
Submitted:
26 March 2025
Posted:
27 March 2025
You are already at the latest version
Abstract
With the first evidence of the association between bats and influenza A viruses, there are some various studies have begun to be generated to understand this interesting and important association around bats conservation, animal health and public health. This study aimed to verify the presence of anti-influenza A vipothesrus antibodies, as well as the molecular identification of these viruses in bats distributed in forest fragments located in southeastern Mexico. Blood samples were obtained from 600 bats belonging to 24 different species, using an enzyme immunoassay to detect antibodies against the nucleoprotein antigen of the avian influenza A virus. Likewise, oropharyngeal swabs, rectal swabs and organs were taken for the molecular diagnosis of these viruses. We were able only six bats (1%) tested positive to serology and molecular tests. Although this suggests a very low prevalence of influenza A viruses in Mexican bats, it is the first study to address this association and, following the precautionary principle, we consider it necessary to establish systematic monitoring of the presence of influenza A in bats, since they are known to harbor infectious agents with zoonotic potential. Furthermore, it is possible that the association of influenza A viruses circulating in Latin American bats has an important coevolutionary component with some bat species with exclusive distribution in the American continent.
Keywords:
Alphainfluenzavirus
; forest bats
; prevalence
; Southeast Mexico
; tropical forests16
Introduction
Given the major impacts that influenza A viruses (Alphainfluenzavirus influenzae) can have on public health, farmers, livelihoods, the poultry industry, and international trade, these viruses have captured increasing attention in the international community [1]. Therefore, identifying the viral reservoirs is urgently needed to better understand the viral evolution and potential cross-species transmissions. Influenza A viruses are known to be present in different domesticated animals (e.g., ducks, chickens, pigs, horses, and dogs). Yet, their presence in wildlife is not as well known, especially in some species-rich and widely distributed taxa, such as bats [2,3].
Bats are known to have multiple associations with several infectious agents because, in general, they have relatively long lifespans, and some species can travel long distances and use anthropogenic landscapes where there may be increased contact (and interaction) between humans, wildlife, and domestic species [2,3]. Bats have been shown to have the potential to host a large proportion of viruses with zoonotic potential [4]. Some infectious agents that bats have been associated with have zoonotic potential, such as Lyssavirus, coronaviruses and members of the influenza A virus [5]. However, the ability of bats to host influenza A viruses and the potential risk of interspecies transmission that this association could trigger remain poorly known.
The great interest in learning more about this virus-bat association arose from recognizing the influenza A virus in fruit bat species in Guatemala and Peru, designated as H17N10 and H18N11, subtypes currently considered exclusive to bats [6,7]. In the case of influenza A viruses related to other host species, exposure and infection by virus subtype H9 have been identified in fruit bats from Africa [8,9]. This evidence allows us to hypothesize that some bat species might have ecological and evolutionary conditions that support their participation in the ecology of avian influenza A viruses.
However, there is still a large information gap regarding the ecology of influenza A viruses in bats and their relationship with other hosts. To fill this gap, our study aimed to verify the prevalence of antibodies against influenza A viruses and identify the infection of these viruses in bats sampled in a human-modified tropical rainforest from southeastern Mexico. This information is highly valuable as previous research on the topic has assessed the presence of viruses with molecular detection of viral RNA. For instance, studies carried out in America [5,6,7,10] and Africa [9] have identified bat species mainly infected by the specific subtypes of this group of mammals or through genetic sequencing have managed to detect an avian subtype. However, the lack of knowledge regarding the development of infection caused by influenza A viruses in bats, and the difficulty of obtaining an adequate amount of blood samples hinders the possibility of detecting individuals exposed to different influenza A subtypes [11]. The use of serological and molecular methods allows us to broaden our understanding of the associations between bat species and influenza A viruses, as well as detect past exposures and infections. Because of this, the purpose of this study was to identify the prevalence of antibodies against influenza A viruses and the molecular prevalence of these viruses in Bats from Southeast Mexico distributed in fragmented tropical rainforests.
Materials and Methods
The fieldwork was authorized by the Secretariat of Environment and Natural Resources (permit number SGPA/DGVS/04241/19) and showed the approval of the Institutional Animal Care and Use Subcommittee with protocol number SICUAE.DC-2021/3-2 of the Postgraduate Program in Animal Production and Health Sciences of the National Autonomous University of Mexico.
The study was carried out from April 2021 to April 2022 in 23 forest sites located within 23 rainforest fragments from the state of Campeche, Mexico (Figure 1a and 1b). Within each forest site, we placed six 12 m x 2.5 m mist nets covering as much area as possible and recorded all bats captured during 14 hours/net per site for two consecutive nights. The networks were opened at sunset and seven hours later they were closed. Once captured, each individual was collected in a separate cloth bag for later identification and sampling. The identification of the bat species was carried out through identification guides and trained personnel. Bats were released after sampling and only some were selected for euthanasia and necropsy to meet other project objectives. The selected individuals were only individuals belonging to species that are not in danger of extinction or threatened, that is, they are not found in any of the risk categories and are in the category of least concern in Mexican standards and their populations are also stable. In addition to the species, only adult females or males and juvenile females or males were selected. No pregnant or lactating females were selected.
The blood samples were collected from the brachial vein, not exceeding more than 10% of their body weight, using filter paper, and then the samples were stored at -20°C until processed [12]. Individuals weighing 10g were not sampled so as not to compromise their lives (pregnant or lactating females or individuals in any physiological state of protected species). Filter paper strips were eluted in 400 µl of phosphate-buffered saline in 1.5 mL microcentrifuge tubes at 4°C for 24 hours. Afterward, the samples underwent serum heat inactivation at 56°C for 30 minutes. Finally, the supernatant was obtained through centrifugation at 3000 rpm for five minutes. Therefore, the serum was obtained with a 1:10 dilution [13]. To identify sera containing influenza A antibodies, we used an enzymatic immunoassay to detect antibodies for avian influenza A virus nucleoprotein antigen (IDEXX Influenza Virus Ab Test Kit). The assay was performed in 96-well plates coated with the viral antigen of influenza A viruses. After incubation of the sample analyzed in the coated wells, the specific antibody against influenza A viruses forms a complex with the antigen. Subsequently, unbound material was removed, and an anti-influenza A monoclonal antibody-enzyme conjugate was added to the wells. Since there are no antibodies against the NP of the influenza A viruses, the conjugate reacts directly with the antigen of the influenza A viruses present on the plate. Conversely, if there are antibodies against the NP of influenza A viruses present in the sample, the anti-influenza A conjugate will not be able to bind to the antigen. The unbound conjugate is removed by washing and an enzyme substrate is added. The color that appears is inversely proportional to the amount of anti-influenza A antibodies in the sample analyzed. To identify seropositive individuals, we considered those with less than 0.7nm sera positive [14].
Positive samples underwent a microneutralization assay for influenza subtypes H1N1, H5N1, H7N3, and H9N2 of the avian origin with an ELISA-based endpoint evaluation as described in the protocol available from the World Health Organization (WHO) [15]. Virus viability was assessed with MDCK cells in 96-well plates using ELISA to identify positive culture wells with infected cells. Subsequently, in the 96-well plate, it was mixed with the serum dilution to be analyzed and left to incubate for one hour at 37°C and then a suspension of MDCK cells was added to each well. At 22 hours post-infection, the cells were fixed with acetone. Virus infection in cells in each well is assessed by ELISA as described in the WHO protocol. The neutralization titer will be considered as the reciprocal of the highest serum dilution.
We also took samples of oropharyngeal and rectal scrapings through swabs that were transported in cryotubes with MEM medium in liquid nitrogen. The samples were stored deep frozen at -70°C until processing. Similarly, samples were taken from the trachea, lungs and intestines of some species of bats whose populations are stable. Euthanasia was performed using an inhalation anesthesia capsule specifically designed for bats where Isoflurane was used at a concentration of 2 to 5% to bring the individuals to a deep anesthetic plane where they are desensitized [16,17]. They were subsequently administered an overdose of ketamine (10 mg/kg of body weight) for euthanasia [18]. Once the death of the individuals was confirmed, a necropsy was performed and samples were taken from the trachea, lung, and small and large intestine (Supplementary Video 1) [10]. These samples were transported in 2 ml cryotubes in liquid nitrogen and stored in the same way as the scraping samples through swabs. Subsequently, the viral RNA was extracted from the oropharyngeal, rectal scraping samples and the organs using the commercial kit "QIAamp® Viral RNA (Qiagen)" and using linear acrylamide as a carrier, following the specifications recommended by the manufacturer. After this, the qRT-PCR (Real-Time Polymerase Chain Reaction) test was performed aimed at the amplification and detection of the M protein (Matrix) with the use of the commercial kit "VetMAX-Gold AIV Detection Kit" (Life Technologies) following the recommendations established by the manufacturer. Positive samples were considered those that obtained a CT value less than 38, suspicious samples were those that obtained a value of 38 to 40, and negative samples were those that did not obtain any value.
Samples that tested positive were processed for whole genome amplification and sequencing. Genome amplification was performed through a multi-segment RT-PCR reaction using universal oligonucleotides. The oligonucleotides used are described in [19,20] and for library construction, following in detail the protocol described in https://dx.doi.org/10.17504/protocols.io.n2bvj8mrxgk5/v1. The RT-PCR reaction products were purified and concentrated using Ampure XP Beads (Beckman, Coulter). The amplified fragments were verified as 1% agarose gel. The amplification products were tagged using the EBLTS (beads) of the COVIDSeq Assay Kit to finally amplify and label the tagged fragments using the Nextera XT Index Kit v2 as specified in the protocol. The sequence was performed on an Illumina NextSeq 500 device in a configuration of 2X150 cycles and 200K reads per sample.
Results
We obtained samples from 600 bats belonging to 24 species and four families. Phyllostomidae was by far the most abundant family, with Artibeus jamaicensis (36% of captured individuals), Dermanura phaeotis (17%), and Artibeus lituratus (13%), being the most abundant species (Supplementary Table1). Only six individuals from three species (i.e., Artibeus jamaicensis, n = four individuals, of which four adult females, one pregnant, one lactating and two post-lactating; Carollia perspicilliata, n = one, adult female; and Glossophaga commisarissi, n = one, pregnant female) presented serological evidence of influenza A antibodies, but it was not possible to identify the subtype to which they were exposed because we obtained negative results in microneutralization for the H1N1, H5N1, H7N3, and H9N2 subtypes of avian origin. Artibeus jamaicensis and Carollia perspicilliata are frugivorous species, and Glossophaga commisarissi is nectarivorous. These individuals were captured in the municipalities of Palizada, Carmen, and Calakmul (Figure 1 panels c, d, and e; Figure 2). Similarly, we identified six RT-PCR positive individuals from five different species (i.e., Artibeus jamaicensis, n = two individuals, of which one is a juvenile male and the other is an adult male; Carollia sowelli, n = one, adult male; Carollia perspicillata, n = one, adult male; Dermanura phaeotis, n = one, juvenile female; and Micronycteris microtis, n = one, adult female. The quantification of the cycle threshold (Ct) ranged between 35.45 and 37.9 (Figure 1 panels c, d and e; Table 1). RT-PCR-positive samples correspond to oropharyngeal and rectal swab samples. The individuals with these positive samples were captured in Palizada, Carmen, Escárcega, and Calakmul. No positive results were obtained from the tracheas, lungs and intestines of the individuals selected for necropsy (n=270 individuals out of 600). RT-PCR-positive samples amplified for all eight segments, but it was not possible to assemble the genome and reach a conclusive result.
Discussion
Bats have been associated with a long list of infectious agents mainly due to their resistance to infections and their great adaptive and co-evolutionary capacity. In the Americas, various species of neotropical bats have been primarily associated with RNA viruses, with notable findings in the families Phyllostomidae, Moormopidae, Molossidae, and Vespertilionidae [21]. Our findings suggest possible exposure and infection of influenza A viruses in neotropical bats associated with tropical forests and human-modified landscapes in southeastern Mexico.
Only six out of 600 bats (1%) tested positive by serology and another six individuals (1%) tested positive by RT-PCR, indicating a very low seroprevalence and molecular prevalence of influenza A viruses in the study region. However, as these are not isolated cases, our findings add to the growing evidence indicating that some bat species may participate in the ecology of influenza A viruses [5,6,7,8,9,10]. Nonetheless, in comparison to other hosts of these viruses such as wild birds, there is still ignorance, in most parts of the world, about the participation of wild mammals, such as bats, in transmission cycles, particularly in the infection and exposure to influenza A viruses.
This information supports the hypothesis of a possible coevolutionary association between the Phyllostomidae family found only in America and the H17N10 and H18N11 subtypes. This is why assays with a higher degree of specificity may reduce the coverage of positive individuals by not having the specific subtypes to identify exposure to and infection with influenza viruses in bats. In this study, we had the limitation of not having the H17 and H18 subtypes and the rest of the H2-H4, H6, and H8-H16 subtypes to carry out the microneutralization tests and obtain more specific information about the association of the bats in our study with different subtypes of influenza A viruses. Likewise, it is important to recognize the limitations of the ELISA assay and over time, to generate panels of relevant HI viral antigens that cover exposure of bats to influenza A viruses in a regional and global context. Likewise, in this study it was not possible to obtain quality sequences that would allow identifying the viral identity because the RNA concentration was very low and it was only possible to amplify the eight viral segments and build the libraries, but it was not possible to assemble the genome.
However, despite the limitations, our molecular results demonstrate molecular evidence of influenza A virus infection in six bats of five of 24 species captured in Campeche. It has been shown that the detection of influenza A viruses in bats is very low, for example, Tong et al. (2012), evidenced for the first time the presence of H17N10 in fruit bats of the species Sturnira lilium in Guatemala with a positivity frequency of 0.94% (3 positive individuals out of 316 total bats) [6]. Similarly, in 2013 the H18N11 virus was identified in Artibeus planirostris from Peru, with a positivity frequency of 0.1% [7]. The same has happened in more recent years, for example in Brazil, in 2019, an H18N11 was demonstrated for the first time in bats of the species Artibeus lituratus with a frequency of 0.4%, that is, 2 individuals out of 533 bats [10]. Under these characteristics, our results show, for the first time in Mexico, serological evidence about the possible exposure of Phyllostomidae bats to influenza A viruses in the neotropical region. However, the number of studies on the association between bats and the influenza A virus is very low compared to the number of studies on other species. This panorama reinforces the importance of exploring this association in greater depth with new research, especially in fruit bats due to the coincidence reported so far.
The findings of this study indicate that the competitive ELISA that we used can be used for seroepidemiological studies of influenza A in species that have been little explored and with difficulty in obtaining quality samples because the objective of the test is to detect the presence of nucleoprotein antibodies of the anti-influenza A virus cross-reacts with influenza A viruses because this region is highly conserved. In such a way that the detection coverage of positive individuals is increased, which on many occasions cannot be detected by other specific tests such as hemagglutination inhibition (HI) due to the specificity of the viral subtype. In this sense, the samples positive to the ELISA were not positive to the microneutralization assay directed at the avian H1N1, H5N1, H7N3, and H9N2 subtypes due to the specificity of the test and the subtypes used. Although these subtypes are of great relevance in the epidemiological context of influenza A viruses due to their impact on animal health and public health, as is the case with the H1N1, H5N1, and H7N3 subtypes, we still do not have sufficient knowledge to determine the participation and relationship of these subtypes with neotropical bats. Likewise, although the H9N2 subtype has already been reported in other parts of the world in Old World bats [9], in our study we did not report seropositivity for this subtype, which coincides with what has been reported up to this point for the American continent.
Frugivorous bats, such as those from the Artibeus genus, can participate in the ecology of influenza A viruses. As mentioned, the presence of influenza A viruses in bats was first confirmed in Sturnira lilium and Artibeus planirostris, particularly the H17N10 and H18N11 subtypes in Guatemala and Peru, respectively [6,7]. Some years later, in Brazil, the isolation and sequencing of the H18N11 virus were reported in the intestines of two Artibeus lituratus individuals from different places [10]. In Colombia, there are also reports of bat samples, Carollia and Artibeus in particular, that tested positive for influenza A viruses [5]. Thus, phyllostomid bats are expected to be positive for influenza A viruses, which allows us to hypothesize that it is probable that, besides being exposed, the bats sampled in our research can get infected with the subtypes reported in other parts of America, such as H17N10 and H18N11, as well as avian subtypes as has happened in other parts of the world [6,7,9,10]. Based on the findings reported in American studies, we could suggest a coevolutive association between the Phyllostomidae family found only in this continent and the H17N10 and H18N11 subtypes as they have not been reported in other parts of the world.
The relatively high prevalence of influenza A viruses in Artibeus jamaicensis may be related to the dominance of this bat species in the study region. Bats from the Artibeus genus tend to be abundant in Neotropical rainforests [22], and A. jamaicensis was the most abundant species in our survey. A. jamaicensis is considered an abundant and proportionally dominant species in some communities [23], and this could lead them to be exposed to a higher degree of contact with other bats and other species. In addition to abundance in ecosystems, the gregarious habits of bats that tested positive could make them prone to greater exposure and possible infection with influenza A viruses, along with other infectious agents. The habit of forming large colonies to rest in caves or other shelters, as well as their social structure based on polygyny and the formation of harems of between four to 18 females, their offspring, one or two dominant males that actively defend the harem from other males [23], fission-fusion behavior and longevity, increases the probability of interspecific contact with bats from southeastern Mexico [24].
All the species that showed evidence of infection by influenza A virus belong to the family Phyllostomidae. This may be since members of the family Phyllostomidae show evidence of interspecific attraction in mixed-species roosting groups of bats in natural roosts, particularly in the case of species of the genus Carollia and Glossophaga [25]. A documented example is the coexistence of bats of the genus Artibeus with bats of the genus Carollia [26]. This coexistence is facilitated by the abundance of food resources that are part of the diet of these species, as well as by the morphological differences that allow differentiation of foraging behavior about fruit consumption, and by the structural characteristics of the vegetation of this type of neotropical forests. These dietary preferences and anatomical differences suggest that other resources such as a preference to rest in caves or certain mechanisms such as predation, parasitosis or the dynamics of certain infectious agents explain their ability to coexist. The availability of areas within forests and fruits used by these species is so great that there is little need for ecological segregation into different niches [26]. This in turn favors greater interaction between individuals of different species that belong to the same trophic guild. Therefore, it is not surprising that in our study there are positive individuals of other species that are not of the genus Artibeus, as is the case of Carollia, Dermanura, Glossophaga, and Micronycteris because these species coexist in the neotropical forests of southeastern Mexico due to the abundance of resources obtained from these diverse forests with great biological complexity. This coexistence may promote the transmission of influenza A viruses between bats of different species that coexist and share ecological niches within Neotropical forests and that, in addition, various species have closer phylogenetic relationships that allow them to be infected by the same viruses, as has been demonstrated in other studies [27].
Understanding the ecoepidemiology of influenza A viruses among bats and between bats and various species is then the challenge of future research to assess the consequences on animal and public health and to determine possible taxonomic jumps of influenza A viruses. Recently, the position proposed by authors such as Ciminski et al, 2019, that influenza A viruses detected in bats are limited to interspecies transmission between bats has been weakened [28]. Experimental conditions have shown that H18N11 subtype viral particles can infect human leukocytes and replicate them easily in human macrophages [29]. Other studies have revealed that bats could play an important role in the ecology of avian influenza A viruses, as epithelial cells in their lungs are susceptible to infection by certain avian-origin viruses and show evidence of avian and human influenza virus compatible sialic acid receptors [30,31]. In bats from Egypt, an influenza A H9N2 virus, phylogenetically distinct from viruses detected in birds, was successfully isolated and, it is considered a virus that recently diverged from birds and effectively adapted to Egyptian bats [32,33]. In addition, there is growing information about their association with a wide variety of infectious agents which can potentially generate serious animal and public health consequences [2].
Conclusions
This study provides this study provides the first findings on the potential interaction and infection of Neotropical bats with influenza A viruses and suggests a potential critical list for the target bat species in the viral ecology of this viruses in southeast Mexico. It is necessary to open new research questions and hypotheses to explore the capacity that Mexican bats must harbor different subtypes of influenza A viruses and to understand their participation in the ecology of influenza A viruses in fragmented tropical forests. Because bats harbor a significantly high proportion of viruses with zoonotic potential, it is essential to carry out serological studies in other areas and the molecular subtyping to broaden substantially our understanding of the origin of influenza A viruses in bats and their ability to participate in the transmission cycles of these viruses. With time, it is possible to generate knowledge as extensive as that which exists for other wild hosts, mainly in bats distributed in tropical ecosystems affected by human activities.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Table 1. Abundance and bats species captured for sampling. Supplementary Video 1. Taking organ samples at a necropsy of selected individuals.
Author Contributions
Brenda Aline Maya-Badillo: Conceptualization, formal analysis, investigation, methodology, project administration, resources, visualization, and writing (original draft preparation). Guillermo Orta-Pineda: Conceptualization, investigation, methodology, resources, visualization, and writing (original draft preparation). Gerardo Suzán: Funding acquisition, resources, supervision, validation, and writing (review and editing). Karen Elizabeth Rivera-Rosas: Investigation and methodology. Diego Zavala-Vasco: Investigation and methodology. Adrián Uribe-Jacinto: Investigation and methodology. Andrea Chaves: Supervision and writing (review and editing). Ricardo Alfredo Grande Cano: Methodology and writing (review and editing). René Segura: Resources and writing (review and editing). José Iván Sánchez-Betancourt: Conceptualization, funding acquisition, project administration, resources, supervision, and validation, writing (review and editing).
Data Availability Statement
The data related of this experimental study are available on request from the corresponding author.
Acknowledgments
The authors thank the PAPIIT IN224820 Project and the PRONAII 303002 Project for funding to carry out the study. The authors also thank Víctor Arroyo-Rodríguez for reviewing the manuscript and all the people including site owners, ranch workers, as well as the families of the workers, and staff of the different institutions in Campeche, Mexico who supported the development and logistics of the fieldwork. Especially Roberto Calzada, Juan Carlos García, Sergio Pech, Jorge Luis Ayala, Isabel Serrano McGregor, Carmelo Pech, Ignacio Seara, Alfonso de la Rosa, Adriana Velázquez Morlet, Lázaro Casado, Alsides Pérez Álvarez, Eulalio López Orama, Otilio Noh Canché, José Puhol, Yaritza Abreu, Yolanda del Rivero, Roberto Lules, Guillermo, Adrián, Ruperto Uribe y Wendy Puhol. Likewise, we thank the Centro de Investigación y Transferencia de Tecnología Forestal El Tormento, the Instituto Nacional de Antropología e Historia, and the "Xpuhil" and "El Hormiguero" archaeological zones and the Comisión Nacional de Áreas Naturales Protegidas. We thank Ana L. Vigueras Galván for her support in the logistics of a field trip and training in obtaining serum from wild animals, and the students who provide social and professional services for their short stays in the field work José E. Guzmán Romero, Diana B. Ramírez Gutiérrez, María E. Meneses González and Jessica G. Conti González. Finally, we thank the Unidad de Investigación and the Laboratorio de Investigación del Departamento de Medicina y Zootecnia de Cerdos of Facultad de Medicina Veterinaria y Zootecnia of the Universidad Nacional Autónoma de México and Laboratorio Mixto Internacional ELDORADO, Patricia Stephany Justo Berrueta, Anita Aguirre Barbosa, and Rodrigo Aparicio of the Instituto de Diagnóstico y Referencia Epidemiológicos, México and the Mario Solís Hernández of the Comisión México-Estados Unidos para la Prevención de la Fiebre Aftosa y otras Enfermedades Exóticas de los Animales for the advice provided for the development of complementary serological tests. We would also like to thank Marco Cuellar and Manuel Saavedra for their technical support in the laboratory for organ processing. We also thank the Programa de Posgrado en Ciencias de la Producción y Salud Animal of the Universidad Nacional Autónoma de México and the Consejo Nacional de Humanidades Ciencias y Tecnologías for the scholarship awarded to doctoral student Brenda Aline Maya-Badillo.
Conflicts of Interest statement
All authors declared no conflict of interest.
References
- Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG, García-Sastre A. Influenza. Nat Rev Dis Primers. 2018;4(1):3. [CrossRef] [PubMed] [PubMed Central]
- Violet-Lozano L, Haach V, Barboza CM, dos Santos J, Gomes BF, de Cassia Pardo de Souza T, et al. No molecular evidence for influenza A virus and coronavirus in bats belonging to the families Phyllostomidae, Vespertilionidae, and Molossidae in the state of São Paulo, Brazil. Brazilian J Microbiol. 2022;(0123456789). Available from: . [CrossRef]
- Nabi G, Wang Y, Lü L, Jiang C, Ahmad S, Wu Y, et al. Bats and birds as viral reservoirs: A physiological and ecological perspective. Sci Total Environ. 2021;754:142372. Available from: . [CrossRef]
- Olival, K., Hosseini, P., Zambrana-Torrelio, C. et al. Host and viral traits predict zoonotic spillover from mammals. Nature. 2017; 546, 646–650. [CrossRef]
- Uribe M, Rodríguez-Posada ME, Ramirez-Nieto GC. Molecular Evidence of Orthomyxovirus Presence in Colombian Neotropical Bats. Front Microbiol. 2022;13(April):1–9. Available from:. [CrossRef]
- Tong S, Li Y, Rivailler P, Conrardy C, Alvarez Castillo DA, Chen LM, et al. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci U S A. 2012;109(11):4269–74. Available from:. [CrossRef]
- Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, et al. New World Bats Harbor Diverse Influenza A Viruses. PLoS Pathog. 2013;9(10). Available from:. [CrossRef]
- Freidl GS, Binger T, Müller MA, De Bruin E, Van Beek J, Corman VM, et al. Serological evidence of influenza a viruses in frugivorous bats from Africa. PLoS One. 2015;10(5):1–7. Available from: . [CrossRef]
- Kandeil A, Gomaa MR, Shehata MM, El Taweel AN, Mahmoud SH, Bagato O, et al. Isolation and Characterization of a Distinct Influenza A Virus from Egyptian Bats. J Virol. 2019;93(2). Available from:. [CrossRef]
- Campos ACA, Góes LGB, Moreira-Soto A, de Carvalho C, Ambar G, Sander AL, et al. Bat influenza a(HL18NL11) virus in fruit bats, Brazil. Emerg Infect Dis. 2019;25(2):333–7. Available from:. [CrossRef]
- Fereidouni S, Kwasnitschka L, Balkema Buschmann A, Müller T, Freuling C, Schatz J, et al. No virological evidence for an influenza A - like virus in European bats. Zoonoses Public Health. 2015;62(3):187–9. Available from:. [CrossRef]
- Villena FE, Gomez-Puerta LA, Jhonston EJ, Del Alcazar OM, Maguiña JL, Albujar C, et al. First report of Trypanosoma cruzi infection in salivary gland of bats from the Peruvian Amazon. Am J Trop Med Hyg. 2018;99(3):723–8. Available from:. [CrossRef]
- Dusek RJ, Hall JS, Nashold SW, Teslaa JL, Ip HS. Evaluation of Nobuto filter paper strips for the detection of avian influenza virus antibody in waterfowl. Avian Dis. 2011;55(4):674–6. Available from:. [CrossRef]
- Shriner SA, VanDalen KK, Root JJ, Sullivan HJ. Evaluation and optimization of a commercial blocking ELISA for detecting antibodies to influenza A virus for research and surveillance of mallards. J Virol Methods. 2016;228:130–4. Available from:. [CrossRef]
- WHO, 2011. (Accessed September 2023). Microsoft Word - Serological diagnosis for web 6_12_2010.doc (who.int). Available in https://iris.who.int/bitstream/handle/10665/44518/9789241548090_eng.pdf?sequence=1.
- Hooper SE and Amelon SK. Handling and blood collection in the little brown bat (Myotis lucifugus). Lab Anim. 2014, (NY). 43, 197–199. [CrossRef]
- Irving AT, Ng JHJ, Boyd V, Dutertre CA, Ginhoux F, Dekkers MH, Meers J, Field HE, Crameri G, Wang LF. Optimizing dissection, sample collection and cell isolation protocols for frugivorous bats. Methods Ecol. Evol. 2020,11, 150–158. [CrossRef]
- Wirawati V, Widiati NDA, Gunawan G, Saragih GR, Hening P, Wihadmadyatami H. The distribution of serotonergic nerve on the hippocampus of the fruit bats (Rousettus amplexicaudatus). Vet. World. 2019, 12, 1460–1466. [CrossRef]
- Zhou B, Wentworth DE. Influenza A Virus Molecular Virology Techniques. In: Kawaoka, Y., Neumann, G. (eds) Influenza Virus. Methods in Molecular Biology. 2012; vol 865. pp.175-192. Humana Press. [CrossRef]
- Zhou B, Lin X, Wang W, Halpin RA, Bera J, Stockwell TB, Barr IG, Wentworth DE. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics, and reverse genetics. J Clin Microbiol. 2014; May;52(5):1330-7. Epub 2014 Feb 5. [CrossRef] [PubMed] [PubMed Central]
- De Oliveira, M.B., and Bonvicino, C.R. Incidence of viruses in neotropical bats. Acta Chiropterol. 2020; 22, 461–489. [CrossRef]
- Da Silva AG, Gaona O, Medellín RA. Diet and trophic structure in a community of fruit-eating bats in Lacandon Forest, México. J Mammal. 2008;89(1):43–9. Available from: . [CrossRef]
- Ortega J, and Arita HT. (2000), Defence of Females by Dominant Males of Artibeus jamaicensis (Chiroptera: Phyllostomidae). Ethology, 106: 395-407. https://doi-org.pbidi.unam.mx:2443/10.1046/j.1439-0310.2000.00557.x.
- Bai Y, Kosoy M, Recuenco S, Alvarez D, Moran D, Turmelle A, Ellison J, Garcia DL, Estevez A, Lindblade K, Rupprecht C. Bartonella spp. in Bats, Guatemala. Emerg Infect Dis. 2011 Jul;17(7):1269-72. [CrossRef] [PubMed] [PubMed Central]
- Kelm DH, Toelch U, Jones MM. Mixed-species groups in bats: non-random roost associations and roost selection in neotropical understory bats. Front Zool 18, 53 (2021). [CrossRef]
- Vleut I, Galindo-González J, de Boer WF, Levy-Tacher SI, Vazquez LB. Niche Differentiation and its Relationship with Food Abundance and Vegetation Complexity in Four Frugivorous Bat Species in Southern Mexico. Biotropica. 2015; 47: 606-615. [CrossRef]
- Jacquot M, Wallace MA, Streicker DG, Biek R. Geographic Range Overlap Rather than Phylogenetic Distance Explains Rabies Virus Transmission among Closely Related Bat Species. Viruses. 2022; 14(11):2399. [CrossRef]
- Ciminski K, Ran W, Gorka M, et al. Bat influenza viruses transmit among bats but are poorly adapted to non-bat species. Nat Microbiol. 2019; 4, 2298–2309. [CrossRef]
- Kessler S, Burke B, Andrieux G, et al. Deciphering bat influenza H18N11 infection dynamics in male Jamaican fruit bats on a single-cell level. Nat Commun. 2024; 15, 4500. [CrossRef]
- Chothe SK, Bhushan G, Nissly RH, et al. Avian and human influenza virus compatible sialic acid receptors in little brown bats. Sci Rep. 2017; 7, 660. [CrossRef]
- Slater T, Eckerle I, and Chang KC. Bat lung epithelial cells show greater host species-specific innate resistance than MDCK cells to human and avian influenza viruses. Virol J. 2018; 15, 68. [CrossRef]
- Halwe NJ, Gorka M, Hoffmann B, Rissmann M, Breithaupt A, Schwemmle M, Beer M, Kandeil A, Ali MA, Kayali G, Hoffmann D, Balkema-Buschmann A. Egyptian Fruit Bats (Rousettus aegyptiacus) Were Resistant to Experimental Inoculation with Avian-Origin Influenza A Virus of Subtype H9N2, But Are Susceptible to Experimental Infection with Bat-Borne H9N2 Virus. Viruses. 2021 Apr 14;13(4):672. [CrossRef] [PubMed] [PubMed Central]
- Kandeil A, Gomaa MR, Shehata MM, El Taweel AN, Mahmoud SH, Bagato O, Moatasim Y, Kutkat O, Kayed AS, Dawson P, Qiu X, Bahl J, Webby RJ, Karesh WB, Kayali G, Ali MA. Isolation and Characterization of a Distinct Influenza A Virus from Egyptian Bats. J Virol. 2019; Jan 4;93(2):e01059-18. [CrossRef] [PubMed] [PubMed Central]
Figure 1.
Location of 23 sampling sites in the state of Campeche, Mexico (panels a and b). The six bats that tested positive for influenza A antibodies were captured at four sites in three different municipalities, shown in red (sites I, XIII, XVIII, and XXI). The six sites where the six molecularly positive bats were captured are shown in orange (sites I, VI, XIV, XVII, XXII, and XXIII). Only site I, indicated with a triangle, had seropositive and molecularly positive individuals (panel c). Positive species are shown in panels d and e; the latter panel shows the two species with the most positive individuals.
Figure 1.
Location of 23 sampling sites in the state of Campeche, Mexico (panels a and b). The six bats that tested positive for influenza A antibodies were captured at four sites in three different municipalities, shown in red (sites I, XIII, XVIII, and XXI). The six sites where the six molecularly positive bats were captured are shown in orange (sites I, VI, XIV, XVII, XXII, and XXIII). Only site I, indicated with a triangle, had seropositive and molecularly positive individuals (panel c). Positive species are shown in panels d and e; the latter panel shows the two species with the most positive individuals.

Figure 2.
Serological evidence of influenza A virus antibodies in bats from Campeche, Mexico. Blue dots represent the sera that obtained values below the cutoff value (0.7nm) represented by the red dotted line; these sera are considered positive for the presence of influenza A virus antibodies.
Figure 2.
Serological evidence of influenza A virus antibodies in bats from Campeche, Mexico. Blue dots represent the sera that obtained values below the cutoff value (0.7nm) represented by the red dotted line; these sera are considered positive for the presence of influenza A virus antibodies.
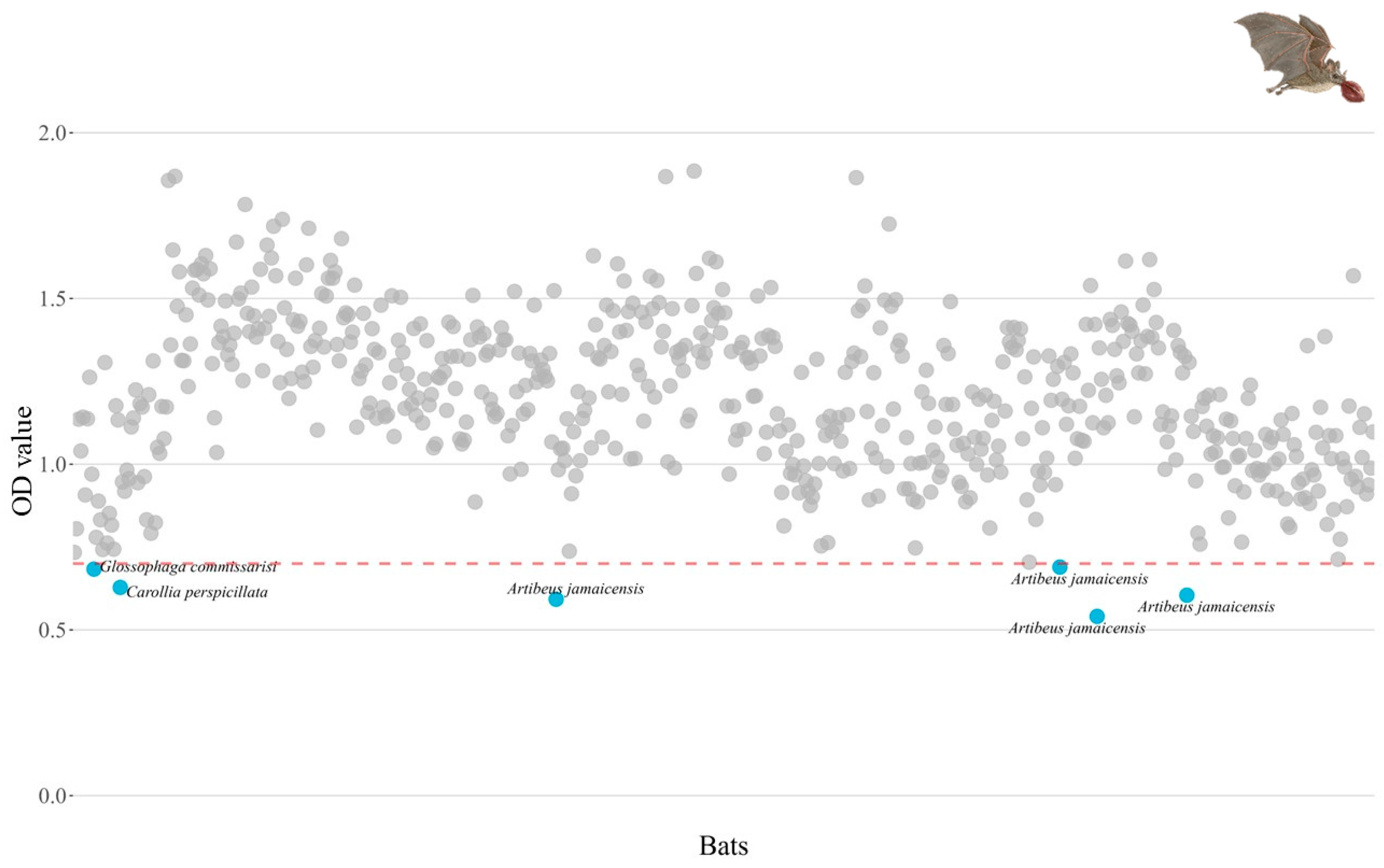

Table 1.
Ct values obtained in bat samples from southeastern Mexico were positive for influenza A virus.
Table 1.
Ct values obtained in bat samples from southeastern Mexico were positive for influenza A virus.
| Bats | Ct | Sample |
|---|---|---|
| Artibeus jamaicensis | 36.25 | Oropharyngeal swab |
| Artibeus jamaicensis | 37.78 | Oropharyngeal swab |
| Carollia sowelli | 37.9 | Oropharyngeal swab |
| Carollia perspicillata | 36.61 | Oropharyngeal swab |
| Dermanura phaeotis | 33.92 | Oropharyngeal swab |
| Micronycteris microtis | 37.55 | Rectal swab |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



