Submitted:
10 April 2025
Posted:
11 April 2025
You are already at the latest version
Abstract
Actinobacterial natural ecological niches are characterized by variations in the availability of nutrients, resulting in a complex metabolism. Their remarkable ability to adapt to fluctuating nutrient conditions is possible through the utilization of large amounts of substrates. Recent discoveries in bacterial metabolism suggested the importance of polyamine metabolism in bacteria, particularly in those of the order Actinomycetales, to survive in their natural habitats. This makes such enzymes promising targets to inhibit their growth. Since the polyamine metabolism of soil bacteria of the genus Streptomyces and the human pathogenic Mycobacteria is surprisingly similar, a target-based drug development in Streptomyces and Mycobacterium spp. is an alternative approach to the classical search of antibiotics. The recent development of drugs to treat epidemic diseases like tuberculosis (TB) has gained attention due to the occurrence of multi-drug-resistant strains. In addition, drug repurposing plays a crucial role in the treatment of various complex diseases, such as malaria. With that notion, the treatment of TB could also benefit from this approach. For example, molecular chaperones, proteins that help other proteins to fold properly are found in almost all living organisms including the causative agents of TB. Therefore, targeting these molecules could help in the treatment of TB. We aim to summarize the knowledge of nitrogen and carbon metabolism of the two closely related actinobacterial genera, Streptomyces and Mycobacterium, and of the identification of new potential drug targets.
Keywords:
nitrogen assimilation
; drug targets metabolism
; Streptomyces
; carbon metabolism
; Mycobacterium
1. Tuberculosis as a Global Treat
1.1. Distribution of Tuberculosis and Its Pathogenesis
Worldwide, tuberculosis (TB) remains the most frequent, infectious, long-persisting, and difficult-to-treat disease with a very high mortality rate. In 2022, about 10.0 million people (range, 9.0–11.1 million) developed TB disease, with an estimated 1.3 million deaths (WHO report 2023). Treatments of MDR-TB infections are of particular concern due to their extended duration, poor safety, high cost, and overall poor efficacy. There is no effective anti-TB vaccine available that could prevent TB in adults. Developed in the 1930s, the bacilli Calmette-Guérin (BCG) vaccine was developed to prevent severe forms of TB in children and is still widely used today. While several antibiotics are effective in treating mycobacterial infections, these drugs target a limited number of essential functions in the cell (Natarajan et al., 2020). Moreover, multidrug-resistant tuberculosis (MDR-TB) was reported on all continents, e.g., about 500 000 people developed rifampicin-resistant TB (RR-TB) in 2022 (WHO Report 2023). Thus, the identification of pathways that are required for mycobacterial growth in vivo would provide new targets for the rational design of more effective anti-TB agents that could be active against MDR-TB (Vasiliu et al., 2024)..
The main agent causing tuberculosis, Mycobacterium tuberculosis (MTB), which belongs to the phylum Actinobacteria, is an example of an intracellular pathogen, which is well adapted to survive and persist within human macrophages (Pai M. et al., 2016; Alsayed & Gunosewoyo, 2023). Bacteria that are able to colonize, survive or manipulate macrophages aside from M. tuberculosis, include Klebsiella pneumonia (Cano et al., 2015), Salmonella typhimurium (Monack et al., 2004), Brucella abortus (Kerrinnes et al., 2018) and Acinetobacter baumannii (Sycz et al., 2021). Recent studies have suggested that intracellular pathogens are able to employ strategies to manipulate human macrophage differentiation, in particular their basic cell metabolism. They can induce a metabolic shift in macrophages from M1 (a kill/inhibit) to M2 (a repair/heal) state during the infection. This causes a time-dependent upregulation of the metabolic regulator (PPARγ) in infected macrophages, resulting in increased expression of M2 markers and down-modulation of the M1 response (Muraille et al., 2014; Sun et al., 2024). PPARγ induces the arginine metabolism leading to the synthesis of spermine from putrescine via spermidine (Hesse et al., 2001). Of all bacteria that colonize the macrophages, M. tuberculosis is the most dangerous infection agent representing. It has been shown that virulent M. tuberculosis strains are able to divert away from the production of nitric oxide while enhancing the synthesis of polyamines, which are necessary for the reparative functions of the host macrophages (Hesse et al., 2001). Even though this elevated polyamine production has been considered beneficial for the host, it may be even more advantageous for the invader, serving as a supply of nutrients and energy to the pathogen (Muraille et al., 2014).
1.2. Common Features of Pathogenic and Non-Pathogenic Actinobacteria
The Actinomycetota (Actinobacteria or Actinomycetes) are a phylum containing a group of terrestrial and aquatic Gram-positive bacteria with high G+C content. This phylum includes bacteria that are of great importance for the environment, industry, and medicine. Representative genera include: Actinomyces, Arthrobacter, Corynebacterium, Frankia, Micrococcus, Micromonospora, Mycobacterium, Nocardia, Propionibacterium and Streptomyces. Some soil Actinobacteria, such as Frankia, are endosymbionts of plants that are able to fix nitrogen in exchange for access to the plant’s saccharides and other nutrients. Other species, such as members of the genera Mycobacterium, Corynebacterium, Nocardia, and Rhodococcus are human pathogens (Siavashifar et al., 2021).
Actinomycetota contains Streptomyces, which is one of the largest of the bacterial genera, containing contributors to the biological buffering of soils and bacteria-producers of many antibiotics and other secondary metabolites. Actinomycetes-derived antibiotics important for medicine include tetracyclines, aminoglycosides, macrolides, anthracyclines, glycopeptides etc (Alam et al., 2022; Krysemko & Wohlleben, 2024). Streptomyces spp. share with pathogens from the genus Mycobacterium, an almost identical core metabolism, which made organisms like S. coelicolor a model system for mycobacterial research and translational medicine (Sherr & Nguyen, 2009).
Actinobacteria live mainly under long-lasting nutrient limitations in constant competition with other organisms in their respective habitats (Hodgson, 2000). These non-motile bacteria have no opportunities to move towards more nutrient-rich ecological niches and therefore they have multiple mechanisms to adapt to their habitats. On the one hand, they can use a variety of nutrient sources, including mono- and polyamines. On the other hand, they possess a variety of adaptation mechanisms for rapid changes in nutrient availability (Krysenko & Wohlleben, 2022).
2. Polyamine Metabolism in M. tuberculosis as a Part of Nitrogen Metabolism for Survival and Pathogenicity
2.1. Nitrogen Assimilation and Its Control in Mycobacterium Tuberculosis
Nitrogen metabolism has been extensively studied in pathogenic Actinobacteria from the genus Mycobacterium, which includes soil bacteria like Mycobacterium smegmatis, as well as mammal and human pathogens like Mycobacterium tuberculosis (causative agent of tuberculosis) and Mycobacterium leprae (caused leprosy). In M. tuberculosis, the glutamine synthetase and nitrogen assimilatory protein GlnA1, has been associated with virulence and pathogenicity (Tullius et al., 2003). Understanding nitrogen assimilation in this bacterium allows us to understand infection mechanisms as well as aid the development of novel therapeutic strategies to control M. tuberculosis and multidrug resistance strains. Although similar to closely related actinobacterial from the genus Streptomyces, M. tuberculosis possesses an ammonium transporter protein AmtB; it does not have an active glutamate dehydrogenase (GDH) enzyme. Thus, GS / GOGAT is the only way to assimilate nitrogen. M. tuberculosis genome contains one glutamine synthetase encoding gene glnA1 (also referred as glnA) and three GS-like enzymes encoded by glnA2, glnA3 and glnA4. The GlnA1 belongs to the GSI (prokaryotic) type of glutamine synthetases (Tullius et al., 2003; Harth, et al., 2005).
Although all GS-like enzymes have been reported to be active in cells, only GSI encoded by glnA1 has been found to be essential for M. tuberculosis growth. The activity of the GlnA1 enzyme can be downregulated under nitrogen excess by bifunctional adenylyl transferase GlnE. Under nitrogen starvation, GlnE deadenylylates GlnA1 and restores its activity. In contrast to E. coli, GlnE activity in M. tuberculosis is not regulated by GlnK and GlnD. At the transcriptional level, GlnR controls the nitrogen assimilation. It is a functional homologue of the global transcriptional regulator GlnR from S. coelicolor. A putative TetR-like transcriptional regulator AmtR has also been found in M. tuberculosis demonstrating only 27.9% amino acid sequence identity to the AmtR protein from C. glutamicum. In M. tuberculosis, global regulator GlnR regulated the transcription of amtB-glnK-glnD, gltBD, and nirBD operons as well as the transcription of glnA (Gouzy et al., 2014).
2.2. GS-like Enzymes GlnA2, GlnA3 and GlnA4 in M. tuberculosis
The function of GS-like enzymes such as GlnA2, GlnA3, and GlnA4 in M. tuberculosis remained not characterized, although reported to be non-essential for cellular growth (Harth, et al., 2005). In silico analysis of glnA-like genes across the actinobacterial genomes revealed that glnA3 and glnA4 genes, encoding proteins that might be involved in the colonization, persistence, and survival in diverse habitats, might have a common glnA ancestor (Krysenko et al., 2021). In M. tuberculosis, in silico homology, protein modeling revealed that the structure of GlnA2Mt, GlnA3Mt, and GlnA4Mt is very similar to the structure of GlnA1Mt, but also respective homologs in S. coelicolor – GlnA2Sc, GlnA3Sc, GlnA4Sc, indicating high confidence for their homology (Krysenko et al., 2025).
In the model actinobacterium S. coelicolor, the central enzyme reported for the γ-glutamylation pathway is the gamma-glutamyl polyamine synthetase GlnA3Sc (Krysenko et al., 2017). The reaction catalyzed by GlnA3 is comparable to the reaction catalyzed by glutamine synthetase (GS). Both enzymes can ligate a glutamyl group from glutamate with an amino group of the specific substrate (polyamine or ammonium, respectively) using energy from ATP. However, GS catalyzes the production of L-glutamine, whereas GlnA3Sc generates gamma-glutamyl polyamine, which is further metabolized via GABA to succinate, of which both enzyme types share structural similarities. (Krysenko et al., 2017).
In a recent study, it was demonstrated that M. tuberculosis growth was inhibited by the polyamine spermine. Using in vitro enzymatic assays, it was determined that GlnA3Mt (Rv1878) possesses genuine gamma-glutamyl spermine synthetase catalytic activity. It was further shown that purified GlnA3Mt preferred spermine as a substrate over putrescine, cadaverine, spermidine, or other monoamines and amino acids, suggesting that GlnA3Mt plays a specific role in the detoxification of the polyamine spermine (Krysenko et al., 2025).
2.3. Polyamine Metabolism in Actinobacteria
Polyamines are positively charged molecules with a hydrocarbon chain and multiple amino groups. Polyamines are small aliphatic polyvalent cations, predominantly derived from amino acids such as ornithine, arginine, and lysine (Miller-Fleming et al., 2015). They are widely distributed in nature and are present in all organisms, with the most common cellular polyamines being putrescine, cadaverine, spermidine, and spermine. These compounds are able to interfere with negatively charged molecules, such as DNA, RNA, polyphosphate, and phospholipids. Intracellular polyamine levels have to be regulated since polyamine imbalance can dramatically change cell homeostasis (Kusano & Suzuki, 2015; Miller-Fleming et al., 2015). Polyamine excess was reported to be toxic for both eukaryotic and prokaryotic organisms, causing apoptosis (Davis et al., 1991). However, polyamine metabolism remained almost uninvestigated in actinobacteria, although de novo biosynthesis of spermine has been reported in Mycobacterium (Paulin et al., 1985; Jain & Tyagi, 1987).
Polyamines have been implicated in a wide range of biological processes, and their intracellular level is elevated predominantly during exposure to several stress conditions (Kusano & Suzuki, 2015; Miller-Fleming et al., 2015; Michael, 2018). Thus, the intracellular polyamine concentrations are tightly regulated by cell metabolic pathways (Kusano & Suzuki, 2015), as polyamine excess is toxic for prokaryotic and eukaryotic organisms and may lead to cell death (Davis & Ristow, 1991; Tome et al., 1997; Pegg et al., 2013, Lasbury et al., 2007). Polyamines can interact with negatively charged molecules like RNA, DNA, proteins, polyphosphate, and phospholipids (Norris et al., 2015). Consequently, an imbalance in polyamine metabolism can deeply affect cellular homeostasis.
Polyamine assimilation is required to utilize them as a C/N source under deficiency conditions and to detoxify under excess. Polyamine catabolism has been studied in some bacterial species, revealing that the polyamine utilization pathway is not universal for all bacteria and is usually interconnected with prior polyamine detoxification. This process has been studied extensively in the Gram-negative bacteria like E. coli and P. aeruginosa POA1 (Kurihara et al., 2008; Yao et al., 2011; Kusano & Suzuki, 2015), which can catabolize polyamines via the aminotransferase pathway (Schneider & Reitzer, 2012) or the γ-glutamylation pathway (Kurihara et al., 2008; Yao et al., 2011; Schneider & Reitzer, 2012) In P. aeruginosa, the direct oxidation pathway for putrescine and the spermine/spermidine dehydrogenase pathway were described; in E. coli and B. subtilis - the acetylation pathway for spermidine (Forouhar et al., 2005; Yao et al., 2011; Foster et al., 2013; Campilongo et al., 2014). In the actinobacterial model organism Streptomyces coelicolor, the γ-glutamylation pathway has also been described (Krysenko et al., 2017). Hardly anything is known about polyamine utilization and its regulation in M. tuberculosis, but it has been hypothesized that it may occur via the γ-glutamylation pathway described in the model actinobacterium S. coelicolor (Krysenko et al., 2021), which is a close relative of M. tuberculosis. Recently, it has been demonstrated that M. tuberculosis growth was inhibited by polyamine spermine (Emani & Reiling, 2024; Krysenko et al., 2025).
3. Current Tuberculosis Drug Targets and Validated Drug Candidates in Actinobacteria
Common drug targets in Actinobacteria include metabolic enzymes (e.g., protein kinases, proteases, esterases, and phosphatases), membrane transport proteins, ion channels, and nucleic acids (Bhat et al., 2018; Shetye et al., 2020; Zhang et al., 2024). Conventional antibiotics function by killing the bacteria or by inhibiting growth. Modern antibiotics affect mostly the same cellular targets as peptidoglycan synthesis, the cytoplasmic membrane, translation, Nucleic acid replication, and transcription (Silver, 2016). The development and widespread of antibacterial compounds resulted in a rapid occurrence of resistance mechanisms of pathogens to existing drugs. Bacterial antibiotic resistance is based on some principal mechanisms, including the production of proteins to inactivate the antibiotic; mutation in the target site protein receptor or the ribosomal subunit leading to ineffective drug binding; changes in transport proteins preventing antibiotic entry as well as active compounds efflux from cells (Silver, 2016; Zhang et al., 2024).
There is thus an urgent need to discover new strategies for developing novel antibacterial drugs. Actinobacteria offer numerous secondary metabolites of enormous importance for the healthcare and food industry (Selim et al., 2021. An effective strategy is the identification of bacterial proteins that may be the drug targets for antibiotics. In this way, essential bacterial proteins or pathways (e.g., key proteins for the viability, growth, replication, or survival of the pathogen) that do not have human homologues can be identified (Sakharkar et al., 2008).
In M. tuberculosis drug discovery, there are several current hot drug targets in the primary carbon, nitrogen, phosphate and sulfur metabolism, in the DNA replication machinery, protein synthesis and others, including such targets as GyrA/B, QcrB, ATP synthase, DprE1, FadD32, Pks13, MmpL3 (Alsayed & Gunosewoyo, 2023) and GlnA3Mt (Krysenko et al., 2025) (Figure 1).
There are molecular targets that are involved in pathways of macromolecular synthesis. From antibiotics of the main classes used in systemic monotherapy, only a few targets belong to essential enzymes. Most systemic mono-therapeutic agents have nonprotein targets and interfere with the ribosomal RNA, membranes, or cell wall synthesis. Up-to-date intracellular enzymes can be targeted by β-lactams and fluoroquinolones. Many antibiotics do target single essential enzymes. Clinical resistance to antibacterial agents is usually caused by horizontal gene transfer with the contribution of the in vitro type of resistance that was observed in a few cases. These investigations led to the multitarget hypothesis. Successful systemic mono-therapeutic agents should have low levels of endogenous single-step resistance. This is due to their targeting of the products of multiple genes or pathways (Brotz-Oesterhelt & Brunner 2008).
3.1. Targeting the DNA Replication and Protein Synthesis (Transcription and Translation)
A well-tested therapeutic approach targets DNA replication and protein synthesis. Many compounds and chemical inhibitors from this category have been used in the first and second line of therapy, e.g., to combat mycobacterial infections. These compounds include streptomycin, amikacin, kanamycin, and capreomycin, which target translation; fluoroquinolones, which target DNA unwinding and replication; rifampin, which targets transcription (Shetye et al., 2020). Only a few of the antibacterials are used in systemic monotherapy. Rifampicin, trimethoprim, and sulfamethoxazole are used in combination (Fernandes 2016). From the single-enzyme inhibitors, only fosfomycin is currently used as monotherapy, but mainly for urinary tract infections (UTIs) (Karageorgopoulos et al. 2012). Some inhibitors targeting the DNA replication, transcription, and translation have been studied in M. tuberculosis: SPR719/SPR720, GSK3036656 (GSK 656, GSK 070), oxazolidinones linezolid, sutezolid, delpazolid, TBI-223 (Shetye et al., 2020).
3.2. Targeting Cell Wall/Peptidoglycan Biosynthesis
The cell wall of pathogenic Actinobacteria, e.g., M. tuberculosis, consists of the mycelial-arabinogalactan-peptidoglycan (mAGP) complex and has been observed as a potential drug target too. Disruption of peptidoglican biosynthesis has been demonstrated in M. tuberculosis MDR-Mtb and XDR-Mtb by cycloserine, a known broad-spectrum antibiotic, which binds to d-alanine:d-alanine ligase (Ddl). Further drugs targeting cell wall biosynthesis include enduracidin, ramoplanin (a lipoglycodepsipeptide), and teixobactin. Combination therapy with clavulanate, carbapenem, and meropenem showed potent activity against M. tuberculosis (Radkov et al., 2018; Boutte et al., 2016; Capela et al., 2023).
DprE1 - decaprenyl phosphoryl-β-D-ribose oxidase - is a key enzyme in mycobacterial cell wall biosynthesis. DprE1 has been identified as the target of a novel class of inhibitors, 1,3-benzothiazin-4-ones (BTZs). DprE1 is a flavoprotein that works in concert with decaprenylphosphoryl-D-2-keto erythropentose reductase (DprE2) to generate an arabinose precursor that plays a fundamental role in the synthesis of the mycobacterial cell wall polysaccharides arabinogalactan and lipoarabinomannan. The DPA biosynthesis was recently shown to take place in the periplasmic space of the mycobacterial cell wall, where DprE1 was also found to be located. The extracytoplasmic localization of DprE1 makes it more accessible to drugs that contribute to its vulnerability. It was demonstrated that inhibiting DprE1 abolishes the formation of DPA, thereby provoking cell lysis and mycobacterial death (Alsayed & Gunosewoyo, 2023).
3.3. Targeting Arabinogalactan
Another potential target is arabinogalactan which is composed of a polysaccharide backbone (galactose and arabinose sugar moieties) and is contently linked to the peptidoglycan layer and the mycolic acid layer. Targeting arabinogalactan biosynthesis by ethambutol, disrupts the arabinogalactan assembly. The fatty acyl-AMP ligase 32 (FAAL32 or FadD32) and polyketide synthase 13 (Pks13) are crucial enzymes in the biosynthetic machinery of MAs, which are the major integral lipid components of the exceptionally fortified waxy cell wall of M. tuberculosis. After the FadD32 and Pks13 crosstalk takes place resulting in the formation of TMM, these MAs precursors are then flipped into periplasm via the inner membrane protein MmpL3. The mycolyl portion gets anchored to arabinogalactan, the major cell wall polysaccharide, which is further linked to peptidoglycan (Alsayed & Gunosewoyo, 2023).
Mycobacterial membrane proteins have been recognized as potential drug targets in M. tuberculosis, inhibition of which can be achieved using the SQ109, NITD-304 & NITD-349 and BM212/ BM635 drugs (Stec et al., 2016). Compounds Isoniazid (INH) and Ethionamide (ETH) have been elucidated to inactivate InhA (enoyl-ACP reductase) involved in mycolic acid biosynthesis. Further antitubercular drugs targeting mycolic acid synthesis include an indazole sulfonamide GSK 724 (DG167 or GSK3011724A) inhibiting β-ketoacyl-ACP synthase (KasA); Delamanid and Pretomanid (Banerjee et al., 1994; Shetve et al., 2020).
3.4. Targeting Bytochrome b Subunit QcrB
The cytochrome b subunit (QcrB) of the cytochrome bc1 complex has recently emerged as an interesting target in M. tuberculosis Campanico et al., 2018. The cytochrome bc1 complex is a key component of the respiratory electron transport chain required for ATP synthesis. Therefore, the inhibition of this complex disrupts the M. tuberculosis’s ability to generate energy. A phenotypic screening of a library encompassing more than 100,000 compounds as antimycobacterial agents led to the identification of imidazopyridine amides (IPAs) as a promising class that blocks the M. tb growth by targeting QcrB. An optimized IPA derivative Q203 showed potent growth inhibition against DS M. tuberculosis H37Rv strain (MIC50 = 2.7 nM) and numerous MDR and XDR M. tuberculosis clinical isolates in vitro (MIC90 < 0.43 nM for most DR strains) (Campanico et al., 2018).
3.5. Targeting Clp Proteases
One of the chemotherapeutic strategies is targeting proteostasis and proteolysis enzymes, e.g., in M. tuberculosis the two ATP-dependent proteases, caseinolytic protease (Clp) and proteasome. ClpP forms a complex with AAA+ ATPase chaperone proteins, such as ClpC1 or ClpX. ClpP proteolytic complex consists of a functional central channel, which is a stacked assembly formed by 2 ClpP heptamers – ClpP1 and ClpP2. The opening of the central channel is capped by AAA+ ATPases, ClpC or ClpX. The ClpC or ClpX maintains chaperone substrate proteins into the ClpP central channel and aligns the catalytic triad for proteolysis (Culp & Wright, 2017; Schmitz & Sauer, 2014).
Disruption of proteolytic complexes (ClpP/ClpC or ClpP/ClpX) can result in inhibition of proteolysis, activation, and uncontrolled proteolysis, decoupling of ClpC’s or ClpX’s chaperone activity from ClpP1P2 proteolysis. Since clpp1p2 genes have been demonstrated to be essential for the M. tuberculosis pathogen and unlike bacterial proteases, ClpP is not cellular in humans but mitochondrial; these proteins become promising drug targets. Currently proposed compounds targeting the ClpP1P2 proteases include peptidyl boronate (bortezomib analogs, which also inhibit Mtb20S proteasome) and β-lactones. Compounds that target ClpX/ClpC include cyclic acyldepsipeptide (ADEP. ClpC can be targeted by Actinomycetes-derived cyclic peptides, such as cyclomarin A, ecumicin, lassomycin, and rufomycin (Shetve et al., 2020).

Table 1.
Proteins in Actinobacteria that are validated drug targets.
| Target | Function | Reference |
|---|---|---|
| Gyrase B, Leucyl tRNA synthetase, rRNA, RNA Polymerase | DNA replication and protein synthesis | Shetye et al., 2020; Alsayed & Gunosewoyo, 2023 |
| MurX, l,d-traspeptidases, l,d-transpeptidases + β lactamase, Lipid II | peptidoglycan biosynthesis | Capela et al., 2023 |
| WecA, DprE1 (Covalent inhibitors), DprE1 (Noncovalent inhibitors) | arabinogalactan biosynthesis | Capela et al., 2023 |
| MmpL3, InhA, β-ketoacyl-ACP synthase (kasA), Inhibition of methoxy and keto mycolic acid (exact target unknown) | mycolic acid biosynthesis | Stec et al., 2016 |
| ATP synthase (AtpE), Cytochrome bc1/aa3 super, NDH-2, MenA, MenG, Isocitrate lyase (ICL) | energy metabolism | Haagsma et al., 2009 |
| ClpC | proteolysis | Culp & Wright, 2017 |
| Glutamine Synthetase GlnA1 | Primary metabolism, glutamine synthesis | Eisenberg et al., 2000 |
| Gamma-Glutamylpolyamine Synthetase GlnA3 | Polyamine metabolism | Krysenko et al., 2025 |
3.6. Targeting Primary Metabolism
3.6.1. Targeting Carbon Metabolism
Central carbon metabolism (CCM) includes the enzyme-mediated transformation of carbon through glycolysis, the tricarboxylic acid (TCA) cycle, and gluconeogenesis to incorporate carbon into the bacterial metabolism; the genes encoding these reactions were found to be conserved (Feist et al., 2009). Recent studies have revealed distinctive elements of Actinobacterial CCM that have helped these organisms survive in specific ecological niches. Especially important is the knowledge of CCM in pathogenic Actinobacterial, such as M. tuberculosis, in which carbon metabolism has evolved to serve interdependent physiologic and pathogenic roles (Rhee et al., 2011).
Carbon metabolism in M. tuberculosis has been extensively investigated in association with mutations of essential CCM enzymes and the opportunity to design inhibitors based on transition state analogs. Recent advances in inhibitor design demonstrated that active sites of enzymes involved in carbon metabolism are often sub-saturated, making them potentially more susceptible to inhibitors. Broad-spectrum antibiotics in widespread clinical use nowadays include fluoroquinolones inhibiting bacterial DNA gyrases and trimethoprim (TMP) targeting dihydrofolate reductases (DHFR). Newer drugs include diarylquinoline TMC207, which inhibits the C subunit of its ATP synthase (Haagsma et al., 2009), rhodanine inhibiting DlaT, triazospirodimethoxybenzoyls targeting Lpd and oxadiazole-2-ones that inhibit the proteasome (Bryk et al., 2010). DNA gyrase GyrA/B is considered to be a hot drug target as a conserved type II topoisomerase enzyme, essential for DNA transcription, replication, and recombination in M. tuberculosis. Inhibition of the DNA gyrase consisting of two sub-units, A and B, results in impaired DNA replication and bacterial death. A promising drug candidate for GyrA/B inhibition is SPR720 (Alsayed & Gunosewoyo, 2023).
The challenge in the validation of CCMs as potential drug targets is that metabolic enzymes can exist at levels higher than those needed to support cell viability (Wei et al., 2011). Most enzymes of carbon metabolism serve diverse, interconnected pathways that are subject to strict regulation. Validation of actinobacterial enzymes of carbon metabolism as potential drug targets required biochemical knowledge of the specific metabolic pathways that define the essentiality of enzymes of interest combined with the quantitative level of inhibition needed to achieve the death of a pathogen (Rhee et al., 2011).
3.6.2. Targeting Sulfur Metabolism
Extracellular sulfated metabolites play a regulatory role in cell-cell and host-pathogen communication (Mougous et al., 2002). Acquisition and metabolism of sulfur is essential for diverse Actinobacterial species, e.g., it is a key determinant in virulence and survival of Mycobacterium spp. (Paritala & Carroll, 2013).
Microbial sulfur metabolic pathways are largely absent in humans. Thus, sulfur metabolism, including amino acid biosynthetic pathways and downstream metabolites, represents a potential and unique target for drug development (Mougous et al., 2002). Sulfur metabolic pathways are required for virulence in different pathogenic actinobacteria, e.g., M. tuberculosis (Haizios & Bertozzi, 2011). Mutants in genes of mycobacterial sulfur metabolism have been demonstrated to have an impaired ability to persist and cause disease (Sassetti et al., 2003). Recent investigations provided molecules that inhibit the PAPS- and substrate–binding domains of Sts, as well as synthetic bi-substrate analogs that have been employed (Rath et al., 2004). Such compounds incorporate elements from the cofactor, PAPS, and the substrate, resulting in specificity via critical interactions within the binding pocket of the enzyme. Inhibitor potency is achieved by linking structures that mimic substrates. For example, bisubstrate-based compounds were identified (Rath et al., 2004) as inhibitors of EST. Other aproach includes the kinase inhibitors. It was proposed that ATP derivatives might also function as ST inhibitors (Bhave et al., 2007). Recent studies identified potent inhibitors of β–arylsulfotransferases (β-AST-IV) (Chapman et al., 2002). Furthermore, isoquinoline sulfonamides have also been tested for inhibitory activity against STs consisting of EST, NodH, GST-2 as well as two inhibitors of Golgi-resident tyrosyl protein ST-2 (TPST-2) (Bhave et al., 2007). Recent advances in structure-based drug design and high-throughput screening will greatly facilitate the discovery of new inhibitors for STs and other sulfonucleotide-binding enzymes.
3.6.3. Targeting Phosphate/ATP Metabolism
Pathogenic Actinobacteria like M. tuberculosis generate ATP via two inter-linked metabolic pathways: oxidative phosphorylation and substrate-level phosphorylation. Since pathogens like M. tuberculosis need higher basal energy, oxidative phosphorylation has proved essential for M. tuberculosis (Cook et al., 2017). The current tuberculosis pipeline has numerous drug candidates with targets in these metabolic pathways (Bald et al., 2017). For targeting oxidative phosphorylation, compounds such as bedaquiline (BDQ) and diarylquinoline blocking ATP synthesis have been reported. Combination therapy of BDQ, linezolid (BpaL), and pretomanid was evaluated in clinical trials and approved for treating XDR-Mtb or MDR-Mtb. Furthermore, BDQ with pretomanid, MOX, and pyrazinamide (PZA) (BPaMZ) has been studied for safety, efficacy, and shortened treatment drug-sensitive (DS) and drug-resistant (DR) tuberculosis (Cook et al., 2014; Shetve et al., 2020; Capela et al., 2023).
The diarylquinoline BDQ was recently approved as an anti-TB drug, and it was found to elicit its activity by inhibiting the c subunit of the mycobacterial ATP synthase enzyme. It disrupts the energy metabolism and decreases intracellular ATP levels in M. tuberculosis. In this respect, two diarylquinolines TBAJ-587 and TBAJ-876 were identified as compounds with anti-TB activity against the H37Rv strain in vitro (Alsayed & Gunosewoyo, 2023).
3.6.4. Targeting Nitrogen and Metabolism of Polyamines
An important drug target evaluated in Actinobacteria is the glutamine synthetase (GS) GlnA. In silico analysis of glnA-like genes across the mycobacterial genomes revealed that glnA1 might be evolved to specialized glnA-like genes, such as GS-like enzyme encoding genes glnA2, glnA3, glnA4 that encode proteins which may be involved in the colonization and survival in many diverse habitats (Krysenko et al., 2021). GS-like genes have been found in actinobacterial pathogens like M. tuberculosis Rhodococcus jostii, as well as plant symbiotic bacteria like Frankia alni or plant pathogens like S. scabies. Validated inhibitors targeting GSs include methionine sulfoximine (MSO), phoshpinothricin (PPT) and some synthetic inhibitors. Recently, the first inhibitor of GS-like proteins has been introduced (Purder et al., 2022) (Table 2). Another group is the larger hydrophobic heterocycles that compete with ATP targeting the nucleotide-binding site, such as purine analogs. Also, GOGAT and GDH in M. tuberculosis have been identified as imported drug targets with the azaserine inhibitor being able to disrupt GOGAT (Krysenko et al., 2024).
Some alternative metabolic pathways were shown to be effectively targeted. These include for instance mono- and polyamine biosynthesis and utilization. A group of inhibitors that target the polyamine biosynthesis has been introduced, including D,L-α-difluoromethylornithine (DFMO), the putrescine analogs 3-aminooxy-1-aminopropane (APA) and 1,4-diamino-2-butanone (DAB), the agmatine analog 1-guanidinooxy-3-aminopropane (GAPA), and the spermine analog MDL 27695 (N,N′-bis(3-((phenylmethyl)amino)propyl)-1,7-diaminoheptane), amongst others. However, the development of drugs targeting monoamine ethanolamine and polyamine utilization is far less investigated. The only drug that is able to target these pathways in Actinobacteria up to date was proposed recently (Purder et al., 2022).
4. Drug Repurposing Targeting Polyamines in Actinobacteria
Drug repurposing that targets polyamines in Actinobacteria is a promising area of research in the fight against tuberculosis (TB). As mentioned, polyamines such as spermidine and spermine are essential for various cellular functions, including growth, motility, and stress response (Krysenko and Wohlleben, 2022). One notable role of polyamines is their regulation of reactive oxygen species (ROS) during stressful conditions, which helps protect cells from oxidative damage. Generally, stress conditions can increase the expression of genes involved in polyamine biosynthesis. This, in turn, elevates polyamine levels, thereby enhancing stress tolerance. Polyamines play a crucial role in complex signaling pathways that activate various stress responses. This includes the interaction of signaling molecules like abscisic acid (ABA) and nitric oxide (NO), which together help organisms respond effectively to stressful conditions. Both genetic engineering and the exogenous application of the polyamines approach in plants have shown that they can mitigate the effects of environmental stresses (Leon et al., 2014; Astier et al., 2018). Taken together, there have been strategies to exploit the molecules that are produced in response to stressful conditions, such as molecular chaperones that are known for their involvement in protein folding and regulations. Some drugs can be used or tested against these molecules to inhibit their activities, effectively shutting down the growth and differentiation of Actinobacteria as a strategy to develop a treatment for tuberculosis (Krysenko et al., 2024).
4.1. Reactive Oxygen Species (ROS) as Drug Targets for TB
One of the key factors in the pathogenesis and survival of Mycobacterium tuberculosis is the presence of highly reactive molecules known as reactive oxygen species (ROS), which are generated from oxygen. These molecules serve two primary roles in biological systems: they act as signaling molecules and can also cause cellular damage. Different types of ROS include superoxide anion, hydrogen peroxide, hydroxyl radical, and singlet oxygen. When present in high concentrations, ROS can induce oxidative stress, resulting in damage to DNA, proteins, and lipids (Bajeli et al., 2020). Complex diseases, such as cancer, cardiovascular diseases, and neurodegenerative disorders, are associated with elevated levels of ROS. Additionally, the malaria-causing parasite Plasmodium falciparum also utilizes these molecules to its advantage. ROS are byproducts of the metabolism of this obligate parasite and can lead to oxidative damage to cellular components. To survive, Plasmodium falciparum has developed strategies to regulate ROS levels effectively. Two important enzymes that are employed by the malarial parasite as antioxidants include superoxide dismutase (SOD) and glutathione peroxidase to detoxify ROS (citation?). Therefore, blocking these enzymes could lead to high levels of ROS that can lead to the death of the parasite. Antimalarial drugs such as artemisinin and its derivatives could contribute to the generation of ROS within the cellular system of the parasite, which could cellular damage and lead to parasite death (Gunjan et al., 2018). Therefore, the drug repurposing approach is very important, and this approach may be used to combat TB too, because the Mycobacterium depends on ROS for its growth. Thus, testing drugs that target ROS could lead to the death of the bacteria.
4.2. The Synergy of Abscisic Acid and Nitric Oxide as a Therapeutic Target
Abscisic acid (ABA) is a plant molecule known for its role in plant stress responses and development. However, these molecules are also produced in different microorganisms, such as actinobacteria, and their role is to influence stress responses and secondary metabolite production (Lievens et al., 2017). On the other hand, nitric oxide (NO) is a versatile molecule that plays a crucial role in the physiological processes of both prokaryotic and eukaryotic organisms. Its role in actinobacteria is regulating secondary metabolism and morphological differentiation. The synergistic interaction between ABA and NO in actinobacteria is an emerging area of research (Cook et al., 2017). However, few studies conducted thus far show that both molecules are involved in stress responses. For example, in plants, ABA can induce NO production and, therefore, mediates different stress responses. It is believed that the same mode of action could exist in actinobacteria, where ABA and NO could cooperate to enhance stress tolerance (Xu et al.,2024). This can be a very good starting point for developing the treatment for TB.
4.3. Molecular Chaperones as Drug Targets
Molecular chaperones or heat shock proteins are found in almost all living organisms, from plants to bacteria. Their role is to help other proteins to fold properly and prevent aggregation (Makhoba, 2025). These molecules are divided into different subclasses, namely, small heat shock proteins and middle and major heat shock proteins with various roles in cellular homeostasis (Table 3).
4.4. Targeting Chaperone Networks
Molecular chaperones function as cooperative partners in the folding of newly synthesized polypeptides. For some, the role is to identify client proteins and activate the ATPase domain of the folding chaperones, thus opening the binding site for the client protein to bind for folding purposes (Figure 2). Almost all living organisms require this system to maintain the balance in cellular function. From plants to bacteria, molecular chaperones are crucial for functional activities in cell signaling and proteostasis (Makhoba, 2025). Therefore, these molecules are ideal drug targets for various diseases like malaria, cancer, and TB. The idea is that, if the molecular chaperone network is targeted, it can lead to cell death, thus eliminating TB.
5. Combination Therapies for Actinobacteria Treatments
Combination therapy has received significant attention in the pharmaceutical industry as a strategy to combat complex diseases. In combating malaria, this approach has been proposed to address the problem of drug resistance, particularly in Sub-Saharan Africa and other regions where malaria is prevalent. Various studies have indicated that combination therapy could serve as a crucial solution not just for malaria, but also for other diseases like tuberculosis (TB) (Fournier and Richet, 2006). In the case of tuberculosis, combination therapy typically involves using multiple antibiotics to effectively treat the infection and prevent the development of drug resistance. For instance, one effective regimen combines rifapentine and moxifloxacin, which is typically administered for four months (Lolans et al., 2006). Recently, it has been demonstrated that the polyamine spermine can enhance the activity of currently available and WHO-approved tuberculosis drugs, such as isoniazid, rifampicin, aminosalicylic acid, and bedaquiline (Sao Emani & Reiling, 2023). Table 4, gives a brief overview of some combination treatments for infections caused by Actinobacteria.
6. Conclusions
Antibacterial agents must meet a variety of criteria to be successful: selectivity, safety, no cross-resistance with existing therapeutics, pharmacological properties, and low propensity for rapid resistance selection. Search for molecular targets/target proteins for new inhibitors and antibiotics requires careful analysis of the target’s role in metabolism and proper inhibitor design. Essentially, lack of close human homologs, lack of target-based cross-resistance, and presence of important pathogens can be predicted based on the target. However, the choice of a single protein or enzyme target may also increase the likelihood of resistance selection. Thus, specificity is of importance to develop a potent drug. These potential problems must be resolved for the success of novel target-based discovery.
Author Contributions
All authors contributed to the writing, editing and review process. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are involved in the study.
Conflicts of Interest
All authors declare no conflict of interest.
References
- WHO. 2023. Global tuberculosis report; Geneva; Licence: CC BY-NC-SA 3.0 IGO.
- Natarajan, A.; Beena, P.; Devnikar, A.V.; Mali, S. A systemic review on tuberculosis. Indian J. Tuberc. 2020, 67, 295–311. [CrossRef]
- Vasiliu, A.; Martinez, L.; Gupta, R.K.; Hamada, Y.; Ness, T.; Kay, A.; Bonnet, M.; Sester, M.; Kaufmann, S.H.; Lange, C.; et al. Tuberculosis prevention: current strategies and future directions. Clin. Microbiol. Infect. 2023, 30, 1123–1130. [CrossRef]
- Gouzy, A.; Poquet, Y.; Neyrolles, O. Nitrogen metabolism in Mycobacterium tuberculosis physiology and virulence. Nat. Rev. Microbiol. 2014, 12, 729–737. [CrossRef]
- Mazlan, M.K.N.; Tazizi, M.H.D.M.; Ahmad, R.; Noh, M.A.A.; Bakhtiar, A.; Wahab, H.A.; Gazzali, A.M. Antituberculosis Targeted Drug Delivery as a Potential Future Treatment Approach. Antibiotics 2021, 10, 908. [CrossRef]
- Capela, R.; Félix, R.; Clariano, M.; Nunes, D.; Perry, M.d.J.; Lopes, F. Target Identification in Anti-Tuberculosis Drug Discovery. Int. J. Mol. Sci. 2023, 24, 10482. [CrossRef]
- Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int. J. Mol. Sci. 2023, 24, 5202. [CrossRef]
- Rodríguez-Bustamante, E.; Gómez-Manzo, S.; Valle, A.D.O.F.d.; Arreguín-Espinosa, R.; Espitia-Pinzón, C.; Rodríguez-Flores, E. New Alternatives in the Fight against Tuberculosis: Possible Targets for Resistant Mycobacteria. Processes 2023, 11, 2793. [CrossRef]
- Sun, F.; Li, J.; Cao, L.; Yan, C. Mycobacterium tuberculosis virulence protein ESAT-6 influences M1/M2 polarization and macrophage apoptosis to regulate tuberculosis progression. Genes Genom. 2023, 46, 37–47. [CrossRef]
- Muraille, E.; Leo, O.; Moser, M. Th1/Th2 Paradigm Extended: Macrophage Polarization as an Unappreciated Pathogen-Driven Escape Mechanism? Front. Immunol. 2014, 5, 603. [CrossRef]
- Krysenko, S.; Emani, C.S.; Bäuerle, M.; Oswald, M.; Kulik, A.; Meyners, C.; Hillemann, D.; Merker, M.; Prosser, G.; Wohlers, I.; et al. GlnA3 Mt is able to glutamylate spermine but it is not essential for the detoxification of spermine in Mycobacterium tuberculosis. J. Bacteriol. 2025, 207, e0043924. [CrossRef]
- Emani, C.S.; Reiling, N. Spermine enhances the activity of anti-tuberculosis drugs. Microbiol. Spectr. 2024, 12, e0356823. [CrossRef]
- Pai M, Behr MA, Dowdy D, Dheda K, Divangahi M, Boehme CC, Ginsberg A, Swaminathan S, Spigelman M, Getahun H. 2016. Tuberculosis. Nat Rev Dis Primers 2:16076. [CrossRef]
- Siavashifar, M.; Rezaei, F.; Motallebirad, T.; Azadi, D.; Absalan, A.; Naserramezani, Z.; Golshani, M.; Jafarinia, M.; Ghaffari, K. Species diversity and molecular analysis of opportunistic Mycobacterium, Nocardia and Rhodococcus isolated from the hospital environment in a developing country, a potential resources for nosocomial infection. Genes Environ. 2021, 43, 1–12. [CrossRef]
- Miller-Fleming, L.; Olin-Sandoval, V.; Campbell, K.; Ralser, M. Remaining mysteries of Molecular Biology: The role of polyamines in the cell. [CrossRef]
- Krysenko, S.; Wohlleben, W. Polyamine and Ethanolamine Metabolism in Bacteria as an Important Component of Nitrogen Assimilation for Survival and Pathogenicity. Med Sci. 2022, 10, 40. [CrossRef]
- Alam, K.; Mazumder, A.; Sikdar, S.; Zhao, Y.-M.; Hao, J.; Song, C.; Wang, Y.; Sarkar, R.; Islam, S.; Zhang, Y.; et al. Streptomyces: The biofactory of secondary metabolites. Front. Microbiol. 2022, 13, 968053. [CrossRef]
- Krysenko, S.; Wohlleben, W. Role of Carbon, Nitrogen, Phosphate and Sulfur Metabolism in Secondary Metabolism Precursor Supply in Streptomyces spp.. Microorganisms 2024, 12, 1571. [CrossRef]
- Hamana K, Matsuzaki S. 1987. Distribution of polyamines in actinomycetes. FEMS Microbiology Letters 41:211-215. [CrossRef]
- Paulin, L.G.; E Brander, E.; Pösö, H.J. Specific inhibition of spermidine synthesis in Mycobacteria spp. by the dextro isomer of ethambutol. Antimicrob. Agents Chemother. 1985, 28, 157–159. [CrossRef]
- Jain, A.; Tyagi, A.K. Role of polyamines in the synthesis of RNA in mycobacteria. Mol. Cell. Biochem. 1987, 78, 3–8. [CrossRef]
- Kusano T, Suzuki H. 2015. Polyamines: a universal molecular nexus for growth, survival, and specialized metabolism. Springer, Tokyo.
- Davis, R.H.; Ristow, J.L. Polyamine toxicity in Neurospora crassa: Protective role of the vacuole. Arch. Biochem. Biophys. 1991, 285, 306–311. [CrossRef]
- Pegg, A.E. Toxicity of Polyamines and Their Metabolic Products. Chem. Res. Toxicol. 2013, 26, 1782–1800. [CrossRef]
- Lasbury, M.E.; Merali, S.; Durant, P.J.; Tschang, D.; Ray, C.A.; Lee, C.-H. Polyamine-mediated Apoptosis of Alveolar Macrophages during Pneumocystis Pneumonia. J. Biol. Chem. 2007, 282, 11009–11020. [CrossRef]
- Tome, E.M.; Fiser, M.S.; Payne, M.C.; Gerner, W.E. Excess putrescine accumulation inhibits the formation of modified eukaryotic initiation factor 5A (eIF-5A) and induces apoptosis. Biochem. J. 1997, 328, 847–854. [CrossRef]
- Norris, V.; Reusch, R.N.; Igarashi, K.; Root-Bernstein, R. Molecular complementarity between simple, universal molecules and ions limited phenotype space in the precursors of cells. Biol. Direct 2015, 10, 1–20. [CrossRef]
- Schneider, B.L.; Reitzer, L. Pathway and Enzyme Redundancy in Putrescine Catabolism in Escherichia coli. J. Bacteriol. 2012, 194, 4080–4088. [CrossRef]
- Kurihara, S.; Oda, S.; Tsuboi, Y.; Kim, H.G.; Oshida, M.; Kumagai, H.; Suzuki, H. γ-Glutamylputrescine Synthetase in the Putrescine Utilization Pathway of Escherichia coli K-12. J. Biol. Chem. 2008, 283, 19981–19990. [CrossRef]
- Yao, X.; He, W.; Lu, C.-D. Functional Characterization of Seven γ-Glutamylpolyamine Synthetase Genes and the bauRABCD Locus for Polyamine and β-Alanine Utilization in Pseudomonas aeruginosa PAO1. J. Bacteriol. 2011, 193, 3923–3930. [CrossRef]
- Foster, A.; Barnes, N.; Speight, R.; Keane, M.A. Genomic organisation, activity and distribution analysis of the microbial putrescine oxidase degradation pathway. Syst. Appl. Microbiol. 2013, 36, 457–466. [CrossRef]
- Forouhar, F.; Lee, I.-S.; Vujcic, J.; Vujcic, S.; Shen, J.; Vorobiev, S.M.; Xiao, R.; Acton, T.B.; Montelione, G.T.; Porter, C.W.; et al. Structural and Functional Evidence for Bacillus subtilis PaiA as a Novel N1-Spermidine/Spermine Acetyltransferase. J. Biol. Chem. 2005, 280, 40328–40336. [CrossRef]
- Campilongo, R.; Di Martino, M.L.; Marcocci, L.; Pietrangeli, P.; Leuzzi, A.; Grossi, M.; Casalino, M.; Nicoletti, M.; Micheli, G.; Colonna, B.; et al. Molecular and Functional Profiling of the Polyamine Content in Enteroinvasive E. coli: Looking into the Gap between Commensal E. coli and Harmful Shigella. PLoS ONE 2014, 9, e106589. [CrossRef]
- Krysenko, S.; Okoniewski, N.; Kulik, A.; Matthews, A.; Grimpo, J.; Wohlleben, W.; Bera, A. Gamma-Glutamylpolyamine Synthetase GlnA3 Is Involved in the First Step of Polyamine Degradation Pathway in Streptomyces coelicolor M145. Front. Microbiol. 2017, 8, 726. [CrossRef]
- Krysenko, S.; Matthews, A.; Busche, T.; Bera, A.; Wohlleben, W. Poly- and Monoamine Metabolism in Streptomyces coelicolor: The New Role of Glutamine Synthetase-Like Enzymes in the Survival under Environmental Stress. Microb. Physiol. 2021, 31, 233–247. [CrossRef]
- Krysenko S, Okoniewski N, Nentwich M, Matthews A, Bäuerle M, Zinser A, Busche T, Kulik A, Gursch S, Kemeny A. 2022. A second gamma-Glutamylpolyamine synthetase, GlnA2, is involved in polyamine catabolism in Streptomyces coelicolor. Int J Mol Sci 23:3752. [CrossRef]
- Purder PL, Meyners C, Krysenko S, Funk J, Wohlleben W, Hausch F. 2022. Mechanism-Based Design of the First GlnA4-Specific Inhibitors. Chembiochem 23:e202200312. [CrossRef]
- Harth, G.; Masleša-Galić, S.; Tullius, M.V.; Horwitz, M.A. All four Mycobacterium tuberculosis glnA genes encode glutamine synthetase activities but only GlnA1 is abundantly expressed and essential for bacterial homeostasis. Mol. Microbiol. 2005, 58, 1157–1172. [CrossRef]
- Eisenberg, D.; Gill, H.S.; Pfluegl, G.M.; Rotstein, S.H. Structure–function relationships of glutamine synthetases. Biochim. et Biophys. Acta (BBA) - Protein Struct. Mol. Enzym. 2000, 1477, 122–145. [CrossRef]
- Michael, A.J. Polyamine function in archaea and bacteria. J. Biol. Chem. 2018, 293, 18693–18701. [CrossRef]
- Bryk, R.; Arango, N.; Venugopal, A.; Warren, J.D.; Park, Y.-H.; Patel, M.S.; Lima, C.D.; Nathan, C. Triazaspirodimethoxybenzoyls as Selective Inhibitors of Mycobacterial Lipoamide Dehydrogenase. Biochemistry 2010, 49, 1616–1627. [CrossRef]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting Energy Metabolism in Mycobacterium tuberculosis, a New Paradigm in Antimycobacterial Drug Discovery. mBio 2017, 8, e00272-17. [CrossRef]
- Rexer, H.U.; Schäberle, T.; Wohlleben, W.; Engels, A. Investigation of the functional properties and regulation of three glutamine synthetase-like genes in Streptomyces coelicolor A3(2). Arch. Microbiol. 2006, 186, 447–458. [CrossRef]
- Harth, G.; Clemens, D.L.; A Horwitz, M. Glutamine synthetase of Mycobacterium tuberculosis: extracellular release and characterization of its enzymatic activity.. Proc. Natl. Acad. Sci. 1994, 91, 9342–9346. [CrossRef]
- Paulin, L.G.; E Brander, E.; Pösö, H.J. Specific inhibition of spermidine synthesis in Mycobacteria spp. by the dextro isomer of ethambutol. Antimicrob. Agents Chemother. 1985, 28, 157–159. [CrossRef]
- Jain, A.; Tyagi, A.K. Role of polyamines in the synthesis of RNA in mycobacteria. Mol. Cell. Biochem. 1987, 78, 3–8. [CrossRef]
- Culp, E.; Wright, G.D. Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 2016, 70, 366–377. [CrossRef]
- Malm, S.; Tiffert, Y.; Micklinghoff, J.; Schultze, S.; Joost, I.; Weber, I.; Horst, S.; Ackermann, B.; Schmidt, M.; Wohlleben, W.; et al. The roles of the nitrate reductase NarGHJI, the nitrite reductase NirBD and the response regulator GlnR in nitrate assimilation of Mycobacterium tuberculosis. Microbiology 2009, 155, 1332–1339. [CrossRef]
- Tiffert, Y.; Supra, P.; Wurm, R.; Wohlleben, W.; Wagner, R.; Reuther, J. The Streptomyces coelicolor GlnR regulon: identification of new GlnR targets and evidence for a central role of GlnR in nitrogen metabolism in actinomycetes. Mol. Microbiol. 2008, 67, 861–880. [CrossRef]
- Gawronski, J.D.; Benson, D.R. Microtiter assay for glutamine synthetase biosynthetic activity using inorganic phosphate detection. Anal. Biochem. 2004, 327, 114–118. [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; De Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [CrossRef]
- Gill, H.S.; Pfluegl, G.M.U.; Eisenberg, D. Multicopy Crystallographic Refinement of a Relaxed Glutamine Synthetase from Mycobacterium tuberculosis Highlights Flexible Loops in the Enzymatic Mechanism and Its Regulation. Biochemistry 2002, 41, 9863–9872. [CrossRef]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; Ahmad, Z. Drug targets exploited in Mycobacterium tuberculosis: Pitfalls and promises on the horizon. Biomed. Pharmacother. 2018, 103, 1733–1747. [CrossRef]
- Mougous, J.D.; E Green, R.; Williams, S.J.; E Brenner, S.; Bertozzi, C.R. Sulfotransferases and Sulfatases in Mycobacteria. Chem. Biol. 2002, 9, 767–776. [CrossRef]
- Hatzios, S.K.; Bertozzi, C.R. The Regulation of Sulfur Metabolism in Mycobacterium tuberculosis. PLOS Pathog. 2011, 7, e1002036. [CrossRef]
- Shetye, G.S.; Franzblau, S.G.; Cho, S. New tuberculosis drug targets, their inhibitors, and potential therapeutic impact. Transl. Res. 2020, 220, 68–97. [CrossRef]
- Brotzoesterhelt, H.; Brunner, N. How many modes of action should an antibiotic have?. Curr. Opin. Pharmacol. 2008, 8, 564–573. [CrossRef]
- Haagsma, A.C.; Abdillahi-Ibrahim, R.; Wagner, M.J.; Krab, K.; Vergauwen, K.; Guillemont, J.; Andries, K.; Lill, H.; Koul, A.; Bald, D. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 2009, 53, 1290–1292. [CrossRef]
- Radkov, A.D.; Hsu, Y.-P.; Booher, G.; VanNieuwenhze, M.S. Imaging Bacterial Cell Wall Biosynthesis. Annu. Rev. Biochem. 2018, 87, 991–1014. [CrossRef]
- Krajewski, W.W.; Jones, T.A.; Mowbray, S.L. Structure of Mycobacterium tuberculosis glutamine synthetase in complex with a transition-state mimic provides functional insights. Proc. Natl. Acad. Sci. 2005, 102, 10499–10504. [CrossRef]
- Nilsson MT, Mowbray SL. 2012. Crystal structure of Mycobacterium tuberculosis glutamine synthethase in complex with imidazopyridine inhibitor ((4-(6-Bromo-3-(Butylamino)imidazo(1,2-A)Pyridin-2-YL)Phenoxy) Acetic acid) and L-Methionine-S-Sulfoximine phosphate. MedChemComm 3:620. [CrossRef]
- Nilsson MT, Mowbray SL. 2012. Crystal structure of Mycobacterium Tuberculosis Glutamine Synthetase in complex with tri-substituted imidazole inhibitor (4-(2-tert-butyl- 4-(6-methoxynaphthalen-2-yl)-1H-imidazol-5-yl)pyridin-2-amine) and L- methionine-S-sulfoximine phosphate. J Med Chem 55.
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [CrossRef]
- Hooft, R.W.; Sander, C.; Vriend, G. Objectively judging the quality of a protein structure from a Ramachandran plot. Bioinformatics 1997, 13, 425–430. [CrossRef]
- Chen, V.B.; Arendall, W.B., III; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21.
- Ladner, J.E.; Atanasova, V.; Dolezelova, Z.; Parsons, J.F. Structure and Activity of PA5508, a Hexameric Glutamine Synthetase Homologue. Biochemistry 2012, 51, 10121–10123. [CrossRef]
- Chanphai, P.; Thomas, T.; Tajmir-Riahi, H. Conjugation of biogenic and synthetic polyamines with serum proteins: A comprehensive review. Int. J. Biol. Macromol. 2016, 92, 515–522. [CrossRef]
- Rhee, K.Y.; de Carvalho, L.P.S.; Bryk, R.; Ehrt, S.; Marrero, J.; Park, S.W.; Schnappinger, D.; Venugopal, A.; Nathan, C. Central carbon metabolism in Mycobacterium tuberculosis: an unexpected frontier. Trends Microbiol. 2011, 19, 307–314. [CrossRef]
- Sakharkar, K. R., Sakharkar, M. K., & Chow, V. T. (2008). Biocomputational strategies for microbial drug target identification. Methods in molecular medicine, 142, 1–9. [CrossRef]
- Schmitz, K.R.; Sauer, R.T. Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol. Microbiol. 2014, 93, 617–628. [CrossRef]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R. inhA, a Gene Encoding a Target for Isoniazid and Ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [CrossRef]
- Bayer, E.; Gugel, K.H.; Hägele, K.; Hagenmaier, H.; Jessipow, S.; König, W.A.; Zähner, H. Stoffwechselprodukte von Mikroorganismen. 98. Phosphinothricin und Phosphinothricyl-Alanyl-Alanin [Metabolic products of microorganisms. 98. Phosphinothricin and phosphinothricyl-alanyl-analine]. Helvetica Chim. Acta 1972, 55, 224–239. [CrossRef]
- Krysenko, S.; Purder, P.; Hausch, F.; Wohlleben, W. Neuartige synthetische Wirkstoffe zur Tuberkulosebekämpfung. BIOspektrum 2024, 30, 819–821. [CrossRef]
- Selim, M.S.M.; Abdelhamid, S.A.; Mohamed, S.S. Secondary metabolites and biodiversity of actinomycetes. J. Genet. Eng. Biotechnol. 2021, 19, 72–13. [CrossRef]
- Parish, T.; Stoker, N.G. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 2000, 146, 1969–1975. [CrossRef]
- Sao Emani C, Williams MJ, Van Helden PD, Taylor MJC, Carolis C, Wiid IJ, Baker B. 2018. Generation and characterization of thiol-deficient Mycobacterium tuberculosis mutants. Sci Data 5:180184.
- De Rossi, E.; Branzoni, M.; Cantoni, R.; Milano, A.; Riccardi, G.; Ciferri, O. mmr, a Mycobacterium tuberculosis Gene Conferring Resistance to Small Cationic Dyes and Inhibitors. J. Bacteriol. 1998, 180, 6068–6071. [CrossRef]
- Zelmer, A.; Carroll, P.; Andreu, N.; Hagens, K.; Mahlo, J.; Redinger, N.; Robertson, B.D.; Wiles, S.; Ward, T.H.; Parish, T.; et al. A new in vivo model to test anti-tuberculosis drugs using fluorescence imaging. J. Antimicrob. Chemother. 2012, 67, 1948–1960. [CrossRef]
- Carroll, P.; Schreuder, L.J.; Muwanguzi-Karugaba, J.; Wiles, S.; Robertson, B.D.; Ripoll, J.; Ward, T.H.; Bancroft, G.J.; Schaible, U.E.; Parish, T. Sensitive Detection of Gene Expression in Mycobacteria under Replicating and Non-Replicating Conditions Using Optimized Far-Red Reporters. PLoS ONE 2010, 5, e9823. [CrossRef]
- Stec, J.; Onajole, O.K.; Lun, S.; Guo, H.; Merenbloom, B.; Vistoli, G.; Bishai, W.R.; Kozikowski, A.P. Indole-2-carboxamide-based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem. 2016, 59, 6232–6247. [CrossRef]
- Boutte, C.C.; E Baer, C.; Papavinasasundaram, K.; Liu, W.; Chase, M.R.; Meniche, X.; Fortune, S.M.; Sassetti, C.M.; Ioerger, T.R.; Rubin, E.J. A cytoplasmic peptidoglycan amidase homologue controls mycobacterial cell wall synthesis. eLife 2016, 5. [CrossRef]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [CrossRef]
- Evans, C.G.T.; Herbert, D.; Tempest, D.W. Chapter XIII The Continuous Cultivation of Micro-organisms. Methods Microbiol. 1970, 2, 277–327. [CrossRef]
- Gust, B.; Challis, G.L.; Fowler, K.; Kieser, T.; Chater, K.F. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. 2003, 100, 1541–1546. [CrossRef]
- Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical streptomyces genetics, vol 291. John Innes Foundation Norwich, Berlin.
- Jumde, R.P.; Guardigni, M.; Gierse, R.M.; Alhayek, A.; Zhu, D.; Hamid, Z.; Johannsen, S.; Elgaher, W.A.M.; Neusens, P.J.; Nehls, C.; et al. Hit-optimization using target-directed dynamic combinatorial chemistry: development of inhibitors of the anti-infective target 1-deoxy-d-xylulose-5-phosphate synthase. Chem. Sci. 2021, 12, 7775–7785. [CrossRef]
- Wallace, R.J.; Nash, D.R.; Steele, L.C.; Steingrube, V. Susceptibility testing of slowly growing mycobacteria by a microdilution MIC method with 7H9 broth. J. Clin. Microbiol. 1986, 24, 976–981. [CrossRef]
- Palomino, J.-C.; Martin, A.; Camacho, M.; Guerra, H.; Swings, J.; Portaels, F. Resazurin Microtiter Assay Plate: Simple and Inexpensive Method for Detection of Drug Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2. [CrossRef]
- Merker, M.; Kohl, T.A.; Roetzer, A.; Truebe, L.; Richter, E.; Rüsch-Gerdes, S.; Fattorini, L.; Oggioni, M.R.; Cox, H.; Varaine, F.; et al. Whole Genome Sequencing Reveals Complex Evolution Patterns of Multidrug-Resistant Mycobacterium tuberculosis Beijing Strains in Patients. PLoS ONE 2013, 8, e82551. [CrossRef]
- Andreu, N.; Zelmer, A.; Fletcher, T.; Elkington, P.T.; Ward, T.H.; Ripoll, J.; Parish, T.; Bancroft, G.J.; Schaible, U.; Robertson, B.D.; et al. Optimisation of Bioluminescent Reporters for Use with Mycobacteria. PLoS ONE 2010, 5, e10777. [CrossRef]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, W252–W258. [CrossRef]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 1–15. [CrossRef]
- Biasini, M.; Schmidt, T.; Bienert, S.; Mariani, V.; Studer, G.; Haas, J.; Johner, N.; Schenk, A.D.; Philippsen, A.; Schwede, T. OpenStructure: an integrated software framework for computational structural biology. Acta Crystallogr. Sect. D Struct. Biol. 2013, 69, 701–709. [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [CrossRef]
- Zheng L, Baumann U, Reymond J-L. 2004. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res 32:e115-e115.
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [CrossRef]
- Latour, Y.L.; Gobert, A.P.; Wilson, K.T. The role of polyamines in the regulation of macrophage polarization and function. Amino Acids 2019, 52, 151–160. [CrossRef]
- Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; Crick, D.C.; Fontes, F.L.; Pethe, K.; Hasenoehrl, E.; et al. Oxidative Phosphorylation as a Target Space for Tuberculosis: Success, Caution, and Future Directions. Microbiol. Spectr. 2017, 5. [CrossRef]
- Zhang, X.; Zhao, R.; Qi, Y.; Yan, X.; Qi, G.; Peng, Q. The progress of Mycobacterium tuberculosis drug targets. Front. Med. 2024, 11, 1455715. [CrossRef]
- Paritala, H.; Carroll, K.S. New Targets and Inhibitors of Mycobacterial Sulfur Metabolism. Infect. Disord. - Drug Targets 2013, 13, 85–115. [CrossRef]
- Rath, V.L.; Verdugo, D.; Hemmerich, S. Sulfotransferase structural biology and inhibitor discovery. Drug Discov. Today 2004, 9, 1003–1011. [CrossRef]
- Bhave, D.P.; Iii, W.B.M.; Carroll, K.S. Drug Targets in Mycobacterial Sulfur Metabolism. Infect. Disord. - Drug Targets 2007, 7, 140–158. [CrossRef]
- Chapman, E.; Ding, S.; Schultz, P.G.; Wong, C.-H. A Potent and Highly Selective Sulfotransferase Inhibitor. J. Am. Chem. Soc. 2002, 124, 14524–14525. [CrossRef]
- Silver, R.F.; Myers, A.J.; Jarvela, J.; Flynn, J.; Rutledge, T.; Bonfield, T.; Lin, P.L. Diversity of Human and Macaque Airway Immune Cells at Baseline and during Tuberculosis Infection. Am. J. Respir. Cell Mol. Biol. 2016, 55, 899–908. [CrossRef]
- Patrick, G.J.; Fang, L.; Schaefer, J.; Singh, S.; Bowman, G.R.; Wencewicz, T.A. Mechanistic Basis for ATP-Dependent Inhibition of Glutamine Synthetase by Tabtoxinine-β-lactam. Biochemistry 2017, 57, 117–135. [CrossRef]
- Hirsch, J.G.; Dubos, R.J. The effect of spermine on tubercle bacilli. J. Exp. Med. 1952, 95, 191–208. [CrossRef]
- Joshi, G.S.; Spontak, J.S.; Klapper, D.G.; Richardson, A.R. Arginine catabolic mobile element encoded speG abrogates the unique hypersensitivity of Staphylococcus aureus to exogenous polyamines. Mol. Microbiol. 2011, 82, 9–20. [CrossRef]
- Tullius, M.V.; Harth, G.; Horwitz, M.A. Glutamine Synthetase GlnA1 Is Essential for Growth of Mycobacterium tuberculosis in Human THP-1 Macrophages and Guinea Pigs. Infect. Immun. 2003, 71, 3927–36. [CrossRef]
- Kruh, N.A.; Troudt, J.; Izzo, A.; Prenni, J.; Dobos, K.M. Portrait of a Pathogen: The Mycobacterium tuberculosis Proteome In Vivo. PLoS ONE 2010, 5, e13938. [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.A.; Garnier, T.; Churcher, C.; Harris, D.R.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., III; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [CrossRef]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [CrossRef]
- Bosch, B.; DeJesus, M.A.; Poulton, N.C.; Zhang, W.; Engelhart, C.A.; Zaveri, A.; Lavalette, S.; Ruecker, N.; Trujillo, C.; Wallach, J.B.; et al. Genome-wide gene expression tuning reveals diverse vulnerabilities of M. tuberculosis. Cell 2021, 184, 4579–4592.e24. [CrossRef]
- De Rossi, E.; Arrigo, P.; Bellinzoni, M.; Silva, P.E.A.; Martin, C.; Aínsa, J.A.; Guglierame, P.; Riccardi, G. The Multidrug Transporters Belonging to Major Facilitator Superfamily (MFS) in Mycobacterium tuberculosis. Mol. Med. 2002, 8, 714–724. [CrossRef]
- Balganesh, M.; Dinesh, N.; Sharma, S.; Kuruppath, S.; Nair, A.V.; Sharma, U. Efflux Pumps of Mycobacterium tuberculosis Play a Significant Role in Antituberculosis Activity of Potential Drug Candidates. Antimicrob. Agents Chemother. 2012, 56, 2643–2651. [CrossRef]
- Dutta, N.K.; Mehra, S.; Kaushal, D. A Mycobacterium tuberculosis Sigma Factor Network Responds to Cell-Envelope Damage by the Promising Anti-Mycobacterial Thioridazine. PLoS ONE 2010, 5, e10069. [CrossRef]
- Rodrigues, L.; Villellas, C.; Bailo, R.; Viveiros, M.; Aínsa, J.A. Role of the Mmr Efflux Pump in Drug Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 751–757. [CrossRef]
- Krysenko, S.; Matthews, A.; Okoniewski, N.; Kulik, A.; Girbas, M.G.; Tsypik, O.; Meyners, C.S.; Hausch, F.; Wohlleben, W.; Bera, A. Initial Metabolic Step of a Novel Ethanolamine Utilization Pathway and Its Regulation in Streptomyces coelicolor M145. mBio 2019, 10, e00326-19. [CrossRef]
- Bullock WO, Fernandez JM, Short JM. 1987. XL1-blue: a high efficiency plasmid transforming recA Escherichia coli strain with beta-galactosidase selection. BioTechniques 5:376.
- Simon, R.; Priefer, U.; Pühler, A. A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Bio/Technology 1983, 1, 784–791. [CrossRef]
- Davanloo, P.; Rosenberg, A.H.; Dunn, J.J.; Studier, F.W. Cloning and expression of the gene for bacteriophage T7 RNA polymerase.. Proc. Natl. Acad. Sci. 1984, 81, 2035–2039. [CrossRef]
- Kubica, G.P.; Kim, T.H.; Dunbar, F.P. Designation of Strain H37Rv as the Neotype of Mycobacterium tuberculosis. Int. J. Syst. Evol. Microbiol. 1972, 22, 99–106. [CrossRef]
- Krysenko S, and Wohlleben W. Polyamine and Ethanolamine Metabolism in Bacteria as an Important Component of Nitrogen Assimilation for Survival and Pathogenicity. Med. Sci. 2022, 10, 40. [CrossRef]
- León, J.; Castillo, M.C.; Coego, A.; Lozano-Juste, J.; Mir, R. Diverse functional interactions between nitric oxide and abscisic acid in plant development and responses to stress. J. Exp. Bot. 2013, 65, 907–921. [CrossRef]
- Astier J, Gross I, Durner J (2018) Nitric oxide production in plants: an update. J. Exp. Bot. 69(14):3401–3411. [CrossRef]
- Bajeli, S.; Baid, N.; Kaur, M.; Pawar, G.P.; Chaudhari, V.D.; Kumar, A. Terminal Respiratory Oxidases: A Targetables Vulnerability of Mycobacterial Bioenergetics?. Front. Cell. Infect. Microbiol. 2020, 10. [CrossRef]
- Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; Crick, D.C.; Fontes, F.L.; Pethe, K.; Hasenoehrl, E.; et al. Oxidative Phosphorylation as a Target Space for Tuberculosis: Success, Caution, and Future Directions. Microbiol. Spectr. 2017, 5. [CrossRef]
- Gunjan, S.; Sharma, T.; Yadav, K.; Chauhan, B.S.; Singh, S.K.; Siddiqi, M.I.; Tripathi, R. Artemisinin Derivatives and Synthetic Trioxane Trigger Apoptotic Cell Death in Asexual Stages of Plasmodium. Front. Cell. Infect. Microbiol. 2018, 8, 256. [CrossRef]
- Lievens, L.; Pollier, J.; Goossens, A.; Beyaert, R.; Staal, J. Abscisic Acid as Pathogen Effector and Immune Regulator. Front. Plant Sci. 2017, 8, 587. [CrossRef]
- Xu, J.; Lu, X.; Liu, Y.; Lan, W.; Wei, Z.; Yu, W.; Li, C. Interaction between ABA and NO in plants under abiotic stresses and its regulatory mechanisms. Front. Plant Sci. 2024, 15, 1330948. [CrossRef]
- Makhoba, X.H. Two sides of the same coin: heat shock proteins as biomarkers and therapeutic targets for some complex diseases. Front. Mol. Biosci. 2025, 12, 1491227. [CrossRef]
- Fournier, P.E.; Richet, H.; Weinstein, R.A. The Epidemiology and Control of Acinetobacter baumannii in Health Care Facilities. Clin. Infect. Dis. 2006, 42, 692–699. [CrossRef]
- Lolans, K.; Rice, T.W.; Munoz-Price, L.S.; Quinn, J.P. Multicity Outbreak of Carbapenem-Resistant Acinetobacter baumannii Isolates Producing the Carbapenemase OXA-40. Antimicrob. Agents Chemother. 2006, 50, 2941–2945. [CrossRef]
Figure 1.
Drugs targets in M. tuberculosis sorted according to their mechanism of action (modified after Alsayed & Gunosewoyo, 2023).
Figure 1.
Drugs targets in M. tuberculosis sorted according to their mechanism of action (modified after Alsayed & Gunosewoyo, 2023).
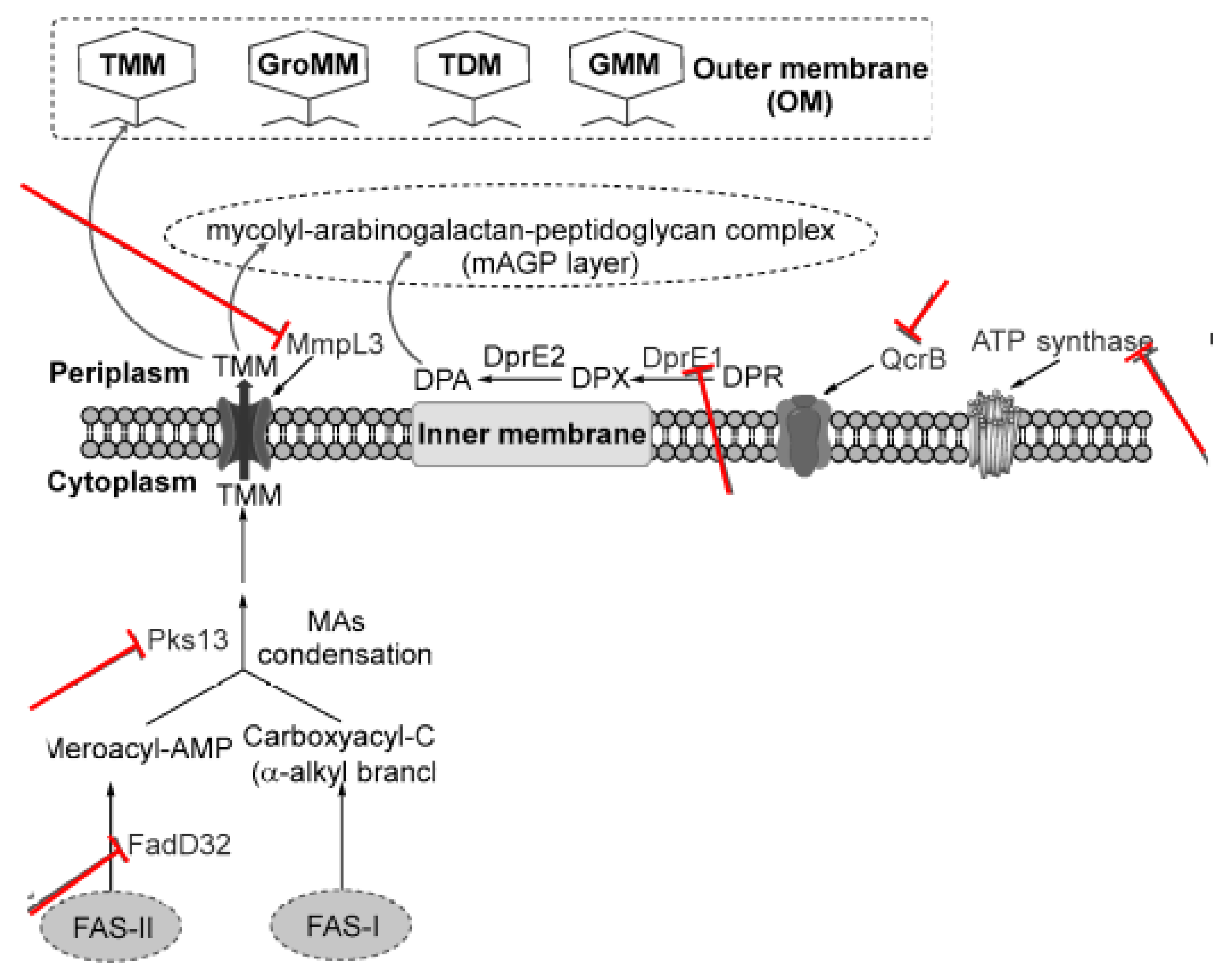
Figure 2.
Summary of Hsp70 reaction. Hsp40, as cochaperones, binds to the client protein or unfolded protein and hands it over to Hsp70 (ATP form) for folding purposes. The nucleotide exchange factor binds to Hsp70, thus catalyzing the dissociation of ADP. Finally, ATP binds to Hsp70, inducing the opening of the lid, thereby enabling substrate release.
Figure 2.
Summary of Hsp70 reaction. Hsp40, as cochaperones, binds to the client protein or unfolded protein and hands it over to Hsp70 (ATP form) for folding purposes. The nucleotide exchange factor binds to Hsp70, thus catalyzing the dissociation of ADP. Finally, ATP binds to Hsp70, inducing the opening of the lid, thereby enabling substrate release.
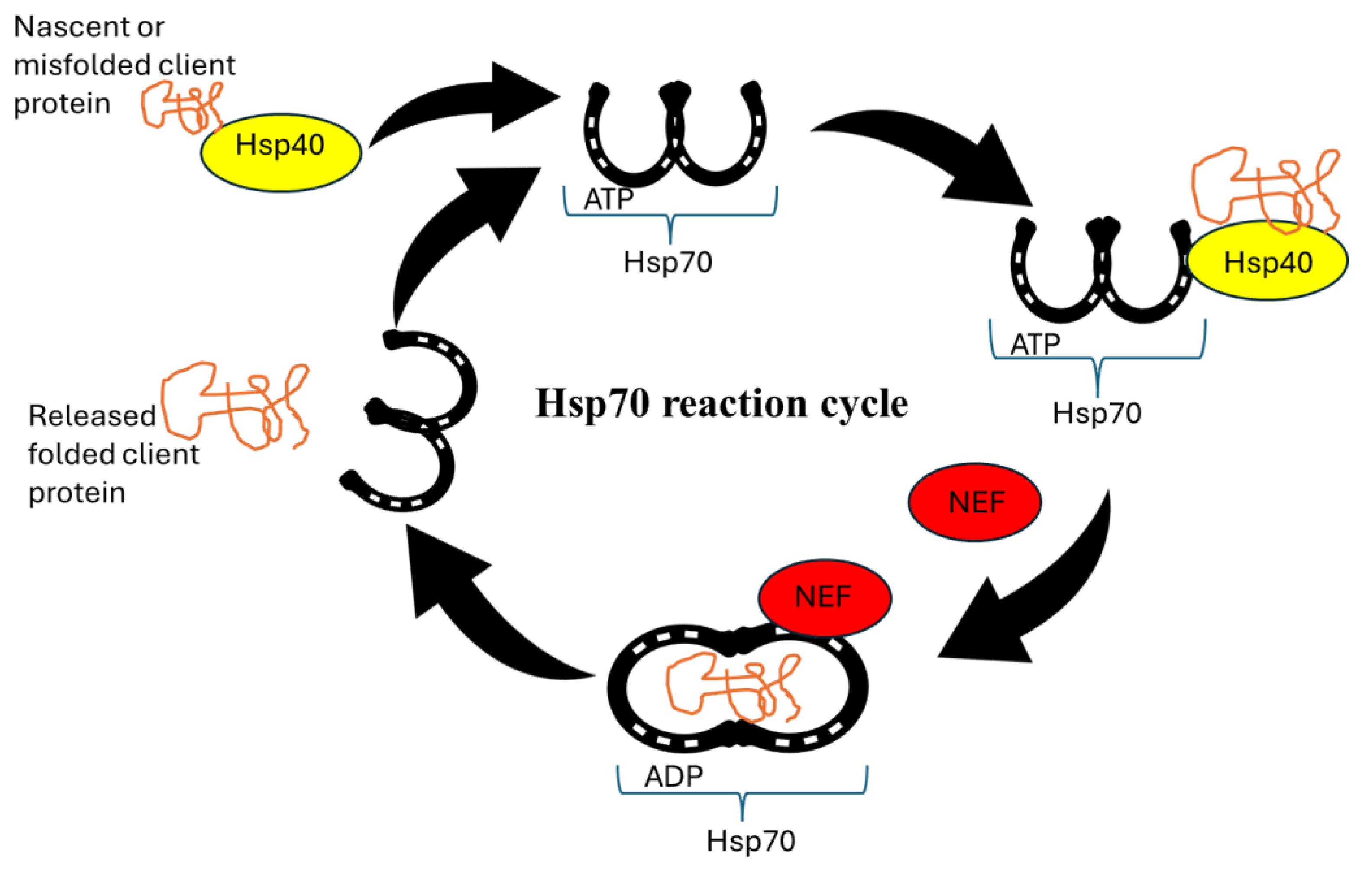
Table 2.
Available GS and GS-like inhibitors targeting Actinobacteria.
| Name | Mode of action | Reference | Structure |
|---|---|---|---|
| Methionine sulfoximine (MetSox/MSO) | Potent ATP-dependent inactivator of GS. | Krajewski et al., 2005 | 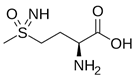 |
| Phosphinothricin (PPT) | Potent ATP-dependent inactivator of GS that is produced as part of a tripeptide antibiotic by Streptomyces viridochromogenes. | Bayer et al., 1972 | 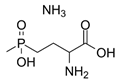 |
| Tabtoxinine β-lactam | Potent ATP-dependent inactivator of GS produced by Pseudomonas pv. tabaci. | Patrick et al., 2018 | 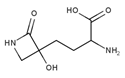 |
| Alanosine | Antibiotic produced by Streptomyces alanosinicus. | Eisenberg et al., 2000 | 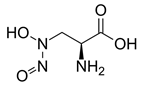 |
| Oxetin | Antibiotic produced by Streptomyces sp., inhibitor of GS | Eisenberg et al., 2000 | 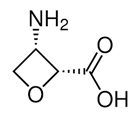 |
| 7b (PPU301) | Synthetic inactivator of the GS-like enzyme GlnA4 from Streptomyces coelicolor | Purder et al., 2022 | 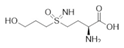 |
| PPU268 | Synthetic inactivator of the GS-like enzyme GlnA2 from Streptomyces coelicolor | Krysenko et al., 2023 | 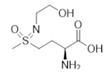 |
Table 3.
Summary of heat shock proteins in Mycobacterium tuberculosis and therapeutic strategies.
| Name | Roles | Inhibitor | Action |
|---|---|---|---|
| Hsp90 (Grp94) | Holdase | Geldanamycin and its derivatives | These compounds bind to the N-terminal ATP-binding domain of Hsp90, inhibiting its chaperone activit |
| Hsp70 (Dnak) | Foldase | VER-155008 | A small molecule inhibitor that binds to the ATPase domain of Hsp70, inhibiting its activity |
| ClpB | Holdase | Ecumicin | A cyclic peptide that targets the ClpC1 ATPase component of the Clp protease complex, enhancing its ATPase activity but preventing proteolysis |
Table 4.
Summary of some common combination therapies for actinobacteria treatment.
| Combination | Mechanism | Use Case |
|---|---|---|
| Colistin + Rifampicin | Synergistic effect | Multidrug-resistant strains |
| Tigecycline + Sulbactam | Enhanced efficacy | Severe infections |
| Meropenem + Polymyxin | Broad-spectrum activity | Carbapenem-resistant strains |
| Beta-lactam + Aminoglycoside | Synergistic effect | General use in resistant infection |
| Fluoroquinolone + Beta-lactam | Targeting different pathways | Reducing resistance development |
| Spermine + isoniazid, rifampicin, aminosalicylic acid, bedaquiline | Enchancing effect of spermine on conventional drugs | General use in resistant infection |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



