Submitted:
11 March 2025
Posted:
13 March 2025
You are already at the latest version
Abstract
This review highlights a paradigm shift in our understanding of hypocalcemia during milk fever by introducing the concept of the Calci-Inflammatory Axis. Traditionally viewed as a pathological deficiency necessitating rapid correction (e.g., through calcium borogluconate infusions or dietary adjustments like dietary cation-anion difference), periparturient hypocalcemia is reinterpreted here as an adaptive, protective response. Within this new framework, reduced circulating calcium levels may help temper systemic inflammation by limiting lipopolysaccharide (LPS) aggregation and curbing excessive macrophage activation. The review discusses how calcium signaling, the cal-cium-sensing receptor (CaSR), and immune cell functions adapt under hypocalcemic conditions to modulate inflammatory processes. This integrated perspective not only redefines the role of hy-pocalcemia but also proposes the Calci-Inflammatory Axis as a novel concept through which we can understand how changes in calcium homeostasis mitigate inflammatory cascades—potentially lowering the incidence of periparturient diseases and enhancing overall cow health and farm productivity. Future research should investigate the long-term effects of hypocalcemia, the envi-ronmental influences on this Calci-Inflammatory Axis, and their collective impact on disease sus-ceptibility and inflammation.
Keywords:
milk fever
; hypocalcemia
; inflammation
; dairy cows
; endotoxemia
1. Introduction
Milk fever, or periparturient hypocalcemia, has long been recognized as a critical metabolic disorder affecting dairy cows during the transition period. Traditionally, this condition has been understood as a direct consequence of a significant drop in blood calcium levels [1,2], leading to the widespread adoption of preventive and therapeutic strategies focused on managing dietary calcium prior to parturition and dietary adjustments. Approaches such as dietary cation-anion difference (DCAD) manipulation [3], calcium boluses, subcutaneous or intravenous calcium borogluconate have been developed to counteract hypocalcemia, reflecting a reductionist view that equates milk fever primarily with calcium deficiency [4].
However, despite the extensive use of these interventions, milk fever continues to pose a persistent challenge in dairy herds in North America [2,5] and globally [6,7], with many cows still experiencing its debilitating effects. The limited success of these tradi-tional strategies prompts a critical reassessment of the underlying assumptions in milk fever management. If correcting calcium deficiency were indeed the central solution, the persistence of the disorder suggests that these strategies may fail to address the full complexity of the condition [2,8].
This paradox highlights a potential gap in our understanding of milk fever, indi-cating that the condition might not be as straightforward as previously thought. Emerging research, notably the hypothesis proposed by Ametaj and his team [9,10,11], challenges the conventional paradigm by suggesting that hypocalcemia during milk fever may not merely represent a pathological deficiency but rather an evolutionary adaptive physiological response aimed at mitigating systemic inflammation. This shift in perspective encour-ages us to reconsider hypocalcemia not as a problem to be corrected but as a potential protective mechanism within the broader context of the cow’s immune response.
Building on this adaptive hypothesis, this review introduces a novel concept—"The Calci-Inflammatory Axis” —which proposes that fluctuations in circulating calcium levels, sensed in part by the calcium-sensing receptor (CaSR) on immune cells, play a pivotal role in orchestrating inflammatory processes. Rather than viewing transient hypocalcemia purely as a deficiency, the Calci-Inflammatory Axis proposes that controlled reductions in calcium concentration can help modulate the intensity of immune activation and cytokine release, potentially averting excessive inflammation. By framing hypocalcemia as a physiological buffer rather than a pathological anomaly, this perspective offers a fresh lens through which to understand how calcium dynamics can influence both metabolic and immunological outcomes during the periparturient period.
In this review, we will revisit the historical context of milk fever management, critically analyzing why conventional approaches have often fallen short in fully resolving the condition. We will then discuss the growing body of evidence supporting the concept that hypocalcemia may play an adaptive, anti-inflammatory role, particularly in the context of endotoxemia and inflammation. By examining the physiological and molecular mechanisms underlying calcium homeostasis and immune regulation, we aim to deepen the understanding of milk fever. Our objective is not to introduce new management strategies but to refine and expand the current knowledge of milk fever from an integrative perspective.
2. Evolving Hypotheses of Milk Fever: A Historical and Contemporary Overview
Milk fever, also known as parturient paresis, has long been recognized as one of the most complex and puzzling diseases in veterinary medicine, particularly affecting dairy cows during the postparturient period. Since its identification in the 18th century, the condition has prompted numerous hypotheses and research endeavors aimed at unraveling its etiology, reflecting ongoing challenges in veterinary science. The evolution of scientific thought surrounding milk fever is well documented in the seminal review by Hibbs in 1954 [12], which provides a comprehensive historical perspective on the different hypotheses proposed until that time. Despite extensive research efforts, many of these early postulates were eventually proven to be incorrect or incomplete, and new hypotheses continue to emerge as our understanding of the disease deepens.
2.1. Historical Context and Early Theories
2.1.1. Hibbs Review and Subsequent Hypotheses
One of the most comprehensive reviews on milk fever was authored by Hibbs [12]. By that time, milk fever had become a significant focus of research, and by the 1950s, around 30 different hypotheses had been proposed to explain the condition. The disease was mentioned for the first time in 1793 by Eberhardt [13]. The proposed hypotheses ranged widely—encompassing general inflammation, nervous system disorders, circulatory issues, cerebral anemia and congestion, apoplexy, thrombosis, fat embolism, spinal trauma, general infections, bacterial infections of uterine or mammary origin, anaphylaxis, mammary-neurasthenia, dehydration, auto-intoxication, impaired tissue oxidation, excess oxytocin post-parturition, faulty protein metabolism, ovarian dysfunction, hyperactivity of the anterior pituitary, disturbed cholesterol metabolism, hyperadrenalinemia, magnesium narcosis, alkalosis, acidosis, auto-asphyxiation, and hypoglycemia [12]. Although diverse, many of these theories have not stood the test of time.
2.1.2. Development of the Hypocalcemia Theory
The first authors to propose a parathyroid deficiency and hypocalcemia in dairy cows affected by milk fever were Dryerre and Greig in 1925 [14]. Based on clinical observations and speculative reasoning, they hypothesized that udder inflation would stimulate secretion of adrenalin to “oxidize toxins.” However, the nature of these toxins was not elaborated, and the hypothesis was soon discarded when experiments by Auger [15] failed to detect increased blood pressure indicative of elevated adrenalin. A pivotal development came with the work of Little and Write (1925) [16], who demonstrated a significant decrease in blood calcium concentrations in cows with clinical milk fever, thereby establishing hypocalcemia as a key feature of the disease. Almost a decade later, Dryerre and Greig (1935) [17] demonstrated that subcutaneous or intramuscular injections of calcium boro-gluconate improved clinical signs, reinforcing that an acute drop in blood calcium—precipitated by the sudden onset of lactation—was central to the pathophysiology. Although these findings dramatically lowered milk fever incidence and led to widely adopted therapeutic measures, the hypocalcemia hypothesis alone could not explain every aspect of the disease, driving further research into endocrine and metabolic imbalances that underlie calcium regulation.
2.1.3. The DCAD and Potassium Hypothesis: A Shift in Thinking
Beginning in the 1950s, the DCAD theory brought fresh insights into milk fever prevention. Researchers such as Ender [18], Dishington [19], and later Block [3] found that balancing dietary cations (e.g., potassium, sodium) and anions (e.g., chloride, sulfur) influences calcium metabolism, protecting cows from hypocalcemia near calving. More specifically, feeding a diet with a negative DCAD value promotes calcium mobilization and supports better serum calcium concentrations in the transition period. Subsequently, Horst et al. [20] highlighted that high dietary potassium could impede calcium absorption, rendering dairy cows more vulnerable to milk fever. They provided the first direct evidence that feeding cows a diet lower in potassium decreased the incidence of milk fever. These discoveries propelled a more comprehensive view of disease management by acknowledging the interplay of dietary factors, metabolic processes, and calcium regulation [20].
2.1.4. The Endotoxin Hypothesis and Systemic Inflammation: A New Perspective
Within the last three decades, the endotoxin hypothesis has garnered attention as a compelling explanation for milk fever’s etiology, proposing that Gram-negative bacterial LPS absorbed from mucosal sites (e.g., the gastrointestinal tract) disrupts calcium homeostasis through systemic inflammation. Aiumlamai et al. [21] initially suggested that endotoxin translocation might aggravate parturient paresis, while Andersen [22] argued that ruminal acidosis allows LPS to breach the rumen barrier, subsequently contributing to laminitis, abomasal displacement, and other production-related diseases. Although the precise mechanisms remain elusive, Ametaj et al. [23,24] advanced the notion that endotoxins may initiate or worsen milk fever and fatty liver, later extending it to multiple periparturient conditions—ranging from ruminal acidosis to retained placenta, mastitis, and downer cow syndrome [10,11,25]. By attributing significant importance to immune-system activation, this new perspective emphasizes that milk fever involves more than just a metabolic issue.
2.1.5. Endotoxins and Systemic Inflammation
Endotoxins strongly stimulate the innate immune response through Toll-like receptor 4 (TLR4), initiating pathways involving nuclear factor-kappa B (NF-κB) and leading to proinflammatory cytokine secretion (tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1) [26]. These events manifest as the acute-phase response (APR), which includes fever, acute-phase proteins, and shifts in plasma ion concentrations [27,28]. Augustine et al. [29] reported that TNF-α suppresses parathyroid hormone (PTH) synthesis and inhibits renal 1a-hydroxylase activity, leading to lower levels of 1,25-dihydroxyvitamin D, thereby compromising calcium regulation. Additionally, endotoxins can modify the expression of renal and intestinal Ca²⁺ transporters [30]. Meanwhile, albumin, lactate, non-esterified fatty acids (NEFA), and certain anions (Cl⁻, SO₄²⁻) bind or chelate calcium (Figure 1), while cells sequester it, collectively exacerbating hypocalcemia just when cows require calcium most. In this context, proinflammatory cytokines further impede bone resorption and calcium mobilization [31]. We are referring to these interconnected metabolic and immune pathways as part of a broader concept of the Calci-Inflammatory Axis that sets the stage for Section 4’s in-depth discussion.
3. Inflammatory Response in Milk Fever Cows
A significant feature of milk fever is the presence of a low-grade or subclinical inflammatory state that both precedes and coincides with clinical hypocalcemia. Elevated concentrations of acute-phase proteins [e.g., haptoglobin, serum amyloid A (SAA)] and proinflammatory cytokines (e.g., TNF-α) indicate that systemic inflammation may be integral to milk fever pathogenesis [25,32]. This aligns with the general trend in periparturient disorders, where immune dysregulation and metabolic strain interact.
Ametaj and collaborators further explored this immune component in various periparturient diseases, including milk fever [9,33]. Their findings propose that the inflammatory response mounted against endotoxins may act as both a pathogenic contributor to hypocalcemia and, paradoxically, a protective mechanism aiming to contain inflammation during the transition period. Such an immune-metabolic synergy offers a more subtle understanding than traditional “calcium deficiency” paradigm alone. The interplay between inflammation and ionized calcium—which Ametaj is proposing to call the Calci-Inflammatory Axis — anticipates the broader re-evaluation of milk fever theories in Section 4.
4. Re-Evaluating Traditional Theories in Light of the Endotoxin Hypothesis: The Calci-Inflammatory Axis
The history of milk fever research demonstrates an evolution from early, often speculative frameworks toward more integrative models. While the 1954 review by Hibbs [12] anchored milk fever in a predominantly metabolic context, subsequent progress with the hypocalcemia theory and the DCAD/potassium hypothesis substantially reduced disease incidence by highlighting dietary and endocrine regulation of calcium [5,6,7,8]. Yet, these metabolic-centered paradigms do not fully account for systemic inflammation and immune activation—especially as emphasized by the endotoxin hypothesis, which proposes that Gram-negative bacterial LPS can trigger an inflammatory cascade that disrupts calcium homeostasis [9,10,11,21,22].
Recent studies have integrated these findings, leading to the proposal of the Calci-Inflammatory Axis, which highlights the reciprocal interactions between inflammation and serum iCa²⁺ levels. At the core of this axis is the observation that endotoxin-induced inflammation can lower blood calcium levels, both by (1) altering hormone signaling (e.g., PTH, 1,25 dihydroxyvitamin D) and (2) sequestering Ca²⁺ into tissues or binding it through acute-phase reactants like albumin or fatty acids. Simultaneously, changes in serum iCa²⁺—particularly moderate hypocalcemia—can reduce certain proinflammatory signaling cascades, sometimes acting as a physiological brake on overactive immune response [34,35].
4.1. Multiple Sources of Endotoxin and Early Immune Activation
Evidence suggests that LPS or endotoxin can translocate from a range of sites, including the rumen—particularly under subacute ruminal acidosis (SARA) conditions [36]—as well as from mastitic udders, metritic uteri, or infected hooves [21,22]. In all these cases, local Gram-negative bacterial growth and tissue damage facilitate LPS leakage into the bloodstream, where it engages Toll-like receptor 4 (TLR4) on immune cells, triggering the release of potent cytokines and acute-phase proteins such as TNF-α, IL-6, and IL-1 and SAA [37,38]. Notably, innate immune activation can be present before the clinical onset of milk fever. Zhang et al. [25] reported increased TNF-α, IL-6, and SAA levels in cows during the dry-off period, well prior to parturition and overt symptoms of milk fever. These findings indicate that a low-grade inflammatory state—potentially arising from subclinical infections or endotoxin translocation—may prime the immune system in advance of calving, setting the stage for subsequent hypocalcemia once lactational calcium demands surge.
4.2. Mechanisms of the Calci-Inflammatory Axis
From an immunophysiological standpoint, macrophages and neutrophils depend critically on Ca²⁺-dependent signaling pathways when activated by LPS. High intracellular Ca²⁺ levels facilitate the activation of various downstream pathways that drive the release of pro-inflammatory cytokines, including TNF-α, IL-1, and IL-6 [39,40]. This robust cytokine response is crucial for an effective defense against pathogens; however, it can also contribute to tissue damage if left unchecked.
A controlled, transient reduction in intracellular Ca²⁺—as observed in the periparturient hypocalcemia of cows—appears to serve as a regulatory brake on this inflammatory cascade. By diminishing Ca²⁺-dependent signaling, the immune cells produce lower quantities of cytokines, thereby potentially mitigating tissue damage that would arise from an overly aggressive inflammatory response [41]. In essence, this temporary hypocalcemic state may act as a natural anti-inflammatory mechanism, preserving tissue integrity during periods of high metabolic and immunological stress.
The Calci-Inflammatory Axis, however, can become dysregulated under conditions of chronic or repeated exposure to LPS. In scenarios such as persistent subacute ruminal acidosis (SARA), mastitis, metritis, or untreated hoof infections, the immune system may remain in a prolonged state of activation. This sustained inflammatory state leads to continuous cytokine production, which in turn interferes with calcium homeostasis by antagonizing PTH actions or by altering vitamin D metabolism. As a result, the restoration of normal calcium levels is delayed, perpetuating a cycle where chronic hypocalcemia further undermines neuromuscular function and metabolic stability, while persistent inflammation continues to disrupt calcium mobilization [42].
Adding to this complexity, the CaSR plays a pivotal role in mediating these interactions. Expressed on both the parathyroid glands and various immune cells, CaSR modulates immune responses based on extracellular Ca²⁺ concentrations. During inflammatory stress, cytokines can downregulate CaSR function, effectively shifting the calcium homeostasis set point and exacerbating hypocalcemia [34,42,43]. Furthermore, the active form of vitamin D, 1,25-dihydroxyvitamin D, may influence both CaSR signaling and immune cell activity, thereby reinforcing the hypocalcemic state under conditions of inflammation [44,45,46].
Collectively, the Calci-Inflammatory Axis is not merely a linear feedback loop but a multifaceted network involving receptors, hormones, transport proteins, and cytokines. This network intricately regulates serum calcium levels in response to both physiological and pathological challenges, highlighting the dual role of calcium as both a metabolic substrate and a key modulator of immune function. Understanding this axis offers new insights into how transient hypocalcemia may serve as an adaptive response, while also underscoring the risks associated with its chronic dysregulation in dairy cows.
4.3. Clinical Consequences
Seen through this lens, milk fever is neither strictly a metabolic deficiency nor purely an inflammatory disease. Rather, it arises at the intersection of mounting metabolic demands for calcium at parturition and an immune system that may already be hyperreactive. Excessive inflammatory signals impede calcium retrieval from bone and gut, perpetuating hypocalcemia. In turn, prolonged or severe hypocalcemia weakens core physiological functions, increasing the risk of muscle weakness, recumbency, and organ dysfunction. The Calci-Inflammatory Axis thus reframes milk fever as a condition wherein systemic inflammation and calcium dysregulation mutually amplify each other’s injurious effects.
4.4. Hypocalcemia in Multiple Periparturient Diseases: The Common Role of Inflammation
Multiple lines of evidence reveal that hypocalcemia is not solely linked to milk fever but frequently occurs in other periparturient diseases, including ketosis, left displaced abomasum (LDA), acute puerperal metritis (APM), and mastitis. In a study by Venjakob et al. [47], cows displaying suboptimal calcium levels around calving showed persistently reduced serum calcium on days 0, 1, and 3 postpartum—an outcome mirrored in these other health conditions. Moreover, ketosis, particularly among first-calf heifers at 3 and 7 days postpartum, consistently features lower calcium concentrations, while cows with LDA are disproportionately prone to developing hypocalcemia [48,49]. Comparable patterns are evident in cows affected by APM, mastitis, retained placenta, dystocia, and left displaced abomasum pointing toward hypocalcemia as both a predisposing factor for these disorders and a by-product of the inflammation they provoke [50,51].
These convergent findings highlight inflammation as a central mechanism linking hypocalcemia with various periparturient diseases. Metabolic disturbances such as ketosis, LDA, APM, and mastitis trigger inflammatory pathways that elevate the release of cytokines like IL-6 and TNF-α, which disrupt calcium equilibrium in several ways. Specifically, cytokines can depress PTH secretion and modify the CaSR, ultimately compromising intestinal calcium absorption, skeletal calcium mobilization, and renal calcium reabsorption [42,52,53]. As inflammation worsens, hypocalcemia intensifies, creating susceptibility to further metabolic or infectious insults and perpetuating a negative feedback loop of immune system impairment and low calcium.
In inflammatory states, the surge in immune-cell activity heightens the need for calcium to fuel fundamental cellular processes. Immunometabolomic research [54] shows that chronic, subclinical inflammation helps sustain this increased calcium requirement. Simultaneously, deepening hypocalcemia weakens neutrophil functionality [55], thereby raising animals’ vulnerability to secondary infections. This two-way interaction magnifies existing metabolic imbalances and promotes a cycle wherein hypocalcemia both stems from and exacerbates ongoing inflammation.
Nutritional factors can compound this cyclical pattern. Conditions like ketosis and mastitis often reduce dry matter intake, limiting dietary calcium precisely when metabolic demands spike. As a consequence, serum calcium decreases even further, impeding the cow’s capability to manage acute-phase responses effectively. Meanwhile, inflammatory mediators impede the usual hormonal processes that restore calcium homeostasis, allowing persistent hypocalcemia to heighten immune dysfunction and metabolic strain.
Central to this cycle is the mutual causation between hypocalcemia and inflammation. Low iCa²⁺ availability reduces immune-cell efficiency—evidenced by poorer neutrophil performance and diminished immune-cell populations [55]—thereby escalating the odds of infection and subsequent inflammatory events. Conversely, infections and endotoxemia stimulate the production of proinflammatory cytokines that interfere with CaSR and other calcium-regulatory pathways [42]. Studies by Zhang et al. [25] and Abuajamieh et al. [56] further confirm that disordered metabolism and inflammation, acting in unison, are key contributors to conditions like milk fever and ketosis. Such a self-perpetuating feedback loop—where hypocalcemia-induced immunosuppression triggers infection or endotoxin influx, which then reinforces inflammatory activity—emphasizes the necessity of a comprehensive strategy for disease prevention and control.
5. A New Hypothesis on Hypocalcemia and Its Role in Inflammation
Ametaj et al. [10,11] have hypothesized that hypocalcemia, frequently observed during milk fever and also during systemic inflammation and endotoxemia, may serve a protective function by helping control LPS bioactivity in the bloodstream and modulating the systemic inflammatory response. According to this hypothesis, moderate or transient reductions in iCa²⁺ might limit excessive calcium-dependent activation of immune cells, providing a physiological brake on inflammation [57,58]. Evidence suggests that rather than being an outright deficiency, this hypocalcemia could be a built-in adaptive response aimed at mitigating harmful effects of endotoxemia and systemic inflammation.
5.1. Fate of Endotoxin in the Circulation
Endotoxin (ET), primarily LPS shed in the gastrointestinal tract (GIT) due to high grain or fat intake, often associates with chylomicrons in epithelial cells, travels via the lymph, and triggers proinflammatory cytokine release by macrophages in the mesenteric lymph node (Figure 1) [59]. Once in the circulation, a portion of ET enters the portal vein and is scavenged by immune cells, Kupffer cells, and hepatocytes in the liver, thereby reducing its systemic levels [60]. However, some ET may bypass hepatic clearance and reach systemic circulation, where it binds to high-density lipoprotein (HDL) and low-density lipoprotein (LDL). This binding is mediated by lipopolysaccharide-binding protein (LBP) and soluble CD14 (sCD14) [61,62,63].
5.2. Calcium’s Role in LPS Aggregation and Immune Activation
Ionized calcium influences how LPS behaves in the bloodstream (Figure 1). Under in vitro conditions, divalent cations such as Ca²⁺ help LPS adopt tightly packed “lamellar” or “multilamellar” structures, generally lowering LPS’s capacity to trigger Toll-like receptor (TLR)-mediated inflammation [64]. Nevertheless, LPS can also occur in various aggregate states, and binding to LBP can rearrange or partially disaggregate LPS [65,66]. When LPS effectively engages macrophages via membrane-bound CD14 (mCD14), a robust cytokine response ensues (e.g., TNF-α, IL-1, IL-6), which is vital for combating infection but detrimental if overactivated [66]. In vivo, elevated intracellular calcium in immune cells can fuel the inflammatory cascade [35,67], potentially aggravating organ damage. Thus, although Ca²⁺ in plasma can stabilize less-inflammatory LPS structures, excessive Ca²⁺-dependent signaling inside macrophages or other immune cells may exacerbate inflammation.
5.3. Hypocalcemia as a Protective Mechanism
Ametaj postulates that hypocalcemia, observed during sepsis and systemic inflammation, may serve as an adaptive mechanism to reduce overactivation of the immune system. By lowering iCa²⁺, the stimulus for calcium-dependent intracellular pathways in macrophages and neutrophils is diminished, limiting the explosive release of proinflammatory cytokines [11]. Indeed, studies show that interventions that reduce iCa²⁺—for instance, through calcium chelation—can curb LPS-induced mortality, hinting that transient hypocalcemia buffers against excessive endotoxemia [35,68]. Rather than simply aggregating LPS into harmless multilamellar structures, this protective mechanism primarily lessens the immune-cell hyperactivation that otherwise leads to systemic inflammation and potential organ failure.
5.4. Lipoproteins and LPS Clearance
Regardless of LPS’s structural form (aggregated or partially disaggregated), HDL and LDL serve as major vehicles for LPS clearance (Figure 1). During systemic inflammation, normal HDL (n-HDL) undergoes compositional changes, with SAA replacing apolipoprotein A-I (apoA-I) and forming acute-phase HDL (ap-HDL) [62,63]. Acute-phase HDL exhibits higher affinity for LPS, promoting its neutralization and hepatic excretion via bile [61,69]. This pathway not only removes LPS from circulation but also prevents excessive immune activation, underscoring the importance of lipoproteins in managing systemic inflammation. In the context of hypocalcemia, dampened immune-cell activity gives lipoprotein-mediated clearance an extended window to operate without provoking large cytokine surges. Thus, the protective advantage of lower iCa²⁺ may lie in controlling macrophage overactivation and forcing LPS into multilamellar forms.
5.5. Modulation of the Inflammatory Response
By containing calcium-dependent signaling in immune cells and enhancing LPS clearance via lipoproteins, mild hypocalcemia can help modulate the inflammatory response, mitigating the overproduction of cytokines. This is especially relevant in conditions such as endotoxemia and sepsis, where an excessive immune response can be detrimental [63,70]. Indeed, multiple studies in humans and animal models report that elevating iCa²⁺ during sepsis may worsen outcomes, whereas interventions that keep iCa²⁺ at lower thresholds can improve survival and reduce organ damage [35]. These findings align with the idea that transient hypocalcemia might function as a physiological safeguard, preventing excessive inflammation until endotoxins are effectively cleared by hepatic macrophages or lipoproteins.
5.6. Interactions of Calcium with Lactate, Fatty Acids, and Albumin
The hypothesis also acknowledges that elevated lactate, NEFA levels, and albumin—commonly observed during inflammatory states—can further modulate iCa²⁺ (Figure 1). These metabolites and proteins bind or chelate ionized calcium, lowering free iCa²⁺ in the bloodstream. Such binding helps reduce the pool of biologically active calcium (and thus calcium-dependent cellular processes), offering another route by which systemic inflammation might be restricted [71,72,73].
5.7. Lactate Binding to Ionized Calcium
Lactate is produced by both aerobic and anaerobic glycolysis and circulates as the lactate anion (C₃H₅O₃⁻) [71]. It can weakly bind iCa²⁺ through electrostatic interactions, forming reversible calcium-lactate complexes [72]. Elevated lactate levels in sepsis or lactic acidosis thus transiently lower free iCa²⁺, potentially affecting nerve function, muscle contraction, blood clotting, and immune-cell activities. The formation of calcium-lactate complexes may help explain the clinical signs of hypocalcemia that appear during severe inflammation or high lactate production [73].
5.8. Non-Esterified Fatty Acids Binding to Ionized Calcium
Non-esterified fatty acids circulate bound to albumin, and their free carboxyl group (-COOH) can also chelate iCa²⁺ (Figure 1) [74]. The binding affinity can vary with fatty-acid chain length and degree of saturation [74,75]. During metabolic stress—such as the periparturient period in dairy cows—elevated NEFA levels can further reduce free iCa²⁺, potentially contributing to hypocalcemia’s clinical manifestations [76]. This decline in iCa²⁺ may help limit some inflammatory processes, although chronic or severe hypocalcemia remains a health risk.
5.9. Albumin Binding to Ionized Calcium
Albumin is the most abundant plasma protein and a key buffer for calcium, as roughly 40–50% of total calcium in the blood is protein-bound (Figure 2) [77]. Negatively charged sites on albumin (carboxyl groups, histidine residues) bind Ca²⁺, forming a calcium-albumin complex [78]. Although traditionally considered a negative acute-phase protein, albumin can sometimes increase in certain inflammatory contexts, enhancing its calcium-binding capacity and thereby reducing free iCa²⁺ [79]. This reduction in biologically active calcium may assist in downregulating processes that depend on Ca²⁺ for inflammatory signaling. Conversely, conditions such as hypoalbuminemia increase the fraction of free iCa²⁺ [80], with potential ramifications for immune-cell activity. Consequently, accurate assessment of calcium status necessitates measuring iCa²⁺ directly or applying albumin correction to total calcium measurements [81].
Overall, emerging data and the Ametaj et al. [10,11] hypothesis propose that hypocalcemia—commonly observed during sepsis, endotoxemia, or the periparturient period—can act as a physiological modulator of inflammation. While adequate Ca²⁺ levels can promote lamellar LPS aggregates that are inherently less inflammatory, excessive Ca²⁺ within immune cells can intensify the inflammatory cascade. By lowering iCa²⁺, hypocalcemia tempers calcium-dependent immune pathways and allows lipoprotein-mediated clearance of LPS to proceed without triggering widespread cytokine surges (Figure 1). Meanwhile, factors like lactate, NEFAs, and albumin further shape iCa²⁺ concentrations, linking metabolic and immune homeostasis. This balanced view of calcium’s dual roles—both in LPS structure and in intracellular signaling—helps clarify why a transient decrease in iCa²⁺ may be protective, even though severe or prolonged hypocalcemia poses its own risks.
6. Calcium Buffering and Sequestration: Distinct Mechanisms and Key Locations in Calcium Homeostasis During Inflammation
Understanding the distinction between calcium buffering and calcium sequestration is essential for grasping calcium homeostasis, particularly in complex physiological states such as systemic inflammation and milk fever in dairy cows. Although these processes are interrelated, they fulfill different roles in regulating the body’s calcium levels. Calcium buffering involves the short-term control of free iCa²⁺ concentrations to prevent rapid fluctuations that could disrupt cellular functions [82]. In contrast, calcium sequestration refers to the storage of calcium ions in specific cellular compartments or structural reservoirs for long-term management and controlled release [83,84]. This section examines both processes and highlights the main anatomical locations involved, emphasizing their importance in maintaining overall calcium balance—especially under inflammatory conditions.
6.1. Calcium Buffering
Calcium buffering relates to the immediate regulation of free calcium ions within biological fluids and cells. By binding free calcium to proteins and other molecules, buffering stabilizes cytosolic and extracellular iCa²⁺ levels, protecting crucial physiological processes such as muscle contraction, nerve impulse transmission, and blood clotting. Key binding proteins in this system include albumin in the bloodstream and calmodulin within cells; they help keep free calcium concentrations within a narrow optimal range [85].
6.1.1. Primary Sites of Calcium Buffering
6.1.1.2. Blood Plasma
In the bloodstream, around 40% of total calcium is bound to plasma proteins, mostly albumin and globulins (Figure 2). This protein-bound fraction acts as a buffer, ensuring stable free calcium levels needed for physiological functions [77]. By binding calcium ions, these proteins prevent significant swings in iCa²⁺, the bioactive form of calcium essential for cardiac function and neural transmission [78].
6.1.1.3. Extracellular Matrix (ECM)
The extracellular matrix (ECM), which comprises proteins like collagen, elastin, and specialized glycoproteins, also participates in buffering extracellular calcium (Figure 2). This buffering is especially important in bone, where the ECM promotes mineralization and contributes to systemic calcium homeostasis [31]. Hydroxyapatite crystals—formed from calcium and phosphate—reside within the collagenous bone matrix, offering both structural integrity and a ready source of calcium. In other tissues, specific ECM molecules (e.g., osteopontin, vitronectin, fibronectin) can bind calcium to stabilize extracellular levels and ensure proper cellular signaling [86].
6.1.1.4. Intracellular Compartments
Within cells, calcium buffering involves calmodulin in the cytosol and storage in organelles such as the endoplasmic reticulum (ER) and sarcoplasmic reticulum (SR) (Figure 3) [87]. Cytosolic calmodulin binds free calcium, regulating key signaling pathways for muscle contraction, secretion, and metabolism. Meanwhile, the ER and SR temporarily retain calcium, releasing it when signals (e.g., action potentials) demand. This controlled release underlies vital processes such as muscle contraction and neurotransmitter exocytosis [88]. By preventing large shifts in intracellular calcium, this buffering system maintains cellular homeostasis.
6.2. Calcium Sequestration
Calcium sequestration involves storing calcium ions in specialized compartments or structural sites for long-term management [89]. This ensures an accessible supply of calcium during times of high demand, such as growth, pregnancy, lactation, or tissue repair, while preventing unnecessary surges in serum calcium. Major sites of sequestration include bones, the liver, the kidneys, and muscles (Figure 2). By allowing the controlled release of stored calcium, sequestration mechanisms help maintain systemic balance, even during inflammation.
6.2.1. Primary Sites of Calcium Sequestration
6.2.1.2. Bones
Bones serve as the principal reservoir of calcium in the body, housing roughly 99% of total calcium in the form of hydroxyapatite within the mineralized matrix (Figure 2) [90]. In response to low serum calcium, bone tissue releases calcium to preserve levels necessary for muscle contraction, nerve signaling, and blood coagulation [31]. Parathyroid hormone and calcitonin tightly regulate this process: PTH stimulates osteoclast activity to free calcium under hypocalcemia, whereas calcitonin inhibits osteoclasts to favor deposition during hypercalcemia [91]. This remodeling cycle sustains skeletal integrity while dynamically buffering systemic calcium levels [92]. In dairy cows, persistent inflammatory or metabolic stresses, like those in periparturient period, can destabilize this remodeling, increasing the likelihood of disorders such as osteoporosis or hypocalcemia [93].
6.2.1.3. Skeletal Muscles (Sarcoplasmic Reticulum)
In skeletal muscle cells, the SR functions as a critical calcium reservoir (Figure 2). It stores large amounts of calcium ions and releases them into the cytosol when muscles receive electrical signals [94]. The liberated calcium binds troponin, initiating the interaction of actin and myosin for muscle contraction. Subsequently, a SERCA pump drives calcium back into the SR, reducing cytosolic calcium and inducing relaxation [95]. Proper cycling of calcium release and uptake maintains effective muscle function, and preventing fatigue [96,97].
6.2.1.4. Liver (Hepatocytes)
In the liver, hepatocytes store calcium within the ER and mitochondria (Figure 2). This storage supports key intracellular signaling pathways necessary for metabolic processes such as protein synthesis and lipid metabolism [98,99]. Mitochondrial calcium buffering supports ATP generation, matching energy supply to cellular demands. Moreover, calcium-dependent signaling drives hepatic detoxification enzyme activity [100]. Consequently, disturbed calcium handling in hepatocytes, whether via inflammation or other stressors, can disrupt metabolic balance and compromise overall health [101]. By functioning as a major intracellular reservoir, the liver complements bone and kidney roles in systemic calcium regulation [102].
6.2.1.5. Kidneys (Renal Tubular Cells)
The kidneys maintain calcium equilibrium by controlling its reabsorption or excretion in the renal tubules (Figure 2). Renal tubular cells sequester calcium to conserve it under low-calcium conditions or excrete it to prevent hypercalcemia. Hormones, especially PTH and active vitamin D, modulate these processes, altering calcium reabsorption rates based on systemic needs [103]. Elevated PTH levels, for instance, enhance calcium reabsorption in the distal tubules, minimizing urinary loss during hypocalcemia, whereas vitamin D amplifies tubular cell expression of calcium transporters [103]. When renal calcium handling weakens, imbalances such as chronic hypocalcemia or hypercalcemia can emerge [42].
6.2.1.5. Brain (Neurons and Glial Cells)
Within the nervous system, neurons and glial cells coordinate calcium sequestration to sustain cellular homeostasis and support synaptic activity (Figure 2). Calcium is primarily stored in the ER and mitochondria, enabling precise regulation of numerous neural processes [104]. In neurons, calcium entering synaptic terminals upon depolarization prompts neurotransmitter release, a crucial step in synaptic plasticity and signal transmission [105]. Additionally, intracellular calcium acts as a second messenger in pathways modulating gene expression, metabolism, and longer-term synaptic adaptations. In glial cells (e.g., astrocytes, microglia), calcium signaling influences neuronal excitability, synaptic strength, and neuroprotection [106]. By buffering excess calcium, glial cells avert excitotoxicity and preserve ionic stability, safeguarding neurons from overstimulation. This intricate neuron–glia interplay is essential for normal brain function, learning, memory, and a healthy neural environment.
6.2.1.6. Mitochondria
Mitochondria fulfill a dual role in calcium sequestration by combining their well-known function as cellular powerhouses with calcium buffering (Figure 2). Mitochondrial calcium uptake involves the mitochondrial calcium uniporter (MCU), which mediates cytosolic calcium entry into the organelle [107]. Once inside, calcium ions stimulate key citric acid cycle enzymes—such as isocitrate dehydrogenase and α-ketoglutarate dehydrogenase—boosting ATP production to meet the cell’s energy requirements [108]. At the same time, mitochondria sequester excess calcium to prevent cytosolic overload and potential cytotoxic effects [109]. During inflammatory episodes (e.g., mastitis or systemic inflammatory response in dairy cows), cytokines like IL-1 and TNF-α can disrupt normal calcium homeostasis, raising cytosolic calcium and heightening the burden on mitochondria [110]. Excessive mitochondrial calcium uptake under prolonged inflammation may impair ATP synthesis and induce oxidative stress, ultimately exacerbating cellular damage. By balancing energy output and calcium buffering, mitochondria play a pivotal role in shielding cells from inflammatory harm and preserving overall homeostasis.
7. Impact of Systemic Inflammation on Calcium Buffering and Sequestration
Systemic inflammation during the periparturient period in dairy cows significantly affects calcium homeostasis, a critical concern given the high metabolic demands of lactation. Proinflammatory mediators have been shown to impair calcium buffering and sequestration in major organs such as the liver, kidneys, and bone. These mediators disrupt key intracellular structures like the ER and mitochondria [111,112], thereby limiting the cow’s ability to maintain adequate intracellular Ca²⁺ levels. This impairment increases the risk of hypocalcemia and related clinical issues, including muscle weakness, reduced appetite, and decreased milk production [8,42].
In the liver, cytokines such as IL-1 and TNF-α compromise calcium balance by reducing the ER’s buffering capacity, which is one of the principal calcium reservoirs, and by lowering mitochondrial calcium uptake [113]. These alterations lead to a cytosolic calcium overload, potentially resulting in cellular dysfunction. Systemically, the hepatic disturbances exacerbate hypocalcemia by impairing the liver’s normal role in stabilizing serum calcium levels, thereby aggravating metabolic imbalances [114].
Inflammation also adversely affects renal calcium handling. In the kidneys, particularly within renal tubular cells, inflammation alters the expression and function of calcium transport proteins, such as transient receptor potential (TRP) channels and various calcium ATPases [115]. These changes impair renal reabsorption of calcium, causing increased urinary losses. The resulting reduction in serum calcium is especially problematic during early lactation when calcium demand is elevated, deepening hypocalcemia and increasing the risk of periparturient disorders [116].
Elevated inflammatory cytokines also impact bone metabolism. Inflammatory mediators stimulate osteoclast activity, leading to accelerated bone resorption and the release of stored calcium [117]. Although this process is an attempt to maintain serum calcium levels, the excessive periparturient demands can overwhelm the bone’s ability to release sufficient calcium. Persistent inflammation ultimately disrupts the balance of bone remodeling, weakening the skeletal structure while failing to meet systemic calcium requirements [118].
Skeletal muscles, particularly through the action of the SR, also suffer under inflammatory stress. In skeletal muscle cells, inflammation can impair calcium handling by the SR, as cytokine-driven increases in calcium demand for immune responses reduce the available pool for muscle contraction [119]. This mechanism contributes to muscle weakness and recumbency, classical signs of severe hypocalcemia. Additionally, oxidative stress induced by inflammation may degrade the SERCA pump, diminishing its ability to re-sequester calcium into the SR [120].
Platelet function is not exempt from the effects of systemic inflammation. Inflammatory mediators can modulate calcium dynamics within platelets; high intracellular calcium levels can intensify platelet activation and clot formation, which serves as a protective mechanism against hemorrhage but also increases the risk of thrombotic complications under chronic inflammatory conditions [121]. Conversely, hypocalcemia can hamper platelet function and delay clot formation, thereby complicating hemostasis and healing outcomes [122].
Red blood cells (RBCs) and immune cells are similarly affected by systemic inflammation. In RBCs, excess intracellular calcium reduces cell deformability, thereby shortening their lifespan and undermining effective oxygen delivery to tissues [123]. In immune cells, disruptions in calcium signaling can amplify inflammatory responses by boosting cytokine release, which further perpetuates inflammation and contributes to conditions such as mastitis and systemic hypocalcemia [124].
The brain is also impacted by these inflammatory processes. Neurons and glial cells experience disrupted calcium buffering under systemic inflammation, contributing to excitotoxicity and neuroinflammatory processes [125]. Such alterations can compromise central regulatory mechanisms for calcium homeostasis, thereby compounding hypocalcemia and impairing the overall physiological response [126].
Vascular endothelial cells are particularly sensitive to inflammatory signals, which provoke increased calcium influx and impair their ability to sequester calcium effectively. This imbalance can alter vascular tone and permeability [127], leading to excessive vasodilation, promotion of leukocyte adhesion, and ultimately vascular instability. Concurrently, low circulating Ca²⁺ levels further worsen perfusion deficits and compromise end-organ function [128].
Inflammation also triggers remodeling of the extracellular matrix (ECM), which can release bound calcium into the circulation [129]. Although this release may briefly boost serum calcium levels, the resulting fluctuations can degrade tissue integrity, promote local injury, and contribute to maladaptive remodeling, such as fibrosis [130].
In vascular smooth muscle cells, inflammation-induced dysregulation of calcium homeostasis can impair blood pressure control [131]. Cytokine-driven modifications of calcium handling in these cells can lead to either hypertension or hypotension, both of which jeopardize tissue perfusion and exacerbate inflammatory complications [132].
The mechanisms underlying hypocalcemia during systemic inflammation are multifactorial. Cytokine-driven changes in calcium handling and tissue-level responses promote intracellular sequestration and binding, reducing free serum calcium. While these processes may serve to protect tissues from calcium overload, the net reduction in available Ca²⁺ diminishes normal cellular functions and further intensifies inflammation and clinical pathology [42,133]. Additionally, inflammatory states drive the formation of insoluble calcium complexes with plasma anions such as lactate, chloride, and phosphate, which further reduces the availability of free Ca²⁺ [134]. Although this reduction can help curb excessive inflammatory signaling, it also compromises critical processes such as muscle contraction and immune function, thereby complicating disease management [135].
Finally, various cell types actively sequester intracellular Ca²⁺ during inflammation to prevent cytotoxicity. Endothelial cells, immune cells, and hepatocytes utilize pumps and transporters—including SERCA, PMCA, MCU, and NCX—to govern intracellular calcium flux. Proper functioning of these transport systems is essential for regulating immune responses and maintaining tissue integrity, thereby preventing a detrimental cycle of sustained inflammation and impaired calcium homeostasis [136].
8. Intracellular Calcium Buffers: Pathways and Components Involved in Calcium Homeostasis
Intracellular calcium is a pivotal second messenger that regulates numerous cellular processes, and its concentration is tightly controlled by an elaborate network of buffering, sequestration, and transport mechanisms (Figure 3). The SR in muscle cells serves as the primary reservoir for Ca²⁺, orchestrating its rapid sequestration and release during muscle contraction and relaxation. Key calcium-binding proteins such as calsequestrin, sarcalumenin, junctate, and triadin facilitate these processes. For instance, calsequestrin binds Ca²⁺ with high capacity, enabling the storage of large quantities of calcium that can be quickly mobilized when required [137,138]. Proteins like sarcalumenin and junctate further modulate calcium dynamics, while triadin is critical for anchoring these proteins to the ryanodine receptor complex, thereby optimizing excitation–contraction coupling [139].
In the cytoplasm, soluble calcium-binding proteins such as calmodulin, calbindin-D28K, calbindin-D9K, and calreticulin transiently bind free Ca²⁺, serving as dynamic buffers that modulate both the amplitude and duration of calcium signals. Calmodulin, in particular, functions as a versatile calcium sensor, interacting with a broad range of enzymes, ion channels, and other proteins to influence diverse processes from metabolism to gene transcription [140,141]. This buffering capacity is crucial for ensuring that cellular calcium levels remain within a narrow range optimal for proper cell function.
Membrane-associated proteins also play an essential role in maintaining intracellular calcium homeostasis (Figure 3). The sarco/endoplasmic reticulum Ca²⁺-ATPase (SERCA) pump is responsible for actively transporting Ca²⁺ from the cytoplasm back into the SR/ER, utilizing ATP to re-establish basal cytosolic calcium levels following cellular signaling events [142]. Additionally, transient receptor potential (TRP) channels facilitate Ca²⁺ entry in response to a variety of stimuli [143], while the sodium-calcium exchanger (NCX) helps extrude Ca²⁺ from cells by exchanging it for sodium ions, thereby contributing to the overall balance of intracellular calcium [144].
The plasma membrane Ca²⁺-ATPase (PMCA) is a critical membrane protein responsible for the active extrusion of Ca²⁺ from the cytosol, thereby maintaining low intracellular calcium levels. Utilizing ATP, PMCA rapidly decreases cytosolic Ca²⁺ following cellular activation, which is essential for terminating calcium signals. Multiple isoforms of PMCA, each with distinct regulatory properties, allow fine-tuning of calcium homeostasis in various tissues [145]. Disruptions in PMCA function have been linked to several pathophysiological conditions, highlighting its vital role in cellular health.
Beyond these classical compartments, lysosomes have emerged as significant secondary Ca²⁺ stores. Calcium exchange between lysosomes and the ER occurs through two-pore calcium channels (TPC1 and TPC2), which enable localized calcium signaling events crucial for processes such as autophagy and vesicular trafficking [146]. Furthermore, the ER-mitochondria junctions serve as specialized sites for efficient calcium transfer. At these junctions, inositol trisphosphate receptors (IP3Rs) on the ER interface with voltage-dependent anion channels (VDACs) on mitochondria, facilitating the transfer of Ca²⁺ into mitochondria via the rapid uptake mode (RaM). This process is essential not only for buffering cytosolic Ca²⁺ but also for supporting key metabolic functions, including ATP production and the regulation of apoptotic pathways [147].
Another critical aspect of calcium homeostasis involves the ER-plasma membrane junction, where store-operated calcium entry (SOCE) replenishes ER calcium stores. SOCE is mediated by the interaction between ORAI channels and stromal interaction molecule 1 (STIM1). When ER calcium levels fall, STIM1 undergoes a conformational change, aggregates near the plasma membrane, and activates ORAI channels to permit Ca²⁺ influx, thereby restoring calcium balance within the ER [148]. The Golgi apparatus further contributes to intracellular calcium buffering by serving as a calcium reservoir that is vital for vesicular trafficking and protein modification. Calcium within the Golgi plays an important role in the sorting and maturation of secretory proteins, thereby aiding in the spatial and temporal regulation of calcium signaling within the cell [149].
Together, these interconnected systems highlight the complexity of intracellular Ca²⁺ regulation. The coordinated activity of the SR/ER, cytoplasmic buffers, membrane transport proteins, lysosomal channels, and inter-organelle communication ensures that cells can respond precisely to physiological stimuli while maintaining calcium homeostasis. A deep understanding of these pathways not only enhances our grasp of fundamental cellular physiology but also holds significant implications for therapeutic strategies in conditions where calcium regulation is disrupted.
9. Mechanistic Insights into the Calci-Inflammatory Axis
9.1. Adaptive Hypocalcemia as a Protective Mechanism
Emerging evidence supports the existence of the Calci-Inflammatory Axis, a dynamic regulatory network in which adaptive hypocalcemia modulates immune responses through finely tuned calcium signaling to mitigate excessive inflammation during critical physiological challenges. Systemic hypocalcemia, traditionally regarded as a pathological condition requiring immediate correction, is now emerging as an adaptive response that modulates inflammation during critical physiological challenges such as endotoxemia. Emerging evidence suggests that during systemic inflammatory states, the reduction in blood calcium levels may serve a protective role by alleviating the inflammatory cascade [74]. In this adaptive scenario, lower extracellular calcium concentrations influence various intracellular signaling mechanisms, thereby modulating the intensity and duration of the inflammatory response. This adaptive hypocalcemia might help prevent the harmful effects of an overactivated immune response, which can lead to tissue damage and organ failure.
9.2. Calcium Signaling Pathways in Inflammation
Calcium ions serve as ubiquitous second messengers that regulate numerous cellular processes. The fine-tuning of intracellular calcium levels is achieved through a complex interplay of channels, pumps, and exchangers located in the plasma membrane and intracellular organelles. The influx of Ca²⁺ into cells initiates signaling cascades that control the activity of enzymes, transcription factors, and other proteins crucial for immune function [150]. These well-coordinated pathways ensure that calcium signals are both spatially and temporally precise, enabling cells to mount an effective yet controlled response during inflammatory challenges.
9.3. The Calcium-Sensing Receptor (CaSR): Function and Mechanisms
Central to calcium homeostasis is the CaSR, a specialized G-protein-coupled receptor that is very sensitive to changes in extracellular calcium concentrations. Expressed in a variety of tissues including the parathyroid glands, kidneys, and immune cells such as macrophages and neutrophils [151,152], CaSR acts as a molecular rheostat. When extracellular Ca²⁺ levels rise, CaSR undergoes a conformational change that activates intracellular signaling cascades, primarily via the phospholipase C (PLC) pathway. This activation leads to the production of secondary messengers such as inositol triphosphate (IP3) and diacylglycerol (DAG), which then stimulate protein kinase C (PKC) and promote further calcium mobilization within the cell [153,154,155]. In the parathyroid glands, this cascade inhibits the secretion of PTH when calcium is abundant, reducing bone resorption and renal calcium reabsorption. Conversely, low extracellular calcium levels relieve this inhibition, allowing PTH secretion to restore calcium balance [156,157].
9.4. CaSR in Immune Modulation
Beyond its classical role in maintaining systemic calcium homeostasis, CaSR has emerged as a key modulator of immune cell function. In immune cells, CaSR detects local fluctuations in extracellular calcium that occur during immune activation and inflammation [158]. When activated, CaSR amplifies pro-inflammatory signaling by engaging not only the PLC-IP3-PKC pathway but also by activating mitogen-activated protein kinases (MAPKs) and NF-κB. This coordinated response leads to increased production of cytokines such as IL-6, TNF-α, and IL-1, which are vital for orchestrating an effective immune response [41,42]. In hypocalcemic conditions, the reduced activation of CaSR in immune cells results in decreased production of these pro-inflammatory mediators, effectively decreasing the inflammatory response. This mechanism suggests that adaptive hypocalcemia may help prevent immune overactivation, thereby lessening tissue damage and reducing the risk of organ failure during severe inflammatory states.
In summary, the interaction between hypocalcemia, calcium signaling pathways, and CaSR function forms a complex regulatory network that modulates inflammation at both molecular and cellular levels. While calcium acts as a key second messenger in the activation of immune responses, the adaptive downregulation of calcium signals through hypocalcemia and diminished CaSR activation appears to be a strategic response to prevent excessive inflammation. This dual role of calcium—as both an activator of immune responses and a modulator of their intensity—highlights the intricate balance required to maintain optimal cellular function and systemic health during inflammatory stress.
10. Evidence Linking Hypocalcemia to Inflammatory Responses in Dairy Cows
10.1. Recent Research in Dairy Cows Supporting the Hypothesis
Recent studies, provide substantial evidence supporting the hypothesis that hypocalcemia serves as an adaptive response to modulate inflammation during endotoxemia. This evidence emphasizes the protective role of hypocalcemia in controlling systemic inflammation and offers new insights into managing periparturient diseases in dairy cows. For instance, Chandler et al. [159] described significant changes in calcium dynamics following an acute intravenous (iv) LPS challenge in early postpartum cows. Their research showed that serum total calcium (tCa) and iCa concentrations decreased following the LPS challenge, supporting the idea that hypocalcemia is a response to inflammation rather than merely a deficiency state. The decrease in calcium levels was not attributable to urinary excretion, suggesting that other mechanisms, such as calcium sequestration in tissues, contribute to hypocalcemia during inflammatory challenges [159].
10.2. Exacerbation of Inflammation by High Calcium Levels
Additional research has demonstrated that maintaining high calcium levels during immune challenges can exacerbate inflammation, confirming that hypocalcemia helps alleviate the inflammatory response. For example, Horst et al. [147] investigated the effects of maintaining eucalcemia in dairy cows subjected to LPS-induced endotoxemia. They found that cows with elevated calcium levels exhibited significantly increased levels of inflammatory markers, such as LBP and SAA, compared to cows with hypocalcemia. This study highlights the aggravation of systemic inflammation due to high calcium levels, supporting the hypothesis that hypocalcemia serves as a protective mechanism [147].
10.3. Experimental Designs Inducing Endotoxemia
Various experimental designs, including the administration of LPS to induce endotoxemia, have consistently shown significant decreases in serum calcium concentrations following immune activation. This decline in calcium levels appears to be dose-dependent and temporally correlated with the inflammatory response. In a study conducted by Waldron et al. [160] different doses of LPS were administered to lactating cows, leading to a dose-dependent decrease in serum calcium levels. The findings indicate that immune activation via LPS infusion disrupts calcium homeostasis, thereby underscoring the role of hypocalcemia in controlling inflammation [160].
10.4. Hypocalcemia as a Response to Endotoxin-Induced Systemic Inflammation
In the experiment conducted by Horst et al. [147] they aimed to evaluate the effects of maintaining eucalcemia on metabolic, immune, and production variables during a LPS challenge in lactating dairy cows. The study involved two groups: cows receiving LPS alone (LPS-Con) and cows receiving LPS along with intravenous calcium to maintain eucalcemia (LPS-Ca). Cows in the LPS-Ca group demonstrated heightened inflammatory responses, with significantly increased levels of acute-phase proteins (LBP and SAA) compared to the LPS-Con group. Maintaining eucalcemia also led to reduced neutrophil counts and an exacerbated febrile response, but neutrophil function remained unaffected.
Interestingly, milk yield decreased more in the LPS-Ca group, and their recovery to baseline dry matter intake (DMI) was delayed, suggesting negative effects of calcium infusion on production performance. The study further highlighted that hypocalcemia, commonly observed during systemic inflammation, may play a role in regulating immune responses by influencing calcium homeostasis and inflammatory signaling pathways. These findings support the hypothesis that hypocalcemia helps relieve inflammation triggered by intravenous endotoxin infusion, serving as a protective mechanism to prevent excessive immune activation.
10.5. Pre-Existing Systemic Inflammation in Cows Prone to Milk Fever
Research focusing on cows susceptible to milk fever has revealed significant alterations in acute phase proteins and pro-inflammatory cytokines before the onset of the disease and during the disease occurrence. Ametaj et al. [33] and Zhang et al. [25] reported that cows prone to developing milk fever exhibited elevated concentrations of pro-inflammatory cytokines, such as TNF-α and acute phase proteins such as SAA, weeks before parturition as well as at parturition week. These findings suggest a pre-existing state of systemic inflammation in cows prone to milk fever, which persists postpartum. Elevated levels of these inflammatory markers indicate that systemic inflammation is a critical factor in the pathogenesis of milk fever [25,33].
In another study by Seminara et al. [161], they provide further evidence that an acute phase response precedes the development of hypocalcemia or calcium dysregulation (i.e., low tCa serum concentrations) in postpartum dairy cows. Their research found that cows that developed dyscalcemia exhibited significant increases in acute phase proteins such as SAA, haptoglobin, and LBP within the first four days postpartum, whereas no such increase was observed in eucalcemic cows. This finding suggests that inflammatory activation is a key early event in cows that later experience calcium dysregulation. Specifically, elevated concentrations of SAA and haptoglobin were detected in dyscalcemic cows, with a notable temporal association indicating that inflammation likely plays a critical role in the onset of dyscalcemia. The data support the hypothesis that postpartum inflammation and calcium dysregulation are interrelated processes, with inflammation occurring first, potentially contributing to the development of calcium dysregulation. This adds to the growing body of evidence linking systemic inflammation to metabolic disturbances like hypocalcemia during the early postpartum period. The study calls for further research to elucidate the underlying mechanisms that connect these processes, which could ultimately improve therapeutic strategies to prevent postpartum calcium-related disorders in dairy cows.
10.6. Calcium Oral Supplementation and Its Role in Postpartum Inflammation in Dairy Cows
Couto Serrenho et al. [162] investigated the effects of postpartum calcium (Ca) supplementation on systemic inflammation in clinically healthy multiparous dairy cows. The study involved 101 cows assigned to either receive two oral Ca boluses (TRT) or no supplementation (CON) within 24 hours of calving. Blood Ca levels and markers of inflammation, including Hp, SAA, and LBP, were measured over eight days postpartum.
The TRT group exhibited transiently higher total Ca levels immediately after supplementation but lower Ca levels by day 2 compared to controls. Concentrations of SAA were significantly higher in the TRT group on day 2, reflecting increased systemic inflammation. Similarly, younger multiparous cows (parity 2) in the TRT group showed higher LBP levels, suggesting heightened inflammatory responses compared to older cows.
Haptoglobin and albumin levels, however, were unaffected by treatment, indicating a selective inflammatory response. These results suggest that supplemental Ca may modestly increase markers of inflammation postpartum, particularly in cows less predisposed to hypocalcemia. The study also highlights the potential for Ca supplementation to disrupt natural postpartum calcium homeostasis.
11. How Hypocalcemia Eases Inflammation: A Mechanistic Overview
Hypocalcemia in dairy cows, often considered detrimental, may instead function as a protective, adaptive mechanism to mitigate systemic inflammation. During inflammatory states or endotoxemia, a decrease in free iCa²⁺ blunts calcium-dependent signaling in immune cells [34]. This attenuation limits the overactivation of macrophages and neutrophils, thereby reducing the release of pro-inflammatory cytokines (e.g., TNF-α, IL-6, and IL-1). Although earlier reports suggested that lower iCa²⁺ “disaggregates” LPS, a stronger consensus from in vitro studies indicates that higher iCa²⁺ stabilizes LPS into multilamellar structures that diminish its toxicity [163,164]. Thus, the primary benefit of moderate or transient hypocalcemia in vivo may not be direct LPS disaggregation but rather the weakening of excessive immune-cell activity via reduced CaSR stimulation, lower intracellular calcium flux, and diminished cytokine release [34]. In this way, hypocalcemia can serve as a short-term defense against uncontrolled inflammatory response.
Beyond controlling immune-cell activation, hypocalcemia may also indirectly influence LPS clearance pathways. Under normocalcemic or mildly hypocalcemic conditions, both macrophage uptake and lipoprotein binding (particularly via HDL) contribute to the removal of LPS from circulation (Figure 1) [165,166]. Macrophages, including Kupffer cells in the liver, efficiently endocytose LPS-containing particles, while HDL shuttles endotoxins to the liver for detoxification [167]. Conversely, if iCa²⁺ remains excessively high during intense inflammation, certain calcium-dependent inflammatory pathways may be overstimulated, leading to increased cytokine production and tissue damage [168]. Consequently, a controlled decrease in iCa²⁺ can act as a “brake” on these pathways until LPS is cleared by hepatic macrophages or sequestered by lipoproteins, thereby preventing severe endotoxemic episodes.
In dairy cows, the anti-inflammatory potential of hypocalcemia becomes especially relevant during the early postpartum period, when both metabolic demands and exposure to bacterial endotoxins are high. By transiently reducing iCa²⁺, the cow’s physiology may alleviate the metabolic burden of severe inflammatory reactions [147]. However, chronic or severe hypocalcemia remains deleterious to overall health, underscoring the importance of balancing sufficient calcium levels for normal physiological functions with the protective, anti-inflammatory effects of transient hypocalcemia. Future work should clarify optimal post-partum calcium supplementation strategies so that cows receive enough calcium for health and production without losing the beneficial immunomodulatory “window” provided by brief, moderate hypocalcemia.
12. The Role of Calcium in LPS Aggregation and Clearance Without Inducing Inflammation
Lipopolysaccharide, a major component of Gram-negative bacterial outer membranes, can trigger potent inflammation via the LBP–CD14–TLR4 axis. Its bioactivity is strongly dictated by its structural conformation [163,165]. Under many in vitro and in vivo conditions, divalent cations—particularly Ca²⁺ and Ba²⁺—promote the transition of LPS from “fluid” or “micellar” aggregate phases into multilamellar aggregates (Redeker et al., 2019). These lamellar forms exhibit reduced fluidity, diminished acyl-chain mobility, and tighter lipid packing, making them less accessible to LBP or CD14 [167]. As a result, multilamellar LPS is less capable of activating TLR4, which translates into reduced production of pro-inflammatory cytokines [168]. Such calcium-induced LPS aggregates are generally recognized as relatively inert particles or “debris” that Kupffer cells and other macrophages can phagocytose without prompting a substantial inflammatory burst [169,170].
The clearance of these aggregated LPS particles proceeds via multiple non-inflammatory routes. Scavenger receptors (e.g., SR-A1, SR-B1) on macrophages readily bind the structurally modified or aggregated endotoxins, promoting their endocytosis and subsequent lysosomal degradation [171,172]. Additionally, plasma lipoproteins and proteins such as HDL and lactoferrin bind lamellar LPS, further reducing its capacity to stimulate TLR4 and directing it toward hepatic clearance [166,173]. Ultimately, Kupffer cells degrade LPS internally and excrete the fragments into bile, from where they are eliminated through the gastrointestinal tract (Figure 1) [171]. In some instances, LPS–calcium complexes may be temporarily stored in adipose tissue, where tissue-resident macrophages gradually remove them [174,175]. Because the multilamellar conformation is less immunogenic, these clearance processes remain largely non-inflammatory [176].
Together, these findings help reconcile the dual roles of calcium. On one hand, elevated extracellular Ca²⁺ promotes the formation of lamellar LPS aggregates that neutralize its toxicity. On the other, transient hypocalcemia in vivo can be advantageous by restraining calcium-dependent pro-inflammatory signaling within immune cells [34,164]. Rather than being contradictory, these phenomena—lamellar neutralization of LPS in the presence of adequate Ca²⁺ and immune diminishing under moderate hypocalcemia—can coexist, each providing protective mechanisms at different phases of the inflammatory response or in distinct physiological compartments. This view emphasizes the delicate balance required: calcium must be available in sufficient amounts to stabilize and inactivate LPS structurally, yet excessive calcium-driven immune stimulation can be harmful if it promotes enhanced cytokine release. Further studies focusing on these dual roles of calcium in LPS conformation and immune-cell activation are needed to optimize preventive and therapeutic strategies for endotoxemia, sepsis, and periparturient diseases in dairy cattle [177].
13. Conclusions
This review redefines our understanding of hypocalcemia in milk fever by demonstrating that fluctuations in calcium levels are not merely pathological deficiency demanding immediate correction, but rather dynamic, adaptive responses that modulate systemic inflammation. Central to this concept is the Calci-Inflammatory Axis, which underscores how calcium oscillations—governed in part by the CaSR on immune cells as well as parathyroid and renal tissues—perform a dual regulatory function. On one side, elevated intracellular calcium aids in the neutralization of LPS and reinforces key immune mechanisms; on the other, transient hypocalcemia helps curb immune overactivity and excessive cytokine release, thereby mitigating tissue damage and preventing organ failure.
Multiple studies, including those by Chandler et al. [159], Horst et al. [147], Waldron et al. [160], Seminara et al. [161], Couto-Serrenho et al. [162], and Ametaj [33] and Zhang [25], offer compelling evidence in support of this interconnected model of calcium homeostasis and inflammatory control. Their work collectively illustrates how adaptive changes in calcium levels are essential for striking a balance between robust host defense and the avoidance of detrimental, hyperinflammatory states. Moreover, CaSR activation in parathyroid and renal tissues regulates the production of PTH and 1,25-dihydroxyvitamin D, further fine-tuning calcium balance and immune modulation.
The ramifications of this emerging framework go well beyond traditional milk fever management, providing novel insights into dairy cow health during the critical periparturient period. Future intervention strategies should strive to harmonize calcium replenishment with the advantageous effects of these controlled fluctuations, ultimately optimizing immune function and overall cow welfare. Ongoing research into the genetic and environmental factors influencing calcium dynamics will be instrumental in developing precision management protocols and targeted therapies, thereby advancing herd health, productivity, and sustainable dairy practices.
Author Contributions
Conceptualization, writing—original draft preparation, writing—review and editing by B.N.A.
Funding
Writing of this review article received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to express my gratitude to PhD graduate student Zohaib Saleem for his invaluable assistance in developing the figures for this review article. I deeply appreciate his diligence, skill, and persistence in mastering the BioRender software, which he used to create all the figures included in this article. His contributions have significantly enhanced the quality and clarity of the article.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Goff, J.P. The monitoring, prevention, and treatment of milk fever and subclinical hypocalcemia in dairy cows. Vet. J. 2008, 176, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, T.A.; Lippolis, J.D.; McCluskey, B.J.; Goff, J.P.; Horst, R.L. Prevalence of subclinical hypocalcemia in dairy herds. J. Dairy Sci. 2011, 94, 2928–2934. [Google Scholar] [CrossRef]
- Block, E. Manipulating dietary anions and cations for prepartum dairy cows to reduce incidence of milk fever. J. Dairy Sci. 1984, 67, 2939–2948. [Google Scholar] [CrossRef]
- Lean, I.J.; DeGaris, P.J.; McNeil, D.M.; Block, E. Hypocalcemia in dairy cows: Pathophysiology and preventive strategies. J. Dairy Sci. 2006, 89, 1094–1108. [Google Scholar]
- Block, E. Manipulation of dietary cation-anion difference on nutritionally related production diseases, productivity, and metabolic responses of dairy cows. J. Dairy Sci. 1994, 77, 1437–1450. [Google Scholar] [CrossRef]
- Venjakob, P.L.; Borchardt, S.; Heuwieser, W. Hypocalcemia—cow-level prevalence and preventive strategies in German dairy herds. J. Dairy Sci. 2017, 100, 9258–9266. [Google Scholar] [CrossRef]
- DeGaris, P.J.; Lean, I.J. Milk fever in dairy cows: A review of pathophysiology and control principles. Vet. J. 2008, 176, 58–69. [Google Scholar] [CrossRef]
- Couto Serrenho, R.; DeVries, T.J.; Duffield, T.F.; LeBlanc, S.J. Graduate student literature review: What do we know about the effects of clinical and subclinical hypocalcemia on health and performance of dairy cows? J. Dairy Sci. 2021, 104, 6304–6326. [Google Scholar] [CrossRef]
- Zebeli, Q.; Beitz, D.C.; Bradford, B.J.; Dunn, S.M.; Ametaj, B.N. Peripartal alterations of calcitonin gene-related peptide and minerals in dairy cows affected by milk fever. Vet. Clin. Pathol. 2013, 42, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Ametaj, B.N.; Zebeli, Q.; Iqbal, S. Nutrition, microbiota, and endotoxin-related diseases in dairy cows. Rev. Bras. Zootec. 2010, 39, 433–444. [Google Scholar] [CrossRef]
- Eckel, E.F.; Ametaj, B.N. Invited review: Role of bacterial endotoxins in the etiopathogenesis of periparturient diseases of transition dairy cows. J. Dairy Sci. 2016, 99, 5967–5990. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, J.W. Milk fever (parturient paresis) in dairy cows: A review. J. Dairy Sci. 1950, 33, 758–789. [Google Scholar]
- Hutyra, F.; Marek, J.; Manniger, R. Special pathology and therapeutics of the diseases of domestic animals, 4th ed.; Greig, J.R., Ed.; 1938.
- Dryerre, H.; Greig, J.R. Milk fever: Its possible association with derangements in the internal secretions. Vet. Rec. 1925, 5, 225–231. [Google Scholar]
- Auger, M.L. Réalisation expérimentale de la fièvre vitulaire. Compt. Rend. 1926, 182, 348–350. [Google Scholar]
- Little, W. L., and N. C. Wright. The aetiology of milk fever in cattle. Br. J. Exp. Pathol. 1925, 6, 129.
- Dryrre, H.; Greig, J.R. The specific chemotherapy of milk fever by the parenteral administration of Ca-boro-gluconate. Vet. Med. 1935, 30, 234–238. [Google Scholar]
- Ender, F.; Dishington, I.W.; Helgebostad, A. Calcium balance studies in dairy cows under experimental induction and prevention of hypocalcaemic paresis puerperalis. The solution of the aetiology and the prevention of milk fever by dietary means. Z. Tierphysiol. Tierernaehr. Futtermittelkd. 1971, 28, 233. [Google Scholar]
- Dishington, I.W. Prevention of milk fever (hypocalcaemia paresis puerperalis) by dietary salt supplements. Acta Vet. Scand. 1975, 16, 503–512. [Google Scholar]
- Horst, R.L.; Goff, J.P.; Reinhardt, T.A.; Buxton, D.R. Strategies for preventing milk fever in dairy cattle. J. Dairy Sci. 1997, 80, 1269–1280. [Google Scholar]
- Aiumlamai, S.; Kindahl, H.; Fredriksson, G.; Edqvist, L.E.; Kulander, L.; Eriksson, O. The role of endotoxins in induced ruminal acidosis in calves. Acta Vet. Scand. 1992, 33, 117–127. [Google Scholar] [CrossRef]
- Andersen, P.H. Bovine endotoxicosis: Some aspects of relevance to production diseases. A review. Acta Vet. Scand. Suppl. 2003, 98, 141–155. [Google Scholar] [PubMed]
- Ametaj, B.N. A new understanding of the causes of fatty liver in dairy cows. Adv. Dairy Technol. 2005, 17, 97–112. [Google Scholar]
- Ametaj, B.N. Strong relationships between mediators of the acute phase response and fatty liver in dairy cows. Can. J. Dairy Sci. 2005, 85, 165–175. [Google Scholar]
- Zhang, G.; Dervishi, E.; Ametaj, B.N. Milk fever in dairy cows is preceded by activation of innate immunity and alterations in carbohydrate metabolism prior to disease occurrence. Res. Vet. Sci. 2018, 117, 167–177. [Google Scholar] [CrossRef]
- Shandilya, U.; Sharma, A.; Mallikarjunappa, S.; Guo, J.; Mao, Y.; Meade, K.; Karrow, N. CRISPR-Cas9-mediated knockout of TLR4 modulates Mycobacterium avium ssp. paratuberculosis cell lysate-induced inflammation in bovine mammary epithelial cells. J. Dairy Sci. 2021. [Google Scholar] [CrossRef]
- Carroll, J.; Reuter, R.; Chase, C.; Coleman, S.; Riley, D.; Spiers, D.; Arthington, J.; Galyean, M. Profile of the bovine acute-phase response following an intravenous bolus-dose lipopolysaccharide challenge. Innate Immun. 2009, 15, 81–89. [Google Scholar] [CrossRef]
- Jermann, P.; Wagner, L.; Fritsche, D.; Gross, J.; Wellnitz, O.; Bruckmaier, R. Acute phase reaction to LPS-induced mastitis in early lactation dairy cows fed nitrogenic, glucogenic or lipogenic diets. J. Dairy Sci. 2023. [CrossRef]
- Augustine, M.; Leonard, M.; Thayu, M.; Baldassano, R.; De Boer, I.; Shults, J.; Denson, L.; DeBoer, M.; Herskovitz, R.; Denburg, M. Changes in vitamin D-related mineral metabolism after induction with anti-tumor necrosis factor-α therapy in Crohn’s disease. J. Clin. Endocrinol. Metab. 2014, 99, E991–E998. [Google Scholar] [CrossRef]
- Meurer, M.; Höcherl, K. Endotoxaemia differentially regulates the expression of renal Ca2+ transport proteins in mice. Acta Physiol. 2018, 225. [Google Scholar] [CrossRef]
- Klein, G. The role of calcium in inflammation-associated bone resorption. Biomolecules 2018, 8. [Google Scholar] [CrossRef]
- Dervishi, E.; Ametaj, B.N. Milk fever: Reductionist versus systems veterinary approach. In: Ametaj, B.N., ed. Periparturient Diseases of Dairy Cows; Springer: Berlin, Germany, 2017; pp. 247–266.
- Ametaj, B.N.; Goff, J.P.; Horst, R.L.; Bradford, B.J.; Beitz, D.C. Presence of acute phase response in normal and milk fever dairy cows around parturition. Acta Vet. Scand. Suppl. 2003, 98, 241. [Google Scholar]
- Thaveeratitham, P.; Khovidhunkit, W.; Patumraj, S. High-density lipoproteins (HDL) inhibit endotoxin-induced leukocyte adhesion on endothelial cells in rats: Effect of the acute-phase HDL. Clin. Hemorheol. Microcirc. 2007, 36, 1–12. [Google Scholar]
- Collage, R.D.; Howell, G.M.; Zhang, X.; Stripay, J.L.; Lee, J.S.; Angus, D.C.; Rosengart, M.R. Calcium supplementation during sepsis exacerbates organ failure and mortality via calcium/calmodulin-dependent protein kinase kinase signaling. Crit. Care Med. 2013, 41, e352–e360. [Google Scholar] [CrossRef]
- Emmanuel, D.G.; Madsen, K.L.; Churchill, T.A.; Dunn, S.M.; Ametaj, B.N. Acidosis and lipopolysaccharide from Escherichia coli B:055 cause hyperpermeability of rumen and colon tissues. J. Dairy Sci. 2007, 90, 5552–5557. [Google Scholar] [CrossRef]
- Su, G.; Klein, R.; Aminlari, A.; Zhang, H.; Steinstraesser, L.; Alarcon, W.; Remick, D.; Wang, S. Kupffer cell activation by lipopolysaccharide in rats: Role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 2000, 31, —. [Google Scholar] [CrossRef]
- Hu, X.; Li, S.; Mu, R.; Guo, J.; Zhao, C.; Cao, Y.; Zhang, N.; Fu, Y. The rumen microbiota contributes to the development of mastitis in dairy cows. Microbiol. Spectr. 2022, 10(1): e0251221. [CrossRef]
- Papaioannou, N.; Voutsas, I.; Samara, P.; Tsitsilonis, O. A flow cytometric approach for studying alterations in the cytoplasmic concentration of calcium ions in immune cells following stimulation with thymic peptides. Cell. Immunol. 2016, 302, 32–40. [Google Scholar] [CrossRef]
- Yang, R.; Mark, M.; Gray, A.; Huang, A.; Xie, M.; Zhang, M.; Goddard, A.; Wood, W.; Gurney, A.; Godowski, P. Toll-like receptor-2 mediates lipopolysaccharide-induced cellular signalling. Nature 1998, 395, 284–288. [Google Scholar] [CrossRef]
- Iamartino, L.; Brandi, M. The calcium-sensing receptor in inflammation: Recent updates. Front. Physiol. 2022, 13. [Google Scholar] [CrossRef]
- Hendy, G.; Canaff, L. Calcium-sensing receptor, proinflammatory cytokines and calcium homeostasis. Semin. Cell Dev. Biol. 2016, 49, 37–43. [Google Scholar] [CrossRef]
- Brown, E.M. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Nat. Rev. Endocrinol. 2013, 9, 391–403. [Google Scholar] [CrossRef]
- Kehrli, M.E. Jr.; Goff, J.P. Periparturient hypocalcemia in cows: Effects on peripheral blood neutrophil and lymphocyte function. J. Dairy Sci. 1989, 72, 1188–1196. [Google Scholar] [CrossRef]
- Kimura, K.; Reinhardt, T.A.; Goff, J.P. Parturition and hypocalcemia blunts calcium signals in immune cells of dairy cattle. J. Dairy Sci. 2006, 89, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Canaff, L.; Hendy, G. Human calcium-sensing receptor gene. J. Biol. Chem. 2002, 277, 30337–30350. [Google Scholar] [CrossRef]
- Venjakob, P.; Staufenbiel, R.; Heuwieser, W.; Borchardt, S. Association between serum calcium dynamics around parturition and common postpartum diseases in dairy cows. J. Dairy Sci. 2020. [CrossRef]
- Venjakob, P.; Staufenbiel, R.; Heuwieser, W.; Borchardt, S. Serum calcium dynamics within the first 3 days in milk and the associated risk of acute puerperal metritis. J. Dairy Sci. 2019. [CrossRef]
- Rodríguez, E.M.; Arís, A.; Bach, A. Associations between subclinical hypocalcemia and postparturient diseases in dairy cows. J. Dairy Sci. 2017, 100, 7427–7434. [Google Scholar] [CrossRef]
- Atalay, H. The effect of serum β-hydroxybutyric acid and calcium levels on left displaced abomasum in Holstein cows on transition period. J. Istanbul Vet. Sci. 2019, 3, 43–48. [Google Scholar] [CrossRef]
- Curtis, C.; Erb, H.; Sniffen, C.; Smith, R.; Powers, P.; Smith, M.; Me, W.; Rb, H.; Pearson, E. Association of parturient hypocalcemia with eight periparturient disorders in Holstein cows. J. Am. Vet. Med. Assoc. 1983, 183, 559–561. [Google Scholar]
- Kantham, L.; Quinn, S.; Egbuna, O.; Baxi, K.; Butters, R.; Pang, J.; Pollak, M.; Goltzman, D.; Brown, E. The calcium-sensing receptor (CaSR) defends against hypercalcemia independently of its regulation of parathyroid hormone secretion. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E915–E923. [Google Scholar] [CrossRef]
- Marmalyuk, D.; Runova, G.; Fadeyev, V. The role of the calcium-sensing receptor in the regulation of parathyroid hormone secretion in physiology and in calcitropic diseases. Osteoporos. Bone Dis. 2024, 13. [Google Scholar] [CrossRef]
- Zhang, B.X.; Huang, B.; Jiang, Q.; Loor, J.; Lv, X.; Zhang, W.; Li, M.; Wen, J.; Yin, Y.; Wang, J.; Yang, W.; Xu, C. Transcriptomics of circulating neutrophils in dairy cows with subclinical hypocalcemia. Front. Vet. Sci. 2022, 9. [Google Scholar] [CrossRef]
- Martinez, N.; Sinedino, L.; Bisinotto, R.; Ribeiro, E.; Gomes, G.; Lima, F.; Greco, L.; Risco, C.; Galvão, K.; Taylor-Rodriguez, D.; Driver, J.; Thatcher, W.; Santos, J. Effect of induced subclinical hypocalcemia on physiological responses and neutrophil function in dairy cows. J. Dairy Sci. 2014, 97, 874–887. [Google Scholar] [CrossRef]
- Abuajamieh, M.; Kvidera, S.K.; Fernandez, M.V.; Nayeri, A.; Upah, N.C.; Nolan, E.A.; Lei, S.M.; DeFrain, J.M.; Green, H.B.; Schoenberg, K.M.; Trout, W.E.; Baumgard, L.H. Inflammatory biomarkers are associated with ketosis in periparturient Holstein cows. Res. Vet. Sci. 2016, 109, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Reinhardt, T.; Goff, J. Parturition and hypocalcemia blunts calcium signals in immune cells of dairy cattle. J. Dairy Sci. 2006, 89, 2588–2595. [Google Scholar] [CrossRef] [PubMed]
- Trebak, M.; Kinet, J. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Ghoshal, S.; Witta, J.; Zhong, J.; de Villiers, W.; Eckhardt, E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J. Lipid Res. 2009, 50, 90–97. [Google Scholar] [CrossRef]
- Trebicka, J.; Krag, A.; Gansweid, S.; Appenrodt, B.; Schiedermaier, P.; Sauerbruch, T.; Spengler, U. Endotoxin and tumor necrosis factor-receptor levels in portal and hepatic vein of patients with alcoholic liver cirrhosis receiving elective transjugular intrahepatic portosystemic shunt. Eur. J. Gastroenterol. Hepatol. 2011, 23, 1218–1225. [Google Scholar] [CrossRef]
- Munford, R.S.; Hall, C.L.; Dietschy, J.M. Binding of Salmonella typhimurium lipopolysaccharides to rat high-density lipoproteins. Infect. Immun. 1981, 34, 835–843. [Google Scholar] [CrossRef]
- Wurfel, M.M.; Wright, S.D. Lipopolysaccharide (LPS) binding protein catalyzes binding of LPS to lipoproteins. Prog. Clin. Biol. Res. 1995, 392, 287–295. [Google Scholar]
- Kitchens, R.L.; Wolfbauer, G.; Albers, J.J.; Munford, R.S. Plasma lipoproteins promote the release of bacterial lipopolysaccharide from the monocyte cell surface. J. Biol. Chem. 1999, 274, 34116–34122. [Google Scholar] [CrossRef]
- Tobias, P.S.; Soldau, K.; Ulevitch, R.J. Identification of a lipid A binding site in the acute phase reactant lipopolysaccharide binding protein. J. Biol. Chem. 1989, 264, 10867–10871. [Google Scholar]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Vogel, S.N. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002, 4, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Zaloga, G.P. Ionized hypocalcemia during sepsis. Crit. Care Med. 2000, 28, 266–268. [Google Scholar] [CrossRef] [PubMed]
- Skarnes, R.C.; Chedid, L. Biological degradation and inactivation of endotoxin (chromate-labeled). In: Landy, M.; Braun, W., Eds. Bacterial Endotoxins; Rutgers Univ. Press: New Brunswick, NJ, USA, 1964; pp. 575–587. Braun, W. Bacterial Endotoxins, Ed.;
- Hotchkiss, R.S.; Karl, I.E. Calcium: A regulator of the inflammatory response in endotoxemia and sepsis. New Horiz. 1996, 4, 58–71. [Google Scholar] [PubMed]
- Feingold, K.R.; Grunfeld, C. Lipids: A key player in the battle between the host and microorganisms. J. Lipid Res. 2012, 53, 2487–2489. [Google Scholar] [CrossRef]
- Gibot, S. On the origins of lactate during sepsis. Crit. Care 2012, 16, 151. [Google Scholar] [CrossRef]
- Toffaletti, J.; Abrams, B. Effects of in vivo and in vitro production of lactic acid on ionized, protein-bound, and complex-bound calcium in blood. Clin. Chem. 1989, 35, 935–938. [Google Scholar]
- Khatua, B.; Yaron, J.R.; El-Kurdi, B.; Kostenko, S.; Papachristou, G.I.; Singh, V.P. Ringer’s lactate prevents early organ failure by providing extracellular calcium. J. Clin. Med. 2020, 9, 263. [Google Scholar] [CrossRef]
- Zaloga, G.P.; Willey, S.; Tomasic, P.; Chernow, B. Free fatty acids alter calcium binding: A cause for misinterpretation of serum calcium values and hypocalcemia in critical illness. J. Clin. Endocrinol. Metab. 1987, 64, 1010–1014. [Google Scholar] [CrossRef]
- Whitsett, J.; Tsang, R.C. In vitro effects of fatty acids on serum-ionized calcium. J. Pediatr. 1977, 91, 233–236. [Google Scholar] [CrossRef]
- Chapinal, N.; Leblanc, S.J.; Carson, M.E.; Leslie, K.E.; Godden, S.; Capel, M.; Santos, J.E.; Overton, M.W.; Duffield, T.F. Herd-level association of serum metabolites in the transition period with disease, milk production, and early lactation reproductive performance. J. Dairy Sci. 2012, 95, 5676–5682. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.O. Protein-bound calcium in human serum: Quantitative examination of binding and its variables by a molecular binding model and clinical chemical implications for measurement of ionized calcium. Scand. J. Clin. Lab. Investig. 1972, 30, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Dimeski, G.; Treacy, O. The influence of albumin and pH on total and ionized calcium and magnesium. Point Care 2018, 17, 123–126. [Google Scholar] [CrossRef]
- Dalal, P.; Muller, W.; Sullivan, D. Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 2019, 189, 1529–1542. [Google Scholar] [CrossRef]
- Ridefelt, P.; Helmersson-Karlqvist, J. Albumin adjustment of total calcium does not improve the estimation of calcium status. Scand. J. Clin. Lab. Investig. 2017, 77, 442–447. [Google Scholar] [CrossRef]
- Buege, M.; Do, B.; Lee, H.; Weber, D.; Horowitz, S.; Feng, L.; Qing, Y.; Shank, B. Corrected calcium versus ionized calcium measurements for identifying hypercalcemia in patients with multiple myeloma. Cancer Treat. Res. Commun. 2019, 21, 100159. [Google Scholar] [CrossRef]
- Gilabert, J. Cytoplasmic calcium buffering: An integrative crosstalk. Adv. Exp. Med. Biol. 2020, 1131, 163–182. [Google Scholar] [CrossRef]
- Kraus-Friedmann, N. Calcium sequestration in the liver. Cell Calcium 1990, 11, 625–640. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Baird, G.S. Ionized calcium. Clin. Chim. Acta 2011, 412, 696–701. [Google Scholar] [CrossRef]
- Contri, M.; Boraldi, F.; Taparelli, F.; Paepe, A.; Ronchetti, I. Matrix proteins with high affinity for calcium ions are associated with mineralization within the elastic fibers of pseudoxanthoma elasticum dermis. Am. J. Pathol. 1996, 148, 569–577. [Google Scholar] [PubMed]
- Hoeflich, K.P.; Ikura, M. Calmodulin in action: Diversity in target recognition and activation mechanisms. Cell 2002, 108, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Bazil, J.N.; Blomeyer, C.A.; Pradhan, R.K.; Camara, A.K.; Dash, R.K. Modeling the calcium sequestration system in isolated guinea pig cardiac mitochondria. J. Bioenerg. Biomembr. 2013, 45, 177–188. [Google Scholar] [CrossRef]
- Vannucci, L.; Fossi, C.; Quattrini, S.; Guasti, L.; Pampaloni, B.; Gronchi, G.; Giusti, F.; Romagnoli, C.; Cianferotti, L.; Marcucci, G.; Brandi, M. Calcium intake in bone health: A focus on calcium-rich mineral waters. Nutrients 2018, 10, 102645. [Google Scholar] [CrossRef]
- Zikan, V.; Stepan, J.J. Marker of bone resorption in acute response to exogenous or endogenous parathyroid hormone. Biomark. Insights 2008, 3, 19–24. [Google Scholar] [CrossRef]
- Cianferotti, L.; Gomes, A.R.; Fabbri, S.; Tanini, A.; Brandi, M.L. The calcium-sensing receptor in bone metabolism: From bench to bedside and back. Osteoporos. Int. 2015, 26, 2055–2071. [Google Scholar] [CrossRef]
- Hatate, K.; Kawashima, C.; Hanada, M.; Kayano, M.; Yamagishi, N. Short communication: Serum osteoprotegerin concentrations in periparturient dairy cows. J. Dairy Sci. 2018, 101, 6622–6626. [Google Scholar] [CrossRef]
- Roux, E.; Marhl, M. Role of sarcoplasmic reticulum and mitochondria in Ca2+ removal in airway myocytes. Biophys. J. 2004, 86, 2583–2595. [Google Scholar] [CrossRef]
- Stammers, A.; Susser, S.; Hamm, N.; Hlynsky, M.; Kimber, D.; Kehler, D.; Duhamel, T. The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can. J. Physiol. Pharmacol. 2015, 93, 843–854. [Google Scholar] [CrossRef]
- Treves, S.; Jungbluth, H.; Muntoni, F.; Zorzato, F. Congenital muscle disorders with cores: The ryanodine receptor calcium channel paradigm. Curr. Opin. Pharmacol. 2008, 8, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Michelucci, A.; García-Castañeda, M.; Boncompagni, S.; Dirksen, R.T. Role of STIM1/ORAI1-mediated store-operated Ca2+ entry in skeletal muscle physiology and disease. Cell Calcium 2018, 76, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Barritt, G. Calcium signalling in liver cells; in Calcium Signalling in Liver Cells; 2000; pp. 73–94. [CrossRef]
- Rieusset, J. Endoplasmic reticulum–mitochondria calcium signaling in hepatic metabolic diseases. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Oliva-Vilarnau, N.; Hankeova, S.; Vorrink, S.U.; Mkrtchian, S.; Andersson, E.R.; Lauschke, V.M. Calcium signaling in liver injury and regeneration. Front. Med. (Lausanne) 2018, 5, 192. [Google Scholar] [CrossRef]
- Jagtap, Y.; Adlakha, N. Simulation of buffered advection diffusion of calcium in a hepatocyte cell. Math. Biol. Bioinform. 2018. [CrossRef]
- Kang, Y.; McKenna, T.; Watson, L.; Williams, R.; Holt, M. Cytochemical changes in hepatocytes of rats with endotoxemia or sepsis: Localization of fibronectin, calcium, and enzymes. J. Histochem. Cytochem. 1988, 36, 665–678. [Google Scholar] [CrossRef]
- Bernardo, J.; Friedman, P. Renal calcium metabolism. In: Renal Physiology; pp. 2225–2247. [CrossRef]
- Lim, D.; Semyanov, A.; Genazzani, A.; Verkhratsky, A. Calcium signaling in neuroglia. Int. Rev. Cell Mol. Biol. 2021, 362, 1–53. [Google Scholar] [CrossRef]
- Wang, F.; Yuan, T.; Pereira, A.; Verkhratsky, A.; Huang, J. Glial cells and synaptic plasticity. Neural Plast. 2016, 2016. [Google Scholar] [CrossRef]
- Clementi, E.; Racchetti, G.; Melino, G.; Meldolesi, J. Cytosolic Ca2+ buffering, a cell property that in some neurons markedly decreases during aging, has a protective effect against NMDA/nitric oxide-induced excitotoxicity. Life Sci. 1996, 59, 389–397. [Google Scholar] [CrossRef]
- Yamada, A.; Watanabe, A.; Yamamoto, T. Regulatory mechanisms of mitochondrial calcium uptake by the calcium uniporter complex. Biophys. Physicobiol. 2023, 20. [Google Scholar] [CrossRef]
- Denton, R. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Evelson, P.; Vanasco, V.; Magnani, N.; Cimolai, M.; Marchini, T. The role of mitochondria in inflammatory syndromes; in Mitochondria in Inflammation; pp. 233–247. [CrossRef]
- Zhang, H.; Kovacs-Nolan, J.; Kodera, T.; Eto, Y.; Mine, Y. γ-Glutamyl cysteine and γ-glutamyl valine inhibit TNF-α signaling in intestinal epithelial cells and reduce inflammation in a mouse model of colitis via allosteric activation of the calcium-sensing receptor. Biochim. Biophys. Acta 2015, 1852, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B.; Michelet, M.; Theurey, P.; Vial, G.; Demion, M.; Blond, E.; Zoulim, F.; Gomez, L.; Vidal, H.; Lacampagne, A.; Ovize, M. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2016, 59, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Guan, Z.; Gao, Y.; Bai, Y.; Zhan, X.; Ji, X.; Xu, J.; Zhou, H.; Rao, Z. ER stress promotes mitochondrial calcium overload and activates the ROS/NLRP3 axis to mediate fatty liver ischemic injury. Hepatol. Commun. 2024, 8. [Google Scholar] [CrossRef]
- Briones-Suarez, L.; Cifuentes, M.; Bravo-Sagua, R. Secretory factors from calcium-sensing receptor-activated SW872 pre-adipocytes induce cellular senescence and a mitochondrial fragmentation-mediated inflammatory response in HepG2 cells. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef]
- Zhou, Y.; Greka, A. Calcium-permeable ion channels in the kidney. Am. J. Physiol. Renal Physiol. 2016, 310, F1157–F1167. [Google Scholar] [CrossRef]
- Woudenberg-Vrenken, T.; Bindels, R.; Hoenderop, J. The role of transient receptor potential channels in kidney disease. Nat. Rev. Nephrol. 2009, 5, 441–449. [Google Scholar] [CrossRef]
- Klein, G.; Castro, S.; Garofalo, R. The calcium-sensing receptor as a mediator of inflammation. Semin. Cell Dev. Biol. 2016, 49, 52–56. [Google Scholar] [CrossRef]
- Zhao, B. Does TNF promote or restrain osteoclastogenesis and inflammatory bone resorption? Crit. Rev. Immunol. 2018, 38, 253–261. [Google Scholar] [CrossRef]
- Tupling, A. The sarcoplasmic reticulum in muscle fatigue and disease: Role of the sarco(endo)plasmic reticulum Ca2+-ATPase. Can. J. Appl. Physiol. 2004, 29, 308–329. [Google Scholar] [CrossRef] [PubMed]
- Qaisar, R.; Bhaskaran, S.; Ranjit, R.; Premkumar, P.; Huseman, K.; Sataranatarajan, K.; Van Remmen, H. Restoration of SERCA ATPase as an intervention to muscle impairment associated with oxidative stress. FASEB J. 2018, 32, —. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; Schmid, J.A. Cell type-specific roles of NF-κB linking inflammation and thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Esmon, C. The impact of the inflammatory response on coagulation. Thromb. Res. 2004, 114, 321–327. [Google Scholar] [CrossRef]
- Mohanty, J.; Barodka, V.; Berkowitz, D.; Rifkind, J. Intracellular calcium mediated stiffness of red blood cells is reversed by hypoxic pre-incubation with nitrite ions. Biophys. J. 2010, 98. [Google Scholar] [CrossRef]
- Lacy, P.; Stow, J. Cytokine release from innate immune cells: Association with diverse membrane trafficking pathways. Blood 2011, 118, 9–18. [Google Scholar] [CrossRef]
- Kirdajova, D.; Kriska, J.; Tureckova, J.; Anděrová, M. Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Arundine, M.; Tymianski, M. Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 2003, 34, 325–337. [Google Scholar] [CrossRef]
- Muller, W. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am. J. Pathol. 2014, 184, 886–896. [Google Scholar] [CrossRef]
- Dalal, P.; Muller, W.; Sullivan, D. Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 2019. [CrossRef]
- Shannon, T.; Ginsburg, K.; Bers, D. Potentiation of fractional sarcoplasmic reticulum calcium release by total and free intra-sarcoplasmic reticulum calcium concentration. Biophys. J. 2000, 78, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Cataldi, A.; Giacomo, V. Extracellular matrix: Immunity and inflammation; in Extracellular Matrix; pp. 83–109. [CrossRef]
- Touyz, R.; Alves-Lopes, R.; Rios, F.; Camargo, L.; Anagnostopoulou, A.; Arner, A.; Montezano, A. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Qu, J.; He, L.; Zhang, F.; Zhou, Z.; Yang, S.; Zhou, Y. Calcium in vascular smooth muscle cell elasticity and adhesion: Novel insights into the mechanism of action. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Carlstedt, F.; Lind, L. Hypocalcemic syndromes. Crit. Care Clin. 2001, 17, 139–153, vii–viii. [CrossRef]
- Klein, G. Phosphate as an adjunct to calcium in promoting coronary vascular calcification in chronic inflammatory states. eLife 2024, 13. [Google Scholar] [CrossRef]
- Edwards, F.; Taheri, A.; Dann, S.; Dye, J. Characterization of cytolytic neutrophil activation in vitro by amorphous hydrated calcium phosphate as a model of biomaterial inflammation. J. Biomed. Mater. Res. Part A 2011, 96, 552–565. [Google Scholar] [CrossRef]
- Chen, J.; Sitsel, A.; Benoy, V.; Sepúlveda, M.; Vangheluwe, P. Primary active Ca2+ transport systems in health and disease. Cold Spring Harb. Perspect. Biol. 2019. [CrossRef]
- Bers, D.M. Cardiac Excitation-Contraction Coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Wong, P.T. Isolation of a High Capacity Ca²⁺-Binding Protein from Sarcoplasmic Reticulum. Nature New Biology 1971, 231, 198–200. [Google Scholar]
- Beard, N.A.; Laver, D.R.; Dulhunty, A.F. Calsequestrin and the Calcium Release Channel of Skeletal and Cardiac Muscle. Prog. Biophys. Mol. Biol. 2004, 85, 33–69. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal Calcium Signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Means, A.R. Calmodulin: A Prototypical Calcium Sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, M.; Kalyanasundaram, A. SERCA Pump Isoforms: Their Role in Calcium Transport and Disease. Muscle Nerve 2007, 35, 430–442. [Google Scholar] [CrossRef]
- Nilius, B.; Voets, T. The Puzzle of TRPV4 Channelopathies. EMBO Rep. 2013, 14, 152–163. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Lederer, W.J. Sodium/Calcium Exchange: Its Physiological Implications. Physiol. Rev. 1999, 79, 763–854. [Google Scholar] [CrossRef]
- Brini, M.; Carafoli, E. The Plasma Membrane Ca²⁺ ATPase and the Plasma Membrane Sodium Calcium Exchanger Cooperate in the Regulation of Cell Calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef]
- Pizzo, P.; Lissandron, V.; Capitanio, P.; Pozzan, T. Ca²⁺ Signalling in the Golgi Apparatus. Cell Calcium 2011, 50, 184–192. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of Intracellular Ca²⁺: Molecular Determinants and Functional Consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef]
- Pizzo, P.; Pozzan, T. Golgi Ca²⁺, Messengers and Function. Cell Calcium 2007, 42, 405–412. [Google Scholar] [CrossRef]
- Feske, S.; Wulff, H.; Skolnik, E. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015, 33, 291–353. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Soto, G.; Rocher, A.; García-Rodríguez, C.; Núñez, L.; Villalobos, C. The calcium-sensing receptor in health and disease. Int. Rev. Cell Mol. Biol. 2016, 327, 321–369. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Suqin, H.; Zhu, W. Calcium sensing receptor: Signaling pathways and physiological functions. Chin. J. Anim. Nutr. 2015, 27, 703–714. [Google Scholar]
- Chavez-Abiega, S.; Mos, I.; Centeno, P.; Elajnaf, T.; Schlattl, W.; Ward, D.; Goedhart, J.; Kallay, E. Sensing extracellular calcium—An insight into the structure and function of the calcium-sensing receptor (CaSR). Adv. Exp. Med. Biol. 2020, 1131, 1031–1063. [Google Scholar] [CrossRef]
- Chakravarti, B.; Chattopadhyay, N.; Brown, E. Signaling through the extracellular calcium-sensing receptor (CaSR). Adv. Exp. Med. Biol. 2012, 740, 103–142. [Google Scholar] [CrossRef]
- Bird, G.; Putney, J. Differential effects of PLC-coupled receptors on intracellular calcium oscillations in HEK293 cells. Biophys. J. 2015, 108. [Google Scholar] [CrossRef]
- Chen, R.; Goodman, W. Role of the calcium-sensing receptor in parathyroid gland physiology. Am. J. Physiol. Renal Physiol. 2004, 286, F1005–F1011. [Google Scholar] [CrossRef]
- Kinoshita, Y. The functions of calcium-sensing receptor in regulating mineral metabolism. Clin. Calcium 2017, 27, 491–497. [Google Scholar]
- Liu, W.; Guo, Y.; Liu, Y.; Sun, J.; Yin, X. Calcium-sensing receptor of immune cells and diseases. 2021, —. [CrossRef]
- Chandler, T.L.; Westhoff, T.A.; Behling-Kelly, E.L.; Sipka, A.S.; Mann, S. Eucalcemia during lipopolysaccharide challenge in postpartum dairy cows: I. Clinical, inflammatory, and metabolic response. J. Dairy Sci. 2023, 106, 3586–3600. [Google Scholar] [CrossRef]
- Horst, E.A.; Mayorga, E.J.; Al-Qaisi, M.; Abeyta, M.A.; Portner, S.L.; McCarthy, C.S.; Goetz, B.M.; Kvidera, S.K.; Baumgard, L.H. Effects of maintaining eucalcemia following immunoactivation in lactating Holstein dairy cows. J. Dairy Sci. 2020, 103, 7472–7486. [Google Scholar] [CrossRef]
- Waldron, M.R.; Nonnecke, B.J.; Nishida, T.; Horst, R.L.; Overton, T.R. Effect of lipopolysaccharide infusion on serum macromineral and vitamin D concentrations in dairy cows. J. Dairy Sci. 2003, 86, 3440–3446. [Google Scholar] [CrossRef] [PubMed]
- Seminara, J.A.; Seely, C.R.; McArt, J.A.A. Acute phase responses in clinically healthy multiparous Holsteins with and without calcium dysregulation during the early postpartum period. J. Dairy Sci. 2024. [CrossRef]
- Couto Serrenho, R.; Morrison, E.; Bruinjé, T.C.; LeBlanc, S.J. Assessment of systemic inflammation following oral calcium supplementation in healthy postpartum multiparous dairy cows—a randomized controlled trial. JDS Commun. 2023, 5, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Garidel, P.; Rappolt, M.; Schromm, A.; Howe, J.; Lohner, K.; Andrä, J.; Koch, M.; Brandenburg, K. Divalent cations affect chain mobility and aggregate structure of lipopolysaccharide from Salmonella minnesota reflected in a decrease of its biological activity. Biochim. Biophys. Acta 2005, 1715, 122–131. [Google Scholar] [CrossRef]
- Redeker, C.; Briscoe, W. Interactions between mutant bacterial lipopolysaccharide (LPS-Ra) surface layers: Surface vesicles, membrane fusion, and effect of Ca2+ and temperature. Langmuir 2019. [CrossRef]
- Ryu, J.K.; Kim, S.J.; Rah, S.H.; Kang, J.I.; Jung, H.E.; Lee, D.; Lee, H.K.; Lee, J.O.; Park, B.S.; Yoon, T.Y.; Kim, H.M. Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity 2017, 46, 38–50. [Google Scholar] [CrossRef]
- Drago-Serrano, M.E.; de la Garza-Amaya, M.; Luna, J.S.; Campos-Rodríguez, R. Lactoferrin-lipopolysaccharide (LPS) binding as key to antibacterial and antiendotoxic effects. Int. Immunopharmacol. 2012, 12, 1–9. [Google Scholar] [CrossRef]
- Wurfel, M.M.; Wright, S.D. Lipopolysaccharide-binding protein and soluble CD14 transfer lipopolysaccharide to phospholipid bilayers: Preferential interaction with particular classes of lipid. J. Immunol. 1997, 158, 3925–3934. [Google Scholar]
- Tsukamoto, H.; Takeuchi, S.; Kubota, K.; Kobayashi, Y.; Kozakai, S.; Ukai, I.; Shichiku, A.; Okubo, M.; Numasaki, M.; Kanemitsu, Y.; Matsumoto, Y.; Nochi, T.; Watanabe, K.; Aso, H.; Tomioka, Y. Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent toll-like receptor 4 internalization and LPS-induced TBK1-IKKϵ-IRF3 axis activation. J. Biol. Chem. 2018, 293, 10186–10201. [Google Scholar] [CrossRef]
- Uhrig, A.; Banafsche, R.; Kremer, M.; Hegenbarth, S.; Hamann, A.; Neurath, M.; Gerken, G.; Limmer, A.; Knolle, P.A. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J. Leukoc. Biol. 2005, 77, 626–633. [Google Scholar] [CrossRef]
- Baranova, I.N.; Vishnyakova, T.G.; Bocharov, A.V.; Leelahavanichkul, A.; Kurlander, R.; Chen, Z.; Souza, A.C.; Yuen, P.S.; Star, R.A.; Csako, G.; Patterson, A.P.; Eggerman, T.L. Class B scavenger receptor types I and II and CD36 mediate bacterial recognition and proinflammatory signaling induced by Escherichia coli, lipopolysaccharide, and cytosolic chaperonin 60. J. Immunol. 2012, 188, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Van Bossuyt, H.; Wisse, E. Structural changes produced in Kupffer cells in the rat liver by injection of lipopolysaccharide. Cell Tissue Res. 1988, 251, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Tamura, Y.; Osuga, J.; Adachi, H.; Tozawa, R.; Takanezawa, Y.; Ohashi, K.; Yahagi, N.; Sekiya, M.; Okazaki, H.; Tomita, S.; Iizuka, Y.; Koizumi, H.; Inaba, T.; Yagyu, H.; Kamada, N.; Suzuki, H.; Shimano, H.; Kadowaki, T.; Tsujimoto, M.; Arai, H.; Yamada, N.; Ishibashi, S. Scavenger receptor expressed by endothelial cells I (SREC-I) mediates the uptake of acetylated low density lipoproteins by macrophages stimulated with lipopolysaccharide. J. Biol. Chem. 2004, 279, 30938–30944. [Google Scholar] [CrossRef] [PubMed]
- König, V.; Hopf, U.; Möller, B.; Lobeck, H.; Assmann, G.; Freudenberg, M.; Galanos, C. The significance of high-density lipoproteins (HDL) in the clearance of intravenously administered bacterial lipopolysaccharides (LPS) in mice. Hepatogastroenterology 1988, 35, 111–115. [Google Scholar]
- Hersoug, L.G.; Møller, P.; Loft, S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: Implications for inflammation and obesity. Obes. Rev. 2016, 17, 297–312. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Kumar, P.; Schroder, E.A.; Rajaram, M.V.S.; Harris, E.N.; Ganesan, L.P. The battle of LPS clearance in host defense vs. inflammatory signaling. Cells 2024, 13, 1590. [Google Scholar] [CrossRef]
- Safavi, K.; Nichols, F. Alteration of biological properties of bacterial lipopolysaccharide by calcium hydroxide treatment. J. Endod. 1994, 20, 127–129. [Google Scholar] [CrossRef]
Figure 1.
Hypocalcemia as an Adaptive Response to Systemic Inflammation. This schematic illustrates how shifts in ionized calcium (iCa²⁺) modulate the clearance and inflammatory potential of lipopolysaccharide (LPS) originating from mucosal tissues such as the mammary gland, gastrointestinal tract, reproductive tract, and infected hooves (1, 2, 3, 4). LPS traverses the epithelial barrier into the bloodstream (5), where it binds to chelators (e.g., albumin, lactate, and NEFA), which reduce free iCa²⁺ (7). Under transient or moderate hypocalcemia (10), lower iCa²⁺ dampens certain calcium-dependent immune pathways, steering LPS toward lipoprotein-mediated clearance via acute-phase high-density lipoproteins containing serum amyloid A (SAA-ap-HDL) (9). Lipopolysaccharide-binding protein (LBP) and CD14 (11) facilitate LPS binding to ap-HDL, directing it for detoxification in hepatocytes (possibly contributing to fatty liver) (12) or temporary storage in adipocytes (13). Apolipoprotein A1, dissociated from n-HDL, also binds LPS for renal excretion (14). During mild hypocalcemia, macrophage activation (15) remains subdued, leading to limited release of pro-inflammatory cytokines (IL-1, IL-6, TNF-α). In contrast, normocalcemia (16) supports lamellar LPS aggregation in the presence of iCa²⁺, enabling macrophages to take up these larger, structurally inert complexes (18). Although macrophages then release cytokines (IL-1, IL-6, TNF-α) (19), the inflammatory response may remain more localized than systemic. Some fraction of LPS is also bound by normal HDL (n-HDL) and cleared by the liver (20). Overall, this figure emphasizes that mild hypocalcemia can serve as an adaptive brake on systemic inflammation by reducing excessive immune-cell activation and directing LPS toward lipoprotein-based clearance, whereas adequate iCa²⁺ levels favor lamellar LPS aggregation and macrophage uptake. Taken together, these complementary pathways underscore the dynamic role of calcium homeostasis and lipoprotein interactions in balancing LPS clearance under varying inflammatory states. [Figure created by Zohaib Saleem and B.N. Ametaj using BioRender].
Figure 1.
Hypocalcemia as an Adaptive Response to Systemic Inflammation. This schematic illustrates how shifts in ionized calcium (iCa²⁺) modulate the clearance and inflammatory potential of lipopolysaccharide (LPS) originating from mucosal tissues such as the mammary gland, gastrointestinal tract, reproductive tract, and infected hooves (1, 2, 3, 4). LPS traverses the epithelial barrier into the bloodstream (5), where it binds to chelators (e.g., albumin, lactate, and NEFA), which reduce free iCa²⁺ (7). Under transient or moderate hypocalcemia (10), lower iCa²⁺ dampens certain calcium-dependent immune pathways, steering LPS toward lipoprotein-mediated clearance via acute-phase high-density lipoproteins containing serum amyloid A (SAA-ap-HDL) (9). Lipopolysaccharide-binding protein (LBP) and CD14 (11) facilitate LPS binding to ap-HDL, directing it for detoxification in hepatocytes (possibly contributing to fatty liver) (12) or temporary storage in adipocytes (13). Apolipoprotein A1, dissociated from n-HDL, also binds LPS for renal excretion (14). During mild hypocalcemia, macrophage activation (15) remains subdued, leading to limited release of pro-inflammatory cytokines (IL-1, IL-6, TNF-α). In contrast, normocalcemia (16) supports lamellar LPS aggregation in the presence of iCa²⁺, enabling macrophages to take up these larger, structurally inert complexes (18). Although macrophages then release cytokines (IL-1, IL-6, TNF-α) (19), the inflammatory response may remain more localized than systemic. Some fraction of LPS is also bound by normal HDL (n-HDL) and cleared by the liver (20). Overall, this figure emphasizes that mild hypocalcemia can serve as an adaptive brake on systemic inflammation by reducing excessive immune-cell activation and directing LPS toward lipoprotein-based clearance, whereas adequate iCa²⁺ levels favor lamellar LPS aggregation and macrophage uptake. Taken together, these complementary pathways underscore the dynamic role of calcium homeostasis and lipoprotein interactions in balancing LPS clearance under varying inflammatory states. [Figure created by Zohaib Saleem and B.N. Ametaj using BioRender].
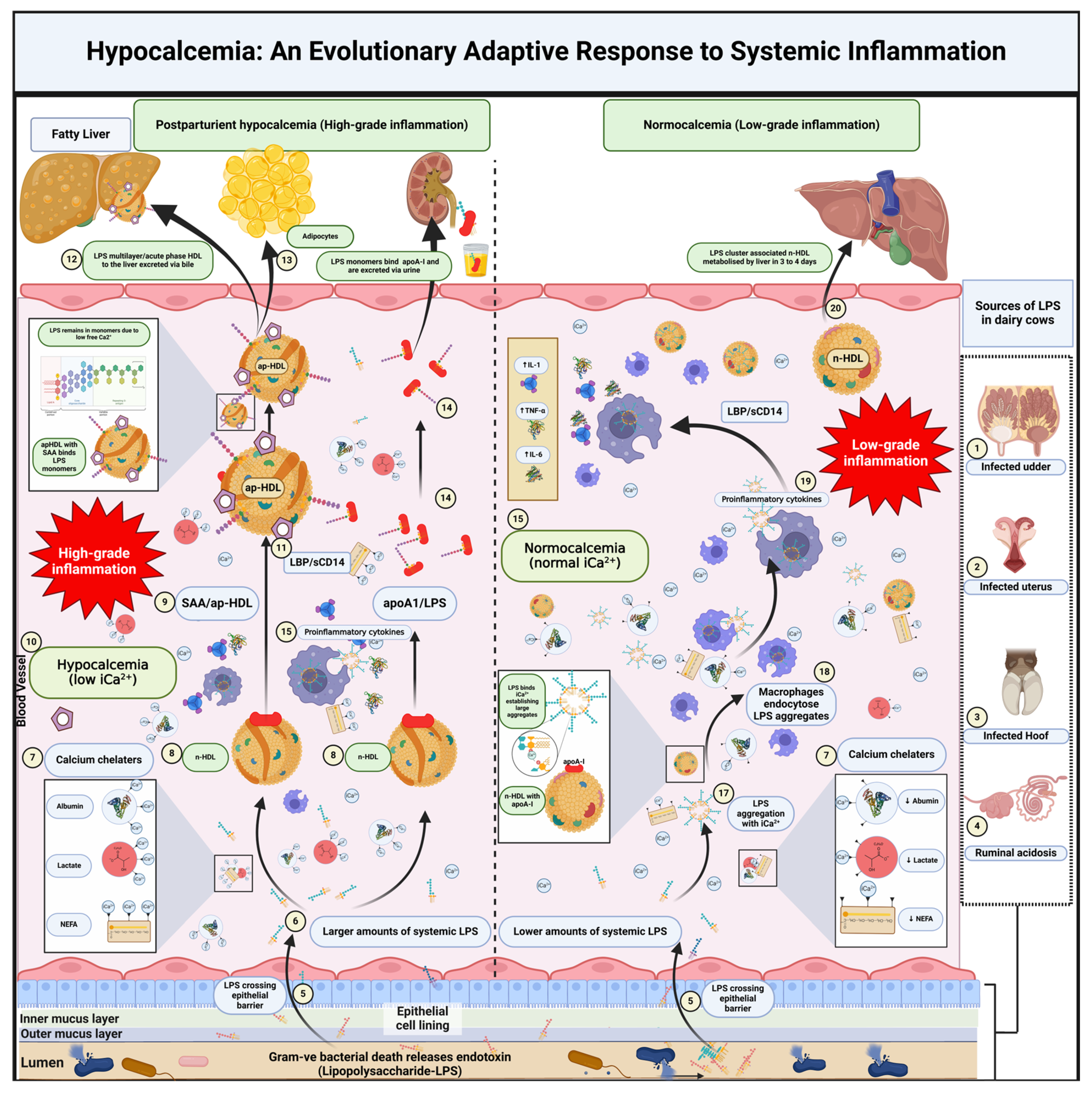
Figure 2.
Tissues and Organs Involved in iCa²⁺ Buffering and Sequestration During Inflammation. Calcium in the bloodstream exists in three primary forms: protein-bound (∼40%), ionized (iCa²⁺, ∼50%), and chelated (∼10%). This delicate balance supports essential physiological processes and adapts to systemic inflammation (1). Platelets sequester and release iCa²⁺ during inflammatory processes to regulate coagulation and immune responses (2). Red blood cells buffer iCa²⁺ to maintain membrane flexibility and oxygen transport, both of which can be compromised under inflammatory conditions (3). Immune cells depend on iCa²⁺ buffering for activation, migration, and cytokine release—functions that intensify during inflammation (4). Skeletal muscle cells store and release iCa²⁺ via the sarcoplasmic reticulum, enabling contractions that may be heightened in inflammatory states (5). Bones act as the largest calcium reservoir, releasing iCa²⁺ into the bloodstream during inflammation-induced hypocalcemia or storing it in periods of hypercalcemia (6). The kidneys regulate iCa²⁺ homeostasis through reabsorption or excretion; however, systemic inflammation can impair renal function, leading to abnormal calcium wasting or retention (7). Hepatocytes buffer iCa²⁺ within the endoplasmic reticulum and mitochondria, playing a critical role in detoxification and metabolic responses to inflammation (8). Neurons rely on iCa²⁺ buffering for synaptic signaling, while glial cells modulate intra- and extracellular calcium levels during neuroinflammatory states (9). Vascular endothelial cells buffer iCa²⁺ in response to inflammatory mediators such as histamine and bradykinin, thereby affecting vascular permeability and tone (10). In the extracellular matrix, iCa²⁺ binds to collagen, elastic fibers, and osteopontin, buffering excess calcium while maintaining tissue integrity under inflammatory stress (11). Finally, smooth muscle cells regulate contraction and relaxation by buffering and releasing iCa²⁺—processes crucial for preserving vascular integrity and blood flow during inflammation (12). [Figure created by Zohaib Saleem and B.N. Ametaj using BioRender].
Figure 2.
Tissues and Organs Involved in iCa²⁺ Buffering and Sequestration During Inflammation. Calcium in the bloodstream exists in three primary forms: protein-bound (∼40%), ionized (iCa²⁺, ∼50%), and chelated (∼10%). This delicate balance supports essential physiological processes and adapts to systemic inflammation (1). Platelets sequester and release iCa²⁺ during inflammatory processes to regulate coagulation and immune responses (2). Red blood cells buffer iCa²⁺ to maintain membrane flexibility and oxygen transport, both of which can be compromised under inflammatory conditions (3). Immune cells depend on iCa²⁺ buffering for activation, migration, and cytokine release—functions that intensify during inflammation (4). Skeletal muscle cells store and release iCa²⁺ via the sarcoplasmic reticulum, enabling contractions that may be heightened in inflammatory states (5). Bones act as the largest calcium reservoir, releasing iCa²⁺ into the bloodstream during inflammation-induced hypocalcemia or storing it in periods of hypercalcemia (6). The kidneys regulate iCa²⁺ homeostasis through reabsorption or excretion; however, systemic inflammation can impair renal function, leading to abnormal calcium wasting or retention (7). Hepatocytes buffer iCa²⁺ within the endoplasmic reticulum and mitochondria, playing a critical role in detoxification and metabolic responses to inflammation (8). Neurons rely on iCa²⁺ buffering for synaptic signaling, while glial cells modulate intra- and extracellular calcium levels during neuroinflammatory states (9). Vascular endothelial cells buffer iCa²⁺ in response to inflammatory mediators such as histamine and bradykinin, thereby affecting vascular permeability and tone (10). In the extracellular matrix, iCa²⁺ binds to collagen, elastic fibers, and osteopontin, buffering excess calcium while maintaining tissue integrity under inflammatory stress (11). Finally, smooth muscle cells regulate contraction and relaxation by buffering and releasing iCa²⁺—processes crucial for preserving vascular integrity and blood flow during inflammation (12). [Figure created by Zohaib Saleem and B.N. Ametaj using BioRender].
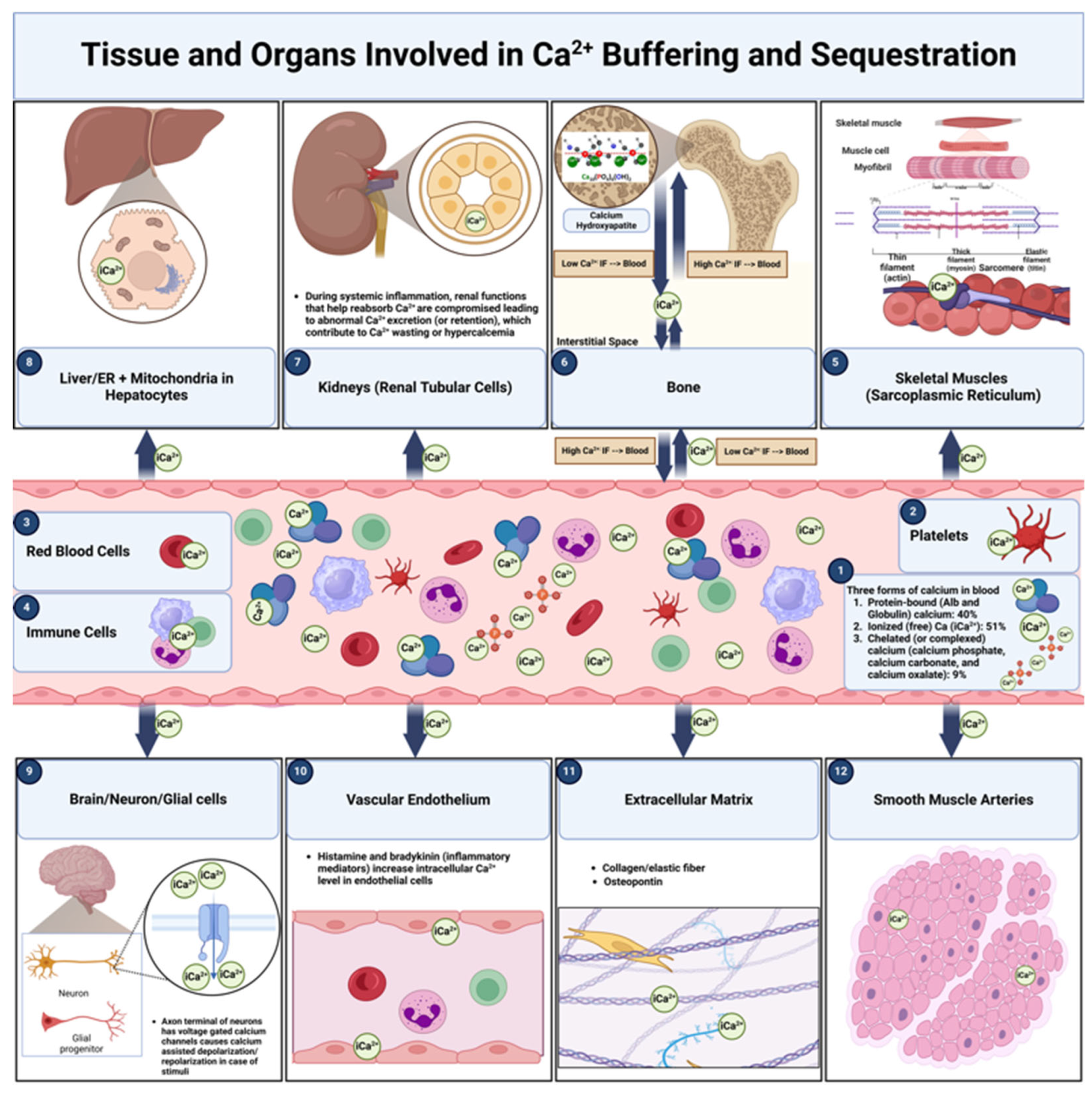
Figure 3.
Intracellular Calcium Buffers. Pathways and components involved in intracellular calcium (Ca²⁺) buffering across various organelles and cellular structures are shown: (1) In muscle cells, the sarcoplasmic reticulum functions as a primary site for Ca²⁺ sequestration and release, mediated by calcium-binding proteins such as calsequestrin, sarcalumenin, junctate, and triadin; (2) In the cytoplasm, soluble calcium-binding proteins—including calmodulin, calbindin-D28K, calbindin-D9K, and calreticulin—transiently bind free Ca²⁺, thereby regulating intracellular levels and signaling; (3) Membrane-associated proteins, such as the sarco/endoplasmic reticulum Ca²⁺-ATPase (SERCA), transient receptor potential (TRP) calcium channels, and the sodium-calcium exchanger (NCX), transport calcium across cellular membranes to maintain overall calcium homeostasis; (4) Calcium exchange between lysosomes and the endoplasmic reticulum occurs via two-pore calcium channels (TPC1/2), supporting both calcium storage and signaling; (5) At the endoplasmic reticulum (ER)-mitochondria junction, calcium transfer is mediated by inositol trisphosphate receptor (IP3R)-VDAC (voltage-dependent anion channel) complexes, with mitochondria buffering calcium through the rapid uptake mode (RaM) to support metabolic functions; (6) At the ER-plasma membrane junction, store-operated calcium entry (SOCE) is controlled by ORAI-STIM1 (Orai-stromal interaction molecule 1) interactions, which replenish ER calcium stores; (7) The Golgi apparatus also serves as a calcium reservoir, contributing to vesicular trafficking and protein modification. Collectively, these systems underscore the intricate and interconnected mechanisms by which calcium is buffered, stored, and mobilized within cells to support vital cellular processes and signaling pathways. [Figure created by Zohaib Saleem and B.N. Ametaj using BioRender].
Figure 3.
Intracellular Calcium Buffers. Pathways and components involved in intracellular calcium (Ca²⁺) buffering across various organelles and cellular structures are shown: (1) In muscle cells, the sarcoplasmic reticulum functions as a primary site for Ca²⁺ sequestration and release, mediated by calcium-binding proteins such as calsequestrin, sarcalumenin, junctate, and triadin; (2) In the cytoplasm, soluble calcium-binding proteins—including calmodulin, calbindin-D28K, calbindin-D9K, and calreticulin—transiently bind free Ca²⁺, thereby regulating intracellular levels and signaling; (3) Membrane-associated proteins, such as the sarco/endoplasmic reticulum Ca²⁺-ATPase (SERCA), transient receptor potential (TRP) calcium channels, and the sodium-calcium exchanger (NCX), transport calcium across cellular membranes to maintain overall calcium homeostasis; (4) Calcium exchange between lysosomes and the endoplasmic reticulum occurs via two-pore calcium channels (TPC1/2), supporting both calcium storage and signaling; (5) At the endoplasmic reticulum (ER)-mitochondria junction, calcium transfer is mediated by inositol trisphosphate receptor (IP3R)-VDAC (voltage-dependent anion channel) complexes, with mitochondria buffering calcium through the rapid uptake mode (RaM) to support metabolic functions; (6) At the ER-plasma membrane junction, store-operated calcium entry (SOCE) is controlled by ORAI-STIM1 (Orai-stromal interaction molecule 1) interactions, which replenish ER calcium stores; (7) The Golgi apparatus also serves as a calcium reservoir, contributing to vesicular trafficking and protein modification. Collectively, these systems underscore the intricate and interconnected mechanisms by which calcium is buffered, stored, and mobilized within cells to support vital cellular processes and signaling pathways. [Figure created by Zohaib Saleem and B.N. Ametaj using BioRender].
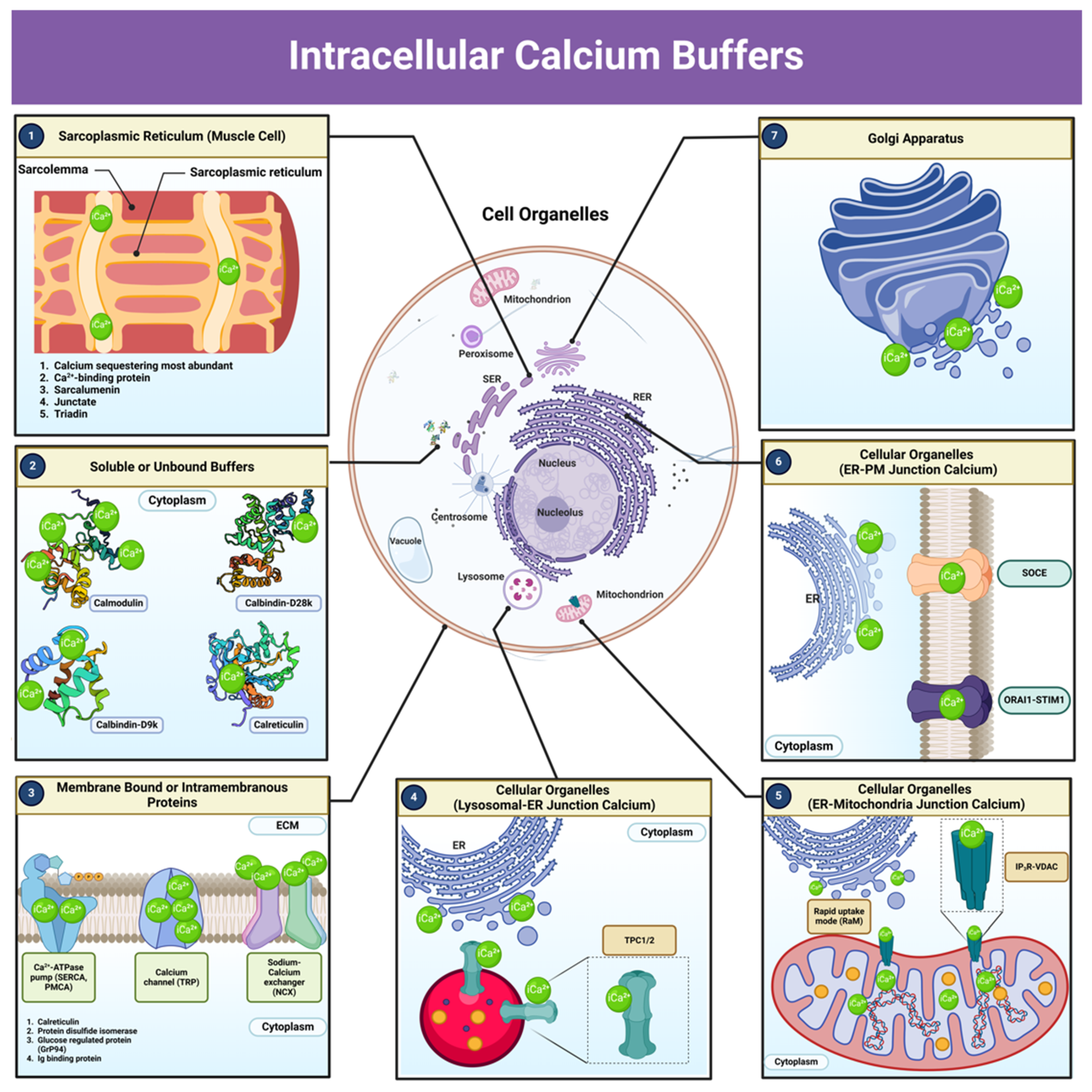
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



