1. Introduction
The taxonomic position of the genus
Leucobacter was formally established in 1996 [
1]. Its naming and classification were grounded in comprehensive phylogenetic studies, with particular emphasis on analyses of 16S rRNA gene sequences and cellular chemical characteristics. Prior to this formal establishment, these bacteria were classified among other related Gram-positive bacteria with high G+C content. However, molecular phylogenetic studies revealed distinct taxonomic features that rendered them unsuitable for inclusion in their original taxonomic groups. Consequently, Takeuchi and Hatano [
1] proposed the creation of the new genus
Leucobacter, which was subsequently recognized as a novel family-level member within the class Actinobacteria.
The genus name
Leucobacter is derived from the Greek words "leucos" (white) and "bacter" (rod), highlighting the characteristic coloration of certain strains on culture media. Members of this genus are typically Gram-positive, non-spore-forming bacteria distinguished by their unique carotenoid pigments. Notably, they demonstrate a remarkable ability to tolerate high concentrations of metal ions and are well-adapted to survive in diverse extreme environments [
2].
C. elegans is widely recognized as an outstanding model organism for the rapid screening of virulence factors and infection strategies employed by a variety of pathogens. Small size, ease of cultivation, and well-characterized genetic background make it an ideal system for studying host-pathogen interactions in real time. For example, pathogens such as
Pseudomonas aeruginosa [
3] and
Bacillus cereus [
4] have been shown to cause nematode death through the secretion of toxins or by disrupting critical host signaling pathways. These pathogens utilize a variety of mechanisms to subvert host defenses, and
C. elegans serves as a powerful tool to identify and characterize these virulence factors.
Despite lacking the complex adaptive immune system seen in higher organisms,
C. elegans relies heavily on its innate immune responses to combat infections. This innate immunity is primarily mediated through highly conserved pathways, many of which are shared with mammals. For instance,
C. elegans modulates the expression of immune-related genes via key signaling pathways such as the p38 MAPK [
5] and DAF-2/DAF-16 [
6] insulin pathways. These pathways regulate the nematode's immune response to bacterial infections, cellular stress, and environmental insults. Previous studies have highlighted the importance of these pathways in mediating resistance to bacterial pathogens, providing valuable insights into the evolution of immune responses across species.
The genus
Leucobacter was first named in 1996. Despite its formal establishment, research on this genus has remained relatively limited. The earliest described species,
Leucobacter komagatae, was defined by Takeuchi and Hatano [
1]. In 2004,
Leucobacter albus [
7] was characterized by Lin et al., notable for its resilience in polluted environments, which underscored the genus's adaptability and biotechnological potential. In the same year, Morais et al. named
Leucobacter chromiireducens and
Leucobacter aridicollis [
8], both of which were isolated from chromium-contaminated environments. The species name chromiireducens reflects its remarkable ability to reduce hexavalent chromium (Cr(VI)) to trivalent chromium (Cr(III)), highlighting its potential for heavy metal bioremediation.Subsequently, in 2006, Jogler et al. described
Leucobacter luti and
Leucobacter alluvii [
9], isolated from activated sludge and river sediments, respectively, both of which were exposed to chromium contamination.In 2011,
Leucobacter celer [
10] and
Leucobacter salsicius [
11] were identified by Shin et al. and Yun et al. in food products, while Behrendt et al. and Her et al. discovered
Leucobacter exalbidus [
12],
Leucobacter tardus [
13] and
Leucobacter humi [
14] in soil environments. These species demonstrate the genus's adaptability to extreme environmental conditions, including high salinity, low temperatures, nutrient limitations, and its ability to thrive in dry habitats.Moreover, within the microenvironment of living organisms,
Leucobacter iarius [
15] and
Leucobacter chromiireducens [
16] were isolated from
C. elegans, suggesting that
Leucobacter species may have a specialized ecological affinity for certain environmental niches [
17].
C. elegans has an exceptionally short lifespan, typically ranging from 2 to 3 weeks under standard laboratory conditions. This short life cycle allows researchers to study multiple generations in a limited time, making it an ideal model for studying the genetic, environmental, and molecular factors that influence aging and longevity [
18,
19]. The ability to quickly assess the effects of genetic modifications, drugs, or environmental changes on the lifespan of
C. elegans significantly reduces the time and resources needed compared to studies on longer-lived organisms.
The aging process in
C. elegans is well-characterized and easily observed [
20]. As the worms age, they exhibit typical signs of aging, such as reduced mobility, reproductive decline, and increased susceptibility to stress. Due to its short lifespan,
C. elegans is also an excellent model for investigating the impact of environmental factors (e.g., diet, toxins, and stress) and lifestyle interventions (e.g., caloric restriction or exercise) on aging.
Despite its simplicity,
C. elegans shares many conserved molecular pathways with humans, making it an ideal model for studying disease mechanisms [
21], drug responses [
22], and host-microbe interactions [
23]. These similarities further highlight its utility in human health research, particularly in understanding aging and age-related diseases.
2. Results
2.1. The Developmental Toxicity of Leucobacter sp. HNU-1 to Caenorhabditis elegans
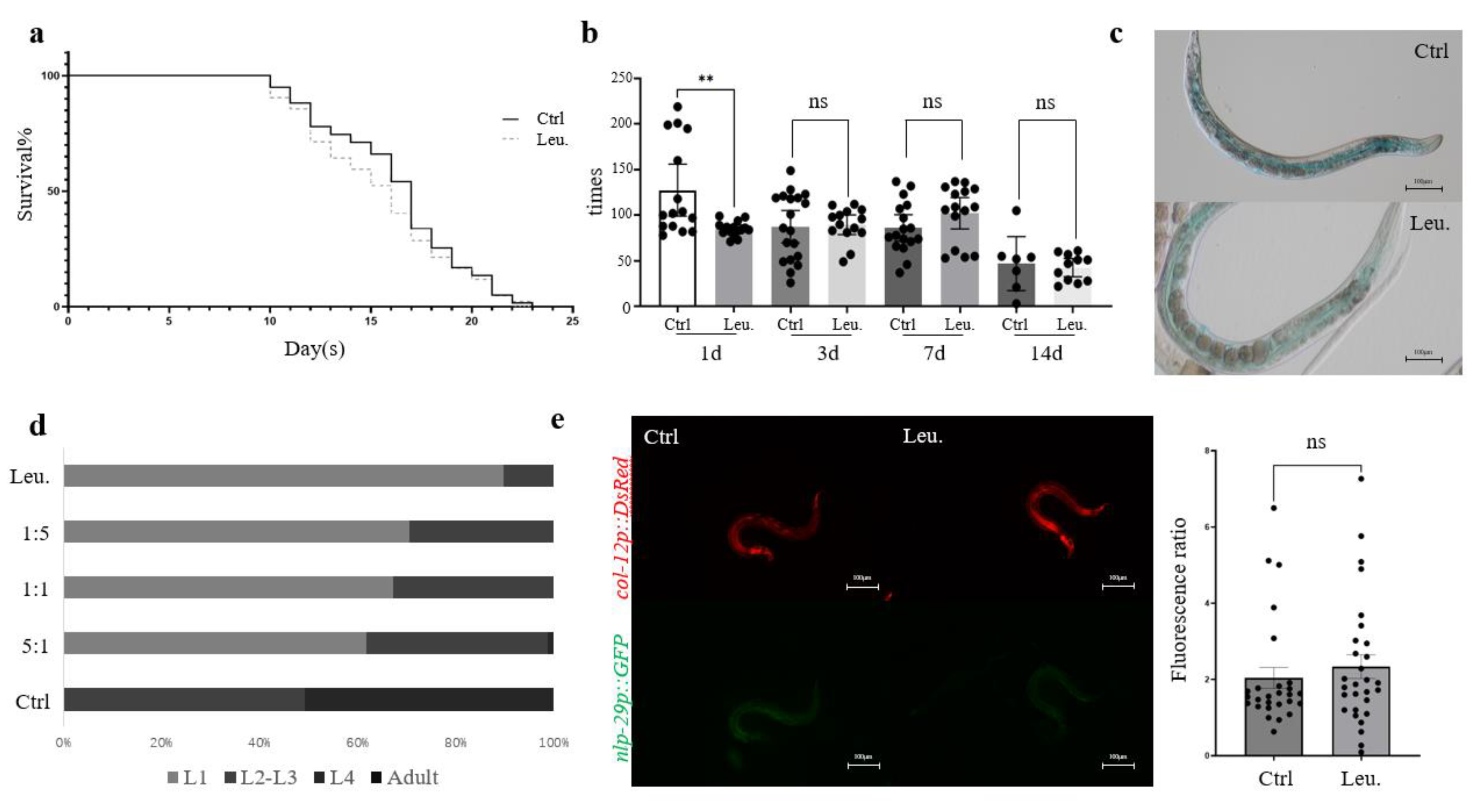
While
C. elegans shows robust immune responses to a variety of pathogens, the ingestion of HNU-1 does not significantly affect the nematode's lifespan (n=60, with three biological replicates,
Figure 1a) or its locomotor activity (n=15, with three biological replicates,
Figure 1b), nor induce detectable intestinal damage (n=20, with three biological replicates,
Figure 1c), despite the fact that many infections originate in the gastrointestinal tract [
24]. This suggests that HNU-1 may not activate the same harmful effects typically observed with other pathogens that directly damage host tissues. When the environment is unfavorable for growth and development,
C. elegans may experience immune responses or stress reactions triggered by bacterial metabolites, which could lead to developmental arrest [
25]. This phenomenon was observed following ingestion of HNU-1 (n=150, with three biological replicates,
Figure 1d), indicating that bacterial toxicity, immune signaling, and host-pathogen interactions may be intricately involved in this process.
To illuminate these potential interactions between C.elegans and HNU-1,
C. elegans IG274, which contains a cuticular collagen
col-12::DsRed reporter and the innate immune gene
nlp-29::GFP, was used to assess immune responses [
26]. The epidermal green fluorescence was activated when infected by
Sporangium or exposure to various stresses, such as epidermal damage or osmotic stress, while DsRed fluorescence serves as a baseline control. The GFP/DsRed ratio is commonly employed as a measure of immune activation or tissue damage in
C. elegans. However, in this study, ingestion of HNU-1 did not significantly alter the GFP/DsRed ratio (n=30, with three biological replicates,
Figure 1e), suggesting that no substantial immune stress response occurred in the nematodes following exposure. This finding further reinforces the notion that
C. elegans can adapt to environmental challenges in ways that might not invoke the typical immune responses associated with pathogen-induced damage. Moreover, this observation provides valuable insights into the dynamic and sometimes subtle nature of host-pathogen interactions, revealing the complexity of the biological relationships between bacteria and their host organisms.
Figure 1.
Toxic effect of HNU to C. elegans. (a) Lifespan of C. elegans fed with E.coli OP50 (a traditional lab food for C. elegans) and Leucobacter sp. HNU-1. (b) Quantification of pharyngeal pumping rates of C. elegans fed with indicated food. Pharynx pumping times per 30 seconds were plotted. (c) Intestinal staining with brilliant blue in C. elegans with indicated bacterial feeding. (d) Developmental defects of C. elegans were resulted from exposure to HNU-1. C. elegans at different developmental stages, i.e. L1, L2, L3, L4 and Adults were quantitated after 48 hours of feeding on OP50 (control group), Leu. (experimental group), or mixtures of OP50 and Leu with varied ratios (1:5, 1:1, or 5:1). (e) Exposure of HNU-1 does not upregulate nlp-29, an innate immune pathway gene. A representative images of worms stably expressing nlp-29p::GFP and col-12p::DsRed, that were fed with OP50 or Leu. Quantitative analysis of GFP/DsRed mean fluorescent intensity (MFI) ratios as a measure of the wound response were shown on the right. Error bars represent Standard Error of mean (SEM), statistic difference were determined by student t test; *: 0.01<p<0.05, **: p<0.01, n.s: not significant.
Figure 1.
Toxic effect of HNU to C. elegans. (a) Lifespan of C. elegans fed with E.coli OP50 (a traditional lab food for C. elegans) and Leucobacter sp. HNU-1. (b) Quantification of pharyngeal pumping rates of C. elegans fed with indicated food. Pharynx pumping times per 30 seconds were plotted. (c) Intestinal staining with brilliant blue in C. elegans with indicated bacterial feeding. (d) Developmental defects of C. elegans were resulted from exposure to HNU-1. C. elegans at different developmental stages, i.e. L1, L2, L3, L4 and Adults were quantitated after 48 hours of feeding on OP50 (control group), Leu. (experimental group), or mixtures of OP50 and Leu with varied ratios (1:5, 1:1, or 5:1). (e) Exposure of HNU-1 does not upregulate nlp-29, an innate immune pathway gene. A representative images of worms stably expressing nlp-29p::GFP and col-12p::DsRed, that were fed with OP50 or Leu. Quantitative analysis of GFP/DsRed mean fluorescent intensity (MFI) ratios as a measure of the wound response were shown on the right. Error bars represent Standard Error of mean (SEM), statistic difference were determined by student t test; *: 0.01<p<0.05, **: p<0.01, n.s: not significant.
2.2. Morphological Identification of Leucobacter
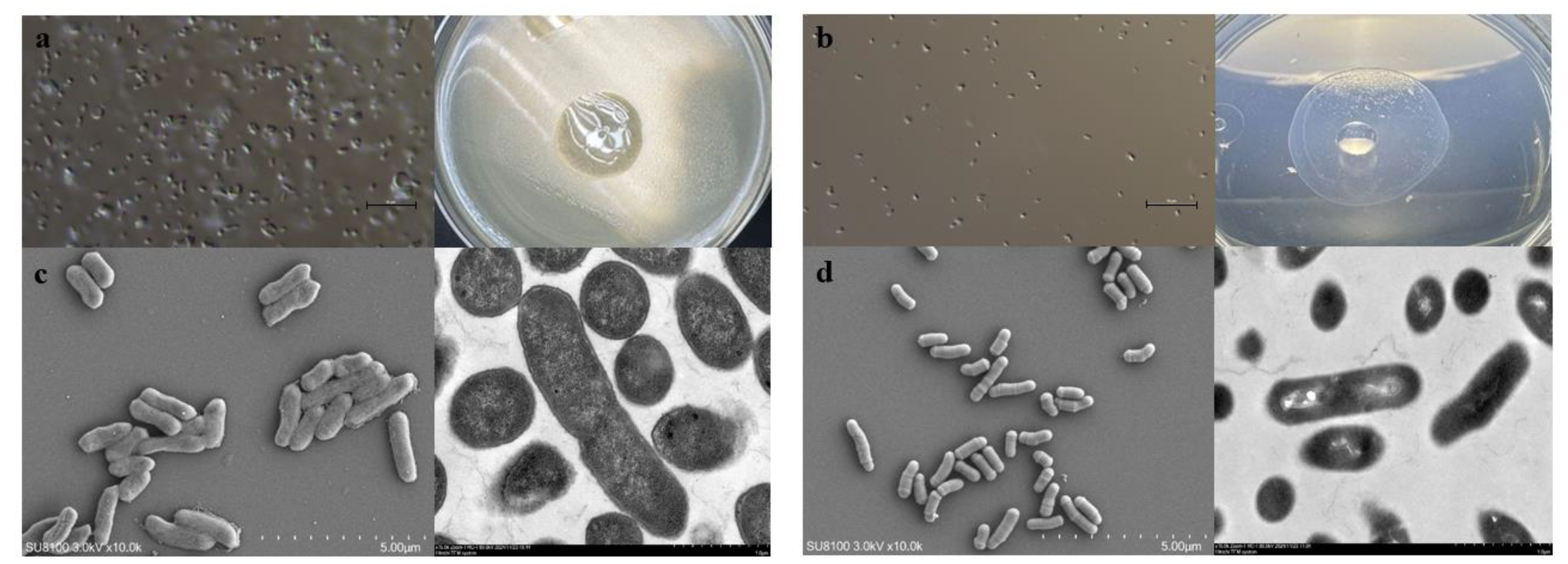
HNU-1 exhibits a distinct growth pattern, characterized by an initial lag phase lasting from 0 to 9 hours, followed by a logarithmic growth phase commencing at 9 hours, and culminating in the stationary phase at 25 hours (
Figure S1) .Morphologically, HNU-1 is a short, rod-shaped bacterium that does not produce spores. It grows slowly on conventional media, forming smooth, regular colonies with well-defined edges and hydrophobic properties (
Figure 2 A–D).
Overall, these findings highlight the remarkable adaptability of C. elegans in the face of environmental stressors and pathogenic challenges. They also underscore the utility of C. elegans as a model organism for studying the molecular and cellular basis of host immune responses, bacterial pathogenesis, and the broader implications of microbial interactions in health and disease.
2.3. Biochemical Tests of HNU-1
A comprehensive biochemical characterization of the isolated
Leucobacter HNU-1 strain was performed to elucidate its metabolic capabilities and physiological properties. Based on the biochemical reaction results (
Table 1), HNU-1 exhibited weak cloud-like diffusion in semi-solid agar, indicating limited motility. Additionally, it was unable to utilize citrate, ornithine, inositol, ribitol, or raffinose, suggesting specific constraints in its carbon and nitrogen metabolism pathways.
Regarding amino acid metabolism, HNU-1 tested positive for lysine decarboxylation, indicating lysine decarboxylase activity. However, it failed to hydrolyze urea, implying the absence of urease activity, and did not produce hydrogen sulfide, suggesting it lacks the ability to reduce thiosulfate to H₂S. In other biochemical assays, HNU-1 tested positive for both the indole and methyl red (MR) tests, demonstrating its capacity to degrade tryptophan into indole and generate stable acidic products through mixed-acid fermentation. In contrast, the Voges-Proskauer (VP) test was negative, indicating the strain does not synthesize acetoin via the butanediol fermentation pathway.
Furthermore, HNU-1 effectively utilized mannitol, sorbitol, and maltose as carbon sources, suggesting the presence of the necessary carbohydrate-metabolizing enzymes. However, it was unable to deaminate phenylalanine, indicating the absence of phenylalanine deaminase activity. These biochemical characteristics highlight the metabolic specificity of HNU-1, providing fundamental insights for its taxonomic classification and functional studies.
Table 1.
Biochemical Test Results for the Isolated Leucobacter sp. HNU-1 Strain.
Table 1.
Biochemical Test Results for the Isolated Leucobacter sp. HNU-1 Strain.
| Biochemical Test |
Result |
| Semi-solid agar motility |
+ |
| Ornithine decarboxylase |
- |
| Lysine decarboxylase |
+ |
| Citrate utilization |
- |
| H₂S production |
- |
| Urease |
- |
| Indole test |
+ |
| Methyl red (MR) test |
+ |
| Voges-Proskauer (VP) test |
- |
| Phenylalanine deamination |
- |
| Mannitol utilization |
+ |
| Inositol utilization |
- |
| Sorbitol utilization |
+ |
| Maltose utilization |
+ |
| Ribitol utilization |
- |
| Raffinose utilization |
- |
2.4. 16S Sequencing, Genome Assembly, and KEGG, GO, and COG Pathway Annotation of HNU-1
Molecular identification was performed using the 16S rDNA sequencing. The 16S rDNA of HNU-1 was amplified by PCR and genomic DNA extracted from HNU-1 as the template (
Figure S2). The purified PCR product, verified through agarose gel electrophoresis, was sequenced to obtain the nucleotide sequence (with three replicates). The resulting 16S rDNA (
Figure S3) has been deposited in the GenBank database under accession number PQ664966. A blastn search against all available 16S rDNA sequences in the NCBI database showed the highest similarity to the genus
Leucobacter. Based on morphological, physiological, and biochemical analyses, HNU-1 was preliminarily identified as a member of the genus
Leucobacter.
In recent years, the misuse and overuse of antibiotics have emerged as critical global public health challenges [
27]. Antibiotics are introduced into the human body through various pathways, including the food chain, via poultry, livestock, crops, and environmental sources such as water and microorganisms. These practices have significantly accelerated the development of antibiotic resistance, undermining the effectiveness of antimicrobial therapies and facilitating the emergence of superbugs and resistant viruses. Genomic sequencing of multidrug-resistant microorganisms is pivotal for elucidating the underlying mechanisms of resistance and developing targeted strategies to combat these pathogens. Addressing these issues is essential to curbing antibiotic misuse.
In this context, the whole-genome sequencing of the
Leucobacter sp. HNU-1 was undertaken to provide comprehensive insights into its multidrug resistance and to support future research aimed at identifying potential therapeutic interventions.The genomic analysis revealed a genome length of 3,375,033 bp, with an average coding gene length of 15,672 bp, a GC content of 70.37%, and a total of 3,270 protein-coding genes, 9 rRNA genes, and 52 tRNA genes (
Figure 3a). Functional annotation of all genes was performed using the KEGG (Kyoto Encyclopedia of Genes and Genomes), GO (Gene Ontology), and COG (Clusters of Orthologous Groups) databases. The KEGG database classifies biological metabolic pathways into six categories [
28]. In HNU-1, 2492 annotated genes, representing 5.01% of the total coding genes, were associated with various functions and metabolic pathways. Of these,80.30% are involved in metabolism, 7.02% in genetic information processing, 4.37% in cellular processes, 2.73% in environmental information processing, 2.24% in human diseases, and 3.33% in organismal systems. Each of these categories contains specific subcategories, allowing for the identification of genes involved in the regulation of target metabolites, either upregulated or downregulated (
Figure 3b). In the GO database, genomic information is classified into three main categories: molecular function (M), biological process (P), and cellular component (C) [
29]. In
Leucobacter sp. HNU-1, 2,082 genes were annotated in the GO database, accounting for 63.67% of the total coding genes, with a predominant involvement in molecular functions. The statistical results are shown in
Figure 4a. The COG database was used to classify the phylogenetic relationships of proteins encoded by the complete genome of HNU-1 [
30]. A total of 2,867 annotated genes, or 87.68% of the total genes, were categorized. Among these, 18.66% (535 genes) are related to signal transduction mechanisms (S), 16.08% to nucleotide transport and metabolism (E), 10.04% (288 genes) to replication, recombination, and repair (K), and 7.99% (229 genes) to the biosynthesis, transport, and catabolism of secondary metabolites (P). Additionally, 2.72% of genes have general functional predictions, and 5.62% have unknown functions, indicating the potential for further functional research (
Figure 4b).
2.5. Carbohydrate-Active Enzymes Pathway Annotation of HNU-1
The functional annotation of
Leucobacter sp. HNU-1 genes in the CAZy (Carbohydrate-Active Enzymes [
31]) database is summarized in
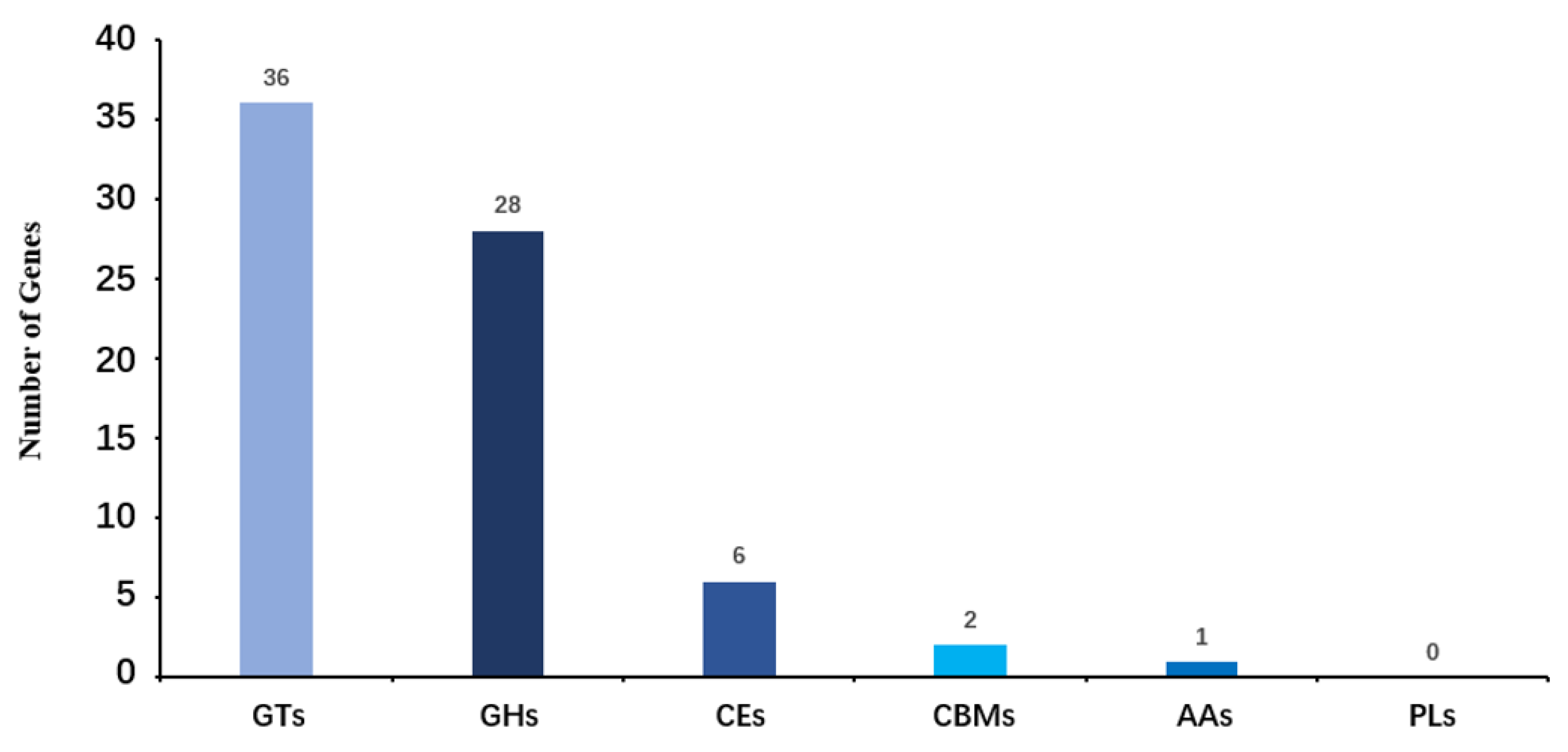
Figure 5. A total of 73 genes encoding carbohydrate-active enzymes were identified, reflecting the bacterium's potential involvement in diverse carbohydrate metabolism processes. Among these, 28 genes were classified as glycoside hydrolases (GHs), which are responsible for breaking glycosidic bonds in complex carbohydrates, facilitating their degradation and utilization. Another 36 genes were annotated as glycosyltransferases (GTs), enzymes essential for the synthesis of glycosidic bonds, suggesting an active role in the biosynthesis of polysaccharides and glycoconjugates.
In addition, two genes were identified as encoding carbohydrate-binding modules (CBMs), which are often associated with other enzyme domains and play a critical role in recognizing and binding specific carbohydrate substrates. Six genes were assigned to the carbohydrate esterase (CE) family, enzymes that act on ester linkages within carbohydrates, contributing to their structural modification. One gene was annotated as an auxiliary activity enzyme (AAs), which typically supports oxidative breakdown of polysaccharides and other carbohydrate polymers.
Interestingly, no genes from the polysaccharide lyase (PL) family were detected in the Leucobacter sp. HNU-1 genome, indicating that enzymatic machinery deficiency for cleaving polysaccharides through a β-elimination mechanism in Leucobacter sp. HNU-1. This absence may suggest a unique adaptation or a limited role in specific polysaccharide degradation pathways compared to other bacteria. Overall, these findings highlight the diverse enzymatic toolkit of HNU-1 for carbohydrate metabolism, providing insights into its ecological functions and metabolic capabilities.
Figure 5.
Functional Annotation and Classification of carbonhydrate-active enzymes (CAZy) in the genome of Leucobacter sp. HNU-1. The horizontal axis represents the classification of enzymes (different colors represent different kinds of enzymes), and the vertical axis represents the number of genes contained in this classification.
Figure 5.
Functional Annotation and Classification of carbonhydrate-active enzymes (CAZy) in the genome of Leucobacter sp. HNU-1. The horizontal axis represents the classification of enzymes (different colors represent different kinds of enzymes), and the vertical axis represents the number of genes contained in this classification.
2.6. CARD Antibiotic Resistance Prediction and K-B Method Antimicrobial Susceptibility Testing of HNU-1
In addition, a comprehensive analysis using the Comprehensive Antibiotic Research Database (CARD) [
32] revealed five antibiotic and secondary metabolite biosynthetic gene clusters within the HNU-1 genome. The identified clusters predominantly include two vancomycin resistance-related biosynthetic genes: the vanY gene in the vanB cluster and the vanY gene in the vanG cluster, both of which are implicated in resistance to this critical glycopeptide antibiotic. Furthermore, a sulfonamide resistance gene (
su11) and an aminoglycoside resistance gene (
aadA2) were also identified, contributing to HNU-1’s resistance against these commonly used antibiotics(
Table 2). These findings suggest that HNU-1 harbors multiple resistance mechanisms, which could influence its response to antibiotic treatments.
In this study, we evaluated the antibiotic susceptibility of an isolated strain using 30 commonly employed antimicrobial agents and the Kirby–Bauer disc diffusion method, in accordance with Clinical and Laboratory Standards Institute (CLSI) guidelines. The results, as shown in
Table 3, revealed that the strain exhibited marked resistance to several classes of antibiotics, including β-lactams (oxacillin [OX], ceftriaxone [CTR], cefoperazone [CPZ]), aminoglycosides (kanamycin [KAN], streptomycin [S]), polymyxin B (PB), lincosamides (clindamycin [CC], lincomycin [MY]), carbapenems (imipenem [IPM]), fluoroquinolones (ciprofloxacin [CIP], norfloxacin [NOR]), and sulfonamides (trimethoprim-sulfamethoxazole [SXT]). Additionally, the strain exhibited intermediate resistance to vancomycin, erythromycin, and tetracycline. Conversely, it was sensitive to selected antibiotics within the same classes, including β-lactams (penicillin, ampicillin, cefuroxime sodium, cefalexin, piperacillin, ceftazidime), tetracyclines (minocycline, doxycycline), chloramphenicol derivatives (chloramphenicol, florfenicol), fluoroquinolones (levofloxacin), aminoglycosides (gentamicin, amikacin), and macrolides (azithromycin). The underlying resistance mechanisms warrant further investigation.
The increasing prevalence of multidrug-resistant bacteria poses significant challenges for clinical treatment. Resistance is mediated by a suite of virulence genes that enhance bacterial persistence and pathogenicity, promoting both colonization and tissue invasion. These findings underscore the broad-spectrum resistance of the HNU-1 strain and its potential implications for clinical infection control and environmental microbiology.
2.7. Construction of the Phylogenetic Tree of HNU-1
A phylogenetic tree of seven
Leucobacter genomes was reconstructed using the Construct/Test Neighbor-Joining Tree method [
33,
34].
Curtobacterium flaccumfaciens strain GBBC 3199 (CP041259.1, 78.19% similarity) from the family
Microbacteriaceae , genus
Curtobacterium and
Microbacterium oxydans strain NBRC 15586 (CP162522.1, 77.35% similarity) from the family
Microbacteriaceae ,genus
Microbacterium were used as outgroups. Phylogenetic analysis revealed that 15
Leucobacter species formed two distinct clades. Among them, five species—
Leucobacter triazinivorans,
Leucobacter luti,
Leucobacter denitrificans,
Leucobacter chinensis, and
Leucobacter komagatae—grouped into a terminal branch, whereas the remaining eight species formed a monophyletic cluster nested within the evolutionary lineage of
Leucobacter sp. (
Figure 6). Notably,
Leucobacter sp. HNU-1 was closely related to
Leucobacter iarius.
2.8. Comparative Genomics of HNU-1 and Six Leucobacter Species, and ANI Analysis of All Leucobacter Genomes Available on NCBI
To further explore genomic relationships, the genome of
Leucobacter sp. HNU-1 was compared with six other reference genomes within the genus available in the NCBI database. Among these genomes,
Leucobacter iarius and
Leucobacter sp. HNU-1 shared similar chromosome lengths, numbers of coding sequences (CDSs), and tRNA counts, all of which were higher than those of three other strains. Their GC content also showed consistent patterns. The number of tRNAs was comparable across the seven strains, averaging around 50 per genome. Notably, the
Leucobacter sp. HNU-1 genome contained six CRISPR structures, whereas
Leucobacter iarius JCM 14736 harbored 13, and the other strains typically contained 1–3 (
Table 4). Average Nucleotide Identity (ANI) [
35] was calculated to assess genetic relatedness at the nucleotide level by comparing homologous genomic segments, as described by Goris et al. in 2007 [
36]. ANI values above 95% generally indicate conspecific genomes, while values below 75% are considered unreliable. ANI analysis of
Leucobacter sp. HNU-1 against 149 known
Leucobacter genomes in the NCBI database revealed that all strains had ANI values exceeding 75%. However, only one strain, GCA_040392405.1, exceeded the 95% threshold, with an ANI of 96.76%. This value was deemed unreliable due to the small genome size (2.2 Mb) of GCA_040392405.1. Consistent with these findings, ANI values below the interspecies threshold of 95% and the independent evolutionary branch observed in the 16S rDNA phylogenetic tree support the classification of
Leucobacter sp. HNU-1 as a distinct variant within the
Leucobacter genus(
Table S1).
Based on the results of phylogenetic tree analysis, six Leucobacter reference genomes Leucobacter iarius JCM 14736, Leucobacter chromiireducens TAN 31504, Leucobacter aridicollis DSM 17380, Leucobacter coleopterorum HDW9A, Leucobacter komagatae DSM 8803, and Leucobacter luti RF6 were selected for synteny analysis with the Leucobacter sp. HNU-1 genome using Mauve. This approach allowed for the comparison of gene order and structural variation between the genomes, shedding light on their evolutionary relationships.
2.9. Bacterial Synteny Analysis of HNU-1 and Six Leucobacter Species
As depicted in
Figure 7,
Leucobacter sp. HNU-1 demonstrated strong synteny with
L. iarius JCM 14736, with substantial conservation of gene order across large genomic regions. However, notable genomic rearrangements, including insertions, deletions, inversions, and translocations, were observed, highlighting significant evolutionary divergence between the two strains.
In contrast, the synteny analysis of
Leucobacter sp. HNU-1 with the other five reference genomes (
L. chromiireducens TAN 31504,
L. aridicollis DSM 17380,
L. coleopterorum HDW9A,
L. komagatae DSM 8803, and
L. luti RF6) revealed considerable differences in the arrangement of Locally Collinear Blocks (LCBs) [
37]. These variations in LCBs suggest that while the
Leucobacter species share a common evolutionary origin, their genomes have diverged significantly over time, likely reflecting adaptations to different ecological niches or functional specializations (
Figure 7). The analysis underscores the complex genomic architecture within the
Leucobacter genus and provides insights into the genomic flexibility that may contribute to the diversity of metabolic and ecological functions observed in this group of bacteria.
Figure 7.
Genome Synteny Analysis of Leucobacter sp. HNU-1 and Related Strains. The synteny of Leucobacter sp. HNU-1 was analyzed in comparison with L. iarius JCM 14736, L. chromiireducens TAN 31504, L. aridicollis DSM 17380, L. coleopterorum HDW9A, L. komagatae DSM 8803, and L. luti RF6. Colored regions in the visualization represent conserved syntenic segments among the genomes.
Figure 7.
Genome Synteny Analysis of Leucobacter sp. HNU-1 and Related Strains. The synteny of Leucobacter sp. HNU-1 was analyzed in comparison with L. iarius JCM 14736, L. chromiireducens TAN 31504, L. aridicollis DSM 17380, L. coleopterorum HDW9A, L. komagatae DSM 8803, and L. luti RF6. Colored regions in the visualization represent conserved syntenic segments among the genomes.
3. Discussion
In this study,
Leucobacter sp. HNU-1 was identified as a bacterium capable of inducing developmental delay formation in
C. elegans without causing significant effects on the nematode’s lifespan, motility, or intestinal morphology. Dauer formation is a specialized developmental state in
C. elegans that typically occurs under unfavorable environmental conditions, such as high population density [
38] or limited food availability [
39]. The ability of HNU-1 to induce this state in the absence of such stressors highlights a novel interaction between the bacterium and its host. Interestingly, this dauer-inducing effect differs from previously reported trauma-like responses [
40], as HNU-1 ingestion did not lead to an increase in IG274 fluorescence, a known marker of immune activation in
C. elegans. This finding indicates that HNU-1 does not trigger the immune response pathways typically associated with bacterial challenge.
Further analysis using electron microscopy revealed that HNU-1 is morphologically distinct from the widely used laboratory food source Escherichia coli OP50. Specifically, HNU-1 cells are smaller in both length and width, lack prominent membrane structures, and exhibit a hydrophobic surface. These physical characteristics may influence interaction between HNU-1 and the intestinal environment of C. elegans. Whole-genome sequencing provided additional insights into the unique features of HNU-1, uncovering a genome enriched with genes associated with molecular synthesis processes, signal transduction pathways, and nucleotide transport and metabolism. These genomic features suggest that HNU-1 has a complex metabolic capacity, which may play a role in its ability to induce developmental delay in C. elegans. Such interactions between bacterial metabolites and host signaling pathways may represent a novel mechanism underlying the developmental delay phenomenon, warranting further molecular investigation.
In addition to its developmental delay properties, HNU-1 was found to harbor multiple antibiotic resistance genes through CARD (Comprehensive Antibiotic Resistance Database) analysis. The presence of these resistance genes raises intriguing questions about the potential role of bacterial antibiotic metabolites in modulating host developmental pathways. While the specific contribution of these resistance-related genes to developmental delay remains unclear, future studies could explore whether bacterial antibiotic production affects the nematode's physiology or behavior.
Comparative genomic analyses highlighted the distinctiveness of HNU-1 when compared to other Leucobacter species with complete genomes available in the NCBI database. Phylogenetic analysis based on the 16S rDNA sequences showed that HNU-1 forms a relatively independent branch within the Leucobacter genus, further supporting its classification as a novel variant species. These findings suggest that HNU-1 is evolutionarily unique and distinct from previously characterized Leucobacter species. The discovery expands understanding of the diversity within Leucobacter genus and underscores the importance of exploring bacterial species in under-studied environments.
The comprehensive genomic and phenotypic analysis of HNU-1 provides a theoretical foundation for further research on antibiotic-resistant bacteria in tropical regions of China. As a bacterium with both unique genomic features and the ability to modulate host development, HNU-1 offers a valuable model for studying bacterial-host interactions and their implications for microbial ecology, evolutionary biology, and medical research. These findings contribute to the growing body of knowledge on the role of environmental bacteria in influencing the behavior and physiology of host organisms.
4. Materials and Methods
4.1. Isolation of the HNU-1 and Extraction of Genomic DNA
HNU-1 was isolated from wastewater in the tropical environment of Hainan, China, using the method of Her et al. for extracting Leucobacter humi from soil14: 1 ml of sewage was inoculated into 100 ml of 1/10 diluted nutrient broth and incubated aerobically in a shaking incubator (150 rpm, 28°C) for 3 days. After enrichment, 100 µl of the sample was spread onto a 1/10 diluted nutrient agar (LA) plate and incubated at 28°C for 2 days. Single colonies were then inoculated into 100 mL of liquid LB medium (containing 1 g of tryptone, 1 g of sodium chloride, and 0.5 g of yeast extract) and cultured at 180 rpm and 37°C for 48 hours. One portion of the bacterial culture was stored at −80°C with 50% glycerol (v/v) for future use, while another portion was used for genomic DNA extraction following the manufacturer's protocol (Shenggong Biological Co., Ltd., Shanghai, China). The quality and concentration of the genomic DNA were assessed by agarose gel electrophoresis and NanoDrop analysis. All culture media and solutions were sterilized at 121°C for 20 minutes. The Leucobacter sp. HNU-1 is currently preserved at Hainan University.
4.2. Characterization and Molecular Identification of Leucobacter sp. HNU-1
The identification of Leucobacter sp. HNU-1 was based on a combination of morphological, physiological, biochemical, and molecular characteristics. Morphological examination was performed using a Nikon Ni-C differential interference fluorescence microscope, a Hitachi Regulus 8100 scanning electron microscope, and a Hitachi HT7800 transmission electron microscope. Hydrophobicity tests were also conducted. For molecular identification, the 16S rDNA gene was amplified using the universal bacterial primers 27F (AGAGTTTGATCCTGGCTCAG) and 1492R (GGTTACCTTGTTACGACTT). The amplified fragment was sequenced, and the obtained sequence was compared with known sequences in GenBank using nucleotide BLAST to identify the bacterial species. Based on these analyses, the strain was classified as Leucobacter sp. HNU-1.
4.3. C. elegans Strains and Maintenance
The C. elegans N2 strain were maintained on nematode growth medium (NGM) plates seeded with bacteria (E. coli OP50) at 20 °C.
The following strains/alleles were obtained from the Caenorhabditis Genetics Center (CGC) or as indicated:
N2 Bristol (wild-type control strain);
IG274:frIs7 [nlp-29p::GFP + col-12p::DsRed] IV.
4.4. Preparation of Leucobacter sp. (HNU-1) and OP50 Mixed Suspension
To prepare Leucobacter sp. (HNU-1), an overnight culture in LB broth (grown at 37°C) was diluted 1:100 in fresh LB broth. Once the culture reached an optical density of OD600 = 0.5, Leucobacter sp. was spread evenly onto NGM plates.
For the preparation of mixed bacterial suspensions, 50 μl of Escherichia coli OP50 and 50 μl of Leucobacter sp. were combined in ratios of 1:5, 1:1, and 5:1. A total of 100 μl of the resulting mixture was then spread uniformly onto NGM plates.
Approximately 100–200 synchronized L1-stage C. elegans were transferred onto the designated plates (containing either Leucobacter sp. alone or a mixture of E. coli OP50 and Leucobacter sp.) and incubated at 20°C to observe and analyze growth phenotypes.
4.5. Lifespan Observation of C. elegans
Synchronized
C. elegans were exposed to NGM medium for 72 hours until reaching the L4 stage, after which they were transferred to NGM medium containing 5-fluorouracil. A total of 20
C. elegans were transferred to each plate. The number of deceased worms was recorded every 24 hours until all individuals had died, and the experiment was repeated three times. A picker was used to gently touch the tails and heads of the worms; individuals that did not respond were considered dead [
41]. The young adult stage of
C. elegans was designated as the initial exposure stage, during which the worms were exposed to the treated NGM medium19. Each experimental group included three biological replicates, with 60
C. elegans per parallel experiment. Experimental data were visualized using GraphPad Prism 9.5.0 software.
4.6. Analysis of the Fluorescence Intensity in Worms
For fluorescence imaging (nlp-29p::GFP + col-12p::DsRed), the worms were anesthetized with 25 mM levamisole, and images were captured using a Nikon Ni-C upright differential interference contrast fluorescence microscope equipped with a CMOS camera. The GFP/DsRed ratio was calculated to quantify fluorescence intensity. The entire worm region was outlined and quantified using ImageJ software, and the fluorescence intensity was normalized for comparison.
4.7. Complete Genome Sequencing and Annotation
The draft genome of HNU-1 was sequenced and annotated by BGI Technology Co., Ltd. (Wuhan, China) using both the DNBSEQ [
42] and Nanopore [
43] platforms. Sequencing was performed on the DNBSEQ-T10×4RS and PromethION instruments. To ensure the accuracy of the data, raw reads were processed to remove low-quality sequences, adapters, and short reads using the default parameters of the Porechop 0.2.4 software. Nanopore reads were assembled with Flye [
44,
45]. Genome polishing was carried out using Pilon 1.22 [
46] with the Quiver algorithm from the Genomic Consensus package. Gene prediction was performed using Prodigal 2.6.3 [
47] with default settings. Transfer RNAs (tRNAs) were identified with the tRNAscan-SE [
48] program, and ribosomal RNAs (rRNAs) were annotated using RNAmmer [
49]. Gene annotation was performed using Diamond 0.8.15 against the database. Functional annotations of the predicted genes were obtained with Blast2Go 2.5 [
49], using the Gene Ontology (GO) database, while pathway annotations were assigned via Blast 2.2.28+ with the KEGG database. Phylogenetic classification of protein-coding genes was carried out using Hmmscan 3.1b2 from the COG database. Enzymes involved in carbohydrate degradation, synthesis, and modification were annotated with Diamond 0.8.15 (E-value 0.00001) to provide catalytic structural and functional details from the CAZy database. Finally, Circos 0.69 software [
50] was used to generate a circular visualization of coding sequences (CDS), non-coding RNAs (ncRNA), GC content, repetitive sequences, and rRNA.
4.8. 16S rDNA Phylogenetic Tree, ANI Analysis, Comparative Genomics, and Synteny Analysis
The phylogenetic relationship of
Leucobacter sp. HNU-1 was determined based on its 16S rDNA sequence. Using BLASTn, 15 complete 16S rDNA sequences were retrieved from the NCBI nucleotide database for phylogenetic analysis.
Leucobacter viscericola HDW9C strain (NR_180647.1, 79.82% similarity) and
Leucobacter triazinivorans JW-1 strain (CP035806.1, 81.58% similarity) were selected as outgroup taxa. Phylogenetic trees were constructed using the Neighbor-Joining method [
33] in MEGA11, employing the p-distance model and performing Bootstrap analysis with 1,000 replications to assess the robustness of the tree.
Average Nucleotide Identity (ANI) [
36] analysis was conducted to evaluate the genomic relatedness between
Leucobacter sp. HNU-1 and other strains. Using the MUMmer alignment method, the average ANI values were calculated for 33 genomes. Genome accession numbers for numerous
Bacillus species are available in GenBank.
Based on the phylogenetic distances inferred from the 16S rDNA tree, complete genome sequences for all
Leucobacter species were downloaded from the NCBI database for comparative genomics analysis. In this analysis, both ANI and dDDH (digital DNA-DNA hybridization) values were used to assess genomic similarity. Strains with an ANI greater than 95% or a dDDH value greater than 70% are considered to belong to the same species [
51,
52]. ANI values for
Leucobacter sp. HNU-1 were calculated using the Kostas Lab ANI Calculator [
53] (
http://enve-omics.ce.gatech.edu/ani/index) and compared with reference strains
Leucobacter iarius JCM 14736,
Leucobacter chromiireducens TAN 31504,
Leucobacter aridicollis DSM 17380,
Leucobacter coleopterorum HDW9A,
Leucobacter komagatae DSM 8803, and
Leucobacter luti RF6. Genomic structure and synteny between
Leucobacter sp. HNU-1 and the reference strains were analyzed using the Mauve program within Geneious R9 software (Align with progressiveMauve) [
37].
4.9. Biochemical Identification of the Isolated Strain Using 16 Biochemical Tests
A total of 16 biochemical characteristics were selected for the biochemical identification of the isolated strain, including the following tests:Semi-solid agar motility, Ornithine decarboxylase, Lysine decarboxylase, itrate utilization, H₂S production, Urease activity, Indole test, Methyl red (MR) test, Voges-Proskauer (VP) test, Phenylalanine deamination, Mannitol utilization, Inositol utilization, Sorbitol utilization, Maltose utilization, Ribitol utilization, Raffinose utilization.For the identification procedure, a single colony of HNU-1, initially isolated on a plate, was picked and suspended in a 0.9% saline solution to achieve the appropriate turbidity. Biochemical tests were then carried out following the guidelines provided by Qingdao Hi-tech Industrial Park Hope Bio-technology Co., Ltd.
4.10. Prediction of Antibiotic Synthesis Gene Clusters and Analysis of Associated Genes
The Comprehensive Antibiotic Research Database (CARD) integrates antibiotic resistance ontology (ARO) with curated sequences of antimicrobial resistance (AMR) genes (ARGs) and resistance-conferring mutations, providing an informatics framework for the annotation and interpretation of resistance gene clusters. Gene cluster prediction for
Leucobacter sp. HNU-1 was performed using the CARD database, which includes both strict and perfect hits, as well as those that are nudged from loose to strict hits. Short contigs were excluded from partial gene predictions. Gene sequences with ≥ 85% similarity to known gene cluster sequences were classified as encoding compounds already identified, while sequences with < 85% similarity were considered to encode potentially novel compounds [
32,
54].
4.11. Antibiotic Susceptibility Testing Using the K-B Disk Diffusion
Antibiotic susceptibility testing was conducted using the Kirby-Bauer disk diffusion method, following the guidelines recommended by the Clinical and Laboratory Standards Institute (CLSI). In a biosafety cabinet, 100 µL of a prepared HNU-1 suspension was evenly spread onto an LB agar plate using a sterile swab. After the inoculum dried, sterile forceps were used to place the antibiotic discs gently onto the inoculated agar surface (care should be taken to avoid puncturing the plate). The plates were then incubated at 37°C for 16 hours. The zone of inhibition diameter was measured with a ruler, and the susceptibility of the HNU-1 strain to each antibiotic was determined based on the criteria provided in the antibiotic disk instructions. Three replicates were performed for each antibiotic.The following 30 antibiotics were tested:Penicillin, Vancomycin, Oxacillin, Levofloxacin, Clindamycin, Erythromycin, Polymyxin B, Gentamicin, Lincomycin, Minocycline, Tetracycline, Chloramphenicol, Imipenem, Doxycycline, Azithromycin, Ceftriaxone, Ceftazidime, Cefoperazone, Ciprofloxacin, Norfloxacin, Florfenicol, Piperacillin, Streptomycin, Compound Sulfonamides, Ampicillin, Kanamycin, Amikacin, Cefuroxime, Cephalexin, Cefamezin. Results were reported based on the criteria of resistance, intermediate, or susceptible.
4.12. Complete Nucleotide Sequence and Strain Accession Numbers
The complete nucleotide sequence of Leucobacter sp. HNU-1 has been deposited in the GenBank database under the accession number PRJNA1137138. The strain is available from the Environmental Health and Public Health Research Laboratory, Hainan University, Haikou, Hainan Province, China, with strain number 570228.
5. Conclusions
Leucobacter sp. HNU-1 significantly affects the morphological and physiological characteristics of Caenorhabditis elegans, particularly in inducing developmental delay. Although HNU-1 does not notably impact the nematode’s lifespan, motility, or intestinal morphology, it induces a developmental delay in the absence of external environmental stress, revealing a novel interaction between the bacterium and its host. Furthermore, the genomic analysis of HNU-1 reveals a complex metabolic capacity, particularly genes associated with molecular synthesis, signal transduction, and nucleotide metabolism, which may contribute to the induction of the developmental delay phenotype. HNU-1 also harbors multiple antibiotic resistance genes, suggesting its potential role in complex bacterial-host interactions. Overall, this study identifies Leucobacter HNU-1 as a novel species within the Leucobacter genus, offering new insights into bacterial-host interactions and providing a novel research direction for exploring non-traditional immune pathways and cellular stress responses triggered by bacteria. These findings have significant biological and medical implications.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: The growth curve of the strain Leucobacter sp. HNU-1.; Figure S2:Agarose gel electrophoresis. (a) PCR product of 16S rDNA of Leucobacter sp. HNU-1.; Figure S3: Nucleotide sequence of 16S rDNA from Leucobacter sp. HNU-1. Table S1: ANI Analysis of All Leucobacter Genomes Available on NCBI.
Author Contributions
H.L. and S.X. designed the experimental scheme. J.J. completed the experiment and wrote the first draft of the paper. X.L. and Z.G. analyzed the experimental data. H.L. and H.Y. revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the Hainan Provincial Natural Science Foundation of China (823QN230).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and analysed during the current study are available in the WGS and Genbank on NCBI repository, PRJNA1137138 and PQ664966.
Acknowledgments
Thanks to the reviewers and editors for their sincere comments.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- TAKEUCHI, M.; WEISS, N.; SCHUMANN, P.; YOKOTA, A.; TAKEUCHI, M.; WEISS, N.; SCHUMANN, P.; YOKOTA, A., Leucobacter komagatae gen. nov., sp. nov., a New Aerobic Gram-Positive, Nonsporulating Rod with 2,4-Diaminobutyric Acid in the Cell Wall. International Journal of Systematic and Evolutionary Microbiology 1996/10/01, 46, (4).
- Bates, K. A.; King, K. C. , <em>Leucobacter</em>. Trends in Microbiology 2021, 29, 1046–1047. [Google Scholar] [PubMed]
- Tan, M.-W.; Rahme, L. G.; Sternberg, J. A.; Tompkins, R. G.; Ausubel, F. M. , Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proceedings of the National Academy of Sciences 1999, 96, 2408–2413. [Google Scholar]
- Gao, H.; Qi, G.; Yin, R.; Zhang, H.; Li, C.; Zhao, X. , Bacillus cereus strain S2 shows high nematicidal activity against Meloidogyne incognita by producing sphingosine. Scientific Reports 2016, 6, 28756. [Google Scholar]
- Dinić, M.; Jakovljević, S.; Đokić, J.; Popović, N.; Radojević, D.; Strahinić, I.; Golić, N. , Probiotic-mediated p38 MAPK immune signaling prolongs the survival of Caenorhabditis elegans exposed to pathogenic bacteria. Scientific Reports 2021, 11, 21258. [Google Scholar]
- Lee, S. S.; Kennedy, S.; Tolonen, A. C.; Ruvkun, G. DAF-16 Target Genes That Control C. elegans Life-Span and Metabolism. Science 2003, 300, 644-647.
- Lin, Y.-C.; Uemori, K.; de Briel, D. A.; Arunpairojana, V.; Yokota, A. Zimmermannella helvola gen. nov., sp. nov., Zimmermannella alba sp. nov., Zimmermannella bifida sp. nov., Zimmermannella faecalis sp. nov. and Leucobacter albus sp. nov., novel members of the family Microbacteriaceae. International Journal of Systematic and Evolutionary Microbiology 2004, 54, 1669-1676.
- Morais, P. V.; Francisco, R.; Branco, R.; Chung, A. P.; da Costa, M. S. Leucobacter chromiireducens sp. nov, and Leucobacter aridicollis sp. nov., Two New Species Isolated From a Chromium Contaminated Environment. Systematic and Applied Microbiology 2004, 27, 646-652.
- Morais, P. V.; Paulo, C.; Francisco, R.; Branco, R.; Paula Chung, A.; da Costa, M. S. Leucobacter luti sp. nov., and Leucobacter alluvii sp. nov., two new species of the genus Leucobacter isolated under chromium stress. Systematic and Applied Microbiology 2006, 29, 414-421.
- Shin, N.-R.; Kim, M.-S.; Jung, M.-J.; Roh, S. W.; Nam, Y.-D.; Park, E.-J.; Bae, J.-W. Leucobacter celer sp. nov., isolated from Korean fermented seafood. International Journal of Systematic and Evolutionary Microbiology 2011, 61, 2353-2357.
- Yun, J.-H.; Roh, S. W.; Kim, M.-S.; Jung, M.-J.; Park, E.-J.; Shin, K.-S.; Nam, Y.-D.; Bae, J.-W. Leucobacter salsicius sp. nov., from a salt-fermented food. International Journal of Systematic and Evolutionary Microbiology 2011, 61, 502-506.
- Ue, H., Leucobacter exalbidus sp. nov., an actinobacterium isolated from a mixed culture from compost. The Journal of General and Applied Microbiology 2011, 57, 27-33.
- Behrendt, U.; Ulrich, A.; Schumann, P.Leucobacter tardus sp. nov., isolated from the phyllosphere of Solanum tuberosum L. International Journal of Systematic and Evolutionary Microbiology 2008, 58, 2574-2578.
- Her, J.; Lee, S.-S. Leucobacterhumi sp. nov., Isolated from Forest Soil. Current Microbiology 2015, 71, 235-242.
- Somvanshi, V. S.; Lang, E.; Schumann, P.; Pukall, R.; Kroppenstedt, R. M.; Ganguly, S.; Stackebrandt, E. Leucobacter iarius sp. nov., in the family Microbacteriaceae. International Journal of Systematic and Evolutionary Microbiology 2007, 57, 682-686.
- Muir, R. E.; Tan, M.-W. Leucobacter chromiireducens subsp. solipictus subsp. nov., a pigmented bacterium isolated from the nematode Caenorhabditis elegans, and emended description of L. chromiireducens. International Journal of Systematic and Evolutionary Microbiology 2007, 57, 2770-2776.
- Percudani, R. A Microbial Metagenome (Leucobacter sp.) in Caenorhabditis Whole Genome Sequences. Bioinformatics and Biology Insights 2013, 7, BBI.S11064.
- Hekimi, S.; Lakowski, B.; Barnes, T. M.; Ewbank, J. J. , Molecular genetics of life span in <em>C. elegans</em>: How much does it teach us? Trends in Genetics 1998, 14, 14–20. [Google Scholar]
- Park, H.-E. H.; Jung, Y.; Lee, S.-J. V. , Survival assays using Caenorhabditis elegans. Molecules and Cells 2017, 40, 90–99. [Google Scholar] [PubMed]
- Franco-Romero, A.; Morbidoni, V.; Milan, G.; Sartori, R.; Wulff, J.; Romanello, V.; Armani, A.; Salviati, L.; Conte, M.; Salvioli, S.; Franceschi, C.; Buonomo, V.; Swoboda, C. O.; Grumati, P.; Pannone, L.; Martinelli, S.; Jefferies, H. B. J.; Dikic, I.; van der Laan, J.; Cabreiro, F.; Millay, D. P.; Tooze, S. A.; Trevisson, E.; Sandri, M. C16ORF70/MYTHO promotes healthy aging in C.elegans and prevents cellular senescence in mammals. The Journal of Clinical Investigation 2024, 134, (15).
- Melentijevic, I.; Toth, M. L.; Arnold, M. L.; Guasp, R. J.; Harinath, G.; Nguyen, K. C.; Taub, D.; Parker, J. A.; Neri, C.; Gabel, C. V.; Hall, D. H.; Driscoll, M., C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 2017, 542, 367–371. [Google Scholar]
- O’Donnell, M. P.; Fox, B. W.; Chao, P.-H.; Schroeder, F. C.; Sengupta, P. , A neurotransmitter produced by gut bacteria modulates host sensory behaviour. Nature 2020, 583, 415–420. [Google Scholar]
- Ortiz, A.; Vega, N. M.; Ratzke, C.; Gore, J. , Interspecies bacterial competition regulates community assembly in the C. elegans intestine. The ISME Journal 2021, 15, 2131–2145. [Google Scholar]
- Liu, H.; Chen, P.; Yang, X.; Hao, F.; Tian, G.; Shan, Z.; Qi, B. Probiotics-sensing mechanism in neurons that initiates gut mitochondrial surveillance for pathogen defense. Cell Reports 2024, 43, (12).
- Wang, C.; Yang, M.; Liu, D.; Zheng, C. Metabolic rescue of α-synuclein-induced neurodegeneration through propionate supplementation and intestine-neuron signaling in C. elegans. Cell Reports 2024, 43, (3).
- Pujol, N.; Cypowyj, S.; Ziegler, K.; Millet, A.; Astrain, A.; Goncharov, A.; Jin, Y.; Chisholm, A. D.; Ewbank, J. J. , Distinct Innate Immune Responses to Infection and Wounding in the C. elegans Epidermis. Current Biology 2008, 18, 481–489. [Google Scholar]
- Ahmed, S. K.; Hussein, S.; Qurbani, K.; Ibrahim, R. H.; Fareeq, A.; Mahmood, K. A.; Mohamed, M. G. Antimicrobial resistance: Impacts, challenges, and future prospects. Journal of Medicine, Surgery, and Public Health 2024, 2, 100081.
- Kanehisa, M.; Goto, S. , KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C. A.; Blake, J. A.; Botstein, D.; Butler, H.; Cherry, J. M.; Davis, A. P.; Dolinski, K.; Dwight, S. S.; Eppig, J. T.; Harris, M. A.; Hill, D. P.; Issel-Tarver, L.; Kasarskis, A.; Lewis, S.; Matese, J. C.; Richardson, J. E.; Ringwald, M.; Rubin, G. M.; Sherlock, G. Gene Ontology: tool for the unification of biology. Nature Genetics 2000, 25, 25–29. [Google Scholar]
- Galperin, M. Y.; Wolf, Y. I.; Makarova, K. S.; Vera Alvarez, R.; Landsman, D.; Koonin, E. V. , COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Research 2021, (D1), D274–D281. [Google Scholar]
- Cantarel, B. L.; Coutinho, P. M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Research 2009, 37, (suppl_1), D233-D238.
- Jia, B.; Raphenya, A. R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K. K.; Lago, B. A.; Dave, B. M.; Pereira, S.; Sharma, A. N.; Doshi, S.; Courtot, M.; Lo, R.; Williams, L. E.; Frye, J. G.; Elsayegh, T.; Sardar, D.; Westman, E. L.; Pawlowski, A. C.; Johnson, T. A.; Brinkman, F. S. L.; Wright, G. D.; McArthur, A. G. , CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Research 2017, (D1), D566–D573. [Google Scholar]
- Saitou, N.; Nei, M. , The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 1987, 4, 406–425. [Google Scholar] [PubMed]
- Nei, M.; Kumar, S. , Molecular Evolution and Phylogenetics. In Oxford University Press: 2000.
- Rodriguez-R, L. M.; Konstantinidis, K. T. , Estimating coverage in metagenomic data sets and why it matters. The ISME Journal 2014, 8, 2349–2351. [Google Scholar] [CrossRef]
- Goris, J.; Konstantinidis, K. T.; Klappenbach, J. A.; Coenye, T.; Vandamme, P.; Tiedje, J. M. , DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. International Journal of Systematic and Evolutionary Microbiology 2007, 57, 81–91. [Google Scholar]
- Darling, A. C. E.; Mau, B.; Blattner, F. R.; Perna, N. T. , Mauve: Multiple Alignment of Conserved Genomic Sequence With Rearrangements. Genome Research 2004, 14, 1394–1403. [Google Scholar]
- Lee, D.; Zdraljevic, S.; Cook, D. E.; Frézal, L.; Hsu, J.-C.; Sterken, M. G.; Riksen, J. A. G.; Wang, J.; Kammenga, J. E.; Braendle, C.; Félix, M.-A.; Schroeder, F. C.; Andersen, E. C. Selection and gene flow shape niche-associated variation in pheromone response. Nature Ecology & Evolution 2019, 3, 1455-1463.
- Kang, W. K.; Florman, J. T.; Araya, A.; Fox, B. W.; Thackeray, A.; Schroeder, F. C.; Walhout, A. J. M.; Alkema, M. J. , Vitamin B12 produced by gut bacteria modulates cholinergic signalling. Nature Cell Biology 2024, 26, 72–85. [Google Scholar]
- Mallick, S.; Mishra, N.; Barik, B. K.; Negi, V. D. , <em>Salmonella</em> Typhimurium <em>fepB</em> negatively regulates <em>C. elegans</em> behavioral plasticity. Journal of Infection 2022, 84, 518–530. [Google Scholar]
- Amrit, F. R. G.; Ratnappan, R.; Keith, S. A.; Ghazi, A. , The C. elegans lifespan assay toolkit. Methods 2014, 68, 465–475. [Google Scholar] [PubMed]
- Naval-Sanchez, M.; Deshpande, N.; Tran, M.; Zhang, J.; Alhomrani, M.; Alsanie, W.; Nguyen, Q.; Nefzger, C. M. Benchmarking of ATAC Sequencing Data From BGI’s Low-Cost DNBSEQ-G400 Instrument for Identification of Open and Occupied Chromatin Regions. Frontiers in Molecular Biosciences 2022, 9.
- Deamer, D.; Akeson, M.; Branton, D. , Three decades of nanopore sequencing. Nature Biotechnology 2016, 34, 518–524. [Google Scholar] [PubMed]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P. A. , Assembly of long, error-prone reads using repeat graphs. Nature Biotechnology 2019, 37, 540–546. [Google Scholar]
- Cosma, B.-M.; Shirali Hossein Zade, R.; Jordan, E. N.; van Lent, P.; Peng, C.; Pillay, S.; Abeel, T. , Evaluating long-read de novo assembly tools for eukaryotic genomes: insights and considerations. GigaScience 2023, 12, giad100. [Google Scholar]
- Walker, B. J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C. A.; Zeng, Q.; Wortman, J.; Young, S. K.; Earl, A. M. , Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE 2014, 9, e112963. [Google Scholar]
- Delcher, A. L.; Bratke, K. A.; Powers, E. C.; Salzberg, S. L. , Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar]
- Lowe, T. M.; Eddy, S. R. , tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Research 1997, 25, 955–964. [Google Scholar]
- Lagesen, K.; Hallin, P.; Rødland, E. A.; Stærfeldt, H.-H.; Rognes, T.; Ussery, D. W. , RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research 2007, 35, 3100–3108. [Google Scholar]
- Chen, C.; Wu, Y.; Xia, R. , A painless way to customize Circos plot: From data preparation to visualization using TBtools. iMeta 2022, 1, e35. [Google Scholar]
- Richter, M.; Rosselló-Móra, R. , Shifting the genomic gold standard for the prokaryotic species definition. Proceedings of the National Academy of Sciences 2009, 106, 19126–19131. [Google Scholar]
- Wayne, L. G.; Brenner, D. J.; Colwell, R. R.; Grimont, P. A. D.; Kandler, O.; Krichevsky, M. I.; Moore, L. H.; Moore, W. E. C.; Murray, R. G. E.; Stackebrandt, E.; Starr, M. P.; Truper, H. G. , Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. International Journal of Systematic and Evolutionary Microbiology 1987, 37, 463–464. [Google Scholar]
- Kook, J.-K.; Park, S.-N.; Lim, Y. K.; Cho, E.; Jo, E.; Roh, H.; Shin, Y.; Paek, J.; Kim, H.-S.; Kim, H.; Shin, J. H.; Chang, Y.-H. , Genome-Based Reclassification of Fusobacterium nucleatum Subspecies at the Species Level. Current Microbiology 2017, 74, 1137–1147. [Google Scholar] [PubMed]
- McArthur Andrew, G.; Waglechner, N.; Nizam, F.; Yan, A.; Azad Marisa, A.; Baylay Alison, J.; Bhullar, K.; Canova Marc, J.; De Pascale, G.; Ejim, L.; Kalan, L.; King Andrew, M.; Koteva, K.; Morar, M.; Mulvey Michael, R.; O'Brien Jonathan, S.; Pawlowski Andrew, C.; Piddock Laura, J. V.; Spanogiannopoulos, P.; Sutherland Arlene, D.; Tang, I.; Taylor Patricia, L.; Thaker, M.; Wang, W.; Yan, M.; Yu, T.; Wright Gerard, D. , The Comprehensive Antibiotic Resistance Database. Antimicrobial Agents and Chemotherapy 2013, 57, 3348–3357. [Google Scholar] [CrossRef]
Figure 2.
Waterproof property of HNU-1 colony and characterization of its morphology. (a,b) E. coli (a) and HNU-1 (b) under optical microscope. Colony of E.coli shows no waterproof property (a, right), while colony of HNU-1 are waterproof (b, right). (c,d) Representative images of E. coli (c) and HNU-1 (d) under scanning electron microscope (SEM, left) and transmit electron microscope (TEM, right). Scale bars represent 5µm for c, and 1µm for d.
Figure 2.
Waterproof property of HNU-1 colony and characterization of its morphology. (a,b) E. coli (a) and HNU-1 (b) under optical microscope. Colony of E.coli shows no waterproof property (a, right), while colony of HNU-1 are waterproof (b, right). (c,d) Representative images of E. coli (c) and HNU-1 (d) under scanning electron microscope (SEM, left) and transmit electron microscope (TEM, right). Scale bars represent 5µm for c, and 1µm for d.
Figure 3.
Nanopore sequencing and assembly of HNU-1 genome. (
a) Chromosomal Genomic Map of
Leucobacter sp. HNU-1. Starting from the outermost ring: Ring 1: GC Skew (The specific formula is G-C/G+C, which measures the relative content of G and C. If G > C, the GC skew value is positive and is represented by the inward pink region; if G < C, it is negative and represented by the outward light green region.) Ring 2: GC Content (The inward red region indicates that the GC content in this area is lower than the average GC content of the entire genome, while the outward green region indicates the opposite. The higher the peak, the greater the difference from the average GC content.)Ring 3: Prokka Annotation (+)Ring 4: CARD RGI Results (+) Ring 5:Backbone (
Leucobacter sp. HNU-1) Ring 6: CARD RGI Results (-) Ring 7: CRISPRCasFinder Annotation (+)Ring 8: CRISPRCasFinder Annotation (-) Ring 9: ORFs (+3) Ring 10: ORFs (+2) Ring 11: ORFs (+1) Ring 12: ORFs (-1) Ring 13: ORFs (-2) Ring 14: ORFs (-3) Ring 15: Prokka Annotation (-) (
b) Details of KEGG of
Leucobacter sp. HNU-1. There are six categories, as shown on the right side of
Figure 2b, each category is divided into secondary classification system, X-axis is the number of genes, Y-axis is biological pathway Secondary classification, different colors are used to distinguish the primary classification of biological pathways.
Figure 3.
Nanopore sequencing and assembly of HNU-1 genome. (
a) Chromosomal Genomic Map of
Leucobacter sp. HNU-1. Starting from the outermost ring: Ring 1: GC Skew (The specific formula is G-C/G+C, which measures the relative content of G and C. If G > C, the GC skew value is positive and is represented by the inward pink region; if G < C, it is negative and represented by the outward light green region.) Ring 2: GC Content (The inward red region indicates that the GC content in this area is lower than the average GC content of the entire genome, while the outward green region indicates the opposite. The higher the peak, the greater the difference from the average GC content.)Ring 3: Prokka Annotation (+)Ring 4: CARD RGI Results (+) Ring 5:Backbone (
Leucobacter sp. HNU-1) Ring 6: CARD RGI Results (-) Ring 7: CRISPRCasFinder Annotation (+)Ring 8: CRISPRCasFinder Annotation (-) Ring 9: ORFs (+3) Ring 10: ORFs (+2) Ring 11: ORFs (+1) Ring 12: ORFs (-1) Ring 13: ORFs (-2) Ring 14: ORFs (-3) Ring 15: Prokka Annotation (-) (
b) Details of KEGG of
Leucobacter sp. HNU-1. There are six categories, as shown on the right side of
Figure 2b, each category is divided into secondary classification system, X-axis is the number of genes, Y-axis is biological pathway Secondary classification, different colors are used to distinguish the primary classification of biological pathways.
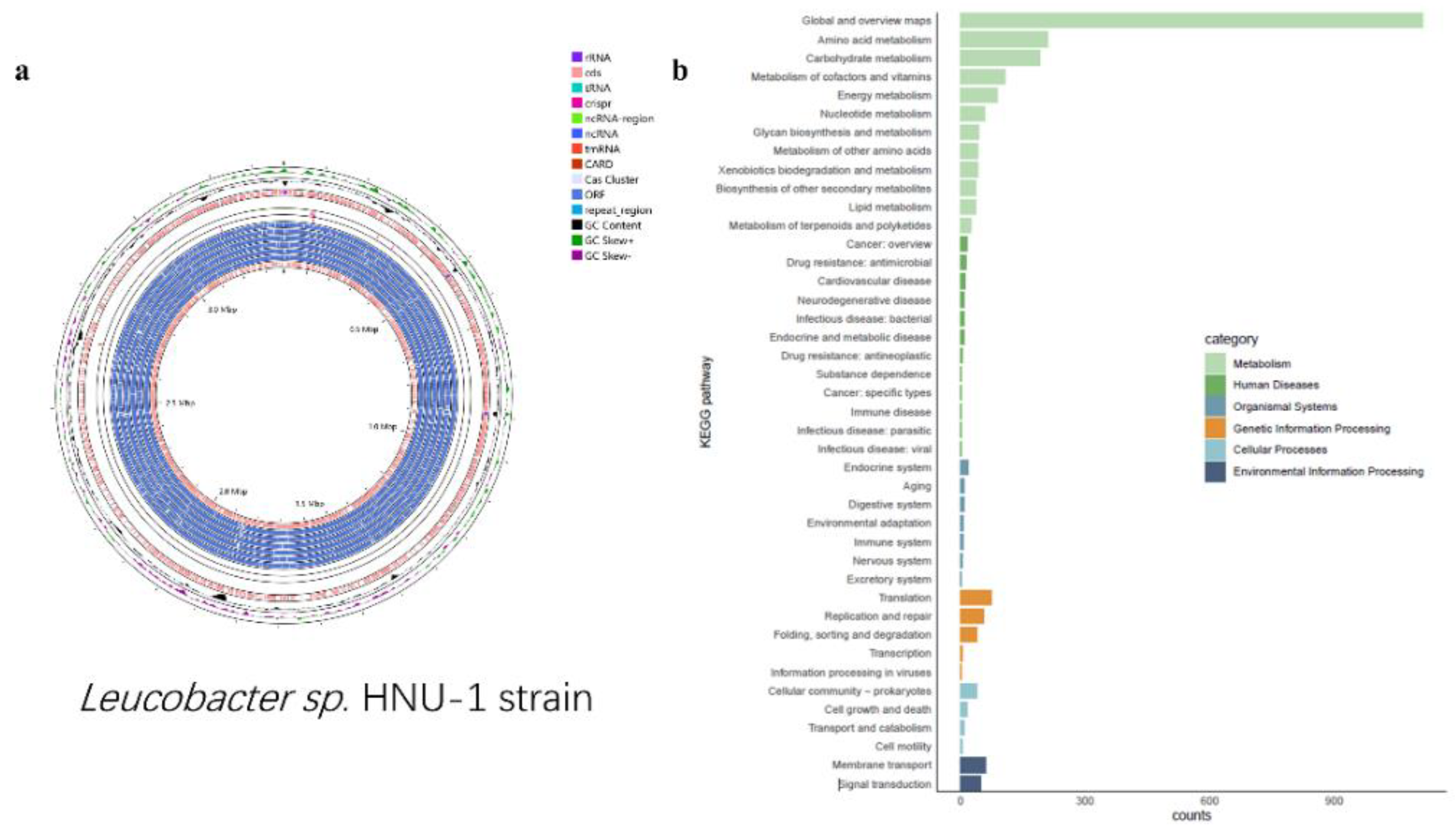
Figure 4.
Gene enrichment analysis of HNU-1 genome. (a) GO function classification of Leucobacter sp. HNU-1. The X-axis is the number of genes, Y-axis is GO term, and different colors are used to distinguish biological processes, cell components, and molecular functions. (b) COG Functional Annotation Classification of Leucobacter sp. HNU-1 A: RNA processing and modification; B: Energy production and conversion; C: Cell cycle control, cell division, and chromosome partitioning; D: Amino acid transport and metabolism;E: Nucleotide transport and metabolism; F: Carbohydrate transport and metabolism;G: Coenzyme transport and metabolism; H: Lipid transport and metabolism; I: Translation, ribosomal structure, and biogenesis; J: Transcription; K: Replication, recombination, and repair;L: Cell wall/membrane/envelope biogenesis; M: Cell motility; N: Posttranslational modification, protein turnover, and chaperones; O: Inorganic ion transport and metabolism; P: Secondary metabolite biosynthesis, transport, and catabolism; Q: General function prediction only;R: Function unknown; S: Signal transduction mechanisms; T: Intracellular trafficking, secretion, and vesicular transport; U: Defense mechanisms; V: Extracellular structures; W: Mobilome: prophages and transposons.
Figure 4.
Gene enrichment analysis of HNU-1 genome. (a) GO function classification of Leucobacter sp. HNU-1. The X-axis is the number of genes, Y-axis is GO term, and different colors are used to distinguish biological processes, cell components, and molecular functions. (b) COG Functional Annotation Classification of Leucobacter sp. HNU-1 A: RNA processing and modification; B: Energy production and conversion; C: Cell cycle control, cell division, and chromosome partitioning; D: Amino acid transport and metabolism;E: Nucleotide transport and metabolism; F: Carbohydrate transport and metabolism;G: Coenzyme transport and metabolism; H: Lipid transport and metabolism; I: Translation, ribosomal structure, and biogenesis; J: Transcription; K: Replication, recombination, and repair;L: Cell wall/membrane/envelope biogenesis; M: Cell motility; N: Posttranslational modification, protein turnover, and chaperones; O: Inorganic ion transport and metabolism; P: Secondary metabolite biosynthesis, transport, and catabolism; Q: General function prediction only;R: Function unknown; S: Signal transduction mechanisms; T: Intracellular trafficking, secretion, and vesicular transport; U: Defense mechanisms; V: Extracellular structures; W: Mobilome: prophages and transposons.
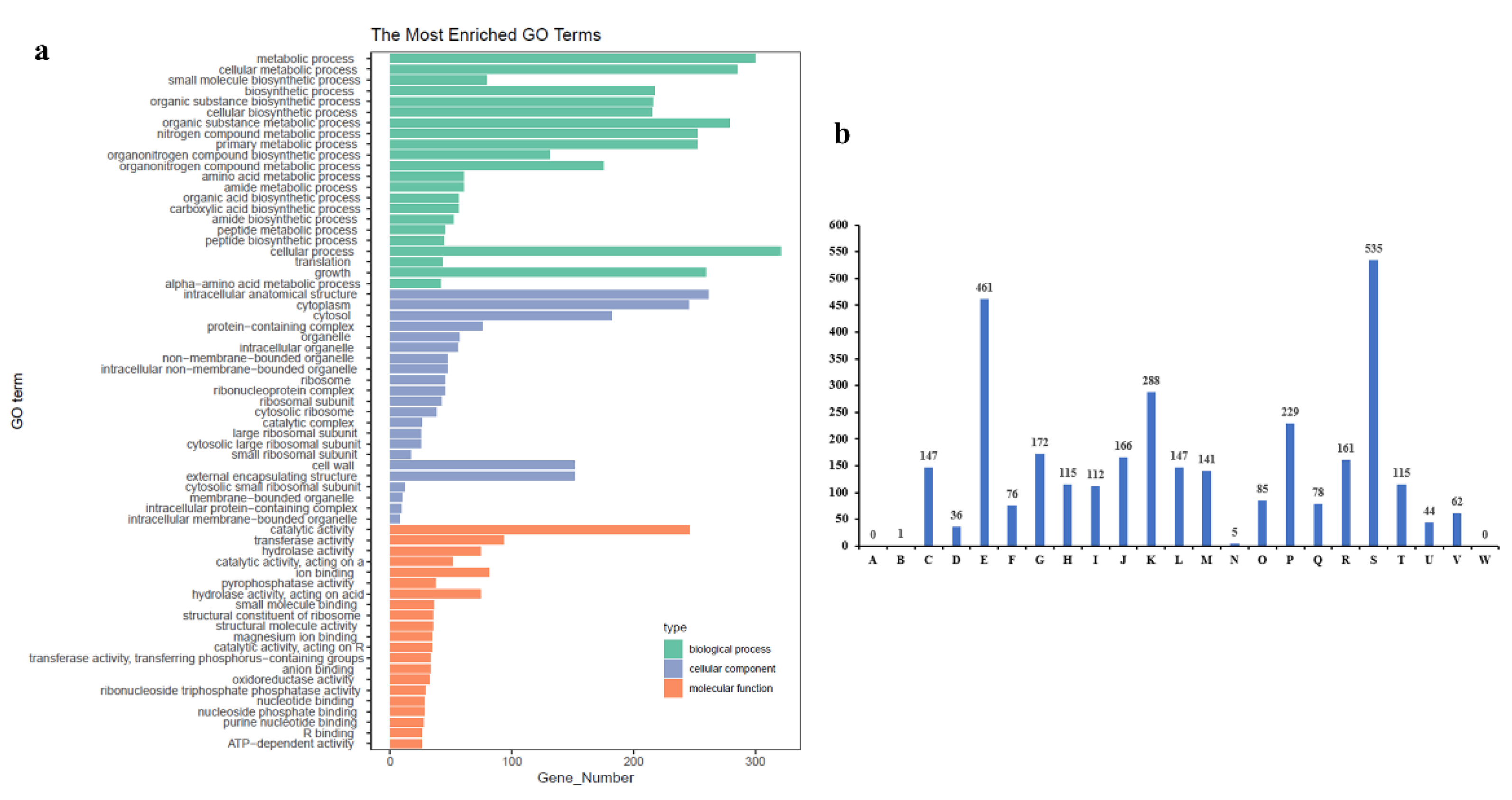
Figure 6.
Phylogenetic placement of HNU-1 among related bacteria. The phylogenetic tree was reconstructed based on the 16S rDNA genomes of 15 strains belonging to the genus Kocuria rhizophila strain NBC_01227 and Microbacterium oxydans strain NBRC 15586 were designated as outgroups.
Figure 6.
Phylogenetic placement of HNU-1 among related bacteria. The phylogenetic tree was reconstructed based on the 16S rDNA genomes of 15 strains belonging to the genus Kocuria rhizophila strain NBC_01227 and Microbacterium oxydans strain NBRC 15586 were designated as outgroups.
Table 2.
Analysis of Antibiotic Synthesis-Related Gene Clusters in Leucobacter sp. HNU-1.
Table 2.
Analysis of Antibiotic Synthesis-Related Gene Clusters in Leucobacter sp. HNU-1.
| Antibiotic |
Start |
Stop |
AMR Gene Family |
Best
Identities |
Orientation |
| vancomycin |
418,532 |
419,158 |
vanY;glycolpeptide resistance gene cluster |
31.71 |
+ |
| vancomycin |
659,487 |
660,260 |
vanY;glycopeptide resistance gene cluster |
38.93 |
+ |
| sulfadiazine |
2,675,593 |
2,676,432 |
sulfonamide resistant sul |
100 |
- |
| spectinomycin |
2,676,937 |
2,677,716 |
ANT(3'') |
100 |
- |
| defensin |
1,174,301 |
1,174,543 |
defensin resistant mprF |
100 |
- |
Table 3.
Antimicrobial Susceptibility Testing Results. According to the guidelines provided by the Clinical and Laboratory Standards Institute (CLSI), antimicrobial susceptibility test results are categorized into three groups:S (Sensitive): The strain is susceptible to the antibiotic, indicating effective inhibition of bacterial growth at standard dosages.I (Intermediate): The strain demonstrates intermediate resistance to the antibiotic, suggesting potential effectiveness under specific clinical conditions, such as high doses or localized treatment.R (Resistant): The strain is resistant to the antibiotic, meaning the antibiotic is generally ineffective at standard dosages.
Table 3.
Antimicrobial Susceptibility Testing Results. According to the guidelines provided by the Clinical and Laboratory Standards Institute (CLSI), antimicrobial susceptibility test results are categorized into three groups:S (Sensitive): The strain is susceptible to the antibiotic, indicating effective inhibition of bacterial growth at standard dosages.I (Intermediate): The strain demonstrates intermediate resistance to the antibiotic, suggesting potential effectiveness under specific clinical conditions, such as high doses or localized treatment.R (Resistant): The strain is resistant to the antibiotic, meaning the antibiotic is generally ineffective at standard dosages.
| Category |
Concentration/disc |
Interpretation Standard (mm) |
Diameter (mm) |
Result |
| Resistant (R) |
Intermediate (I) |
Sensitive (S) |
| Penicillin |
10U |
≤10 |
11-16 |
≥17 |
26.74 |
S |
| Vancomycin |
30µg |
≤14 |
15-16 |
≥17 |
15.9 |
I |
| Oxacillin |
1µg |
≤10 |
11-12 |
≥13 |
3 |
R |
| Levofloxacin |
5µg |
≤13 |
14-17 |
≥18 |
18.26 |
S |
| Clindamycin |
2µg |
≤14 |
15-19 |
≥20 |
12.1 |
R |
| Erythromycin |
15µg |
≤13 |
14-22 |
≥23 |
15.22 |
I |
| Polymyxin B |
300IU |
≤8 |
9-11 |
≥12 |
6.42 |
R |
| Gentamicin |
10µg |
≤12 |
13-14 |
≥15 |
18.72 |
S |
| Lincomycin |
2µg |
≤14 |
15-20 |
≥21 |
3 |
R |
| Minocycline |
30µg |
≤15 |
16-18 |
≥19 |
32.16 |
S |
| Tetracycline |
30µg |
≤11 |
12-14 |
≥15 |
12.26 |
I |
| Chloramphenicol |
30µg |
≤12 |
13-17 |
≥18 |
29.56 |
S |
| Imipenem |
10µg |
≤19 |
20-22 |
≥23 |
17.06 |
R |
| Doxycycline |
30µg |
≤12 |
13-15 |
≥16 |
17.8 |
S |
| Azithromycin |
15µg |
≤17 |
18-19 |
≥20 |
25.48 |
S |
| Ceftriaxone |
30µg |
≤19 |
20-22 |
≥23 |
18.82 |
R |
| Ceftazidime |
30µg |
≤14 |
15-17 |
≥18 |
19.72 |
S |
| Cefoperazone |
75µg |
≤22 |
23-25 |
≥26 |
8 |
R |
| Ciprofloxacin |
5µg |
≤15 |
16-20 |
≥21 |
12 |
R |
| Norfloxacin |
10µg |
≤12 |
13-16 |
≥17 |
6.44 |
R |
| Florfenicol |
30µg |
≤12 |
13-17 |
≥18 |
31.56 |
S |
| Piperacillin |
100µg |
≤17 |
18-20 |
≥21 |
29.32 |
S |
| Streptomycin |
10µg |
≤6 |
7-9 |
≥10 |
3 |
R |
Compound-
Sulfonamides |
25µg |
≤10 |
11-15 |
≥16 |
3 |
R |
| Ampicillin |
10µg |
≤13 |
14-16 |
≥17 |
24.14 |
S |
| Kanamycin |
30µg |
≤13 |
14-17 |
≥18 |
3 |
R |
| Amikacin |
30µg |
≤14 |
15-16 |
≥17 |
17.98 |
S |
| Cefuroxim |
30µg |
≤14 |
15-17 |
≥18 |
18.48 |
S |
| Cephalexin |
30µg |
≤14 |
15-17 |
≥18 |
22.64 |
S |
| Cefamezin |
30µg |
≤14 |
15-17 |
≥18 |
24.42 |
S |
Table 4.
Comparison of Sequence Characteristics Between Leucobacter sp. HNU-1 and Six Other Species in the Leucobacter Genus.
Table 4.
Comparison of Sequence Characteristics Between Leucobacter sp. HNU-1 and Six Other Species in the Leucobacter Genus.
| Item |
Leucobacter sp. HNU-1 |
Leucobacter iarius JCM 14736 |
Leucobacter chromiireducens TAN 31504 |
Leucobacter aridicollis DSM 17380 |
Leucobacter coleopterorum HDW9A |
Leucobacter komagatae DSM 8803 |
Leucobacter luti RF6 |
| GeneBank assembly |
PRJNA1137138 |
GCA_039530105.1 |
GCA_016758195.1 |
GCA_013409595.1 |
GCA_011382985.1 |
GCA_006716085.1 |
GCA_004217175.1 |
| Length of chromosome /bp |
3,375,033 |
3,524,626 |
3,537,946 |
3,573,416 |
3,215,551 |
3,752,337 |
3,618,231 |
| GC content/% |
70.37 |
70.57 |
68.93 |
67.32 |
60.32 |
66.63 |
69.46 |
| Number of CDSs |
3,268 |
3,188 |
3,096 |
3,257 |
2,980 |
3,375 |
3,089 |
| Number of rRNAs |
9 |
3 |
3 |
9 |
6 |
9 |
5 |
| Number of tRNAs |
52 |
53 |
54 |
52 |
45 |
50 |
46 |
| Number of CRISPRS |
6 |
13 |
1 |
2 |
2 |
2 |
3 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).