
Submitted:
07 March 2025
Posted:
07 March 2025
You are already at the latest version
Abstract
Ubiquitination is dynamic and tightly regulated post-translational modifications essential for modulating protein stability, trafficking, and function to preserve cellular homeostasis. This process is orchestrated through a hierarchical enzymatic cascade involving three key enzymes: the E1 ubiquitin-activating enzyme, the E2 ubiquitin-conjugating enzyme, and the E3 ubiquitin ligase. The final step of ubiquitination is catalyzed by E3 ubiquitin ligase, which facilitates the transfer of ubiquitin from the E2 enzyme to the substrate, thereby dictating which proteins undergo ubiquitination. Emerging evidence underscores the critical roles of ubiquitin ligases in neurodevelopment, regulating fundamental processes such as neuronal polarization, axonal outgrowth, synaptogenesis, and synaptic function. Mutations in genes encoding ubiquitin ligases and the consequent dysregulation of these pathways have been increasingly implicated in a spectrum of neurodevelopmental disorders, including autism spectrum disorder, intellectual disability, and attention-deficit/hyperactivity disorder. This review synthesizes current knowledge on the molecular mechanisms underlying neurodevelopment regulated by Cullin-RING ubiquitin ligases—the largest subclass of ubiquitin ligases—and their involvement in the pathophysiology of neurodevelopmental disorders. A deeper understanding of these mechanisms holds significant promise for informing novel therapeutic strategies, ultimately advancing clinical outcomes for individuals affected by neurodevelopmental disorders.
Keywords:
Cullin-Ring Ubiquitin Ligase
; Neurodevelopment
; Neurodevelopmental disorders
1. Introduction
Ubiquitination is dynamic post-translational modifications that play critical roles in maintaining cellular function and homeostasis by modulating protein stability, activity, and localization [1,2]. Ubiquitination refers to the covalent attachment of ubiquitin molecules to a substrate protein. This process is carried out through a sequential enzymatic cascade involving three key enzymes. First, E1 ubiquitin-activating enzyme activates ubiquitin in an ATP-dependent manner, forming a high-energy thioester bond. Next, the activated ubiquitin is transferred to E2 ubiquitin-conjugating enzyme. Finally, E3 ubiquitin ligase facilitates the transfer of ubiquitin from the E2 enzyme to the target protein, determining the substrate specificity [3]. The transfer of ubiquitin to the substrate is orchestrated by either a RING (really interesting new gene) finger domain, a HECT (homologous to the E6-AP carboxyl terminus) domain, or a RBR (RING-between-RING) domain, leading to the classification of ubiquitin ligases into RING-type, HECT-type or BRB-type based on the domain they possess [4,5]. Ubiquitin ligases function as either single-protein entities which contain both E2- and substrate-binding domains, or multi-subunit complexes which harbor a protein responsible for binding E2, and a substrate receptor protein.
The largest subclass of ubiquitin ligases is Cullin-RING ubiquitin ligase (CRL) in which Cullin protein serves as a scaffold to bind both RING domain protein ROC1/2 (also called RBX1/2), and substrate receptor protein directly or indirectly through adaptor protein [6,7]. Mammalian cells contain eight (CUL1, 2, 3, 4A, 4B, 5, 7, 9) cullin proteins, each of which builds their specific ubiquitin ligase complexes[6,7]. For example, CUL1-based ubiquitin ligase uses F-box proteins as a substrate receptor which is linked to CUL1 via SKP1 adaptor protein [8], while CUL4A or CUL4B employs DDB1 adaptor protein which recruits DCAF substrate receptor proteins [9,10] (Figure 1). In contrast to CUL1, CUL2, CUL3, CUL4A, CUL4B, and CUL5, recent structural investigations have demonstrated that CUL7 and CUL9 directly interact with their substrates, with the ubiquitylation module being supplied by CUL1 in the case of CUL7 [11]. CUL9, on the other hand, employs its intrinsic RBR domain to help catalyze ubiquitylation [12]. All Cullin proteins are thought to use ROC1 as a RING domain protein, except for CUL5 which uses ROC2 [13] (Figure 1). In addition to these Cullin-based complexes, APC/C (anaphase promoting complex/cyclosome) ubiquitin ligase constitutes a similar structure in which APC2 serves as a main scaffold, APC11 as a RING domain protein, and CDC20 or CDH1 as substrate receptor [14] (Figure 1). Due to this structural similarity and its significant functions in neurodevelopment [15], we include and discuss APC/C ubiquitin ligase in this review.
In neurodevelopment, ubiquitination has been demonstrated to play a significant role in neurodevelopmental processes such as neural stem cell proliferation/differentiation, axonal/dendritic growth, migration, synaptogenesis, and synaptic function (Figure 2). Thus, dysregulation of these processes has been linked to several neurodevelopmental disorders (NDDs), including autism spectrum disorder (ASD), intellectual disability (ID), and attention-deficit/hyperactivity disorder (ADHD). Focusing on CRLs, this review aims to explore the role of ubiquitination in neurodevelopment and their implications for NDDs, providing insights into their potential as therapeutic targets.
2. Human Genetics of CRL in NDD Patients
Exome sequencing and chromosomal microarray testing of NDD patients identified de novo heterozygous loss-of-function mutations in CUL3 on chromosome 2 [16,17], homozygous or compound heterozygous loss-of-function mutations in CUL7 on chromosome 6, and hemizygous loss-of-function mutations in CUL4B on X chromosome [18,19] in NDD patients. Furthermore, variants of genes encoding CRL1 substrate receptors FBXO10 [20], FBXO11[21,22], FBXO28 [22,23], FBXO31 [24], FBXO47 [25], FBXL3 [26], FBXL4 [27,28], FBXL10 (also known as KDM2B) [29], β-TrCP1 (also known as FBXW1) [30,31,32], β-TrCP2 (also known as FBXW11) [31], FBXW7 [33,34,35], as well as CRL3 substrate receptors KLHL15, KLHL17 (also known as actinfilin) [36], KLHL20 [37], KCTD7 [38], KCTD13 [39], and CRL4 substrate receptors DCAF1 (also known as VprBP) [40], DCAF14 (also known as PHIP, BRWD2 and RepID) [41,42], COP1 [43], and CRBN [44,45], and APC/C substrate receptor CDH1 [46] have been implicated in NDD patients. These findings suggest the involvement of these genes in NDDs, thereby prompting further research to elucidate how these mutations contribute to the pathogenesis of NDDs.
3. Behavioral Phenotypes of Mice with CRL Mutations
Mouse models have been developed to investigate the mechanisms underlying the pathogenesis of NDDs [47,48,49]. To assess the suitability of these models for NDD research, various behavioral evaluations have been designed and conducted [50,51]. For instance, communication deficits are quantified through open field and three-chamber social interaction tests, while impairments in learning and memory are assessed using a range of maze and contextual fear conditioning paradigms [50,51]. Additionally, increased distance traveled serves as an indicator of hyperactivity, and anxiety is primarily evaluated through open field and elevated plus-maze tests [50,51]. Several CRL mutant mouse models have been shown to exhibit these NDD-associated behaviors, as summarized in Table 1. These findings suggest that mutations in these genes are most likely causal, rather than merely correlative, in the etiology of NDDs.
4. CRLs in Neural Stem Cell Proliferation and Differentiation
In the following sections, we will provide an overview of the current understanding of the molecular mechanisms underlying neurodevelopment and NDDs, as derived from studies on CRLs.
Neural stem cells (NSCs) are multipotent progenitor cells that give rise to neurons, astrocytes, and oligodendrocytes during brain development [62,63]. NSCs first give rise to radial glial cells (RGCs), which serve dual roles as neural progenitors and as scaffolds that guide migrating neurons. RGCs divide asymmetrically, producing either neurons directly or intermediate progenitors (IPs) as an intermediary step in neurogenesis. IPs undergo a limited number of divisions, amplifying the production of neurons to meet developmental demands. Mature neurons are generated from the differentiation of IPs or directly from RGCs in a highly regulated process. In the early stages of development, RGCs primarily produce neurons directly; however, as development progresses, they increasingly generate IPs to enhance neuronal numbers. This sequential progression from NSCs to RGCs, then to IPs, and finally to mature neurons ensures the proper formation and organization of the nervous system. By maintaining the progenitor pool, NSCs and RGCs regulate the balance between self-renewal and differentiation, while IPs contribute to amplifying neurogenesis without depleting the pool of progenitors. This hierarchical relationship is essential for the precise development of neural structures and circuits [64,65,66]. The tightly regulated processes of NSC proliferation and differentiation are fundamental for proper neurodevelopment and rely heavily on the dynamic control of protein expression and degradation. CRLs play a pivotal role in these processes by regulating key signaling pathways and transcription factors (Figure 3).
NSC proliferation is governed by the precise regulation of cell cycle progression, which is controlled by cyclins, cyclin-dependent kinases (CDKs), and their inhibitors. Ubiquitination ensures the timely degradation of these cell cycle regulators, thus maintaining proper cell cycle transitions. For example, APC/C ubiquitin ligase recognizes mitotic regulators such as cyclin B through CDH1 substrate receptor (APC/CCDH1), and targets them for ubiquitination, promoting the progression of mitosis [67,68]. In Cdh1-deficient neural stem cells, aberrantly elevated cyclin B-associated CDK activity induces DNA damage-mediated apoptotic cell death [68], potentially by disrupting the precise regulation of S phase timing, thereby eliciting DNA replication stress [67]. Another key regulator is the CUL1-based ubiquitin ligase complex (CRL1, also known as SKP1-CUL1-F-box SCF ubiquitin ligase), which ubiquitinates cyclin E for degradation with the use of FBXW7 substrate receptor (CRL1FBXW7). ASPM suppresses this SCFFBXW7-mediated ubiquitination, thereby stabilizing cyclin E. This stabilization facilitates the shortening of the G1 phase in the cell cycle, promoting NSC proliferation [69].
NSC differentiation requires the fine-tuned suppression of self-renewal programs and the activation of lineage-specific transcriptional networks. Ubiquitination modulates this process by targeting signaling pathway components and transcription factors that drive differentiation. For instance, the Notch signaling pathway, which maintains NSC and RGC self-renewal, is also regulated by the ubiquitin ligase CRL1FBXW7, which ubiquitinates and targets Notch intracellular domain (NICD) for proteasomal degradation [70]. This degradation facilitates the transition from NSC maintenance to astrocytic differentiation [70]. APC/C also plays a critical role in NSC differentiation. APC/CCDH1 ubiquitinates and facilitates the degradation of the CDK activator CDC25A and SKP2, a substrate receptor of SCF ubiquitin ligase that targets CDK inhibitors for degradation [68]. This process prevents cell cycle re-entry, thereby promoting neuronal differentiation [68]. Similarly, the Wnt/β-catenin signaling pathway, a critical regulator of NSC maintenance, is modulated by ubiquitination. The E3 ligase CRL1β-TrCP ubiquitinates β-catenin, leading to its degradation and attenuation of Wnt signaling [71,72].
In addition to cell cycle and signaling pathways, ubiquitination directly regulates transcription factors critical for NSC identity and differentiation. SOX2, a core transcription factor that maintains NSC pluripotency, is subject to ubiquitination. In human pluripotent stem cell-derived NSC, CUL4A-based Cullin-RING ubiquitin ligase with DET1-COP1 heterodimer as a substrate receptor (CRL4ADET1-COP1) ubiquitinates SOX2 for degradation, leading to neural differentiation [73].
Dysregulation of ubiquitination in NSC proliferation and differentiation has profound implications for NDDs. Mutations in genes encoding CRL-related proteins (please refer to section 2) and FBXW7 regulator ASPM [74,75] are identified in NDD patients and these could be caused by aberrant NSC proliferation of differentiation.
5. CRLs in Neuronal Polarization
Immature neurons extend processes known as neurites, which subsequently differentiate into axon, the intracellular signal-transmitting structure, or dendrites, the primary signal-receiving components of neurons. The developmental process that defines this distinction is known as neuronal polarization, signifying the establishment of distinct subcellular compartments to enable precise signal transmission and reception [76]. Neuronal polarization is initiated by extracellular symmetry-breaking cues, followed by cytoskeletal rearrangements [77], with actin filaments (F-actin) guiding axon/dendrite extension and microtubules playing an essential role in axon stabilization [78,79]. By modulating protein functions, CRLs ensure the spatial and temporal coordination of neuronal polarization (Figure 4).
The protein kinase AKT facilitates axon formation by phosphorylating GSK-3β to negate its function, leading to the activation of microtubule-binding proteins and subsequent stabilization of microtubules [80]. AKT activity is negatively regulated by PTEN [81], necessitating PTEN inactivation within the axon. Notably, the HECT-type ubiquitin ligase NEDD4 is reported to facilitate PTEN degradation through ubiquitination, thereby promoting neurite outgrowth [82] and axon branching [83]. However, the decrease in ubiquitination and accumulation of PTEN were not detected in NEDD4-deficient neurons [84], indicating that the E3 ubiquitin ligase for PTEN in developing axon awaits to be revealed. Although not in neuronal cells, CRL4BDCAF13 [85,86] and SCFFBXO22 [87] are reported to ubiquitinate PTEN for degradation, providing the possibility that Cullin-RING ubiquitin ligases are involved in axon formation through PTEN regulation.
The activity of a small G protein RhoA is critical for dendrite formation through promoting actin arc formation, which prevents microtubule protrusion and axon growth [88]. RhoA is stabilized in dendrites by the ubiquitin ligase APC/CCDH1 that ubiquitinates the RhoA targeting HECT-type ubiquitin ligase SMURF1 for degradation [89]. In the axon, the activity of RhoA is downregulated by a mechanism in which RhoA activator PDZ-RhoGEF is targeted for degradation by the ubiquitin ligase CUL3 with KLHL20 serving as a substrate receptor (CRL3KLHL20) [90]. A symmetry-breaking signaling by BDNF induces phosphorylation of PDZ-RhoGEF, potentiating its ubiquitination and subsequent degradation [90], facilitating the neuronal polarization.
The interplay between F-actin and microtubules, mediated by coupling proteins, is also implicated in axon and dendrite formation [91]. One such coupling protein, DCX, is subject to negative regulation by CRL3KLHL15 [92], CRL4ACRBN [93], and CRL4BCRBN [93]. The ubiquitination and subsequent proteasomal degradation of DCX by these CRLs have been shown to attenuate axonal and dendritic complexity and length [92,93].
An additional regulatory layer in the process of neuronal polarization is provided by transcriptional mechanisms, whose activities are also modulated by ubiquitination. For instance, APC/CCDH1 facilitates the ubiquitination of SnoN [94], a transcription factor that promotes the expression of positive regulators of axon growth, and ID2 [95], an inhibitor of axon growth repressors, thereby suppressing axon extension. We have also demonstrated that the ubiquitin ligase CUL4B regulates NGF-induced neurite extension via transcriptional modulation [96,97]. CUL4B mediates the ubiquitination and subsequent degradation of WDR5 [96], a key component of the histone H3 lysine 4 methyltransferase transcriptional complex [98], in rat neuroblastoma PC12 cells, downregulating the neuronal gene expression [99]. These findings underscore the critical role of transcriptional regulation in neuronal process formation.
Dysregulation of ubiquitination in neuronal polarization has also profound implications for NDDs. Mutations in genes encoding CRL-related proteins (please refer to section 2), as well as DCX [100,101], SMURF1 [36,102], and PTEN [103,104] are identified in NDD patients and these could be caused by aberrant neuronal polarization.
6. CRLs in Neuronal Migration
NSCs are generated and remain adjacent to the brain ventricle, while differentiating neurons migrate towards the cortical surface, with later-differentiated neurons passing by earlier-generated neurons during corticogenesis [66,105]. This inside-out layering of the cortex is guided by various mechanisms, some of which are regulated by CRLs (Figure 5).
The most well-characterized regulator of neuronal migration is Reelin/DAB1 signaling [106]. Reelin binds to its receptors and activates tyrosine kinases, leading to the phosphorylation of DAB1 [107]. Phosphorylated DAB1 then interacts with several proteins, including LIS1, a regulator of microtubule-based dynein-dynactin motor proteins, which facilitates cytoskeletal reorganization essential for migration [108]. CUL5 with SOCS proteins as substrate receptors (CRL5SOCS) targets phosphorylated DAB1 for ubiquitination and degradation, thus preventing DAB1 hyperactivation [109]. CUL5 knockdown-induced accumulation of phosphorylated DAB1 results in excessive migration and abnormal superficial positioning [109]. Furthermore, CUL4B was shown to bind to LIS1 [110], suggesting that CRL4B may also play a role in regulating neuronal migration through Reelin signaling.
In contrast, heterozygous deletion of Cul3, a condition that mirrors NDD in humans, leads to impaired migration of embryonic neurons and cortical lamination abnormalities in mice [53]. Proteomic analysis has identified PLS3, an actin-bundling protein, as a critical substrate of CUL3, and the accumulation of PLS3 caused by Cul3 haploinsufficiency disrupts actin filament organization and results in abnormal adhesion with impaired migration [53]. The substrate receptor responsible for PLS3 recognition remains unidentified.
The generation of the cerebellum and hippocampus also depends on proper neuronal migration [111,112]. FBXO41, a neuron-specific substrate receptor of CRL1 ubiquitin ligase, has been shown to promote neuronal migration in the cerebellum (from the external to the internal granule layer) [113], and hippocampus (from the hilus to the granule cell layer) [114]. It localizes to the centrosome and disassembles primary cilia, an antenna-like structure that receives several signaling ligands [115]. However, the involvement of primary cilia and the substrates of SCFFBXO41 in neuronal migration remain unclear.
7. CRLs in Synaptogenesis and Synaptic Function
Elongating axons extend towards the dendrites of target neurons, where synapses are established [120,121]. At the presynaptic terminal, neurons secrete neurotransmitters such as glutamate, dopamine, acetylcholine (ACh), and gamma-aminobutyric acid (GABA), which bind to their corresponding receptors in the specialized postsynaptic density (PSD) region of the postsynaptic neurons [122], thereby enabling communication and the transmission of information. CLRs play a significant role in synaptogenesis and synaptic function (Figure 6).
Presynaptic development is transcriptionally regulated by APC/CCDC20, in which the ubiquitination and degradation of the transcription factor NeuroD2 leads to the downregulation of Complexin II expression, a negative regulator of presynaptic differentiation, thus promoting the formation of presynaptic structures [123]. Both the presynaptic active zones and PSDs are supported by scaffolding proteins, the abundance of which is frequently regulated by ubiquitin ligases [124]. For example, FBXL20 (also known as SCRAPPER), a distinctive F-box protein characterized by a C-terminal CAXX motif essential for membrane localization, localizes at the presynaptic terminal, where it targets RIM1, a Ca²⁺-sensing synaptic vesicle regulator, for ubiquitination and degradation, ensuring the proper release of glutamate [125].
Glutamate stimulates postsynaptic neurons by activating four types of ion channels -AMPA, kainate, NMDA, and GluD receptors- [126], and the G-protein-coupled metabotropic receptor (mGluR) [127]. Chronic synaptic activity triggers the degradation of the AMPA receptor, a mechanism critical for preventing toxic hyperactivation of postsynaptic neurons, known as excitotoxicity. APC/CCDH1 plays a pivotal role in the downregulation of the AMPA receptor by targeting the GluA1 (also known as GluR1) subunit for ubiquitination and degradation in response to synaptic stimuli [128]. Periodic synaptic activation induces long-term potentiation (LTP) or long-term depression (LTD) mediated through glutamate receptors, which are the foundations of synaptic plasticity essential for learning and memory and processes frequently dysregulated in NDD patients [129]. APC/CCDH1 is involved in mGluR-mediated LTD by synaptic stimulus-dependent ubiquitination of FMRP1, a negative regulator of LTD, leading to its degradation [130]. Accordingly, loss of CDH1 in excitatory neurons of the mouse brain impairs LTD, resulting in sustained cell surface expression of the AMPA receptor and defective degradation of FMRP1 [130]. The levels of the NMDA receptor are also regulated by CRL3KCTD13 which targets GluN1 for ubiquitination and degradation [131]. In patients with epileptic seizures, the expression of KCTD13 is diminished, likely leading to heightened activation of excitatory synaptic transmission and an increased susceptibility to epilepsy [131].
Dysfunction of the dopamine system is implicated in the phenotypes associated with NDDs [132,133]. Consistently, mice with a heterozygous Cul3 deletion in DA neurons exhibit hyperactivity of DA neurons accompanied by the behavioral abnormalities such as increased locomotion, impaired working memory, and deficits in sensorimotor gating (please refer to Table 1), all of which were reversed by the forced inactivation of DA neuron [55]. The hyperexcitability of DA neurons was shown to be caused by the accumulation of HCN2 channels, a substrate of CUL3 [55]. These findings confirm the critical role of elevated DA activity in NDDs.
Potassium channels are essential for maintaining neuronal activity by regulating membrane potential [134]. The potassium channel Kv10.1 (also known as EAG1 or KCNH1), whose gene is mutated in NDDs [135,136], undergoes ubiquitination-mediated degradation, a process driven by CRL7FBXW8 [137]. Consequently, overexpression of CUL7 diminishes potassium currents, whereas reduced CUL7 expression leads to enhanced potassium currents in non-neuronal cells that exogenously express Kv10.1 [137]. The extent to which these findings can be extrapolated to neuronal function requires further investigation.
The postsynaptic sites in dendrites often form specialized protruded structures known as spines [138]. KLHL17 has been shown to enlarge spines and facilitate synaptic activity. Since this function requires the BTB domain, which is essential for association with CUL3, CRL3KLHL17 likely contributes to this process, although the specific substrate remains to be identified [57]. Dendrites are dynamic structures that must be maintained to ensure the proper functioning of the brain. One destabilizing factor of dendrites is ROCK2, whose levels are regulated by APC/CCDH1 [61]. Therefore, loss of CDH1 leads to the accumulation of ROCK2, resulting in dendritic spine disruption and learning deficits, which can be rescued by a ROCK inhibitor [61].
8. Therapeutic Implications and Future Directions
The critical roles of Cullin-RING ubiquitin ligases (CRLs) in neurodevelopment and their involvement in NDDs, as summarized in Table 2, highlight the potential for therapeutic targeting. Given that mutations in genes encoding CRL-related proteins have been identified in NDD patients (as discussed in Section 2), modulating ubiquitination pathways represents a promising approach to mitigate disease phenotypes. One potential therapeutic strategy involves the use of small-molecule inhibitors to regulate CRL activity. For instance, MLN4924, a NEDD8-activating enzyme (NAE) inhibitor disrupts all CRL functions, leading to the stabilization of CRL substrates [139]. Given the role of CRLs in neuronal differentiation and synaptic function, controlled inhibition of specific CRLs may hold therapeutic potential for certain NDDs. However, systemic CRL inhibition may lead to broad cellular dysfunction, necessitating the development of highly selective inhibitors targeting specific CRL complexes or substrate receptors.
Gene-editing technologies, including CRISPR/Cas9 and base editing, provide potential avenues for correcting pathogenic mutations in CRL-associated genes. For example, CRISPR-mediated correction of loss-of-function mutations could restore normal ubiquitination processes, thereby ameliorating NDD-related phenotypes. Additionally, targeted modulation of gene expression using CRISPR activation (CRISPRa) or inhibition (CRISPRi) may help fine-tune the activity of specific CRL components without permanent genomic alterations [140]. A particularly intriguing target is the CRL1FBXW7 and APC/CCDH1 complexes, the best characterized CRLs in relation to tumorigenesis, which also regulates key neurodevelopmental factors. Pharmacological stabilization of FBXW7 or its substrates may provide therapeutic avenues for NDDs characterized by disrupted Notch or Wnt signaling. Modulating APC/CCDH1 activity could influence synaptic function and plasticity, with potential implications for learning and memory disorders [130].
Not all of the CRL-related proteins discussed in this review have been identified as mutated, but the increasing sample sizes may reveal mutations in these genes. In relation, patient-specific genetic and molecular profiling can guide therapeutic interventions for NDDs. Whole-genome, transcriptome, and proteomic analyses could aid in identifying individual CRL-related mutations, enabling the selection of personalized therapeutic strategies. In addition, patient-derived induced pluripotent stem cells (iPSCs) offer an invaluable platform for modeling disease mechanisms and testing novel therapeutics in a personalized context [141].
Despite significant progress in understanding the role of CRLs in neurodevelopment, several key questions remain unanswered. Future research should focus on (1) identifying substrate specificity: Many CRL substrate receptors remain uncharacterized. Understanding their target specificity is essential for developing selective therapeutic interventions; (2) developing selective CRL modulators: current pharmacological tools lack specificity for individual CRL complexes, though CUL-level inhibitors are emerging, as exemplified by DI-1548 and DI-1859 for CUL3 [142], and 33-11 and KH-4-43 for CUL4 [143]. Advances in structure-based drug design may enable the development of highly selective CRL inhibitors or activators; (3) exploring non-degradative ubiquitination: while ubiquitination is often associated with protein degradation, non-degradative ubiquitin modifications also play critical roles in signaling pathways. Investigating these roles could reveal novel therapeutic targets.
9. Concluding Remarks
The ubiquitination pathway, particularly through the action of CRLs, plays a fundamental role in neurodevelopment and the pathophysiology of NDDs. Advances in molecular and pharmacological approaches have provided promising avenues for targeting ubiquitin signaling in therapeutic contexts. However, further research is needed to refine these strategies, ensuring both efficacy and safety in clinical applications. As our understanding of ubiquitin-mediated neurodevelopmental regulation continues to expand, new opportunities for treating NDDs are likely to emerge, bringing us closer to precision medicine-based interventions for these complex disorders.
Author Contributions
Conceptualization, T.N. and M.N.; investigation, H.A., T.N., M.N.; writing—original draft preparation, H.A. and T.N.; writing—review and editing, M.N. and T.H.; supervision, T.N.; project administration, T.N.; funding acquisition, T.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by KAKENHI (grant number: 23K06367).
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
The authors acknowledge Keiichi I. Nakayama, Keiko Nakayama, and Yue Xiong for guiding and supporting for our research on CRLs. We would like to also thank lab members for the productive discussions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Glickman, M.H. and A. Ciechanover, The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev, 2002. 82(2): p. 373-428. [CrossRef]
- Chen, Z.J. and L.J. Sun, Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell, 2009. 33(3): p. 275-86. [CrossRef]
- Hershko, A. and A. Ciechanover, The ubiquitin system. Annu Rev Biochem, 1998. 67: p. 425-79.
- Metzger, M.B., V.A. Hristova, and A.M. Weissman, HECT and RING finger families of E3 ubiquitin ligases at a glance. J Cell Sci, 2012. 125(Pt 3): p. 531-7. [CrossRef]
- Uchida, C. and M. Kitagawa, RING-, HECT-, and RBR-type E3 Ubiquitin Ligases: Involvement in Human Cancer. Curr Cancer Drug Targets, 2016. 16(2): p. 157-74.
- Petroski, M.D. and R.J. Deshaies, Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol, 2005. 6(1): p. 9-20.
- Harper, J.W. and B.A. Schulman, Cullin-RING Ubiquitin Ligase Regulatory Circuits: A Quarter Century Beyond the F-Box Hypothesis. Annu Rev Biochem, 2021. 90: p. 403-429.
- Skaar, J.R., J.K. Pagan, and M. Pagano, Mechanisms and function of substrate recruitment by F-box proteins. Nat Rev Mol Cell Biol, 2013. 14(6): p. 369-81.
- Jackson, S. and Y. Xiong, CRL4s: the CUL4-RING E3 ubiquitin ligases. Trends Biochem Sci, 2009. 34(11): p. 562-70.
- Nakagawa, M. and T. Nakagawa, CUL4-Based Ubiquitin Ligases in Chromatin Regulation: An Evolutionary Perspective. Cells, 2025. 14(2). [CrossRef]
- Hopf, L.V.M., et al., Structure of CRL7(FBXW8) reveals coupling with CUL1-RBX1/ROC1 for multi-cullin-RING E3-catalyzed ubiquitin ligation. Nat Struct Mol Biol, 2022. 29(9): p. 854-862.
- Horn-Ghetko, D., et al., Noncanonical assembly, neddylation and chimeric cullin-RING/RBR ubiquitylation by the 1.8 MDa CUL9 E3 ligase complex. Nat Struct Mol Biol, 2024. 31(7): p. 1083-1094.
- Kamura, T., et al., VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev, 2004. 18(24): p. 3055-65. [CrossRef]
- Yamano, H., APC/C: current understanding and future perspectives. F1000Res, 2019. 8.
- Fuchsberger, T., A. Lloret, and J. Viña, New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer's Disease. Int J Mol Sci, 2017. 18(5).
- O'Roak, B.J., et al., Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science, 2012. 338(6114): p. 1619-22.
- Sanders, S.J., et al., Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron, 2015. 87(6): p. 1215-1233.
- Tarpey, P.S., et al., Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet, 2007. 80(2): p. 345-52. [CrossRef]
- Zou, Y., et al., Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet, 2007. 80(3): p. 561-6.
- O'Roak, B.J., et al., Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 2012. 485(7397): p. 246-50.
- Gregor, A., et al., De Novo Variants in the F-Box Protein FBXO11 in 20 Individuals with a Variable Neurodevelopmental Disorder. Am J Hum Genet, 2018. 103(2): p. 305-316.
- Gregor, A., et al., De novo missense variants in FBXO11 alter its protein expression and subcellular localization. Hum Mol Genet, 2022. 31(3): p. 440-454.
- Schneider, A.L., et al., FBXO28 causes developmental and epileptic encephalopathy with profound intellectual disability. Epilepsia, 2021. 62(1): p. e13-e21.
- Mir, A., et al., Truncation of the E3 ubiquitin ligase component FBXO31 causes non-syndromic autosomal recessive intellectual disability in a Pakistani family. Hum Genet, 2014. 133(8): p. 975-84.
- Harripaul, R., et al., Mapping autosomal recessive intellectual disability: combined microarray and exome sequencing identifies 26 novel candidate genes in 192 consanguineous families. Mol Psychiatry, 2018. 23(4): p. 973-984.
- Ansar, M., et al., Biallelic variants in FBXL3 cause intellectual disability, delayed motor development and short stature. Hum Mol Genet, 2019. 28(6): p. 972-979. [CrossRef]
- Bonnen, P.E., et al., Mutations in FBXL4 cause mitochondrial encephalopathy and a disorder of mitochondrial DNA maintenance. Am J Hum Genet, 2013. 93(3): p. 471-81.
- Gai, X., et al., Mutations in FBXL4, encoding a mitochondrial protein, cause early-onset mitochondrial encephalomyopathy. Am J Hum Genet, 2013. 93(3): p. 482-95.
- Charng, W.L., et al., Exome sequencing in mostly consanguineous Arab families with neurologic disease provides a high potential molecular diagnosis rate. BMC Med Genomics, 2016. 9(1): p. 42.
- Ruzzo, E.K., et al., Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell, 2019. 178(4): p. 850-866.e26.
- Holt, R.J., et al., De Novo Missense Variants in FBXW11 Cause Diverse Developmental Phenotypes Including Brain, Eye, and Digit Anomalies. Am J Hum Genet, 2019. 105(3): p. 640-657.
- Chau, K.K., et al., Full-length isoform transcriptome of the developing human brain provides further insights into autism. Cell Rep, 2021. 36(9): p. 109631.
- Stephenson, S.E.M., et al., Germline variants in tumor suppressor FBXW7 lead to impaired ubiquitination and a neurodevelopmental syndrome. Am J Hum Genet, 2022. 109(4): p. 601-617.
- Meier-Abt, F., et al., Further evidence that the neurodevelopmental gene FBXW7 predisposes to Wilms tumor. Am J Med Genet A, 2024. 194(6): p. e63528.
- Wang, Y., et al., Case report: A novel FBXW7 gene variant causes global developmental delay. Front Genet, 2024. 15: p. 1436462.
- De Rubeis, S., et al., Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 2014. 515(7526): p. 209-15.
- Sleyp, Y., et al., De novo missense variants in the E3 ubiquitin ligase adaptor KLHL20 cause a developmental disorder with intellectual disability, epilepsy, and autism spectrum disorder. Genet Med, 2022. 24(12): p. 2464-2474. [CrossRef]
- Mastrangelo, M., et al., Progressive myoclonus epilepsy and ceroidolipofuscinosis 14: The multifaceted phenotypic spectrum of KCTD7-related disorders. Eur J Med Genet, 2019. 62(12): p. 103591.
- Golzio, C., et al., KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature, 2012. 485(7398): p. 363-7.
- Clothier, J.L., et al., Identification of DCAF1 by Clinical Exome Sequencing and Methylation Analysis as a Candidate Gene for Autism and Intellectual Disability: A Case Report. J Pers Med, 2022. 12(6).
- Webster, E., et al., De novo PHIP-predicted deleterious variants are associated with developmental delay, intellectual disability, obesity, and dysmorphic features. Cold Spring Harb Mol Case Stud, 2016. 2(6): p. a001172.
- Jansen, S., et al., A genotype-first approach identifies an intellectual disability-overweight syndrome caused by PHIP haploinsufficiency. Eur J Hum Genet, 2018. 26(1): p. 54-63.
- Glessner, J.T., et al., Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature, 2009. 459(7246): p. 569-73.
- Higgins, J.J., et al., A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology, 2004. 63(10): p. 1927-31.
- Sheereen, A., et al., A missense mutation in the CRBN gene that segregates with intellectual disability and self-mutilating behaviour in a consanguineous Saudi family. J Med Genet, 2017. 54(4): p. 236-240.
- Rodríguez, C., et al., A novel human Cdh1 mutation impairs anaphase promoting complex/cyclosome activity resulting in microcephaly, psychomotor retardation, and epilepsy. J Neurochem, 2019. 151(1): p. 103-115. [CrossRef]
- Gonzalez-Sulser, A., Rodent genetic models of neurodevelopmental disorders and epilepsy. Eur J Paediatr Neurol, 2020. 24: p. 66-69.
- Silverman, J.L., et al., Reconsidering animal models used to study autism spectrum disorder: Current state and optimizing future. Genes Brain Behav, 2022. 21(5): p. e12803.
- Damianidou, E., L. Mouratidou, and C. Kyrousi, Research models of neurodevelopmental disorders: The right model in the right place. Front Neurosci, 2022. 16: p. 1031075.
- Takao, K., N. Yamasaki, and T. Miyakawa, Impact of brain-behavior phenotypying of genetically-engineered mice on research of neuropsychiatric disorders. Neurosci Res, 2007. 58(2): p. 124-32.
- Huang, L., et al., Behavioral tests for evaluating the characteristics of brain diseases in rodent models: Optimal choices for improved outcomes (Review). Mol Med Rep, 2022. 25(5).
- Amar, M., et al., Autism-linked Cullin3 germline haploinsufficiency impacts cytoskeletal dynamics and cortical neurogenesis through RhoA signaling. Mol Psychiatry, 2021. 26(7): p. 3586-3613.
- Morandell, J., et al., Cul3 regulates cytoskeleton protein homeostasis and cell migration during a critical window of brain development. Nat Commun, 2021. 12(1): p. 3058.
- Dong, Z., et al., CUL3 Deficiency Causes Social Deficits and Anxiety-like Behaviors by Impairing Excitation-Inhibition Balance through the Promotion of Cap-Dependent Translation. Neuron, 2020. 105(3): p. 475-490.e6.
- Gao, N., et al., Deficiency of Cullin 3, a Protein Encoded by a Schizophrenia and Autism Risk Gene, Impairs Behaviors by Enhancing the Excitability of Ventral Tegmental Area (VTA) DA Neurons. J Neurosci, 2023. 43(36): p. 6249-6267.
- Chen, C.Y., et al., Rescue of the genetically engineered Cul4b mutant mouse as a potential model for human X-linked mental retardation. Hum Mol Genet, 2012. 21(19): p. 4270-85.
- Hu, H.T., T.N. Huang, and Y.P. Hsueh, KLHL17/Actinfilin, a brain-specific gene associated with infantile spasms and autism, regulates dendritic spine enlargement. J Biomed Sci, 2020. 27(1): p. 103. [CrossRef]
- Arbogast, T., et al., Kctd13-deficient mice display short-term memory impairment and sex-dependent genetic interactions. Hum Mol Genet, 2019. 28(9): p. 1474-1486.
- Rajadhyaksha, A.M., et al., Behavioral characterization of cereblon forebrain-specific conditional null mice: a model for human non-syndromic intellectual disability. Behav Brain Res, 2012. 226(2): p. 428-34.
- Li, M., et al., The adaptor protein of the anaphase promoting complex Cdh1 is essential in maintaining replicative lifespan and in learning and memory. Nat Cell Biol, 2008. 10(9): p. 1083-9.
- Bobo-Jiménez, V., et al., APC/C(Cdh1)-Rock2 pathway controls dendritic integrity and memory. Proc Natl Acad Sci U S A, 2017. 114(17): p. 4513-4518.
- Navarro Negredo, P., R.W. Yeo, and A. Brunet, Aging and Rejuvenation of Neural Stem Cells and Their Niches. Cell Stem Cell, 2020. 27(2): p. 202-223.
- Alonso, M., A.C. Petit, and P.M. Lledo, The impact of adult neurogenesis on affective functions: of mice and men. Mol Psychiatry, 2024. 29(8): p. 2527-2542.
- Florio, M. and W.B. Huttner, Neural progenitors, neurogenesis and the evolution of the neocortex. Development, 2014. 141(11): p. 2182-94.
- Taverna, E., M. Götz, and W.B. Huttner, The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol, 2014. 30: p. 465-502.
- Zhou, Y., H. Song, and G.L. Ming, Genetics of human brain development. Nat Rev Genet, 2024. 25(1): p. 26-45.
- Pines, J., Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol, 2011. 12(7): p. 427-38.
- Delgado-Esteban, M., et al., APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat Commun, 2013. 4: p. 2879. [CrossRef]
- Capecchi, M.R. and A. Pozner, ASPM regulates symmetric stem cell division by tuning Cyclin E ubiquitination. Nat Commun, 2015. 6: p. 8763.
- Matsumoto, A., et al., Fbxw7-dependent degradation of Notch is required for control of "stemness" and neuronal-glial differentiation in neural stem cells. J Biol Chem, 2011. 286(15): p. 13754-64.
- MacDonald, B.T., K. Tamai, and X. He, Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell, 2009. 17(1): p. 9-26.
- Liu, J., et al., Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct Target Ther, 2022. 7(1): p. 3.
- Cui, C.P., et al., Dynamic ubiquitylation of Sox2 regulates proteostasis and governs neural progenitor cell differentiation. Nat Commun, 2018. 9(1): p. 4648.
- Bond, J., et al., Protein-truncating mutations in ASPM cause variable reduction in brain size. Am J Hum Genet, 2003. 73(5): p. 1170-7.
- Létard, P., et al., Autosomal recessive primary microcephaly due to ASPM mutations: An update. Hum Mutat, 2018. 39(3): p. 319-332.
- Arimura, N. and K. Kaibuchi, Neuronal polarity: from extracellular signals to intracellular mechanisms. Nat Rev Neurosci, 2007. 8(3): p. 194-205.
- Yogev, S. and K. Shen, Establishing Neuronal Polarity with Environmental and Intrinsic Mechanisms. Neuron, 2017. 96(3): p. 638-650. [CrossRef]
- Jung, M., D. Kim, and J.Y. Mun, Direct Visualization of Actin Filaments and Actin-Binding Proteins in Neuronal Cells. Front Cell Dev Biol, 2020. 8: p. 588556.
- Meka, D.P., et al., Centrosome-dependent microtubule modifications set the conditions for axon formation. Cell Rep, 2022. 39(3): p. 110686.
- Yoshimura, T., N. Arimura, and K. Kaibuchi, Signaling networks in neuronal polarization. J Neurosci, 2006. 26(42): p. 10626-30.
- Lee, Y.R., M. Chen, and P.P. Pandolfi, The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol, 2018. 19(9): p. 547-562.
- Christie, K.J., J.A. Martinez, and D.W. Zochodne, Disruption of E3 ligase NEDD4 in peripheral neurons interrupts axon outgrowth: Linkage to PTEN. Mol Cell Neurosci, 2012. 50(2): p. 179-92.
- Drinjakovic, J., et al., E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron, 2010. 65(3): p. 341-57.
- Hsia, H.E., et al., Ubiquitin E3 ligase Nedd4-1 acts as a downstream target of PI3K/PTEN-mTORC1 signaling to promote neurite growth. Proc Natl Acad Sci U S A, 2014. 111(36): p. 13205-10.
- Chen, Z., et al., MicroRNA-300 Regulates the Ubiquitination of PTEN through the CRL4B(DCAF13) E3 Ligase in Osteosarcoma Cells. Mol Ther Nucleic Acids, 2018. 10: p. 254-268.
- Zhang, J., et al., The CRL4-DCAF13 ubiquitin E3 ligase supports oocyte meiotic resumption by targeting PTEN degradation. Cell Mol Life Sci, 2020. 77(11): p. 2181-2197.
- Ge, M.K., et al., FBXO22 degrades nuclear PTEN to promote tumorigenesis. Nat Commun, 2020. 11(1): p. 1720. [CrossRef]
- Dupraz, S., et al., RhoA Controls Axon Extension Independent of Specification in the Developing Brain. Curr Biol, 2019. 29(22): p. 3874-3886.e9.
- Kannan, M., et al., The E3 ligase Cdh1-anaphase promoting complex operates upstream of the E3 ligase Smurf1 in the control of axon growth. Development, 2012. 139(19): p. 3600-12.
- Lin, M.Y., et al., PDZ-RhoGEF ubiquitination by Cullin3-KLHL20 controls neurotrophin-induced neurite outgrowth. J Cell Biol, 2011. 193(6): p. 985-94.
- Dogterom, M. and G.H. Koenderink, Actin-microtubule crosstalk in cell biology. Nat Rev Mol Cell Biol, 2019. 20(1): p. 38-54.
- Song, J., et al., The X-linked intellectual disability gene product and E3 ubiquitin ligase KLHL15 degrades doublecortin proteins to constrain neuronal dendritogenesis. J Biol Chem, 2021. 296: p. 100082.
- Shim, T., et al., Cullin-RING E3 ubiquitin ligase 4 regulates neurite morphogenesis during neurodevelopment. iScience, 2024. 27(2): p. 108933.
- Stegmüller, J., et al., Cell-intrinsic regulation of axonal morphogenesis by the Cdh1-APC target SnoN. Neuron, 2006. 50(3): p. 389-400.
- Lasorella, A., et al., Degradation of Id2 by the anaphase-promoting complex couples cell cycle exit and axonal growth. Nature, 2006. 442(7101): p. 471-4.
- Nakagawa, T. and Y. Xiong, X-linked mental retardation gene CUL4B targets ubiquitylation of H3K4 methyltransferase component WDR5 and regulates neuronal gene expression. Mol Cell, 2011. 43(3): p. 381-91.
- Nakagawa, T. and Y. Xiong, Chromatin regulation by CRL4 E3 ubiquitin ligases: CUL4B targets WDR5 ubiquitylation in the nucleus, in Cell Cycle. 2011: United States. p. 4197-8.
- Shilatifard, A., The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem, 2012. 81: p. 65-95.
- Green, E.M. and O. Gozani, CUL4B: trash talking at chromatin. Mol Cell, 2011. 43(3): p. 321-3.
- des Portes, V., et al., A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell, 1998. 92(1): p. 51-61.
- Gleeson, J.G., et al., Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell, 1998. 92(1): p. 63-72.
- Al-Sarraj, Y., et al., The genetic landscape of autism spectrum disorder in the Middle Eastern population. Front Genet, 2024. 15: p. 1363849.
- Butler, M.G., et al., Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet, 2005. 42(4): p. 318-21.
- Tan, M.H., et al., A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet, 2011. 88(1): p. 42-56.
- Agirman, G., L. Broix, and L. Nguyen, Cerebral cortex development: an outside-in perspective. FEBS Lett, 2017. 591(24): p. 3978-3992.
- Jossin, Y., Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules, 2020. 10(6).
- Joly-Amado, A., N. Kulkarni, and K.R. Nash, Reelin Signaling in Neurodevelopmental Disorders and Neurodegenerative Diseases. Brain Sci, 2023. 13(10).
- Gao, Z. and R. Godbout, Reelin-Disabled-1 signaling in neuronal migration: splicing takes the stage. Cell Mol Life Sci, 2013. 70(13): p. 2319-29.
- Feng, L., et al., Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev, 2007. 21(21): p. 2717-30.
- Stier, A., et al., The CUL4B-based E3 ubiquitin ligase regulates mitosis and brain development by recruiting phospho-specific DCAFs. Embo j, 2023. 42(17): p. e112847.
- Butts, T., M.J. Green, and R.J. Wingate, Development of the cerebellum: simple steps to make a 'little brain'. Development, 2014. 141(21): p. 4031-41.
- Cossart, R. and R. Khazipov, How development sculpts hippocampal circuits and function. Physiol Rev, 2022. 102(1): p. 343-378.
- Mukherjee, C., et al., Loss of the neuron-specific F-box protein FBXO41 models an ataxia-like phenotype in mice with neuronal migration defects and degeneration in the cerebellum. J Neurosci, 2015. 35(23): p. 8701-17.
- Quadros, A., et al., Neuronal F-Box protein FBXO41 regulates synaptic transmission and hippocampal network maturation. iScience, 2022. 25(4): p. 104069.
- King, C.R., et al., Fbxo41 Promotes Disassembly of Neuronal Primary Cilia. Sci Rep, 2019. 9(1): p. 8179.
- Persico, A.M., et al., Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol Psychiatry, 2001. 6(2): p. 150-9.
- Skaar, D.A., et al., Analysis of the RELN gene as a genetic risk factor for autism. Mol Psychiatry, 2005. 10(6): p. 563-71.
- Nawa, Y., et al., Rare single-nucleotide DAB1 variants and their contribution to Schizophrenia and autism spectrum disorder susceptibility. Hum Genome Var, 2020. 7(1): p. 37.
- Saillour, Y., et al., LIS1-related isolated lissencephaly: spectrum of mutations and relationships with malformation severity. Arch Neurol, 2009. 66(8): p. 1007-15.
- Südhof, T.C., Towards an Understanding of Synapse Formation. Neuron, 2018. 100(2): p. 276-293.
- Qi, C., et al., Molecular mechanisms of synaptogenesis. Front Synaptic Neurosci, 2022. 14: p. 939793.
- Kaizuka, T. and T. Takumi, Postsynaptic density proteins and their involvement in neurodevelopmental disorders. J Biochem, 2018. 163(6): p. 447-455.
- Yang, Y., et al., A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science, 2009. 326(5952): p. 575-8.
- Kawabe, H. and J. Stegmüller, The role of E3 ubiquitin ligases in synapse function in the healthy and diseased brain. Mol Cell Neurosci, 2021. 112: p. 103602.
- Yao, I., et al., SCRAPPER-dependent ubiquitination of active zone protein RIM1 regulates synaptic vesicle release. Cell, 2007. 130(5): p. 943-57.
- Hansen, K.B., et al., Structure, Function, and Pharmacology of Glutamate Receptor Ion Channels. Pharmacol Rev, 2021. 73(4): p. 298-487.
- Niswender, C.M. and P.J. Conn, Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol, 2010. 50: p. 295-322.
- Fu, A.K., et al., APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat Neurosci, 2011. 14(2): p. 181-9.
- Malenka, R.C. and M.F. Bear, LTP and LTD: an embarrassment of riches. Neuron, 2004. 44(1): p. 5-21.
- Huang, J., et al., A Cdh1-APC/FMRP Ubiquitin Signaling Link Drives mGluR-Dependent Synaptic Plasticity in the Mammalian Brain. Neuron, 2015. 86(3): p. 726-39.
- Gu, J., et al., KCTD13-mediated ubiquitination and degradation of GluN1 regulates excitatory synaptic transmission and seizure susceptibility. Cell Death Differ, 2023. 30(7): p. 1726-1741.
- Mandic-Maravic, V., et al., Dopamine in Autism Spectrum Disorders-Focus on D2/D3 Partial Agonists and Their Possible Use in Treatment. Front Psychiatry, 2021. 12: p. 787097.
- Pavăl, D., The dopamine hypothesis of autism spectrum disorder: A comprehensive analysis of the evidence. Int Rev Neurobiol, 2023. 173: p. 1-42.
- Maljevic, S. and H. Lerche, Potassium channels: a review of broadening therapeutic possibilities for neurological diseases. J Neurol, 2013. 260(9): p. 2201-11.
- Simons, C., et al., Mutations in the voltage-gated potassium channel gene KCNH1 cause Temple-Baraitser syndrome and epilepsy. Nat Genet, 2015. 47(1): p. 73-7.
- Kortüm, F., et al., Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet, 2015. 47(6): p. 661-7.
- Hsu, P.H., et al., Cullin 7 mediates proteasomal and lysosomal degradations of rat Eag1 potassium channels. Sci Rep, 2017. 7: p. 40825.
- Rochefort, N.L. and A. Konnerth, Dendritic spines: from structure to in vivo function. EMBO Rep, 2012. 13(8): p. 699-708.
- Soucy, T.A., et al., An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature, 2009. 458(7239): p. 732-6.
- Li, T., et al., CRISPR/Cas9 therapeutics: progress and prospects. Signal Transduct Target Ther, 2023. 8(1): p. 36.
- Shi, Y., et al., Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov, 2017. 16(2): p. 115-130.
- Zhou, H., et al., Selective inhibition of cullin 3 neddylation through covalent targeting DCN1 protects mice from acetaminophen-induced liver toxicity. Nat Commun, 2021. 12(1): p. 2621.
- Wu, K., et al., Inhibitors of cullin-RING E3 ubiquitin ligase 4 with antitumor potential. Proc Natl Acad Sci U S A, 2021. 118(8).
Figure 1.
The composition of CRL ubiquitin ligases. Cullin proteins (red and purple) serve to link the substrate binding module with the ubiquitylation module. CUL1, CUL2, CUL4A, CUL4B, CUL5, and APC2 interact with substrate receptors (blue) through adaptor proteins (green), whereas CUL3 directly associates with substrate receptors. In contrast, CUL7 and CUL9 appear to bypass substrate receptors, binding directly to their substrates. CUL7 recruits CRL1 as a ubiquitylation module, whereas CUL9 uniquely harbors an RBR domain, which facilitates the catalysis of ubiquitylation through a ROC1-based ubiquitylation module. EB, Elongin B; EC, Elongin C; APCs, APC proteins.
Figure 1.
The composition of CRL ubiquitin ligases. Cullin proteins (red and purple) serve to link the substrate binding module with the ubiquitylation module. CUL1, CUL2, CUL4A, CUL4B, CUL5, and APC2 interact with substrate receptors (blue) through adaptor proteins (green), whereas CUL3 directly associates with substrate receptors. In contrast, CUL7 and CUL9 appear to bypass substrate receptors, binding directly to their substrates. CUL7 recruits CRL1 as a ubiquitylation module, whereas CUL9 uniquely harbors an RBR domain, which facilitates the catalysis of ubiquitylation through a ROC1-based ubiquitylation module. EB, Elongin B; EC, Elongin C; APCs, APC proteins.
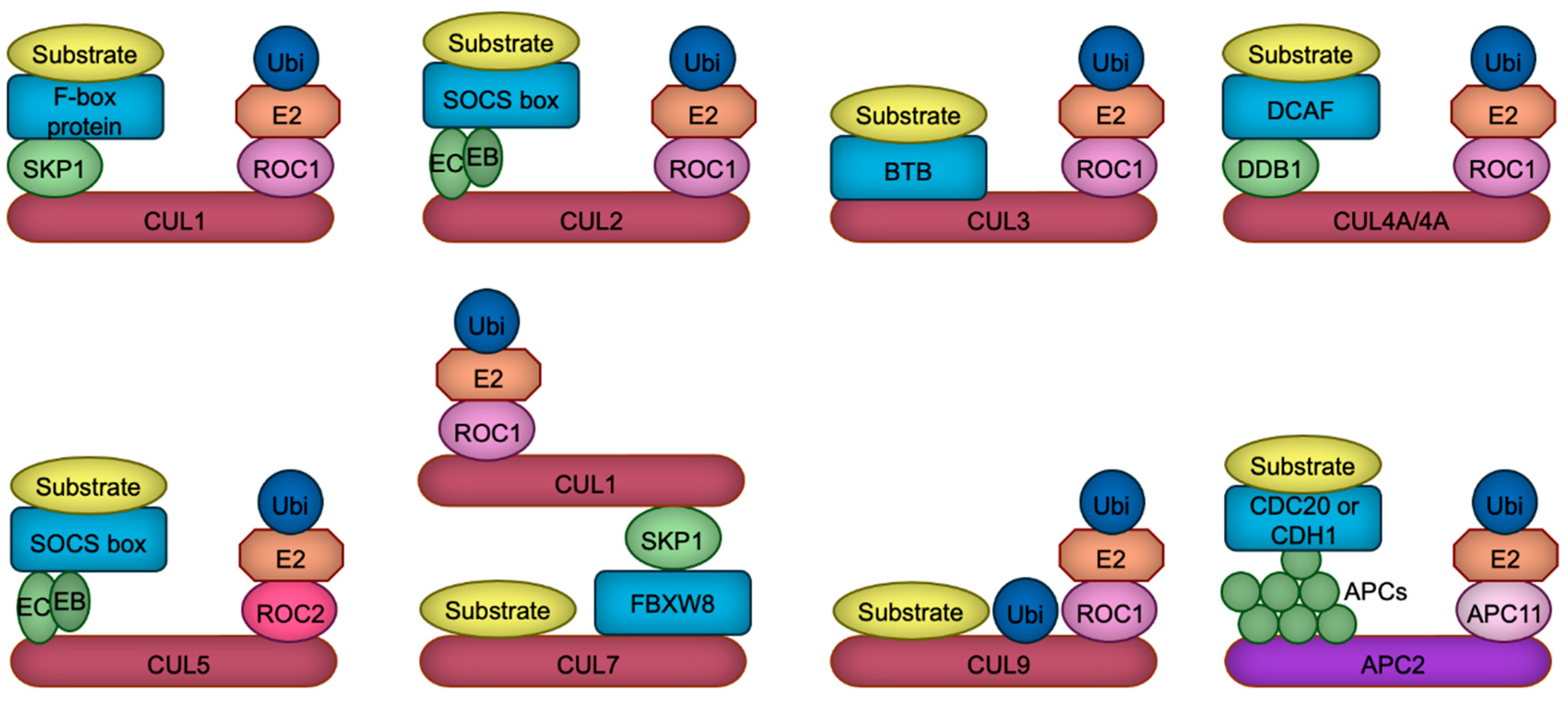
Figure 2.
Schematic depiction of neuronal differentiation processes. Neural stem cells undergo proliferation and self-renewal. Upon initiating differentiation, these cells develop axons and establish synaptic connections with other neurons, culminating in the assembly of neural circuits essential for processing information in response to both environmental and endogenous stimuli. Created in BioRender. Nakagawa, T. (2025) https://BioRender.com/t69e513.
Figure 2.
Schematic depiction of neuronal differentiation processes. Neural stem cells undergo proliferation and self-renewal. Upon initiating differentiation, these cells develop axons and establish synaptic connections with other neurons, culminating in the assembly of neural circuits essential for processing information in response to both environmental and endogenous stimuli. Created in BioRender. Nakagawa, T. (2025) https://BioRender.com/t69e513.
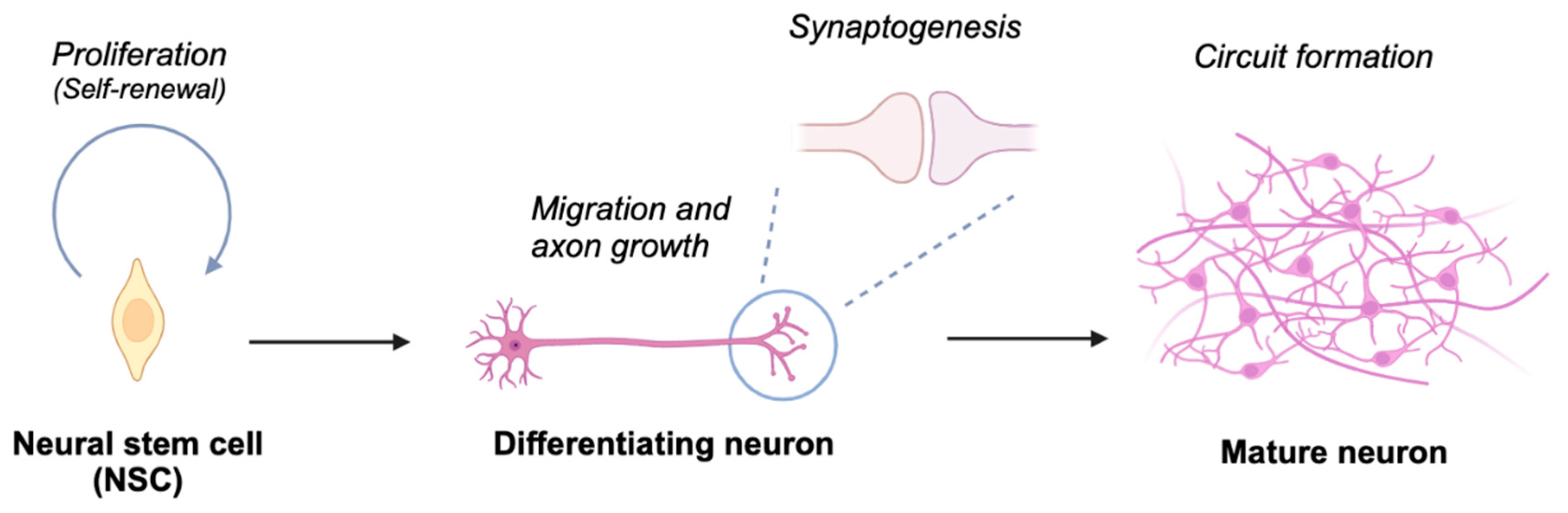
Figure 3.
CRLs in neural stem cell (NSC) proliferation and differentiation. The transcription factor β-catenin and cyclins are targeted for degradation by CRL1 or APC/C to regulate NSC proliferation. NSC differentiation is precisely regulated through CRL1, CRL4A, or APC/C-mediated degradation of key cell cycle regulators, including SKP2 and CDC25A, as well as transcription factors such as SOX2 and NICD, thereby ensuring the timely generation of neurons or glial cells.
Figure 3.
CRLs in neural stem cell (NSC) proliferation and differentiation. The transcription factor β-catenin and cyclins are targeted for degradation by CRL1 or APC/C to regulate NSC proliferation. NSC differentiation is precisely regulated through CRL1, CRL4A, or APC/C-mediated degradation of key cell cycle regulators, including SKP2 and CDC25A, as well as transcription factors such as SOX2 and NICD, thereby ensuring the timely generation of neurons or glial cells.
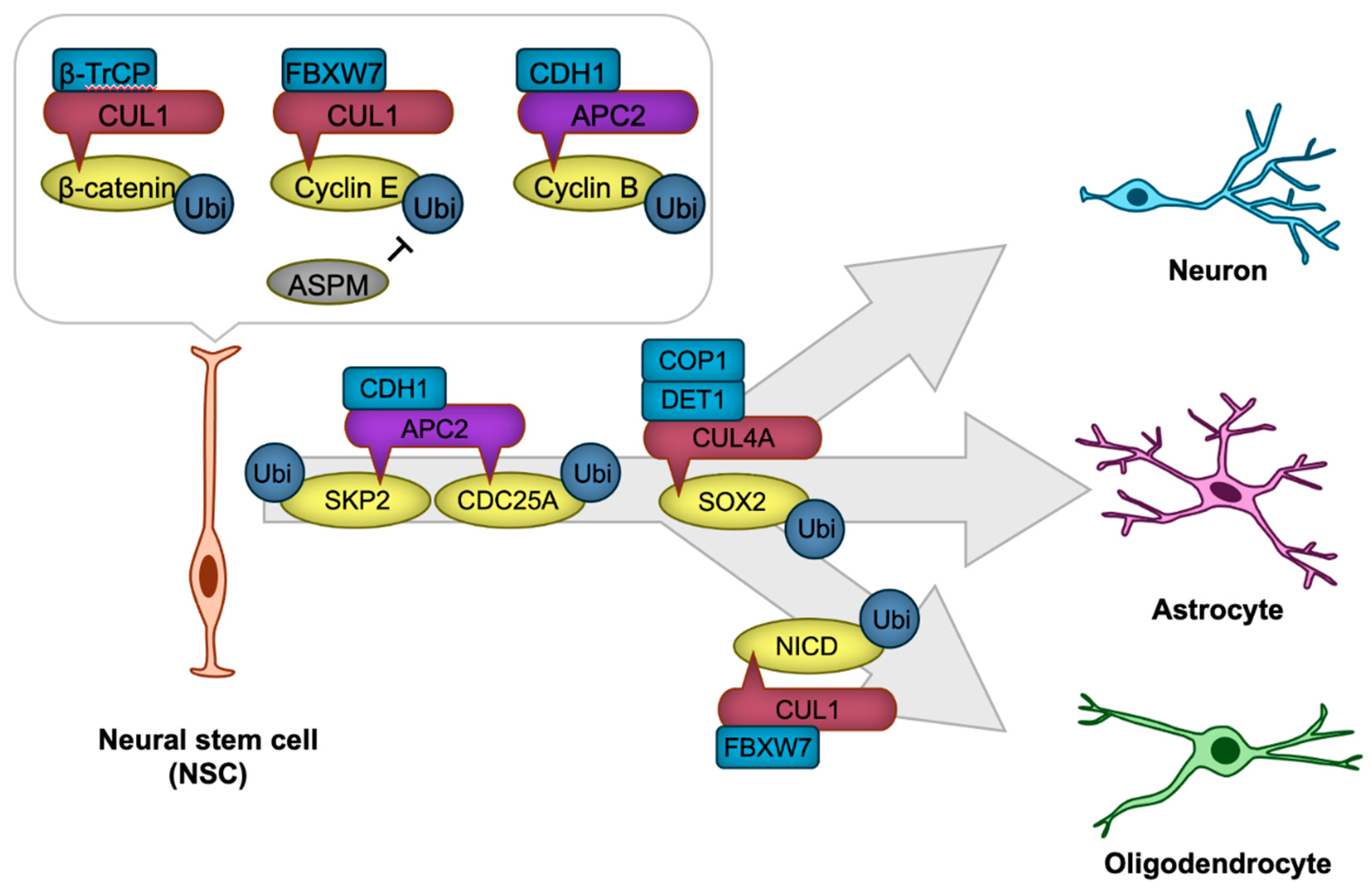
Figure 4.
CRLs in neuronal polarization. APC/C promotes dendrite formation by targeting SMURF1, a negative regulator of RhoA, along with transcription factors SnoN and ID2, positive regulators of axon formation, for ubiquitylation and subsequent degradation. RhoA, essential for dendrite formation, is also regulated through the degradation of its activator, PDZ-RhoGEF, by CRL3. BDNF enhances this degradation, thereby accelerating axon formation. CUL4B-mediated degradation of WDR5 further promotes neurite growth. PTEN, a negative regulator of axonogenesis, is maintained at low levels in axons through ubiquitylation-mediated degradation. While the specific ubiquitin ligases remain unidentified, CRL4B and CRL1 are known to ubiquitylate PTEN in non-neuronal cancer cells, suggesting that these ligases may also regulate PTEN in developing neurons. DCX, which facilitates axonal and dendritic complexity, is negatively regulated by CRL3 and CRL4A/B. BDNF, Brain-derived neurotrophic factor.
Figure 4.
CRLs in neuronal polarization. APC/C promotes dendrite formation by targeting SMURF1, a negative regulator of RhoA, along with transcription factors SnoN and ID2, positive regulators of axon formation, for ubiquitylation and subsequent degradation. RhoA, essential for dendrite formation, is also regulated through the degradation of its activator, PDZ-RhoGEF, by CRL3. BDNF enhances this degradation, thereby accelerating axon formation. CUL4B-mediated degradation of WDR5 further promotes neurite growth. PTEN, a negative regulator of axonogenesis, is maintained at low levels in axons through ubiquitylation-mediated degradation. While the specific ubiquitin ligases remain unidentified, CRL4B and CRL1 are known to ubiquitylate PTEN in non-neuronal cancer cells, suggesting that these ligases may also regulate PTEN in developing neurons. DCX, which facilitates axonal and dendritic complexity, is negatively regulated by CRL3 and CRL4A/B. BDNF, Brain-derived neurotrophic factor.

Figure 5.
CRLs in neuronal migration. RGCs, the first differentiated cells derived from neural stem cells, generate IPs that migrate towards the cortical surface, with later-differentiated neurons passing by earlier-generated neurons. CRL3, CRL4B, and CRL5 regulate neuronal migration by ubiquitylating key migration regulatory proteins, including PLS3, LIS1, and DAB1, respectively. RGC, radial glial cell; IP, intermediate progenitor; VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone; CP, cortical plate.
Figure 5.
CRLs in neuronal migration. RGCs, the first differentiated cells derived from neural stem cells, generate IPs that migrate towards the cortical surface, with later-differentiated neurons passing by earlier-generated neurons. CRL3, CRL4B, and CRL5 regulate neuronal migration by ubiquitylating key migration regulatory proteins, including PLS3, LIS1, and DAB1, respectively. RGC, radial glial cell; IP, intermediate progenitor; VZ, ventricular zone; SVZ, subventricular zone; IZ, intermediate zone; CP, cortical plate.
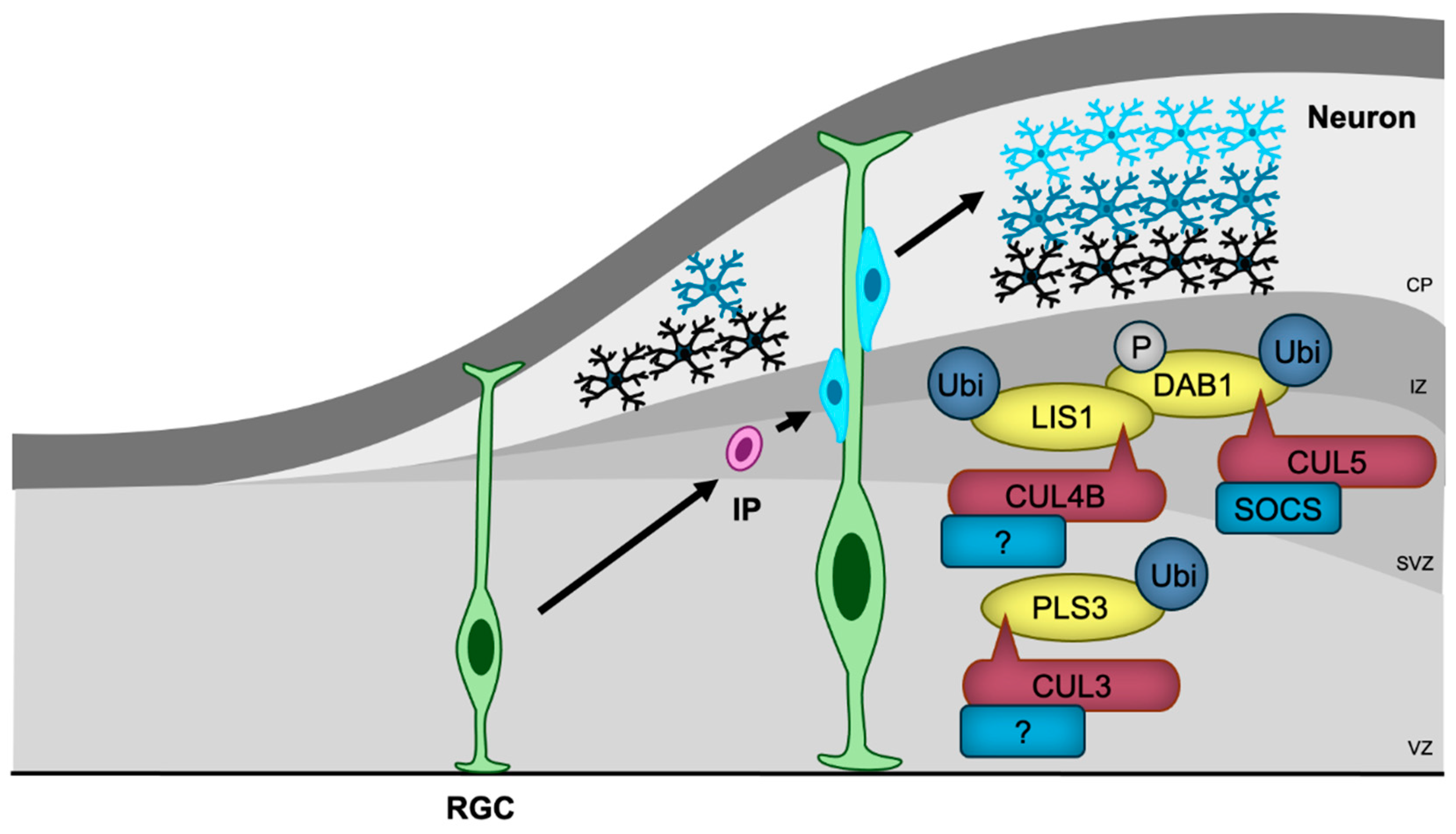
Figure 6.
CRLs in synaptogenesis and synaptic function. CRL1 and APC/C regulate presynaptic terminal functions, while CRL3, CRL7, and APC/C control postsynaptic functions through the ubiquitylation of the indicated substrates. Notably, KLHL17-loaded CRL3 has been shown to promote spine enlargement and enhance synaptic activity, although the specific substrate responsible for this function remains unidentified.
Figure 6.
CRLs in synaptogenesis and synaptic function. CRL1 and APC/C regulate presynaptic terminal functions, while CRL3, CRL7, and APC/C control postsynaptic functions through the ubiquitylation of the indicated substrates. Notably, KLHL17-loaded CRL3 has been shown to promote spine enlargement and enhance synaptic activity, although the specific substrate responsible for this function remains unidentified.
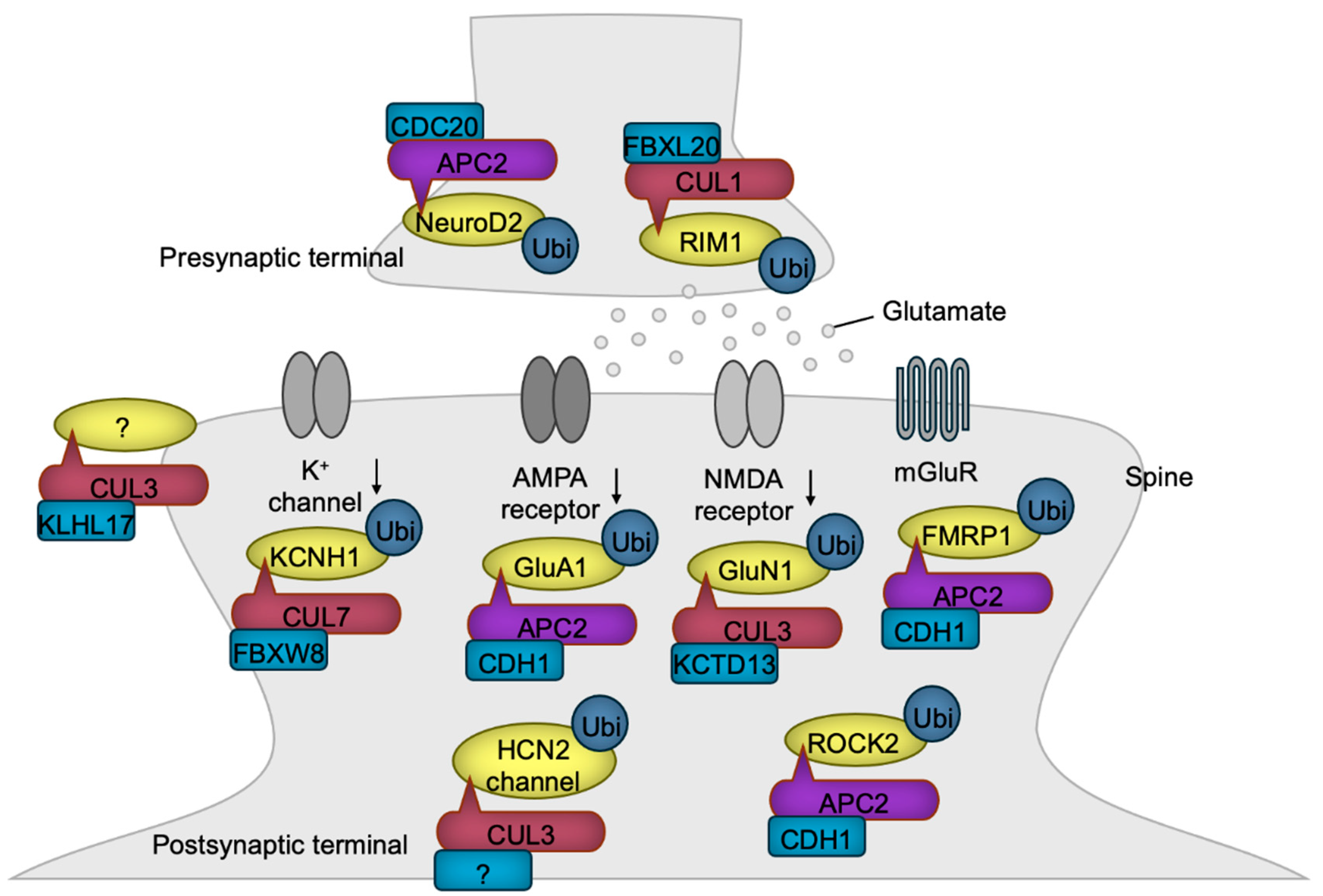

Table 1.
NDD-related behavioral abnormalities exhibited by CRL-related gene mutations in mice. This is a table. A check mark denotes the presence, while a minus sign in parentheses indicates the absence of the behavioral abnormalities. *, these mice exhibit hypoactivity. DA, dopaminergic; Glu, glutamatergic.
Table 1.
NDD-related behavioral abnormalities exhibited by CRL-related gene mutations in mice. This is a table. A check mark denotes the presence, while a minus sign in parentheses indicates the absence of the behavioral abnormalities. *, these mice exhibit hypoactivity. DA, dopaminergic; Glu, glutamatergic.
| Target disrupted | Behavioral abnormality | Reference | ||||
| Gene | Cell type | Communication | Learning and memory | Hyperactivity | Anxiety | |
| Cul3 | Whole body | ✔ | ✔ | ✔ | (-) | [52,53] |
| Cul3 | Neurons and astrocytes | ✔ | (-) | (-) | ✔ | [54] |
| Cul3 | DA neurons | N.T. | ✔ | ✔ | (-) | [55] |
| Cul4b | Whole body | (-) | ✔ | (-) | (-) | [56] |
| Klhl17 | Whole body | ✔ | N.T. | ✔ | (-) | [57] |
| Kctd13 | Whole body | (-) | ✔ | (-) | (-) | [58] |
| Crbn | Forebrain Glu neurons | N.T. | ✔ | (-) | (-) | [59] |
| Cdh1 | Whole body | N.T. | ✔ | N.T. | N.T. | [60] |
| Cdh1 | Forebrain Glu neurons | N.T. | ✔ | (-)* | ✔ | [61] |
Table 2.
CRL scaffolds, substrate receptors, their substrates, and functions of ubiquitination discussed in this article. *, CUL4B does not require substrate receptor to bind to WDR5. NSC, neural stem cell; LTD, long-term depression.
Table 2.
CRL scaffolds, substrate receptors, their substrates, and functions of ubiquitination discussed in this article. *, CUL4B does not require substrate receptor to bind to WDR5. NSC, neural stem cell; LTD, long-term depression.
| CRL scaffold | Substrate receptor | Substrate | Function | Reference |
| APC/C | CDH1 | Cyclin B | Promotion of NSC proliferation | [68] |
| CUL1 | FBXW7 | Cyclin E | Inhibition of NSC proliferation | [69] |
| CUL1 | FBXW7 | NICD | Promotion of NSC differentiation | [70] |
| APC/C | CDH1 | CDC25A | Promotion of NSC differentiation | [68] |
| APC/C | CDH1 | SKP2 | Promotion of NSC differentiation | [68] |
| CUL1 | β-TrCP | β-Catenin | Promotion of NSC differentiation | [72] |
| CUL4A | DET1-COP1 | SOX2 | Promotion of NSC differentiation | [73] |
| APC/C | CDH1 | SMURF1 | Promotion of dendritogenesis | [89] |
| CUL3 | KLHL20 | PDZ-RhoGEF | Inhibition of axon formation | [90] |
| CUL3 | KLHL15 | DCX | Inhibition of axon/dendrite complexity | [92] |
| CUL4A | CRBN | DCX | Inhibition of axon/dendrite complexity | [93] |
| CUL4B | CRBN | DCX | Inhibition of axon/dendrite complexity | [93] |
| APC/C | CDH1 | SnoN | Inhibition of axon formation | [94] |
| APC/C | CDH1 | ID2 | Inhibition of axon formation | [95] |
| CUL4B | (-)* | WDR5 | Promotion of neurite extension | [96] |
| CUL5 | SOCS | DAB1 | Inhibition of neuronal migration | [109] |
| CUL3 | Unknown | PLS3 | Promotion of neuronal migration | [53] |
| APC/C | CDC20 | NeuroD2 | Promotion of presynapse formation | [123] |
| CUL1 | FBXL20 | RIM1 | Inhibition of postsynaptic function | [125] |
| APC/C | CDH1 | GluA1 | Inhibition of excess synaptic activity | [128] |
| APC/C | CDH1 | FMRP1 | Promotion of LTD | [130] |
| CUL3 | KCTD13 | GluN1 | Inhibition of excess synaptic activity | [131] |
| CUL3 | Unknown | HCN2 | Inhibition of excess synaptic activity | [55] |
| CUL7 | FBXW8 | Kv10.1 | Inhibition of potassium current | [137] |
| CUL3 | KLHL17 | Unknown | Promotion of synaptic activity | [57] |
| APC/C | CDH1 | ROCK2 | Destabilization of dendritic spines | [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



