brieler J, Breeden MA, Tucker J. Cardiomyopathy: An Overview. Am Fam Physician. 2017;96(10):640-646.
- Ciarambino T, Menna G, Sansone G, Giordano M. Cardiomyopathies: An Overview. Int J Mol Sci. 2021;22(14):7722. [CrossRef]
- McKenna WJ, Maron BJ, Thiene G. Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ Res. 2017;121(7):722-730. [CrossRef]
- Ommen SR, Ho CY, Asif IM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation. 2024;149(23):e1239-e1311. [CrossRef]
- Austin KM, Trembley MA, Chandler SF, et al. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2019;16(9):519-537. [CrossRef]
- van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123(1):37-45. [CrossRef]
- Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res. 2011;109(1):86-96. [CrossRef]
- Pinilla-Vera M, Hahn VS, Kass DA. Leveraging Signaling Pathways to Treat Heart Failure With Reduced Ejection Fraction. Circ Res. 2019;124(11):1618-1632. [CrossRef]
- Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339(6121):826-830. [CrossRef]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786-791. [CrossRef]
- Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142-1149. [CrossRef]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science. 2013;341(6152):1390-1394. [CrossRef]
- Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21(9):548-569. [CrossRef]
- Zhang X, Wu J, Du F, et al. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014;6(3):421-430. [CrossRef]
- Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5(214):ra20. [CrossRef]
- Zhang C, Shang G, Gui X, Zhang X, Bai XC, Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. 2019;567(7748):394-398. [CrossRef]
- Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017;113(4):389-398. [CrossRef]
- Shi S, Chen Y, Luo Z, Nie G, Dai Y. Role of oxidative stress and inflammation-related signaling pathways in doxorubicin-induced cardiomyopathy. Cell Commun Signal CCS. 2023;21(1):61. [CrossRef]
- Harding D, Chong MHA, Lahoti N, et al. Dilated cardiomyopathy and chronic cardiac inflammation: Pathogenesis, diagnosis and therapy. J Intern Med. 2023;293(1):23-47. [CrossRef]
- Nunes JPS, Roda VM de P, Andrieux P, Kalil J, Chevillard C, Cunha-Neto E. Inflammation and mitochondria in the pathogenesis of chronic Chagas disease cardiomyopathy. Exp Biol Med Maywood NJ. 2023;248(22):2062-2071. [CrossRef]
- Luo W, Zou X, Wang Y, et al. Critical Role of the cGAS-STING Pathway in Doxorubicin-Induced Cardiotoxicity. Circ Res. 2023;132(11):e223-e242. [CrossRef]
- Lei Y, VanPortfliet JJ, Chen YF, et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell. 2023;186(14):3013-3032.e22. [CrossRef]
- Lei C, Tan Y, Ni D, Peng J, Yi G. cGAS-STING signaling in ischemic diseases. Clin Chim Acta Int J Clin Chem. 2022;531:177-182. [CrossRef]
- Heymans S, Lakdawala NK, Tschöpe C, Klingel K. Dilated cardiomyopathy: causes, mechanisms, and current and future treatment approaches. Lancet Lond Engl. 2023;402(10406):998-1011. [CrossRef]
- Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: A translational review of current literature. J Intern Med. 2019;286(4):362-372. [CrossRef]
- Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet Lond Engl. 2017;390(10092):400-414. [CrossRef]
- Peters S, Johnson R, Birch S, Zentner D, Hershberger RE, Fatkin D. Familial Dilated Cardiomyopathy. Heart Lung Circ. 2020;29(4):566-574. [CrossRef]
- McNally EM, Mestroni L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ Res. 2017;121(7):731-748. [CrossRef]
- Chen SN, Sbaizero O, Taylor MRG, Mestroni L. Lamin A/C Cardiomyopathy: Implications for Treatment. Curr Cardiol Rep. 2019;21(12):160. [CrossRef]
- Bhide S, Chandran S, Rajasekaran NS, Melkani GC. Genetic and Pathophysiological Basis of Cardiac and Skeletal Muscle Laminopathies. Genes. 2024;15(8):1095. [CrossRef]
- Donnaloja F, Carnevali F, Jacchetti E, Raimondi MT. Lamin A/C Mechanotransduction in Laminopathies. Cells. 2020;9(5):1306. [CrossRef]
- Murray-Nerger LA, Cristea IM. Lamin post-translational modifications: emerging toggles of nuclear organization and function. Trends Biochem Sci. 2021;46(10):832-847. [CrossRef]
- Jiang Y, Ji JY. Understanding lamin proteins and their roles in aging and cardiovascular diseases. Life Sci. 2018;212:20-29. [CrossRef]
- Wong X, Melendez-Perez AJ, Reddy KL. The Nuclear Lamina. Cold Spring Harb Perspect Biol. 2022;14(2):a040113. [CrossRef]
- Qiu H, Sun Y, Wang X, et al. Lamin A/C deficiency-mediated ROS elevation contributes to pathogenic phenotypes of dilated cardiomyopathy in iPSC model. Nat Commun. 2024;15(1):7000. [CrossRef]
- Captur G, Arbustini E, Bonne G, et al. Lamin and the heart. Heart Br Card Soc. 2018;104(6):468-479. [CrossRef]
- Ito K, Patel PN, Gorham JM, et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc Natl Acad Sci U S A. 2017;114(29):7689-7694. [CrossRef]
- Wang Y, Elsherbiny A, Kessler L, et al. Lamin A/C-dependent chromatin architecture safeguards naïve pluripotency to prevent aberrant cardiovascular cell fate and function. Nat Commun. 2022;13(1):6663. [CrossRef]
- Cheedipudi SM, Asghar S, Marian AJ. Genetic Ablation of the DNA Damage Response Pathway Attenuates Lamin-Associated Dilated Cardiomyopathy in Mice. JACC Basic Transl Sci. 2022;7(12):1232-1245. [CrossRef]
- En A, Bogireddi H, Thomas B, et al. Pervasive nuclear envelope ruptures precede ECM signaling and disease onset without activating cGAS-STING in Lamin-cardiomyopathy mice. Cell Rep. 2024;43(6):114284. [CrossRef]
- Zuela-Sopilniak N, Morival J, Lammerding J. Multi-level transcriptomic analysis of LMNA -related dilated cardiomyopathy identifies disease-driving processes. BioRxiv Prepr Serv Biol. Published online June 13, 2024:2024.06.11.598511. [CrossRef]
- Feurle P, Abentung A, Cera I, et al. SATB2-LEMD2 interaction links nuclear shape plasticity to regulation of cognition-related genes. EMBO J. 2021;40(3):e103701. [CrossRef]
- Thanisch K, Song C, Engelkamp D, et al. Nuclear envelope localization of LEMD2 is developmentally dynamic and lamin A/C dependent yet insufficient for heterochromatin tethering. Differ Res Biol Divers. 2017;94:58-70. [CrossRef]
- Vargas JD. The Role of the LEMD2 p.L13R Mutation in Dilated Cardiomyopathy. Circ Res. 2023;132(2):185-186. [CrossRef]
- Caravia XM, Ramirez-Martinez A, Gan P, et al. Loss of function of the nuclear envelope protein LEMD2 causes DNA damage-dependent cardiomyopathy. J Clin Invest. 2022;132(22):e158897. [CrossRef]
- Abdelfatah N, Chen R, Duff HJ, et al. Characterization of a Unique Form of Arrhythmic Cardiomyopathy Caused by Recessive Mutation in LEMD2. JACC Basic Transl Sci. 2019;4(2):204-221. [CrossRef]
- Chen R, Buchmann S, Kroth A, et al. Mechanistic Insights of the LEMD2 p.L13R Mutation and Its Role in Cardiomyopathy. Circ Res. 2023;132(2):e43-e58. [CrossRef]
- Lorenzo-Almorós A, Cepeda-Rodrigo JM, Lorenzo Ó. Diabetic cardiomyopathy. Rev Clin Esp. 2022;222(2):100-111. [CrossRef]
- Dillmann WH. Diabetic Cardiomyopathy. Circ Res. 2019;124(8):1160-1162. [CrossRef]
- Zhao X, Liu S, Wang X, et al. Diabetic cardiomyopathy: Clinical phenotype and practice. Front Endocrinol. 2022;13:1032268. [CrossRef]
- Khan S, Ahmad SS, Kamal MA. Diabetic Cardiomyopathy: From Mechanism to Management in a Nutshell. Endocr Metab Immune Disord Drug Targets. 2021;21(2):268-281. [CrossRef]
- Peterson LR, Gropler RJ. Metabolic and Molecular Imaging of the Diabetic Cardiomyopathy. Circ Res. 2020;126(11):1628-1645. [CrossRef]
- Ma XM, Geng K, Law BYK, et al. Lipotoxicity-induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS-STING pathway in obesity-related diabetes. Cell Biol Toxicol. 2023;39(1):277-299. [CrossRef]
- Yan M, Li Y, Luo Q, et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS-STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 2022;8(1):258. [CrossRef]
- Lu QB, Ding Y, Liu Y, et al. Metrnl ameliorates diabetic cardiomyopathy via inactivation of cGAS/STING signaling dependent on LKB1/AMPK/ULK1-mediated autophagy. J Adv Res. 2023;51:161-179. [CrossRef]
- Lu L, Shao Y, Xiong X, et al. Irisin improves diabetic cardiomyopathy-induced cardiac remodeling by regulating GSDMD-mediated pyroptosis through MITOL/STING signaling. Biomed Pharmacother Biomedecine Pharmacother. 2024;171:116007. [CrossRef]
- Chen Z, Lai X, Li J, et al. BRG1 Deficiency Promotes Cardiomyocyte Inflammation and Apoptosis by Activating the cGAS-STING Signaling in Diabetic Cardiomyopathy. Inflammation. Published online June 13, 2024. [CrossRef]
- Huang Q, Chen T, Li J, et al. IL-37 ameliorates myocardial fibrosis by regulating mtDNA-enriched vesicle release in diabetic cardiomyopathy mice. J Transl Med. 2024;22(1):494. [CrossRef]
- Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017;121(7):784-802. [CrossRef]
- Smith ED, Lakdawala NK, Papoutsidakis N, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141(23):1872-1884. [CrossRef]
- Gacita AM, McNally EM. Genetic Spectrum of Arrhythmogenic Cardiomyopathy. Circ Heart Fail. 2019;12(3):e005850. [CrossRef]
- Lombardi R, Marian AJ. Arrhythmogenic right ventricular cardiomyopathy is a disease of cardiac stem cells. Curr Opin Cardiol. 2010;25(3):222-228. [CrossRef]
- Desai YB, Parikh VN. Genetic Risk Stratification in Arrhythmogenic Left Ventricular Cardiomyopathy. Card Electrophysiol Clin. 2023;15(3):391-399. [CrossRef]
- Kang H, Lee CJ. Transmembrane proteins with unknown function (TMEMs) as ion channels: electrophysiological properties, structure, and pathophysiological roles. Exp Mol Med. 2024;56(4):850-860. [CrossRef]
- Matos J, Helle E, Care M, et al. Cardiac MRI and Clinical Outcomes in TMEM43 Arrhythmogenic Cardiomyopathy. Radiol Cardiothorac Imaging. 2023;5(6):e230155. [CrossRef]
- Rouhi L, Cheedipudi SM, Chen SN, et al. Haploinsufficiency of Tmem43 in cardiac myocytes activates the DNA damage response pathway leading to a late-onset senescence-associated pro-fibrotic cardiomyopathy. Cardiovasc Res. 2021;117(11):2377-2394. [CrossRef]
- Almajidi YQ, Kadhim MM, Alsaikhan F, et al. Doxorubicin-loaded micelles in tumor cell-specific chemotherapy. Environ Res. 2023;227:115722. [CrossRef]
- Carvalho C, Santos RX, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16(25):3267-3285. [CrossRef]
- Sheibani M, Azizi Y, Shayan M, et al. Doxorubicin-Induced Cardiotoxicity: An Overview on Pre-clinical Therapeutic Approaches. Cardiovasc Toxicol. 2022;22(4):292-310. [CrossRef]
- Kong CY, Guo Z, Song P, et al. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int J Biol Sci. 2022;18(2):760-770. [CrossRef]
- Rawat PS, Jaiswal A, Khurana A, Bhatti JS, Navik U. Doxorubicin-induced cardiotoxicity: An update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed Pharmacother Biomedecine Pharmacother. 2021;139:111708. [CrossRef]
- Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ Res. 2020;126(7):926-941. [CrossRef]
- Chen Y, Shi S, Dai Y. Research progress of therapeutic drugs for doxorubicin-induced cardiomyopathy. Biomed Pharmacother Biomedecine Pharmacother. 2022;156:113903. [CrossRef]
- de Baat EC, Mulder RL, Armenian S, et al. Dexrazoxane for preventing or reducing cardiotoxicity in adults and children with cancer receiving anthracyclines. Cochrane Database Syst Rev. 2022;9(9):CD014638. [CrossRef]
- Rahimi P, Barootkoob B, ElHashash A, Nair A. Efficacy of Dexrazoxane in Cardiac Protection in Pediatric Patients Treated With Anthracyclines. Cureus. 2023;15(4):e37308. [CrossRef]
- Xiao Z, Yu Z, Chen C, Chen R, Su Y. GAS-STING signaling plays an essential pathogenetic role in Doxorubicin-Induced Cardiotoxicity. BMC Pharmacol Toxicol. 2023;24(1):19. [CrossRef]
- Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18(11):1639-1642. [CrossRef]
- Srzić I, Nesek Adam V, Tunjić Pejak D. SEPSIS DEFINITION: WHAT’S NEW IN THE TREATMENT GUIDELINES. Acta Clin Croat. 2022;61(Suppl 1):67-72. [CrossRef]
- Jacobi J. The pathophysiology of sepsis - 2021 update: Part 2, organ dysfunction and assessment. Am J Health-Syst Pharm AJHP Off J Am Soc Health-Syst Pharm. 2022;79(6):424-436. [CrossRef]
- Bleakley G, Cole M. Recognition and management of sepsis: the nurse’s role. Br J Nurs Mark Allen Publ. 2020;29(21):1248-1251. [CrossRef]
- Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol. 2021;18(6):424-434. [CrossRef]
- L’Heureux M, Sternberg M, Brath L, Turlington J, Kashiouris MG. Sepsis-Induced Cardiomyopathy: a Comprehensive Review. Curr Cardiol Rep. 2020;22(5):35. [CrossRef]
- Li N, Zhou H, Wu H, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215. [CrossRef]
- Kong C, Ni X, Wang Y, et al. ICA69 aggravates ferroptosis causing septic cardiac dysfunction via STING trafficking. Cell Death Discov. 2022;8(1):187. [CrossRef]
- Liu H, Hu Q, Ren K, Wu P, Wang Y, Lv C. ALDH2 mitigates LPS-induced cardiac dysfunction, inflammation, and apoptosis through the cGAS/STING pathway. Mol Med Camb Mass. 2023;29(1):171. [CrossRef]
- Bremer J. Carnitine--metabolism and functions. Physiol Rev. 1983;63(4):1420-1480. [CrossRef]
- Scholte HR, Rodrigues Pereira R, de Jonge PC, et al. Primary carnitine deficiency. J Clin Chem Clin Biochem Z Klin Chem Klin Biochem. 1990;28(5):351-357.
- Berg SM, Beck-Nielsen H, Færgeman NJ, Gaster M. Carnitine acetyltransferase: A new player in skeletal muscle insulin resistance? Biochem Biophys Rep. 2017;9:47-50. [CrossRef]
- Song MJ, Park CH, Kim H, et al. Carnitine acetyltransferase deficiency mediates mitochondrial dysfunction-induced cellular senescence in dermal fibroblasts. Aging Cell. 2023;22(11):e14000. [CrossRef]
- Mao H, Angelini A, Li S, et al. CRAT links cholesterol metabolism to innate immune responses in the heart. Nat Metab. 2023;5(8):1382-1394. [CrossRef]
- Echavarría NG, Echeverría LE, Stewart M, Gallego C, Saldarriaga C. Chagas Disease: Chronic Chagas Cardiomyopathy. Curr Probl Cardiol. 2021;46(3):100507. [CrossRef]
- Santos É, Menezes Falcão L. Chagas cardiomyopathy and heart failure: From epidemiology to treatment. Rev Port Cardiol. 2020;39(5):279-289. [CrossRef]
- Nunes MCP, Beaton A, Acquatella H, et al. Chagas Cardiomyopathy: An Update of Current Clinical Knowledge and Management: A Scientific Statement From the American Heart Association. Circulation. 2018;138(12):e169-e209. [CrossRef]
- Choudhuri S, Garg NJ. PARP1-cGAS-NF-κB pathway of proinflammatory macrophage activation by extracellular vesicles released during Trypanosoma cruzi infection and Chagas disease. PLoS Pathog. 2020;16(4):e1008474. [CrossRef]
- Medina de Chazal H, Del Buono MG, Keyser-Marcus L, et al. Stress Cardiomyopathy Diagnosis and Treatment: JACC State-of-the-Art Review. J Am Coll Cardiol. 2018;72(16):1955-1971. [CrossRef]
- Amin HZ, Amin LZ, Pradipta A. Takotsubo Cardiomyopathy: A Brief Review. J Med Life. 2020;13(1):3-7. [CrossRef]
- Dawson DK. Acute stress-induced (takotsubo) cardiomyopathy. Heart Br Card Soc. 2018;104(2):96-102. [CrossRef]
- Ravindran J, Brieger D. Clinical perspectives: Takotsubo cardiomyopathy. Intern Med J. 2024;54(11):1785-1795. [CrossRef]
- Wang Y, Tang X, Cui J, et al. Ginsenoside Rb1 mitigates acute catecholamine surge-induced myocardial injuries in part by suppressing STING-mediated macrophage activation. Biomed Pharmacother Biomedecine Pharmacother. 2024;175:116794. [CrossRef]
- Chaffin M, Papangeli I, Simonson B, et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature. 2022;608(7921):174-180. [CrossRef]
- Jenson JM, Li T, Du F, Ea CK, Chen ZJ. Ubiquitin-like conjugation by bacterial cGAS enhances anti-phage defence. Nature. 2023;616(7956):326-331. [CrossRef]
- Rodgers JL, Jones J, Bolleddu SI, et al. Cardiovascular Risks Associated with Gender and Aging. J Cardiovasc Dev Dis. 2019;6(2):19. [CrossRef]
- Tahsili-Fahadan P, Geocadin RG. Heart-Brain Axis: Effects of Neurologic Injury on Cardiovascular Function. Circ Res. 2017;120(3):559-572. [CrossRef]
Figure 1.
Schematic illustration of the cGAS/STING pathway. Cyclic guanosine monophosphate adenosine monophosphate synthase (cGAS), which is freely present in the cytosol, binds to both exogenous and endogenous double-stranded DNA (dsDNA). Viruses and bacteria can be sources of exogenous dsDNA, while endogenous dsDNA mainly originates from cell apoptosis, auto cellular death as well as mitochondrial damage. After binding to dsDNA, cGAS catalyzes a reaction between ATP and GTP to form 2‘,3’-cGAMP. 2‘,3’-cGAMP then binds to the ‘stimulator of interferon genes’ (STING), located on the membrane of the endoplasmic reticulum (ER), triggers a conformational change and subsequently activates STING. The activated STING is transported to the Golgi apparatus via the coatomer protein complex I (COP I), where it phosphorylates TANK-binding kinase 1 (TBK1) to form a tetramer. TBK1 then phosphorylates interferon regulatory factor 3 (IRF3), which dimerizes and enters the nucleus together with nuclear factor kappa B (NFκB) to induce the expression of type 1 interferons as well as other proinflammatory cytokines. Many chemical compounds are known to modulate the cGAS/STING signaling pathway (see green and red box).
Figure 1.
Schematic illustration of the cGAS/STING pathway. Cyclic guanosine monophosphate adenosine monophosphate synthase (cGAS), which is freely present in the cytosol, binds to both exogenous and endogenous double-stranded DNA (dsDNA). Viruses and bacteria can be sources of exogenous dsDNA, while endogenous dsDNA mainly originates from cell apoptosis, auto cellular death as well as mitochondrial damage. After binding to dsDNA, cGAS catalyzes a reaction between ATP and GTP to form 2‘,3’-cGAMP. 2‘,3’-cGAMP then binds to the ‘stimulator of interferon genes’ (STING), located on the membrane of the endoplasmic reticulum (ER), triggers a conformational change and subsequently activates STING. The activated STING is transported to the Golgi apparatus via the coatomer protein complex I (COP I), where it phosphorylates TANK-binding kinase 1 (TBK1) to form a tetramer. TBK1 then phosphorylates interferon regulatory factor 3 (IRF3), which dimerizes and enters the nucleus together with nuclear factor kappa B (NFκB) to induce the expression of type 1 interferons as well as other proinflammatory cytokines. Many chemical compounds are known to modulate the cGAS/STING signaling pathway (see green and red box).
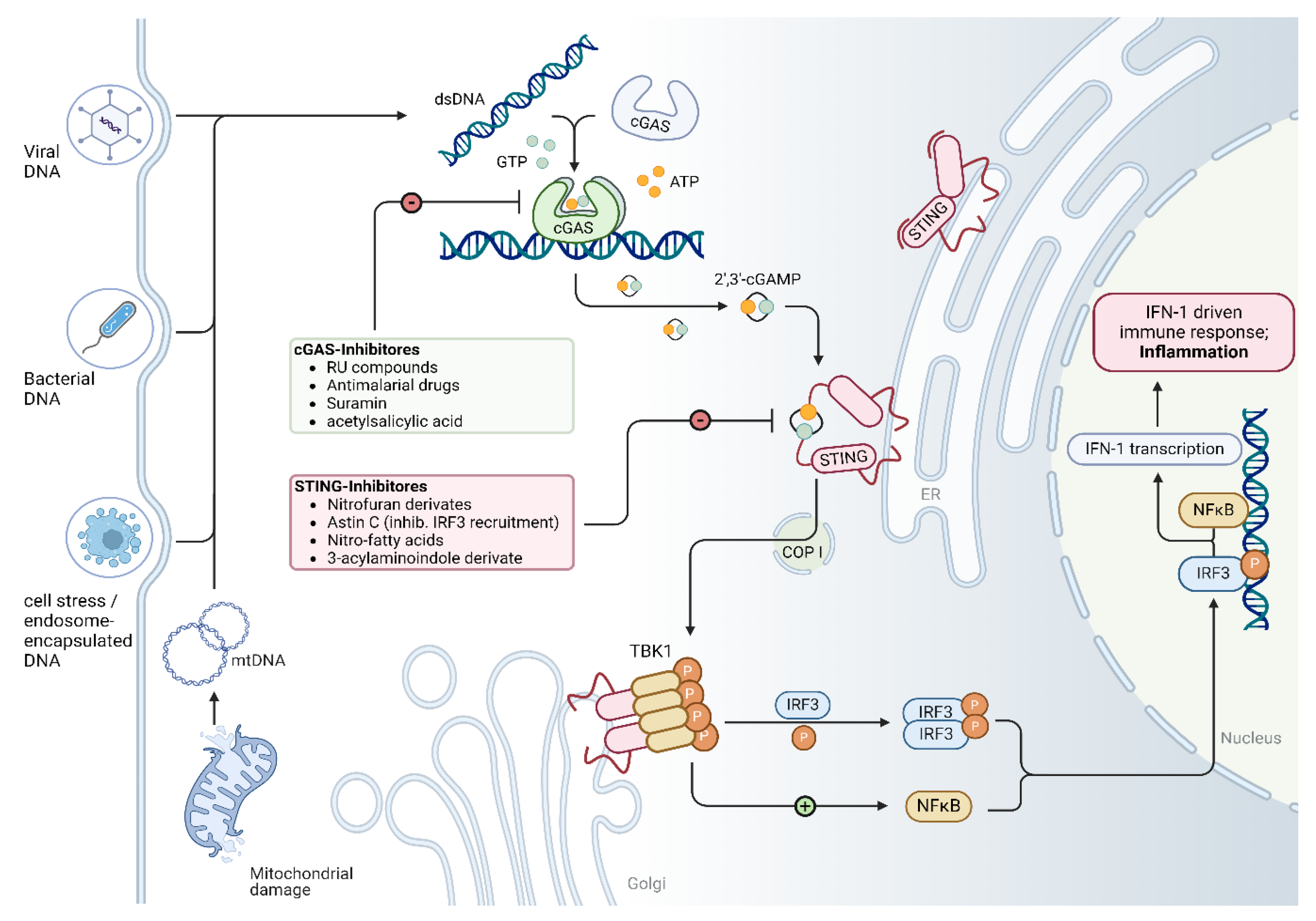
Figure 2.
Mitochondrial alterations in human cardiomyopathy. (a) Heatmap displays the normalized expression of mitochondrial genes in cardiac muscle cells, which are averaged across individuals. (b) Violin plots of mitochondrial genes across different disease groups showing normalized expression value. Violin plots are centered around the mean value with standard deviation, and the shape represents sample distribution. P values are determined by Mann–Whitney U test (only significant P values are shown, *P < 0.05, **P < 0.01, ***P < 0.001). NF, non-failing hearts; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy.
Figure 2.
Mitochondrial alterations in human cardiomyopathy. (a) Heatmap displays the normalized expression of mitochondrial genes in cardiac muscle cells, which are averaged across individuals. (b) Violin plots of mitochondrial genes across different disease groups showing normalized expression value. Violin plots are centered around the mean value with standard deviation, and the shape represents sample distribution. P values are determined by Mann–Whitney U test (only significant P values are shown, *P < 0.05, **P < 0.01, ***P < 0.001). NF, non-failing hearts; DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy.
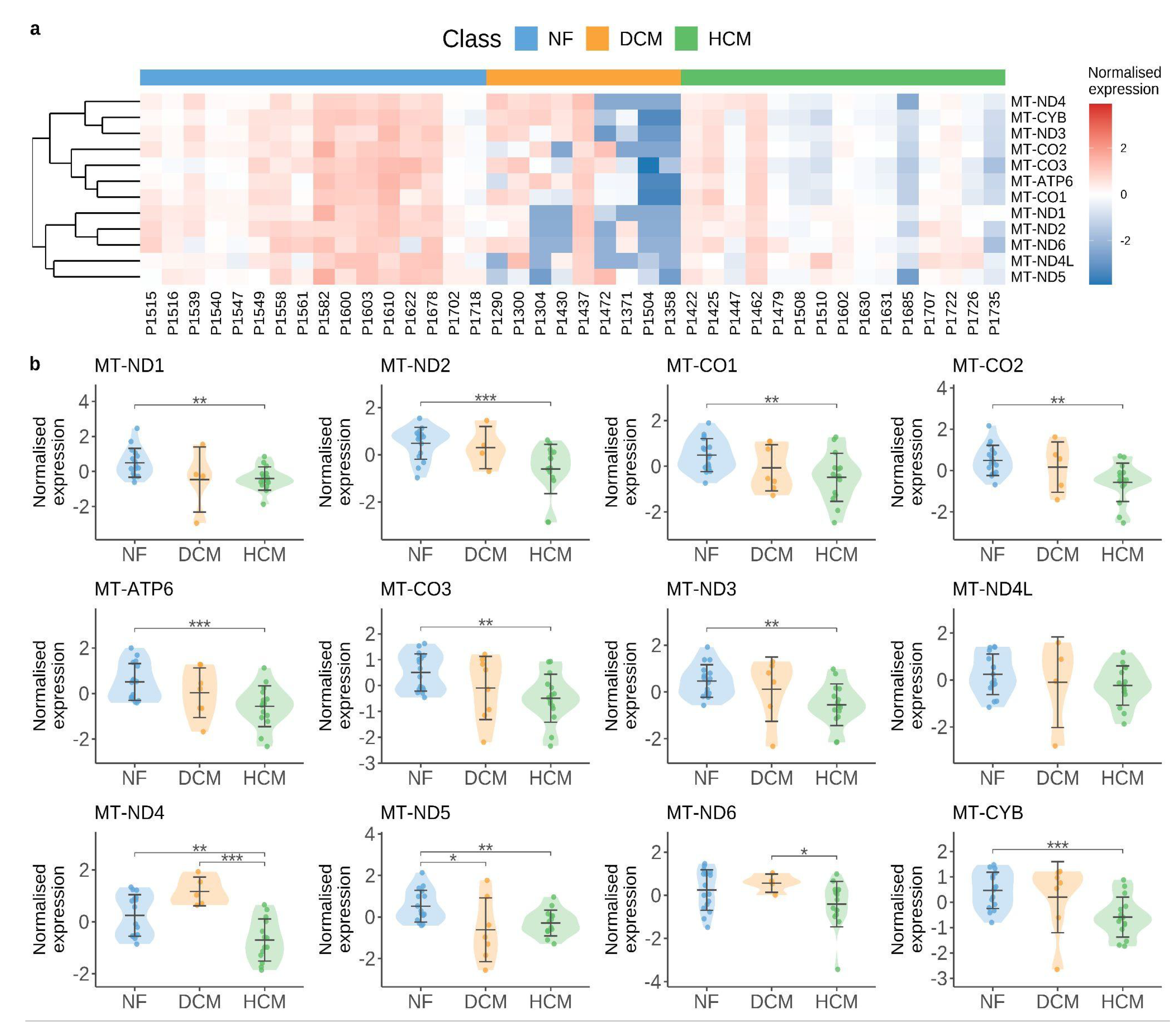

Table 1.
Main studies on cGAS/STING pathway in cardiomyopathy.
| Cardiomyopathy | Year | Model | Methods | Conclusion | Ref |
|---|---|---|---|---|---|
| Sepsis-induced cardiomyopathy (SIC) | 2019 | ∙ In vivo: Male mouse injected with LPS ∙ In vitro: Neonatal rat cardiomyocytes (NRCMs); H9C2 cells |
∙ IB ∙ IF ∙ Real time RT-qPCR |
∙ cGAS/STING is activated in LPS-treated heart tissues and cardiomyocytes. ∙ Activated molecules: NLRP3, IRF3, IL-1β, TNF-α, MCP-1, HMGBA, caspase-1, IL-18 ∙ STING knockdown inhibits LPS-induced phosphorylation and nuclear translocation of IRF3, suppresses inflammation, apoptosis and pyroptosis, improves cardiac function and survival. |
[1] |
| 2022 | ∙ Human blood samples ∙ In vivo: Male mouse injected with LPS ∙ In vitro: RAW 264.7 macrophages. H9C2 myofibroblasts |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR |
∙ STING is activated in LPS-treated cardiac tissues. ∙ STING is increased in the peripheral blood samples of septic patients. ∙ Activated molecules: TNF-α, IL-1β, IL-6, COX2. ∙ ICA69 knockout inhibits STING-mediated inflammation and ferroptosis. |
[2] | |
| 2023 | ∙ In vivo: LPS-treated mouse ∙ In vitro: LPS-stimulated H9C2 cells (rat cardiomyocytes) |
∙ IB ∙ ELISA ∙ IF/IHC |
∙ cGAS/STING is activated in both heart tissue and H9C2 cells following LPS treatment. ∙ Activated molecules: IRF3, TBK1, IL-6, IL-1β, TNF-α. ∙ Knocking down cGAS in H9C2 cardiomyocytes alleviates cardiac inflammation and apoptosis induced by LPS. ∙ ALDH2 inhibits cGAS/STING signaling both in vivo and in vitro. |
[3] | |
| Dilated cardiomyopathy | 2023 | ∙ In vivo: Myh6-Cre:LmnaF/F:Crat-/- mouse ∙ In vitro: Neonatal rat ventricular myocytes (NRVMs) |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR ∙ RNA-Seq ∙ scRNA-Seq |
∙ cGAS and type I interferon responses are activated in CRAT-deficient NRVMs. ∙ Activated molecules: IL-1β, IL-6, TNF-α ∙ Knockdown of cGAS abrogates interferon-stimulated gene expression. |
[4] |
| 2023 | ∙ Human hearts with DCM | ∙ IB ∙ RNA-Seq |
∙ cGAS is increased in human heart samples from patients with primary DCM. ∙ STING1 and phospho-STING1 (serine residue 365) are unchanged. ∙ Activated molecules: TBK1. |
[5] | |
| 2022 | ∙ In vivo (LMNA–DCM model): Myh6-Cre:LmnaF/F mouse; Myh6-Cre:LmnaF/F:Mb21d1-/- mouse |
∙ IB ∙ IF |
∙ cGAS/STING is activated in LMNA-DCM whole heart tissue. ∙ Activated molecules: ATM, H2AFX, p-TP53, total TP53, CDKN1A. ∙ Knockout of CGAS prolonged survival, improved cardiac function, partially restored levels of molecular markers of heart failure, and attenuated myocardial apoptosis and fibrosis in the LMNA-deficient mice. |
[6] | |
| 2024 | ∙ In vivo (LMNA–DCM model): LmnaF/F;Myh6-MerCreMer mouse |
∙ IF/IHC ∙ snRNA-seq |
∙ cGAS/STING-related transcription is not activated in cardiomyocytes. ∙ CGAS or STING knockout does not rescue the phenotypes of LMNA-DCM. |
[7] | |
| Diabetic cardiomyopathy | 2022 | ∙ In vivo: Male db/db and db/+ mouse ∙ In vitro: Palmitic acid (PA)-treated H9C2 cells (rat cardiomyocytes) |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR |
∙ Mitochondria-derived mtDNA can activate cGAS/STING pathway in cardiomyocytes. ∙ Activated molecules: IRF3, NF-κB, IL-18, IL-1β. ∙ Knockdown of STING in H9C2 cardiomyocytes and inhibition of STING with C176 can ameliorate myocardial inflammation and apoptosis. |
[8] |
| 2022 | ∙ In vivo: Male mouse ∙ In vitro: Palmitic acid (PA)-treated H9C2 cells (rat cardiomyocytes) |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR |
∙ Cytosolic mtDNA activates cGAS/STING in DCM hearts and H9C2 cells. ∙ Activated molecules: p-TBK1, p-IRF3, NLRP3, caspase-1, GSDMD, TNF-α, INF-β, IL-1β, IL-18. ∙ cGAS or STING knockdown inhibits cardiomyocyte pyroptosis and inflammation. |
[9] | |
| 2023 | ∙ In vivo: STZ-treated mouse; db/db mouse ∙ In vitro: Neonatal rat cardiomyocytes (NRCMs); cultured cardiac fibroblasts; endothelial cells |
∙ IB ∙ IF ∙ TUNEL ∙ Real time RT-qPCR |
∙ cGAS/STING is activated by ULK1 in cardiomyocytes. ∙ Metrnl downregulation exacerbates high glucose-elicited hypertrophy, apoptosis, and oxidative damage in neonatal rat cardiomyocytes. ∙ Metrnl activates the autophagy pathway and inhibits the cGAS/STING signaling in a LKB1/AMPK/ULK1-dependent mechanism in cardiomyocytes. |
[10] | |
| 2023 | ∙ In vivo: STZ-treated and HFD-fed mouse ∙ In vitro: Neonatal rat cardiomyocytes (NRCMs) |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR ∙ TUNEL |
∙ cGAS/STING is activated in cardiomyocytes both in vivo and in vitro. ∙ Activated molecules: p-TBK, p-NF-κB, IL-1β, Caspase-3. ∙ BRG1 deficiency results in the accumulation of dsDNA and triggers cGAS/STING, exacerbating cardiomyocyte inflammation and apoptosis induced by hyperglycemia and hyperlipidemia. |
[11] | |
| 2024 | ∙ In vivo: Human blood samples; STZ-treated and HFD-fed mouse ∙ In vitro: |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR ∙ TUNEL |
∙ cGAS/STING is activated in fibroblasts which engulf released extracellular vesicles containing mtDNA from cardiomyocytes. ∙ Activated molecules: p-TBK1, p-IRF3, p-p65, IL-37. ∙ IL-37 ameliorates mitochondrial injury, reduces the release of mtDNA-enriched vesicles, which attenuates the progression of DCM. |
[12] | |
| 2024 | ∙ In vivo: STZ-treated and HFD-fed mouse ∙ In vitro: HG/HF-treated H9C2 cells |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR |
∙ cGAS/STING is activated in myocardium and H9C2 cells. ∙ Activated molecules: MITOL, NLRP3, Caspase 1, IL-1β, GSDMD. ∙ Irisin and MITOL administration alleviates cardiac dysfunction via inhibition of the cGAS/STING pathway. |
[13] | |
| Other cardiomyopathies | 2021 | TMEM43 arrhythmogenic cardiomyopathy ∙ In vivo: Myh6-Cre:Tmem43W/F mice |
∙ IB ∙ IF ∙ bulk RNA-Seq |
∙ cGAS/STING is activated in cardiomyocytes at later stage. ∙ Activated molecules: pATM, ATM, pH2AFX, LGALS3, VCAN, GDF15, TGFβ1. |
[14] |
| 2020 | Chagas cardiomyopathy ∙ In vivo: T. cruzi trypomastigotes infected mice ∙ In vitro: Murine bone marrow cells; RAW 264.7 macrophages; C2C12 mouse myoblast cells |
∙ IHC ∙ Real time RT-qPCR |
∙ cGAS/STING is the early responder in recognizing T.cruzi-induced extracellular vesicles stimulus and signaling proinflammatory cytokine gene expression in macrophages. ∙ Activated molecules: NF-κB, IL-6, IL-1β, TNF-α. ∙ PARP1 synergizes with cGAS in signaling the NF-κB transcriptional activity in macrophages; inhibition of PARP1 reduces myocardial inflammatory infiltrates and improves the left ventricular function. |
[15] | |
| 2024 | Stress cardiomyopathy ∙ In vivo: Ovariectomized mice treated with isoproterenol; ∙ In vitro: RAW 264.7 macrophages |
∙ IF/IHC ∙ bulk RNA-seq |
∙ STING is activated in macrophages. ∙ Activated molecules: TBK1, TNF, IL6, CCL2, IFN-β. ∙ Ginsenoside Rb1 suppresses DNA-stimulated STING-mediated proinflammatory activation of macrophages. |
[16] | |
| 2023 | LEMD2 arrhythmogenic cardiomyopathy ∙ In vivo: Lemd2 p.L13R knock-in mouse ∙ In vitro: HeLa LEMD2 p.L13R KI; HeLa LEMD2 DEL; HEK293 |
∙ IF ∙ bulk RNA-seq |
∙ cGAS is recruited to the nuclear envelope rupture sites and micronuclei, subsequently activating cGAS/STING/IFN pathway in mutant LEMD2 cell lines. ∙ Activated molecules: H2AFX, SASPs (Gdf15, Tgfβ2 and Edn3) |
[17] | |
| 2023 | Doxorubicin-induced cardiomyopathy ∙ In vivo: Male mouse treated with doxorubicin for acute injury |
∙ IB ∙ IF/IHC ∙ Real time RT-qPCR ∙ TUNEL |
∙ cGAS/STING is activated in myocardium. ∙ Activated molecules: p-IRF3, p-p65, p-TBK1. ∙ STING knockdown reduces vacuolization and myofibril loss, suppresses inflammation and apoptosis, improves survival and cardiac function. |
[18] | |
| 2023 | Doxorubicin-induced cardiomyopathy ∙ In vivo: Mouse treated with low-dose doxorubicin for chronic injury ∙ In vitro: human cardiac microvascular endothelial cells (HCMECs) |
∙ IB ∙ IF ∙ Real time RT-qPCR ∙ bulk RNA-Seq |
∙ cGAS/STING is activated in cardiac endothelial cells. ∙ Activated molecules: p-TBK1, p-IRF3 ∙ Global cGAS, Sting, and Irf3 deficiency ameliorates DIC. ∙ Endothelial cell-specific Sting deficiency prevents DIC and endothelial dysfunction. |
[19] |
Table 2.
Molecular intervention of cGAS/STING pathway in cardiomyopathy.
| Target | Compound / Drug | Mode of action | Effects on signalling cascades and in animal models | Testing systems | Disease to be investigated in the animal model: | Ref |
|---|---|---|---|---|---|---|
| cGAS | RU-compounds (RU.365, RU.521) |
catalytic site inhibitor | reduced expression levels of Ifnb1 mRNA in Trex knockout mice (which constitutively activate cGAS) ∙ ↓ IL-1β, ↓ cleaved caspase-3 ∙ ↓ apoptosis |
In vivo: Trex1−/− mice In vitro: Neonatal rat cardiomyocytes (NRCMs) |
multi-organ inflammation Diabetic cardiomyopathy |
[20] |
| Antimalarial drugs (i.e. Hydroxychloroquine, Quinacrine) |
disrupting dsDNA binding | Hydroxychloroquine and Quinacrine inhibit dsDNA binding to cGAS in vitro: ↓ IFN-β expression In vivo: ↓ early IFN-1 response in Hydroxycloroquine-treated mice |
In vitro: THP1-Dual cells In vivo: C57BL/6 mice UVB inflammation model |
[21] | ||
| Suramin | disrupting dsDNA binding | suramin inhibits dsDNA binding to cGAS in vitro THP1-Dual cells: ↓ IFN-β expression (mRNA and protein) |
In vitro: THP1-Dual cells |
[22] | ||
| Acetylsalicylic acid | cGAS acetylation and inhibition | ↓ IFN-production in vitro (THP-1 cells) and ↓ expression of interferon-stimulated genes (ISG) Trex1–/– bone marrow cells; ↓ ISG expression in the hearts of Trex1–/– mice |
In vitro: - THP-1 cells - Trex1–/– bone marrow In vivo: Trex1–/– mice |
multi-organ inflammation | [23] | |
| STING and TBK1 | Astin C | STING inhibition- targeting the cyclic dinucleotide binding site | ↓ expression of Ifnb, Cxcl10, Isg15, Isg56 and Tnf mRNA in the heart of Trex1-/- mice (in vivo); ↓ expression of type 1 interferone in Trex1−/− bone marrow cells (in vitro) |
In vivo: Trex1−/− mice In vitro: Trex1−/− bone marrow cells |
multi-organ inflammation | [24] |
| Nitrofuran derivatives - C176 and C178 |
STING inhibition – Covalent binding to cysteine residue 91, inhibiting palmitoylation and activation of STING |
↓ serum levels of type I interferons and IL-6 in Trex1−/− mice |
In vivo: Trex1−/− mice |
multi-organ inflammation |
[25] | |
| ↓ phosphorylation of p65 ↑ improve diastolic cardiac function ↑ Partially improve myocardial hypertrophy |
In vivo: db/db mice In vitro: H9C2 rat cardiomyocytes |
Diabetic cardiomyopathy | [26] | |||
| ↓ cardiac IRF3 phosphorylation, IRF3 nuclear translocation and CD38 expression. ↑ cardiomyocyte NAD levels, mitochondrial function and ↑ left ventricular systolic function. ↓ cardiomyocyte apoptosis. ↓ antitumor effects of doxorubicin |
In vivo: Tumor free doxorubicin treated mice |
Doxorubicin-induced cardiomyopathy (DIC) | [27] | |||
| ∙ ↓ IL-1β, cleaved caspase-3; ∙ no effect on γ-H2AX; ∙ ↓ apoptosis |
∙ In vitro: Neonatal rat cardiomyocytes (NRCMs) |
Diabetic cardiomyopathy | [28] | |||
| Amlexanox | TBK1 inhibitor | Same effect as C176 |
In vivo: Tumor free doxorubicin treated mice |
Doxorubicin-induced cardiomyopathy (DIC) | [29] | |
| 3-acylaminoindole derivative - H-151 |
STING inhibition – blocking the activation-induced palmitoylation and clustering of STING |
↓ calf thymus DNA-induced production of TNF in a dose-dependent manner |
In vitro: calf thymus DNA-stimulated RAW264.7 cells (DMXAA stimulated – STING activator) |
Stress cardiomyopathy (SCM) | [30] | |
| ↓ reduced IFN-β levels in a dose-dependent manner |
In vitro: RAW264.7 cells stimulated with recombinant murine (rm) CIRP |
[31] | ||||
| Ginsenoside Rb1 | major chemical constituent of ginseng; suppressing the activation of STING |
↓ STING-mediated proinflammatory activation of macrophages. ↓ myocardial fibrosis and inflammatory responses in the heart. ↓ DNA-triggered proinflammatory activation of macrophages. ↓ DNA-triggered whole-genome gene expression alterations in macrophages; |
In vivo: OVX-ISO mice; In vitro: calf thymus DNA-stimulated RAW264.7 cells (DMXAA stimulated – STING activator) |
Stress cardiomyopathy (SCM) | [32] | |
| DMXAA | STING agonist | ↑STING phosphorylation. ↑TNF, IL6, CCL2, IFN-β; |
∙ In vitro: RAW264.7 cells |
Stress cardiomyopathy (SCM) | [33] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



