Submitted:
07 April 2025
Posted:
07 April 2025
You are already at the latest version
Abstract
Three new amide-preformed triphenylamine-diamine monomers, namely 4,4’-bis(p-aminobenzamido)triphenylamine (4), 4,4’-bis(p-aminobenzamido)-4”-methoxytriphenylamine (MeO-4) and 4,4’-bis(p-aminobenzamido)-4”-tert-butyltriphenylamine (t-Bu-4), were synthesized and led to three series of electroactive aromatic poly(amide-imide)s (PAIs) by the two-step polycondensation reactions with commercially available tetracarboxylic dianhydrides. Strong and flexible PAI films could be obtained by solution casting of the poly(amic acid) films followed by thermal imidization or direct solution casting from the organosoluble PAI samples. The PAIs had high glass-transition temperatures of 296−355 oC and showed no significant decomposition before 500 oC. The PAIs based on diamines MeO-4 and t-Bu-4 showed a high electrochemical redox stability, accompanied by strong color changes upon oxidation. For the PAIs derived from diamine 4, the TPA radical cation formed in situ during the electro-oxidative process could dimerize to a tetraphenylbenzidine structure, which led to an additional oxidation state and color change. These PAIs exhibited increased solubility, lowered oxidation potentials, and enhanced redox stability as compared to their corresponding polyimide analogs.
Keywords:
Triphenylamine
; Poly(amide-imide)s
; Electrochemistry
; Electrochromism
; Redox-active polymers
1. Introduction
Electrochromic materials can show a reversible optical change in absorption or transmittance upon electrochemically oxidized or reduced. The main kinds of electrochromic materials include metal oxides (such as WO3), metal hexacyanoferrates, organic small molecules (such as viologens) and organic polymers (e.g., polyanilines, polythiophenes, and polypyrroles) [1,2]. Among these, organic polymers have several advantages such as high coloration efficiency, fast response time, can be fabricated into flexible devices, and easy color tuning [3,4,5,6]. This technology can be applied to smart windows, anti-glare rear-view mirrors, displays, and eye-wear. Electronically dimmable windows developed by Gentex have been housed on the Boeing 787 Dreamliner.
Triphenylamine (TPA) derivatives are well known for photo and electroactive properties that have found optoelectronic applications as photoconductors, hole-transporters, and light-emitters [7,8,9]. TPAs can be easily oxidized to form stable radical cations, and the oxidation process is always associated with a noticeable change of coloration. In early 1990s, the synthesis and characterization of polyimides and polyamides containing TPA unit were first reported by Imai group [10,11]. Since 2005, the Liou’s group and our group disclosed the interesting electrochromic properties of high performance polymers, such as aromatic polyamides and polyimides, carrying the TPA unit as an electrochromic functional moiety [12,13,14]. Then, many TPA-based electrochromic polymers have been reported in the literature [15,16,17,18,19]. Yen and Liou have published some comprehensive review papers on the TPA-based electrochromic polymers [20,21,22]. The introduction of bulky groups such as tert-butyl group [23,24,25] or the electron-donating substituents such as methoxy groups [26,27,28] onto the active sites of triarylamine units reduced the oxidation potential and allowed to obtain more stable radical cations preventing dimerization reactions at these positions.
It has been demonstrated that TPA-based polyimides generally exhibited poor electrochemical and electrochromic stability as compared with their polyamide analogs because of the strong electron-withdrawing imide group, which increases the oxidation potential of the TPA unit and destabilizes the resultant amino radical cation upon oxidation. Incorporating a spacer between the TPA core and the imide ring may improve the elec trochemical and elcetrochromic stability of this kind of electroactive polymers. Thus, three new amide-preformed triphenylamine-diamine monomers, namely 4,4’-bis(p-aminobenzamido)triphenylamine (4), 4,4’-bis(p-aminobenzamido)-4”-methoxytriphenylamine (MeO-4) and 4,4’-bis(p-aminobenzamido)-4”-tert-butyltriphenylamine (t-Bu-4), were synthesized and a series of poly(amide-imide)s (PAIs) with main-chain TPA and benzamide moieties were prepared from these diamide-preformed diamine monomers with the corresponding tetracarboxylic dianhydrides. By the incorporation of the benzamide spacer between the TPA and imide units, the resulting PAIs are expected to exhibit enhanced electrochemical and electrochromic properties.
2. Materials and Methods
Aniline (Acros), p-anisidine (Acros), p-fluoronitrobenzene (Acros), p-tert-butylaniline (AKSci), p-nitrobenzoyl chloride (Acros), cesium fluoride (CsF, Acros), hydrazine monohydrate (Alpha), 10% palladium on activated carbon (Pd/C, Fluka), dimethyl sulfoxide (DMSO, Tedia), and acetic anhydride (Acros) were used as received from commercial sources. N-Methyl-2-pyrrolidone (NMP, Tedia) was dried over calcium hydride for 24 h, distilled under reduced pressure, and stored over 4 Å molecular sieves in sealed bottles. The aromatic tetracarboxylic dianhydrides including pyromellitic dianhydride (PMDA; 5a, TCI), 3,3’,4,4’-biphenyltetracarboxylic dianhydride (BPDA; 5b Oxychem), 3,3’,4,4’-benzophenonetetracarboxylic dianhydride (BTDA; 5c Oxychem), 4,4’-oxydiphthalic anhydride (ODPA; 5d, Oxychem), 3,3’,4,4’-diphenylsulfonetetracarboxylic dianhydride (DSDA; 5e, Oxychem), 2,2-bis(3,4-dicarboxyphenyl)hexafluoropropane dianhydride (6FDA; 5f, Hoechst Celanese) were purified by dehydration at 250 oC in vacuum for 3 h. Other reagents and solvents were used as received from commercial sources. According to a reported synthetic procedure [14], 4,4’-diaminotriphenylamine (2), 4,4’-diamino-4”-methoxytriphenylamine (MeO-2), and 4,4’-diamino-4”-tert-butyltriphenylamine (t-Bu-2) were synthesized via the fluoro-displacement of p-fluoronitrobenzene with aniline, p-anisidine and p-tert-butylaniline in the presence of CsF in DMSO, followed by Pd/C-catalyzed hydrazine reduction of the intermediate dinitro compounds 4,4’-dinitrotriphenylamine (1), 4-methoxy-4’,4”-dinitrotriphenylamine (MeO-1), and 4-tert-butyl-4’,4”-dinitrotriphenylamine (t-Bu-1) in ethanol, respectively.
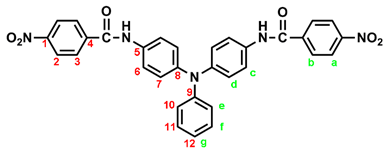
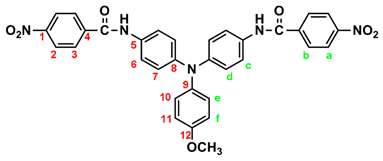
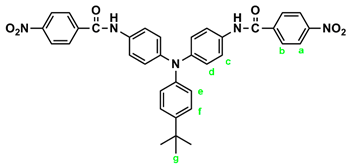
2.1. Synthesis of 4,4’-bis(p-nitrobenzamido)triphenylamine (3), 4,4’-bis(p-nitrobenzamido)-4”-methoxytriphenylamine (MeO-3) and 4,4’-bis(p-nitrobenzamido)-4”-tert-butyltriphenylamine (t-Bu-3)
In a 250 mL round-bottom flask equipped with a stirring bar, 2.75 g (0.01 mol) of 4,4’-diaminotriphenylamine and 3 mL pyridine were dissolves in 30 mL DMF. A solution of 4.53 g (0.024 mol) of p-nitrobenzoyl chloride in 20 mL DMF was added. The reaction mixture was stirred for 3 h and then poured into 600 mL of mixture of methanol and water (2:1). The precipitated solid was collected by filtration and recrystallized from ethanol, and dried to give 4.36 g (76 %) of diamide-dinitro compound 3 as dark red crystals with a melting point of 237~239 oC (by DSC). IR (KBr): 3282 cm–1 (amide N–H stretch), 1651 cm–1 (amide C=O stretch), 1595, 1350 cm–1 (nitro –NO2 stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 7.00 (m, 3H, Hg + He), 7.05 (d, J = 8.9 Hz, 4H, Hd), 7.30 (t, J = 7.8 Hz, 2H, Hf), 7.73 (d, J = 8.9 Hz, 4H, Hc), 8.18 (d, J = 8.9 Hz, 4H, Hb), 8.35 (d, J = 8.9 Hz, 4H, Ha), 10.55 (s, 2H, amide N–H). 13C NMR (150 MHz, DMSO-d6, δ, ppm): 163.57 (amide carbon), 149.10 (C1), 147.35 (C9), 143.36 (C8), 140.61 (C5), 133.99 (C4), 129.43 (C11), 129.12 (C3), 124.22 (C7), 123.53 (C2), 122.65 (C10), 122.27 (C12), 121.50 (C6).
 |
By a similar procedure, diamide-dinitro compound MeO-3 was synthesized from the condensation of 3.05 g (0.01 mol) of 4,4’-diamino-4”-methoxytriphenylamine (MeO-2) and 4.53 g (0.024 mol) of p-nitrobenzoyl chloride as orange powder (4.23 g; 70 % yield) with a melting point of 219~221 oC. IR (KBr): 3286 cm–1 (amide N–H stretch), 1651 cm–1 (amide C=O stretch), 1600, 1350 cm–1 (nitro –NO2 stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 3.75 (s, 3H, methoxy), 6.94 (d, J = 8.9 Hz, 2H, He), 6.96 (d, J = 8.9 Hz, 4H, Hd), 7.03 (d, J = 8.9 Hz, 2H, Hf), 7.68 (d, J = 8.9 Hz, 4H, Hc), 8.18 (d, J = 8.8 Hz, 4H, Hb), 8.36 (d, J = 8.8 Hz, 4H, Ha), 10.05 (s, 2H, amide N–H). 13C NMR (150 MHz, DMSO-d6, δ, ppm): 163.44 (amide carbon), 155.71 (C12), 149.06 (C1), 143.96 (C8), 140.64 (C5), 140.09 (C9), 133.05 (C4), 129.08 (C3), 126.45 (C11), 123.51 (C2), 122.60 (C7), 121.73 (C6), 115.01 (C10), 55.24 (–OCH3).
 |
Similarly, diamide-dinitro compound t-Bu-3 was synthesized from the condensation of 3.31 g (0.01 mol) of 4,4’-diamino-4”-tert-butyltriphenylamine (t-Bu-2) and 4.53 g (0.024 mol) of p-nitrobenzoyl chloride as dark-red crystals (5.35 g; 85 % yield) with a melting point of 223~224 oC. IR (KBr): 3300 cm–1 (amide –N–H stretch), 1655 cm–1 (amide C=O stretch), 1596, 1350 cm–1 (nitro –NO2 stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 1.27 (s, 9H, Hg), 6.94 (d, J = 6.8 Hz, 2H, He), 7.02 (d, J = 8.9 Hz, 4H, Hd), 7.32 (d, J = 6.8 Hz, 2H, Hf), 7.71 (d, J = 8.9 Hz, 4H, Hc), 8.18 (d, J = 7.8 Hz, 4H, Hb), 8.36 (d, J = 7.9 Hz, 4H, Ha), 10.53 (s, 2H, amide N–H).
 |
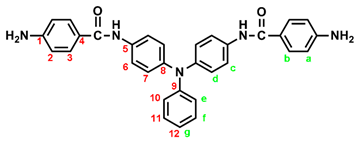
2.2. Synthesis of 4,4’-bis(p-aminobenzamido)triphenylamine (4), 4,4’-bis(p-aminobenzamido)-4”methoxytriphenylamine (MeO-4) and 4,4’-bis(p-aminobenzamido)-4”-tert-butytriphenylamine (t-Bu-4)
In a 100 mL three-neck round-bottom flask equipped a stirring bar, 1.72 g (0.003 mol) of diamide-dinitro compound 3 and 0.15 g of 10% Pd/C were dispersed in 50 mL of ethanol. The equivalent amount of hydrazine was added to the mixture and the solution was stirred at a reflux temperature for about 6 h. The solution was then filtered to remove Pd/C, and the filtrate was concentrated in a rotatory evaporator and cooled to precipitate the product that was collected by filtration and dried in vacuum to give 1.08 g (70 %) of diamide-diamino compound 4 with a melting point of 261~264 oC. IR (KBr): 3350, 3300 cm–1 (amide and amino N–H stretch), 1630 cm–1 (amide C=O stretch). 1H NMR (600 MHz, DMSO-d6, δ, ppm): 5.72 (s, 4H, –NH2), 6.60 (d, J = 8.7 Hz, 4H, Ha), 6.92-6.94 (two overlapped doublets, 3H, Hg + He), 6.98 (d, J = 8.9 Hz, 4H, Hd), 7.24 (t, J = 7.8 Hz, 2H, Hf), 7.68 (d, J = 8.9 Hz, 4H, Hc), 7.70 (d, J = 8.7 Hz, 4H, Hb), 9.74 (s, 2H, amide N–H). 13C NMR (150 MHz, DMSO-d6, δ, ppm): 165.04 (amide carbon), 152.03 (C1), 147.76 (C9), 142.29 (C8), 135.23 (C5), 129.23 (C3,C11), 124.41 (C7), 121.68 (C10), 121.43 (C12), 121.38 (C6), 121.12 (C4), 112.51 (C2).
 |
By a similar procedure, diamide-diamines MeO-4 and t-Bu-4 were synthesized by hydrazine Pd/C-catalyzed reduction of diamide-dinitro compounds MeO-3 and t-Bu-3, respectively. The spectroscopic data are shown below.
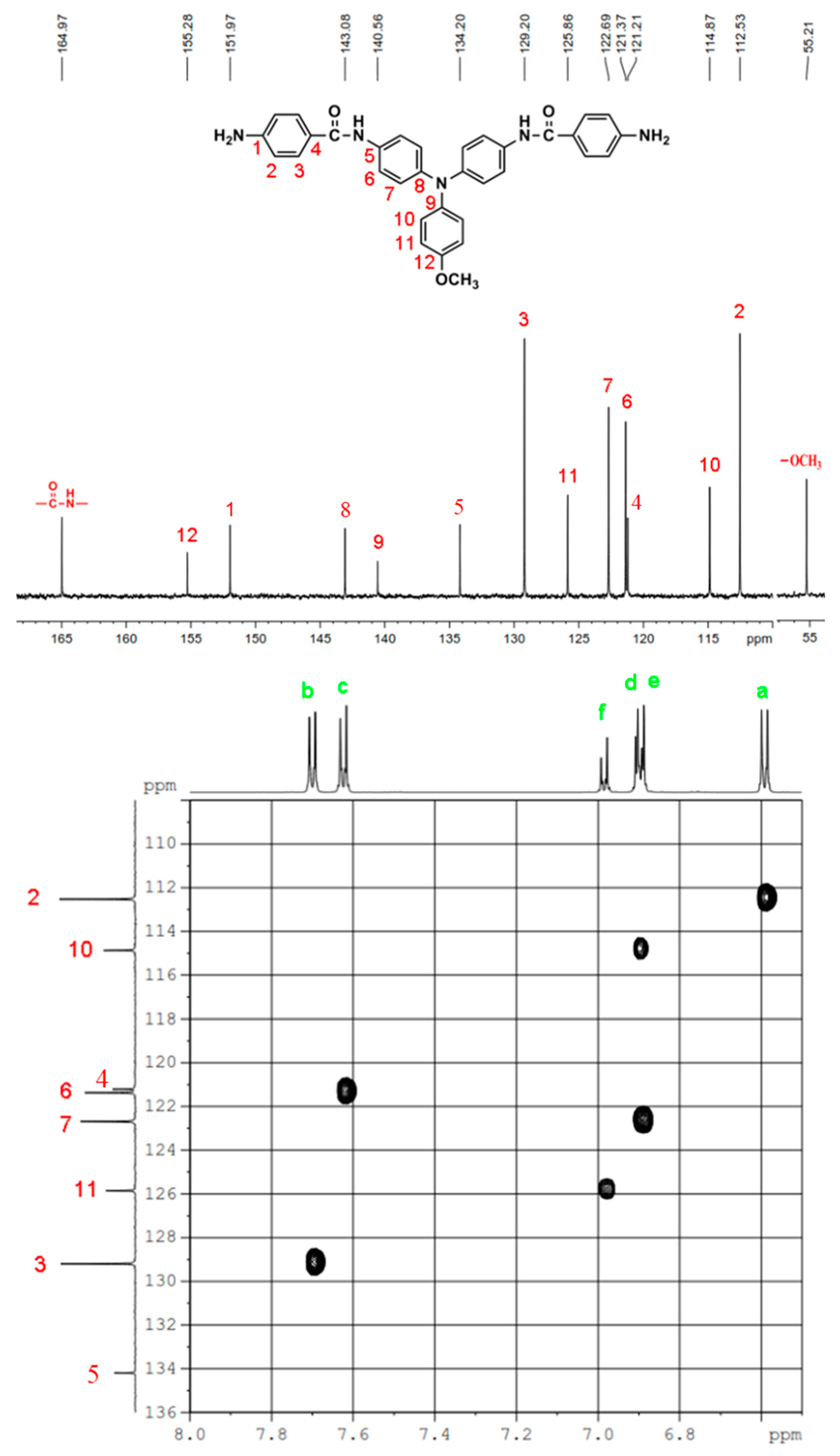
IR for MeO-4 (KBr): 3500~3300 cm–1 (amine and amide N–H stretch), 1623 cm–1 (amide C=O stretch). 1H NMR for MeO-4 (600 MHz, DMSO-d6, δ, ppm): 3.75 (s, 3H, methoxy), 5.70 (s, 4H, –NH2), 6.59 (d, J = 7.8 Hz, 4H, Ha), 6.90 (two overlapped doublets, 6H, Hd + He), 6.98 (d, J = 9.0 Hz, 4H, Hf), 7.62 (d, J = 6.9 Hz, 4H, Hc), 7.70 (d, J = 7.8 Hz, 4H, Hb), 9.69 (s, 2H, amide N–H). 13C NMR for MeO-4 (150 MHz, DMSO-d6, δ, ppm): 164.97 (amide carbon), 155.26 (C12), 151.97 (C1), 143.08 (C8), 140.56 (C9), 134.20 (C5), 129.20 (C3), 125.86 (C11), 122.69 (C7), 121.37 (C6), 121.21 (C4), 114.87 (C10), 112.53 (C2), 55.21 (–OCH3)
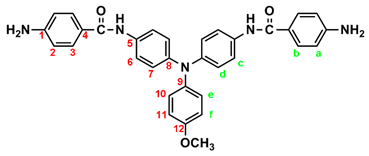 |
IR for t-Bu-4 (KBr): 3500~3300 cm–1 (amine and amide N–H stretch), 1623 cm–1 (amide C=O stretch). 1H NMR for t-Bu-4 (600 MHz, DMSO-d6, δ, ppm): 1.27 (s, 9H, Hg), 5.72 (s, 4H, –NH2), 6.60 (d, J = 8.6 Hz, 4H, Ha), 6.89 (d, J = 8.7 Hz, 2H, He), 6.95 (d, J = 8.9 Hz, 4H, Hd), 7.28 (d, J = 8.7 Hz, 2H, Hf), 7.66 (d, J = 8.9 Hz, 4H, Hc), 7.70 (d, J = 8.6 Hz, 4H, Hb), 9.72 (s, 2H, amide N–H).
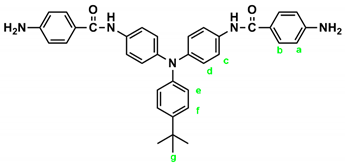 |
2.3. Synthesis of Poly(amide-imide)s
The PAIs were prepared from various tetracarboxylic dianhydrides (PMDA, BPDA, BTDA, ODPA, DSDA, and 6FDA) with amide-performed diamino compounds 4, MeO-4 and t-Bu-4, respectively, by a conventional two-step method via thermal or chemical imidization reaction. The synthesis of PAI 6f is described as an example. Into a solution of amide-performed diamino compound 4 (0.5362 g; 1.04 mmol) in 9.5 mL anhydrous DMAc in a 50 mL round-bottom flask, 0.4638 g (1.04 mmol) of 6FDA (5f) was added in one portion. Thus, the solid content of the solution is approximately 10 wt%. The mixture was stirred at room temperature for 6 h to yield a viscous poly(amide-amic acid) (PAA) solution with an inherent viscosity of 0.68 dL/g, measured in DMAc at concentration of 0.50 g/dL at 30 oC. The poly(amide-amic acid) film was obtained by casting from the reaction polymer solution onto a glass Petri dish and drying at 90 oC overnight. The poly(amide-amic acid) in the form of solid film was converted to the PAI film by successive heating at 150 oC for 30 min, 200 oC for 30 min, and 250 oC for 1 h. For chemical imidization method, 2 mL of acetic anhydride and 1mL of pyridine were added to the PAA solution obtained by a similar process as above, and the mixture was heated at 100 oC for 1 h to effect a complete imidization. The homogenous polymer solution was poured slowly into an excess of methanol giving rise to a precipitate that was collected by filtration, washed thoroughly with hot water and methanol, and dried. The inherent viscosity of the resulting PAI 6f was 0.64 dL/g, measured in DMAc at concentration of 0.50 g/dL at 30 oC. A polymer solution was made by the dissolution of about 0.5 g of the PAI sample in 4 mL of hot DMAc. The homogeneous solution was poured into a glass Petri dish, which was placed in a 90 oC oven overnight for slow release of solvent, and then the film was stripped off from the glass substrate and further dried in vacuum at 160 oC for 6 h. The IR spectrum of 6f (film) exhibited characteristic imide and amide absorption bands at 1786 cm–1 (asymmetrical imide C=O stretch), 1725 cm–1 (symmetrical imide stretch), and 1650 cm–1 (amide C=O stretch).
2.4. Measurements
Infrared (IR) spectra were recorded on a Horiba FT-720 FT-IR spectrometer. 1H spectra were measured on a Bruker Avance III HD-600 MHz NMR spectrometer. The inherent viscosities were determined with a Cannon-Fenske viscometer at 30 oC. Gel permeation chromatography (GPC) analysis was carried out on a Waters chromatography unit interfaced with a Waters 2410 refractive index detector at 3 mg/mL concentration. Two Waters 5 μm Styragel HR-2 and HR-4 columns (7.8 mm I. D. x 300 mm) were connected in series with NMP as the eluent at a flow rate of 0.6 mL/min at 50 oC and were calibrated with polystyrene standards. Thermogravimetric analysis (TGA) was performed with a Perkin-Elmer Pyris 1 TGA. Experiments were carried out on approximately 3−5 mg of polymer film samples heated in flowing nitrogen or air (flow rate = 40 cm3/min) at a heating rate of 20 oC/min. DSC analyses were performed on a Perkin-Elmer DSC 4000 at a scan rate of 20 oC/min in flowing nitrogen. Electrochemistry was performed with a CHI 750A electrochemical analyzer. Voltammograms are presented with the positive potential pointing to the left and with increasing anodic currents pointing downwards. Cyclic voltammetry was conducted with the use of a three-electrode cell in which ITO (polymer films area about 0.5 cm × 2.0 cm) was used as a working electrode. All cell potentials were taken with the use of a home-made Ag/AgCl, KCl (sat.) reference electrode. Ferrocene was used as an external reference for calibration (+0.44 V vs. Ag/AgCl). Spectroelectrochemistry analyses were carried out with an electrolytic cell, which was composed of a 1 cm cuvette, ITO as a working electrode, a platinum wire as an auxiliary electrode, and a home-made Ag/AgCl, KCl (sat.) reference electrode. Absorption spectra in the spectroelectrochemical experiments were also measured with an Agilent 8453 UV-visible photodiode array spectrophotometer.
3. Results and Discussion
3.1. Monomer Synthesis
The target TPA-based diamide-diamine monomers 4, MeO-4 and t-Bu-4 were synthesized by a four-step reaction sequence as shown in Scheme 1. According to a well-known synthetic method [14], CsF-assisted N,N-diarylation of aniline, p-anisidine and p-tert-butylaniline, respectively, with two equivalent amount of p-fluoronitrobenzene in DMSO gave the TPA-dinitro compounds 4,4-dinitrotriphenylamine (1), 4-methoxy-4’,4”-dinitrotriphenylamine (MeO-1), 4-tert-butyl-4’,4”-dinitrotriphenylamine (t-Bu-1), which were subsequently converted to the TPA-diamines 4,4’-diaminotriphenylamine (2), 4,4’-diamino-4”-methoxytriphenylamine (MeO-2) and 4,4’-diamino-4”-tert-butyl-triphenylamine (t-Bu-2) by hydrazine Pd/C-catalyzed reduction. The targeted diamide-diamine monomers 4,4’-bis(p-aminobenzamido)triphenylamine (4), 4-bis(p-aminobenzamido)-4”-methoxytriphenylamine (MeO-4) and 4,4-bis(p-aminobenzamido)-4”-tert-butyltriphenylamine (t-Bu-4) were prepared by condensation of 2, MeO-2 and t-Bu-2 with two equivalent amount of p-nitrobenzoyl chloride and followed by hydrazine Pd/C-catalyzed reduction of the intermediate diamide-dinitro compounds 3, MeO-3 and t-Bu-3. All the synthesized compounds were characterized by FT-IR and 1H NMR spectroscopy. The diamide-dinito and diamide-diamino compounds were also characterized by 13C NMR spectroscopy.
Figure S1 in the Supplemental Materials illustrates FT-IR spectra of compounds 1-4. The nitro groups of 1 show the characteristic absorptions at 1579 cm−1 and 1350 cm−1 (−NO2 asymmetric and symmetric stretching). After reduction, the characteristic absorptions of nitro group disappear and 2 shows the typical −NH2 stretching absorption pair at 3421 cm−1 and 3341 cm−1. Compound 3 show the characteristic amide absorption at 3282 cm−1 (N−H stretching) and 1651 cm-1 (amide C=O stretching) and show the characteristic absorptions at 1595 cm−1 and 1350 cm−1 (−NO2 asymmetric and symmetric stretching). After reduction, the characteristic absorptions of nitro group disappear and 4 shows the typical −NH2 stretching absorption band at 3300 and 3350 cm−1 (amide and amine N−H stretching) and 1630 cm-1 (amide C=O stretching). The IR spectra of diamide-diamines MeO-4 and t-Bu-4 and their precursor compounds are also included in the Supplemental Materials (Figure S2).
The molecular structures of all the TPA-based diamide-dinitro and diamide-diamino compounds were also confirmed by 1H and 13C NMR spectra. As a typical example, the 1H NMR and H-H COSY of diamide-diamine monomer MeO-4 in DMSO-d6 are illustrated in Figure 1, and its 13C NMR and C-H HMQC are shown in Figure 2. The assignments of resonance peaks were assisted by 2D NMR spectra, and all the NMR spectra are in good agreement with the molecular structure of MeO-4. The NMR spectra of the other synthesized compounds are summarized in the Supplemental Materials Figures S3 to S7. Thus, the IR and NMR spectra confirmed all the compounds reported herein have been successfully synthesized.
3.2. Synthesis of Poly(amide-imide)s (PAIs)
Three series of aromatic PAIs (6a-f, MeO-6a-f and t-Bu-6a-f) with TPA units were prepared in conventional two-step method by the reaction of equal molar amount of diamine; 4, MeO-4 and t-Bu-4, respectively, with various aromatic dianhydrides (5a-5f) to form poly(amide-amic acid)s, followed by the thermal or chemical cycloehydration (Scheme 2). As shown in Table 1, the poly(amide-amic acid) precursors had inherent viscosities in the range of 0.29-0.91 dL/g. The molecular weights of these poly(amide-amic acid)s were sufficiently high to permit the casting of flexible and tough poly(amide-amic acid) films, which were subsequently converted into tough PAI films by stage-by-stage heating to elevated temperatures. The inherent viscosities of these thermally imidixed PAIs were recorded in the range of 0.35-0.88 dL/g, as measured at a concentration of 0.5 dL/g in DMAc at 30 oC. The transformation from poly(amide-amic acid) to a PAI could also be carried out via chemical cyclodehydration by using acetic anhydride and pyridine.
The structures of the PAIs were confirmed by IR and NMR spectroscopy. A typical pair of the IR spectra of PAI 6f and its poly(amide-amic acid) precursor are illustrated in Figure S8. PAI 6f exhibited characteristic imide group absorptions around 1786 and 1725 cm-1 (typical of imide carbonyl asymmetrical and symmetrical stretch). Proton NMR spectra of PAIs 6f, MeO-6f and t-Bu-6f in DMSO-d6 are illustrated in Figure S9, and the resonance peaks are well assigned to the repeating structure of the polymer backbone. All the aromatic protons in TPA moiety resonated in the region of δ 7.62-6.91 ppm, the protons in 6FDA component appeared at 8.22-7.77 ppm, and protons on the benzamide unit appeared at 8.07-7.74 ppm.
3.3. Solubility and Thermal Properties
The qualitative solubility properties of the polymers in several organic solvents at 10 % (w/v) and the inherent viscosities are summarized in Table 1. Most of the PAIs were easily soluble in polar organic solvents such as NMP, DMAc, DMF and DMSO at room temperature or on heating. Some of them were even soluble in less polar m-cresol upon heating at 60 oC. Therefore, they could be easily solution-cast into flexible and tough films.
Thermal properties of the polymers were investigated by DSC and TGA. The relevant data are summarized in Table 2. DSC measurements were conducted with a heating rate of 20 oC/min under a nitrogen flow. Quenching from an elevated temperature of about 400 oC to 50 oC gave predominantly amorphous samples so that the glass-transition temperature (Tg) of these PAIs could be easily measured in the second heating traces of DSC. Typical DSC traces of the 6 series PAIs are shown in Figure S10. Glass transition temperature (Tg) is defined as the temperature at the midpoint of the baseline shift. These PAIs exhibited moderately high Tg ranging from 286 to 355°C. The PAIs derived from ODPA (5d) possessed the lowest Tg among each series ones because of the presence of flexible ether linkage in the dianhydride component. The thermal stability of the polymers was evaluated by TGA in both air and nitrogen atmospheres. TGA curves of PAI 6f measured in nitrogen and in air are illustrated in Figure S10. These polymers exhibited reasonable thermal stability without significant weight loss up to 500 oC under nitrogen or air atmosphere. The decomposition temperatures (Td) at 5 % and 10 % weight losses in nitrogen and air atmospheres taken from the original TGA thermograms are given in Table 2.
3.4. Electrochemical Properties
The electrochemical behavior of the polymer was investigated by cyclic voltammetry (CV) conducted for the cast film on an ITO-coated glass substrate as working electrode in dry acetonitrile (MeCN) containing 0.1 M of tetrabutylammonium perchlorate (TBAP; Bu4NClO4) as the supporting electrolyte and saturated Ag/AgCl as the reference electrode under nitrogen atmosphere. The derived oxidation potentials are summarized in Table 3. As illustrated in Figure S11, all of the PAIs 6a-6f with non-substituted TPA show a reversible oxidation process in the initial CV scans with oxidation peak potentials (Epa) at about 0.90-0.98 V and onset potentials (Eonset) at 0.70-0.72 V that corresponds to the TPA oxidation. In contrast, the MeO-6 and t-Bu-6 series PAIs with methoxy or tert-butyl-substituted TPA, CV diagrams shown in Figure S13 and S15, have lower oxidation peak and onset potentials (Eonset at 0.56-0.59V and Epa at about 0.78-0.88 V of methoxy-substituted polymer; Eonset at 0.63-0.68 V and Epa at about 0.86-0.97 V of tert-butyl-substituted polymer) than non-substituted polymers. Because of the electron-donating property of methoxy and tert-butyl groups, the nitrogen center of TPA can be oxidized easily. The MeO-6 series PAIs have the lowest oxidation potentials due to the strong electron-donating property. Repetitive scans between 0 and 1.2 V, the redox waves of the non-substituted PAIs slightly broadened due to coupling reaction of the TPA units (see Figure S12). The CV-scanned films of the 6 series PAIs was essentially insoluble in sulfuric acid or NMP. In the other hand, the methoxy or t-butyl-substituted polymers displayed a very stable electrochemical stability. After 50 repeated cycling, their CV diagrams are almost the same with the first scanned ones (Figure S14 and S16). The energy levels of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of the corresponding polymers were estimated from the E1/2Ox values. The redox potentials and energy levels of all the polymers are summarized in Table 3. Assuming that the HOMO energy level for the ferrocene/ferrocenium (Fc/Fc+) standard is 4.80 eV with respect to the zero vacuum level, the HOMO energy levels for the PAIs were calculated (from E1/2Ox values) to be in the range of 5.07−5.20 eV. The LUMO/HOMO energy gaps estimated from the absorption spectra were then used to obtain the LUMO energy levels.
The effect of inserting benzamide unit on the electrochemical stability of the polymer can be seen from Figure 3. The polymer without benzamide like MeO-6’d shows a higher oxidation onset potential at 0.84 V and anodic peak potential at 1.08 V. After fifty repeated cycling, the CV diagram of MeO-6d has little change, implying a very stable electrochemical stability. In contrast, MeO-6’d which without the benzamide spacer shows unstable electrochemical stability upon repeated cycling. Therefore, inserting the benzamide spacer to separate TPA and imide ring can improve the electrochemical stability of these PAIs.
3.5. Spectroelectrochemical Properties
Spectroelectrochemical measurements were performed on films of polymers drop-coated onto ITO-coated glass slides immerged in an electrolyte solution. The electrode preparations and solution conditions were identical to those used in the CV experiments. During the test, a three-electrode configuration was used for applying potential to the polymer films in a 0.1 M Bu4NClO4/CH3CN electrolyte solution. When the films were electrochemically oxidized, a strong color change of them was observed.
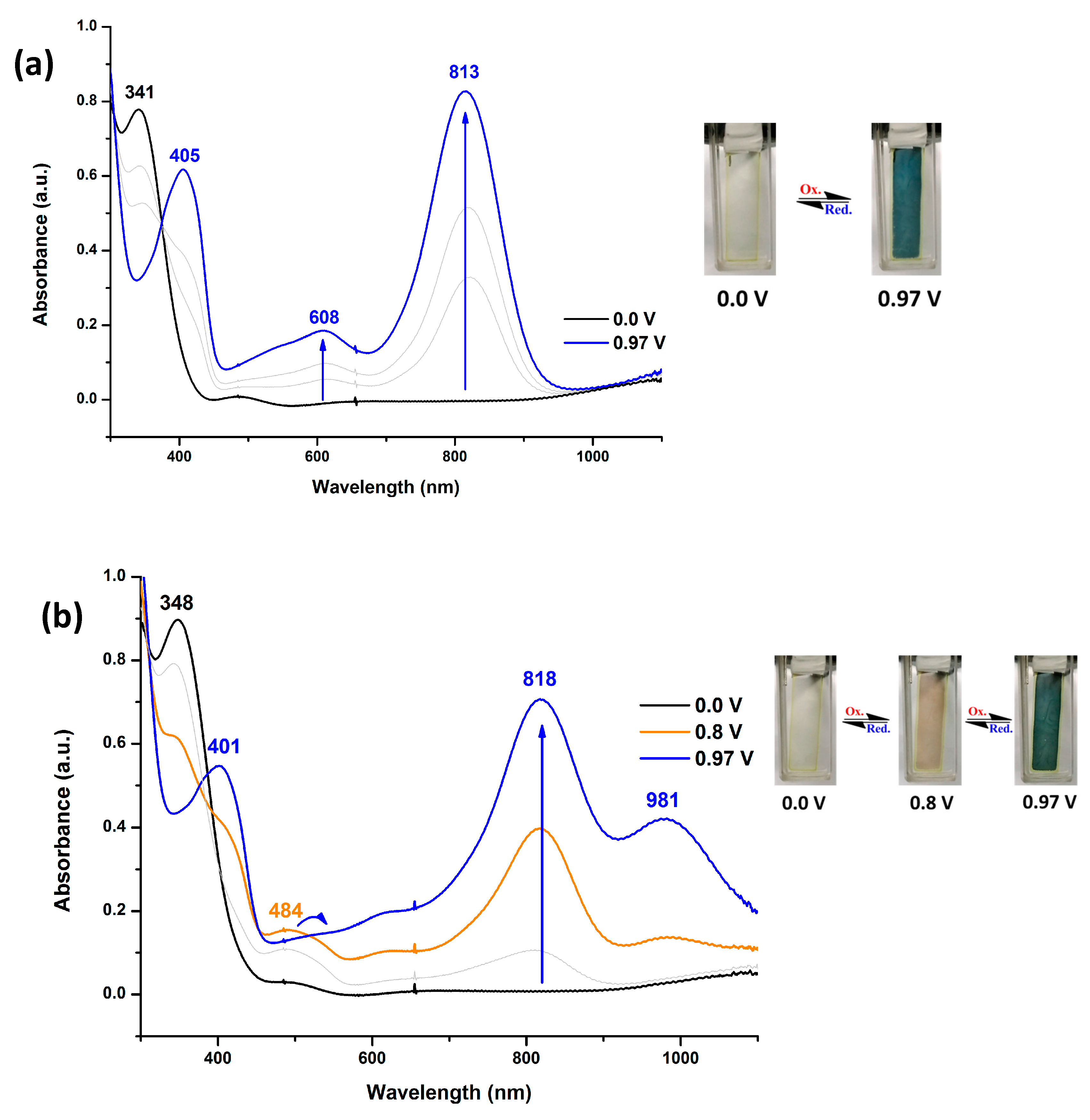
As shown in Figure 4, the film of PAI 6d with non-substituted TPA exhibited strong absorption at wavelength around 341 nm in the neutral form. Upon oxidation, two main bands at 405 and 813 nm appeared, while the main band of neutral state around 341 nm gradually decreases in intensity. The long wavelength absorption band which appears upon oxidation is characteristic of the cation-radical form. In the same time, the film turned from colorless (L*: 74; a*: 0; b*: 7) into blue-green (L*: 38; a*: −12; b*: 6) with increasing applied voltages. If this polymer was repeatedly scanned for fifty cycles, we found when the voltage was set at 0.8 V, a new peak around 484 nm appeared, and the film changed color to yellowish orange (L*: 63; a*: 4; b*: 17). When the applied voltage increased, new absorption bands around 818 and 981 nm appeared and the color of the film changed to blue-green. The new orange color state and the different absorption spectra implied that TPA underwent coupling reaction to form the tetraphenylbenzidine (TPB) moiety as shown in Scheme 3. Figure 5 depicts the spectral change of PAI 6d after various scanning cycles at 1.0 V. As the number of scanning cycle increased, the absorption peak at 981 nm intensified and the peak at 813 nm gradually shifted to 818 nm. The other 6 series PAIs showed a similar spectra and color change as that of 6d.
On the other hand, the methoxy or t-butyl-substituted TPA PAIs MeO-6d and t-Bu-6d exhibited strong absorption at wavelength around 340 nm in the neutral form as shown in Figure 6 and Figure S17, respectively. Upon oxidation, two main bands at 338-341 and 779-802 nm appears, while the main band of neutral state around 340 nm gradually decreases in intensity. The long wavelength absorption band which appears upon oxidation is characteristic of the TPA cation-radical form. In the same time, the films turned from colorless (MeO-6d: L*: 67; a*: 0; b*: 8, t-Bu-6d: L*: 67; a*: 1; b*: 4) into green (MeO-6d: L*: 44; a*: −24; b*: 10, t-Bu-6d: L*: 56; a*: -8; b*: 1) with increasing applied voltages. The methoxy or t-butyl-substituted PAIs revealedalmost the same absorption profile and color change as the initial ones after 50 repeated CV cycles. No orange coloring state was observed. Therefore, substitution at the para position of TPA with methoxy or t-butyl can hinder the coupling reaction. All the other MeO-6 and t-Bu-6 series PAIs showed a similar electrochromic behavior as those of MeO-6d and t-Bu-6d.
3.6. Electrochromic Switching Properties
Electrochromic switching studies for the polymers were performed to monitor the % transmittance (%T) as a function of time at their absorption maximum (λmax) and to determine the response time by stepping potential repeatedly between the neutral and oxidized states. The active area of the polymer film on ITO-glass is approximately 1 cm2. As a typical example, Figure 7 depicts the %T changes of PAIs 6d and MeO-6d as a function of time at its long-wavelength absorption maximum of 813 nm and 778 nm by applying square wave potential steps (between 0 and 1.05 V for 6d and between 0 and 0.85 V for MeO-6d) with a residence time of 10 s and 12 s, respectively. The optical contrast measured as Δ%T between neutral and oxidized state was found to be 82 % for 6d and 85% for MeO-6d in the first switching cycle. The PAI film of 6d showed a slight optical contrast loss after 50 full switches from 82% to 69%; however, MeO-6d showed almost no optical contrast loss during the first 50 switching cycles.
The response time was calculated at 90% of the full-transmittance change, because it is difficult to perceive any further color change with naked eye beyond this point. As shown in Figure 7(a), PAI 6d attained 90% of a complete coloring and bleaching in 2.9 and 1.4 s, respectively. Figure 7(b) indicates that the PAI MeO-6d attained 90% of a complete coloring and bleaching in 3.5 and 1.5s, respectively. The electrochromic coloring efficiency (CE) for the blue coloring (ΔOD813/Q) of the PAI 6d was estimated to be 157 cm2/C, and that for the green coloring (ΔOD778/Q) of the PAI MeO-6d was estimated to be 195 cm2/C. Figure S18 depicts the optical transmittance at 802 nm as a function of time by applying square-wave potential steps between 0 and 0.95 V for a resident time of 15 s for PAI t-Bu-6d. In general, most of the MeO-6 and t-Bu-6 series PAIs exhibited high electrochromic stability in the first fifty switching cycles. The electrochromic properties of all the polymer films during electro-oxidation processes are summarized in Table 4.
4. Conclusions
Three benzamide-containing diamine monomers 4,4’-bis(p-aminobenzamido)triphenylamine (4), 4,4’-bis(p-aminobenzamido)-4”-methoxytriphenylamine (MeO-4) and 4,4’-bis(p-aminobenzamido)-4”-tert-butyltriphenylamine (t-Bu-4) were synthesized and led to a series electroactive aromatic PAIs. Insertion of the benzamide spacer between imide ring and TPA unit can decrease the oxidation potential and enhance good electrochemical and electrochromic stability of the polymers. For the PAIs derived from diamine monomer 4, a coupling reaction between TPA units occurred during the oxidative process of the polymers because no protecting substituent is located at the p-position of the TPA pendent phenyl group. In contrast, the polymers with methoxy or tert-butyl-substituted TPA can hinder the coupling reaction. The PAIs show high electrochemical and electrochromic stability and strong color changes with high contrast ratio upon electro-oxidation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: IR spectra of diamine monomer 4 and its precursor compounds. Figure S2: IR spectra of diamine monomers MeO-4 and t-Bu-4 and their precursor compounds. Figure S3: 1H NMR, 13C NMR, H-H COSY, and C-H HMQC NM spectra of diamide-dinitro compound 3 in DMSO-d6. Figure S4: 1H NMR, 13C NMR, H-H COSY, and C-H HMQC NMR spectra of diamide-diamine 4 in DMSO-d6. Figure S5: 1H NMR, 13C NMR, H-H COSY, and C-H HMQC NMR spectra of diamide-dinitro compound MeO-3 in DMSO-d6. Figure S6: 1H NMR, 13C NMR, H-H COSY, and C-H HMQC NMR spectra of diamide-diamine MeO-4 in DMSO-d6. Figure S7: 1H NMR spectra of diamide-dinitro compound t-Bu-3 and diamide-diamine t-Bu-4 in DMSO-d6. Figure S8: IR spectra of PAI 6f and its poly(amide-amic acid) precursor. Figure S9: Proton NMR spectra of (a) PAI 6f, (b) PAI MeO-6f, and (c) PAI t-Bu-6f in DMSO-d6. Figure S10: DSC curves of the 6 series PAIs and typical TGA curves of PAI 6f. Figure S11: CV scans of the cast films of PAIs (a) 6a, (b) 6b, (c) 6c, (d) 6d, (e) 6e, and (f) 6f on an ITO-coated glass substrate in 0.1 M Bu4NClO4/MeCN solutions at a scan rate of 50 mV/s. Figure S12: Repetitive CV scans of the cast films of PAIs (a) 6a, (b) 6b, (c) 6c, (d) 6d, (e) 6e, and (f) 6f on an ITO-coated glass substrate in 0.1 M Bu4NClO4/MeCN solutions at a scan rate of 50 mV/s. Figure S13: CV scans of the cast films of PAIs (a) MeO-6a, (b) MeO-6b, (c) MeO-6c, (d) MeO-6d, (e) MeO-6e, and (f) MeO-6f on an ITO-coated glass substrate in 0.1 M Bu4NClO4/MeCN solutions at a scan rate of 50 mV/s. Figure S14: Repetitive CV scans of the cast films of PAIs (a) MeO-6a, (b) MeO-6b, (c) MeO-6c, (d) MeO-6d, (e) MeO-6e, and (f) MeO-6f on an ITO-coated glass substrate in 0.1 M Bu4NClO4/MeCN solutions at a scan rate of 50 mV/s. Figure S15: CV scans of the cast films of PAIs (a) t-Bu-6a, (b) t-Bu-6b, (c) t-Bu -6c, (d) t-Bu-6d, (e) t-Bu-6e, and (f) t-Bu-6f on an ITO-coated glass substrate in 0.1 M Bu4NClO4/MeCN solutions at a scan rate of 50 mV/s. Figure S16: Repetitive CV scans of the cast films of PAIs (a) t-Bu-6a, (b) t-Bu-6b, (c) t-Bu-6c, (d) t-Bu-6d, (e) t-Bu-6e, and (f) t-Bu-6f on an ITO-coated glass substrate in 0.1 M Bu4NClO4/MeCN solutions at a scan rate of 50 mV/s. Figure S17: Spectroelectrograms and color changes of PAI t-Bu-6d on an ITO-glass slide in 0.1 M Bu4NClO4/MeCN at various applied voltages (a) first cycle and (b) 50th cycle. Figure S18: Potential step absorptiometry of the cast film PAI t-Bu-6d of on ITO-glass slide (in MeCN with 0.1 M Bu4NClO4 as a supporting electrolyte) by applying a potential step between 0 and 0.95 V for a resident time of 15s at 802 nm.
Author Contributions
Investigation, Zong-De Ni; supervision, S.H. Hsiao; writing—original draft preparation, Zong-De Ni; writing—review and editing, S.H. Hsiao. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministry of Science and Technology, Taiwan.
Acknowledgments
The authors are grateful for the financial support from the Ministry of Science and Technology, Taiwan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rosseinsky, D.R.; Mortimer, R.J. Electrochromic Systems and the Prospects for Devices. Adv. Mater. 2001, 13, 783–793. [Google Scholar] [CrossRef]
- Monk, P.M.S.; Mortimer, R.J.; Rosseinsky, D.R. Electrochromism and Electrochromic Devices; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Beaujuge, P.M.; Reynolds, J.R. Color Control in π-Conjugated Organic Polymers for Use in Electrochromic Devices. Chem. Rev. 2010, 110, 268–320. [Google Scholar] [CrossRef]
- Gunbas, G.; Toppare, L. Electrochromic Conjugated Polyheterocycles and Derivatives—Highlights from the Last Decade towards Realization of Long Lived Aspirations. Chem. Commun. 2012, 48, 1083–1101. [Google Scholar] [CrossRef] [PubMed]
- Neo, W.; Ye, Q.; Chua, S.-J.; Xu, J. Conjugated Polymer-Based Electrochromics: Materials, Device Fabrication and Application Prospects. J. Mater. Chem. C 2016, 4, 7364–7376. [Google Scholar] [CrossRef]
- Lv, X.; Li, W.; Ouyang, M.; Zhang, Y.; Wright, D.S.; Zhang, C. Polymeric Electrochromic Materials with Donor-Acceptor Structures. J. Mater. Chem. C 2017, 5, 12–28. [Google Scholar] [CrossRef]
- Shirota, Y. Organic Materials for Electronic and Optoelectronic Devices, J. Mater. Chem. 2000, 10, 1–25. [Google Scholar] [CrossRef]
- Thelakkat, M. Star-Shaped, Dendrimeric and Polymeric Triarylamines as Photoconductors and Hole Transport Materials for Electro-optical Applications. Macromol. Mater. Eng. 2002, 287, 442–461. [Google Scholar] [CrossRef]
- Shirota, Y. Photo- and Electroactive Amorphous Molecular Materials—Molecular Design, Syntheses, Reactions, Properties, and Applications. J. Mater. Chem. 2005, 15, 75–93. [Google Scholar] [CrossRef]
- Oishi, Y.; Takado, H.; Yoneyama, M.; Kakimoto, M.; Imai, Y.; Kurosaki, T. Preparation and Properties of New Aromatic Polyamides from 4,4′-Diaminotriphenylamine and Aromatic Dicarboxylic Acids. J. Polym. Sci. Part A: Polym. Chem. 1990, 30, 1763–1769. [Google Scholar] [CrossRef]
- Oishi, Y.; Ishida, M.; Kakimoto, M.; Imai, Y.; Kurosaki, T. Preparation and Properties of Novel Soluble Aromatic Polyimides from 4,4′-Diaminotriphenylamine and Aromatic Tetracarboxylic Dianhydrides. J. Polym. Sci. Part A: Polym. Chem. 1992, 30, 1027–1035. [Google Scholar] [CrossRef]
- Cheng, S.-H.; Hsiao, S.-H.; Su, T.-H.; Liou, G.-S. Novel Aromatic Poly(amine-imide)s Bearing a Pendent Triphenylamine Group: Synthesis, Thermal, Photophysical, Electrochemical, and Electrochromic Characteristics. Macromolecules 2005, 38, 307–316. [Google Scholar] [CrossRef]
- Liou, G.-S.; Hsiao, S.-H.; Chen, H.-W. Novel High Tg Poly(amine-imide)s Bearing Pendent N-phenylcarbazole Units: Synthesis and Photophysical, Electrochemical and Electrochromic Properties. J. Mater. Chem. 2006, 16, 1831–1842. [Google Scholar] [CrossRef]
- Chang, C.-W.; Liou, G.-S.; Hsiao, S.-H. Highly Stable Anodic Green Electrochromic Aromatic Polyamides: Synthesis and Electrochromic Properties. J. Mater. Chem. 2007, 17, 1007–1015. [Google Scholar] [CrossRef]
- Li, S.; Liu, G.; Ju, X.; Zhang, Y.; Zhao, J. Synthesis, Characterization and Application of Four Novel Electrochromic Materials Employing Nitrotriphenylamine Unit as the Acceptor and Different Thiophene Derivatives as the Donor. Polymers 2017, 9, 173. [Google Scholar] [CrossRef]
- Gao, Z.; Zhao, F.; Ming, S.; Zhang, Y.; Zhao, J. Donor-acceptor Type Conjugated Porous Polymers Based on Triphenylamine and Benzothiadiazole Units as Ambipolar Electrochromic Materials. Polymer 2023, 274, 125908. [Google Scholar] [CrossRef]
- Lu, Q.; Cai, W.; Niu, H.; Wang, W.; Bai, X.; Hou, Y. Novel Polyamides with 5H-Dibenzo[b,f]azepin-5-yl-Substituted Triphenylamine: Synthesis and Visible-NIR Electrochromic Properties. Polymers 2017, 9, 543. [Google Scholar] [CrossRef]
- Li, D.; Shi, X.; Cai, W.; Niu, H. Fluorescence Switching of Triarylamine-based Polyamides with Pendant Pyrene Units and its Application in Electrochromic Smart Displays. Electrochim. Acta 2024, 508, 145277. [Google Scholar] [CrossRef]
- Wu, T.-Y.; Chung, H.-H. Applications of Tris(4-(thiophen-2-yl)phenyl)amine and Dithienylpyrrole-based Conjugated Copolymers in High-Contrast Electrochromic Devices. Polymers 2016, 8, 206. [Google Scholar] [CrossRef]
- Yen, H.-J.; Liou, G.-S. Solution-processable Triarylamine-based Electroactive High Performance Polymers for Anodically Electrochromic Applications. Polym. Chem. 2012, 3, 255–264. [Google Scholar] [CrossRef]
- Yen, H.-J.; Liou, G.-S. Recent Advances in Triphenylamine-based Electrochromic Derivatives and Polymers. Polym. Chem. 2018, 9, 3001–3018. [Google Scholar] [CrossRef]
- Yen, H.-J.; Liou, G.-S. Design and Preparation of Triphenylamine-based Polymeric Materials towards Emergent Optoelectronic Applications. Prog. Polym. Sci. 2019, 89, 250–287. [Google Scholar] [CrossRef]
- Wang, H.-M.; Hsiao, S.-H.; Liou, G.-S. Highly Stable Electrochromic Polyamides Based on N,N-Bis(4-aminophenyl)-N’,N’-bis(4-tert-butylphenyl)-1,4-phenylenediamine. J. Polym. Sci. Part A: Polym. Chem. 2009, 47, 2330–2343. [Google Scholar]
- Wang, H.-M.; Hsiao, S.-H. Electrochemically and Electrochromically Stable Polyimides Bearing tert-butyl-blocked N,N,N′,N′-Tetraphenyl-1,4-phenylenediamine units. Polymer 2009, 50, 1692–1699. [Google Scholar] [CrossRef]
- Hsiao, S.-H.; Wang, H.-M.; Liao, S.-H. Redox-stable and Visible/near-infrared Electrochromic Aramids with Main-chain Triphenylamine and Pendent 3,6-Di-tert-butylcarbazole Units. Polym. Chem. 2014, 5, 2473–2483. [Google Scholar] [CrossRef]
- Wang, H.-M.; Hsiao, S.-H. Enhancement of Redox Stability and Electrochromic Performance of Aromatic Polyamides by Incorporation of (3,6-Dimethoxycarbazol-9-yl)triphenylamine units. J. Polym. Sci. Part A: Polym. Chem. 2014, 52, 272–286. [Google Scholar] [CrossRef]
- Wang, H.-M.; Hsiao, S.-H. Substituent Effects on Electrochemical and Electrochromic Properties of Aromatic Polyimides with 4-(Carbazol-9-yl)triphenylamine Moieties. J. Polym. Sci. Part A: Polym. Chem. 2014, 52, 1172–1184. [Google Scholar] [CrossRef]
- Chern, Y.-T.; Yen, C.-C.; Wang, J.-M.; Lu, I.-S.; Huang, B.-W.; Hsiao, S.-H. Redox-Stable and Multicolor Electrochromic Polyamides with Four Triarylamine Cores in the Repeating Unit. Polymers 2024, 16, 1644. [Google Scholar] [CrossRef]
Scheme 1.
Synthetic routes to TPA-based diamide-diamine monomers 4, MeO-4 and t-Bu-4.
Figure 1.
1H NMR and H-H COSY of diamide-diamine MeO-4 in DMSO-d6.
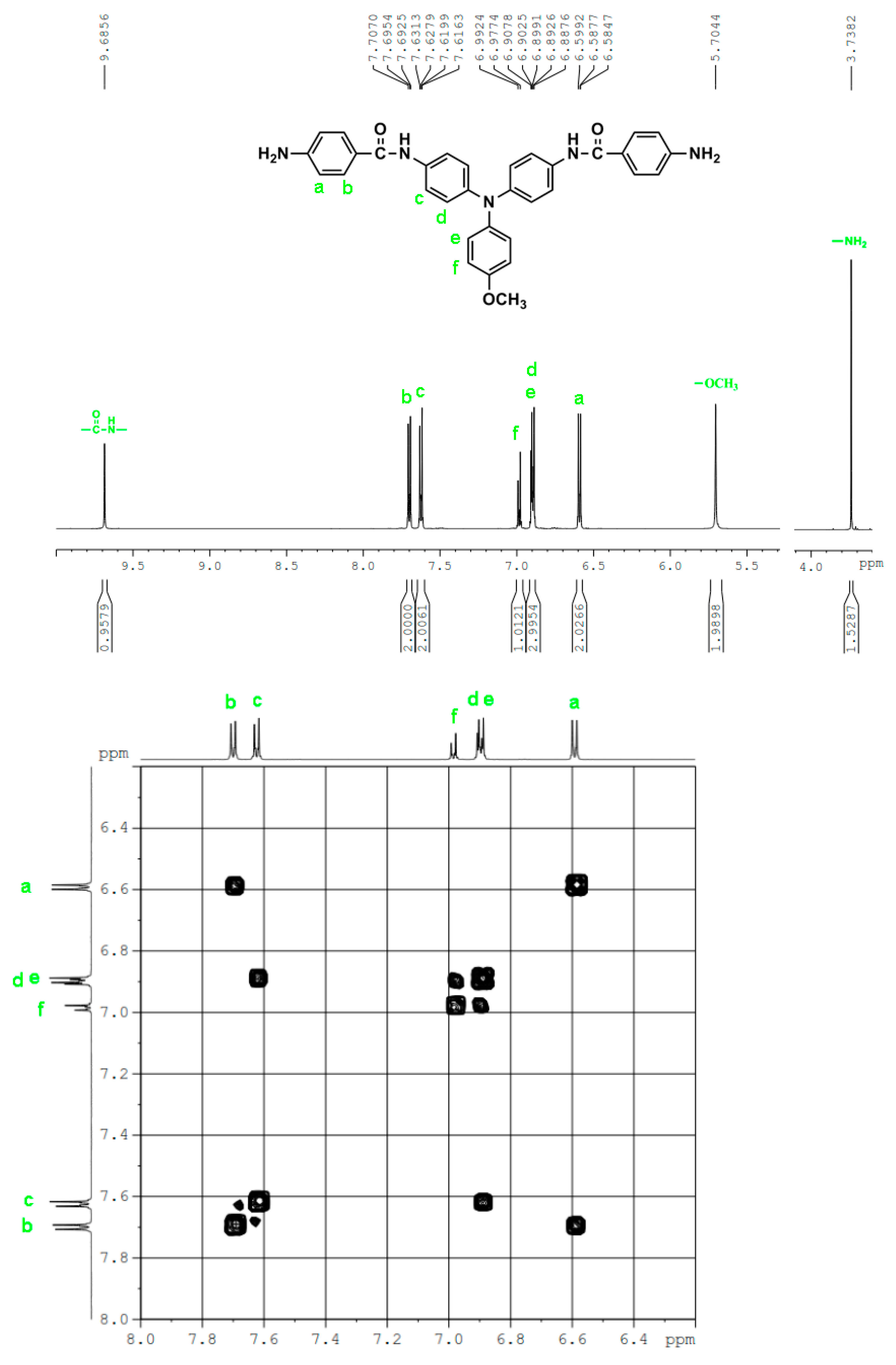
Figure 2.
13C NMR and C-H HMQC of diamide-diamine MeO-4 in DMSO-d6.
Scheme 2.
Synthesis of poly(amide-imide)s.
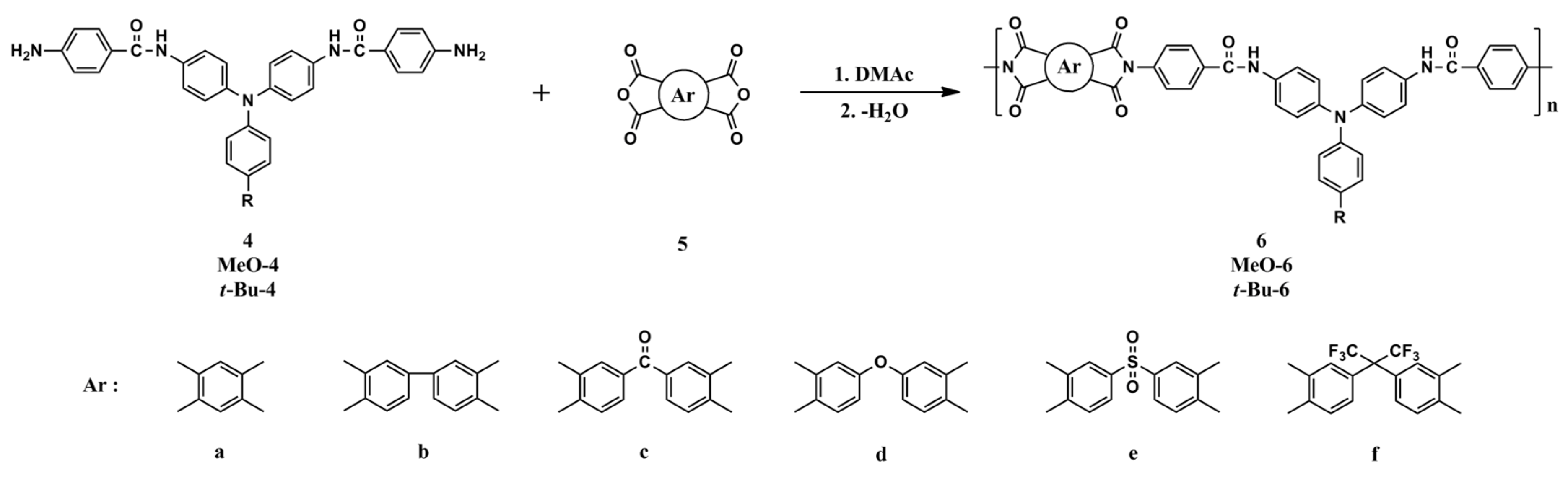
Figure 3.
CV diagrams of PAIs (a) MeO-6d and (b) MeO-6’d on an ITO-glass slide in MeCN (0.1 M Bu4NClO4) at a scan rate 50 mV/s.
Figure 3.
CV diagrams of PAIs (a) MeO-6d and (b) MeO-6’d on an ITO-glass slide in MeCN (0.1 M Bu4NClO4) at a scan rate 50 mV/s.
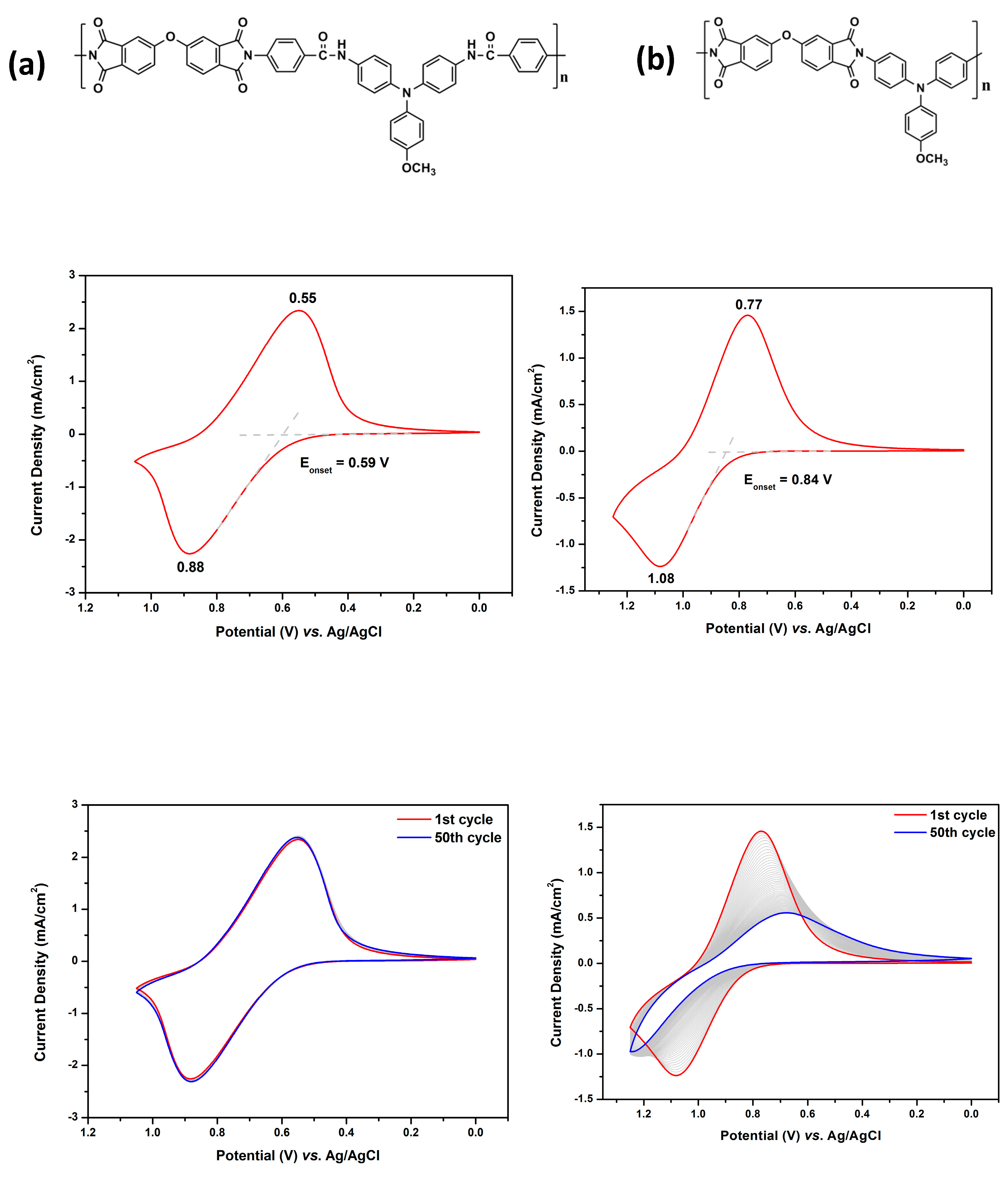
Figure 4.
Spectroelectrograms and color changes of PAI 6d on an ITO-glass slide in 0.1 M Bu4NClO4MeCN at various applied voltages (a) first cycle and (b) after 50 cycling.
Figure 4.
Spectroelectrograms and color changes of PAI 6d on an ITO-glass slide in 0.1 M Bu4NClO4MeCN at various applied voltages (a) first cycle and (b) after 50 cycling.
Scheme 3.
The proposed coupling reaction of TPA units of PAI 6d during applied voltage cycling between 0 and 1.0 V.
Scheme 3.
The proposed coupling reaction of TPA units of PAI 6d during applied voltage cycling between 0 and 1.0 V.
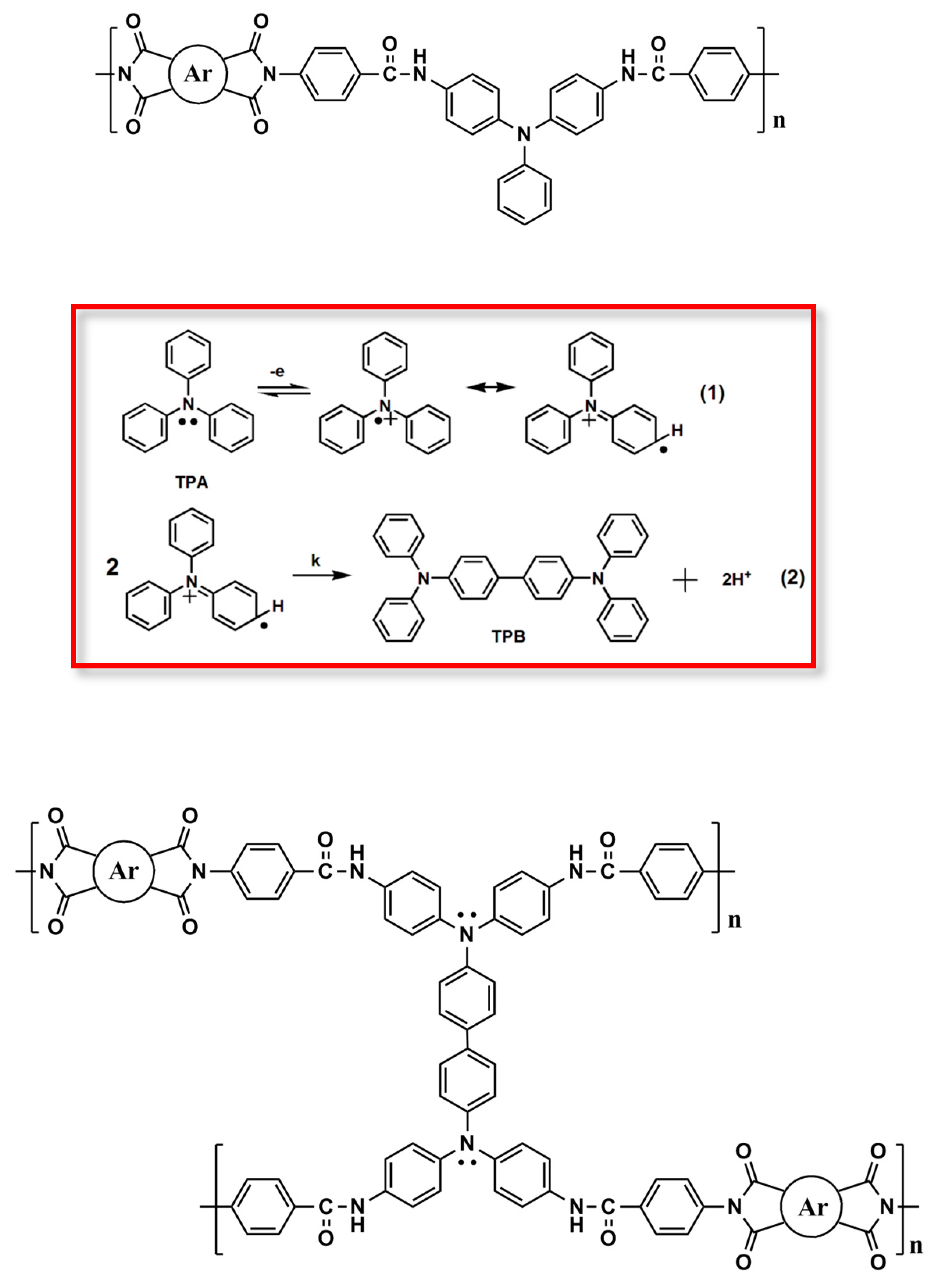
Figure 5.
Spectral changes of polymer film 6d on an ITO-glass slide in 0.1 M Bu4NClO4/MeCN after various scanning cycles at applied voltage of 1.0 V.
Figure 5.
Spectral changes of polymer film 6d on an ITO-glass slide in 0.1 M Bu4NClO4/MeCN after various scanning cycles at applied voltage of 1.0 V.
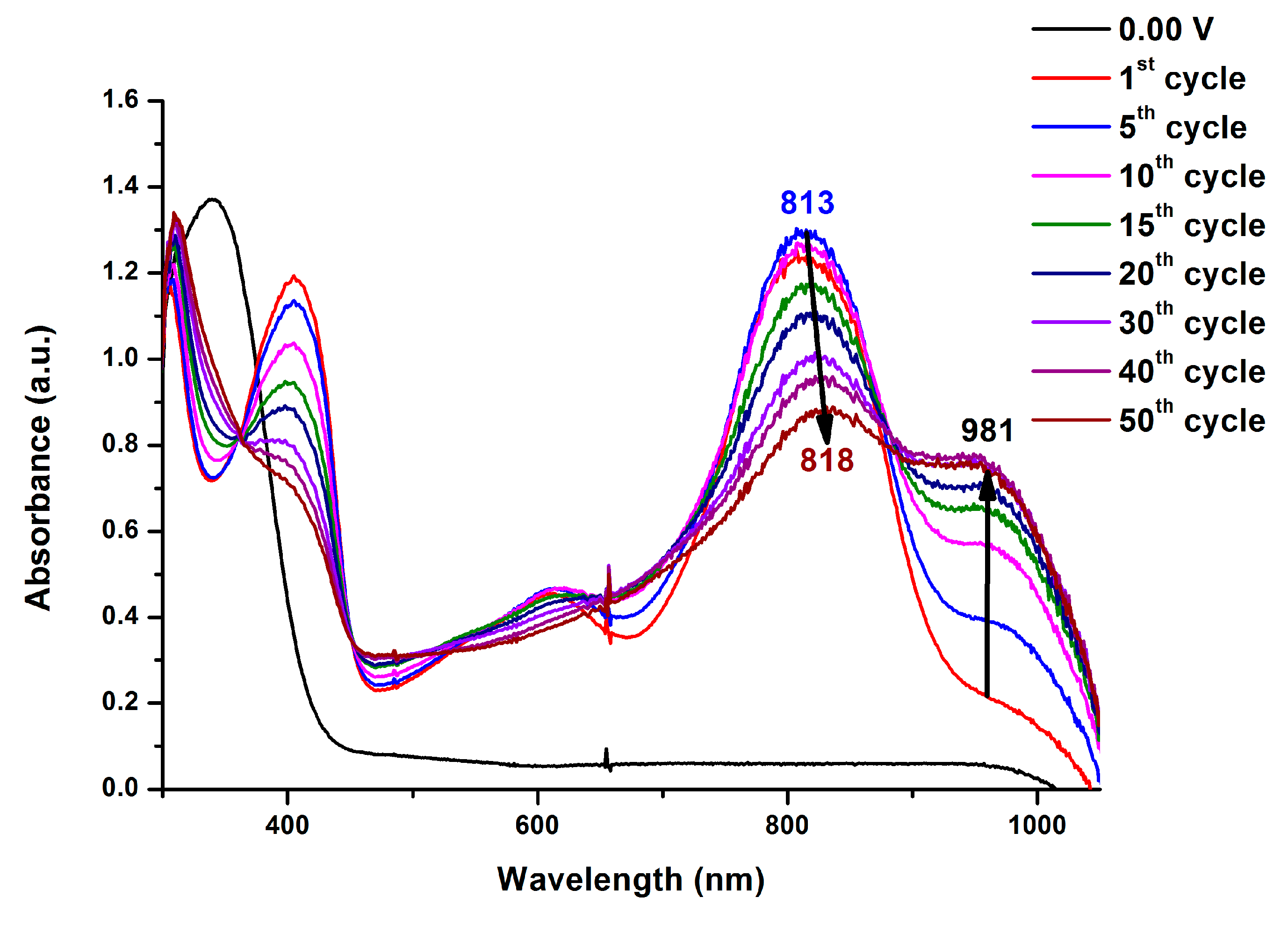
Figure 6.
Spectroelectrograms and color changes of PAI MeO-6d on an ITO-glass slide in 0.1 M Bu4NClO4/MeCN at various applied voltages (a) first cycle and (b) 50th cycle.
Figure 6.
Spectroelectrograms and color changes of PAI MeO-6d on an ITO-glass slide in 0.1 M Bu4NClO4/MeCN at various applied voltages (a) first cycle and (b) 50th cycle.
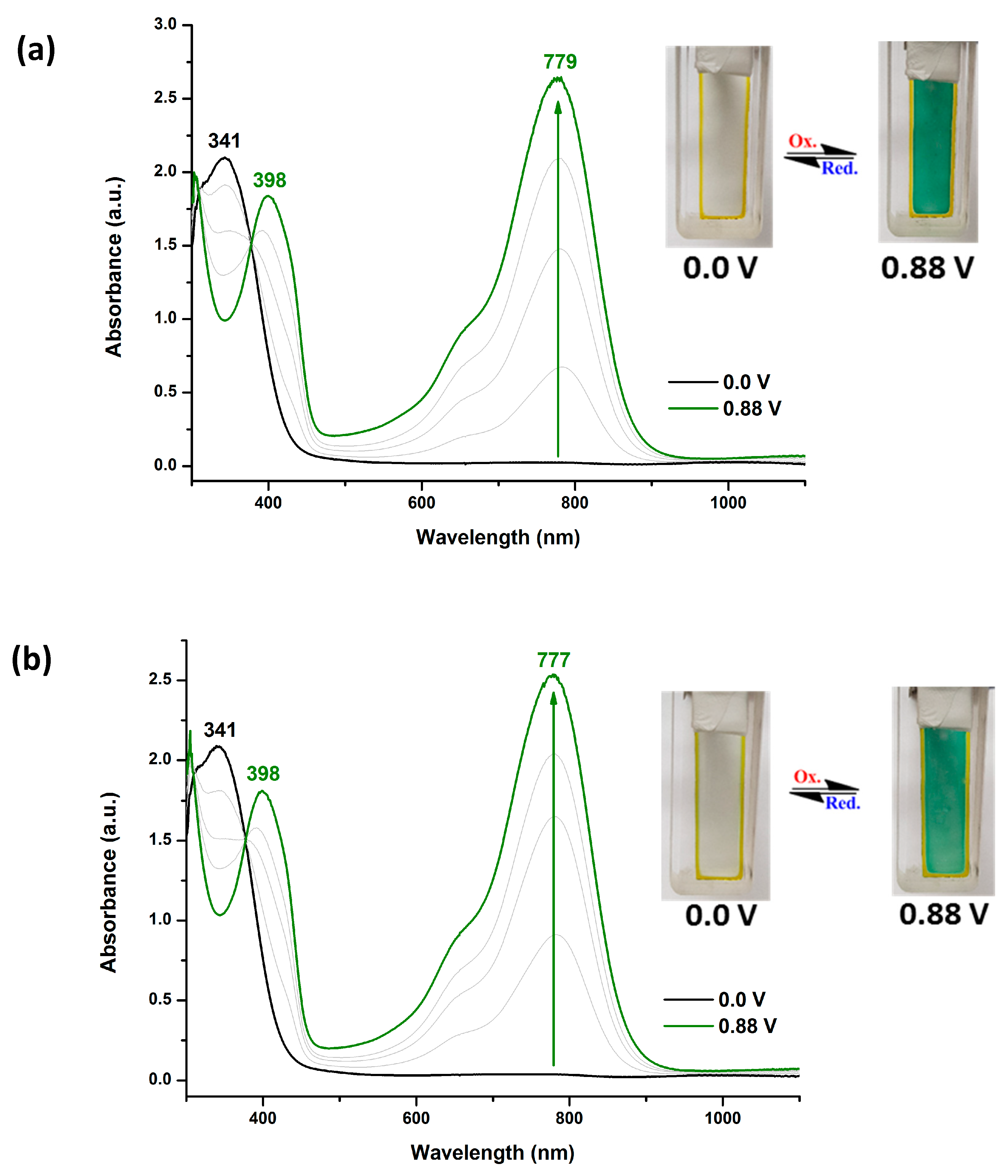
Figure 7.
Potential step absorptiometry of the cast film on ITO-glass slide (in MeCN with 0.1 M Bu4NClO4 as a supporting electrolyte) by applying a potential step: (a) optical switching for 6d at potential between 0.0 V and 1.05 V and residence time 10 s, monitored at λmax = 813 nm; (b) MeO-6d at potential between 0.0 V and 0.85 V and residence time 12 s, monitored at λmax = 778 nm.
Figure 7.
Potential step absorptiometry of the cast film on ITO-glass slide (in MeCN with 0.1 M Bu4NClO4 as a supporting electrolyte) by applying a potential step: (a) optical switching for 6d at potential between 0.0 V and 1.05 V and residence time 10 s, monitored at λmax = 813 nm; (b) MeO-6d at potential between 0.0 V and 0.85 V and residence time 12 s, monitored at λmax = 778 nm.

Table 1.
Inherent viscosity and solubility behavior of PAIs.
| Polymer code | ηinh (dL/g)a | Solventsb,c | |||||||
| PAA | PAI | NMP | DMAc | DMF | DMSO | m-Cresol | THF | ||
| 6a | 0.69 | ‒ | +‒ | +‒ | +‒ | +‒ | ‒ | ‒ | |
| 6b | 0.91 | ‒ | +‒ | +‒ | +‒ | +‒ | ‒ | ‒ | |
| 6c | 0.67 | 0.66 | + | + | +‒ | + | +‒ | ‒ | |
| 6d | 0.50 | 0.49 | + | + | +‒ | + | + | ‒ | |
| 6e | 0.29 | 0.35 | + | + | + | + | +‒ | ‒ | |
| 6f | 0.68 | 0.64 | + | + | + | + | + | ‒ | |
| MeO-6a | 0.74 | 0.77 | + | + | + | + | +- | ‒ | |
| MeO-6b | 0.87 | 0.88 | + | + | +- | + | +- | ‒ | |
| MeO-6c | 0.58 | 0.58 | + | + | + | + | + | ‒ | |
| MeO-6d | 0.70 | 0.70 | + | + | +- | + | + | ‒ | |
| MeO-6e | 0.59 | 0.57 | ++ | ++ | ++ | ++ | + | ‒ | |
| MeO-6f | 0.92 | 0.56 | ++ | ++ | ++ | ++ | + | +‒ | |
| t-Bu-6a | 0.40 | 0.80 | + | + | + | + | + | ‒ | |
| t-Bu-6b | 0.45 | 0.65 | + | + | + | + | + | ‒ | |
| t-Bu-6c | 0.65 | 0.64 | + | + | + | + | + | ‒ | |
| t-Bu-6d | 0.39 | 0.58 | + | + | + | + | + | ‒ | |
| t-Bu-6e | 0.46 | 0.43 | ++ | ++ | ++ | ++ | + | ‒ | |
| t-Bu-6f | 0.48 | 0.50 | ++ | ++ | ++ | ++ | + | +‒ | |
a Inherent viscosity measured at a concentration of 0.5 dL/g in DMAc at 30 oC. b The qualitative solubility was tested with 10 mg of a sample in 1 mL of stirred solvent. ++: soluble at room temperature; +: soluble on heating; +‒ : partially soluble; ‒ : insoluble even on heating. c Solvent: NMP: N-methyl-2-pyrrolidone; DMAc: N,N-dimethylacetamide; DMF: N,N-dimethylformamide; DMSO: dimethyl sulfoxide; THF: tetrahydrofuran.
Table 2.
Thermal properties of PAIs.
| Polymer code |
Tg (℃)a | Td at 5% Weight Loss (℃)b | Td at 10% Weight Loss (℃)b | Char Yield (wt %)c | ||
| N2 | air | N2 | air | |||
| 6a | 355 | 525 | 537 | 553 | 575 | 58 |
| 6b | 326 | 545 | 550 | 578 | 600 | 60 |
| 6c | 304 | 532 | 539 | 576 | 589 | 55 |
| 6d | 296 | 504 | 488 | 541 | 549 | 58 |
| 6e | 331 | 496 | 514 | 523 | 559 | 58 |
| 6f | 326 | 549 | 535 | 585 | 576 | 62 |
| MeO-6a | 325 | 464 | 438 | 495 | 469 | 68 |
| MeO-6b | 323 | 483 | 482 | 519 | 520 | 66 |
| MeO-6c | 299 | 500 | 476 | 532 | 522 | 50 |
| MeO-6d | 286 | 501 | 473 | 539 | 509 | 60 |
| MeO-6e | 300 | 444 | 447 | 562 | 478 | 52 |
| MeO-6f | 326 | 480 | 473 | 522 | 511 | 55 |
| t-Bu-6a | 350 | 527 | 515 | 560 | 554 | 58 |
| t-Bu-6b | 304 | 513 | 522 | 549 | 556 | 58 |
| t-Bu-6c | 309 | 479 | 473 | 512 | 522 | 55 |
| t-Bu-6d | 289 | 501 | 509 | 532 | 554 | 56 |
| t-Bu-6e | 313 | 423 | 437 | 448 | 471 | 50 |
| t-Bu-6f | 322 | 511 | 486 | 552 | 525 | 58 |
a Midpoint temperature of the baseline shift on the second DSC heating trace (rate = 20 oC/min) after quenching from 400 to 50 oC in nitrogen. b Decomposition temperature at which a 5 % or 10 % weight loss was recorded by TGA at a heating rate of 20 oC/min and a gas flow rate of 20 cm3/min. c Residual weight % at 800 oC at a scan rate 20 oC/min in nitrogen.
Table 3.
Optical and electrochemical properties of the polymers.
| Polymer code |
Thin film (nm) | Oxidation potential (V)a |
Egopt (eV)b |
HOMO (eV)c | LUMO (eV)d | ||
| λmax | λonset | Eonset | E1/2Ox | ||||
| 6a | 330 | 420 | 0.70 | 0.84 | 2.95 | 5.20 | 2.25 |
| 6b | 338 | 407 | 0.71 | 0.83 | 3.05 | 5.19 | 2.14 |
| 6c | 346 | 418 | 0.70 | 0.83 | 2.97 | 5.19 | 2.22 |
| 6d | 341 | 411 | 0.70 | 0.82 | 3.02 | 5.18 | 2.16 |
| 6e | 344 | 409 | 0.70 | 0.82 | 3.03 | 5.18 | 2.15 |
| 6f | 342 | 406 | 0.72 | 0.83 | 3.05 | 5.19 | 2.14 |
| MeO-6a | 343 | 425 | 0.56 | 0.71 | 2.92 | 5.07 | 2.15 |
| MeO-6b | 344 | 413 | 0.59 | 0.71 | 3.00 | 5.07 | 2.07 |
| MeO-6c | 347 | 422 | 0.59 | 0.72 | 2.94 | 5.08 | 2.14 |
| MeO-6d | 341 | 425 | 0.59 | 0.72 | 2.92 | 5.08 | 2.16 |
| MeO-6e | 348 | 425 | 0.59 | 0.71 | 2.92 | 5.07 | 2.15 |
| MeO-6f | 346 | 419 | 0.59 | 0.72 | 2.96 | 5.08 | 2.12 |
| t-Bu-6a | 333 | 421 | 0.67 | 0.82 | 2.94 | 5.18 | 2.24 |
| t-Bu-6b | 336 | 406 | 0.64 | 0.78 | 3.05 | 5.14 | 2.09 |
| t-Bu-6c | 332 | 421 | 0.63 | 0.77 | 2.95 | 5.13 | 2.18 |
| t-Bu-6d | 339 | 414 | 0.68 | 0.80 | 3.00 | 5.16 | 2.16 |
| t-Bu-6e | 341 | 421 | 0.65 | 0.78 | 2.95 | 5.14 | 2.19 |
| t-Bu-6f | 345 | 413 | 0.66 | 0.79 | 3.00 | 5.15 | 2.15 |
a vs. Ag/AgCl in CH3CN. E1/2 = average potential of redox couple peaks. b Bandgap calculated from absorption edge of the polymer film. Energy gap = 1240/λonset. c The HOMO energy levels were calculated from E1/2Ox values of CV curves and were referenced to ferrocene (4.8 eV relative to the vacuum energy level). d LUMO = HOMO ‒ Egopt.
Table 4.
Electrochromic properties of PAIs.
| Polymer code | λmaxa (nm) | Δ%T | Response time b | ΔODc | Qdd (mC/cm2) | CEe (cm2/C) | |
| tc (s) | tb (s) | ||||||
| 6a | 804 | 77 | 3.7 | 2.5 | 1.35 | 16.42 | 82 |
| 6b | 818 | 73 | 4.8 | 2.3 | 0.85 | 9.29 | 91 |
| 6c | 818 | 52 | 2.9 | 1.1 | 0.33 | 1.82 | 182 |
| 6d | 813 | 82 | 3.0 | 1.4 | 0.73 | 4.63 | 157 |
| 6e | 810 | 56 | 3.2 | 1.0 | 0.38 | 2.85 | 131 |
| 6f | 809 | 89 | 6.3 | 3.2 | 1.36 | 9.36 | 145 |
| MeO-6a | 779 | 74 | 4.4 | 2.0 | 0.88 | 6.71 | 131 |
| MeO-6b | 779 | 69 | 2.6 | 1.4 | 0.77 | 5.07 | 153 |
| MeO-6c | 780 | 80 | 5.4 | 3.1 | 0.63 | 4.55 | 140 |
| MeO-6d | 779 | 85 | 3.5 | 1.5 | 0.74 | 4.34 | 195 |
| MeO-6e | 780 | 86 | 3.2 | 1.7 | 1.06 | 7.17 | 148 |
| MeO-6f | 775 | 79 | 2.2 | 3.0 | 1.31 | 9.65 | 136 |
| t-Bu-6a | 791 | 97 | 2.7 | 2.0 | 1.63 | 10.39 | 157 |
| t-Bu-6b | 779 | 88 | 3.2 | 1.6 | 0.92 | 5.23 | 176 |
| t-Bu-6c | 799 | 77 | 3.1 | 1.4 | 0.63 | 4.63 | 136 |
| t-Bu-6d | 802 | 88 | 3.1 | 1.4 | 0.96 | 6.62 | 144 |
| t-Bu-6e | 795 | 72 | 2.9 | 1.4 | 0.57 | 4.42 | 128 |
| t-Bu-6f | 796 | 62 | 3.1 | 1.2 | 0.43 | 5.73 | 75 |
a Wavelength of absorption maximum. b Time for 90% of the full-transmittance change. c Optical Density (ΔOD) = log[Tbleached/Tcolored], where Tcolored and Tbleached are the maximum transmittance in the oxidized and neutral states, respectively. d Qd is ejected charge, determined from the in situ experiments. e Coloration efficiency (CE) = ΔOD/Qd.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



