Submitted:
13 May 2025
Posted:
19 May 2025
You are already at the latest version
Abstract
Chronic stress is a significant public health concern, with occupational stress being a predominant global source. It is increasingly recognized for its role in metabolic disorders, including obesity, insulin resistance, and hyperuricemia, through complex neuroendocrine, inflammatory, and behavioral mechanisms. This review explores the interplay between chronic stress and metabolic dysfunction, focusing on the dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis and sympathetic nervous system (SNS), and how stress-induced changes contribute to metabolic syndrome and hyperuricemia. Prolonged cortisol and catecholamine release contributes to insulin resistance, visceral adiposity, and systemic inflammation, while stress-induced behavioral changes, such as poor diet and physical inactivity, exacerbate metabolic disturbances. Additionally, emerging evidence highlights the roles of oxidative stress, mitochondrial dysfunction, and caveolae impairment in stress-related metabolic diseases. The bidirectional relationship between stress and metabolic disorders further complicates disease progression, as metabolic dysfunction itself amplifies stress responses. Future research should prioritize biomarker discovery, epigenetic influences, and personalized interventions, including both pharmacological and lifestyle-based strategies. Public health policies and workplace interventions are also essential to mitigate stress-induced metabolic risks. This review underscores the need for a multidisciplinary approach to address the growing burden of stress-related metabolic diseases.
Keywords:
Chronic stress
; Metabolic syndrome
; HPA axis dysregulation
; Hyperuricemia
; Caveolae
1. Introduction
The detrimental effects of stress on both emotional and physical health [1] are well-documented and increasingly recognized as significant public health concerns. Occupational stress, in particular, represents the predominant source of chronic stress globally and has steadily intensified over recent decades [2]. Elevated levels of job-related stress, characterized by high demands coupled with a perceived lack of control, have been consistently associated with heightened risks of cardiovascular events (e.g., myocardial infarction, hypertension), metabolic disorders (e.g., obesity), substance-use-disorders, anxiety, depression, and other mental and physical health conditions [3]. According to Gallup's State of the Global Workplace 2023 report, 41% of global and 52% of U.S. or East Asian employees reported experiencing significant stress on the preceding day, underscoring the widespread prevalence of this issue (https://www.gallup.com/workplace/349484/state-of-the-global-workplace.aspx).
Among the various health consequences, stress-induced metabolic disorders have emerged as a critical area of concern. These disorders result from the interplay of physiological, behavioral, and molecular mechanisms. Stress is fundamentally a state of threatened homeostasis that elicits adaptive physiological responses, centrally regulated by the brain. The neuroendocrine systems of the hypothalamic-pituitary-adrenal (HPA) axis [4] and the sympathetic nervous system (SNS) play a central role in mediating these responses [5]. The key stress hormones cortisol and the catecholamines (e.g., adrenaline and noradrenaline) are essential for short-term adaptation to stress. However, chronic activation of these systems can override their protective functions. Sustained elevations in cortisol and catecholamines contribute to systemic dysregulation and are implicated in the pathogenesis of a range of health conditions, including metabolic syndrome [6], obesity [7], cancer [8], mental health disorders [9], hyperuricemia [10], cardiovascular disease [11], and increased susceptibility to infections [12].
Stress rapidly reprograms hepatic energy metabolism, with effects that persist beyond the period of exposure [13]. The HPA axis responds almost immediately to stress, triggering a cascade of physiological responses [14]. Chronic psychosocial stress is associated with disrupted sleep and impaired metabolic health and may contribute to the increasing global prevalence of subclinical hypothyroidism [14]. Prolonged stress, such as war-related stress, can disrupt systemic homeostasis, affecting metabolic processes, neuroendocrine regulation, and the function of the cardiovascular and respiratory systems [15]. In experimental models, chronic restraint stress alters hepatic metabolomic profiles, particularly the betaine metabolism pathway, and modulates critical metabolic signaling pathways, including INSR/PI3K/AKT/AMPK [16]. It also impacts the gut microbiome, altering its diversity, composition, and metabolic output [17], thereby influencing host systemic metabolism.
Stress-induced metabolic disorders result from a complex interplay of hormonal dysregulation, chronic inflammation, behavioral changes, molecular disturbances, and structural alterations on the cellular level, such as those affecting caveolae. These interconnected mechanisms create a self-perpetuating cycle that progressively impairs metabolic health. Understanding the biological pathways that link chronic stress to metabolic dysfunction is therefore essential.
Given the widespread prevalence of both chronic stress and metabolic diseases in modern society, their potential pathophysiological interaction represents a significant public health concern. Effective prevention and management require a comprehensive approach that includes stress reduction, nutritional interventions, and regular physical activity.
This review aims to examine the key mechanisms underlying the relationship between chronic stress and metabolic disorders, with a particular focus on metabolic syndrome and hyperuricemia.
2. Mechanisms Underlying Stress-Induced Metabolic Disorders
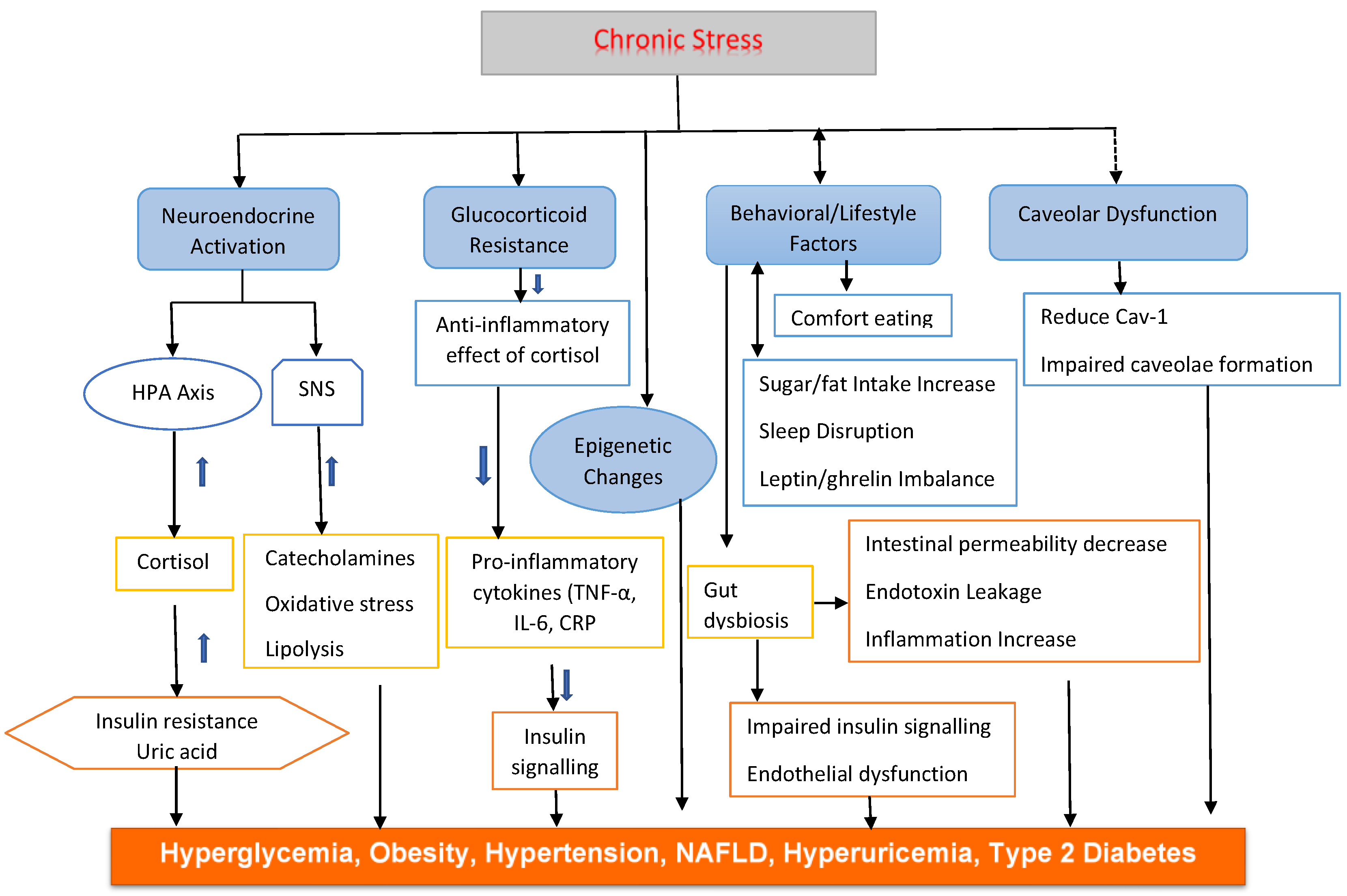
Chronic psychological stress disrupts metabolic balance through neuroendocrine, immune, and behavioral mechanisms [18,19]. Persistent activation of the HPA axis elevates cortisol, leading to insulin resistance, abdominal obesity, and other metabolic syndrome features. The glucocorticoids (GCs) and the catecholamines act synergistically to raise blood glucose levels [20] as does the ramp-up of cardiovascular output by the catecholamines [21]. Concurrent stimulation of the SNS raises catecholamine levels, further impairing metabolism and promoting inflammation. Behavioral changes such as poor diet, inactivity, and inadequate sleep exacerbate these effects. On a cellular level, stress-related hormones and cytokines hinder insulin signaling, damage mitochondria, and elevate oxidative stress [22]. These processes contribute to metabolic disorders, including hyperuricemia, highlighting the complex and multifactorial nature of stress-induced metabolic dysfunction (Figure 1).
2.1. Hormonal Imbalances via HPA Axis and SNS Activation
Chronic stress activates the HPA axis, leading to sustained cortisol secretion. Cortisol promotes gluconeogenesis and lipolysis, increasing blood glucose and free fatty acids. Over time, this contributes to insulin resistance and hyperglycemia [23], and hypertension [24], hallmarks of metabolic syndrome. Cortisol may be associated with uric acid levels under physical stress [25].
SNS activation releases catecholamines, which enhance lipolysis and gluconeogenesis [26], and mobilize glucose and fatty acids, exacerbating oxidative stress and inflammation. Prolonged SNS activity elevates blood pressure and free fatty acids, exacerbating hypertension and insulin resistance [27], and hyperuricemia [28].
HPA activity promotes visceral fat storage, which is metabolically active and secretes pro-inflammatory cytokines [29]. This chronic low-grade inflammation impairs insulin signaling, linking obesity to insulin resistance and metabolic syndrome [30].
Cortisol and catecholamines increase uric acid levels through enhanced purine degradation and reduced renal excretion [31]. Cortisol contributes via protein catabolism and insulin resistance, while catecholamines impair renal clearance through vasoconstriction and altered tubular handling.
2.2. Glucocorticoid Resistance
Chronic stress contributes to metabolic dysfunction through the development of GC resistance, which diminishes cortisol’s anti-inflammatory effects [32]. In glucocorticoid resistance, despite reduced GC signaling in some tissues, visceral adipose tissue often remains sensitive, contributing to the redistribution of fat toward visceral depots, increasing cardiovascular risk [33]. This resistance allows inflammation to persist, particularly within visceral adipose tissue, where immune cells actively contribute to tissue damage, atherosclerosis, and insulin resistance. Inflammatory cytokines, such as TNF-α, IL-6, and CRP, further disrupt insulin receptor signaling, impairing glucose uptake [34] in the liver and skeletal muscles. Concurrently, elevated gluconeogenesis and increased levels of free fatty acids exacerbate hepatic insulin resistance and contribute to the development of non-alcoholic fatty liver disease (NAFLD) [35]. Persistent insulin resistance places chronic demand on pancreatic β-cells, which may ultimately lead to β-cell exhaustion and the progression of type 2 diabetes. Additionally, chronic stress increases the production of reactive oxygen species (ROS) [36], causing mitochondrial damage and further impairing insulin signaling pathways [37]. Together, these stress-induced inflammatory and oxidative processes play a central role in the pathogenesis of insulin resistance and related metabolic disorders.
2.3. Behavioral and Lifestyle Factors
Beyond hormonal dysregulation, stress significantly influences behavioral patterns that contribute to metabolic disorders. Stress-induced comfort eating leads to increased consumption of high-calorie, sugar- and fat-rich foods, a behavior mediated through hypothalamic reward pathways [38] and associated with weight gain. Additionally, stress-related sleep disturbances disrupt the balance of leptin (a satiety hormone) and ghrelin (a hunger hormone), enhancing appetite and food cravings [39]. Reduced physical activity due to stress exacerbates energy imbalance [40], accelerating obesity and metabolic complications.
2.4. Gut Dysbiosis
2.5. Epigenetic Changes
Chronic stress may induce epigenetic changes (e.g., DNA methylation) in genes regulating glucose/lipid metabolism [45], predisposing individuals to metabolic diseases.
2.6. Oxidative Stress and Mitochondrial Dysfunction
Chronic stress amplifies oxidative stress, contributing to mitochondrial dysfunction and impaired insulin signaling. Stress-induced TNF-α, IL-6, and CRP promote insulin resistance and endothelial dysfunction [46]. Oxidative stress from prolonged cortisol exposure damages mitochondria, impairing energy metabolism [47].
2.7. Caveolar Dysfunction
Psychological stress exerts its effects indirectly, but meaningfully, on caveolar function through systemic pathways. Caveolae are small plasma membrane invaginations that serve as critical platforms for cellular adaptation to various stressors [48]. Their structure and function are deeply influenced by mechanical, oxidative, and metabolic stress, with widespread implications for metabolic diseases [49]. Caveolae are critical regulators of cellular responses to stress. Dysfunction of caveolae is linked to a range of diseases, including cardiovascular disorders, metabolic syndrome, and hyperuricemia, highlighting their potential as therapeutic targets for enhancing cellular resilience [44,50].
Mechanical stress induces caveolae formation [51]. Caveolae flatten in response to membrane tension, acting as protective buffers against mechanical damage [52]. This mechanoprotective function buffers cells against rupture and damage. It relies on the caveolin-1 (Cav1) protein for maintaining caveolar structure. Deficiency or dysfunction in caveolae increases susceptibility to diseases like muscular dystrophy, pulmonary fibrosis, and atherosclerosis [53].
Caveolae compartmentalize key components of redox signaling by localizing ROS-producing enzymes and antioxidant systems, maintaining redox homeostasis [54]. They also regulate key signaling pathways (e.g., MAPK, AKT) involved in cell survival and apoptosis. Disruption of these roles contributes to oxidative stress and metabolic dysfunction [55].
Caveolae are central to lipid, eNOS, uric acid and glucose metabolism [56,57]. They facilitate cholesterol uptake, insulin receptor organization, and glucose or uric acid transporter function. Metabolic stressors, resulting from excess nutrients such as hyperlipidaemia and diabetes, impair these processes, promoting insulin resistance and metabolic syndrome [49]. Caveolae can help endothelial cells adapt to shear stress from blood flow, and loss of caveolae can disrupt vascular tone and promotes hypertension [58].
3. Clinical Manifestations of Metabolic Diseases under Chronic Stress
Chronic stress is a risk factor for the development of metabolic diseases. Meta-analysis links anxiety/stress to 7–14% higher odds of metabolic syndrome [59]. Chronic stress disrupts energy homeostasis, promoting metabolic diseases, and can exacerbate existing conditions, making them harder to manage. Patients under chronic stress might have poorer outcomes, and managing stress could be part of treatment plans. Furthermore, the interplay between psychological factors and physiological changes, such as poor sleep diet, and sedentary lifestyle, crease additional risk factors for metabolic disorders. Inflammation can link stress with metabolic diseases. These interconnected mechanisms culminate in glucose metabolism, visceral adiposity, inflammation, and behaviors that worsen metabolic health. The cluster of conditions, including insulin resistance, obesity, and dyslipidemia, resulting from systemic dysregulation may also exert synergistic effects on the body's response to stress.
3.1. Hyperglycemia and Diabetes
The effects of stress on type I diabetes remain contradictory. Some retrospective human studies suggest that psychological stress may precipitate type I diabetes as various stressors can either trigger or prevent the onset in experimental diabetes animal models [60]. Chronic stress impairs GLUT4 translocation and promotes hepatic gluconeogenesis [61], and reliably produces hyperglycemia which induces type II diabetes. At the cellular level, both environmental and internal stressors contribute to insulin resistance and β-cell dysfunction. These stressors activate molecular pathways that intensify endoplasmic reticulum (ER) stress, the integrated stress response, oxidative stress, and impair autophagy [62]. Although these stress-responsive pathways are interconnected, their individual roles in maintaining glucose homeostasis and preserving β-cell function remain under investigation [63]. Hyperinsulinism itself can cause elevated ER luminal hydrogen peroxide (H₂O₂) production, leading to mild ER stress and reduced cell viability, through additional harmful factors beyond H₂O₂ are involved in β-cell dysfunction [64]. Other stress induced pathologies that can drive diabetes progression include dysregulated lipid signaling, mitochondrial oxidative stress, ER stress, and localized inflammation [65]. Catecholamines are the primary hormonal mediators of the stress response. Although they do not typically cause adverse effects in the acute phase, prolonged exposure can disrupt glucose homeostasis, contributing to chronic hyperglycemia, insulin resistance, and the eventual development of type II diabetes [66].
In skeletal muscle, GCs inhibit the insulin-induced translocation of GLUT4 to the cell membrane, reducing glucose uptake and increasing blood glucose levels [67]. In white adipose tissue, GCs promote lipolysis, generating glycerol (a gluconeogenic substrate) and leading to the accumulation of nonesterified fatty acids in muscle cells [68]. These fatty acids impair insulin signaling, further diminishing glucose uptake and perpetuating a hyperglycemic state. Additionally, corticosteroids inhibit pancreatic β-cells from producing and secreting insulin [69].
Interestingly, acute hyperglycemia during stress may serve as an adaptive mechanism. It provides readily available energy to the brain and immune system during injury, infection, or stress, functioning as part of an evolutionary survival response [70]. However, when stress becomes chronic, persistent hyperglycemia contributes to insulin resistance and eventually type II diabetes. Additionally, diabetes may also cause abnormalities in the regulation of these stress hormones [35].
3.2. Obesity
Stress and obesity are two increasingly common health issues that are intricately connected through multiple pathways. Firstly, stress can lead to poor decision-making related to food choices and lifestyle habits [71]. Secondly, stress influences behavior by promoting overeating, particularly of high-calorie, high-fat, and high-sugar foods, while simultaneously reducing physical activity and shortening sleep duration, all of which contribute to weight gain [72,73].
On a physiological level, stress activates the HPA axis and alters reward processing in the brain [74]. It may also influence the gut microbiome [75], further impacting metabolic health. Additionally, stress stimulates the release of hormones and peptides including leptin, ghrelin, and neuropeptide Y [76,77], all of which play key roles in appetite regulation and energy balance.
Obesity itself can also become a source of chronic stress due to widespread weight stigma [78], exacerbating the cycle. Occupational stress has been linked to lipid disturbances through HPA axis dysregulation, influencing lipid intake and metabolism [79]. Chronic stress elevates cortisol levels, which in turn increases GC synthesis and glucose availability, promotes visceral fat accumulation, enhances lipolysis, and elevates circulating fatty acids, leading to dyslipidemia and contributing further to obesity [80].
3.3. Hypertension
Stress-induced hypertension refers to elevated blood pressure triggered or worsened by psychological or physical stress. Acute stress activates the SNS and the HPA axis, leading to the release of stress hormones such as adrenaline and cortisol. These hormones increase heart rate, constrict blood vessels, and raise blood pressure as part of the body's "fight or flight" response [81]. While this response is adaptive in short-term situations, chronic stress can result in persistent activation of these systems, leading to sustained hypertension [82]. Repeated exposure to stress may also contribute to unhealthy behaviors like poor diet, lack of exercise, smoking, and disrupted sleep, further increasing blood pressure. In addition, stress alters vascular tone, endothelial function, and kidney activity, all of which play important roles in blood pressure regulation. Stress-induced hypertension, dyslipidemia, and endothelial dysfunction accelerate atherosclerosis. Managing stress through lifestyle changes, relaxation techniques, regular physical activity, and psychological support is essential in preventing and controlling stress-related hypertension. β-blockers can mitigate stress-driven vascular damage [83].
3.4. Hyperuricemia
Stress has been shown to induce hyperuricemia [84], a condition characterized by elevated levels of uric acid in the blood. Under restraint stress, there is a simultaneous increase in plasma uric acid levels and ROS generation, primarily due to xanthine oxidoreductase (XOR) activation in visceral adipose tissue (VAT), liver, and intestine. This stress-induced oxidative stress is further amplified by upregulation of NADPH oxidase (NOX) subunits and a reduction in antioxidant enzyme activities in VAT. In addition to oxidative stress, stress also triggers lipolysis and inflammation in adipose tissue, decreases insulin sensitivity, and promotes a prothrombotic state [85]. These changes contribute to a metabolic environment that favors the development of hyperuricemia and related complications.
Hyperuricemia has been shown to disrupt normal cortisol metabolism [86]. In this condition, the adrenal glands become less responsive to adrenocorticotropic hormone (ACTH), leading to reduced cortisol production, while corticosterone levels remain unaffected. This is linked to decreased mRNA expression of key cortisol-synthesizing enzymes, including aldosterone synthase, 11β-hydroxylase, and 3β-hydroxysteroid dehydrogenase 1 [87]. Additionally, the reduced expression of hepatic 5α-reductase and renal 11β-hydroxysteroid dehydrogenase 2 further impairs cortisol clearance. Together, these disturbances constitute a cortisol metabolism disorder associated with hyperuricemia [86].
3.5. Bidirectional Relationship
Metabolic disorders, the components of metabolic syndrome including obesity, type II diabetes mellitus, hypertension, and dyslipidemia, are intricately linked with both physiological and psychological stress [88]. These conditions are not only influenced by chronic stress but also act as significant contributors to stress-related pathologies, establishing a bidirectional and self-perpetuating cycle. Chronic emotional or occupational stress has been shown to increase the risk of developing metabolic syndrome [89,90]. In turn, the presence of metabolic dysfunctions can exacerbate stress responses by disrupting immune regulation and altering neurochemical pathways in the brain [91], thereby heightening stress sensitivity. Thus, stress serves both as a precursor to and a consequence of metabolic disease, reinforcing the complexity of their interrelationship.
Metabolic dysfunction disrupts the body’s internal balance, activating the HPA axis, raising cortisol levels [92] that further aggravate metabolic disturbances by boosting blood sugar and fat storage. Living with a metabolic disorder often leads to psychological stress [93], driven by health concerns such as diabetic complications, restrictive lifestyle changes, and body image dissatisfaction. This chronic stress elevates levels of cortisol and catecholamines, which in turn promote maladaptive behaviors like overeating, disrupted sleep, and reduced physical activity. These behaviors exacerbate insulin resistance and contribute to further weight gain, reinforcing the cycle of metabolic dysfunction. Obesity, in particular, is strongly linked to poorer mental health outcomes, including depression and subclinical depressive symptoms. Although the relationship is bidirectional, evidence suggests that increased body weight more commonly leads to psychological distress rather than the reverse [94].
4. Prospects
The increasing recognition of chronic stress as a critical contributor to metabolic dysfunction has spurred a growing interest in uncovering its underlying mechanisms, improving early detection, and developing targeted therapeutic strategies. Future research directions are expected to focus on the integration of molecular, behavioral, and systemic approaches to prevent and manage stress-induced metabolic diseases.
A central area of investigation involves the dysregulation of the HPA axis and heightened SNS activity, both of which are implicated in the pathogenesis of conditions such as obesity, insulin resistance, and cardiovascular disease. Identifying reliable biomarkers, such as pro-inflammatory cytokines (e.g., interleukin-6), acute phase reactants (e.g., C-reactive protein), and cortisol secretion patterns, may enhance the early prediction and risk stratification of stress-related metabolic disorders [95]. Psychological stress influences caveolar function (which can induce metabolic syndrome and hyperuricemia [44,49]) in indirect but significant ways through systemic pathways. As our understanding of the mind-body connection deepens, this area is becoming an increasingly important focus of research at the cellular level [96].
Emerging research on epigenetic modifications and mitochondrial dysfunction suggests that chronic stress may induce long-term changes in metabolic regulation, potentially predisposing individuals to disease later in life. In particular, epigenetic regulation of genes involved in glucose metabolism and mitochondrial efficiency could serve as a mechanistic link between psychological stress and metabolic impairment [97].
Given the interindividual variability in stress response, personalized medicine represents a promising frontier. Future studies may focus on resilience profiling by identifying genetic variants (e.g., GC receptor polymorphisms), behavioral traits, and environmental factors that confer protection against stress-induced metabolic disturbances. In this context, digital health technologies, such as wearable devices and mobile applications, offer innovative tools for real-time monitoring of stress indicators (e.g., heart rate variability, salivary cortisol) and for delivering personalized, adaptive stress management strategies [98,99].
Psychological interventions such as cognitive-behavioral therapy (CBT) and mindfulness-based stress reduction have shown efficacy in attenuating stress-induced inflammation and improving metabolic outcomes [100,101]. These approaches hold promise for integration into preventive and therapeutic frameworks.
Given that inflammation is estimated to mediate approximately 61.5% of the association between stress and metabolic syndrome [95], anti-inflammatory strategies warrant particular attention. Targeted therapies, including cytokine inhibitors (e.g., IL-1β antagonists), may be beneficial for individuals with prolonged exposure to psychosocial stress [102,103]. Nutritional interventions, such as diets rich in omega-3 fatty acids and polyphenols, can further mitigate oxidative stress and inflammation [104].
Socioeconomic and occupational stressors also play a significant role in the development of metabolic disorders. Future strategies should include workplace-level interventions, such as flexible scheduling and organizational stress reduction programs, aimed at lowering stress-related metabolic risk. In parallel, public health policies addressing broader social determinants, such as income inequality and neighborhood disadvantage, are critical to reducing chronic stress on a population level [105,106].
From a clinical perspective, a multidisciplinary and integrative approach is essential. Healthcare providers should routinely assess stress exposure in patients with metabolic disorders and incorporate stress management into treatment plans. This may involve combining pharmacologic interventions (e.g., β-blockers to reduce SNS overactivity) with lifestyle modifications [107], including exercise, nutritional guidance, and sleep hygiene.
In summary, the interplay between stress and metabolic health represents a vital area for ongoing scientific and clinical exploration. Advancing our understanding of biological pathways, enhancing personalized care, and enacting systemic changes are essential for addressing the rising burden of stress-induced metabolic diseases. With the support of emerging technologies and integrative healthcare models, more effective and sustainable strategies for prevention and treatment are on the horizon.
5. Conclusions
Chronic stress plays a critical role in metabolic dysfunction through sustained activation of the HPA axis and SNS, and indirectly through disruption of caveolae, leading to hormonal imbalances, inflammation, and insulin resistance. These physiological effects are intensified by unhealthy behaviors such as poor diet, inactivity, and sleep disruption. The relationship between stress and metabolic disorders is bidirectional, forming a self-perpetuating cycle reinforced by cellular dysfunction, gut dysbiosis, and epigenetic changes. Addressing this complex interaction requires a comprehensive approach, integrating early biomarker detection, psychological and pharmacological therapies, and public health strategies targeting social and occupational stressors. Ultimately, interdisciplinary efforts are essential to disrupt this cycle and improve metabolic and mental health outcomes at both individual and societal levels.
Funding
Not applicable.
Conflicts of interest
The authors declare no conflicts of interest.
References
- Shaw W, Labott-Smith S, Burg M, et al. Stress effects on the body. Am Psychol Assoc. 2018;12.
- Gunasekra KA, Perera BAKS. Defining occupational stress: A systematic literature review. FARU J. 2023;10(1). [CrossRef]
- Hanxiao Luo # 1 2, Linlin Xing # 3, Tongtong Fu 1, Shiqi Xiao # 4 LF# 5 6. Occupational stress trajectories and metabolic dysfunction-associated steatotic liver disease among female nurses: a prospective Cohort Study. MC Public Heal. 2024;16(24):3188. 10.1186/s12889-024-20742-z.
- Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. 2006;8(4). [CrossRef]
- Gupta S, Bharatha A, Cohall D, Rahman S, Haque M, Azim Majumder MA. Aerobic Exercise and Endocannabinoids: A Narrative Review of Stress Regulation and Brain Reward Systems. Cureus. Published online 2024. [CrossRef]
- Volarić M, Šojat D, Majnarić LT, Vučić D. The Association between Functional Dyspepsia and Metabolic Syndrome—The State of the Art. Int J Environ Res Public Health. 2024;21(2). [CrossRef]
- Piątkowska-Chmiel I, Krawiec P, Ziętara KJ, et al. The Impact of Chronic Stress Related to COVID-19 on Eating Behaviors and the Risk of Obesity in Children and Adolescents. Nutrients. 2024;16(1). [CrossRef]
- McCollum SE, Shah YM. Stressing Out Cancer: Chronic Stress Induces Dysbiosis and Enhances Colon Cancer Growth. Cancer Res. 2024;84(5). [CrossRef]
- Shin HS, Lee SH, Moon HJ, et al. Prolonged stress response induced by chronic stress and corticosterone exposure causes adult neurogenesis inhibition and astrocyte loss in mouse hippocampus. Brain Res Bull. 2024;208. [CrossRef]
- Ma Y, Chen H. Relationship between life events and serum uric acid in patients with hyperuricemia. Chinese J Endocrinol Metab. 2020;36(2). [CrossRef]
- Ajibewa TA, Kershaw KN, Carr JJ, et al. Chronic Stress and Cardiovascular Events: Findings From the CARDIA Study. Am J Prev Med. 2024;67(1). [CrossRef]
- Wang T, Li L, Li S, et al. Clostridium butyricum relieve the visceral hypersensitivity in mice induced by Citrobacter rodentium infection with chronic stress. PeerJ. Published online 2021. [CrossRef]
- Nikolic A, Fahlbusch P, Riffelmann NK, et al. Chronic stress alters hepatic metabolism and thermodynamic respiratory efficiency affecting epigenetics in C57BL/6 mice. iScience. 2024;27(3). [CrossRef]
- Vyunytska L V., Yuzvenko TY, Dashuk TI, Nikonov V V., Vasyuk VL, Korotchuk N V. Stress-induced urgent conditions in endocrinology. Miznarodnij Endokrinol Z. 2024;20(1). [CrossRef]
- Pisaruk A, Asanov E, Naskalova S, et al. Effects of war-related stress on the cardiovascular system, metabolism and the rate of ageing in women. Ageing Longev. 2024;(1 2024). [CrossRef]
- Zhang S, Liu B, Huang L, Zhang R, An L, Liu Z. Metabolomics reveals that chronic restraint stress alleviates carbon tetrachloride-induced hepatic fibrosis through the INSR/PI3K/AKT/AMPK pathway. J Mol Med. 2024;102(1). [CrossRef]
- Zhao L, Hou X, Feng Y, et al. A chronic stress-induced microbiome perturbation, highly enriched in Ruminococcaceae_UCG-014, promotes colorectal cancer growth and metastasis. Int J Med Sci. 2024;21(5). [CrossRef]
- Lu C, Xu J, Li K, et al. Chronic Stress Blocks the Endometriosis Immune Response by Metabolic Reprogramming. Int J Mol Sci. 2024;25(1). [CrossRef]
- Pace SA, Myers B. Hindbrain Adrenergic/Noradrenergic Control of Integrated Endocrine and Autonomic Stress Responses. Endocrinol (United States). 2024;165(1). [CrossRef]
- Kuo T, McQueen A, Chen TC, Wang JC. Regulation of glucose homeostasis by glucocorticoids. Adv Exp Med Biol. 2015;872. [CrossRef]
- Petrák O, Krátká Z, Holaj R, et al. Cardiovascular Complications in Pheochromocytoma and Paraganglioma: Does Phenotype Matter? Hypertension. 2024;81(3). [CrossRef]
- Sarwar H, Rafiqi SI, Ahmad S, et al. Hyperinsulinemia Associated Depression. Clin Med Insights Endocrinol Diabetes. 2022;15. [CrossRef]
- Khani S, Tayek JA. Cortisol increases gluconeogenesis in humans: Its role in the metabolic syndrome. Clin Sci. 2001;101(6). [CrossRef]
- Lee WH, Larsson SC, Wood A, et al. Genetically predicted plasma cortisol and common chronic diseases: A Mendelian randomization study. Clin Endocrinol (Oxf). 2024;100(3). [CrossRef]
- Acevedo AM, Fortier MA, Campos B, Brown YC, Riis J. Salivary uric acid reactivity and baseline associations with physiological stress response. Psychoneuroendocrinology. 2022;146. [CrossRef]
- Nedosugova L V. Role of the endocrine system in maintaining glucose homeostasis in health and disease. Russ Med Inq. 2021;5(9). [CrossRef]
- Hu Y, Bao J, Gao Z, Ye L, Wang L. Sodium–Glucose Cotransporter Protein 2 Inhibitors: Novel Application for the Treatment of Obesity-Associated Hypertension. Diabetes, Metab Syndr Obes. 2024;17. [CrossRef]
- Yonetani Y, Ishii M, Ogawa Y. Catecholamine-induced hyperuricemia : Potentiation of catecholamine action in DOCA-treated animals. Jpn J Pharmacol. 1977;27. [CrossRef]
- Careaga MBL, Wu TJ. Chronically stressed male and female mice show a similar peripheral and central pro-inflammatory profile after an immune challenge. PLoS One. 2024;19(2 February). [CrossRef]
- Shively CA, Register TC, Clarkson TB. Social stress, visceral obesity, and coronary artery atherosclerosis in female primates. Obesity. 2009;17(8). [CrossRef]
- Shibutani Y, Ueo T, Takahashi S, Moriwaki Y, Yamamoto T. Effect of ACTH on renal excretion of purine bases in a patient with isolated ACTH deficiency. Clin Chim Acta. 2000;294(1-2). [CrossRef]
- Ruijters EJB, Haenen GRMM, Willemsen M, Weseler AR, Bast A. Food-derived bioactives can protect the anti-inflammatory activity of cortisol with antioxidant-dependent and -independent mechanisms. Int J Mol Sci. 2016;17(2). [CrossRef]
- Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta - Mol Basis Dis. 2014;1842(3). [CrossRef]
- Omran F, Christian M. Inflammatory Signaling and Brown Fat Activity. Front Endocrinol (Lausanne). 2020;11. [CrossRef]
- Onyango AN. Excessive gluconeogenesis causes the hepatic insulin resistance paradox and its sequelae. Heliyon. 2022;8(12). [CrossRef]
- Ko SY, Wang SE, Lee HK, et al. Transient receptor potential melastatin 2 governs stress-induced depressive-like behaviors. Proc Natl Acad Sci U S A. 2019;116(5). [CrossRef]
- Chang LY, Chao YL, Chiu CC, Chen PL, Lin HYH. Mitochondrial Signaling, the Mechanisms of AKI-to-CKD Transition and Potential Treatment Targets. Int J Mol Sci. 2024;25(3). [CrossRef]
- Calcaterra V, Rossi V, Magenes VC, et al. Dietary habits, depression and obesity: an intricate relationship to explore in pediatric preventive strategies. Front Pediatr. 2024;12. [CrossRef]
- Kullik L, Stork M, Kiel A, Kellmann M, Jakowski S. The prevalence of menstrual cycle symptoms and their association with mental health and sleep in German exercising women and athletes. J Sci Med Sport. 2024;27(6). [CrossRef]
- Ricart W, Crujeiras AB, Mateos A, Castells-Nobau A, Fernández-Real JM. Is obesity the next step in evolution through brain changes? Neurosci Appl. 2024;3. [CrossRef]
- Yiming Zhang, Xiaoang Li, Siqi Lu, Huaizhu Guo, Zhuangyi Zhang, Haonan Zheng, Cunzheng Zhang, Jindong Zhang, Kun Wang FP& LD. Stress triggers gut dysbiosis via CRH-CRHR1-mitochondria pathway. npj Biofilms Microbiomes. 2024;10(93). doi.org/10.1038/s41522-024-00571-z.
- Sun L, Ni C, Zhao J, Wang G, Chen W. Probiotics, bioactive compounds and dietary patterns for the effective management of hyperuricemia: a review. Crit Rev Food Sci Nutr. 2024;64(7). [CrossRef]
- Rodrigues e-Lacerda R, Fang H, Robin N, Bhatwa A, Marko DM, Schertzer JD. Microbiota and Nod-like receptors balance inflammation and metabolism during obesity and diabetes. Biomed J. 2023;46(5). [CrossRef]
- Zhang Wei zheng. Hyperuricemia: Current State and Prospects. Explor Res Hypothesis Med. 2025;10(1):49-55. 10.14218/ERHM.2024.00199.
- Arzate-Mejia RG, Carullo NVN, Mansuy IM. The epigenome under pressure: On regulatory adaptation to chronic stress in the brain. Curr Opin Neurobiol. 2024;84. [CrossRef]
- Marsland AL, Walsh C, Lockwood K, John-Henderson NA. The effects of acute psychological stress on circulating and stimulated inflammatory markers: A systematic review and meta-analysis. Brain Behav Immun. 2017;64. [CrossRef]
- Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the “gluc” back in glucocorticoids. Nat Rev Endocrinol. 2014;10(5). [CrossRef]
- Shu Y, Jin S. Caveolin-1 in endothelial cells: A potential therapeutic target for atherosclerosis. Heliyon. 2023;9(8). [CrossRef]
- Zhang Wei zheng. An association of metabolic syndrome constellation with cellular membrane caveolae. Pathobiol Aging Age-related Dis. 2014;4(1). [CrossRef]
- Wei-zheng Zhang. Pharmacologically targeting caveolae in metabolic diseases. In: Pharmacology Targets in Metabolic Diseases, Accepted for Publication. Elsevier Academic Press. Elsevier Academic Press.; 2025.
- Boyd NL, Park H, Yi H, et al. Chronic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells. Am J Physiol - Hear Circ Physiol. 2003;285(3 54-3). [CrossRef]
- Del Pozo MA, Lolo FN, Echarri A. Caveolae: Mechanosensing and mechanotransduction devices linking membrane trafficking to mechanoadaptation. Curr Opin Cell Biol. 2021;68. [CrossRef]
- Hnasko R, Lisanti MP. The biology of caveolae: lessons from caveolin knockout mice and implications for human disease. Mol Interv. 2003;3(8). [CrossRef]
- Mineo C, Shaul PW. Regulation of eNOS in caveolae. Adv Exp Med Biol. 2012;729. [CrossRef]
- Wu Y, Lim YW, Stroud DA, et al. Caveolae sense oxidative stress through membrane lipid peroxidation and cytosolic release of CAVIN1 to regulate NRF2. Dev Cell. 2023;58(5). [CrossRef]
- Higuchi Y, Ogata T, Nakanishi N, et al. Requirement of Cavin-2 for the expression and stability of IRβ in adequate adipocyte differentiation. Mol Metab. 2022;55. [CrossRef]
- Zhang W zheng. Uric acid en route to gout. In: Advances in Clinical Chemistry. ; 2023. [CrossRef]
- Lian X, Matthaeus C, Kaßmann M, Daumke O, Gollasch M. Pathophysiological Role of Caveolae in Hypertension. Front Med. 2019;6. [CrossRef]
- Ji S, Chen Y, Zhou Y, et al. Association between anxiety and metabolic syndrome: An updated systematic review and meta-analysis. Front Psychiatry. 2023;14. [CrossRef]
- N SRSSMSFM. Stress hyperglycemia in acute pancreatitis: From mechanisms to prognostic implications. Diabetes Care. 1992;15(10):1413. 10.2337/diacare.15.10.1413.
- Yajurvedi H. Stress and Glucose metabolism: A Review. Imaging J Clin Med Sci. Published online 2018. [CrossRef]
- Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8). [CrossRef]
- Kulkarni A, Muralidharan C, May SC, Tersey SA, Mirmira RG. Inside the β Cell: Molecular Stress Response Pathways in Diabetes Pathogenesis. Endocrinol (United States). 2023;164(1). [CrossRef]
- Vidrio-Huerta B, Plötz T, Lortz S. Oxidative and ER stress by elevated insulin biosynthesis and palmitic acid in insulin-producing cells. J Mol Endocrinol. 2024;72(2). [CrossRef]
- Xourafa G, Korbmacher M, Roden M. Inter-organ crosstalk during development and progression of type 2 diabetes mellitus. Nat Rev Endocrinol. 2024;20(1). [CrossRef]
- Sharma K, Akre S, Chakole S, Wanjari MB. Stress-Induced Diabetes: A Review. Cureus. Published online 2022. [CrossRef]
- Meng Z, Yu Y, Zhang Y, et al. Highly bioavailable berberine formulation improves glucocorticoid receptor-mediated insulin resistance via reduction in association of the glucocorticoid receptor with phosphatidylinositol-3-kinase. Int J Biol Sci. 2020;16(14). [CrossRef]
- Mir N, Chin SA, Riddell MC, Beaudry JL. Genomic and non-genomic actions of glucocorticoids on adipose tissue lipid metabolism. Int J Mol Sci. 2021;22(16). [CrossRef]
- Campbell JE, Newgard CB. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat Rev Mol Cell Biol. 2021;22(2). [CrossRef]
- 1 YG, GL, FT, C XW, D JS, G QH. Stress hyperglycemia in acute pancreatitis: From mechanisms to prognostic implications. Life Sci. 2025;365:123469. 10.1016/j.lfs.2025.123469.
- Shields GS, Sazma MA, Yonelinas AP. The effects of acute stress on core executive functions: A meta-analysis and comparison with cortisol. Neurosci Biobehav Rev. 2016;68. [CrossRef]
- Goens D, Virzi NE, Jung SE, Rutledge TR, Zarrinpar A. Obesity, Chronic Stress, and Stress Reduction. Gastroenterol Clin North Am. 2023;52(2). [CrossRef]
- Papatriantafyllou E, Efthymiou D, Zoumbaneas E, Popescu CA, Vassilopoulou E. Sleep Deprivation: Effects on Weight Loss and Weight Loss Maintenance. Nutrients. 2022;14(8). [CrossRef]
- Novick AM, Levandowski ML, Laumann LE, Philip NS, Price LH, Tyrka AR. The effects of early life stress on reward processing. J Psychiatr Res. 2018;101. [CrossRef]
- Desiree R. Delgadillo, Jessica L. Borelli, Emeran A. Mayer, Jennifer S. Labus MPC& SDP. Biological, environmental, and psychological stress and the human gut microbiome in healthy adults. Sci Rep. 2025;15(262). [CrossRef]
- Huang Y, Lin X, Lin S. Neuropeptide Y and Metabolism Syndrome: An Update on Perspectives of Clinical Therapeutic Intervention Strategies. Front Cell Dev Biol. 2021;9. [CrossRef]
- Yamada C. Involvement of ghrelin dynamics in stress-induced eating disorder: Effects of sex and aging. Int J Mol Sci. 2021;22(21). [CrossRef]
- A. Janet Tomiyama. Stress and Obesity. Annu Rev Psychol. 2019;70:703-718. [CrossRef]
- Chida Y, Steptoe A. Cortisol awakening response and psychosocial factors: A systematic review and meta-analysis. Biol Psychol. 2009;80(3). [CrossRef]
- Zhang H, Shao MM, Lin X Da, et al. A cross-sectional survey on occupational stress and associated dyslipidemia among medical staff in tertiary public hospitals in Wenzhou, China. Brain Behav. 2021;11(3). [CrossRef]
- Ayada C, Toru, Korkut Y. The relationship of stress and blood pressure effectors. Hippokratia. 2015;19(2).
- Bos-Roubos AG, Wingbermühle E, Giesen M, Kersseboom R, De Graaff LCG, Egger JIM. Hypertension with hidden causes: the cognitive and behavioral profile of an adult female with chronic stress and 16p11.2 microdeletion. J Hypertens. 2024;42(1). [CrossRef]
- Kim JH, Almuwaqqat Z, Hammadah M, et al. Peripheral Vasoconstriction During Mental Stress and Adverse Cardiovascular Outcomes in Patients With Coronary Artery Disease. Circ Res. 2019;125(10). [CrossRef]
- Bauer CM, Oranges MA, Firempong G, Romero LM. Corticosterone Alters Body Weight, but Not Metabolites, during Chronic Stress. Physiol Biochem Zool. 2022;95(6). [CrossRef]
- Yisireyili M, Hayashi M, Wu H, et al. Xanthine oxidase inhibition by febuxostat attenuates stress-induced hyperuricemia, glucose dysmetabolism, and prothrombotic state in mice. Sci Rep. 2017;7(1). [CrossRef]
- Bao R, Chen B, Pan J, et al. Pseudohypoadrenalism, a subclinical cortisol metabolism disorder in hyperuricemia. Front Endocrinol (Lausanne). 2023;14. [CrossRef]
- Molenaar N, Bijkerk RM, Beishuizen A, et al. Steroidogenesis in the adrenal dysfunction of critical illness: Impact of etomidate. Crit Care. 2012;16(4). [CrossRef]
- Janssen JAMJL. New Insights into the Role of Insulin and Hypothalamic-Pituitary-Adrenal (HPA) Axis in the Metabolic Syndrome. Int J Mol Sci. 2022;23(15). [CrossRef]
- Zhang M, Liu B, Ke W, et al. Correlation analysis between occupational stress and metabolic syndrome in workers of a petrochemical enterprise: based on two assessment models of occupational stress. BMC Public Health. 2024;24(1). [CrossRef]
- Chico-Barba G, Jiménez-Limas K, Sánchez-Jiménez B, et al. Burnout and metabolic syndrome in female nurses: An observational study. Int J Environ Res Public Health. 2019;16(11). [CrossRef]
- Aleksandar Sic, Kiana Cvetkovic, Eshanika Manchanda 1 NNK. Neurobiological Implications of Chronic Stress and Metabolic Dysregulation in Inflammatory Bowel Diseases. Diseases. 2024;18(12):22. 10.3390/diseases12090220.
- Barrea L, Verde L, Camajani E, et al. Effects of very low-calorie ketogenic diet on hypothalamic–pituitary–adrenal axis and renin–angiotensin–aldosterone system. J Endocrinol Invest. 2023;46(8). [CrossRef]
- Lin Z, Li LY, Chen L, et al. Lonicerin promotes wound healing in diabetic rats by enhancing blood vessel regeneration through Sirt1-mediated autophagy. Acta Pharmacol Sin. 2024;45(4). [CrossRef]
- Steptoe A, Frank P. Obesity and psychological distress. Philos Trans R Soc B Biol Sci. 2023;378(1888). [CrossRef]
- Jurgens SM, Prieto S, Hayes JP. Inflammatory biomarkers link perceived stress with metabolic dysregulation. Brain, Behav Immun - Heal. 2023;34. [CrossRef]
- Wu Y, Lim YW, Parton RG. Caveolae and the oxidative stress response. Biochem Soc Trans. 2023;51(3). [CrossRef]
- Ashwani, Anjali Sharma, Mayank Kumar Choudhary, Dalapathi Gugulothu, Deepti Pandita, Surajpal Verma, Lalitkumar K. Vora DKK& DG. Epigenetic and Mitochondrial Metabolic Dysfunction in Multiple Sclerosis: A Review of Herbal Drug Approaches and Current Clinical Trials. Mol Neurobiol. Published online 2025. [CrossRef]
- Pinge A, Sen APGGJJGG, Sen S. Detection and monitoring of stress using wearables: a systematic review. Front Comput Sci. 2024;6. [CrossRef]
- Sandra Cano, Claudio Cubillos, Rodrigo Alfaro, Andrés Romo 1, Matías García and Fernando Moreira O. Wearable Solutions Using Physiological Signals for Stress Monitoring on Individuals with Autism Spectrum Disorder (ASD): A Systematic Literature Review. Sensors. 2024;24(24):8137. [CrossRef]
- Saban KL, Collins EG, Mathews HL, et al. Impact of a Mindfulness-Based Stress Reduction Program on Psychological Well-Being, Cortisol, and Inflammation in Women Veterans. J Gen Intern Med. 2022;37. [CrossRef]
- Lindsay EK, Marsland AL, Cole SW, et al. Mindfulness-Based Stress Reduction Reduces Proinflammatory Gene Regulation but Not Systemic Inflammation among Older Adults: A Randomized Controlled Trial. Psychosom Med. 2024;86(5). [CrossRef]
- Jones ME, Lebonville CL, Barrus D, Lysle DT. The Role of Brain Interleukin-1 in Stress-Enhanced Fear Learning. Neuropsychopharmacology. 2015;40. [CrossRef]
- Yin W, Godbout JP, Sheridan JF. Interleukin-1 beta in psychosocial stress. In: Stress: Immunology and Inflammation: Handbook of Stress Series Volume 5. Vol 5. ; 2023. [CrossRef]
- Siroma TK, Machate DJ, Zorgetto-Pinheiro VA, et al. Polyphenols and ω-3 PUFAs: Beneficial Outcomes to Obesity and Its Related Metabolic Diseases. Front Nutr. 2022;8. [CrossRef]
- Pampel FC, Krueger PM, Denney JT. Socioeconomic disparities in health behaviors. Annu Rev Sociol. 2010;36. [CrossRef]
- Lead A. Reducing income inequality to advance health. (2018, January 18). American Public Health Association. https://www.apha.org/policy-and-advocacy/public-health-policy-briefs/policy-database/2018/01/18/reducing-income-inequality-to-advance-health.
- Akram M Eraky, Yashwanth Yerramalla, Adnan Khan, Yasser Mokhtar 3, Mostafa Alamrosy, Amr Farag, Alisha Wright, Matthew Grounds NMG. Beta-Blockers as an Immunologic and Autonomic Manipulator in Critically Ill Patients: A Review of the Recent Literature. Int J Mol Sci. 2024;25(15):8058. [CrossRef]
Figure 1.
Caption.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



