
Submitted:
22 March 2025
Posted:
24 March 2025
You are already at the latest version
Abstract
The crystal structures of naphthalene dicarboxamides, namely 1,4-naphthalene dicarboxamide (1,4-NDA), 2,6-naphthalene dicarboxamide (2,6-NDA), and 2,7-naphthalene dicarboxamide (2,7-NDA), are presented for the first time along with an analysis of their supramolecular organization. The compounds, obtained in single-crystalline form via solvothermal crystallization from methanol, are stable in air to near 350 °C and have melting points above 300 °C. In their densely packed structures (r = 1.43-1.47 cm3g-1) the combination of C11(4) chains and R22(8) rings give one-dimensional hydrogen-bonded ladders with the additional R42(8) pattern. The amide groups and the naphthalene rings form dihedral angles between 22° and 40°. Neighboring H-bond ladders run parallel in 1,4-NDA and 2,6-NDA and are connected by means of the naphthalenedyil cores along, so that two-dimensional (2D) H bonded sheets are obtained Except for a weak intra-sheet π-π stacking in 1,4-NDA there are no π-π stacking and C–H⋯π interactions. The R22(8) rings act as four-connected nodes, leading to the formation of two-dimensional H-bonded planar sheets with sql topology for the nearly linear dicarboxamides 1,4-NDA and 2,6-NDA and cds topology for the angular 2,7-NDA. Hirshfeld surface analysis and NCI plots provide additional insight into the H-bonding interactions.
Keywords:
H-bonding
; naphthalene dicarboxamides
; amide
; ladder
1. Introduction
The important role of hydrogen-bonding to sustain the functioning of complex biological systems has inspired first the Fischer’s lock-and-key model [1] and eventually led to the establishment of supramolecular chemistry as a core field for rational design of self-assembled materials [2,3]. Over the years, perspectives on self-assembly have evolved significantly. Initially, researchers assumed its principles could be universally applied across disciplines, from biology and engineering to materials science. [4,5,6]. Now however, machine learning could be effective in identifying patterns and correlations within complex datasets, making it a valuable tool for targeted structure prediction in self-assembly [7,8,9,10].
The amide group (RCO-NR'R'') is fundamental in the context of hydrogen-bonded solids, due to its high frequency in biological systems, its synthetic affordability, and proven practical utility in material science, with polyamide materials, e.g., Kevlar®, being one of the most recognizable examples [10,11,12]. Additionally, the hydrogen-bonding properties of amides have found extensive application in pharmaceuticals, where they are used to enhance drug efficacy [13,14,15,16,17,18,19].
The self-assembly patterns of secondary, N-monosubstituted amides (RCO-NHR′), having one NH hydrogen bond donor and one CO hydrogen bond acceptor (1:1 ratio), are relatively predictable. It usually leads to the formation of (8) homodimeric rings (Figure 1) as described by Etter’s notation. The hydrogen-bonded (8) bis-amide synthon with syn-NH involved is similar to the (8) ring for carboxylic acids. When the other packing constraints are not favorable, (4) chains with H-bonds from anti-NH to the O atoms are formed (Figure 1) [20,21,22,23]. The primary amides (RCO-NH2) demonstrate a more complex and less predictable self-assembly due to two NH2 hydrogen-bond donors over only one CO acceptor (2:1 ratio). This leads to a larger number of assembly variants, which can include branched hydrogen-bonding or involvement of terminating hydrogen-bond acceptor molecules, typically solvents. Unlike cyclic dicarboxylic acids, which consistently form predictable hydrogen-bonded structures (e.g (8)), primary amides behave less predictably. Therefore, their use in rational design requires a precise understanding of the structural context [24].
Successful supramolecular design with amide groups requires accounting for steric effects and secondary interactions that also influence the resulting network. In this context, our previous study demonstrated how heteroatoms, such as nitrogen, sulfur, and oxygen, present in pyridine, thiophene, and furan rings, significantly affected the hydrogen-bonding motifs in dicarboxamides. The results indicated that both the position and basicity of the heteroatom are crucial in determining the hydrogen-bonded packing arrangement [25].
This study investigates the relative (linear vs. angular) orientation of two carboxamide groups with a naphthalene linker (Figure 2) on the hydrogen-bonding motifs and crystal packing. The naphthalene core would have the additional possibility to engage in π-π stacking and C–H⋯π interactions which are important interactions in the supramolecular chemistry of aromatic ring structures [26,27,28,29,30,31,32,33]. The π-π stacking interactions are typically in the energy range of 2 kJ mol⁻¹, whereas hydrogen bonds range from weak (4 kJ mol⁻¹) to strong (45 kJ mol⁻¹) [34]. Both influence molecular packing and conformational dynamics in various systems, from biological macromolecules, such as protein folding and nucleobase stacking [35,36,37,38] to applications in materials science and drug design [39,40,41,42,43]. Hydrogen-bonded organic frameworks can have functionality in the form of improved conductivity and magnetism, making such materials interesting in the context of organic electronics, molecular magnetism, and spintronics [44,45,46].
2. Materials and Methods
All chemicals were purchased commercially and used without further purification (see Supplementary Materials, Section S1). Deionized water was used in all cases where water is mentioned. Single-crystal X-ray diffraction (SCXRD) data were collected using a Rigaku XtaLAB Synergy S diffractometer (Rigaku, Tokyo, Japan) equipped with a hybrid pixel array detector and a PhotonJet copper X-ray source (λ = 1.54184 Å). Suitable crystals were selected under a Leica M80 polarized-light micro-scope (Leica, Wetzlar, Germany) and mounted on a cryo-loop in oil. The data were processed using CrysAlisPro, which includes cell refinement, reduction, and absorption correction. The structure was solved and refined with Olex2, using SHELXT for structure solution and SHELXL for refinement [47,48,49]. The molecular graphics were created using Diamond 5 software [50]. The X-ray powder diffraction (PXRD) patterns were recorded on a Rigaku Mini-Flex600 diffractometer (Rigaku, Tokyo, Japan) (600 W, 40 kV, 15 mA) at room temperature using Cu-Kα radiation (λ = 1.54184 Å). The PXRD patterns were normalized based on the intensity of the highest peak. The simulation of the PXRD data as well as the graph set analysis were performed using MERCURY 2020.3.0 software based on the single-crystal XRD data [51]. The Hirshfeld surface analysis was carried out using the CrystalExplorer software [52], while the ”Atoms in Molecules” (AIM) and scatterplot analyses were performed using MultiWFN 3.8 with .wfn files generated from crystal structure data via DFT (B3LYP/6-31G**) in Gaussian16 [53] (note the precision limitations due to reasonable, but somewhat limited accounting electron-correlation, important for weaker interactions, by the LYP correlation functional). AIM analysis was conducted on molecular coordinates from single-crystal structures, and energy values were calculated for each distinct hydrogen bond between crystallographically independent molecules. The NCI figures and scatterplots were prepared in VMD 1.9.4 and gnuplot 5.4, respectively [54,55].
Fourier-transform infrared (FT-IR) spectra were measured between 500 and 4000 cm-1 using a Bruker TENSOR 37 IR spectrometer (Bruker, Billerica, MA, USA) in ATR mode (Platinum ATR-QL, Diamond). Nuclear magnetic resonance (NMR) spectra were ac-quired on a Bruker Avance III—300 spectrometer (Bruker, Billerica, MA, USA) with the operating frequency of 300 MHz for 1H-NMR. Electron impact (EI) mass spectra were recorded on a Thermo Finnigan Trace DSQ spectrometer (Thermo Fisher Scientific).
Thermogravimetric analysis (TGA) was performed under air using a Netzsch TG209 F3 Tarsus (Netzsch, Selb, Germany) with a heating rate of 10 K min-1 up to 1000 °C. Melting points were determined using a Büchi Melting Point B-540 apparatus in an open capillary (Büchi Labortechnik AG, Flawil, Switzerland).
2.1. Synthesis of the Dicarboxamides
The synthesis of the naphthalene-based dicarboxamides was conducted using a variant of a classical method, involving the conversion of the commercially available carboxylic acids to chloroanhydrides followed by the subsequent transformation to amides [56]. The amount of 2.16 g (10.00 mmol) of the respective naphthalenedicarboxylic acid was dissolved in 25 mL of thionyl chloride and subjected to reflux at 90 °C for a duration of 12 hours, in the presence of a catalytic amount (1 mmol) of dimethylformamide (DMF). After this time, the excess thionyl chloride was distilled off, and the resulting residue washed with hexane (3 × 15 mL) followed by drying under vacuum (~10-2 Torr, room temperature). Subsequently, 5 mL of a 25% aqueous ammonia solution was added dropwise while stirring in an ice bath. The reaction mixture was maintained under stirring on an ice bath for one hour, after which the solvents were removed under reduced pressure. The obtained solid was thoroughly washed with water and ethanol (3×15 mL) before being dried under vacuum to yield 1.88 g (88%) for 1,4-NDA, 1.92 g (90%) for 2,6-NDA, and 1.96 g (91%) for 2,7-NDA.
The crystals, suitable for SCXRD studies, were grown by slow cooling of a methanolic solution. A clear solution, nearly saturated at ~100 °C, in a sealed culture tube was cooled with a rate of 0.8 °C h-1 to room temperature to yield colorless single crystals. The experimentally obtained powder X-ray diffraction patterns matched well with the simulated patterns derived from the crystal structures. Thereby, the phase purities of the bulk crystalline products were confirmed (see Supplementary Materials, Section S6).
3. Results and Discussion
The dicarboxamides 1,4-naphthalenedicarboxamide (1,4-NDA), 2,6-naphthalenedicarboxamide (2.6-NDA) and 2,7-naphthalenedicarboxamide (2,7-NDA) were synthesized from the corresponding dicarboxylic acids via conversion to acyl chloride by thionyl chloride, followed by reaction with aqueous ammonia under Shotten-Baumann reaction conditions [56]. The isolated products were characterized and their purity confirmed by 1H-NMR, IR, and mass spectrometry (see Supplementary Materials, Sections S3-S5).
Successful crystallization of all the three compounds was achieved from methanol. The latter was chosen due to sufficient solubilizing capability at elevated temperatures combined with moderate-to-low probability of the non-desirable incorporation into the structure as a solvent of crystallization.
3.1. Crystal Structures of the Dicarboxamides 1,4-NDA, 2,6-NDA and 2,7-NDA
The molecular structures within the asymmetric unit are illustrated in Figure 3. Compounds 1,4-NDA and 2,6-NDA crystallize in the triclinic system, 2,7-NDA in monoclinic system. While both 1,4-NDA and 2,6-NDA are assigned to the space group P, their asymmetric units feature different content. The asymmetric unit of 1,4-NDA contains a single molecule (Z = 2, Z' = 1), while that of 2,6-NDA consists of half a molecule in the asymmetric unit (Z = 4, Z' = 0.5). In contrast, 2,7-NDA crystallizes in the C2/c space group, with a quarter of the molecule representing the asymmetric unit. The hydrogen-bond graph-set notation depicts only the basic units with the shortest rings and chains as shown in Figure 1d and calculated by Mercury [51].
In 1,4-NDA the Cnaph–Camide bond vectors are close to parallel to each other (acute angle of 10.6°), the dihedral angle between the amide planes defined by the N-C-O atoms is 14.2°, not far from coplanarity. In 2,6 NDA the Cnaph–Camide bond vectors are exactly parallel, the amide planes exactly coplanar by the centrosymmetry of the molecule. In 2,7 NDA the Cnaph–Camide bond vectors form an angle of 64.8°, the amide N-C-O planes a dihedral angle of 43.6°.
3.1.1. Hydrogen Bonding in 1,4-NDA
The basic H-bonded self-assembly of the structure of 1,4-NDA is a combination of (4) chains and (8) rings which generates a one-dimensional (1D) almost straight ladder parallel to the a axis with its additional (8) pattern (Figure 4, cf. Figure 1, Table 1). This 1D H-bond ladder is common for all reported structures here. Neighboring ladders run parallel and are connected by means of the naphthalene-1,4-dyil molecular cores along the b axis, so that two-dimensional H bonded sheets in the ab plane are obtained (Figure 4). If the H-bonded bis-amide (8) rings are viewed as four-connected nodes – with two opposite edges defined by the naphthalene connectors and the other two the side-connections via the (8) ring to the neighboring (8) rings – then the topology of the sheets corresponds to a square planar net, sql (see Section 3.1.4).

Table 1.
Crystal data for 1,4-NDA, 2,6-NDA and 2,7-NDA.
| 1,4-NDA | 2,6-NDA | 2,7-NDA | |
|---|---|---|---|
| empirical formula | C12H10N2O2 | C12H10N2O2 | C12H10N2O2 |
| mol wt (g mol–1) | 214.22 | 214.22 | 214.22 |
| temperature (K) | 150 | 150 | 150 |
| crystal system | triclinic | triclinic | monoclinic |
| space group | P | P | C2/c |
| a (Å) | 5.0306 (1) | 5.0058 (3) | 6.9974 (2) |
| b (Å) | 9.9799 (3) | 7.2347 (7) | 7.2089 (2) |
| c (Å) | 10.0706 (3) | 7.4349 (7) | 19.4417 (5) |
| α (deg) | 98.620 (2) | 68.216 (9) | 90.00 |
| β (deg) | 92.433 (2) | 82.306 (7) | 98.110 (2) |
| γ (deg) | 96.459 (2) | 80.076 (7) | 90.00 |
| Volume, V (Å3) | 495.80 (2) | 245.57 (4) | 970.90 (5) |
| Z, Z’ | Z = 2, Z´=1 | Z = 1, Z´=0.5 | Z = 4, Z´= 0.5 |
| Dcalc (g/cm3) | 1.435 | 1.449 | 1.466 |
| μ (mm–1) | 0.823 | 0.830 | 0.840 |
| F(000) | 224 | 112 | 448 |
| crystal size [mm3] | 0.65 × 0.05 × 0.02 | 0.13 × 0.07 × 0.04 | 0.13 × 0.07 × 0.05 |
| wavelength (Å) | 1.54184 | 1.54184 | 1.54184 |
| No. of unique reflections | 2112 | 938 | 1042 |
| No. of total reflections | 19080 | 2180 | 5554 |
| No. of parameters | 161 | 73 | 82 |
| Rint | 0.0509 | 0.0304 | 0.0245 |
| R1[F2 > 2σ(F2)] (a) | 0.0486 | 0.0539 | 0.0358 |
| wR1[F2 > 2σ(F2)] (a) | 0.1438 | 0.1547 | 0.1006 |
| R2, wR2(F2) [all data](a) | 0.0535, 0.1492 | 0.0609, 0.1604 | 0.0414, 0.1059 |
| S [all data] (a) | 1.045 | 1.134 | 1.095 |
| Δρmax, Δρmin (e·Å−3) (b) | 0.320, -0.215 | 0.279, -0.207 | 0.158, -0.193 |
| CCDC no. | 2431507 | 2431506 | 2431508 |
(a) R1 = [Σ(||Fo| − |Fc||)/Σ|Fo|]; wR2 = [Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]]1/2. Goodness-of-fit S =[Σ[w(Fo2 − Fc2)2]/(n − p)]1/2. (b) Largest difference peak and hole.
Table 2.
The parameters of the H-bonded interactions in the crystal structure of 1,4-NDA.
| D—H···A(a)(a) | D—H [ Å] | H···A [ Å] | D···A [ Å] | D—H···A [deg] | Bond Energy [kcal/mol] |
| N1—H1A···O2iii | 0.90 (2) | 2.07 (2) | 2.9601 (16) | 170.8 (17) | − 16 |
| N1—H1B···O1iv | 0.93 (2) | 1.96 (2) | 2.8105 (16) | 152.4 (18) | −17 |
| N2—H2A···O1i | 0.92 (2) | 2.01 (2) | 2.9255 (16) | 177.5 (16) | − 16 |
| N2—H2B···O2ii | 0.92 (2) | 1.98 (2) | 2.8386 (16) | 153.2 (19) | − 19 |
(a) Symmetry codes: (i) x, y+1, z; (ii) x+1, y, z; (iii) x, y-1, z; (iv) x-1, y, z.
The amide group rotates out of the aromatic plane, with a dihedral angle ϕ between the planes of the naphthalene core and the amide moiety close to 40° (Figure 5, Table 3). The two amide groups in a 1,4-NDA molecule are close to coplanar, which leads to the parallel orientation of their respective 1D H-bond ladders. The deplanarization of the naphthalenedicarboxamide molecules is a prerequisite for the 1D-ladder structure where otherwise the (4) edge of the(8) ring (Figure 4) would be shorter than the length of naphthalene moiety, making such a ladder impossible.
At the same time the naphthalene cores participate in a weak π-π interaction (dintc ≈ 3.8 Å inter-centroid distance) along the chains and within the 2D layer. The optimization of the π-π interaction also influences the ϕ rotation angle. Accordingly, the annulated C6 rings of the 1,4-NDA molecules are oriented to one side of the 2D sheet. There are no inter-sheet π-π stacking and C–H⋯π interactions.
3.1.2. Hydrogen Bonding in 2,6-NDA
As in 1,4-NDA from the combination of (4) chains and (8) rings, 1D H-bonded ladders, albeit now kinked, are generated with the additional (8) pattern. Again, the 1D ladders run parallel to each other and parallel to the a axis. The bridging action of the naphthalene-2,6-diyl core takes place along the bc diagonal, so that two-dimensional H-bonded sheets parallel to the {0 1 -1} (or {0 -1 1} planes are obtained (Figure 6, Table 4). As for 1,4-NDA the topology of the sheets corresponds to a square planar net, sql (see Section 3.1.4). The 2,6-NDA molecule shows a dihedral angle ϕ = 29° between the naphthalene core and the amide group (Figure 5, Table 3). The two amide groups in a 2,6-NDA molecule are coplanar by the centrosymmetry of the molecule, which leads to the parallel orientation of their respective 1D H-bond ladders. There are no inter-sheet π-π stacking or C–H⋯π interactions between the naphthalene rings in 2,6-NDA.
3.1.3. Hydrogen Bonding in 2,7-NDA
The combination of (4) chains and (8) rings lead again to 1D H-bonded ladders with the additional (8) pattern. The ladders are kinked as in 2,6-NDA. Each amide group of 2,7-NDA is part of a 1D ladder but different to 1,4-NDA and 2,6-NDA the ladders of the two amide groups from a 2,7-NDA molecule neither run parallel to each other nor parallel to a crystallographic axis (Figure 7, Table 5). This non-parallel and almost perpendicular orientation of the H-bond ladders at an angle of 88.2° is due to the angular disposition of the amide groups in 2,7-NDA. Both the angle of 64.8° between the Cnaph–Camide bond vectors and the dihedral angle of 43.6° between the amide N-C-O planes are far off co-linearity and co-planarity, respectively as in 1,4-NDA and 2,6-NDA
While the co-linear and co-planar amide arrangement in 1,4-NDA and 2,6-NDA yielded parallel 1D H-bond ladders arranged in 2D sheets, the angular disposition of the amide groups in 2,7-NDA yields 1D-ladders close to perpendicular to each other and, thereby, arranged in a 3D H-bond network. On the level of the 4-connected bis-amide (8) node, the topology is then a CdSO4 (cds) net. The latter is the primary alternative to the square planar net (sql), when two out of the four neighboring nodes in a 4-c net are situated off-plane (see Section 3.1.4). There are again no π-π stacking or C–H⋯π interactions between the naphthalene rings in 2,7-NDA.
3.1.4. Comparative Topological Presentation
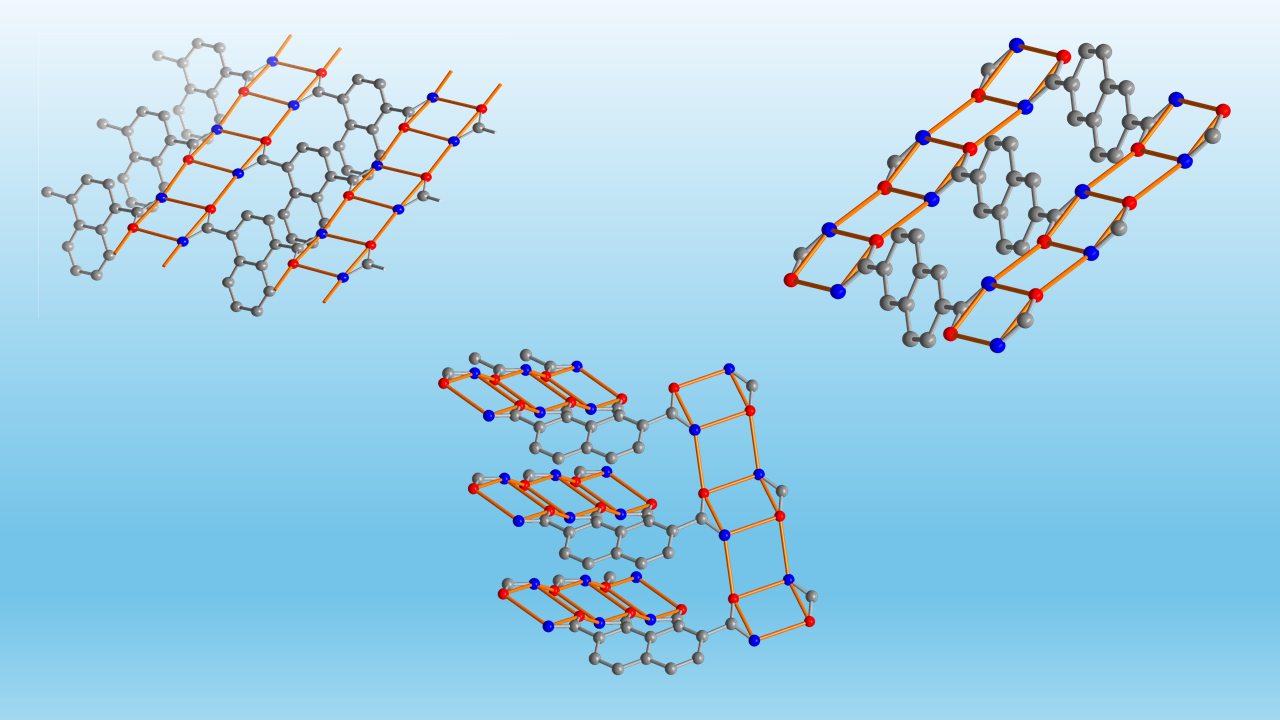
A direct comparison of the linear 1D H-bond ladders and their relative parallel orientation in 2D sheets in 1,4-NDA and 2,6-NDA versus an almost perpendicular orientation in a 3D net is given in Figure 8.
As for the topology analysis of the structures, in all of them the bis-amide (8) rings are taken as four-connected nodes. The H-bonds connect these bis-amide rings in one direction in 1D ladders, while the naphthalene cores then connect these 1D ladders.
In the structure of 1,4-NDA and 2,6-NDA then sheets with sql topology are formed, with an inter-sheet distance of 4.1 Å and 4.2 Å, respectively. Adjacent, neighboring sheets are arranged in a staggered fashion, with the naphthalene units of one sheet positioned above the 1D H-bond ladders of the next sheet (Figure 9a,b).
The topology in 2,7-NDA differs from the previous cases of 2D structures, because of the nearly perpendicular directions of the 1D H-bond ladder propagation. Thus, 2,7-NDA features a 3D network structure (Figure 9c). The resulting 3D CdSO4 (cds) topology is typical for the case, when the connectivity of the neighboring square-planar nodes proceeds no longer close to parallel but off-plane or more perpendicular to each other. It is worth recalling, that there are no H-bonds, no π-π stacking or C–H⋯π interactions between the naphthalene rings between the sheets in both 1,4-NDA and 2,6-NDA or within the network of 2,7-NDA. Relevant π-π stacking interactions require centroid-centroid contacts of less than 3.8 Å, near parallel ring planes, small slip angles and small vertical displacements (slippage <1.5 Å) which would translate into a sizable overlap of the aryl-plane areas [33]. Significant intermolecular C-H··· π contacts are below 2.7 Å for the (C-)H···ring centroid distances with H-perp also below 2.6-2.7 Å and C-H·· centroid > 145° [67,68].
3.2. Hirshfeld Analysis
The analysis of the Hirshfeld surface, which defines the boundaries of each molecular entity based on the electron density distribution, is used here as a practical visualization tool for the strength of the intermolecular interactions with di and de, the distances from the point on the Hirshfeld surface to the nearest atom in the neighbor- and in the same molecule respectively, being the measures of the interaction strengths via their comparison with the typical van der Waals radii by means of a normalized distance function. The latter, dnorm, is equal to (di-dvdwi)/dvdwi + (di-dvdwe)/dvdwe – where dvdwi and dvdwe are the van der Waals radii of the closest atoms of the inner and outer molecules – and describe the deflection from the average van der Waals distances.
The Hirshfeld surfaces, derived from electron density distributions, enable a depiction of intermolecular contacts (Figure 10). In the two-dimensional de and di contact distance fingerprint plots (Figure 11) a visualization of the N-H···O intermolecular interactions is depicted by the spikes directed towards the lower left corner. The individual O···H, N···H, C···H, C···C, and H···H interaction sub-plots distinguish between the respective interactions and are provided in the Supplementary Materials, Section S7.
All examined dicarboxamides display characteristic spikes in the Hirshfeld 2D plots, indicating the formation of N-H···O hydrogen bonds (Figures S22, S27 and S33). These spikes extend for 1,4-NDA and 2,7-NDA to (di, de) ≈ (<0.8, 1.1) and correspond to H···O distances of 1.9–2.0 Å. For 2,6-NDA, the spikes are slightly shorter (di, de) ≈ (0.8, 1.2), consistent with slightly longer hydrogen bonds (Table 2, Table 4, and Table 5). The O···H contacts for the classic strong N-H···O hydrogen bonds have nearly the same share of 25-28% of all intermolecular contacts for the three NDA structures. Broad wings at di + de ≈ 2.8 Å represent C···H interactions (Figure S21), while H···H interactions dominate the remaining interactions (Figure S22). The distinctly green colored zones around di = de ≈ 1.9 Å are associated primarily with C···C contacts (Figure S20). The majority of the intermolecular interactions are represented by the combined weak C···H, H···H and C···C van der Waals contacts with a total of 66-69%, which is typical for organic structures [57,58,59,60]).
The contribution of the C···C contacts is slightly higher in the 1,4-NDA structure (8.6% compared to 5.4% and 5.1%), because of the weak intra-ladder π-π interactions, seen in this structure.
An interesting small detail on the 2D fingerprint plot for 2,7-NDA is the presence of an additional green spike (in-between the characteristic hydrogen bond spikes for the strong hydrogen bonds) extending to di = de ≈ 1.7 Å. This spike corresponds to somewhat stronger H···H interactions.
3.3. NCI Plots
The Non-Covalent Interaction (NCI) plot method graphically represents non-covalent interactions in real space using color-coded 3D isosurfaces: blue for attractive, green for van der Waals interactions, and red for steric repulsions, highlighting their spatial distribution within molecular systems (Figure 12). These 3D isosurfaces can also be visualized in NCI scattering plots, which use the Reduced Density Gradient (RDG) and (sign (λ2)ρ) to characterize interactions. Similar to the 3D plot, scattering plots of RDG vs. (sign (λ2)ρ) show strong attractions (blue) at lower density (ρ < 0.01 a.u.), repulsive interactions (red) at higher density (ρ > 0.01 a.u.), and weak van der Waals interactions (green) in the intermediate range [61,62,63]. The position and intensity of the spikes provide insights into non-covalent interactions. However, NCI analysis remains qualitative and does not quantify individual energy contributions from Coulombic and dispersive interactions. While the Hirshfeld analysis provides a global and semi-quantitative assessment of intermolecular contacts, the NCI plot offers a local and rather qualitative analysis of individual non-covalent interactions; together, they complement each other to provide a comprehensive interaction profile.
For the NDA structures, strong non-covalent interactions appear as pronounced downward blue spikes in the scattering plot, corresponding to small blue isosurfaces of N-H···O hydrogen bonds. Additionally, intense green spikes with a significant fraction are observed in the attractive interaction region, representing the weak C···H, H···H and C···C van der Waals contacts. A deeper look into the green spike region in the scattering plots reveals subtle structural differences. Notably, in 1,4-NDA, the most intense green spike splits downward, with its largest contribution occurring at a lower ρ critical value sign (λ2)ρ < − 0.01 a.u., indicating stronger π···π interactions (vide supra). In contrast, for the other structures, the green spike is divided into three components, with the most prominent part of peaks appearing at a higher ρ critical value sign (λ2)ρ > − 0.01 a.u., suggesting no π···π interactions compared to 1,4-NDA.
3.4. Thermogravimetric Analysis and Melting Points
The temperature stability of the compounds was assessed by thermogravimetric analysis (TGA) (Figure 13).
TGA reveals that all investigated dicarboxamides exhibit very similar thermal behavior. Both 1,4-NDA and 2,6-NDA show an oxidative degradation starting at around 305 °C with the oxidation finished at approximately 360 °C. For 2,7-NDA, a comparable degradation pattern is observed, but the oxidation starts slightly earlier at 290 °C and is nearly complete at around 345 °C, leaving a small amount of residue, which completely burns off only at ~480° (a possible hypothesis is that those are residues reach in triazine, which are the condensation products of the amides. They form at elevated temperatures and oxidize less eagerly).
The melting points were measured using the open capillary method (1.15 mm diameter), where the temperature gradually increased while visually monitoring the samples. Notably, all NDA compounds melt within a specific temperature range before undergoing decomposition. The recorded melting intervals were 325–330 °C for 1,4-NDA, 322–330 °C for 2,6-NDA, and 300–312 °C for 2,7-NDA, aligning with the onset of decomposition observed in the TGA analysis. Additionally, all samples changed color to brown and shrank before reaching their melting points, indicating partial decomposition before complete melting.
4. Conclusions
The structural investigation of three primary naphthalenedicarboxamide opens up an interesting uncharted territory of supramolecular periodic structures of dicarboxamides with a non-heterocyclic annulated aromatic core. While primary amides are among the most important functionalities regarding supramolecular chemistry of both molecular and periodic H-bonded solids, the structural chemistry of annulated aromatic amides of is nearly uncharted. The structures of the unsubstituted 1-naphthaleneamide and 2-naphthaleneamide are unknown, the only related reported structure is those of 9-anthraceneamide [64] in addition to 6-bromo-2-naphthaleneamide [65] and 5,6,7,8-tetrafluoro-2-naphthaleneamide [66]. The structures of primary bis- or polyamides of naphthalenes or larger non-heterocyclic annulated aromatic derivatives are not reported at all.
It was shown that in all three investigated naphthalenedicarboxamide structures a highly favorable one-dimensional (1D) H-bonded amide ladder motif is formed from the combination of (4) chains and (8) as well as (8) pattern. In 1,4-NDA and in 2,6-NDA, where the amide groups are arranged close to linear, the parallel 1D hydrogen-bonded ladders assemble into two-dimensional (2D) sheets exhibiting sql topology. In 2,7-NDA, the bend or angular amide disposition results in an almost perpendicular arrangement of the amide ladders. Consequently, the hydrogen-bonded network extends into a three-dimensional (3D) cds topology. There are no π-π stacking or C–H···π interactions of the napthalene rings between the 2D sheets in 1,4-NDA and in 2,6-NDA or in the 3D network of 2,7-NDA.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Section S1: Chemicals used; Section S2: Preparation of dicarboxamides; Section S3: NMR spectra of the dicarboxamides; Section S4: IR spectra of the dicarboxamides; Section S5: Mass spectra of the dicarboxamides; Section S6: PXRD spectra of the dicarboxamides; Section S7: 2D Hirshfeld plots of the dicarboxamides.
Author Contributions
Conceptualization, A.M. and C.J.; methodology, A.M.; software, A.M.; T.S. validation, A.M., I.B.; formal analysis, A.M., I.B., N.R., P.S., T.S and C.J.; investigation, A.M. and N.R.; resources, C.J.; data curation, A.M., P.S, T.S. and N.R.; writing—original draft preparation, A.M., I.B.; writing—review and editing, C.J.; visualization, A.M., I.B. and C.J.; supervision, I.B. and C.J.; project administration, C.J.; funding acquisition, C.J. All authors have read and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Deutsche Forschungsgemeinschaft (DFG) under grant 440366605 (for the Rigaku diffractometer).
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The CCDC numbers 2431506-2431508 for compounds 2,6-NDA, 1,4-NDA and 2,7-NDA, respectively, contain the supplementary crystallographic data reported in this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (accessed on 15 March 2025).
Acknowledgments
The authors would like to thank Birgit Tommes for the IR measurements. We also thank the Center for Molecular and Structural Analytics at Heinrich Heine University (CeMSA@HHU) for recording the mass spectrometric and NMR-spectrometric data.
Conflicts of Interest
The authors declare no conflict of interest.”.
References
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme (Influence of configuration on the action of enzymes). Ber. Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Pauling, L. The nature of the chemical bond. II. The one-electron bond and the three-electron bond. J. Am. Chem. Soc. 1931, 53, 3225–3237. [Google Scholar] [CrossRef]
- Lugger, S.J.D.; Houben, S.J.A.; Foelen, Y.; Debije, M.G.; Schenning, A.P.; Mulder, D.J. Hydrogen-bonded supramolecular liquid crystal polymers: smart materials with stimuli-responsive, self-healing, and recyclable properties. Chem. Rev. 2021, 122, 4946–4975. [Google Scholar] [CrossRef]
- Stang, P.J.; Olenyuk, B. Self-assembly, symmetry, and molecular architecture: Coordination as the motif in the rational design of supramolecular metallacyclic polygons and polyhedra. Acc. Chem. Res. 1997, 30, 502–518. [Google Scholar] [CrossRef]
- Keller, E.F. Self-organization, self-assembly, and the origin of life. Mapping the Future of Biology: Evolving Concepts and Theories; Springer, 2009; pp. 131–140. [Google Scholar] [CrossRef]
- Palma, C.-A.; Cecchini, M.; Samorì, P. Predicting self-assembly: from empirism to determinism. Chem. Soc. Rev. 2012, 41, 3713–3730. [Google Scholar] [CrossRef]
- Dijkstra, M.; Luijten, E. From predictive modelling to machine learning and reverse engineering of colloidal self-assembly. Nat. Mater. 2021, 20, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Han, J.; Cao, T.; Lam, W.; Fan, B.; Tang, W.; Chen, S.; Fok, K.L.; Li, L. Design of self-assembly dipeptide hydrogels and machine learning via their chemical features. Proc. Natl. Acad. Sci. 2019, 116, 11259–11264. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, R.; Yan, X.; Fan, K. Machine learning facilitating the rational design of nanozymes. J. Mater. Chem. B 2023, 11, 6466–6477. [Google Scholar] [CrossRef] [PubMed]
- Gardin, A.; Perego, C.; Doni, G.; Pavan, G.M. Classifying soft self-assembled materials via unsupervised machine learning of defects. Commun. Chem. 2022, 5, 82. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, X.; Shen, J.; Gao, C.; van der Bruggen, B. The potential of Kevlar aramid nanofiber composite membranes. J. Mater. Chem. A 2020, 8, 7548–7568. [Google Scholar] [CrossRef]
- Prasad, V.V.; Talupula, S. A review on reinforcement of basalt and aramid (Kevlar 129) fibers. Mater. Today: Proc. 2018, 5, 5993–5998. [Google Scholar] [CrossRef]
- Hubbard, R.E.; Haider, M.K. Hydrogen bonds in proteins: role and strength. Encycl. Life Sci. 2010, 1, 1–6. [Google Scholar] [CrossRef]
- Dong, Y.; Yao, C.; Zhu, Y.; Yang, L.; Luo, D.; Yang, D. DNA functional materials assembled from branched DNA: design, synthesis, and applications. Chem. Rev. 2020, 120, 9420–9481. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, K.M. Functional materials based on molecules with hydrogen-bonding ability: applications to drug co-crystals and polymer complexes. R. Soc. Open Sci. 2018, 5, 180564. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Deng, S.-S.; Pan, S.-Y.; Wu, Z.-Y.; Hu, Y.-Y.; Li, K.; Zhou, Y.; Li, J.-T.; Huang, L.; Sun, S.-G. Multivalent amide-hydrogen-bond supramolecular binder enhances the cyclic stability of silicon-based anodes for lithium-ion batteries. ACS Appl. Mater. Interfaces. 2021, 13, 22567–22576. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, J.; Wang, K.; Huang, W. Recent structural engineering of polymer semiconductors incorporating hydrogen bonds. Adv. Mater. 2022, 34, 2110639. [Google Scholar] [CrossRef]
- Galuska, L.A.; Ocheje, M.U.; Ahmad, Z.C.; Rondeau-Gagné, S.; Gu, X. Elucidating the role of hydrogen bonds for improved mechanical properties in a high-performance semiconducting polymer. Chem. Mat. 2022, 34, 2259–2267. [Google Scholar] [CrossRef]
- Yu, X.; Li, C.; Gao, C.; Zhang, X.; Zhang, G.; Zhang, D. Incorporation of hydrogen-bonding units into polymeric semiconductors toward boosting charge mobility, intrinsic stretchability, and self-healing ability. SmartMat 2021, 2, 347–366. [Google Scholar] [CrossRef]
- Leiserowitz, L.; Schmidt, G.M. Molecular packing modes. Part III. Primary amides. J. Chem. Soc. A 1969, 2372–2382. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C. Aggregate structures of carboxylic acids and amides. Isr. J. Chem. 1985, 25, 312–319. [Google Scholar] [CrossRef]
- Etter, M.C. Hydrogen bonds as design elements in organic chemistry. J. Phys. Chem. 1991, 95, 4601–4610. [Google Scholar] [CrossRef]
- Leiserowitz, L. Molecular packing modes. Carboxylic acids. Acta Crystallogr. B: Structural Crystallography and Crystal Chemistry 1976, 32, 775–802. [Google Scholar] [CrossRef]
- Mohabbat, A.; Salama, J.; Seiffert, P.; Boldog, I.; Janiak, C. Single-Crystal Structure Analysis of Dicarboxamides: Impact of Heteroatoms on Hydrogen Bonding of Carboxamide Groups. Crystals 2024, 14, 811. [Google Scholar] [CrossRef]
- Shishkina, S.V.; Konovalova, I.S.; Kovalenko, S.M.; Trostianko, P.V.; Geleverya, A.O.; Bunyatyan, N.D. Hydrogen bonding vs. stacking interaction in the crystals of the simplest coumarin derivatives: a study from the energetic viewpoint. CrystEngComm 2019, 21, 6945–6957. [Google Scholar] [CrossRef]
- Niu, D.; Jiang, Y.; Ji, L.; Ouyang, G.; Liu, M. Self-assembly through coordination and π-stacking: controlled switching of circularly polarized luminescence. Angew. Chem. 2019, 131, 6007–6011. [Google Scholar] [CrossRef]
- Li, G.-L.; Zhuo, Z.; Wang, B.; Cao, X.-L.; Su, H.-F.; Wang, W.; Huang, Y.-G.; Hong, M. Constructing π-stacked supramolecular cage based hierarchical self-assemblies via π··· π stacking and hydrogen bonding. J. Am. Chem. Soc. 2021, 143, 10920–10929. [Google Scholar] [CrossRef]
- Zhuang, W.-R.; Wang, Y.; Cui, P.-F.; Xing, L.; Lee, J.; Kim, D.; Jiang, H.-L.; Oh, Y.-K. Applications of π-π stacking interactions in the design of drug-delivery systems. J. Control. Release 2019, 294, 311–326. [Google Scholar] [CrossRef]
- Huang, C.-W.; Mohamed, M.G.; Zhu, C.-Y.; Kuo, S.-W. Functional supramolecular polypeptides involving π–π stacking and strong hydrogen-bonding interactions: a conformation study toward carbon nanotubes (CNTs) dispersion. Macromolecules 2016, 49, 5374–5385. [Google Scholar] [CrossRef]
- Yagai, S. Supramolecularly engineered functional π-assemblies based on complementary hydrogen-bonding interactions. Bull. Chem. Soc. Jpn. 2015, 88, 28–58. [Google Scholar] [CrossRef]
- Burattini, S.; Greenland, B.W.; Merino, D.H.; Weng, W.; Seppala, J.; Colquhoun, H.M.; Hayes, W.; Mackay, M.E.; Hamley, I.W.; Rowan, S.J. A healable supramolecular polymer blend based on aromatic π− π stacking and hydrogen-bonding interactions. J. Am. Chem. Soc. 2010, 132, 12051–12058. [Google Scholar] [CrossRef]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc., Dalton Trans. 2000, 3885–3896. [Google Scholar] [CrossRef]
- Calhorda, M.J. Weak hydrogen bonds: theoretical studies. Chem. Commun. 2000, 801–809. [Google Scholar] [CrossRef]
- Bogdanov, A.M.; Acharya, A.; Titelmayer, A.V.; Mamontova, A.V.; Bravaya, K.B.; Kolomeisky, A.B.; Lukyanov, K.A.; Krylov, A.I. Turning on and off photoinduced electron transfer in fluorescent proteins by π-stacking, halide binding, and Tyr145 mutations. J. Am. Chem. Soc. 2016, 138, 4807–4817. [Google Scholar] [CrossRef]
- Ardejani, M.S.; Powers, E.T.; Kelly, J.W. Using cooperatively folded peptides to measure interaction energies and conformational propensities. Acc. Chem. Res. 2017, 50, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Beall, E.; Ulku, S.; Liu, C.; Wierzbinski, E.; Zhang, Y.; Bae, Y.; Zhang, P.; Achim, C.; Beratan, D.N.; Waldeck, D.H. Effects of the backbone and chemical linker on the molecular conductance of nucleic acid duplexes. J. Am. Chem. Soc. 2017, 139, 6726–6735. [Google Scholar] [CrossRef]
- Heller, B.A.; Gindt, Y.M. A biochemical study of noncovalent forces in proteins using phycocyanin from Spirulina. J. Chem. Educ. 2000, 77, 1458. [Google Scholar] [CrossRef]
- Wang, S.; Guo, G.; Lu, X.; Ji, S.; Tan, G.; Gao, L. Facile soaking strategy toward simultaneously enhanced conductivity and toughness of self-healing composite hydrogels through constructing multiple noncovalent interactions. ACS Appl. Mater. Interfaces. 2018, 10, 19133–19142. [Google Scholar] [CrossRef]
- Meli, M.; Engel, H.; Laor, D.; Gazit, E.; Colombo, G. Mechanisms of metabolite amyloid formation: computational studies for drug design against metabolic disorders. ACS Med. Chem. Lett. 2019, 10, 666–670. [Google Scholar] [CrossRef]
- Yang, D.; Gao, S.; Fang, Y.; Lin, X.; Jin, X.; Wang, X.; Ke, L.; Shi, K. The π–π stacking-guided supramolecular self-assembly of nanomedicine for effective delivery of antineoplastic therapies. Nanomed. 2018, 13, 3159–3177. [Google Scholar] [CrossRef]
- Wei, X.; Wang, Y.; Xiong, X.; Guo, X.; Zhang, L.; Zhang, X.; Zhou, S. Codelivery of a π–π stacked dual anticancer drug combination with nanocarriers for overcoming multidrug resistance and tumor metastasis. Adv. Funct. Mater. 2016, 26, 8266–8280. [Google Scholar] [CrossRef]
- Li, P.; Ryder, M.R.; Stoddart, J.F. Hydrogen-bonded organic frameworks: a rising class of porous molecular materials. Acc. Mater. 2020, 1, 77–87. [Google Scholar] [CrossRef]
- Ueda, A.; Hatakeyama, A.; Enomoto, M.; Kumai, R.; Murakami, Y.; Mori, H. Modulation of a molecular π-electron system in a purely organic conductor that shows hydrogen-bond-dynamics-based switching of conductivity and magnetism. Chem. Eur. J. 2015, 21, 15020–15028. [Google Scholar] [CrossRef]
- Ren, X.M.; Nishihara, S.; Akutagawa, T.; Noro, S.; Nakamura, T. Design of a Magnetic Bistability Molecular System Constructed by H-Bonding and π···π-Stacking Interactions. Inorg. Chem. 2006, 45, 2229–2234. [Google Scholar] [CrossRef] [PubMed]
- Ueda, A.; Yamada, S.; Isono, T.; Kamo, H.; Nakao, A.; Kumai, R.; Nakao, H.; Murakami, Y.; Yamamoto, K.; Nishio, Y. Hydrogen-bond-dynamics-based switching of conductivity and magnetism: a phase transition caused by deuterium and electron transfer in a hydrogen-bonded purely organic conductor crystal. J. Am. Chem. Soc. 2014, 136, 12184–12192. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond, Version 5.0.0; Crystal and Molecular Structure Visualization, Crystal Impact; K. Brandenburg & H. Putz Gbr: Bonn, Germany, 1997-2023. [Google Scholar]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lu, T.; Chen, Q. Interaction region indicator: a simple real space function clearly revealing both chemical bonds and weak interactions. Chem.-Methods 2021, 1, 231–239. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Meng, M.; Wang, G.Y.; Feng, P.; Liu, C.Y. Optically probing the localized to delocalized transition in Mo2–Mo2 mixed-valence systems. Chem. Commun. 2017, 53, 3030–3033. [Google Scholar] [CrossRef]
- Al-Resayes, S.I.; Azam, M.; Trzesowska-Kruszynska, A.; Kruszynski, R.; Soliman, S.M.; Mohapatra, R.K.; Khan, Z. Structural and theoretical investigations, Hirshfeld surface analyses, and cytotoxicity of a naphthalene-based chiral compound. ACS omega 2020, 5, 27227–27234. [Google Scholar] [CrossRef] [PubMed]
- Karmakar, A.; Oliver, C.L.; Platero-Prats, A.E.; Laurila, E.; Öhrström, L. Crystal structures and hydrogen bond analysis of five amino acid conjugates of terephthalic and benzene-1, 2, 3-tricarboxylic acids. CrystEngComm 2014, 16, 8243–8251. [Google Scholar] [CrossRef]
- Hachuła, B.; Polasz, A.; Książek, M.; Kusz, J.; Kozik, V.; Matussek, M.; Pisarski, W. Insight into hydrogen bonding of terephthalamides with amino acids: Synthesis, structural and spectroscopic investigations. Tetrahedron 2017, 73, 2901–2912. [Google Scholar] [CrossRef]
- Martin, A.D.; Hartlieb, K.J.; Sobolev, A.N.; Raston, C.L. Hirshfeld surface analysis of substituted phenols. Cryst. Growth Des. 2010, 10, 5302–5306. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Boto, R.A.; Izquierdo-Ruiz, F.; Reva, I.; Woller, T.; Alonso, M. A benchmark for the non-covalent interaction (NCI) index or… is it really all in the geometry? Theor. Chem. Acc. 2016, 135, 1–14. [Google Scholar] [CrossRef]
- Venkataramanan, N.S.; Suvitha, A. Nature of bonding and cooperativity in linear DMSO clusters: a DFT, AIM and NCI analysis. J. Mol. Graph. 2018, 81, 50–59. [Google Scholar] [CrossRef]
- Heller, E.; Schmidt, G.M. Topochemistry. Part XXXIII. The Solid-State Photochemistry of Some Anthracene Derivatives. Isr. J. Chem. 1971, 9, 449–462. [Google Scholar] [CrossRef]
- Ossowski, J.; Wächter, T.; Silies, L.; Kind, M.; Noworolska, A.; Blobner, F.; Gnatek, D.; Rysz, J.; Bolte, M.; Feulner, P. Thiolate versus selenolate: Structure, stability, and charge transfer properties. ACS Nano 2015, 9, 4508–4526. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, F.; Bacchi, S.; Filippini, G.; Pilati, T.; Gavezzotti, A. Competition between hydrogen bonding and arene–perfluoroarene stacking. X-Ray diffraction and molecular simulation on 5,6,7,8-tetrafluoro-2-naphthoic acid and 5,6,7,8-tetrafluoro-2-naphthamide crystals. CrystEngComm 2009, 11, 1122–1127. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π Hydrogen Bond in Chemistry. Conformation, Supramolecules, Optical Resolution and Interactions Involving Carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π Hydrogen Bonds in Organic and Organometallic Chemistry. CrystEngComm 2009, 11, 1757. [Google Scholar] [CrossRef]
Figure 1.
(a) Differentiation of the anti- and syn-H atom in primary amides. Classical hydrogen-bonding interactions between amide groups with the Etter notation for a (b) (8) ring, (c) (4) chain motif and the combination of (4) and (8) in a (d) ladder which generates a new (8) pattern. In the Etter notation the subscript stands for the number of H-bond donors, the superscript for the number of H-Bond acceptors and the number in parentheses for the number of atoms involved in the motif [20,21,22,23].
Figure 1.
(a) Differentiation of the anti- and syn-H atom in primary amides. Classical hydrogen-bonding interactions between amide groups with the Etter notation for a (b) (8) ring, (c) (4) chain motif and the combination of (4) and (8) in a (d) ladder which generates a new (8) pattern. In the Etter notation the subscript stands for the number of H-bond donors, the superscript for the number of H-Bond acceptors and the number in parentheses for the number of atoms involved in the motif [20,21,22,23].
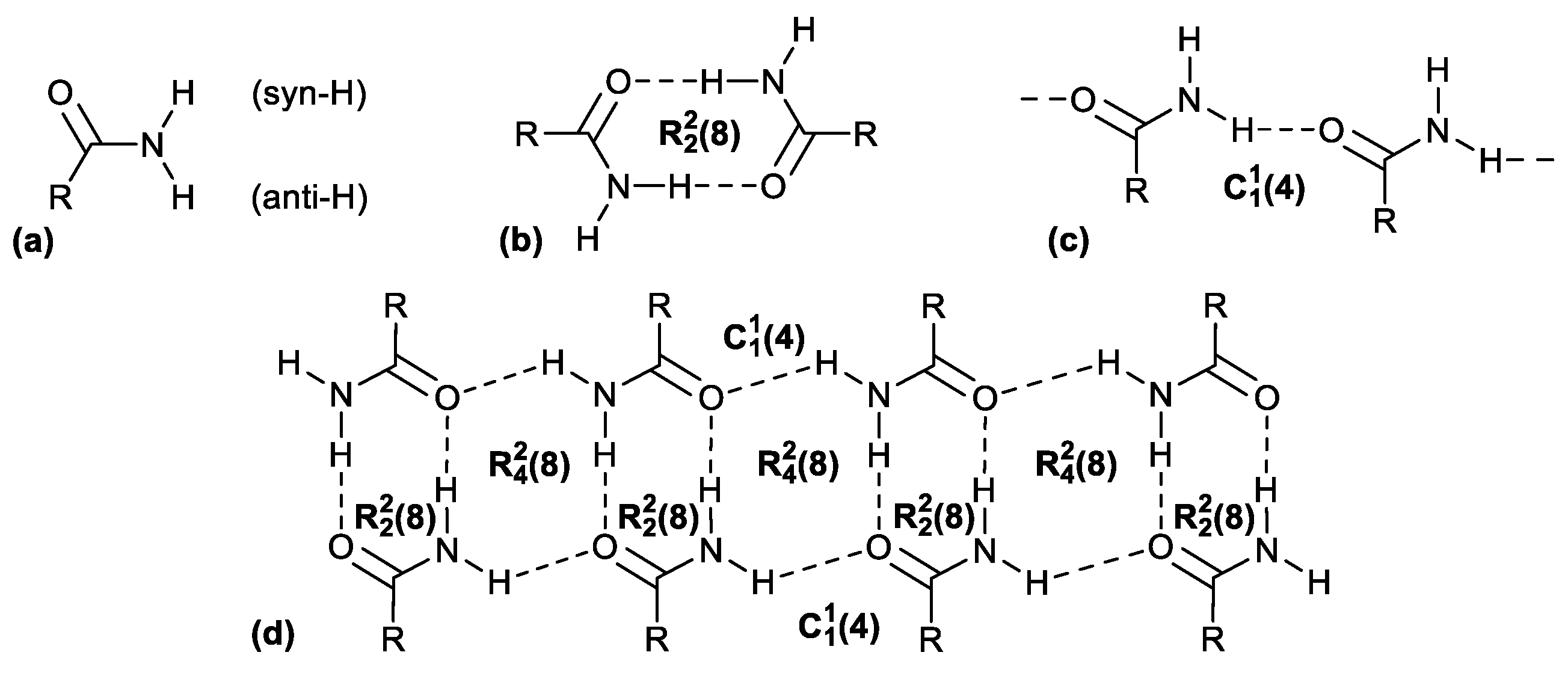
Figure 2.
The dicarboxamides studied in this work: 1,4-naphthalene dicarboxamide (1,4-NDA), 2,6-naphthalene dicarboxamide (2,6-NDA), and 2,7-naphthalene dicarboxamide (2,7-NDA) and the syn/anti amide-CO (in red) and amide-NH2 (in blue) orientation observed in the crystal structures (see below).
Figure 2.
The dicarboxamides studied in this work: 1,4-naphthalene dicarboxamide (1,4-NDA), 2,6-naphthalene dicarboxamide (2,6-NDA), and 2,7-naphthalene dicarboxamide (2,7-NDA) and the syn/anti amide-CO (in red) and amide-NH2 (in blue) orientation observed in the crystal structures (see below).
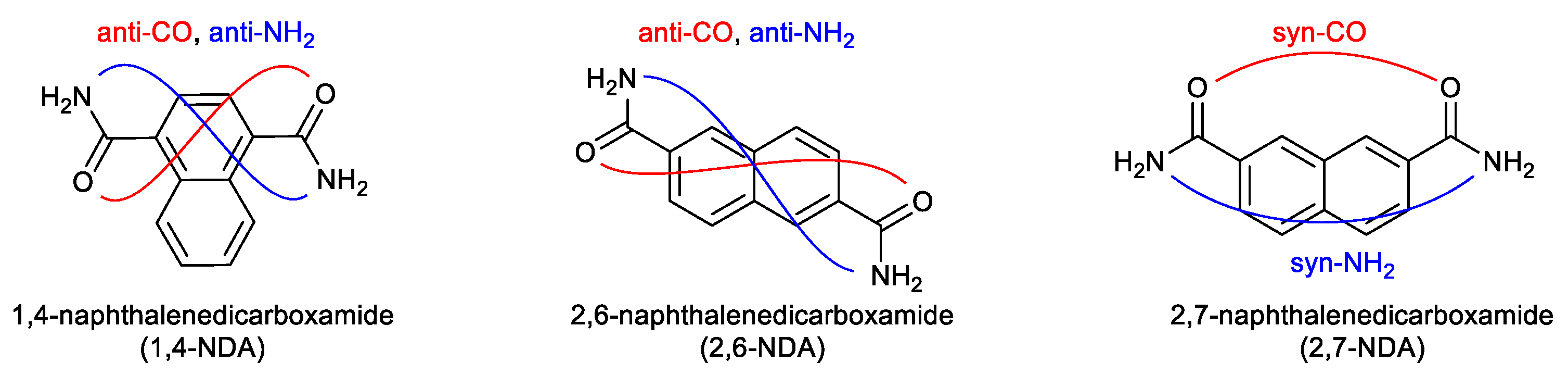
Figure 3.
Expanded asymmetric units for (a) 1,4-NDA; (b) 2,6 NDA, symmetry code: 1-x, 1-y, 1-z; (c) 2,7-NDA, symmetry code: 1-x, y, 3/2-z.
Figure 3.
Expanded asymmetric units for (a) 1,4-NDA; (b) 2,6 NDA, symmetry code: 1-x, 1-y, 1-z; (c) 2,7-NDA, symmetry code: 1-x, y, 3/2-z.
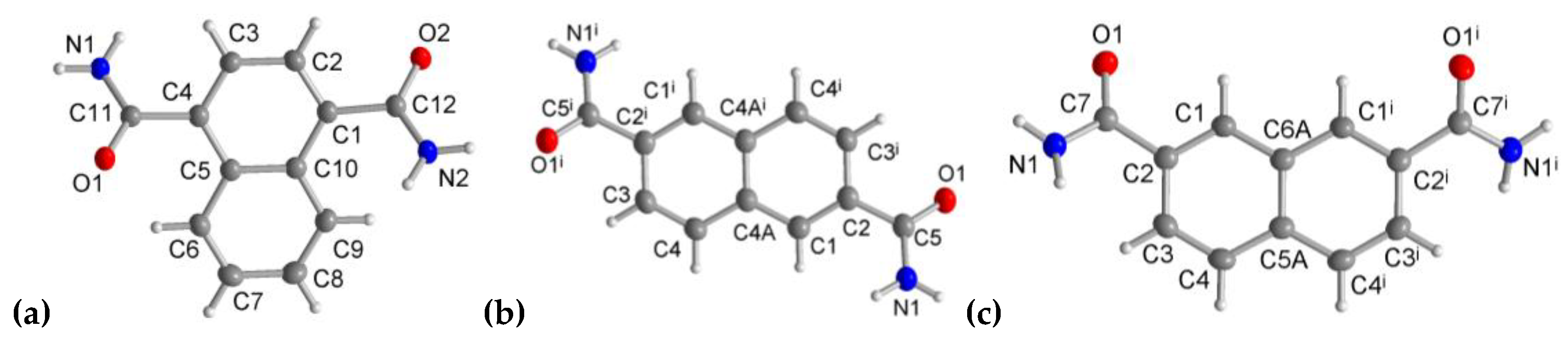
Figure 4.
Section of the 2D H-bonded sheet of 1,4-NDA with graph-set patterns. Symmetry codes: (i) x, y+1, z; (ii) x+1, y, z; (iii) x, y-1, z; (iv) x-1, y, z.
Figure 4.
Section of the 2D H-bonded sheet of 1,4-NDA with graph-set patterns. Symmetry codes: (i) x, y+1, z; (ii) x+1, y, z; (iii) x, y-1, z; (iv) x-1, y, z.
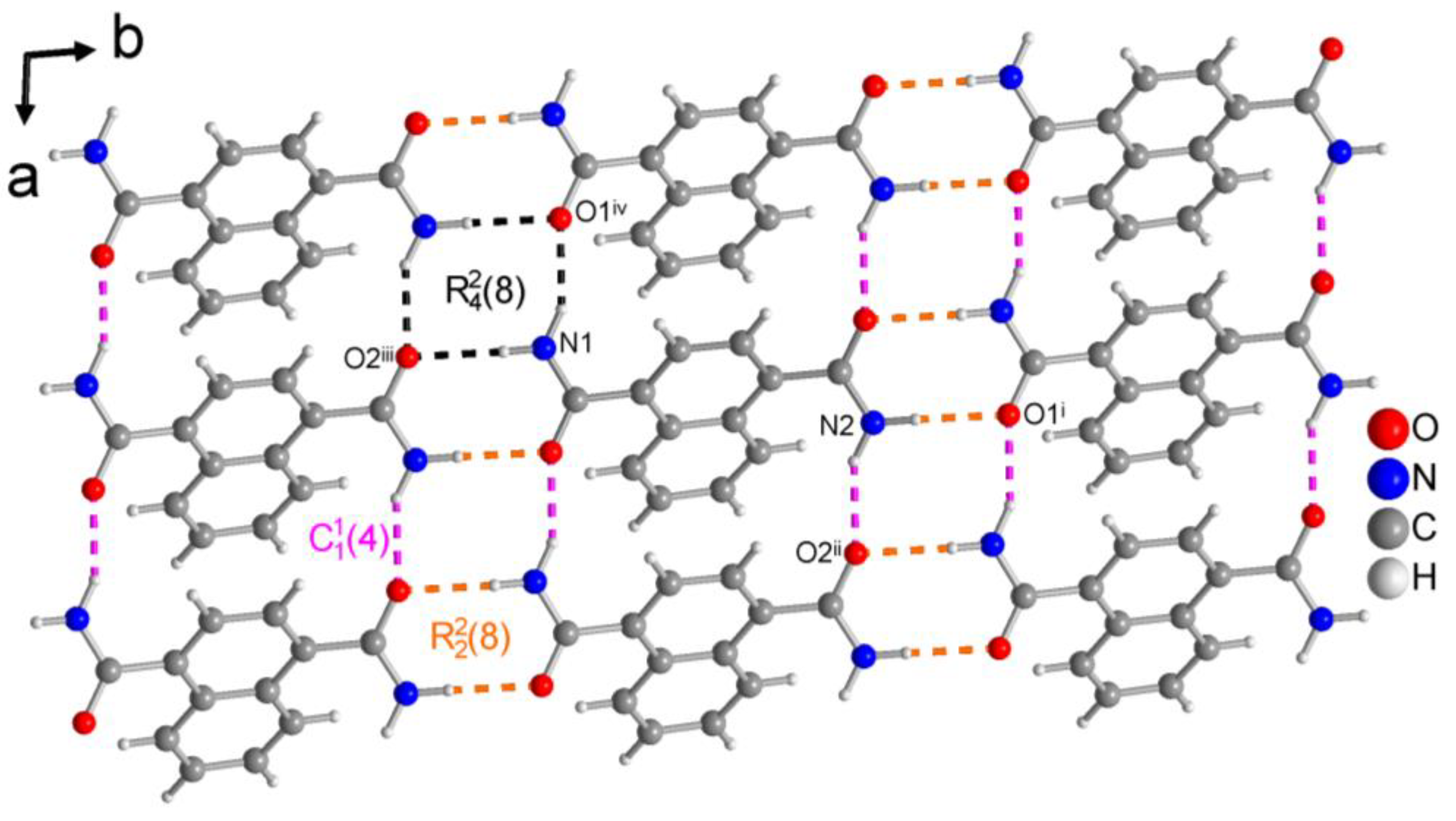
Figure 5.
Non-planar conformation of the dicarboxamides molecules exemplified by 1,4-NDA, with the dihedral angle ϕ between the naphthalene core and the amide group.
Figure 5.
Non-planar conformation of the dicarboxamides molecules exemplified by 1,4-NDA, with the dihedral angle ϕ between the naphthalene core and the amide group.
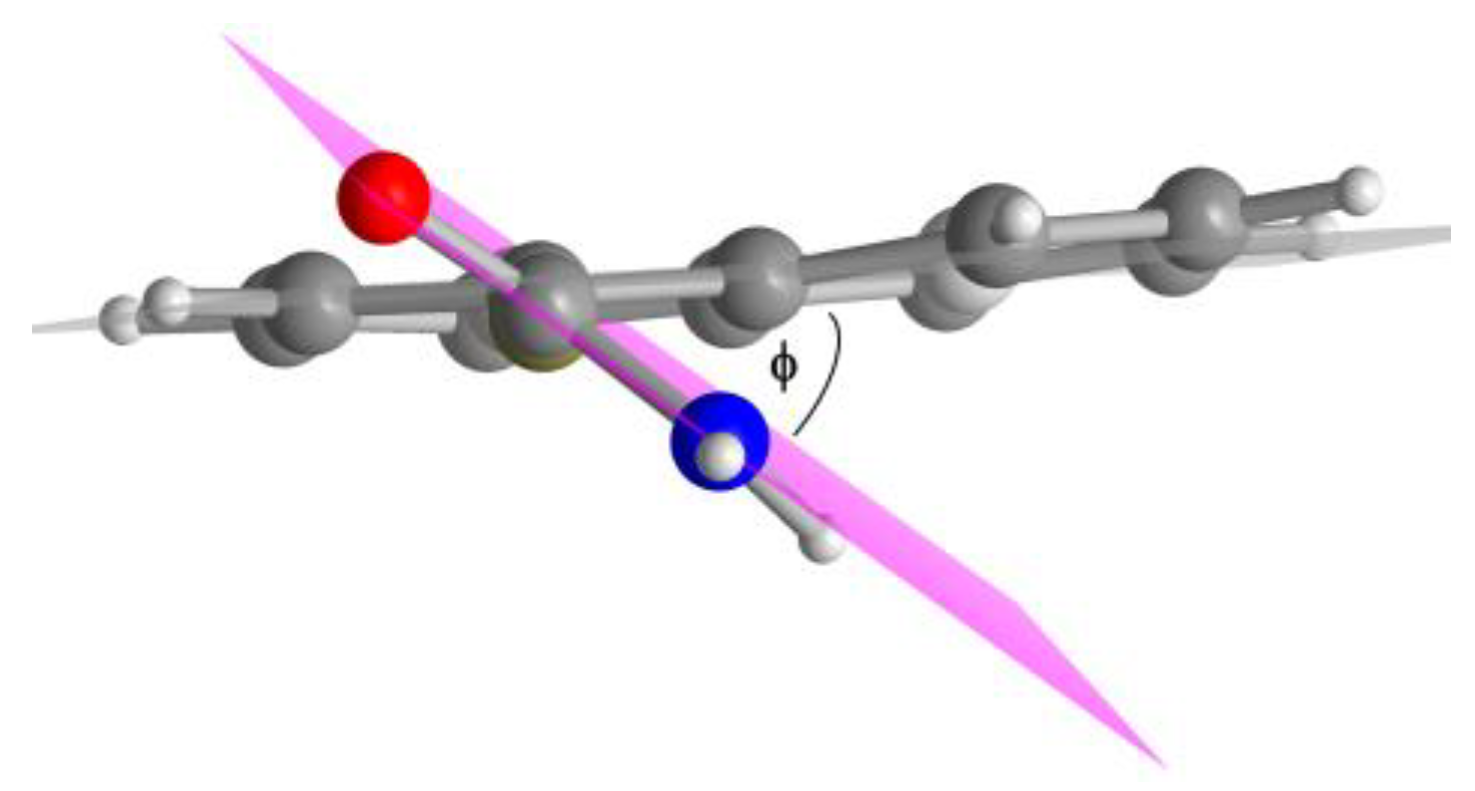
Figure 6.
Section of the 2D H-bonded sheet of 2,6-NDA with graph-set patterns. Symmetry codes: (ii) -x+1, -y, -z; (iii) x+1, y, z.
Figure 6.
Section of the 2D H-bonded sheet of 2,6-NDA with graph-set patterns. Symmetry codes: (ii) -x+1, -y, -z; (iii) x+1, y, z.
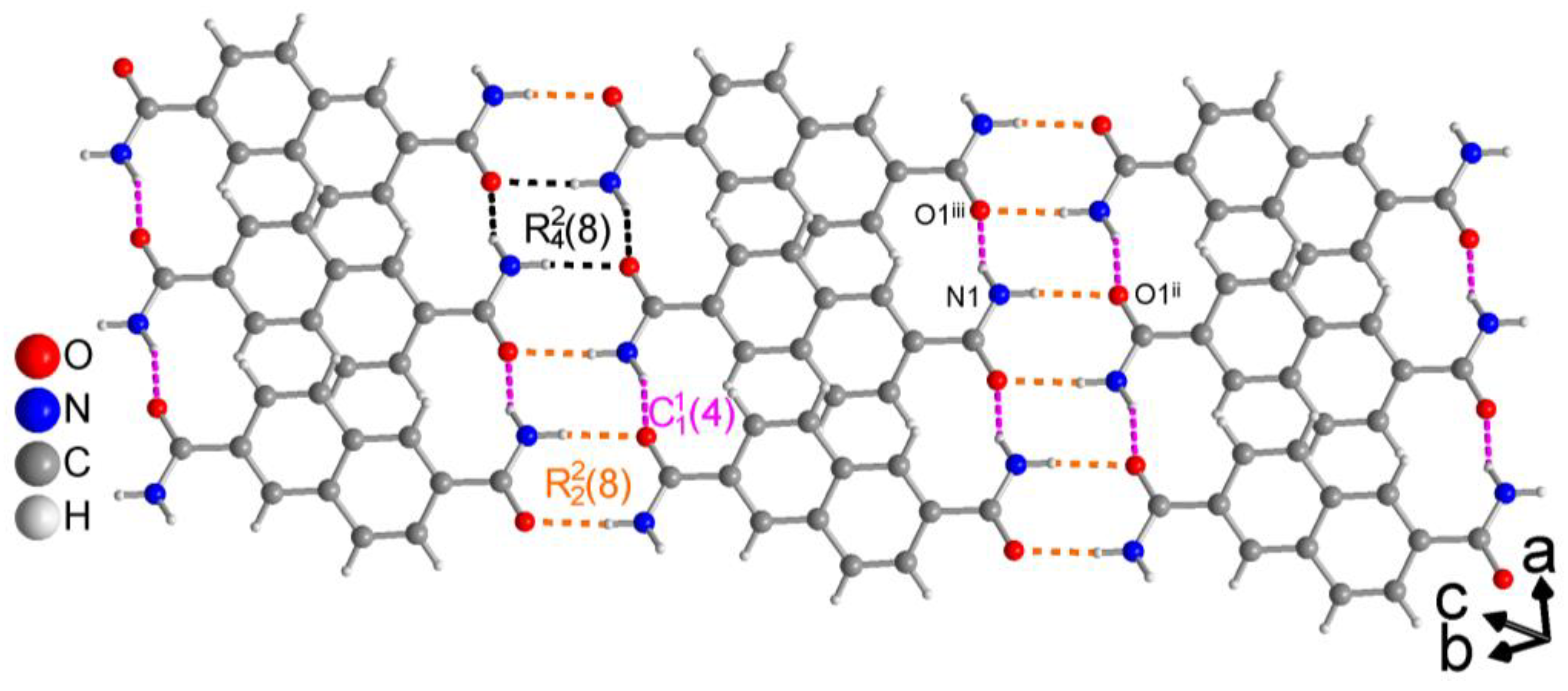
Figure 7.
Section of 3D H-bonded network of 2,7-NDA with graph-set pattern. Symmetry codes: (ii) -x+3/2, -y+1/2, -z+1; (iii) x+1/2, y+1/2, z.
Figure 7.
Section of 3D H-bonded network of 2,7-NDA with graph-set pattern. Symmetry codes: (ii) -x+3/2, -y+1/2, -z+1; (iii) x+1/2, y+1/2, z.
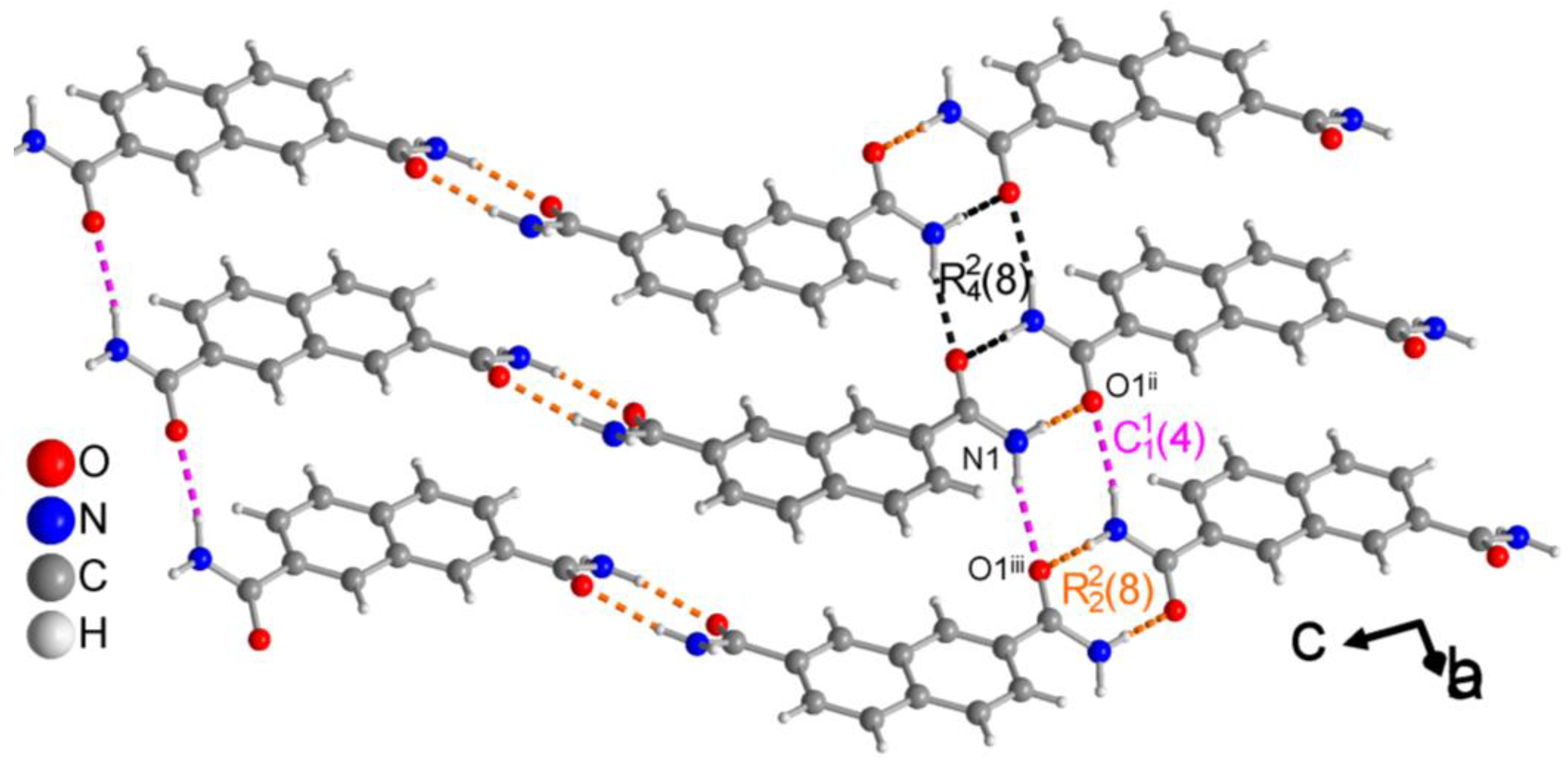
Figure 8.
H-bonded ladder arrangements in (a) 1,4 NDA, (b) 2,6-NDA molecules into 2D networks and for (c) 2,7-NDA into a 3D network. For the structure of 1,4-NDA also the weak intra-ladder π-π stacking with a centroid-centroid distance of 3.80 Å is indicated (black dashed line).
Figure 8.
H-bonded ladder arrangements in (a) 1,4 NDA, (b) 2,6-NDA molecules into 2D networks and for (c) 2,7-NDA into a 3D network. For the structure of 1,4-NDA also the weak intra-ladder π-π stacking with a centroid-centroid distance of 3.80 Å is indicated (black dashed line).
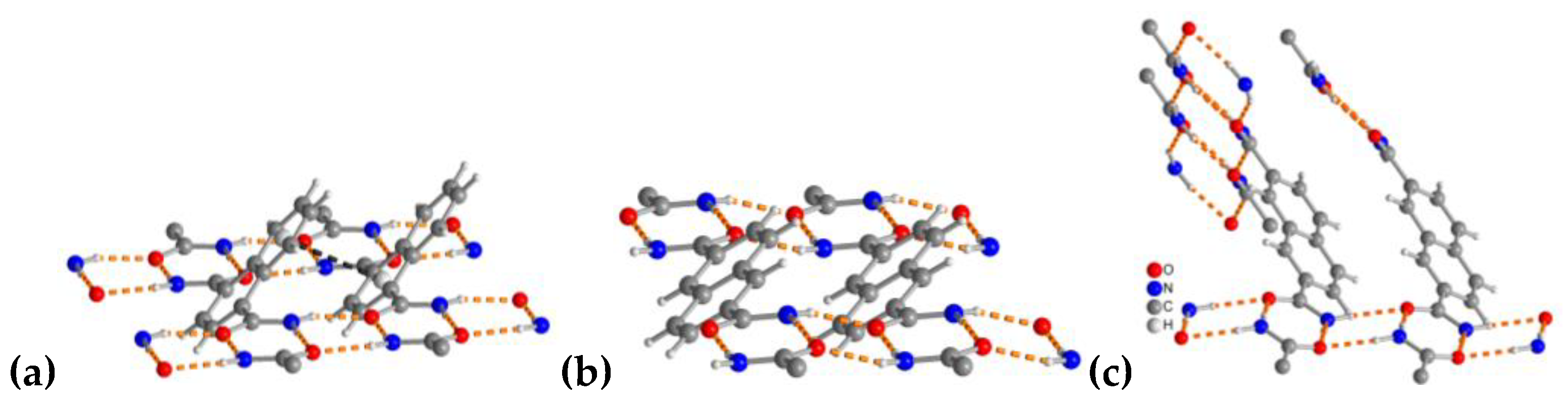
Figure 9.
Topological representation of the propagation of the (8) bis-amide rings in 1D ladders (orange lines) which are arranged into 2D staggered planar sheets of sql topology for (a) 1,4-NDA and (b) 2,6-NDA and into a 3D network of cds topology for (c) 2,7-NDA. For clarity, only the naphthalene carbon framework is shown, with propagating bis-amides represented by orange lines passing through the center of the dimeric (8) bis-amide rings.
Figure 9.
Topological representation of the propagation of the (8) bis-amide rings in 1D ladders (orange lines) which are arranged into 2D staggered planar sheets of sql topology for (a) 1,4-NDA and (b) 2,6-NDA and into a 3D network of cds topology for (c) 2,7-NDA. For clarity, only the naphthalene carbon framework is shown, with propagating bis-amides represented by orange lines passing through the center of the dimeric (8) bis-amide rings.
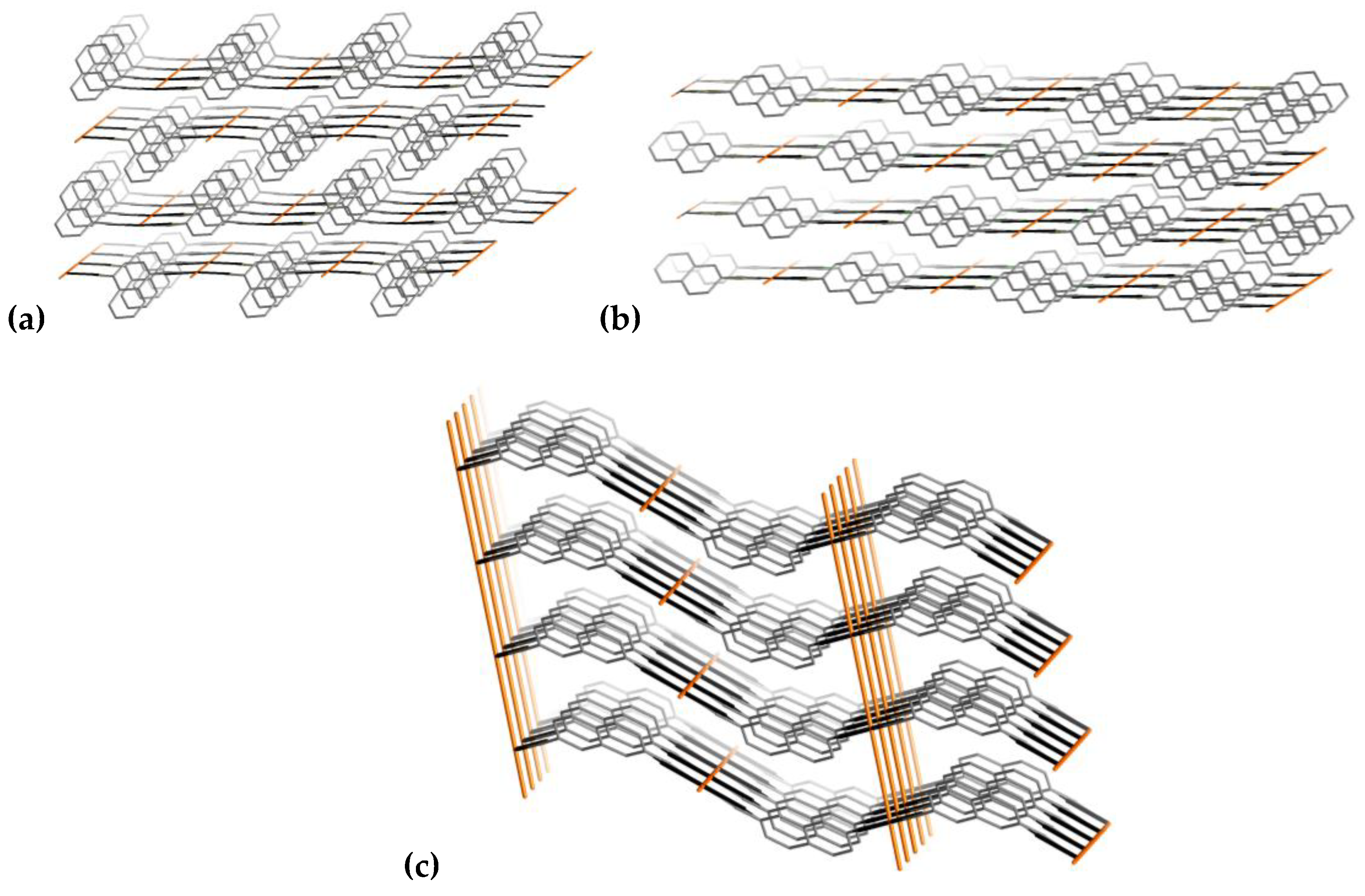
Figure 10.
3D Hirshfeld surface representation of the dicarboxamides plotted over dnorm for (a) 1,4-NDA, (b) 2,6-NDA and (c) 2,7-NDA. (dnorm < 0, red regions, indicate strong attractive interactions to an adjacent molecule; dnorm > 0, blue regions, indicate interactions weaker than the van der Waals interactions.).
Figure 10.
3D Hirshfeld surface representation of the dicarboxamides plotted over dnorm for (a) 1,4-NDA, (b) 2,6-NDA and (c) 2,7-NDA. (dnorm < 0, red regions, indicate strong attractive interactions to an adjacent molecule; dnorm > 0, blue regions, indicate interactions weaker than the van der Waals interactions.).
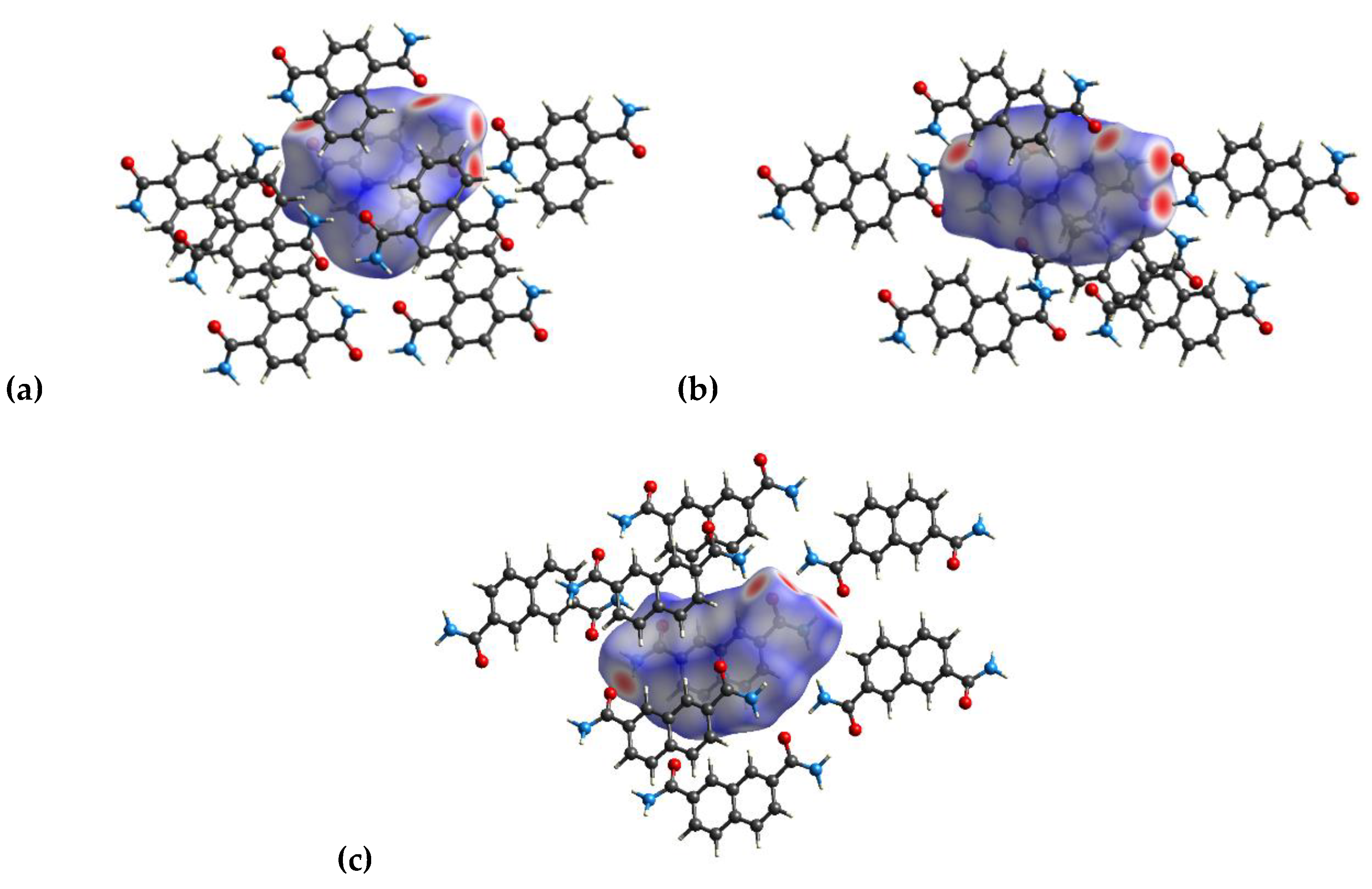
Figure 11.
2D fingerprint plots and the percentage contribution of specific non-covalently bound atomic contacts to the Hirshfeld surface for (a) 1,4-NDA, (b) 2,6-NDA, and (c) 2,7-NDA. The spikes, representing the shortest intermolecular distances, directed towards the lower left corner correspond to the N-H···O hydrogen bonds.
Figure 11.
2D fingerprint plots and the percentage contribution of specific non-covalently bound atomic contacts to the Hirshfeld surface for (a) 1,4-NDA, (b) 2,6-NDA, and (c) 2,7-NDA. The spikes, representing the shortest intermolecular distances, directed towards the lower left corner correspond to the N-H···O hydrogen bonds.
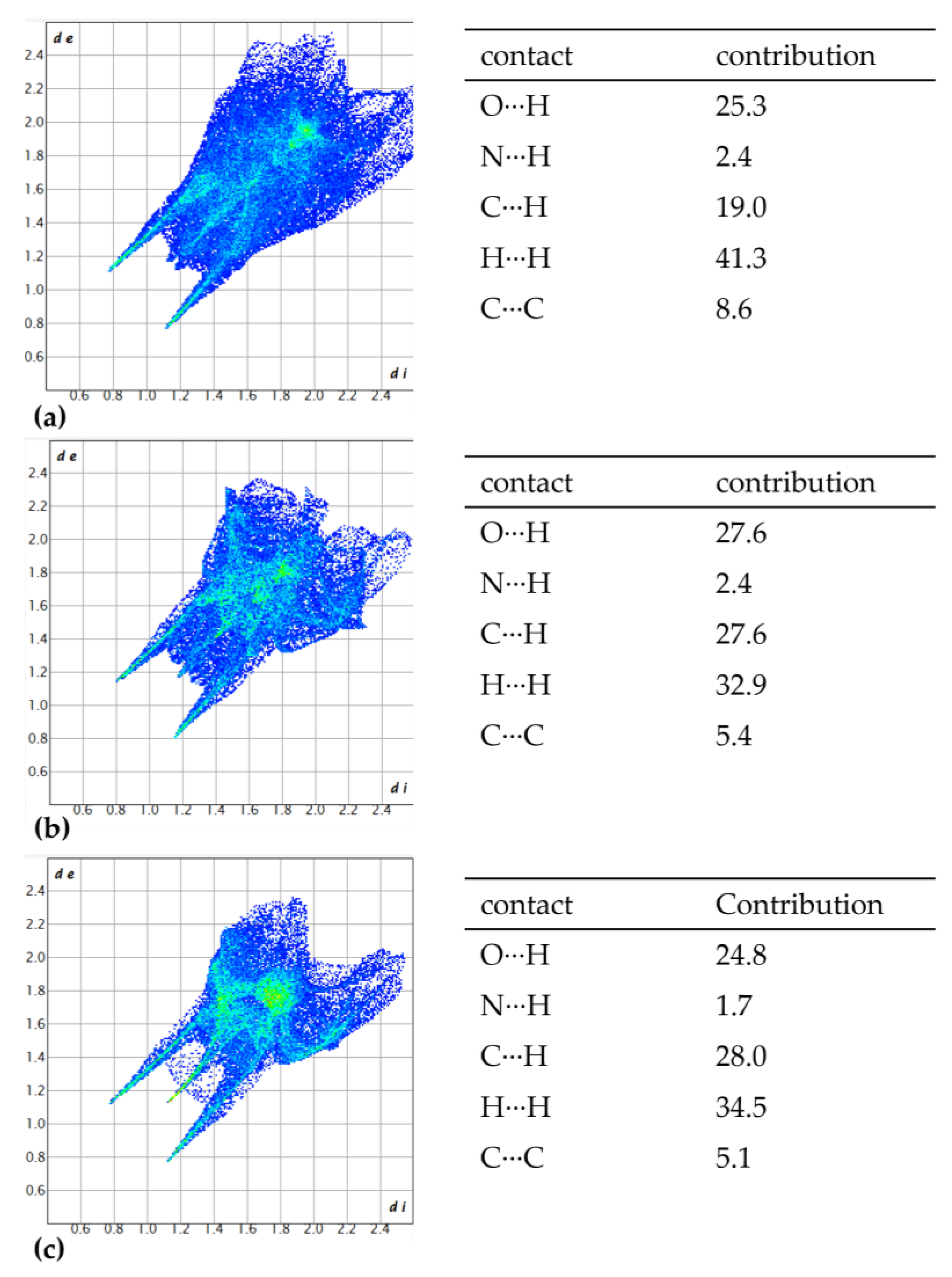
Figure 12.
NCI-based two-dimensional scatterplot (left) and 3D iso-surface (right) for (a) 1,4-NDA, (b) 2,6-NDA and (c) 2,7-NDA.
Figure 12.
NCI-based two-dimensional scatterplot (left) and 3D iso-surface (right) for (a) 1,4-NDA, (b) 2,6-NDA and (c) 2,7-NDA.
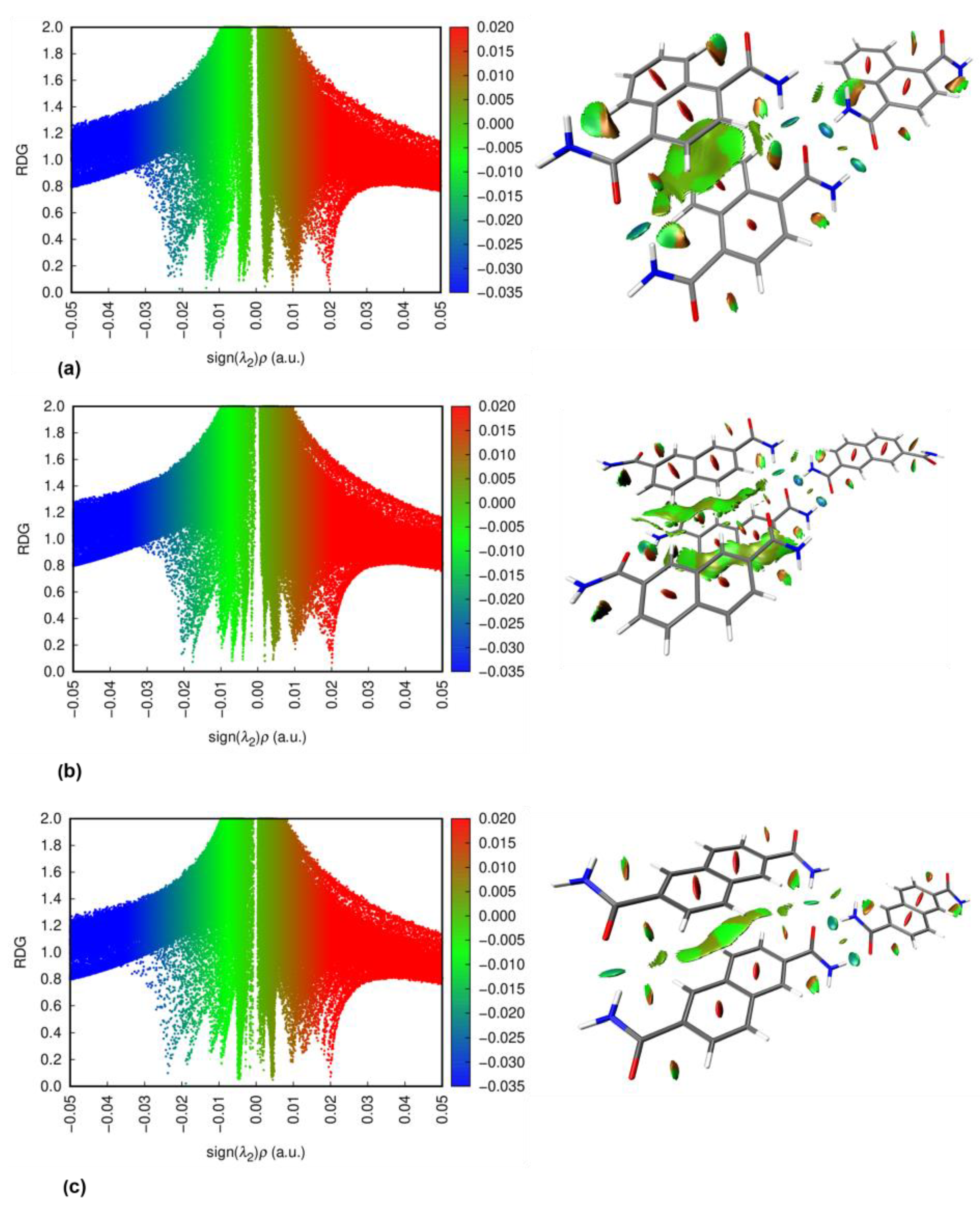
Figure 13.
TGA curves for the reported dicarboxamides (synthetic air, heating rate 10 K min–1).
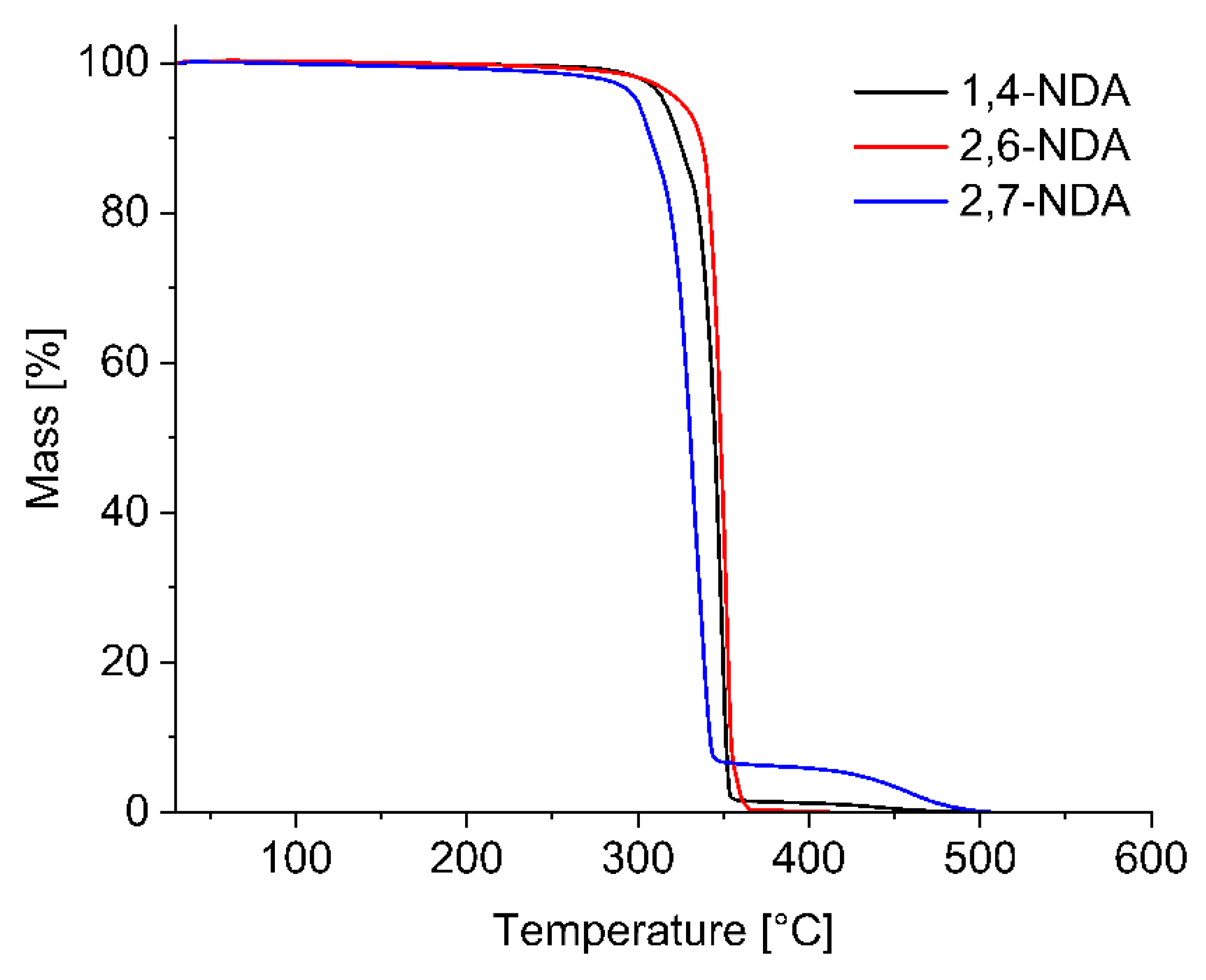
Table 3.
Dihedral angles ϕ between the naphthalene core and the amide group for 1,4-NDA, 2,6-NDA and 2,7-NDA.
Table 3.
Dihedral angles ϕ between the naphthalene core and the amide group for 1,4-NDA, 2,6-NDA and 2,7-NDA.
| Compound | Dihedral angle ϕ [°] |
| 1,4-NDA | 40 |
| 2,6-NDA | 29 |
| 2,7-NDA | 22 |
Table 4.
The parameters of the H-bonded interactions in the structure of 2,6-NDA.
| D—H···A(a)(a) | D—H [Å] | H···A [Å] | D···A [Å] | D—H···A [°] | Bond Energy [kcal/mol] |
| N1—H1A···O1ii | 0.89 (3) | 2.04 (3) | 2.864 (2) | 154 (2) | − 16 |
| N1—H1B···O1iii | 0.88 (3) | 2.07 (3) | 2.942 (2) | 173 (2) | − 16 |
(a) Symmetry codes: (ii) -x+1, -y, -z; (iii) x+1, y, z.
Table 5.
The parameters of the H-bonded interactions in the crystal structure 2,7-NDA.
| D—H···A(a) | D—H [ Å] | H···A [ Å] | D···A [ Å] | D—H···A [deg] | Bond Energy [kcal/mol] |
| N1—H1A···O1ii | 0.906 (18) | 1.998 (19) | 2.8983 (14) | 171.9 (15) | − 19 |
| N1—H1B···O1iii | 0.924 (18) | 2.074 (18) | 2.9133 (13) | 150.4 (14) | − 14 |
(a) Symmetry codes: (ii) -x+3/2, -y+1/2, -z+1; (iii) x+1/2, y+1/2, z.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



