
Submitted:
26 March 2025
Posted:
27 March 2025
You are already at the latest version
Abstract
Evidence supporting the clinical use of neuroprotective drugs for cerebral ischemia remains limited. Spatial and temporal disorientation, along with cognitive dysfunction, are among the most prominent long-term consequences of hippocampal neurodegeneration following cerebral ischemia. Cannabigerol (CBG), a non-psychotomimetic constituent of Cannabis sativa, has demonstrated neuroprotective effects in experimental models of cerebral injury. This study investigated the neuroprotective mechanisms of CBG in mitigating memory impairments caused by transient global cerebral ischemia in C57BL/6 mice using the bilateral common carotid artery occlusion (BCCAO) model. Mice underwent sham or BCCAO surgeries and received intraperitoneal (i.p.) injections of either a vehicle or CBG (1, 5, or 10 mg/Kg), starting 1 h post-surgery and continuing daily for 7 days. Spatial memory performance and depression-like behaviors were assessed using the object location test (OLT) and tail suspension test (TST), respectively. Additional analyses examined neuronal degeneration, neuroinflammation, and neuronal plasticity markers in the hippocampus. CBG attenuated ischemia-induced memory deficits, reduced neuronal loss in the hippocampus, and enhanced neuronal plasticity. These findings suggest that CBG’s neuroprotective effects against BCCAO-induced memory impairments may be mediated by reductions in neuroinflammation and modifications in neuroplasticity within the hippocampus.
Keywords:
Bilateral common carotid artery occlusion
; Glial response
; Cognition
; Neuroprotection
; Cannabigerol
1. Introduction
Interest in the pharmacotherapeutic potential of Cannabis sp. and its constituents has grown significantly in recent years worldwide [1]. Among more than 100 psychoactive compounds isolated from Cannabis sativa, cannabigerol (CBG) has stood out as a promising phytocannabinoid, due to its lack of psychotomimetic effects and its demonstrated anti-inflammatory, antioxidant and potentially neuroprotective properties [for review see 1,2]. In particular, CBG is also highly lipophilic and easily crosses the blood brain barrier (BBB), making it an attractive compound with plausible positive effects in central nervous system (CNS) diseases [3].
The pharmacological effects of CBG have been associated with the reduction of neuroinflammation and oxidative stress. In vitro, CBG defended neuroblastoma spinal cord (NSC)-34 motor neurons from the toxicity induced by lipopolysaccharide (LPS)-stimulated RAW 264.7, reduced apoptosis, and increased anti-apoptosis protein BcL-2 expression. In the same model, CBG pre-treatment reduced the interleukin (IL)-1β, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and peroxisome proliferator-activated receptor (PPAR)-γ protein levels [4]. CBG exerted antioxidant effects and reduced apoptosis in rat CTX-TNA2 astrocytes exposed to hydrogen peroxide (H2O2) and restored the serotonin level depleted by neurotoxic stimuli in isolated cortexes [5]. In an in vivo model of Huntington’s disease, CBG improved motor deficits, conferred neuroprotection and attenuated microgliosis [6]. Furthermore, a recent study has revealed that CBG promotes gene expression in initiating cytoskeletal remodeling and axon guidance [7]. All these findings indicate that CBG has potential as a neuroprotective agent. However, few studies using in vivo models of CNS injuries have been conducted using CBG.
The bilateral common carotid artery occlusion (BCCAO) is an animal model of transient global cerebral ischemia (TGCI) and has been used to study neuroprotective therapies for cerebral ischemic conditions [8,9,10]. BCCAO triggers multiple and interconnected pathological events, including increased oxidative stress, neuroinflammation, BBB disruption, and white matter (WM) injury [8,9,11]. A few minutes of BCCAO can produce extensive neuronal loss and impact synaptic plasticity in vulnerable areas of the brain such as the hippocampus [8,9,12]. Alongside neurodegenerative processes, however, restorative mechanisms such as reperfusion, vasculogenesis, neurogenesis, and dendritic remodeling may occur to protect the brain tissue and enable recovery [13]. From a functional point of view, BCCAO in mice results in cognitive deficits and increased anxiety- and depression-like behaviors [8,9,14,15,16]. To the best of our knowledge, no study to date has examined the effects of CBG on the cognitive and pathological responses induced by cerebral ischemia, including the BCCAO model.
The present study aimed to investigate the effects of treatment with CBG on the functional and morphological consequences of TGCI using the BCCAO model in mice. The effects of CBG treatment on behavioral performance were first assessed using the object location test (OLT) and the tail suspension test (TST). Subsequently, we evaluated the impact of the CBG on the expression of proteins associated with neuroinflammation, reflected by microglial and astrocytic activation, using ionized calcium-binding adaptor molecule 1 (Iba-1) and glial fibrillary acidic protein (GFAP), respectively. Glial precursor cells were analyzed through the expression of neural/glial antigen 2 (NG2). We also examined the effects of CBG in ischemic mice on markers of neuronal integrity, including neuronal nuclear protein (NeuN) and microtubule-associated protein 2 (MAP-2), as well as markers of neuroplasticity, such as doublecortin (DCX) and brain-derived neurotrophic factor (BDNF).
2. Results
2.1. CBG Improves Memory Impairment Induced by BCCAO in Mice
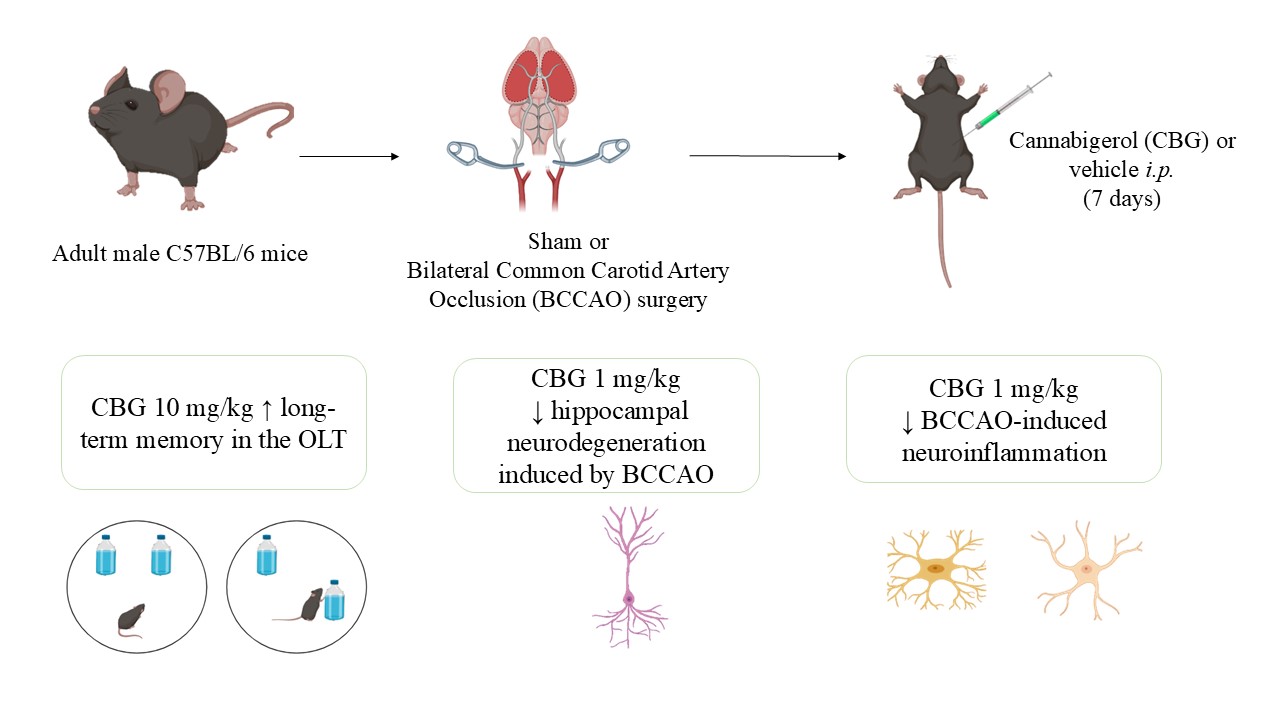
Figure 1 show the effects of BCCAO on the behavior of animals subjected to OLT and TST. In the OLT (Figure 1a), a significant main effect of group was found at 24-h interval (F4, 53 = 3.50; P = 0.013) but not at 1-h and 4-h (F4, 53 – 57 = 0.90 – 1.95; P > 0.05) intervals. At 24-h interval, the BCCAO + vehicle (Veh) group presented the lowest D2 scores compared with the sham + Veh group (p = 0.016), indicating impairment in long-term spatial memory. This memory impairment was significantly inhibited by CBG at a dose of 10 mg/Kg (CBG10) in comparation with the BCCAO + Veh (p = 0.038). When the D2 index in the 24-h interval was analyzed within the group, it was demonstrated to differentiate from zero in the sham + Veh and BCCAO + CBG10 groups (t = 2.32 – 2.99; P = 0.013 – 0.048), indicating that the spatial memory was recovered by CBG10 to the level of sham. The same did not occur for the groups BCCAO treated with the Veh, CBG at a dose of 1 mg/Kg (CBG1) or CBG at a dose of 5 mg/Kg (CBG5) (t = 0.90 – 1.85; P = 0.084 – 0.380). In the TST (Figure 1b), no between-group differences were detected in the latency for the first episode of immobility (F4, 55 = 1.332; P = 0.27) and the immobility time (F4, 55 = 0.943; P = 0.446).
2.2. CBG Decreases Hippocampal Neurodegeneration Induced by BCCAO
Hippocampal damage was evaluated by analyzing NeuN-immunoreactive (NeuN-IR) and MAP-2-immunoreactive (MAP-2-IR) in the CA1 and CA3 subfields 14 days after sham or BCCAO surgery (Figure 2). A main effect of group was found for NeuN-IR measurements in the CA1 and CA3 subfields (F4, 23 = 5.86 – 11.18; P < 0.0001 – 0.01), indicating the loss of adult pyramidal neurons (Figure 2c and 2d). Compared to the sham group, these neurons were significantly reduced in the CA1 and CA3 subfields of the ischemic, Veh-treated group (p < 0.001). CBG1 and CBG5 decreased this neurodegenerative effect of BCCAO in the CA1 subfield (p < 0.01). In the CA3 subfield, however, the neuronal rescue by CBG did not reach statistical significance at 5% level (p > 0.05).
As shown in Figure 2e and 2f, a main effect of group for the integrated optical density (IOD) of MAP-2-IR neurons was found in the stratum radiatum of the CA1 subfield (F4, 23 = 13.13; P < 0.0001), but not in the stratum lucidem of the CA3 subfield (F4, 24 = 1.397; P = 0.265). In the stratum radiatum, the IOD of MAP-2-IR was significantly reduced in the BCCAO + Veh group compared with the sham + Veh group (p = 0.0004), indicating dendritic degeneration. This degeneration and neurohistological sequelae were prevented of by CBG1 (p = 0.020 vs. Veh). The other two doses had no effect (p > 0.05).
2.3. CBG Reduces BCCAO-Induced Neuroinflammation
Neuroinflammation was assessed by analyzing the expression of Iba-1 (microglia), GFAP (astrocytes), and NG2 (glial cell precursor) in the CA1 and CA3 subfields of the hippocampus (Figure 3). The analysis of variance (ANOVA) revealed a main effect of group for all these three markers, in both the CA1 (F4, 23 – 24 = 5.38 – 8.92; P < 0.001 – 0.01) and CA3 (F4, 21 – 24 = 3.60 – 15.56; P < 0.0001 – 0.05). Compared to sham surgery, the BCCAO surgery increased the Iba-1-immunoreactive (Iba-1-IR) and the GFAP-immunoreactive (GFAP-IR) in both the CA1 (p < 0.001 - 0.01; Figure 3d and 3f) and CA3 subfields (p < 0.001 - 0.05; Figure 3e and 3g). CBG1 prevented these effects of ischemia in these hippocampal subfields (p < 0.01 – 0.05). In contrast to the Iba-1-IR and GFAP-IR, the NG2-immunoreactive (NG2-IR) cells were reduced in the BCCAO + Veh group, also in the CA1 and CA3 regions (p < 0.001 – 0.01; Figure 3h and 3i). This effect of ischemia on NG2-IR in the CA1 and CA3 hippocampus was abolished by treatment with CBG1 (p < 0.001 - 0.05).
2.4. The Impact of CBG on Ischemia-Induced Neuroplasticity
Neuronal plasticity in the hippocampus was assessed based on the quantity of newly generated neurons expressing DCX and the concentrations of BDNF and its precursor, proBDNF (Figure 4). The ANOVA revealed a main effect of group on the number of DCX-immunoreactive (DCX-IR) neurons within the subgranular zone (SGZ) and granular cell layer (GCL) regions of the dentate gyrus (F4, 21 = 9.67; P < 0.001) (Figure 4b). BCCAO increased the number of DCX-IR neurons (p < 0.01 vs. sham), indicating a compensatory response to ischemia. This effect was not altered by CBG (p > 0.05 vs. Veh). A main effect of group was also observed for BDNF (F4, 19 = 8.614; P < 0.001), but not for the proBDNF levels (F4, 23 = 2.31; P > 0.05) (Figure 4d and 4e). In animals subjected to BCCAO + Veh, BDNF levels were increased compared to the sham + Veh group (p < 0.01). CBG1, CBG5, and CBG10 prevented the increase in BDNF levels when compared with the BCCAO + Veh group (p < 0.01). When considering the ratio between BDNF and proBDNF levels (Figure 4f), there was a significant difference among groups (F4,23 = 3.274; P = 0.029). Although there was an increase in the ratio in BCCAO + Veh group in comparison to sham + Veh group, this difference did not reach statistical significance (p = 0.091). CBG10 reduced the ratio when compared to BCCAO + Veh group (p = 0.022).
3. Discussion
This study evaluates the neuroprotective effects of CBG on behavioral, histological, and biochemical outcomes in a murine model of TGCI induced by BCCAO. To our knowledge, this is the first in vivo investigation demonstrating CBG’s protective properties in ischemic brain injury. BCCAO caused memory deficits, hippocampal neurodegeneration and neuroinflammation. As a compensatory response, BCCAO also increased the expression of BDNF and DCX in the hippocampus of ischemic animals. CBG attenuated memory deficits and neuroinflammation in the hippocampus. Additionally, CBG protected neurons against BCCAO-induced neurodegeneration and restored neuroplasticity markers in the hippocampus.
When tested in the OLT, sham-operated mice successfully discriminated novel from familiar locations at 1, 4, and 24 h post-training (D2 index > 0), reflecting intact spatial memory. In contrast, the BCCAO + Veh group exhibited memory impairment (D2 index < 0), reaching statistical significance only at the 24-h interval. These results differ from previous studies, where BCCAO-induced deficits were observed at 1 and 4 h but not at 24 h [12,16,17]. Although BCCAO did not significantly affect memory at earlier intervals in the present study, the D2 index approached zero, suggesting impaired discrimination in ischemic mice. The discrepancy between studies may stem from differences in stress induced by intraperitoneal (i.p.) injections. In previous studies, injections were administered daily from surgery [12] or pre-surgery [16] until 21–27 days post-ischemia, whereas in the present study, injections ceased on day 7, avoiding overlap with behavioral assessments. This procedural variation may account for differences in ischemia-induced memory deficits. CBG10 significantly mitigated BCCAO-induced memory impairment in the OLT at 24 h post-training, whereas lower doses (CBG1 and CBG5) had no effect. This neuroprotective role of CBG has not been previously documented in in vivo models of cerebral ischemia. Accordingly, CBG produced motor improvements and striatal neuron preservation in a Huntington’s disease model induced by 3-nitropropionate [6]. Moreover, Fleisher-Berkovich et al. (2023) demonstrated reduced neurological deficits attenuation of neuronal loss in a multiple sclerosis model [18]. Collectively, these findings suggest CBG may contribute to functional recovery following brain injury.
BCCAO did not induce significant behavioral changes in mice when assessed in the TST 14 days post-insult. No comparable studies were available, but Hu et al. (2023) reported increased immobility in a BCCAO model followed by 14-day chronic unpredictable stress [19]. In that study, the immobility time increased significantly in comparison to the sham-operated mice, indicating a depressive-like behavior. However, their study lacked a BCCAO-only group for direct comparison.
BCCAO induced significant neuronal loss in the CA1 and CA3 hippocampal subfields, as evidenced by NeuN-IR. CBG10 alleviated BCCAO-induced memory deficits, although this effect was absent at lower doses (CBG1 and CBG5). However, neuroprotection in CA1 neurons was observed at CBG1 and CBG5 but not at CBG10. CBG-mediated neuronal preservation in CA1 was not detected in the CA3 region. The reason for this selective protection in different hippocampal subfields is unknown. In fact, neurons in the CA1 subfield are more vulnerable to the effects of ischemia [20]. However, establishing a direct relationship between hippocampal damage and cognitive impairment or between neuronal rescue and functional recovery, following global ischemia remains challenging [21]. This complexity likely stems from dysfunction and recovery occurring at subcellular, synaptic, or electrophysiological levels rather than solely from morphological changes measurable by cell counts [22].
Neuroinflammation is a central aspect of the brain ischemia injury that includes the glial activation and rapid synthesis of cytokines and chemokines, which are closely associated with ischemia-induced neurodegeneration [8,23,24,25]. Our analysis demonstrated a persistent glial response following BCCAO in mice as shown by an increase in the Iba-1-IR and GFAP-IR in the hippocampus 14 days post-BCCAO. CBG1 prevented both astroglial and microglial responses in both CA1 and CA3 hippocampal subfields of BCCAO mice. Since CBG was administered in the first 7 days after BCCAO, the findings indicate that the neuroprotective effect of CBG occurred in the acute/initial phase of ischemia and that it promoted a sustained anti-inflammatory action.
Previous evidence of CBG-mediated neuroprotection has been predominantly derived from in vitro studies, including models such oxygen-glucose deprivation-induced injury in BBB cell cultures to simulate stroke-like conditions [2], lipopolysaccharide-induced neurotoxicity in spinal cord motor neuron-derived neuroblastoma cells, and excitotoxicity induced by MTT assay [4]. CBG’s anti-inflammatory effects may also stem from its antioxidant activity, countering ischemia-induced oxidative stress [26,27]. It inhibits H₂O₂-induced stress in murine macrophages by downregulating oxidative signals, such as inducible nitric oxide synthase (iNOS), nitrotyrosine, PPAR-1, blocking nuclear factor kappa B (NF-κB) activation, and modulating the mitogen-activated protein kinase (MAPK) pathway. Additionally, CBG enhances antioxidant defense by regulating superoxide dismutase 1 (SOD-1), preventing cell death [28].
In this study, BCCAO decrease the expression of NG-2-positive cells in the hippocampus of ischemic mice. NG2 glial cells proliferate and generate oligodendrocytes throughout life [29]. While mainly oligodendrocyte precursors, some studies suggest their potential differentiation into astrocytes or neurons [30,31]. Sugimoto et al. (2014) reported Iba-1 plus NG2-positive cells accumulation at the peri-infarct/core lesion boundary in a rat stroke model, highlighting NG2-positive cells' diverse roles [32]. We observed reduced NG2-IR in CA1 and CA3 of BCCAO mice 14 days post-ischemia, aligning with studies showing an initial NG2 increase followed by a decline after 2–3 weeks [33,34]. Herein, CBG1 restored NG2 expression to sham levels, possibly linked to GFAP changes, suggesting NG2-positive cell recruitment for astrocyte/oligodendrocyte differentiation. Indeed, a transient astrocyte-like NG2 glia subpopulation may emerge after focal cerebral ischemia, resembling cortical astrocytes and potentially contributing to glial scar formation and repair [35]. Whether CBG’s anti-inflammatory effects modulates this process in the BCCAO model, warranting further investigation.
Hippocampal neuroplasticity is a fundamental mechanism of learning and memory. For example, following a cerebral ischemic episode, hippocampal neurogenesis occurs to compensate for damaged cells and neural pathways, typically resulting in the emergence of new neurons in the hippocampal dentate gyrus [36]. In line, ischemic mice showed increased DCX-IR in the SGZ and GCL of the dentate gyrus 14 days post-BCCAO, suggesting compensatory neurogenesis. BCCAO animals also showed elevated BDNF expression, potentially linked to increase of newborn neurons identified as DCX-positive cells [37]. CBG reduced BDNF expression to baseline despite the neurogenesis increase, highlighting the need for further investigation.
MAP-2, crucial for dendritic structure and function, is also a marker of neuronal integrity [38,39]. Its reduction in cerebral ischemia indicates neurodegeneration [40], while increased expression weeks later suggests dendritic recovery and neuroplasticity, linked to memory improvement despite neuronal loss [41]. MAP-2-IR decreased in the CA1 hippocampal subfield of BCCAO mice, but CBG1 significantly attenuated this loss. Moreover, CBG1 provided qualitative dendritic protection, suggesting CBG may prevent degeneration and/or promote regeneration.
One limitation of this study is the lack of investigation into the pharmacological mechanisms underlying CBG's effects in mice subjected to BCCAO. However, CBG appears to act through multiple pharmacological targets. CBG may function as a weak or partial agonist of cannabinoid receptors CB1 and CB2 [42], an agonist of PPARɣ receptors [43], and/or an agonist of transient receptor potential (TRP) channels [44]. Additionally, it acts as an antagonist of the serotonin 1A receptor (5-HT1A) and an agonist of the α2-adrenergic receptor [45]. The fact that CBG treatment had protective effects lasting 14 days after BCCAO implies a promising therapeutic action of this compound against the long-term consequences of cerebral ischemia. Many studies still need to be performed to better understand the biological properties of CBG on other pathophysiological events of cerebral ischemia, in addition to its mechanisms of action.
4. Materials and Methods
4.1. Animals
Male C57BL/6 mice with 2- to 3-months-old (approximately 30 g) were used. The animals were maintained under standard housing conditions with a 12-hour light/dark cycle, controlled temperature (22 ± 1°C), and had free access to water and commercial chow diet (Nutrilab-CR1®; Nuvital Nutrients, Curitiba, Brazil). All experimental procedures were conducted under the approval of the Animal Ethics Committee of the State University of Maringá (CEUA Nº 4320080223).
4.2. Drugs
CBG (PurMed Global, USA) was dissolved in 3% Tween 80 (Synth, Diadema, Brazil) and 3% dimethyl sulfoxide (DMSO, Synth, Diadema, Brazil) in sterile saline (Veh). The animals were randomly assigned to receive i.p. injections of Veh, CBG1, CBG5 or CBG10 for 7 days following BCCAO.
4.3. Bilateral Common Carotid Arteries Occlusion (BCCAO)
The surgeries were performed as previously reported [8]. Mice were anesthetized with a mixture of isoflurane/oxygen (1.3%-1.5% isoflurane in 100% oxygen, Isoforine®, Cristália) via a universal vaporizer (Oxigel, São Paulo, Brazil). A ventral neck incision was made to expose the common carotid arteries, which were occluded for 20 min with aneurysm clips (ADCA, Barbacena, Brazil). After the occlusion period, the clips were carefully removed, and the arteries were visually inspected for reperfusion. The incision was then closed with sutures. During the first 3 h post-reperfusion, the animals were kept in a heating box (internal temperature of 30 ± 1°C) to prevent ischemia-induced cerebral hypothermia [46]. The animals were then returned to their cages with free access to water and food. Some mice underwent exposure of the carotid arteries without occlusion and served as non-ischemic controls (sham). All efforts were made to minimize animal suffering.
4.4. Experimental Design
All the animals were operated on and tested in sequence (Figure 5). C57BL/6 mice were distributed into the following experimental groups: sham + Veh (n = 11), BCCAO + Veh (n = 17), BCCAO + CBG1 (n = 12), BCCAO + CBG5 (n = 11) and BCCAO + CBG10 (n = 11). The pharmacological treatment started 1 h after BCCAO, i.e., at 1 h after reperfusion. Veh or CBG was injected i.p. daily, between 2:00 p.m. and 3:00 p.m., for 7 days. The behavioral tests began 9 days after sham or BCCAO surgery and were conducted during the light phase (between 8:00 a.m. and 1:00 p.m.) in a sound-attenuated experimental room kept at 22 ± 2°C. The mice were acclimatized to the experimental room for 30 min before each test. On days 9-12th the animals were tested in OLT and on day 14th in the TST. To mitigate potential bias resulting from residual odors left by previous animals, each apparatus was thoroughly cleaned with 70% ethanol followed by water before testing a new mouse. The test sessions were recorded and analyzed using a contrast-sensitive video tracking system (ANYmaze, Stoelting, Wood Dale, IL, USA). After the behavioral evaluation (day 14th), the animals were euthanized with an excessive dose of sodium thiopental i.p. (Thiopentax®, Cristalia, São Paulo, Brazil) and had their brains appropriately removed for histological and molecular analysis.
4.5. Behavioral Testing
4.5.1. Object Location Test (OLT)
The OLT is designed to evaluate spatial memory and discrimination, in rodent models of CNS disorders. This test leverages rodents' natural tendency to detect changes in the location of objects [47]. It was conducted in an open-field arena where animals were first habituated. The apparatus consisted of a circular arena with a 43 cm diameter and 40 cm high transparent polyvinyl chloride walls. Three distinct sets of objects are used, each one available in triplicate. These objects include: (1) an aluminum cube with a tapering top (4.5 cm × 4.5 cm × 8.5 cm), (2) a 200 mL glass bottle filled with water (5.5 cm in diameter, 15.0 cm in height), and (3) a porcelain cube (9.5 cm × 6.5 cm × 6.5 cm). The objects are securely fixed within the arena to prevent movement by the mice. One week before the BCCAO procedure, the animals were introduced to the OLT. Initially, they can explore the empty arena for two consecutive days (3 min per day) to familiarize themselves with the environment. During the next four days, the mice undergo training with the objects until they achieve consistent performance, demonstrating reliable discrimination at a 1-h interval.
Following BCCAO surgery, the OLT was conducted on days 9-12 post-surgery at 24-h, 4-h, and 1-h intervals between the training phase and the test phase. Each test session consists of two trials, each lasting 3 min. In the first trial (T1), two identical objects are placed in the arena, and the mouse was allowed to explore them. After a set time interval (24 h, 4 h and 1 h), the mouse was returned for a second trial (T2), where one of the objects had been relocated. The amount of time the mouse spent exploring each object during both trials was recorded by an observer that was blinded to the treatment conditions. A discrimination index (D2) was then calculated, reflecting the animal's ability to perceive the object relocation, i.e., spatial memory. The formula was as follows: D2 = (time spent exploring the novel location – time spent exploring the familiar location) / (total exploration time). This measure corrects for total exploration time during T2, ensuring comparability even if treatment affects overall exploration. Exploration is defined as the mouse directing its nose within 1 cm of the object or touching it with its nose, whereas sitting on the object is not considered exploration.
To reduce potential biases, the order of objects, the identity of the relocated object, and the position of relocation are balanced across the experiment and between groups.
4.5.2. Tail Suspension Test (TST)
The TST was used to assess the increased depression-like behaviors. The animals were positioned in the wall of the box with a white background with the aim of adhesive tape. The parameters analyzed were the latency for the first episode of immobility (latency) and the immobility time in the last 4 min of the testing [48]. The camera was positioned in front of the box used to record the animal's behavior. The test lasted 6 min.
4.6. Western Blot
For western blotting analysis, 5-6 animals from each experimental group were randomly taken and anesthetized with sodium thiopental i.p. (Thiopentax®, Cristalia, São Paulo, Brazil). The hippocampus was carefully removed and dissected using spatulas, tweezers, and a table magnifying glass [49]. Each tissue was transferred to an Eppendorf containing homogenization buffer (10% glycerol, NaCl; 137mM, 20mM Tris HCl, pH7.5) with protease inhibitor (Sigma-Aldrich, St. Louis, MO, USA). The material was centrifuged in a refrigerated centrifuge at 12,000 rpm for 10 min. Samples were diluted to reach protein concentrations equal to 3 µg/µL. 30-60 μg of proteins were separated by electrophoresis in 12% SDS-polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane and incubated for 45 min in Tris buffer (25 mM Tris base, 192 mM Glycine, 200 mL methanol, pH 8). Blocking was performed with 3% dehydrated milk in tris buffer saline-tween (TBS-T) at room temperature for 1 h. Then, the membranes were incubated with primary antibody in TBS-T (Table 1) overnight at 4°C. After that, the membranes were incubated for two hours with the horseradish peroxidase (HRP) conjugated rabbit anti-mouse secondary antibody (1:1000, Cat# ab6728, Abcam, Massachusetts, USA). All blots were stripped with harsh stripping buffer (20% SDS 10%, 12.5% Tris HCl 0.5M, and 0.8% β-mercaptoethanol in H2O), to assess the protein control β-actin. The revelation was performed with an ECLplus® chemiluminescence kit (Invitrogen, Carlsbad, CA, USA) and the bands were visualized with the aid of Chemi-doc (Bio-Rad Laboratories Inc., Hercules, USA). Specific band intensities were quantified using ImageJ (NIH, Bethesda, MD, USA) and normalized to β-actin protein levels. Results are expressed as the mean ± SEM of the protein level.
4.7. Immunohistochemistry
For immunohistochemistry assays, 5-6 animals of each experimental group were deeply anesthetized with sodium thiopental i.p. (Thiopentax®; Cristália, São Paulo, Brazil) and transcardially perfused with 0.01 M phosphate-buffered saline (PBS) (2 min; 22 mL/min) followed by 4% paraformaldehyde (PFA) in 0.2 M phosphate buffer (PB) (3 min; 22 mL/min). The brains were post-fixed in the same fixative for 2 h and cryoprotected by immersion in a gradient of sucrose solution in 0.01 M PBS (20% and 30%) for 24 h each at 4 ºC. The brains were then quickly frozen in liquid nitrogen and stored at -80°C. Frozen brains were sectioned coronally at 40 μm using a cryostat (Criocut 1800, Reichert-Jung, Heidelberg, Germany). Thirty-six sections of the hippocampus were collected in six alternating tubes with cryoprotectant solution (15% sucrose and 30% ethylene glycol in PB) and stored at -24°C until further processing. The sections corresponded to the stereotaxic coordinates (-1.70 to -2.46 mm from Bregma), according to the atlas of Paxinos and Franklin (2004) [50].
Sections were washed 3 times in PBS to remove cryoprotectant. Endogenous peroxidase activity was blocked in 1% H2O2 (Merck, Darmstadt, Germany) in PBS for 30 min. After washing in PBS with 0.3% Triton X-100 (PBST), sections were incubated with 2% bovine serum albumin (BSA; Sigma-Aldrich, Darmstadt, Germany) in PBST for 60 min at room temperature. Following 3 PBST washes, sections were incubated overnight under constant stirring at 4°C with primary antibodies (Table 1) overnight. Sections were then incubated with biotinylated anti-rabbit secondary antibody (1:500, Cat# sc-2491, Santa Cruz Biotechnology, California, USA) for 2 h, followed by avidin–biotin complex solution (ABC Kit, Vector Laboratories, California, USA) for 2 h at room temperature. Peroxidase color reaction was developed using 0.025% solution of 3,3'-diaminobenzidine (DAB; Sigma-Aldrich, Darmstadt, Germany) and 0.05% H2O2 with NiCl2 for enhanced contrast. The sections were properly washed in 0.01 M PBS and mounted on previously gelatinized slides. Dehydration and diaphanization processes with xylene were performed previously to cover the slides with Permount® (Fisher Scientific, New Jersey, USA) and coverslips.
Quantitative analysis was performed using a Leica DM 2500 microscope with a Leica DF345 FX camera. The number of DCX-IR neurons was manually quantified in the SGZ and GCL of the dentate gyrus in both the right and left brain hemispheres. The results are expressed as the mean ± SEM of 3-4 sections per animal. For NeuN-IR, MAP-2-IR, Iba-1-IR, GFAP-IR, and NG2-IR, digital microscopic areas were captured bilaterally for the CA1 (0.12 mm2) and CA3 (0.15 mm2) hippocampal subfields in 3-4 sections per animal. Lighting conditions and magnifications were kept constant to avoid signal saturation. ImageJ software was used to obtain IOD [51,52]. The selected images were converted to 16-bit grayscale and the background was subtracted. The threshold for a positive signal was predefined and the IOD was calculated. IOD values were calculated and presented as mean ± SEM of the IOD/area for each experimental group, expressed as a percentage of the sham group.
4.8. Statistical Analysis
Statistical analyses were performed using the GraphPad Prism 8.0.2 sotfware. Both the behavioral, immunohistochemistry, and western blot data fit the assumptions of normal distribution (D'Agostino & Pearson or Shapiro–Wilk tests) and homoscedasticity (Bartlett’s test). Therefore, the one-way analysis of variance (ANOVA) followed by Sidak’s post hoc test were used for between-group comparisons. Additionally, in the TLO test, the paired one-sample t-test was used for within-group evaluation of spatial memory by computing the D2 discrimination index values that differ significantly from 0. Statistical significance was set at p < 0.05.
Author Contributions
NA Kohara performed the animals’ surgeries, CBG treatment, behavioral testing, and western blot analysis. JGP Carrasco, LFF Miranda and PP Quini assisted with animal care and histological procedures. EDB Guimarães provided the CBG and analyzed the histological data. H Milani analyzed the data and assisted with statistical analysis and manuscript correction. RMW Oliveira designed the experiments, planned the study, and assisted with manuscript correction. CC Bacarin performed the animals’ surgeries, immunohistochemistry analysis, processed the data, and wrote the first draft of the manuscript. All authors contributed to the discussion of results and provided feedback on the manuscript.
Funding
This research was funded by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação Araucária - NAPI, and State University of Maringá (UEM).
Institutional Review Board Statement
The animal study protocol was approved by Ethics Committee of State University of Maringá (protocol code 4320080223, approved on February 17, 2023).
Acknowledgments
The authors gratefully acknowledge the Dr. Elaine Del Bel Guimarães to provide the CBG to conduct this study.
Conflicts of Interest
There is no conflict of interest in this study.
Abbreviations
The following abbreviations are used in this manuscript:
| 5-HT1A ANOVA BBB BDNF BCCAO BSA CNS CBG CBG1 CBG5 CBG10 |
Serotonin 1A receptor Analysis of variance Blood brain barrier Brain-derived neurotrophic factor Common carotid artery occlusion Bovine serum albumin Central nervous system Cannabigerol CBG at a dose of 1 mg/Kg CBG at a dose of 5 mg/Kg CBG at a dose of 10 mg/Kg |
| DAB DCX DMSO GCL GFAP IFN IL Iba-1 iNOS IOD i.p. IR H2O2 HRP PPAR PBS PBST PB PFA MAP-2 MAPK NaCl NeuN NiCl2 NF-κB NG2 NSC OLT SGZ SOD-1 TBST-T TGCI |
3,3'-diaminobenzidine Doublecortin Dimethyl sulfoxide Granular cell layer Glial fibrillary acidic protein Interferon Interleukin Ionized calcium-binding adaptor molecule 1 Inducible nitric oxide synthase Integrated optical density Intraperitoneal Immunoreactive Hydrogen peroxide Horseradish peroxidase Peroxisome proliferator-activated receptor Phosphate-buffered saline PBS with 0.3% Triton X-100 Phosphate buffer Paraformaldehyde Microtubule-associated protein 2 Mitogen-activated protein kinase Sodium chloride Neuronal nuclear protein Nickel chloride Nuclear factor kappa B Neural/glial antigen 2 Neuroblastoma spinal cord Object location test Subgranular zone Superoxide dismutase 1 Tris buffer saline-tween Transient global cerebral ischemia |
| TNF | Tumor necrosis factor |
| TRP TST Veh WM |
Transient receptor potential Tail suspension test Vehicle White matter |
References
- Calapai, F.; Cardia, L.; Esposito, E.; Ammendolia, I.; Mondello, C.; Lo Giudice, R.; Gangemi, S.; Calapai, G.; Mannucci, C. Pharmacological Aspects and Biological Effects of Cannabigerol and Its Synthetic Derivatives. Evid. Based Complement. Alternat. Med. 2022, 3336516. [CrossRef]
- Stone, N.L.; England, T.J.; O'Sullivan, S.E. Protective Effects of Cannabidivarin and Cannabigerol on Cells of the Blood-Brain Barrier Under Ischemic Conditions. Cannabis Cannabinoid Res. 2021, 6(4), 315-326. [CrossRef]
- Deiana, S.; Watanabe, A.; Yamasaki, Y.; Amada, N.; Arthur, M.; Fleming, S.; Woodcock, H.; Dorward, P.; Pigliacampo, B.; Close, S.; Platt, B.; Riedel, G. Plasma and Brain Pharmacokinetic Profile of Cannabidiol (CBD), Cannabidivarine (CBDV), Δ⁹-Tetrahydrocannabivarin (THCV) and Cannabigerol (CBG) in Rats and Mice Following Oral and Intraperitoneal Administration and CBD Action on Obsessive-Compulsive Behaviour. Psychopharmacology (Berl.) 2012, 219, 859–873. https://10.1007/s00213-011-2415-0.
- Gugliandolo, A.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. In Vitro Model of Neuroinflammation: Efficacy of Cannabigerol, a Non-Psychoactive Cannabinoid. Int. J. Mol. Sci. 2018, 19, 1992. https://10.3390/ijms19071992.
- di Giacomo, V.; Chiavaroli, A.; Recinella, L.; Orlando, G.; Cataldi, A.; Rapino, M.; Di Valerio, V.; Ronci, M.; Leone, S.; Brunetti, L.; Menghini, L.; Zengin, G.; Ak, G.; Abdallah, H. H.; Ferrante, C. Antioxidant and Neuroprotective Effects Induced by Cannabidiol and Cannabigerol in Rat CTX-TNA2 Astrocytes and Isolated Cortexes. Int. J. Mol. Sci. 2020, 21, 3575. https://10.3390/ijms21103575.
- Valdeolivas, S.; Navarrete, C.; Cantarero, I.; Bellido, M.; Muñoz, E.; Sagredo, O. Neuroprotective Properties of Cannabigerol in Huntington’s Disease: Studies in R6/2 Mice and 3-Nitropropionate-Lesioned Mice. Neurotherapeutics 2015, 12, 185-199. [CrossRef]
- Anchesi, I.; Betto, F.; Chiricosta, L.; Gugliandolo, A.; Pollastro, F.; Salamone, S.; Mazzon, E. Cannabigerol Activates Cytoskeletal Remodeling via Wnt/PCP in NSC-34: An In Vitro Transcriptional Study. Plants 2023, 12(1), 193. [CrossRef]
- Mori, M.A.; Meyer, E.; Soares, L.M.; Milani, H.; Guimarães, F.S.; de Oliveira, R.M.W. Cannabidiol Reduces Neuroinflammation and Promotes Neuroplasticity and Functional Recovery After Brain Ischemia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 94-105. [CrossRef]
- Washida, K.; Hattori, Y.; Ihara, M. Animal Models of Chronic Cerebral Hypoperfusion: From Mouse to Primate. Int. J. Mol. Sci. 2019, 20(24), 6176. [CrossRef]
- León-Moreno, L.C.; Castañeda-Arellano, R.; Rivas-Carrillo, J.D.; Dueñas-Jiménez, S.H. Challenges and Improvements of Developing an Ischemia Mouse Model Through Bilateral Common Carotid Artery Occlusion. J. Stroke Cerebrovasc. Dis. 2020, 29(5), 104773. [CrossRef]
- Rahmati, H.; Momenabadi, S.; Vafaei, A.A.; Bandegi, A.Z.; Mazaheri, Z.; Vakili, A. Probiotic Supplementation Attenuates Hippocampus Injury and Spatial Learning and Memory Impairments in a Cerebral Hypoperfusion Mouse Model. Mol. Biol. Rep. 2019, 46, 4985–4995. [CrossRef]
- Soares, L.M.; Meyer, E.; Milani, H.; Steinbusch, H.W.; Prickaerts, J.; de Oliveira, R.M. The Phosphodiesterase Type 2 Inhibitor BAY 60-7550 Reverses Functional Impairments Induced by Brain Ischemia by Decreasing Hippocampal Neurodegeneration and Enhancing Hippocampal Neuronal Plasticity. Eur. J. Neurosci. 2016a, 45, 510–520. http://dx.doi.org/10.1111/EJN.13461.
- Dirnagl, U. Pathobiology of Injury After Stroke: The Neurovascular Unit and Beyond. Ann. N.Y. Acad. Sci. 2012, 1268, 21-25. [CrossRef]
- Jiwa, N.S.; Garrard, P.; Hainsworth, A.H. Experimental Models of Vascular Dementia and Vascular Cognitive Impairment: A Systematic Review. J. Neurochem. 2010, 115(4), 814-828. [CrossRef]
- Soares, L.M.; Schiavon, A.P.; Milani, H.; de Oliveira, R.M. Cognitive Impairment and Persistent Anxiety-Related Responses Following Bilateral Common Carotid Artery Occlusion in Mice. Behav. Brain Res. 2013, 249, 28–37. [CrossRef]
- Aguiar, R.P.; Soares, L.M.; Meyer, E.; da Silveira, F.C.; Milani, H.; Newman-Tancredi, A.; Varney, M.; Prickaerts, J.; Oliveira, R.M.W. Activation of 5-HT1A Postsynaptic Receptors by NLX-101 Results in Functional Recovery and an Increase in Neuroplasticity in Mice with Brain Ischemia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 99, 109832. [CrossRef]
- Soares, L.M.; De Vry, J.; Steinbusch, H.W.; Milani, H.; Prickaerts, J.; Weffort de Oliveira, R.M. Rolipram Improves Cognition, Reduces Anxiety- and Despair-Like Behaviors and Impacts Hippocampal Neuroplasticity After Transient Global Cerebral Ischemia. Neuroscience 2016b, 326, 69-83. http://dx.doi.org/10.1016/j.neuroscience.2016.03.062.
- Fleisher-Berkovich, S.; Ventura, Y.; Amoyal, M.; Dahan, A.; Feinshtein, V.; Alfahel, L.; Israelson, A.; Bernstein, N.; Gorelick, J.; Ben-Shabat, S. Therapeutic Potential of Phytocannabinoid Cannabigerol for Multiple Sclerosis: Modulation of Microglial Activation In Vitro and In Vivo. Biomolecules 2023, 13, 376. [CrossRef]
- Hu, G.; Zhou, C.; Wang, J.; Ma, X.; Ma, H.; Yu, H.; Peng, Z.; Huang, J.; Cai, M. Electroacupuncture Treatment Ameliorates Depressive-Like Behavior and Cognitive Dysfunction via CB1R Dependent Mitochondria Biogenesis After Experimental Global Cerebral Ischemic Stroke. Front. Cell Neurosci. 2023, 17, 1135227. http://dx.doi.org/10.3389/fncel.2023.1135227.
- Bueters, T.; von Euler, M.; Bendel, O.; von Euler, G. Degeneration of Newly Formed CA1 Neurons Following Global Ischemia in the Rat. Exp. Neurol. 2008, 209(1), 114-124. [CrossRef]
- Bachevalier, J.; Meunier, M. Cerebral Ischemia: Are the Memory Deficits Associated with Hippocampal Cell Loss? Hippocampus 1996, 6, 553–560. https://10.1002/(SICI)1098-1063(1996)6:5<553::AID-HIPO8>3.0.CO;2-J.
- Aronowski, J.; Samways, E.; Strong, R.; Rhoades, H.M.; Grotta, J.C. An Alternative Method for the Quantitation of Neuronal Damage After Experimental Middle Cerebral Artery Occlusion in Rats: Analysis of Behavioral Deficit. J. Cereb. Blood Flow Metab. 1996, 16, 705–713. https://10.1097/00004647-199607000-00022.
- Gehrmann, J.; Bonnekoh, P.; Miyazawa, T.; Hossmann, K.A.; Kreutzberg, G.W. Immunocytochemical Study of an Early Microglial Activation in Ischemia. J. Cereb. Blood Flow Metab. 1992, 12, 257–269. [CrossRef]
- Stoll, M.; Capper, D.; Dietz, K.; Warth, A.; Schleich, A.; Schlaszus, H.; Meyermann, R.; Mittelbronn, M. Differential Microglial Regulation in the Human Spinal Cord under Normal and Pathological Conditions. Neuropathol. Appl. Neurobiol. 2006, 32, 650–661. https://10.1111/j.1365-2990.2006.00774.x.
- Yasuda, Y.; Shimoda, T.; Uno, K.; Tateishi, N.; Furuya, S.; Tsuchihashi, Y.; et al. Temporal and Sequential Changes of Glial Cells and Cytokine Expression During Neuronal Degeneration After Transient Global Ischemia in Rats. J. Neuroinflammation 2011, 8, 70. [CrossRef]
- Collino, M.; Aragno, M.; Mastrocola, R.; Benetti, E.; Gallicchio, M.; Dianzani, C.; Danni, O.; Thiemermann, C.; Fantozzi, R. Oxidative Stress and Inflammatory Response Evoked by Transient Cerebral Ischemia/Reperfusion: Effects of the PPAR-Alpha Agonist WY14643. Free Radic. Biol. Med. 2006, 41, 579–589. https:// 10.1016/j.freeradbiomed.2006.04.030.
- Chehaibi, K.; Trabelsi, I.; Mahdouani, K.; Slimane, M.N. Correlation of Oxidative Stress Parameters and Inflammatory Markers in Ischemic Stroke Patients. J. Stroke Cerebrovasc. Dis. 2016, 25, 2585–2593. [CrossRef]
- Giacoppo, S.; Gugliandolo, A.; Trubiani, O.; Pollastro, F.; Grassi, G.; Bramanti, P.; Mazzon, E. Cannabinoid CB₂ Receptors Are Involved in the Protection of RAW264.7 Macrophages Against Oxidative Stress: An In Vitro Study. Eur. J. Histochem. 2017, 61, 2749. https://10.4081/ejh.2017.2749.
- Hill, R.A.; Nishiyama, A. NG2 Cells (Polydendrocytes): Listeners to the Neural Network with Diverse Properties. Glia 2014, 62(8), 1195–1210. [CrossRef]
- Vigano, F.; Dimou, L. The Heterogeneous Nature of NG2-Glia. Brain Res. 2016, 1638, 129–137. [CrossRef]
- Kirdajova, D.; Anderova, M. NG2 Cells and Their Neurogenic Potential. Curr. Opin. Pharmacol. 2020, 50, 53–60. [CrossRef]
- Sugimoto, K.; Nishioka, R.; Ikeda, A.; Mise, A.; Takahashi, H.; Yano, H.; Kumon, Y.; Ohnishi, T.; Tanaka, J. Activated Microglia in a Rat Stroke Model Express NG2 Proteoglycan in Peri-Infarct Tissue Through the Involvement of TGF-β1. Glia 2014, 62, 185–198. https://10.1002/glia.22598.
- Jin, X.; Riew, T.; Kim, S.; Kim, H.L.; Lee, M. Spatiotemporal Profile and Morphological Changes of NG2 Glia in the CA1 Region of the Rat Hippocampus After Transient Forebrain Ischemia. Exp. Neurobiol. 2020, 29, 50–69. [CrossRef]
- Steliga, A.; Lietzau, G.; Wójcik, S.; Kowiański, P. Transient Cerebral Ischemia Induces the Neuroglial Proliferative Activity and the Potential to Redirect Neuroglial Differentiation. J. Chem. Neuroanat. 2023, 127, 102192. [CrossRef]
- Kirdajova, D.; Valihrach, L.; Valny, M.; Kriska, J.; Krocianova, D.; Benesova, S.; Abaffy, P.; Zucha, D.; Klassen, R.; Kolenicova, D.; Honsa, P.; Kubista, M.; Anderova, M. Transient Astrocyte-Like NG2 Glia Subpopulation Emerges Solely Following Permanent Brain Ischemia. Glia 2021, 69(11), 2658–2681. [CrossRef]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell 2008, 132, 645–660. [CrossRef]
- Feng, B.; Jia, S.; Li, L.; Wang, J.; Zhou, F.; Gou, X.; Wang, Q.; Xiong, L.; Zeng, Y.; Zhong, H. TAT-LBD-Ngn2-Improved Cognitive Functions After Global Cerebral Ischemia by Enhancing Neurogenesis. Brain Behav. 2023, 13(1), e2847. [CrossRef]
- Conde, C.; Caceres, A. Microtubule Assembly, Organization and Dynamics in Axons and Dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [CrossRef]
- DeGiosio, R.A.; Grubisha, M.J.; MacDonald, M.L.; McKinney, B.C.; Camacho, C.J.; Sweet, R.A. More Than a Marker: Potential Pathogenic Functions of MAP2. Front. Mol. Neurosci. 2022, 15, 974890. [CrossRef]
- Matesic, D.; Lin, R. Microtubule-Associated Protein 2 as an Early Indicator of Ischemia-Induced Neurodegeneration in the Gerbil Forebrain. J. Neurochem. 1994, 63. [CrossRef]
- Bacarin, C.C.; Godinho, J.; de Oliveira, R.M.W.; Matsushita, M.; Gohara, A.K.; Cardozo Filho, L.; Lima, J.C.; Previdelli, I.S.; Melo, S.R.; Ribeiro, M.H.M.; Milani, H. Postischemic Fish Oil Treatment Restores Long-Term Retrograde Memory and Dendritic Density: An Analysis of the Time Window of Efficacy. Behav. Brain Res. 2016, 31, 425–439. [CrossRef]
- Navarro, G.; Varani, K.; Reyes-Resina, I.; Sánchez de Medina, V.; Rivas-Santisteban, R.; Sánchez-Carnerero Callado, C.; Vincenzi, F.; Casano, S.; Ferreiro-Vera, C.; Canela, E. I.; Borea, P. A.; Nadal, X.; Franco, R. Cannabigerol Action at Cannabinoid CB₁ and CB₂ Receptors and at CB₁-CB₂ Heteroreceptor Complexes. Front. Pharmacol. 2018, 9, 632. https://10.3389/fphar.2018.00632.
- O’Sullivan, S.E. An Update on PPAR Activation by Cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [CrossRef]
- Müller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2019, 11, 487. [CrossRef]
- Cascio, M.G.; Gauson, L.A.; Stevenson, L.A.; Ross, R.A.; Pertwee, R.G. Evidence that the Plant Cannabinoid Cannabigerol is a Highly Potent Alpha2-Adrenoceptor Agonist and Moderately Potent 5HT1A Receptor Antagonist. Br. J. Pharmacol. 2010, 159(1), 129-141. [CrossRef]
- Seif el Nasr, M.; Nuglisch, J.; Krieglstein, J. Prevention of Ischemia-Induced Cerebral Hypothermia by Controlling the Environmental Temperature. J. Pharmacol. Toxicol. Methods 1992, 27, 23–26. [CrossRef]
- Denninger, J.K.; Smith, B.M.; Kirby, E.D. Novel Object Recognition and Object Location Behavioral Testing in Mice on a Budget. J. Vis. Exp. 2018, 141, 10.3791/58593. [CrossRef]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The Tail Suspension Test: A New Method for Screening Antidepressants in Mice. Psychopharmacology 1985, 85, 367–370. [CrossRef]
- Anuncibay-Soto, B.; Pérez-Rodríguez, D.; Santos-Galdiano, M.; Font-Belmonte, E.; Ugidos, I. F.; Gonzalez-Rodriguez, P.; Regueiro-Purriños, M.; Fernández-López, A. Salubrinal and Robenacoxib Treatment After Global Cerebral Ischemia: Exploring the Interactions Between ER Stress and Inflammation. Biochemical Pharmacology 2018, 151, 26-37. [CrossRef]
- Paxinos, G., & Franklin, K. B. J. (2004). The Mouse Brain in Stereotaxic Coordinates, (2nd ed.); Elsevier Academic Press.
- Haley, M. J.; Lawrence, C. B. The Blood-Brain Barrier After Stroke: Structural Studies and the Role of Transcytotic Vesicles. J. Cereb. Blood Flow Metab. 2017, 37 (2), 456–470. [CrossRef]
- Kho, A. R.; Choi, B. Y.; Lee, S. H.; Hong, D. K.; Lee, S. H.; Jeong, J. H.; Park, K. H.; Song, H. K.; Choi, H. C.; Suh, S. W. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. Int. J. Mol. Sci. 2018, 19 (5), 1420. [CrossRef]
Figure 1.
CBG improves memory impairment induced by BCCAO in mice. (a) Spatial learning and memory performance was analyzed in the OLT using the D2 exploration index at 1, 4, and 24-h intervals. (b) Depression-like behavior was evaluated in the TST by measuring the latency for the first episode of immobility (latency) and the immobility time. The bars represent the mean ± SEM in the different groups (n = 11-17/group). *p < 0.05 vs sham + Veh; #p < 0.05 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
Figure 1.
CBG improves memory impairment induced by BCCAO in mice. (a) Spatial learning and memory performance was analyzed in the OLT using the D2 exploration index at 1, 4, and 24-h intervals. (b) Depression-like behavior was evaluated in the TST by measuring the latency for the first episode of immobility (latency) and the immobility time. The bars represent the mean ± SEM in the different groups (n = 11-17/group). *p < 0.05 vs sham + Veh; #p < 0.05 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
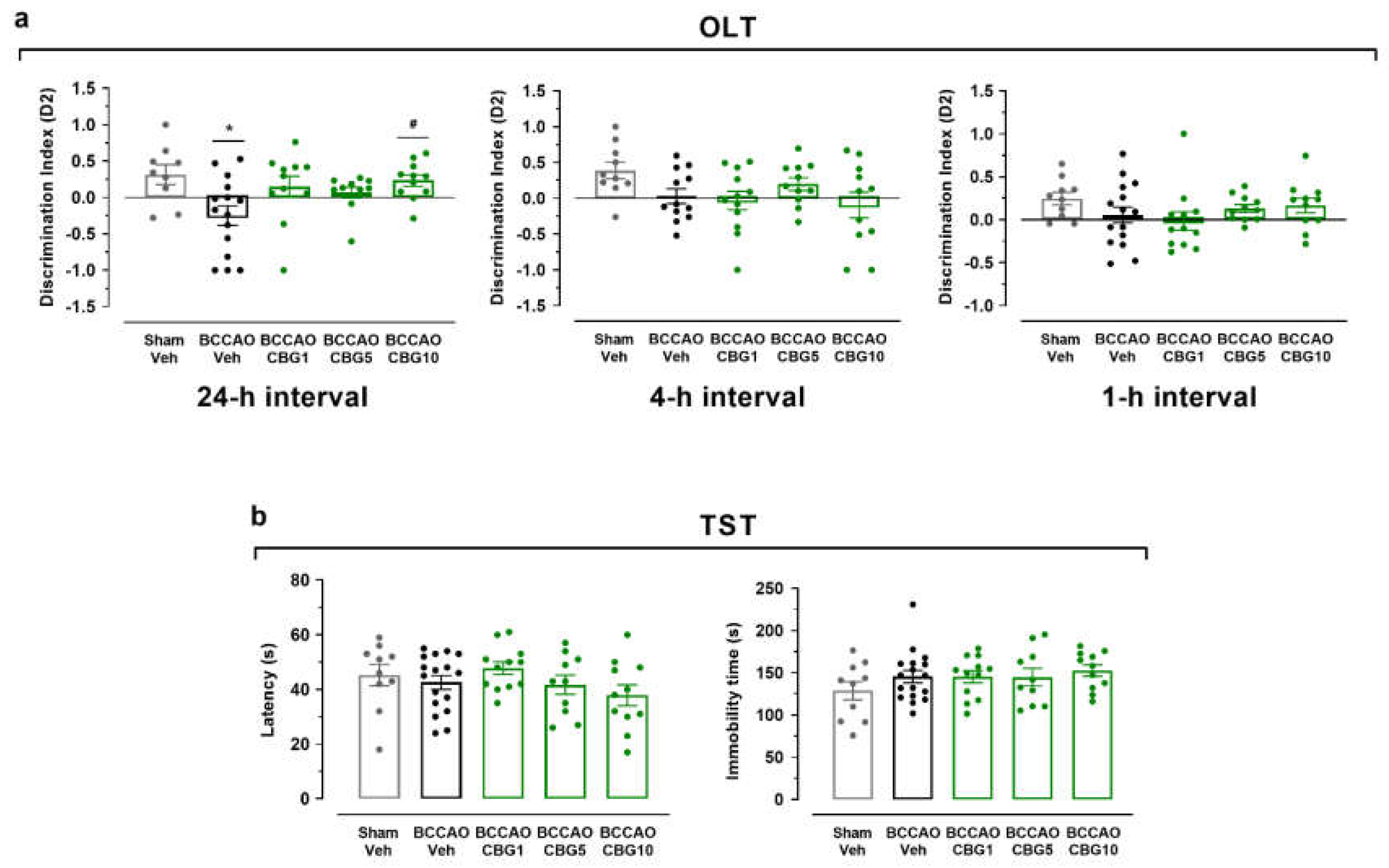
Figure 2.
CBG decreases hippocampal neurodegeneration induced by BCCAO. (a) Representative photomicrographs of the CA1 and CA3 hippocampal subfields (NeuN-IR), indicating intact-appearing neurons (arrows). (b) Representative photomicrographs of MAP-2-IR cells in the stratum radiatum (CA1 subfield) and stratum lucidem (CA3 subfield). The dashed lines delimit the layers of neuronal bodies. (c and d) IOD of the neurons in the CA1 and CA3 subfields of the hippocampus (NeuN-IR). (e and f) IOD of the dendrites (MAP-2-IR) in the CA1/stratum radiatum and CA3/stratum lucidem. The bars represent the mean ± SEM in the different groups (n = 5-6/group). ***p < 0.001 vs sham + Veh; #p < 0.05, ##p < 0.01 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
Figure 2.
CBG decreases hippocampal neurodegeneration induced by BCCAO. (a) Representative photomicrographs of the CA1 and CA3 hippocampal subfields (NeuN-IR), indicating intact-appearing neurons (arrows). (b) Representative photomicrographs of MAP-2-IR cells in the stratum radiatum (CA1 subfield) and stratum lucidem (CA3 subfield). The dashed lines delimit the layers of neuronal bodies. (c and d) IOD of the neurons in the CA1 and CA3 subfields of the hippocampus (NeuN-IR). (e and f) IOD of the dendrites (MAP-2-IR) in the CA1/stratum radiatum and CA3/stratum lucidem. The bars represent the mean ± SEM in the different groups (n = 5-6/group). ***p < 0.001 vs sham + Veh; #p < 0.05, ##p < 0.01 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
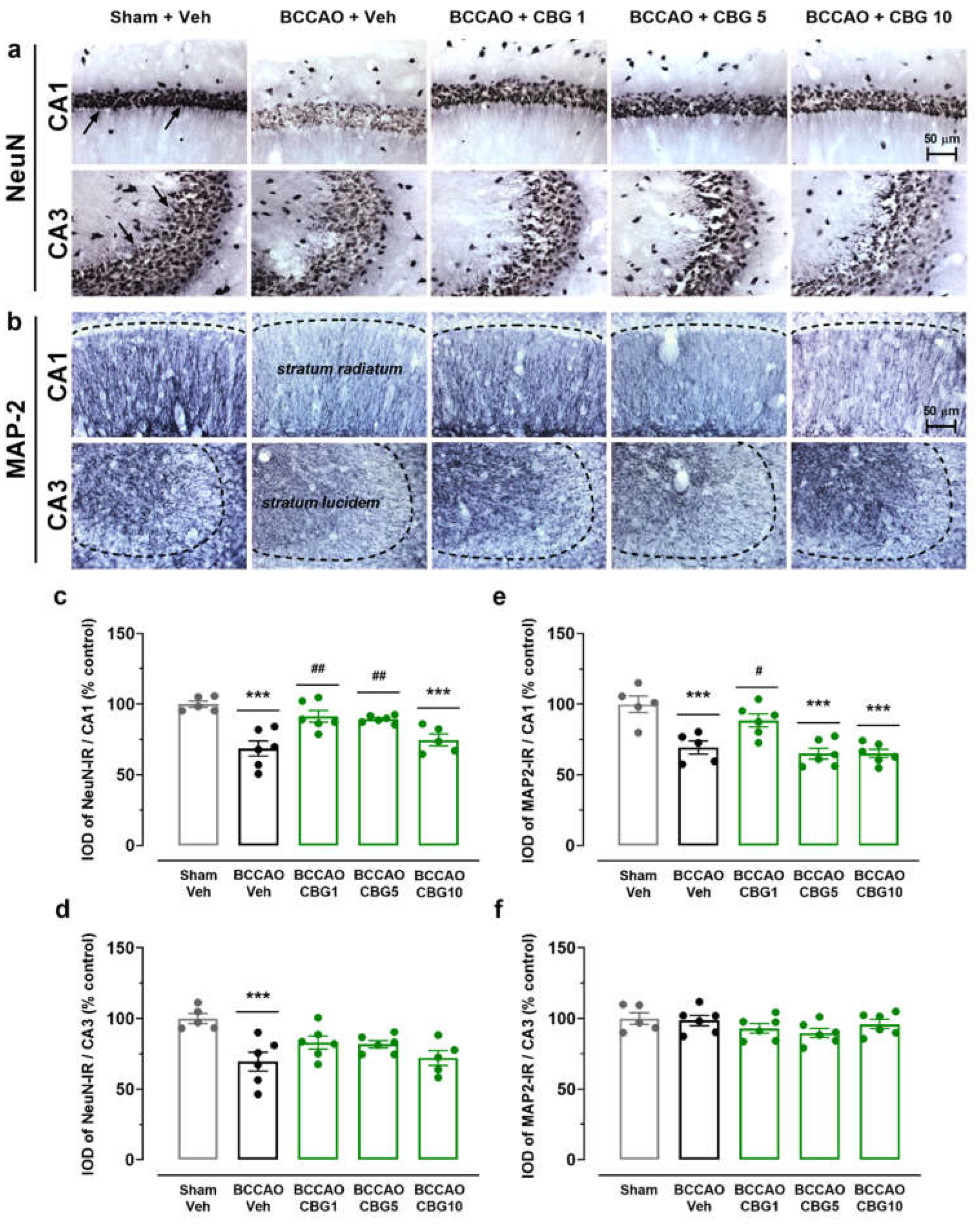
Figure 3.
CBG reduces BCCAO-induced neuroinflammation in the hippocampus. (a, b, and c) Representative photomicrographs of Iba-1-IR, GFAP-IR, and NG2-IR cells in the CA1 and CA3 subfields of the hippocampus. The dashed lines delimit the layers of neuronal bodies. (d and e) IOD of the microglia (Iba-1-IR) in the CA1 and CA3 subfields of the hippocampus. (f and g) IOD of the astrocytes (GFAP-IR) in the CA1 and CA3 subfields of the hippocampus. (h and i) IOD of the glial cell precursor (NG2-IR) in the CA1 and CA3 subfields of the hippocampus. The bars represent the mean ± SEM in the different groups (n = 5-6/group). *p < 0.05, **p < 0.01, ***p < 0.001 vs sham + Veh; #p < 0.05, ##p < 0.01, ###p < 0.001 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
Figure 3.
CBG reduces BCCAO-induced neuroinflammation in the hippocampus. (a, b, and c) Representative photomicrographs of Iba-1-IR, GFAP-IR, and NG2-IR cells in the CA1 and CA3 subfields of the hippocampus. The dashed lines delimit the layers of neuronal bodies. (d and e) IOD of the microglia (Iba-1-IR) in the CA1 and CA3 subfields of the hippocampus. (f and g) IOD of the astrocytes (GFAP-IR) in the CA1 and CA3 subfields of the hippocampus. (h and i) IOD of the glial cell precursor (NG2-IR) in the CA1 and CA3 subfields of the hippocampus. The bars represent the mean ± SEM in the different groups (n = 5-6/group). *p < 0.05, **p < 0.01, ***p < 0.001 vs sham + Veh; #p < 0.05, ##p < 0.01, ###p < 0.001 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
Figure 4.
The impact of CBG on ischemia-induced neuroplasticity in the hippocampus. (a) Representative photomicrographs of DCX-IR neurons in the GCL and SGZ of the hippocampal dentate gyrus. (b) Number of DCX-IR neurons in the SGZ and GCL of the dentate gyrus. (c) Representative western blot bands of BDNF, proBDNF, and β-actin. (d and e) BDNF and proBDNF protein levels in the hippocampus. (f) ratio between BDNF and proBDNF levels. The bars represent the mean ± SEM in the different groups (n = 5-7/group). **p < 0.01 vs sham + Veh; #p < 0.05, ##p < 0.01, ###p < 0.001 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
Figure 4.
The impact of CBG on ischemia-induced neuroplasticity in the hippocampus. (a) Representative photomicrographs of DCX-IR neurons in the GCL and SGZ of the hippocampal dentate gyrus. (b) Number of DCX-IR neurons in the SGZ and GCL of the dentate gyrus. (c) Representative western blot bands of BDNF, proBDNF, and β-actin. (d and e) BDNF and proBDNF protein levels in the hippocampus. (f) ratio between BDNF and proBDNF levels. The bars represent the mean ± SEM in the different groups (n = 5-7/group). **p < 0.01 vs sham + Veh; #p < 0.05, ##p < 0.01, ###p < 0.001 vs BCCAO + Veh (one-way ANOVA followed by Sidak's test).
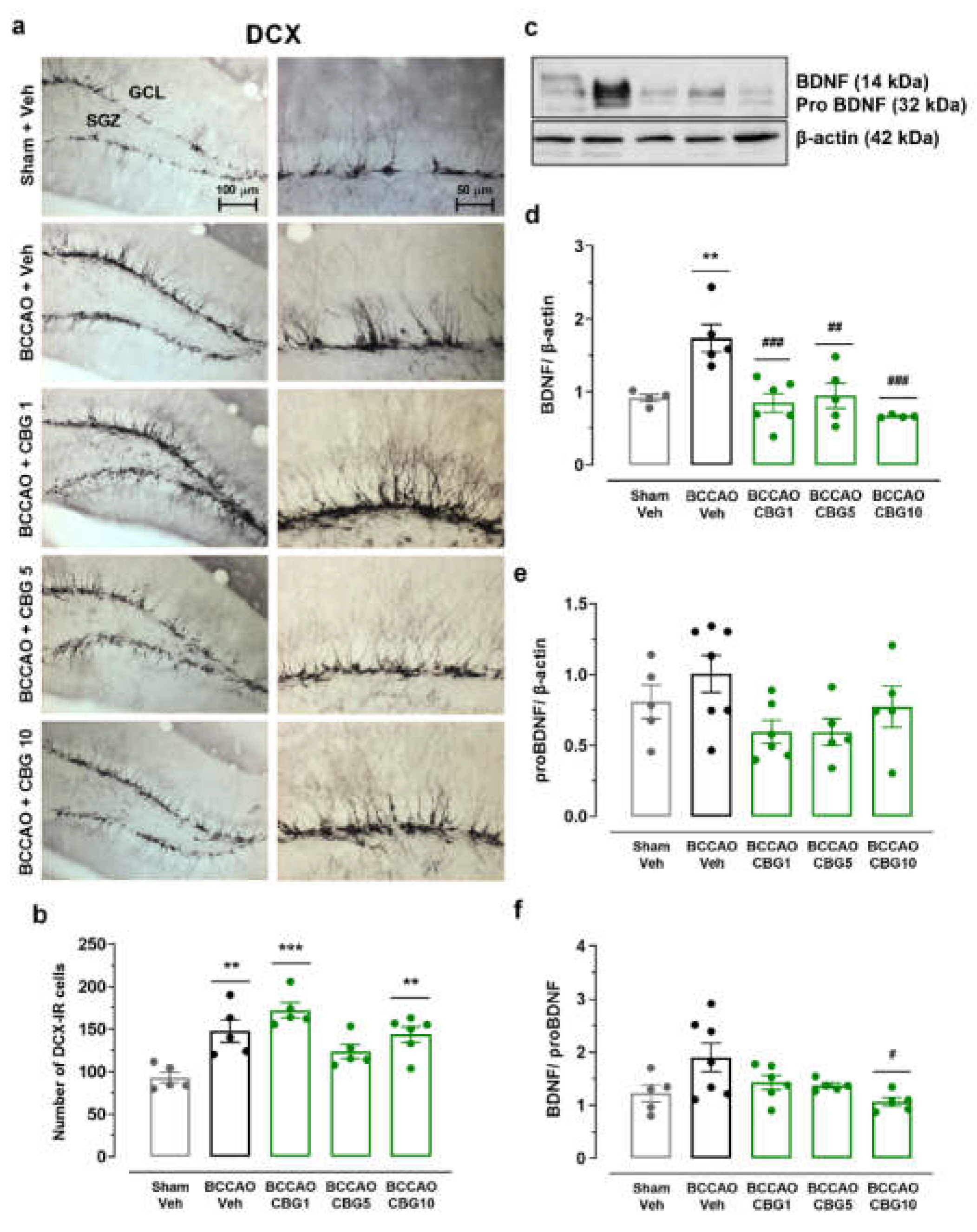
Figure 5.
Experimental design. From day -7 to day -1 the animals were familiarized with the OLT environment. In the day 1 occurred sham or BCCAO surgery and Veh, CBG1, CBG5 or CBG10 was administered i.p. during 7 days. Behavioral testing was performed from day 9 to day 14 after BCCAO. Immediately after the last behavioral test, the animals were euthanized, and their brains were processed for immunohistochemical (IHC) and western blot (WB) analysis. OLT, object location test; TST, tail suspension test.
Figure 5.
Experimental design. From day -7 to day -1 the animals were familiarized with the OLT environment. In the day 1 occurred sham or BCCAO surgery and Veh, CBG1, CBG5 or CBG10 was administered i.p. during 7 days. Behavioral testing was performed from day 9 to day 14 after BCCAO. Immediately after the last behavioral test, the animals were euthanized, and their brains were processed for immunohistochemical (IHC) and western blot (WB) analysis. OLT, object location test; TST, tail suspension test.
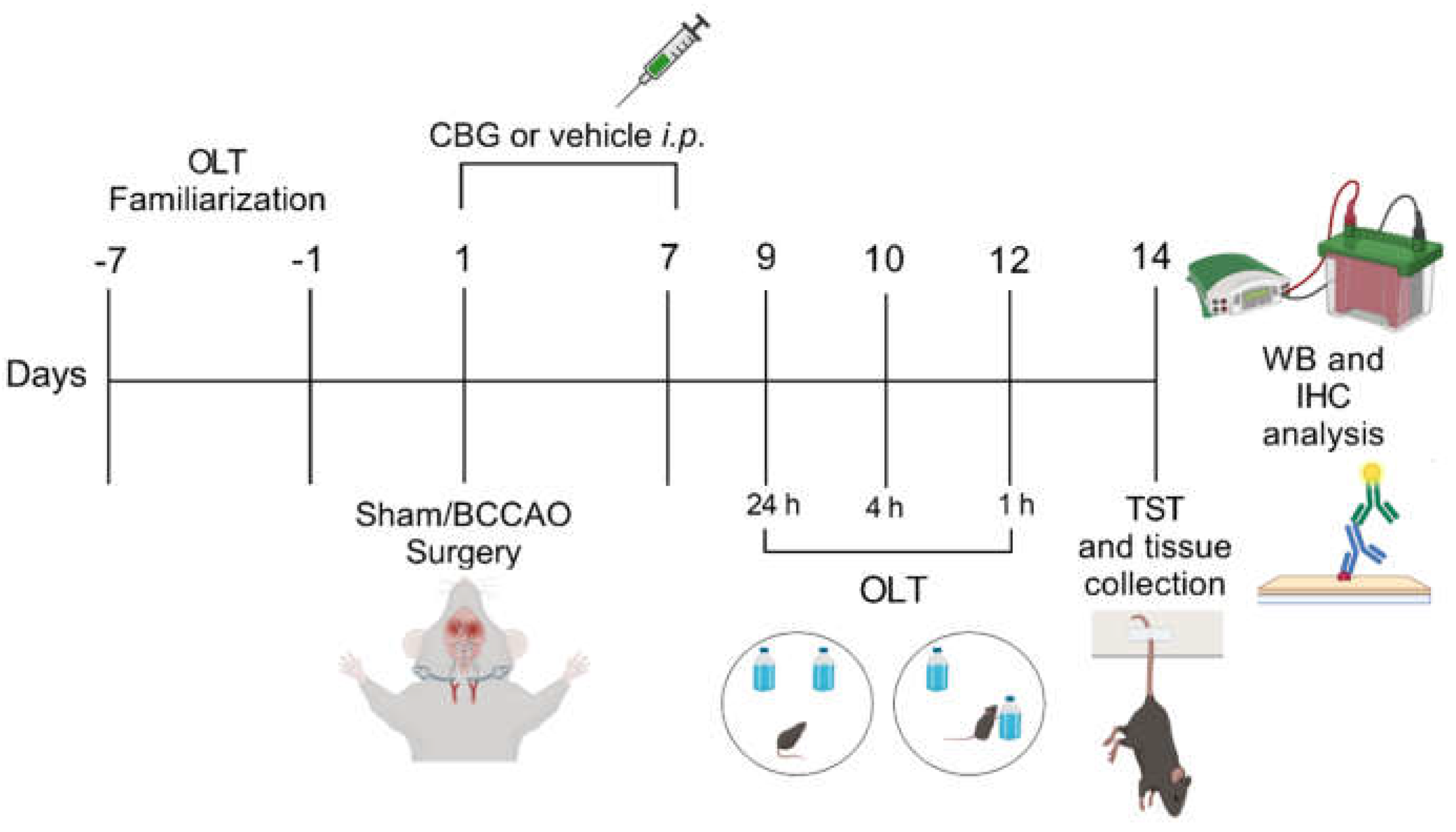

Table 1.
List of primary antibodies used in immunohistochemistry and western blot assays.
| Antibodies (dilution) | Company | Code |
|---|---|---|
| Rabbit anti-Iba-1 (1:1500) | Wako Chemicals | 019-19741 |
| Rabbit anti-GFAP (1:2000) Rabbit anti-NG2 (1:200) Rabbit anti-NeuN (1:500) Rabbit anti-DCX (1:1000) Mouse anti-pBDNF (1:300) Rabbit anti-MAP-2 (1:500) Rabbit anti-β-actin (1:5000) |
Abcam Merck Millipore Abcam Cell Signaling Technology Santa Cruz Biotechnology Sigma-Aldrich Abcam |
Ab7260 AB5320 Ab177487 4604S Sc65514 M3696 Ab227387 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



