
Submitted:
06 April 2025
Posted:
07 April 2025
You are already at the latest version
Abstract
Prenatal hypoxia (PH) disrupts fetal heart development and induces persistent cardiovascular dysfunction in offspring. The nitric oxide (NO) system is pivotal in regulating myocardial function, influencing vascular tone, contractility, and endothelial integrity during development. PH alters NO signaling, leading to endothelial dysfunction, mitochondrial damage, and oxidative stress, which exacerbate myocardial impairment and increase cardiomyocyte apoptosis. Elevated nitrosative stress, through excessive reactive nitrogen species, further exacerbates these effects, promoting inflammation and cellular injury. The interaction between NO and hypoxia-inducible factor (HIF) also mediates adaptive responses to PH. Additionally, NO modulates the expression of heat shock protein 70 (HSP70), providing cellular protection under stress. This review highlights the role of NO in PH-induced cardiovascular damage and evaluates the therapeutic potential of NO modulators—Angiolin, Tiotriazoline, Mildronate, and L-arginine—as promising agents for cardioprotection. These compounds mitigate oxidative stress, improve endothelial function, and counteract the adverse effects of PH on the heart, suggesting novel therapeutic strategies for preventing cardiovascular dysfunction in offspring exposed to prenatal hypoxia.
Keywords:
prenatal hypoxia
; cardiomyopathy
; endothelial dysfunction
; mitochondrial dysfunction
; oxidative stress
; nitrosative stress
; cardiomyocyte apoptosis
; NO modulators
; HSP70
1. Introduction
Introduction. Post-hypoxic disorders of the cardiovascular system are among the leading causes of morbidity in newborns, occurring in 40–70% of children who have experienced prenatal hypoxia, according to various sources. These disorders are the starting point for many often serious diseases in both children and adults. To this day, the mechanisms of post-hypoxic cardiac disturbances remain poorly understood, making it a relevant issue in pediatric cardiology [1,2,3,4,5,6,7,8]. The clinical symptoms of this pathology in the acute period are polymorphic, often masking other diseases, and differential diagnosis with congenital heart defects, congenital myocarditis, and cardiomyopathies is often required. To this day, there is no consensus on the step-by-step comprehensive therapy for post-hypoxic cardiac disorders [9,10]. Therefore, identifying new structural, molecular, and biochemical features of post-hypoxic cardiovascular disorders in newborns and developing pharmacotherapy strategies based on these findings is of scientific interest. According to current understanding, endothelial dysfunction and the associated disturbances in the NO system are fundamental to the development of many cardiovascular diseases [11,12]. Under the influence of hypoxia, infection, and other damaging factors, the functioning of the nitric oxide system is disrupted, leading to the development of pathology in various organs and systems, including the cardiovascular system [13,14]. However, the literature concerning the role of the NO system in the development of cardiovascular pathology in newborns and the potential cardioprotective effects of its modulators is quite limited. Several studies have identified cardio- and endothelial-protective properties of drugs that can both increase NO synthesis and enhance the bioavailability of this messenger [15,16,17,18,19].
1. Prenatal Hypoxia and Its Impact on Cardiovascular Development
1.1. Prenatal Hypoxia and Its Consequences
Hypoxic changes in the myocardial energy metabolism lead to a rapid decrease in its contractile function (Figure 1). This is facilitated by certain anatomical and physiological characteristics of newborns, such as the diffuse type of coronary arteries with numerous anastomoses between the right and left coronary arteries, their small diameter, as well as the predominance of sympathetic nervous system influence, the tone of which is maintained by the preceding hypoxic condition of the central nervous system (CNS), known as the cerebrocardiac syndrome [20,21,22].
Fetal hypoxia leads to a disruption of the vegetative regulation of coronary vessels, impairing energy metabolism and causing a sharp decrease in the formation of macroergic compounds in the mitochondria of cardiomyocytes [23,24,25,26]. Acidosis, hypercatecholaminemia, hypoglycemia, and the deterioration of the blood's rheological properties are key factors in the pathogenesis of hypoxic damage to the conduction cardiomyocytes in newborns and are the cause of various types of arrhythmias [27,28]. A well-known role in the development of post-hypoxic cardiac rhythm disturbances is played by disruptions in vegetative regulation [29,30]. The connection between hypoxic myocardial damage and various disturbances in cardiac rhythm and conductivity is evidenced by morphological and ultrastructural studies [31,32].
In the conduction system of the heart after prenatal hypoxia, signs of apoptosis and dystrophy are observed, with a certain correlation between the severity of morphological changes and clinically identified rhythm and conductivity disturbances. The final morphological outcome of hypoxic myocardial damage can be focal dystrophy, which has two possible outcomes: either complete resolution and restoration of function, or the formation of focal cardiosclerosis [33,34,35,36,37]. Currently, cardiomyopathies are understood as diseases of unclear etiology, primarily affecting the myocardium, where the contractile proteins of the heart muscle lose some of their properties, resulting in an insufficiently effective contraction of the heart muscle [38,39,40,41].
This, in turn, negatively impacts the entire circulatory system of the child – symptoms of heart failure arise and gradually worsen, accompanied by blood shunting from one circulatory circuit to another in the absence of morphological signs of active inflammation. Cardiomyopathies are classified into primary (idiopathic) and secondary types [34,42,43]. Transient post-hypoxic myocardial ischemia is classified as a secondary cardiomyopathy and is predominantly observed in the first hours and days of a newborn's life. Among the hemodynamic factors, transient pulmonary hypertension, increased blood pressure, and the closure of fetal communications play a significant role in the development of post-hypoxic cardiomyopathy, as they create additional workload on the myocardium with reduced functional capacity [44,45,46].
Since at the time of birth, and depending on its outcome, the level and degree of cardiovascular system damage will vary: neonatal pulmonary hypertension, persistence of fetal communications, myocardial dysfunction with chamber dilation, myocardial ischemia, and disturbances in heart rhythm and conductivity [47,48,49,50,51,52,53]. Hypoxia leads to an increased workload on the heart, as the child experiences narrowing of blood vessels in both the pulmonary and systemic circulations, which is a result of the release of catecholamines and the direct effect of carbon dioxide [23,26,54,55]. Blood return to the heart increases, raising the pressure in the right ventricle, which may become equal to the systemic arterial pressure. Myocardial blood flow is unable to fully supply the cardiomyocytes with oxygen, and consequently, the demand for oxygen rises. This leads to the development of coronary insufficiency and myocardial ischemia [34,56,57,58,59].
In children who have undergone both chronic intrauterine and perinatal hypoxia, a cardiovascular system maladaptation syndrome is observed during the neonatal period, accompanied by a prolonged (up to several months of life) increase in the activity of cardiac-specific enzymes. This period can be considered transitional in terms of myocardial metabolism in newborns affected by hypoxia. In children who underwent chronic intrauterine hypoxia, the cardiovascular maladaptation syndrome is of a transient, benign nature, with rapid reversal of clinical symptoms and almost complete absence of residual phenomena. One in three children who experienced perinatal hypoxia have residual symptoms, such as minimal signs of pulmonary hypertension. Valve insufficiency and reduced contractile ability of the ventricular myocardium are identified much less frequently. This dictates the need for prolonged outpatient monitoring of this group of children and appropriate medical interventions.
Identifying the type of autonomic reactivity helps determine the leading direction for corrective measures, preventing the formation of functional heart pathology in these patients in the future. Prenatal hypoxia leads to an increased workload on the heart, as vasoconstriction of the vessels in both the systemic and pulmonary circulations occurs in the child, due to disruptions in the nitric oxide system, nitrosative stress, and endothelial dysfunction [23,60,61,62,63,64,65,66,67,68,69,70,71]. All of this leads to the circulatory system failing to perform its function of providing oxygen to the working heart, resulting in myocardial ischemia.
On average, 30% of children who have experienced intrauterine hypoxia retain residual phenomena, such as minimal signs of pulmonary hypertension, reduced heart pump function, and disturbances in the autonomic regulation of cardiac activity. Prenatal hypoxia negatively impacts the morphological and functional characteristics of the cardiovascular system at all stages of ontogenesis and may lead to the formation of a disproportionate development pattern of the heart, as well as morphological and functional disturbances in the conduction system.
The functional changes in the cardiovascular system seen in the post-hypoxic maladaptation syndrome are based on impaired neurohumoral regulation of vascular tone, transient neonatal pulmonary hypertension, prolonged persistence of fetal communications (PFC), and a delay in the formation of the mature type of cardiomyocyte metabolism [6,8,33,72,73,74,75]. Disorders of the cardiovascular system are already detected during the initial examination of the newborn, including incomplete block of the right bundle branch of the His bundle, extrasystole, and signs of subendocardial ischemia. Therefore, post-hypoxic cardiomyopathy is considered one of the risk factors for the development of cardiovascular pathology (such as rhythm disturbances, vascular dystonias, and others) in later stages of life [6,76,77,78].
We have established that modeling prenatal hypoxia in rats leads to a reduced heart rate and a significant dominance of parasympathetic innervation in the regulation of the heart's electrical activity. The decrease in heart rate after experiencing hypoxia could be caused by sinus block, which may also reflect parasympathetic regulation of the heart instead of sympathetic control of electrical activity under normal conditions. The development of disturbances in the bioelectrical activity of the heart after PH led to an extension of the electrical systole of the ventricles, which may have been caused by impaired myocardial conductivity in the ventricles. Under these conditions, the power of ventricular electrical repolarization increased 5.5 times, indicating significant issues with the restoration of the membrane potential in ventricular cardiomyocytes.
Fetal hypoxia leads to a disruption in the autonomic regulation of coronary vessels, deterioration of energy metabolism, and damage to the ultrastructure of mitochondria, both in cardiomyocytes and in the cells of the conduction system, which may be a possible cause of reduced myocardial contractility and impaired normal functioning of the sinoatrial node [79]. In the contractile myocardium and conduction system during the post-hypoxic period, cells with signs of apoptosis and dystrophy are observed, with a certain correlation between the severity of morphological changes and the bioelectrical disturbances in rhythm and conductivity. The result of hypoxic myocardial damage can be focal dystrophy, which, if not adequately treated, may lead to focal cardiosclerosis [80,81].
To summarize the above, it can be said that PH is a powerful damaging factor that triggers mechanisms such as oxidative and nitrosative stress, mitochondrial dysfunction, disruption of energy supply to the heart, and apoptosis. Research into the molecular and biochemical mechanisms of post-hypoxic cardiomyopathy in newborns has shown that many of these processes are dependent on nitric oxide (NO). All of this makes the NO system an attractive area for study from the perspective of fundamental medicine and biology, as well as a promising target for pharmacological intervention.
1.2. The Role of NO in Heart Regulation
The role of NO in regulating various processes in the cardiovascular system is well known. Endothelial cells synthesize and release NO, which mediates a variety of effects, including vascular tone, hemostasis, blood pressure, and vascular remodeling [82,83,84,85]. The importance of NO for the function of cardiomyocytes is well known, with its role in the regulation of ion channels, Ca²⁺ homeostasis, contractility, energy metabolism, proliferation, and the formation of resistance to hypoxia. NO exerts its metabolotropic, physiological, and other effects through various mechanisms. For instance, NO can post-translationally modify target proteins, primarily by adding a nitroso group to the thiol side chain of cysteine, a process known as S-nitrosylation, which leads to the acquisition of new properties by the protein [86,87,88,89]. The effects of direct modification of target proteins by NO are limited by the relatively short diffusion distance of the molecule. Molecules of cellular carriers, such as S-nitrosoglutathione, mediate more distant NO signal transmission, acting as a carrier and donor, transferring NO to more distant targets [90,91,92]. Exogenous administration of glutathione exacerbates ventricular arrhythmias induced by stretch, which is believed to be a potential link between the cyclicity of NO and the mechanical response [93,94].
NO also activates the cGMP/protein kinase G (PKG)-dependent phosphorylation pathway. Activation of this pathway leads to the phosphorylation of target proteins, inhibition of the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK1/2/ERK1/2) pathway, and activation of the c-Jun N-terminal kinase (JNK)1, 2, and 3 pathways. The ultimate result of this process is cardioprotection and the suppression of genes involved in hypertrophy, as well as the regulation of genes involved in apoptosis [95,96,97]. Data has been obtained indicating that in the myocardium, the cGMP nitrosylation pathway is mediated by eNOS (endothelial nitric oxide synthase) [89,98,99]. In the hearts of healthy newborn rats, NO regulates the integrin complex. The integrin complex is a group of cytoskeletal proteins essential not only for facilitating cellular adhesion but also for the perception and integration of mechanical signals. Integrins are heterodimeric transmembrane receptors that bind the extracellular matrix to the actin cytoskeleton, thereby transmitting mechanical signals to the cytoskeleton [100,101,102,103]. In cardiomyocytes of healthy individuals, integrins contribute to the maintenance of heart function and have the ability to modulate mechanical and electrical coupling in the heart. In cardiomyocytes of healthy newborn rats, integrins, with the participation of NO, promote the release of Ca²⁺ from the sarcoplasmic reticulum. In addition to integrins modulating Ca²⁺ homeostasis through NO signaling, it has been reported that the stimulating effects of NO directly regulate integrin expression through cGMP signaling [86,104,105]. The functional properties of certain ion channels in the myocardium can be regulated by NO through the nitrosylation of protein fragments of the channel (nitrosylation of cysteine fragments). This nitrosylation can either enhance or inhibit channel activity, depending on the specific ion channel involved. In particular, there are several channels that are both mechanically sensitive and modulated by NO through the nitrosylation of thiol groups. NO is released in cardiomyocytes in response to mechanical stimuli and can regulate conductivity by modulating the activity of ion channels [106,107,108,109].
1.3. The Role of NO in the Heart During Fetal Development
The cardioprotective and endothelial-protective role of NO, produced by eNOS, is well known. NO provides protection against myocardial reperfusion injury, regulates the conduction system, participates in the synchronization of heart contraction and relaxation, activates compensatory energy shunts, modulates platelet aggregation, regulates vascular tone, and inhibits the proliferation of smooth muscle cells in blood vessels [110,111,112,113,114,115,116].
However, the global effects of NO on the developing cardiovascular system are not fully understood. It is known that NO influences the early migration of cardiac progenitor cells and vasculogenesis [117,118]. NO activates soluble guanylate cyclase and catalyzes the synthesis of cGMP, a secondary messenger that targets a number of proteins, including bone morphogenetic protein-4 (BMP4). BMP4 plays a role in determining the positioning of the heart during embryonic cardiovascular development. It is involved in the migration of cardiac progenitor cells to the left side of the embryo [117,119]. NO can influence BMP4 signaling by generating reactive nitrogen species, such as peroxynitrite. NO causes organ transposition by altering the migration of cardiac progenitor cells from blood islands. NO regulates the expression of heart-specific genes and also affects apoptotic signaling. NO promotes cardiac differentiation by both switching towards a cardiac phenotype and inducing apoptosis in cells not committed to cardiac differentiation [120,121,122].
It is also known that iNOS and, especially, eNOS are significantly expressed during the early stages of cardiomyogenesis. Reduced expression of eNOS inhibits the maturation of terminally differentiated cardiomyocytes [123]. NO positively regulates the expression of genes involved in heart morphogenesis, as well as controlling heart contraction, cardiac cell development, calcium signaling, and the structure and development of the heart in the embryo[124,125,126].
1.4. Changes in the Nitric Oxide System in the Heart of Offspring After PH
It is known that alterations in the NO levels during pregnancy lead to the onset of classic symptoms of eclampsia, placental development disorders, changes in placental circulation, embryopathy and fetopathy, intrauterine growth restriction, and fetal death [127]. NO can play a role both as a factor in pathogenesis and as a cyto- and organoprotector (cardio- and endothelioprotective) [128,129]. NO plays a central role in endothelial growth and functions as a critical regulator of the vascular endothelial growth factor (VEGF) family of proteins, including placental growth factor (PGF), angiopoietins (ANG-1 and ANG-2), and their corresponding soluble receptors (SFLT-1 and STIE-2). The VEGF protein family is essential for proper placental vascularization, blood vessel growth, and remodeling during pregnancy [130,131,132].
NO production increases pro-angiogenic VEGF-A and PGF in human trophoblast cultures, whereas inhibition of NO synthesis leads to elevated SFLT-1 levels and hypertensive reactions in pregnant mice [133,134,135]. NO also reduces the expression of endothelial adhesion receptors and pro-inflammatory cytokines and is capable of rapid HIF-1α expression [136,137,138]. Under conditions of prenatal hypoxia, on the one hand, NO production increases, while on the other hand, the synthesis of essential factors for the preservation and transport of this molecule decreases, leading to NO deficiency in the heart and blood vessels. During prolonged prenatal hypoxia, ROS can affect NO bioavailability [139]. An increase in ROS during prenatal hypoxia and an imbalance in the ROS/NO ratio in the fetus enhance peripheral vasoconstriction, leading to hypoxia in vital organs such as the heart and brain [140].
PH also enhances the expression of iNOS mRNA and increases iNOS protein levels in the ventricles of the fetal heart [141]. These findings were confirmed by clinical studies, which demonstrated that hypoxia reduces eNOS activity and gene expression in the cardiac tissue of patients with cyanotic congenital heart defects. In contrast, iNOS activity and expression are increased in cyanotic children [142]. PH increases the expression of NADPH oxidase 1 homolog, which stimulates the production of superoxide. This, in turn, can react with NO to form a stable peroxynitrite anion, reducing the bioavailability of NO [143]. The persistent disturbances in the nitric oxide system of the heart after experimental PH are characterized by the suppression of eNOS expression and eNOS mRNA, increased expression of iNOS and iNOS mRNA, reduced NO bioavailability, and activation of nitrosative stress [144]. This is also confirmed by other studies, which show a decrease in eNOS mRNA and protein in the cardiomyocytes of adult animals exposed to PH [145]. Apparently, there is a reduction in NO production by endothelial nitric oxide synthase, while the NO produced during iNOS expression activation is converted into peroxynitrite. The obtained results indicate significant disruptions in the NO system of the myocardium in rat pups after PH – changes in the expression pattern of NOS, reduced NO bioavailability, and activation of nitrosative stress.
NO deficiency leads to a number of serious disturbances in the offspring’s organism after PH [11]. Such changes in the nitric oxide system of the myocardium in the offspring after PH align with current views on the mechanisms of myocardial damage in ischemia and hypoxia, as developed through experimental studies and clinical observations [142,146]. It is well known that PH impairs the heart's tolerance to ischemia/reperfusion, damages endothelial-dependent mechanisms of vasodilation/vasoconstriction, and further contributes to the development of cardiovascular pathology such as hypertension, atherosclerotic vascular diseases, and congestive heart failure, which occur in the presence of NO deficiency [147].
There is evidence of a reduced expression and activity of eNOS in cardiomyocytes and endothelium, as well as an increased risk of endothelial dysfunction following intrauterine hypoxia. The disruption of eNOS activity can be explained by changes in its interaction with regulatory partner proteins such as caveolin-1, calmodulin, and Hsp90. Alterations in the phosphorylation and dephosphorylation of key serine and threonine residues in eNOS may also be involved in the dysfunction of eNOS activity [1,148]. Disruptions in endothelial-dependent vasodilation have been identified in the coronary arteries of both male and female offspring exposed to PH at the ages of 4 and 9.5 months, against the background of reduced eNOS and impaired function of SKCa and IKC channels [26]. Low levels of eNOS lead to the disruption of NO-dependent regulation of glutathione synthesis and a reduced resistance to oxidative stress [149]. The decrease in eNOS may occur due to a deficiency of HIF-1α, as this factor activates eNOS expression by phosphorylating the serine residue [150].
Following a decrease in eNOS after PH, there is an elevated expression of iNOS, which serves to compensate for the reduced NO production [151]. PH increases the expression of inducible nitric oxide synthase (iNOS) mRNA and iNOS protein levels in the ventricles of the fetal guinea pig heart [141]. High iNOS activity may be associated with a cofactor deficiency and the release of superoxide and other reactive forms of NO [152]. However, under conditions of reduced reduced thiol antioxidants, this leads to the formation of cytotoxic NO derivatives in "parasitic reactions."
Similarly, other researchers have established that in preeclampsia, low endothelial NO synthesis activity and redox-dependent transformation of NO into peroxynitrite result in a decrease in blood NO levels [153]. Such reactions can occur in conditions of L-arginine deficiency, antioxidant insufficiency, mitochondrial dysfunction, and increased iNOS expression. Uncontrolled formation of cytotoxic NO derivatives leads to the nitration of the most active sites in protein structures, ion channels, receptors, transmembrane pores, and signaling molecules, i.e., the development of nitrosative stress.
An equally important consequence of myocardial ischemia is the loss of NO-mediated effects, such as the inhibition of cell proliferation, platelet aggregation, and, most importantly, the suppression of monocyte activation by so-called adhesion molecules [154]. Nitrosative stress also leads to a deficiency of HSP70 in the cell against the backdrop of glutathione pathway deprivation in the thiol-disulfide system. Cytotoxic forms of NO not only modify (both reversibly and irreversibly) macromolecules, including HSP70 itself, but also reduce the expression activity of genes encoding its synthesis [155,156]. The role of NO derivatives in suppressing gene activity and reducing the levels of various transcription factors has been demonstrated [157]. Apparently, the excess of nitrogen oxide forms such as peroxynitrite and the nitrosonium ion initially nitrates the thiol–redox-dependent regions of these genes, and then, with increasing concentration, oxidizes them [158].
2. Mechanisms of Cardiovascular Dysfunction After Prenatal Hypoxia
2.1. Disruption of Energy Metabolism in the Myocardium and Mitochondrial Dysfunction in the Offspring After PH
The structural integrity and functional activity of myocardial mitochondria ensure its main role as the cellular "power station" – producing energy to sustain cardiac function [159,160,161]. Disruption of mitochondrial functional activity leads to various functional and pathological shifts in heart function (Figure 2). The concept of mitochondrial dysfunction has become a general pathological term. Many cardiovascular diseases are linked to both primary and secondary mitochondrial dysfunctions. Mitochondrial dysfunction is associated with hypertrophic cardiomyopathy, reperfusion injury after myocardial ischemia, diastolic dysfunction leading to heart failure, early development of heart failure, and hypertension in women during pregnancy [162,163,164,165]. A decrease in the functional activity of myocardial mitochondria leads to ATP deficiency and disruption of the heart's energy metabolism. Impaired ATP transport results in a deficiency of creatine phosphate, suppression of the creatine phosphate shuttle mechanism, and disruption of the energy capacity required for rapid support of heart contraction. All of this forms the basis for the development of heart failure [115,166,167,168,169].
It is known that in the heart of adult offspring after PH, there is a persistent disruption in the activity of enzymes and the expression of proteins in mitochondrial complexes of oxidative phosphorylation under hypoxia [170]. The succinate dehydrogenase complex of the mitochondria, associated with the Krebs cycle, is the most sensitive to PH. Changes in the programming of heart development caused by PH may lead to a disruption of the complex succinate-linked pathway in female fetuses and may persist as long-term changes in mitochondrial heart function in young adult female offspring [171].
It is also known that PH leads to a disruption in the electron transfer from cytochrome c to oxygen in the myocardial mitochondria, resulting in mitochondrial dysfunction and associated heart impairments, such as reduced stroke volume and cardiac output in male offspring [172]. PH reduces the expression of mitochondrial transcripts of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), cytochrome c oxidase subunit II (COXII), and uncoupling proteins, leading to a decrease in the mitochondrial respiratory activity of the offspring's heart [173]. It is known that NO can serve as a natural short-term regulator of mitochondrial physiology, enhancing the efficiency of oxidative phosphorylation in redox processes by reducing errors and failures in proton pumps [174]. Therefore, its deficiency due to PH may lead to disruptions in oxidative processes in mitochondria.
Cytotoxic products of nitric oxide metabolism play a direct role in the formation of mitochondrial dysfunction under PH conditions – reducing the energy-producing function of mitochondria, inhibiting complexes I/IV, and progressing the decline in energy metabolism [175]. These data are consistent with studies showing that NO, produced by iNOS, is converted into peroxynitrite and inhibits oxidative phosphorylation in the mitochondria. Peroxynitrite irreversibly inhibits the enzymes of the electron transport chain by nitrosylating them and removing iron. Suppression of mitochondrial respiration leads to a decrease in membrane potential, which can result in mitochondrial dysfunction and initiate the apoptotic process [176,177].
There is also evidence of direct activation of the opening of the mitochondrial megachannel by nitric oxide, leading to the release of cytochrome c and the initiation of the caspase cascade. NO and its derivatives (peroxynitrite, nitrosonium ion) can cause oxidation of the thiol groups of mitochondrial membrane proteins, which also leads to the release of apoptotic factors into the cytosol [11,178,179,180]. Suppression of protein synthesis in mitochondria after hypoxia is associated with an increased susceptibility of cells to apoptotic stimuli mediated by NO [181,182,183].
In cases of insufficient antioxidant system activity, a vicious cycle may form between the production of ROS by the mitochondrial electron transport chain and the mutational process in mitochondrial DNA. The consequence of this is the progression of mitochondrial dysfunction after PH [184,185,186]. Disruption of any mitochondrial function — energy-producing, death-inducing — or activation of ROS production by mitochondria can serve as a cause for the development of functional and morphological heart abnormalities in the offspring after PH [171,184,187].
The mitochondrial apparatus of ventricular cardiomyocytes in rats after PH is characterized by pronounced degradation processes in the organelles of the subsarcollemmal zone, swelling of "high-energy" mitochondria in the intermyofibrillar zones, and "low-energy" mitochondria in the perinuclear zone of cardiomyocytes, with a reduction in the number of associations between mitochondria. Myofibrils appeared fragmented, and mitochondria varied in size. Lipid inclusions were found in the sarcoplasm. In the contractile cardiomyocytes after PH, areas of myofibrillar apparatus over-contraction, known as "rigor," were observed. Ischemic but minimally altered heart cells are often referred to in the literature as "oscillating," as they are in a state of electrical instability.
The main ultrastructural manifestation of this is the phenomenon of myofibril contracture, recorded during electron microscopy studies of experimental animals after PH, which is an undeniable confirmation of both hypoxic and ischemic cell damage. The latter is a pathognomonic sign of disrupted sarcolemma permeability and the movement of Ca2+ ions from the intercellular space into the cardiomyocyte, indicating the development of ion imbalance. Evidence of this is also the appearance of electron-dense inclusions in the mitochondria and relative expansion of the sarcoplasmic reticulum. These cells can serve as a source of rhythm disturbances [79,172,188,189,190,191].
Moreover, mitochondrial dysfunction can lead to the activation of the immune system. The production of ROS from dysfunctional mitochondria can result in damage to lipids and proteins, which may activate inflammatory pathways [192,193,194,195]. Dysfunctional mitochondria can release damage-associated molecular patterns, including cardiolipin, N-formyl peptides, ROS, and mtDNA, which can activate the inflammasome [196,197,198]. Activation of the immune system and increased expression of pro-inflammatory cytokines lead to further upregulation of iNOS expression, increasing the production of NO and ROS, which can result in further detrimental effects on already dysfunctional mitochondria [199,200,201].
2.2. Nitrosative Stress in the Heart of Offspring After PH
Several studies have shown that experimental PH leads to dysfunction of the nitric oxide system in the myocardium of both the fetus and the offspring. The authors have demonstrated that PH suppresses the expression and activity of endothelial nitric oxide synthase (eNOS) and significantly increases the inducible form (iNOS), along with increased NO production. In this situation, NO loses its physiologically "beneficial" properties and is converted into peroxynitrite and other cytotoxic forms. Excessive generation of NO can induce nitrosative stress, leading to the formation of peroxynitrite, which may increase the expression of matrix metalloproteinases (MMPs). PH causes oxidative-nitrosative stress and alters the expression of extracellular matrix proteins through the regulation of the iNOS pathway in the fetal heart ventricles. This characterizes NO, produced by iNOS, as a key stimulus for initiating the adverse effects of peroxynitrite in the fetal heart [141,202,203,204].
PH increases peroxynitrite in the hearts of offspring through the synthesis of NO produced by iNOS. Peroxynitrite stimulates the translocation of nuclear transcription factors (nuclear factor kappa B and activator protein-1) to the promoter site of matrix metalloproteinase (MMP), thereby increasing MMP expression. The MMP gene family consists of several subtypes of proteins that contribute to the modulation of the extracellular matrix (ECM), with MMP2 and MMP9 playing key roles in cardiac tissue. The role of MMPs as modulators of the ECM in the myocardium is well-known in cardiac pathologies (e.g., heart failure, postnatal hypoxic cardiomyopathy, ischemia-reperfusion injury, and contractile dysfunction of the myocardium) [205,206,207].
Increased expression and activation of MMP2, MMP9, and MMP13 under the influence of reactive nitrogen species stimulate collagen synthesis, disrupting the balance between synthesis and degradation, which leads to collagen accumulation and cardiac fibrosis [208,209,210]. It has been shown that peroxynitrite is a highly reactive molecule that has numerous downstream effects, such as gene transcription, oxidation and nitration of proteins, DNA damage, and lipid peroxidation [211,212,213]. It has also been shown that peroxynitrite increases the activity of MMP9 through autolytic cleavage of the cysteine thiol group and enhances MMP9 expression through transcriptional regulation via nuclear factor kappa B and activator protein-1 [214,215,216].
Currently, there is a generalized concept of "nitrosative stress." As a result of the action of reactive forms of NO, either nitrosylative stress (formation of nitrosamines, S-nitrosothiols, deamination of DNA bases) or oxidative stress develops. NO forms reactive intermediates such as nitrosonium (NO⁺), nitroxyl (NO⁻), and peroxynitrite (ONOO⁻). Most of the cytotoxic effects of NO actually belong to ONOO⁻, which is formed in a reaction with superoxide (O₂⁻). Peroxynitrite is significantly more active, it intensely nitrosylates proteins and can be a source of the highly toxic hydroxyl radical (·OH). NO⁺ is a powerful nitrosylating agent, and its targets can be nucleophilic groups of active thiols, amines, carboxyls, hydroxyls, and aromatic rings. NO⁺ is formed from excess NO with the participation of divalent iron and oxygen. NO⁻ has reducing properties and exerts positive inotropic and lusitropic effects on the myocardium. In ischemia or hypoxia, NO⁻ in the conditions of developing lactate acidosis exhibits pro-oxidant properties towards thiols and amines. It has been shown that NO⁻ decreases glutathione levels and disrupts electrical activity, inhibiting sodium channel activity in the heart [11,217,218,219,220,221].
Apparently, the dual effect of NO⁻ is related to its concentration, as an increase in its levels leads to the formation of the toxic nitrite anion. N₂O₃, being a source of NO⁺, exhibits strong nitrosylating properties, interacting with aliphatic and aromatic amines to form N-nitrosamines. Nitrosamines, and specifically their conversion products under the action of P450 enzymes (such as diazonium ions and formaldehyde), are alkylating agents for nucleic acids, deaminating purines, inhibiting O⁶-methylguanine-DNA methyltransferase, and increasing the formation of 8-hydroxyguanine. N₂O₃ interacts with cysteine to form S-nitrosocysteine and with glutathione to form S-nitroglutathione. S-nitroglutathione is the main transport molecule for NO transfer [222,223,224,225,226].
Some studies have established that the transport of NO occurs through the formation of N₂O₃, which then nitrosylates thiols. Subsequently, with the involvement of disulfide isomerase, NO is released [227,228,229]. There is also a mechanism for the release of NO from S-nitrosoglutathione involving glutamyltranspeptidase, leading to the formation of S-nitrosocysteinylglycine, from which NO is released. Cystine participates in the transport of S-nitrosoglutathione, being reduced to cysteine, which then reacts with S-nitrosoglutathione to form S-cysteine. S-cysteine plays a role in rapid signal transmission, forming adaptive responses to hypoxia. These reactions are regulated by glutathione reductase and glutathione transferase. When these enzymes are inhibited under ischemic conditions, oxidative modification of low molecular weight thiols occurs, leading to the formation of homocysteine and, consequently, disruption of NO transport, resulting in the formation of its cytotoxic derivatives that further enhance thiol oxidation [230,231,232,233,234,235].
The presence of a sufficiently active thiol antioxidant system in the cell, capable of regulating NO transport, ensures the cell's resistance to nitrosative stress. NO inhibits oxidative phosphorylation in the mitochondria of target cells by reversibly binding to cytochrome C oxidase in the mitochondria. Suppression of electron transport in the mitochondria leads to the generation of superoxide, and as a result, the formation of ONOO⁻ (peroxynitrite) [236,237,238]. The synthesis of peroxynitrite is observed in cells with high activity of iNOS and enzymes that produce ROS (xanthine oxidase, NADH oxidoreductase, cyclooxygenase, lipoxygenase, and enzymes of the electron transport chain). The targets of the nitrosative attack of peroxynitrite are thiols, CO₂, metalloproteins, nucleic acids, metabolite-specific transmitters, and lipids [239,240].
Peroxynitrite, being a relatively stable compound, quickly protonates when the pH shifts to acidic conditions, forming the main product—the nitrate anion—along with the hydroxyl radical and nitrogen dioxide, which accounts for its oxidative properties. Peroxynitrite inhibits the activity of the interacting metabolic cycles of methionine and cysteine by suppressing key enzymes that regulate cysteine levels, thereby increasing the formation of homocysteine. Peroxynitrite also reacts with the metabolite-specific transmitter CO₂, forming a strong nitrosylating agent—nitrosoperoxicarbonate. An important mechanism of the neurotoxic action of peroxynitrite is its reaction with tyrosine, leading to the formation of nitrotyrosine. Peroxynitrite significantly inhibits the activity of Cu-Zn-SOD and Mn-SOD by nitrating the 34th tyrosine residue and by binding to copper, altering its valence [241,242,243,244,245].
Peroxynitrite is a specific agent that irreversibly inhibits mitochondrial respiration during ischemia by directly interacting with the iron centers of key enzyme active sites, as well as nitrosylating the S-, N-, and O-elements of thiol, phenolic, hydroxyl, and amine groups in the protein components of these enzymes. In cases of more pronounced nitrosative stress, it irreversibly oxidizes them. The spectrum of peroxynitrite activity also includes the nitrosylation of guanine and DNA strand breaks, which can lead to mutations or trigger apoptosis. In relation to genomic damage, another effect of NO is known: products of its reaction with O₂ inhibit enzymes responsible for DNA repair. Depending on the source (different NO donors), the effects of NO on alkyltransferase, formamidopyrimidine-DNA glycosylase, and ligase have been shown. It is also known that NO can activate PARP- and ADP-ribosylation, possibly due to DNA breaks, but this more likely leads to necrosis due to depletion of NAD and ATP pools [246,247,248,249,250,251].
There is evidence of the direct activation of the opening of the mitochondrial permeability transition pore (MPTP) by nitric oxide (NO), leading to the release of cytochrome c and the initiation of the caspase cascade. These data were obtained by exposing mitochondria to cytotoxic NO derivatives such as peroxynitrite and the nitrosonium ion, in which the mechanism involves the modification of thiol proteins in the mitochondrial pore. NO and its derivatives can induce peroxidative oxidation of phospholipids. Under the influence of cytotoxic NO derivatives and the hydroxyl radical, mitochondrial pores open, leading to the expression and release of pro-apoptotic proteins into the cytosol. The opening of these pores occurs due to the oxidation or nitrosylation of thiol groups in the cysteine-dependent site of the inner mitochondrial membrane protein (ATP/ADP antiporter), which converts it into a permeable, non-specific channel or pore. The opening of the pores transforms mitochondria from "powerhouses" to "furnaces" for substrate oxidation without ATP production.
We have established a significant disruption of the nitric oxide system in the myocardium of rats after PH, with a disturbance in the eNOS/iNOS expression ratio, accompanied by NO deficiency and an increase in nitrotyrosine levels [11,115,144,252,253], amid the inhibition of glutathione-dependent enzymes GPX1 and GPX4 activity [254]. Equally important consequence of hypoxia and activation of nitrosative stress for the myocardium is the loss of NO-mediated effects such as inhibition of cell proliferation, platelet aggregation, and, most importantly, suppression of monocyte activation by so-called adhesion molecules [154]. Nitrosative stress also leads to a deficiency of HSP70 in the cell against the background of glutathione depletion in the thiol-disulfide system. Cytotoxic forms of NO not only modify (both reversibly and irreversibly) macromolecules, including HSP70 itself, but also reduce the expression activity of genes encoding its synthesis [155,156]. We have established that PH leads to a decrease in HSP70 expression against the background of suppressed eNOS expression and a significant increase in nitrotyrosine concentration [255]. The role of NO derivatives in the suppression of gene activity and the reduction of various transcription factor levels has been demonstrated [157]. Apparently, an excess of such forms of nitric oxide as peroxynitrite and the nitrosonium ion initially nitrosylate thiol-redox-dependent regions of these genes, and then, with increasing concentration, oxidize them [158].
2.3. NO-Dependent Mechanisms of Endothelial Dysfunction After PH
Cardiovascular disorders caused by hypoxia are a major cause of illness in newborns, with studies showing that 40–70% of children who have undergone intrauterine hypoxia experience these issues. These disorders play a significant role in the development of numerous, often severe, diseases in both children and adults [1,4,256]. The mechanisms behind post-hypoxic heart disorders are not yet fully understood, making them a significant issue in pediatric cardiology. The clinical symptoms of this pathology during the acute phase are polymorphic, often resembling other diseases, and there is frequently a need for differential diagnosis with congenital heart defects, congenital carditis, and cardiomyopathies [33,257].
In current perspectives, endothelial dysfunction and the associated disturbances in the NO system lay the foundation for the development of numerous cardiovascular diseases [12,148]. Prenatal hypoxia contributes to asymmetric growth restriction of the fetus, hypertrophic growth of the heart and aorta, changes in heart function, and sympathetic hyperinnervation of peripheral resistive arteries in newborns. In adulthood, prenatal hypoxia plays a role in the development of hypertension, ischemic heart disease, heart failure, metabolic syndrome, and increased vulnerability to ischemic damage in individuals [23,258]. The presence of endothelial dysfunction mechanisms has been identified in cardiovascular pathology following PH. Clinical manifestations of impaired functional state and maladaptation of the cardiovascular system after prenatal hypoxia directly correlated with signs of endothelial dysfunction (changes in the production of endothelin-1, NO, VEGF, circulating desquamated endothelial cells) in both newborns and in later stages of life [60,259,260,261].
Endothelial dysfunction and cardiovascular pathology, including those following prenatal hypoxia, are partially attributed to disturbances in the nitric oxide system. Studies have shown that prenatal hypoxia can alter both the production and bioavailability of NO. During prenatal hypoxia, elevated concentrations of superoxide radicals and other reactive oxygen species can lead to oxidative modification of NO, converting it into peroxynitrite, which negatively affects the fetal organs [23,260,262]. Hypoxia reduces the expression of eNOS and can also affect its enzymatic activity by modulating its post-translational modifications. Under hypoxic conditions, when there is a deficiency of L-arginine, eNOS may produce superoxide radicals instead of NO. Such dysfunctions in eNOS activity are considered a primary cause of endothelial dysfunction observed in cardiovascular diseases [11,60].
We have identified a decrease in eNOS expression alongside a significant increase in nitrotyrosine levels in both 1-month-old and 2-month-old rats after prenatal hypoxia. This can be explained by the increased ROS production during prenatal hypoxia, which leads to reduced NO bioavailability and suppression of eNOS expression [255]. The excess of NADPH during prenatal hypoxia is the cause of the formation of ROS, which can react with NO to form the stable peroxynitrite anion, reducing the bioavailability of NO [247].
PH significantly reduces NO-dependent vasodilation mediated by acetylcholine in the thoracic aortic rings of both fetal and adult offspring. In the offspring after PH, there is reduced expression of eNOS, primarily due to increased expression of NADPH oxidase type 2 and high levels of ROS production against the background of elevated miR-155-5 levels in endothelial cells of blood vessels [263,264]. NADPH oxidase type 2 is a key component in the generation of ROS after PН, which makes NO more susceptible to oxidation, reducing its stability and initiating the first step toward the formation of endothelial dysfunction [82,265].
The excess of ROS during hypoxia removes NO produced by eNOS in endothelial cells, thereby limiting the bioavailability of NO. ROS impair NO-mediated dilation of coronary microvessels by increasing the activity of arginase. The formed peroxynitrite oxidizes tetrahydrobiopterin (BH4), a cofactor required for eNOS, leading to eNOS uncoupling. Additionally, oxidative stress disrupts the balance between L-arginine and asymmetric dimethylarginine (ADMA) [60]. The expression of arginase in endothelial cells during hypoxia is induced by the activated ROS through the RhoA/Rho kinase pathway [266].
Chronic prenatal hypoxia leads to a decrease in the expression of HIF-1 mRNA in cells of other organs in rats [267], and, in our opinion, this may indicate the exhaustion of compensatory-adaptive responses after intrauterine hypoxia. Hypoxia-inducible factors (HIF) act as transcription factors and regulate the expression of genes encoding the synthesis of proteins involved in the physiological response to hypoxia/ischemia [269]. HIF exhibit cytoprotective properties under hypoxic conditions, stimulate reparative processes, and increase the concentration of free radical scavengers (heme oxygenase-1, heme oxygenase-1, VEGF, angiopoietin) [270]. HIF-1 under hypoxic conditions affects energy metabolism by regulating compensatory ATP synthesis shunts, increases glutathione synthesis, and enhances cell resistance to oxidative stress. It is known that HSP70 prolongs the "lifespan" of HIF-1. We have established that the suppression of HIF-1 mRNA expression after intrauterine hypoxia occurs against the background of HSP70 deficiency.
A sufficient number of studies have shown divergent changes in the concentration of HIF-1 and its forms under different types of hypoxia, its duration, and in different organs [271]. Under nitrosative stress and increased levels of cytotoxic products resulting from NO and ATP deficiency in tissues, there is a decrease in HIF, associated with the activation of the ubiquitin-independent degradation pathway of oxidatively modified HIF-1α and the suppression of its synthesis at the ATP deficiency stage. The regulatory role of NO in the regulation of HIF-1α mRNA expression is well known [138].
We obtained convincing results that modeled PH leads to significant cardiovascular system dysfunction in offspring (1- and 2-month-old rats). In the myocardium of rats that underwent PH, we observed an increase in the endothelial dysfunction marker – sEPCR, against the background of a decrease in Tie-2 and VEGF-B, which perform protective functions, as well as antioxidant deficiency (decrease in Cu/ZnSOD and GPX) [254]. The identified disruptions in the nitric oxide system after PH, against the background of increased specific proteins, suggest a disturbance in the heart's tolerance to ischemia/reperfusion, endothelial-dependent mechanisms of vasodilation/vasoconstriction, and contribute to the further development of endothelial dysfunction after intrauterine hypoxia.
Endothelial dysfunction formation after PH occurs against a background of HIF-1α deficiency (a factor that activates eNOS expression through serine phosphorylation) and nitrosative stress, which leads to HSP70 deficiency, deprivation of the glutathione component of the thiol-disulfide system, reduced NO bioavailability, and suppression of gene transcription by cytotoxic NO products [144,156,255]. The data we obtained [144,254,255], show that prenatal hypoxia (PH) leads to pathological changes in the cardiovascular system of newborns and the formation of endothelial dysfunction. It is known that EPCR is elevated on endothelial cells during post-ischemic neovascularization. It is important to note that the exogenous addition of NO significantly enhanced the formation of endothelial angiogenic sprouts from aortic rings and primary endothelial cells isolated from mice with a PAR1 mutation. Thus, maintaining the bioavailability of NO during angiogenic processes is a key function of endothelial signaling through the EPCR-PAR1 pathway [272,273].
The reduced expression of key antioxidant enzymes that we established, against the previously identified increase in nitrotyrosine in the myocardium of 1- and 2-month-old rats after PH [144]. This indicates a significant activation of oxidative stress after PH. Oxidative stress in the heart and vascular system of the fetus underlies the mechanism through which prenatal hypoxia programs cardiovascular pathology and endothelial dysfunction at later ages [258]. Our data are consistent with other researchers who have shown that PH contributed to aortic thickening with increased nitrotyrosine staining and enhanced expression of cardiac HSP70, as well as noticeable impairment of NO-dependent relaxation in arteries and increased myocardial contractility with sympathetic dominance [33].
The most important enzyme for protecting cells during oxidative stress is GPX-4, which directly reduces lipid hydroperoxides, even when they are incorporated into membranes and lipoproteins. GPX-4 can also reduce fatty acid hydroperoxides, cholesterol hydroperoxides, and thymine hydroperoxides. It plays a key role in protecting cells from oxidative damage by preventing lipid peroxidation in membrane lipids. GPX-4 is required to protect cells from ferroptosis, a non-apoptotic form of cell death resulting from iron-dependent accumulation of lipid reactive oxygen species [274,275].
GPx4 is required to prevent mitochondrial cell death by mediating the reduction of cardiolipin hydroperoxide. GPx4 is involved in the direct detoxification of lipid peroxides in the cell membrane and acts as an inhibitor of ferroptosis caused by lipid peroxidation. The cytosolic isoform of GPx4 plays a key role in inhibiting ferroptosis in somatic cells, while the mitochondrial isoform of GPx4 (mGPx4) may play a role in reducing the risks of mitochondrial dysfunction [276]. It has been discovered for the first time that PH can lead to ferroptosis of human trophoblast cells, which may subsequently cause miscarriage. This underscores the importance of GPX-4 [277]. GPx-1 is an intracellular antioxidant enzyme that enzymatically reduces H2O2 to H2O to limit its harmful effects, as well as regulates H2O2-dependent signaling mechanisms mediated by growth factors, mitochondrial function, and maintenance of normal thiol redox balance. The reduction in GPx-1 expression in the hearts of rats after PH that we identified may be associated with an excess of cytotoxic forms of NO in the context of high iNOS expression [278]. what we identified in our previous study [144]. GPx-1 plays an important role in maintaining endothelial function and NO bioavailability [278] GPx-1 deficiency leads to marked vasoconstriction and forms endothelial dysfunction [279].
SODs are typically classified into four groups: manganese SOD (MnSOD), copper-zinc SOD (Cu/ZnSOD), iron SOD (FeSOD), and nickel SOD (NiSOD). Cu/ZnSOD and MnSOD are localized in the cytoplasm and serve as the main scavengers of radicals within the intracellular environment, attracting considerable attention due to their physiological function and therapeutic potential [280]. Our studies, which show low concentrations of Cu/ZnSOD in the cytosol of rats after PH, are supported by other research, which demonstrates that PH reduces the expression of Cu/ZnSOD at both the transcriptional and posttranslational levels. Additionally, PH decreases the activity of Cu/ZnSOD and may serve as a cause of subsequent cardiovascular diseases [281] and endothelial dysfunction [282].
There is compelling evidence of a proven link between reduced antioxidant enzyme activity and adverse pregnancy outcomes, as oxidative stress has a detrimental impact on the physiology of the mother, the course of pregnancy, and fetal development. It disrupts placental function, impairs the delivery of oxygen and nutrients to the developing fetus, and contributes to cardiovascular system dysfunction, particularly cardiomyopathy and endothelial dysfunction [283].
2.4. NO and Cardiomyocyte Apoptosis After PH
Since hypoxia is a powerful stress factor, many researchers show that perinatal hypoxia induces cell death by activating both apoptotic and necrotic pathways, depending on the cell type [284,285,286,287]. The study of molecular mechanisms of apoptosis in various forms of heart pathology is one of the pressing issues in medical science. For a long time, apoptosis was considered atypical for highly differentiated tissues. However, in recent years, cardiomyocyte apoptosis after PH has been identified. At the same time, the specific features of the induction and course of cardiomyocyte apoptosis during and after intrauterine hypoxia are not yet fully understood.
It is known that apoptosis is programmed cell death, which, unlike necrosis, is an active and highly regulated process. It involves a cascade of specific signaling and effector molecules that interact with each other with a high degree of selectivity and sequence [288,289,290,291,292,293]. As a result, cell and nuclear shrinkage occurs, along with DNA fragmentation, chromatin condensation, and the subsequent formation of "apoptotic bodies," which are membrane-bound clusters of condensed cellular contents that the cell breaks down into during apoptosis. These "apoptotic bodies" are either phagocytosed or degrade with subsequent breakdown (secondary necrosis). However, in both cases, an inflammatory response does not develop [289,294,295,296].
Programmed cell death plays a role in postnatal morphogenesis of the heart's conduction system: the sinoatrial and atrioventricular nodes, as well as the His bundle [297,298]. Apoptosis of pacemaker cells may play a role in the development of paroxysmal arrhythmias, conduction disturbances, and the genesis of sudden coronary death. For cells that have reached terminal differentiation, such as cardiomyocytes, apoptosis is not typically characteristic. However, in cardiomyopathies, myocardial hypertrophy, and chronic heart failure of various etiologies, there is often a progressive decline in the contractile ability of the left ventricle. This process frequently occurs in the absence of any signs of myocardial ischemia. Therefore, apoptosis of cardiomyocytes has been used as a working hypothesis to explain the mechanism of heart failure development, which is supported by several experiments [299,300,301,302,303,304].
At early stages of ischemia, apoptosis is the predominant form of cardiomyocyte death in newborns. A typical response of apoptosis in hypoxic cardiomyopathies and congenital heart defects is the intensification of its mitochondrial pathway. Heart failure resulting from perinatal hypoxia is accompanied by the activation of all pathways of programmed cell death, with the extent of apoptosis induction depending on the stage of circulatory failure. The dynamics of apoptosis markers—lymphocytes with activated CD95+ expression in the blood of patients with chronic heart failure—can be used as a criterion for evaluating the effectiveness of therapy.
The initiation of apoptosis in cardiomyocytes under hypoxia can be triggered by a variety of stimuli. However, all these activation pathways converge on the activation of the aspartate-specific cysteine protease system, known as caspases, which are constitutionally expressed in cells as inactive zymogens. Once, under the influence of apoptosis inducers, caspases undergo dimerization or specific proteolysis, they become active and, through a cascade of proteolytic reactions, initiate all the biochemical and morphological changes that constitute the apoptosis process [305,306,307,308,309,310].
The increase in apoptosis caused by PH is supported by studies indicating that prenatal hypoxia enhances death signaling through increased activity of caspase 3 and Fas mRNA, while suppressing survival pathways through reduced expression of Bcl-2 and Hsp70 in the hearts of the fetus [311]. The reduction in the bioavailability of NO and the increase in its cytotoxic forms enhance the sensitivity of cells to signals transmitted through Fas receptors. Studies demonstrate the role of peroxynitrite in triggering the process of apoptotic cell death in the context of decreased CuZn-SOD levels [312]. Our recent studies have confirmed that modeling PH leads to a significant reduction in the expression of CuZn-SOD in the cytosol of the heart in 1- and 2-month-old offspring, against the backdrop of increased nitrotyrosine levels and a disproportionate expression of iNOS/eNOS [254].
The extrinsic mechanism of apoptosis begins with the binding of specific ligands, known as "death ligands" (such as FasL), to specific transmembrane receptors, such as Fas/CD95/Apo1, or the binding of tumor necrosis factor alpha (TNF-α) to its receptor. In endothelial cells after hypoxia, H2O2 stimulates the activity of iNOS, which contributes to oxidative cell damage and apoptosis [149,313,314]. NO and its cytotoxic forms lead to the opening of the mitochondrial permeability transition pore (mPTP), which results in the release of cytochrome c and the initiation of the caspase cascade of apoptosis [315,316].
NO and its derivatives can cause peroxidative oxidation of phospholipids and oxidation of thiol groups in mitochondrial membrane proteins, which also leads to the release of apoptotic factors into the cytosol [115,317,318]. Data have also been obtained confirming the direct action of excess NO on the induction of apoptosis in a cGMP-dependent manner in isolated cardiomyocytes [319]. In fetuses subjected to PH, a decrease in the expression of Bcl-2 mRNA, an anti-apoptotic protein, was observed in the left ventricle [320]. Physiological concentrations of NO, produced in vivo by eNOS, can activate cGMP-dependent protein kinases, which in turn influence the proteins involved in apoptotic cascades (such as Bcl-2). A reduction in eNOS activity and NO deficiency may impact the expression of Bcl-2. Bcl-2 can inhibit apoptosis mediated by NO and its derivatives, as well as the cleavage of poly(ADP-ribose) polymerase [321].
Excess NO due to iNOS activity neutralizes the anti-apoptotic members of the BCL-2 family by activating the ASK1-JNK1 pathway, leading to BAX/BAK-dependent cell death. NO and its cytotoxic forms can cause direct S-nitrosylation of cysteine residues in thioredoxin, thereby releasing ASK1 to induce cell death. The mechanism through which NO activates the ASK1-JNK1 axis to initiate BAX/BAK-dependent cell death involves the generation of reactive oxygen species (ROS) and is associated with the formation of peroxynitrite [322,323,324].
3. Molecular Mechanisms and Stress Responses
3.1. The Interaction Between NO and HIF in the Myocardium After PH
Hypoxia-induced factors (HIF) act as transcription factors and regulate the expression of genes that encode proteins involved in the physiological response to hypoxia/ischemia [325]. HIF exhibit cytoprotective properties under hypoxic conditions, stimulate reparative processes, and increase the concentration of "traps" for free radicals (heme oxygenase-1, heme-oxygenase-1), VEGF, and angiopoietin [269,271]. HIF-1 under hypoxic conditions affects energy metabolism by regulating compensatory ATP synthesis shunts, increases glutathione synthesis, and enhances the cells' resistance to oxidative stress [326].
A sufficient amount of research has shown that the concentration of HIF-1 and its forms varies in different types of hypoxia, its duration, and across various organs [327,328,329,330,331]. Under conditions of increased nitrosative stress and elevated levels of cytotoxic NO products, along with ATP depletion in tissues, a decrease in HIF is observed, associated with the activation of the ubiquitin-independent degradation pathway of oxidatively modified HIF-1α and the suppression of its synthesis during ATP deficiency [255,332,333].
The activity of HIF is regulated in a complex manner through both oxygen-dependent and oxygen-independent mechanisms targeting the HIFα subunit. Oxygen-independent mechanisms include the regulation of HIFA gene expression by transcription factors such as nuclear factor-kappa B (NF-κB), specificity protein 1 (SP1), and NF-E2 related factor 2 (NRF2), which can, in turn, be regulated by active ROS, cytokines, and/or lipopolysaccharide (LPS)-dependent signaling through pathways involving protein kinase C (PKC), inhibitor of NF-κB kinase (IKK), and/or phosphoinositide 3-kinase (PI3K).
The levels of HIFA mRNA and/or translation can be regulated by microRNAs (miRNA), long non-coding RNAs (lncRNA), and/or angiotensin II-mediated signaling involving PI3K. The stability and/or activity of HIFα can be modulated by various oxygen-independent post-translational modifications, including phosphorylation, sumoylation (SUMO), acetylation (Ac), and S-nitrosylation via NO [334,335,336,337,338]. We have established the suppression of HIF-1 mRNA expression in the heart of 1- and 2-month-old animals after PH, against the background of reduced expression of eNOS and NO metabolites. In this same study, we also found that the suppression of HIF-1 mRNA expression after intrauterine hypoxia occurs in the context of HSP70 deficiency [255].
It is known that HSP70 prolongs the "lifespan" of HIF-1 under hypoxic conditions [339,340]. The reduction in HIF-1 mRNA expression in the hearts of rats after prolonged perinatal hypoxia has also been confirmed by other authors [267,268]. And, in our opinion, this may indicate the exhaustion of compensatory-adaptive responses. The regulatory role of NO in the regulation of HIF-1α mRNA expression is well known. Physiological concentrations of NO caused a faster but temporary accumulation of HIF-1α compared to higher doses of the same NO donor. Cytotoxic forms of NO suppressed the expression of HIF-1α. The regulation of HIF-1α by NO is an additional important mechanism through which NO can modulate cellular responses to hypoxia in mammalian cells. NO not only modulates the HIF-1 response under hypoxic conditions but also functions as an inducer of HIF-1 [341,342].
3.2. NO and Inflammation After PH
NO is a signaling molecule that plays a key role in the pathogenesis of inflammation. It exerts anti-inflammatory effects under normal physiological conditions. On the other hand, NO is considered a pro-inflammatory mediator that induces inflammation due to excessive production in abnormal situations. NO is synthesized and released by endothelial cells via nitric oxide synthases (NOS), which convert arginine into citrulline, producing NO in the process. Oxygen and NADPH are essential cofactors in this conversion. NO is believed to cause vasodilation in the cardiovascular system and, in addition, it plays a role in immune responses of activated cells. NO participates in the pathogenesis of inflammatory diseases [11,343].
Cytotoxic forms of NO and ROS in the hypoxia of newborns can trigger the production of oxygen stress-induced high-mobility group box-1 (HMGB-1), an endogenous protein of molecular structures associated with damage (DAMPs), which is linked to toll-like receptor (TLR)-4. This activation leads to the stimulation of nuclear factor kappa B (NF-κB), resulting in the production of an excessive amount of inflammatory mediators. Peroxynitrite, ROS, and inflammatory mediators are produced not only in activated inflammatory cells but also in non-immune cells, such as endothelial cells and cardiomyocytes [344].
Excessive inflammation during hypoxia in newborns and after PН exacerbates tissue/organ damage through the expression of genes and proteins. Elevated cytokines activate various inflammatory immune cells through receptors, including toll-like receptors (TLRs), further increasing the production of cytokines and other inflammatory mediators. For example, the concentration of cytokines such as interleukin (IL)-1β, IL-6, IL-8, and tumor necrosis factor (TNF)-α has increased [345,346,347,348]. Hypoxia-induced IL-1β also increases the expression of the NF-κB gene. Thus, both ROS and hypoxia can activate the NF-κB pathway.
The activated NF-κB pathway, in turn, increases the expression of inflammatory mediator genes and regulates RNS, including the synthesis of iNOS and the production of NO. During inflammation, the increase in HIF-1 induces iNOS and reduces L-arginine, leading to an increase in the production of ONOO and hydroxyl radicals, further reactivating nitrosative stress [349,350,351,352,353].
3.3. NO and HSP70 After PH
Recently, a number of studies have focused on the role of heat shock proteins (HSPs) in hypoxia, particularly prenatal hypoxia [354,355,356,357]. HSPs are synthesized in cells of all living organisms in response to various stress factors, including cerebral ischemia. However, the genes for these proteins are activated not only under stress conditions but also during the normal life processes of the cell, including proliferation, differentiation, and apoptosis [358,359,360]. Proteins of this class are involved in all life processes of tissues, organs, and the entire organism.
The most studied protein of this family is the 70 kDa molecular mass protein, HSP70. HSP70 is an inducible representative of the heat shock protein family [361,362,363]. HSP70 is the first protein to be called a chaperone. The function of chaperones in the cell is to bind to damaged or newly synthesized polypeptides and assist them in adopting their native conformation; chaperones also participate in the delivery of proteins to specific organelles [364,365,366]. Chaperones are capable of identifying hydrophobic regions in target polypeptides that are exposed in damaged proteins or may open up in normal, mature cellular proteins during conformational changes. Such conformational changes occur, for example, as a result of cascade modifications of proteins during the process of cellular signal transduction [367,368,369].
Proteins from the HSP70 family are some of the main components of the cellular protein quality control system. Chaperone activity is generally associated with the protective function of HSP70. The fact that the chaperone protects cells from a wide range of factors, including those inducing apoptosis, in an ATP-mediated manner, and removes irreparable proteins through the proteasomal machinery, has been confirmed in numerous in vitro and in vivo experiments using a variety of experimental models. Additionally, much evidence supports the protective effect of HSP70 [115,370,371,372,373].
The results of experiments have highlighted several directions for the practical use of the protective properties of HSP70. First, the organism's resistance to stress conditions can be increased by enhancing the intracellular content of HSP70 [374,375,376]. Secondly, a number of studies show interest in utilizing the protective properties of extracellular HSP70. Once outside the cell, HSP70 likely interacts with neighboring cells and protects them from cell death. Thus, exogenous HSP70 has demonstrated protective properties similar to those of the intracellular chaperone [377,378,379,380].
Recently, there have been data regarding the regulatory effect of heat shock proteins on mitochondrial dysfunction, which develops during brain ischemia, myocardial ischemia, and prenatal hypoxia, as a result of the pathobiochemical cascade of events [340,381,382,383].
Thus, it would be reasonable to assume that HSP70 is involved in the regulation of signaling pathways in the cell's response to hypoxia at the level of HIF-1α regulation [339,384,385]. The cytoprotective effect of HSP70 under hypoxic conditions is realized through its anti-apoptotic and mitochondria-protective activities. It is well-known that depending on the concentration of ROS, oxidative stress ultimately leads to either necrosis or apoptosis [340,386,387]. A high level of ROS causes significant damage to proteins, lipids, and nucleic acids, leading to necrosis. Moderate oxidative stress results in programmed cell death – apoptosis. HSP70 and HIF-1α, through their prolonged action on the synthesis of antioxidant enzymes, chaperone activity, and stabilization of active filaments, prevent the development of necrosis [388,389].
A number of authors indicate that an increase in HSP70 levels leads to the normalization of the glutathione linkage in the thiol-disulfide system and enhances the resistance of cells to ischemia [115,340,390,391]. The introduction of exogenous HSP70 leads to an increase in the functional activity of the glutathione system [356,392,393]. In other words, it has been shown that HSP70, proteins with pronounced cytoprotective properties, under hypoxic conditions, mobilize antioxidant resources, particularly increasing the levels of both cytosolic and mitochondrial glutathione, which prevents the development of oxidative stress [386,394].
Moreover, it is known that by modulating the level of endogenous reduced glutathione, the expression of heat shock proteins can be regulated within the cell [356]. The deficit of HSP70 in the neuron, in the case of reduced glutathione, is, in our opinion, associated with the hyperproduction of ROS and cytotoxic forms of nitric oxide, which lead not only to the modification (reversible and irreversible) of macromolecules, including HSP70 itself, but also to a decrease in the expression activity of genes encoding its synthesis. By stabilizing oxidatively damaged macromolecules, HSP70 is able to prevent the opening of the mitochondrial pore, thereby blocking the release of cytochrome C from the mitochondria, thus exerting a direct anti-apoptotic effect [11,395,396].
HSP70 plays an important role in preventing oxidative stress. However, a sudden depletion of endogenous glutathione can reduce the expression of HSP70 in tissues/organs under hypoxia, which leads to increased oxidative damage to macromolecules and an enhancement of hypoxic changes [397,398,399,400]. It has been shown that under in vitro conditions, HSP70 is capable of preventing the aggregation of oxidatively damaged citrate synthase, glutathione-S-transferase, glutathione reductase, superoxide dismutase, lactate dehydrogenase, malate dehydrogenase, and regulating the thiol-disulfide balance [356,357].
Moreover, one of the main functions of HSP70 is the induction and prolongation of the stable form of HIF-1α, which triggers further adaptive responses in the cell. We have established that HSP70 "prolongs" the activity of HIF-1α and also independently maintains the expression of NAD-MDH-MH, thereby sustaining the activity of the compensatory ATP production mechanism – the malate-aspartate shuttle mechanism – for an extended period [401].
Thus, it can be concluded that HSP70 is an inevitable companion of the pathobiochemical reactions that develop as a result of PH. The current level of knowledge of the pathophysiological and pathobiochemical processes occurring during hypoxia (ischemia) allows for the consideration of pathogenetic correction of metabolic and morphofunctional changes using pharmaceuticals, for which HSP70 will be the target of action.
We have established that modeling PH leads to a deficiency of HSP70 in the 1- and 2-month-old offspring against the background of glutathione deficiency and the expression of glutathione-dependent enzymes (GPX1 and GPX4) [254,255]. We have shown that significant activation of nitrosative stress leads to a decrease in HSP70 concentration. The modeling of nitrosative stress in vitro was conducted by introducing a dinitrosyl iron complex (DNIC) into the neuron suspension at a cytotoxic concentration of 250 µmol. DNIC is an unstable complex of divalent iron, nitric oxide, and ligands [355,402].
Being a stronger nitrating agent, DNIC interacts with -SH groups, glutathione, cysteine residues of proteins, enzymes, transcription factors, and DNA, forming S-nitrosothiols and N-nitrosothiols [403]. After existing for several minutes, DNIC decomposes, releasing a large amount of nitric oxide and free iron. Free iron catalyzes the Haber-Weiss reaction, leading to the formation of hydroxyl radicals, which are capable of oxidizing proteins, degrading cellular membrane lipids, and damaging DNA. In turn, the large amount of nitric oxide released from the complex exhibits its toxic properties in the form of dinitrogen trioxide (N₂O₃) and peroxynitrite (ONOO⁻), which, when excessively synthesized in vivo, lead the organism into a state of nitrosative and oxidative stress [404,405,406].
At the 60th minute of observation, the HSP70 protein content decreased by 51.4% (p < 0.05) compared to the intact group. The results of our research show that the decrease in GSH levels at the 60th minute of observation was accompanied by a low level of HSP70 protein, which is confirmed by the strong correlation between GSH and HSP70 (Pearson's multiple correlation coefficient (R = 0.94678)). We also established a strong negative correlation between HSP70 and the marker of nitrosative stress, nitrotyrosine (Pearson's multiple correlation coefficient (R = -0.8899)) [407].
It can be assumed that intrauterine hypoxia leads to a decrease in HSP70, which plays a role as an endogenous cytoprotective factor, through nitrosative stress in response to the disruption of the nitroxidative system.
3.4. Oxidative Stress in Myocardial Damage After PH
Modeling PH leads to the development of postnatal heart dysfunction. In our previous study, it was established that the use of this PH model results in a decrease in myocardial contractile activity and dysfunction of the sinus node. In the contractile myocardium and conduction system, cells with signs of apoptosis and dystrophy are observed, with a certain correlation between the severity of morphological changes and bioelectrical disturbances in rhythm and conduction [79].The final result of hypoxic heart injury is focal dystrophy [23]. This was confirmed by molecular methods, specifically the increase in the concentration of ST2 in the blood of animals after PH. ST2 is a highly sensitive marker of myocardial remodeling and the risk of heart failure development. It can be elevated in cases of prenatal hypoxia and preeclampsia [408].
Our data on the increase in nitrotyrosine levels after PH are consistent with other studies regarding the elevation of cardiac oxidative stress markers following intrauterine hypoxia, both in male and female rats exposed to intrauterine hypoxia [149,409]. Increased oxidative stress is closely associated with cardiovascular diseases such as hypertension and coronary artery disease. It leads to hypertrophy, fibrosis, and apoptosis, which result in impaired heart function [410,411,412].
The characteristic feature of myocardial damage after PH, according to many authors, is the prenatal damage to myocardial mitochondria caused by hypoxia. This makes the mitochondria a source of reactive oxygen species and pro-apoptotic proteins. Against the background of impaired energy production (decreased ATP), this leads to significant activation of oxidative stress and apoptosis [172,173,413,414,415].
The data obtained by several researchers demonstrate that in rats, after intrauterine hypoxia, there is an increase in the mitochondrial protein cytochrome C in the blood, against the background of a decrease in the average mitochondrial density and the density of cristae, as well as a reduction in the expression of mitochondrial Mn-SOD [416,417,418]. Also, several studies have established that intrauterine hypoxia alters the expression profile of 48 genes related to metabolic and oxidative stress, such as the subunit of glutathione-S-transferase and cytochrome C oxidase [204,419,420,421].
The effect of intrauterine hypoxia on the expression of eNOS depends on the duration of hypoxia, as the increased production of reactive oxygen species (ROS) can impair the bioavailability of NO and suppress the expression of eNOS [23,60]. The balance between ROS and NO, in the context of sufficient endogenous reduced thiol compounds, determines vascular tone. This is because hypoxia-induced increases in ROS, low NO production, and a deficiency of reduced low-molecular thiol compounds in the fetus lead to the formation of cytotoxic derivatives of nitric oxide, which enhances peripheral vasoconstriction and exacerbates myocardial ischemia. An excess of NADPH during prenatal hypoxia is a key factor in the formation of ROS, which can react with NO to form the stable peroxynitrite anion, thereby reducing the bioavailability of NO [422,423,424,425].
4. Cardioprotection and Therapeutic Approaches
4.1. Cardioprotection After PH
According to several researchers, when modeling hypoxia-ischemia-reoxygenation conditions, the oxygen transport compound — perfluorane emulsion — demonstrated a high degree of pharmacological cardioprotection efficacy. Solutions of adenosine triphosphate (ATP), cocarboxylase, magnesium sulfate, riboxin, solcoseryl, cytochrome C, and essentiale showed a medium degree of efficacy. Low efficacy was observed with ascorbic acid solution and L-carnitine chloride solution (levocarnitine) [426,427,428,429,430,431,432,433,434].
For a long time, the most significant group of substances that could be classified as regulatory antihypoxants consisted of nonspecific activators of enzymatic and coenzyme systems. They were considered the only clinically available drugs of this kind. These include B vitamins, thiol derivatives, and pyrimidine derivatives. However, even those that were once extremely popular, with numerous publications on their successful clinical application, are unlikely to remain in the arsenal of resuscitation specialists, although the theoretical grounds for their implementation in practice were promising. For example, the conversion of nicotinamide to NAD requires a series of directed synthesis reactions, which are inhibited by oxygen deficiency, and therefore the final antihypoxic effect of nicotinamide is small and unstable [435,436,437,438].
Recently, antihypoxants have been discovered that do not lower body temperature or oxygen consumption, do not stimulate gluconeogenesis, almost halve glycolysis, and do not possess antioxidant properties. The mechanism of their action is unknown and cannot yet be linked to any of the previously discussed mechanisms, but their protective effect significantly exceeds that of "first" and "second" generation antihypoxants. One such drug is meldonium (Grindex), synthesized in the early 1980s at the Institute of Organic Synthesis in Latvia. Structurally, meldonium is a synthetic analogue of the precursor in carnitine biosynthesis – gamma-butyrobetaine. It has been established that, like carnitine, it participates in the energy metabolism of cells [439,440,441,442]. Thus, it prevents the activation of glycolysis reactions, which dominate under tissue hypoxia conditions, and therefore exhibits cytoprotective effects.
The anti-ischemic and cytoprotective effects of meldonium on the postoperative course after open-heart or brain surgery interventions have been demonstrated in various studies [443,444,445]. Its effects are especially pronounced when its use is started 2–3 days before the surgery and continued afterward. With a single dose, the drug had an optimizing effect on the cerebral circulation system when there were significant impairments in the reactivity of the cerebral blood vessels. At the same time, with unstable hemodynamics after a single intravenous administration of Meldonium, a clear increase in systemic blood pressure was observed, and the quality characteristics of circulation improved [446,447].
It has been shown that the prophylactic administration of succinic acid, aminothiol antihypoxants such as gutimine and amtyzol, as well as succinate-containing aminothiol antihypoxants like gutimine succinate and amtyzol succinate, exhibits pronounced antihypoxic and antioxidant effects [448]. There is an antihypoxant called Lipin. Lipin, a modified egg phosphatidylcholine (lecithin), exhibits antihypoxic effects, promotes the increased diffusion rate of oxygen from the lungs into the blood and from the blood into tissues, normalizes tissue respiration processes, restores the functional activity of endothelial cells, and stimulates the synthesis and secretion of endothelial relaxation factor. It also improves microcirculation and the rheological properties of blood. Lipin inhibits lipid peroxidation processes in the blood and tissues, supports the activity of the body’s antioxidant systems, exhibits a membrane-protective effect, functions as a nonspecific detoxifying agent, and enhances nonspecific immunity. When administered via inhalation, it has a positive effect on lung surfactant, improves pulmonary and alveolar ventilation, and increases the rate of oxygen transport through biological membranes [449,450,451].
Thus, in the hearts of newborn rats under conditions of intrauterine hypoxia, reversible changes develop, and in severe cases, irreversible changes occur both in cardiomyocytes (conducting and contractile) and in the vessels of the hemomicrocirculatory bed. This is a manifestation not only of hypoxic damage to the heart muscle but also evidence of an ischemic nature of the myocardial damage that has developed. Therefore, studying the pathways for correcting antenatal hypoxia remains a relevant issue, within which it is advisable to study the effects of modern antihypoxants on the myocardium in experimental settings, as well as the possibilities for their combination to enhance the effect.
4.2.NO Modulators – Promising Cardioprotectors After PH
The literature data regarding the role of the NO system in the development of cardiovascular pathology in newborns and the potential cardioprotective effects of its modulators is quite limited. Several studies have established the cardio- and endothelial-protective properties of drugs that are capable of both increasing the synthesis of NO and enhancing the bioavailability of this messenger [114,254,452,453]. In this regard, substances such as L-arginine, tiotriazoline, angiollin, and meldonium are of interest.
L-arginine is a substrate for the formation of NO in endothelial cells of blood vessels, exhibiting antioxidant, cytoprotective, antihypoxic, and membrane-stabilizing properties [454]. Tiotriazoline (morpholinium 3-methyl-1,2,4-triazolyl-5-thioacetate) is a specific scavenger of NO and its cytotoxic forms, enhancing the bioavailability of NO by protecting it from ROS. In ischemic conditions, tiotriazoline strengthens the compensatory activation of the malate-aspartate shuttle mechanism, reduces the inhibition of oxidation processes in the Krebs cycle while preserving intracellular ATP levels, and demonstrates hepatoprotective, cardioprotective, anti-ischemic, and antioxidant properties [455].
Angiollin ([S]-2,6-diaminohexane acid 3-methyl-1,2,4-triazolyl-5-thioacetate) increases the expression of VEGF and the density of proliferating endothelial cells in muscular-type blood vessels and the microcirculatory bed. It enhances the bioavailability of NO, preserves the ultrastructure of mitochondria during ischemia, and also boosts ATP production in ischemic conditions by activating the compensatory malate-aspartate shuttle mechanism. Angiollin increases the expression of eNOS and exhibits endothelial, cardioprotective, neuroprotective, antioxidant, and anti-ischemic properties [356].
Meldonium reduces the formation of carnitine from its precursor, gamma-butyrobetaine, and the accumulation of the latter stimulates NO synthesis. Meldonium decreases carnitine-mediated transport of long-chain fatty acids across mitochondrial membranes without affecting the metabolism of short-chain fatty acids and activates an alternative energy production system—glucose oxidation [456]. Meldonium exhibits anti-ischemic and cardioprotective properties.
In our studies using the PH model, the cardioprotective effects of NO modulators—L-arginine, tiotriazoline, angiollin, and meldonium—were demonstrated for the first time, with varying degrees of expression. The presented drugs reduced the concentration of ST2 protein, normalized the expression of iNOS mRNA and eNOS mRNA, as well as iNOS and eNOS proteins, increased the concentration of HSP70 and HIF-1, and decreased the marker of nitrosative stress—nitrotyrosine—in the blood and myocardium of 1- and 2-month-old offspring.
Two drugs—Angiollin and Tiotriazoline—were able to have a full impact on endothelial dysfunction indicators after PH (reducing sEPCR with increased Tie-2, VEGF-B, and Cu/ZnSOD, GPX), which perform protective and antioxidant functions [144,254]. The cardioprotective effect of NO modulators was also manifested in the improvement of the electrophysiological properties of the heart in the offspring after PН. The most pronounced therapeutic effect was observed with Angiollin and Tiotriazoline, which contributed to almost complete normalization of heart rate, while Angiollin also restored the neurogenic control of the sinus node's automaticity. The obtained results allowed us to rank the therapeutic efficacy of the used drugs in descending order: Angiollin > Tiotriazoline > Meldonium > L-Arginine in eliminating disturbances in the electrical activity of the heart [79].
4.2.1. Angiolin ((S)-2,6-Diaminohexanoic acid 3-methyl-1,2,4-triazolyl-5-thioacetate)
Angiolin exhibits pronounced endothelial-protective, neuroprotective, cardioprotective, energotropic, antioxidant, anti-ischemic, and anti-inflammatory properties.
Angiolin acts as an anti-ischemic and antioxidant agent with a significant effect on the endothelium of the brain and heart vessels and metabolism. Its neuroprotective properties are due to the conversion of L-lysine into pipecolic acid, which enhances the affinity of the GABA-benzodiazepine receptor complex, thereby reducing manifestations of glutamate excitotoxicity. The drug significantly reduces neuronal death in ischemic and hemorrhagic strokes, normalizes the functioning of the compensatory GABA-shunt, and increases the ATP level in brain tissues. Under acute ischemic conditions in the brain, the drug preserves the functional activity of neuronal mitochondria.
The drug exhibits pronounced antioxidant properties, activates the glutathione branch of the thiol-disulfide system, increases the activity of glutathione peroxidase and glutathione transferase, reduces the formation of reactive oxygen species, and decreases the accumulation of markers of oxidative and nitrosative stress. The endothelial-protective effect of the drug in cerebrovascular disorders and hypertension is due to its ability to regulate NO production, reduce the formation of peroxynitrite and homocysteine, increase the activity of superoxide dismutase and NO-synthase, and preserve reduced thiol groups and L-arginine. The drug increases the bioavailability of NO and can improve its transport to target cells when endothelial function is impaired in brain blood vessels.
In cerebrovascular disorders and vascular surgeries, the drug preserves the morphofunctional parameters of endothelial cells in muscle-type vessels and capillaries of the brain, increases RNA content in the nuclei and cytoplasm of endothelial cells, activates their proliferation, and increases the binding coefficient of vascular endothelial growth factor (VEGF) with the endothelium. The drug demonstrates anti-inflammatory properties, reducing the expression of the pro-inflammatory cytokine IL-1β.
Cardioprotective properties of the drug are aimed at increasing the survival rate of cardiomyocytes during acute myocardial ischemia, improving ECG parameters. The drug improves overall and cardiodynamics during acute myocardial ischemia—normalizing systolic blood pressure, reducing ischemic left ventricular dysfunction, increasing left ventricular pressure, and increasing working and systolic heart indices, while lowering overall peripheral vascular resistance. In angina and myocardial infarction, Angiollin improves myocardial energy metabolism by intensifying aerobic ATP formation reactions and activates the compensatory malate-aspartate shuttle for ATP production [356,457,458,459]. When administered parenterally, the average time for Angiollin's presence in the body is 20 minutes. The distribution coefficient in the body is 464 µg/g/min, indicating a rapid reach of the drug to target organs and tissues. 17% of the administered dose of the drug binds to plasma proteins. Within the first 3–5 minutes, 34.7% of the administered dose of Angiollin reaches the myocardium. The time to reach the maximum concentration in the myocardium is 7.8 minutes. The maximum concentration in the myocardium (Cmax cor) is 16.7 µg/g. The clearance rate from the myocardium for Angiollin is 324 µg/min. The drug is excreted via the kidneys, with 62% of the dose eliminated in unchanged form, and its renal clearance is 3.6 mg/min.
According to acute toxicity studies of the drug "ANGIOLIN" (mice, rats, rabbits), it was classified as a V class of toxicity (practically non-toxic substances with no cumulative properties according to the accumulation index). The drug "ANGIOLIN" does not cause skin irritation on undamaged rat skin, does not have a local irritant effect on the undamaged conjunctiva of guinea pigs' eyes, does not cause allergic reactions in guinea pigs, and does not have ulcerogenic effects in rats. In a 180-day intragastric administration study of "ANGIOLIN" in doses of 100, 500, and 1000 mg/kg, it was found that the drug does not cause structural-functional changes, does not lead to dystrophic or hemodynamic disorders, nor does it provoke destructive reactions in the studied tissues of animals [460].
The investigated drug "ANGIOLIN" has a good safety profile, has passed the first phase of clinical trials, and, by approval from the State Expert Center of the Ministry of Health of Ukraine, has been authorized for the second phase of clinical trials. The primary cardioprotective action of Angiollin, when administered after intrauterine hypoxia, with sustained effects even after a one-month cessation, can be explained by the following properties:
In conditions of acute brain ischemia, Angiollin exhibits pronounced endothelial-protective properties—it maintains endothelial cell density, increases RNA concentration in cell nuclei, boosts the density of proliferating endothelial cells (BrdU test), enhances the utilization of endogenous L-arginine, and increases the expression of vascular endothelial growth factor (VEGF) and eNOS. Additionally, the presence of divalent sulfur in its structure gives it the property of scavenging NO. Angiollin, together with vitamin C, forms L-carnitine and normalizes mitochondrial function.
Our studies have shown that Angiollin improves the ultrastructure of neurons in the CA1 zone of the hippocampus in chronic cerebral ischemia (reducing crystal destruction, uneven electron density of the matrix, and increasing mitochondrial density). It also lowers intra-mitochondrial iNOS levels and raises the concentration of cytosolic and intra-mitochondrial HSP70. Our work has demonstrated that Angiollin can activate the malate-aspartate shuttle mechanism in the myocardium during ischemia. The positive effect on the NO system and the reduction of oxidative stress, along with the increase in mRNA of HIF-1 and HSP70, seem to provide Angiollin with its cardioprotective effect after intrauterine hypoxia.
It is noteworthy that the cardioprotective effect of Angiollin in offspring after PH was sustained even one month after the discontinuation of the drug. Experimental studies have shown that Angiollin increases the expression of eNOS mRNA and eNOS activity in ischemic myocardium in rats. Angiollin also enhances the expression of VEGF and the binding coefficient of VEGF to endothelial cells, as well as the density of endothelial cells and proliferating endothelial cells in the capillary network and vessel walls. It increases RNA concentration in endothelial cells under hypoxic and circulatory ischemic conditions [461].
Angiolin exhibits protective action towards NO and also increases its bioavailability. NO is an unstable, short-lived radical, and to prolong its "life," it forms more stable S-nitrosyl complexes with low-molecular compounds (such as glutathione, cysteine). In the absence of these compounds, the bioavailability of NO decreases sharply. When low-molecular thiols are deficient, NO, under the influence of ROS, transforms into peroxynitrite, which can trigger the initiation of nitrosative stress [462]. Angiolin, due to its chemical structure, acts as a spin trap and can form a complex with NO. Angiolin positively affects the state of the nitric oxide system in the myocardium under experimental ischemia – it increases NO synthesis by enhancing eNOS expression, increases NO bioavailability, and reduces parasitic reactions by decreasing iNOS hyperactivity. The mechanism of its effect on eNOS expression can be explained by Angiolin's influence on HSP70 and HIF-1α. Angiolin prolongs the "lifetime" of HIF-1α through HSP70 mechanisms. Angiolin also has a positive effect on the glutathione thiol-disulfide system, which is coupled with the NO system. It has been found that during experimental myocardial ischemia, Angiolin increases the activity of glutathione reductase, glutathione peroxidase, and raises the concentration of reduced glutathione in the myocardium cytosol of rats [340]. As is well known, HIF-1α increases the expression of eNOS and VEGF during hypoxia and ischemia [463].
Angiolin, due to its positive effect on the NO system, positively influenced cardio- and hemodynamics during experimental myocardial ischemia. Administration of Angiolin to rabbits with occlusion of the descending coronary artery led to the restoration of left ventricular dysfunction, which was expressed in an increase in the left ventricular work index and left ventricular stroke work index, an increase in left ventricular pressure, and a decrease in total peripheral vascular resistance [461]. We have shown that Angiolin can have a significant impact on endothelial dysfunction markers after PH (decrease in sEPCR along with an increase in Tie-2, VEGF-B, Cu/ZnSOD, GPX), which perform protective and antioxidant functions. Angiolin (S)-2,6-diaminohexanoic acid 3-methyl-1,2,4-triazolyl-5-thioacetate, which has scavenger properties for NO, with fragments of its chemical structure participating in this process, can form nitrothiols and enhance the bioavailability of NO. Angiolin normalizes the expression of eNOS/iNOS. Studies on a rat model of cerebral ischemia have demonstrated the endothelial-protective activity of Angiolin, including increased endothelial cell density in muscular-type vessels and the microcirculatory bed, an increase in the density of proliferating endothelial cells, as well as an increase in the expression of vascular endothelial growth factor (VEGF) and its receptor binding coefficient [11,461].
There is data indicating that VEGF enhances the regulation of the enzyme eNOS and induces a biphasic stimulation of endothelial NO production [464]. This suggests the possibility of eNOS expression being mediated through VEGF under the influence of Angiolin. Angiolin may affect the expression of endothelial-specific factors and antioxidant enzymes by influencing the thiol-disulfide system through the enhancement of glutathione levels and regulating post-translational mechanisms. Based on the conducted studies, the experimental justification for further preclinical investigation of Angiolin as a promising cardioprotective agent after PH has been established.
4.2.2. Tiothiazoline (Morpholine 3-methyl-1,2,4-triazolyl-5-thioacetate; Morpholine Thiazotate)
The history of tiothiazoline dates back to the 1960s. Preclinical studies, conducted according to the requirements of the Pharmacological Committee of the Ministry of Health of the USSR and the State Expert Center of the Ministry of Health of Ukraine, have shown that tiothiazoline exhibits high antioxidant, anti-ischemic, cardioprotective, and anti-hypoxic properties, surpassing reference drugs in terms of strength. It has been established that tiothiazoline has low toxicity when administered through various routes to four types of animals, meaning the drug belongs to Class V of toxicity (practically non-toxic substances). Tiothiazoline does not have cumulative properties, does not exhibit skin-irritating effects on intact skin, does not cause allergic reactions, and does not have ulcerogenic or immunotoxic effects.
Toxicological analysis, along with comprehensive behavioral, physiological, biochemical studies, and pathological examination of the internal organs of white rats and dogs, has shown that intraperitoneal, intravenous, and intragastric administration of tiothiazoline at therapeutic, intermediate, and sub-toxic doses over 180 days does not cause structural changes, does not lead to dystrophic and hemodynamic disorders, nor does it cause destructive reactions in the examined tissues of the animals. The administration of the drug does not result in irreversible changes in biochemical markers of liver and kidney functional status. Research has also established that tiothiazoline does not exhibit teratogenic, embryotoxic, mutagenic, or carcinogenic effects [465,466,467].
Tiothiazoline exhibits scavenger properties for cytotoxic forms of NO and exerts protective effects on the transport of NO by positively influencing the thiol-disulfide equilibrium and increasing the levels of reduced thiols and glutathione. Furthermore, we hypothesize that tiothiazoline itself may act as a carrier for NO, forming stable S-nitrosyl complexes with it [356,468]. Thiotriazoline is capable of increasing the bioavailability of NO in the presence of excessive ROS. It acts as an antioxidant, a scavenger of ROS and NO, enhancing the activity of glutathione-dependent enzymes and increasing the levels of reduced glutathione during myocardial ischemia. Thiotriazoline (10-5–10-7 M) in vitro reduced the levels of superoxide radical and peroxynitrite due to the presence of a thiol group in its structure. Thiotriazoline prevents the irreversible inactivation of the transcription factor NF-kB, protecting cysteine residues (Cys 252, Cys 154, and Cys 61) in its DNA-binding domains, which are sensitive to excess ROS. Thiotriazoline may participate in the restoration of these groups during reversible inactivation, acting as a Redox Factor-1. It enhances the activation of the expression of redox-sensitive genes necessary to protect cells from oxidative stress. Additionally, tiothiazoline reduces the intensity of nitrosative stress and increases eNOS (endothelial nitric oxide synthase) activity [356,469,470,471].
Thiotriazoline exhibits cardioprotective effects by positively influencing energy metabolism in the ischemic myocardium. It increases ATP levels during ischemia and hypoxia by normalizing the Krebs cycle, enhancing glucose utilization, and promoting the oxidation of free fatty acids [472]. Thiotriazoline stimulates lactate dehydrogenase, promoting the conversion of lactate to pyruvate. This not only eliminates lactate acidosis and normalizes intracellular pH but also stimulates the Krebs cycle by increasing the pyruvate levels. Additionally, thiotriazoline demonstrates antioxidant properties. Numerous studies have established its ability to reduce the formation of end products of oxidative and nitrosative stress, while enhancing the activity of Cu/ZnSOD, GPX1, and GPX4 in the liver, heart, and brain of animals with various experimental pathologies [356]. It is known that Tiotriazoline also demonstrates a cardioprotective effect and enhances the endurance of animals under working hypoxia by increasing HIF-1 and preserving mitochondrial ultrastructure. Due to its antioxidant action, Tiotriazoline maintains receptor sensitivity thresholds, preserves membrane fluidity, and protects phospholipids from oxidation. Tiotriazoline also enhances the effectiveness of arginine when used in combination. The pharmacological effect of this combination is attributed to the positive impact on the synthesis, transport, and bioavailability of NO, as well as the physiological functions of this molecular messenger [473]. In rats with modeling isadrin-pituitrin myocardial infarction Thiotriazolin stimulated LDH in the direction of the formation of pyruvate from lactate, which eliminated lactic acidosis and normalized intracellular pH and stimulated the Krebs cycle by increasing pyruvate [474].
In the same experimental mode Thiotriazolin activated the malate-aspartate shunt in the myocardium in the acute period of myocardial infarction [455]. The study involving 8,298 patients with various cardiovascular diseases, including 5,700 patients with different forms of coronary artery disease, deserves attention. Proven effects of tiotriazoline include a significant reduction in the number of ventricular arrhythmias and correction of rhythm disorders (ectopic beats, paroxysmal atrial fibrillation, sinus node dysfunction). The drug also has an application in myocardial dystrophies, as it works under both oxygen deficiency and sufficient oxygen conditions. Furthermore, tiotriazoline improves the metabolism not only of cardiomyocytes but also of cells in the CNS, liver, etc. Importantly, tiotriazoline had a positive effect on quality of life, as assessed by classical methods using the Minnesota questionnaire and the Nottingham health profile: improvement in overall quality of life indicators, increased physical activity, and enhanced emotional well-being [475]. Thiotriazolin administration (600 mg/day) to 8298 patients with II–III class stable angina pectoris reduced the number of weekly angina attacks by 46.32 %; in the control group - by 33.24 % (p = 0.028), respectively (p = 0.031), and increased exercise tolerance [476]. Our research has revealed that Tiotriazoline exhibits a significant cardioprotective and endothelialotropic effect after PH, which is implemented through the modulation of NO [114,254]. The obtained results justify the potential for conducting additional preclinical and clinical studies of Tiotriazoline (as an approved drug) as a treatment for cardiovascular system pathologies following intrauterine hypoxia.
4.2.3. Mildronate
Mildronate (3-(2,2,2-trimethylhydrazine) propionate) reversibly blocks gamma-butyrobetaine hydroxylase, which catalyzes the conversion of gamma-butyrobetaine into carnitine, thus significantly inhibiting the intake of carnitine, which is responsible for the transport of fatty acids through the membrane into muscle cells. This effect of Mildronate is accompanied by a reduction in carnitine-dependent oxidation of free fatty acids (FFAs) and, as a result, leads to the activation of glucose oxidation, which is more energy-efficient under ischemic conditions. Treatment with Mildronate is accompanied by a compensatory increase in the expression of a number of genes in the myocardium that code for enzymes involved in lipid metabolism—lipoprotein lipase, fatty acid translocase, carnitine palmitoyltransferase I, and enzymes involved in triglyceride synthesis [456,477,478].
Mildronate is capable of improving the contractile function of the myocardium, enhancing hexokinase activity, and modulating the ATP/ADP/AMP ratio by activating AMP-activated protein kinase (AMPK), which helps restore ATP levels [479]. An important feature of Mildronate's action, which distinguishes it from other drugs affecting myocardial metabolism, is the absence of accumulation of underoxidized fatty acids within the mitochondria and the increase in nitric oxide (NO) production. This occurs because Mildronate inhibits the hydroxylation of γ-butyrobetaine and increases the intracellular pool of γ-butyrobetaine, which, through esterification, exhibits cholinomimetic properties. The esters of γ-butyrobetaine can activate NOS via acetylcholine receptors on endothelial cells. Mildronate improves exercise tolerance and the quality of life in patients, positively affecting the functional parameters of the heart, including ejection fraction and systolic volume, in cases of myocardial ischemia [446].
Our research has confirmed the anti-hypoxic activity of Mildronate during PH. However, no significant positive effect of Mildronate on the NO system indicators in the myocardium of animals that underwent PН was observed. It was found that Mildronate reliably increased the mRNA expression of iNOS while significantly reducing the concentration of nitrotyrosine, which more strongly indicates its antioxidant properties [480].
4.2.4. L-arginine
L-arginine is a substrate for the production of nitric oxide (NO) in the endothelial cells of blood vessels, which is a factor responsible for the dilation of peripheral vessels. The NO produced from arginine reduces the overall peripheral vascular resistance and blood pressure, alleviates oxygen deprivation, particularly in the heart tissues [481]. L-arginine exerts antioxidant effects by participating in the transamination cycle and the elimination of nitrogenous waste products from the body, including ammonia, urea, and uric acid, which are by-products of protein metabolism. The ability to synthesize urea and eliminate protein waste depends on the efficiency of the ornithine-citrulline-L-arginine cycle. However, oxidative stress reduces the clinical effectiveness of L-arginine [482].
L-arginine plays an important role in protein synthesis, increasing muscle mass, improving muscle recovery after physical exertion, accelerating wound healing, eliminating waste, optimizing immune system function, and enhancing the production of growth hormone [483]. L-arginine is a common substrate for both NO (nitric oxide) and polyamines (putrescine, spermidine, and spermine). NO and polyamines play an important role in reproduction, embryogenesis, reducing neonatal mortality, and embryonic angiogenesis. NO regulates gene expression, protein synthesis, and facilitates the proliferation, growth, and differentiation of the fetus [484]. Therefore, researchers and clinicians have focused on the use of the NO substrate, L-arginine, to reduce the negative consequences of PH [485].
Our studies have revealed the positive effect of L-arginine on the NO system indicators in the hearts of 1- and 2-month-old rats after PH. However, in terms of effectiveness, L-arginine was less potent than the new molecules—Tiotriazoline and Angiolin. Nevertheless, a certain positive influence of L-arginine on molecular indicators of endothelial dysfunction in the hearts of rats after PH was identified.
The weaker effect of L-arginine compared to Angiolin and Tiotriazoline can be explained in terms of the lifespan of NO under ischemic and hypoxic conditions, which are accompanied by oxidative stress. The "newborn" NO is immediately at risk of being "bitten" by the superoxide radical and converted into the harmful peroxynitrite [11,486]. Only combinations of L-arginine with SH-group donors or antioxidants (cysteine, glutathione, tiotriazoline, angiolin) are capable of enhancing its NO-modulating activity [356]. The combined action of L-arginine and tiotriazoline is aimed at the synthesis, stabilization, and enhancement of the bioavailability of NO [340,487,488]. The combined action of L-arginine and tiotriazoline can be used to correct disorders caused by NO deficiency.
4.2.5. Repurposing Pharmacological Agents for Cardiovascular Protection in Prenatal Hypoxia, Comorbid Conditions, and Long-term Consequences
Contemporary pharmacological approaches are investigating the repurposing of established drugs to address cardiovascular complications arising from PH, which impact both neonates and adults, with long-term sequelae of hypoxia persisting into adulthood [489,490]. Widely used in the management of type 2 diabetes, metformin exhibits cardioprotective properties through the activation of AMP-activated protein kinase (AMPK) [491,492,493], stimulation of endothelial nitric oxide synthase, and reduction of mitochondrial oxidative stress [494,495,496], which may be beneficial in offspring exposed to PH [497,498]. Antiviral agents developed for the treatment of COVID-19 have shown potential in attenuating systemic inflammation and stress responses [499], which is particularly relevant for individuals with comorbidities [500,501,502]. Emerging evidence suggests that prenatal hypoxia and viral exposure may interact with genetically regulated pathways—including those associated with interferon signaling, nitric oxide metabolism, and hypoxia-inducible factor-mediated mechanisms—thereby amplifying the risk of cardiovascular dysfunction in the offspring [503,504,505]. A potent antioxidant, alpha-lipoic acid can enhance endothelial function, decrease reactive oxygen species levels, and restore cellular redox balance, making it a promising candidate for managing PH-induced nitrosative dysfunction [506,507]. Additionally, natural compounds such as melatonin, resveratrol, and curcumin have demonstrated the ability to modulate NO and hypoxia-inducible factor signaling pathways, thereby reducing inflammation and cardiomyocyte apoptosis [508,509,510]. In patients with comorbid conditions, the inclusion of such agents may offer therapeutic advantages due to their multimodal mechanisms of action [511,512]. Further preclinical and clinical studies are required to comprehensively assess the efficacy, safety, optimal treatment regimens, and long-term outcomes of these repurposed agents in the context of prenatal hypoxia, associated comorbidities, and long-term consequences.
5. Conclusions
Thus, the search and development of approaches to pharmacotherapy for prenatal myocardial injury based on the modulation of the NO system is a relevant task in modern pharmacology. The above theoretically substantiates the promising study of NO system modulators with various mechanisms of action – L-arginine, tiotriazoline, angiolin, and mildronate – as agents for cardioprotection of post-hypoxic cardiovascular disorders in newborns.
Author Contributions
Conceptualization, O.P., I.B., N.B., V.R.; validation, N.G., V.O. and O.K writing—review and editing, I.B., O.P., N.B.; visualization, O.P., V.R., N.G., V.O. and O.K.; supervision I.B., O.P., N.B., V.R., N.G., V.O. and O.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Patterson, A.J.; Zhang, L.; Zhang, L. Hypoxia and Fetal Heart Development. Curr. Mol. Med. 2010, 10, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Ream, M.; Ray, A.M.; Chandra, R.; Chikaraishi, D.M. Early fetal hypoxia leads to growth restriction and myocardial thinning. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R583–R595. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P. Effects of Chronic Hypoxia on Fetal Coronary Responses. High Alt. Med. Biol. 2003, 4, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. Prenatal Hypoxia and Cardiac Programming. J. Soc. Gynecol. Investig. 2005, 12, 2–13. [Google Scholar] [CrossRef]
- Narohan, M.V.; Bazhenova, L.K.; Kapranova, E.I.; Melnikova, E.V.; Belousova, N.A. Posthypoxic Dysfunction of the Cardiovascular System in Newborns. Curr. Issues Pediatr. 2007, 6:42–46. (In Ukranian).
- Zhao, Y.; Xiong, W.; Li, C.; Zhao, R.; Lu, H.; Song, S.; Zhou, Y.; Hu, Y.; Shi, B.; Ge, J. Hypoxia-induced signaling in the cardiovascular system: pathogenesis and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 431. [Google Scholar] [CrossRef]
- Lazzarin, T.; Tonon, C.R.; Martins, D.; Fávero, E.L.; Baumgratz, T.D.; Pereira, F.W.L.; Pinheiro, V.R.; Ballarin, R.S.; Queiroz, D.A.R.; Azevedo, P.S.; et al. Post-Cardiac Arrest: Mechanisms, Management, and Future Perspectives. J. Clin. Med. 2022, 12, 259. [Google Scholar] [CrossRef]
- Rojas, E.Y.L.G.; Villanueva, C.; Bond, R.A. Hypoxia Inducible Factors as Central Players in the Pathogenesis and Pathophysiology of Cardiovascular Diseases. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Sun, R.; Liu, M.; Lu, L.; Zheng, Y.; Zhang, P. Congenital Heart Disease: Causes, Diagnosis, Symptoms, and Treatments. Cell Biochem. Biophys. 2015, 72, 857–860. [Google Scholar] [CrossRef]
- Chlif, M.; Ammar, M.M.; Ben Said, N.; Sergey, L.; Ahmaidi, S.; Alassery, F.; Hamam, H. Mechanism of Dyspnea during Exercise in Children with Corrected Congenital Heart Disease. Int. J. Environ. Res. Public Heal. 2021, 19, 99. [Google Scholar] [CrossRef]
- Belenichev, I.; Popazova, O.; Bukhtiyarova, N.; Savchenko, D.; Oksenych, V.; Kamyshnyi, O. Modulating Nitric Oxide: Implications for Cytotoxicity and Cytoprotection. Antioxidants 2024, 13, 504. [Google Scholar] [CrossRef]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Endothelial Dysfunction in Cardiovascular Diseases: The Link Between Inflammation and Hydrogen Sulfide. Front. Pharmacol. 2020, 10, 1568. [Google Scholar] [CrossRef] [PubMed]
- Bhutta, B.S.; Alghoula, F.; Berim, I. Hypoxia. [Updated 2024 Mar 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025, Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482316.
- Okada, Y.; Paton, J.F.R.; López-Barneo, J.; Wilson, R.J.A.; Marina, N.; Pokorski, M. Editorial: Hypoxia and Cardiorespiratory Control. Front. Physiol. 2021, 12, 820815. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Bak, P.G.; Popazova, O.O.; Bukhtiyarova, N.V.; Yadlovsky, O.E. Nitric oxide-dependent mechanism of endothelial dysfunction formation is a promising target link for pharmacological management. Biopolym. Cell 2022, 38, 145–157. [Google Scholar] [CrossRef]
- Belenichev, I.; Kucherenko, L.; Pavlov, S.; Bukhtiyarova, N.; Popazova, O.; Derevianko, N.; Nimenko, G. Therapy of post-COVID-19 syndrome: improving the efficiency and safety of basic metabolic drug treatment with tiazotic acid (thiotriazoline). Pharmacia 2022, 69, 509–516. [Google Scholar] [CrossRef]
- Kiani, A.K.; Bonetti, G.; Medori, M.C.; Caruso, P.; Manganotti, P.; Fioretti, F.; Nodari, S.; Connelly, S.T.; Bertelli, M. Dietary supplements for improving nitric-oxide synthesis. J Prev Med Hyg. 2022, 63, E239–E245. [Google Scholar] [CrossRef]
- Bryan, N.S. Nitric Oxide Enhancement Strategies. Futur. Sci. OA 2015, 1, FSO48. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Napoli, C.; Loscalzo, J. Nitric Oxide Donors and Cardiovascular Agents Modulating the Bioactivity of Nitric Oxide: an overview. Circ. Res. 2002, 90, 21–28. [Google Scholar] [CrossRef]
- Gandoy-Fieiras, N.; Gonzalez-Juanatey, J.R.; Eiras, S. Myocardium Metabolism in Physiological and Pathophysiological States: Implications of Epicardial Adipose Tissue and Potential Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 2641. [Google Scholar] [CrossRef]
- Leone, A. Morphology of coronary arteries in relation to ischemic heart disease. J. Cardiol. Curr. Res. 2021, 14, 27–2. [Google Scholar] [CrossRef]
- Peng, X.; Du, J.; Wang, Y. Metabolic signatures in post-myocardial infarction heart failure, including insights into prediction, intervention, and prognosis. Biomed. Pharmacother. 2024, 170, 116079. [Google Scholar] [CrossRef]
- Sutovska, H.; Babarikova, K.; Zeman, M.; Molcan, L. Prenatal Hypoxia Affects Foetal Cardiovascular Regulatory Mechanisms in a Sex- and Circadian-Dependent Manner: A Review. Int. J. Mol. Sci. 2022, 23, 2885. [Google Scholar] [CrossRef] [PubMed]
- Frasch, M.G.; Giussani, D.A. Impact of Chronic Fetal Hypoxia and Inflammation on Cardiac Pacemaker Cell Development. Cells 2020, 9, 733. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zeng, H.; Liu, J.; Sun, M. Effects of Prenatal Hypoxia on Nervous System Development and Related Diseases. Front. Neurosci. 2021, 15, 755554. [Google Scholar] [CrossRef]
- Hula, N.; Liu, R.; Spaans, F.; Pasha, M.; Quon, A.; Kirschenman, R.; Cooke, C.-L.M.; Davidge, S.T. The Long-Term Effects of Prenatal Hypoxia on Coronary Artery Function of the Male and Female Offspring. Biomedicines 2022, 10, 3019. [Google Scholar] [CrossRef]
- Salameh, A.; Zöbisch, H.; Schröder, B.; Vigelahn, J.; Jahn, M.; Abraham, G.; Seeger, J.; Dähnert, I.; Dhein, S. Effects of Hypoxia and Acidosis on Cardiac Electrophysiology and Hemodynamics. Is NHE-Inhibition by Cariporide Still Advantageous? Front. Physiol. 2020, 11, 224. [Google Scholar] [CrossRef]
- Neubert, E.; Rassler, B.; Hoschke, A.; Raffort, C.; Salameh, A. Effects of Normobaric Hypoxia and Adrenergic Blockade over 72 h on Cardiac Function in Rats. Int. J. Mol. Sci. 2023, 24, 11417, Erratum in: Int J Mol Sci. 2024, Aug 01;25(15):8408. doi: 10.3390/ijms25158408. [Google Scholar] [CrossRef]
- Autonomic dysfunction syndromes after acute brain injury. Handb Clin Neurol. 2015, 128:539-51. [CrossRef]
- Mehra, R.; Tjurmina, O.A.; Ajijola, O.A.; Arora, R.; Bolser, D.C.; Chapleau, M.W.; Chen, P.-S.; Clancy, C.E.; Delisle, B.P.; Gold, M.R.; et al. Research Opportunities in Autonomic Neural Mechanisms of Cardiopulmonary Regulation: A Report From the National Heart, Lung, and Blood Institute and the National Institutes of Health Office of the Director Workshop. JACC: Basic Transl. Sci. 2022, 7, 265–293. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grune, J.; Yamazoe, M.; Nahrendorf, M. Electroimmunology and cardiac arrhythmia. Nat. Rev. Cardiol. 2021, 18, 547–564. [Google Scholar] [CrossRef]
- Dwyer, K.D.; Snyder, C.A.; Coulombe, K.L.K. Cardiomyocytes in Hypoxia: Cellular Responses and Implications for Cell-Based Cardiac Regenerative Therapies. Bioengineering 2025, 12, 154. [Google Scholar] [CrossRef]
- Giussani, D.A.; Camm, E.J.; Niu, Y.; Richter, H.G.; Blanco, C.E.; Gottschalk, R.; Blake, E.Z.; Horder, K.A.; Thakor, A.S.; Hansell, J.A.; et al. Developmental Programming of Cardiovascular Dysfunction by Prenatal Hypoxia and Oxidative Stress. PLOS ONE 2012, 7, e31017. [Google Scholar] [CrossRef]
- Hutter, D.; Kingdom, J.; Jaeggi, E. Causes and Mechanisms of Intrauterine Hypoxia and Its Impact on the Fetal Cardiovascular System: A Review. Int. J. Pediatr. 2010, 2010, 401323. [Google Scholar] [CrossRef] [PubMed]
- Nerheim, P.; Krishnan, S.C.; Olshansky, B.; Shivkumar, K. APOPTOSIS IN THE GENESIS OF CARDIAC RHYTHM DISORDERS. Cardiol. Clin. 2001, 19, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, E9–E68. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Zhu, X.; Jiao, Y.; Liu, H.; Huang, Z.; Pei, J.; Xu, Y.; Yang, Y.; Ren, K. Cardiac cell senescence: molecular mechanisms, key proteins and therapeutic targets. Cell Death Discov. 2024, 10, 78. [Google Scholar] [CrossRef]
- Sisakian, H. Cardiomyopathies: Evolution of pathogenesis concepts and potential for new therapies. World J. Cardiol. 2014, 6, 478–94. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Yamada, T.; Nomura, S. Recent Findings Related to Cardiomyopathy and Genetics. Int. J. Mol. Sci. 2021, 22, 12522. [Google Scholar] [CrossRef]
- Ahluwalia, M.; Kpodonu, J.; Agu, E. Risk Stratification in Hypertrophic Cardiomyopathy: Leveraging Artificial Intelligence to Provide Guidance in the Future. JACC: Adv. 2023, 2, 100562. [Google Scholar] [CrossRef]
- Ducsay, C.A.; Goyal, R.; Pearce, W.J.; Wilson, S.; Hu, X.-Q.; Zhang, L. Gestational Hypoxia and Developmental Plasticity. Physiol. Rev. 2018, 98, 1241–1334. [Google Scholar] [CrossRef]
- McCartan, C.; Mason, R.; Jayasinghe, S.R.; Griffiths, L.R. Cardiomyopathy Classification: Ongoing Debate in the Genomics Era. Biochem. Res. Int. 2012, 2012, 796926. [Google Scholar] [CrossRef]
- Oh, K.S.; Bender, T.M.; Bowen, A.; Godine, L.; Park, S.C. Transient myocardial ischemia of the newborn infant. Pediatr. Radiol. 1985, 15, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Flores-Nava, G.; Echevarría-Ybargüengoitia, J.L.; Navarro-Barrón, J.L.; García-Alonso, A. Isquemia miocárdica transitoria en el recién nacido con asfixia perinatal (miocardiopatía hipóxica) [Transient myocardial ischemia in newborn babies with perinatal asphyxia (hypoxic cardiomyopathy)]. Bol Med Hosp Infant Mex. 1990, 47(12):809-14. Spanish. [PubMed]
- Posthypoxic myocardial ischemia in newborn: diagnosis and treatment of severe type. Anesteziol Reanimatol. 2012, (1):65-8. Russian. [PubMed]
- Ovali, F. Hemodynamic changes and evaluation during hypoxic-ischemic encephalopathy and therapeutic hypothermia. Early Hum. Dev. 2022, 167, 105563. [Google Scholar] [CrossRef] [PubMed]
- Davey, B.; Szwast, A.; Rychik, J. Diagnosis and management of heart failure in the fetus. Minerva Pediatr. 2012, 64, 471–92. [Google Scholar] [PubMed]
- Ding, H.; Luo, Y.; Hu, K.; Huang, H.; Liu, P.; Xiong, M.; Zhu, L.; Yi, J.; Xu, Y. Hypoxia in utero increases the risk of pulmonary hypertension in rat offspring and is associated with vasopressin type-2 receptor upregulation. Mol. Med. Rep. 2020, 22, 4173–4182. [Google Scholar] [CrossRef]
- Nguyen, T.-T.N.; Lewis, C.V.; Hidalgo, D.C.; Posey, J.N.; Jordan, M.; Porfilio, T.E.; Grayck, M.R.; Wright, C.J.; Delaney, C.; Nozik, E.S. A maternal hypoxia mouse model to study the effect of late gestational hypoxia on offspring lung outcomes. Front. Physiol. 2025, 16, 1513703. [Google Scholar] [CrossRef]
- Bhasin, H.; Kohli, C. Myocardial dysfunction as a predictor of the severity and mortality of hypoxic ischaemic encephalopathy in severe perinatal asphyxia: a case–control study. Paediatr. Int. Child Health. 2019, 39, 259–264. [Google Scholar] [CrossRef]
- Neary, M.T.; Breckenridge, R.A. Hypoxia at the heart of sudden infant death syndrome? Pediatr. Res. 2013, 74, 375–379. [Google Scholar] [CrossRef]
- Joglar, J.A.; Kapa, S.; Saarel, E.V.; Dubin, A.M.; Gorenek, B.; Hameed, A.B.; de Melo, S.L.; Leal, M.A.; Mondésert, B.; Pacheco, L.D.; et al. 2023 HRS expert consensus statement on the management of arrhythmias during pregnancy. Hear. Rhythm. 2023, 20, e175–e264. [Google Scholar] [CrossRef]
- Waypa, G.B.; Schumacker, P.T. Hypoxia-induced changes in pulmonary and systemic vascular resistance: Where is the O2 sensor? Respir. Physiol. Neurobiol. 2010, 174, 201–211. [Google Scholar] [CrossRef]
- Liang, Y.; Ruan, W.; Jiang, Y.; Smalling, R.; Yuan, X.; Eltzschig, H.K. Interplay of hypoxia-inducible factors and oxygen therapy in cardiovascular medicine. Nat. Rev. Cardiol. 2023, 20, 723–737. [Google Scholar] [CrossRef]
- Giussani, D.A. Breath of Life: Heart Disease Link to Developmental Hypoxia. Circulation 2021, 144, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Bo, B.; Li, S.; Zhou, K.; Wei, J. The Regulatory Role of Oxygen Metabolism in Exercise-Induced Cardiomyocyte Regeneration. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Hula, N.; Spaans, F.; Vu, J.; Quon, A.; Kirschenman, R.; Cooke, C.-L.M.; Phillips, T.J.; Case, C.P.; Davidge, S.T. Placental treatment improves cardiac tolerance to ischemia/reperfusion insult in adult male and female offspring exposed to prenatal hypoxia. Pharmacol. Res. 2021, 165, 105461. [Google Scholar] [CrossRef] [PubMed]
- Rock, C.R.; Miller, S.L.; Allison, B.J. The Use of Antioxidants for Cardiovascular Protection in Fetal Growth Restriction: A Systematic Review. Antioxidants 2024, 13, 1400. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Siekierzycka, A.; Płoska, A.; Dobrucki, I.T.; Kalinowski, L. Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules 2021, 11, 982. [Google Scholar] [CrossRef]
- Roy, R.; Wilcox, J.; Webb, A.J.; O’gallagher, K. Dysfunctional and Dysregulated Nitric Oxide Synthases in Cardiovascular Disease: Mechanisms and Therapeutic Potential. Int. J. Mol. Sci. 2023, 24, 15200. [Google Scholar] [CrossRef]
- Niewinski, P.; Tubek, S.; Josiak, K.; Nowak, K.; Ponikowski, P. Cardiac parasympathetic denervation reduces hypoxic tachycardia, baroreflex sensitivity and heart rate variability in humans. Sci. Rep. 2025, 15, 6633. [Google Scholar] [CrossRef]
- Mitrică, M.; Lorusso, L.; Badea, A.-A.; Sîrbu, C.-A.; Pleșa, A.; Stănescu, A.-M.A.; Pleșa, F.C.; Sîrbu, O.M.; Munteanu, A.E. The Hidden Heart: Exploring Cardiac Damage Post-Stroke: A Narrative Review. Medicina 2024, 60, 1699. [Google Scholar] [CrossRef]
- Horenstein, M.S.; Diaz-Frias, J.; Guillaume, M. Tetralogy of Fallot. [Updated 2024 Dec 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513288.
- D'Cunha, C.; Sankaran, K. Persistent fetal circulation. Paediatr. Child Heal. 2001, 6, 744–750. [Google Scholar] [CrossRef]
- Malhotra, A.; Allison, B.J.; Castillo-Melendez, M.; Jenkin, G.; Polglase, G.R.; Miller, S.L. Neonatal Morbidities of Fetal Growth Restriction: Pathophysiology and Impact. Front. Endocrinol. 2019, 10, 55. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J.; Zhuravin, I.A. Role of Prenatal Hypoxia in Brain Development, Cognitive Functions, and Neurodegeneration. Front. Neurosci. 2018, 12, 825. [Google Scholar] [CrossRef] [PubMed]
- Romanowicz, J.; Guerrelli, D.; Dhari, Z.; Mulvany, C.; Reilly, M.; Swift, L.; Vasandani, N.; Ramadan, M.; Leatherbury, L.; Ishibashi, N.; et al. Chronic perinatal hypoxia delays cardiac maturation in a mouse model for cyanotic congenital heart disease. Am. J. Physiol. Circ. Physiol. 2021, 320, H1873–H1886. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Liu, Y.; Zhang, H. Adaptive Cardiac Metabolism Under Chronic Hypoxia: Mechanism and Clinical Implications. Front. Cell Dev. Biol. 2021, 9, 625524. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, K.M.; McGovern, P.E.; Mejaddam, A.; Rossidis, A.C.; Baumgarten, H.; Kim, A.; Grinspan, J.B.; Licht, D.J.; Didier, R.A.; Vossough, A.; et al. Chronic intrauterine hypoxia alters neurodevelopment in fetal sheep. J. Thorac. Cardiovasc. Surg. 2019, 157, 1982–1991. [Google Scholar] [CrossRef]
- Bhatt, A.B.; Foster, E.; Kuehl, K.; Alpert, J.; Brabeck, S.; Crumb, S.; Davidson, W.R.; Earing, M.G.; Ghoshhajra, B.B.; Karamlou, T.; et al. Congenital Heart Disease in the Older Adult: a scientific statement from the American Heart Association. Circulation 2015, 131, 1884–1931, Epub 2015 Apr 20. Erratum in: Circulation. 2015, May 26;131(21):e510. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, S.; Yang, G.; Wang, X.; Liu, F.; Li, Y.; Chen, Y.; Zhou, T.; Xie, D.; Liu, Y.; et al. Energy metabolism: A critical target of cardiovascular injury. Biomed. Pharmacother. 2023, 165, 115271. [Google Scholar] [CrossRef]
- Elia, A.; Fossati, S. Autonomic nervous system and cardiac neuro-signaling pathway modulation in cardiovascular disorders and Alzheimer’s disease. Front. Physiol. 2023, 14, 1060666. [Google Scholar] [CrossRef]
- Reece, E.A.; Hobbins, J.C. Clinical obstetrics : the fetus & mother 3rd ed. 2007 by Blackwell Publishing Ltd https://onlinelibrary.wiley.com/doi/pdf/10.1002/9780470753293.answ1.
- Szklarz, M.; Gontarz-Nowak, K.; Matuszewski, W.; Bandurska-Stankiewicz, E. Can Iron Play a Crucial Role in Maintaining Cardiovascular Health in the 21st Century? Int. J. Environ. Res. Public Heal. 2022, 19, 11990. [Google Scholar] [CrossRef]
- Floria, M.; Parteni, A.N.; Neagu, I.A.; Sascau, R.A.; Statescu, C.; Tanase, D.M.; Romania, I.; Popa, G.T. ; University of Medicine and Pharmacy Incomplete right bundle branch block: Challenges in electrocardiogram diagnosis. Anatol. J. Cardiol. 2021, 25, 380–384. [Google Scholar] [CrossRef]
- Doundoulakis, I.; Tsiachris, D.; Kordalis, A.; Soulaidopoulos, S.; Arsenos, P.; Xintarakou, A.; Koliastasis, L.; Vlachakis, P.K.; Tsioufis, K.; A Gatzoulis, K.; et al. Management of Patients With Unexplained Syncope and Bundle Branch Block: Predictive Factors of Recurrent Syncope. Cureus 2023, 15, e35827. [Google Scholar] [CrossRef]
- Sun, Y.; Deng, X.-M.; Cai, Y.; Shen, S.-E.; Dong, L.-Y. Post-cardiopulmonary bypass hypoxaemia in paediatric patients undergoing congenital heart disease surgery: risk factors, features, and postoperative pulmonary complications. BMC Cardiovasc. Disord. 2022, 22, 430. [Google Scholar] [CrossRef] [PubMed]
- Popazova, O.; Belenichev, I.; Abramov, A.; Bukhtiyarova, N.; Chereshniuk, I.; Skoryna, D. Indicators of Bioelectrical Activity of the Rat Heart After Prenatal Hypoxia and Pharmacological Correction. Innov. Biosyst. Bioeng. 2023, 6, 148–160. [Google Scholar] [CrossRef]
- Li, T.; Li, K.; Zhang, S.; Wang, Y.; Xu, Y.; Cronin, S.J.F.; Sun, Y.; Zhang, Y.; Xie, C.; Rodriguez, J.; et al. Overexpression of apoptosis inducing factor aggravates hypoxic-ischemic brain injury in neonatal mice. Cell Death Dis. 2020, 11, 77. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Galli, G.; Wang, Y.; Fan, Q.; Wang, Z.; Wang, X.; Xiao, W. Novel Therapeutic Targets for Hypoxia-Related Cardiovascular Diseases: The Role of HIF-1. Front. Physiol. 2020, 11, 774. [Google Scholar] [CrossRef]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Andrabi, S.M.; Sharma, N.S.; Karan, A.; Shahriar, S.M.S.; Cordon, B.; Ma, B.; Xie, J. Nitric Oxide: Physiological Functions, Delivery, and Biomedical Applications. Adv. Sci. 2023, 10, e2303259. [Google Scholar] [CrossRef]
- Kurhaluk, N.; Tkaczenko, H. L-Arginine and Nitric Oxide in Vascular Regulation—Experimental Findings in the Context of Blood Donation. Nutrients 2025, 17, 665. [Google Scholar] [CrossRef]
- Boycott, H.E.; Nguyen, M.-N.; Vrellaku, B.; Gehmlich, K.; Robinson, P. Nitric Oxide and Mechano-Electrical Transduction in Cardiomyocytes. Front. Physiol. 2020, 11, 606740. [Google Scholar] [CrossRef]
- Huang, C.L.-H.; Lei, M. Cardiomyocyte electrophysiology and its modulation: current views and future prospects. Philos. Trans. R. Soc. B: Biol. Sci. 2023, 378, 20220160. [Google Scholar] [CrossRef]
- Chemin, J.; Girard, C.; Duprat, F.; Lesage, F.; Romey, G.; Lazdunski, M. Mechanisms underlying excitatory effects of group I metabotropic glutamate receptors via inhibition of 2P domain K+ channels. EMBO J. 2003, 22, 5403–5411. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Qiu, H. Post-Translational S-Nitrosylation of Proteins in Regulating Cardiac Oxidative Stress. Antioxidants 2020, 9, 1051. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E. Nitric oxide signaling in health and disease. Cell 2022, 185, 2853–2878. [Google Scholar] [CrossRef] [PubMed]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef]
- Yang, N.J.; Hinner, M.J. Getting across the cell membrane: an overview for small molecules, peptides, and proteins. Methods Mol Biol. 2015, 1266, 29–53. [Google Scholar] [CrossRef]
- Lkhagva, B.; Lee, T.-W.; Lin, Y.-K.; Chen, Y.-C.; Chung, C.-C.; Higa, S.; Chen, Y.-J. Disturbed Cardiac Metabolism Triggers Atrial Arrhythmogenesis in Diabetes Mellitus: Energy Substrate Alternate as a Potential Therapeutic Intervention. Cells 2022, 11, 2915. [Google Scholar] [CrossRef]
- Woo, S.-H.; Kim, J.-C.; Eslenur, N.; Trinh, T.N.; Do, L.N.H. Modulations of Cardiac Functions and Pathogenesis by Reactive Oxygen Species and Natural Antioxidants. Antioxidants 2021, 10, 760. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D. cGMP-Dependent Protein Kinases and cGMP Phosphodiesterases in Nitric Oxide and cGMP Action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, J.; Fontes, S.K.; Bautista, E.N.; Cheng, Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc. Res. 2022, 118, 386–398. [Google Scholar] [CrossRef]
- Li, Y.; Li, W.-J.; Du, J.-M.; Wu, H.-H.; Zhou, S.-Y.; Li, M.; Li, Y.-Y.; Wang, S.-Y.; Wang, H.-Y.; Zheng, Y.; et al. Downregulation of PGAM2 alleviates angiotensin II-induced cardiac hypertrophy by destabilizing HSP90 and inactivating the mTOR/IKKα signaling pathway. Int. J. Biol. Sci. 2025, 21, 1308–1321. [Google Scholar] [CrossRef]
- Guo, B.; Li, Y.; Jin, X.; Liu, S.; Miao, C. Nitric oxide/cyclic GMP pathway mediates the endothelin-1-upregulation of adiponectin expression in rat cardiomyocytes. Biomed. Rep. 2017, 7, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Kayki-Mutlu, G.; Koch, W.J. Nitric Oxide and S-Nitrosylation in Cardiac Regulation: G Protein-Coupled Receptor Kinase-2 and β-Arrestins as Targets. Int. J. Mol. Sci. 2021, 22, 521. [Google Scholar] [CrossRef] [PubMed]
- Doul, J.; Minaříková, M.; Charvátová, Z.; Maxová, H. Nitric oxide is involved in the cardioprotection of neonatal rat hearts, but not in neonatal ischemic postconditioning. Physiol. Rep. 2024, 12, e16147. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, L.G.; Wright, C.J.; Kinsella, J.P.; Delaney, C. Inhaled nitric oxide use in neonates: Balancing what is evidence-based and what is physiologically sound. Nitric Oxide 2020, 95, 12–16. [Google Scholar] [CrossRef]
- Uray, I.P.; Uray, K. Mechanotransduction at the Plasma Membrane-Cytoskeleton Interface. Int. J. Mol. Sci. 2021, 22, 11566. [Google Scholar] [CrossRef]
- Katoh, K. Signal Transduction Mechanisms of Focal Adhesions: Src and FAK-Mediated Cell Response. Front. Biosci. 2024, 29, 392. [Google Scholar] [CrossRef]
- Ławkowska, K.; Bonowicz, K.; Jerka, D.; Bai, Y.; Gagat, M. Integrins in Cardiovascular Health and Disease: Molecular Mechanisms and Therapeutic Opportunities. Biomolecules 2025, 15, 233. [Google Scholar] [CrossRef]
- Zhang, F.; Xia, Y.; Su, J.; Quan, F.; Zhou, H.; Li, Q.; Feng, Q.; Lin, C.; Wang, D.; Jiang, Z. Neutrophil diversity and function in health and disease. Signal Transduct. Target. Ther. 2024, 9, 343. [Google Scholar] [CrossRef]
- Spiers, J.G.; Steinert, J.R. Nitrergic modulation of ion channel function in regulating neuronal excitability. Channels 2021, 15, 666–679. [Google Scholar] [CrossRef]
- Alwatban, A.Z.; Orfali, R.S.; Lau, L.; Chea, N.; Alotaibi, A.M.; Nam, Y.-W.; Zhang, M. Oxidative stress and ion channels in neurodegenerative diseases. Front. Physiol. 2024, 15, 1320086. [Google Scholar] [CrossRef]
- Kalinina, E.V.; Novichkova, M.D. S-Glutathionylation and S-Nitrosylation as Modulators of Redox-Dependent Processes in Cancer Cell. Biochem. (Moscow) 2023, 88, 924–943. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Hui, W.; Huang, M.; Guo, Y.; Yu, M.; Yang, X.; Liu, Y.; Chen, X. Dynamic Changes in Ion Channels during Myocardial Infarction and Therapeutic Challenges. Int. J. Mol. Sci. 2024, 25, 6467. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S.M. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: in Physiology and in Disease States. Am J Biomed Sci Res. 2022, 15, 153–177. [Google Scholar] [PubMed]
- Wang, W.-L.; Ge, T.-Y.; Chen, X.; Mao, Y.; Zhu, Y.-Z. Advances in the Protective Mechanism of NO, H2S, and H2 in Myocardial Ischemic Injury. Front. Cardiovasc. Med. 2020, 7, 588206. [Google Scholar] [CrossRef]
- Wei, X.; Yohannan, S.; Richards, J.R. Physiology, Cardiac Repolarization Dispersion and Reserve. [Updated 2023 Apr 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025, Available from: https://www.ncbi.nlm.nih.gov/books/NBK537194.
- Pappas, G.; Wilkinson, M.L.; Gow, A.J. Nitric oxide regulation of cellular metabolism: Adaptive tuning of cellular energy. Nitric Oxide 2023, 131, 8–17. [Google Scholar] [CrossRef]
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429. [Google Scholar] [CrossRef]
- Belenichev, I.; Popazova, O.; Bukhtiyarova, N.; Ryzhenko, V.; Pavlov, S.; Suprun, E.; Oksenych, V.; Kamyshnyi, O. Targeting Mitochondrial Dysfunction in Cerebral Ischemia: Advances in Pharmacological Interventions. Antioxidants 2025, 14, 108. [Google Scholar] [CrossRef]
- Chirkov, Y.Y.; Nguyen, T.H.; Horowitz, J.D. Impairment of Anti-Aggregatory Responses to Nitric Oxide and Prostacyclin: Mechanisms and Clinical Implications in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 1042. [Google Scholar] [CrossRef]
- Siamwala, J.H.; Kumar, P.; Veeriah, V.; Muley, A.; Rajendran, S.; Konikkat, S.; Majumder, S.; Mani, K.P.; Chatterjee, S. Nitric Oxide Reverses the Position of the Heart during Embryonic Development. Int. J. Mol. Sci. 2019, 20, 1157. [Google Scholar] [CrossRef]
- Dougherty, J.A.; Patel, N.; Kumar, N.; Rao, S.G.; Angelos, M.G.; Singh, H.; Cai, C.; Khan, M. Human Cardiac Progenitor Cells Enhance Exosome Release and Promote Angiogenesis Under Physoxia. Front. Cell Dev. Biol. 2020, 8, 130. [Google Scholar] [CrossRef]
- Tsaytler, P.; Liu, J.; Blaess, G.; Schifferl, D.; Veenvliet, J.V.; Wittler, L.; Timmermann, B.; Herrmann, B.G.; Koch, F. BMP4 triggers regulatory circuits specifying the cardiac mesoderm lineage. Development 2023, 150, dev201450. [Google Scholar] [CrossRef]
- Kovács, T.; Halasy, V.; Pethő, C.; Szőcs, E.; Soós, Á.; Dóra, D.; Barbara, P.d.S.; Faure, S.; Stavely, R.; Goldstein, A.M.; et al. Essential Role of BMP4 Signaling in the Avian Ceca in Colorectal Enteric Nervous System Development. Int. J. Mol. Sci. 2023, 24, 15664. [Google Scholar] [CrossRef] [PubMed]
- Govier-Cole, A.E.; Wood, R.J.; Fletcher, J.L.; Gonsalvez, D.G.; Merlo, D.; Cate, H.S.; Murray, S.S.; Xiao, J. Inhibiting Bone Morphogenetic Protein 4 Type I Receptor Signaling Promotes Remyelination by Potentiating Oligodendrocyte Differentiation. eneuro 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Pirahanchi, Y.; Dimri, M. Biochemistry, Guanylate Cyclase. [Updated 2023 Jul 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK53715).
- Bloch, W.; Fleischmann, B.; Lorke, D.; Andressen, C.; Hops, B.; Hescheler, J.; Addicks, K. Nitric oxide synthase expression and role during cardiomyogenesis. Cardiovasc. Res. 1999, 43, 675–684. [Google Scholar] [CrossRef]
- Kumar, P.; Ghosh, A.; Sundaresan, L.; Kathirvel, P.; Sankaranarayanan, K.; Chatterjee, S. Ectopic release of nitric oxide modulates the onset of cardiac development in avian model. Vitr. Cell. Dev. Biol. - Anim. 2020, 56, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Du, J.; Deng, S.; Liu, B.; Jing, X.; Yan, Y.; Liu, Y.; Wang, J.; Zhou, X.; She, Q. The molecular mechanisms of cardiac development and related diseases. Signal Transduct. Target. Ther. 2024, 9, 1–60. [Google Scholar] [CrossRef]
- Mensah, I.K.; Gowher, H. Epigenetic Regulation of Mammalian Cardiomyocyte Development. Epigenomes 2024, 8, 25. [Google Scholar] [CrossRef]
- Filho, B.J.A.; da Costa, B.E.P.; Ogando, P.B.; Vieira, M.C.; Antonello, I.C.; Poli-De-Figueiredo, C.E. Serum nitrate and NOx levels in preeclampsia are higher than in normal pregnancy. Hypertens. Pregnancy 2016, 35, 226–233. [Google Scholar] [CrossRef]
- Khetsuriani, T.; Chabashvili, N.; Sanikidze, T. [Role of endothelin-1 and nitric oxide level in pathogenesis preeclampsia]. Georgian Med News. 2006, 17–21. [Google Scholar]
- Vural, P. Nitric oxide/endothelin-1 in preeclampsia. Clin. Chim. Acta 2002, 317, 65–70. [Google Scholar] [CrossRef]
- Guo, X.; Yi, H.; Li, T.C.; Wang, Y.; Wang, H.; Chen, X. Role of Vascular Endothelial Growth Factor (VEGF) in Human Embryo Implantation: Clinical Implications. Biomolecules 2021, 11, 253. [Google Scholar] [CrossRef] [PubMed]
- Ivanina, A.V.; Borah, B.; Rimkevicius, T.; Macrander, J.; Piontkivska, H.; Sokolova, I.M.; Beniash, E. The Role of the Vascular Endothelial Growth Factor (VEGF) Signaling in Biomineralization of the Oyster Crassostrea gigas. Front. Mar. Sci. 2018, 5, 309. [Google Scholar] [CrossRef]
- Boldeanu, L.; Dijmărescu, A.l.; Radu, M.; Siloşi, C.A.; Popescu-Drigă, M.V.; Poenariu, I.S.; Siloşi, I.; Boldeanu, M.V.; Novac, M.B.; Novac, L.V. The role of mediating factors involved in angiogenesis during implantation. Romanian J. Morphol. Embryol. 2020, 61, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Groesch, K.; Torry, R.; Wilber, A.; Abrams, R.; Bieniarz, A.; Guilbert, L.; Torry, D. Nitric oxide generation affects pro- and anti-angiogenic growth factor expression in primary human trophoblast. Placenta 2011, 32, 926–931. [Google Scholar] [CrossRef]
- Opichka, M.A.; Rappelt, M.W.; Gutterman, D.D.; Grobe, J.L.; McIntosh, J.J. Vascular Dysfunction in Preeclampsia. Cells 2021, 10, 3055. [Google Scholar] [CrossRef]
- Rana, S.; Burke, S.D.; Karumanchi, S.A. Imbalances in circulating angiogenic factors in the pathophysiology of preeclampsia and related disorders. Am. J. Obstet. Gynecol. 2022, 226, S1019–S1034. [Google Scholar] [CrossRef]
- Liu, Q.; Guan, C.; Liu, C.; Li, H.; Wu, J.; Sun, C. Targeting hypoxia-inducible factor-1alpha: A new strategy for triple-negative breast cancer therapy. Biomed. Pharmacother. 2022, 156, 113861. [Google Scholar] [CrossRef]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef]
- Olson, N.; van der Vliet, A. Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide 2011, 25, 125–137. [Google Scholar] [CrossRef]
- Rood, K.; Lopez, V.; La Frano, M.R.; Fiehn, O.; Zhang, L.; Blood, A.B.; Wilson, S.M. Gestational Hypoxia and Programing of Lung Metabolism. Front. Physiol. 2019, 10, 1453. [Google Scholar] [CrossRef]
- Kane, A.; Kothmann, E.; Giussani, D. Detection and response to acute systemic hypoxia. BJA Educ. 2020, 20, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.C.; Liu, H.; Pinkas, G.A.; Thompson, L.P. Chronic hypoxia increases peroxynitrite, MMP9 expression, and collagen accumulation in fetal guinea pig hearts. Pediatr. Res. 2012, 71, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, C.R.; Chagas, A.C.P.; Carvalho, M.H.C.; Dantas, A.P.; Jatene, M.B.; de Souza, L.C.B.; da Luz, P.L. Influence of Hypoxia on Nitric Oxide Synthase Activity and Gene Expression in Children With Congenital Heart Disease: a novel pathophysiological adaptive mechanism. Circulation 2001, 103, 2272–2276. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Góngora, M.C.; Harrison, D.G.; Lambeth, J.D.; Dikalov, S.; Griendling, K.K. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am. J. Physiol. Circ. Physiol. 2010, 299, H673–H679. [Google Scholar] [CrossRef]
- Popazova, O.; Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Oksenych, V.; Kamyshnyi, A. Cardioprotective Activity of Pharmacological Agents Affecting NO Production and Bioavailability in the Early Postnatal Period after Intrauterine Hypoxia in Rats. Biomedicines 2023, 11, 2854. [Google Scholar] [CrossRef]
- Dong, Y.; Thompson, L.P. Differential Expression of Endothelial Nitric Oxide Synthase in Coronary and Cardiac Tissue in Hypoxic Fetal Guinea Pig Hearts. J. Soc. Gynecol. Investig. 2006, 13, 483–490. [Google Scholar] [CrossRef]
- Liu, J.; Gao, Y.; Negash, S.; Longo, L.D.; Raj, J.U. Long-term effects of prenatal hypoxia on endothelium-dependent relaxation responses in pulmonary arteries of adult sheep. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L547–L554. [Google Scholar] [CrossRef]
- Niu, Y.; Kane, A.D.; Lusby, C.M.; Allison, B.J.; Chua, Y.Y.; Kaandorp, J.J.; Nevin-Dolan, R.; Ashmore, T.J.; Blackmore, H.L.; Derks, J.B.; et al. Maternal Allopurinol Prevents Cardiac Dysfunction in Adult Male Offspring Programmed by Chronic Hypoxia During Pregnancy. Hypertension 2018, 72, 971–978. [Google Scholar] [CrossRef]
- Hauton, D.; Ousley, V. Prenatal hypoxia induces increased cardiac contractility on a background of decreased capillary density. BMC Cardiovasc. Disord. 2009, 9, 1–1. [Google Scholar] [CrossRef]
- De Pascali, F.; Hemann, C.; Samons, K.; Chen, C.-A.; Zweier, J.L. Hypoxia and Reoxygenation Induce Endothelial Nitric Oxide Synthase Uncoupling in Endothelial Cells through Tetrahydrobiopterin Depletion and S-Glutathionylation. Biochemistry 2014, 53, 3679–3688. [Google Scholar] [CrossRef]
- Zhang, Z.; Yao, L.; Yang, J.; Wang, Z.; Du, G. PI3K/Akt and HIF-1 signaling pathway in hypoxia-ischemia (Review). Mol. Med. Rep. 2018, 18, 3547–3554. [Google Scholar] [CrossRef] [PubMed]
- Jung, F.; Palmer, L.A.; Zhou, N.; Johns, R.A. Hypoxic Regulation of Inducible Nitric Oxide Synthase via Hypoxia Inducible Factor-1 in Cardiac Myocytes. Circ. Res. 2000, 86, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S. Redox signaling: Nitrosylation and related target interactions of nitric oxide. Cell 1994, 78, 931–936. [Google Scholar] [CrossRef]
- Khetsuriani, T.; Chabashvili, N.; Sanikidze, T. [Role of endothelin-1 and nitric oxide level in pathogenesis preeclampsia]. Georgian Med News. 2006, 17–21. [Google Scholar]
- Pérez-Torres, I.; Manzano-Pech, L.; Rubio-Ruíz, M.E.; Soto, M.E.; Guarner-Lans, V. Nitrosative Stress and Its Association with Cardiometabolic Disorders. Molecules 2020, 25, 2555. [Google Scholar] [CrossRef]
- Dattilo, S.; Mancuso, C.; Koverech, G.; Di Mauro, P.; Ontario, M.L.; Petralia, C.C.; Petralia, A.; Maiolino, L.; Serra, A.; Calabrese, E.J.; et al. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing 2015, 12, 20. [Google Scholar] [CrossRef]
- Aengwanich, W.; Wandee, J. The effect of increased ambient temperature on Hsp70, superoxide dismutase, nitric oxide, malondialdehyde, and caspase activity in relation to the intrinsic and extrinsic apoptosis pathway of broiler blood cells. J. Therm. Biol. 2022, 105, 103211. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the regulation of gene expression. Trends Cell Biol. 2001, 11, 66–75. [Google Scholar] [CrossRef]
- Kröncke, K.-D. Nitrosative Stress and Transcription. Biol. Chem. 2003, 384, 1365–77. [Google Scholar] [CrossRef]
- Sun, X.; Alford, J.; Qiu, H. Structural and Functional Remodeling of Mitochondria in Cardiac Diseases. Int. J. Mol. Sci. 2021, 22, 4167. [Google Scholar] [CrossRef]
- Nguyen, B.Y.; Ruiz-Velasco, A.; Bui, T.; Collins, L.; Wang, X.; Liu, W. Mitochondrial function in the heart: the insight into mechanisms and therapeutic potentials. Br. J. Pharmacol. 2019, 176, 4302–4318. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.F.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Paraskevaidis, I.; Kourek, C.; Farmakis, D.; Tsougos, E. Mitochondrial Dysfunction in Cardiac Disease: The Fort Fell. Biomolecules 2024, 14, 1534. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Jiang, T.; Hua, J.; Xiong, Z.; Dai, K.; Chen, H.; Li, L.; Peng, J.; Peng, X.; Zheng, Z.; et al. PINK1/Parkin-mediated mitophagy in cardiovascular disease: From pathogenesis to novel therapy. Int. J. Cardiol. 2022, 361, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.M.; Tate, M.; Prakoso, D.; Deo, M.; Willis, A.M.; Nash, D.M.; Donner, D.G.; Crawford, S.; Kiriazis, H.; Granata, C.; et al. Characterisation of the Myocardial Mitochondria Structural and Functional Phenotype in a Murine Model of Diabetic Cardiomyopathy. Front. Physiol. 2021, 12, 672252. [Google Scholar] [CrossRef]
- Ravindran, S.; Rau, C.D. The multifaceted role of mitochondria in cardiac function: insights and approaches. Cell Commun. Signal. 2024, 22, 1–32. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. J. Clin. Investig. 2018, 128, 3716–3726. [Google Scholar] [CrossRef]
- Chang, X.; Liu, R.; Li, R.; Peng, Y.; Zhu, P.; Zhou, H. Molecular Mechanisms of Mitochondrial Quality Control in Ischemic Cardiomyopathy. Int. J. Biol. Sci. 2023, 19, 426–448. [Google Scholar] [CrossRef]
- McClellan, G.; Weisberg, A.; Winegrad, S. Energy transport from mitochondria to myofibril by a creatine phosphate shuttle in cardiac cells. Am. J. Physiol. Physiol. 1983, 245, C423–C427. [Google Scholar] [CrossRef]
- Schlattner, U.; Tokarska-Schlattner, M.; Wallimann, T. Mitochondrial creatine kinase in human health and disease. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2006, 1762, 164–180. [Google Scholar] [CrossRef]
- Xin, Y.; Zhang, X.; Li, J.; Gao, H.; Li, J.; Li, J.; Hu, W.; Li, H. New Insights Into the Role of Mitochondria Quality Control in Ischemic Heart Disease. Front. Cardiovasc. Med. 2021, 8, 774619. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Holody, C.D.; Kirschenman, R.; Graton, M.E.; Spaans, F.; Phillips, T.J.; Case, C.P.; Bourque, S.L.; Lemieux, H.; Davidge, S.T. Sex-Specific Effects of Prenatal Hypoxia and a Placental Antioxidant Treatment on Cardiac Mitochondrial Function in the Young Adult Offspring. Int. J. Mol. Sci. 2023, 24, 13624. [Google Scholar] [CrossRef]
- Thompson, L.P.; Chen, L.; Polster, B.M.; Pinkas, G.; Song, H. Prenatal hypoxia impairs cardiac mitochondrial and ventricular function in guinea pig offspring in a sex-related manner. Am. J. Physiol. Integr. Comp. Physiol. 2018, 315, R1232–R1241. [Google Scholar] [CrossRef]
- Al-Hasan, Y.M.; Pinkas, G.A.; Thompson, L.P. Prenatal Hypoxia Reduces Mitochondrial Protein Levels and Cytochrome c Oxidase Activity in Offspring Guinea Pig Hearts. Reprod. Sci. 2014, 21, 883–891. [Google Scholar] [CrossRef]
- Clerc, P.; Rigoulet, M.; Leverve, X.; Fontaine, E. Nitric oxide increases oxidative phosphorylation efficiency. J. Bioenerg. Biomembr. 2007, 39, 158–166. [Google Scholar] [CrossRef]
- Song, H.; Telugu, B.P.; Thompson, L.P. Sexual dimorphism of mitochondrial function in the hypoxic guinea pig placenta†. Biol. Reprod. 2019, 100, 208–216. [Google Scholar] [CrossRef]
- Shao, Y.; Li, X.; Wood, J.W.; Ma, J.-X. Mitochondrial dysfunctions, endothelial progenitor cells and diabetic retinopathy. J. Diabetes Its Complicat. 2018, 32, 966–973. [Google Scholar] [CrossRef]
- Hüttemann, M.; Helling, S.; Sanderson, T.H.; Sinkler, C.; Samavati, L.; Mahapatra, G.; Varughese, A.; Lu, G.; Liu, J.; Ramzan, R.; et al. Regulation of mitochondrial respiration and apoptosis through cell signaling: Cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2012, 1817, 598–609. [Google Scholar] [CrossRef]
- Shemarova, I.; Nesterov, V.; Emelyanova, L.; Korotkov, S. Mitochondrial mechanisms by which gasotransmitters (H2S, NO and CO) protect cardiovascular system against hypoxia. Front. Biosci. 2021, 13, 105–130. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Moellering, D.R.; Ceaser, E.; Shiva, S.; Xu, J.; Darley-Usmar, V. Inhibition of mitochondrial protein synthesis results in increased endothelial cell susceptibility to nitric oxide-induced apoptosis. Proc. Natl. Acad. Sci. 2002, 99, 6643–6648. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; Lerner, A. Nutraceuticals Targeting Generation and Oxidant Activity of Peroxynitrite May Aid Prevention and Control of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3624. [Google Scholar] [CrossRef]
- Batty, M.; Bennett, M.R.; Yu, E. The Role of Oxidative Stress in Atherosclerosis. Cells 2022, 11, 3843. [Google Scholar] [CrossRef]
- Hu, X.-Q.; Zhang, L. Hypoxia and Mitochondrial Dysfunction in Pregnancy Complications. Antioxidants 2021, 10, 405. [Google Scholar] [CrossRef]
- Gasparre, G.; Porcelli, A.M.; Lenaz, G.; Romeo, G. Relevance of Mitochondrial Genetics and Metabolism in Cancer Development. Cold Spring Harb. Perspect. Biol. 2013, 5, a011411–a011411. [Google Scholar] [CrossRef]
- Xia, D.; Liu, Y.; Wu, P.; Wei, D. Current Advances of Mitochondrial Dysfunction and Cardiovascular Disease and Promising Therapeutic Strategies. Am. J. Pathol. 2023, 193, 1485–1500. [Google Scholar] [CrossRef]
- Vangrieken, P.; Al-Nasiry, S.; Bast, A.; Leermakers, P.A.; Tulen, C.B.M.; Janssen, G.M.J.; Kaminski, I.; Geomini, I.; Lemmens, T.; Schiffers, P.M.H.; et al. Hypoxia-induced mitochondrial abnormalities in cells of the placenta. PLOS ONE 2021, 16, e0245155. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Energy metabolism design of the striated muscle cell. Physiol. Rev. 2021, 101, 1561–1607. [Google Scholar] [CrossRef]
- Tverdokhleb, I.V. Heterogeneity of Mitochondrial Apparatus of Myocardium and Mechanisms of Its Formation in Early Ontogenesis of Rats. Tsitologiya i Genetika 1998, 32, 8–12 [In Russian]. [Google Scholar]
- Leitan, E.B. Pathomorphology and Ultrastructure of Myocardium in Fetuses and Newborns with Asphyxia. Author's Abstract of Dissertation... Cand. Med. Sci.; Novosibirsk, 1981, 16 p. [In Russian].
- Song, H.; Thompson, L.P. Effects of Gestational Hypoxia on PGC1α and Mitochondrial Acetylation in Fetal Guinea Pig Hearts. Reprod. Sci. 2023, 30, 2996–3009. [Google Scholar] [CrossRef] [PubMed]
- Orekhov, A.N.; Summerhill, V.I.; Khotina, V.A.; Popov, M.A.; Uzokov, J.K.; Sukhorukov, V.N. Role of Mitochondria in the Chronification of Inflammation: Focus on Dysfunctional Mitophagy and Mitochondrial DNA Mutations. Gene Expr. 2023, 22, 329–344. [Google Scholar] [CrossRef]
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898. [Google Scholar] [CrossRef] [PubMed]
- Nesci, S.; Spagnoletta, A.; Oppedisano, F. Inflammation, Mitochondria and Natural Compounds Together in the Circle of Trust. Int. J. Mol. Sci. 2023, 24, 6106. [Google Scholar] [CrossRef]
- Li, P.; Zhou, M.; Wang, J.; Tian, J.; Zhang, L.; Wei, Y.; Yang, F.; Xu, Y.; Wang, G. Important Role of Mitochondrial Dysfunction in Immune Triggering and Inflammatory Response in Rheumatoid Arthritis. J. Inflamm. Res. 2024, 17, 11631–11657. [Google Scholar] [CrossRef]
- Nakahira, K.; Hisata, S.; Choi, A.M. The Roles of Mitochondrial Damage-Associated Molecular Patterns in Diseases. Antioxidants Redox Signal. 2015, 23, 1329–1350. [Google Scholar] [CrossRef]
- Cruz, C.S.D.; Kang, M.-J. Mitochondrial dysfunction and damage associated molecular patterns (DAMPs) in chronic inflammatory diseases. Mitochondrion 2018, 41, 37–44. [Google Scholar] [CrossRef]
- Vringer, E.; Tait, S.W.G. Mitochondria and cell death-associated inflammation. Cell Death Differ. 2023, 30, 304–312. [Google Scholar] [CrossRef]
- Fischer, R.; Maier, O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxidative Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Schulz, R.; Liaudet, L.; Szabó, C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol. Sci. 2005, 26, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hutchins, J.B.; Rhodes, P.G. Intrauterine hypoxia–ischemia alters nitric oxide synthase expression and activity in fetal and neonatal rat brains. Dev. Brain Res. 1998, 109, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Calcaterra, V.; Pelizzo, G.; Bramanti, P.; Mazzon, E. Prenatal Hypoxia and Placental Oxidative Stress: Insights from Animal Models to Clinical Evidences. Antioxidants 2020, 9, 414. [Google Scholar] [CrossRef]
- Matsubara, K.; Higaki, T.; Matsubara, Y.; Nawa, A. Nitric Oxide and Reactive Oxygen Species in the Pathogenesis of Preeclampsia. Int. J. Mol. Sci. 2015, 16, 4600–4614. [Google Scholar] [CrossRef]
- Migita, K.; Maeda, Y.; Abiru, S.; Komori, A.; Yokoyama, T.; Takii, Y.; Nakamura, M.; Yatsuhashi, H.; Eguchi, K.; Ishibashi, H. Peroxynitrite-mediated matrix metalloproteinase-2 activation in human hepatic stellate cells. FEBS Lett. 2005, 579, 3119–3125. [Google Scholar] [CrossRef]
- Vincenti, M.P.; Brinckerhoff, C.E. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. Ther. 2002, 4, 157. [Google Scholar] [CrossRef]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Quintero-Fabián, S.; Arreola, R.; Becerril-Villanueva, E.; Torres-Romero, J.C.; Arana-Argáez, V.; Lara-Riegos, J.; Ramírez-Camacho, M.A.; Alvarez-Sánchez, M.E. Role of Matrix Metalloproteinases in Angiogenesis and Cancer. Front. Oncol. 2019, 9, 1370. [Google Scholar] [CrossRef]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef]
- Islam, B.U.; Habib, S.; Ahmad, P.; Allarakha, S.; Moinuddin; Ali, A. Pathophysiological Role of Peroxynitrite Induced DNA Damage in Human Diseases: A Special Focus on Poly(ADP-ribose) Polymerase (PARP). Indian J. Clin. Biochem. 2015, 30, 368–385. [Google Scholar] [CrossRef] [PubMed]
- de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. The Nitration of Proteins, Lipids and DNA by Peroxynitrite Derivatives-Chemistry Involved and Biological Relevance. Stresses 2022, 2, 53–64. [Google Scholar] [CrossRef]
- Arunachalam, G.; Samuel, S.M.; Ding, H.; Triggle, C.R. Peroxynitrite Biology. In: Laher, I. (eds) Systems Biology of Free Radicals and Antioxidants. Springer, 2014, Berlin, Heidelberg. [CrossRef]
- Bencsik, P.; Kupai, K.; Giricz, Z.; Görbe, A.; Pipis, J.; Murlasits, Z.; Kocsis, G.F.; Varga-Orvos, Z.; Puskás, L.G.; Csonka, C.; et al. Role of iNOS and peroxynitrite–matrix metalloproteinase-2 signaling in myocardial late preconditioning in rats. Am. J. Physiol. Circ. Physiol. 2010, 299, H512–H518. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, E.; White, V.; Sosa, M.; Di Marco, I.; Basualdo, M.N.; Faingold, M.C.; Jawerbaum, A. Regulation of Matrix Metalloproteinases 2 and 9 Activities by Peroxynitrites in Term Placentas From Type 2 Diabetic Patients. Reprod. Sci. 2012, 19, 814–822. [Google Scholar] [CrossRef]
- Hadler-Olsen, E.; Fadnes, B.; Sylte, I.; Uhlin-Hansen, L.; Winberg, J. Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 2010, 278, 28–45. [Google Scholar] [CrossRef]
- Membrino, V.; Di Paolo, A.; Di Crescenzo, T.; Cecati, M.; Alia, S.; Vignini, A. Effects of Animal-Based and Plant-Based Nitrates and Nitrites on Human Health: Beyond Nitric Oxide Production. Biomolecules 2025, 15, 236. [Google Scholar] [CrossRef]
- A Heinrich, T.; da Silva, R.S.; Miranda, K.M.; Switzer, C.H.; A Wink, D.; Fukuto, J.M. Biological nitric oxide signalling: chemistry and terminology. Br. J. Pharmacol. 2013, 169, 1417–1429. [Google Scholar] [CrossRef]
- Rubbo, H.; Radi, R.; Trujillo, M.; Telleri, R.; Kalyanaraman, B.; Barnes, S.; Kirk, M.; A Freeman, B. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem. 1994, 269, 26066–26075. [Google Scholar] [CrossRef]
- Jourd'heuil, D.; Jourd'heuil, F.L.; Kutchukian, P.S.; Musah, R.A.; Wink, D.A.; Grisham, M.B. Reaction of superoxide and nitric oxide with peroxynitrite. Implications for peroxynitrite-mediated oxidation reactions in vivo. J Biol Chem. 2001, 276, 28799–805. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2017, 118, 1253–1337. [Google Scholar] [CrossRef]
- Wichitnithad, W.; Nantaphol, S.; Noppakhunsomboon, K.; Rojsitthisak, P. An update on the current status and prospects of nitrosation pathways and possible root causes of nitrosamine formation in various pharmaceuticals. Saudi Pharm. J. 2023, 31, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Christmann, M. DNA Alkylation Damage by Nitrosamines and Relevant DNA Repair Pathways. Int. J. Mol. Sci. 2023, 24, 4684. [Google Scholar] [CrossRef]
- Grossi, L.; Montevecchi, P.C. S-nitrosocysteine and cystine from reaction of cysteine with nitrous acid. A kinetic investigation. J Org Chem. 2002, Nov 29;67(24):8625-30. [CrossRef]
- Abalenikhina, Y.V.; Shchulkin, A.V.; Suchkova, O.N.; Ananyeva, P.D.; Mylnikov, P.Y.; Yakusheva, E.N.; Suchkov, I.A.; Kalinin, R.E. S-Nitrosoglutathione Is Not a Substrate of OATP1B1, but Stimulates Its Expression and Activity. Biomolecules 2025, 15, 428. [Google Scholar] [CrossRef] [PubMed]
- Broniowska, K.A.; Diers, A.R.; Hogg, N. S-Nitrosoglutathione. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2013, 1830, 3173–3181. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Gorbacheva, S.V.; Demchenko, A.V.; Bukhtiyarova, N.V. The thiol-disulfide balance and the nitric oxide system in the brain tissue of rats subjected to experimental acute impairment of cerebral blood flow: The therapeutic effects of nootropic drugs. Neurochem. J. 2014, 8, 24–27. [Google Scholar] [CrossRef]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef]
- Salvagno, M.; Sterchele, E.D.; Zaccarelli, M.; Mrakic-Sposta, S.; Welsby, I.J.; Balestra, C.; Taccone, F.S. Oxidative Stress and Cerebral Vascular Tone: The Role of Reactive Oxygen and Nitrogen Species. Int. J. Mol. Sci. 2024, 25, 3007. [Google Scholar] [CrossRef]
- Hogg, N.; Singh, R.J.; Konorev, E.; Joseph, J.; Kalyanaraman, B. S-Nitrosoglutathione as a substrate for γ-glutamyl transpeptidase. Biochem. J. 1997, 323, 477–481. [Google Scholar] [CrossRef]
- Zhang, Y.; Hogg, N. The mechanism of transmembraneS-nitrosothiol transport. Proc. Natl. Acad. Sci. 2004, 101, 7891–7896. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Boutin, C.; Clément, C.; Rivoal, J. Post-Translational Modifications to Cysteine Residues in Plant Proteins and Their Impact on the Regulation of Metabolism and Signal Transduction. Int. J. Mol. Sci. 2024, 25, 9845. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.-M.; Holdship, P.; To, T.Q.; Ratcliffe, P.J.; Keeley, T.P. Comparative analysis of N-terminal cysteine dioxygenation and prolyl-hydroxylation as oxygen-sensing pathways in mammalian cells. J. Biol. Chem. 2023, 299, 105156. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Kumar, G.; Chhabra, A.; Sethy, N.K.; Jain, N.; Meena, R.N.; Tulsawani, R.; Prasad, D.N.; Kumar, B.; Sharma, M. Cysteine becomes conditionally essential during hypobaric hypoxia and regulates adaptive neuro-physiological responses through CBS/H2S pathway. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2020, 1866, 165769. [Google Scholar] [CrossRef]
- Erusalimsky, J.D.; Moncada, S. Nitric Oxide and Mitochondrial Signaling: From physiology to pathophysiology. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2524–2531. [Google Scholar] [CrossRef]
- Brown, G.C. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2001, 1504, 46–57. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19–19. [Google Scholar] [CrossRef]
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642. [Google Scholar] [CrossRef]
- Weidinger, A.; Kozlov, A.V. Biological Activities of Reactive Oxygen and Nitrogen Species: Oxidative Stress versus Signal Transduction. Biomolecules 2015, 5, 472–484. [Google Scholar] [CrossRef]
- Abu-Soud, H.M.; Camp, O.G.; Ramadoss, J.; Chatzicharalampous, C.; Kofinas, G.; Kofinas, J.D. Regulation of nitric oxide generation and consumption. Int. J. Biol. Sci. 2025, 21, 1097–1109. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a Stealthy Biological Oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef]
- Koppenol, W.H. Chemistry of peroxynitrite and its relevance to biological systems. Met Ions Biol Syst. 1999, 36:597-619. [PubMed]
- Makarov, S.V.; Horváth, A.K.; Makarova, A.S. Reactivity of Small Oxoacids of Sulfur. Molecules 2019, 24, 2768. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.; Merényi, G. The chemistry of peroxynitrite: implications for biological activity. Methods Enzymol. 2008, 436:49-61. [CrossRef] [PubMed]
- Radi, R.; Cassina, A.; Hodara, R.; Quijano, C.; Castro, L. Peroxynitrite reactions and formation in mitochondria. Free. Radic. Biol. Med. 2002, 33, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Pérez-Torres, I.; Soto, M.E.; Castrejón-Tellez, V.; Rubio-Ruiz, M.E.; Pech, L.M.; Guarner-Lans, V. Oxidative, Reductive, and Nitrosative Stress Effects on Epigenetics and on Posttranslational Modification of Enzymes in Cardiometabolic Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 719. [Google Scholar] [CrossRef]
- Alnajjar, K.S.; Sweasy, J.B. A new perspective on oxidation of DNA repair proteins and cancer. DNA Repair 2019, 76, 60–69. [Google Scholar] [CrossRef]
- Gilboa, R.; Zharkov, D.O.; Golan, G.; Fernandes, A.S.; Gerchman, S.E.; Matz, E.; Kycia, J.H.; Grollman, A.P.; Shoham, G. Structure of Formamidopyrimidine-DNA Glycosylase Covalently Complexed to DNA. J. Biol. Chem. 2002, 277, 19811–19816. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA glycosylases: in DNA repair and beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef]
- Rodrigues, F.P.; Pestana, C.R.; Polizello, A.C.; Pardo-Andreu, G.L.; Uyemura, S.A.; Santos, A.C.; Alberici, L.C.; da Silva, R.S.; Curti, C. Release of NO from a nitrosyl ruthenium complex through oxidation of mitochondrial NADH and effects on mitochondria. Nitric Oxide 2012, 26, 174–181. [Google Scholar] [CrossRef]
- Murray, J.; Taylor, S.W.; Zhang, B.; Ghosh, S.S.; Capaldi, R.A. Oxidative Damage to Mitochondrial Complex I Due to Peroxynitrite: identification of reactive tyrosines by mass spectrometry. J. Biol. Chem. 2003, 278, 37223–37230. [Google Scholar] [CrossRef]
- Belenichev, I.; Popazova, O.; Yadlovskyi, O.; Bukhtiyarova, N.; Ryzhenko, V.; Pavlov, S.; Oksenych, V.; Kamyshnyi, O. Possibility of Using NO Modulators for Pharmacocorrection of Endothelial Dysfunction After Prenatal Hypoxia. Pharmaceuticals 2025, 18, 106. [Google Scholar] [CrossRef]
- Popazova, O.; Belenichev, I.; Yadlovskyi, O.; Oksenych, V.; Kamyshnyi, A. Altered Blood Molecular Markers of Cardiovascular Function in Rats after Intrauterine Hypoxia and Drug Therapy. Curr. Issues Mol. Biol. 2023, 45, 8704–8715. [Google Scholar] [CrossRef] [PubMed]
- Narohan, M.V.; Bazhenova, L.K.; Kapranova, E.I.; Melnikova, E.V.; Belousova, N.A. Posthypoxic Dysfunction of the Cardiovascular System in Newborns. Curr. Issues Pediatr. 2007, 6, 42–46 [In Ukranian]. [Google Scholar]
- E Lipshultz, S.; Wilkinson, J.D.; E Messiah, S.; Miller, T.L. Clinical research directions in pediatric cardiology. Curr. Opin. Pediatr. 2009, 21, 585–593. [Google Scholar] [CrossRef]
- Giussani, D.A.; Niu, Y.; Herrera, E.A.; Richter, H.G.; Camm, E.J.; Thakor, A.S.; Kane, A.D.; Hansell, J.A.; Brain, K.L.; Skeffington, K.L.; Itani, N.; Wooding, F.B.; Cross, C.M.; Allison, B.J. Heart disease link to fetal hypoxia and oxidative stress. Adv Exp Med Biol. 2014, 814, 77–87. [Google Scholar] [CrossRef]
- Kornacki, J.; Gutaj, P.; Kalantarova, A.; Sibiak, R.; Jankowski, M.; Wender-Ozegowska, E. Endothelial Dysfunction in Pregnancy Complications. Biomedicines 2021, 9, 1756. [Google Scholar] [CrossRef]
- McElwain, C.J.; Tuboly, E.; McCarthy, F.P.; McCarthy, C.M. Mechanisms of Endothelial Dysfunction in Pre-eclampsia and Gestational Diabetes Mellitus: Windows Into Future Cardiometabolic Health? Front. Endocrinol. 2020, 11, 655. [Google Scholar] [CrossRef]
- Allbritton-King, J.D.; García-Cardeña, G. Endothelial cell dysfunction in cardiac disease: driver or consequence? Front. Cell Dev. Biol. 2023, 11, 1278166. [Google Scholar] [CrossRef]
- Zhang, C.; Guo, Y.; Yang, Y.; Du, Z.; Fan, Y.; Zhao, Y.; Yuan, S. Oxidative stress on vessels at the maternal-fetal interface for female reproductive system disorders: Update. Front. Endocrinol. 2023, 14, 1118121. [Google Scholar] [CrossRef] [PubMed]
- Paz, A.A.; Jiménez, T.A.; Ibarra-Gonzalez, J.; Astudillo-Maya, C.; Beñaldo, F.A.; Figueroa, E.G.; Llanos, A.J.; Gonzalez-Candia, A.; Herrera, E.A. Gestational hypoxia elicits long-term cardiovascular dysfunction in female guinea pigs. Life Sci. 2025, 361, 123282. [Google Scholar] [CrossRef]
- Zhao, M.; Lei, J.; Deng, F.; Zhao, C.; Xu, T.; Ji, B.; Fu, M.; Wang, X.; Sun, M.; Zhang, M.; et al. Gestational Hypoxia Impaired Endothelial Nitric Oxide Synthesis Via miR-155-5p/NADPH Oxidase/Reactive Oxygen Species Axis in Male Offspring Vessels. J. Am. Hear. Assoc. 2024, 13, e032079. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Drummond, G.R.; Sobey, C.G.; De Silva, T.M.; Kemp-Harper, B.K. The opposing roles of NO and oxidative stress in cardiovascular disease. Pharmacol. Res. 2017, 116, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Romero, M.; Shatanawi, A.; Alkilany, A.; Caldwell, R. Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br. J. Pharmacol. 2012, 165, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged Hypoxia Differentially Regulates Hypoxia-inducible Factor (HIF)-1α and HIF-2α Expression in Lung Epithelial Cells: implication of natural antisense HIF-1alpha. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Kurihara, T.; Westenskow, P.D.; Friedlander, M. Hypoxia-inducible factor (HIF)/vascular endothelial growth factor (VEGF) signaling in the retina. Adv. Exp. Med. Biol. 2014, 801:275-81. [CrossRef]
- Ziello, J.E.; Jovin, I.S.; Huang, Y. Hypoxia-Inducible Factor (HIF)-1 Regulatory Pathway and its Potential for Therapeutic Intervention in Malignancy and Ischemia. Yale J. Biol. Med. 2007, 80, 51–60. [Google Scholar]
- Bochenek, M.L.; Gogiraju, R.; Großmann, S.; Krug, J.; Orth, J.; Reyda, S.; Georgiadis, G.S.; Spronk, H.M.; Konstantinides, S.; Münzel, T.; et al. EPCR-PAR1 biased signaling regulates perfusion recovery and neovascularization in peripheral ischemia. J. Clin. Investig. 2022, 7, e157701. [Google Scholar] [CrossRef]
- Ireland, H.; Konstantoulas, C.; Cooper, J.; Hawe, E.; Humphries, S.; Mather, H.; Goodall, A.; Hogwood, J.; Juhanvague, I.; Yudkin, J.; et al. EPCR Ser219Gly: Elevated sEPCR, prothrombin F1+2, risk for coronary heart disease, and increased sEPCR shedding in vitro. Atherosclerosis 2005, 183, 283–292. [Google Scholar] [CrossRef]
- Arai, M.; Imai, H.; Koumura, T.; Yoshida, M.; Emoto, K.; Umeda, M.; Chiba, N.; Nakagawa, Y. Mitochondrial Phospholipid Hydroperoxide Glutathione Peroxidase Plays a Major Role in Preventing Oxidative Injury to Cells. J. Biol. Chem. 1999, 274, 4924–4933. [Google Scholar] [CrossRef]
- Roveri, A.; Casasco, A.; Maiorino, M.; Dalan, P.; Calligaro, A.; Ursini, F. Phospholipid hydroperoxide glutathione peroxidase of rat testis. Gonadotropin dependence and immunocytochemical identification. J. Biol. Chem. 1992, 267, 6142–6146. [Google Scholar] [CrossRef]
- Oh, S.-J.; Ikeda, M.; Ide, T.; Hur, K.Y.; Lee, M.-S. Mitochondrial event as an ultimate step in ferroptosis. Cell Death Discov. 2022, 8, 414. [Google Scholar] [CrossRef] [PubMed]
- Tian, P.; Xu, Z.; Guo, J.; Zhao, J.; Chen, W.; Huang, W.; Wang, M.; Mi, C.; Zhang, Y.; Yang, Y.; et al. Hypoxia causes trophoblast cell ferroptosis to induce miscarriage through lnc-HZ06/HIF1α-SUMO/NCOA4 axis. Redox Biol. 2024, 70, 103073. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxidants Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Forgione, M.A.; Weiss, N.; Heydrick, S.; Cap, A.; Klings, E.S.; Bierl, C.; Eberhardt, R.T.; Farber, H.W.; Loscalzo, J. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am. J. Physiol. Circ. Physiol. 2002, 282, H1255–H1261. [Google Scholar] [CrossRef]
- Kong, X.; Qiao, D.; Zhao, X.; Wang, L.; Zhang, J.; Liu, D.; Zhang, H. The molecular characterizations of Cu/ZnSOD and MnSOD and its responses of mRNA expression and enzyme activity to Aeromonas hydrophila or lipopolysaccharide challenge in Qihe crucian carp Carassius auratus. Fish Shellfish. Immunol. 2017, 67, 429–440. [Google Scholar] [CrossRef]
- Giles, B.-L.; Suliman, H.; Mamo, L.B.; Piantadosi, C.A.; Oury, T.D.; Nozik-Grayck, E. Prenatal hypoxia decreases lung extracellular superoxide dismutase expression and activity. Am. J. Physiol. Cell. Mol. Physiol. 2002, 283, L549–L554. [Google Scholar] [CrossRef]
- Sherlock, L.G.; Trumpie, A.; Hernandez-Lagunas, L.; McKenna, S.; Fisher, S.; Bowler, R.; Wright, C.J.; Delaney, C.; Nozik-Grayck, E. Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia. Antioxidants 2018, 7, 42. [Google Scholar] [CrossRef]
- Grzeszczak, K.; Łanocha-Arendarczyk, N.; Malinowski, W.; Ziętek, P.; Kosik-Bogacka, D. Oxidative Stress in Pregnancy. Biomolecules. 2023 Dec 9;13(12):1768. [CrossRef]
- Luo, Z.; Tian, M.; Yang, G.; Tan, Q.; Chen, Y.; Li, G.; Zhang, Q.; Li, Y.; Wan, P.; Wu, J. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal Transduct. Target. Ther. 2022, 7, 218. [Google Scholar] [CrossRef]
- Malhotra, R.; Lin, Z.; Vincenz, C.; Brosius, F.C. Hypoxia induces apoptosis via two independent pathways in Jurkat cells: differential regulation by glucose. Am. J. Physiol. Physiol. 2001, 281, C1596–C1603. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Kari, S.; Subramanian, K.; Altomonte, I.A.; Murugesan, A.; Yli-Harja, O.; Kandhavelu, M. Programmed cell death detection methods: a systematic review and a categorical comparison. Apoptosis 2022, 27, 482–508. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–16. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Ren, D.; Li, F.; Gao, A.; Cao, Q.; Liu, Y.; Zhang, J. RETRACTED ARTICLE: Hypoxia-induced apoptosis of cardiomyocytes is restricted by ginkgolide B-downregulated microRNA-29. Cell Cycle 2020, 19, 1067–1076. [Google Scholar] [CrossRef]
- Ren, D.; Li, F.; Gao, A.; Cao, Q.; Liu, Y.; Zhang, J. Statement of Retraction: Hypoxia-induced apoptosis of cardiomyocytes is restricted by ginkgolide B-downregulated microRNA-29. Cell Cycle 2022, 21, 757–757. [Google Scholar] [CrossRef]
- Qiu, Z.; Wang, Y.; Liu, W.; Li, C.; Zhao, R.; Long, X.; Rong, J.; Deng, W.; Shen, C.; Yuan, J.; et al. CircHIPK3 regulates the autophagy and apoptosis of hypoxia/reoxygenation-stimulated cardiomyocytes via the miR-20b-5p/ATG7 axis. Cell Death Discov. 2021, 7, 64. [Google Scholar] [CrossRef]
- Mustafa, M.; Ahmad, R.; Tantry, I.Q.; Ahmad, W.; Siddiqui, S.; Alam, M.; Abbas, K.; Moinuddin; Hassan, I.; Habib, S.; et al. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells 2024, 13, 1838. [Google Scholar] [CrossRef]
- Kholodenko, I.V.; Kholodenko, R.V.; Majouga, A.G.; Yarygin, K.N. Apoptotic MSCs and MSC-Derived Apoptotic Bodies as New Therapeutic Tools. Curr. Issues Mol. Biol. 2022, 44, 5153–5172. [Google Scholar] [CrossRef]
- Miller, M.A.; Zachary, J.F. Mechanisms and Morphology of Cellular Injury, Adaptation, and Death 11 For a glossary of abbreviations and terms used in this chapter see E-Glossary 1-1. Pathol. Basis Vet. Dis. 2017, 2–43.e19. [CrossRef]
- Geatrell, J.C.; Gan, P.M. (.; Mansergh, F.C.; Kisiswa, L.; Jarrin, M.; Williams, L.A.; Evans, M.J.; Boulton, M.E.; Wride, M.A. Apoptosis gene profiling reveals spatio-temporal regulated expression of the p53/Mdm2 pathway during lens development. Exp. Eye Res. 2009, 88, 1137–1151. [Google Scholar] [CrossRef]
- James, T.N. Normal and abnormal consequences of apoptosis in the human heart. From postnatal morphogenesis to paroxysmal arrhythmias. Circulation 1994, 90, 556–573. [Google Scholar] [CrossRef]
- Oh, Y.; Abid, R.; Dababneh, S.; Bakr, M.; Aslani, T.; Cook, D.P.; Vanderhyden, B.C.; Park, J.G.; Munshi, N.V.; Hui, C.-C.; et al. Transcriptional regulation of the postnatal cardiac conduction system heterogeneity. Nat. Commun. 2024, 15, 6550. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Indolfi, C.; Nadal-Ginard, B. Generation of new cardiomyocytes after injury: de novo formation from resident progenitors vs. replication of pre-existing cardiomyocytes. Ann Transl Med. 2015, 3, S8. [Google Scholar] [CrossRef] [PubMed]
- James, T.N. Normal and abnormal consequences of apoptosis in the human heart: from postnatal morphogenesis to paroxysmal arrhythmias. Trans Am Clin Climatol Assoc. 1994, 105, 145–178. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Deng, J.; Jiang, Y.; Chen, Z.B.; Rhee, J.-W.; Deng, Y.; Wang, Z.V. Mitochondrial Dysfunction in Cardiac Arrhythmias. Cells 2023, 12, 679. [Google Scholar] [CrossRef]
- Yehya, A.; Azar, J.; Al-Fares, M.; Boeuf, H.; Abou-Kheir, W.; Zeineddine, D.; Hadadeh, O. Cardiac differentiation is modulated by anti-apoptotic signals in murine embryonic stem cells. World J. Stem Cells 2024, 16, 551–559. [Google Scholar] [CrossRef]
- Bennett, M.R. APOPTOSIS IN THE CARDIOVASCULAR SYSTEM. Heart 2002, 87, 480–487. [Google Scholar] [CrossRef]
- Van Empel, V.P.; Bertrand, A.T.; Hofstra, L.; Crijns, H.J.; Doevendans, P.A.; De Windt, L.J. Myocyte apoptosis in heart failure. Cardiovasc. Res. 2005, 67, 21–29. [Google Scholar] [CrossRef]
- Kim, N.-H.; Kang, P.M. Apoptosis in Cardiovascular Diseases: Mechanism and Clinical Implications. Korean Circ. J. 2010, 40, 299–305. [Google Scholar] [CrossRef]
- Krijnen, P.A.J.; Nijmeijer, R.; Meijer, C.J.L.M.; A Visser, C.; E Hack, C.; Niessen, H.W.M. Apoptosis in myocardial ischaemia and infarction. J. Clin. Pathol. 2002, 55, 801–811. [Google Scholar] [CrossRef]
- Crow, M.T.; Mani, K.; Nam, Y.-J.; Kitsis, R.N. The Mitochondrial Death Pathway and Cardiac Myocyte Apoptosis. Circ. Res. 2004, 95, 957–970. [Google Scholar] [CrossRef]
- Koehler, R.C.; Yang, Z.-J.; Lee, J.K.; Martin, L.J. Perinatal hypoxic-ischemic brain injury in large animal models: Relevance to human neonatal encephalopathy. J. Cereb. Blood Flow Metab. 2018, 38, 2092–2111. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Yu, X.; Herndon, J.M.; Green, D.R.; A Ferguson, T. CD95-Induced Apoptosis of Lymphocytes in an Immune Privileged Site Induces Immunological Tolerance. Immunity 1996, 5, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Le Hingrat, Q.; Sereti, I.; Landay, A.L.; Pandrea, I.; Apetrei, C. The Hitchhiker Guide to CD4+ T-Cell Depletion in Lentiviral Infection. A Critical Review of the Dynamics of the CD4+ T Cells in SIV and HIV Infection. Front. Immunol. 2021, 12, 695674. [Google Scholar] [CrossRef]
- Bae, S.; Xiao, Y.; Li, G.; Casiano, C.A.; Zhang, L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am. J. Physiol. Circ. Physiol. 2003, 285, H983–H990. [Google Scholar] [CrossRef]
- Troy, C.; Derossi, D.; Prochiantz, A.; Greene, L.; Shelanski, M. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J. Neurosci. 1996, 16, 253–261. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef]
- Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia 2022, 2, 1624–1636. [Google Scholar] [CrossRef]
- Crimi, M.; Degli Esposti, M. Apoptosis-induced changes in mitochondrial lipids. Biochim. et Biophys. Acta (BBA) - Mol. Cell Res. 2011, 1813, 551–557. [Google Scholar] [CrossRef]
- Marí, M.; Morales, A.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Mitochondrial glutathione: Features, regulation and role in disease. Biochim. et Biophys. Acta (BBA) - Gen. Subj. 2013, 1830, 3317–3328. [Google Scholar] [CrossRef]
- Korotkov, S.M. Mitochondrial Oxidative Stress Is the General Reason for Apoptosis Induced by Different-Valence Heavy Metals in Cells and Mitochondria. Int. J. Mol. Sci. 2023, 24, 14459. [Google Scholar] [CrossRef] [PubMed]
- Taimor, G.; Hofstaetter, B.; Piper, H. Apoptosis induction by nitric oxide in adult cardiomyocytes via cGMP-signaling and its impairment after simulated ischemia. Cardiovasc. Res. 2000, 45, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Botting, K.J.; McMillen, I.C.; Forbes, H.; Nyengaard, J.R.; Morrison, J.L. Chronic Hypoxemia in Late Gestation Decreases Cardiomyocyte Number but Does Not Change Expression of Hypoxia-Responsive Genes. J. Am. Hear. Assoc. 2014, 3, e000531. [Google Scholar] [CrossRef]
- Mělková, Z.; Lee, S.B.; Rodriguez, D.; Esteban, M. Bcl-2 prevents nitric oxide-mediated apoptosis and poly(ADP-ribose) polymerase cleavage. FEBS Lett. 1997, 403, 273–278. [Google Scholar] [CrossRef]
- Gotoh, Y.; Cooper, J.A. Reactive Oxygen Species- and Dimerization-induced Activation of Apoptosis Signal-regulating Kinase 1 in Tumor Necrosis Factor-α Signal Transduction. J. Biol. Chem. 1998, 273, 17477–17482. [Google Scholar] [CrossRef]
- Mendelson, K.G.; Contois, L.-R.; Tevosian, S.G.; Davis, R.J.; Paulson, K.E. Independent regulation of JNK/p38 mitogen-activated protein kinases by metabolic oxidative stress in the liver. Proc. Natl. Acad. Sci. USA 1996, 93, 12908–12913. [Google Scholar] [CrossRef]
- Ing, D.J.; Zang, J.; Dzau, V.J.; Webster, K.A.; Bishopric, N.H. Modulation of Cytokine-Induced Cardiac Myocyte Apoptosis by Nitric Oxide, Bak, and Bcl-x. Circ. Res. 1999, 84, 21–33. [Google Scholar] [CrossRef]
- Yfantis, A.; Mylonis, I.; Chachami, G.; Nikolaidis, M.; Amoutzias, G.D.; Paraskeva, E.; Simos, G. Transcriptional Response to Hypoxia: The Role of HIF-1-Associated Co-Regulators. Cells 2023, 12, 798. [Google Scholar] [CrossRef]
- Taylor, C.T.; Scholz, C.C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 2022, 18, 573–587. [Google Scholar] [CrossRef]
- Jaśkiewicz, M.; Moszyńska, A.; Króliczewski, J.; Cabaj, A.; Bartoszewska, S.; Charzyńska, A.; Gebert, M.; Dąbrowski, M.; Collawn, J.F.; Bartoszewski, R. The transition from HIF-1 to HIF-2 during prolonged hypoxia results from reactivation of PHDs and HIF1A mRNA instability. Cell. Mol. Biol. Lett. 2022, 27, 109. [Google Scholar] [CrossRef]
- Vranesić-Bender, D. The role of nutraceuticals in anti-aging medicine. Acta Clin Croat. 2010, 49, 537–44. [Google Scholar] [PubMed]
- Xu, L.; Song, H.; Qiu, Q.; Jiang, T.; Ge, P.; Su, Z.; Ma, W.; Zhang, R.; Huang, C.; Li, S.; et al. Different Expressions of HIF-1α and Metabolism in Brain and Major Visceral Organs of Acute Hypoxic Mice. Int. J. Mol. Sci. 2021, 22, 6705. [Google Scholar] [CrossRef] [PubMed]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: a novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Ke, Q.; Costa, M. Hypoxia-inducible factor-1 (HIF-1). Mol. Pharmacol. 2006, 70, 1469–1480. [Google Scholar] [CrossRef]
- Madai, S.; Kilic, P.; Schmidt, R.M.; Bas-Orth, C.; Korff, T.; Büttner, M.; Klinke, G.; Poschet, G.; Marti, H.H.; Kunze, R. Activation of the hypoxia-inducible factor pathway protects against acute ischemic stroke by reprogramming central carbon metabolism. Theranostics 2024, 14, 2856–2880. [Google Scholar] [CrossRef]
- Mijatović, S.; Savić-Radojević, A.; Plješa-Ercegovac, M.; Simić, T.; Nicoletti, F.; Maksimović-Ivanić, D. The Double-Faced Role of Nitric Oxide and Reactive Oxygen Species in Solid Tumors. Antioxidants 2020, 9, 374. [Google Scholar] [CrossRef]
- Brocato, J.; Chervona, Y.; Costa, M. Molecular Responses to Hypoxia-Inducible Factor 1α and Beyond. Mol. Pharmacol. 2014, 85, 651–657. [Google Scholar] [CrossRef]
- McGettrick, A.F.; O’neill, L.A. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Zhang, P.; Yao, Q.; Lu, L.; Li, Y.; Chen, P.-J.; Duan, C. Hypoxia-Inducible Factor 3 Is an Oxygen-Dependent Transcription Activator and Regulates a Distinct Transcriptional Response to Hypoxia. Cell Rep. 2014, 6, 1110–1121. [Google Scholar] [CrossRef]
- Zhang, J.; Yao, M.; Xia, S.; Zeng, F.; Liu, Q. Systematic and comprehensive insights into HIF-1 stabilization under normoxic conditions: implications for cellular adaptation and therapeutic strategies in cancer. Cell. Mol. Biol. Lett. 2025, 30, 2. [Google Scholar] [CrossRef]
- Toth, R.K.; Warfel, N.A. Strange Bedfellows: Nuclear Factor, Erythroid 2-Like 2 (Nrf2) and Hypoxia-Inducible Factor 1 (HIF-1) in Tumor Hypoxia. Antioxidants (Basel) 2017, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.-Y.; Wang, J.-Q.; Liu, C.; Gao, X.-M.; Zhang, Y.-B.; Ding, J.; Hou, C.-C.; Zhu, J.-Q.; Lou, B.; Shen, W.-L.; et al. Hif-1α/Hsf1/Hsp70 signaling pathway regulates redox homeostasis and apoptosis in large yellow croaker (Larimichthys crocea) under environmental hypoxia. Zool. Res. 2021, 42, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Aliyeva, O.G.; Popazova, O.O.; Bukhtiyarova, N.V. Involvement of heat shock proteins HSP70 in the mechanisms of endogenous neuroprotection: the prospect of using HSP70 modulators. Front. Cell. Neurosci. 2023, 17, 1131683. [Google Scholar] [CrossRef]
- Agani, F.H.; Puchowicz, M.; Chavez, J.C.; Pichiule, P.; LaManna, J. Role of nitric oxide in the regulation of HIF-1α expression during hypoxia. Am. J. Physiol. Physiol. 2002, 283, C178–C186. [Google Scholar] [CrossRef]
- Sandau, K.B.; Fandrey, J.; Brüne, B. Accumulation of HIF-1α under the influence of nitric oxide. Blood 2001, 97, 1009–1015. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Okazaki, K.; Nakamura, S.; Koyano, K.; Konishi, Y.; Kondo, M.; Kusaka, T. Neonatal asphyxia as an inflammatory disease: Reactive oxygen species and cytokines. Front. Pediatr. 2023, 11, 1070743. [Google Scholar] [CrossRef]
- Serdar, M.; Walther, K.-A.; Gallert, M.; Kempe, K.; Obst, S.; Labusek, N.; Herrmann, R.; Herz, J.; Felderhoff-Müser, U.; Bendix, I. Prenatal inflammation exacerbates hyperoxia-induced neonatal brain injury. J. Neuroinflammation 2025, 22, 57. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Manik, M.; Singh, R.K. Role of toll-like receptors in modulation of cytokine storm signaling in SARS-CoV-2-induced COVID-19. J. Med Virol. 2022, 94, 869–877. [Google Scholar] [CrossRef]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The interplay between cytokines, inflammation, and antioxidants: mechanistic insights and therapeutic potentials of various antioxidants and anti-cytokine compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.C.; Cavadas, M.A.S.; Tambuwala, M.M.; Hams, E.; Rodríguez, J.; von Kriegsheim, A.; Cotter, P.; Bruning, U.; Fallon, P.G.; Cheong, A.; et al. Regulation of IL-1β–induced NF-κB by hydroxylases links key hypoxic and inflammatory signaling pathways. Proc. Natl. Acad. Sci. 2013, 110, 18490–18495. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-κB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701 https://. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Brüne, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; Von Knethen, A.; Weigert, A. Redox Control of Inflammation in Macrophages. Antioxid. Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef]
- D’oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxidative Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Stetler, R.A.; Gan, Y.; Zhang, W.; Liou, A.K.; Gao, Y.; Cao, G.; Chen, J. Heat shock proteins: Cellular and molecular mechanisms in the central nervous system. Prog. Neurobiol. 2010, 92, 184–211. [Google Scholar] [CrossRef]
- Baird, N.A.; Turnbull, D.W.; Johnson, E.A. Induction of the Heat Shock Pathway during Hypoxia Requires Regulation of Heat Shock Factor by Hypoxia-inducible Factor-1. J. Biol. Chem. 2006, 281, 38675–38681. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Cherniy, V.I.; Nagornaya, E.A.; Bukhtiyarova, N.V.; Kucherenko, V.I. Neuroprotection and neuroplasticity. Kiev: Logos,2015, 510.
- Belenichev, I.F.; Kolesnik, Y.M.; Pavlov, S.V.; Sokolik, E.P.; Bukhtiyarova, N.V. Disturbance of HSP70 chaperone activity is a possible mechanism of mitochondrial dysfunction. Neurochem. J. 2011, 5, 251–256. [Google Scholar] [CrossRef]
- Hu, C.; Yang, J.; Qi, Z.; Wu, H.; Wang, B.; Zou, F.; Mei, H.; Liu, J.; Wang, W.; Liu, Q. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. Medcomm 2022, 3, e161. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, J.W.; Yenari, M.A. Heat shock protein signaling in brain ischemia and injury. Neurosci. Lett. 2020, 715, 134642–134642. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, Y.; Bao, E.; Tang, S. The Protective Role of Heat Shock Proteins against Stresses in Animal Breeding. Int. J. Mol. Sci. 2024, 25, 8208. [Google Scholar] [CrossRef] [PubMed]
- Kiang, J. Heat Shock Protein 70 kDa Molecular Biology, Biochemistry, and Physiology. Pharmacol. Ther. 1998, 80, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.K.; Shin, Y.; Ju, S.; Han, S.; Choe, W.; Yoon, K.-S.; Kim, S.S.; Kang, I. Heat Shock Response and Heat Shock Proteins: Current Understanding and Future Opportunities in Human Diseases. Int. J. Mol. Sci. 2024, 25, 4209. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Cheng, S.-Y.; Liu, C.-H.; Tsai, W.-C.; Wu, H.-H.; Huang, M.-D. Exploration of the truncated cytosolic Hsp70 in plants - unveiling the diverse T1 lineage and the conserved T2 lineage. Front. Plant Sci. 2023, 14, 1279540. [Google Scholar] [CrossRef]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Kudryavtseva, S.S.; Leisi, E.V.; Kurochkina, L.P.; Barinova, K.V.; Schmalhausen, E.V. Regulation by Different Types of Chaperones of Amyloid Transformation of Proteins Involved in the Development of Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 2747. [Google Scholar] [CrossRef]
- Storey, J.M.; Storey, K.B. Chaperone proteins: universal roles in surviving environmental stress. Cell Stress Chaperon- 2023, 28, 455–466. [Google Scholar] [CrossRef]
- Díaz-Villanueva, J.F.; Díaz-Molina, R.; García-González, V. Protein Folding and Mechanisms of Proteostasis. Int. J. Mol. Sci. 2015, 16, 17193–17230. [Google Scholar] [CrossRef]
- Verghese, J.; Abrams, J.; Wang, Y.; Morano, K.A. Biology of the Heat Shock Response and Protein Chaperones: Budding Yeast (Saccharomyces cerevisiae) as a Model System. Microbiol. Mol. Biol. Rev. 2012, 76, 115–158. [Google Scholar] [CrossRef]
- Hu, S.; Tan, J.; Qin, L.; Lv, L.; Yan, W.; Zhang, H.; Tang, B.; Wang, C. Molecular chaperones and Parkinson's disease. Neurobiol. Dis. 2021, 160, 105527. [Google Scholar] [CrossRef] [PubMed]
- Serlidaki, D.; van Waarde, M.A.W.; Rohland, L.; Wentink, A.S.; Dekker, S.L.; Kamphuis, M.J.; Boertien, J.M.; Brunsting, J.F.; Nillegoda, N.B.; Bukau, B.; et al. Functional diversity between HSP70 paralogs caused by variable interactions with specific co-chaperones. J. Biol. Chem. 2020, 295, 7301–7316. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic Reticulum Chaperone Protein GRP78 Protects Cells from Apoptosis Induced by Topoisomerase Inhibitors: Role Of ATP Binding Site In Suppression Of Caspase-7 Activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed]
- Beere, H.M.; Wolf, B.B.; Cain, K.; Mosser, D.D.; Mahboubi, A.; Kuwana, T.; Tailor, P.; Morimoto, R.I.; Cohen, G.M.; Green, D.R. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000, 2, 469–475. [Google Scholar] [CrossRef]
- Zia, A.; Pourbagher-Shahri, A.M.; Farkhondeh, T.; Samarghandian, S. Molecular and cellular pathways contributing to brain aging. Behav. Brain Funct. 2021, 17, 1–30. [Google Scholar] [CrossRef]
- Nagai, M.; Kaji, H. Thermal Effect on Heat Shock Protein 70 Family to Prevent Atherosclerotic Cardiovascular Disease. Biomolecules 2023, 13, 867. [Google Scholar] [CrossRef]
- Niinuma, S.A.; Lubbad, L.; Lubbad, W.; Moin, A.S.M.; Butler, A.E. The Role of Heat Shock Proteins in the Pathogenesis of Polycystic Ovarian Syndrome: A Review of the Literature. Int. J. Mol. Sci. 2023, 24, 1838. [Google Scholar] [CrossRef]
- Evgen'Ev, M.B.; Onikienko, S.B.; Chuvakova, L.N.; Garbuz, D.G.; Zatsepina, O.G. The Role of Hsp70 in Adaptation to Adverse Conditions and Its Possible Medical Application. Front. Biosci. 2023, 28, 25. [Google Scholar] [CrossRef]
- Hulina, A.; Rajković, M.G.; Despot, D.J.; Jelić, D.; Dojder, A.; Čepelak, I.; Rumora, L. Extracellular Hsp70 induces inflammation and modulates LPS/LTA-stimulated inflammatory response in THP-1 cells. Cell Stress Chaperon- 2018, 23, 373–384. [Google Scholar] [CrossRef]
- nbsp; Alberti, G.; Paladino, L.; Vitale, A.; Caruso Bavisotto, C.; Conway de Macario, E.; Campanella, C.; Macario, A.; Marino Gammazza, A. Functions and Therapeutic Potential of Extracellular Hsp60, Hsp70, and Hsp90 in Neuroinflammatory Disorders. Appl. Sci. 2021, 11, 736. [Google Scholar] [CrossRef]
- Shevtsov, M.A.; Komarova, E.Y.; Meshalkina, D.A.; Bychkova, N.V.; Aksenov, N.D.; Abkin, S.V.; Margulis, B.A.; Guzhova, I.V. Exogenously delivered heat shock protein 70 displaces its endogenous analogue and sensitizes cancer cells to lymphocytes-mediated cytotoxicity. Oncotarget 2014, 5, 3101–3114. [Google Scholar] [CrossRef] [PubMed]
- Demyanenko, S.V.; Kalyuzhnaya, Y.N.; Bachurin, S.S.; Khaitin, A.M.; Kunitsyna, A.E.; Batalshchikova, S.A.; Evgen'Ev, M.B.; Garbuz, D.G. Exogenous Hsp70 exerts neuroprotective effects in peripheral nerve rupture model. Exp. Neurol. 2024, 373, 114670. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.; Ryzhenko, V.; Popazova, O.; Bukhtiyarova, N.; Gorchakova, N.; Oksenych, V.; Kamyshnyi, O. Optimization of the Search for Neuroprotectors among Bioflavonoids. Pharmaceuticals 2024, 17, 877. [Google Scholar] [CrossRef] [PubMed]
- Aliyeva, O.; Belenichev, I.; Bukhtiyarova, N.; Semenov, D.; Voloshchuk, S. Comparative Assessment of the Effectiveness of HSP70 / HIF-1α System Modulators after Prenatal Hypoxia. Biomed. Pharmacol. J. 2024, 17, 223–233. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Aliyeva, E.G.; Kamyshny, O.M.; Bukhtiyarova, N.V.; Ryzhenko, V.P.; Gorchakova, N.O. Pharmacological Modulation of Endogenous Neuroprotection after Experimental Prenatal Hypoxia. Neurochem. J. 2022, 16, 68–75. [Google Scholar] [CrossRef]
- Cao, J.; Yang, L.; Wang, L.; Zhao, Q.; Wu, D.; Li, M.; Mu, Y. Heat shock protein 70 attenuates hypoxia-induced apoptosis of pulmonary microvascular endothelial cells isolated from neonatal rats. Mol. Med. Rep. 2021, 24, 690. [Google Scholar] [CrossRef]
- Fu, H.; Li, Y.; Tian, J.; Yang, B.; Li, Y.; Li, Q.; Liu, S. Contribution of HIF-1α to Heat Shock Response by Transcriptional Regulation of HSF1/HSP70 Signaling Pathway in Pacific Oyster, Crassostrea gigas. Mar. Biotechnol. 2023, 25, 691–700. [Google Scholar] [CrossRef]
- Ikwegbue, P.C.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Roles of Heat Shock Proteins in Apoptosis, Oxidative Stress, Human Inflammatory Diseases, and Cancer. Pharmaceuticals 2017, 11, 2. [Google Scholar] [CrossRef]
- Sulzbacher, M.M.; Ludwig, M.S.; Heck, T.G. Oxidative stress and decreased tissue HSP70 are involved in the genesis of sepsis: HSP70 as a therapeutic target. Rev. Bras. de Ter. Intensiv. 2020, 32, 585–591. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive Oxygen Species: Drivers of Physiological and Pathological Processes. J. Inflamm. Res. 2020, ume 13, 1057–1073. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Simoes, I.C.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria And Reactive Oxygen Species In Aging And Age-Related Diseases. Int Rev Cell Mol Biol. 2018; 340:209-344. [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; White, C.W.; Chang, L.-Y.; Schneider, B.K.; Allen, C.B. Glutamine protects mitochondrial structure and function in oxygen toxicity. Am. J. Physiol. Cell. Mol. Physiol. 2001, 280, L779–L791. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Testa, G.; Ravagna, A.; Bates, T.E.; Stella, A.M.G. HSP70 Induction in the Brain Following Ethanol Administration in the Rat: Regulation by Glutathione Redox State. Biochem. Biophys. Res. Commun. 2000, 269, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Kaur, G. HSP70 induction and oxidative stress protection mediated by a subtoxic dose of NMDA in the retinoic acid-differentiated C6 glioma cell line. Brain Res. Bull. 2006, 69, 37–47. [Google Scholar] [CrossRef]
- Kawana, K.-I.; Miyamoto, Y.; Tanonaka, K.; Han-No, Y.; Yoshida, H.; Takahashi, M.; Takeo, S. Cytoprotective Mechanism of Heat Shock Protein 70 against Hypoxia/Reoxygenation Injury. J. Mol. Cell. Cardiol. 2000, 32, 2229–2237. [Google Scholar] [CrossRef]
- Zhang, H.; Gong, W.; Wu, S.; Perrett, S. Hsp70 in Redox Homeostasis. Cells 2022, 11, 829. [Google Scholar] [CrossRef]
- Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells 2020, 9, 2020. [Google Scholar] [CrossRef]
- Guo, S.; Wharton, W.; Moseley, P.; Shi, H. Heat shock protein 70 regulates cellular redox status by modulating glutathione-related enzyme activities. Cell Stress Chaperon- 2007, 12, 245–254. [Google Scholar] [CrossRef]
- Chen, S.; Wu, S.; Lin, B. The potential therapeutic value of the natural plant compounds matrine and oxymatrine in cardiovascular diseases. Front. Cardiovasc. Med. 2024, 11, 1417672. [Google Scholar] [CrossRef]
- Urban-Chmiel, R.; Puchalski, A.; Wernicki, A.; Dec, M.; Paluch, E. Characterization of Hsp70 proteins in bovine leukocytes induced by the temperature 41 degrees C. Pol J Vet Sci. 2009, 12, 323–8. [Google Scholar]
- Ferreira-Cravo, M.; Moreira, D.C.; Hermes-Lima, M. Glutathione Depletion Disrupts Redox Homeostasis in an Anoxia-Tolerant Invertebrate. Antioxidants 2023, 12, 1197. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Kolesnik, Y.M.; Pavlov, S.V.; Sokolik, E.P.; Bukhtiyarova, N.V. Malate-aspartate shunt in neuronal adaptation to ischemic conditions: Molecular-biochemical mechanisms of activation and regulation. Neurochem. J. 2012, 6, 22–28. [Google Scholar] [CrossRef]
- Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Makyeyeva, L.; Morozova, O.; Oksenych, V.; Kamyshnyi, O. Methodological Approaches to Experimental Evaluation of Neuroprotective Action of Potential Drugs. Int. J. Mol. Sci. 2024, 25, 10475. [Google Scholar] [CrossRef]
- Vanin, A.F. Dinitrosyl iron complexes with thiol-containing ligands as a “working form” of endogenous nitric oxide. Nitric Oxide 2016, 54, 15–29. [Google Scholar] [CrossRef]
- Henard, C.A.; Vázquez-Torres, A. Nitric Oxide and Salmonella Pathogenesis. Front. Microbiol. 2011, 2, 84. [Google Scholar] [CrossRef]
- Thompho, S.; Fritzsche, S.; Chokbunpiam, T.; Remsungnen, T.; Janke, W.; Hannongbua, S. Adsorption and the Chemical Reaction N2O4 ↔ 2NO2 in the Presence of N2 in a Gas Phase Connected with a Carbon Nanotube. ACS Omega 2021, 6, 17342–17352. [Google Scholar] [CrossRef]
- Möller, M.N.; Vitturi, D.A. The chemical biology of dinitrogen trioxide. Redox Biochem. Chem. 2024, 8. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Shah, F.; Chekman, I.S.; Nagornaya, E.A.; Gorbacheva, S.V.; Gorchakova, N.A.; Bukhtiyarova, N.V. Thiol-Disulfide System: Role In Endogenous Cyto- And Organoprotection, Pathways Of Pharmacological Modulation. Kyiv: Yuston Publishing House, 2020, 232.
- Granne, I.; Southcombe, J.H.; Snider, J.V.; Tannetta, D.S.; Child, T.; Redman, C.W.G.; Sargent, I.L. ST2 and IL-33 in Pregnancy and Pre-Eclampsia. PLOS ONE 2011, 6, e24463. [Google Scholar] [CrossRef]
- Shah, A.; Reyes, L.M.; Morton, J.S.; Fung, D.; Schneider, J.; Davidge, S.T. Effect of resveratrol on metabolic and cardiovascular function in male and female adult offspring exposed to prenatal hypoxia and a high-fat diet. J. Physiol. 2016, 594, 1465–1482. [Google Scholar] [CrossRef]
- Qian, S.; Chen, G.; Li, R.; Ma, Y.; Pan, L.; Wang, X.; Wang, X. Disulfide stress and its role in cardiovascular diseases. Redox Biol. 2024, 75, 103297. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative Stress in Cardiovascular Diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef] [PubMed]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, P.; Krych, S.; Kramkowski, K.; Jęczmyk, A.; Hrapkowicz, T. Effect of Oxidative Stress on Mitochondrial Damage and Repair in Heart Disease and Ischemic Events. Int. J. Mol. Sci. 2024, 25, 12467. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tian, Y.; Liu, N.; Chen, Y.; Chen, X.; Yuan, G.; Chang, A.; Chang, X.; Wu, J.; Zhou, H. Mitochondrial Stress as a Central Player in the Pathogenesis of Hypoxia-Related Myocardial Dysfunction: New Insights. Int. J. Med Sci. 2024, 21, 2502–2509. [Google Scholar] [CrossRef]
- Guo, Z.; Tian, Y.; Gao, J.; Zhou, B.; Zhou, X.; Chang, X.; Zhou, H. Enhancement of Mitochondrial Homeostasis: A Novel Approach to Attenuate Hypoxic Myocardial Injury. Int. J. Med Sci. 2024, 21, 2897–2911. [Google Scholar] [CrossRef]
- Song, H.; Polster, B.M.; Thompson, L.P. Chronic hypoxia alters cardiac mitochondrial complex protein expression and activity in fetal guinea pigs in a sex-selective manner. Am. J. Physiol. Integr. Comp. Physiol. 2021, 321, R912–R924. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; Clair, D.S.; Batinic-Haberle, I. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2012, 1822, 794–814. [Google Scholar] [CrossRef]
- Yuen, R.K.; Chen, B.; Blair, J.D.; Robinson, W.P.; Nelson, D.M. Hypoxia alters the epigenetic profile in cultured human placental trophoblasts. Epigenetics 2013, 8, 192–202. [Google Scholar] [CrossRef]
- Yan, X.; Fang, Y.; Yuan, Y.; Ding, Y.; Yu, H.; Li, Y.; Shi, Q.; Gao, Y.; Zhou, X.; Zhang, D.; et al. Combined analysis of the effects of hypoxia and oxidative stress on DNA methylation and the transcriptome in HTR-8/SVneo trophoblast cells. J. Cell. Mol. Med. 2024, 28, e18469. [Google Scholar] [CrossRef]
- Leonard, M.O.; Cottell, D.C.; Godson, C.; Brady, H.R.; Taylor, C.T. The Role of HIF-1α in Transcriptional Regulation of the Proximal Tubular Epithelial Cell Response to Hypoxia. J. Biol. Chem. 2003, 278, 40296–40304. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, E.N.; Ciriolo, M.R.; Ciccarone, F. Hypoxia-Induced Reactive Oxygen Species: Their Role in Cancer Resistance and Emerging Therapies to Overcome It. Antioxidants 2025, 14, 94. [Google Scholar] [CrossRef] [PubMed]
- Giussani, D.A. The fetal brain sparing response to hypoxia: physiological mechanisms. J. Physiol. 2016, 594, 1215–1230. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yu, X.; Liu, Y.; Lu, L.; Zhu, D.; Zhang, Y.; Li, L.; Zhang, P.; Gao, Q.; Lu, X.; et al. Prenatal hypothyroidism diminished exogenous NO-mediated diastolic effects in fetal rat thoracic aorta smooth muscle via increased oxidative stress. Reprod. Toxicol. 2022, 113, 52–61. [Google Scholar] [CrossRef]
- Korge, P.; Ping, P.; Weiss, J.N. Reactive Oxygen Species Production in Energized Cardiac Mitochondria During Hypoxia/Reoxygenation: Modulation By Nitric Oxide. Circ. Res. 2008, 103, 873–880. [Google Scholar] [CrossRef]
- Spiess, B.D. Perfluorocarbon Emulsions, Platelet Counts, and Inflammation. Shock 2019, 52, 13–18. [Google Scholar] [CrossRef]
- Gale, S.C.; Gorman, G.D.; Copeland, J.G.; McDonagh, P.F. Perflubron Emulsion Prevents PMN Activation and Improves Myocardial Functional Recovery After Cold Ischemia and Reperfusion. J. Surg. Res. 2007, 138, 135–140. [Google Scholar] [CrossRef]
- Krafft, M.; Riess, J. Corrigendum to “Therapeutic oxygen delivery by perfluorocarbon-based colloids'” [Advances in Colloid and Interface Science 294 (2021) 102407]. Adv. Colloid Interface Sci. 2021, 295, 102505. [Google Scholar] [CrossRef]
- Janjua, B.M.; Makepeace, C.M.; Anastacio, M.M.; Schuessler, R.B.; Nichols, C.G.; Lawton, J.S. Cardioprotective Benefits of Adenosine Triphosphate-Sensitive Potassium Channel Opener Diazoxide Are Lost with Administration after the Onset of Stress in Mouse and Human Myocytes. J. Am. Coll. Surg. 2014, 219, 803–813. [Google Scholar] [CrossRef]
- Ahmad, T.; Wang, J.; Velez, A.K.; Suarez-Pierre, A.; Clement, K.C.; Dong, J.; Sebestyen, K.; Canner, J.K.; Murphy, M.P.; Lawton, J.S. Cardioprotective mechanisms of mitochondria-targeted S-nitrosating agent and adenosine triphosphate-sensitive potassium channel opener are mutually exclusive. JTCVS Open 2021, 8, 338–354. [Google Scholar] [CrossRef]
- Larrieu, A.; Yazdanfar, S.; Redovan, E.; Eftychiadis, A.; Kao, R.; Silver, J.; Ghosh, S. Beneficial-effects of cocarboxylase in the treatment of experimental myocardial-infarction IN dogs. Am Surg. 1987, 53, 721–725. [Google Scholar] [PubMed]
- Bondarenko, I.P. Effect of riboxin on hemodynamics and microcirculation in patients with chronic ischemic heart disease. Vrach Delo. 1983, 15–8. [Google Scholar]
- Dunaev, V.V.; Tishkin, V.S.; Evdokimov, E.I.; Belaĭ, I.M. The Mechanism Of Action Of Riboxin. Farmakol Toksikol. 1989, Nov-Dec; 52(6):56-58. Russian. [PubMed]
- Mazur, I.A.; Chekman, I.S.; Belenichev, I.F. Metabolitotropnye Preparaty. Zaporozhye, 2007, 309 p ISBN 5-225-04144-2. https://www.researchgate.net/publication/326369488_Metabolitotropnye_preparaty.
- Novikov, V.E.; Levchenkova, O.S.; Ivantsova, E.N.; Vorobieva, V.V.; Егoрoвич, Н.В.; Сергеевна, Л.О.; Никoлаевна, И.Е.; Владимирoвна, В.В. Mitochondrial dysfunctions and antihypoxants. Rev. Clin. Pharmacol. Drug Ther. 2019, 17, 31–42. [Google Scholar] [CrossRef]
- Pospelova, M.L.; Barnaulov, O.D. The antihypoxant and antioxidant effects of medicinal plants as the basis for their use in destructive diseases of the brain. Hum. Physiol. 2000, 26, 86–91. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Domann, F.E. Regulatory crosstalk and interference between the xenobiotic and hypoxia sensing pathways at the AhR-ARNT-HIF1α signaling node. Chem. Interactions 2014, 218, 82–88. [Google Scholar] [CrossRef]
- Dow, L.F.; Case, A.M.; Paustian, M.P.; Pinkerton, B.R.; Simeon, P.; Trippier, P.C. The evolution of small molecule enzyme activators. RSC Med. Chem. 2023, 14, 2206–2230. [Google Scholar] [CrossRef]
- Weinberg, J.; Liu, K.H.; Lee, C.-M.; Crandall, W.J.; Cuevas, A.R.; Druzak, S.A.; Morgan, E.T.; Jarrell, Z.R.; Ortlund, E.A.; Martin, G.S.; et al. Mammalian hydroxylation of microbiome-derived obesogen, delta-valerobetaine, to homocarnitine, a 5-carbon carnitine analog. J. Biol. Chem. 2025, 301, 108074. [Google Scholar] [CrossRef]
- Stoyanova, S.; Bogdanov, M.G. Rational Design, Synthesis, and In Vitro Activity of Heterocyclic Gamma-Butyrobetaines as Potential Carnitine Acetyltransferase Inhibitors. Molecules 2025, 30, 735. [Google Scholar] [CrossRef]
- Gerashchenko, A.D.; Pozdnyakov, D.I.; Voronkov, A.V. Study of dose-dependent actoprotective effect of ATACL on physical performancend psychoemotional status of animals under exhausting exercise. Res. Results Pharmacol. 2022, 8, 13–22. [Google Scholar] [CrossRef]
- Liepinsh, E.; Vilskersts, R.; Skapare, E.; Svalbe, B.; Kuka, J.; Cirule, H.; Pugovics, O.; Kalvinsh, I.; Dambrova, M. Mildronate decreases carnitine availability and up-regulates glucose uptake and related gene expression in the mouse heart. Life Sci. 2008, 83, 613–619. [Google Scholar] [CrossRef]
- Grigoryan, S.V.; Hazarapetyan, L.G.; Stepanyan, A.A. An Experience of Meldonium Use in Patients with Ventricular Arrhythmias of Ischemic Genesis. Kardiologiia 2019, 59, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Sjakste, N.; Gutcaits, A.; Kalvinsh, I. Mildronate: An Antiischemic Drug for Neurological Indications. CNS Drug Rev. 2005, 11, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Bellman, V. Unlocking the Potential of Meldonium: From Performance Enhancement to Therapeutic Insights. Psychoactives 2024, 3, 235–247. [Google Scholar] [CrossRef]
- Vilskersts, R.; Kigitovica, D.; Korzh, S.; Videja, M.; Vilks, K.; Cirule, H.; Skride, A.; Makrecka-Kuka, M.; Liepinsh, E.; Dambrova, M. Protective Effects of Meldonium in Experimental Models of Cardiovascular Complications with a Potential Application in COVID-19. Int. J. Mol. Sci. 2022, 23, 45. [Google Scholar] [CrossRef]
- Dambrova, M.; Makrecka-Kuka, M.; Vilskersts, R.; Makarova, E.; Kuka, J.; Liepinsh, E. Pharmacological effects of meldonium: Biochemical mechanisms and biomarkers of cardiometabolic activity. Pharmacol. Res. 2016, 113, 771–780. [Google Scholar] [CrossRef]
- Zarubina, I.V.; Lukk, M.V.; Shabanov, P.D. Antihypoxic and Antioxidant Effects of Exogenous Succinic Acid and Aminothiol Succinate-Containing Antihypoxants. Bull. Exp. Biol. Med. 2012, 153, 336–339. [Google Scholar] [CrossRef]
- Zhao, F.; Li, R.; Liu, Y.; Chen, H. Perspectives on lecithin from egg yolk: Extraction, physicochemical properties, modification, and applications. Front. Nutr. 2023, 9, 1082671. [Google Scholar] [CrossRef]
- Tanaka-Kanegae, R.; Kimura, H.; Hamada, K. Oral Administration of Egg- and Soy-Derived Lysophosphatidylcholine Mitigated Acetylcholine Depletion in the Brain of Scopolamine-Treated Rats. Nutrients 2023, 15, 3618. [Google Scholar] [CrossRef]
- Onaolapo, M.C.; Alabi, O.D.; Akano, O.P.; Olateju, B.S.; Okeleji, L.O.; Adeyemi, W.J.; Ajayi, A.F. Lecithin and cardiovascular health: a comprehensive review. Egypt. Hear. J. 2024, 76, 92. [Google Scholar] [CrossRef]
- Tuteja, N.; Chandra, M.; Tuteja, R.; Misra, M.K. Nitric Oxide as a Unique Bioactive Signaling Messenger in Physiology and Pathophysiology. BioMed Res. Int. 2004, 2004, 227–237. [Google Scholar] [CrossRef]
- Cannavo, A.; Koch, W.J. GRK2 as negative modulator of NO bioavailability: Implications for cardiovascular disease. Cell. Signal. 2018, 41, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jiang, Q.; Zhang, X.; Zhu, X.; Dong, X.; Shen, L.; Zhang, S.; Niu, L.; Chen, L.; Zhang, M.; et al. l-Arginine Alleviates LPS-Induced Oxidative Stress and Apoptosis via Activating SIRT1-AKT-Nrf2 and SIRT1-FOXO3a Signaling Pathways in C2C12 Myotube Cells. Antioxidants 2021, 10, 1957. [Google Scholar] [CrossRef] [PubMed]
- Bielenichev, I.F.; Vіzіr, V.A.; Mamchur, V.Y.; Kuriata, O.V. Place of tiotriazoline in the gallery of modern metabolitotropic medicines. Zaporozhye Med J. 2019, 21. [Google Scholar] [CrossRef]
- Gureev, A.P.; Nesterova, V.V.; Babenkova, P.I.; Ivanov, M.E.; Plotnikov, E.Y.; Silachev, D.N. L-Carnitine and Mildronate Demonstrate Divergent Protective Effects on Mitochondrial DNA Quality Control and Inflammation Following Traumatic Brain Injury. Int. J. Mol. Sci. 2025, 26, 2902. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Mazur, I.A.; Chekman, I.S.; et al. Report on Preclinical Study of Specific Biological Activity (Anti-Ischemic, Endotelioprotective) of the "Liziniy" Preparation upon Parenteral Administration. Zaporizhzhya, 2011, 87 p.
- Belenichev, I.F.; Mazur, I.A.; Chekman, I.S.; et al. Report on Preclinical Study of Specific Pharmacological Activity (Cardioprotective, Neuroprotective, Endotelioprotective) of "Angiolin" Tablets. Zaporizhzhya, 2015, 167 p.
- Abramov, A.V.; et al. Report on Blind Study of Cardioprotective and Endotelioprotective Effects of New "Angiolin" Injection Solution, 25 mg/mL (OJSC "NPO "Farmatron", Ukraine) Compared with "Tiotriazoline" Injection Solution, 25 mg/mL (JSC "Galichfarm" with Participation of OJSC "NPO "Farmatron", Ukraine) and "Mildronate" Injection Solution, 100 mg/mL (JSC "Grindex", Latvia) upon Intraperitoneal Administration in Pituitrin-Isadrin Myocardial Infarction Model in Laboratory Rats. Zaporizhzhya, 2013, 25 p.
- Mamchur, V.I.; Belenichev, I.F.; Mazur, I.A. Report on Pharmacological Expertise (Acute and Chronic Toxicity) of (S)-2,6-Diaminogexanoic Acid 3-Methyl-1,2,4-Triazolyl-5-Thioacetate ("Liziniy") and Its 2.5% Solution for Parenteral Use. Dnipropetrovsk, 2011, 139 p.
- Belenichev, I.F.; Mazur, I.A.; Abramov, A.V.; Kucherenko, L.I.; Bukhtiyarova, N.V.; Egorov, A.A.; Belenicheva, O.I.; Polyakova, E.N. The endothelium-protective effect of 3-methyl-1,2,4-triazolyl-5-thioacetate (S)-2,6-diaminohexanic acid (lysinium): Effects on the expression of vascular endothelial growth factor (VEGF) and the characteristics of the endotheliocytes of the cerebral vessels of animals with cerebral ischemia. Neurochem. J. 2013, 7, 296–302. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Rodriguez-Miguelez, P.; Lima-Cabello, E.; Martínez-Flórez, S.; Almar, M.; Cuevas, M.J.; González-Gallego, J. Hypoxia-inducible factor-1 modulates the expression of vascular endothelial growth factor and endothelial nitric oxide synthase induced by eccentric exercise. J. Appl. Physiol. (1985) 2015, 118, 1075–1083. [Google Scholar] [CrossRef]
- Hood, J.D.; Meininger, C.J.; Ziche, M.; Granger, H.J. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. Am. J. Physiol. Circ. Physiol. 1998, 274, H1054–H1058. [Google Scholar] [CrossRef]
- Tishkin, V.S.; Belenichev, I.F.; Serikov, V.I. Report on Study of General Toxic Effects of "Thiotriazoline Injection Solution 2.5% for Intravenous Administration". Zaporizhzhya, 1990, 41 p.
- Reshetilov, V.I.; Karzov, M.V.; Knysh, E.G. Report on Study of Embryotoxic Properties of New Drug "Thiotriazoline" and Its Pharmaceutical Forms. Zaporizhzhya, 1994, 66 p.
- Mazur, I.A.; Voloshin, N.A.; Knysh, E.G.; et al. Report on Study of Mutagenic Properties of New Drug "Thiotriazoline" and Its Pharmaceutical Forms. Zaporizhzhya, 1994, 123 p.
- Mazur, I.; Belenichev, I.; Kucherenko, L.; Siusiuka, V.; Reznichenko, N.; Kyryliuk, A.; Ryzhenko, V.; Shevchenko, A.; Khromylova, O. Thiotriazoline and Its Today. Monograph. Dnipro: Jurfond, 2025, 384 p. ISBN 978-966-934-648-3.
- Belenichev, I.F.; et al. Thiol-Disulfide System: Role in Endogenous Cytoprotection and Organoprotection, Paths of Pharmacological Modulation. Monograph. Kyiv: LLC "Poligraph Plus", 2020, 232 p. ISBN 978-617-7854-05-9.
- Zvyagintseva, T.V.; Myronchenko, S.I.; Kytsyuk, N.I.; Naumova, O.V. The Effect Of Ointments With Antioxidant Activity On The Morphological Structures Of Skin Of Guinea Pigs Exposed To Local Ultraviolet Irradiation. Precarpathian Bull Shevchenko Sci Soc Pulse. 2019, 6, 64–72. [Google Scholar] [CrossRef]
- Manischenkova, Y.; Shkala, L.; Dudich, T.; Litvinova, M.; Ivanova, M. Oxidative Stress In Patients With Diabetes Mellitus Type 2 Associated With Exacerbation Of Chronic Pyelonephritis. Int J Endocrinol (Ukraine). 2022, 3, 19–22. [Google Scholar] [CrossRef]
- Mukhina, I.V. Report on Independent Preclinical Assessment of Specific Activity of Thiotriazoline Pharmaceutical Forms (2.5% Injection Solution and Tablets). Nizhny Novgorod, 2009, 57 p.
- Kucherenko, L.I.; Hromyleva, O.V.; Mazur, I.A.; Shishkina, S.V. Theoretical study about L-arginine complexes formation with thiotriazolin. Zaporozhye Med J. 2017, 19, 108–112. [Google Scholar] [CrossRef]
- Kastanayan, A.A.; Kartashova, E.A.; Zheleznyak, E.I. THE EFFECT OF THIOTRIAZOLINE ON ENERGY PRODUCTION IN CONDITIONS OF CHRONIC MYOCARDIAL ISCHEMIA. South Russ. J. Ther. Pr. 2020, 1, 84–90. [Google Scholar] [CrossRef]
- Dzyak, G.V. Report on Clinical Study: "Double-Blind, Multicenter, Randomized Study in Parallel Groups to Evaluate the Efficacy and Tolerability of Thiotriazoline". Kyiv, 2010.
- Kadin, D.K.; Chumak, B.C.; Filippov, A.F.; Shustov, S.S.; Academy, S.P.S.K.S.P.M.M. Thiotriazoline in the Treatment of Stable Angina II–III Functional Class. Kardiologiia 2015, 55, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Kuka, J.; Vilskersts, R.; Cirule, H.; Makrecka, M.; Pugovics, O.; Kalvinsh, I.; Dambrova, M.; Liepinsh, E. The Cardioprotective Effect of Mildronate is Diminished After Co-Treatment With l-Carnitine. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 215–222. [Google Scholar] [CrossRef]
- Liepinsh, E.; Vilskersts, R.; Loca, D.; Kirjanova, O.; Pugovichs, O.; Kalvinsh, I.; Dambrova, M. Mildronate, an Inhibitor of Carnitine Biosynthesis, Induces an Increase in Gamma-Butyrobetaine Contents and Cardioprotection in Isolated Rat Heart Infarction. J. Cardiovasc. Pharmacol. 2006, 48, 314–319. [Google Scholar] [CrossRef]
- Berlato, D.G.; de Bairros, A.V. Meldonium: Pharmacological, toxicological, and analytical aspects. Toxicol. Res. Appl. 2020, 4. [Google Scholar] [CrossRef]
- Gureev, A.P.; Sadovnikova, I.S.; Shaforostova, E.A.; Starkov, A.A.; Popov, V.N. Mildronate protects heart mtDNA from oxidative stress toxicity induced by exhaustive physical exercise. Arch. Biochem. Biophys. 2021, 705, 108892. [Google Scholar] [CrossRef]
- Bielenichev, I.F.; Gorchakova, N.A.; Doroshenko, E.Y.; Samura, I.B.; Ryzhenko, V.P.; Bukhtiiarova, N.V. Use of metabolites, metabolithotropic agents and nutritional supplements in sports and sports medicine: a modern view on the problem. Mod. Med Technol. 2023, 76–88. [Google Scholar] [CrossRef]
- Abel, T.; Knechtle, B.; Perret, C.; Eser, P.; von Arx, P.; Knecht, H. Influence of Chronic Supplementation of Arginine Aspartate in Endurance Athletes on Performance and Substrate Metabolism‒A Randomized, Double-Blind, Placebo-Controlled Study. Int. J. Sports Med. 2005, 26, 344–349. [Google Scholar] [CrossRef]
- Castell, L.M.; Stear, S.J.; Burke, L.M. (Eds.) Nutritional Supplements In Sport, Exercise And Health: An A-Z Guide. London: Routledge. [CrossRef]
- Hsu, C.-N.; Tain, Y.-L. Impact of Arginine Nutrition and Metabolism during Pregnancy on Offspring Outcomes. Nutrients 2019, 11, 1452. [Google Scholar] [CrossRef]
- Bednov, A.; Espinoza, J.; Betancourt, A.; Vedernikov, Y.; Belfort, M.; Yallampalli, C. l-arginine prevents hypoxia-induced vasoconstriction in dual-perfused human placental cotyledons. Placenta 2015, 36, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Suvorava, T.; Nagy, N.; Pick, S.; Lieven, O.; Rüther, U.; Dao, V.T.-V.; Fischer, J.W.; Weber, M.; Kojda, G. Impact of eNOS-Dependent Oxidative Stress on Endothelial Function and Neointima Formation. Antioxidants Redox Signal. 2015, 23, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Parkhomenko, D.; Belenichev, I.; Bukhtiyarova, N.; Kuchkovskyi, O.; Gorchakova, N.; Diachenko, V.; Fedotov, Е. Pharmacocorrection of Disturbances in the no System in Experimental Chronic Generalized Periodontitis. Open Access Maced. J. Med Sci. 2023, 11, 47–52. [Google Scholar] [CrossRef]
- Maslennikov, S.O.; Belenichev, I.F.; Golovakha, M.L.; Makyeyeva, L.V. Regenerative Properties of Polypropylene Mesh Coated with Thiotriazoline and L-arginine. Biomed. Pharmacol. J. 2022, 15, 1985–1993. [Google Scholar] [CrossRef]
- Malla, R.; Viswanathan, S.; Makena, S.; Kapoor, S.; Verma, D.; Raju, A.A.; Dunna, M.; Muniraj, N. Revitalizing Cancer Treatment: Exploring the Role of Drug Repurposing. Cancers 2024, 16, 1463. [Google Scholar] [CrossRef]
- Cousins, H.C.; Nayar, G.; Altman, R.B. Computational Approaches to Drug Repurposing: Methods, Challenges, and Opportunities. Annu. Rev. Biomed. Data Sci. 2024, 7, 15–29. [Google Scholar] [CrossRef]
- Goel, S.; Singh, R.; Singh, V.; Singh, H.; Kumari, P.; Chopra, H.; Sharma, R.; Nepovimova, E.; Valis, M.; Kuca, K.; et al. Metformin: Activation of 5′ AMP-activated protein kinase and its emerging potential beyond anti-hyperglycemic action. Front. Genet. 2022, 13, 1022739. [Google Scholar] [CrossRef]
- Halabitska, I.; Petakh, P.; Lushchak, O.; Kamyshna, I.; Oksenych, V.; Kamyshnyi, O. Metformin in Antiviral Therapy: Evidence and Perspectives. Viruses 2024, 16, 1938. [Google Scholar] [CrossRef]
- Petakh, P.; Kamyshna, I.; Oksenych, V.; Kainov, D.; Kamyshnyi, A. Metformin Therapy Changes Gut Microbiota Alpha-Diversity in COVID-19 Patients with Type 2 Diabetes: The Role of SARS-CoV-2 Variants and Antibiotic Treatment. Pharmaceuticals 2023, 16, 904. [Google Scholar] [CrossRef]
- Halabitska, I.; Babinets, L.; Oksenych, V.; Kamyshnyi, O. Diabetes and Osteoarthritis: Exploring the Interactions and Therapeutic Implications of Insulin, Metformin, and GLP-1-Based Interventions. Biomedicines 2024, 12, 1630. [Google Scholar] [CrossRef]
- Petakh, P.; Griga, V.; Mohammed, I.; Loshak, K.; Poliak, I.; Kamyshnyiy, A. Effects of Metformin, Insulin on Hematological Parameters of COVID-19 Patients with Type 2 Diabetes. Med Arch. 2022, 76, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Petakh, P.; Kamyshna, I.; Kamyshnyi, A. Effects of metformin on the gut microbiota: A systematic review. Mol. Metab. 2023, 77, 101805. [Google Scholar] [CrossRef] [PubMed]
- Triggle, C.R.; Mohammed, I.; Bshesh, K.; Marei, I.; Ye, K.; Ding, H.; MacDonald, R.; Hollenberg, M.D.; Hill, M.A. Metformin: Is it a drug for all reasons and diseases? Metabolism 2022, 133, 155223. [Google Scholar] [CrossRef]
- Petakh, P.; Kamyshna, I.; Oksenych, V.; Kamyshnyi, O. Metformin Alters mRNA Expression of FOXP3, RORC, and TBX21 and Modulates Gut Microbiota in COVID-19 Patients with Type 2 Diabetes. Viruses 2024, 16, 281. [Google Scholar] [CrossRef]
- Buchynskyi, M.; Kamyshna, I.; Lyubomirskaya, K.; Moshynets, O.; Kobyliak, N.; Oksenych, V.; Kamyshnyi, A. Efficacy of interferon alpha for the treatment of hospitalized patients with COVID-19: A meta-analysis. Front. Immunol. 2023, 14, 1069894. [Google Scholar] [CrossRef]
- Kamyshnyi, A.; Koval, H.; Kobevko, O.; Buchynskyi, M.; Oksenych, V.; Kainov, D.; Lyubomirskaya, K.; Kamyshna, I.; Potters, G.; Moshynets, O. Therapeutic Effectiveness of Interferon-α2b against COVID-19 with Community-Acquired Pneumonia: The Ukrainian Experience. Int. J. Mol. Sci. 2023, 24, 6887. [Google Scholar] [CrossRef]
- Buchynskyi, M.; Oksenych, V.; Kamyshna, I.; Vorobets, I.; Halabitska, I.; Kamyshnyi, O. Modulatory Roles of AHR, FFAR2, FXR, and TGR5 Gene Expression in Metabolic-Associated Fatty Liver Disease and COVID-19 Outcomes. Viruses 2024, 16. [Google Scholar] [CrossRef]
- Buchynskyi, M.; Oksenych, V.; Kamyshna, I.; Budarna, O.; Halabitska, I.; Petakh, P.; Kamyshnyi, O. Genomic insight into COVID-19 severity in MAFLD patients: a single-center prospective cohort study. Front. Genet. 2024, 15, 1460318. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Sheida, A.; Taghizadieh, M.; Memar, M.Y.; Hamblin, M.R.; Baghi, H.B.; Nahand, J.S.; Asemi, Z.; Mirzaei, H. Paxlovid (Nirmatrelvir/Ritonavir): A new approach to Covid-19 therapy? Biomed. Pharmacother. 2023, 162, 114367. [Google Scholar] [CrossRef]
- Kamyshna, I.I.; Pavlovych, L.B.; Maslyanko, V.A.; Kamyshnyi, A.M. Analysis of the transcriptional activity of genes of neuropeptides and their receptors in the blood of patients with thyroid pathology. J. Med. Life 2021, 14, 243–249. [Google Scholar] [CrossRef]
- Topol, I.; Kamyshny, A. Study of expression of TLR2, TLR4 and transckription factor NF-kB structures of galt of rats in the conditions of the chronic social stress and modulation of structure of intestinal microflora. Georgian Med. News 2013, 225, 115–22. [Google Scholar]
- Halabitska, I.; Oksenych, V.; Kamyshnyi, O. Exploring the Efficacy of Alpha-Lipoic Acid in Comorbid Osteoarthritis and Type 2 Diabetes Mellitus. Nutrients 2024, 16, 3349. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Andrabi, S.S.; Mansoor, S.; Kuang, Y.; Kwon, B.K.; Labhasetwar, V. Antioxidant Therapy in Oxidative Stress-Induced Neurodegenerative Diseases: Role of Nanoparticle-Based Drug Delivery Systems in Clinical Translation. Antioxidants 2022, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Halabitska, I.; Petakh, P.; Kamyshna, I.; Oksenych, V.; Kainov, D.E.; Kamyshnyi, O. The interplay of gut microbiota, obesity, and depression: insights and interventions. Cell. Mol. Life Sci. 2024, 81, 443. [Google Scholar] [CrossRef]
- Ghareghomi, S.; Rahban, M.; Moosavi-Movahedi, Z.; Habibi-Rezaei, M.; Saso, L.; Moosavi-Movahedi, A.A. The Potential Role of Curcumin in Modulating the Master Antioxidant Pathway in Diabetic Hypoxia-Induced Complications. Molecules 2021, 26, 7658. [Google Scholar] [CrossRef]
- Zhang, X.; Zheng, Y.; Wang, Z.; Gan, J.; Yu, B.; Lu, B.; Jiang, X. Melatonin as a therapeutic agent for alleviating endothelial dysfunction in cardiovascular diseases: Emphasis on oxidative stress. Biomed. Pharmacother. 2023, 167, 115475. [Google Scholar] [CrossRef]
- Sydorchuk, L.; Dzhuryak, V.; Sydorchuk, A.; Levytska, S.; Petrynych, V.; Knut, R.; Kshanovska, A.; Iftoda, O.; Tkachuk, O.; Kyfiak, P.; et al. The cytochrome 11B2 aldosterone synthase gene rs1799998 single nucleotide polymorphism determines elevated aldosterone, higher blood pressure, and reduced glomerular filtration, especially in diabetic female patients. Endocr. Regul. 2020, 54, 217–226. [Google Scholar] [CrossRef]
- Strumillo, J.; Nowak, K.E.; Krokosz, A.; Rodacka, A.; Puchala, M.; Bartosz, G. The role of resveratrol and melatonin in the nitric oxide and its oxidation products mediated functional and structural modifications of two glycolytic enzymes: GAPDH and LDH. Biochim. et Biophys. Acta (BBA) Gen. Subj. 2018, 1862, 877–885. [Google Scholar] [CrossRef]
Figure 1.
Prenatal hypoxia and its effects on the cardiovascular system of the fetus and offspring. PH causes disruption in the nitric oxide (NO) system, leading to NO deficiency, increased reactive oxygen species (ROS) and their cytotoxic forms, and the activation of NO- dependent molecular-biochemical mechanisms that lead to apoptosis, endothelial dysfunction, mitochondrial dysfunction, inflammation, oxidative stress, and nitrosative stress. This initiates the foundation for the development of cardiovascular diseases in adulthood (chronic heart failure (СHF), arterial hypertension (AH), cerebrovascular accident (CVA), ischemic heart disease (IHD)).
Figure 1.
Prenatal hypoxia and its effects on the cardiovascular system of the fetus and offspring. PH causes disruption in the nitric oxide (NO) system, leading to NO deficiency, increased reactive oxygen species (ROS) and their cytotoxic forms, and the activation of NO- dependent molecular-biochemical mechanisms that lead to apoptosis, endothelial dysfunction, mitochondrial dysfunction, inflammation, oxidative stress, and nitrosative stress. This initiates the foundation for the development of cardiovascular diseases in adulthood (chronic heart failure (СHF), arterial hypertension (AH), cerebrovascular accident (CVA), ischemic heart disease (IHD)).
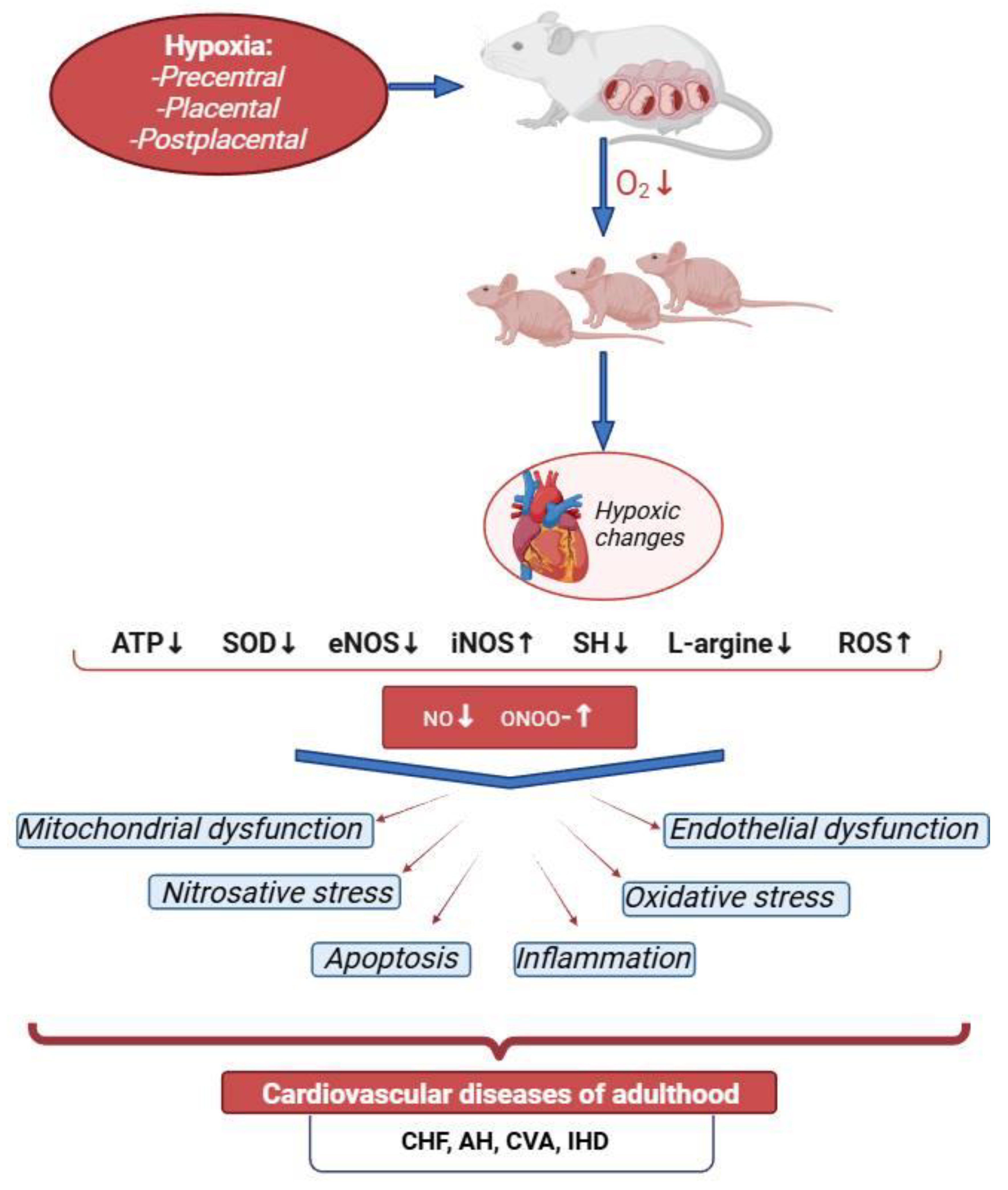
Figure 2.
NO-dependent mechanisms of mitochondrial dysfunction formation after prenatal hypoxia.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



