
Submitted:
27 March 2025
Posted:
28 March 2025
You are already at the latest version
Abstract
Podoplanin (PDPN), also referred to as T1a/Aggrus, is a type I transmembrane sialoglycoprotein that plays a crucial role in invasiveness, stemness, and epithelial-to-mesenchymal transition, all of which contribute to malignant progression of tumors. Therefore, monoclonal antibody (mAb) against PDPN has been evaluated in preclinical models as a promising tumor therapy strategy. However, PDPN plays an essential role in normal development, such as in the development of the lungs. The on-target toxicity by anti-PDPN mAbs to normal cells should be avoided to minimize adverse effects. A cancer-specific mAb (CasMab) against PDPN, PMab-117 (rat IgM, kappa) was previously established. This study engineered the humanized IgG1 version (humPMab-117) to investigate antitumor activity. Flow cytometry analysis confirmed that humPMab-117 recognized PDPN-overexpressed glioma LN229 (LN229/PDPN) cells as well as PDPN-positive PC-10 (human lung squamous cell carcinoma) and LN319 (human glioblastoma) cells. In contrast, humPMab-117 did not react with normal epithelial cells from the lung bronchus, gingiva, mammary gland, corneal, and normal kidney podocytes, suggesting that humPMab-117 retains the cancer-specific reactivity. Furthermore, humPMab-117 effectively induced antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity against LN229/PDPN, PC-10, and LN319 cells. In the xenograft tumor models, humPMab-117 demonstrated strong antitumor efficacy. These results suggest the potential of humPMab-117 as a therapeutic antibody for treating PDPN-positive malignant tumors.
Keywords:
podoplanin
; cancer-specific monoclonal antibody
; ADCC
; CDC
; antitumor activity
1. Introduction
The validation of adequate antigenic targets is essential for the development of monoclonal antibodies (mAb)-based tumor therapy[1]. To achieve an acceptable therapeutic index with low on-target toxicity, targets highly expressed in tumors and little or no expression in normal tissues are considered ideal antigenic targets. However, the number of ideal antigenic targets is limited, which is a significant problem for developing therapeutic mAbs for tumors.
To solve the problem, we have developed cancer-specific mAbs (CasMabs) for various antigens and revealed the cancer-specific epitope and the recognition structure. In the anti-human epidermal growth factor receptor 2 (HER2) CasMab development, we established more than three hundred anti-HER2 mAb clones by immunization of mice with cancer cell-expressed HER2. These mAbs were screened by the reactivity to HER2-expressed tumor and normal cells by flow cytometry [2]. Among them, H2Mab-250/H2CasMab-2 recognized HER2 in breast cancer cells but not in normal epithelial cells from the mammary gland, lung bronchus, colon, and kidney proximal tubule [2]. Epitope analysis identified Trp614 in the extracellular domain 4 of HER2 as a critical residue for H2Mab-250 recognition [2].
Furthermore, mouse IgG2a type or humanized H2Mab-250 exhibited antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and in vivo antitumor efficacy against human breast cancer xenografts [3,4,5]. A single chain variable fragment of H2Mab-250 was further developed to chimeric antigen receptor (CAR)-T cell therapy and showed cancer-specific recognition and cytotoxicity [6]. A phase I clinical trial is underway for patients with HER2-positive advanced solid tumors in the US (NCT06241456). Therefore, selecting CasMab and identifying the cancer-specific epitopes are essential strategies for developing therapeutic mAbs and modalities.
Podoplanin (PDPN) (also known as T1α, PA2.26 antigen, E11 antigen, and Aggrus) is a heavily glycosylated type I transmembrane protein, which has an N-terminal extracellular domain, a transmembrane domain, and a short intracellular domain [7,8]. The N-terminal extracellular domain possesses platelet aggregation-stimulating (PLAG) domains, which have a consensus repeat sequence of EDxxVTPG [9]. The O-glycosylation sites at Thr52 in PLAG3 or a PLAG-like domain (PLD, also named PLAG4) have been reported to be crucial for the interaction of PDPN to C-type lectin-like receptor 2 (CLEC-2), which is essential for platelet aggregation and hematogenous metastasis to lung [10,11].
PDPN is involved in the malignant progression of tumors by promoting invasiveness and metastasis. PDPN-expressing tumor cells show a diverse pattern of invasion [12], including the epithelial-to-mesenchymal transition-like pattern in various tumors [13,14], collective invasion in squamous cell carcinomas (SCCs) [15], and ameboid invasion in melanoma [16]. The intracellular domain of PDPN possesses basic residues as binding sites for ezrin, radixin, and moesin proteins [17], which mediates Rho GTPase activity and regulate the diverse pattern of invasiveness [18,19]. Furthermore, PDPN binds to matrix metalloproteinases [20] and a hyaluronan receptor CD44 [21], which mediate the invadopodia formation and extracellular matrix degradation. In the clinic, high PDPN expression was associated with shortened overall survival in patients with gliomas, head and neck SCC, esophageal SCC, gastric adenocarcinomas, and mesotheliomas [22,23,24,25]. Therefore, PDPN has been considered a promising target of mAb-based therapy. However, PDPN also plays an essential role in normal cells, such as lung alveolar type I cells [26] and kidney podocytes [27,28]. Therefore, cancer-specific reactivity is required to reduce adverse effects on normal cells.
We have developed CasMabs against PDPN by selecting the cancer-specific reactivity in flow cytometry and immunohistochemistry. LpMab-2 [29] and LpMab-23 [30] were obtained by immunization of mice with PDPN-overexpressed glioblastoma LN229 (LN229/PDPN). LpMab-2 recognizes a glycopeptide structure (Thr55-Leu64) of PDPN [29]. In contrast, LpMab-23 recognizes a naked peptide structure (Gly54–Leu64) of PDPN [31]. Mouse-human chimeric LpMab-2 and LpMab-23 (chLpMab-2 and chLpMab-23, respectively) exhibited the ADCC activity and antitumor effect in human tumor xenograft models [31,32]. Furthermore, we obtained another CasMab against PDPN (PMab-117) by immunization of LN229/PDPN with a rat. In flow cytometry, PMab-117 showed the reactivity to PDPN expressing tumor PC-10 and LN319. PMab-117 did not react with normal kidney podocytes and normal epithelial cells from the mammary gland, lung bronchus, and cornea. In contrast, NZ-1, one of the non-CasMabs against PDPN, exhibited high reactivity to both tumor and normal cells [33]. PMab-117 recognizes the glycopeptide structure of PDPN (Ile78-Thr85) within PLD, including O-glycosylated Thr85 [7].
This study evaluates the effects of the humanized version of PMab-117 (humPMab-117) on the ADCC, CDC, and antitumor activity.
2. Results
2.1. Production of Humanized Anti-PDPN mAb, humPMab-117
We previously established an anti-PDPN CasMab (PMab-117; rat IgM, kappa) by immunization of LN229/PDPN. PMab-117 was shown to recognize cancer cell-expressed PDPN, but not normal cell-expressed PDPN in flow cytometry [33]. In this study, we engineered a humanized PMab-117 (humPMab-117) by fusing the VH and VL CDRs of PMab-117 with the CH and CL chains of human IgG1, respectively (Figure 1A). Under reduced conditions, we confirmed the purity of original and recombinant mAbs by SDS-PAGE (Figure 1B). As shown in Figure 1C, humPMab-117 reacted with LN229/PDPN, PC-10, and LN319, but not with PDPN-negative LN229 and PDPN-knockout LN319 (BINDS-55). NZ-16, a rat-human chimeric anti-PDPN mAb [34], showed a higher reactivity to those cancer cell lines.
We next compared the reactivity of humPMab-117 and NZ-16 to a TERT-expressed normal kidney podocyte, PODO/TERT256, and TERT-expressed normal epithelial cell lines, HBEC3-KT (lung bronchus), hTERT-TIGKs (gingiva), hTERT-HME1 (mammary gland), and hTCEpi (cornea). As shown in Figure 1D, humPMab-117 did not show reactivity to PODO/TERT256, HBEC3-KT, hTERT-TIGKs, hTERT-HME1, and hTCEpi. In contrast, NZ-16 showed reactivity to those normal cells. These results indicated that humPMab-117 retains cancer-specific reactivity.
The KD values for the interaction of humPMab-117 and NZ-16 with LN229/PDPN were determined by flow cytometry. The KD values for humPMab-117 and NZ-16 with LN229/PDPN cells were 5.4 × 10-7 M and 8.6 × 10-9 M, respectively (Figure 2). These results indicated that humPMab-117 possesses approximately 60-fold lower affinity to LN229/PDPN than NZ-16.
2.2. ADCC by humPMab-117 Against PDPN-Positive Cells
We next investigated whether humPMab-117 exhibits ADCC activity against PDPN-positive cells. As shown in Figure 3, humPMab-117 induced significant ADCC against LN229/PDPN cells (18.7% cytotoxicity; p < 0.05) compared to the control human IgG1 9.8% cytotoxicity). Furthermore, humPMab-117 elicited ADCC against endogenous PDPN expressing tumor PC-10 (23.4% cytotoxicity; p < 0.05) more effectively than the control human IgG1 13.0% cytotoxicity). Additionally, humPMab-117 also showed ADCC against endogenous PDPN expressing tumor LN319 (4.5% cytotoxicity; p < 0.05) more effectively than the control human IgG1 (2.4% cytotoxicity).
2.3. CDC by humPMab-117 Against PDPN-Positive Cells
We next examined the CDC activity of humPMab-117 against PDPN-positive cells. As shown in Figure 4, humPMab-117 induced significant CDC against LN229/PDPN cells (25.1% cytotoxicity; p < 0.01) compared to the control human IgG1 (4.5% cytotoxicity). Furthermore, humPMab-117 elicited CDC against PC-10 (17.2% cytotoxicity; p < 0.05) more effectively than the control human IgG1 5.8% cytotoxicity). In addition, humPMab-117 also showed CDC against LN319 (6.6% cytotoxicity; p < 0.05) more effectively than the control human IgG1 (2.1% cytotoxicity).
2.4. Antitumor Effects of humPMab-117 Against PDPN-Positive Cells in Mouse Xenograft Models
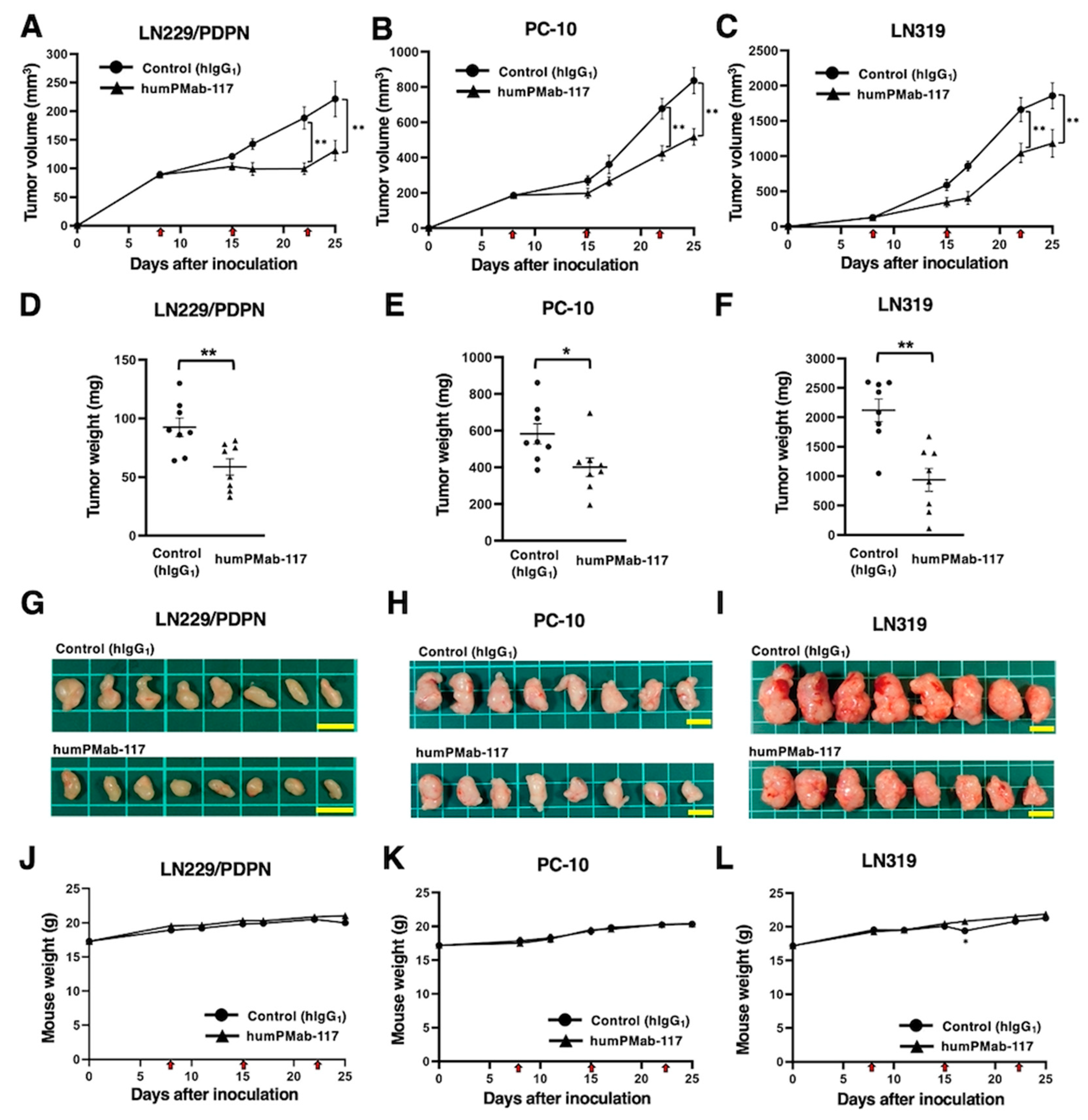
After the inoculation of LN229/PDPN, PC-10, or LN319 at the left flanks of BALB/c nude mice, humPMab-117 or human IgG1 was intraperitoneally injected into the xenograft-bearing mice on days 8, 15, and 22. The tumor volume was measured on the indicated days. The humPMab-117 administration resulted in a significant reduction in LN229/PDPN xenografts on days 22 (p < 0.01) and 25 (p < 0.01) compared with that of control human IgG1 (Figure 5A). In the PC-10 xenograft, a significant reduction was observed on days 22 (p < 0.01) and 25 (p < 0.01) (Figure 5B). In the LN319 xenograft, a significant reduction was also observed on days 22 (p < 0.01) and 25 (p < 0.01) (Figure 5C).
In the xenograft weight, humPMab-117 also showed the reduction in LN229/PDPN (36% reduction; p < 0.01; Figure 5D), PC-10 (31% reduction; p < 0.05; Figure 5E), and LN319 (56% reduction; p < 0.01; Figure 5F) compared with control human IgG1. The resected LN229/PDPN, PC-10, and LN319 tumors on day 25 are shown in Figure 5G–I. The xenograft-bearing mice did not lose body weight with the humPMab-117 treatment (Figure 5J–L). The mice on day 25 are shown in Supplementary Figure S1.
3. Discussion
This study evaluated the in vitro and in vivo antitumor effects of a novel CasMab against PDPN. The human IgG1 type PMab-117 (humPMab-117) retained the reactivity to the PDPN-expressing tumor cells but not to normal epithelial cells or kidney podocytes in flow cytometry (Figure 1). Furthermore, humPMab-117 exerted ADCC (Figure 3), CDC (Figure 4), and antitumor effects in LN229/PDPN, PC-10, and LN319 xenografts (Figure 5).
Several preclinical studies have evaluated the antitumor activities of humanized or chimeric anti-PDPN mAbs. The anti-PDPN mAb NZ-1 (a non-CasMab, rat IgG2a), recognizes the PLAG2/3 domain, has a neutralizing activity to the PDPN–CLEC-2 interaction and inhibits PDPN-induced platelet aggregation and hematogenous lung metastasis [35,36]. NZ-16, a rat-human chimeric anti-PDPN mAb derived from NZ-1, was developed for alpha-radioimmunotherapy. 225Ac-labeled NZ-16 showed antitumor efficacy against human malignant pleural mesothelioma xenografts and prolonged survival without apparent adverse effect [34].
Another non-CasMab, PG4D2, recognizes the PLD of PDPN and possesses neutralizing activity against the PDPN–CLEC-2 interaction and platelet aggregation [11]. The humanized PG4D2 (AP201) is a human IgG4 mAb, which does not have ADCC and CDC activity. Nevertheless, AP201 suppressed not only osteosarcoma hematogenous lung metastasis but also xenograft growth [37]. The authors proposed the possibility that platelet-derived growth factors from activated platelet support the osteosarcoma proliferation, which AP201 inhibits. PMab-117 also recognizes the PLD of PDPN [7] and humPMab-117 (human IgG1) exerted ADCC and CDC activity against PDPN-expressing tumors (Figure 3 and Figure 4). Therefore, it is worthwhile to investigate the platelet aggregation-inhibitory effect of humPMab-117 to clarify its contribution to antitumor efficacy.
Although anti-PDPN CasMabs, LpMab-2 and LpMab-23, do not possess neutralizing activity to the PDPN–CLEC-2 interaction, human IgG1 mAbs, including chLpMab-2, chLpMab-23, and a humanized LpMab-23 (humLpMab-23), exhibited the antitumor effect against human tumor xenograft through ADCC and CDC activity [31,32,38]. These results suggest that neutralizing activity is not essential for antitumor efficacy. However, whether these mAbs affect the normal tissue in vivo is a concern. We previously evaluated the toxicity of mouse-human chimeric LpMab-23 (20 mg/kg) against cynomolgus monkeys, and no toxicity was observed [31]. A similar analysis is required to prove the safety of humPMab-117.
The diagnosis to determine the PDPN-positive tumor is also essential for the selection of the patients [39]. LpMab-2, LpMab-23, and PMab-117 recognize different epitopes of PDPN [7]. LpMab-2 and LpMab-23 retain the cancer-specific reactivity in immunohistochemistry [29,30]. In contrast, PMab-117 is not suitable for immunohistochemistry. Although the reactivity of humPMab-117 was much weaker than NZ-16 in flow cytometry (Figure 1), humPMab-117 demonstrated significant antitumor activities (Figure 5). It is possible that humPMab-117 may recognize a part of cancer-specific aberrant-structured PDPN. Further studies are required to clarify the mechanism of recognition by humPMab-117 and the distribution of PMab-117-positive tumors.
CAR-T cell therapy has achieved significant success in the treatment of hematopoietic malignancies [40]. However, the strategy has not been fully translated to solid tumors [41]. The most crucial problem of CAR-T cell therapy against solid tumors is a lack of tumor-specific antigens. Most therapeutic targets such as epidermal growth factor receptor [42], HER2 [43], and MUC1 [44] are expressed on normal cells, which leads to on-target off-tumor toxicity due to the targeting of normal cells. Since the dosage of CAR-T cells is limited, the reactivity of CAR to normal cells should be minimized. In that sense, our CasMabs against PDPN are suitable for CAR selection. The humanized NZ-1 and LpMab-2-based CAR-T have been evaluated in preclinical models and showed the significant antitumor efficacy [45,46]. As shown in Figure 2, humPMab-117 has approximately 60-fold lower affinity (KD: 5.4 × 10−7 M) than NZ-16 (KD: 8.6 × 10−9 M). The KD values of LpMab-2 and chLpMab-23 were previously determined as 5.7 × 10−9 M and 1.2 × 10−8 M, respectively [29,31]. These CasMabs have different binding affinities ranging from 10−7 M to 10−9 M. Recently, low affinity and avidity CAR-T cell therapy has exhibited enhanced cytotoxicity [47,48,49], elevated expansion [48,50], better trafficking [47], and increased selectivity [49]. Furthermore, low affinity and avidity CAR-T cells have been shown to decrease exhaustion [51] and mitigate trogocytosis [50,52]. It is necessary to explore the cancer-specific reactivity of PMab-117 single-chain Fv for CAR-T cell therapy. It is essential to compare the antitumor activity of three cancer-specific anti-PDPN CAR-T therapies in the future.
4. Materials and Methods
4.1. Cell Lines
PODO/TERT256 and hTCEpi were purchased from EVERCYTE (Vienna, Austria). LN229, HBEC3-KT, hTERT-HME1, and hTERT TIGKs were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Human glioblastoma LN319 cells were purchased from Addexbio Technologies (San Diego, CA, USA). Human lung SCC PC-10 cells were purchased from Immuno-Biological Laboratories Co., Ltd. (Gunma, Japan).
LN229/PDPN cells were established as previously described [29]. LN229, LN229/PDPN, and LN319 cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) [Nacalai Tesque, Inc. (Nacalai), Kyoto, Japan]. PC-10 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Nacalai). These media were supplemented with 10% heat-inactivated fetal bovine serum [FBS; Thermo Fisher Scientific, Inc. (Thermo), Waltham, MA, USA], 0.25 μg/mL amphotericin B, 100 μg/mL streptomycin, and 100 units/mL penicillin (Nacalai). ExpiCHO-S and Fut8-deficient ExpiCHO-S (BINDS-09) cells were cultured as described previously [38].
Immortalized normal epithelial cell lines were maintained as follows: PODO/TERT256, MCDB131 (Pan Biotech, Bayern, Germany) supplemented with GlutaMAXTM-I (Thermo), Bovine Brain Extract (9.6 μg/mL, Lonza, Basel, Switzerland), EGF [8 ng/mL, Sigma-Aldrich Corp. (Sigma), St. Louis, MO, USA], Hydrocortisone (20 ng/mL, Sigma), 20% FBS (Sigma), and G418 (100 μg/mL, InvivoGen, San Diego, CA); HBEC3-KT, Airway Epithelial Cell Basal Medium and Bronchial Epithelial Cell Growth Kit (ATCC); hTERT TIGKs, Dermal Cell Basal Medium and Keratinocyte Growth Kit (ATCC); hTERT-HME1, Mammary Epithelial Cell Basal Medium BulletKitTM without GA-1000 (Lonza); hTCEpi, KGMTM-2 BulletKitTM (Lonza).
All cell lines were cultured at 37°C in a humidified atmosphere with 5% CO2 and 95% air.
4.2. Animals
The Institutional Committee for Experiments of the Institute of Microbial Chemistry (Numazu, Japan) authorized animal studies for the antitumor efficacy of humPMab-117 (approval number: 2024-076). The animal studies followed the NIH (National Research Council) Guide for the Care and Use of Laboratory Animals. Humane objectives for euthanasia were established as a loss of original body weight to a point >25% and/or a maximal tumor size >3,000 mm3.
4.3. Antibodies
To generate a humanized anti-human PDPN mAb (humPMab-117), the CDRs of PMab-117 VH and VL were cloned into human IgG1 and human kappa chain expression vectors [53], respectively. We transfected the antibody expression vectors of humPMab-117 into BINDS-09 (fucosyltransferase 8-knockout ExpiCHO-S) cells using the ExpiCHO-S Expression System (Thermo). As a control human IgG1 mAb, humCvMab-62 was produced from CvMab-62 (an anti-SARS-CoV-2 spike protein S2 subunit mAb) using the abovementioned method. NZ-16, a rat-human chimeric anti-PDPN mAb, was previously described [34]. humCvMab-62, humPMab-117, and NZ-16 were purified using Ab-Capcher (ProteNova Co., Ltd., Kagawa, Japan). To confirm the purity of mAbs, they were treated with sodium dodecyl sulfate sample buffer containing 2-mercaptoethanol, separated on 5%–20% polyacrylamide gel (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan), and stained by Bio-Safe CBB G-250 (Bio-Rad Laboratories, Inc., Berkeley, CA).
4.4. Flow Cytometry
Cells were harvested using 0.25% trypsin and 1 mM ethylenediaminetetraacetic acid (EDTA; Nacalai). Subsequently, they were washed with 0.1% bovine serum albumin (Nacalai) in phosphate-buffered saline (PBS), followed by treatment with humPMab-117 or NZ-16 for 30 minutes at 4°C. Then, the cells were treated with fluorescein isothiocyanate (FITC)-conjugated anti-human IgG (1:2000; Sigma) for 30 minutes at 4°C. Fluorescence data were collected using the SA3800 Cell Analyzer (Sony Corp., Tokyo, Japan) and analyzed with FlowJo software (BD Biosciences, Franklin Lakes, NJ, USA).
4.5. ADCC
Human NK cells were purchased from Takara Bio, Inc. (Shiga, Japan) and were used as effector cells immediately after thawing as follows. Target cells (LN229/PDPN, PC-10, and LN319) were labeled with 10 µg/mL of Calcein AM (Thermo). The target cells were plated in 96-well plates at a density of 5 × 103 cells/well and combined with effector cells (effector-to-target ratio, 50:1) and 100 μg/mL of either control human IgG1 or humPMab-117. After incubating for 4.5 hours, the calcein released into the supernatant was measured as described previously [4].
4.6. CDC
The target cells labeled with Calcein AM (LN229/PDPN, PC-10, and LN319) were seeded and combined with rabbit complement (final concentration 10%, Low-Tox-M Rabbit Complement; Cedarlane Laboratories, Hornby, ON, Canada) along with 100 μg/mL of either control human IgG1 or humPMab-117. After a 4.5-hour incubation at 37°C, the amount of calcein released into the medium was measured as described previously [4].
4.7. Antitumor Activities of humPMab-117 in Xenografts of Human Tumors
LN229/PDPN, PC-10, and LN319 were mixed with DMEM and Matrigel Matrix Growth Factor Reduced (BD Biosciences). Subcutaneous injections were then given to the left flanks of BALB/c nude mice. On the eighth post-inoculation day, 100 µg of control human IgG1 (n = 8) or humPMab-117 (n = 8) in 100 µL PBS were administered intraperitoneally. Additional antibody injections were given on days 15 and 22. The tumor diameter was assessed on days 8, 15, 17, 22, and 25 after the tumor cell implantation. Tumor volumes were calculated in the same manner as previously stated. The weight of the mice was also assessed on days 8, 11, 15, 17, 22, and 25 following breast tumor cell inoculation. When the observations were finished on day 25, the mice were sacrificed, and tumor weights were assessed following tumor excision.
4.8. Statistical Analyses
All data are represented as mean ± standard error of the mean (SEM). A two-tailed unpaired t-test was conducted to measure ADCC activity, CDC activity, and tumor weight. ANOVA with Sidak’s post hoc test was performed for tumor volume and mouse weight. GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA, USA) was used for all calculations. p < 0.05 was considered to indicate a statistically significant difference.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Figure S1: Body appearance in LN229/PDPN (A), PC-10 (B), and LN319 (C) xenografts-implanted mice treated with control hIgG1 or humPMab-117 on day 25 after cell inoculation.
Author Contributions
Conceptualization, Y.K.; methodology, T.O., M.K.; formal analysis, T.T.; investigation, H.S., T.O., and T.T.; data curation, H.S.; writing—original draft preparation, H.S.; writing—review and editing, Y.K.; project administration, Y.K.; funding acquisition, H.S., T.T., and Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported in part by Japan Agency for Medical Research and Development (AMED) under Grant Numbers: JP24am0521010 (to Y.K.), JP24ama121008 (to Y.K.), JP24ama221339 (to Y.K.), JP24bm1123027 (to Y.K.), and JP24ck0106730 (to Y.K.), and by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) grant nos. 22K06995 (to H.S), 24K18268 (to T.T.), and 22K07224 (to Y.K.).
Institutional Review Board Statement
The Institutional Committee for Experiments of the Institute of Microbial Chemistry approved animal experiments (approval no. 2024-076).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and supplementary material.
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- Paul, S.; Konig, M.F.; Pardoll, D.M.; et al. Cancer therapy with antibodies. Nat Rev Cancer 2024;24(6): 399-426.
- Kaneko, M.K.; Suzuki, H.; Kato, Y. Establishment of a Novel Cancer-Specific Anti-HER2 Monoclonal Antibody H(2)Mab-250/H(2)CasMab-2 for Breast Cancers. Monoclon Antib Immunodiagn Immunother 2024;43(2): 35-43.
- Kaneko, M.K.; Suzuki, H.; Ohishi, T.; et al. Antitumor Activities of a Humanized Cancer-Specific Anti-HER2 Monoclonal Antibody, humH(2)Mab-250 in Human Breast Cancer Xenografts. Int J Mol Sci 2025;26(3). [CrossRef]
- Suzuki, H.; Ohishi, T.; Tanaka, T.; Kaneko, M.K.; Kato, Y. Anti-HER2 Cancer-Specific mAb, H(2)Mab-250-hG(1), Possesses Higher Complement-Dependent Cytotoxicity than Trastuzumab. Int J Mol Sci 2024;25(15). [CrossRef]
- Kaneko, M.K.; Suzuki, H.; Ohishi, T.; et al. A Cancer-Specific Monoclonal Antibody against HER2 Exerts Antitumor Activities in Human Breast Cancer Xenograft Models. Int J Mol Sci 2024;25(3). [CrossRef]
- Hosking, M.; Shirinbak, S.; Omilusik, K.; et al. 268 Development of FT825/ONO-8250: an off-the-shelf CAR-T cell with preferential HER2 targeting and engineered to enable multi-antigen targeting, improve trafficking, and overcome immunosuppression. Journal for ImmunoTherapy of Cancer 2023;11(Suppl 1): A307-A307.
- Suzuki, H.; Kaneko, M.K.; Kato, Y. Roles of Podoplanin in Malignant Progression of Tumor. Cells 2022;11(3). [CrossRef]
- Quintanilla, M.; Montero-Montero, L.; Renart, J.; Martín-Villar, E. Podoplanin in Inflammation and Cancer. Int J Mol Sci 2019;20(3).
- Kato, Y.; Fujita, N.; Kunita, A.; et al. Molecular identification of Aggrus/T1alpha as a platelet aggregation-inducing factor expressed in colorectal tumors. J Biol Chem 2003;278(51): 51599-51605. [CrossRef]
- Kaneko, M.K.; Kato, Y.; Kameyama, A.; et al. Functional glycosylation of human podoplanin: glycan structure of platelet aggregation-inducing factor. FEBS Lett 2007;581(2): 331-336. [CrossRef]
- Sekiguchi, T.; Takemoto, A.; Takagi, S.; et al. Targeting a novel domain in podoplanin for inhibiting platelet-mediated tumor metastasis. Oncotarget 2016;7(4): 3934-3946. [CrossRef]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol Oncol 2017;11(1): 5-27. [CrossRef]
- Lambert, A.W.; Weinberg, R.A. Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat Rev Cancer 2021;21(5): 325-338. [CrossRef]
- Astarita, J.L.; Acton, S.E.; Turley, S.J. Podoplanin: emerging functions in development, the immune system, and cancer. Front Immunol 2012;3: 283. [CrossRef]
- Wicki, A.; Lehembre, F.; Wick, N.; et al. Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006;9(4): 261-272. [CrossRef]
- de Winde, C.M.; George, S.L.; Crosas-Molist, E.; et al. Podoplanin drives dedifferentiation and amoeboid invasion of melanoma. iScience 2021;24(9): 102976. [CrossRef]
- Martín-Villar, E.; Megías, D.; Castel, S.; et al. Podoplanin binds ERM proteins to activate RhoA and promote epithelial-mesenchymal transition. J Cell Sci 2006;119(Pt 21): 4541-4553. [CrossRef]
- Pecora, A.; Laprise, J.; Dahmene, M.; Laurin, M. Skin Cancers and the Contribution of Rho GTPase Signaling Networks to Their Progression. Cancers (Basel) 2021;13(17). [CrossRef]
- Zhang, Z.; Liu, M.; Zheng, Y. Role of Rho GTPases in stem cell regulation. Biochem Soc Trans 2021. [CrossRef]
- Li, Y.Y.; Zhou, C.X.; Gao, Y. Podoplanin promotes the invasion of oral squamous cell carcinoma in coordination with MT1-MMP and Rho GTPases. Am J Cancer Res 2015;5(2): 514-529.
- Martín-Villar, E.; Fernández-Muñoz, B.; Parsons, M.; et al. Podoplanin associates with CD44 to promote directional cell migration. Mol Biol Cell 2010;21(24): 4387-4399. [CrossRef]
- Friedman, G.; Levi-Galibov, O.; David, E.; et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4(+) and PDPN(+) CAFs to clinical outcome. Nat Cancer 2020;1(7): 692-708. [CrossRef]
- Hirayama, K.; Kono, H.; Nakata, Y.; et al. Expression of podoplanin in stromal fibroblasts plays a pivotal role in the prognosis of patients with pancreatic cancer. Surg Today 2018;48(1): 110-118. [CrossRef]
- Liu, X.; Cao, Y.; Lv, K.; et al. Tumor-infiltrating podoplanin(+) cells in gastric cancer: clinical outcomes and association with immune contexture. Oncoimmunology 2020;9(1): 1845038. [CrossRef]
- Wang, H.; Hu, C.; Song, X.; et al. Expression of Podoplanin in Sinonasal Squamous Cell Carcinoma and Its Clinical Significance. Am J Rhinol Allergy 2020;34(6): 800-809. [CrossRef]
- Ramirez, M.I.; Millien, G.; Hinds, A.; et al. T1alpha, a lung type I cell differentiation gene, is required for normal lung cell proliferation and alveolus formation at birth. Dev Biol 2003;256(1): 61-72. [CrossRef]
- Koop, K.; Eikmans, M.; Wehland, M.; et al. Selective loss of podoplanin protein expression accompanies proteinuria and precedes alterations in podocyte morphology in a spontaneous proteinuric rat model. Am J Pathol 2008;173(2): 315-326. [CrossRef]
- Ijpelaar, D.H.; Schulz, A.; Koop, K.; et al. Glomerular hypertrophy precedes albuminuria and segmental loss of podoplanin in podocytes in Munich-Wistar-Frömter rats. Am J Physiol Renal Physiol 2008;294(4): F758-767. [CrossRef]
- Kato, Y.; Kaneko, M.K. A cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci Rep 2014;4: 5924. [CrossRef]
- Yamada, S.; Ogasawara, S.; Kaneko, M.K.; Kato, Y. LpMab-23: A Cancer-Specific Monoclonal Antibody Against Human Podoplanin. Monoclon Antib Immunodiagn Immunother 2017;36(2): 72-76. [CrossRef]
- Kaneko, M.K.; Nakamura, T.; Kunita, A.; et al. ChLpMab-23: Cancer-Specific Human-Mouse Chimeric Anti-Podoplanin Antibody Exhibits Antitumor Activity via Antibody-Dependent Cellular Cytotoxicity. Monoclon Antib Immunodiagn Immunother 2017;36(3): 104-112. [CrossRef]
- Kaneko, M.K.; Yamada, S.; Nakamura, T.; et al. Antitumor activity of chLpMab-2, a human-mouse chimeric cancer-specific antihuman podoplanin antibody, via antibody-dependent cellular cytotoxicity. Cancer Med 2017;6(4): 768-777.
- Tanaka, T.; Suzuki, H.; Ohishi, T.; Kaneko, M.K.; Kato, Y. A Cancer-Specific Anti-Podoplanin Monoclonal Antibody, PMab-117-mG(2a) Exerts Antitumor Activities in Human Tumor Xenograft Models. Cells 2024;13(22).
- Sudo, H.; Tsuji, A.B.; Sugyo, A.; et al. Preclinical Evaluation of Podoplanin-Targeted Alpha-Radioimmunotherapy with the Novel Antibody NZ-16 for Malignant Mesothelioma. Cells 2021;10(10). [CrossRef]
- Kato, Y.; Kaneko, M.K.; Kunita, A.; et al. Molecular analysis of the pathophysiological binding of the platelet aggregation-inducing factor podoplanin to the C-type lectin-like receptor CLEC-2. Cancer Sci 2008;99(1): 54-61. [CrossRef]
- Kato, Y.; Kaneko, M.K.; Kuno, A.; et al. Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem Biophys Res Commun 2006;349(4): 1301-1307. [CrossRef]
- Takemoto, A.; Takagi, S.; Ukaji, T.; et al. Targeting Podoplanin for the Treatment of Osteosarcoma. Clin Cancer Res 2022;28(12): 2633-2645. [CrossRef]
- Suzuki, H.; Ohishi, T.; Kaneko, M.K.; Kato, Y. A Humanized and Defucosylated Antibody against Podoplanin (humLpMab-23-f) Exerts Antitumor Activities in Human Lung Cancer and Glioblastoma Xenograft Models. Cancers (Basel) 2023;15(20). [CrossRef]
- Zhou, Y.; Tao, L.; Qiu, J.; et al. Tumor biomarkers for diagnosis, prognosis and targeted therapy. Signal Transduct Target Ther 2024;9(1): 132.
- First-Ever CAR T-cell Therapy Approved in U.S. Cancer Discov 2017;7(10): Of1.
- Kakarla, S.; Gottschalk, S. CAR T cells for solid tumors: armed and ready to go? Cancer J 2014;20(2): 151-155.
- Feng, K.; Guo, Y.; Dai, H.; et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci 2016;59(5): 468-479. [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 2017;3(8): 1094-1101.
- Mei, Z.; Zhang, K.; Lam, A.K.; et al. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med 2020;9(2): 640-652. [CrossRef]
- Chalise, L.; Kato, A.; Ohno, M.; et al. Efficacy of cancer-specific anti-podoplanin CAR-T cells and oncolytic herpes virus G47Δ combination therapy against glioblastoma. Mol Ther Oncolytics 2022;26: 265-274. [CrossRef]
- Shiina, S.; Ohno, M.; Ohka, F.; et al. CAR T Cells Targeting Podoplanin Reduce Orthotopic Glioblastomas in Mouse Brains. Cancer Immunol Res 2016;4(3): 259-268. [CrossRef]
- Castellarin, M.; Sands, C.; Da, T.; et al. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI Insight 2020;5(14). [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med 2019;25(9): 1408-1414. [CrossRef]
- Arcangeli, S.; Rotiroti, M.C.; Bardelli, M.; et al. Balance of Anti-CD123 Chimeric Antigen Receptor Binding Affinity and Density for the Targeting of Acute Myeloid Leukemia. Mol Ther 2017;25(8): 1933-1945. [CrossRef]
- Olson, M.L.; Mause, E.R.V.; Radhakrishnan, S.V.; et al. Low-affinity CAR T cells exhibit reduced trogocytosis, preventing rapid antigen loss, and increasing CAR T cell expansion. Leukemia 2022;36(7): 1943-1946. [CrossRef]
- Caraballo Galva, L.D.; Jiang, X.; Hussein, M.S.; et al. Novel low-avidity glypican-3 specific CARTs resist exhaustion and mediate durable antitumor effects against HCC. Hepatology 2022;76(2): 330-344. [CrossRef]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019;568(7750): 112-116. [CrossRef]
- Suzuki, H.; Ohishi, T.; Tanaka, T.; Kaneko, M.K.; Kato, Y. A Cancer-Specific Monoclonal Antibody against Podocalyxin Exerted Antitumor Activities in Pancreatic Cancer Xenografts. Int J Mol Sci 2023;25(1). [CrossRef]
Figure 1.
Production of humPMab-117 and reactivity to cancer cells, normal kidney podocytes, and epithelial cells. (A) Human IgG1 mAb, humPMab-117, was generated from PMab-117 (rat IgM). (B) MAbs were treated with sodium dodecyl sulfate sample buffer containing 2-mercaptoethanol. Proteins were separated on polyacrylamide gel. Bio-Safe CBB G-250 Stain stained the gel. (C) Flow cytometry using humPMab-117 (10 μg/mL; Red line), NZ-16 (10 μg/mL; Red line), or buffer control (Black line) against LN229, LN229/PDPN, PC-10, LN319, and PDPN-knockout LN319 (BINDS-55). (D) Flow cytometry using humPMab-117 (10 μg/mL; Red line), NZ-16 (10 μg/mL; Red line) or buffer control (Black line) against PODO/TERT256 (kidney podocyte), HBEC3-KT (lung bronchus epithelial cell), hTERT-TIGKs (gingiva), hTERT-HME1 (mammary gland epithelial cell), and hTCEpi (corneal epithelial cell). The cells were treated with FITC-conjugated anti-human IgG. Fluorescence data were analyzed using the SA3800 Cell Analyzer.
Figure 1.
Production of humPMab-117 and reactivity to cancer cells, normal kidney podocytes, and epithelial cells. (A) Human IgG1 mAb, humPMab-117, was generated from PMab-117 (rat IgM). (B) MAbs were treated with sodium dodecyl sulfate sample buffer containing 2-mercaptoethanol. Proteins were separated on polyacrylamide gel. Bio-Safe CBB G-250 Stain stained the gel. (C) Flow cytometry using humPMab-117 (10 μg/mL; Red line), NZ-16 (10 μg/mL; Red line), or buffer control (Black line) against LN229, LN229/PDPN, PC-10, LN319, and PDPN-knockout LN319 (BINDS-55). (D) Flow cytometry using humPMab-117 (10 μg/mL; Red line), NZ-16 (10 μg/mL; Red line) or buffer control (Black line) against PODO/TERT256 (kidney podocyte), HBEC3-KT (lung bronchus epithelial cell), hTERT-TIGKs (gingiva), hTERT-HME1 (mammary gland epithelial cell), and hTCEpi (corneal epithelial cell). The cells were treated with FITC-conjugated anti-human IgG. Fluorescence data were analyzed using the SA3800 Cell Analyzer.
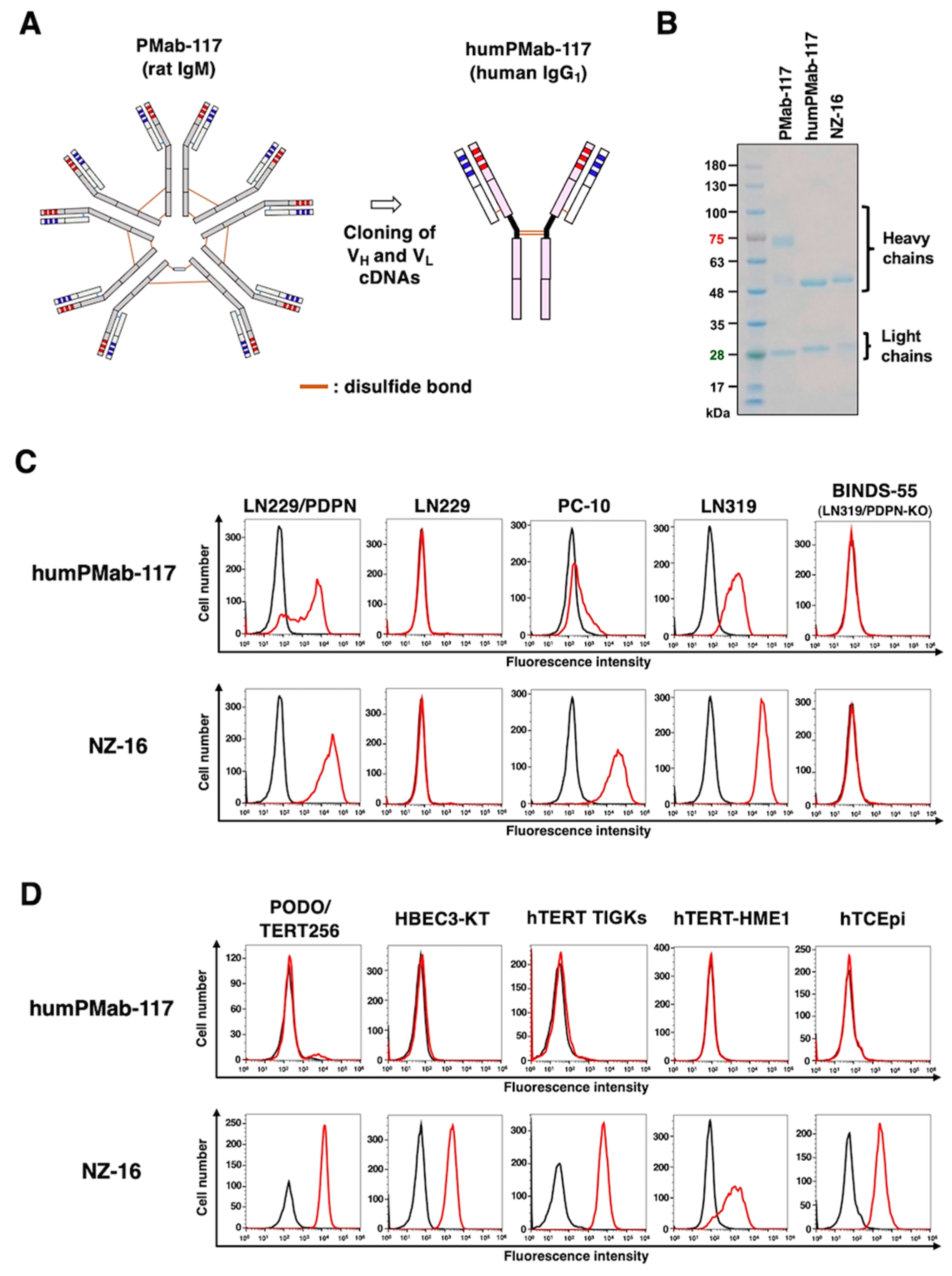
Figure 2.
Determination of the binding affinity of humPMab-117 and NZ-16 using flow cytometry. LN229/PDPN cells were suspended in humPMab-117 (A) or NZ-16 (B) at indicated concentrations, followed by treatment with FITC-conjugated anti-human IgG. The SA3800 Cell Analyzer was used to analyze fluorescence data. The dissociation constant (KD) values were determined using GraphPad Prism 6.
Figure 2.
Determination of the binding affinity of humPMab-117 and NZ-16 using flow cytometry. LN229/PDPN cells were suspended in humPMab-117 (A) or NZ-16 (B) at indicated concentrations, followed by treatment with FITC-conjugated anti-human IgG. The SA3800 Cell Analyzer was used to analyze fluorescence data. The dissociation constant (KD) values were determined using GraphPad Prism 6.
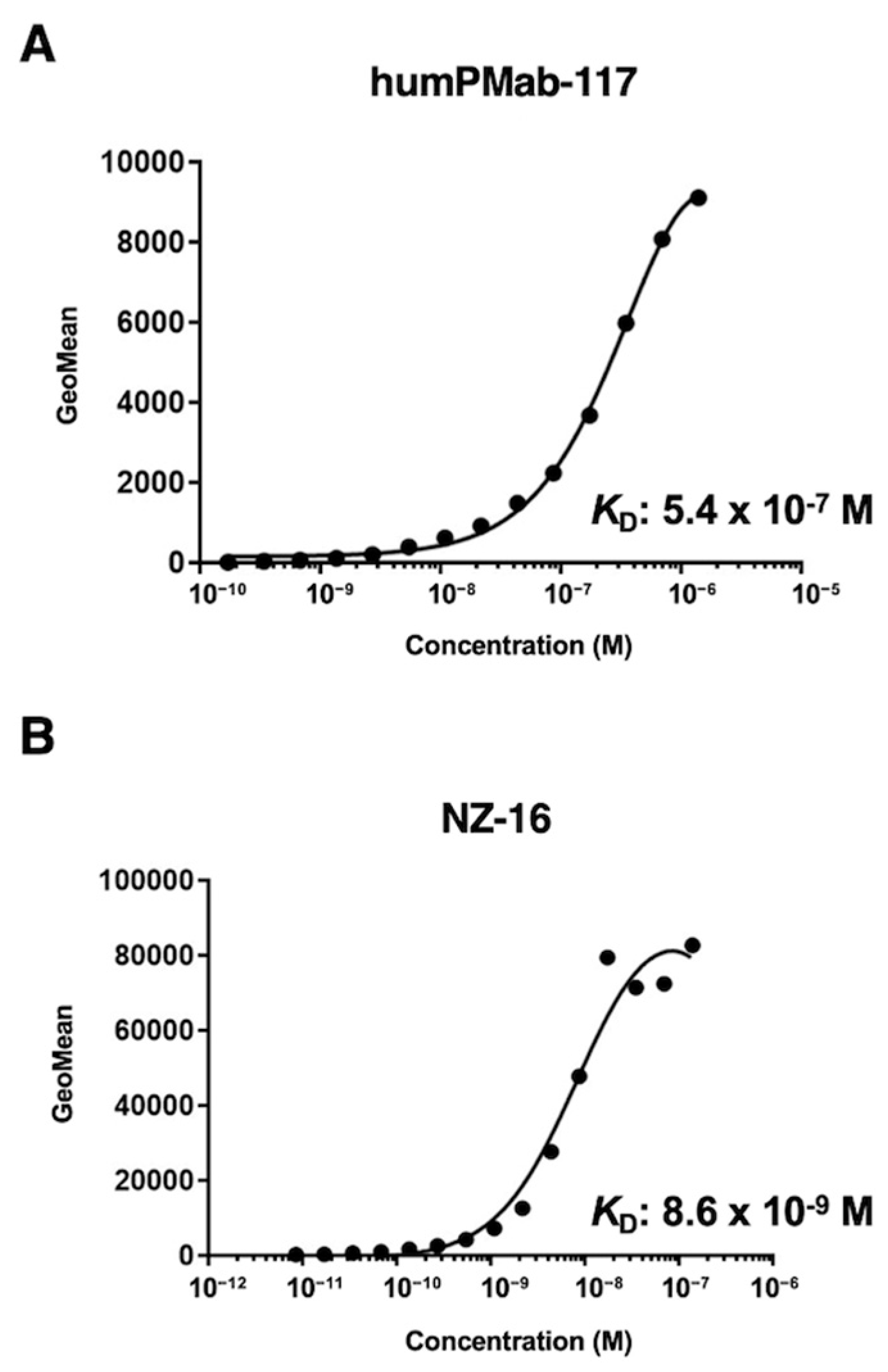
Figure 3.
ADCC activity by humPMab-117 against PDPN-positive cells. The target cells labeled with Calcein AM (LN229/PDPN, PC-10, and LN319) were incubated with human NK cells in the presence of humPMab-117 or control human IgG1 (hIgG1). The ADCC activities against LN229/PDPN (A), PC-10 (B), and LN319 (C) cells were determined by the calcein release. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (* p < 0.05; two-tailed unpaired t-test).
Figure 3.
ADCC activity by humPMab-117 against PDPN-positive cells. The target cells labeled with Calcein AM (LN229/PDPN, PC-10, and LN319) were incubated with human NK cells in the presence of humPMab-117 or control human IgG1 (hIgG1). The ADCC activities against LN229/PDPN (A), PC-10 (B), and LN319 (C) cells were determined by the calcein release. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (* p < 0.05; two-tailed unpaired t-test).
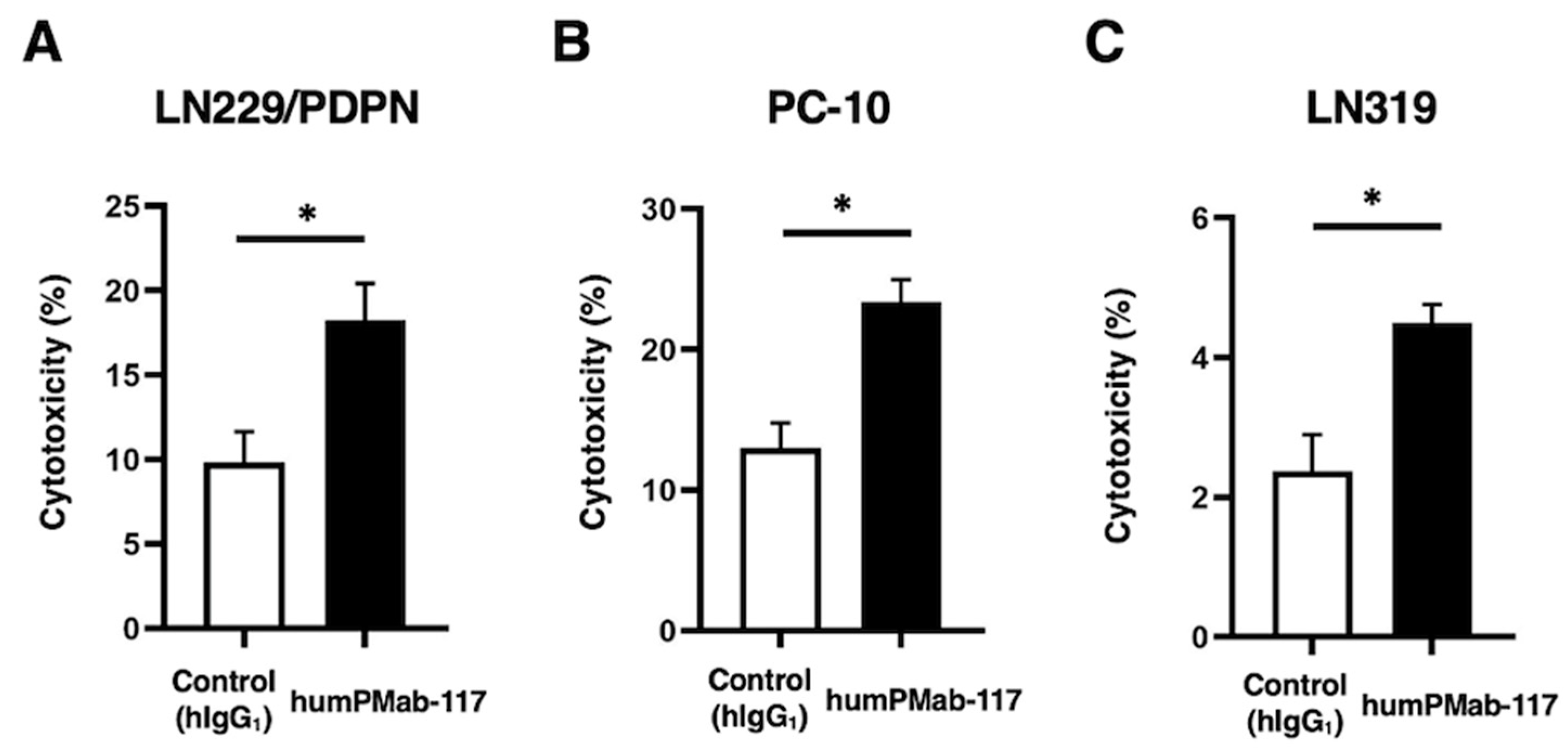
Figure 4.
CDC activity by humPMab-117 against PDPN-positive cells. The target cells labeled with Calcein AM (LN229/PDPN, PC-10, and LN319) were incubated with rabbit complement in the presence of humPMab-117 or control human IgG1 (hIgG1). The CDC activities against LN229/PDPN (A), PC-10 (B), and LN319 (C) cells were determined by the calcein release. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (** p < 0.01 and * p < 0.05; two-tailed unpaired t-test).
Figure 4.
CDC activity by humPMab-117 against PDPN-positive cells. The target cells labeled with Calcein AM (LN229/PDPN, PC-10, and LN319) were incubated with rabbit complement in the presence of humPMab-117 or control human IgG1 (hIgG1). The CDC activities against LN229/PDPN (A), PC-10 (B), and LN319 (C) cells were determined by the calcein release. Values are shown as the mean ± SEM. Asterisks indicate statistical significance (** p < 0.01 and * p < 0.05; two-tailed unpaired t-test).
Figure 5.
Antitumor activity of humPMab-117 against human tumor xenografts. (A–C) LN229/PDPN (A), PC-10 (B), and LN319 (C) cells were subcutaneously injected into BALB/c nude mice (day 0). humPMab-117 (100 μg) or control human IgG1 (hIgG1, 100 μg) were intraperitoneally injected into each mouse on days 8, 15, and 22 (arrows). The tumor volume is represented as the mean ± SEM. ** p < 0.01 (two-way ANOVA with Sidak’s post hoc test). (D–F) After cell inoculation, the mice were euthanized on day 25. The tumor weights of LN229/PDPN (D), PC-10 (E), and LN319 (F) xenografts were measured. Values are presented as the mean ± SEM. ** p < 0.01 and * p < 0.05 (two-tailed unpaired t-test). (G–I) LN229/PDPN (G), PC-10 (H), and LN319 (I) xenograft tumors on day 25 (scale bar, 1 cm). (J–L) Body weights of LN229/PDPN (J), PC-10 (K), and LN319 (L) xenograft-bearing mice treated with control hIgG1 or humPMab-117. The body weight is represented as the mean ± SEM. * p < 0.05 (two-way ANOVA with Sidak’s post hoc test).
Figure 5.
Antitumor activity of humPMab-117 against human tumor xenografts. (A–C) LN229/PDPN (A), PC-10 (B), and LN319 (C) cells were subcutaneously injected into BALB/c nude mice (day 0). humPMab-117 (100 μg) or control human IgG1 (hIgG1, 100 μg) were intraperitoneally injected into each mouse on days 8, 15, and 22 (arrows). The tumor volume is represented as the mean ± SEM. ** p < 0.01 (two-way ANOVA with Sidak’s post hoc test). (D–F) After cell inoculation, the mice were euthanized on day 25. The tumor weights of LN229/PDPN (D), PC-10 (E), and LN319 (F) xenografts were measured. Values are presented as the mean ± SEM. ** p < 0.01 and * p < 0.05 (two-tailed unpaired t-test). (G–I) LN229/PDPN (G), PC-10 (H), and LN319 (I) xenograft tumors on day 25 (scale bar, 1 cm). (J–L) Body weights of LN229/PDPN (J), PC-10 (K), and LN319 (L) xenograft-bearing mice treated with control hIgG1 or humPMab-117. The body weight is represented as the mean ± SEM. * p < 0.05 (two-way ANOVA with Sidak’s post hoc test).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



