Submitted:
31 March 2025
Posted:
31 March 2025
You are already at the latest version
Abstract
The growing threat of antimicrobial resistance has driven the search for new bioactive compounds in extreme environments such as Antarctica. Streptomyces fildesensis So13.3, isolated from Antarctic soil, has been shown to contain a biosynthetic gene cluster associated with producing actinomycin D, an antibiotic with therapeutic potential. In this study, we analysed the regulatory role of TetR/AcrR family transcription factors present within this BGC, focusing on their activation under different nutritional conditions and their structural scharacterisation using bioinformatics tools and molecular dynamics simulations. The results showed that TetR/AcrR expression increased significantly in ISP4 and IMA media, suggesting their involvement in nutrient-dependent regulation of the cluster. At the structural level, two TetR proteins (TetR-206 and TetR-279) were modelled, with the latter standing out due to a C-terminal tetracycline repressor-like domain. A 200 ns molecular dynamics simulation was performed in GROMACS to evaluate the stability and flexibility of TetR-279, including analysis of point mutations (S166P, V167A, V167I). The S166P mutation significantly impacted structural flexibility, while V167A and V167I caused only minor alterations. This work demonstrates the value of integrating omics approaches, structural modelling, and gene editing with CRISPR to study and potentially activate silent BGCs in non-model bacteria such as Antarctic Streptomyces. In particular, the targeted inhibition of TetR-279 may trigger metabolic rewiring and facilitate the expression of novel antibiotics encoded in cryptic biosynthetic gene clusters.
Keywords:
Antarctic Streptomyces
; Biosynthetic gene clusters
; CRISPR-based activation
; Molecular dynamics simulation
1. Introduction
The high prevalence of antimicrobial resistance is perceived as an imminent crisis, and the World Health Organization (WHO) has stated that several bacterial pathogens are considered "critical priorities." [1,2]. In line with this concern, the WHO has included mitigating antimicrobial resistance as a major focus in its 2019-2023 work program, highlighting the urgency to address this global challenge [3]. Owing to the urgency of this emergency, there is an increasing need for new sources and molecules with antimicrobial properties. Considering that traditional methods often lead to the rediscovery of known compounds and diminish the chances of uncovering new bioactive compounds is that research in poorly explored or extreme environments's, such as those with inhospitable conditions, offers new opportunities for discovering novel antimicrobial compounds [4,5]. A prime example of this extreme environment is the Antarctic Territory, characterised by a cold, dry and windy climate with high exposure to UV radiation, low presence of organic matter and high salt concentration in soils [6,7]. In this region, Actinobacteria considered a rare phylum among bacteria, have been studied as they could likely offer novel bioactive molecules due to their adaptation to those extreme conditions involving uncommon metabolic pathways and compounds, which can be deciphered through metabolomic or genomic approaches [8,9]. However, advances in sequencing technologies have revealed new precursors in bacterial genomes, offering the possibility of synthesising novel compounds [10,11]. Nevertheless, the expression of most of these biosynthetic gene's clusters (BGCs) is either silenced or barely expressed under conventional laboratory conditions, which has motivated the exploration of new strategies to activate them [12,13]
Núñez-Montero et al. [14] conducted a study with a Streptomyces fildesensis So. 13.3 strain isolated in Antarctica. Sequencing and analysis of the complete 9.47 Mb genome included 42 predicted BGCs and 56 putative clusters, constituting approximately 22% of the total genome content. Notably, many of these clusters (11 out of 42 BGCs and 40 out of 56 putative BGCs) showed no similarity to other known BGCs. In a subsequent study conducted by our group, Núñez-Montero et al [15], strain So13.3 was grown under various nutrient and elicitation conditions, such as the addition of lipopolysaccharide (LPS), sodium nitroprusside (SNP), and co-culture. Metabolomes obtained by HPLC-QTOF-MS/MS were analysed by molecular networks linking BGCs identified in the genome with metabolites detected by MS/MS. The results revealed the presence of an actinomycin BGC in strain So13.3, which was corroborated by structural matches with actinomycin D according to genomic analysis. These findings provide valuable information on the complexity of metabolic networks and how strain So13.3 responds to pleiotropic stimuli. Using omics techniques allowed us to identify more general response pathways that present themselves as candidates for disruption, like actinomycin D cluster [16].
Genetic analysis of the antinomycin D cluster reveals the presence of different regulatory genes [17]. Notably, the TetR/AcrR family transcriptional regulator is among them. TetR/AcrR family members are two-domain proteins that contain an N-terminal DNA-binding motif (HTH) and a C-terminal ligand recognition domain [18]. The C-terminal ligand-recognition domain can be used to identify compounds like those transferred by their target transporters [19]. TetR proteins act as chemical sensors, monitoring the dynamics of the cellular environment and regulating genes associated with various events, such as antibiotic production, osmotic stress, efflux pumps, multidrug resistance, metabolic modulation, and pathogenesis [20]. Although there are over 200,000 probable sequences in public databases, and the structure of approximately 200 has been solved, most sequences remain uncharacterised [21]. Actinobacteria, especially Streptomyces coelicolor and Streptomyces avermitilis, showed a high presence of these regulators, reflecting the complexity of their morphological differentiation and secondary metabolism [22,23]. For this reason, it is interesting to note that inhibition of the TetR/AcrR family member factors in the actinomycin D cluster could contribute to the metabolic modification of So13.3 bacteria to increase the generation of new secondary metabolites with antibiotic application [24]. The CRISPR/Cas9 gene editing method, specifically the pCRISPR-Cas9-scalingD system described by Tong [25], could be used for this inhibition. However, due to the limited information on the TetR/AcrR family within the actinomycin D cluster and its potential influence on cluster regulation, this study aims to analyse the impact of CRISPR-induced mutations before in vitro or in vivo assays, focusing on the structural configuration dynamics and overall stability of TetR/AcrR and its impact as a functional unit.
2. Results
2.1. Structural Characterisation and Refinement of TetR Transcription Factors in the Actinomycin D Biosynthetic Cluster of Streptomyces fildesensis
In the genome of Streptomyces fildesensis (GenBank accession: CP048835.1), a BGC potentially linked to Actinomycin D production was discovered using antiSMASH. This cluster contains two genes that encode putative transcription factors (TFs) of the TetR family, specifically the AccR subtype: TetR-206 (Protein ID: QNA70898.1) with 206 amino acids, and TetR-279 (Protein ID: QNA70921.1) with 279 amino acids. To investigate the structural features of these TFs, BLAST searches were conducted against the PDB, but no crystallised structures were identified. As a result, homology modeling was performed using AlphaFold, which retrieved UniProt entries A0A5R9MB50 for TetR-206 and A0A5R9M760 for TetR-279. Structural models were created, and their confidence scores were evaluated to pinpoint areas of low structural quality needing refinement. Further inspection of protein features depicts that, despite being both TFs annotated as TetR/AcrR, TetR-206 presents only an HTH-DNA binding motif, common for TetR transcription factors and other TFs families. At the same time, TetR-279 has a specific Tetraciclyn repressor-like C terminal domain, as depicted by InterProScan in their individual UniProt entries. Previous findings highlight the potential of TetR-279 as the primary regulator of this Actinomycin D BGC regulation as its C-terminal domain couldind and respond to metabolic stimuli as required.
To improve the structural reliability of these regions, loop refinement was performed using the GalaxyWeb Server, resulting in optimised models. In both cases, the low-confidence regions—according to AlphaFold's scoring system—were primarily loops near each protein's N-terminal. The refined structures were used as initial conformations for all-atom MD simulations with GROMACS. Furthermore, to assess whether these transcription factors have membrane-associated functions, SignalP 5.0 was employed to predict the presence of signal peptides. The analysis confirmed the absence of signal sequences, indicating that both proteins are likely cytoplasmic and not directly involved in membrane interactions. Considering that TetR-279 was functionally annotated as having just a C-terminal domain related to ligand binding, and no HTH-DNA binding motif was reported, we assessed its ability to bind DNA using its sequence features using DPBind webserver (https://lcg.rit.albany.edu/dp-bind), were it shown that this TF has several predicted DNA-binding related residues despite not being classified with a N-terminal DNA-binding region by InterProScan (Figure 1A and Figure 1B).
2.2. Nutrient-Dependent Regulation of TetR-279 Expression in the Actinomycin D Gene Cluster
Gene expression analysis for the TetR-279 transcription factor within the actinomycin D cluster showed notable variations based on the culture medium employed. There was a marked increase in TetR-279 expression in IMA and ISP4 media compared to the control condition (p<0.05), indicating that this transcription factor is actively responsive to particular nutritional environments (Figure 2A). Conversely, gene expression did not significantly differ from the control condition in the YES medium, suggesting that this medium does not trigger the activation of the actinomycin D cluster via TetR-279 (Figure 2B and Table 1). s adenine phosphoribosyltransferase (APRT) gene served as an internal control for data normalisation. Furthermore, the results were derived from three biological replicates and two replicates for each experimental condition. These findings imply that TetR-279 might be crucial in regulating actinomycin D biosynthesis under specific nutritional conditions.
2.3. Structural Effects of Point Mutations in TetR-279 via Molecular Dynamics Simulation
After structural refinement, given that TetR-279 was highlighted as a putative ligand-dependent regulator of the actinomycin D BGC, it was chosen for MD simulations using GROMACS. A biological system was set up using Periodic Boundary Conditions (PBC) in a dodecahedron TIP3P water box, and a 200-ns simulation was executed at 288.15 K, as this temperature resembles the Streptomyces strain environment. The Root Mean Square Deviation (RMSD) analysis showed that the structure reached stability after about 125 ns, indicating a conformationally stable state. To evaluate the uniformity of conformational states during the stabilisation phase, clustering analysis was conducted using the GROMOS method with a 0.30 nm cutoff. This analysis identified a predominant cluster representing stabilised conformations, extending from 122.5 ns to 200 ns of the simulation. Following this, Root Mean Square Fluctuation (RMSF) analysis was performed on the stabilised cluster to pinpoint regions with high flexibility. The findings revealed that the C-terminal region had the highest fluctuations, likely linked to structural compaction. This observation was further corroborated by Solvent Accessible Surface Area (SASA) analysis, which showed a gradual reduction in accessible surface area over time, indicating a progressive compaction of the structure. a PROCHECK analysis was conducted. It verified the representative model's structural integrity, which verified high structural quality with only two residues found in disallowed regions of the Ramachandran plot.
Based on the parameters defined in CRISPy-web and the protocol described by Tong, these correspond to the top-ranked random mutations, although their structural impact remains unknown. Three mutation models were developed to evaluate the impact of specific point mutations on TetR-279 stability based on insights from MD simulations: S166P: A substitution introducing a proline, potentially affecting flexibility. V167A: A minor volume change, maintaining hydrophobicity. V167I: A conservative change affecting residue volume but preserving hydrophobicity. RMSD analysis (Figure 3A) showed that S166P caused the most significant perturbation, although the structure stabilised after 75 ns.
In contrast, V167A and V167I showed minimal disturbances, with clustering analysis unable to distinguish them as distinct conformational states when compared with its starting point, suggesting these mutations did not significantly impact structural dynamics. Additionally, RMSF analysis (Figure 3B) indicated that S166P caused significant fluctuations, particularly in the N-terminal region. Similarly, V167A, despite not having major structural changes, presents a significant impact near the 100-residue region. However, the V167I mutation resulted in only minor deviations compared to the other two. The SASA analysis (Figure 3C) confirmed that V167A mutations did not notably affect structural stability as the accessible area did not change significantly, whereas S166P led to substantial reorganisation, especially in the N-terminal region, as indicated by RMSF fluctuations. This effect aligns with the physical-chemical changes introduced by the proline substitution, which reduces localised backbone flexibility and causes global structural reordering. Based on these findings and reference data on CRISPR-based gene editing, three stabilised mutant models were proposed for experimental validation, aiming to assess the structural consequences of TetR-279 mutations and their potential impact on its regulatory function in the Actinomycin D biosynthetic pathway.
3. Discussion
The production of actinomycin has been traced to various Streptomyces species, revealing a core structure that is conserved but exhibits species-specific variations [26]. These differences underscore the evolutionary adaptability of secondary metabolite pathways and their functional diversification across ecological settings Streptomyces antibioticus contains a 20-gene BGC homologous to part of the S. chrysomallus cluster. This gene set includes orthologs involved in actinomycin biosynthesis and modification, such as cytochrome P450 enzymes, suggesting a shared biosynthetic origin with genomic streamlining [27]. Streptomyces costaricanus SCSIO ZS0073, found in a mangrove habitat, has a 39.8 kb actinomycin D cluster with 25 ORFs, including NRPS genes, biosynthetic genes for 4-MHA, and four positive regulatory genes (acnWU4RO), indicating potential for enhanced metabolic control and suitability for genetic engineering [28]. Marine-derived Streptomyces griseus SCSIO PteL053 contains NRPS-type BGCs for actinomycin D and Xoβ among 28 total secondary metabolite clusters in its genome, suggesting strong capacity for metabolic diversity in marine environments [29]. These findings show that while the actinomycin BGC is widely conserved across Streptomyces species, it is also structurally dynamic. This genomic diversity supports the wide range of actinomycin analogues found in nature and highlights the potential of these clusters as targets for metabolic engineering and new antibiotic discovery [30] . The findings validate the biosynthetic capabilities of Streptomyces fildesensis So13.3, demonstrated by activating the actinomycin D cluster in reaction to nutritional triggers [31]. This discovery not only underscores the metabolic intricacy of Antarctic strains but also indicates the potential to activate dormant BGCs using tools like CRISPR, particularly in non-model strains with pharmaceutical promise [32,33].
Although components of the TetR/AcrR TF family have been identified within the actinomycin D biosynthetic gene cluster, no reports currently confirm their direct role in regulating this cluster. Instead, in Streptomyces antibioticus ZS, a cluster-situated regulator, ActO, has been characterised and shown to belong to the LmbU family. ActO functions as a positive regulator of the entire actinomycin (act) cluster. Its deletion resulted in the complete loss of actinomycin D and V production, while overexpression increased their yields by 4.4-fold and 2.6-fold, respectively. Although ActO is not a TetR/AcrR family memberits function underscores the importance of cluster-specific regulatory elements in actinomycin biosynthesis [34]. A comprehensive review of the TetR family of regulators in actinobacteria has highlighted their diverse roles in the control of antibiotic biosynthesis, drug resistance, primary metabolism, and activation of silent biosynthetic gene clusters. While actinomycin D was not directly addressed, the review emphasises the broad functional relevance of TetR-like regulators in Streptomyces and their potential application in enhancing metabolite production or activating cryptic BGCs [35,36].
Furthermore, a TetR-like regulator (AalR) has been identified within the biosynthetic gene cluster for actinoallolides, macrolides with anti-trypanosomal activity produced by actinobacteria. Functional validation of AalR via heterologous expression confirmed its regulatory role, demonstrating that TetR-type regulators can be integral components of functional biosynthetic clusters in Streptomyces species [37]. The successful application of CRISPR/Cas9 to mutate TetR-family regulators demonstrates its potential for enhancing antibiotic production in Streptomyces. In Streptomyces rimosus M527, deletion of the TetR24 gene using CRISPR resulted in a 38% increase in rimocidin production, while overexpression reduced it by 40%. EMSA and DNase I footprinting confirmed that TetR24 binds directly to promoter regions of key biosynthetic genes (rimA and rimR2), acting as a direct repressor [38]. This study highlights the feasibility of using CRISPR to manipulate TetR regulators and opens the door for similar strategies in other biosynthetic clusters, including actinomycin D. Although these results were obtained from a well-studied bacterium, there are no reports describing the role of this TetR/AcrR transcription factor in Antarctic bacteria, nor evidence on whether its mutation effectively leads to protein activation and subsequent biosynthetic cluster expression.
Integrating bioinformatic tools into CRISPR-based genome editing processes has become crucial for predicting, validating, and enhancing genetic alterations, especially in complex organisms like Streptomyces [39]. A significant innovation is the creation of CRISPR-BEST, a base-editing system that circumvents DNA double-strand breaks. This system has been effectively utilised in Streptomyces collinus to introduce specific point mutations within the Kirromycin BGC. The method was confirmed through sequence-based bioinformatic analyses, including off-target prediction and mutation modelling, demonstrating its high precision and functional reliability [40]. Streptomyces fradiae, a highly efficient CRISPR/Cas9n system, was developed for targeted genome editing. Using high-fidelity Cas9 variants reduced cytotoxicity and enhanced editing specificity [41]. In addition to these methods, the EXPosition platform offers a new approach to designing CRISPR guide RNAs by predicting cutting efficiency and subsequent effects on gene expression, such as impacts on transcription, splicing, and translation. Although it has not yet been applied in Streptomyces, this strategy widely applies to bacteria with well-annotated genomes, providing valuable insights into how mutations might influence protein function [42]. Bioinformatic prediction tools were used to minimise and validate off-target effects, ensuring safe and precise modification of genes involved in neomycin biosynthesis [43]. Although the EXPosition platform boasts an innovative design, its predictive accuracy heavily relies on the presence of high-quality genome annotations. This is generally adequate for model organisms; however, numerous Streptomyces strains still lack comprehensive or consistent genomic annotations, especially in non-coding regions and regulatory elements [44]. This shortcoming can greatly affect the tool's capacity to predict splicing, transcriptional, and translational outcomes accurately. Additionally, EXPosition does not conduct structural or functional simulations of the resulting proteins, which limits its ability to predict phenotypic or biochemical effects of CRISPR-induced mutations. Therefore, while the tool shows promise, its current version may offer limited predictive value for organisms with complex, partially annotated genomes like Streptomyces.
To fully grasp the biological significance of CRISPR-induced mutations, assessing their effects at the protein level is essential. Although many prediction tools focus on gene expression or sequence disruption, structural bioinformatics offers a more profound understanding of how mutations affect protein function [45]. Research by Chellapandi et al. [46] has demonstrated that even minor structural mutations can greatly impact enzymatic activity and virulence through molecular dynamics and modelling techniques. Likewise, Pires et al. [47] showed that structural simulations could predict over 80% of the phenotypic effects observed from mutagenesis, underscoring their role in connecting genotype to function. Consistent with these findings, our research employed GROMACS molecular dynamics to examine the TetR-279 protein, utilising RMSD, RMSF, SASA, and conformational clustering analyses. These methods enabled us to evaluate the stability and flexibility of the mutated protein and the importance of structural simulations in understanding CRISPR modifications. Amino acid changes like S166P, V167A, and V167I can profoundly impact protein structure and function by modifying essential physicochemical characteristics such as flexibility, volume, and hydrophobicity. The S166P change introduces proline, a residue known for its structural rigidity and tendency to disrupt α-helices by creating kinks. Yu et al. [48] demonstrated in their study on Photinus pyralis luciferase that inserting proline (D476P and H489P) in flexible regions increased thermostability but also risked impairing catalytic function due to limited backbone flexibility. This indicates that while the S166P mutation might enhance structural rigidity, it could also adversely affect dynamic regions crucial for protein activity.
On the other hand, the V167A and V167I mutations are conservative changes that preserve hydrophobicity but modify side chain volume. Matreyek et al. [49] found through large-scale mutagenesis across 14 proteins that such substitutions, especially involving residues like valine, alanine, and isoleucine, can subtly affect protein stability and folding, largely depending on the local structural context. While valine-to-alanine substitutions decrease side chain bulk, valine-to-isoleucine increases it, potentially influencing local packing interactions without significantly disrupting the hydrophobic core [50]. These findings highlight that even conservative or seemingly minor substitutions can lead to functionally significant outcomes. Their effects rely not only on the inherent properties of the substituted residues but also on their positional context within secondary and tertiary structural elements, underscoring the importance of combining mutagenesis with structural and dynamic analyses to predict phenotypic consequences. These structural insights suggest that inhibition of TetR-279 through CRISPR-induced mutations or regulatory interference could release metabolic constraints imposed on the actinomycin cluster and potentially activate alternative or silent pathways involved in secondary metabolite production. This strategy could represent a novel route to induce biosynthetic diversity in extremophilic bacteria.
4. Materials and Methods
4.1. Identification and Structural Analysis of the Protein
In the genome of Streptomyces sp. So13.3 (CP048835.1), a BGC associated with the production of actinomycin D was discovered using AntiSMASH. This cluster includes two genes that code for TFs belonging to the TetR AcrR subtype family: TetR-206 (Protein ID: QNA70898.1), a protein with 206 amino acids, and TetR-279 (Protein ID: QNA70921.1), a protein consisting of 279 amino acids. A BLAST search was conducted against the Protein Data Bank (PDB) to check for available crystallised structures, but none were found [51]. Consequently, homology modeling was carried out using AlphaFold [52], with the following UniProt identifiers: TetR-206 (UniProt: A0A5R9MB50) and TetR-279 (UniProt: A0A5R9M760). Furthermore, SignalP 5.0 [53] was utilized utilised to evaluate the presence of signal peptides and their potential function in subcellular localisation. DP-Bind webserver (https://lcg.rit.albany.edu/dp-bind/help.html) was used to assess putative non-reported DNA binding motifs in TetR-279.
4.2. Cultivation Conditions of Streptomyces fildesensis So13.3
The strain Streptomyces fildesensis So13.3 was previously isolated from Antarctic soil samples and characterised through morphological and molecular analyses, as reported by Nuñez et al. [24]. For the current research, stock cultures of Streptomyces fildesensis So13.3 were initially cultivated on M2 agar plates, pH of 7.0, at a temperature of 15 °C for one week. These cultures were then utilised in subsequent experimental assays, which included analysing growth under various nutritional conditions and evaluating gene expression (Table 2).
4.3. Gene Expression Analysis of TetR-279 Under Different Nutritional Conditions
To assess TetR-279 expression under varying nutritional conditions, bacterial cultures of Streptomyces fildesensis So13.3 were cultivated in three different media: IMA, ISP4, and YES, with M2 medium used as a control. Each bacterial strain was started from a single colony and inoculated into 1.5 mL of the respective media using a micro bioreactor system from Applikon Biotechnology. For each type of medium, an uninoculated well served as a negative control. The cultures were incubated at 15 °C with shaking at 190 rpm for 7 days or until they reached optimal growth [54]. After incubation, total RNA was extracted using Trizol (Invitrogen) following the manufacturer's instructions. Quantitative real-time PCR (qPCR) was conducted using the Power SYBR® Green RNA-to-CT™ 1-Step Kit (4389986, ThermoFisher) on a LightCycler 96 system (Roche). Three biological replicates were examined, with each sample having two technical replicates. The relative expression levels of TetR-279 were calculated using the ΔΔCt method [55], with APRT (adenine phosphoribosyltransferase) mRNA serving as the internal reference gene for normalisation [56]. Specific primers were designed and used to amplify the tetR-279 gene and the internal reference gene APRT. For tetR-279, the primers were Fw1 "CGAGTACACGCAGTTGGAGA", Rv1: "GATGACACGACGGACCTTCA". For APRT, the primers used were: Fw2: "GGAGCTCGGCTTCCTCAAG", Rv2: "ACGATGATCAGTGCGTCCAG".
4.4. sgRNA Design Targeting Transcriptional Regulators of the Actinomycin D Cluster
The sgRNA was designed by adhering to the approach outlined by Tong et al. [25]. To achieve this, genomic information from Streptomyces sp. So13.3, accessed via CRISPy web [57], was utilised alongside antiSMASH [58] to pinpoint the segment containing the actinomycin D biosynthetic gene cluster, situated between coordinates 461,186 and 529,693. The gene coding for the TetR/AcrR-type regulator (C8250_002330) was identified within this segment. Five sgRNAs were crafted, and the sequence deemed most appropriate was: "AGAGCACGGAGATGACACGA", with the associated PAM site "CGG".
4.5. Homology Modeling, Refinement, and Molecular Dynamics Simulations
The models generated by AlphaFold were evaluated by examining their confidence scores to pinpoint regions of low resolution. Low confidence structural loops (following pLDDT Alphafold internal score) were found between residues 1-14 in TetR-206 and 18-37 in TetR-279. These areas were refined using GalaxyWebServer [59], resulting in models used as starting structures for MD simulations. TetR-279 was chosen for MD simulations with GROMACS [60] due to having a predicted C-terminal tetracyclin repressor-like motif that can modulate this TF response via ligand bindings, as reported for other members of the TetR TF family, and the system was set up with PBC in a dodecahedron box. The GROMACS 2019 software suite was used for molecular dynamics simulations, adhering to the methodology established by Berendsen [60]. The energy minimisation procedure consisted of 5,000 steps with an energy tolerance (emtol) value of 1000 kJ/mol/nm. Equilibration steps were divided into NVT and NPT equilibration, with temperature coupling to T=288.15K using the Berendsen thermostat. In the NpT equilibration, the pressure coupling utilised the Berendsen barostat with an isotropic coupling scheme with a compressibility value of 4.5 x 10-5. Additionally, a restraint of 1000 kj/mol/nm was applied to all alpha carbons along the x, y and z axis to restrain their position during equilibration steps. Non-bonded interactions were computed using Lennard-Jones potentials for van der Waals forces, and long-range electrostatics were calculated using the Particle Mesh Ewald (PME) method with a cutoff of 1.0 nm. For the production run, electrostatic and nonbonded interactions were computed using the equilibration method, but temperature and pressure coupling schemes were changed to V-rescale and Parrinello-Rahman, respectively. LINCS constraints for H-bonds were applied at each step, and a step of 2 femtoseconds was used. Visualisation of the simulations was performed using the VMD software. [61] Protein structure images were obtained using PyMOL Molecular Graphics System, Version 1.1 Schrödinger, LLC and plots were generated using ggplot2 R package [62]. An RMSD analysis was carried out to assess the structural stability over time. Clustering Analysis was performed using the GROMOS method with a cutoff of 0.30 nm to identify dominant conformational states. The resulting clusters were examined to select the representative model of the stabilised conformations. To pinpoint regions with significant flexibility, a RMSF analysis was carried out on the stabilised cluster, obtaining data related to each cluster timeframe. Furthermore, a SASA analysis was performed to evaluate the degree of structural compaction and explore its possible relationship with RMSF variations.
5. Conclusions
This study provides substantial evidence of the biotechnological potential of Streptomyces fildesensis So13.3, an Antarctic strain containing a biosynthetic gene cluster (BGC) related to the production of actinomycin D. Through an integrative approach combining gene expression analysis, structural modelling, molecular dynamics, and bioinformatic prediction, the role of TetR/AcrR family transcription factors as potential cluster regulators was thoroughly characterised. The differential expression of these regulators under specific nutritional conditions, particularly in ISP4 and IMA media, suggests that the metabolic environment may strongly influence cluster regulation. Structural analysis revealed key functional differences between the two identified TetR regulators, highlighting TetR-279 due to its C-terminal tetracycline-repressor-like domain, which may modulate its activity through interactions with metabolic ligands. Molecular dynamics simulations confirmed the overall structural stability of TetR-279 but also revealed significant alterations caused by point mutations—especially S166P, which notably affected the flexibility of the N-terminal region. This indicates that even conservative mutations like V167A and V167I can lead to localised structural effects with potential functional implications. These findings underscore the importance of performing structural validation before CRISPR-based gene editing, allowing for better anticipation of the functional impact of mutations. Altogether, this work lays the groundwork for future strategies aimed at rational activating silent biosynthetic clusters in extremophile bacteria, expanding the search for novel antimicrobial compounds. Notably, the targeted inhibition of TetR-279 emerges as a promising approach to induce metabolic rewiring and unlock the expression of cryptic antibiotic pathways in Streptomyces fildesensis So13.3.
Author Contributions
K.L; Conceptualisation, methodology, writing—original draft preparation J.M.; Conceptualisation, formal analysis, data curation, writing—original draft preparation. M.P; methodology and proofreading of original draft. A.G; Revision, editing, discussion, and proofreading of the original draft. H.G and M.J.C; Revision and editing of the original draft; K.Ñ-M; Writing—review of the original draft. L.B; supervises funding acquisition, revising, and editing the original draft. All authors have read and agreed to the published version of the manuscript.".
Funding
The authors sincerely acknowledge the financial support from ANID: L.B. Fondecyt Regular N°1210563; K.Ñ.-M. Fondecyt Iniciación N°11230475; M.J.C Fondecyt Iniciación N°11240632.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| BGC: | Biosynthetic Gene Clusters |
| CRISPR: | Clustered Regularly Interspaced Short Palindromic Repeats |
| MD: | Molecular Dynamics |
| RMSD: | Root Mean Square Deviation |
| RMSF: | Root Mean Square Fluctuation |
| SASA: | Solvent Accessible Surface Area |
| TF: | Transcription Factor |
| ISP4: | International Streptomyces Project Medium 4 |
| YES: | Yeast Extract Sucrose Medium |
| HTH: | Helix-Turn-Helix |
| SNP: | Sodium Nitroprusside |
| LPS: | Lipopolysaccharide |
| qPCR: | Quantitative Polymerase Chain Reaction |
| PDB: | Protein Data Bank |
| pLDDT: | Predicted Local Distance Difference Test |
| VMD: | Visual Molecular Dynamics |
| PBC: | Periodic Boundary Conditions |
| PME: | Particle Mesh Ewald |
| NVT: | Constant Number, Volume, Temperature Ensemble |
| NPT: | Constant Number, Pressure, Temperature Ensemble |
| LINCS: | Linear Constraint Solver |
| EMSA: | Electrophoretic Mobility Shift Assay |
| DSB: | Double-Strand Break |
| NRPS: | Non-Ribosomal Peptide Synthetase |
| CRISPR-BEST: | CRISPR Base Editing SysTem |
| EXPosition: | EXpression Prediction from Sequence Mutations |
References
- Govindaraj Vaithinathan and A. Vanitha, "WHO global priority pathogens list on antibiotic resistance: an urgent need for action to integrate One Health data," Perspect Public Health, vol. 138, no. 2, pp. 87–88, Mar. 2018. [CrossRef]
- T. T. Nisa, D. Nakatani, F. Kaneko, T. Takeda, and K. Nakata, "Antimicrobial resistance patterns of WHO priority pathogens isolated in shospitalised patients in Japan: A tertiary center observational study," PLoS One, vol. 19, no. 1 January, Jan. 2024. [CrossRef]
- S. Choudhury, A. Medina-Lara, and R. Smith, "Antimicrobial resistance and the COVID-19 pandemic," Bull World Health Organ, vol. 100, no. 5, pp. 295–295, May 2022. [CrossRef]
- K. Núñez-Montero and L. Barrientos, "Advances in Antarctic Research for Antimicrobial Discovery: A Comprehensive Narrative Review of Bacteria from Antarctic Environments as Potential Sources of Novel Antibiotic Compounds Against Human Pathogens and Microorganisms of Industrial Importance," Antibiotics 2018, Vol. 7, Page 90, vol. 7, no. 4, p. 90, Oct. 2018. [CrossRef]
- T. P. Thompson and B. F. Gilmore, "Exploring halophilic environments as a source of new antibiotics," Crit Rev Microbiol, vol. 50, no. 3, pp. 341–370, 2024. [CrossRef]
- D. J. Lugg and C. R. Roy, "Ultraviolet radiation and health effects in the Antarctic," Polar Res, vol. 18, no. 2, pp. 353–359, Jan. 1999. [CrossRef]
- D. Coppola et al., "Biodiversity of UV-Resistant Bacteria in Antarctic Aquatic Environments," J Mar Sci Eng, vol. 11, no. 5, May 2023. [CrossRef]
- L. J. Silva et al., "Actinobacteria from Antarctica as a source for anticancer discovery," Sci Rep, vol. 10, no. 1, Dec. 2020. [CrossRef]
- N. Benaud et al., "Antarctic desert soil bacteria exhibit high novel natural product potential, evaluated through long-read genome sequencing and comparative genomics.," Environ Microbiol, vol. 23, no. 7, pp. 3646–3664, Jul. 2021. [CrossRef]
- Russell, R. Lacret, and A. Truman, "Genome-led discovery of novel microbial natural products," Access Microbiol, vol. 1, no. 1A, Mar. 2019. [CrossRef]
- J. Lebedeva, G. Jukneviciute, R. Čepaitė, V. Vickackaite, R. Pranckutė, and N. Kuisiene, "Genome Mining and Characterization of Biosynthetic Gene Clusters in Two Cave Strains of Paenibacillus sp.," Front Microbiol, vol. 11, Jan. 2021. [CrossRef]
- D. Mao, B. K. Okada, Y. Wu, F. Xu, and M. R. Seyedsayamdost, "Recent advances in activating silent biosynthetic gene clusters in bacteria.," Curr Opin Microbiol, vol. 45, pp. 156–163, Oct. 2018. [CrossRef]
- D. Montiel, H. S. Kang, F. Y. Chang, Z. Charlop-Powers, and S. F. Brady, "Yeast homologous recombination-based promoter engineering for the activation of silent natural product biosynthetic gene clusters," Proceedings of the National Academy of Sciences, vol. 112, no. 29, pp. 8953–8958, Jul. 2015. [CrossRef]
- K. Núñez-Montero et al., "Antarctic Streptomyces fildesensis So13.3 strain as a promising source for antimicrobials discovery," Scientific Reports 2019 9:1, vol. 9, no. 1, pp. 1–13, May 2019. [CrossRef]
- K. Núñez-Montero, D. Quezada-Solís, Z. G. Khalil, R. J. Capon, F. D. Andreote, and L. Barrientos, "Genomic and Metabolomic Analysis of Antarctic Bacteria Revealed Culture and Elicitation Conditions for the Production of Antimicrobial Compounds," Biomolecules, vol. 10, no. 5, May 2020. [CrossRef]
- Y. Lin et al., “Multi-Omics Analysis Reveals Anti-Staphylococcus aureus Activity of Actinomycin D Originating from Streptomyces parvulus,” Int J Mol Sci, vol. 22, no. 22, Nov. 2021. [CrossRef]
- M. Liu et al., “Identification of the Actinomycin D Biosynthetic Pathway from Marine-Derived Streptomyces costaricanus SCSIO ZS0073,” Mar Drugs, vol. 17, no. 4, 2019. [CrossRef]
- W. Deng, C. Li, and J. Xie, "The underling mechanism of bacterial TetR/AcrR family transcriptional repressors.," Cell Signal, vol. 25 7, no. 7, pp. 1608–13, 2013. [CrossRef]
- J. Filipek et al., "Comprehensive structural overview of the C-terminal ligand-binding domains of the TetR family regulators.," J Struct Biol, vol. 216, no. 2, Jun. 2024. [CrossRef]
- J. Krushkal, S. Sontineni, C. Leang, Y. Qu, R. M. Adkins, and D. R. Lovley, "Genome diversity of the TetR family of transcriptional regulators in a metal-reducing bacterial family Geobacteraceae and other microbial species.," OMICS, vol. 15 7–8, no. 7–8, pp. 495–506, Jul. 2011. [CrossRef]
- L. Cuthbertson and J. R. Nodwell, "The TetR Family of Regulators," Microbiology and Molecular Biology Reviews, vol. 77, no. 3, pp. 440–475, Sep. 2013. [CrossRef]
- L. Liu et al., "AveI, an AtrA homolog of Streptomyces avermitilis, controls avermectin and oligomycin production, melanogenesis, and morphological differentiation," Appl Microbiol Biotechnol, vol. 103, no. 20, pp. 8459–8472, Oct. 2019. [CrossRef]
- D. Xu, P. Waack, Q. Zhang, S. Werten, W. Hinrichs, and M. J. Virolle, "Structure and regulatory targets of SCO3201, a highly promiscuous TetR-like regulator of Streptomyces coelicolor M145.," Biochem Biophys Res Commun, vol. 450 1, no. 1, pp. 513–8, Jul. 2014. [CrossRef]
- K. Núñez-Montero et al., "Antarctic Streptomyces fildesensis So13.3 strain as a promising source for antimicrobials discovery," Scientific Reports 2019 9:1, vol. 9, no. 1, pp. 1–13, May 2019. [CrossRef]
- Y. Tong, C. M. Whitford, K. Blin, T. S. Jørgensen, T. Weber, and S. Y. Lee, "CRISPR–Cas9, CRISPRi and CRISPR-BEST-mediated genetic manipulation in streptomycetes," Nature Protocols 2020 15:8, vol. 15, no. 8, pp. 2470–2502, Jul. 2020. [CrossRef]
- U. Keller, M. Lang, I. Crnovcic, F. Pfennig, and F. Schauwecker, "The Actinomycin Biosynthetic Gene Cluster of Streptomyces chrysomallus: a Genetic Hall of Mirrors for Synthesis of a Molecule with Mirror Symmetry," J Bacteriol, vol. 192, no. 10, pp. 2583–2595, May 2010. [CrossRef]
- Crnovčić, C. Rückert, S. Semsary, M. Lang, J. Kalinowski, and U. Keller, "Genetic interrelations in the actinomycin biosynthetic gene clusters of <em>Streptomyces antibioticus</em> IMRU 3720 and <em>Streptomyces chrysomallus</em> ATCC11523, producers of actinomycin X and actinomycin C," Advances and Applications in Bioinformatics and Chemistry, vol. 10, no. 1, pp. 29–46, Apr. 2017. [CrossRef]
- M. Liu et al., “Identification of the Actinomycin D Biosynthetic Pathway from Marine-Derived Streptomyces costaricanus SCSIO ZS0073,” Mar Drugs, vol. 17, no. 4, 2019. [CrossRef]
- G. Govindarajan et al., “Genome Sequencing of Streptomyces griseus SCSIO PteL053, the Producer of 2,2′-Bipyridine and Actinomycin Analogs, and Associated Biosynthetic Gene Cluster Analysis,” J Mar Sci Eng, vol. 11, no. 2, Feb. 2023. [CrossRef]
- U. Keller, M. Lang, I. Crnovcic, F. Pfennig, and F. Schauwecker, "The Actinomycin Biosynthetic Gene Cluster of Streptomyces chrysomallus: a Genetic Hall of Mirrors for Synthesis of a Molecule with Mirror Symmetry," J Bacteriol, vol. 192, no. 10, pp. 2583–2595, May 2010. [CrossRef]
- Núñez-Montero, D. Quezada-Solís, Z. G. Khalil, R. J. Capon, F. D. Andreote, and L. Barrientos, "Genomic and Metabolomic Analysis of Antarctic Bacteria Revealed Culture and Elicitation Conditions for the Production of Antimicrobial Compounds," Biomolecules 2020, Vol. 10, Page 673, vol. 10, no. 5, p. 673, Apr. 2020. [CrossRef]
- Ke et al., "CRAGE-CRISPR facilitates rapid activation of secondary metabolite biosynthetic gene clusters in bacteria.," Cell Chem Biol, vol. 29, no. 4, pp. 696-710.e4, Apr. 2022. [CrossRef]
- Ameruoso, M. C. V. Kcam, K. P. Cohen, and J. Chappell, "Activating natural product synthesis using CRISPR interference and activation systems in Streptomyces," Nucleic Acids Res, vol. 50, no. 13, pp. 7751–7760, Jul. 2022. [CrossRef]
- Y. Liang et al., "ActO, a positive cluster-situated regulator for actinomycins biosynthesis in Streptomyces antibioticus ZS.," Gene, vol. 933, Jan. 2025. [CrossRef]
- Y. Lei, S. Asamizu, T. Ishizuka, and H. Onaka, “Regulation of Multidrug Efflux Pumps by TetR Family Transcriptional Repressor Negatively Affects Secondary Metabolism in Streptomyces coelicolor A3(2),” Appl Environ Microbiol, vol. 89, no. 3, Mar. 2023. [CrossRef]
- Y. Xu et al., "TetR family regulator AbrT controls lincomycin production and morphological development in Streptomyces lincolnensis," Microb Cell Fact, vol. 23, no. 1, pp. 1–13, Dec. 2024. [CrossRef]
- Y. Inahashi et al., "Identification and heterologous expression of the actinoallolide biosynthetic gene cluster," J Antibiot (Tokyo), vol. 71, no. 8, pp. 749–752, Aug. 2018. [CrossRef]
- D. Yu, H. Lin, A. Bechthold, X. Yu, and Z. Ma, "RS24090, a TetR family transcriptional repressor, negatively affects the rimocidin biosynthesis in Streptomyces rimosus M527.," Int J Biol Macromol, vol. 285, Jan. 2025. [CrossRef]
- Naeem and O. S. Alkhnbashi, "Current Bioinformatics Tools to Optimise CRISPR/Cas9 Experiments to Reduce Off-Target Effects," Int J Mol Sci, vol. 24, no. 7, Apr. 2023. [CrossRef]
- Y. Tong et al., "Highly efficient DSB-free base editing for streptomycetes with CRISPR-BEST," Proc Natl Acad Sci U S A, vol. 116, no. 41, pp. 20366–20375, 2019. [CrossRef]
- M.-S. Liu, S. Gong, H. Yu, K. Jung, K. Johnson, and D. W. Taylor, "Basis for discrimination by engineered CRISPR/Cas9 enzymes," bioRxiv, 2019. [CrossRef]
- S. Cohen, S. Bergman, N. Lynn, and T. Tuller, "A tool for CRISPR-Cas9 gRNA evaluation based on computational models of gene expression," bioRxiv, p. 2024.06.08.598047, Jun. 2024. [CrossRef]
- S. Yuan, "Mitigating the Off-target Effects in CRISPR/Cas9-mediated Genetic Editing with Bioinformatic Technologies," Transactions on Materials, Biotechnology and Life Sciences, vol. 3, pp. 318–326, Mar. 2024. [CrossRef]
- S. Hwang et al., "Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome," Nucleic Acids Res, vol. 47, no. 12, pp. 6114–6129, 2019. [CrossRef]
- Z. A. Hussein and A. A. Al-Kazaz, "BIOINFORMATICS EVALUATION OF CRISP2 GENE SNPs AND THEIR IMPACTS ON PROTEIN," IRAQI JOURNAL OF AGRICULTURAL SCIENCES, vol. 54, no. 2, pp. 369–377, 2023. [CrossRef]
- P. Chellapandi, "Structural-functional integrity of hypothetical proteins identical to ADPribosylation superfamily upon point mutations.," Protein Pept Lett, vol. 21 8, no. 8, pp. 722–35, Sep. 2014. [CrossRef]
- D. E. V. Pires, J. Chen, T. L. Blundell, and D. B. Ascher, "In silico functional dissection of saturation mutagenesis: Interpreting the relationship between phenotypes and changes in protein stability, interactions and activity," Sci Rep, vol. 6, Jan. 2016. [CrossRef]
- H. Yu, Y. Zhao, C. Guo, Y. Gan, and H. Huang, "The role of proline substitutions within flexible regions on thermostability of luciferase.," Biochim Biophys Acta, vol. 1854 1, no. 1, pp. 65–72, 2015. [CrossRef]
- Atsavapranee, F. Sunden, D. Herschlag, and P. M. Fordyce, "Quantifying protein unfolding kinetics with a high-throughput microfluidic platform," Jan. 2025. [CrossRef]
- J. T. Kellis, K. Nyberg, and A. R. Fersht, "Energetics of complementary side-chain packing in a protein hydrophobic core.," Biochemistry, vol. 28 11, no. 11, pp. 4914–22, 1989. [CrossRef]
- H. M. Berman et al., "The Protein Data Bank," Nucleic Acids Res, vol. 28, no. 1, pp. 235–242, Jan. 2000. [CrossRef]
- J. Jumper et al., "Highly accurate protein structure prediction with AlphaFold," Nature 2021 596:7873, vol. 596, no. 7873, pp. 583–589, Jul. 2021. [CrossRef]
- J. J. Almagro Armenteros et al., "SignalP 5.0 improves signal peptide predictions using deep neural networks," Nature Biotechnology 2019 37:4, vol. 37, no. 4, pp. 420–423, Feb. 2019. [CrossRef]
- K. Núñez-Montero, D. Rojas-Villalta, R. Hernández-Moncada, A. Esquivel, and L. Barrientos, “Genome Sequence of Pseudomonas sp. Strain So3.2b, Isolated from a Soil Sample from Robert Island (Antarctic Specially Protected Area 112), Antarctic," Microbiol Resour Announc, vol. 12, no. 3, Mar. 2023. [CrossRef]
- K. J. Livak and T. D. Schmittgen, "Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method," Methods, vol. 25, no. 4, pp. 402–408, Dec. 2001. [CrossRef]
- S. Lin, Z. Zou, C. Zhou, H. Zhang, and Z. Cai, "Transcriptome Analysis Reveals the Molecular Mechanisms Underlying Adenosine Biosynthesis in Anamorph Strain of Caterpillar Fungus," Biomed Res Int, vol. 2019, 2019. [CrossRef]
- K. Blin, L. E. Pedersen, T. Weber, and S. Y. Lee, "CRISPy-web: An online resource to design sgRNAs for CRISPR applications," Synth Syst Biotechnol, vol. 1, no. 2, pp. 118–121, Jun. 2016. [CrossRef]
- K. Blin et al., "antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline," Nucleic Acids Res, vol. 47, no. W1, pp. W81–W87, Jul. 2019. [CrossRef]
- E. Afgan et al., "The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update," Nucleic Acids Res, vol. 46, no. W1, pp. W537–W544, Jul. 2018. [CrossRef]
- H. J. C. Berendsen, D. van der Spoel, and R. van Drunen, "GROMACS: A message-passing parallel molecular dynamics implementation," Comput Phys Commun, vol. 91, no. 1–3, pp. 43–56, Sep. 1995. [CrossRef]
- W. Humphrey, A. Dalke, and K. Schulten, "VMD: Visual molecular dynamics," J Mol Graph, vol. 14, no. 1, pp. 33–38, Feb. 1996. [CrossRef]
- H. Wickham, "ggplot2," 2016. [CrossRef]
Figure 1.
Three-dimensional representative structure of the putative TetR/AcrR protein after 200 ns of molecular dynamics. 1A. 3D configuration of TetR-279; pink structures mark putative DNA-binding regions. 1B. DP-Bind results indicating predicted DNA-interacting residues.
Figure 1.
Three-dimensional representative structure of the putative TetR/AcrR protein after 200 ns of molecular dynamics. 1A. 3D configuration of TetR-279; pink structures mark putative DNA-binding regions. 1B. DP-Bind results indicating predicted DNA-interacting residues.
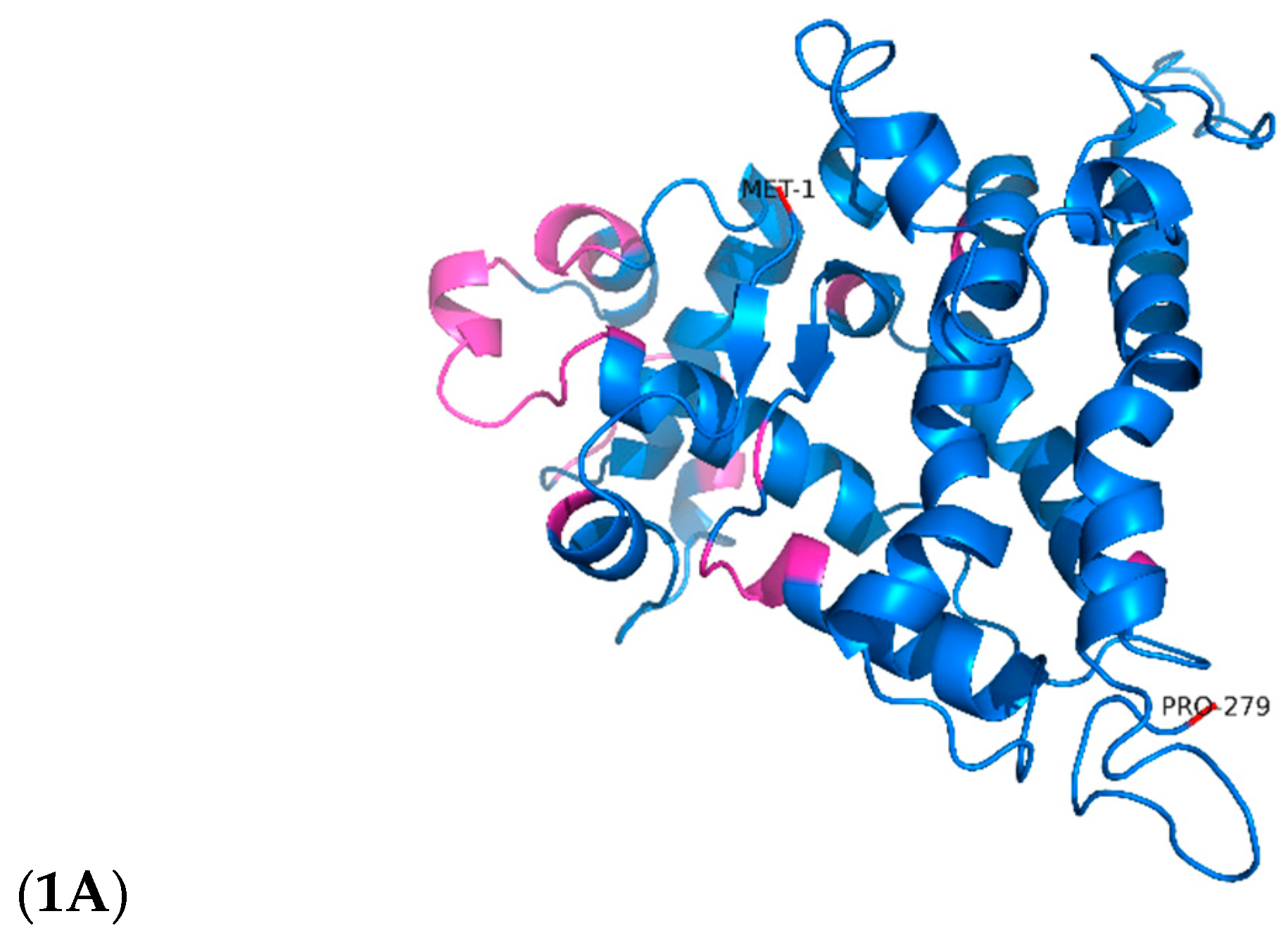
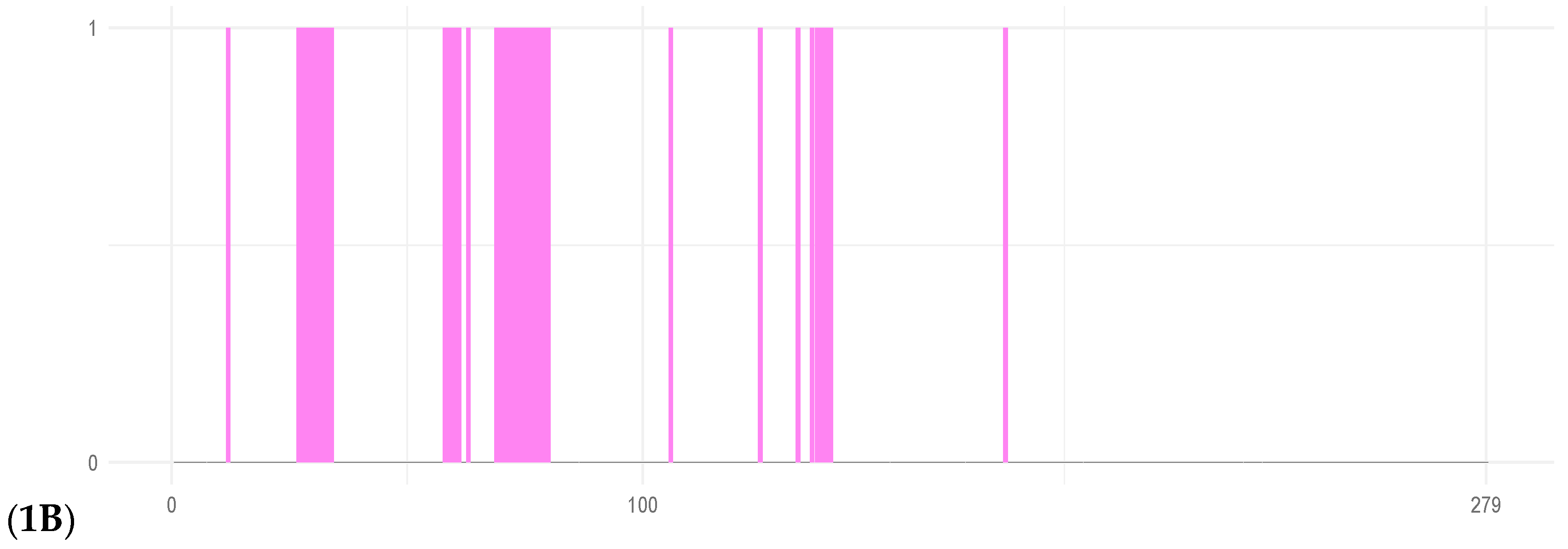
Figure 2.
Gene expression analysis revealed a significant upregulation of TetR-279 in IMA and ISP4 media, indicating nutrient-dependent activation of the actinomycin D cluster. 2A. Illustrates the relative expression levels of the TetR/AcrR transcription factor within the actinomycin D cluster across three different nutritional environments: IMA, ISP4, and YES. The findings indicate a notable rise in gene expression in both IMA and ISP4 media compared to the control group (p<0.05). The APRT gene served as an internal reference for normalisation purposes. Furthermore, the analysis was conducted using three biological replicates and two analytical methods for each condition. 2B. summarises the TF identified within the actinomycin D biosynthetic gene cluster. Both proteins belong to the TetR/AcrR family, which is commonly associated with transcriptional repression in response to environmental or metabolic signals.
Figure 2.
Gene expression analysis revealed a significant upregulation of TetR-279 in IMA and ISP4 media, indicating nutrient-dependent activation of the actinomycin D cluster. 2A. Illustrates the relative expression levels of the TetR/AcrR transcription factor within the actinomycin D cluster across three different nutritional environments: IMA, ISP4, and YES. The findings indicate a notable rise in gene expression in both IMA and ISP4 media compared to the control group (p<0.05). The APRT gene served as an internal reference for normalisation purposes. Furthermore, the analysis was conducted using three biological replicates and two analytical methods for each condition. 2B. summarises the TF identified within the actinomycin D biosynthetic gene cluster. Both proteins belong to the TetR/AcrR family, which is commonly associated with transcriptional repression in response to environmental or metabolic signals.
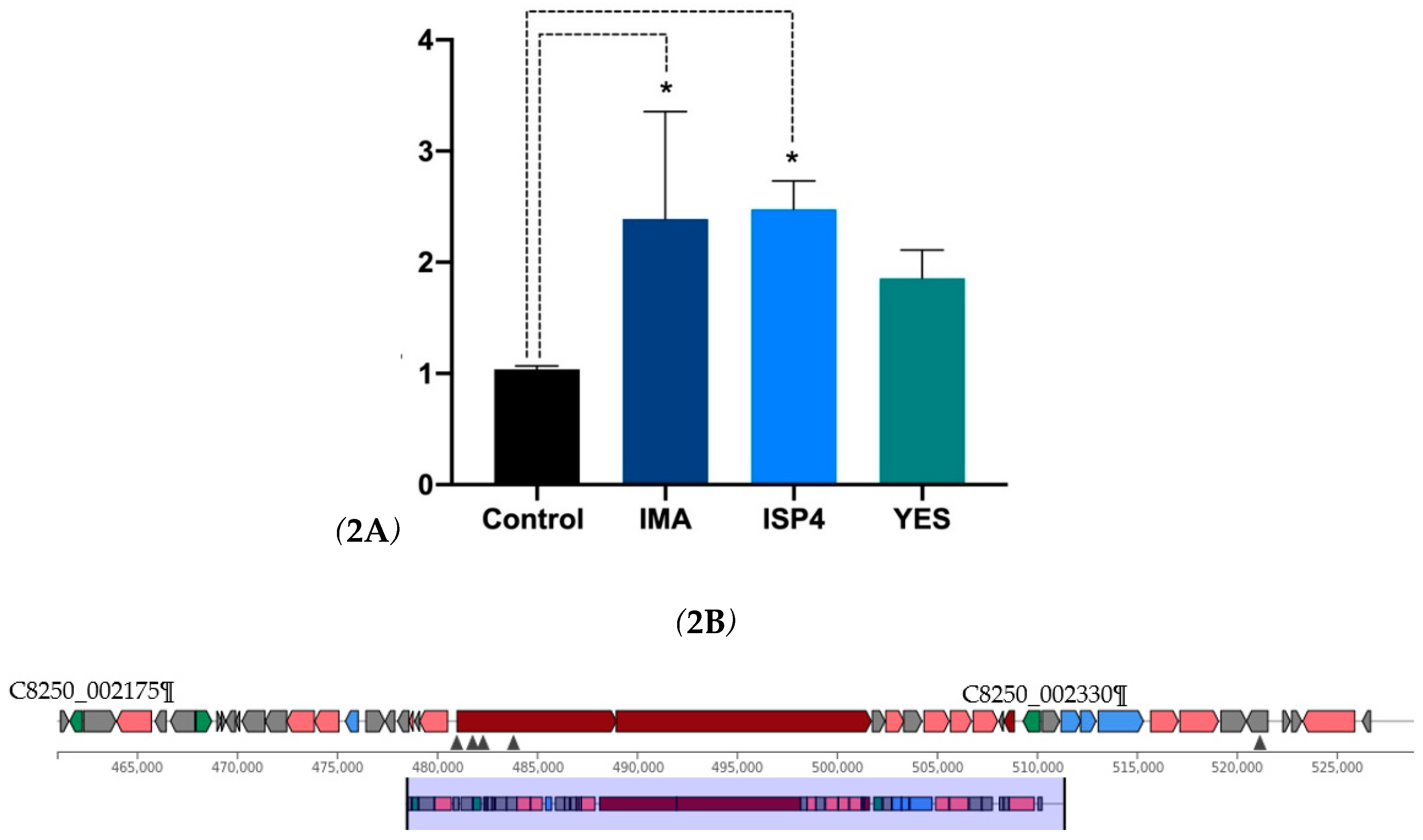
Figure 3.
Molecular dynamics revealed that the S166P mutation significantly altered TetR-279 structure and flexibility, while V167A and V167I had minimal impact. 3A. Molecular dynamic analysis for overall conformational changes (backbone RMSD) and residue fluctuation in the putative TetR-279 protein. 3A. The RMSD plot of the alpha carbons (Cα) as a function of time during a 200 ns molecular dynamics simulation for different variants of the TetR-279 protein: native, S166P, V167A, and V167I. 3B. The root mean square fluctuation as a function of residue number for different variants of TetR-279 protein: native, S166P, V167A, and V167I. RMSF measures the flexibility of each residue in the protein structure over time during a molecular dynamic' simulation. 3C. The figure shows the evolution of SASA over time (in nanoseconds) for different protein variants throughout a 200 ns molecular dynamics simulation. The figure indicates that the S166P mutation has a more significant effect on the protein's conformation, reducing its solvent-accessible surface area after compensating for proline residue-residue clashes. In contrast, the V167A and V167I mutations have less impact on the overall protein structure.
Figure 3.
Molecular dynamics revealed that the S166P mutation significantly altered TetR-279 structure and flexibility, while V167A and V167I had minimal impact. 3A. Molecular dynamic analysis for overall conformational changes (backbone RMSD) and residue fluctuation in the putative TetR-279 protein. 3A. The RMSD plot of the alpha carbons (Cα) as a function of time during a 200 ns molecular dynamics simulation for different variants of the TetR-279 protein: native, S166P, V167A, and V167I. 3B. The root mean square fluctuation as a function of residue number for different variants of TetR-279 protein: native, S166P, V167A, and V167I. RMSF measures the flexibility of each residue in the protein structure over time during a molecular dynamic' simulation. 3C. The figure shows the evolution of SASA over time (in nanoseconds) for different protein variants throughout a 200 ns molecular dynamics simulation. The figure indicates that the S166P mutation has a more significant effect on the protein's conformation, reducing its solvent-accessible surface area after compensating for proline residue-residue clashes. In contrast, the V167A and V167I mutations have less impact on the overall protein structure.
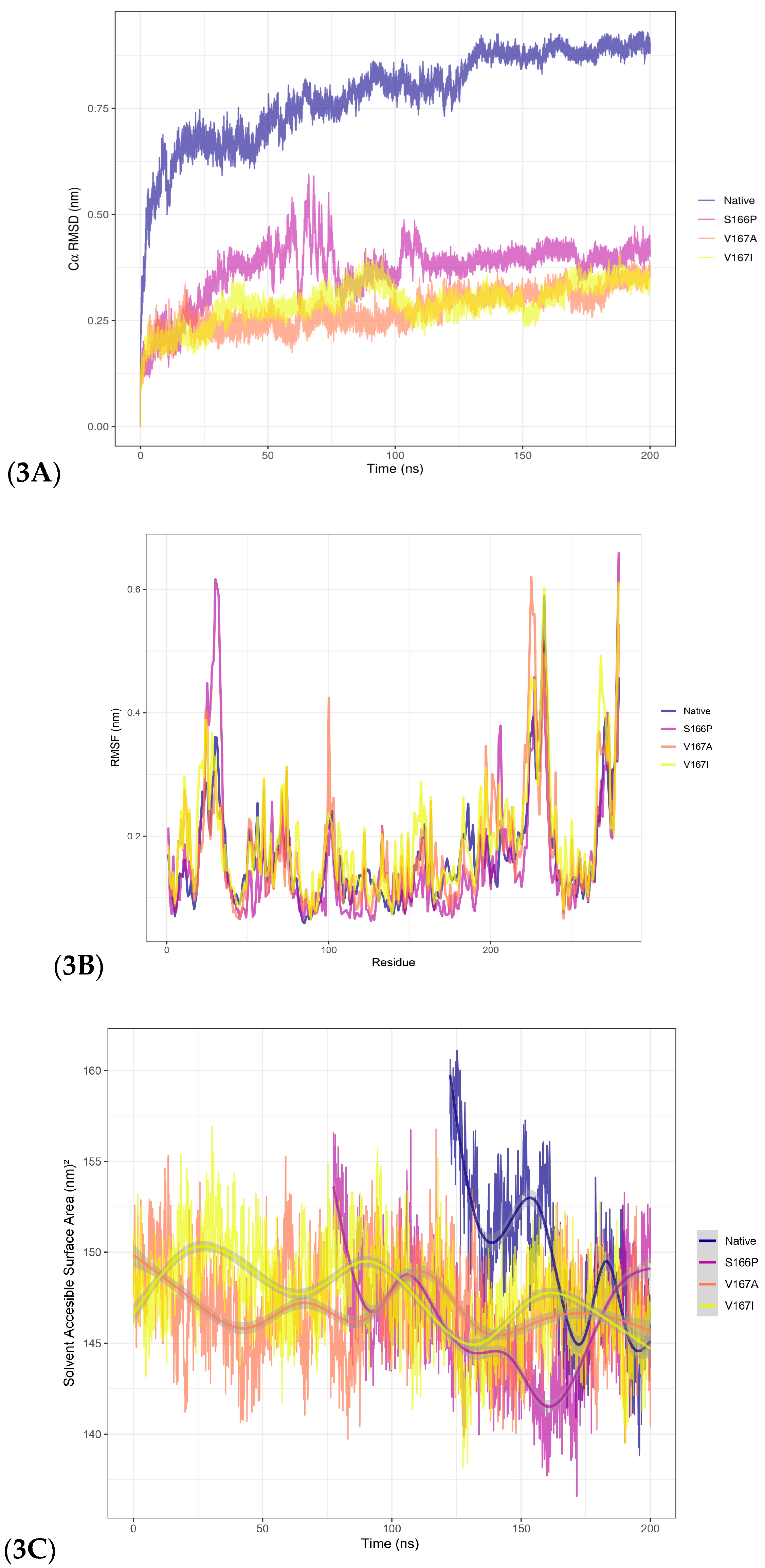

Table 1.
Regulatory Proteins of the TetR/AcrR Family Identified in the Actinomycin D Biosynthetic Cluster.
Table 1.
Regulatory Proteins of the TetR/AcrR Family Identified in the Actinomycin D Biosynthetic Cluster.
| Locus tag | Product | Length | Function | |
|---|---|---|---|---|
| NT | AA | |||
| C8250_002175 | TetR/AcrR family transcriptional regulator | 621 | 206 | Regulatory |
| C8250_002330 | TetR/AcrR family transcriptional regulator | 840 | 279 | Regulatory |
*NT: nucleotides; *AA: Amino acids.
Table 2.
Culture media and nutritional composition. .
| Medium | Components (per litre) |
|---|---|
| IMA | Yeast extract (4 g), Malt extract (10 g), Glucose (4 g), Mannitol (40g) |
| YES | Sucrose (150 g), Yeast extract (20 g), MgSO₄·7H₂O (0.5 g), ZnSO₄·7H₂O (0.01 g), CuSO₄·5H₂O (0.005 g) |
| ISP-4 | Starch (10 g), CaCO₃ (2 g), (NH₄) ₂SO₄ (2 g), K₂HPO₄ (1 g), MgSO₄·7H₂O (1 g), NaCl (1 g), FeSO₄·7H₂O (1 mg), MnCl₂·7H₂O (1 mg), ZnSO₄·7H₂O (1 mg) |
| M2 | Mannitol (40 g), Maltose (40 g), Yeast extract (10 g), K₂HPO₄ (2 g), MgSO₄·7H₂O (0.5 g), FeSO₄·7H₂O (0.01 g) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



