
Submitted:
03 April 2025
Posted:
07 April 2025
You are already at the latest version
Abstract
Generative artificial intelligence (AI) has emerged as a disruptive paradigm in molecular science, enabling algorithmic navigation and construction of chemical and proteomic spaces through data-driven modeling. This review systematically delineates the theoretical underpinnings, algorithmic architectures, and translational applications of deep generative models—including variational autoencoders (VAEs), generative adversarial networks (GANs), autoregressive transformers, and score-based denoising diffusion probabilistic models (DDPMs)—in the rational design of bioactive small molecules and functional proteins. We examine the role of latent space learning, probabilistic manifold exploration, and reinforcement learning in inverse molecular design, focusing on optimization of pharmacologically relevant objectives such as ADMET profiles, synthetic accessibility, and target affinity. Furthermore, we survey advancements in graph-based molecular generative frameworks, LLM-guided protein sequence modeling, and diffusion-based structural prediction pipelines (e.g., RFdiffusion, FrameDiff), which have demonstrated state-of-the-art performance in de novo protein engineering and conformational sampling. Generative AI is also catalyzing a paradigm shift in structure-based drug discovery via AI-augmented molecular docking (e.g., DiffDock), end-to-end binding affinity prediction, and quantum chemistry-informed neural potentials. We explore the convergence of generative models with Bayesian retrosynthesis planners, self-supervised pretraining on ultra-large chemical corpora, and multimodal integration of omics-derived features for precision therapeutics. Finally, we discuss translational milestones wherein AI-designed ligands and proteins have progressed to preclinical and clinical validation, and speculate on the synthesis of generative AI, closed-loop automation, and quantum computing in future autonomous molecular design ecosystems.
Keywords:
Generative AI
; Molecular Design
; Protein Engineering
; Diffusion Models
; Drug Discovery
1. Introduction
Drug discovery is traditionally costly, slow, and failure-prone (1). Preclinical discovery takes over five years, consuming one-third of total costs (2). With fewer than 10% of candidates succeeding, R&D expenditure per new drug exceeds $2 billion, mainly due to failures (3,4). Many failures stem from safety/efficacy issues emerging late (5). AI has recently accelerated in silico modeling across the pipeline, improving QSAR-based virtual screening and ML-driven protein engineering (6).
Historically, rule-based de novo drug design (e.g., LUDI, PRO_LIGAND) explored limited chemical space due to human bias (6,7,8). Generative AI overcomes this by learning molecular patterns and creating novel compounds (10). Unlike classical methods that recombine known motifs, it explores uncharted chemical space (11). Given an estimated >1060 drug-like molecules, AI efficiently samples viable candidates via chemical manifolds (12). AI designs millions of molecules in the time it takes for manual design, optimizing multiple properties simultaneously (13). Recent pipelines enhance synthetic feasibility and drug-likeness.
AI-driven discovery integrates generative models with computational chemistry, transitioning from empirical screening to rational design (14). Mid-20th-century drug discovery relied on trial-and-error; later, structure-based (X-ray) and ligand-based (pharmacophore) methods emerged (15). The 2000s saw docking and QSAR improvements (16). AI now creates novel molecules, with the first AI-designed drug (DSP-1181) entering trials in 2020 (17).
In AI workflows, generative models design de novo molecules, filtered via predictive models (binding affinity, ADMET) (18,19). Top hits undergo docking, synthesis planning, and wet-lab validation. Retrosynthesis AI suggests lab synthesis routes, while experimental feedback refines models (20). AI continuously self-improves, navigating chemical space intelligently. Future directions include autonomous discovery, quantum computing, and regulatory frameworks (21).
2. Deep Generative Models: Core Architectures
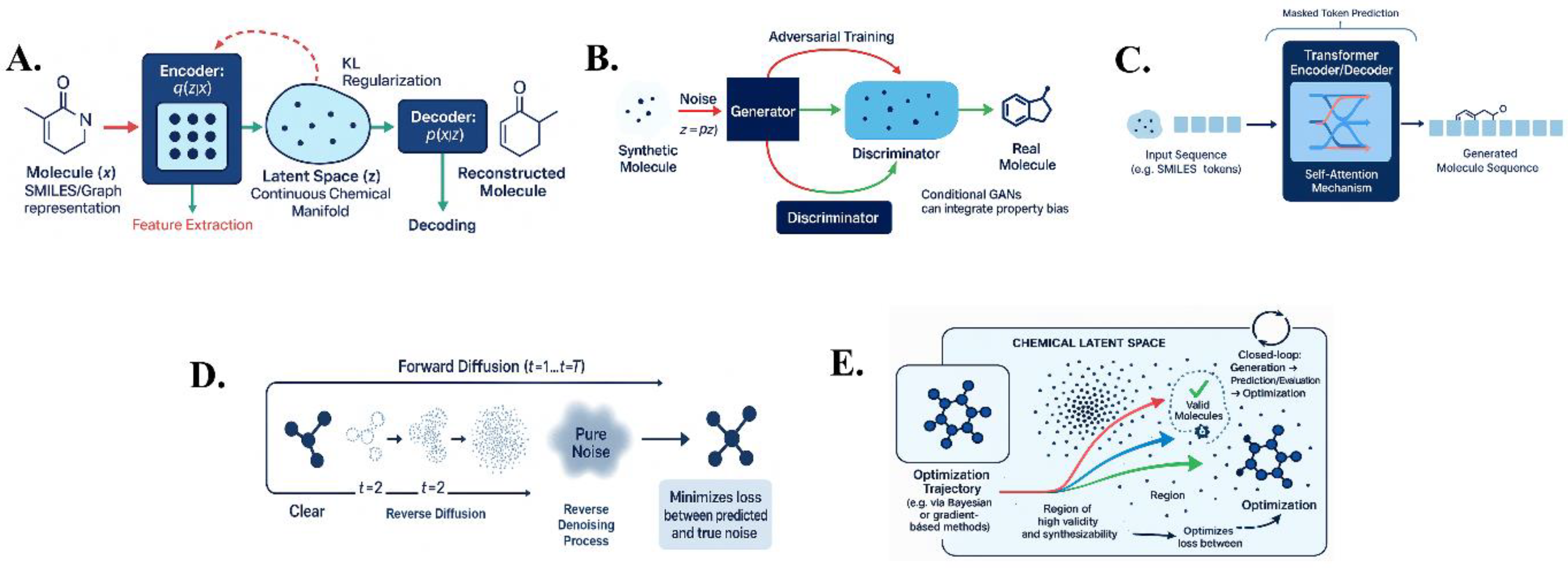
Deep generative models enable de novo molecular design by learning statistical patterns in chemical datasets to generate novel, valid compounds. The five principal architectures and optimization strategies are illustrated in Figure 1, providing a conceptual overview of their roles in molecular generation
2.1. Variational Autoencoders (VAEs)
A VAE comprises an encoder that compresses a molecule (typically represented as a SMILES string or molecular graph) into a continuous latent vector, and a decoder that reconstructs the molecule from this vector (22). Trained on large compound datasets, the encoder maps structurally similar molecules to proximate points in latent space, effectively learning a continuous chemical manifold (23). The training objective is to maximize data reconstruction likelihood while enforcing the latent vectors to follow a smooth, multi-dimensional Gaussian distribution using Kullback–Leibler (KL) regularization, enabling meaningful interpolation (24). The model optimizes the evidence lower bound (ELBO):
Where is the encoder (approximate posterior), and is the decoder (likelihood). Once trained, a random vector can be decoded into a novel molecule. Demonstrated for drug-like molecules by Gómez-Bombarelli et al. (2016–2017), VAEs enabled direct optimization in latent space to search for molecules with improved properties (25). The continuous latent space allows smooth interpolation between compounds (chemical morphing) to explore analogs (26). However, early SMILES-based VAEs often generated invalid or implausible structures (27). To address this, chemically informed decoders like Junction-Tree VAE were introduced, generating molecules as a tree of substructures, ensuring valency constraints are satisfied (28). VAEs have also been extended to 3D molecular conformations (23).
VAEs provide a principled framework grounded in Bayesian inference, ensuring training stability and interpretability (29). Despite limitations in output validity and reconstruction bias, VAEs are frequently combined with latent space optimization techniques such as Bayesian optimization or gradient-based methods to identify latent vectors yielding molecules with desirable properties (30). While their abstract latent space can hinder direct property optimization, VAEs remain foundational in molecular generation (31).
2.2. Generative Adversarial Networks (GANs)
GANs approach generation differently. Instead of modeling data likelihood explicitly, GANs set up a two-player game between a generator and a discriminator (32). The generator creates molecules from random noise, while the discriminator distinguishes real from generated samples. Adversarial training forces the generator to produce realistic molecules (33). Applied to molecules in 2018 (e.g., ORGAN by Insilico Medicine), GANs used an RNN generator to output SMILES strings and a discriminator that rewarded drug-like outputs (34). Conditional GANs guide generation towards desired properties (e.g., target binding) by conditioning on context (35).
Figure 1.
Architectures of deep generative models and latent space optimization in molecular design. (A) Variational Autoencoders (VAEs), (B) Generative Adversarial Networks (GANs), (C) Transformer-based models, and (D) Denoising Diffusion Models generate molecules using distinct mechanisms. (E) Latent space optimization explores continuous chemical manifolds to design molecules with desired properties.
Figure 1.
Architectures of deep generative models and latent space optimization in molecular design. (A) Variational Autoencoders (VAEs), (B) Generative Adversarial Networks (GANs), (C) Transformer-based models, and (D) Denoising Diffusion Models generate molecules using distinct mechanisms. (E) Latent space optimization explores continuous chemical manifolds to design molecules with desired properties.
GANs face challenges in molecular domains due to discrete outputs: the generator’s character-sequence output is non-differentiable. Solutions include policy gradient reinforcement learning and differentiable relaxations. Insilico’s Adversarial Threshold Neural Computer integrated GANs with reinforcement learning, using a differentiable neural computer as the generator and providing external rewards based on pharmacological properties (36). This hybrid generated a high percentage of valid, unique, and property-optimized molecules, while also incorporating synthesizability constraints (37). MolGAN, another milestone, generated molecular graphs (atom and bond matrices) directly. It achieved nearly 100% validity, improved synthetic accessibility, and solubility profiles compared to ORGAN (38).
Despite these advances, GANs may suffer from mode collapse and training instability. Their learned distribution might not cover the full chemical space (39). However, conditional GANs remain powerful for generating analogs of lead compounds (40). Overall, GANs introduce adversarial learning into molecular design, emphasizing realistic outputs and targeted objectives, though maintaining output diversity and validity requires care.
2.3. Transformer-Based Models
Transformer-based models, originally developed for NLP, are also applied to molecular science (41). Their self-attention mechanism captures long-range dependencies in sequences. For small molecules (SMILES or SELFIES) and protein sequences, transformers treat chemistry as a language. For example, ChemBERTa, trained on millions of SMILES strings using masked token prediction, produces rich, label-free molecular embeddings suitable for downstream tasks such as property prediction or generative modelling (42,43).
Transformers can generate molecules token-by-token (GPT-style), learning chemical syntax analogous to grammar. Such models can be conditioned to bias outputs toward specific property profiles (44). In lead optimization, transformers have proposed structural analogs based on known series (45). Protein language models (e.g., ProGen with 1.2B parameters trained on ~280M protein sequences) treat amino acid sequences like sentences. ProGen has generated functional enzymes with catalytic activity comparable to natural lysozymes, despite ~30% sequence identity. X-ray crystallography confirmed correct folds and active-site geometries (46,47). Transformers also support sequence-to-sequence tasks like codon optimization or property prediction (48).
These models bridge sequence and structure, enabling protein and molecule generation via attention-based encoding of complex dependencies. They leverage large unlabeled datasets for self-supervised learning, yielding representations useful for property prediction, structure generation, and analog design (49,50).
2.4. Denoising Diffusion Models (DDPMs)
DDPMs represent the latest generative modeling wave. They iteratively corrupt data with Gaussian noise and learn to reverse this process. A forward Markov chain adds noise over T steps until the sample becomes pure noise. A neural network then learns to reverse the corruption by predicting denoised data at each step. Training minimizes a reweighted variational bound, typically reducing to the loss between predicted and true noise, equivalent to learning the score function (51).
Generation begins from random noise and progressively reconstructs data, enabling generation in the original space (e.g., 3D coordinates of atoms) rather than latent space. This supports highly diverse and high-quality outputs (52,53). Diffusion models have been applied to 2D molecular graphs, 3D conformations, and protein structures (54). For instance, graph diffusion models like GeoDiff (55) and RFdiffusion (56) add noise to adjacency and node feature matrices or 3D coordinates and reconstruct valid molecular structures, preserving symmetry and chemical rules. DiffDock (57), using SE(3)-equivariant diffusion, generates ligand poses in binding sites by diffusing atomic positions.
Mathematically, as , the model can approximate any data distribution, offering theoretical guarantees absent in VAEs or GANs (58). Though generation is slow due to multiple neural evaluations, recent innovations like DDIMs have reduced required steps. Diffusion models enable unconditional generation with high validity and conditional generation guided by context (e.g., pharmacophores, protein pockets). RFdiffusion can be prompted with a protein backbone motif to generate a full structure incorporating it, resulting in functional de novo binders (54,56).
VAEs, GANs, transformers, and diffusion models each offer a distinct lens on learning and sampling chemical space (59). VAEs provide continuous latent embeddings and stable training. GANs deliver adversarial realism and property-driven design (60). Transformers model long-range dependencies in molecular/protein sequences, leveraging large datasets. Diffusion models refine samples from noise with high fidelity, especially in complex structured outputs. Modern workflows often combine models: e.g., using a transformer or VAE to generate candidates, then refining with a Diffusion model, or using a GAN to further optimize properties. Hybrid architectures (e.g., Diffusion models using transformers, or VAEs with GAN-style discriminators) are increasingly common (60–64).
2.5. Theoretical Considerations in Chemical Space Exploration
2.5.1. Latent Space Optimization and Chemical Manifolds
Generative models operate in high-dimensional chemical spaces and must ensure output validity and synthesizability, while efficiently identifying rare, high-quality candidates (14). VAEs and certain autoregressive models learn latent chemical manifolds, where distances correspond to structural similarity. In theory, optimizing in this latent space (via Bayesian or gradient methods) enables design of potent analogs near known actives (65–67). Yet, not all latent directions map to valid molecules. Some lead off the learned manifold, producing invalid or strange outputs (68). Techniques like property-conditioned latent spaces and validity filters help mitigate this (69). Alternatively, methods like PASITHEA invert differentiable property predictors to optimize input molecules directly (70).
2.5.2. Validity and Synthesizability Constraints
A well-trained generative model maps continuous latent vectors to discrete molecular space (71). Ensuring this mapping is smooth and chemically realistic remains an open challenge (72). Validity and synthesizability are essential. Early SMILES generators often violated valency rules (73). Modern models use graph-based construction, fragment-based methods (e.g., junction trees), or validity filters, achieving >95% valid molecules (28). Synthesizability remains difficult to measure. Some models use synthetic accessibility scores or predicted retrosynthesis steps as proxies (74). Reinforcement learning agents can be rewarded for generating molecules requiring fewer synthesis steps (75). TRACER, a conditional transformer, generates both molecules and plausible reaction paths using learned transformations, ensuring synthetic feasibility (76).
2.5.3. High-Dimensional Chemical Space
Exploring high-dimensional chemical space is computationally demanding. A typical drug-like molecule has 20–70 heavy atoms, leading to enormous combinatorial possibilities (77). Generative models, trained on bioactive molecules, bias outputs toward favorable motifs (78). From a theoretical view, this is importance sampling: the model learns a distribution q(x) focused on regions where the true utility distribution p(x) is high.
Techniques like reinforcement learning (RL) and Monte Carlo tree search (MCTS) efficiently guide search (79). In RL, the model acts as an agent adding atoms or groups, receiving rewards for desirable properties (e.g., potency, low toxicity), enabling targeted exploration (80). For example, a DDR1 kinase inhibitor was discovered within 21 days using RL-guided generative models (81). Genetic algorithms (GAs), which evolve molecular populations using crossover and mutation, also explore chemical space (82). Modern GAs use neural networks to bias mutations or select crossover points (83).
Exploration balances exploitation (refining known scaffolds) with discovering novel ones. This balance is adjustable via model hyperparameters (e.g., softmax temperature, diffusion noise variance) (84). Some models explicitly incorporate exploration-exploitation tradeoffs, using strategies like Thompson sampling or multi-objective optimization (85).
Generative models must also contend with the curse of dimensionality: high-dimensional property landscapes are complex, with many local optima (86). Generative models, trained on real data, implicitly learn some of this structure. But they rely on property predictors or experiments to evaluate novel molecules. Thus, the best strategy is integrating generation, prediction, and optimization. This closed loop, used in Bayesian optimization and active learning, iteratively improves candidates with fewer evaluations (73,78,87,88).
Navigating chemical space with AI requires smooth, chemically realistic mappings, valid and synthesizable outputs, and strategic exploration (89). Embedding generative models within predictive-evaluative frameworks enables discovery of novel bioactives that would be infeasible by brute force (90). Future models may guarantee validity and bound synthetic complexity, further expanding the reach of AI-driven drug design (91).
3. Generative AI for Molecular Structure Prediction and Optimization
With the foundations in place, we turn to the practical applications of generative AI in designing new molecular structures and optimizing them for drug-like properties. These applications fall into two categories: small molecule design (drug-like compounds) and macromolecule/protein design (biologics, enzymes, antibodies). Generative models are used in conjunction with other AI techniques (self-supervised pretraining or reinforcement learning) to achieve specific goals, such as improving a lead compound’s potency or inventing a protein that binds a given target (14). This section discusses molecular optimization (Section 3.1) and protein design (Section 3.2), highlighting representative methods.
With the foundations in place, we turn to the practical applications of generative AI in designing new molecular structures and optimizing them for drug-like properties (Figure 2).
3.1. AI-Driven Small Molecule Design
Designing small-molecule drugs is a multi-objective optimization problem: achieving potency against the target while satisfying other criteria (selectivity, pharmacokinetics, safety, etc.). Three approaches in AI-driven molecule design are: self-supervised learning of molecular representations, reinforcement learning for goal-directed optimization, and graph-based generative models.
3.1.1. Self-Supervised Learning for Molecular Representations
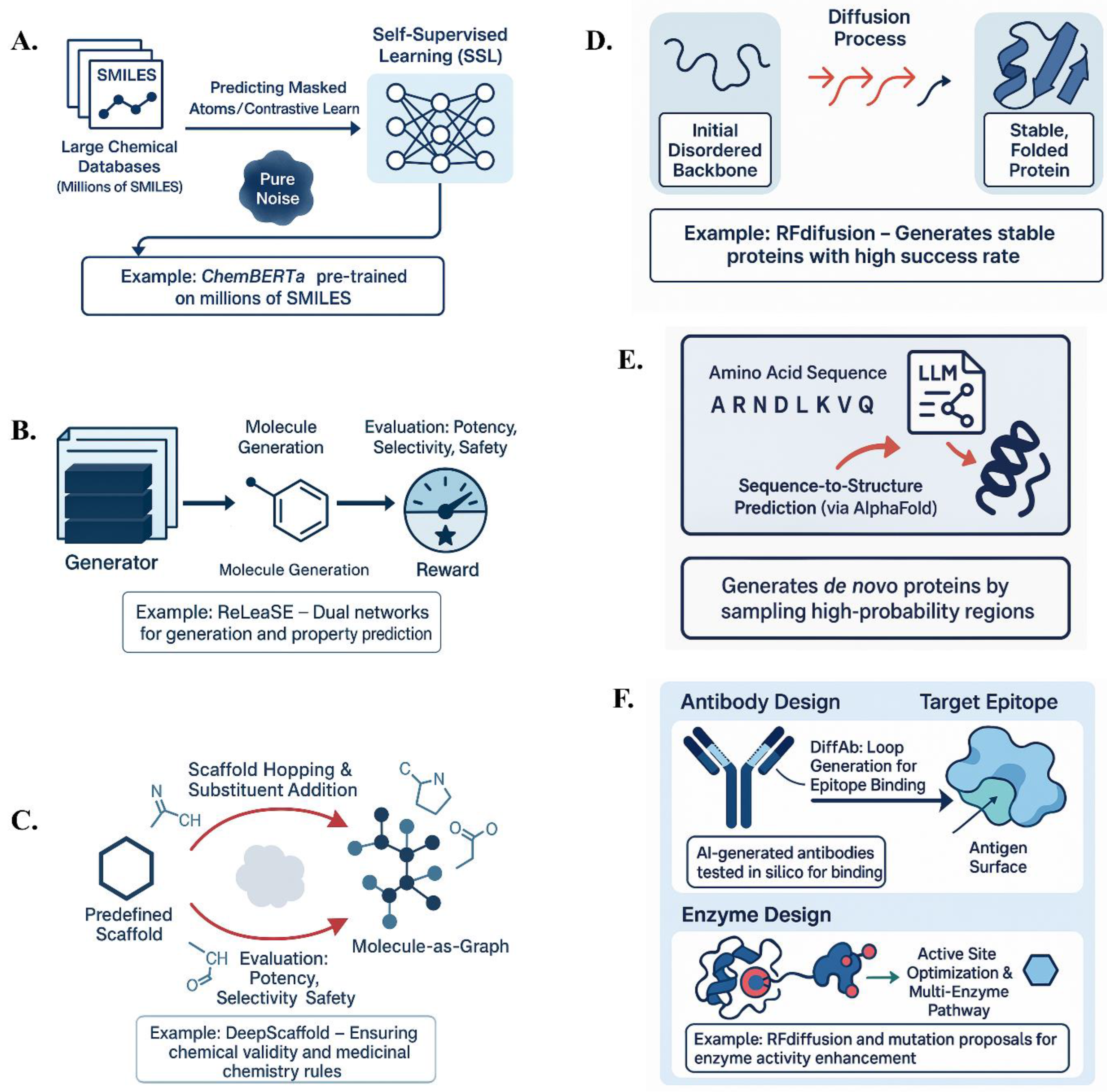
A critical aspect of molecular optimization is having a rich representation of molecules. Self-supervised learning (SSL) trains models on large chemical databases using tasks like predicting masked atoms or contrastive learning between molecule augmentations (92–94). Models such as ChemBERTa were trained to learn chemical context through a masked token prediction task on millions of SMILES. The resulting models can predict properties or initialize generative tasks (42). For example, transformer models pre-trained to predict missing atoms can generate complete molecules from partial fragments (95). Denoising autoencoders, trained to reconstruct a molecule from a corrupted version, can propose modifications to lead compounds, such as filling in missing parts (96).
Figure 2.
Generative AI strategies for molecular and protein design. (A–C) Approaches for small molecule optimization using self-supervised learning (ChemBERTa), reinforcement learning (ReLeaSE), and graph-based models (DeepScaffold). (D–F) Protein design methods, including diffusion models (RFdiffusion), large language models for sequence generation, and applications in antibody and enzyme engineering.
Figure 2.
Generative AI strategies for molecular and protein design. (A–C) Approaches for small molecule optimization using self-supervised learning (ChemBERTa), reinforcement learning (ReLeaSE), and graph-based models (DeepScaffold). (D–F) Protein design methods, including diffusion models (RFdiffusion), large language models for sequence generation, and applications in antibody and enzyme engineering.
3.1.2. Reinforcement Learning (RL) for Molecular Optimization:
Generative models can be coupled with reinforcement learning (RL) to optimize objectives (97). Generation of a molecule is treated as a sequential decision process, where a generative model (e.g., an RNN or transformer) chooses actions like which atom to add at each step. A reward function reflects design goals, e.g., high reward for molecules predicted to bind a target and low reward for those likely to be toxic (14,28). ReLeaSE (Reinforcement Learning for Structural Evolution) uses two neural networks—one for generation and one for predicting properties like biological activity (98). RL algorithms like policy gradients or Q-learning guide the model towards molecules with better scores. RL can discover non-intuitive modifications to improve a molecule’s profile (99,100).
3.1.3. Graph-Based Generative Models:
Since molecules are naturally graphs, generative models often use graph neural networks (GNNs). In graph grammar-based approaches, molecules are built by adding atoms or larger fragments to a partial graph (101,102). DeepScaffold uses CNN-based GNNs to add substituents to a predefined scaffold. These models ensure that molecules remain valid and incorporate medicinal chemistry rules (103). Some models allow scaffold hopping, generating a different core structure that still satisfies activity requirements (104). Combined with reinforcement learning, graph-based design is particularly useful in lead optimization, generating analogs of a given lead compound that improve properties (98).
3.2. AI-Driven Protein Design
Designing proteins with specific structures or functions is a grand challenge, and AI is revolutionizing this process. Unlike small molecules, proteins are large macromolecules with complex folding patterns and vast design spaces. Generative AI tackles these problems using techniques like diffusion models for protein structures, large language models for protein sequences, and specialized models for antibody or enzyme design.
3.2.1. Diffusion Models for Protein Folding & Stability Prediction:
Diffusion models, like RFdiffusion, treat the 3D coordinates of a protein backbone as data to be diffused. Starting from a random initial backbone, the model iteratively refines it into a physically plausible structure (56,105). RFdiffusion generates novel protein structures that are computationally predicted to be stable and experimentally verified to fold and function. This model excels at designing symmetric protein assemblies and enzyme active site scaffolds. The success rate of generating foldable proteins was significantly higher than prior methods. Diffusion models incorporate physical and evolutionary constraints, learning rules of protein folding that guide the generation process, leading to more stable and functional designs.
3.2.2. LLMs for De Novo Protein Sequence Generation:
Sequence-based generative models, particularly large language models (LLMs), have opened new pathways in protein engineering (106,107). Trained on large sequence databases (e.g., Uniprot), these models capture evolutionary patterns like motifs and domains that relate to protein function. LLMs can generate protein variants and rank them by their likelihood of being functional. Sampling in high-probability regions produces novel proteins that might not exist in nature. LLMs complement structure-first methods by generating sequences that can be predicted to fold into desired structures using tools like AlphaFold. This reduces the need for extensive wet-lab testing by filtering out likely failures in silico (107–111).
3.2.3. Antibody and Enzyme Design Using AI:
Two significant application areas are antibody design and enzyme design, where generative AI proves highly effective.
- Antibody Design: AI can design antibodies by generating complementarity-determining region (CDR) sequences likely to bind a target antigen or by generating 3D conformations of antibody loops that complement antigen surfaces (112). DiffAb, a diffusion model, generates antibody structures conditioned on the 3D structure of the target antigen’s epitope, effectively growing an antibody loop to fit into the epitope pocket (113). The success of AbSci’s model in creating functional antibodies in silico indicates that these methods can produce viable therapeutic candidates (114).
- Enzyme and Biocatalyst Design: Enzymes catalyze chemical reactions, and AI is transforming enzyme design by improving active site modeling and exploring backbone arrangements. RFdiffusion has been used to design enzyme active sites, with some designs showing promising activity. AI can also optimize existing enzymes by proposing mutations that stabilize them or alter their substrate scope. Generative models can propose multi-enzyme pathways for synthetic routes, offering a new approach to metabolic network design (115–117).
In both antibody and enzyme design, integrating experimental feedback accelerates the process. AI-generated designs are tested through high-throughput experiments, and the resulting data refines the models. This experiment-AI loop is becoming more efficient with automated laboratories that integrate robotics and AI for real-time analysis (118,119).
4. Computational Strategies for AI-Guided Drug–Target Interactions
A critical aspect of drug design is not just proposing molecules, but understanding and predicting how those molecules will interact with biological targets (proteins, nucleic acids, etc.). This involves docking (predicting the binding pose of a ligand in a protein’s active site), scoring and predicting binding affinity, and efficiently searching through large libraries for those interactions (virtual screening). Traditional computational chemistry methods like molecular docking programs and physics-based scoring functions have been standard for decades, but they have limitations in accuracy and speed. AI-driven approaches are now enhancing or outright replacing these steps: diffusion models are redefining molecular docking, deep neural networks are predicting binding affinities with high accuracy, and generative models are enabling ultra-large virtual screens by focusing on the most promising candidates. In this section, we explore how generative AI and related models contribute to drug–target interaction prediction, covering DiffDock and modern docking, binding affinity prediction, and large-scale virtual screening.
4.1. DiffDock and Beyond: AI in Molecular Docking
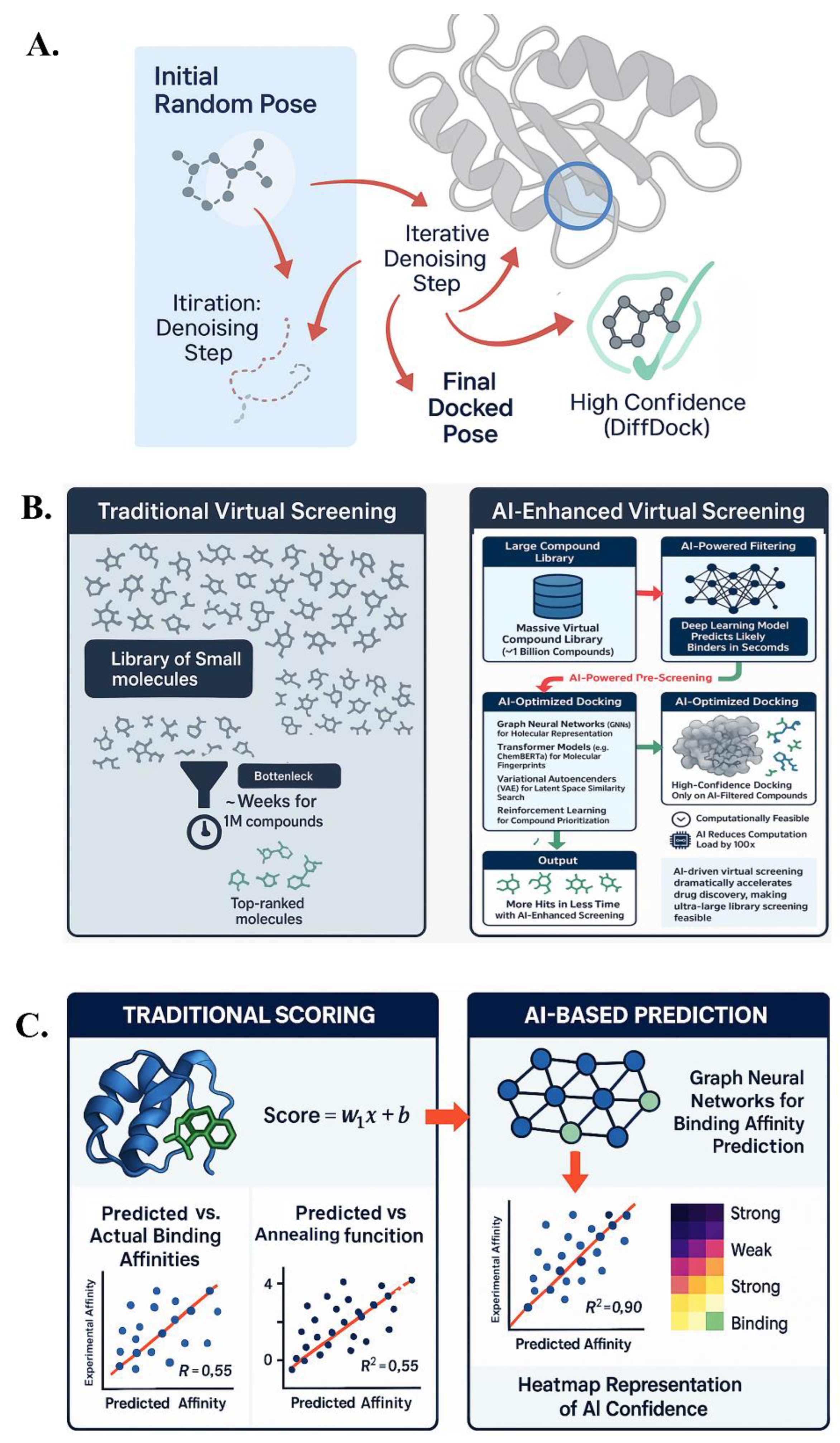
Molecular docking is the computational prediction of a ligand’s preferred orientation (pose) and position when bound to a target protein, typically an early step in in silico drug screening. Classical docking programs (AutoDock, DOCK, Glide, etc.) use physics-inspired scoring functions to evaluate many possible poses but often treat the protein as rigid and use approximations that can mis-rank good binders. Enter AI: methods like DiffDock have reframed docking as a generative modeling problem. DiffDock uses a 3D diffusion model to generate candidate ligand poses in a given protein binding site. It starts with random orientations and positions, then iteratively “denoises” its translation and rotation, guided by a learned scoring potential to bring the ligand toward likely binding modes.
DiffDock doesn’t use a traditional scoring function; instead, it was trained on a large dataset of known protein–ligand complexes, learning an implicit representation of shape complementarity and interactions. On standard benchmarks, DiffDock significantly outperformed traditional docking tools. For example, at a 2 Å RMSD threshold, DiffDock placed ~22% of predictions within that range, more than double the success rate of traditional methods, which often hovered around ~10%. It also maintained strong performance on difficult cases where other methods failed. This success is attributed to DiffDock’s ability to implicitly account for protein flexibility, learning a distribution of likely poses that might correspond to slight protein side-chain movements, something rigid docking struggles with.
DiffDock includes a confidence model that estimates the reliability of its predicted pose. This confidence score correlates well with pose accuracy, helping to prioritize high-confidence predictions. DiffDock also provides speed combined with accuracy, enabling high-throughput docking campaigns. Additionally, as a generative model, DiffDock produces multiple plausible poses, reflecting possible binding modes or tautomeric states of the ligand, giving medicinal chemists a richer view of ligand binding. The DiffDock approach, along with AI-enhanced virtual screening and affinity prediction strategies, is illustrated in Figure 3.
Beyond DiffDock, other AI methods like EquiBind (120), a one-shot GNN-based method, also show promise, though DiffDock’s diffusion approach is more accurate for many targets. Another extension, DiffDock-PP (121), applies diffusion to protein–protein docking, which is a more complex scenario of two flexible bodies coming together, and has shown promising results.
AI in docking also integrates with scoring refinement. Once DiffDock places a ligand, one can use a brief physics-based minimization or a neural network rescoring model to refine the pose and binding score, further improving accuracy. This synergy of AI-guided generation and traditional force-field refinement can deliver both speed and data-driven insights, plus fine-detail adjustment from physics. DiffDock’s ability to learn a statistical potential for interactions from data also implicitly captures difficult-to-model effects like entropy and solvation, helping it outperform hand-crafted scoring functions (122,123).
What does this mean for drug discovery? In practice, DiffDock accelerates drug target identification. Researchers can now screen a library of compounds by docking them with DiffDock to a target, triaging huge libraries in a day. DiffDock also supports polypharmacology studies, screening a drug against many proteins in silico to predict off-targets or new uses (124). The model can help elucidate mechanisms of action for novel phenotypic screening hits by docking them to panels of protein structures.
Figure 3.
AI-driven strategies for drug–target interaction prediction. (A) DiffDock uses diffusion models for pose generation and refinement. (B) AI-enhanced virtual screening accelerates compound prioritization via deep learning and optimized docking. (C) AI-based models, such as GNNs, outperform traditional scoring in binding affinity prediction.
Figure 3.
AI-driven strategies for drug–target interaction prediction. (A) DiffDock uses diffusion models for pose generation and refinement. (B) AI-enhanced virtual screening accelerates compound prioritization via deep learning and optimized docking. (C) AI-based models, such as GNNs, outperform traditional scoring in binding affinity prediction.
4.2. Protein–Ligand Binding Affinity Prediction
Accurately predicting the binding affinity (e.g., Kd or IC50) of a small molecule to its target protein is crucial for lead optimization. Traditional methods like scoring functions or physics-based free energy calculations can be unreliable or slow. AI-driven models now provide data-driven predictions that can quickly estimate binding affinities with high accuracy.
End-to-end deep learning models have been developed that take protein–ligand complexes as input (either 3D coordinates or interaction lists) and output binding affinities or scores. These models often use 3D convolutional neural networks or graph neural networks (GNNs) that treat the protein–ligand pair as a combined graph (125,126). GNN models represent protein residues and ligand atoms as nodes, with edges representing interactions (contacts, hydrogen bonds, etc.), learning to predict affinity (127,128). Some deep models have achieved a Pearson correlation of ~0.8 on benchmarks like PDBBind (129), significantly outperforming traditional methods.
Quantum + AI hybrid models are also emerging, combining quantum mechanics with AI to improve binding predictions, particularly in cases where electronic effects or polarization are critical. These hybrid models use quantum mechanical descriptors as inputs to machine learning models or even quantum circuits to represent parts of the model, potentially improving predictions of subtle electronic interactions. While quantum methods are still in the early stages, quantum + AI combinations are showing promise for more accurate binding affinity predictions (130–133).
Binding affinity prediction also embraces multi-task and multi-modal learning. A single model can be trained to predict not just affinity, but also other experimental readouts like activity in a cell assay or entropy of binding. This allows the model to be more robust through shared representations. Additionally, coupling binding prediction with generative design is powerful: generative models propose analogs, and deep affinity predictors quickly estimate their potency, enabling thousands of designs to be tested in seconds (134,135).
4.3. Large-Scale Virtual Screening
Virtual screening (VS) evaluates large compound libraries to identify hits for a target, often using docking or pharmacophore matching. With AI and improved prediction models, VS is evolving to handle ultra-large libraries and AI-guided combinatorial library generation.
Recent efforts have led to the creation of vast purchasable libraries, like Enamine REAL, containing >1 billion compounds. Screening these with traditional docking is impractical, but deep learning models can predict docking scores or binding likelihood for all compounds, rapidly reducing the library size to a manageable set for further evaluation.
Another approach uses similarity in latent space: if one has a known active ligand, one can encode the compounds into a learned embedding space and do a nearest-neighbor search to find those most similar in relevant ways, faster than traditional docking. AI can also generate focused libraries on the fly, sampling virtual compounds biased toward predicted binders and screening them for efficacy, blurring the line between virtual screening and de novo design. This approach, demonstrated during the COVID-19 pandemic, has the potential to vastly increase the scale of virtual screening.
AI-guided combinatorial chemistry further enhances screening by intelligently selecting which combinations of building blocks to synthesize. AI models evaluate subsets of possible products, learning which parts contribute to desired activity, and pruning the search space to focus on promising combinations.
5. AI-Driven Synthesis Planning and Retrosynthesis
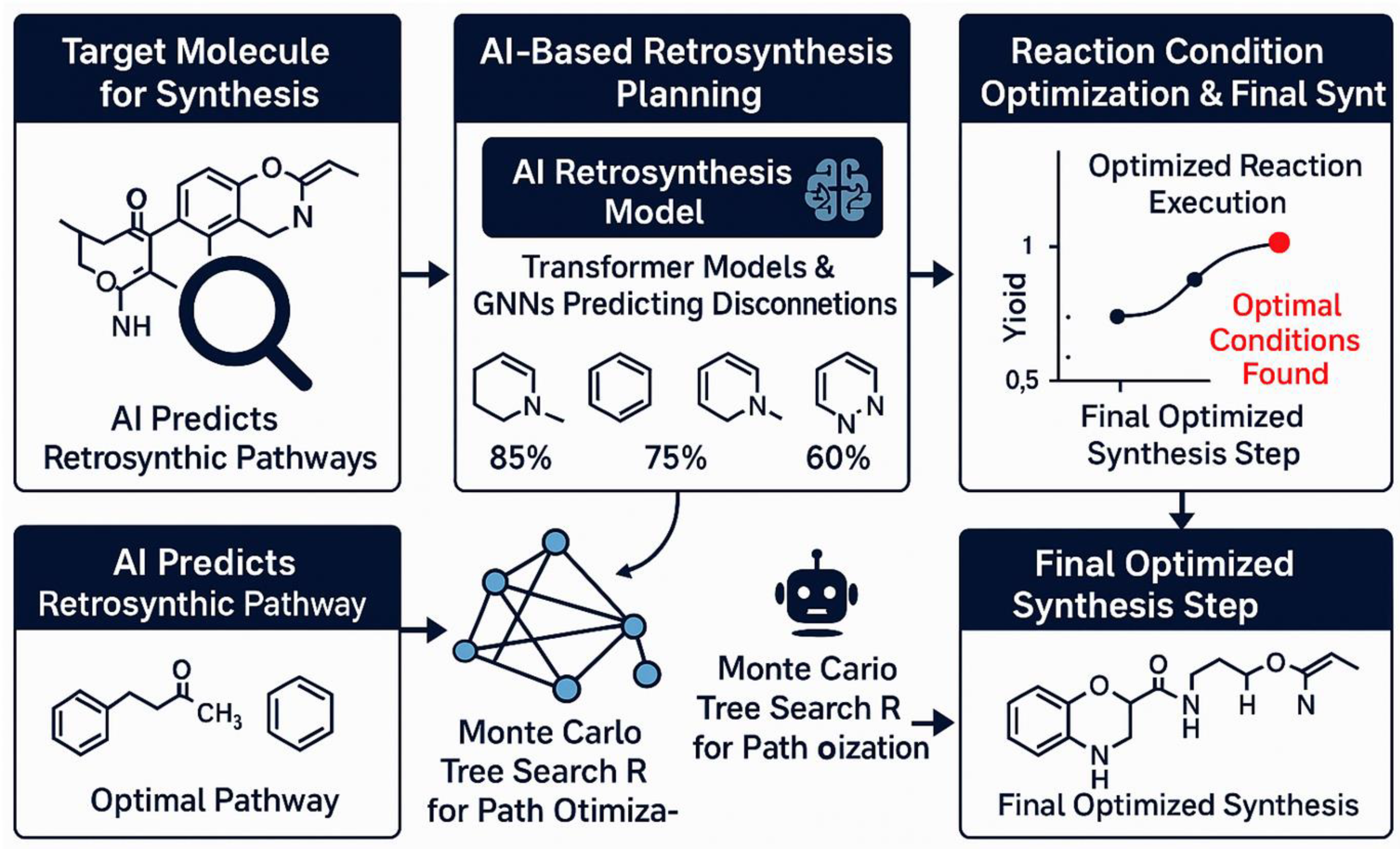
Designing a promising molecule is only half the battle – one must also be able to make that molecule efficiently. Retrosynthesis planning is the process of identifying a sequence of chemical reactions to synthesize a target molecule from available starting materials. Historically tackled by expert chemists and rule-based software (like E.J. Corey’s LHASA or Synthia), AI is now playing a major role in retrosynthesis and synthesis planning, offering data-driven predictions and creative route suggestions. Key contributions of AI include predicting feasible reactions, using reinforcement learning to navigate possible routes, and employing Bayesian optimization to propose optimal reaction conditions or pathways (Figure 4).
Predicting synthetic feasibility: AI helps evaluate a molecule’s structure and suggests possible retrosynthetic disconnections. Transformer models and GNNs trained on millions of reactions can predict reaction patterns (20). For instance, IBM’s RXN for Chemistry uses a sequence-to-sequence transformer to predict reactants given a product. These models output multiple disconnections, which can be recursively applied to break the molecule down stepwise (136). AI retrosynthesis produces a retrosynthetic tree or network of possible routes, each step predicted with a confidence score. Early deep learning models, like RetroTransformer, have achieved success rates comparable to expert chemists and sometimes uncover routes human chemists might overlook (137).
Figure 4.
AI-driven synthesis planning pipeline for retrosynthesis and reaction optimization. The process begins with a target molecule (top left), where AI models predict retrosynthetic disconnections. Transformer models and graph neural networks (GN004Es) are trained on reaction databases to identify viable bond disconnections, yielding confidence scores for each prediction. Monte Carlo Tree Search (MCTS) is then employed to optimize synthetic pathways by evaluating and pruning possible routes. After selecting an optimal pathway, AI-based Bayesian optimization algorithms identify optimal reaction conditions to maximize yield. The entire process culminates in an experimentally feasible and optimized synthesis route.
Figure 4.
AI-driven synthesis planning pipeline for retrosynthesis and reaction optimization. The process begins with a target molecule (top left), where AI models predict retrosynthetic disconnections. Transformer models and graph neural networks (GN004Es) are trained on reaction databases to identify viable bond disconnections, yielding confidence scores for each prediction. Monte Carlo Tree Search (MCTS) is then employed to optimize synthetic pathways by evaluating and pruning possible routes. After selecting an optimal pathway, AI-based Bayesian optimization algorithms identify optimal reaction conditions to maximize yield. The entire process culminates in an experimentally feasible and optimized synthesis route.
However, AI doesn’t fully replace human planning; it acts as a powerful assistant. The model proposes several routes, and a chemist reviews and refines them. A limitation is that AI models are trained on known reaction data, making it difficult for them to suggest truly novel chemistry (138).
Reinforcement Learning for Retrosynthesis: The space of possible synthetic routes is vast, resembling a game with many reactions as possible moves. AI uses methods like Monte Carlo Tree Search (MCTS) guided by learned policies to explore the retrosynthesis tree efficiently (139,140). Deep reinforcement learning (RL) has been applied, with an RL agent proposing retrosynthesis steps and receiving rewards when reaching purchasable building blocks. This approach minimizes the number of steps, rediscovering many known strategies (141). AI-guided search prunes unlikely paths, making it more efficient than traditional rule-based programs. A challenge is ensuring that the predicted steps are not only theoretically plausible but also practically executable.
Reaction condition optimization: Once a route is chosen, AI/ML techniques like Bayesian optimization automate reaction condition optimization. Bayesian optimization treats reaction yield as a function of conditions and selects which conditions to try next. A cost-aware Bayesian optimizer can factor in the time/resource cost of experiments, focusing on cost-effective routes (142–146).
Integration of synthesis planning in design: Generative models can guide design toward more synthesizable regions of chemical space in real-time. Combined design-synthesis optimization frameworks, like TRACER and Syn-MolOpt, optimize both molecular properties and synthetic accessibility (147). For example, a complex molecule predicted to be difficult to synthesize can be deprioritized in favor of a more synthesizable alternative, ensuring a balance between potency and ease of synthesis (148).
AI-driven synthesis planning is narrowing the gap between the molecules we can design and synthesize. By predicting synthesis pathways and optimizing reaction conditions, generative pipelines focus on candidates that are both innovative and realizable. Reinforcement learning and search algorithms enable retrosynthesis tools to handle complex targets. This fusion of design and synthesis planning accelerates the drug discovery cycle and minimizes the risk of pursuing infeasible designs.
6. AI for Pharmacokinetics and Toxicity Prediction
While potency and synthesizability are crucial, a successful drug must also possess suitable pharmacokinetic (PK) and safety profiles. This includes absorption (can it get into the bloodstream?), distribution (does it reach the target tissue?), metabolism (is it broken down too quickly or into toxic metabolites?), excretion (can it be eliminated from the body?), and toxicity (does it harm cells or organs, or cause side effects?). These properties are encapsulated in the acronym ADME/Tox. Generative AI models and predictive machine learning are being used to evaluate and optimize these factors early in the design process, aiming to produce drug candidates that are not only effective but also drug-like and safe.
Figure 5.
AI-driven strategies for optimizing pharmacokinetics, toxicity, and personalized drug discovery. (A) AI predicts ADME/Tox properties to guide early drug optimization. (B) Generative models balance potency with ADME/toxicity profiles. (C) AI leverages multi-omics data for patient-specific drug design in precision medicine.
Figure 5.
AI-driven strategies for optimizing pharmacokinetics, toxicity, and personalized drug discovery. (A) AI predicts ADME/Tox properties to guide early drug optimization. (B) Generative models balance potency with ADME/toxicity profiles. (C) AI leverages multi-omics data for patient-specific drug design in precision medicine.
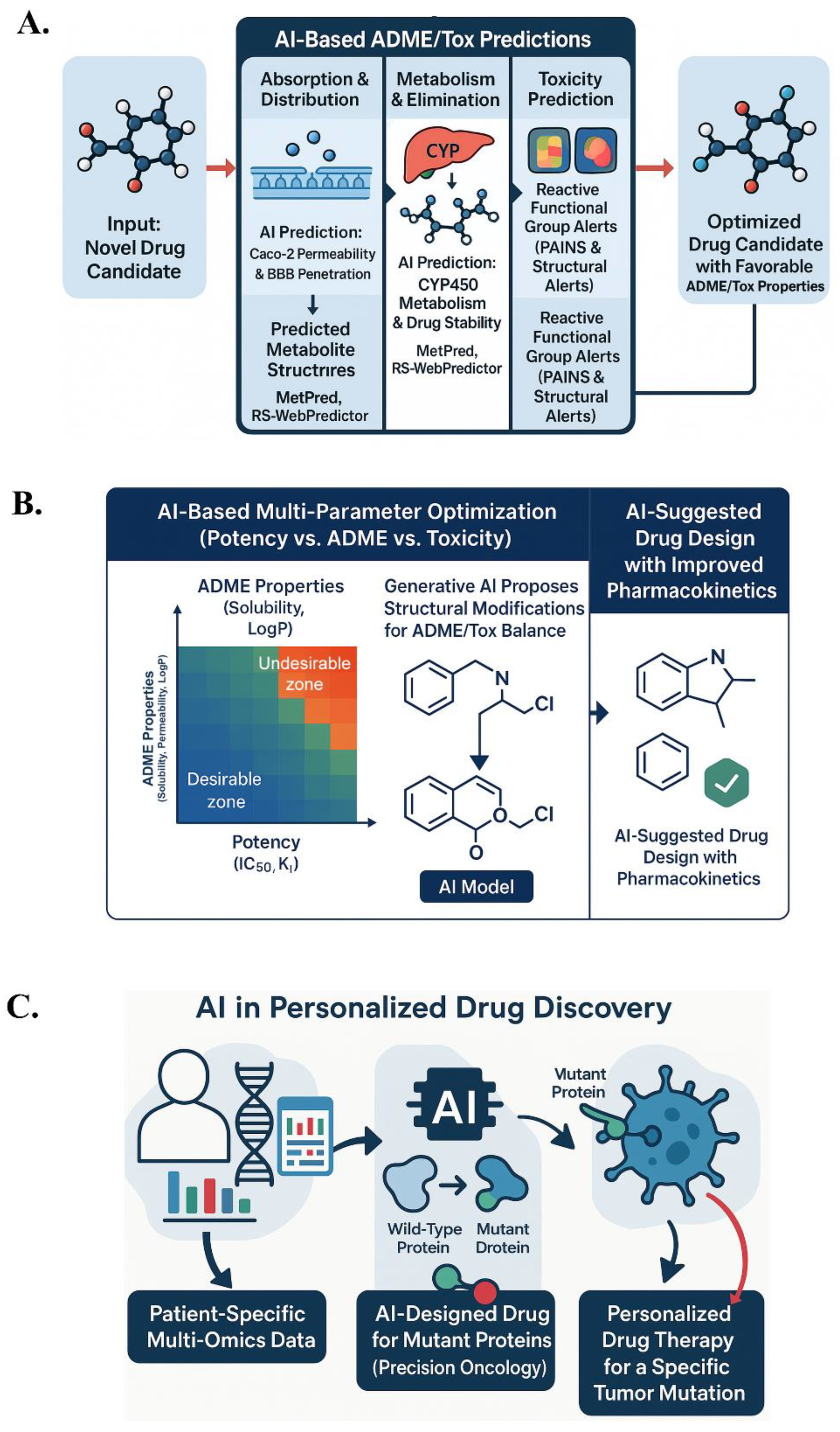
6.1. ADME/Tox Predictions
Drug-likeness constraints: Medicinal chemists apply rules (like Lipinski’s Rule of 5) to ensure oral bioavailability. AI can learn nuanced drug-likeness patterns from large datasets of known drugs and failed compounds. Models like neural networks and ensemble methods (random forests, gradient boosting) distinguish drug vs non-drug molecules, capturing subtle features. Generative models incorporate drug-likeness as part of their scoring function, co-optimizing for favorable ADME properties.
Absorption and distribution: AI models predict permeability, solubility, and plasma protein binding. Deep learning regression models predict Caco-2 cell permeability or blood-brain barrier penetration based on molecular structure. Models for human intestinal absorption can classify compounds as high vs low absorption, guiding early elimination of very polar compounds.
Metabolism and elimination: AI methods (MetPred, RS-WebPredictor) predict metabolic stability and sites of transformation (e.g., CYP450 enzymes). More advanced models predict metabolite structures using sequence-to-sequence learning. Models predict P450 inhibition to avoid drug-drug interactions, penalizing molecules likely to inhibit major isoforms like CYP3A4.
Toxicity and off-target effects: AI predicts various toxicities:
- In vitro cytotoxicity using Tox21 challenge data.
- Organ toxicity (hepatotoxicity, cardiotoxicity), including hERG channel inhibition, predicted by ML models.
- Genotoxicity and carcinogenicity predictions using Ames test data or animal studies.
- Reactive functional group alerts: AI identifies substructures causing nonspecific reactivity or toxicity, learning broader patterns of reactivity beyond known PAINS.
In practice, AI-driven ADMET tools are applied in lead optimization, predicting properties like logP, solubility, permeability, clearance, and hERG risk. Multi-parameter optimization (MPO) frameworks balance potency and ADMET properties. AI helps navigate trade-offs; for instance, improving solubility might reduce CNS toxicity but also lower permeability. AI proposes modifications to improve one property without overly harming others (149,150). By identifying ADME/Tox issues early, AI saves time and cost by avoiding failure due to pharmacokinetic issues or toxicity.
Predicting off-target interactions: AI models trained on bioactivity databases predict unwanted off-target interactions, guiding generative design. Multi-task neural networks like prOCTOR predict activity across multiple off-targets, enabling in silico “safety pharmacology” panels. Generative design can penalize compounds with high affinity for undesirable anti-targets, actively minimizing off-target effects (151).
Modern AI-driven drug design optimizes multi-factor properties (potency, ADME, toxicity), ensuring compounds have a balanced profile. This approach embodies “fail fast, fail cheap” by identifying potential failures early, reducing costly animal studies.
6.2. Personalized Drug Discovery
AI is paving the way for precision medicine, tailoring drug discovery to individual patient data (e.g., genomic, multi-omic). Unlike traditional drug discovery, AI in personalized medicine aims to design drugs for specific subpopulations or individual patients. Generative AI can leverage multi-omics datasets (genomics, transcriptomics, proteomics) to discover novel therapeutic strategies or patient-specific drug candidates.
In oncology, AI models design molecules targeting mutant proteins without affecting normal variants. For example, AI could propose a drug combination for a tumor with specific oncogene dependencies. These applications are illustrated in Figure 5, which highlights AI-driven strategies for ADME/Tox prediction, multi-parameter optimization, and personalized drug design. Generative AI optimizes drugs to reverse a disease-specific expression signature in transcriptomic-driven drug design. AI models predict gene expression changes based on structure and optimize accordingly.
Multi-omics-based generation: AI analyzes rich patient data to identify novel targets or pathways. For example, AI might stabilize an atypical protein conformation in a tumor, thereby blocking its function. AI can also design personalized vaccines, creating neoantigens optimized for an individual’s HLA type. This was successfully demonstrated with AI-generated therapeutic vaccines tailored to a patient’s tumor mutations.
Generative AI also aids in rare disease drug discovery. For example, AI could design a pharmacological chaperone for a unique pathogenic mutation. Moreover, AI suggests drug repurposing for patients with specific gene expression signatures, identifying existing drugs with profiles opposite to the disease state.
While personalized generative drug design is emerging, AI can integrate patient data to suggest personalized therapeutic molecules. This could lead to AI-designed drugs tested on a patient’s cells or organoids, with the potential for rapid, precise treatments. While challenges exist, AI in precision drug discovery promises the long-envisioned goal of “the right drug for the right patient at the right time.”
7. Experimental Validation and AI-Augmented Pipelines
No matter how powerful our in silico methods are, experimental validation is the ultimate proving ground for any AI-designed molecule or protein (Figure 6). In this section, we discuss how AI-designed candidates are being validated in the lab (and some notable success stories), as well as how experiments themselves are becoming more integrated with AI (creating a closed-loop discovery pipeline). We cover wet lab validation of AI-designed drugs (7.1) and how AI assists in protein engineering and biotechnology (7.2), including real-world examples where AI-designed proteins have been synthesized and tested.
7.1. Wet Lab Validation: Case Studies of AI-Designed Drugs and Challenges in Translation
Over the past few years, we’ve seen AI-designed molecules advancing into experimental and clinical stages. A landmark in 2020 was the first fully AI-designed drug (DSP-1181 for OCD, designed by Exscientia) entering Phase I clinical trials (152). This small molecule, optimized for activity on a GPCR target, went from concept to clinic in 12 months, instead of the usual 4–5 years. Similarly, Insilico Medicine’s AI-discovered drug for idiopathic pulmonary fibrosis entered Phase I trials in 2022, reducing time and cost compared to traditional programs.
Another exciting case is AbSci’s 2023 AI-designed de novo antibody, which was synthesized and confirmed to bind and neutralize its target. The FDA also granted Orphan Drug Designation to an Insilico-designed drug for a rare disease in 2023, further validating AI’s role in drug development (153).
Figure 6.
AI-augmented pipelines in drug discovery and biotechnology. (A) AI accelerates drug discovery through molecular design, high-throughput screening, and iterative validation. (B) AI enables de novo protein design, enzyme engineering, and synthetic biology applications, enhancing experimental efficiency and precision.
Figure 6.
AI-augmented pipelines in drug discovery and biotechnology. (A) AI accelerates drug discovery through molecular design, high-throughput screening, and iterative validation. (B) AI enables de novo protein design, enzyme engineering, and synthetic biology applications, enhancing experimental efficiency and precision.
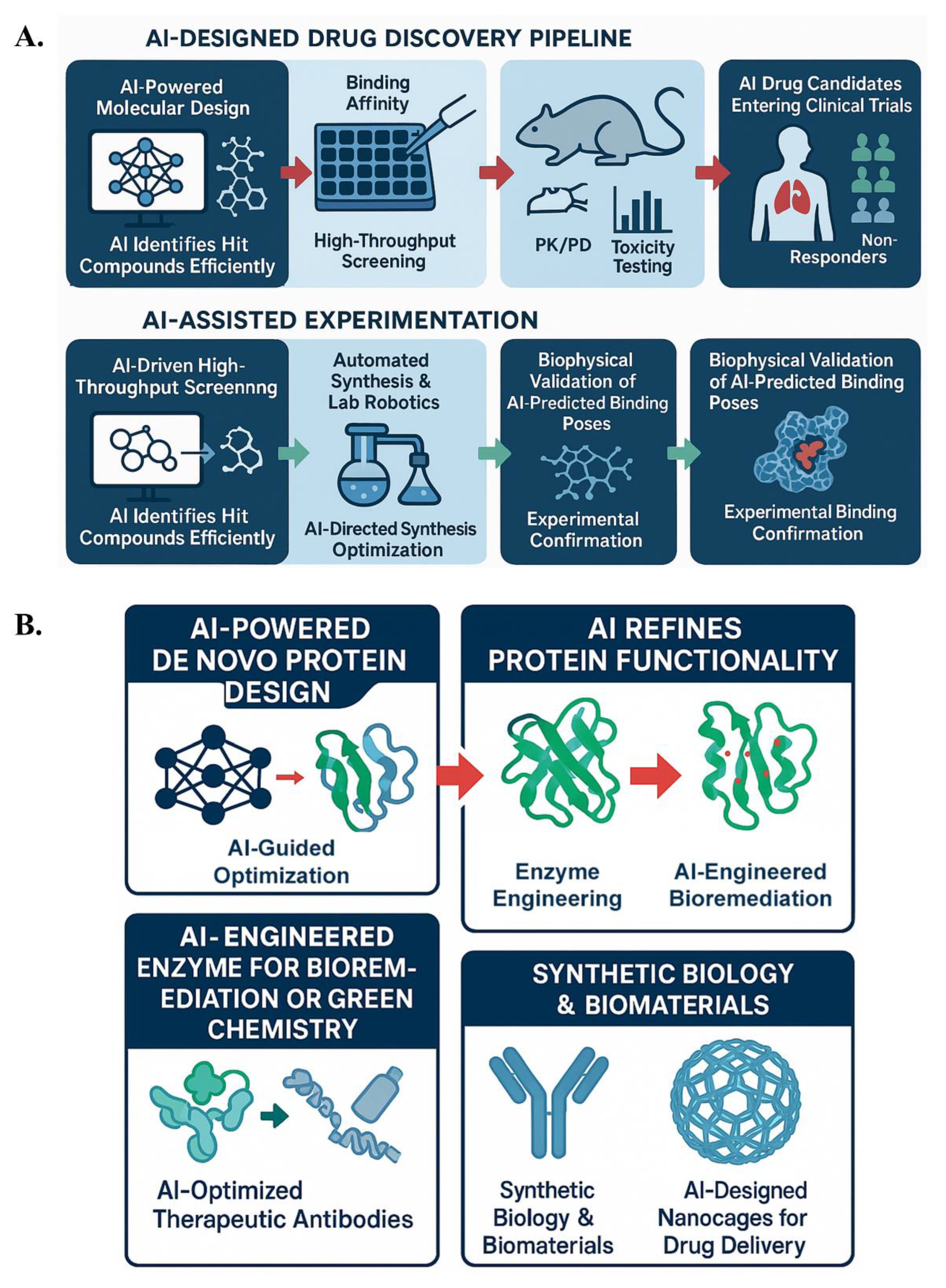
However, not all AI-designed candidates succeed. Some molecules have failed to meet efficacy endpoints or faced unforeseen issues, such as one report where AI-derived molecules did not outperform traditional leads. These instances highlight that while AI expedites clinical candidate development, rigorous experimental validation is essential. AI predictions can be wrong, as compounds predicted to be non-toxic may show toxicity due to overlooked factors, like rare metabolic byproducts.
To mitigate risks, AI-driven projects adopt a fail-fast approach: generating multiple top candidates, testing them in vitro, and iterating. For instance, if AI yields five candidates with similar profiles, all might be tested for potency, solubility, metabolic stability, and toxicity (e.g., hERG patch-clamp assay). Insilico’s fibrosis drug underwent ~6 AI design iterations, testing dozens of compounds, before identifying the clinical candidate.
AI augmenting experiments: AI also aids in experiment planning and analysis. In high-throughput screening, AI can detect patterns in assay readouts, identifying hits that work via desired mechanisms and distinguishing false positives. In robotics and automation, AI directs experiments like flow chemistry setups to optimize reaction conditions, updating the model in real-time. In microfluidics, AI designs experiments, executes them, and analyzes the data with minimal human intervention (154–158).
Challenges in translation: A major issue is the predictive gap. AI models may fail to account for real-world variables, such as molecule instability or dynamic protein structures. Verifying binding through biophysical methods like X-ray crystallography is crucial. Some AI-designed ligands have matched their predicted binding poses with targets, reinforcing confidence in the design (159–163).
Chemical novelty vs synthetic familiarity is another challenge. AI sometimes proposes novel structures that present synthetic difficulties or unexpected reactivity. Medicinal chemists often apply a “chemical intuition filter” to make these designs more practical.
Despite these challenges, each successful case of an AI-designed drug reaching clinical trials validates the approach. By 2024, over 15 AI-designed molecules were in clinical trials, suggesting that in the next decade, many new clinical candidates may involve AI (164).
Experimental validation is essential for testing AI-designed solutions. Proof-of-concept that AI-designed molecules can become real drugs and proteins function as intended marks a significant achievement. The challenges are addressed through iterative testing and improved models, and with advances in AI and laboratory automation, the gap between design and validation will continue to narrow.
7.2. Protein Engineering in Biotechnology: AI-Augmented Enzyme and Pathway Design
Generative AI is profoundly impacting protein engineering and biotechnology. AI is being used to design industrial enzymes, optimize metabolic pathways, and create synthetic biological parts. AI-designed enzymes and proteins, as discussed in previous sections, are having significant applications.
Enzyme design and metabolic engineering: AI is enabling the de novo design of enzymes with functions not found in nature. For example, a de novo enzyme was designed to hydrolyze organophosphates, showing measurable activity in breaking them down—useful in bioremediation. AI-designed enzymes often function without experimental optimization, which was rare with traditional methods (165,166).
In metabolic pathway engineering, AI identifies enzyme variants that improve pathway efficiency, specificity, or by-product formation. For example, AI may suggest enzyme variants for a rate-limiting step or redesign enzymes to improve specificity.
Synthetic biology and novel protein functions: AI is also used to design transcription factors, DNA-binding proteins, and self-assembling peptides. RFdiffusion, for instance, was used to design symmetric nanocages, confirmed by electron microscopy. These can be applied in drug delivery, vaccines, or biomaterials. AI can also design multi-enzyme complexes that streamline metabolic flux by channeling intermediates, reducing the need for separate enzymes (167–169).
From AI design to biotech product: While AI-designed proteins still require substantial lab work, the models help reduce the number of variants to test. AI aids in designing proteins that fluoresce at specific wavelengths, expanding cell biology imaging capabilities.
Real-world impact: AI is helping design solutions in environmental, industrial, agricultural, and medical applications. For example, AI has assisted in designing enzymes to degrade plastic waste, replacing traditional catalysts in pharmaceutical synthesis, and creating pest-resistant proteins in agriculture. AI is also enhancing therapeutics by designing proteins with fewer side effects by altering their surfaces to avoid undesired interactions.
In the future, AI-augmented protein engineering will enable the rapid creation of custom enzymes or proteins on demand. Early results, such as AI-designed proteins binding insulin receptors or enzymes accelerating novel reactions, indicate that AI will revolutionize biotechnology by enabling tailored solutions for various challenges.
8. Future Perspectives: AI-Designed Medicines & Autonomous Discovery
AI has initiated a shift in molecular science, promising more integration into drug and protein discovery, potentially leading to fully autonomous discovery pipelines. These systems could generate hypotheses, test them (virtually or physically via robotics), learn from outcomes, and improve with minimal human intervention. Existing components, like generative models proposing molecules and automated labs synthesizing and testing them, hint at what broader autonomous discovery could achieve. For example, a project used a flow chemistry robot and an AI planner to autonomously synthesize and test hundreds of analogs, improving target activity ten-fold without human chemists deciding each step (170).
A particularly exciting prospect is self-driving AI that not only designs molecules but refines itself by learning from outcomes. An AI could design a drug, test it, adjust its parameters, and generate new hypotheses. These AI agents could handle data crunching and routine decision-making, leaving scientists to focus on higher-level strategy and creative insights.
8.1. Fusion with Quantum Computing
AI combined with quantum computing could revolutionize drug design by solving quantum mechanical problems that classical computers struggle with, like binding free energies or reaction pathways. Quantum machine learning algorithms could operate in chemical Hilbert space, enabling simulations of large biomolecules or materials beyond classical reach. Companies are exploring quantum-enhanced generative models (like quantum GANs for molecules), which may improve proposal quality and diversity. Quantum algorithms might also generate reaction pathways, aiding retrosynthesis. While practical quantum computing is emerging, it promises breakthroughs in complex systems like large drug-targets (171–173).
8.2. Ethics and Regulation of AI-Designed Drugs
As AI plays a larger role, ethical and regulatory questions arise. A key concern is accountability if an AI-designed drug causes adverse effects. Regulators might require additional validation steps and transparency in AI model decisions. Research is ongoing to make AI models interpretable, e.g., highlighting molecular substructures linked to low toxicity. Moreover, AI must avoid generating harmful compounds; an example showed an AI generative model could design chemical weapons if misdirected. Safeguards, such as filtering toxic outputs, are necessary. The FDA has approved AI-designed drugs for trials, and regulators may soon require AI design methodology in submission dossiers, ensuring AI’s role is validated with empirical evidence for safety and efficacy (174–178).
8.3. Personalized Drug Design Ethics
In personalized drug design, regulators must address N-of-1 trials or adaptive trial designs. Equity concerns will arise, ensuring AI-designed therapies are accessible globally, not just to wealthy individuals. Automating the design process and reducing costs will make personalized treatments more accessible (179).
The future of AI in molecular science promises transformative advances. AI will not replace humans but work alongside them, expanding creativity and accelerating innovation in drug discovery and protein engineering. With careful oversight and regulation, AI will help create cures at unprecedented speeds, addressing unmet medical needs, including for rare diseases and personalized therapies (180).
9. Conclusions
Generative AI has revolutionized drug discovery and protein design, shifting from rule-based, labor-intensive methods to AI-driven processes. Deep generative models, including VAEs, GANs, transformers, and diffusion models, enable the creation of novel molecular structures and protein sequences with desired properties. This addresses challenges in early-stage drug discovery: navigating vast chemical space, optimizing multiple parameters, and overcoming human bias.
AI-designed molecules have advanced from models to clinical trials, and AI-generated proteins now perform valuable functions. AI optimizes for multiple metrics simultaneously, producing balanced candidates less likely to fail. The future holds autonomous discovery systems where AI designs molecules and controls robotic experimentation, compressing the time from target identification to preclinical candidate.
However, ethical and regulatory challenges remain. AI can generate harmful molecules, requiring safeguards and human oversight. Regulatory bodies must adapt, evaluating AI-designed drugs with predictive modeling results and ensuring safety and efficacy.
Generative AI is transforming molecular science, uniting computational chemistry, structural biology, and systems biology. Advances in deep generative models, AI-guided docking like DiffDock, and diffusion models for protein design demonstrate rapid field progress. AI promises more effective, personalized medicines, biotech solutions, and faster responses to emerging health threats. Responsible integration will enhance the discovery of cures and engineered biomolecules.
Funding
None.
Author Contributions
U. Das: Writing - Original Draft, Writing - Review & Editing, Visualization; Conceptualization, Validation.
Conflicts of Interest
The author(s) report no conflict of interest.
Declaration of generative AI and AI-assisted technologies in the writing process
The writing of this review paper involved the use of generative AI and AI-assisted technologies only to enhance the clarity, coherence, and overall quality of the manuscript. The authors acknowledges the contributions of AI in the writing process while ensuring that the final content reflects the author’s own insights and interpretations of the literature. All interpretations and conclusions drawn in this manuscript are the sole responsibility of the author.
References
- Hinkson IV, Madej B, Stahlberg EA. Accelerating Therapeutics for Opportunities in Medicine: A Paradigm Shift in Drug Discovery. Front Pharmacol. 2020 Jun 30;11:770. [CrossRef]
- Vijayan RSK, Kihlberg J, Cross JB, Poongavanam V. Enhancing preclinical drug discovery with artificial intelligence. Drug Discov Today. 2022 Apr;27(4):967–84. [CrossRef]
- Sun D, Gao W, Hu H, Zhou S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm Sin B. 2022 Jul;12(7):3049–62. [CrossRef]
- Das U, Banerjee S, Sarkar M. Bibliometric analysis of circular RNA cancer vaccines and their emerging impact. Vacunas. 2025 Mar;500391. [CrossRef]
- Boyd NK, Teng C, Frei CR. Brief Overview of Approaches and Challenges in New Antibiotic Development: A Focus On Drug Repurposing. Front Cell Infect Microbiol. 2021;11:684515. [CrossRef]
- Singh S, Gupta H, Sharma P, Sahi S. Advances in Artificial Intelligence (AI)-assisted approaches in drug screening. Artif Intell Chem. 2024 Jun;2(1):100039. [CrossRef]
- Mouchlis VD, Afantitis A, Serra A, Fratello M, Papadiamantis AG, Aidinis V, et al. Advances in de Novo Drug Design: From Conventional to Machine Learning Methods. Int J Mol Sci. 2021 Feb 7;22(4):1676. [CrossRef]
- Das U, Chanda T, Kumar J, Peter A. Discovery of Natural MCL1 Inhibitors using Pharmacophore modelling, QSAR, Docking, ADMET, Molecular Dynamics, and DFT Analysis [Internet]. 2024 [cited 2025 Jan 9]. Available from: http://biorxiv.org/lookup/doi/10.1101/2024.10.14.618373. [CrossRef]
- Das U, Chandramouli L, Uttarkar A, Kumar J, Niranjan V. Discovery of natural compounds as novel FMS-like tyrosine kinase-3 (FLT3) therapeutic inhibitors for the treatment of acute myeloid leukemia: An in-silico approach. Asp Mol Med. 2025 Jun;5:100058. [CrossRef]
- Gangwal A, Ansari A, Ahmad I, Azad AK, Kumarasamy V, Subramaniyan V, et al. Generative artificial intelligence in drug discovery: basic framework, recent advances, challenges, and opportunities. Front Pharmacol. 2024;15:1331062. [CrossRef]
- Mroz AM, Posligua V, Tarzia A, Wolpert EH, Jelfs KE. Into the Unknown: How Computation Can Help Explore Uncharted Material Space. J Am Chem Soc. 2022 Oct 19;144(41):18730–43. [CrossRef]
- Han R, Yoon H, Kim G, Lee H, Lee Y. Revolutionizing Medicinal Chemistry: The Application of Artificial Intelligence (AI) in Early Drug Discovery. Pharm Basel Switz. 2023 Sep 6;16(9):1259. [CrossRef]
- Ivanenkov YA, Polykovskiy D, Bezrukov D, Zagribelnyy B, Aladinskiy V, Kamya P, et al. Chemistry42: An AI-Driven Platform for Molecular Design and Optimization. J Chem Inf Model. 2023 Feb 13;63(3):695–701. [CrossRef]
- Zeng X, Wang F, Luo Y, Kang S gu, Tang J, Lightstone FC, et al. Deep generative molecular design reshapes drug discovery. Cell Rep Med. 2022 Dec;3(12):100794. [CrossRef]
- Giordano D, Biancaniello C, Argenio MA, Facchiano A. Drug Design by Pharmacophore and Virtual Screening Approach. Pharm Basel Switz. 2022 May 23;15(5):646. [CrossRef]
- Çatalkaya S, Sabancı N, Yavuz SÇ, Sarıpınar E. The effect of stereoisomerism on the 4D-QSAR study of some dipeptidyl boron derivatives. Comput Biol Chem. 2020 Feb;84:107190. [CrossRef]
- Farghali H, Kutinová Canová N, Arora M. The potential applications of artificial intelligence in drug discovery and development. Physiol Res. 2021 Dec 30;70(Suppl4):S715–22. [CrossRef]
- Loeffler HH, He J, Tibo A, Janet JP, Voronov A, Mervin LH, et al. Reinvent 4: Modern AI–driven generative molecule design. J Cheminformatics. 2024 Feb 21;16(1):20. [CrossRef]
- Das U, Banerjee S, Sarkar M, Muhammad L F, Soni TK, Saha M, et al. Circular RNA vaccines: Pioneering the next-gen cancer immunotherapy. Cancer Pathog Ther. 2024 Dec;S2949713224000892. [CrossRef]
- Jiang Y, Yu Y, Kong M, Mei Y, Yuan L, Huang Z, et al. Artificial Intelligence for Retrosynthesis Prediction. Engineering. 2023 Jun;25:32–50.
- Ananikov VP. Top 20 influential AI-based technologies in chemistry. Artif Intell Chem. 2024 Dec;2(2):100075. [CrossRef]
- Liu Y, Yang Z, Yu Z, Liu Z, Liu D, Lin H, et al. Generative artificial intelligence and its applications in materials science: Current situation and future perspectives. J Materiomics. 2023 Jul;9(4):798–816. [CrossRef]
- Ochiai T, Inukai T, Akiyama M, Furui K, Ohue M, Matsumori N, et al. Variational autoencoder-based chemical latent space for large molecular structures with 3D complexity. Commun Chem. 2023 Nov 16;6(1):249. [CrossRef]
- Asperti A, Trentin M. Balancing Reconstruction Error and Kullback-Leibler Divergence in Variational Autoencoders. IEEE Access. 2020;8:199440–8. [CrossRef]
- Zheng W, Li J, Zhang Y. Desirable molecule discovery via generative latent space exploration. Vis Inform. 2023 Dec;7(4):13–21. [CrossRef]
- Abram KJ, McCloskey D. In Search of Disentanglement in Tandem Mass Spectrometry Datasets. Biomolecules. 2023 Sep 4;13(9):1343. [CrossRef]
- Sousa T, Correia J, Pereira V, Rocha M. Generative Deep Learning for Targeted Compound Design. J Chem Inf Model. 2021 Nov 22;61(11):5343–61. [CrossRef]
- Yang N, Wu H, Zeng K, Li Y, Bao S, Yan J. Molecule generation for drug design: A graph learning perspective. Fundam Res. 2024 Dec;S2667325824005259. [CrossRef]
- Vafaii H, Yates JL, Butts DA. Hierarchical VAEs provide a normative account of motion processing in the primate brain [Internet]. 2023 [cited 2025 Mar 30]. Available from: http://biorxiv.org/lookup/doi/10.1101/2023.09.27.559646. [CrossRef]
- Jang H, Seo S, Park S, Kim BJ, Choi GW, Choi J, et al. De novo drug design through gradient-based regularized search in information-theoretically controlled latent space. J Comput Aided Mol Des. 2024 Dec;38(1):32, s10822-024-00571–3. [CrossRef]
- Zhang Y, Li J, Chao X. ChemNav: An interactive visual tool to navigate in the latent space for chemical molecules discovery. Vis Inform. 2024 Dec;8(4):60–70. [CrossRef]
- Sharma P, Kumar M, Sharma HK, Biju SM. Generative adversarial networks (GANs): Introduction, Taxonomy, Variants, Limitations, and Applications. Multimed Tools Appl. 2024 Mar 26;83(41):88811–58. [CrossRef]
- Wu B, Li L, Cui Y, Zheng K. Cross-Adversarial Learning for Molecular Generation in Drug Design. Front Pharmacol. 2022 Jan 21;12:827606. [CrossRef]
- Tripathi S, Augustin AI, Dunlop A, Sukumaran R, Dheer S, Zavalny A, et al. Recent advances and application of generative adversarial networks in drug discovery, development, and targeting. Artif Intell Life Sci. 2022 Dec;2:100045. [CrossRef]
- Kucera T, Togninalli M, Meng-Papaxanthos L. Conditional generative modeling for de novo protein design with hierarchical functions. Wren J, editor. Bioinformatics. 2022 Jun 27;38(13):3454–61. [CrossRef]
- Putin E, Asadulaev A, Vanhaelen Q, Ivanenkov Y, Aladinskaya AV, Aliper A, et al. Adversarial Threshold Neural Computer for Molecular de Novo Design. Mol Pharm. 2018 Oct 1;15(10):4386–97. [CrossRef]
- Feng Y, Yang Y, Deng W, Chen H, Ran T. SyntaLinker-Hybrid: A deep learning approach for target specific drug design. Artif Intell Life Sci. 2022 Dec;2:100035. [CrossRef]
- De Cao N, Kipf T. MolGAN: An implicit generative model for small molecular graphs. 2018 [cited 2025 Mar 31]; Available from: https://arxiv.org/abs/1805.11973.
- Iglesias G, Talavera E, Díaz-Álvarez A. A survey on GANs for computer vision: Recent research, analysis and taxonomy. Comput Sci Rev. 2023 May;48:100553. [CrossRef]
- Méndez-Lucio O, Baillif B, Clevert DA, Rouquié D, Wichard J. De novo generation of hit-like molecules from gene expression signatures using artificial intelligence. Nat Commun. 2020 Jan 3;11(1):10. [CrossRef]
- Jiang J, Ke L, Chen L, Dou B, Zhu Y, Liu J, et al. Transformer technology in molecular science. WIREs Comput Mol Sci. 2024 Jul;14(4):e1725. [CrossRef]
- Chithrananda S, Grand G, Ramsundar B. ChemBERTa: Large-Scale Self-Supervised Pretraining for Molecular Property Prediction [Internet]. arXiv; 2020 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2010.09885.
- Mswahili ME, Jeong YS. Transformer-based models for chemical SMILES representation: A comprehensive literature review. Heliyon. 2024 Oct;10(20):e39038. [CrossRef]
- Luong KD, Singh A. Application of Transformers in Cheminformatics. J Chem Inf Model. 2024 Jun 10;64(11):4392–409. [CrossRef]
- Yoshimori A, Bajorath J. DeepAS – Chemical language model for the extension of active analogue series. Bioorg Med Chem. 2022 Jul;66:116808. [CrossRef]
- Madani A, Krause B, Greene ER, Subramanian S, Mohr BP, Holton JM, et al. Large language models generate functional protein sequences across diverse families. Nat Biotechnol. 2023 Aug;41(8):1099–106. [CrossRef]
- Sumida KH, Núñez-Franco R, Kalvet I, Pellock SJ, Wicky BIM, Milles LF, et al. Improving Protein Expression, Stability, and Function with ProteinMPNN. J Am Chem Soc. 2024 Jan 24;146(3):2054–61. [CrossRef]
- Chandra A, Tünnermann L, Löfstedt T, Gratz R. Transformer-based deep learning for predicting protein properties in the life sciences. eLife. 2023 Jan 18;12:e82819. [CrossRef]
- Cerchia C, Lavecchia A. New avenues in artificial-intelligence-assisted drug discovery. Drug Discov Today. 2023 Apr;28(4):103516. [CrossRef]
- Ramos MC, Collison CJ, White AD. A review of large language models and autonomous agents in chemistry. Chem Sci. 2025;16(6):2514–72. [CrossRef]
- Parigi M, Martina S, Caruso F. Quantum-Noise-Driven Generative Diffusion Models. Adv Quantum Technol. 2024 Jul 15;2300401. [CrossRef]
- Soleymani F, Paquet E, Viktor HL, Michalowski W. Structure-based protein and small molecule generation using EGNN and diffusion models: A comprehensive review. Comput Struct Biotechnol J. 2024 Dec;23:2779–97. [CrossRef]
- Xu C, Liu R, Yao Y, Huang W, Li Z, Luo HB. 3D-EDiffMG: 3D equivariant diffusion-driven molecular generation to accelerate drug discovery. J Pharm Anal. 2025 Mar;101257. [CrossRef]
- Alakhdar A, Poczos B, Washburn N. Diffusion Models in De Novo Drug Design. J Chem Inf Model. 2024 Oct 14;64(19):7238–56. [CrossRef]
- Xu M, Yu L, Song Y, Shi C, Ermon S, Tang J. GeoDiff: a Geometric Diffusion Model for Molecular Conformation Generation [Internet]. arXiv; 2022 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2203.02923.
- Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. De novo design of protein structure and function with RFdiffusion. Nature. 2023 Aug 31;620(7976):1089–100. [CrossRef]
- Corso G, Stärk H, Jing B, Barzilay R, Jaakkola T. DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking [Internet]. arXiv; 2022 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2210.01776.
- Wei YH. VAEs and GANs: Implicitly Approximating Complex Distributions with Simple Base Distributions and Deep Neural Networks -- Principles, Necessity, and Limitations [Internet]. arXiv; 2025 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2503.01898.
- Wu AN, Stouffs R, Biljecki F. Generative Adversarial Networks in the built environment: A comprehensive review of the application of GANs across data types and scales. Build Environ. 2022 Sep;223:109477. [CrossRef]
- Jiang J, Chen L, Ke L, Dou B, Zhang C, Feng H, et al. A review of transformers in drug discovery and beyond. J Pharm Anal. 2024 Aug;101081. [CrossRef]
- Chen M, Mei S, Fan J, Wang M. Opportunities and challenges of diffusion models for generative AI. Natl Sci Rev. 2024 Nov 14;11(12):nwae348. [CrossRef]
- Gupta R, Tiwari S, Chaudhary P. Generative AI Techniques and Models. In: Generative AI: Techniques, Models and Applications [Internet]. Cham: Springer Nature Switzerland; 2025 [cited 2025 Mar 31]. p. 45–64. (Lecture Notes on Data Engineering and Communications Technologies; vol. 241). Available from: https://link.springer.com/10.1007/978-3-031-82062-5_3.
- Li C, Zhang T, Du X, Zhang Y, Xie H. Generative AI models for different steps in architectural design: A literature review. Front Archit Res. 2025 Jun;14(3):759–83. [CrossRef]
- Shu D, Li Z, Barati Farimani A. A physics-informed diffusion model for high-fidelity flow field reconstruction. J Comput Phys. 2023 Apr;478:111972. [CrossRef]
- Connor MC, Canal GH, Rozell CJ. Variational Autoencoder with Learned Latent Structure [Internet]. arXiv; 2020 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2006.10597.
- Chen N, Klushyn A, Ferroni F, Bayer J, van der Smagt P. Learning Flat Latent Manifolds with VAEs. 2020 [cited 2025 Mar 31]; Available from: https://arxiv.org/abs/2002.04881.
- Chandra R, Horne RI, Vendruscolo M. Bayesian Optimization in the Latent Space of a Variational Autoencoder for the Generation of Selective FLT3 Inhibitors. J Chem Theory Comput. 2024 Jan 9;20(1):469–76. [CrossRef]
- Yang X, Wang Y, Byrne R, Schneider G, Yang S. Concepts of Artificial Intelligence for Computer-Assisted Drug Discovery. Chem Rev. 2019 Sep 25;119(18):10520–94. [CrossRef]
- Trunz E, Weinmann M, Merzbach S, Klein R. Efficient structuring of the latent space for controllable data reconstruction and compression. Graph Vis Comput. 2022 Dec;7:200059. [CrossRef]
- Shen C, Krenn M, Eppel S, Aspuru-Guzik A. Deep molecular dreaming: inverse machine learning for de-novo molecular design and interpretability with surjective representations. Mach Learn Sci Technol. 2021 Sep 1;2(3):03LT02. [CrossRef]
- Prykhodko O, Johansson SV, Kotsias PC, Arús-Pous J, Bjerrum EJ, Engkvist O, et al. A de novo molecular generation method using latent vector based generative adversarial network. J Cheminformatics. 2019 Dec;11(1):74. [CrossRef]
- Rossi E, Wheeler JM, Sebastiani M. High-speed nanoindentation mapping: A review of recent advances and applications. Curr Opin Solid State Mater Sci. 2023 Oct;27(5):101107. [CrossRef]
- Bilodeau C, Jin W, Jaakkola T, Barzilay R, Jensen KF. Generative models for molecular discovery: Recent advances and challenges. WIREs Comput Mol Sci. 2022 Sep;12(5):e1608. [CrossRef]
- Guo J, Schwaller P. Directly optimizing for synthesizability in generative molecular design using retrosynthesis models. Chem Sci. 2025;10.1039.D5SC01476J. [CrossRef]
- Wang J, Zhu F. ExSelfRL: An exploration-inspired self-supervised reinforcement learning approach to molecular generation. Expert Syst Appl. 2025 Jan;260:125410. [CrossRef]
- Nakamura S, Yasuo N, Sekijima M. Molecular optimization using a conditional transformer for reaction-aware compound exploration with reinforcement learning. Commun Chem. 2025 Feb 8;8(1):40. [CrossRef]
- Korn M, Ehrt C, Ruggiu F, Gastreich M, Rarey M. Navigating large chemical spaces in early-phase drug discovery. Curr Opin Struct Biol. 2023 Jun;80:102578. [CrossRef]
- Anstine DM, Isayev O. Generative Models as an Emerging Paradigm in the Chemical Sciences. J Am Chem Soc. 2023 Apr 26;145(16):8736–50. [CrossRef]
- Świechowski M, Godlewski K, Sawicki B, Mańdziuk J. Monte Carlo Tree Search: a review of recent modifications and applications. Artif Intell Rev. 2023 Mar;56(3):2497–562. [CrossRef]
- Park J, Ahn J, Choi J, Kim J. Mol-AIR: Molecular Reinforcement Learning with Adaptive Intrinsic Rewards for Goal-Directed Molecular Generation. J Chem Inf Model. 2025 Mar 10;65(5):2283–96. [CrossRef]
- Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol. 2019 Sep;37(9):1038–40. [CrossRef]
- Greenstein BL, Elsey DC, Hutchison GR. Determining best practices for using genetic algorithms in molecular discovery. J Chem Phys. 2023 Sep 7;159(9):091501. [CrossRef]
- McCall J. Genetic algorithms for modelling and optimisation. J Comput Appl Math. 2005 Dec;184(1):205–22. [CrossRef]
- Kim M, Gu J, Yuan Y, Yun T, Liu Z, Bengio Y, et al. Offline Model-Based Optimization: Comprehensive Review [Internet]. arXiv; 2025 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2503.17286.
- Schulam P, Muslea I. Improving the Exploration/Exploitation Trade-Off in Web Content Discovery. In: Companion Proceedings of the ACM Web Conference 2023 [Internet]. Austin TX USA: ACM; 2023 [cited 2025 Mar 31]. p. 1183–9. Available from: https://dl.acm.org/doi/10.1145/3543873.3587574.
- Gupta P, Ding B, Guan C, Ding D. Generative AI: A systematic review using topic modelling techniques. Data Inf Manag. 2024 Jun;8(2):100066. [CrossRef]
- Abeer ANMN, Urban NM, Weil MR, Alexander FJ, Yoon BJ. Multi-objective latent space optimization of generative molecular design models. Patterns. 2024 Oct;5(10):101042. [CrossRef]
- Menon D, Ranganathan R. A Generative Approach to Materials Discovery, Design, and Optimization. ACS Omega. 2022 Aug 2;7(30):25958–73. [CrossRef]
- Aal E Ali RS, Meng J, Khan MEI, Jiang X. Machine learning advancements in organic synthesis: A focused exploration of artificial intelligence applications in chemistry. Artif Intell Chem. 2024 Jun;2(1):100049.
- Vogt M. Exploring chemical space — Generative models and their evaluation. Artif Intell Life Sci. 2023 Dec;3:100064. [CrossRef]
- Rehman AU, Li M, Wu B, Ali Y, Rasheed S, Shaheen S, et al. Role of Artificial Intelligence in Revolutionizing Drug Discovery. Fundam Res. 2024 May;S266732582400205X. [CrossRef]
- Magar R, Wang Y, Barati Farimani A. Crystal twins: self-supervised learning for crystalline material property prediction. Npj Comput Mater. 2022 Nov 10;8(1):231. [CrossRef]
- Wang J, Guan J, Zhou S. Molecular property prediction by contrastive learning with attention-guided positive sample selection. Wren J, editor. Bioinformatics. 2023 May 4;39(5):btad258. [CrossRef]
- Yang X, Wang Y, Lin Y, Zhang M, Liu O, Shuai J, et al. A Multi-Task Self-Supervised Strategy for Predicting Molecular Properties and FGFR1 Inhibitors. Adv Sci. 2025 Feb 8;2412987. [CrossRef]
- Cafiero M. Transformer-Decoder GPT Models for Generating Virtual Screening Libraries of HMG-Coenzyme A Reductase Inhibitors: Effects of Temperature, Prompt Length, and Transfer-Learning Strategies. J Chem Inf Model. 2024 Nov 25;64(22):8464–80. [CrossRef]
- Chen S, Guo W. Auto-Encoders in Deep Learning—A Review with New Perspectives. Mathematics. 2023 Apr 7;11(8):1777. [CrossRef]
- Korshunova M, Huang N, Capuzzi S, Radchenko DS, Savych O, Moroz YS, et al. Generative and reinforcement learning approaches for the automated de novo design of bioactive compounds. Commun Chem. 2022 Oct 18;5(1):129. [CrossRef]
- Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv. 2018 Jul 6;4(7):eaap7885. [CrossRef]
- Tan RK, Liu Y, Xie L. Reinforcement learning for systems pharmacology-oriented and personalized drug design. Expert Opin Drug Discov. 2022 Aug;17(8):849–63. [CrossRef]
- Dodds M, Guo J, Löhr T, Tibo A, Engkvist O, Janet JP. Sample efficient reinforcement learning with active learning for molecular design. Chem Sci. 2024;15(11):4146–60. [CrossRef]
- Reiser P, Neubert M, Eberhard A, Torresi L, Zhou C, Shao C, et al. Graph neural networks for materials science and chemistry. Commun Mater. 2022 Nov 26;3(1):93. [CrossRef]
- Abate C, Decherchi S, Cavalli A. Graph neural networks for conditional de novo drug design. WIREs Comput Mol Sci. 2023 Jul;13(4):e1651. [CrossRef]
- Zheng S, Lei Z, Ai H, Chen H, Deng D, Yang Y. Deep scaffold hopping with multimodal transformer neural networks. J Cheminformatics. 2021 Nov 13;13(1):87. [CrossRef]
- Hu C, Li S, Yang C, Chen J, Xiong Y, Fan G, et al. ScaffoldGVAE: scaffold generation and hopping of drug molecules via a variational autoencoder based on multi-view graph neural networks. J Cheminformatics. 2023 Oct 4;15(1):91. [CrossRef]
- Wu KE, Yang KK, Van Den Berg R, Alamdari S, Zou JY, Lu AX, et al. Protein structure generation via folding diffusion. Nat Commun. 2024 Feb 5;15(1):1059. [CrossRef]
- Sarumi OA, Heider D. Large language models and their applications in bioinformatics. Comput Struct Biotechnol J. 2024 Dec;23:3498–505. [CrossRef]
- Valentini G, Malchiodi D, Gliozzo J, Mesiti M, Soto-Gomez M, Cabri A, et al. The promises of large language models for protein design and modeling. Front Bioinforma. 2023;3:1304099. [CrossRef]
- Nana Teukam YG, Kwate Dassi L, Manica M, Probst D, Schwaller P, Laino T. Language models can identify enzymatic binding sites in protein sequences. Comput Struct Biotechnol J. 2024 Dec;23:1929–37. [CrossRef]
- Liu J, Yang M, Yu Y, Xu H, Wang T, Li K, et al. Advancing bioinformatics with large language models: components, applications and perspectives. ArXiv. 2025 Jan 31;arXiv:2401.04155v2.
- Bzdok D, Thieme A, Levkovskyy O, Wren P, Ray T, Reddy S. Data science opportunities of large language models for neuroscience and biomedicine. Neuron. 2024 Mar;112(5):698–717. [CrossRef]
- Hie BL, Shanker VR, Xu D, Bruun TUJ, Weidenbacher PA, Tang S, et al. Efficient evolution of human antibodies from general protein language models. Nat Biotechnol. 2024 Feb;42(2):275–83. [CrossRef]
- Kim J, McFee M, Fang Q, Abdin O, Kim PM. Computational and artificial intelligence-based methods for antibody development. Trends Pharmacol Sci. 2023 Mar;44(3):175–89. [CrossRef]
- Luo S, Su Y, Peng X, Wang S, Peng J, Ma J. Antigen-Specific Antibody Design and Optimization with Diffusion-Based Generative Models for Protein Structures [Internet]. 2022 [cited 2025 Mar 31]. Available from: http://biorxiv.org/lookup/doi/10.1101/2022.07.10.499510.
- Dewaker V, Morya VK, Kim YH, Park ST, Kim HS, Koh YH. Revolutionizing oncology: the role of Artificial Intelligence (AI) as an antibody design, and optimization tools. Biomark Res. 2025 Mar 29;13(1):52. [CrossRef]
- Yang J, Li FZ, Arnold FH. Opportunities and Challenges for Machine Learning-Assisted Enzyme Engineering. ACS Cent Sci. 2024 Feb 28;10(2):226–41. [CrossRef]
- Zhou J, Huang M. Navigating the landscape of enzyme design: from molecular simulations to machine learning. Chem Soc Rev. 2024;53(16):8202–39. [CrossRef]
- Orsi E, Schada Von Borzyskowski L, Noack S, Nikel PI, Lindner SN. Automated in vivo enzyme engineering accelerates biocatalyst optimization. Nat Commun. 2024 Apr 24;15(1):3447. [CrossRef]
- Baum ZJ, Yu X, Ayala PY, Zhao Y, Watkins SP, Zhou Q. Artificial Intelligence in Chemistry: Current Trends and Future Directions. J Chem Inf Model. 2021 Jul 26;61(7):3197–212. [CrossRef]
- Arya SS, Dias SB, Jelinek HF, Hadjileontiadis LJ, Pappa AM. The convergence of traditional and digital biomarkers through AI-assisted biosensing: A new era in translational diagnostics? Biosens Bioelectron. 2023 Sep;235:115387. [CrossRef]
- Stärk H, Ganea OE, Pattanaik L, Barzilay R, Jaakkola T. EquiBind: Geometric Deep Learning for Drug Binding Structure Prediction. 2022 [cited 2025 Mar 31]; Available from: https://arxiv.org/abs/2202.05146.
- Ketata MA, Laue C, Mammadov R, Stärk H, Wu M, Corso G, et al. DiffDock-PP: Rigid Protein-Protein Docking with Diffusion Models [Internet]. arXiv; 2023 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2304.03889.
- Yang C, Chen EA, Zhang Y. Protein-Ligand Docking in the Machine-Learning Era. Mol Basel Switz. 2022 Jul 18;27(14):4568. [CrossRef]
- Cao D, Chen M, Zhang R, Wang Z, Huang M, Yu J, et al. SurfDock is a surface-informed diffusion generative model for reliable and accurate protein–ligand complex prediction. Nat Methods. 2025 Feb;22(2):310–22. [CrossRef]
- B Fortela DL, Mikolajczyk AP, Carnes MR, Sharp W, Revellame E, Hernandez R, et al. Predicting Molecular Docking of Per- and Polyfluoroalkyl Substances to Blood Protein Using Generative Artificial Intelligence Algorithm Diffdock. BioTechniques. 2024 Jan;76(1):14–26. [CrossRef]
- Wang Y, Jiao Q, Wang J, Cai X, Zhao W, Cui X. Prediction of protein-ligand binding affinity with deep learning. Comput Struct Biotechnol J. 2023;21:5796–806. [CrossRef]
- Wang DD, Wu W, Wang R. Structure-based, deep-learning models for protein-ligand binding affinity prediction. J Cheminformatics. 2024 Jan 3;16(1):2. [CrossRef]
- Zhang S, Jin Y, Liu T, Wang Q, Zhang Z, Zhao S, et al. SS-GNN: A Simple-Structured Graph Neural Network for Affinity Prediction. ACS Omega. 2023 Jun 27;8(25):22496–507. [CrossRef]
- Wang H. Prediction of protein–ligand binding affinity via deep learning models. Brief Bioinform. 2024 Jan 22;25(2):bbae081. [CrossRef]
- Wang R, Fang X, Lu Y, Wang S. The PDBbind Database: Collection of Binding Affinities for Protein−Ligand Complexes with Known Three-Dimensional Structures. J Med Chem. 2004 Jun 1;47(12):2977–80. [CrossRef]
- Weidman JD, Sajjan M, Mikolas C, Stewart ZJ, Pollanen J, Kais S, et al. Quantum computing and chemistry. Cell Rep Phys Sci. 2024 Sep;5(9):102105. [CrossRef]
- Morawietz T, Artrith N. Machine learning-accelerated quantum mechanics-based atomistic simulations for industrial applications. J Comput Aided Mol Des. 2021 Apr;35(4):557–86. [CrossRef]
- Doga H, Raubenolt B, Cumbo F, Joshi J, DiFilippo FP, Qin J, et al. A Perspective on Protein Structure Prediction Using Quantum Computers. J Chem Theory Comput. 2024 May 14;20(9):3359–78. [CrossRef]
- How ML, Cheah SM. Forging the Future: Strategic Approaches to Quantum AI Integration for Industry Transformation. AI. 2024 Jan 29;5(1):290–323. [CrossRef]
- Liu X, Jiang S, Duan X, Vasan A, Liu C, Tien C chan, et al. Binding Affinity Prediction: From Conventional to Machine Learning-Based Approaches [Internet]. arXiv; 2024 [cited 2025 Mar 31]. Available from: https://arxiv.org/abs/2410.00709.
- Yan J, Ye Z, Yang Z, Lu C, Zhang S, Liu Q, et al. Multi-task bioassay pre-training for protein-ligand binding affinity prediction. Brief Bioinform. 2023 Nov 22;25(1):bbad451. [CrossRef]
- Schwaller P, Gaudin T, Lányi D, Bekas C, Laino T. “Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem Sci. 2018;9(28):6091–8. [CrossRef]
- Jackson I, Jesus Saenz M, Ivanov D. From natural language to simulations: applying AI to automate simulation modelling of logistics systems. Int J Prod Res. 2024 Feb 16;62(4):1434–57. [CrossRef]
- Sinha S, Lee YM. Challenges with developing and deploying AI models and applications in industrial systems. Discov Artif Intell. 2024 Aug 16;4(1):55. [CrossRef]
- Hong S, Zhuo HH, Jin K, Shao G, Zhou Z. Retrosynthetic planning with experience-guided Monte Carlo tree search. Commun Chem. 2023 Jun 10;6(1):120. [CrossRef]
- Lai H, Kannas C, Hassen AK, Granqvist E, Westerlund AM, Clevert DA, et al. Multi-objective synthesis planning by means of Monte Carlo Tree search. Artif Intell Life Sci. 2025 Jun;7:100130. [CrossRef]
- Terven J. Deep Reinforcement Learning: A Chronological Overview and Methods. AI. 2025 Feb 24;6(3):46. [CrossRef]
- Nambiar AMK, Breen CP, Hart T, Kulesza T, Jamison TF, Jensen KF. Bayesian Optimization of Computer-Proposed Multistep Synthetic Routes on an Automated Robotic Flow Platform. ACS Cent Sci. 2022 Jun 22;8(6):825–36. [CrossRef]
- Schilter O, Gutierrez DP, Folkmann LM, Castrogiovanni A, García-Durán A, Zipoli F, et al. Combining Bayesian optimization and automation to simultaneously optimize reaction conditions and routes. Chem Sci. 2024;15(20):7732–41. [CrossRef]
- Tachibana R, Zhang K, Zou Z, Burgener S, Ward TR. A Customized Bayesian Algorithm to Optimize Enzyme-Catalyzed Reactions. ACS Sustain Chem Eng. 2023 Aug 21;11(33):12336–44. [CrossRef]
- Omotehinwa TO, Lawrence MO, Oyewola DO, Dada EG. Bayesian optimization of one-dimensional convolutional neural networks (1D CNN) for early diagnosis of Autistic Spectrum Disorder. J Comput Math Data Sci. 2024 Dec;13:100105. [CrossRef]
- Kwon Y, Lee D, Kim JW, Choi YS, Kim S. Exploring Optimal Reaction Conditions Guided by Graph Neural Networks and Bayesian Optimization. ACS Omega. 2022 Dec 13;7(49):44939–50. [CrossRef]
- Parrot M, Tajmouati H, Da Silva VBR, Atwood BR, Fourcade R, Gaston-Mathé Y, et al. Integrating synthetic accessibility with AI-based generative drug design. J Cheminformatics. 2023 Sep 19;15(1):83. [CrossRef]
- Retchin M, Wang Y, Takaba K, Chodera JD. DrugGym: A testbed for the economics of autonomous drug discovery [Internet]. 2024 [cited 2025 Mar 31]. Available from: http://biorxiv.org/lookup/doi/10.1101/2024.05.28.596296.
- D. Segall M. Multi-Parameter Optimization: Identifying High Quality Compounds with a Balance of Properties. Curr Drug Metab. 2012 Mar 1;18(9):1292–310.
- Wager TT, Hou X, Verhoest PR, Villalobos A. Central Nervous System Multiparameter Optimization Desirability: Application in Drug Discovery. ACS Chem Neurosci. 2016 Jun 15;7(6):767–75. [CrossRef]
- Joshi-Barr S, Wampole M. Artificial Intelligence for Drug Toxicity and Safety. In: Hock FJ, Pugsley MK, editors. Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays [Internet]. Cham: Springer International Publishing; 2024 [cited 2025 Mar 31]. p. 2637–71. Available from: https://link.springer.com/10.1007/978-3-031-35529-5_134.
- Burki T. A new paradigm for drug development. Lancet Digit Health. 2020 May;2(5):e226–7. [CrossRef]
- Shanehsazzadeh A, McPartlon M, Kasun G, Steiger AK, Sutton JM, Yassine E, et al. Unlocking de novo antibody design with generative artificial intelligence [Internet]. 2023 [cited 2025 Mar 31]. Available from: http://biorxiv.org/lookup/doi/10.1101/2023.01.08.523187.
- Visan AI, Negut I. Integrating Artificial Intelligence for Drug Discovery in the Context of Revolutionizing Drug Delivery. Life Basel Switz. 2024 Feb 7;14(2):233. [CrossRef]
- Guan S, Wang G. Drug discovery and development in the era of artificial intelligence: From machine learning to large language models. Artif Intell Chem. 2024 Jun;2(1):100070. [CrossRef]
- Schneider G. Automating drug discovery. Nat Rev Drug Discov. 2018 Feb;17(2):97–113. [CrossRef]
- Atomwise AIMS Program. AI is a viable alternative to high throughput screening: a 318-target study. Sci Rep. 2024 Apr 2;14(1):7526.
- Dhudum R, Ganeshpurkar A, Pawar A. Revolutionizing Drug Discovery: A Comprehensive Review of AI Applications. Drugs Drug Candidates. 2024 Feb 13;3(1):148–71. [CrossRef]
- Qiu X, Li H, Ver Steeg G, Godzik A. Advances in AI for Protein Structure Prediction: Implications for Cancer Drug Discovery and Development. Biomolecules. 2024 Mar 12;14(3):339. [CrossRef]
- Qin Y, Chen Z, Peng Y, Xiao Y, Zhong T, Yu X. Deep learning methods for protein structure prediction. MedComm – Future Med. 2024 Sep;3(3):e96. [CrossRef]
- Xu Y, Liu X, Cao X, Huang C, Liu E, Qian S, et al. Artificial intelligence: A powerful paradigm for scientific research. The Innovation. 2021 Nov;2(4):100179. [CrossRef]
- Sliwoski G, Kothiwale S, Meiler J, Lowe EW. Computational methods in drug discovery. Pharmacol Rev. 2014;66(1):334–95. [CrossRef]
- Khakzad H, Igashov I, Schneuing A, Goverde C, Bronstein M, Correia B. A new age in protein design empowered by deep learning. Cell Syst. 2023 Nov;14(11):925–39. [CrossRef]
- Fu C, Chen Q. The future of pharmaceuticals: Artificial intelligence in drug discovery and development. J Pharm Anal. 2025 Feb;101248. [CrossRef]
- Wang X, Xu K, Tan Y, Liu S, Zhou J. Possibilities of Using De Novo Design for Generating Diverse Functional Food Enzymes. Int J Mol Sci. 2023 Feb 14;24(4):3827. [CrossRef]
- Bhisetti G, Fang C. Artificial Intelligence–Enabled De Novo Design of Novel Compounds that Are Synthesizable. In: Heifetz A, editor. Artificial Intelligence in Drug Design [Internet]. New York, NY: Springer US; 2022 [cited 2025 Mar 31]. p. 409–19. (Methods in Molecular Biology; vol. 2390). Available from: https://link.springer.com/10.1007/978-1-0716-1787-8_17.
- Shi Y, Hu H. AI accelerated discovery of self-assembling peptides. Biomater Transl. 2023;4(4):291–3. [CrossRef]
- Ding N, Yuan Z, Ma Z, Wu Y, Yin L. AI-Assisted Rational Design and Activity Prediction of Biological Elements for Optimizing Transcription-Factor-Based Biosensors. Mol Basel Switz. 2024 Jul 26;29(15):3512. [CrossRef]
- Divine R, Dang HV, Ueda G, Fallas JA, Vulovic I, Sheffler W, et al. Designed proteins assemble antibodies into modular nanocages. Science. 2021 Apr 2;372(6537):eabd9994. [CrossRef]
- Tom G, Schmid SP, Baird SG, Cao Y, Darvish K, Hao H, et al. Self-Driving Laboratories for Chemistry and Materials Science. Chem Rev. 2024 Aug 28;124(16):9633–732. [CrossRef]
- Blunt NS, Camps J, Crawford O, Izsák R, Leontica S, Mirani A, et al. Perspective on the Current State-of-the-Art of Quantum Computing for Drug Discovery Applications. J Chem Theory Comput. 2022 Dec 13;18(12):7001–23. [CrossRef]
- Ur Rasool R, Ahmad HF, Rafique W, Qayyum A, Qadir J, Anwar Z. Quantum Computing for Healthcare: A Review. Future Internet. 2023 Feb 27;15(3):94. [CrossRef]
- Outeiral C, Strahm M, Shi J, Morris GM, Benjamin SC, Deane CM. The prospects of quantum computing in computational molecular biology. WIREs Comput Mol Sci. 2021 Jan;11(1):e1481. [CrossRef]
- Serrano DR, Luciano FC, Anaya BJ, Ongoren B, Kara A, Molina G, et al. Artificial Intelligence (AI) Applications in Drug Discovery and Drug Delivery: Revolutionizing Personalized Medicine. Pharmaceutics. 2024 Oct 14;16(10):1328. [CrossRef]
- Cheong BC. Transparency and accountability in AI systems: safeguarding wellbeing in the age of algorithmic decision-making. Front Hum Dyn. 2024 Jul 3;6:1421273. [CrossRef]
- Choudhury A, Asan O. Role of Artificial Intelligence in Patient Safety Outcomes: Systematic Literature Review. JMIR Med Inform. 2020 Jul 24;8(7):e18599. [CrossRef]
- Alizadehsani R, Oyelere SS, Hussain S, Jagatheesaperumal SK, Calixto RR, Rahouti M, et al. Explainable Artificial Intelligence for Drug Discovery and Development: A Comprehensive Survey. IEEE Access. 2024;12:35796–812. [CrossRef]
- Kapustina O, Burmakina P, Gubina N, Serov N, Vinogradov V. User-friendly and industry-integrated AI for medicinal chemists and pharmaceuticals. Artif Intell Chem. 2024 Dec;2(2):100072. [CrossRef]
- Taherdoost H, Ghofrani A. AI’s role in revolutionizing personalized medicine by reshaping pharmacogenomics and drug therapy. Intell Pharm. 2024 Oct;2(5):643–50. [CrossRef]
- Saini JPS, Thakur A, Yadav D. AI-driven innovations in pharmaceuticals: optimizing drug discovery and industry operations. RSC Pharm. 2025;10.1039.D4PM00323C. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



