
Submitted:
11 April 2025
Posted:
11 April 2025
You are already at the latest version
Abstract
Conus venoms are both highly powerful and complex exhibiting a remarkably intriguing molecular variability. The biologic reasons behind such astonishing molecular diversity are yet to be fully understood. We hypothesized that the current knowledge has been hampered by a lack of studies targeting the whole Conus genus backed by a feeding habit analysis, as opposed to the abundant studies focused on single species or at the individual level. We aim to enlighten the understanding of the remarkable venom variability in cone snails while pushing to deliver novel peptides for biomedical applications through a broad transcriptomics approach. Here, we assessed 76 publicly available venom-related and unrelated transcriptomes from a total of 20 different Conus species. The shared transcriptomic repertoire discovered revealed several gene variations in accordance with predatory diets (e.g., gene loss in piscivorous species), indicating that feeding habit largely influences venom evolution. Furthermore, evidences of ubiquitous symbiotic relationships within the venom organs were depicted, as biological processes alien to Conus species (e.g., Sorocarp morphogenesis) were found in all analyzed transcriptomes. Moreover, 88 potential anti-microbial peptides were bioinformatically detected, including one showing similarity with the human ACE2 receptor. Our study highlights the im-portance of in-depth comparative transcriptomic analyses, fostering cross-field synergic assessments by relying on informatic, biologic and pharmacologic resources.
Keywords:
Conus
; Transcriptomes
; Venom
; Anti-microbial peptides
; Feeding habit
; Evolution
; ACE2
1. Introduction
Animal natural venoms are being studied for more than a century, but a particular group emerges entrancingly in this research field [1,2,3]. The highly diverse Conus genus has more than 700 species living in tropical and subtropical regions worldwide [4,5,6,7,8]. As predators, they have three main diets, consisting of either fishes, molluscs or worms, which they catch with their neuroactive venom amidst rocky shores and coral reefs [9,10]. Their predatory venom contains about two hundred toxins known as conotoxins [11]. Synthesized as precursors, these toxins undergo a series of post-transcriptional modifications (PTMs) enabling them to interact specifically with their target, which includes the presynaptic membrane calcium channel, potassium and calcium channels, the receptors of serotonin and adrenal hormone, among others [11,12,13,14,15,16,17,18]. Nowadays, more than 8,000 conopeptides have been discovered out of 50,000 to 1 million estimated to exist [19,20]. For predation, generally compounds from A, M, O1 and T gene superfamilies account for the bulk of venom cocktails, but there is a surprisingly large variation not only between species but also between individuals [1,2,15,21]. Additionally, venom profiles change throughout different venom discharges, as while peptides from the O-superfamily are prominent in most profiles, peptides of the M- and T-superfamilies are only prevalent in subsequent injections [1,2,6,15,22]. This variability is reflected in the conotoxin genes, as there is a discrepancy between their number and their transcripts. Perhaps due to natural variation coupled with PTMs, highly expressed transcripts are similar, but variation of less expressed sequences is also unusually large [1,2,21,23]. The main evolutionary drive behind the hyper inter and intra-specific variations remains unclear, but the conotoxin genes are proposed to develop by gene duplication events, followed by either positive selection resulting in the neofunctionalization of physiological genes in venom-related organs, or by a neutral force of sub-functionalization and restriction to venom-related organs [23,24,25,26,27,28,29]. Indeed, some venom-related proteins have much higher expression levels in the foot than in the venom gland, possibly showing the endogenous nature of these proteins [2]. There are also findings of toxin genes being expressed in venom-unrelated tissues in snakes and in platypus [24,30,31]. After duplication events, venom potency is enhanced via increased dosage and replenishment capabilities, inclusively allowing for greater target diversity [32,33]. Counter-intuitively, there are cases in which it is gene loss the main evolutionary trait resulting in specialization at the cost of reduced fitness [34,35]. Such is the case of rattlesnakes, whose loss of toxin genes constitutes the main force behind evolutionary changes in venom function [36].
In order to increase the comprehension of the variability of Conus venoms, we declined the previous overwhelming focus on single species and single individuals analyses and replace it with an innovative strategy consisting in gathering the greatest possible Conus dataset for searching similar aspects connecting the whole genus. Such aim could provide valuable insights on venom evolution, potentially helping unveil trends behind venom variability. A transcriptomic approach was chosen given its capability of indicating actively expressed genes as well as the functions of the proteins they encode at great speed and scale. These features are particularly informative for functional genomics research, especially in species like Conus, with still limited genetic resources available [37,38]. We conducted in-depth transcriptomic analyses (76 transcriptomes) with the aim of finding shared genetic signatures among the representatives of the 20 cone snail species studied. Informative patterns of gene variation (e.g., gene loss and reduced similarity) were revealed in accordance with predatory diet. This suggests that cone snail venom variability and evolutionary traits are better elucidated through the scope of a contextual feeding habit evaluation. Furthermore, the surprising discovery of shared transcripts unfamiliar to Conus genome supports the existence of extensive symbiose relationships [39,40,41]. Finally, we explored in silico the therapeutic potential of conopeptides by investigating their anti-microbial capabilities. Presently, several studies have been successful in inspecting various marine-derived peptides from venomous species including, for instance, cephalopods and cnidarians [42,43]. Here, our performed analysis relying on our vast library of annotations predicted 88 putative anti-microbial peptides (AMPs) in Conus species. Altogether, our study extended the current understanding of the Conus venom’s evolution and functionalities.
2. Materials and Methods
This investigation made use of data acquired (from NCBI), polished (with Trimmomatic and Cutadapt), assembled (de novo generated using Trinity) and annotated (with GO Ontology) based on a previous work from our group [44]. The dataset consists of 76 transcriptomes from 20 different species (Conus marmoreus, C. betulinus, C. consors, C. coronatus, C. ebraeus, C. ermineus, C. imperialis, C. judaeus, C. lenavati, C. literatus, C. lividus, C.magus, C. miliaris, C. quercinus, C. rattus, C. sponsalis, C. tribblei. C. varius, C. ventricosus, and C. virgo): 69 are from venom duct and gland of 20 species, while the remaining 7 come from 5 other tissues (nervous ganglions, salivary glands, osphradium, foot and proboscis) of 2 species (C. consors and C. ventricosus).
In summary (Figure 1), the whole dataset was divided in two groups: one with transcriptomes from venom-related organs and another with transcriptomes from other body tissues. For annotation, the Swiss-Prot and Tox-Prot databases were used for peptide and toxin identification, respectively [38]. Afterwards, alignment and clustering resulted in creation of three phylogenetic trees (illustrating venom clusters). In the end, we evaluated the biomedical potential of conopeptides by targeting the venom-related and unrelated transcriptomes, as well as from sequences with 14 of the 29 annotations shared by all Conus species but exclusive to venom organs.
The 69 venom-related and 7 venom-unrelated transcriptomes were annotated with Swiss-Prot and Tox-Prot (using Diamond BLASTX and MMseqs2, v15.6f452), for peptide and toxin identification, respectively [45,46]. From the list of 29 shared annotations, 14 were chosen due to higher venom-like specificity or possible symbiont origin (list in Appendix A).
Sequence alignment was built by Mafft (v7.490) with default parameters, then processed by UGENE [47,48]. Venom clustering processes were carried out with VSEARCH (v2.29.1 [49]) on the venom-related dataset according to two criteria, set as follows:
- -
- Criteria A: 100 sequences from at least 30 transcriptomes; the selected sequences would be further annotated according to the database of Tox-Prot;
- -
- Criteria B: 50 sequences from at least 10 transcriptomes; the selected sequences would be further annotated according to the database of UniProt-Trembl.
Next, phylogeny trees were elaborated with iTOL (https://itol.embl.de/) utilizing previous annotation lists with transcriptome number and its corresponding origin species, tissue, diet and blast target [50].
The biomedical prediction model utilized was AMPfinder, a bioinformatic tool which employed with its corresponding database is used to identify antimicrobial peptides (AMPs) [51]. This database is primarily based on experimentally verified AMPs collected from various public repositories, scientific literature, and computational analyses. To test the effectivity of a dedicated AMP finder and the advantages of using one, the first phase of AMP prediction targeted smaller the group of peptides with the chosen 14 annotations. Afterwards, anti-microbial potential was assessed in the whole transcriptomic dataset.
The R software was used to generate UpSet plots with the shared venom-related and unrelated annotations, which allow the visualization of all the intersections in a clean matrix layout [52]. In addition, the R software was used to visualize the AMP distribution across the different feeding habits through a violin plot (Appendix B) and a Principal Component analysis (Appendix C). All data was normalized before being sorted according to the following “Min-Max” normalization formula: normalized data (X’) = . This formula helps reducing distribution issues, ensuring reliability in comparing variables across the different diets [53].
3. Results
3.1. General Transcriptomes Annotation and Biomedical Predictions
The annotation revealed a cumulative total of 553,878 peptides recognized by Swiss-Prot in the venom-related transcriptomes, with 5.2% of them (28,805) being toxin sequences recognized in the Tox-Prot database. In comparison, there were 108,466 cumulative peptide sequence annotations from transcriptomes coming from venom-unrelated tissues, with 1.75% of them (1,902) also being toxins (from the salivary glands’ transcriptome). In addition, a total of 88 potential AMPs were predicted, 15 unique to the venom-related transcriptomes (including 7 from the chosen 14 annotations), 17 unique to venom-unrelated transcriptomes and 56 shared between them. The Table 1 resumes the annotation results.
3.2. Shared Transcriptomic Repertoire
The 7 transcriptomes from nervous ganglions, salivary glands, osphradium, foot and proboscis share 5,913 annotations (GO IDs) in common, which are perceived to represent housekeeping genes (Figure 2 – A). Regarding the 69 venom-related files, they share 2,104 annotations (Figure 2 – B), most of which are also present in other tissues. The removal of the endogenous annotations from the group of shared annotations in the venom-related transcriptomes resulted in an assemblage of 29 shared but venom-exclusive annotations. They are related to six main aspects: DNA replication, metabolism, blood pressure, ion channel and transport regulation, PTMs and seemingly symbiotic beings (Figure 3). The latter is demonstrated by three peculiar annotations: GO:0031288, GO:1903665, and GO:1903669. In Figure 3 it is revealed that most of the annotations are related to a biological metabolism process, showing the backbone of conserved genomic expression is related to the logistical processes behind venom synthesis
3.3. Phylogeny
Following criteria A, two Trees were created: Tree A with 271 branches (Figure 4) and Tree B with 117 branches (Figure 5). Both of these trees consist of venom duct sequences from a total of 12 species while the 21 different blast targets present an array of conotoxin peptides, as expected from the Tox-Prot database. All of these are conotoxins of the M-superfamily, which is the most structurally varied of the identified superfamilies and was detected in every species studied to date [54,55]. M-superfamily peptides are categorized into five branches, designated M1-5, based on the number of residues present in the third cystine loop situated between the fourth and fifth cysteine residues. These branches are further divided into two subgroups, on account of the number of residues in the mature peptide: Mini-M (1-3) and Maxi-M (4-5).
The conotoxins demonstrated in Tree A and B are mostly from the M-1 branch (CM31_CONAE [A], CM35_CONTS [A], CM315_CONBU [A], CM31_CONLE [B], CM302_CONTS [A,B], CM32_CONPE [A,B], CM32_CONLE [A,B], CM33_CONLT [A,B], CM38_CONMR [A,B], CM34_CONLT [A,B], M3A_CONTE [A,B], CM3E_CONMR [A,B], CM3F_CONMR [A,B], CM31_CONPE [A,B], M32_CONTE [A,B], CM36_CONPL [A,B], and M3H_CONTE [A,B]), with additional three from the M-3 branch (CM31_CONQU [A], CM11_CONTS [A,B], and CM336_CONVE [A,B]), and one without attributed branch (CM401_CONAE [B]). The Mini-M group is characterized for containing fewer than 22 residues. It shows unparalleled structural diversity and cysteine connectivity, exhibiting distinct features such as unique patterns of disulphide bond connectivity, variability in loop sizes, and specialized 3D folding structures, even within the same branch. Until now, only members of the M-4 and M-5 branches have identified target receptors. Although little is understood about the biological receptors associated with Mini-M peptides, it is known they target receptors distinct from those of the Maxi-M group [56]. Given that most species here studied are vermivorous, this could mean that the mysterious target receptors of the conotoxins from the Mini-M group are more prominently present in worms, and studying them could reveal more about the nature of these conotoxins. Briefly, the 21 blast targets from the first two phylogeny trees belong to the most diverse and one of the most ubiquitous, yet enigmatic of the conotoxin superfamilies.
Both trees have 9 species, but while C. betulinus, C. coronatus, C. ebraeus, C. literatus, C. miliaris and C. sponsalis are common in A and B, C. ermineus, C. rattus and C. virgo are exclusive to Tree A, while C. judaeus. C. marmoreus and C. ventricosus are exclusive to Tree B. The species from Tree A are all vermivorous, with one exception, C. ermineus, which is piscivorous (Figure 4). The lack of inclusion of the rest of the piscivorous species besides C. ermineus is debated in Section 4.2. On the other hand, Tree B includes vermivorous species with the only molluscivorous, C. marmoreus (Figure 5). This tree (B) is constructed with 17 blast targets, having 2 less and sharing 15 with the previous one (A). Overall, these two phylogeny trees show the most shared, conserved sequences of conotoxins among vermivorous and molluscivorous species, which have potentially been lost for most of the piscivorous species during evolution.
Because neither Tree A nor B have incorporated all 3 diets, criteria B was carried out and the result was a single cluster with 53 branches. A total of 10 cone species are identified in this Tree C: C. betulinus, C. coronatus, C. ermineus, C. literatus, C. marmoreus, C. miliaris, C. quercinus, C. tribblei, C. ventricosus and C. virgo (Figure 6). All seven different blast targets on this tree are plasma-membrane zinc ion-binding proteins, contrasting with the previous trees where the blast targets are actual conotoxins. From the eight vermivorous snails the blast targets very widely, from another mollusc, an owl limpet (V3ZVY9_LOTGI), to polychaete sea worms (R7UA33_CAPTE, R7UQB9_CAPTE), shelled brachiopods (A0A2R2MJ28_LINUN), and interestingly even a coral species (A0A2B4RU89_STYPI). Then, for the one molluscivorous species, C. marmoreus, we got a metalloendopeptidase originating from a fellow marine invertebrate mollusc, a blue mussel (A0A8S3SD11_MYTED). Finally, for the piscivorous C. ermineus the blast target is from a white-tufted-ear marmoset (U3E5A6_CALJA). Although remaining a metalloendopeptidase, its vertebrate origin mirrors the before mentioned affinity of the fish-eating cone snails’ venom to vertebrates, showcasing the reason of its deadliness in humans, for example. Altogether, this phylogeny tree shows the regulatory peptidase sequences expressed in the venoms of cone snails and conserved in all feeding habits.
3.4. AMP Prediction Results
3.4.1. AMP Potential of the Shared Venom Transcripts
A total of 16 AMP sequences belonging to 7 potential AMP kinds were observed, as resumed in Table 2. The first AMP sequence comes from a C. betulinus matching with a pulmonary surfactant-associated protein B precursor. Next, another single sequence from a C. ebraeus shows similarity with a ribonuclease. From C. ermineus, 4 sequences show anti-microbial potential, all related to an antimicrobial NK-lysin precursor. Finally, the last AMP comes from 10 different sequences of venom-related transcriptomes of 2 different Conus species which show similarity with the human ACE2 protein. Of these 10 sequences, coming from the venom duct we found 4 sequences from 2 transcriptomes of C. literatus, 2 sequences from 2 transcriptomes of C. miliaris, another 2 sequences but from just 1 transcriptome of C. imperialis, and finally 1 sequence from a transcriptome of C. ventricosus and another from a C. quercinus. As these AMP sequences are not related to conotoxins, they are likely from other kinds of venom peptides.
3.4.2. Broad Transcriptomes AMP Potential
A total of 88 different potential AMPs were predicted with AMPfinder (Appendix E) by targeting all 76 transcriptomes. Among them, 15 were exclusively present in the venom-related ones, 17 were exclusive to other tissues, and 56 can be found in both categories. Sorting the dataset considering the significance of transcriptome size in venom-related transcriptomes (Figure 7) revealed a general but erratic trend of more AMPs predicted in assemblies larger than 60 megabytes (Mb).
However, since cone snails are specialized in hunting either fish, molluscs or worms, the transcriptomic data was grouped according to predatory diet while still sorted by transcriptome assembly size (Figure 8). Fortunately, there are representative species of all feeding habits, although just one molluscivorous. Starting with the mollusc diet, the results are very modest, but provide a starting context for what could be expected with such assembly size. Next, the results show a very clear peak of AMPs predicted in fish eating Conus transcriptomes, from 83 in the start, to 128 then reaching the high towering numbers of 432 and 456 in transcriptomes of C. consors. The following feeding habit to the right, the vermivorous one, possesses the greatest number of exemplars (55 transcriptomes). Despite sudden heights and drops, a trend of increasing AMP number along growing assembly size can be noticed, especially in the transcriptomes larger than 60 Mb, although not reaching the same levels verified in the fish-eating diet.
4. Discussion
4.1. Symbiosis in Venom Organs
Sorocarp morphogenesis (GO:0031288), negative regulation of asexual reproduction (GO:1903665), and – admittedly to a lesser extent – positive regulation of chemorepellent activity (GO:1903669) are indications of some kind of sporulating bodies existing specifically in the venom organs of all 20 Conus species analysed. These annotations are clearly unrelated to any biological process or molecular function of cone snails, indeed being described in the Gene Ontology knowledgebase for amoebas (e.g., Dictyostelium discoideum) and fungal organisms.
The suspicion of cone snails hosting some kind of symbiosis specifically in the venom organs is not new. Indeed, in 2009 a close symbiotic relationship in the venom duct of three cone snail species was revealed [39]. After exposing the existence of a thriving symbiotic actinomycete community, its results suggested that certain species of symbionts may be hosted in all species of cone snail, while other groups may be specific to their host. In 2017, another study rather unequivocally demonstrated that “cone snails have a distinct and consistent bacterial community” [40]. This research found diverse gene sequences closely matching Stenotrophomonas bacteria in all venom ducts examined. Such results were based on a worldwide variety of Conus species and showed that the 16S rRNA sequences retrieved represented specific symbiotic bacteria habiting the venom duct precisely where bioactive venom peptides are produced. More recently in 2023, it has been study hypothesised the existence of cone snail symbiosis with a seemingly new worm species found inside the egg capsules of C. ermineus [41]. Among several discoveries, the novel annelid species, Dimorphilus oophagus sp. nov. is contemplated as possible symbiont on account of isolated specimens not surviving more than two days in captivity. While the reason could be related to a normal short life cycle (also seemingly typical in species of the Dinophilidae family), it could also be the case that these specimens cannot survive outside its host egg masses because of their hypothetical parasite, commensal, or mutualist symbiotic nature.
Conclusively, studying more cone snails is essential to test, confirm and explore these symbiotic hypotheses. However, the discovery of GO:0031288, GO:1903665, and GO:1903669 in all transcriptomes of venom-related tissues assumes paramount importance in serving as compelling evidence to support the proposition of symbiotic microorganisms in the venom apparatus of all Conus species.
4.2. Predation Impact on Venom Evolution
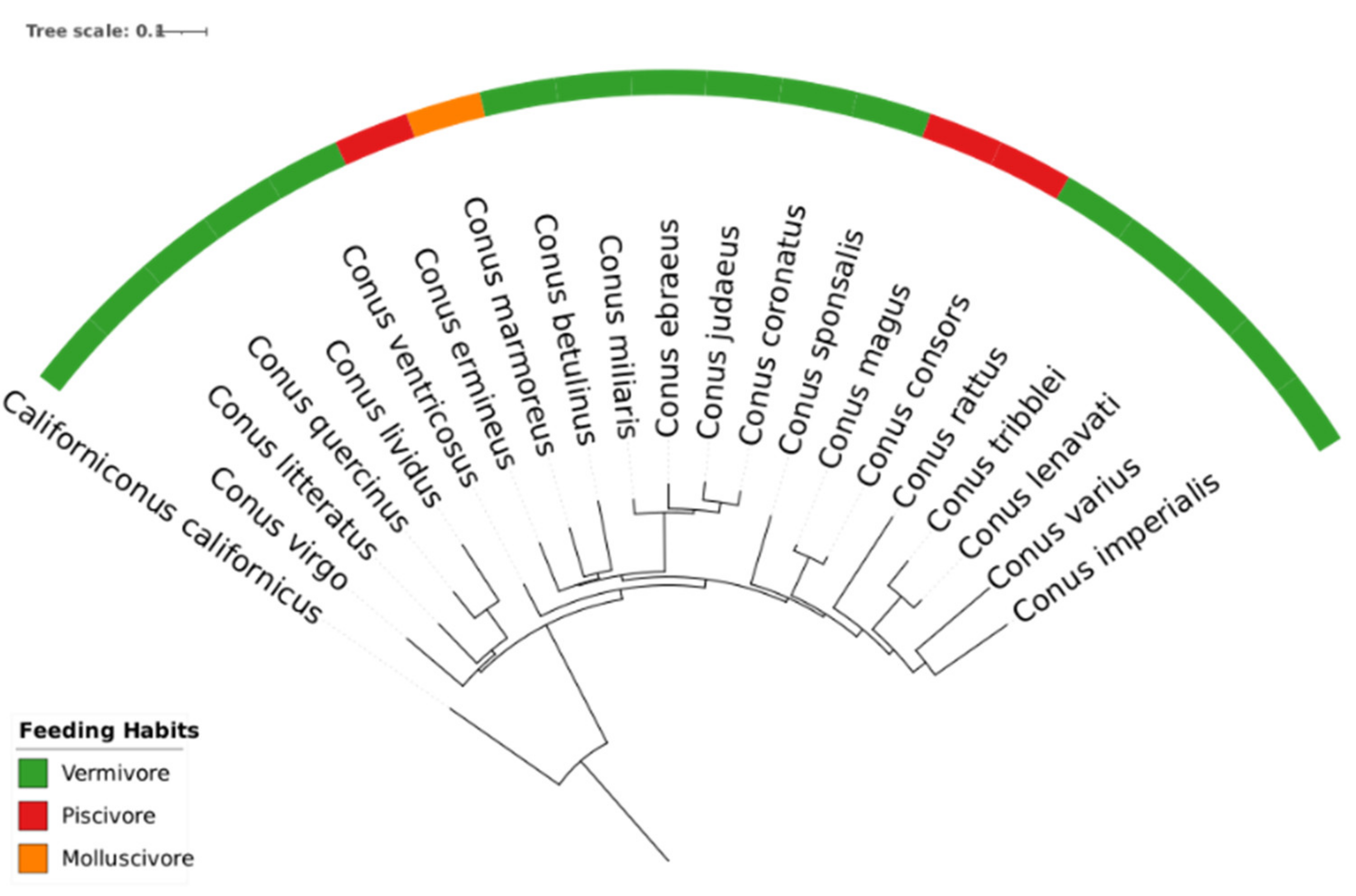
The present study focuses on 20 cone snail species, a selection representing approximately 2% of the estimated total genus diversity (exceeding 800 recognized species). Despite the limited sampling size, there is a comprehensive representation all major phylogenetical clades, providing a good overall coverage of this genus’ evolutionary diversity. This is supported by comparing our phylogeny tree (elaborated for the 20 species using 16S, see Appendix F) with a more extensive phylogenetic analysis involving over 350 Conus (elaborated with barcode sequences, including 12S, 16S and COI) [57].
The first clustering experiment (following criteria A) failed to generate a single tree, instead producing two trees (A and B, Figure 4 and 5, respectively) with an impressive array of toxins belonging to the most diverse and ubiquitous superfamily of conotoxins. This result prompted the lowering of the overall similarity threshold (criteria B), resulting in the creation of Tree C (Figure 6). The reason for criteria A to consistently produce two trees instead of one can only be understood when the predatory diet of each species is considered, as Tree A clusters together vermivorous and a piscivorous species and Tree B clusters the vermivorous and molluscivorous species. While B and even C trees connected harmoniously the vermivorous and molluscivorous species, there was a general failure to incorporate piscivorous species. Interestingly, the only piscivorous cone snail in both trees A and C is C. ermineus, not C. consors or C. magus, and we argue that is not a coincidence. Based on genetic studies performed in this work (see Appendix F) and by our group in the past, C. ermineus does not seem to share any immediate cladistical link with other piscivorous cone snails, in fact being more closely related to vermivorous cone snails [57]. Moreover, despite being classified as piscivore, C. ermineus’ feeding habit remains somewhat confusing [58]. Its inclusion in the cluster with other vermivorous species indicates that it might still be in the process of piscivory specialization, convergently evolving a piscivorous diet similar to other fish-eating Conus [59]. A surprising recent discovery showed that even established piscivorous cone snails (like C. magus) feed on sea worms in its early juvenile age, further illustrating the higher complexity of the piscivorous diet and possibly increasing variation in venom gene expression during the development stage [60]. Following the second clustering attempt resulting in the C tree, there is a noticeable complexity drop in the blasted targets: from entire conotoxins in the first two clusters to smaller, ubiquitous zinc proteases in the last cluster with the three types of diets. The absence of more specialized piscivorous species in Tree A combined with the complexity drop of the Tree C points to a significant transcriptomic gap between the venoms of piscivorous species and the others. Indeed, the first clusters seem to indicate a whole group of genes conserved in the vermivorous and molluscivorous diets but mostly absent from the piscivorous one, excepting the transitory species C. ermineus. The reason for this might be connected with greater predatory advantages for worm-hunting rather than fish-hunting, resulting in specialization through the gradual loss of these genes (or their silencing after juvenile stage). The phenomenon of gene loss in venom evolution is described in various fellow venomous species, such as the previously mentioned rattlesnakes [34,35,36]. The notable distance in the transcriptomic content of piscivorous cone snail’ venoms might also have direct implications in the previously mentioned deadliness to vertebrates and especially to humans [61]. Another important factor in the evolution of piscivorous venoms is the reported recruitment of the presumably more powerful defensive toxins into the offensive predatory venom arsenal [62]. This behaviour could exacerbate significantly the difference between the piscivorous species and the others, helping explain the absence of more specialized fish hunting snails in our results.
In essence, these results suggest that venom evolution and broad variability is directly influenced by preying selections. Recently, an article expanded on the issue of venom evolution shifts in these marine snails by analysing the structure of about 3,000 toxin genes and the overall venom composition from 42 species of cone snails [63]. Such research reached the conclusion that prey-specialization is a crucial factor driving the variability of the venom, especially in piscivorous species. Thus, the feeding habit emerges as a crucial clue to understand the seeming irregular variability in these gastropods’ venom.
4.3. Biomedical Findings in Conus Venoms
A prediction of 88 potential AMPs (see Appendix E) is an indication that the Conus venom might be an interesting source of these peptides, but the results show a numerically biased distribution towards piscivorous species and larger transcriptomes (data size above 60 Mb) (Figure 8). Comparatively, vermivorous species also show an increasing number of potential AMPs in larger transcriptomes, but the trend shows inconsistencies and the overall number of AMPs is lower than in fish-eating Conus. As there is only one mollusc-eating species present in this study, any picture regarding the reliability of AMP finding in Conus with this specific diet is difficult to draw. However, greater numbers of piscivorous and molluscivorous species (and transcriptomes) need to be evaluated for a stronger (in the former) and statistically meaningful (in the latter) AMP prediction.
The reason for the greater number of AMPs predicted in piscivorous species might be connected to their large and good-quality transcriptomes (above 100 Mb) (see Appendix G). Indeed, two out of four of these transcriptomes possess the highest and third highest transcriptome sizes of the whole dataset (167 and 139 megabytes, respectively). However, the deadlier nature of fish-eating Conus venoms to vertebrates could also be influencing the results, as AMPfinder’s database more prominently integrates peptides which directly target other vertebrates (75% of the AMPs in the database of AMPfinder originate from animals that hunt other vertebrates) [61,64,65]. This fact may influence the counting of AMPs as this tool’s This may eventually create a bias for similar stats of peptide length, electrical charge, hydrophobicity and other characteristics such as structural features. Thus, as the predatory venom from molluscivorous and vermivorous species is harmless to humans, it could be safer to develop clinical applications products such as AMPs from the conopeptides of non-fish-eating species.
4.4. ACE2 similarity
We identified a short transcriptomic sequence with AMP potential (see Table 2) exhibiting remarkable similarity to the human angiotensin-converting enzyme 2 (ACE2) protein, plus other sequences showing some analogy with ACE protein from a variety of animals such as the crown-of-thorns starfish and the Golden apple snail (see Appendix D). Although significantly shorter than the full-length ACE2 gene (36 amino acids in our result to 805 amino acids of the ACE2 gene), the sequence homology suggests a potential structural convergence or similar receptor-binding domains. The ACE2 protein is a critical component in human physiology and virology, being a key enzyme in the renin-angiotensin system (RAS) [66]. However, this finding is particularly intriguing given the fact that ACE2 is the primary cellular receptor for SARS-CoV-2, the virus responsible for COVID-19 [67,68].
As previous studies demonstrate the furin cleavage site of the viral Spike protein and certain conotoxins share similarities, we hypothesize that the short sequences we found may encode protein fragments that mimic a portion of the ACE2 receptor binding domain, which could potentially interfere with viral attachment and entry into host cells [69,70]. Further studies should be conducted on the functional significance of these short sequences. Bioinformatic analysis can help identify conserved domains and motifs, while protein structure prediction can provide insights into the three-dimensional conformation and potential binding sites. Functional validation experiments, including in vitro binding assays and viral infection models, are crucial to determine if these fragments can indeed antagonize the Spike protein and inhibit viral entry. The potential implications of this discovery extend beyond our current understanding of cone snail venom. These short sequences, if proven to interfere with viral-ACE2 interactions, could serve as promising scaffolds for the development of novel therapeutic agents against SARS-CoV-2 and potentially other viruses that utilize ACE2 for host cell entry. Furthermore, the structural similarity between these fragments and ACE2 could be harnessed for the design of biosensors for rapid and specific viral detection.
5. Conclusions
In conclusion, our novel strategy in the study of Conus species combined a broad transcriptomics approach which encapsulated all publicly available data with a biomedical prediction model, contributing to further comprehend and unveil hidden potentials behind the complex predatory venom of these fascinating marine gastropods. In summary, our findings not only shed light on venom evolutionary traits and variability, but also hold promise for the development of novel antimicrobial therapeutics.
This research revealed an intrinsic link between venom evolution and predatory diet by showing that the main conopeptides conserved in vermivorous and molluscivorous species are absent in specialized piscivorous species. Their absence manifests the evolutionary distance and the probably more sophisticated nature of the piscivorous species venom compared to the others. Furthermore, transcriptomic evidence of symbiotic relationships within the venom organs was detected with the surprising findings of shared transcripts related to the symbiont’s biology. On account of their presence in all venom transcriptomes, this discovery points to the existence of ubiquitous symbiotic microorganisms in cone snails. Secondly, by applying a comparative bioinformatic model to the venom-transcriptomes, a total of 88 potential AMPs were predicted. This suggests that the Conus marine venoms may present a viable source of AMPs, much sought for to counter the current growing challenge of drug resistant trend inflammatory pathogens. In addition, the finding of ACE2-like sequences opens an exciting door for future research.
For forthcoming investigations on the biomedical potential of conopeptides, the methodology should include machine learning prediction models beyond the comparative database bioinformatic method presented here. The employment of such in-silico mechanisms would both increase the range and confidence of the prediction result. In addition to AMPs, prediction of potential anti-cancer peptides could be targeted as well, given the identical methodology between the two objectives. Regarding Conus venom variability and evolution, we propose the standardization of a feeding habit context, not only in Conus, but also in other groups encompassing closely related species that exhibit different feeding habits.
Author Contributions
Conceptualization, J.M., Y.Z. and A.A.; methodology, J.M. and Y.Z.; software, J.M. and Y.Z.; validation, A.A. and L.H.; formal analysis, J.M. and Y.Z.; investigation, J.M. and Y.Z.; resources, A.A. and L.H.; data curation, J.M. and Y.Z.; writing—original draft preparation, J.M. and Y.Z.; writing—review and editing, A.A., L.H., and Y.Z.; visualization, A.A., L.H., J.M. and Y.Z.; supervision, A.A.; project administration, A.A.; funding acquisition, A.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the national funds from Fundação para a Ciência e a Tecnologia (FCT) under Grant Nos. 2023.03371.BD, UIDB/04423/2020, and UIDP/04423/2020; the European Regional Development Fund (ERDF) through the framework of the program PT 2020; the European Structural and Investment Funds (ESIF) via the Competitiveness and Internationalization Operational Program (COMPETE 2020); and the National Natural Science Foundation of China (Grant No.42077212);
Data Availability Statement
The original transcriptomic data supporting the retrieved results is publicly available.
Acknowledgments
We thank the Evolutionary Genomics and Bioinformatics (EGB) group for transcriptomics guidance and the College of Chemistry and Chemical Engineering, Tianjin University of Technology for technical support in the field of microbiology.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACE2 | Angiotensin-Converting Enzyme 2 |
| AMP(s) | Anti-Microbial Peptide(s) |
| Mb(s) | Megabyte(s) |
| PTM(s) | Post-Transcriptional Modification(s) |
Appendix A

Table A1.
The 14 GO IDs chosen from the 29 shared exclusively in the venom-related transcriptomes.
| GO ID | GO Category | GO Term |
|---|---|---|
| GO:0003081 | Biological process | Regulation of systemic arterial blood pressure by renin-angiotensin |
| GO:0003084 | Biological process | Positive regulation of systemic arterial blood pressure |
| GO:0019882 | Biological process | Antigen processing and presentation |
| GO:0030212 | Biological process | Hyaluronan metabolic process |
| GO:0031288 | Biological process | Sorocarp morphogenesis |
| GO:0071577 | Biological process | Zinc ion transmembrane transport |
| GO:0097067 | Biological process | Cellular response to thyroid hormone stimulus |
| GO:0140206 | Biological process | Dipeptide import across plasma membrane |
| GO:1903052 | Biological process | Positive regulation of proteolysis involved in protein catabolic process |
| GO:1903665 | Biological process | Negative regulation of asexual reproduction |
| GO:1903669 | Biological process | Positive regulation of chemorepellent activity |
| GO:0016532 | Molecular function | Superoxide dismutase copper chaperone activity |
| GO:0016671 | Molecular function | Oxidoreductase activity; acting on a sulphur group of donors; disulphide as acceptor |
| GO:0031545 | Molecular function | Peptidyl-proline 4-dioxygenase activity |
Appendix B
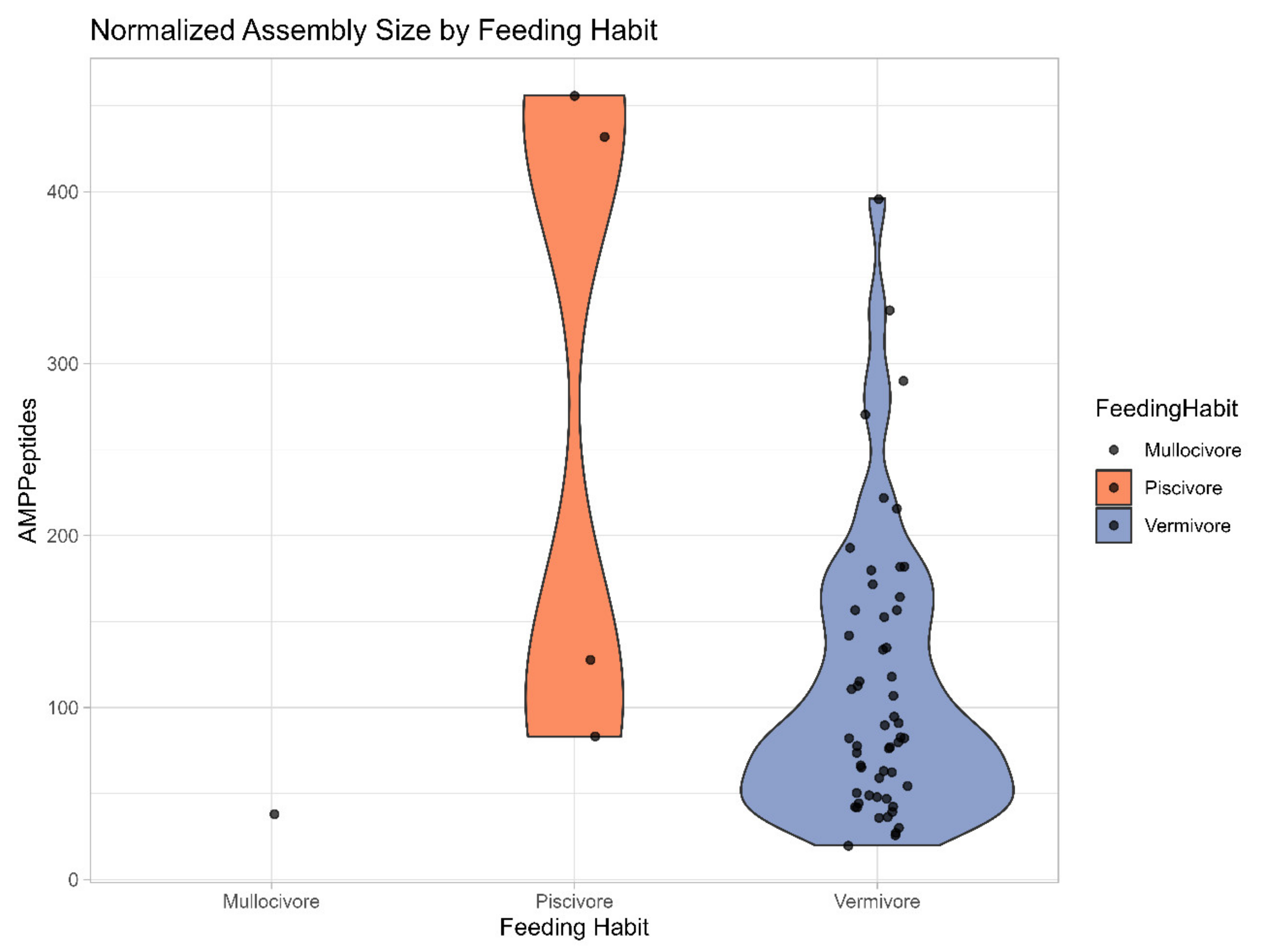
Figure A1.
Violin Plot generated with R software, showing the distribution of AMPs grouped by feeding habit (molluscivore on the left, piscivore in the middle and vermivore on the right).
Figure A1.
Violin Plot generated with R software, showing the distribution of AMPs grouped by feeding habit (molluscivore on the left, piscivore in the middle and vermivore on the right).
Appendix C
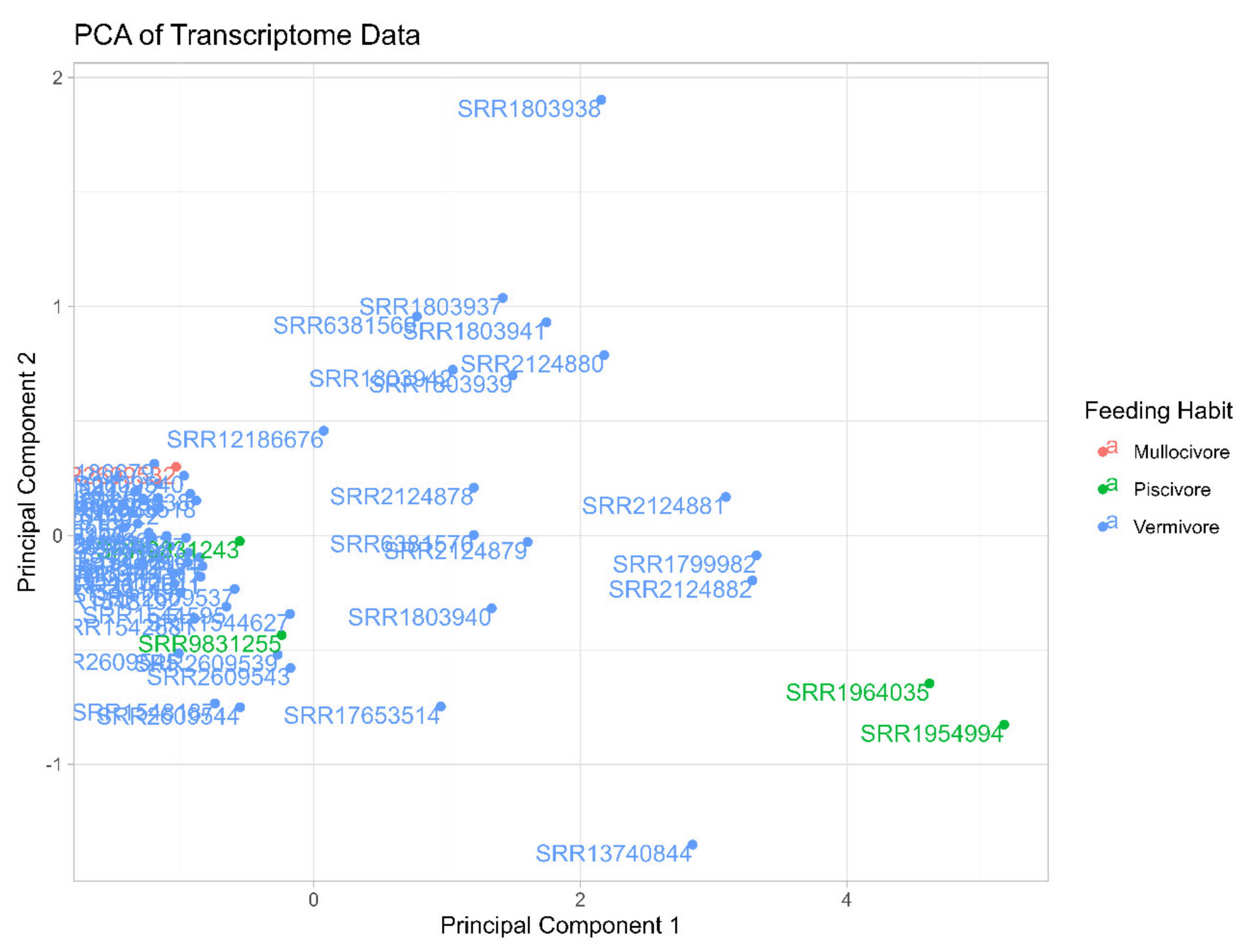
Figure A2.
Principal Component Analysis (PCA) plot demonstrating the different distributions of transcriptomes according to their AMP number and coloured by predatory diet (pink for molluscivore, green for piscivore and blue for vermivore).
Figure A2.
Principal Component Analysis (PCA) plot demonstrating the different distributions of transcriptomes according to their AMP number and coloured by predatory diet (pink for molluscivore, green for piscivore and blue for vermivore).
Appendix D
Table A2.
11 Conus transcriptomic sequences with great similarity with the Angiotensin-Converting Enzyme gene.
Table A2.
11 Conus transcriptomic sequences with great similarity with the Angiotensin-Converting Enzyme gene.
| Sequences | Percentage of Identity |
|---|---|
| >SRR1544119_TRINITY_DN22969_c0_g1_i1 | 51,5% LOC106073291 Biomphalaria glabrata (Bloodfluke planorb) (Freshwater snail) |
| CGGCACTGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTAGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCAAGTGTGCTTTACGGAAGGCAACTGTATCTCCCTGGATCCAGACCTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGACTGAAGCCTGGGAGCTGTGGAGAAAGGCCACTGGAGGAAAAATGAAAGCCCTGTACGAGGAGTATGTTGAGCTGGCGAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGATGACTACGAAGACGAA | |
| >SRR1544120_TRINITY_DN2907_c0_g1_i1 | 50% LOC106073291 Biomphalaria glabrata (Bloodfluke planorb) (Freshwater snail) |
| TTGCCAACGCCACCCAGCGGCGCCTGCTGAAGCAAATCCGCAAGATCGGCACGGCGGCACAGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTAGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCAGGTGTGCTTTACGGAAGACAACTGTATCTCCCTGGATCCAGACCTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGACTGAAGCCTGGGATCTGTGGAGAAAGGCCACTGGAGGGAAAATGAAAGCCCTGTACGAGGAGTATGTTGAGCTGGCGAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGATGACTACGAAGACGAA | |
| >SRR1544137_TRINITY_DN16819_c0_g1_i1 | 51,5% LOC106073291 Biomphalaria glabrata (Bloodfluke planorb) (Freshwater snail) |
| CGGCACTGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTAGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCAAGTGTGCTTTACGGAAGGCAACTGTATCTCCCTGGATCCAGACCTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGACTGAAGCCTGGGAGCTGTGGAGAAAGGCCACTGGAGGAAAAATGAAAGCCCTGTACGAGGAGTATGTTGAGCTGGCGAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGATGACTACGAAGACGAA | |
| >SRR1544140_TRINITY_DN13380_c0_g1_i1 | 52,9% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| AGAACCTGGCGAGGAGCTGGCTACAGAAATACAACCAAGAGCACAAAGACATTTTCTCCAAGTCCTCAGAAATGACCTGGAACTACGCTACCAACGTCACGGACGAAATTCAACAAAAACAAGTGAATGCAGAGCTGCAGGTGGCGCAATGGCAACAGGAGAAGGCAGCAGAAGTGGAACATTACGACTGGGAGCATTTTTCCGACAGCAGTCTTGTACGTCAGTTCCGTTTTGCGAGGAATATAGGCACGTCTGCCATG | |
| >SRR1544595_TRINITY_DN5707_c0_g1_i1 | 61,9% C0Q70_16356 Pomacea canaliculata (Golden apple snail) |
| CTGAGGCTGGACACAAACTGAGGGCCATGCTGTCCAAAGGATCGTCTGAGGTGTGGACAGTACCATTCCAGGCCCTGACAGGACAGACCAAGATGAGCGCACAATCACTGATCCAGTACTTCCAGCCCCTCATGGACTACCTGGAGCAGTACACCAAGGACCACGGCGTGGAGGTTGGGTGGAAGGAGGAGTGTTCT | |
| >SRR1544600_TRINITY_DN9395_c0_g1_i1 | 56,3% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| TCGTTTTTCATGGCAGACGTGCCTATATTCCTTGCAAAACGGAACTGACGTACAAGACTGCTGTCGGAAAAATGCTCCCAGTCGTAATGTTCCACTTCCGCTGCCTTCCTCTGTTGCCATTGCGCCACCTGCAGCTCTGCATTCACTTGTTTTTGTTGATTTTCGTCCGTGACGTTGGTAGCGTAGTTCCAGGTCATTTCTGAGGACTTGGAGACAATGTCTTTGTGCTCTTGGTTGTATTTCTGTAGCCAGCTCCTCGCCAGGTTCTC | |
| >SRR1544692_TRINITY_DN15875_c0_g1_i1 | 55,8% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| TCGTTGCTCAGCGCCACGAACTCCTCGTAATCGCTCTTCATCAGAGGCCCTGTGACGTCACGCCACTCCTTCCACGCCATCAGCAGTTCGTCATAGTCACGCGATGACGCCATCAGTTTGGTCAGTTCAGGATCCAGATTCAAAATGGCCCCGGTCTTTGGATCCTTCACTTTAGCTTTGGCGTAGATACCTTCAATGGTCGACTGTAGCTCTTTTAGCTCTTTCAGTTTAGTTTCGTTTTTCATGGCAGACGTGCCTATATTCCTTGCAAAACGGAACTGACGTACAAGACTGCTGTCGGAAAAATGCTCCCAGTCGTAATGTTCCACTTCCGCTGCCTTCCTCTGTTGCCATTGCGCCACCTGCAGCTCTGCATTCACTTGTTTTTGTTGATTTTCGTCCGTGACGTTGGTAGCGTAGTTCCAGGTCATTTCTGAGGACTTGGAGATAATGTCTTTGTGCTCTTTGTTGTATTTCTGTAGCCAGCTCCTCGCCAGGTTCTC | |
| >SRR1544692_TRINITY_DN2117_c0_g1_i1 | 68,3% LOC110975981 Acanthaster planci (Crown-of-thorns starfish) |
| TCCTCACTCCAGCCAACGGGCTGGCCGGCGTTCTGCTCCTCCAGCCAATCCTGAAGCGGCCTGAAGTACTCCAGCAGCGGCCTCACGTCCATGTGTCTGGTGCCCGTGATCTGCTCCAGGGCCTC | |
| >SRR6983166_TRINITY_DN44676_c0_g1_i1 | 56,8% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| ACAGCACCGCTCGAGTGTGCTTTACGGAAGACAACTGTATTCCCCTGGACCCGGACGTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGGCTGAAGCCTGGGATCTGTGGAGAGAGGCCACTGGAGGAGAAATGAAAGCCCTGTACGAGGAGTATGTTCAGCTGGGAAACGAAGGAGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGACGAGTATGAAGACGAA | |
| >SRR6983168_TRINITY_DN6724_c0_g2_i1 | 49% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| CGGACAGCGAGGCAGTGGAAGCCTTCTTGGAGACGCACGACAAGGAGACTAAGAAGAAGCATGAAAAGTACGAGATTCTGTCCTGGAATCACGAAACCAATATCACCGACTACAATCAGGAGCTGAAGGTCAACTACAGCGTAGAGATGTCAGAATTTGCTAAAGAGCACGCCAGGCAGTCGGCCATGTTTGACCTTGATCACCTTGCCAACGCCACCCAGCGGCGCCTGCTGAAGAAAATCGGCAAAATCGGCACGGCGGCACAGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTGGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCGAGTGTGCTTTACGGAAGACAACTGTATTCCCCTGGACCCGGACGTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGGCTGAAGCCTGGGATCTGTGGAGAGAGGCCACTGGAGGAGAAATGAAAGCCCTGTACGAGGAGTATGTTCAGCTGGGAAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGACGAGTATGAAGACGAAAATCTGCAAGAAGAGTTAGCGGCCCTGATGGAACAGCTCCGTCCTTTGTACGTGAAGCTCCAC | |
| >SRR6983169_TRINITY_DN17529_c0_g1_i1 | 51% C0Q70_02791 Pomacea canaliculata (Golden apple snail) |
| TTCTGTCCTGGAATCACGAAACCAATATCACCGACTACAATCAGGAGCTGAAGGTCAACTACAGCGTAGAGATGTCAGAATTTGCTAAAGAGCACGCCAGGCAGTCGGCCATGTTTGACCTTGATCACCTTGCCAACGCCACCCAGCGGCGCCTGCTGAAGAAAATCGGCAAAATCGGCACGGCGGCACAGAAGGACGAGAACAAAGTGGCACGGTTTAACGAGCTGGTGGCCAAGATGTCGGAGATCTACAGCACCGCTCGAGTGTGCTTTACGGAAGACAACTGTATTCCCCTGGACCCGGACGTCAAACGGCTCTTTGAACACAGCAGAAACTACACATTGCTGGCTGAAGCCTGGGATCTGTGGAGAGAGGCCACTGGAGGAGAAATGAAAGCCCTGTACGAGGAGTATGTTCAGCTGGGAAACGAAGGCGTTCAGGAACTGGGTTTCAACGACATGGGAGAGTACTGGCGAGACGAGTATGAAGACGAA |
Appendix E
Table A3.
AMPfinder software prediction results; each AMP is unique, attesting to its AMP database ID; the Protein ID columns show the function of the sequence hit obtained and the Abundance columns show the number of each particular AMP; the colours represent the tissue origin of each AMP: green for AMPs coming from the venom-related transcriptomes, yellow for venom-unrelated transcriptomes and red for those present in both groups.
Table A3.
AMPfinder software prediction results; each AMP is unique, attesting to its AMP database ID; the Protein ID columns show the function of the sequence hit obtained and the Abundance columns show the number of each particular AMP; the colours represent the tissue origin of each AMP: green for AMPs coming from the venom-related transcriptomes, yellow for venom-unrelated transcriptomes and red for those present in both groups.
| Protein ID | AMP ID | Abundance | Protein ID | AMP ID | Abundance |
|---|---|---|---|---|---|
| P83578|IKP1_PHYSA | AMP_11771 | 7 | P20160|CAP7_HUMAN | AMP_14282 | 20 |
| P61095|SFI1_SEGFL | AMP_15699 | 4 | P0DKT2|TU92_GEMSO | AMP_25599 | 20 |
| Q90WJ8|AJL2_ANGJA | AMP_00722 | 3 | P20160|CAP7_HUMAN | AMP_14283 | 16 |
| P39060|COIA1_HUMAN | AMP_14664 | 3 | P00738|HPT_HUMAN | AMP_04692 | 15 |
| P01038|CYT_CHICK | AMP_23459 | 2 | P05484|O17A_CONMA | AMP_18775 | 11 |
| P0C2D2|OXLA_CRODC | AMP_00139 | 1 | P05484|O17A_CONMA | AMP_18778 | 11 |
| Q9JHY3|WFD12_MOUSE | AMP_02769 | 1 | P05484|O17A_CONMA | AMP_18779 | 11 |
| P22887|NDKC_DICDI | AMP_09422 | 1 | P05484|O17A_CONMA | AMP_18780 | 11 |
| P22355|PSPB_RAT | AMP_00195 | 1 | P05484|O17A_CONMA | AMP_18781 | 11 |
| Q29075|NKL_PIG | AMP_03456 | 1 | P06702|S10A9_HUMAN | AMP_09443 | 6 |
| Q29075|NKL_PIG | AMP_03457 | 1 | P06702|S10A9_HUMAN | AMP_11387 | 6 |
| Q29075|NKL_PIG | AMP_04409 | 1 | P05484|O17A_CONMA | AMP_27220 | 6 |
| Q29075|NKL_PIG | AMP_21444 | 1 | P05484|O17A_CONMA | AMP_27221 | 6 |
| Q8TDE3|RNAS8_HUMAN | AMP_05220 | 1 | P05484|O17A_CONMA | AMP_27222 | 6 |
| A0A7I2V2E9|A0A7I2V2E9_HUMAN | AMP_19944 | 10 | P83952|WAPA_OXYMI | AMP_04982 | 4 |
| P00974|BPT1_BOVIN | AMP_10608 | 76 | B5G6G7|WAPB_OXYMI | AMP_04983 | 4 |
| I2G9B4|VKT_MACLN | AMP_15720 | 76 | P83240|NDB31_PANIM | AMP_02393 | 2 |
| P20160|CAP7_HUMAN | AMP_04915 | 62 | P83240|NDB31_PANIM | AMP_02394 | 2 |
| P20160|CAP7_HUMAN | AMP_04916 | 62 | P83240|NDB31_PANIM | AMP_02395 | 2 |
| P20160|CAP7_HUMAN | AMP_04917 | 62 | A0A0C4G5K0|NDB3_HETSP | AMP_02396 | 2 |
| P20160|CAP7_HUMAN | AMP_04918 | 60 | P82427|WTX1E_NEOGO | AMP_02397 | 2 |
| P08311|CATG_HUMAN | AMP_04624 | 56 | P0DJ02|NDB49_HETPE | AMP_08335 | 2 |
| P86810|OXLA_SIGCA | AMP_11257 | 50 | A0A0C4G5K0|NDB3_HETSP | AMP_17099 | 2 |
| P86810|OXLA_SIGCA | AMP_11258 | 50 | P83240|NDB31_PANIM | AMP_22673 | 2 |
| Q4JHE1|OXLA_PSEAU | AMP_00142 | 48 | P83240|NDB31_PANIM | AMP_22674 | 2 |
| F8S0Z5|OXLA2_CROAD | AMP_00224 | 48 | P83240|NDB31_PANIM | AMP_22676 | 2 |
| P04284|PR06_SOLLC | AMP_07416 | 45 | P83240|NDB31_PANIM | AMP_22677 | 2 |
| P04284|PR06_SOLLC | AMP_10197 | 45 | P13487|KAX11_LEIHE | AMP_01368 | 1 |
| P81382|OXLA_CALRH | AMP_00135 | 44 | A9XE60|KBX11_MESEU | AMP_02691 | 1 |
| Q6TGQ8|OXLA_BOTMO | AMP_00137 | 44 | P82656|NDB21_HOFAZ | AMP_03049 | 1 |
| Q90W54|OXLA_GLOBL | AMP_00138 | 44 | F1AWB0|NDB27_VAEME | AMP_03190 | 1 |
| Q6STF1|OXLA_GLOHA | AMP_00422 | 44 | F1AWB0|NDB27_VAEME | AMP_03191 | 1 |
| Q6STF1|OXLA_GLOHA | AMP_00423 | 44 | P83239|NDB23_PANIM | AMP_03236 | 1 |
| Q6TGQ9|OXLA1_BOTJR | AMP_08879 | 43 | P83313|NDB24_OPICA | AMP_03238 | 1 |
| Q9U8W7|TL5B_TACTR | AMP_01246 | 42 | P83313|NDB24_OPICA | AMP_03239 | 1 |
| B5AR80|OXLA_BOTPA | AMP_00127 | 41 | P83314|NDB2S_OPICA | AMP_04307 | 1 |
| B5AR80|OXLA_BOTPA | AMP_00128 | 41 | A0A0C4G489|NDB2_HETSP | AMP_04308 | 1 |
| Q9U8W8|TL5A_TACTR | AMP_10191 | 41 | P0C2F4|KBX3_HETLA | AMP_04346 | 1 |
| P00734|THRB_HUMAN | AMP_09738 | 39 | P56972|KBX3_PANIM | AMP_04348 | 1 |
| P00734|THRB_HUMAN | AMP_09739 | 39 | Q0GY40|KBX3_HOFGE | AMP_04390 | 1 |
| P20160|CAP7_HUMAN | AMP_14281 | 29 | Q5WR03|KBX31_OPICA | AMP_05482 | 1 |
| Q1PHZ4|VM3B1_BOTJR | AMP_01520 | 27 | P13487|KAX11_LEIHE | AMP_10082 | 1 |
| Q6IWZ0|OXLA_APLCA | AMP_01042 | 26 | P0C2F4|KBX3_HETLA | AMP_17928 | 1 |
| Q6IWZ0|OXLA_APLCA | AMP_01043 | 26 | A9XE60|KBX11_MESEU | AMP_23221 | 1 |
Appendix F
Figure A3.
Phylogeny tree of 21 Conus species using two rRNA 16S genes for each species. The colours represent feeding habits: green for vermivorous, red for piscivorous, and orange for molluscivorous; Conus californicus in the root serves as the reference outgroup species.
Figure A3.
Phylogeny tree of 21 Conus species using two rRNA 16S genes for each species. The colours represent feeding habits: green for vermivorous, red for piscivorous, and orange for molluscivorous; Conus californicus in the root serves as the reference outgroup species.
Appendix G
Table A4.
BUSCO assessment performed against BUSCO’s Metazoa set for all 76 transcriptome assemblies.
Table A4.
BUSCO assessment performed against BUSCO’s Metazoa set for all 76 transcriptome assemblies.
| Species and Tissue | Completed - Total | Complete - Single Copy | Complete - Duplicated | Fragmented | Missing |
|---|---|---|---|---|---|
| C. miliaris Venom duct | 110 | 94 | 16 | 75 | 769 |
| C. miliaris Venom duct | 122 | 102 | 20 | 85 | 747 |
| C. coronatus Venom duct | 122 | 108 | 14 | 74 | 758 |
| C. miliaris Venom duct | 128 | 109 | 19 | 98 | 728 |
| C. miliaris Venom duct | 128 | 110 | 18 | 98 | 728 |
| C. miliaris Venom duct | 142 | 119 | 23 | 116 | 696 |
| C. miliaris Venom duct | 153 | 129 | 24 | 108 | 693 |
| C. miliaris Venom duct | 153 | 129 | 24 | 106 | 695 |
| C. miliaris Venom duct | 166 | 135 | 31 | 118 | 670 |
| C. miliaris Venom duct | 161 | 136 | 25 | 98 | 695 |
| C. miliaris Venom duct | 174 | 138 | 36 | 113 | 667 |
| C. miliaris Venom duct | 164 | 140 | 24 | 138 | 652 |
| C. coronatus Venom duct | 166 | 144 | 22 | 122 | 666 |
| C. miliaris Venom duct | 174 | 145 | 29 | 95 | 685 |
| C. miliaris Venom duct | 180 | 151 | 29 | 133 | 641 |
| C. miliaris Venom duct | 176 | 151 | 25 | 104 | 674 |
| C. miliaris Venom duct | 191 | 161 | 30 | 135 | 628 |
| C. miliaris Venom duct | 191 | 162 | 29 | 135 | 628 |
| C. miliaris Venom duct | 190 | 163 | 27 | 111 | 653 |
| C. consors Foot | 244 | 163 | 81 | 129 | 581 |
| C. coronatus Venom duct | 192 | 166 | 26 | 128 | 634 |
| C. sponsalis Venom duct | 203 | 167 | 36 | 127 | 624 |
| C. miliaris Venom duct | 196 | 173 | 23 | 115 | 643 |
| C. miliaris Venom duct | 210 | 176 | 34 | 163 | 581 |
| C. miliaris Venom duct | 230 | 189 | 41 | 133 | 591 |
| C. imperialis Venom duct | 220 | 190 | 30 | 114 | 620 |
| C. virgo Venom duct | 210 | 191 | 19 | 150 | 594 |
| C. imperialis Venom duct | 227 | 192 | 35 | 162 | 565 |
| C. coronatus Venom duct | 222 | 193 | 29 | 114 | 618 |
| C. miliaris Venom duct | 241 | 196 | 45 | 148 | 565 |
| C. ebraeus Venom duct | 229 | 200 | 29 | 180 | 545 |
| C. imperialis Venom duct | 240 | 207 | 33 | 148 | 566 |
| C. ermineus Venom duct | 256 | 214 | 42 | 111 | 587 |
| C. lividus Venom duct | 259 | 215 | 44 | 189 | 506 |
| C. ermineus Venom duct | 256 | 219 | 37 | 184 | 514 |
| C. imperialis Venom duct | 251 | 222 | 29 | 125 | 578 |
| C. marmoreus Venom duct | 247 | 222 | 25 | 151 | 556 |
| C. miliaris Venom duct | 268 | 224 | 44 | 184 | 502 |
| C. ermineus Venom duct | 271 | 228 | 43 | 110 | 573 |
| C. ebraeus Venom duct | 286 | 232 | 54 | 86 | 582 |
| C. rattus Venom duct | 282 | 234 | 48 | 186 | 486 |
| C. quercinus Venom duct | 267 | 237 | 30 | 161 | 526 |
| C. ermineus Venom duct | 301 | 249 | 52 | 130 | 523 |
| C. varius Venom duct | 297 | 250 | 47 | 163 | 494 |
| C. ermineus Venom duct | 294 | 251 | 43 | 175 | 485 |
| C. tribblei Venom duct | 307 | 253 | 54 | 281 | 366 |
| C. tribblei Venom duct | 313 | 256 | 57 | 264 | 377 |
| C. magus Venom duct | 315 | 258 | 57 | 106 | 533 |
| C. tribblei Venom duct | 318 | 267 | 51 | 256 | 380 |
| C. lenavati Venom duct | 343 | 274 | 69 | 262 | 349 |
| C. lenavati Venom duct | 334 | 274 | 60 | 207 | 413 |
| C. ermineus Venom duct | 356 | 274 | 82 | 104 | 494 |
| C. magus Venom duct | 344 | 279 | 65 | 113 | 497 |
| C. lenavati Venom duct | 365 | 284 | 81 | 253 | 336 |
| C. betulinus Venom duct | 355 | 286 | 69 | 263 | 336 |
| C. ermineus Venom duct | 364 | 287 | 77 | 103 | 487 |
| C. betulinus Venom duct | 388 | 301 | 87 | 268 | 298 |
| C. ermineus Venom duct | 390 | 314 | 76 | 116 | 448 |
| C. judaeus Venom duct | 421 | 337 | 84 | 123 | 410 |
| C. litteratus Venom duct | 453 | 357 | 96 | 230 | 271 |
| C. litteratus Venom duct | 500 | 371 | 129 | 218 | 236 |
| C. ventricosus Foot | 471 | 382 | 89 | 121 | 362 |
| C. imperialis Venom duct | 460 | 385 | 75 | 170 | 324 |
| C. ventricosus Foot | 544 | 424 | 120 | 120 | 290 |
| C. ermineus Venom duct | 574 | 429 | 145 | 133 | 247 |
| C. consors Nervous ganglions | 771 | 446 | 325 | 94 | 89 |
| C. consors Venom duct | 750 | 458 | 292 | 90 | 114 |
| C. ventricosus Venom gland | 584 | 463 | 121 | 124 | 246 |
| C. betulinus Venom duct | 568 | 464 | 104 | 208 | 178 |
| C. consors Osphradium | 836 | 465 | 371 | 65 | 53 |
| C. consors Salivary glands | 694 | 480 | 214 | 130 | 130 |
| C. betulinus Venom duct | 662 | 485 | 177 | 154 | 138 |
| C. consors Proboscis | 826 | 489 | 337 | 63 | 65 |
| C. consors Venom bulb | 751 | 489 | 262 | 99 | 104 |
| C. betulinus Venom duct | 600 | 517 | 83 | 205 | 149 |
| C. tribblei Venom duct | 667 | 542 | 125 | 165 | 122 |
References
- C. H. Y. B. C. L. J. L. J. Z. K. Y. X. L. Z. H. Y. C. J. a. L. Y. Peng, “The first Conus genome assembly reveals a primary genetic central dogma of conopeptides in C. betulinus,” Cell Discov., vol. 7, nº 11, 2021.
- J. I. I. A. S. A. C. T. M. a. Z. R. Pardos-Blas, “The genome of the venomous snail Lautoconus ventricosus sheds light on the origin of conotoxin diversity,” Gigascience, vol. 10, 2021.
- J. R. P.-B. C. M. L. A. M. J. T. R. Z. Ana Herráez-Pérez, “Chromosome-level genome of the venomous snail Kalloconus canariensis: a valuable model for venomics and comparative genomics,” GigaScience, vol. 12, 2023.
- T. F. A. J. K. a. S. R. P. Duda Jr, “Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods,” Biol. J. Linn. Soc., vol. 73, p. 391–409, 2001.
- N. D. T. F. M. C. O. B. M. &. B. P. Puillandre, “One, four or 100 genera? A new classification of the cone snails,” J. Molluscan Stud., vol. 81, p. 1–23, 2015.
- B. P. C. Y. J. Y. Y. Z. J. &. S. Q. Gao, “Cone snails: A big store of conotoxins for novel drug discovery,” Toxins, vol. 9, nº 397, 2017.
- J. R. S. D. A. H. J. V. L. B. H. F. C. C. J. G. D. J. V. P. F. A. a. R. J. L. Prashanth, “The role of defensive ecological interactions in the evolution of conotoxins,” Mol. Ecol., vol. 25, p. 598–615, 2016.
- L. Nguyen, D. Craik e Kaas, “Bibliometric Review of the Literature on Cone Snail Peptide Toxins from 2000 to 2022,” Mar. Drugs, vol. 21, p. 154, 2023.
- M. R. Richard Georges, Panorama sur La Diversite des Conidae 110 Espèces Prédatrices des Plus Efficaces, 2021.
- R. a. C. D. Endean, “The venom apparatus of Conus magus,” Toxicon, vol. 4, p. 275–284., 1967.
- A.-H. M. M. S. D. S. W. A. H. Q. K. D. J. C. R. J. L. a. P. F. A. Jin, “Conotoxins: Chemistry and Biology,” Chem. Rev., nº 119, p. 11510–11549, 2019.
- K. B. M. M. S. D. Q. K. D. J. C. R. J. L. a. P. F. A. Akondi, “Discovery, synthesis, and structure: Activity relationships of conotoxins,” Chem. Rev., vol. 114, p. 5815–5847, 2014.
- O. G. B. a. B. M. O. Buczek, “Conotoxins and the posttranslational modification of secreted gene products,” Cell. Mol. Life Sci., vol. 62, p. 3067–3079, 2005.
- J. L. Z. L. J. S. I. A. A. V. V. B. M. O. a. E. W. S. Neves, “Small Molecules in the Cone Snail Arsenal,” vol. 17, p. 4933–4935, 2015.
- L. Y. S. X. a. C. W. Lu, “Various Conotoxin Diversifications Revealed by a Venomic Study of Conus flavidus,” Mol. Cell. Proteom., vol. 13, p. 105–118., 2014.
- A.-H. S. D. Q. K. V. L. P. K. R. J. L. a. P. F. A. Jin, “Transcriptomic messiness in the venom duct of Conus miles contributes to conotoxin diversity,” Mol. Cell. Proteom., vol. 12, p. 3824–3833, 2013.
- J. A. W. P. K. a. J. V. S. Jakubowski, “Screening for post-translational modifications in conotoxins using liquid chromatography/mass spectrometry: An important component of conotoxin discovery,” Toxicon, vol. 47, p. 688–699, 2006.
- Q. J.-C. W. a. D. J. C. Kaas, “Conopeptide characterization and classifications: An analysis using ConoServer,” Toxicon, vol. 55, p. 1491–1509., 2010.
- K. D. K. L. L. F. S. R. R. M. Laht S, “Identification and classification of conopeptides using profile Hidden Markov Models,” Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, vol. 1824, nº 3, pp. 488-492, 2012.
- Q. Y. R. J. A. D. S. a. C. D. Kaas, “ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database,” Nucleic Acids Res., vol. 40, p. D325–D330, 2011.
- J. C. H. a. M. F. Rivera-Ortiz, “Intraspecies variability and conopeptide profiling of the injected venom of Conus ermineus,” Peptides, vol. 32, p. 306–316, 2011.
- C. M. K. a. S. J. Prator, “Venom variation during prey capture by the cone snail, Conus textile,” PLoS ONE, vol. 9, 2014.
- D. S. D. M. L. V. J. A. L. R. a. A. Jin, “Transcriptomic-Proteomic Correlation in the Predation-Evoked Venom of the Cone Snail, Conus imperialis,” Marine drugs, vol. 17, nº 3, p. p.177, 2019.
- B. C. H. P. L. M. Y. N. a. C. N. Junqueira-de-Azevedo, “Venom-related transcripts from Bothrops jararaca tissues provide novel molecular insights into the production and evolution of snake venom,” Mol Biol Evol, vol. 32, p. 754–66, 2015.
- C. D. A. A. S. K. A. R. S. D. C. N. M. S. a. C. T. Reyes-Velasco, “Expression of venom gene homologs in diverse python tissues suggests a new model for the evolution of snake venom,” Mol Biol Evol, vol. 32, p. 173–83, 2015.
- S. M. H. M. L. D. a. M. J. Hargreaves, “Restriction and recruitment—gene duplication and the origin and evolution of snake venom toxins,” Genome Biol Evol, vol. 6, p. 2088– 95, 2014.
- S. R. P. Thomas F. Duda, “Molecular genetics of ecological diversification: Duplication and rapid evolution of toxin genes of the venomous gastropod Conus,” Biological Sciences, vol. 96, nº 12, pp. 6820-6823, 1999.
- N. W. M. O. B. Puillandre, “Evolution of Conus Peptide Genes: Duplication and Positive Selection in the A-Superfamily,” J Mol Evol, vol. 70, p. 190–202, 2010.
- T. F. D. J. Dan Chang, “Extensive and Continuous Duplication Facilitates Rapid Evolution and Diversification of Gene Families,” Molecular Biology and Evolution, vol. 29, nº 8, p. 2019–2029, 2012.
- a. B. K. Whittington, “Platypus venom genes expressed in non-venom tissues,” Aust J Zool, vol. 57, p. 199–202, 2009.
- E. A. U. S. A. A. T. N. J. J. D. H. S. T. R. D. M. L. C. D. W. R. N. L. v. d. W. N. V. K. R. I. H. S. P. Bryan G. Fry, “Squeezers and Leaf-cutters: Differential Diversification and Degeneration of the Venom System in Toxicoferan Reptiles,” Molecular & Cellular Proteomics, vol. 12, nº 7, pp. 1881-1899, 2013.
- R. D. E. C. H. S. J. D. T. G. F. K. T. J. N. J. A. N. R. J. L. R. S. N. C. R. a. R. C. R. d. l. V. Bryan G. Fry1, “The Toxicogenomic Multiverse: Convergent Recruitment of Proteins Into Animal Venoms,” Annual Review of Genomics and Human Genetics, vol. 10, pp. 483-511, 2009.
- K. B. Emily S.W. Wong, “Venom evolution through gene duplications,” Gene, vol. 496, nº 1, pp. 1-7, 2012.
- K. V. L. V. T. S. M. T. K. J. V. R. J. Jana Helsen, “Gene Loss Predictably Drives Evolutionary Adaptation,” Mol Biol Evol, vol. 37, nº 10, p. 2989–3002, 2020.
- H. J. G. R. L. F. B. E. L. &. M. H. Virag Sharma, “A genomics approach reveals insights into the importance of gene losses for mammalian adaptations,” Nature Communications, vol. 9, p. 1215, 2018.
- Casewell, “Venom Evolution: Gene Loss Shapes Phenotypic Adaptation,” Current Biology, vol. 26, nº 18, pp. R849-R851, 2016.
- Von Reumont, “Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide,” Toxins, vol. 10, nº 7, p. p.292, 2018.
- v. R. B. A. G. C. F. C. M. F. J. H. E. H. B. I. M. J. F. M. P. d. F. T. M. M. M. Y. N. A. P. J. T. A. T. F. V. R. Z. M. A. A. Zancolli G, “Web of venom: exploration of big data resources in animal toxin research,” GigaScience, p. giae054, 2024.
- B. J. H. R. L. A. C. G. O. B. S. E. Peraud O, “Microhabitats within venomous cone snails contain diverse actinobacteria,” Appl Environ Microbiol, vol. 75, nº 21, pp. 6820-6826, 2009.
- J. P. e. a. Torres, “Stenotrophomonas-Like Bacteria Are Widespread Symbionts in Cone Snail Venom Ducts,” Applied and environmental microbiology, vol. 83, nº 23, 2017.
- F. S.,. M. W.,. a. B. O. Matías L. Giglio, “Insights into a putative polychaete-gastropod symbiosis from a newly identified annelid worm that predates upon Conus ermineus eggs,” Contributions to Zoology, vol. 92, nº 2, pp. 97-111, 2023.
- D.-P. Y. M.-P. K. C.-M. A. A. Guillermin Agüero-Chapin, “Unveiling Encrypted Antimicrobial Peptides from Cephalopods’ Salivary Glands: A Proteolysis-Driven Virtual Approach,” ACS Omega, vol. 43, p. 43353–43367, 2024.
- G. A.-C. R. S. Y. M.-P. A. A. Ricardo Alexandre Barroso, “Unlocking Antimicrobial Peptides: In Silico Proteolysis and Artificial Intelligence-Driven Discovery from Cnidarian Omics,” Molecules, vol. 3, nº 550, 2025.
- J. Morim, “Deciphering the transcriptomics of the Conus species’ natural venoms [Master’s Dissertation, University of Porto],” 2022.
- J. S. Martin Steinegger, “MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets,” Nature Biotechnology, vol. 35, p. 1026–1028, 2017.
- R. K. D. H. Buchfink B, “Sensitive protein alignments at tree-of-life scale using DIAMOND,” Nature Methods, vol. 18, p. 366–368, 2021.
- G. O. F. M. Okonechnikov K, “Unipro UGENE: a unified bioinformatics toolkit,” Bioinformatics, vol. 8, p. 1166–1167, 2012.
- J. R. K. D. Y. Kazutaka Katoh, “MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization,” Briefings in Bioinformatics, vol. 20, nº 4, p. 1160–1166, 2019.
- T. N. B. Q. C. M. F. Rognes T, “VSEARCH: a versatile open source tool for metagenomics,” PeerJ, vol. 4, 2016.
- B. Ivica Letunic, “Interactive Tree of Life (iTOL) v6: recent updates to the phylogenetic tree display and annotation tool,” Nucleic Acids Research, vol. 52, pp. 78-82, 2024.
- Z. Y.,. X. N. Sen Yang, “AMPFinder: A computational model to identify antimicrobial peptides and their functions based on sequence-derived information,” Analytical Biochemistry, vol. 673, 2023.
- L. N. G. Jake R Conway, “UpSetR: an R package for the visualization of intersecting sets and their properties,” Bioinformatics, vol. 33, nº 18, p. 2938–2940, 2017.
- K. K. S. S. Gopal Krishna Patro, “Normalization: A Preprocessing Stage,” arXiv, 2015.
- T. M. O. A. P. C. McDougal OM, “Three-dimensional structure of conotoxin tx3a: an m-1 branch peptide of the M-superfamily.,” Biochemistry, vol. 47, p. 2826–2832, 2008.
- M. O. Jacob RB, “The M-superfamily of conotoxins: a review,” Cell Mol Life Sci, vol. 67, nº 1, pp. 17-27, 2010.
- J. R. J. E. W. M. W. C. C. C. G. J. M. O. L. W. G. W. H. D. R. J. M. J. C. L. O. B. Corpuz GP, “Definition of the M-conotoxin superfamily: characterization of novel peptides from molluscivorous Conus venoms,” vol. 44, p. 8176–8186, 2005.
- Y. a. A. A. Zhao, “Biomedical Potential of the Neglected Molluscivorous and Vermivorous Conus Species,” Marine drugs, vol. 20, nº 2, p. p.105, 2022.
- B. M. S. J. H. M. P. &. F. A. E. Olivera, “Prey-Capture Strategies of Fish-Hunting Cone Snails: Behavior, Neurobiology and Evolution,” Brain Behav Evol, vol. 86, nº 1, pp. 58-74, 2015.
- S. M. J. T. C. M. A. a. R. Z. Abalde, “Conotoxin Diversity in Chelyconus ermineus (Born, 1778) and the Convergent Origin of Piscivory in the Atlantic and Indo-Pacific Cones,” Genome biology and evolution, vol. 10, nº 10, pp. 2643-2662, 2018.
- S. W. A. H. R. J. L. Aymeric Rogalski, “Coordinated adaptations define the ontogenetic shift from worm- to fish-hunting in a venomous cone snail,” Nat Commun., vol. 14, nº 1, p.:3287, 2023.
- S. &. L. R. Dutertre, Snails: Biology, Ecology and Conservation, N. Gotsiridze-Columbus, Ed., Nova Science Publishers, 2011, pp. 85-104.
- S. J. A. V. I. H. B. S. K. L. V. D. V. F. B. A. A. V. D. a. A. P. Dutertre, “Evolution of separate predation- and defence-evoked venoms in carnivorous cone snails,” Nat. Commun., vol. 5, nº 3521, 2014.
- S. D. R. P. F. S. K. C. J. B. A. E. F. M. Y. B. M. O. H. S.-H. Thomas Lund Koch, “Prey Shifts Drive Venom Evolution in Cone Snails,” Molecular Biology and Evolution, vol. 41, nº 8, p. msae120, 2024.
- J. S. P. R. a. W. S. Kohn, “Preliminary studies on the venom of the marine snail Conus,” Ann N Y Acad Sci, vol. 90, pp. 706-725, 1960.
- L. X. W. Z. Wang G, “APD3: the antimicrobial peptide database as a tool for research and education,” Nucleic Acids Res, vol. 44, p. 1087–1093, 2016.
- R. H. K. T. A.-I. Seema Patel, “Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies,” Biomedicine & Pharmacotherapy, vol. 94, pp. 317-325, 2017.
- I. A. B. I. C. L. G. Z. S. D. G. T. S. R. A. R. a. M. A. Oliveira, “A potential interaction between the SARS-CoV-2 spike protein and nicotinic acetylcholine receptors,” Biophysical journal, vol. 120, nº 6, pp. 983-993, 2021.
- J. W. X. Lan J., “Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor,” Nature, vol. 581, pp. 215-220, 2020.
- J. A. Z. R. F. a. M. M. Changeux, “A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications,” Comptes Rendus. Biologies, vol. 343, nº 1, pp. 33-39, 2020.
- U. S. C. J. Camacho, “Omicron and Alpha P680H block SARS-CoV2 spike protein from accessing cholinergic inflammatory pathway via α9-nAChR mitigating the risk of MIS-C,” bioRxiv, 2022.
Figure 1.
Methodology workflow.
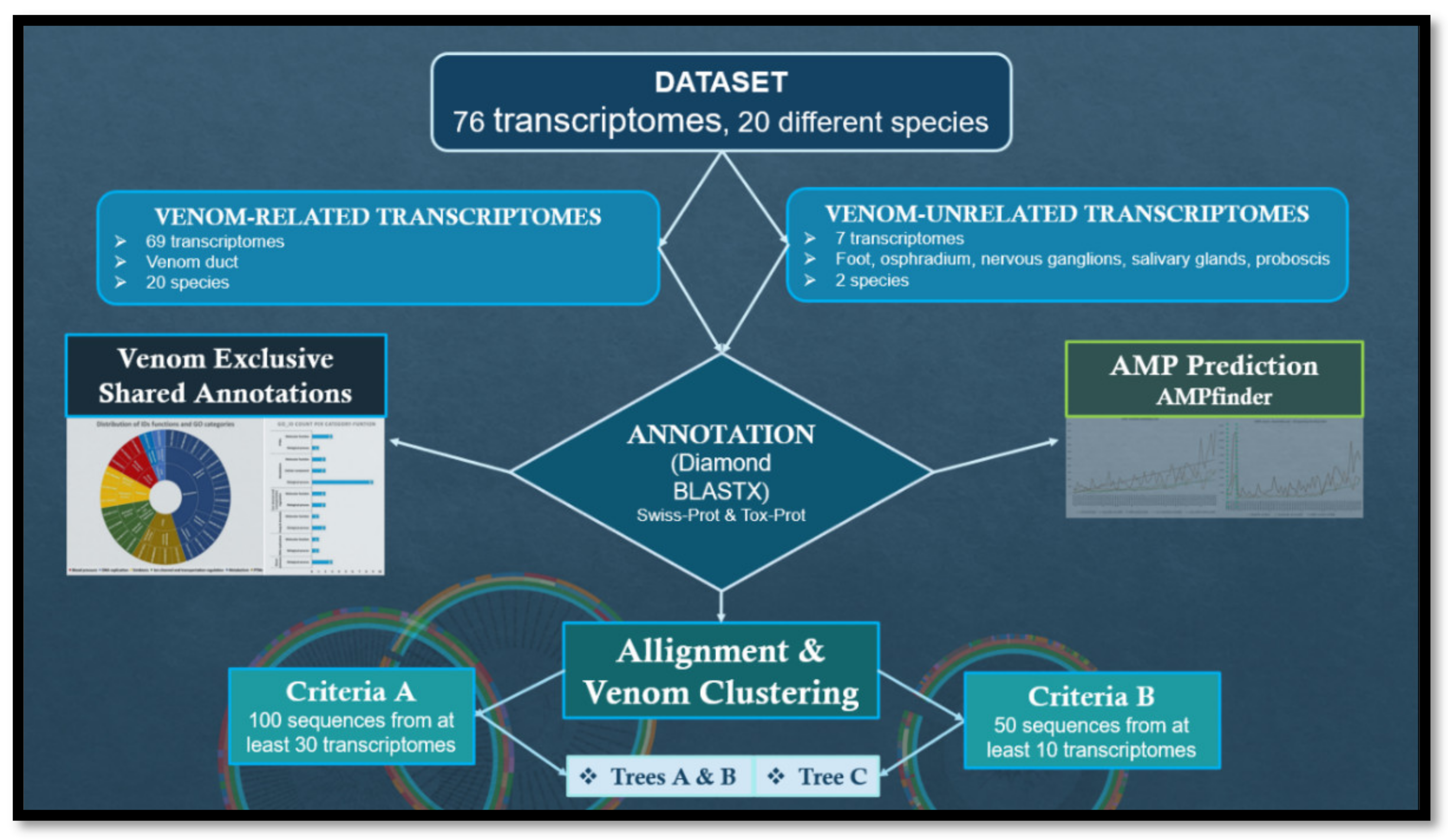
Figure 2.
Shared annotations (GO IDs) in the venom-unrelated (A) and in the venom-related (B) transcriptomes. The blue vertical line intersecting all files indicates the shared nature of the annotations: 5,913 in A and 2,104 in B. The side black bars indicate the total annotations attributed to each transcriptome individually.
Figure 2.
Shared annotations (GO IDs) in the venom-unrelated (A) and in the venom-related (B) transcriptomes. The blue vertical line intersecting all files indicates the shared nature of the annotations: 5,913 in A and 2,104 in B. The side black bars indicate the total annotations attributed to each transcriptome individually.
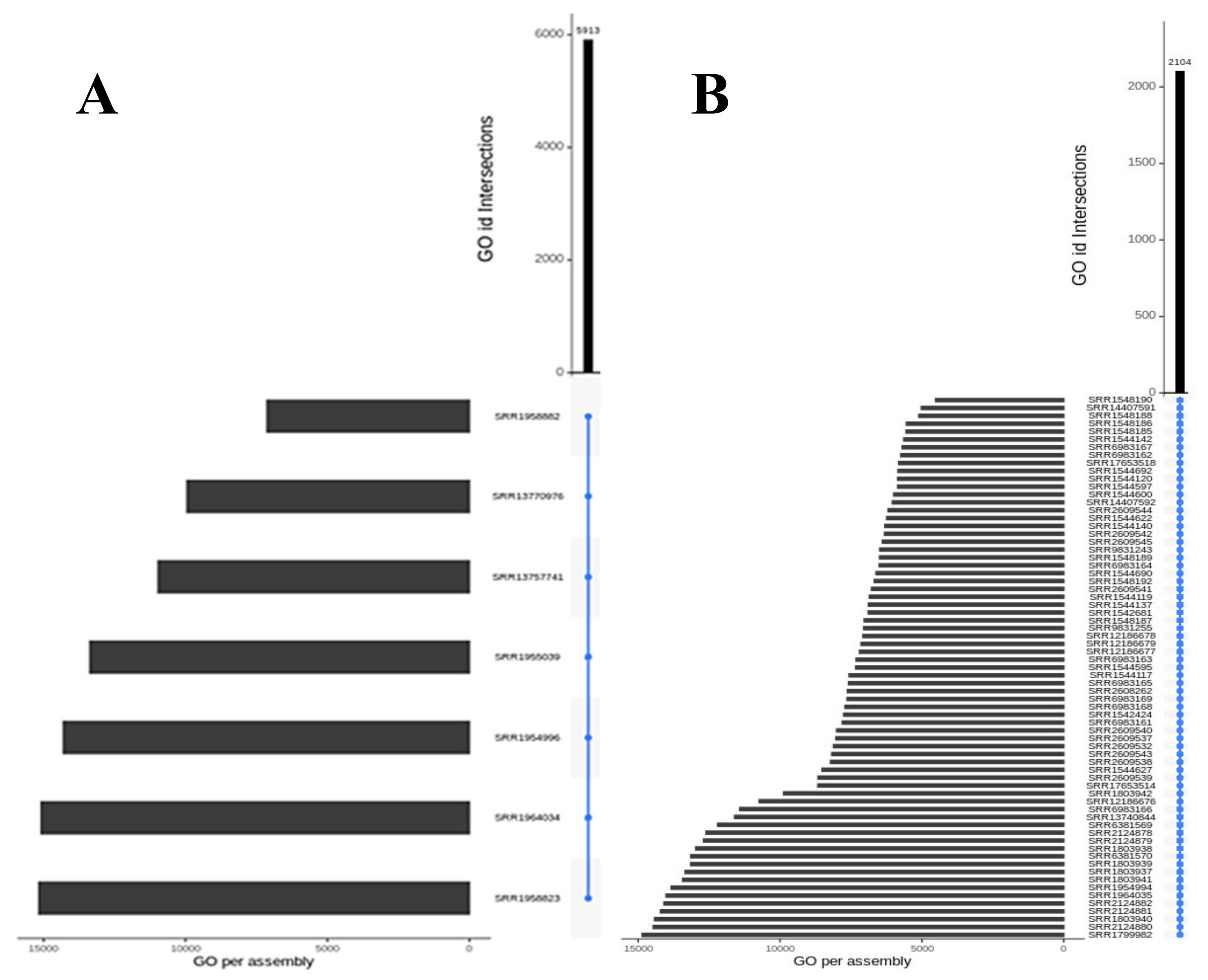
Figure 3.
The left, circular graph shows the distribution of the 29 GO IDs according to their six main functions and their GO category (e.g., “Biological Process” or “Molecular Function”). The bar graph on the right indicates ID count by GO category and designated major function.
Figure 3.
The left, circular graph shows the distribution of the 29 GO IDs according to their six main functions and their GO category (e.g., “Biological Process” or “Molecular Function”). The bar graph on the right indicates ID count by GO category and designated major function.
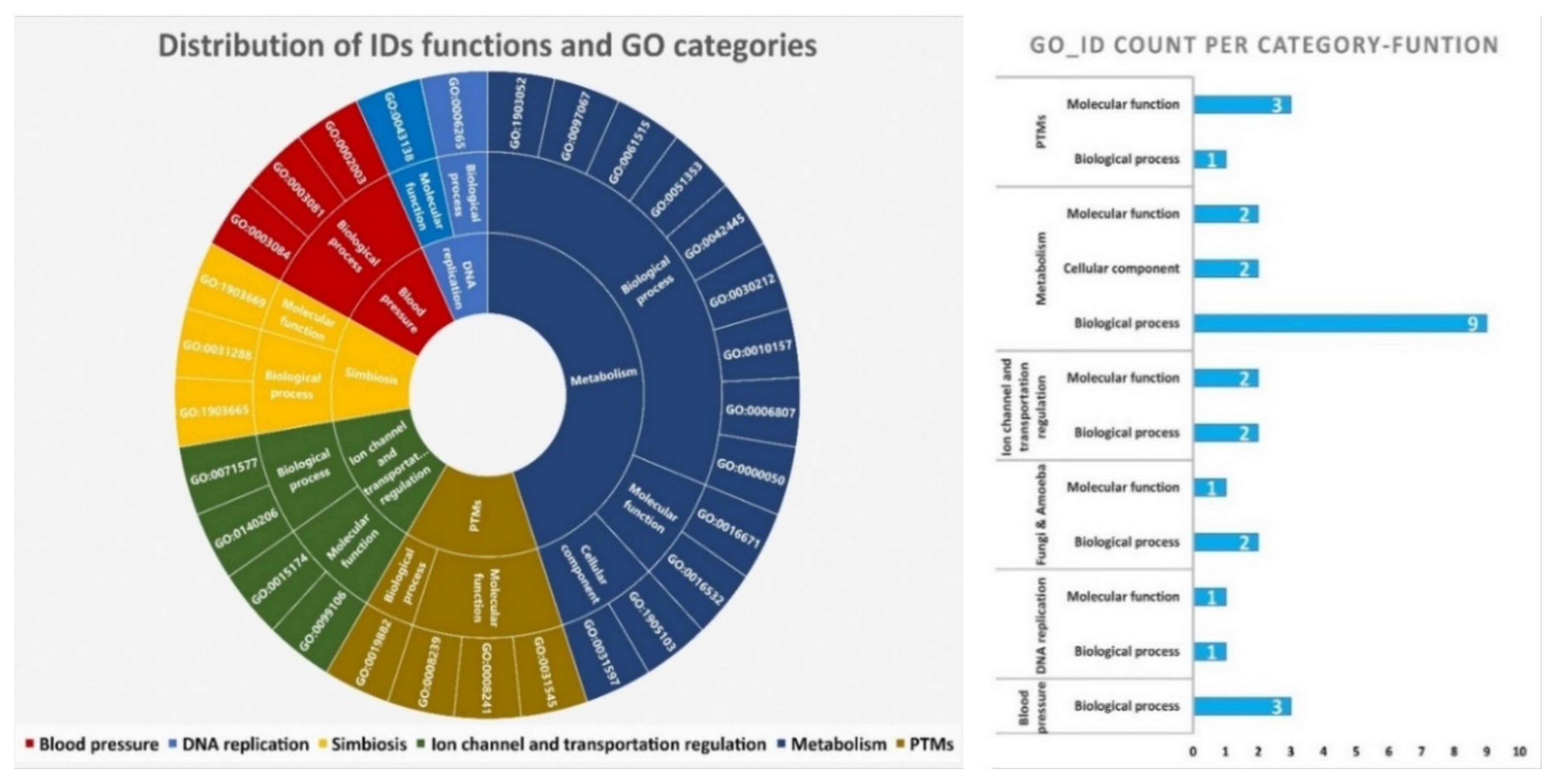
Figure 4.
Phylogeny Tree A of 9 Conus species (C. betulinus, C. coronatus, C. ebraeus, C. ermineus, C. literatus, C. miliaris, C. rattus, C. sponsalis, C. virgo) illustrating the clustering of 271 sequences (blasted with Tox-Prot database) from the vermivorous and piscivorous predatory diets. The annotation colour from the inner circle to the outer is the following: organ, diet, species and Blast target.
Figure 4.
Phylogeny Tree A of 9 Conus species (C. betulinus, C. coronatus, C. ebraeus, C. ermineus, C. literatus, C. miliaris, C. rattus, C. sponsalis, C. virgo) illustrating the clustering of 271 sequences (blasted with Tox-Prot database) from the vermivorous and piscivorous predatory diets. The annotation colour from the inner circle to the outer is the following: organ, diet, species and Blast target.
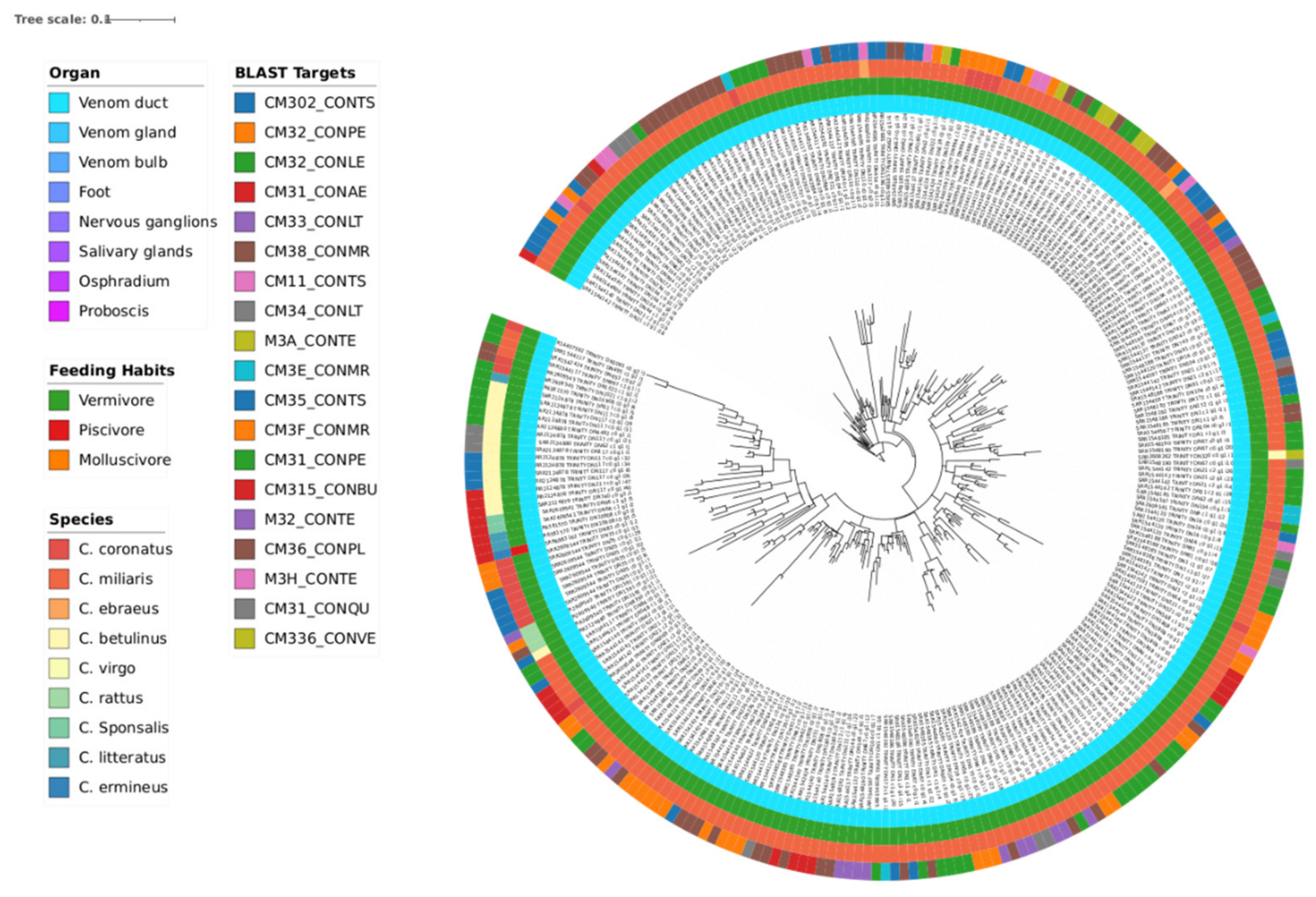
Figure 5.
Phylogeny Tree B of 9 Conus species (C. betulinus, C. coronatus, C. ebraeus, C. judaeus, C. literatus, C. marmoreus, C. miliaris, C. sponsalis, C. ventricosus) illustrating the clustering of 117 sequences (blasted with Tox-Prot database) which encompasses the numerous vermivorous and the rarest (in this dataset) molluscivorous predatory diets. The annotation colour from the inner circle to the outer is the following: organ, diet, species and Blast target.
Figure 5.
Phylogeny Tree B of 9 Conus species (C. betulinus, C. coronatus, C. ebraeus, C. judaeus, C. literatus, C. marmoreus, C. miliaris, C. sponsalis, C. ventricosus) illustrating the clustering of 117 sequences (blasted with Tox-Prot database) which encompasses the numerous vermivorous and the rarest (in this dataset) molluscivorous predatory diets. The annotation colour from the inner circle to the outer is the following: organ, diet, species and Blast target.
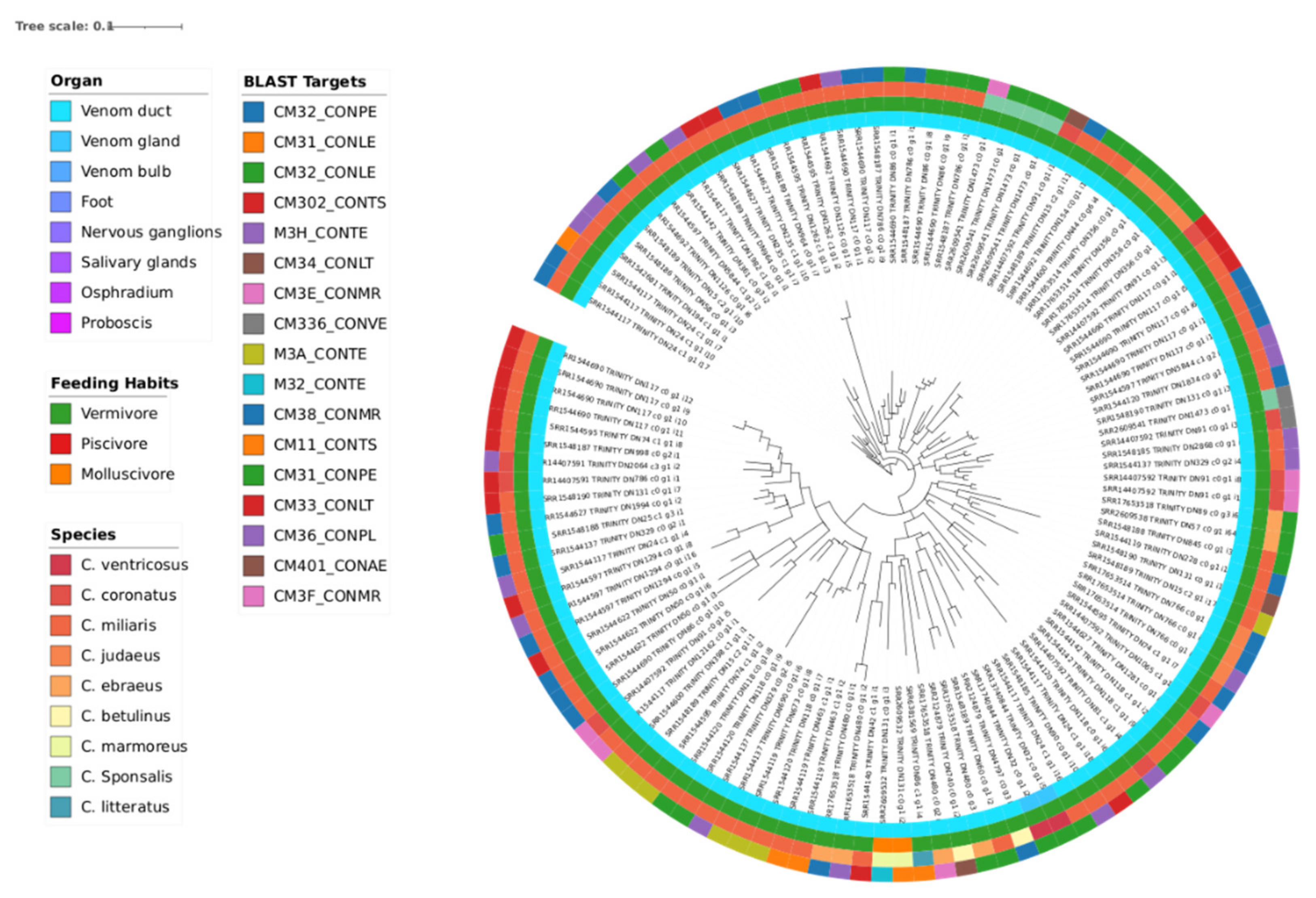
Figure 6.
Phylogeny Tree C of 10 Conus species (C. betulinus, C. coronatus, C. ermineus, C. literatus, C. marmoreus, C. miliaris, C. quercinus, C. tribblei, C. ventricosus, C. virgo) illustrating the clustering of 53 sequences (blasted with UniProt Trembl database) which encompasses all three different feeding habits. The annotation colour from the inner circle to the outer is the following: organ, diet, species and Blast target.
Figure 6.
Phylogeny Tree C of 10 Conus species (C. betulinus, C. coronatus, C. ermineus, C. literatus, C. marmoreus, C. miliaris, C. quercinus, C. tribblei, C. ventricosus, C. virgo) illustrating the clustering of 53 sequences (blasted with UniProt Trembl database) which encompasses all three different feeding habits. The annotation colour from the inner circle to the outer is the following: organ, diet, species and Blast target.
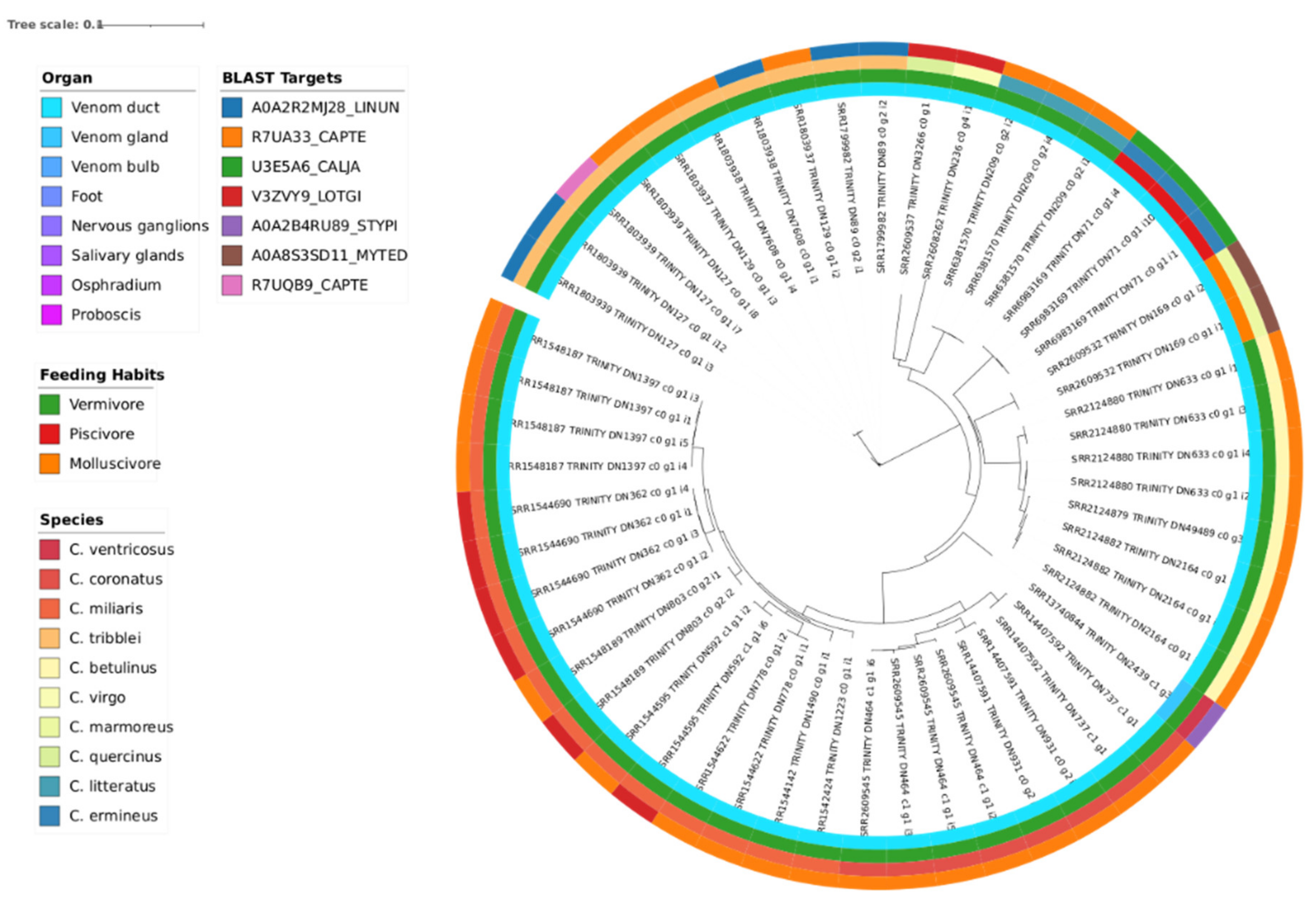
Figure 7.
The linear graph illustrates the general trend of peptide content (Pfam peptide count, in orange) and AMP number (brown) per transcriptome in crescent order of assembly size (green) from left to right; linear tendency lines illustrate the increase of peptide content and corresponding AMP number but the erratic sudden drops and heights suggest an undesignated variable not considered.
Figure 7.
The linear graph illustrates the general trend of peptide content (Pfam peptide count, in orange) and AMP number (brown) per transcriptome in crescent order of assembly size (green) from left to right; linear tendency lines illustrate the increase of peptide content and corresponding AMP number but the erratic sudden drops and heights suggest an undesignated variable not considered.
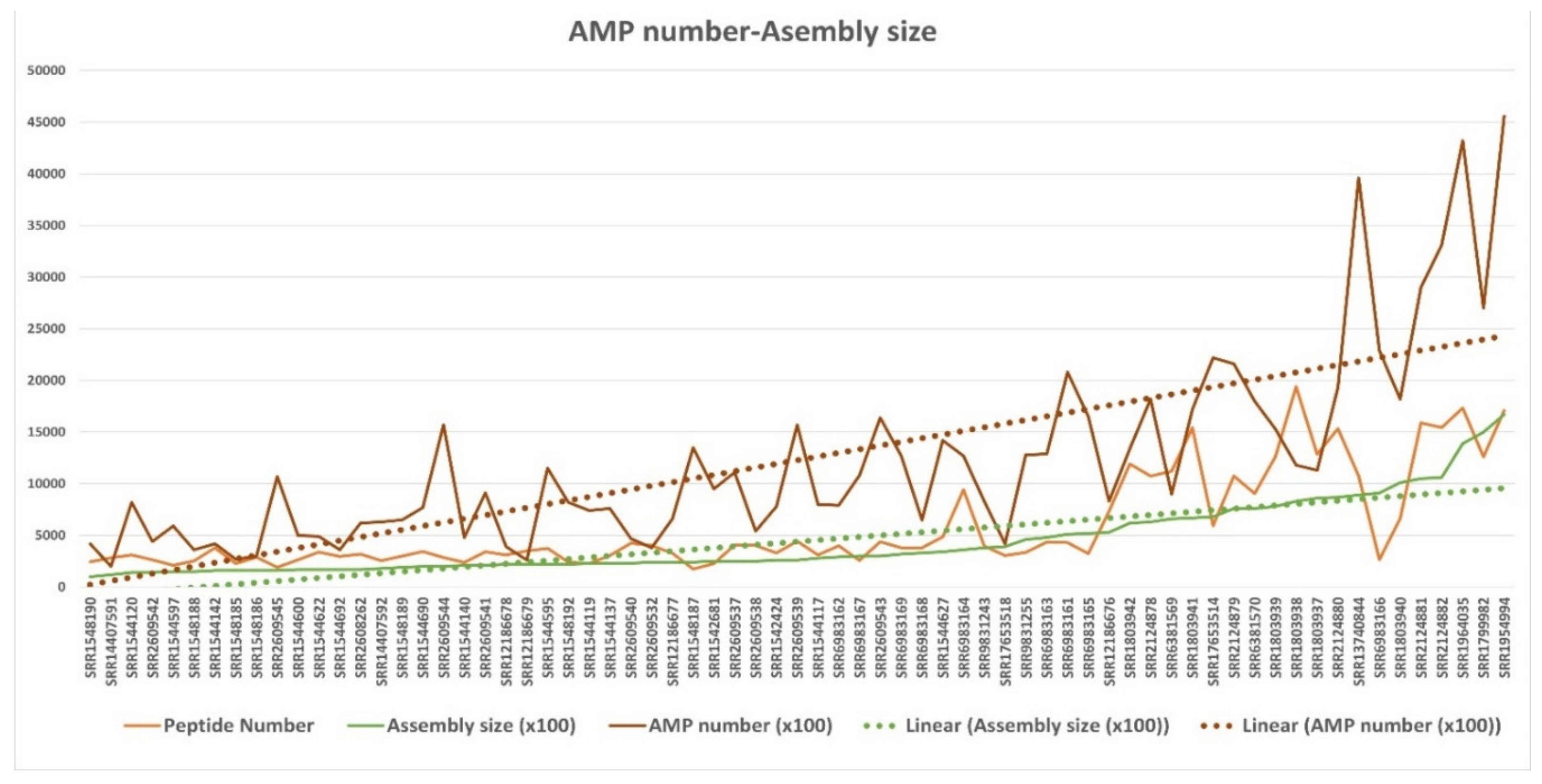
Figure 8.
The linear graph illustrates the general trend of peptide content (orange) and AMP number (brown) per transcriptome grouped by feeding habits and in crescent order of assembly size (green); the vertical dark-yellow dotted lines separate the feeding habit grouping, from left to right: molluscivorous in the left, piscivorous in between and vermivorous to the right.
Figure 8.
The linear graph illustrates the general trend of peptide content (orange) and AMP number (brown) per transcriptome grouped by feeding habits and in crescent order of assembly size (green); the vertical dark-yellow dotted lines separate the feeding habit grouping, from left to right: molluscivorous in the left, piscivorous in between and vermivorous to the right.
Table 1.
AMP findings and transcriptome annotation summary including the cumulative number of peptides and toxins.
Table 1.
AMP findings and transcriptome annotation summary including the cumulative number of peptides and toxins.
| Transcriptomic group | Number of peptides | Number of Toxins | AMP predictions |
|---|---|---|---|
| Venom-related | 553,878 | 28,805 | 71 (15 unique) |
| Venom-unrelated | 108,466 | 1,902 | 73 (17 unique) |
Table 2.
Summary of AMP prediction from the sequences with the 14 chosen exclusively venom-shared annotations.
Table 2.
Summary of AMP prediction from the sequences with the 14 chosen exclusively venom-shared annotations.
| Data index | File |
Species (diet) |
Abundance | Data base hit | Description |
|---|---|---|---|---|---|
| AMP_00195 | SRR2124881 | C. betulinus vermivorous | 1 | AGANDLCQECEDIVHLLTKMTKEDAFQDTIRKFLEQECDILPLKLLVPRCRQVLDVYLPLVIDYFQGQIKPKAICSHVGLC | Pulmonary protein B precursor |
| AMP_03456 | SRR6983168 | C. ermineus piscivorous | 1 | GLICESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKEKT | Antimicrobial peptide NK-lysin precursor |
| AMP_03457 | SRR6983168 | 1 | GLICESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKEKTGLI | ||
| AMP_04409 | SRR6983168 | 1 | GYFCESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKE | ||
| AMP_21444 | SRR6983168 | C. ermineus piscivorous | 1 | YFCESCRKIIQKLEDMVGPQPNEDTVTQAASQVCDKLKILRGLCKKIMRSFLRRISWDILTGKKPQAICVDIKICKE | |
| AMP_05220 | SRR17653518 | C. ebraeus vermivorous | 1 | KPKDMTSSQWFKTQHVQPSPQACNSAMSIINKYTERCKDLNTFLHEPFSSVAITCQTPNIACKNSCKNCHQSHGPMSLTMGELTSGKYPNCRYKEKHLNTPYIVACDPPQQGDPGYPLVPVHLDKVV | Ribonuclease, RNase A family, 8 |
| AMP_19944 | SRR6381569 SRR6381570 |
C. literatus vermivorous |
4 | EEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEE (See Appendix D for more transcripts sharing similarities with ACE protein) |
ACE2 |
| SRR1542424 SRR1544142 | C. miliaris vermivorous | 2 | |||
| SRR12186679 | C. imperialis vermivorous | 2 | |||
| SRR13740844 |
C. ventricosus vermivorous |
1 | |||
| SRR2609537 |
C. quercinus vermivorous |
1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



