Submitted:
28 March 2025
Posted:
31 March 2025
You are already at the latest version
Abstract
Background: Microbial contamination is a significant challenge for the pharmaceutical sector, especially for heat-sensitive sterile products. This contamination can alter the physical and chemical properties of pharmaceutical products, compromising quality and safety. Correct bacterial identification is essential for tracing sources of contamination and implementing preventive and corrective measures. Matrix-Assisted Laser Desorp-tion Ionization-Time of Flight/Mass Spectrometry (MALDI-TOF MS) technology has revolutionized microbial identification, but there are limitations in the databases, re-quiring additional analyses, such as sequencing of the 16S rRNA and housekeeping genes and/or whole genome sequencing. Objectives: This review explores the challenges of identifying bacterial contaminants in the pharmaceutical industry using 16S rRNA gene sequences. Additional sequencing of housekeeping genes can be useful for differ-entiating bacteria at the species level. Advances in DNA sequencing technology have expanded genomic taxonomy, allowing for more accurate bacterial classification. This study aims to demonstrate how the combination of these methods increases the accuracy of bacterial identification. Conclusions and future directions: MALDI-TOF MS is widely used in the pharmaceutical sector for bacterial identification. A public database of spectral profiles of environmental bacterial isolates would be essential for bacterial identification and information exchange between institutions. Complementary methods, such as 16S rRNA and housekeeping genes sequencing, can provide reliable bacterial identification and allow the expansion of the MALDI-TOF MS database. Advances in genomic taxonomy have enabled the development of genomic taxonomy tools to im-prove bacterial identification. The combination of multiple methods provides greater precision and overcomes the limitations of single-gene approaches.
Keywords:
16S rRNA gene sequencing
; housekeeping genes
; MALDI-TOF MS
; genomic taxonomy analysis
; environmental bacterial isolates
1. Background
Microbial contamination is one of the biggest obstacles facing the pharmaceutical industry, especially for heat-sensitive sterile products such as immunobiologicals that cannot be terminally sterilized. Microbial contamination can alter the physical and chemical properties of pharmaceutical products and excipients, affecting product quality and consumer safety. Therefore, the performance of microbiological testing is essential for the quality control of these products [1,2]. Good Manufacturing Practices (GMPs) must be followed to reduce the risk of microbial contamination in pharmaceutical production environments, in order to ensure that biological products meet, among other parameters, acceptable limits for microorganisms [3].
Several groups of bacteria may be present as contaminants in clean areas, but aerobic endospore-forming bacteria have been described as one of the most important groups of bacteria isolated from these environments, due to their ability to produce spores that are resistant to temperature variations and to the sanitizers used in industry, allowing them to persist in the environment for long periods [1,3]. Identification of the detected microbiome is essential to investigate the source of contamination and consequently to take preventive and corrective measures [2,4,5].
A bacterial species is characterized as a group of strains, including the type strain, that share more than 70% similarity in DNA-DNA hybridization (DDH), maximum of 2% G+C span, values above 98.7% in 16S rRNA gene sequence identity, and distinct chemotaxonomic and phenotypic characteristics [6]. Several identification methods are used in clinical or industrial laboratories to identify the bacterial species, which are described in more detail below.
Matrix-Assisted Laser Desorption Ionization–Time of Flight/Mass Spectrometry (MALDI-TOF MS) technology has been widely used to identify microorganisms contaminating pharmaceutical production environments because it offers several advantages over other identification methods, particularly in terms of speed and greater specificity and sensitivity [1,3,7]. Therefore, if the MALDI-TOF database fails to identify a bacterial isolate, it is necessary to sequence the 16S rRNA gene and some housekeeping genes according to the suspected species or genera, or even the whole genome to perform genomic taxonomy analyses [1,7,8]. The biggest challenge is when the strain is a potential new species and the genes and/or genomes of all the closest species in the genus are not deposited in the databases for the purpose of comparing similarities and reconstructing the phylogenetic tree.
This review discusses the difficulties encountered in identifying environmental bacterial isolates from pharmaceutical facilities using MALDI-TOF MS technology, 16S rRNA and housekeeping gene sequence analyses, which often require genomic taxonomy analyses using genome sequencing data.
2. Phenotypic Identification
Phenotypic identification using commercial biochemical systems can be performed before the bacterial isolate is submitted for identification by MALDI-TOF MS or gene sequencing. Biochemical identification of environmental isolates using commercial biochemical systems such as API® sytem (bioMérieux, Craponne, France) and VITEK® 2 Compact System (bioMérieux, Craponne, France) is still used in the pharmaceutical industry [9,10,11].
The API® system consists of a series of biochemical tests based on the fermentation of sugars (carbohydrates), assimilation of other carbon sources and the production of unique enzymes and metabolites. These tests are used to identify Gram-positive, Gram-negative bacteria and yeasts. The type of kit is selected based on colony morphology and staining results. The API profiles obtained are identified using the APIWEBTM database [12,13].
The VITEK® 2 system is used for microbial identification and antimicrobial susceptibility testing. It consists of a card filling/sealing system, an incubator/reader, coupled to a computer. Microbial identification is performed using cards containing dehydrated biochemical substrates that require no additional reagents. As with the API® system, card selection is based on colony morphology and staining results. VITEK® identification includes microbial species of clinical and industrial importance [14].
Both systems have potential limitations, such as (i) the difficulty in determining phenotypic variation between strains; (ii) some may show different results in repeated tests; (iii) limited databases; (iv) small changes in test performance can give false results. In addition, non-fermenting bacteria can be problematic due to their phenotypic variations and slower growth rates [15].
For microorganisms of environmental origin and pharmaceutical products, some studies show the need for molecular methods to complete the identification of different bacterial groups. It is important to note that despite the ineffectiveness of these methods in phenotypic identification, the results of biochemical assays can be useful in differentiating some species [10,11].
3. MALDI-TOF MS
MALDI-TOF MS is considered a high-throughput technology, based on the acquisition of unique molecular signatures that are representative of a wide range of proteins and can clearly distinguish the differences between two closely related species. Its database contains spectra of microorganisms that are compared to the mass spectrum of the microorganism to be identified to find a closer match. Regardless of which system is used, be it the MALDI Biotyper (Bruker Daltonics, France) or the Vitek MS (bioMerieux, France), each of them requires different preparations and has different databases and algorithms [16,17].
This method has undoubtedly revolutionized the microbial identification system, reducing the time and cost of identification. Accuracy and speed in reporting results are crucial for batch release of pharmaceutical products. It is important to note that the MALDI-TOF MS databases were initially created using mainly clinically relevant strains for species identification. The application of technology for the identification of bacterial isolates from other sources, such as soil, industry, freshwater, etc., required new databases with relevant corresponding species. Due to its rapid identification method and low cost per isolate, the technology attracted the interest of several specialist groups who expanded the MALDI-TOF MS databases. Species not detected by MALDI-TOF MS, but identified by 16S rRNA gene sequences, were added to the database to study microbial communities from poorly understood locations, making this technology more widely used beyond clinical laboratories [18]. Costa et al. [1] added 24 Bacillus spp. strains and related genera from the pharmaceutical industry to the MALDI-TOF MS database, after a careful genotypic characterization of the strains using 16S rRNA and rpoB gene sequences. Miranda et al. [10] reported for the first time the isolation of six Sutcliffiella horikoshii strains from an immunobiological pharmaceutical facility that were not identified by MALDI-TOF MS. After gene sequencing, the MALDI-TOF MS database was expanded, and the strains were correctly identified as S. horikoshii by MALDI-TOF MS. In 2017, an aerobic, spore-forming Gram-positive rod isolated from an air monitoring sample from an immunobiological production unit was not identified by MALDI-TOF MS. After physiological and genotypic characterization and biochemical tests, the Bacillus lumeideiriae species was described and the MALDI-TOF database was expanded with the spectra profiles of the proposed new species [8].
It is possible to increase the identification capacity of MALDI-TOF by including the spectral profiles in the database [19]. For this purpose, the bacterial isolate must first be identified by analysis of the 16S ribosomal gene sequence at the genus level, since this gene has limitations in differentiating species. Sequences of genes encoding highly conserved proteins, called housekeeping genes, can be used in conjunction with 16S rRNA gene sequences to try to reach the species level [1,19].
It is important to mention that there is no public database of environmental bacterial isolates for comparing spectra obtained with MALDI-TOF MS, which makes bacterial identification and the exchange of information between researchers and institutions difficult. For highly pathogenic bacteria (biosafety level 3), such as Bacillus anthracis, Yersinia pestis, Burkholderia mallei, Burkholderia pseudomallei and Francisella tularensis, as well as their related species, the Robert Koch German Institute has developed a database with MALDI-TOF MS mass spectra, which serves as a reference for diagnosing these bacteria using microbial identification software. The spectra are available in a zip file containing the original mass spectra in the data format used by Bruker Daltonics [20].
4. Analysis of the 16S Ribosomal Gene Sequences
Sequencing of the 16S ribosomal gene is widely used in bacterial identification [21]. The 16S rRNA gene is approximately 1500 base pairs (bp) in size and is located in the small subunit (30S) of the prokaryotic ribosome [22] (Figure 1). Some species may have shorter or longer sequences. The 16S rRNA gene does not encode proteins, but in addition to having a structural role, it is crucial for protein synthesis. Although rare, horizontal transfer of the 16S rRNA gene can also occur, but only at the intragenus or intraspecific level [23,24].
The 16S rRNA gene is considered an important molecular marker, present in all members of Bacteria and Archaea, highly conserved in bacteria and evolving slowly, it is the target widely used for phylogenetic studies of bacteria. In this sense, its sequences are well used for taxonomic classification at the genus level. The presence of multiple copies in the bacterial genome with sequence differences and low polymorphism are some of the limitations that must be considered for classification at the species level [25].
The 16S rRNA gene contains highly conserved, variable and hypervariable regions that are unevenly distributed, being nine hypervariable regions, designated V1 to V9, that vary in length, position and taxonomic discrimination. Such variation is conducive to infer phylogenetic relationships between phyla, while also being used in comparison of interests [23,24,26].
Although considered the gold standard for bacterial identification, amplification of the 16S rRNA gene is still costly and impractical in the routine of some laboratories [27]. For more accurate molecular identification, complete sequencing of the gene (~1500 bp) is required [25]. The 16S rRNA gene sequences should be compared with available databases such as Basic Local Alignment Search Tool (Blast, https://blast.ncbi.nlm.nih.gov/Blast.cgi) or EzBioCloud (https://www.ezbiocloud.net/). If a new bacterial species is suspected, it is important to limit the comparison of similarities to the sequences of type strains [28].
Figure 1.
Structure of the prokaryotic ribosome. Image adapted from NIAID Visual & Medical Arts, 2024..
Figure 1.
Structure of the prokaryotic ribosome. Image adapted from NIAID Visual & Medical Arts, 2024..
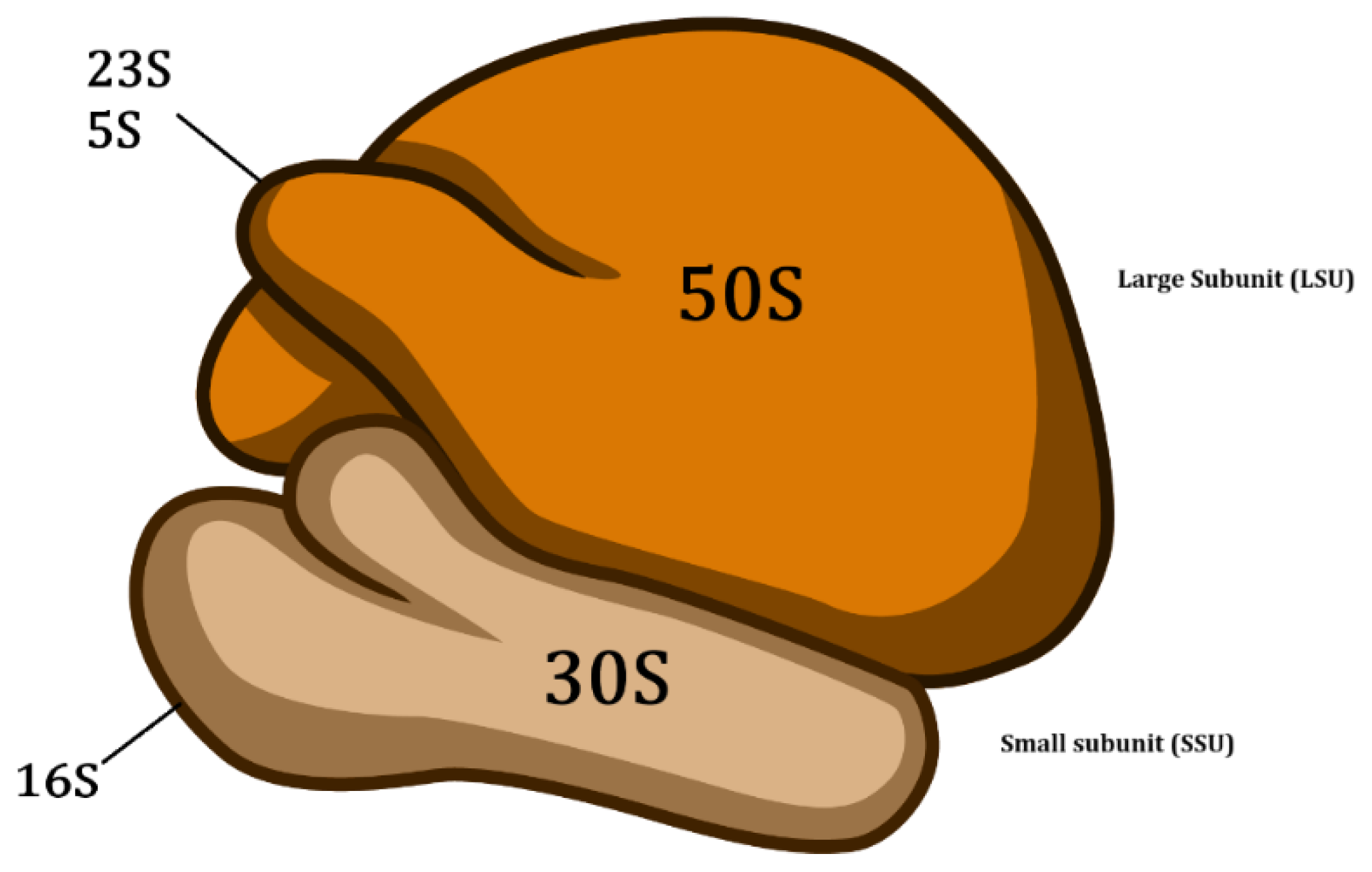
Previously, to be considered from the same species, bacterial isolates should share a similarity in the 16S rRNA gene of more than 97%, based on a relationship with 70% DDH, which is considered the gold standard method for delimiting bacterial species [30]. According to Stackebrandt & Ebers (2006), the similarity criterion for the 16S rRNA gene would be greater than 98.7%. Identity values in the 16S rRNA gene sequence below 95% with the phylogenetically closest species with a validated name may indicate that the isolate is a representative of a new genus [31].
Although they provide valuable phylogenetic information, 16S ribosomal gene sequences are not always useful for distinguishing closely related species due to their highly conserved nature. When several species of the same genus share >98.7% identity in the 16S gene sequence, the sequencing of other genes, called essential or constitutive, is recommended [8,22].
5. Analysis of the Housekeeping Gene Sequences
Housekeeping genes, also called essential genes, encode enzymes that are essential for maintaining cellular function. Examples include the recA gene, which encodes a recombinase protein; rpoB, which encodes the beta subunit of RNA polymerase; and gyrB, which encodes the B subunit of the DNA gyrase protein. Housekeeping genes are highly conserved and accumulate mutations more rapidly than rRNA genes, making them useful for differentiating bacteria at the species level, since the 16S rRNA gene shows high genetic homology within certain genera and may not be useful alone for distinguishing closely related species [21,24,31,32,33].
The analysis of the sequences of a certain number of housekeeping genes, called multilocus sequence analysis (MLSA), incorporates similarity values to differentiate species, and is considered a phylogenetic tool to support and clarify the delimitation of bacterial species with a higher resolution than studies based on 16S rRNA genes. The gene sequences are used to construct a phylogenetic tree to infer phylogenies [34,35]. MLSA is based on multilocus sequence typing (MLST), a method for typing pathogenic bacteria for epidemiological and population genetic purposes, first described by Maiden et al, 1998 [35,36].
The choice of genes for each taxon analyzed is critical to the reliability of the analysis. Housekeeping genes should be considered because they are more stable in relation to rapid genetic change and are present in all species of a genus. It is also recommended that they be single-copy genes, distributed throughout the entire genome. The ad hoc Committee recommends the use of at least five housekeeping genes for the re-evaluation of the species definition in bacteriology, although most studies use seven genes. Some species require the use of more genes for better differentiation [35,37,38]. Before amplifying and sequencing of the selected housekeeping genes of interest for comparison and construction of a phylogenetic tree, it is very important to consult the different sequence databases, such as National Center for Biotechnology Information (NCBI, https://www.ncbi.nlm.nih.gov/), European Molecular Biology Laboratory’s – European Bioinformatics Institute (EMBL-EBI, https://www.ebi.ac.uk/), DNA Data Bank of Japan (DDBJ, https://www.ddbj.nig.ac.jp/index-e.html), Bacterial Diversity Metadatabase (BacDive, https://bacdive.dsmz.de/), Joint Genome Institute (JGI, https://genome.jgi.doe.gov/portal/), among others. If the sequences of the housekeeping genes of interest or the complete genome of the closest type strain are not available, an alternative would be to purchase the type strain from a collection, such as the American Type Culture Collection (ATCC) or the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), for example, and perform the sequencing yourself. Some sequences may be in the possession of researchers who have not yet published their data. Contacting these researchers may also be an alternative.
Gene sequencing for MLSA involves time-consuming and laborious steps. With the advancement of high-throughput sequencing and bioinformatics tools in recent years, MLSA can be performed in silico due to the substantial increase of whole-genome data in public databases, allowing gene sequences to be extracted directly from genomes [38].
6. Genomic Taxonomy Tools
Improvements in DNA sequencing technologies have resulted in a significant increase in the amount of genomic data generated, combined with a reduction in the cost of sequencing [34]. Whole genome sequencing (WGS) data and the development of bioinformatics tools have allowed the establishment of taxonomic schemes based on evolutionary information contained in genome sequences, such as digital DNA-DNA hybridization (dDDH), Average Amino Acid Identity (AAI), supertrees, and others [39]. Such taxonomic schemes, which are described in more detailed below, can be used not only to describe a new bacterial species, but also to confirm the identification of the bacterial isolate, especially when it comes to environmental isolates belonging to the genus Bacillus and/or related genera, which have a large number of described species, and genotypic characterization by sequencing the 16S rRNA and housekeeping genes is not always possible.
Genomic taxonomy is defined based on an integrated comparative genomics approach with the goal of extracting taxonomic information from genomes. The main goal of genomic taxonomy is to extract taxonomic information from genomes that can be used to provide a solid framework for identifying and classifying prokaryotic species and even populations. These new tools mentioned above have led to new understanding of genetic relationships that the 16S rRNA gene can only approximate [6,39,40].
DDH indirectly measures the degree of genetic similarity between two genomes, one of which is the genome of the type strain, and has been the “gold standard” for bacterial species delimitation [41]. In brief, the heated DNA strands are dissociated and immediately reassociated, and hybridization occurs. The degree of relationship between the two genomes is verified, and the two genomes are considered to belong to the same species if the DDH value is greater than 70% [34,38]. However, this value is not sufficient to distinguish, for example, the species Rickettsia rickettsii, Rickettsia conorii, Rickettsia sibirica and Rickettsia montanensis, in other words, the DDH limit used is not applicable to all genera [31]. Few laboratories in the world perform this methodology because it is a laborious, slow, and expensive technique that requires specialized personnel. Another disadvantage is that the results may vary depending on the protocol used, which can lead to experimental errors and the comparison of results obtained by different methodologies is not recommended [30,31,38].
With the advent of high-throughput DNA sequencing and the various genomes deposited in public repositories, dDDH was then proposed [34,42], and some authors suggested replacing DDH as the “gold standard” in prokaryotic taxonomy by pairwise genomic sequence-derived similarity [42,43]. The analysis of dDDH consists of the local alignment between two genomes and intergenomic correspondences are generated, which are later used to calculate the distance matrix, whose values are analogous to DDH (> 70%) [38]. The Genome-to-Genome Distance Calculator (GGDC) is one of the most popular online tools for calculating in silico DDH values, provided free of charge by the German bacterial collection DSMZ. The GGDC allows the comparison of bacterial genomes to determine their similarity on the same scale as the DDH, aiding in the identification and classification of bacterial species. Moreover, the GGDC reports the difference in G+C content, which can be used for species delineation [43,44].
Over the years, there has been an increasing use of the Average Nucleotide Identity (ANI) tool for the classification and identification of bacterial species [45,46], which shows a strong correlation with the DDH [47]. ANI was proposed in 2005 and consisted in the local alignment between two genomes, calculating the average identity of the nucleotides of the shared open reading frames (ORFs) instead of the whole genome [43]. In 2007, the comparison between two whole genomes was implemented, allowing ANI results to be directly compared with the DDH [48]. Two prokaryotic genomes can be considered to belong to the same species if they share an ANI value ≥ 96% [31]. Furthermore, ANI shows a strong correlation with 16S rRNA gene sequence similarity [49]. Although a robust tool for bacterial species delimitation, ANI has been shown to have low resolution at higher taxonomic levels. Therefore, other metrics should be used, such as AAI [50,51], another widely used tool for bacterial species classification and identification, which is based on the calculation of conserved protein-coding genes between a pair of genomes determined by pairwise comparison of whole genomes using the BLAST algorithm. Briefly, all protein-coding genes in one genome are searched for in relation to all protein-coding genes in the other genome. The AAI of all conserved genes between the pair of genomes is then measured from the genetic relatedness between a pair of genomes and a value of ~95% to 96% was established, with a strong correlation to the similarity of the 16S rRNA gene sequence [38,39,52]. The AAI has been shown to be a very useful metric for genera delimitation (>60-65%) [49]. Based on the discontinuous distribution of its AAI values, several studies have developed boundaries to delimit genera, such as Chryseobacterium [53], Prochlorococcus [54] and Lactobacillus [55].
Complex phylogenetic relationships between different taxa can be captured by genome-based phylogenies, or phylogenomics. In 2019, the online platform TYGS (Type Strain Genome Server) was developed by the German collection DSMZ for the classification and identification of microorganisms based on their genomes, combining genomic data with robust phylogenetic approaches to provide accurate and reproducible classification. The Type Strain Genome Server (TYGS) contains a comprehensive database that is constantly updated and revised to ensure accuracy and reliability, and is maintained by the Leibniz Institute/DSMZ. This server allows the analysis of dDDH indices, and the construction of phylogenies based on the 16S rRNA gene and the Genome BLAST Distance Phylogeny (GBDP) method, using as reference the next closest phylogenetic matches previously identified by Mash genomic distances and 16S rRNA gene data. In addition, TYGS is integrated with the database that powers the List of Prokaryotic Names with Standing in Nomenclature (LPSN), providing information on the most recent updates in nomenclature and taxonomic literature. The platform also performs a preliminary species-level classification to assist in the identification of possible new species [56,57].
Another computational tool that supports the phylogenomic classification of bacteria based on genomic data is the Genome Taxonomy Database Toolkit (GTDB-Tk), which compares genomes to the Genome Taxonomy Database (GTDB) taxonomy using a pre-calculated phylogenomic tree and metrics such as ANI and Relative Evolutionary Divergence (RED). GTDB-Tk allows the assignment of taxa to new genomes, facilitating the identification of new species and the reclassification of organisms within a standardized phylogenomic taxonomy. GTDB-Tk is an independent tool linked to GTDB that inserts the genome into a previously calculated multigene-based phylogenies (MBP) phylogenomic tree, calculates RED indices and performs species assignment based on ANI, when viable. This process allows the identification of new taxa, both at the species level and in broader taxonomic categories. The GTDB and backbone trees are updated regularly, including a major annual revision [56,58].
Another approach worth mentioning is Ribosomal Multilocus Sequence Typing (rMLST, https://pubmlst.org/species-id), which was proposed to overcome the limitations of current methods for bacterial typing and phylogenetic reconstruction by using 53 ribosomal genes (rps genes) that are present in all bacteria and distributed along bacterial chromosomes. They also encode ribosomal proteins that are highly conserved. This technique provides greater phylogenetic resolution than traditional methods, such as using the 16S rRNA gene, and allows bacteria to be accurately classified at multiple taxonomic levels, including domain, phylum, class, order, family, genus, and species. The rMLST database is an extensible, web-accessible database containing complete genomic data from thousands of bacterial isolates, enabling rapid and computationally efficient identification of the phylogenetic position of any bacterial sequence at multiple taxonomic levels [59].
7. Conclusions and Future Directions
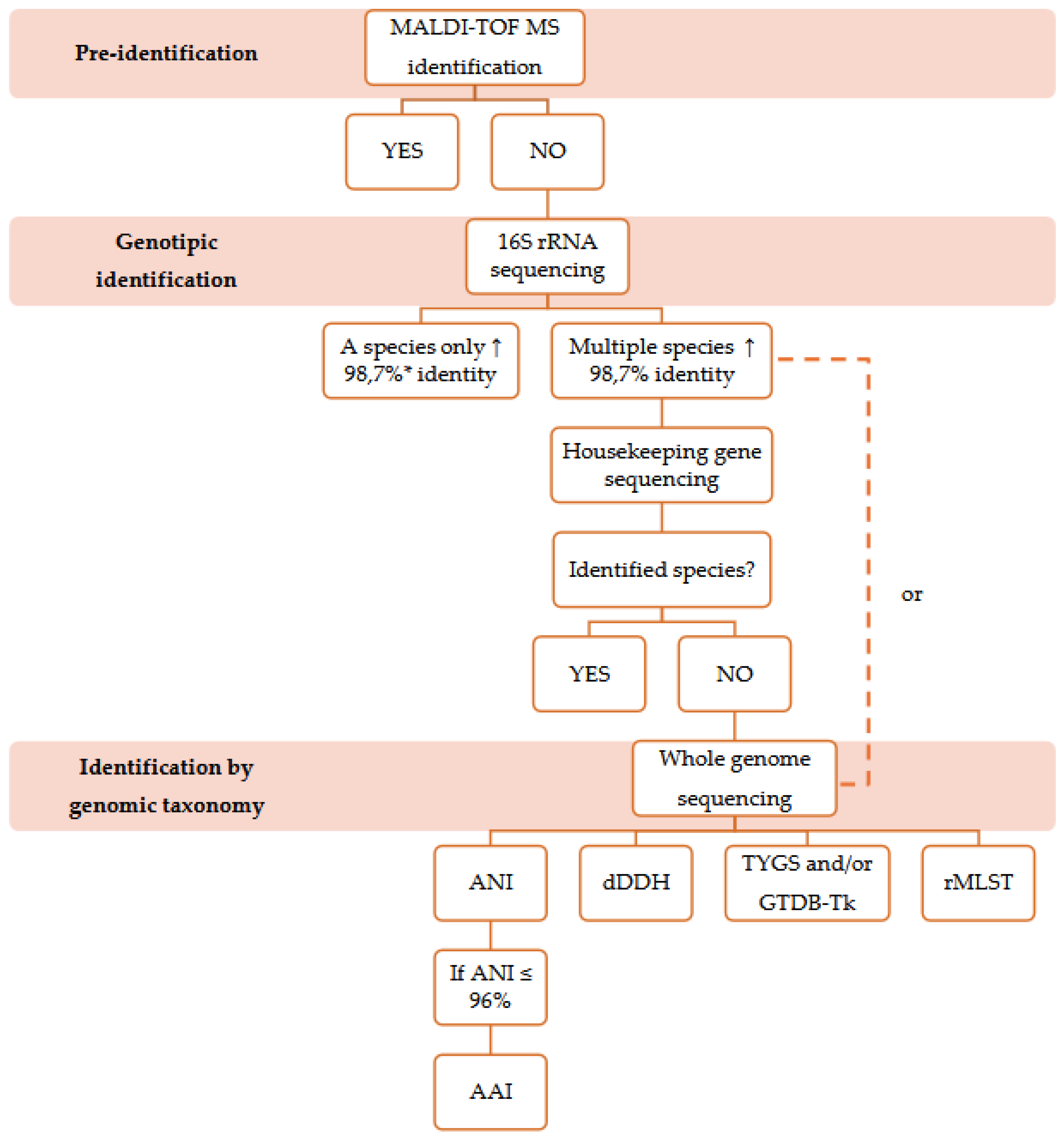
Many laboratories in the pharmaceutical industry use MALDI-TOF MS for bacterial identification because of its rapid results and low cost, which are essential for batch release of pharmaceutical products. Here, we suggest some methods for the identification of environmental bacteria, from MALDI-TOF MS to genomic taxonomy analysis, and the advantages and disadvantages of each method (Figure 2). Initially, MALDI-TOF MS databases focused on clinically relevant strains, but their application has expanded to environmental and industrial samples, requiring new references. Species not detectable by MALDI-TOF MS have been included after identification by 16S rRNA and housekeeping genes sequencing. Identification can be improved by adding spectral profiles to the database.
The most commonly used method to assess the phylogenetic position of a prokaryote is the comparison of the 16S rRNA gene sequence. Sequencing of the 16S rRNA gene is widely used for bacterial identification due to its universal presence in Bacteria and Archaea, high conservation and low rate of evolution, being an essential marker for phylogenetic studies. Nucleotide variations within multiple rRNA operons in a single genome and the possibility of 16S rRNA genes being derived from horizontal gene transfer can distort relationships between taxa in phylogenetic trees. Despite its relevance, the 16S rRNA gene does not always distinguish closely related species due to its high conservation; its phylogeny is robust at the genus level and above, making it necessary to complement the analysis with the sequencing of housekeeping genes, which have been used in MLSA as an alternative for more accurate identification of microorganisms.
Advances in DNA sequencing technologies have increased the availability of genomic data and reduced their costs, allowing the creation of taxonomic schemes based on in silico analysis of genomes. Genomic taxonomy uses tools such as dDDH, ANI and AAI to identify and classify bacterial species. The traditional DDH method, previously considered the gold standard for species delimitation, has been replaced by computational methods, such as the GGDC, which calculates the genomic similarity between organisms. ANI, which compares the average nucleotide identity between two genomes is widely used to define species with values ≥96%, while AAI, based on the identity of conserved coding proteins, helps to delimit genera (>60-65%). In addition, phylogenomic tools such as TYGS and GTDB-Tk provide robust approaches for microbial classification using continuously updated databases and phylogenetic analyses based on multiple genes. In addition to all the bioinformatic tools described above for the identification of the bacterial isolate, we also suggest the rMLST approach, which is a web-accessible database with support for an online database hosted by PubMLST. The approach is based on the analysis of 53 highly conserved ribosomal genes for the identification and classification of a wide range of bacterial species, and can be applied to poorly characterized or undescribed species. The rMLST database is compatible with whole-genome sequencing data or metagenomes, facilitating the analysis of complex microbial communities.
Approaches that combine multiple analyses and metrics are strongly encouraged, as they provide greater accuracy in bacterial identification and overcome the limitations of 16S rRNA gene sequencing in distinguishing closely related species.
Figure 2.
Steps of identification of environmental bacterial isolates, from MALDI-TOF MS to genomic taxonomy analyses. * According to Stackebrandt, Ebbers (2006).
Figure 2.
Steps of identification of environmental bacterial isolates, from MALDI-TOF MS to genomic taxonomy analyses. * According to Stackebrandt, Ebbers (2006).
Author Contributions
Conceptualization, M.L.L.B.; formal analysis, J.N.R., L.V.C., V.V.V. and M.L.L.B.; data curation, L.V.C. and M.L.L.B.; writing-original draft preparation, J.N.R.; writing-review and editing, J.N.R., L.V.C., V.V.V. and M.L.L.B.; supervision, M.L.L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro – FAPERJ (E-26/200.546/2025). The funding body played no role in the design of the study and writing the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AAI | Average Amino Acid Identity |
| ANI | Average Nucleotide Identity |
| ATCC | American Type Culture Collection |
| BacDive | Bacterial Diversity Metadatabase |
| BLAST | Basic Local Alignment Search tool |
| bp | Base pairs |
| DDBJ | DNA Data Bank of Japan |
| dDDH | digital DNA-DNA hybridization |
| DDH | Hibridization DNA-DNA |
| DSMZ | Deutsche Sammlung von Mikroorganismen und Zellkulturen |
| EMBL-EBI | European Molecular Biology Laboratory – European Bioinformatics Institute |
| G+C | Guanine and cytosine |
| GBDP | Genome BLAST Distance Phylogeny |
| GGDC | Genome-to-Genome Distance Calculator |
| GMPs | Good manufacturing practices |
| GTDB | Genome Taxonomy Database |
| GTDB-Tk | Genome Taxonomy Database Toolkit |
| JGI | Joint Genome Institute |
| LPSN | List of Prokatiotic Names with Standing in Nomenclature |
| MALDI-TOF MS | Matrix-Assisted Laser Desorption Ionization–Time of Flight/Mass Spectrometry |
| MBP | multigene-based phylogenies |
| MLSA | multilocus sequence analysis |
| MLST | multilocus sequence typing |
| NCBI | National Center for Biotechnology Information |
| ORFs | open reading frames |
| RED | Relative Evolutionary Divergence |
| rMLST | Ribosomal Multilocus Sequence Typing |
| rRNA | Ribosomal RNA |
| TYGS | Type Strain Genome Server |
| WGS | Whole-genome sequencing |
References
- Costa, L.V. da; Miranda, R.V. da S. L. de; Reis, C.M.F. dos; Andrade, J.M. de; Cruz, F.V.; Frazão, A.M.; Fonseca, E.L. da; Ramos, J.N.; Brandão, M.L.L.; Vieira, V.V. MALDI-TOF MS Database Expansion for Identification of Bacillus and Related Genera Isolated from a Pharmaceutical Facility. J Microbiol Methods 2022, 203. [Google Scholar] [CrossRef]
- United States Pharmacopeial Convention. In The United States Pharmacopeia 43rd ed; The United States Pharmacopeia.
- Caldeira, N.G.S.; de Souza, M.L.S.; de Miranda, R.V.d.S. L.; da Costa, L.V.; Forsythe, S.J.; Zahner, V.; Brandão, M.L.L. Characterization by MALDI-TOF MS and 16S RRNA Gene Sequencing of Aerobic Endospore-Forming Bacteria Isolated from Pharmaceutical Facility in Rio de Janeiro, Brazil. Microorganisms 2024, 12, 724. [Google Scholar] [CrossRef]
- Food and Drug Administration - FDA. Guidance for Industry Sterile Drug Products Produced by Aseptic Processing —Current Good Manufacturing Practice,. ed by Aseptic Processing —Current Good Manufacturing Practice,.
- European Medicines Agency. The Rules Governing Medicinal Products in the European Union. Volume 4: European Union Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1: Manufacture of Sterile Medicinal Products.. The Rules Governing Medicinal Products in the European Union. Volume 4: European Union Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1: Manufacture of Sterile Medicinal Products.
- Thompson, C.C.; Vidal, L.; Salazar, V.; Swings, J.; Thompson, F.L. Microbial Genomic Taxonomy. In Trends in the systematics of bacteria and fungi; CABI: UK, 2021; pp. 168–178. [Google Scholar] [CrossRef]
- Moreira, F.M.; Pereira, P. de A.; Miranda, R.V. da S. L. de; Reis, C.M.F. dos; Braga, L.M.P. da S.; de Andrade, J.M.; do Nascimento, L.G.; Mattoso, J.M.V.; Forsythe, S.J.; da Costa, L.V.; et al. Evaluation of MALDI-TOF MS, Sequencing of D2 LSU RRNA and Internal Transcribed Spacer Regions (ITS) for the Identification of Filamentous Fungi Isolated from a Pharmaceutical Facility. J Pharm Biomed Anal 2023, 234, 115531. [Google Scholar] [CrossRef]
- Costa, L.V. da; Ramos, J.N.; Albuquerque, L. de S.; Miranda, R.V. da S. L. de; Valadão, T.B.; Veras, J.F.C.; Vieira, E.M.D.; Forsythe, S.; Brandão, M.L.L.; Vieira, V.V. Bacillus Lumedeiriae Sp. Nov., a Gram-Positive, Spore-Forming Rod Isolated from a Pharmaceutical Facility Production Environment and Added to the MALDI Biotyper® Database. Microorganisms 2024, 12, 2507. [Google Scholar] [CrossRef]
- Obasi, A.; Nwachukwu, S.; Ugoji, E.; Kohler, C.; Göhler, A.; Balau, V.; Pfeifer, Y.; Steinmetz, I. Extended-Spectrum β-Lactamase-Producing Klebsiella Pneumoniae from Pharmaceutical Wastewaters in South-Western Nigeria. Microbial Drug Resistance 2017, 23, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- da Silva Lage de Miranda, R.V.; da Costa, L.V.; de Sousa Albuquerque, L.; dos Reis, C.M.F.; da Silva Braga, L.M.P.; de Andrade, J.M.; Ramos, J.N.; Mattoso, J.M.V.; Forsythe, S.J.; Brandão, M.L.L. Identification of Sutcliffiella Horikoshii Strains in an Immunobiological Pharmaceutical Industry Facility. Lett Appl Microbiol 2023, 76. [Google Scholar] [CrossRef]
- Costa, L.V. da; Miranda, R.V. da S. L. de; Fonseca, E.L. da; Gonçalves, N.P.; Reis, C.M.F. dos; Frazão, A.M.; Cruz, F.V.; Brandão, M.L.L.; Ramos, J.N.; Vieira, V.V. Assessment of VITEK® 2, MALDI-TOF MS and Full Gene 16S RRNA Sequencing for Aerobic Endospore-Forming Bacteria Isolated from a Pharmaceutical Facility. J Microbiol Methods 2022, 194, 106419. [Google Scholar] [CrossRef]
- Sala-Comorera, L.; Vilaró, C.; Galofré, B.; Blanch, A.R.; García-Aljaro, C. Use of Matrix-Assisted Laser Desorption/Ionization–Time of Flight (MALDI–TOF) Mass Spectrometry for Bacterial Monitoring in Routine Analysis at a Drinking Water Treatment Plant. Int J Hyg Environ Health 2016, 219, 577–584. [Google Scholar] [CrossRef]
- Vithanage, N.R.; Yeager, T.R.; Jadhav, S.R.; Palombo, E.A.; Datta, N. Comparison of Identification Systems for Psychrotrophic Bacteria Isolated from Raw Bovine Milk. Int J Food Microbiol 2014, 189, 26–38. [Google Scholar] [CrossRef]
- Biomérieux. VITEK® 2. VITEK® 2. Fully integrated Identification and Antimicrobial Susceptibility Testing.
- Bosshard, P.P.; Zbinden, R.; Abels, S.; Böddinghaus, B.; Altwegg, M.; Böttger, E.C. 16S RRNA Gene Sequencing versus the API 20 NE System and the VITEK 2 ID-GNB Card for Identification of Nonfermenting Gram-Negative Bacteria in the Clinical Laboratory. J Clin Microbiol 2006, 44, 1359–1366. [Google Scholar] [CrossRef]
- Seuylemezian, A.; Aronson, H.S.; Tan, J.; Lin, M.; Schubert, W.; Vaishampayan, P. Development of a Custom MALDI-TOF MS Database for Species-Level Identification of Bacterial Isolates Collected From Spacecraft and Associated Surfaces. Front Microbiol 2018, 9. [Google Scholar] [CrossRef]
- Zasada, A.A.; Mosiej, E. Contemporary Microbiology and Identification of Corynebacteria Spp. Causing Infections in Human. Lett Appl Microbiol 2018, 66, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.N.; Shah, A.J.; Belgacem, O.; Ward, M.; Dekio, I.; Selami, L.; Duncan, L.; Bruce, K.; Xu, Z.; Mkrtchyan, H.V.; et al. MALDI-TOF MS and Currently Related Proteomic Technologies in Reconciling Bacterial Systematics. In Trends in the systematics of bacteria and fungi; CABI: UK, 2021; pp. 93–118. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Ebers, J. Taxonomic Parameters Revisited: Tarnished Gold Standards – ScienceOpen. Microbial Today. 2006, p. 152. Available online: https://www.scienceopen.com/document?vid=0cf4b084-5683-4ef4-a80c-c0df44a135dc (accessed on 17 March 2025).
- Lasch, P.; Stämmler, M.; Schneider, A. A MALDI-TOF Mass Spectrometry Database for Identification and Classification of Highly Pathogenic Microorganisms from the Robert Koch-Institute (RKI). [CrossRef]
- Caamaño-Antelo, S.; Fernández-No, I.C.; Böhme, K.; Ezzat-Alnakip, M.; Quintela-Baluja, M.; Barros-Velázquez, J.; Calo-Mata, P. Genetic Discrimination of Foodborne Pathogenic and Spoilage Bacillus Spp. Based on Three Housekeeping Genes. Food Microbiol 2015, 46, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Rajendhran, J.; Gunasekaran, P. Microbial Phylogeny and Diversity: Small Subunit Ribosomal RNA Sequence Analysis and Beyond. Microbiol Res 2011, 166, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Church, D.L.; Cerutti, L.; Gürtler, A.; Griener, T.; Zelazny, A.; Emler, S. Performance and Application of 16S rRNA Gene Cycle Sequencing for Routine Identification of Bacteria in the Clinical Microbiology Laboratory. Clin Microbiol Rev 2020, 33, e00053–19. [Google Scholar] [CrossRef]
- Madigan, M.; Martinko, J.; Bender, K.; Buckley, D.; Stahl, D. Brock Biology of Microorganisms, 14th ed.; Benjamin Cummings, 2015.
- Mahato, N.K.; Gupta, V.; Singh, P.; Kumari, R.; Verma, H.; Tripathi, C.; Rani, P.; Sharma, A.; Singhvi, N.; Sood, U.; et al. Microbial Taxonomy in the Era of OMICS: Application of DNA Sequences, Computational Tools and Techniques. Antonie Van Leeuwenhoek 2017, 110, 1357–1371. [Google Scholar] [CrossRef]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A Comprehensive Benchmarking Study of Protocols and Sequencing Platforms for 16S RRNA Community Profiling. BMC Genomics 2016, 17, 55. [Google Scholar] [CrossRef]
- Rodrigues, N.M.B.; Bronzato, G.F.; Santiago, G.S.; Botelho, L.A.B.; Moreira, B.M.; Coelho, I. da S.; Souza, M.M.S. de; Coelho, S. de M. de O. The Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry (MALDI-TOF MS) Identification versus Biochemical Tests: A Study with Enterobacteria from a Dairy Cattle Environment. Brazilian Journal of Microbiology 2016, 48, 132. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Mondotte, J.A.; Fazio, L.L.; Jetten, M. Authors Need to Be Prudent When Assigning Names to Microbial Isolates. Curr Microbiol 2021, 78, 4005–4008. [Google Scholar] [CrossRef]
- NIAID Visual & Medical Arts. Ribosome, NIAID NIH BIOART Source.
- Tindall, B.J.; Rosselló-Móra, R.; Busse, H.J.; Ludwig, W.; Kämpfer, P. Notes on the Characterization of Prokaryote Strains for Taxonomic Purposes. Int J Syst Evol Microbiol 2010, 60 Pt 1, 249–266. [Google Scholar] [CrossRef]
- Sentausa, E.; Fournier, P.E. Advantages and Limitations of Genomics in Prokaryotic Taxonomy. Clinical Microbiology and Infection 2013, 19, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Vlach, J.; Javůrková, B.; Karamonová, L.; Blažková, M.; Fukal, L. Novel PCR-RFLP System Based on RpoB Gene for Differentiation of Cronobacter Species. Food Microbiol 2017, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.W.; Vandamme, P.; Morgan, S.H.; LiPuma, J.J.; Coenye, T.; Weightman, A.J.; Jones, T.H.; Mahenthiralingam, E. Development of a RecA Gene-Based Identification Approach for the Entire Burkholderia Genus. Appl Environ Microbiol 2005, 71, 3917–3927. [Google Scholar] [CrossRef]
- Chun, J.; Rainey, F.A. Integrating Genomics into the Taxonomy and Systematics of the Bacteria and Archaea. Int J Syst Evol Microbiol 2014, 64, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Glaeser, S.P.; Kämpfer, P. Multilocus Sequence Analysis (MLSA) in Prokaryotic Taxonomy. Syst Appl Microbiol 2015, 38, 237–245. [Google Scholar] [CrossRef]
- Maiden, M.C.J.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus Sequence Typing: A Portable Approach to the Identification of Clones within Populations of Pathogenic Microorganisms. Proc Natl Acad Sci U S A 1998, 95, 3140–3145. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Frederiksen, W.; Garrity, G.M.; Grimont, P.A.D.; Kämpfer, P.; Maiden, M.C.J.; Nesme, X.; Rosselló-Mora, R.; Swings, J.; Trüper, H.G.; et al. Report of the Ad Hoc Committee for the Re-Evaluation of the Species Definition in Bacteriology. Int J Syst Evol Microbiol 2002, 52, 1043–1047. [Google Scholar] [CrossRef]
- Hayashi Sant’Anna, F.; Bach, E.; Porto, R.Z.; Guella, F.; Hayashi Sant’Anna, E.; Passaglia, L.M.P. Genomic Metrics Made Easy: What to Do and Where to Go in the New Era of Bacterial Taxonomy. Crit Rev Microbiol 2019, 45, 182–200. [Google Scholar] [CrossRef]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F.L. Microbial Genomic Taxonomy. BMC Genomics 2013, 14. [Google Scholar] [CrossRef]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 Years of Bacterial Genome Sequencing. Funct Integr Genomics 2015, 15, 141. [Google Scholar] [CrossRef]
- Wayne, L.G.; Brenner, D.J.; Colwell, R.R.; Grimont, P.A.D.; Kandler, O.; Krichevsky, M.I.; Moore, L.H.; Moore, W.E.C.; Murray, R.G.E.; Stackebrandt, E.; et al. Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int J Syst Evol Microbiol 1987, 37, 463–464. [Google Scholar] [CrossRef]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.W.; De Meyer, S.; et al. Proposed Minimal Standards for the Use of Genome Data for the Taxonomy of Prokaryotes. Int J Syst Evol Microbiol 2018, 68, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, S.; Fullmer, M.S.; Feng, Y.; Gogarten, J.P. Improving Phylogenies Based on Average Nucleotide Identity, Incorporating Saturation Correction and Nonparametric Bootstrap Support. Syst Biol 2022, 71, 396–409. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res 2022, 50, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; ran, Q.; Du, X.; Wu, S.; Wang, J.; Sheng, D.; Chen, Q.; Du, Z.; Li, Y. zhong. Two New Polyangium Species, P. Aurulentum Sp. Nov. and P. Jinanense Sp. Nov., Isolated from a Soil Sample. Syst Appl Microbiol 2021, 44. [Google Scholar] [CrossRef] [PubMed]
- Cuny, H.; Offret, C.; Boukerb, A.M.; Parizadeh, L.; Lesouhaitier, O.; Le Chevalier, P.; Jégou, C.; Bazire, A.; Brillet, B.; Fleury, Y. Pseudoalteromonas Ostreae Sp. Nov., a New Bacterial Species Harboured by the Flat Oyster Ostrea Edulis. Int J Syst Evol Microbiol 2021, 71. [Google Scholar] [CrossRef]
- Colston, S.M.; Fullmer, M.S.; Beka, L.; Lamy, B.; Peter Gogarten, J.; Graf, J. Bioinformatic Genome Comparisons for Taxonomic and Phylogenetic Assignments Using Aeromonas as a Test Case. mBio 2014, 5. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA Hybridization Values and Their Relationship to Whole-Genome Sequence Similarities. Int J Syst Evol Microbiol 2007, 57 Pt 1, 81–91. [Google Scholar] [CrossRef]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic Insights That Advance the Species Definition for Prokaryotes. Proc Natl Acad Sci U S A 2005, 102, 2567–2572. [Google Scholar] [CrossRef]
- Qin, Q.L.; Xie, B. Bin; Zhang, X.Y.; Chen, X.L.; Zhou, B.C.; Zhou, J.; Oren, A.; Zhang, Y.Z. A Proposed Genus Boundary for the Prokaryotes Based on Genomic Insights. J Bacteriol 2014, 196, 2210. [Google Scholar] [CrossRef]
- Kim, D.; Park, S.; Chun, J. Introducing EzAAI: A Pipeline for High Throughput Calculations of Prokaryotic Average Amino Acid Identity. J Microbiol 2021, 59, 476–480. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Konstantinidis, K.T. Bypassing Cultivation To Identify Bacterial Species: Culture-Independent Genomic Approaches Identify Credibly Distinct Clusters, Avoid Cultivation Bias, and Provide True Insights into Microbial Species. Microbe Magazine 2014, 9, 111–118. [Google Scholar] [CrossRef]
- Nicholson, A.C.; Gulvik, C.A.; Whitney, A.M.; Humrighouse, B.W.; Bell, M.E.; Holmes, B.; Steigerwalt, A.G.; Villarma, A.; Sheth, M.; Batra, D.; et al. Division of the Genus Chryseobacterium: Observation of Discontinuities in Amino Acid Identity Values, a Possible Consequence of Major Extinction Events, Guides Transfer of Nine Species to the Genus Epilithonimonas, Eleven Species to the Genus Kaistella, and Three Species to the Genus Halpernia Gen. Nov., with Description of Kaistella Daneshvariae Sp. Nov. and Epilithonimonas Vandammei Sp. Nov. Derived from Clinical Specimens. Int J Syst Evol Microbiol 2020, 70, 4432–4450. [Google Scholar] [CrossRef]
- Walter, J.M.; Coutinho, F.H.; Dutilh, B.E.; Swings, J.; Thompson, F.L.; Thompson, C.C. Ecogenomics and Taxonomy of Cyanobacteria Phylum. Front Microbiol 2017, 8, 2132. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.A.P.; Harris, H.M.B.; Mattarelli, P.; O’toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A Taxonomic Note on the Genus Lactobacillus: Description of 23 Novel Genera, Emended Description of the Genus Lactobacillus Beijerinck 1901, and Union of Lactobacillaceae and Leuconostocaceae. Int J Syst Evol Microbiol 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Riesco, R.; Trujillo, M.E. Update on the Proposed Minimal Standards for the Use of Genome Data for the Taxonomy of Prokaryotes. Int J Syst Evol Microbiol 2024, 74. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat Commun 2019, 10, 2182. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An Ongoing Census of Bacterial and Archaeal Diversity through a Phylogenetically Consistent, Rank Normalized and Complete Genome-Based Taxonomy. Nucleic Acids Res 2022, 50, D785–D794. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bliss, C.M.; Bennett, J.S.; Bratcher, H.B.; Brehony, C.; Colles, F.M.; Wimalarathna, H.; Harrison, O.B.; Sheppard, S.K.; Cody, A.J.; et al. Ribosomal Multilocus Sequence Typing: Universal Characterization of Bacteria from Domain to Strain. Microbiology (Reading) 2012, 158 Pt 4, 1005–1015. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



