
Submitted:
23 April 2025
Posted:
23 April 2025
You are already at the latest version
Abstract
Glucocorticoid therapy, using agents like dexamethasone (Dexa), often leads to muscle atrophy by increasing protein degradation via the ubiquitin-proteasome system, frequently involving FoxO3 activation and upregulation of MAFbx and MuRF1. Stigmasterol, a phytosterol with known bioactivities, has an unexplored role in muscle atrophy. This study investigated stigmasterol's protective effects against Dexa-induced muscle atrophy and its impact on the FoxO3 signaling pathway. Differentiated C2C12 myotubes were treated with Dexa (50 µM) ± stigmasterol (10 µM), assessing morphology, viability, and FoxO3/MuRF1/MAFbx protein levels. C57BL/6 mice received Dexa (20 mg/kg/day i.p.) ± stigmasterol (3 mg/kg/day oral) for 21 days, measuring body/muscle mass, BMD, fiber CSA, and muscle protein expression. Stigmasterol (10 µM) was non-toxic and attenuated Dexa-induced reductions in myotube diameter and fusion in vitro, concurrent with suppressing Dexa-induced upregulation of FoxO3, MuRF1, and MAFbx proteins. In vivo, stigmasterol mitigated Dexa-induced losses in body weight, muscle mass, BMD, and fiber CSA. This protection was associated with attenuated upregulation of FoxO3 and MAFbx proteins in muscle tissue. Stigmasterol protects against Dexa-induced muscle atrophy in vitro and in vivo, linked to modulation of the FoxO3-MAFbx catabolic pathway. These findings suggest stigmasterol inhibits excessive glucocorticoid-induced muscle protein breakdown and warrants further investigation as a potential therapeutic agent for glucocorticoid myopathy.
Keywords:
Stigmasterol
; Muscle atrophy
; Dexamethasone
; C2C12
; FoxO3
; MuRF1
; MAFbx
; Ubiquitin-proteasome system
; Phytosterol
1. Introduction
Sarcopenia is a syndrome characterized by the progressive, age-related loss of skeletal muscle mass, strength, and function [1,2]. Its prevalence increases significantly with age, impacting a substantial portion of the elderly population [3]. Furthermore, sarcopenia is associated with a range of adverse health outcomes, including functional decline, increased risk of falls and fractures, and higher mortality rates, highlighting its significance as a public health concern [2,4]. Muscle mass is regulated by a delicate balance between muscle protein synthesis (anabolism) and muscle protein breakdown (catabolism). Sarcopenia and muscle atrophy resulting from various causes occur when this balance shifts towards excessive breakdown [5].
A major mechanism underlying muscle atrophy is the activation of protein degradation via the ubiquitin-proteasome system (UPS). Playing pivotal roles in this pathway are the muscle-specific E3 ubiquitin ligases, MAFbx (muscle atrophy F-box, also known as Atrogin-1) and MuRF1 (muscle RING finger 1) [6,7]. The expression of these genes is regulated by various upstream signaling pathways. Notably, the Insulin-like Growth Factor-1 (IGF-1)/Akt pathway promotes muscle growth and inhibits protein breakdown. IGF-1/Akt signaling suppresses the transcription of MAFbx and MuRF1 by phosphorylating Forkhead box O (FOXO) family transcription factors (mainly FoxO1 and FoxO3), retaining them in the cytoplasm [8]. Conversely, under atrophic conditions such as starvation, immobilization, inflammation, or excess glucocorticoids, Akt activity is reduced. This allows dephosphorylated FOXO proteins to translocate into the nucleus, where they increase the expression of MAFbx and MuRF1, thereby promoting protein degradation [9]. Additionally, activation of the energy sensor AMPK (AMP-activated protein kinase) can also influence protein breakdown pathways [10].
Stigmasterol is a phytosterol (plant sterol) found in various plant sources, including soybeans, nuts, seeds, and several vegetable oils [11,12]. It has been reported to possess diverse biological activities, such as anti-inflammatory, antioxidant, cholesterol-lowering, and anti-osteoarthritic effects [13,14]. Structurally, stigmasterol is very similar to β-sitosterol, differing primarily by an additional double bond in the side chain. While β-sitosterol has demonstrated anti-inflammatory properties and the ability to modulate muscle protein homeostasis by reducing markers of muscle atrophy, such as MuRF1 and MAFbx, and improving grip strength and myofiber thickness in animal models [15]. On the other hand, stigmasterol has been studied for its anti-inflammatory and metabolic benefits, including lipid metabolism regulation and bile acid modulation [16,17]. However, no studies have directly linked stigmasterol to muscle mass regulation or function.
Synthetic glucocorticoids, such as dexamethasone, are widely used for treating inflammatory conditions but can cause severe muscle atrophy as a side effect, particularly with long-term or high-dose use [18]. Glucocorticoid-induced muscle atrophy is primarily mediated by increased protein breakdown, specifically through the upregulation of MAFbx and MuRF1 expression via FoxO transcription factors [18]. Despite the various known bioactivities of stigmasterol, its specific impact on muscle mass loss and functional decline, especially under conditions inducing atrophy like glucocorticoid treatment, remains poorly understood. Furthermore, there is a lack of research on its mechanism of action concerning the regulation of muscle protein breakdown pathways.
Therefore, this study aimed to investigate the potential protective effects of stigmasterol against dexamethasone-induced muscle atrophy using both in vitro (C2C12 myotubes) and in vivo (mouse) models. We specifically sought to elucidate the underlying mechanism by examining the impact of stigmasterol on the FoxO3-mediated ubiquitin-proteasome degradation pathway, focusing on the regulation of MuRF1 and MAFbx expression.
2. Materials and Methods
2.1. Chemicals and Reagents
Stigmasterol (Cat. No. S0088) was purchased from Tokyo Chemical Industry (Tokyo, Japan), and dexamethasone (Dexa) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), horse serum (HS), penicillin-streptomycin, and trypsin-EDTA were obtained from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). The following primary antibodies were used: MuRF-1 (Cat# sc-398608; Santa Cruz Biotechnology, Dallas, TX, USA), MAFbx (Cat# sc-166806; Santa Cruz Biotechnology), FoxO3 (Cat# 2497; Cell Signaling Technology, Danvers, MA, USA), and β-actin (Cat# A5441; Sigma-Aldrich, St. Louis, MO, USA). Appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA) were also used. Giemsa and May-Grunwald staining solutions were sourced from Merck (Darmstadt, Germany). All other chemicals and reagents were of analytical grade and obtained from standard commercial suppliers.
2.2. Cell Culture and Treatment
2.2.1. Cell Maintenence and Differentiation
C2C12 mouse myoblast cells (ATCC, Manassas, VA, USA) were cultured in DMEM supplemented with 10% FBS and 1% penicillin-streptomycin at 37°C in a humidified 5% CO₂ atmosphere. For differentiation into myotubes, cells were grown to 80% confluence and then switched to differentiation medium (DMEM with 2% HS) for 5 days, with the medium changed every 48 h.
2.2.2. Dose-Response Study
To assess the effect of stigmasterol on C2C12 myoblast viability, cells were seeded in 96-well plates at a density of 5 × 10³ cells/well and treated with stigmasterol at concentrations of 0, 1, 5, 10, and 20 μM for 24 h. Subsequently, each well was treated following the protocol provided with the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan). Specifically, 10 µL of the CCK-8 reagent was added to each well, and the plate was incubated at 37°C for 1 h in a humidified incubator with 5% CO₂. The absorbance at 450 nm, corresponding to the amount of formazan produced by cellular dehydrogenases, was then measured using a microplate reader (Molecular Devices, San Jose, CA, USA). Data were expressed as a percentage of the untreated control.
2.2.3. Dexamethasone-Induced Atrophy and Stigmasterol Treatment
Differentiated C2C12 myotubes were divided into four groups: (1) Control (CTL), treated with vehicle (0.1% DMSO); (2) Dexa, treated with 50 μM dexamethasone; (3) Stigmasterol (S), treated with 10 μM stigmasterol; and (4) Dexa + Stigmasterol (DS), treated with 50 μM dexamethasone and 10 μM stigmasterol. Treatments were applied for 24 h. The concentration of stigmasterol (10 μM) was selected based on the dose-response study (Figure 1B), which showed mild significant toxicity at this level.
2.2.4. Giemsa and May-Grunwald Staining
Myotubes were fixed with 4% paraformaldehyde for 15 min, stained with Giemsa and May-Grunwald solutions according to the manufacturer’s instructions. Images were acquired from four randomly selected fields of the stained cells using a camera-equipped microscope (Eclipse 80i; Nikon, Tokyo, Japan). Using NIS Elements software (NIS-Elements Advanced Research, Melville, NY, USA), myotubes within each field were randomly selected to measure their diameter.
2.2.4. Fusion Index Quantification
Myotubes were stained with DAPI (4′,6-diamidino-2-phenylindole) to visualize nuclei. The fusion index was calculated as the percentage of nuclei within multinucleated myotubes (containing ≥3 nuclei) relative to the total number of nuclei, as described previously [19]. At least 10 fields per group were analyzed using a fluorescence microscope (Nikon, Tokyo, Japan).
2.3. Western Blot Analysis
C2C12 myotubes and mouse muscle tissues were lysed in RIPA buffer (Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitors (Roche, Basel, Switzerland). Protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of protein (30 μg) were separated by 10% SDS-PAGE and transferred to NC membranes (Millipore, Burlington, MA, USA). Membranes were blocked with 5% skim milk in TBST (Tris-buffered saline with 0.1% Tween-20) for 1 hour at room temperature, then incubated overnight at 4°C with primary antibodies against MuRF1 (1:1000), MAFbx (1:1000), FoxO3 (1:1000), and β-actin (1:5000). After washing with TBST, membranes were incubated with HRP-conjugated secondary antibodies (1:5000) for 1 h at room temperature. Protein bands were visualized using the Clarity Western ECL Substrate (Bio-Rad Laboratories, Inc., Berkeley, CA, USA) and quantified by densitometry using the ChemiDocTM Touch Imaging System (Bio-Rad Laboratories, Inc). Expression levels were normalized to β-actin as a loading control.
2.4. Animal Model and Experimental Design
2.4.1. Animals
Male C57BL/6 mice with an average body weight of 22 g (6 weeks old) were obtained from Core Tech Co., Ltd., (Seoul, Korea) and housed under controlled conditions (22 ± 2°C, 12 h light/dark cycle) with ad libitum access to food and water. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Gyeongsang National University (GNU-230719-M0156-01), in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.4.2. Induction of Muscle Atrophy and Treatment
Mice were randomly divided into three groups (n = 8 per group): (1) Control (CTL), receiving vehicle (saline with 0.1% DMSO); (2) Dexa, receiving 20 mg/kg/day dexamethasone i.p. and 3 mg/kg/day stigmasterol by oral administration. Treatments were administered daily for 21 days. The dose of dexamethasone was selected based on its established ability to induce muscle atrophy in mice [18]. The stigmasterol dose (3 mg/kg/day) was determined based on preliminary studies indicating that this dosage demonstrated efficacy against muscle atrophy indicators while showing no apparent toxicity in the mouse model.
2.4.3. Body Weight and Muscle Mass Measurements
Body weight was recorded weekly throughout the 21-day experimental period. At the end of the experiment, mice were euthanized by CO₂ inhalation, and the gastrocnemius (GA), tibialis anterior (TA), and extensor digitorum longus (EDL) muscles were dissected and weighed immediately to assess muscle mass.
2.4.4. Bone Mineral Density (BMD) Measurement
BMD was measured using a dual-energy X-ray absorptiometry (DXA) scanner (OsteoSys, Seoul, Korea). Mice were anesthetized with 2% isoflurane during the procedure, and BMD was calculated for the whole body excluding the head.
2.4.5. Immunofluorescence and Cross-Sectional Area (CSA) Analysis
GA and TA muscles were harvested, embedded in OCT compound (Sakura Finetek, Torrance, CA, USA), and snap-frozen in liquid nitrogen. Transverse sections (5 μm) were cut using a cryostat (Leica CM1950; Heidelberg, Germany) and stained with wheat germ agglutinin (WGA) conjugated to Alexa Fluor 488 (W11261; Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA) to visualize muscle fiber boundaries. Images were captured using a fluorescence microscope (Nikon Eclipse ni DSRi2; Nikon, Tokyo, Japan) at 20× magnification. Fiber cross-sectional area (CSA) was quantified using MyoVision (University of Kentucky, Lexington, KY, USA) by measuring at least 200 fibers per muscle per mouse [20].
2.5. Statistical Analysis
All data are presented as mean ± standard deviation (SD) from at least three independent experiments (for in vitro studies) and as mean ± standard error of the mean (SEM) from eight mice per group (for in vivo studies). Statistical significance was determined using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple group comparisons. For the body weight time course, two-way ANOVA with repeated measures was used. p < 0.05 was considered statistically significant. Analyses were performed using GraphPad Prism version 9.0 (GraphPad Software, San Diego, CA, USA).
3. Results
3.1. Stigmasterol Exhibits Minimal Toxicity on C2C12 Myoblasts at Low Concentrations
To evaluate the potential cytotoxicity of stigmasterol on C2C12 myoblasts, we performed a dose-response study by treating cells with increasing concentrations of stigmasterol (0, 1, 5, 10, and 20 μM) for 24 h (Figure 1B). At concentrations of 1, 5, and 10 μM, stigmasterol had no significant effect on cell viability, with relative survival rates remaining above 95% compared to the untreated control (0 μM). However, at 20 μM, stigmasterol significantly reduced cell viability to 93.8 ± 1.1% (p < 0.001), and a more pronounced decrease was observed at higher concentrations (data not shown). These results indicate that stigmasterol is well-tolerated by C2C12 myoblasts at concentrations up to 10 μM, which was selected for subsequent experiments to ensure minimal toxicity while maximizing potential therapeutic effects.
3.2. Stigmasterol Mitigates Dexamethasone-Induced Atrophy in C2C12 Myotubes
We next investigated whether stigmasterol could protect C2C12 myotubes from dexamethasone-induced atrophy. Giemsa and May-Grunwald staining revealed significant morphological changes in myotubes treated with 50 μM dexamethasone (Dexa group), including reduced myotube diameter and disrupted structural integrity compared to the control (CTL) group (Figure 2A). Treatment with 10 μM stigmasterol alone (S group) did not alter myotube morphology, while co-treatment with dexamethasone and stigmasterol (DS group) partially restored myotube structure, suggesting a protective effect.
Quantitative analysis of myotube diameter confirmed these observations (Figure 2B). Dexamethasone treatment significantly reduced myotube diameter to 9.4 ± 0.3 μm compared to 15.7 ± 0.6 μm in the control group (**** p < 0.0001). Co-treatment with stigmasterol increased the diameter to 14.1 ± 0.2 μm in the DS group (####p < 0.0001 vs. Dexa), indicating that stigmasterol effectively counteracts dexamethasone-induced atrophy. Stigmasterol alone had no significant effect on myotube diameter (15.0 ± 0.8 μm) compared to the control.
We also assessed the fusion index as a measure of myoblast differentiation and fusion (Figure 2C). Dexamethasone treatment reduced the fusion index to 0.50 ± 0.04 compared to 0.73 ± 0.04 in the control group (*** p < 0.001). Co-treatment with stigmasterol restored the fusion index to 0.62 ± 0.02 in the DS group (## p < 0.01 vs. Dexa), while stigmasterol alone slightly increased the fusion index to 0.69 ± 0.05 (not significant vs. control). These findings suggest that stigmasterol not only prevents dexamethasone-induced atrophy but also promotes myoblast differentiation and fusion.
3.3. Stigmasterol Downregulates the FoxO3-MuRF1-MAFbx Pathway in Dexamethasone-Treated C2C12 Myotubes
To elucidate the molecular mechanism underlying stigmasterol’s protective effects, we examined the expression of atrophy-related proteins in C2C12 myotubes using western blot analysis (Figure 3). Dexamethasone treatment significantly upregulated the expression of MuRF1, MAFbx, and FoxO3 compared to the control group (** p < 0.01 for all). Specifically, MuRF1 expression increased by 1.67-fold, MAFbx by 3.38-fold, and FoxO3 by 2.02-fold in the Dexa group. Treatment with stigmasterol alone had no significant effect on the expression of these proteins compared to the control. However, in the DS group, stigmasterol co-treatment significantly reduced the dexamethasone-induced upregulation of MuRF1 (0.72-fold, # p < 0.05 vs. Dexa), MAFbx (0.68-fold, ## p < 0.01 vs. Dexa), and FoxO3 (0.79-fold, ## p < 0.01 vs. Dexa). These results indicate that stigmasterol mitigates dexamethasone-induced atrophy by suppressing the FoxO3-MuRF1-MAFbx signaling pathway, a key regulator of the ubiquitin-proteasome system.
3.4. Stigmasterol Prevents Dexamethasone-Induced Muscle Atrophy in Mice
We further evaluated stigmasterol’s protective effects in a mouse model of dexamethasone-induced muscle atrophy. Body weight was monitored over 21 days (Figure 4A). The Dexa group exhibited a significant reduction in body weight starting at day 7, reaching 21.2 ± 0.9 g by day 21 compared to 234.8 ± 1.4 g in the control group (**** p < 0.0001). Co-treatment with stigmasterol (DS group) attenuated this weight loss, with a final body weight of 22.5 ± 1.0 g (#### p < 0.0001 vs. Dexa).
Final body weight measurements at day 21 (Figure 4B) confirmed these trends, with the DS group showing a significant improvement over the Dexa group (#p < 0.05). Bone mineral density (BMD) was also assessed (Figure 4C). Dexamethasone treatment reduced BMD to 0.063 ± 0.002 g/cm² compared to 0.064 ± 0.001 g/cm² in the control group (* p < 0.05). Co-treatment with stigmasterol restored BMD to 0.065 ± 0.001 g/cm² in the DS group (## p < 0.01 vs. Dexa).
Muscle mass of the gastrocnemius (GA), tibialis anterior (TA), and extensor digitorum longus (EDL) muscles was measured (Figure 4D). Dexamethasone significantly reduced muscle mass in all three muscles (GA: 0.10 ± 0.01 g, TA: 0.03 ± 0.007 g, EDL: 0.013 ± 0.003 g) compared to the control group (GA: 0.14 ± 0.01 g, TA: 0.05 ± 0.004 g, EDL: 0.021 ± 0.001 g; *** p < 0.001 for EDL, **** p < 0.0001 for GA and TA vs. CTL). Co-treatment with stigmasterol increased muscle mass in the DS group (GA: 0.104 ± 0.002 g, TA: 0.034 ± 0.005 g, EDL: 0.017 ± 0.003 g; # p < 0.05 for all vs. Dexa). These results demonstrate that stigmasterol effectively prevents dexamethasone-induced muscle loss and preserves BMD in vivo.
3.5. Stigmasterol Restores Muscle Fiber Cross-Sectional Area in Dexamethasone-Treated Mice
To further assess stigmasterol’s impact on muscle morphology, we analyzed the cross-sectional area (CSA) of muscle fibers in the GA and TA muscles using immunofluorescence (Figure 5). Dexamethasone treatment significantly reduced fiber CSA in both muscles (GA: 1258 ± 176 μm², TA: 1431 ± 380 μm²) compared to the control group (GA: 1677 ± 197 μm², TA: 1786 ± 286 μm²; *** p < 0.0001 for both). Co-treatment with stigmasterol increased fiber CSA in the DS group (GA: 1607 ± 343 μm², TA: 1781 ± 258 μm²; ## p < 0.0001 vs. Dexa for both). Histogram analysis of fiber CSA distribution confirmed that stigmasterol shifted the distribution toward larger fiber sizes in the DS group compared to the Dexa group. These findings indicate that stigmasterol preserves muscle fiber size in the presence of dexamethasone, further supporting its protective role against muscle atrophy.
3.6. Stigmasterol Reduces Expression of Atrophy-Related Proteins in Mouse Muscle Tissues
Western blot analysis of GA and TA muscle tissues revealed that dexamethasone significantly upregulated the expression of MAFbx and FoxO3 in both muscles compared to the control group (Figure 6). In the GA muscle, MuRF1 expression decreased to 0.85-fold, whereas MAFbx expression increased by 3.82-fold and FoxO3 by 1.26-fold in the Dexa group. Similar trends were observed in the TA muscle (MuRF1: 0.87-fold, MAFbx: 1.42-fold, FoxO3: 1.34-fold). Co-treatment with stigmasterol in the DS group significantly reduced these levels (GA: MuRF1 0.92-fold, MAFbx 0.57-fold, FoxO3 0.55-fold; TA: MuRF1 0.98-fold, MAFbx 0.56-fold, FoxO3 0.74-fold; * p < 0.05, ** p < 0.01, and *** p < 0.001 vs. CTL; # p < 0.05 vs. Dexa). These results confirm that stigmasterol’s protective effects in vivo are mediated by the downregulation of the FoxO3-MAFbx pathway, consistent with our in vitro findings.
4. Discussion
This study demonstrates that Stigmasterol, a naturally occurring phytosterol, exerts significant protective effects against dexamethasone-induced muscle atrophy both in vitro in C2C12 myotubes and in vivo in a mouse model. The key findings indicate that Stigmasterol treatment ameliorates the loss of myotube size and fusion (Figure 2), prevents reductions in body weight and skeletal muscle mass (Figure 4), and preserves muscle fiber cross-sectional area (Figure 5) in the context of dexamethasone exposure. Mechanistically, these protective effects are strongly associated with the downregulation of the critical ubiquitin-proteasome system components, specifically the E3 ubiquitin ligases MuRF1 and MAFbx, and their upstream transcriptional regulator, FoxO3 (Figure 3, Figure 6).
The observed mitigation of dexamethasone-induced atrophy by Stigmasterol highlights its potential to counteract excessive muscle protein breakdown. Glucocorticoids like dexamethasone are well-established inducers of muscle wasting, primarily by enhancing protein catabolism through the activation of the ubiquitin-proteasome system [18]. Central to this process is the activation of Forkhead box O (FOXO) transcription factors, which drive the expression of MuRF1 and MAFbx [6,8,9]. The FOXO signaling pathway is recognized as playing a crucial role in the pathogenesis of skeletal muscle atrophy [21]. Our in vitro results showed that dexamethasone treatment robustly increased FoxO3, MuRF1, and MAFbx expression in C2C12 cells (Figure 3), confirming the activation of this catabolic pathway. In vivo, dexamethasone also significantly upregulated FoxO3 and MAFbx protein levels in mouse skeletal muscle (Figure 6). However, under our experimental conditions, dexamethasone treatment did not result in a significant increase in MuRF1 protein expression in vivo (Figure 6). This discrepancy might reflect the complexities of the in vivo environment, potential time-dependent regulation, or differential sensitivity of MuRF1 versus MAFbx regulation in this specific model. Indeed, studies using knockout mice have shown functional divergence between MuRF1 and MAFbx in response to glucocorticoids in vivo, suggesting their roles are not entirely redundant in this context [22]. Furthermore, the regulation of these atrogenes can be influenced by other factors such as genetic background, potentially leading to varied responses [23]. Crucially, stigmasterol co-treatment significantly attenuated the dexamethasone-induced upregulation of FoxO3 and MAFbx in vivo (Figure 6). While MuRF1 protein expression was suppressed by stigmasterol in vitro (Figure 3), its expression was not significantly increased by dexamethasone treatment in vivo under our experimental conditions (Figure 6), differing from the in vitro findings. This highlights MAFbx as a key downstream target modulated by stigmasterol in vivo in this model. The divergence between MuRF1 protein levels in vitro and in vivo could stem from the complex systemic environment, differing time-course responses, or specific regulatory mechanisms prevailing in vivo under 21-day dexamethasone exposure. Although functional studies like Baehr et al. demonstrated the necessity of MuRF1 for the full atrophic response using knockout mice [22], our protein expression data at this time point most strongly link stigmasterol’s protective effect in vivo to the attenuation of the FoxO3-MAFbx axis. Therefore, our findings provide a direct molecular mechanism for stigmasterol’s muscle-protective effects – primarily involving the suppression of the FoxO3-MAFbx signaling axis in vivo, as depicted in our proposed mechanism (Figure 7). The concordance between our in vitro findings for FoxO3 and MAFbx (Figure 3) and the corresponding in vivo findings (Figure 6) strengthens the validity of this aspect of the conclusion.
Stigmasterol belongs to the phytosterol family, plant-derived compounds known for various bioactivities [11,24]. It shares structural similarity with β-sitosterol [24,25]. Interestingly, a recent study demonstrated that β-sitosterol also attenuates dexamethasone-induced muscle atrophy in vitro and in vivo, with evidence suggesting involvement of FoxO signaling regulation [15]. The current study highlights the modulation of FoxO3 by stigmasterol in mitigating dexamethasone effects. While both structurally similar phytosterols appear protective and converge on modulating FoxO transcription factors, further investigation is warranted to fully elucidate the specificities and potential overlap in their regulation of different FoxO isoforms (including FoxO1 and FoxO3) in the context of muscle atrophy. Beyond direct modulation of atrophy signaling, Stigmasterol possesses known anti-inflammatory and antioxidant properties [13,26]. Chronic inflammation and oxidative stress contribute significantly to muscle wasting pathologies, including glucocorticoid-induced myopathy. Stigmasterol’s anti-inflammatory effects, potentially mediated via NF-κB inhibition [13], and its antioxidant actions could create a more favorable environment for muscle preservation, complementing its effects on the FoxO pathway [11].
The precise mechanism by which Stigmasterol inhibits FoxO3 activation or expression requires further elucidation. Glucocorticoids can suppress the pro-survival Akt signaling pathway, which normally phosphorylates and inhibits FoxO transcription factors [9,27]. It is plausible that Stigmasterol may counteract Dexa’s effect on Akt or act downstream or parallel to it to reduce FoxO3 activity. Our study focused on the key catabolic regulators identified; however, exploring the effects of Stigmasterol on upstream kinases like Akt or other relevant pathways (e.g., AMPK, protein synthesis pathways like mTOR) would provide a more comprehensive mechanistic picture.
This study possesses several strengths, including the consistent demonstration of Stigmasterol’s efficacy in both cell culture (Figures 2, 3) and animal models (Figure 4, Figure 5 and Figure 6), coupled with the investigation of a specific, relevant molecular mechanism (Figures 3, 6, 7). However, certain limitations must be acknowledged. The findings are currently limited to the dexamethasone-induced atrophy model, a specific form of toxic myopathy [12]; its applicability to other prevalent conditions like age-related sarcopenia or disuse atrophy remains to be determined. Furthermore, while muscle mass and fiber size were preserved (Figure 4, Figure 5), direct assessments of muscle function (e.g., strength, endurance) were not included. Evaluating functional outcomes is critical for establishing physiological relevance. Additionally, our mechanistic focus was on protein catabolism via the FoxO3 axis; potential effects on protein synthesis pathways were not investigated. Finally, further studies are needed to determine the optimal dosage, administration route, and long-term bioavailability and safety of Stigmasterol for muscle protection in vivo. Moreover, the in vivo study did not include a group treated with stigmasterol alone; while preliminary studies suggested minimal independent effects of stigmasterol at the tested dose, the inclusion of this group would have allowed for a direct assessment of its baseline effects.
Despite these limitations, the results carry potentially significant implications. Glucocorticoid-induced myopathy remains a challenging clinical problem [28]. Our findings identify Stigmasterol as a potential therapeutic candidate derived from natural sources that could be explored to mitigate this adverse effect. Given the increasing interest in nutritional interventions for muscle health, particularly concerning sarcopenia which affects functionality and quality of life in the elderly [29], Stigmasterol could also hold promise as a nutraceutical ingredient, although specific studies in aging models are needed.
Future research should focus on addressing the current study’s limitations. Testing Stigmasterol’s efficacy in models of sarcopenia and disuse atrophy, incorporating comprehensive muscle function tests, is paramount. Mechanistic studies should expand to include upstream signaling pathways (e.g., Akt/mTOR) regulating both protein synthesis and degradation. Investigating potential synergistic effects with exercise or other nutritional compounds could also yield valuable insights. Finally, detailed pharmacokinetic, dose-optimization, and long-term safety studies are essential before considering any potential clinical translation.
5. Conclusions
This study provides compelling evidence that Stigmasterol effectively ameliorates dexamethasone-induced muscle atrophy in vitro and in vivo. This protection is mediated, at least significantly, through the downregulation of the FoxO3-MuRF1/MAFbx catabolic signaling pathway. These findings highlight Stigmasterol as a promising natural compound for combating muscle wasting, particularly associated with glucocorticoid use, and strongly warrant further investigation into its broader therapeutic and preventative potential for maintaining muscle health.
Author Contributions
Conceptualization, S.-J.K. and Y.-S.H.; methodology, Y.-H.J.; software, S.-J.L.; validation, S.-J.L. and J.H.; investigation, S.-J.K. and Y.-H.J.; resources, J.H. and J.-I.Y.; data curation, Y.-H.J. and S.-J.L.; writing—original draft preparation, S.-J.K. and Y.-S.H.; writing—review and editing, Y-T.J., H-G.K., S.-J.L. and J.-I.Y.; supervision, Y.-H.J.; project administration, J.-I.Y.; funding acquisition, S.-J.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (RS-2022-NR073471).
Institutional Review Board Statement
All animal experiments were performed after obtaining approval from the Institutional Animal Care and Use Committee (IACUC) of Gyeongsang National University (GNU-230719-M0156-01).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cruz-Jentoft, A.J. , et al., Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing, 2010. 39(4): p. 412-23.
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Choo, Y.J.; Chang, M.C. Prevalence of Sarcopenia Among the Elderly in Korea: A Meta-Analysis. J. Prev. Med. Public Heal. 2021, 54, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Beaudart, C.; Zaaria, M.; Pasleau, F.; Reginster, J.-Y.; Bruyère, O. Health Outcomes of Sarcopenia: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0169548. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat. Cell Biol. 2003, 5, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.-M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. 2001, 98, 14440–14445. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt Pathway Prevents Expression of Muscle Atrophy-Induced Ubiquitin Ligases by Inhibiting FOXO Transcription Factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Bakrim, S.; Benkhaira, N.; Bourais, I.; Benali, T.; Lee, L.-H.; El Omari, N.; Sheikh, R.A.; Goh, K.W.; Ming, L.C.; Bouyahya, A. Health Benefits and Pharmacological Properties of Stigmasterol. Antioxidants 2022, 11, 1912. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, P.; Fang, H.; Zou, M.; Yin, J.; Zhong, W.; Luo, Z.; Hu, C.; He, D.; Wang, X. High-oleic rapeseed oil quality indicators and endogenous antioxidant substances under different processing methods. Food Chem. X 2023, 19, 100804. [Google Scholar] [CrossRef] [PubMed]
- Gabay, O.; Sanchez, C.; Salvat, C.; Chevy, F.; Breton, M.; Nourissat, G.; Wolf, C.; Jacques, C.; Berenbaum, F. Stigmasterol: a phytosterol with potential anti-osteoarthritic properties. Osteoarthr. Cartil. 2010, 18, 106–116. [Google Scholar] [CrossRef]
- Panda, S.; Jafri, M.; Kar, A.; Meheta, B. Thyroid inhibitory, antiperoxidative and hypoglycemic effects of stigmasterol isolated from Butea monosperma. Fitoterapia 2009, 80, 123–126. [Google Scholar] [CrossRef]
- Hah, Y.-S.; Lee, W.K.; Lee, S.; Kim, E.J.; Lee, J.H.; Lee, S.-J.; Ji, Y.H.; Kim, S.G.; Lee, H.-H.; Hong, S.Y.; et al. β-Sitosterol Attenuates Dexamethasone-Induced Muscle Atrophy via Regulating FoxO1-Dependent Signaling in C2C12 Cell and Mice Model. Nutrients 2022, 14, 2894. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Dai, Z.; Liu, A.B.; Huang, J.; Narsipur, N.; Guo, G.; Kong, B.; Reuhl, K.; Lu, W.; Luo, Z.; et al. Intake of stigmasterol and β-sitosterol alters lipid metabolism and alleviates NAFLD in mice fed a high-fat western-style diet. Biochim. et Biophys. Acta (BBA) - Mol. Cell Biol. Lipids 2018, 1863, 1274–1284. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, Y.; Jiang, J.; Cui, X.; Cheng, S.; Liu, L.; Huang, Z.; Liao, R.; Zhao, P.; Yu, J.; et al. Stigmasterol attenuates hepatic steatosis in rats by strengthening the intestinal barrier and improving bile acid metabolism. npj Sci. Food 2022, 6, 1–14. [Google Scholar] [CrossRef]
- Schakman, O.; Kalista, S.; Barbé, C.; Loumaye, A.; Thissen, J. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef] [PubMed]
- Cadot, B.; Gache, V.; Vasyutina, E.; Falcone, S.; Birchmeier, C.; Gomes, E.R. Nuclear movement during myotube formation is microtubule and dynein dependent and is regulated by Cdc42, Par6 and Par3. Embo Rep. 2012, 13, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Murach, K.A.; Vechetti, I.J., Jr.; Fry, C.S.; Vickery, C.; Peterson, C.A.; McCarthy, J.J.; Campbell, K.S. MyoVision: software for automated high-content analysis of skeletal muscle immunohistochemistry. J. Appl. Physiol. 2018, 124, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Gao, P.; Li, Z.; Dai, A.; Yang, M.; Chen, S.; Su, J.; Deng, Z.; Li, L. Forkhead Box O Signaling Pathway in Skeletal Muscle Atrophy. Am. J. Pathol. 2022, 192, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar] [CrossRef] [PubMed]
- Seto, J.T.; Roeszler, K.N.; Meehan, L.R.; Wood, H.D.; Tiong, C.; Bek, L.; Lee, S.F.; Shah, M.; Quinlan, K.G.R.; Gregorevic, P.; et al. ACTN3 genotype influences skeletal muscle mass regulation and response to dexamethasone. Sci. Adv. 2021, 7, eabg0088. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Quispe, C.; Sharifi-Rad, J.; Cruz-Martins, N.; Nigam, M.; Mishra, A.P.; Konovalov, D.A.; Orobinskaya, V.; Abu-Reidah, I.M.; Zam, W.; et al. Phytosterols: From Preclinical Evidence to Potential Clinical Applications. Front. Pharmacol. 2021, 11, 599959. [Google Scholar] [CrossRef] [PubMed]
- Valitova, J.; Renkova, A.; Beckett, R.; Minibayeva, F. Stigmasterol: An Enigmatic Plant Stress Sterol with Versatile Functions. Int. J. Mol. Sci. 2024, 25, 8122. [Google Scholar] [CrossRef]
- Zhao, X.; Li, F.; Wen, A.; Yu, X.; Xu, X.; Wan, C.; Cao, Y.; Xin, G.; Huang, W. Elucidating the mechanism of stigmasterol in acute pancreatitis treatment: insights from network pharmacology and in vitro/in vivo experiments. Front. Pharmacol. 2024, 15, 1485915. [Google Scholar] [CrossRef]
- Qin, W.; Pan, J.; Wu, Y.; Bauman, W.A.; Cardozo, C. Protection against dexamethasone-induced muscle atrophy is related to modulation by testosterone of FOXO1 and PGC-1α. Biochem. Biophys. Res. Commun. 2010, 403, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Liu, C.; Sun, D. Glucocorticoid-Induced Myopathy: Typology, Pathogenesis, Diagnosis, and Treatment. Horm. Metab. Res. 2024, 56, 341–349. [Google Scholar] [CrossRef]
- Dominguez, L.J.; Veronese, N.; Smith, L.; Ragusa, F.S.; Schirò, P.; Di Bella, G.; Barbagallo, M. Associations Between Adherence to the Mediterranean Diet and Incident Sarcopenia in Prospective Cohort Studies. Nutrients 2025, 17, 313. [Google Scholar] [CrossRef]
Figure 1.
Chemical structure of stigmasterol and its effect on C2C12 myoblast viability. (A) Chemical structure of stigmasterol, a plant-derived sterol used in this study. (B) Dose-response effect of stigmasterol on C2C12 myoblast viability. Cells were treated with stigmasterol at concentrations of 0, 1, 5, 10, and 20 μM for 24 h, and cell viability was assessed using the CCK-8 assay. Data are presented as mean ± standard deviation (SD) from three independent experiments (n = 4). Statistical significance compared to the control (0 μM) is indicated as follows: *** p < 0.001 (one-way ANOVA with Tukey’s post hoc test).
Figure 1.
Chemical structure of stigmasterol and its effect on C2C12 myoblast viability. (A) Chemical structure of stigmasterol, a plant-derived sterol used in this study. (B) Dose-response effect of stigmasterol on C2C12 myoblast viability. Cells were treated with stigmasterol at concentrations of 0, 1, 5, 10, and 20 μM for 24 h, and cell viability was assessed using the CCK-8 assay. Data are presented as mean ± standard deviation (SD) from three independent experiments (n = 4). Statistical significance compared to the control (0 μM) is indicated as follows: *** p < 0.001 (one-way ANOVA with Tukey’s post hoc test).
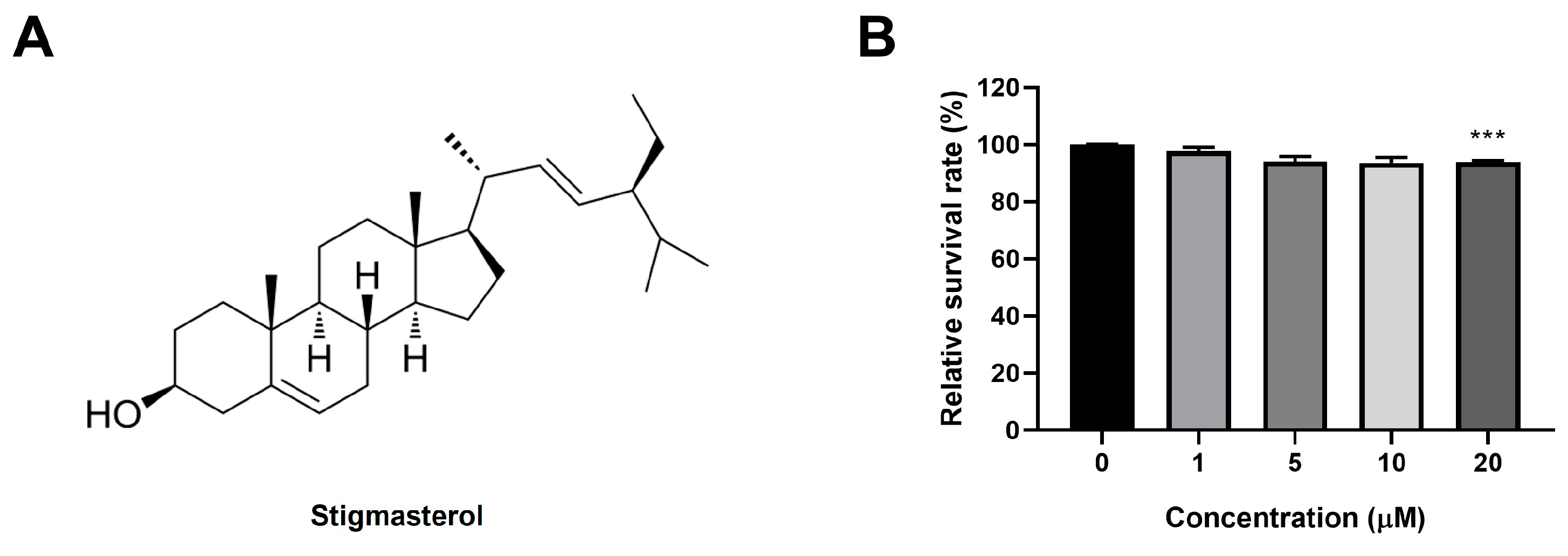
Figure 2.
Stigmasterol attenuates dexamethasone-induced atrophy in C2C12 myotubes. (A) Representative images of C2C12 myotubes stained with Giemsa and May-Grunwald to assess morphological changes. Myotubes were treated for 24 h under the following conditions: control (CTL, vehicle), dexamethasone (Dexa, 50 μM), stigmasterol (S, 10 μM), or a combination of dexamethasone and stigmasterol (DS, 50 μM Dexa + 10 μM S). Scale bar: 250 μm. (B) Quantification of myotube diameter in the four treatment groups. (C) Fusion index, calculated as the percentage of nuclei in multinucleated myotubes (≥3 nuclei) relative to the total number of nuclei, reflecting myoblast differentiation and fusion. Data in (B) and (C) are presented as mean ± SD from three independent experiments (n = 4). Statistical significance is indicated as follows: **** p < 0.0001 vs. CTL; ## p < 0.01, #### p < 0.0001 vs. Dexa (one-way ANOVA with Tukey’s post hoc test). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol.
Figure 2.
Stigmasterol attenuates dexamethasone-induced atrophy in C2C12 myotubes. (A) Representative images of C2C12 myotubes stained with Giemsa and May-Grunwald to assess morphological changes. Myotubes were treated for 24 h under the following conditions: control (CTL, vehicle), dexamethasone (Dexa, 50 μM), stigmasterol (S, 10 μM), or a combination of dexamethasone and stigmasterol (DS, 50 μM Dexa + 10 μM S). Scale bar: 250 μm. (B) Quantification of myotube diameter in the four treatment groups. (C) Fusion index, calculated as the percentage of nuclei in multinucleated myotubes (≥3 nuclei) relative to the total number of nuclei, reflecting myoblast differentiation and fusion. Data in (B) and (C) are presented as mean ± SD from three independent experiments (n = 4). Statistical significance is indicated as follows: **** p < 0.0001 vs. CTL; ## p < 0.01, #### p < 0.0001 vs. Dexa (one-way ANOVA with Tukey’s post hoc test). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol.
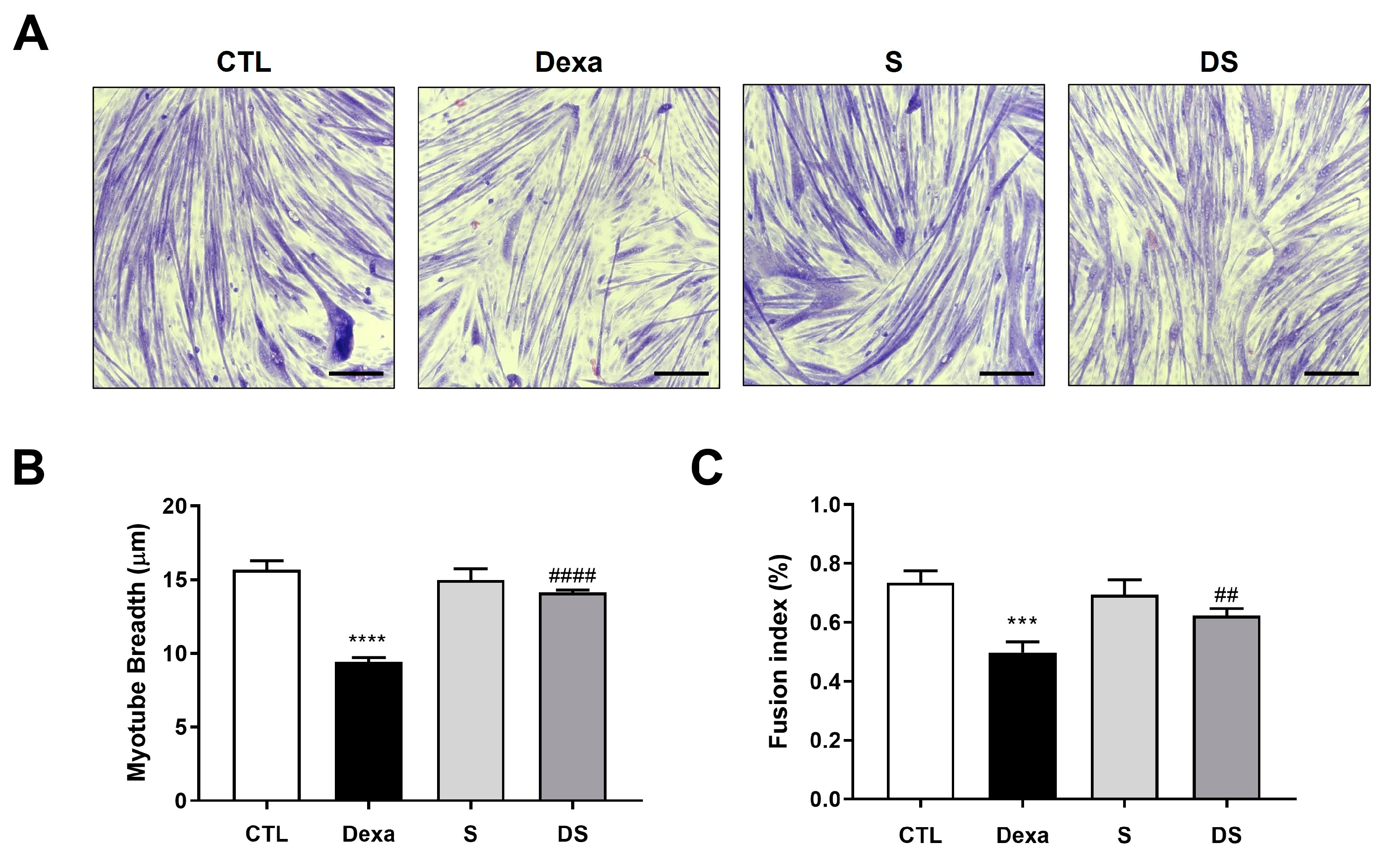
Figure 3.
Stigmasterol suppresses the FoxO3-MuRF1-MAFbx signaling pathway in dexamethasone-treated C2C12 myotubes. (Upper panel) Western blot analysis of MuRF1, MAFbx, and FoxO3 protein expression in C2C12 myotubes treated for 24 h under the following conditions: control (CTL, vehicle), dexamethasone (Dexa, 50 μM), stigmasterol (S, 10 μM), or a combination of dexamethasone and stigmasterol (DS, 50 μM Dexa + 10 μM S). β-actin was used as a loading control. (Lower panel) Densitometric quantification of MuRF1, MAFbx, and FoxO3 expression levels, normalized to β-actin. Data are presented as mean ± SD from three independent experiments. Statistical significance is indicated as follows: ** p < 0.01 vs. CTL; # p < 0.05, ## p < 0.01 vs. Dexa (one-way ANOVA with Tukey’s post hoc test). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; IB, immunoblot.
Figure 3.
Stigmasterol suppresses the FoxO3-MuRF1-MAFbx signaling pathway in dexamethasone-treated C2C12 myotubes. (Upper panel) Western blot analysis of MuRF1, MAFbx, and FoxO3 protein expression in C2C12 myotubes treated for 24 h under the following conditions: control (CTL, vehicle), dexamethasone (Dexa, 50 μM), stigmasterol (S, 10 μM), or a combination of dexamethasone and stigmasterol (DS, 50 μM Dexa + 10 μM S). β-actin was used as a loading control. (Lower panel) Densitometric quantification of MuRF1, MAFbx, and FoxO3 expression levels, normalized to β-actin. Data are presented as mean ± SD from three independent experiments. Statistical significance is indicated as follows: ** p < 0.01 vs. CTL; # p < 0.05, ## p < 0.01 vs. Dexa (one-way ANOVA with Tukey’s post hoc test). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; IB, immunoblot.
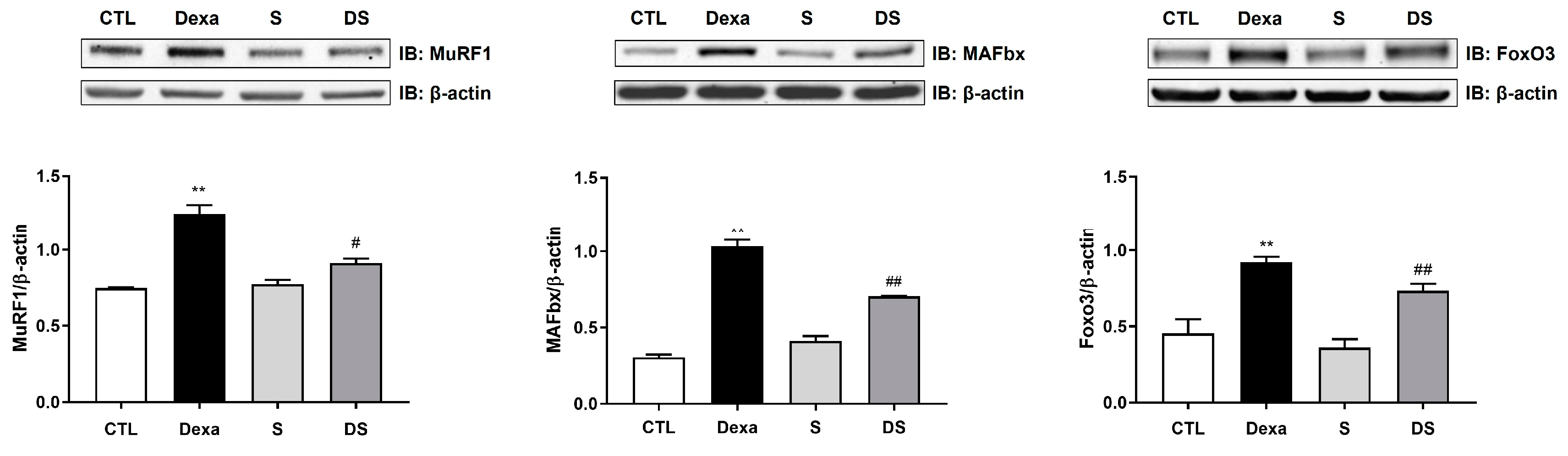
Figure 4.
Stigmasterol ameliorates dexamethasone-induced muscle atrophy in mice. (A) Body weight changes over 21 days in mice treated with vehicle (CTL), dexamethasone (Dexa, 20 mg/kg/day), stigmasterol (S, 3 mg/kg/day), or a combination of dexamethasone and stigmasterol (DS, 20 mg/kg/day Dexa + 3 mg/kg/day S). (B) Final body weight at day 21. (C) Bone mineral density (BMD) measured by dual-energy X-ray absorptiometry (DXA). (D) Muscle mass of the gastrocnemius (GA), tibialis anterior (TA), and extensor digitorum longus (EDL) muscles at day 21. Data in (A) to (D) are presented as mean ± standard error of the mean (SEM) (n = 8 mice per group). Statistical significance is indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001 vs. CTL; # p < 0.05, ## p < 0.01 vs. Dexa (two-way ANOVA with repeated measures for A; one-way ANOVA with Tukey’s post hoc test for B–D). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; GA, gastrocnemius; TA, tibialis anterior; EDL, extensor digitorum longus.
Figure 4.
Stigmasterol ameliorates dexamethasone-induced muscle atrophy in mice. (A) Body weight changes over 21 days in mice treated with vehicle (CTL), dexamethasone (Dexa, 20 mg/kg/day), stigmasterol (S, 3 mg/kg/day), or a combination of dexamethasone and stigmasterol (DS, 20 mg/kg/day Dexa + 3 mg/kg/day S). (B) Final body weight at day 21. (C) Bone mineral density (BMD) measured by dual-energy X-ray absorptiometry (DXA). (D) Muscle mass of the gastrocnemius (GA), tibialis anterior (TA), and extensor digitorum longus (EDL) muscles at day 21. Data in (A) to (D) are presented as mean ± standard error of the mean (SEM) (n = 8 mice per group). Statistical significance is indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001 vs. CTL; # p < 0.05, ## p < 0.01 vs. Dexa (two-way ANOVA with repeated measures for A; one-way ANOVA with Tukey’s post hoc test for B–D). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; GA, gastrocnemius; TA, tibialis anterior; EDL, extensor digitorum longus.
Figure 5.
Stigmasterol preserves muscle fiber cross-sectional area in dexamethasone-treated mice. (A) Representative immunofluorescence images of gastrocnemius (GA) and tibialis anterior (TA) muscle cross-sections stained with wheat germ agglutinin (WGA) to visualize fiber boundaries. Mice were treated for 21 days with vehicle (CTL), dexamethasone (Dexa, 20 mg/kg/day), stigmasterol (S, 3 mg/kg/day), or a combination of dexamethasone and stigmasterol (DS, 20 mg/kg/day Dexa + 3 mg/kg/day S). Scale bar: 50 μm. (B) Histogram distribution of fiber cross-sectional area (CSA) in GA and TA muscles, showing the percentage of fibers within specified CSA ranges. Data are presented as mean ± SEM (n = 8 mice per group). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; GA, gastrocnemius; TA, tibialis anterior.
Figure 5.
Stigmasterol preserves muscle fiber cross-sectional area in dexamethasone-treated mice. (A) Representative immunofluorescence images of gastrocnemius (GA) and tibialis anterior (TA) muscle cross-sections stained with wheat germ agglutinin (WGA) to visualize fiber boundaries. Mice were treated for 21 days with vehicle (CTL), dexamethasone (Dexa, 20 mg/kg/day), stigmasterol (S, 3 mg/kg/day), or a combination of dexamethasone and stigmasterol (DS, 20 mg/kg/day Dexa + 3 mg/kg/day S). Scale bar: 50 μm. (B) Histogram distribution of fiber cross-sectional area (CSA) in GA and TA muscles, showing the percentage of fibers within specified CSA ranges. Data are presented as mean ± SEM (n = 8 mice per group). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; GA, gastrocnemius; TA, tibialis anterior.
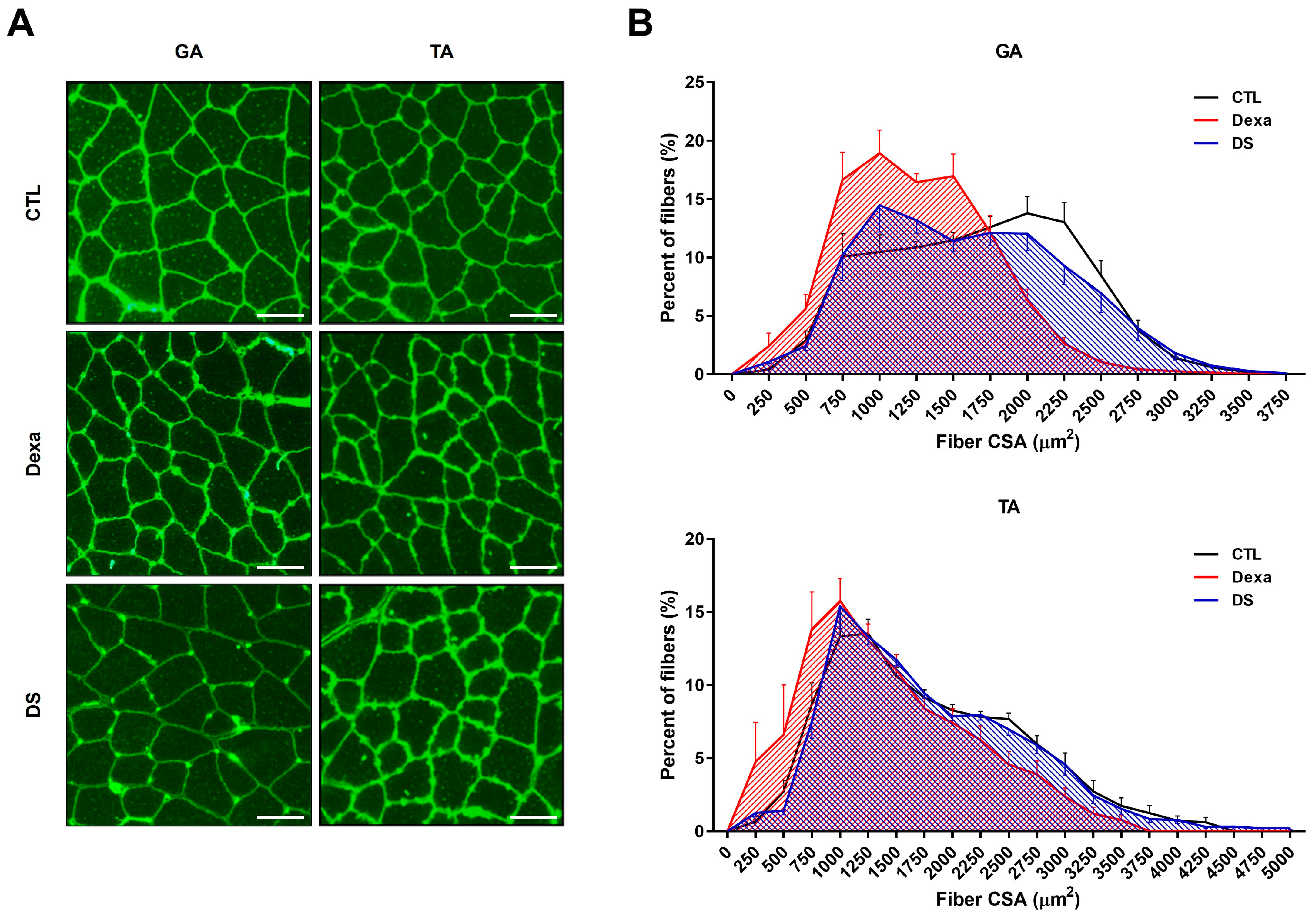
Figure 6.
Stigmasterol reduces expression of atrophy-related proteins in mouse muscle tissues. (A) Western blot analysis of MuRF1, MAFbx, and FoxO3 protein expression in gastrocnemius (GA) and tibialis anterior (TA) muscles from mice treated for 21 days with vehicle (CTL), dexamethasone (Dexa, 20 mg/kg/day), stigmasterol (S, 3 mg/kg/day), or a combination of dexamethasone and stigmasterol (DS, 20 mg/kg/day Dexa + 3 mg/kg/day S). GAPDH was used as a loading control. (B) Densitometric quantification of MuRF1, MAFbx, and FoxO3 expression levels, normalized to GAPDH. Data are presented as mean ± SEM (n = 8 mice per group). Statistical significance is indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001 vs. CTL; # p < 0.05 vs. Dexa (one-way ANOVA with Tukey’s post hoc test). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; GA, gastrocnemius; TA, tibialis anterior; IB, immunoblot.
Figure 6.
Stigmasterol reduces expression of atrophy-related proteins in mouse muscle tissues. (A) Western blot analysis of MuRF1, MAFbx, and FoxO3 protein expression in gastrocnemius (GA) and tibialis anterior (TA) muscles from mice treated for 21 days with vehicle (CTL), dexamethasone (Dexa, 20 mg/kg/day), stigmasterol (S, 3 mg/kg/day), or a combination of dexamethasone and stigmasterol (DS, 20 mg/kg/day Dexa + 3 mg/kg/day S). GAPDH was used as a loading control. (B) Densitometric quantification of MuRF1, MAFbx, and FoxO3 expression levels, normalized to GAPDH. Data are presented as mean ± SEM (n = 8 mice per group). Statistical significance is indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001 vs. CTL; # p < 0.05 vs. Dexa (one-way ANOVA with Tukey’s post hoc test). CTL, control; Dexa, dexamethasone; S, stigmasterol; DS, Dexa + stigmasterol; GA, gastrocnemius; TA, tibialis anterior; IB, immunoblot.
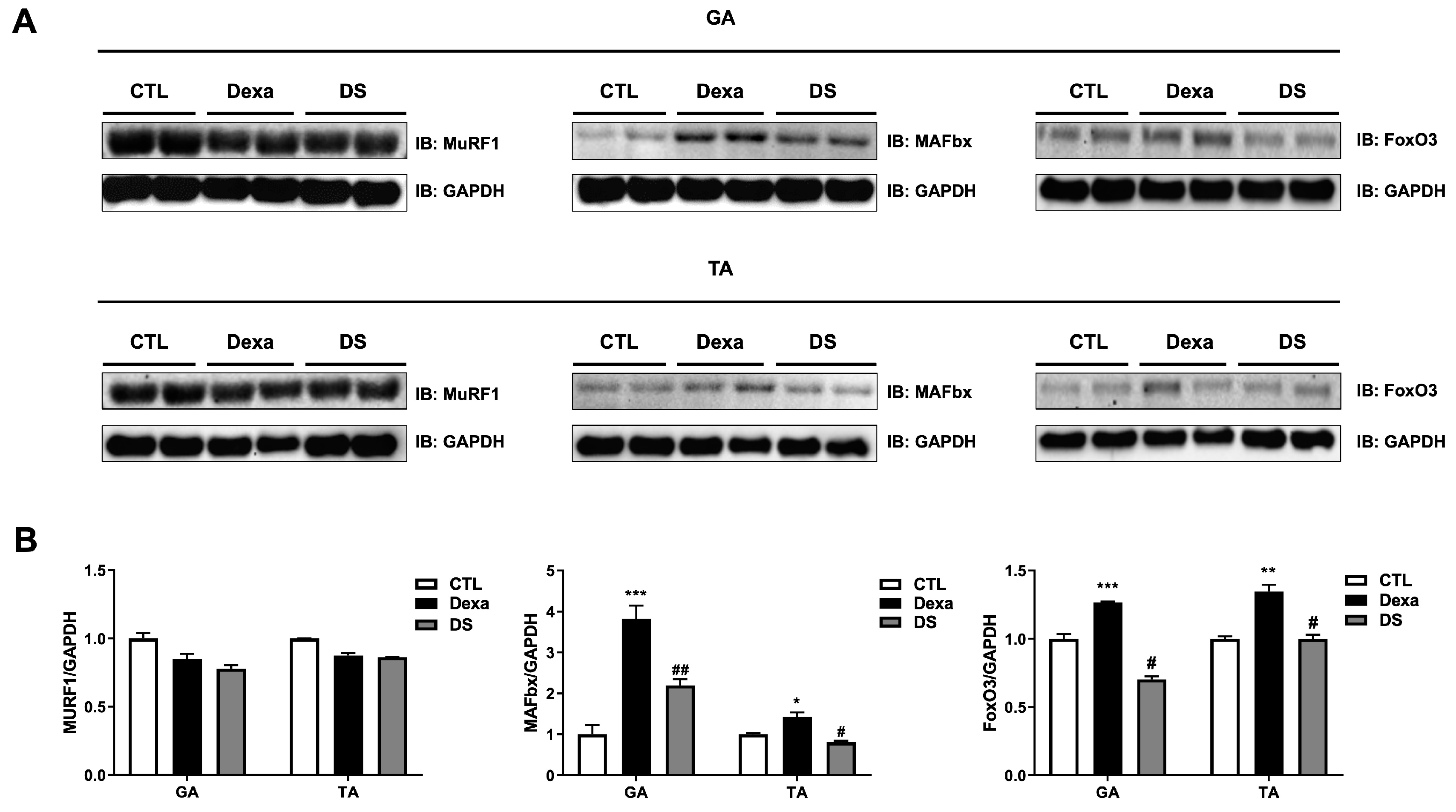
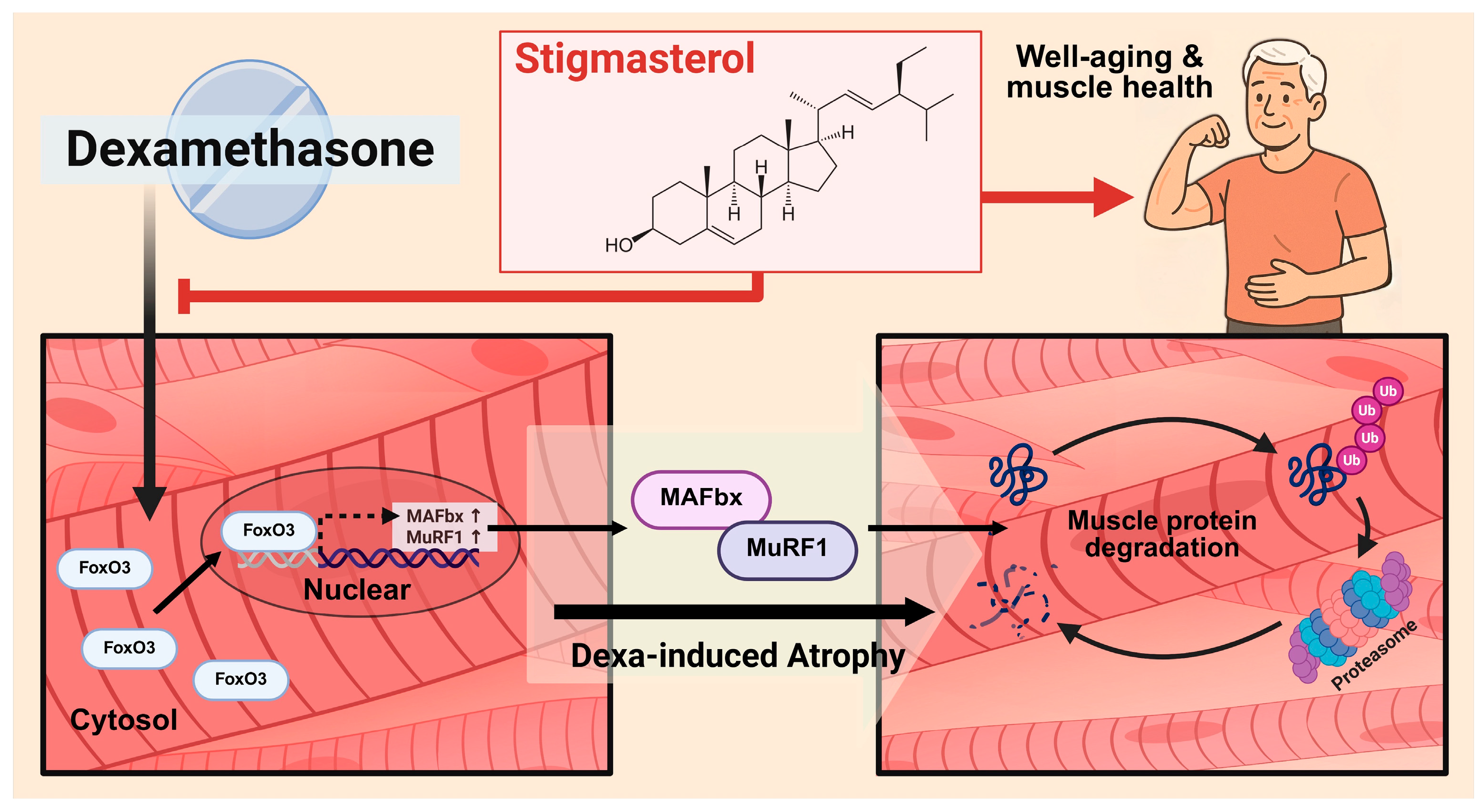
Figure 7.
Proposed mechanism for the protective effect of Stigmasterol against dexamethasone-induced muscle atrophy. Schematic diagram illustrating the hypothesized mechanism. Stigmasterol is proposed to inhibit the activation or nuclear translocation of the transcription factor FoxO3 induced by dexamethasone. This leads to reduced transcription and expression of the E3 ubiquitin ligase MAFbx, a key component of the ubiquitin-proteasome system consistently downregulated by stigmasterol in vivo in this study. While MuRF1 was also downregulated in vitro, its in vivo protein expression was not significantly increased by dexamethasone or reduced by stigmasterol under these conditions. Consequently, the downregulation of the FoxO3-MAFbx axis contributes significantly to decreased muscle protein degradation, thereby ameliorating muscle atrophy induced by dexamethasone.
Figure 7.
Proposed mechanism for the protective effect of Stigmasterol against dexamethasone-induced muscle atrophy. Schematic diagram illustrating the hypothesized mechanism. Stigmasterol is proposed to inhibit the activation or nuclear translocation of the transcription factor FoxO3 induced by dexamethasone. This leads to reduced transcription and expression of the E3 ubiquitin ligase MAFbx, a key component of the ubiquitin-proteasome system consistently downregulated by stigmasterol in vivo in this study. While MuRF1 was also downregulated in vitro, its in vivo protein expression was not significantly increased by dexamethasone or reduced by stigmasterol under these conditions. Consequently, the downregulation of the FoxO3-MAFbx axis contributes significantly to decreased muscle protein degradation, thereby ameliorating muscle atrophy induced by dexamethasone.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



