
Submitted:
28 February 2025
Posted:
05 March 2025
You are already at the latest version
Abstract
Porcine reproductive and respiratory syndrome (PRRS) is a pig respiratory disease threating the global swine industry. It is necessary for the effective diagnostic detection of antibody including neutralizing antibody against porcine reproductive and respiratory syndrome virus (PRRSV), especially the currently prevalent NADC30-like PRRSV in China. In this study, we prepared three monoclonal antibodies (mAbs) against NADC30-like PRRSV glycoprotein 5 (GP5) protein, and identified two corresponding precise epitopes (155WR156 and 196QWGRP200). In neutralization test, 196QWGRP200 recognizing GP5 mAbs (11E6 and 12D1) exhibited obvious neutralizing activity, whereas the 155WR156 recognizing mAb (3A8) showed low neutralizing activity. Based on the two antigenic peptides, the peptide-based ELISA was developed to detect antibodies against PRRSV, presenting high specificity, sensitivity and repeatability. The concordance rate of the peptide-based ELISA and commercial IDEXX PRRSV X3 Ab ELISA in detection of 81 clinical samples was 82.7%. In conclusion, the GP5 peptide-based ELISA can be used for the detection of neutralizing antibodies against NADC30-like PRRSV, providing a rapid and reliable method for monitoring PRRSV infection.
Keywords:
NADC30-like PRRSV
; GP5 protein
; epitope
; peptide-based ELISA
; neutralizing antibody
Introduction
Porcine reproductive and respiratory syndrome (PRRS) is one of the most important infectious viral diseases in pig herds, caused by the porcine reproductive and respiratory syndrome virus (PRRSV) and characterized by reproductive disorders in sows and respiratory distress in piglets [1]. PRRSV can be divided into two species, the Betaarterivirus suid 1 (PRRSV-1, European-type PRRSV, prototype Lelystad strain) and the Betaarterivirus suid 2 (PRRSV-2, North American-type PRRSV, prototype VR-2332 strain) [2]. PRRSV-2 has often been regarded as the epidemiologically predominant strains in China [3]. In 2006, the highly pathogenic PRRSV-2 (HP-PRRSV) characterized by high mortality, fever, and abortion rates first appeared in southern China and devastated the Chinese swine industry [4]. In recent years, NADC30-like PRRSV-2 and NADC34-like PRRSV-2, which show much less pathogenic to pigs than HP-PRRSV, have been circulating in China and become the dominant strains in field [5].
PRRSV is an enveloped, single-stranded, positive-sense RNA virus, which belongs to genus Betaarterivirus, family Arteriviridae, order Nidovirales [6]. It has approximately 15 kb genome containing at least 10 known open reading frames (ORFs) [7]. The ORF1a and ORF1b encode two polyproteins, pp1a and pp1ab, which can be processed into at least 16 mature nonstructural proteins (NSPs) by viral proteases that play a major role in virus replication [8]. The remaining ORFs (ORF2-7) encode structural proteins including GP2a, GP2b, GP3-GP5, GP5a, M, and N proteins [8]. GP5 protein, a glycosylated envelope protein, is encoded by the PRRSV ORF5 gene with the molecular weight of approximately 25 kD [9]. GP5 protein is the most mutable structural protein and is thus often used for PRRSV phylogenetic analyses [10,11]. Among different viral structural proteins, GP5 is the major virulence determinant and plays an important role in virus infection [12,13]. Moreover, GP5 contains important immune domains associated with virus neutralization, serving as a good target antigen for developing new vaccines [9,14].
The effective diagnosis of PRRS is important to control and prevention [15]. The detection of antibody including neutralizing antibody against PRRSV not only promotes diagnosis of PRRS, but also helps assess the immune protection status of pig herds [15,16]. Currently, several methods have been applied for detection of PRRS antibodies, including immunoperoxidase monolayer assay, indirect fluorescent assay, serum neutralization test and enzyme-linked immunosorbent assay (ELISA) [15]. The immunoperoxidase monolayer assay and indirect fluorescent assay are subjective, laborious and expensive, mainly suitable for laboratory test [15]. Serum neutralization test can examine neutralizing antibodies produced by PRRSV infection in pigs, nevertheless it is not suitable for diagnosis of acute infection, and it is also laborious [17]. ELISA is a popular method for the detection of PRRSV antibodies because of its strong specificity, high sensitivity, and low cost [15]. Several indirect ELISA detecting PRRSV GP5 antibodies have been established [18,19,20]; however, the antigen preparation for the ELISA takes a long time and lacks a precision.
In the current study, we generated three mAbs specific for NADC30-like PRRSV GP5 and identified two precise antigenic epitopes that mediate virus neutralization. Based on the two antigenic epitopes, the peptide-based ELISA was developed to detect neutralizing antibodies against NADC30-like PRRSV.
Results
- Expression and purification of the recombinant NADC30-like PRRSV GP5 truncated protein
Bioinformatics analysis revealed that the GP5 protein of NADC30-like PRRSV SD17-38 strain is a multiple transmembrane protein, with the transmembrane (TM) regions located at 10-32, 66-88, and 103-125 amino acids (aa), respectively (Figure 1A), which exhibits the high hydrophobicity (Figure 1B). Both the extramembrane region (89-102 aa) and intramembrane regions (33-65, 126-200 aa) contain the B cells antigenic epitopes highlighted in yellow color (Figure 1C). The signal peptide was predicted to be 1-31 aa, with an accurate cleavage site between residues 31 and 32 (Figure 1D). Given that intramembrane regions (33-65, 126-200 aa) exhibited higher hydrophilicity (Figure 1B) and contain more antigenicity (Figure 1C), the recombinant prokaryotic GP5 plasmid expressing two fused intramembrane regions (33-65, 126-200 aa) was constructed and the plasmid transformed E. coli cells was induced by IPTG to express the GP5 fused truncation protein. The recombinant NADC30-like PRRSV GP5 protein was determined to mainly express in the form of inclusion body at 37°C for 12 h by SDS-PAGE (Figure 1E). The expressed GP5 protein, present about 15~25 kD as anticipated, was confirmed in Western blotting (WB) by both anti-HA mAb (Figure 1F) and anti-His mAb (Figure 1G). Further, the purified GP5 protein exhibited high purity and amount in SDS-PAGE (Figure 1H). The data indicated that the purified recombinant GP5 truncation protein was suitable for immunization.
- Preparation and identification of GP5 protein specific mAbs
Via cell fusion, we obtained three hybridoma cell clones (3A8, 11E6 and 12D1) with notably high positive readings after screening and subcloning thrice in GP5 coated indirect ELISA (Figure 2A). The ascites titers of hybridoma cell clones 3A8, 11E6 and 12D1 were measured using the indirect ELISA to be 1: 409600, 1: 819200, and 1: 819200, respectively (Figure 2B). The isotypes of three mAbs (3A8, 11E6 and 12D1) were determined to be IgG1, IgG2b, and IgG2a, respectively (Figure S1). WB indicated that three mAbs could specifically recognize exogenous GP5 protein in HEK293T cells transfected with pCAGGS-GP5-HA (Figure 2C) and endogenous GP5 protein in CD163-3D4/21 cells infected with NADC30-like PRRSV SD17-38 strain (Figure 2D). In both cases, the detected GP5 proteins had the molecular weight of 25 kD as expected (Figure 2C and 2D). The similar results from Immunofluorescence assay (IFA) further confirmed that three mAbs exhibited strong specificity for both exogenous GP5 (Figure 3) and endogenous GP5 (Figure 4). In both cases, the detected GP5 proteins were located in the cytoplasm (Figure 3 and 4). These results implied that three mAbs (3A8, 11E6 and 12D1) displayed specific reaction with PRRSV and its GP5 protein, recognizing linear epitopes of GP5. Moreover, these mAbs could be used for different immune assays (ELISA, WB and IFA), signifying the application value.
- Precise mapping and identification of linear B cell epitopes recognized by GP5 mAbs
To identify the precise linear B cell epitopes recognized by the three mAbs, the different truncated GP5 fragments were designed and their reactions with three mAbs (3A8, 11E6 and 12D1) were identified by WB (Figure 5). Based on the reactivity of various GP5 fragments with the three GP5 mAbs, we determined the critical amino acids at both N-terminal and C-terminal ends of PRRSV GP5 protein, and defined the precise antigenic epitopes recognized by the three mAbs (Figure 5 and Figure S2). Taking the above-described results together, the 3A8 mAb recognized minimal linear antigenic epitope 155WR156 (Figure 5A), whereas 11E6 and 12D1 mAbs recognized the same minimal linear antigenic peptide 196QWGRP200 (Figure 5B).
- Conservation analysis of the identified antigenic epitopes across different PRRSV strains
To perform bioinformatics analysis of the identified GP5 antigenic epitopes from NADC30-like PRRSV strain, the GP5 protein sequences of different PRRSV strains were downloaded from GenBank and aligned with the two identified GP5 epitope sequences (Figure 6 and Table S1). The results showed that the epitope 1 155WR156 and epitope 2 196QWGRP200 are relatively conserved across different PRRSV strains, with the epitope 1 more conserved than epitope 2 in all PRRSV-2 strains (Figure 6 and Table S1). Further, the three NADC30-like PRRSV GP5 mAbs were examined by WB for the reactivity with different PRRSV-1 and PRRSV-2 strains, including PRRSV-1 SD1291, PRRSV-1 HLJB1, NADC34-like PRRSV-2 Anheal-1, Classic PRRSV-2 VR-2332-like R98, Classic PRRSV-2 vaccine CH-1R, HP-PRRSV-2 virulent XJ17-5 and avirulent JSTZ1712-12 (Figure 7). The 3A8 mAb was verified to specifically react all tested PRRSV-2 strains but neither tested PRRSV-1 strains (Figure 7A), whereas 11E6 and 12D1 mAbs exhibited reactivity towards NADC30-like PRRSV-2 SD17-38, NADC34-like PRRSV-2 Anheal-1, Classic PRRSV-2 VR-2332-like R98, and Classical PRRSV-2 vaccine CH-1R (Figure 7B and 7C), Together, the results reflected the relative conservation of both epitope 1 155WR156 and epitope 2 196QWGRP200 across different PRRSV strains.
- Neutralizing activity of three GP5 mAbs and development of the peptide-based ELISA detecting PRRSV antibody
We purified the three GP5 ascite mAb IgG as illustrated by the SDS-PAGE analysis (Figure 8A). The purified GP5 mAb IgGs plus PRRSV N mAbs we purified previously [21] were used in virus neutralization test to estimate their neutralizing activity against different PRRSV strains. For NADC30-like PRRSV-2 SD17-38, the GP5 mAbs 11E6 and 12D1 could protected 50% cells from viral N protein expression in IFA with the dilutions up to 1:148 and 1:165, respectively, whereas 3A8 mAb had the same performance with the dilution only up to 1:10 (Figure 8B). Additionally, neither of the three GP5 mAbs exhibited 50% protection to NADC34-like PRRSV-2 Anheal-1, Classic PRRSV-2 VR-2332-like R98, and HP-PRRSV-2 virulent XJ17-5 (Figure 8C-8E). As the controls, all four PRRSV N mAbs exhibited no protection against all tested PRRSV strains (Figure 8B-8E). These results suggested that the GP5 mAbs 11E6 and 12D1 have obvious neutralizing activity, whereas GP5 mAb 3A8 has low neutralizing activity against NADC30-like PRRSV.
Next, the epitope peptide-based ELISA was developed to detect PRRSV antibody including neutralization antibody. The optimal ratio of two synthetic peptides P1 (153 to 158 aa) and P2 (196 to 200 aa) was firstly determined to be 1:3, presenting the highest P/N value (Figure 9A). The checkerboard titration assay identified that optimal working concentration of synthetic peptides was 0.5 ng/μL and optimal pig serum dilution was 1:10 (Figure 9B). Based on the determined synthetic peptides and serum dilution, the optimal coating of peptides was at 4 °C overnight and blocking was in 2% BSA buffer for 2 h incubation (Figure 9C). The optimized incubation times for serum, secondary antibody and substrate were determined to be 90 min (Figure 9D), 75 min (Figure 9E) and 15 min (Figure 9F), respectively.
- Specificity, sensitivity and repeatability of the peptide-based ELISA
Using 30 negative pig sera, the ELISA cutoff values of the developed peptide-based ELISA were calculated to be 0.227-0.240 based on the OD450nm values of 30 sera (Table S2). The OD450nm values > 0.240 were regarded as positive, whereas OD450nm values < 0.227 were regarded as negative (Table S2). The specificity of the peptide-based ELISA was examined with different positive sera of PRRSV, African swine fever virus (ASFV), swine influenza virus (SIV), and porcine epidemic diarrhea virus (PEDV) (Figure 10A). The peptide-based ELISA reacted only with the PRRSV serum, but not with other sera, demonstrating the specificity of the peptide-based ELISA for PRRSV (Figure 10A). To test the sensitivity of the peptide-based ELISA, PRRSV positive serum was subjected to serial dilutions, followed by detection of peptide-based ELISA. The maximal dilution of the PRRSV positive serum detectable by peptide-based ELISA was 1:640 (Figure 10B). To evaluate the repeatability of the peptide-based ELISA, the intra- and inter-assay coefficient of variations (CVs) with five positive samples and one negative sample were calculated. As a result, the intra-assay CV was found to range from 1.4 to 3.3% (Figure 10C), while the inter-assay CV ranged from 4.3 to 8.4% (Figure 10D). Both CV values were less than 10%, suggesting that the established peptide-based ELISA for PRRSV antibody detection exhibited excellent repeatability.
- Applicability of the peptide-based ELISA for detection of clinical samples
To assess applicability of the established peptide-based ELISA, a total of 81 clinical pig serum samples were tested by the peptide-based ELISA. The results showed that, of the 81 samples, only 19 were positive tested in the peptide-based ELISA, whereas the other 62 were negative (Table 1). For comparison, the 81 samples were simultaneously detected using the commercial IDEXX PRRSV X3 Ab ELISA. In the IDEXX ELISA, 31 samples were positive and 50 samples were negative (Table 1). Among the 81 samples, 18 were positive by both the peptide-based ELISA and the commercial ELISA, 1 by the peptide-based ELISA only, and 13 by the commercial ELISA only (Table 1 and Table S3). The concordance rate between the peptide-based ELISA and the commercial ELISA was 82.7%, suggesting the applicability of the peptide-based ELISA for detection of clinical samples.
Discussion
Currently, NADC30-like PRRSV has been prevalent and become the dominant strains in China [5,22,23]. Although the NADC30-like PRRSV is less virulent than the HP-PRRSV, it can still cause abortions in sows and respiratory diseases in fattening pigs [5]. Therefore, it is necessary to investigate the prevention and control of NADC30-like PRRSV epidemic. GP5 is a glycosylated envelope protein, which plays a key role in viral virulence and immune response [9,24]. In this study, we developed three NADC30-like PRRSV GP5 specific mAbs, revealed two GP5 antigenic neutralizing epitopes, and established the peptide-based ELISA for detecting NADC30-like PRRSV neutralizing antibody.
GP5 has been considered as the most relevant antigen for neutralizing antibodies in PRRSV [9,16,25]. Accordingly, the three GP5 mAbs we generated in this study exhibited neutralizing activity; specifically, the mAbs 11E6 and 12D1 possessed obvious neutralizing activity, whereas the mAb 3A8 had low neutralizing activity. The neutralizing activity of these mAbs was only for NADC30-like PRRSV but not for other types of PRRSV strains, suggesting the specific feature of NADC30-like PRRSV and also reflecting the hyper-variability of GP5 protein.
The PRRSV GP5 N-terminal ectodomain region (1-61 aa) was determined to contain a neutralizing epitope in the middle (37-45 aa) and surrounding immune dominant non-neutralizing epitopes [14,26,27]. Additional studies have identified more antigenic epitopes of GP5, including 1-15 aa, 31-45 aa, 187-200 aa [28], 7-22 aa, 60-66 aa, 76-91 aa, 95-110 aa, 133-140 aa, 154-161 aa, 185-194 aa [29], 152-156 aa, 169-178 aa, and 196-200 aa [30]. In this study, two GP5 precise antigenic epitopes were identified that are 155WR156 with the mAb 3A8, and 196QWGRP200 with both mAbs 11E6 and 12D1. Interestingly, these two GP5 C-terminal epitopes are consistent with previous identified minimal epitopes 152-156 aa and 196-200 aa [30], demonstrating the relative conservation of the antigenicity in PRRSV-2. Importantly, these two C-terminal GP5 epitopes was shown as the neutralizing epitopes, suggesting multiple neutralization antigenic epitopes existed across GP5 protein [31,32]. The viral protein antigenic epitopes play an important role in protein structure and antigenic properties [33]. Here, precise identification of neutralizing epitopes 155WR156 and 196QWGRP200 opens new insights on the structure and antigenicity of the NADC30-like PRRSV GP5 protein.
ELISA is a feasible, sensitive, rapid and large-scale method for detecting antibody [15]. The peptide-based ELISA has been popular due to its low cost, easy preparation, and no biosafety concerns [34,35]. Moreover, compared to ELISA coated with whole antigen or virus, it is targeted at a single or multiple epitopes contained in the synthetic peptide, thus significantly reducing the nonspecific reaction [34,35]. In this study, we established a peptide-based ELISA based on the identified GP5 epitopes 155WR156 and 196QWGRP200, detecting antibody including neutralizing antibody against NADC30-like PRRSV, which displayed high specificity, sensitivity and repeatability.
In parallel detection of 81 clinical samples, the peptide-based ELISA showed an 82.7% compatibility with the commercial IDEXX ELISA. Among these samples, 18 were positive in both the peptide-based ELISA and the commercial ELISA, whereas 1 positive in the peptide-based ELISA only and 13 positive in the commercial ELISA only. It would not be unexpected that significant variation existed between the peptide-based ELISA and commercial ELISA, because both were detecting antibodies to different antigens. For the 1 positive serum in the peptide-based ELISA only, the coated N protein of PRRSV in IDEXX ELISA kit may present several epitope regions; however, not all of those epitopes may be recognized by PRRSV-positive pig serum antibodies. For the 13 positive in the commercial ELISA only, it is understandable that the commercial ELISA is coated with the most immunogenic and stable N protein of PRRSV, whereas the peptide-based ELISA is coated with synthetic peptides, which were located in the variable GP5 protein.
In conclusion, three mAbs 3A8, 11E6, and 12D1 specific for NADC30-like PRRSV GP5 were successfully generated and used for various immune assays. The 11E6 and 12D1 mAbs exhibited obvious neutralizing activity against NADC30-like PRRSV-2 strain SD17-38, whereas 3A8 mAb showed low neutralizing activity. Two precise linear neutralizing epitopes of GP5 (155WR156 and 196QWGRP200) were identified. The peptide-based ELISA was developed using two synthetic GP5 antigenic peptides, and can be used for the detection neutralizing antibodies against NADC30-like PRRSV, which will contribute to the diagnosis, prevention and control of the pig disease caused by NADC30-like PRRSV.
Materials and Methods
- Cells, viruses and mice
3D4/21-CD163 cells [36] were grown in Roswell Park Memorial Institute 1640 (RPMI-1640; Hyclone Laboratories, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS, Eallbio, Beijing, China), whereas myeloma cells SP2/0, Marc-145, and HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies Corp., Grand Island, NY, USA) containing 10% FBS at 37°C in 5% CO2. Primary porcine alveolar macrophages (PAMs) were prepared using regular bronchoalveolar lavage from 2-month-old domestic pigs, and cultured in RPMI-1640 medium supplemented with 10% FBS.
PRRSV strains used in this study were all stored in our laboratory, including NADC30-like PRRSV-2 strain SD17-38 (lineage 1, GenBank: MH068878.1) [37], NADC34-like PRRSV-2 strain Anheal-1 (lineage 1, GenBank: MH370474.1, a courtesy from Dr. Xizhao Chen at Beijing Anheal Laboratories Co. Ltd.), Classic PRRSV-2 VR-2332-like strain R98 (lineage 5, GenBank: DQ355796.1), classical PRRSV-2 vaccine strain CH-1R (lineage 8, GenBank: EU807840.1), HP-PRRSV-2 virulent strain XJ17-5 (lineage 8, GenBank: MK759853.1) and avirulent strain JSTZ1712-12 (lineage 8, GenBank: MK906026.1) [38], PRRSV-1 strain SD1291 [39] and PRRSV-1 strain HLJB1 (GenBank: KT224385.1) [40]. The 3D4/21-CD163 cells were infected with SD17-38 (0.1 MOI) or Anheal-1 (0.1 MOI) for 72 h; Marc-145 cells were infected with R98 (0.1 MOI), CH-1R (0.1 MOI), XJ17-5 (0.1 MOI), or JSTZ1712-12 (0.1 MOI) for 72 h; primary PAMs were infected with HLJB1 (0.1 MOI) or SD1291 (0.1 MOI) for 72 h.
BALB/c mice were purchased from Yangzhou University animal facility, and this study was approved by Guide for the Care and Use of Laboratory Animals and Yangzhou University (SYXK(JS)-2021-0026).
- Bioinformatics analysis of GP5 protein and construction of recombinant plasmids
The NADC30-like PRRSV-2 SD17-38 strain (GenBank: MH068878.1) GP5 protein was analyzed for prediction of transmembrane (TM) by the online software TMHMM-2.0 (https://services.healthtech.dtu.dk/services/TMHMM-2.0/). The hydrophobicity and hydrophilicity scales of GP5 were analyzed through the online software ProtScale (https://web.expasy.org/protscale/). The B cell antigenic epitopes in the GP5 protein were predicted using the online tool (http://tools.iedb.org/main/bcell/). The signal peptide in the GP5 protein was predicted via the online tool (https://services.healthtech.dtu.dk/services/SignalP-5.0/).
Based on the ORF5 (GP5) sequence of the NADC30-like PRRSV-2 SD17-38 strain and bioinformatics analysis, two pairs of specific PCR primers were designed and used for amplification of two GP5 intramembrane regions (33-65, 126-200 aa), separately. These two overlapping PCR products were cloned into the SalI/EcoRV sites of the Gateway entry vector pENTER4-MCS-2HA by ClonExpress Ultra One Step Cloning Kit (Vazyme, Nanjing, China). By LR recombination (Gateway LR Clonase™ II Enzyme mix, ThermoFisher Scientific, Shanghai, China), the ORF5 (GP5) fused truncation fragment was transferred from the pENTER4-2HA vector to the Destination vector pDEST527 (Addgene) to obtain the recombinant prokaryotic pDEST527-GP5-2HA. The eukaryotic plasmid expressing HA-tagged full length GP5 was made by cloning the ORF5 (GP5) sequence into the EcoRI/EcoRV sites of vector pCAGGS-2HA using seamless cloning (2 × MultiF Seamless Assembly Mix, Abclonal, Wuhan, China). The eukaryotic plasmids expressing GFP-tagged GP5 and its truncation derivatives were made by cloning the corresponding gene fragments into the XholI/EcoRI sites of eukaryotic vector pEGFP-N1 using seamless cloning. The cloning PCR primers used for the study were all listed in Table S4. All recombinant plasmids were confirmed by Sanger DNA sequencing.
- Expression and purification of recombinant GP5 truncation protein
The recombinant pDEST527-GP5-2HA was transformed into E. coli BL21 (DE3) competent cells. After induction in 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), the recombinant GP5 protein was expressed and then subjected into purification. The expressed GP5 protein in the form of inclusion body was solubilized by 8 M urea solution, renatured by a stepwise dialysis (6M, 4M, 2M, 1M urea) and finally dissolved in phosphate buffered saline (PBS). The identification of both expressed and purified GP5 proteins was performed by SDS-PAGE with Coomassie brilliant blue staining. The expressed GP5 protein was also detected using Western blotting with the His mAb and HA mAb (each 1:1000, TransGen Biotech, Beijing, China), respectively.
- Mice immunization, monoclonal antibody (mAb) production and purification
The 6-week-old BALB/c mice were received an initial immunization with 100 μg of purified GP5 protein emulsified 10:1 with Montanide gel (SEPPIC SA, Cedex, France) and injected subcutaneously on the back. Two weeks later, 50 μg of purified GP5 protein was administered via intrasplenic injection to booster immunization. One week later, the spleen cells from the immunized mice were harvested to fuse with myeloma cells SP2/0 using PEG1500 following the routine procedure. The hybridoma supernatants were collected for GP5 antibody detection by GP5 coated indirect ELISA. The hybridoma cells tested positive were subcloned three times by limiting dilution. The supernatants from these hybridoma cells were further confirmed by Western blotting and indirect immunofluorescence assay for secreting GP5 specific mAb. Ascites containing GP5 mAbs were collected from the mice with intraperitoneal injection of sterile liquid paraffin and subsequent hybridoma cells. The GP5 mAbs of ascites were purified by Protein A/G Agarose column (Santa Cruz Biotechnology, Dallas, TX, USA).
- GP5 indirect enzyme-linked immunosorbent assay (ELISA)
The indirect ELISA was used to screen hybridoma cell clones secreting antibodies against the GP5 protein or to measure the titration of ascites mAbs. Briefly, 96-well plates were coated with 0.625 μg/mL recombinant GP5 truncation protein (100 μL/well) overnight at 4 °C. The plates were washed three times with PBST (1×phosphate-buffered saline containing 0.05% Tween-20) and blocked with 5% skim milk for 2 h. Then, hybridoma culture supernatants (100 μL/well) were added to each well and incubated for 1 h at 37 °C. After washed three times, horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (1:10,000 dilution, 100 μL/well, TransGen Biotech, Beijing, China) was added and incubated for 1 h at 37 °C. The substrate 3,3’,5,5’-tetramethylbenzidine (TMB) (50 μL/well, Beyotime, Beijing, China) was added and incubated for 15 min in the dark. Finally, 2M H2SO4 (50 μL/well) was added to terminate the reaction, followed by reading of the absorbance at 450 nm (OD450) with a spectrophotometer (ALL-SHENG, Hangzhou, China). The ratios of hybridoma culture supernatants to negative SP2/0 supernatant (P/N) were calculated, with P/N ≥ 2.1 as positive.
- Western blotting (WB)
Cells were lysed in radio-immunoprecipitation assay (RIPA) buffer and protein samples were harvested by centrifugation. The protein samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with 5% skim milk solution at room temperature (RT) for 1 h, membranes were incubated with appropriate primary antibodies (1:1000) at 4 °C overnight and then with secondary antibody HRP-conjugated goat anti-mouse IgG (1:10000, TransGen Biotech, Beijing, China) at 37 °C for 1 h. Finally, the membrane was exposed with enhanced chemiluminescence (ECL) substrate (Tanon, Shanghai, China). Images were taken by imaging system (Tanon, Shanghai, China).
- Immunofluorescence assay (IFA)
3D4/21-CD163 cells seeded in 12-well plates were infected with SD17-38 strain at 0.1 multiplicity of infection (MOI) for 72 h, whereas HEK293T cells grown in 12-well plates were transfected with eukaryotic pGFP-N1-GP5 plasmid for 24 h. Cells were washed twice with PBS, fixed with 4% paraformaldehyde (Beyotime Biotech, Shanghai, China) for 10 min, and permeabilized with 0.1% Triton X-100 for 10 min at RT. After washing, cells were blocked with 1% bovine serum albumin (BSA) at 37 °C for 1 h and incubated with the GP5 mAbs (1:200) at 4 °C overnight. After three PBS washes, the goat anti-mouse IgG (H+L) secondary antibody DyLight™ 594 (1:800, Thermo Fisher Scientific, Sunnyvale, CA, USA) was added to continue incubation away from light for 1 h. Cell nuclei were stained with 4’, 6-diamidino-2-phenylindole (DAPI, ThermoFisher Scientific) for 10 min in the dark. Signals were observed under a fluorescence microscope (Leica, SPE, Buffalo Grove, IL, USA).
- Precise identification of the mAb recognizing epitopes and the epitope conservation analysis
To identify the B-cell epitope, GP5 protein was first divided into three overlapping fragments. The reactivity of the overlapping fragments with GP5 mAbs was examined by WB. Subsequently, the reacted fragment was subjected to fine progressive truncation at both N- and C-terminal ends, and the reactivity of these truncated fragments with GP5 mAbs was examined by WB. As such, the critical amino acids at both N and C ends were identified, respectively, for reactivity with GP5 mAbs. The precise epitope sequences could be deduced and defined based on the critical amino acids at both ends. All the truncated GP5 fragments were ligated into the vector pEGFP-N1 using seamless cloning and fused with the GFP-tag. The recombinant vectors were transfected into HEK293T cells for expression of different truncated fragments. The fragment reacting with both GP5 mAb and anti-GFP mAb (TransGen Biotech) was determined as the epitope containing region.
To assess the conservation of epitopes recognized by the GP5 mAb among different PRRSV-1 and PRRSV-2 strains, the GP5 protein sequences of all PRRSV-1 and PRRSV-2 were downloaded from GenBank, and aligned with the mAb recognizing epitope using the BioEdit software. The peptides (P1, 153YRWRSP158; P2, 196QWGRP200) were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China).
- Virus neutralization test by mAbs
The purified GP5 mAbs and N mAb in our lab were all normalized to 0.1 mg/mL and used for virus neutralization test. The 10 fold serial diluted purified GP5 mAbs were mixed with an equal volume of 200 TCID50/0.1 mL virus for 1 h at 37 °C in 5% CO2. Then, the mAb treated and PBS treated viruses were added to a 96-well plate containing 3D4/21-CD163 or Marc-145 cells. At 2 h post infection, the cells were changed with media and further incubated for 70 h. The inhibition of virus infections were calculated by counting the IFA positive spots in SD17-38 and Anheal-1 infected 3D4/21-CD163 cells or PRRSV-specific cytopathic effect (CPE) in R98 and XJ17-5 infected Marc-145 cells. The fifty percent end points of neutralization titers were calculated and applied [25].
- Development and optimization of GP5 peptide-based ELISA
The ELISA plate wells were coated with 100 μL synthetic GP5 antigenic peptides (0.25, 0.5, 1, 2, 4, 8 ng/μL) in 0.05 M carbonate and bicarbonate buffer (pH 9.6) for overnight at 4 °C. After washing with PBST, the wells were blocked with 5% skim milk for 2 h, and incubated with the diluted PRRSV-positive pig serum samples (1:10-1:1280, 100 μL/well) for 1 h at 37 °C. Normal healthy porcine serum was used as the negative control. The secondary antibody HRP-conjugated goat anti-swine IgG (1:10,000, Proteintech, Wuhan, China) was added at 100 μL/well and incubated at 37 °C for 1 h. Then, 50 μL of 3,3’,5,5’-tetramethylbenzidine (TMB) was added into each wells and incubated for 15 min in the dark, followed by adding 50 μL of 2M H2SO4 per well to terminate the reaction. The optical density (OD) value was determined at 450 nm by a spectrophotometer (ALL-SHENG, Hangzhou, China).
To achieve good peptide-based ELISA performance, various experimental conditions were optimized, including the coating conditions of synthetic peptide, blocking conditions, dilution of serum, serum incubation time, secondary antibody incubation time, and developing time. The optimal conditions were determined while the largest ratios of OD450 values of the positive sera relative to negative serum (P/N) were achieved.
- Cut-off values, specificity and sensitivity of the peptide-based ELISA
The mean of the OD450 values and standard deviations (SD) of thirty PRRSV-negative pig serum samples were calculated, and the cut-off values of peptide-based ELISA were determined to be between mean + 2SD and mean + 3SD. The specificity of peptide-based ELISA was evaluated using positive pig serum samples, including PRRSV, African swine fever virus (ASFV), swine influenza virus (SIV) and porcine epidemic diarrhea virus (PEDV). The sensitivity of the peptide-based ELISA was determined by testing 2-fold serial dilutions of PRRSV-positive pig serum. All the pig serum samples were stored in our laboratory.
- Repeatability and application of the peptide-based ELISA
The repeatability of the peptide-based ELISA was assessed using five PRRSV positive serum samples and one negative serum sample. To test intra-assay repeatability, three replicates of each sample were examined using the same batch of pre-coated plates. To test the inter-assay repeatability, three batches of pre-coated ELISA plates were used to detect each samples. Based on three replications of each test, the coefficient of variation (CV) was calculated as CV = (SD/Mean) × 100%. The intra- or inter-assay repeatability of the peptide-based ELISA was evaluated according to the CV.
The 81 clinical serum samples from PRRSV-negative pigs and PRRSV-positive pigs were tested by the developed peptide-based ELISA and IDEXX PRRSV X3 Ab ELISA (IDEXX, Westbrook, ME, USA). The overall agreement between the peptide-based ELISA and IDEXX PRRSV X3 Ab ELISA was evaluated.
- Statistical analysis
The data were expressed as means ± SD (n=3). Results were analyzed by GraphPad Prism 8.0 software (San Diego, CA, USA). Statistical analyses were performed by Student t test. A p value less than 0.05 was considered statistically significant.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. Isotype identification of GP5-specific mAbs. The commercial mouse monoclonal antibody isotype identification kit (CELLWAY-LAB, Luoyang China) was used to perform isotype determination of mAbs 3A8, 11E6, and 12D1. Briefly, 50 µL each mAb solutions mixed with 50 µL sample diluent from the kit was dispensed to the detection plate wells, with each mAb 6 replicate wells (100 µL/well). In parallel, the positive and negative solutions from the kit were also dispensed, with each 6 replicates. After incubating at 37°C for 30 min, the plate was washed 5 times and the 6 types of enzyme-linked secondary antibodies were added into the wells of each samples, accordingly (100 µL/well). After incubating at 37°C for 30 min, the plate was washed, and the substrates A and B from the kit were added (50 µL each) for the chromogenic reaction at 37°C for 20 min in the dark. The reaction was stopped by the stopping solution (50 µL/well) and the optical density at 450 nm (OD450) was measured by a spectrophotometer (ALL-SHENG, Hangzhou, China). The mAb isotype was determined based on the higher OD450 value of the exact type of 6 secondary antibodies. The color reaction image was taken (A) and the OD450 values was shown (B). Based on the reaction results, the isotypes of GP5 mAbs 3A8, 11E6, and 12D1 were IgG1, IgG2b, and IgG2a, respectively. Figure S2. Precise identification of the linear B cell epitopes recognized by the GP5 mAbs. The reactions of mAbs 3A8, 11E6, and 12D1 with the indicated GP5 truncated proteins beyond the Figure 5 were determined by Western blotting. The mAb reacted GP5 protein bands were marked with red stars underneath.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Acknowledgement
This work was partly supported by the Natural Science Foundation of China (32473040; 32172867), the 111 Project under Grant D18007, and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author contribution statement
JZ.Z conceived and designed the experiments; S.S, K.Z, JJ.Z, P.Z, P.H and D.D performed the experiments; S.J, W.Z, N.C and J.B provided the resources; S.S and JZ.Z wrote the paper. All authors contributed to the article and approved the submitted version.
References
- Montaner-Tarbes, S.; Del Portillo, H. A.; Montoya, M.; Fraile, L. Key Gaps in the Knowledge of the Porcine Respiratory Reproductive Syndrome Virus (PRRSV). Front Vet Sci 2019, 6, 38. [Google Scholar] [PubMed]
- Brinton, M. A.; Gulyaeva, A. A.; Balasuriya, U. B. R.; Dunowska, M.; Faaberg, K. S.; Goldberg, T.; Leung, F. C. C.; Nauwynck, H. J.; Snijder, E. J.; Stadejek, T.; Gorbalenya, A. E., ICTV Virus Taxonomy Profile: 2021. J Gen Virol 2021, 102, (8).
- Guo, Z. H.; Chen, X. X.; Li, R.; Qiao, S. L.; Zhang, G. P. The prevalent status and genetic diversity of porcine reproductive and respiratory syndrome virus in China: a molecular epidemiological perspective. Virol J 2018, 15. [Google Scholar]
- Tian, K.; Yu, X.; Zhao, T.; Feng, Y.; Cao, Z.; Wang, C.; Hu, Y.; Chen, X.; Hu, D.; Tian, X.; Liu, D.; Zhang, S.; Deng, X.; Ding, Y.; Yang, L.; Zhang, Y.; Xiao, H.; Qiao, M.; Wang, B.; Hou, L.; Wang, X.; Yang, X.; Kang, L.; Sun, M.; Jin, P.; Wang, S.; Kitamura, Y.; Yan, J.; Gao, G. F. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PLoS One 2007, 2(6), e526. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Q.; Cao, Z.; Tang, Y. D.; Xia, D.; Wang, G.; Shan, H. Recent Advances in Porcine Reproductive and Respiratory Syndrome Virus NADC30-Like Research in China: Molecular Characterization, Pathogenicity, and Control. Front Microbiol 2021, 12, 791313. [Google Scholar] [CrossRef]
- Adams, M. J.; Lefkowitz, E. J.; King, A. M. Q.; Harrach, B.; Harrison, R. L.; Knowles, N. J.; Kropinski, A. M.; Krupovic, M.; Kuhn, J. H.; Mushegian, A. R.; Nibert, M.; Sabanadzovic, S.; Sanfacon, H.; Siddell, S. G.; Simmonds, P.; Varsani, A.; Zerbini, F. M.; Gorbalenya, A. E.; Davison, A. J. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch Virol 2017, 162(8), 2505–2538. [Google Scholar]
- Han, M.; Yoo, D., Engineering the PRRS virus genome: updates and perspectives. Vet Microbiol 2014, 174, (3-4), 279-295.
- Kappes, M. A.; Faaberg, K. S., PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 2015, 479-480, 475-86.
- Luo, Q.; Zheng, Y.; Zhang, H.; Yang, Z.; Sha, H.; Kong, W.; Zhao, M.; Wang, N., Research Progress on Glycoprotein 5 of Porcine Reproductive and Respiratory Syndrome Virus. Animals (Basel) 2023, 13, (5).
- Johnson, C. R.; Griggs, T. F.; Gnanandarajah, J.; Murtaugh, M. P. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J Gen Virol 2011, 92, 1107–1116. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, Z. W.; Xu, T.; Zhou, Y. C.; Li, J. L.; Deng, H. D.; Li, F. Q.; Xu, L.; Sun, X. A.; Zhu, L., Molecular Characterization of the Nsp2 and ORF5s of PRRSV Strains in Sichuan China during 2012-2020. Animals-Basel 2022, 12, (23).
- Do, V. T.; Dao, H. T.; Hahn, T. W., Generation of a cold-adapted PRRSV with a nucleotide substitution in the ORF5 and numerous mutations in the hypervariable region of NSP2. J Vet Sci 2020, 21, (6).
- Liu, X. W.; Liu, X.; Bai, J.; Gao, Y. N.; Song, Z. B.; Nauwynck, H.; Wang, X. W.; Yang, Y. Q.; Jiang, P., Glyceraldehyde-3-Phosphate Dehydrogenase Restricted in Cytoplasmic Location by Viral GP5 Facilitates Porcine Reproductive and Respiratory Syndrome Virus Replication via Its Glycolytic Activity. J Virol 2021, 95, (18).
- Popescu, L. N.; Trible, B. R.; Chen, N. H.; Rowland, R. R. R. GP5 of porcine reproductive and respiratory syndrome virus (PRRSV) as a target for homologous and broadly neutralizing antibodies. Veterinary Microbiology 2017, 209, 90–96. [Google Scholar]
- Pan, J.; Zeng, M.; Zhao, M.; Huang, L. Research Progress on the detection methods of porcine reproductive and respiratory syndrome virus. Front Microbiol 2023, 14, 1097905. [Google Scholar]
- Lopez, O. J.; Osorio, F. A. Role of neutralizing antibodies in PRRSV protective immunity. Vet Immunol Immunopathol 2004, 102(3), 155–63. [Google Scholar] [CrossRef]
- Su, J.; Zhou, L.; He, B.; Zhang, X.; Ge, X.; Han, J.; Guo, X.; Yang, H. Nsp2 and GP5-M of Porcine Reproductive and Respiratory Syndrome Virus Contribute to Targets for Neutralizing Antibodies. Virol Sin 2019, 34(6), 631–640. [Google Scholar]
- Z.J., L.; J.F., T.; F., L.; Z.Y., W.; Y.R., D., Expression of GP5 protein of porcine reproductive and respiratory syndrome virus and establishment of indirect ELISA for antibody detection. Chin. J. Prev. Vet. Med. 2018, 40:, 24–28.
- H.F., H.; X.W., L.; J., B.; Z.B., S.; P., J., Establishment and application of indirect ELISA for detection of GP5 protein antibody of porcine reproductive and respiratory syndrome virus. Chin. Vet. Sci. 2018, 48, 1086–1092.
- Chen, Y.; Tian, H.; He, J. H.; Wu, J. Y.; Shang, Y. J.; Liu, X. T. Indirect ELISA with recombinant GP5 for detecting antibodies to porcine reproductive and respiratory syndrome virus. Virol Sin 2011, 26(1), 61–6. [Google Scholar]
- Sun, S.; Zhang, K.; Zhang, J.; He, P.; Zhang, P.; Deng, D.; Chi, C.; Jiang, S.; Zheng, W.; Chen, N.; Zhu, J. A nucleocapsid monoclonal antibody based sandwich ELISA for the general detection of both PRRSV-2 and PRRSV-1. Vet Microbiol 2025, 302, 110399. [Google Scholar]
- Chang, H.; Gao, X.; Wu, Y.; Wang, F.; Lai, M.; Zheng, J.; Qiu, Y.; He, Y.; Liang, X.; Yuan, K.; Lin, L.; Zhao, H.; Zhang, G.; Li, Q.; Sun, Y. Genomic and pathogenicity analysis of two novel highly pathogenic recombinant NADC30-like PRRSV strains in China, in 2023. Microbiol Spectr 2024, 12(10), e0036824. [Google Scholar]
- Wu, Z. Y.; Chang, T.; Wang, D. C.; Zhang, H. L.; Liu, H. Z.; Huang, X. Y.; Tian, Z. J.; Tian, X. X.; Liu, D.; An, T. Q.; Yan, Y., Genomic surveillance and evolutionary dynamics of type 2 porcine reproductive and respiratory syndrome virus in China spanning the African swine fever outbreak. Virus Evol 2024, 10, (1).
- Zhang, L.; Feng, Y.; Martin, D. P.; Chen, J.; Ma, S.; Xia, P.; Zhang, G. Genetic diversity and phylogenetic analysis of the ORF5 gene of PRRSV from central China. Res Vet Sci 2017, 115, 226–234. [Google Scholar] [PubMed]
- Tian, H.; Cheng, Y.; Wu, J. Y.; He, J. H.; Shang, Y. J.; Liu, X. T. Application of GP5 protein to develop monoclonal antibody against porcine reproductive and respiratory syndrome virus. Virol Sin 2011, 26(4), 267–72. [Google Scholar]
- Plagemann, P. G. The primary GP5 neutralization epitope of North American isolates of porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol 2004, 102(3), 263–75. [Google Scholar]
- Ostrowski, M.; Galeota, J. A.; Jar, A. M.; Platt, K. B.; Osorio, F. A.; Lopez, O. J. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J Virol 2002, 76(9), 4241–50. [Google Scholar]
- de Lima, M.; Pattnaik, A. K.; Flores, E. F.; Osorio, F. A. Serologic marker candidates identified among B-cell linear epitopes of Nsp2 and structural proteins of a North American strain of porcine reproductive and respiratory syndrome virus. Virology 2006, 353(2), 410–21. [Google Scholar]
- Liu, D.; Chen, Y. Epitope screening and vaccine molecule design of PRRSV GP3 and GP5 protein based on immunoinformatics. J Cell Mol Med 2024, 28(3), e18103. [Google Scholar]
- Zhou, Y. J.; Yu, H.; Tian, Z. J.; Liu, J. X.; An, T. Q.; Peng, J. M.; Li, G. X.; Jiang, Y. F.; Cai, X. H.; Xue, Q.; Wang, M.; Wang, Y. F.; Tong, G. Z., Monoclonal antibodies and conserved antigenic epitopes in the C terminus of GP5 protein of the North American type porcine reproductive and respiratory syndrome virus. Vet Microbiol 2009, 138, (1-2), 1-10.
- Fan, B.; Liu, X.; Bai, J.; Zhang, T.; Zhang, Q.; Jiang, P. The amino acid residues at 102 and 104 in GP5 of porcine reproductive and respiratory syndrome virus regulate viral neutralization susceptibility to the porcine serum neutralizing antibody. Virus Res 2015, 204, 21–30. [Google Scholar]
- Wang Yanmei, G. J., Lei Liancheng, Feng Xin, Sun Changjiang, Han Wenyu, Construction and induced neutralizing antibody level by recombinant phage expression carboxy terminal of GP5 protein from porcine reproductive and respiratory syndrome virus. Chin. J. Vet. Sci. 2019, 39, (5), 830-834.
- Graham, B. S.; Gilman, M. S. A.; McLellan, J. S. Structure-Based Vaccine Antigen Design. Annu Rev Med 2019, 70, 91–104. [Google Scholar] [PubMed]
- Ren, D.; Zhang, X.; Zhang, W.; Lian, M.; Meng, X.; Li, T.; Xie, Q.; Shao, H.; Wan, Z.; Qin, A.; Gao, W.; Ye, J. A peptide-based ELISA for detection of antibodies against novel goose astrovirus type 1. J Virol Methods 2023, 312, 114646. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Li, J.; Zhang, J.; Zhang, Q.; Ma, L.; Lu, J.; Li, T.; Xie, Q.; Wan, Z.; Qin, A.; Ye, J. Research Note: A novel peptide-based ELISA for efficient detection of antibody against chicken infectious anemia virus. Poult Sci 2023, 102(1), 102284. [Google Scholar] [PubMed]
- Xu, Y.; Ye, M.; Sun, S.; Cao, Q.; Luo, J.; Wang, Y.; Zheng, W.; Meurens, F.; Chen, N.; Zhu, J., CD163-Expressing Porcine Macrophages Support NADC30-like and NADC34-like PRRSV Infections. Viruses 2022, 14, (9).
- Chen, N.; Ye, M.; Li, S.; Huang, Y.; Zhou, R.; Yu, X.; Tian, K.; Zhu, J. Emergence of a novel highly pathogenic recombinant virus from three lineages of porcine reproductive and respiratory syndrome virus 2 in China 2017. Transbound Emerg Dis 2018, 65(6), 1775–1785. [Google Scholar]
- Chen, N.; Ye, M.; Huang, Y.; Li, S.; Xiao, Y.; Li, X.; Li, S.; Li, X.; Yu, X.; Tian, K.; Zhu, J., Identification of Two Porcine Reproductive and Respiratory Syndrome Virus Variants Sharing High Genomic Homology but with Distinct Virulence. Viruses 2019, 11, (9).
- Li, C.; Qiu, M.; Li, S. B.; Sun, Z.; Huang, Z. T.; Qi, W. H.; Qiu, Y. J.; Li, J. X.; Feng, B. H.; Zhao, D. S.; Lin, H.; Zheng, W. L.; Yu, X. L.; Tian, K. G.; Fan, K. W.; Zhu, J. Z.; Chen, N. H., Metagenomic and Pathogenic Assessments Identify a Pathogenic Porcine Reproductive and Respiratory Syndrome Virus 1 with New Deletions from Adult Slaughter Pig in 2022. Transboundary and Emerging Diseases 2023, 2023.
- Chen, N.; Liu, Q.; Qiao, M.; Deng, X.; Chen, X.; Sun, M. Whole genome characterization of a novel porcine reproductive and respiratory syndrome virus 1 isolate: Genetic evidence for recombination between Amervac vaccine and circulating strains in mainland China. Infect Genet Evol 2017, 54, 308–313. [Google Scholar]
Figure 1.
Expression and purification of recombinant PRRSV GP5 truncation protein (A-C) Analysis of the PRRSV GP5 protein transmembrane regions (A), hydrophilicity (B), and B cell antigenic epitopes colored in yellow (C) using online tools, as described in Methods. The higher the score, the higher probability in A-C. (D) Signal peptide was predicted by the SignalP-5.0. SP, signal peptide. CS, cleavage site position. OTHER, no signal peptide at all. (E-G) The bacterial lysate proteins were detected by SDS-PAGE (E), by Western blotting with anti-HA mAb (F) and anti-His mAb (G). (H) The purified GP5 protein was verified by SDS-PAGE different amounts of BSA as the controls. Lanes 1: pDEST527 vector transformed BL21; lanes 2: pDEST527-GP5 transformed BL21; lanes 3, 6, 9: pDEST527-GP5 transformed BL21 induced by IPTG at 16 °C for 12 h; lanes 4, 7, 10: pDEST527-N transformed BL21 induced by IPTG at 16 °C for 24 h; lanes 5, 8, 11: pDEST527-N transformed BL21 induced by IPTG at 37 °C for 12 h. Lanes 1-5: whole bacterial lysates; lanes 6-8, the supernatants of lysed bacteria; lanes 9-11, the precipitates of lysed bacteria. Lane 12: purified recombinant GP5 protein. M: protein marker. The greater expressed GP5 in the inclusion body and the purified GP5 are red box marked.
Figure 1.
Expression and purification of recombinant PRRSV GP5 truncation protein (A-C) Analysis of the PRRSV GP5 protein transmembrane regions (A), hydrophilicity (B), and B cell antigenic epitopes colored in yellow (C) using online tools, as described in Methods. The higher the score, the higher probability in A-C. (D) Signal peptide was predicted by the SignalP-5.0. SP, signal peptide. CS, cleavage site position. OTHER, no signal peptide at all. (E-G) The bacterial lysate proteins were detected by SDS-PAGE (E), by Western blotting with anti-HA mAb (F) and anti-His mAb (G). (H) The purified GP5 protein was verified by SDS-PAGE different amounts of BSA as the controls. Lanes 1: pDEST527 vector transformed BL21; lanes 2: pDEST527-GP5 transformed BL21; lanes 3, 6, 9: pDEST527-GP5 transformed BL21 induced by IPTG at 16 °C for 12 h; lanes 4, 7, 10: pDEST527-N transformed BL21 induced by IPTG at 16 °C for 24 h; lanes 5, 8, 11: pDEST527-N transformed BL21 induced by IPTG at 37 °C for 12 h. Lanes 1-5: whole bacterial lysates; lanes 6-8, the supernatants of lysed bacteria; lanes 9-11, the precipitates of lysed bacteria. Lane 12: purified recombinant GP5 protein. M: protein marker. The greater expressed GP5 in the inclusion body and the purified GP5 are red box marked.
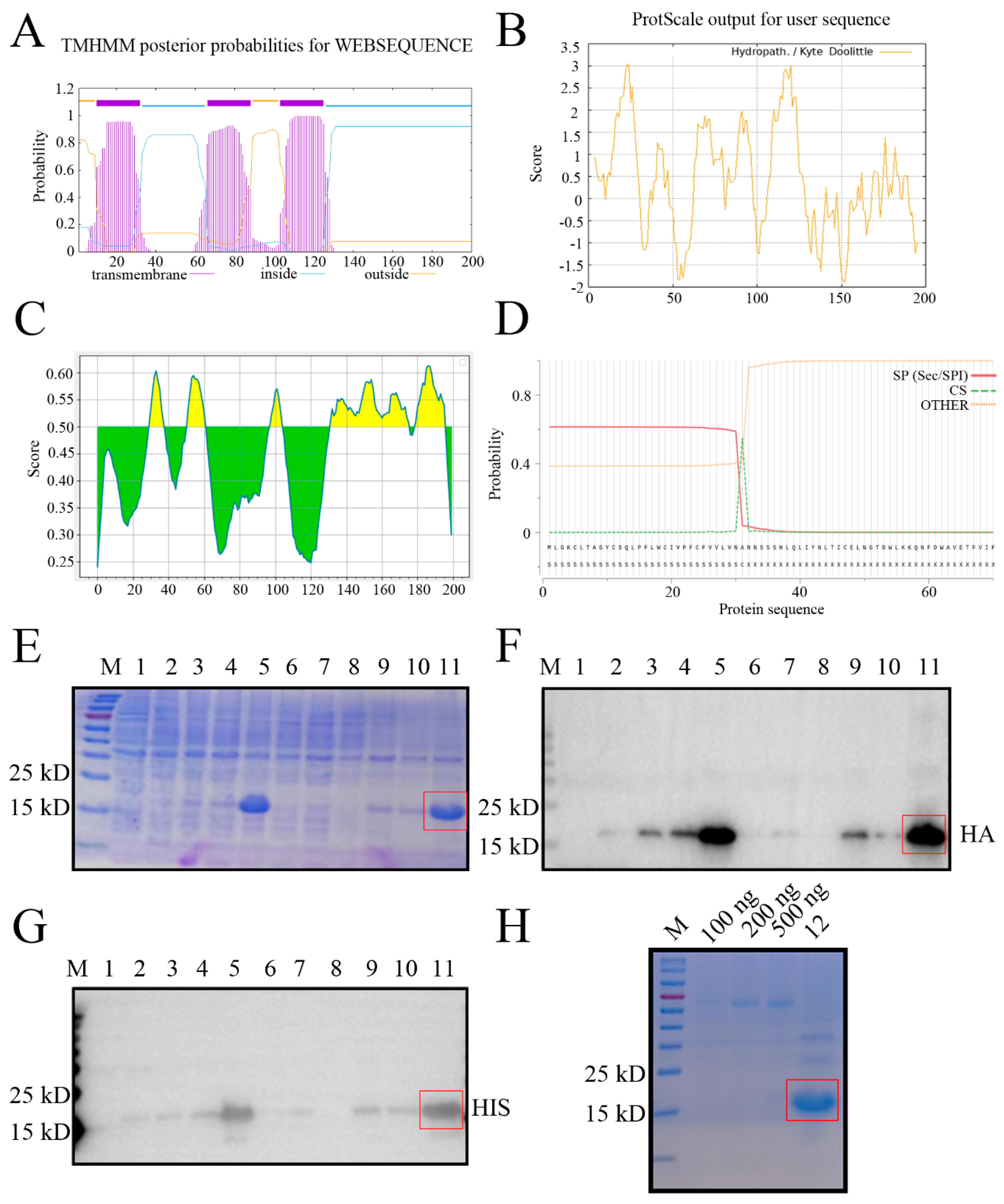
Figure 2.
Identification of GP5 specific mAbs by Western blotting (WB) (A) Detection of GP5 antibodies in supernatants from the hybridoma cell clones (3A8, 11E6 and 12D1) by indirect ELISA. Serum from immunized mice was used as the positive control and SP2/0 cell supernatant was used as the negative control. ***, p < 0.001 vs negative control. (B) Determination of the titers of ascite mAbs (3A8, 11E6 and 12D1) through GP5 indirect ELISA. The ascite fluids were subjected to 2-fold serial dilutions, and the normal mouse serum was used as negative control. (C-D) WB was used to confirm the reactivity of GP5 mAbs (3A8, 11E6 and 12D1) with GP5 in HEK293T cells transfected with pCAGGS-GP5-HA or pCAGGS-HA vector control (C), and to verify the reactivity of GP5 in with 3D4/21-CD163 cells mock infected or infected with 0.1 MOI SD17-38 (D).
Figure 2.
Identification of GP5 specific mAbs by Western blotting (WB) (A) Detection of GP5 antibodies in supernatants from the hybridoma cell clones (3A8, 11E6 and 12D1) by indirect ELISA. Serum from immunized mice was used as the positive control and SP2/0 cell supernatant was used as the negative control. ***, p < 0.001 vs negative control. (B) Determination of the titers of ascite mAbs (3A8, 11E6 and 12D1) through GP5 indirect ELISA. The ascite fluids were subjected to 2-fold serial dilutions, and the normal mouse serum was used as negative control. (C-D) WB was used to confirm the reactivity of GP5 mAbs (3A8, 11E6 and 12D1) with GP5 in HEK293T cells transfected with pCAGGS-GP5-HA or pCAGGS-HA vector control (C), and to verify the reactivity of GP5 in with 3D4/21-CD163 cells mock infected or infected with 0.1 MOI SD17-38 (D).
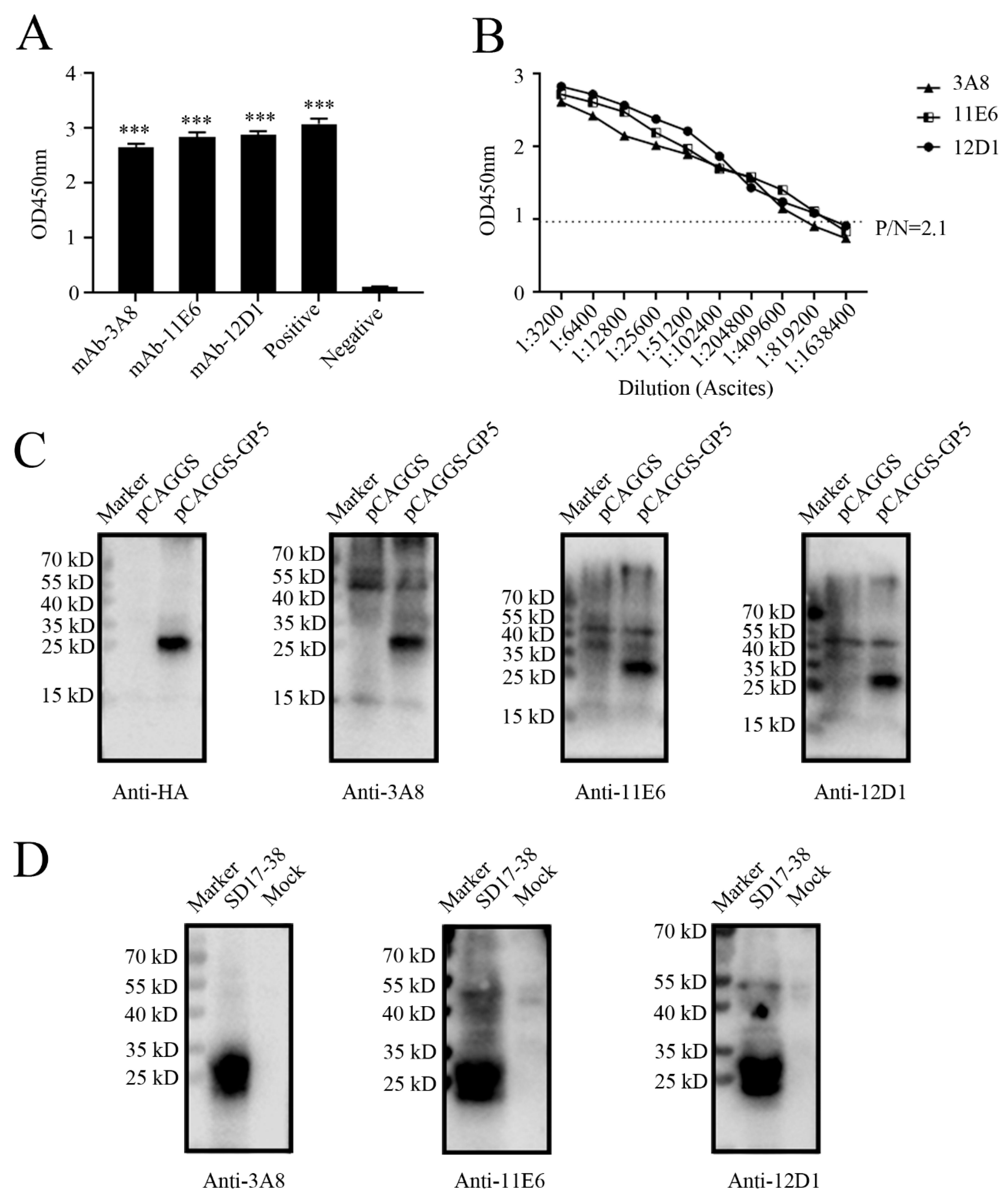
Figure 3.
Specific reactivity of GP5 mAbs with the ectopic GP5 in IFA. HEK293T cells were transfected with pEGFP-N1-GP5 or pEGFP-N1 vector for 24 h. After fixation, cells were stained with GP5 mAbs 3A8 (A), 11E6 (B), and 12D1 (C), together with anti-mouse IgG (H+L) secondary antibody DyLight™ 594 (red). Cell nuclei were counterstained with DAPI (blue). The boxed areas are magnified to place on the upper-right corners of the merged images, clearly illustrating that GP5 protein is located in the cytoplasm. Scar bar, 100 μm.
Figure 3.
Specific reactivity of GP5 mAbs with the ectopic GP5 in IFA. HEK293T cells were transfected with pEGFP-N1-GP5 or pEGFP-N1 vector for 24 h. After fixation, cells were stained with GP5 mAbs 3A8 (A), 11E6 (B), and 12D1 (C), together with anti-mouse IgG (H+L) secondary antibody DyLight™ 594 (red). Cell nuclei were counterstained with DAPI (blue). The boxed areas are magnified to place on the upper-right corners of the merged images, clearly illustrating that GP5 protein is located in the cytoplasm. Scar bar, 100 μm.
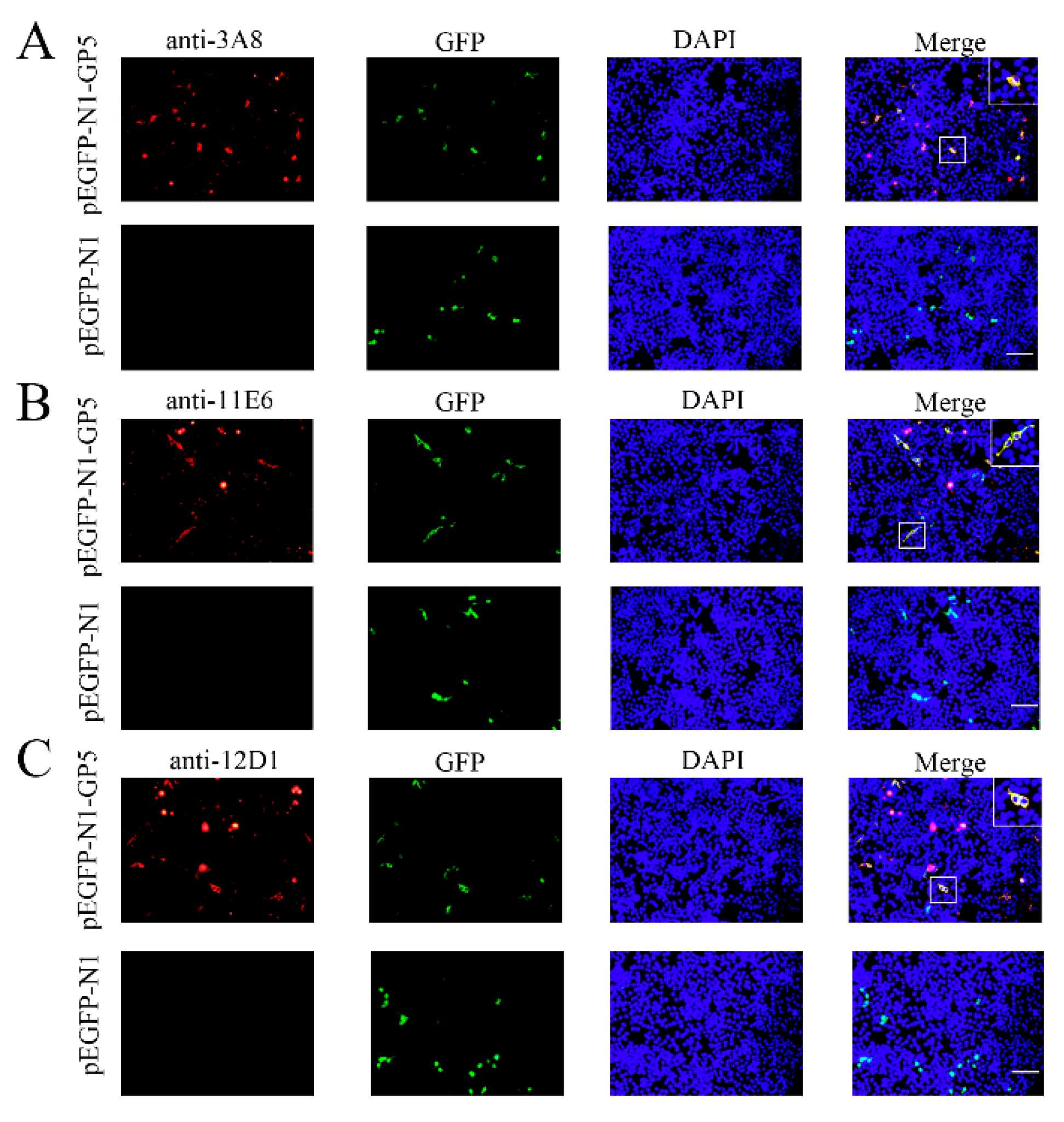
Figure 4.
Specific reactivity of GP5 mAbs with PRRSV expressed GP5 in IFA 3D4/21-CD163 cells were mock infected or infected with 0.1 MOI PRRSV-2 SD17-38 for 72 h. After fixation, the cells were stained with GP5 mAbs 3A8 (A), 11E6 (B), and 12D1 (C), together with anti-mouse IgG (H+L) secondary antibody DyLight™ 594 (red). Cell nuclei were counterstained with DAPI (blue). The boxed areas are magnified to place on the upper-right corners of the merged images, clearly illustrating that GP5 protein is located in the cytoplasm. Scar bar, 100 μm.
Figure 4.
Specific reactivity of GP5 mAbs with PRRSV expressed GP5 in IFA 3D4/21-CD163 cells were mock infected or infected with 0.1 MOI PRRSV-2 SD17-38 for 72 h. After fixation, the cells were stained with GP5 mAbs 3A8 (A), 11E6 (B), and 12D1 (C), together with anti-mouse IgG (H+L) secondary antibody DyLight™ 594 (red). Cell nuclei were counterstained with DAPI (blue). The boxed areas are magnified to place on the upper-right corners of the merged images, clearly illustrating that GP5 protein is located in the cytoplasm. Scar bar, 100 μm.
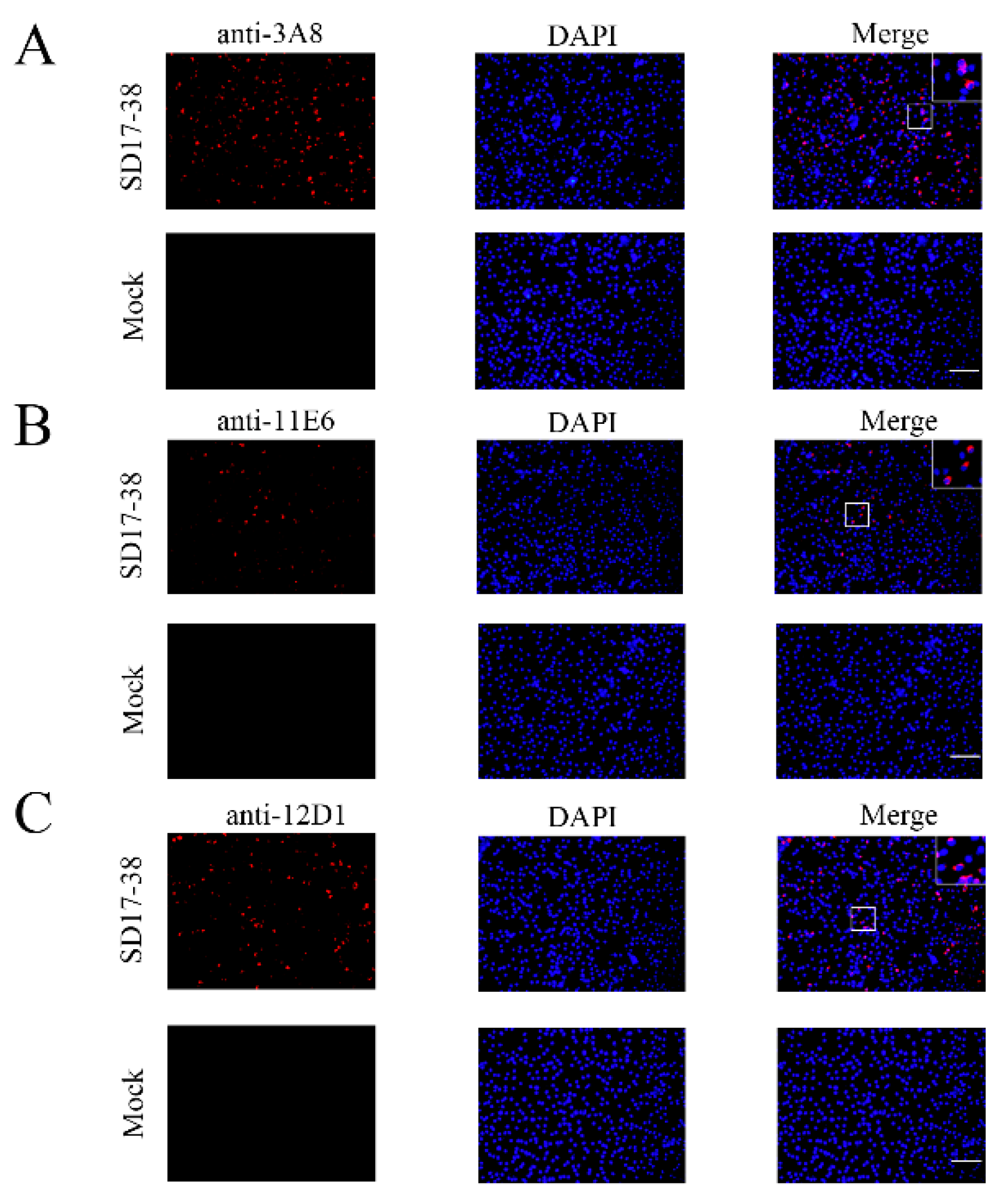
Figure 5.
Precise identification of linear B cell epitopes recognized by GP5 mAbs (A) Schematic diagram of linear B cell epitope precise mapping by GP5 mAb 3A8. The precise epitope (155WR156) recognized by GP5 mAb was shown. WB detected the reactivity of GP5 mAb 3A8 with GP5 protein with and without the critical amino acids at both N and C terminal ends. (B) Schematic diagram of linear B cell epitope precise mapping by GP5 mAbs 11E6 and 12D1. The precise epitope (196QWGRP200) recognized by both GP5 mAbs was shown. WB detected the reactivity of GP5 mAbs 11E6 and 12D1 with GP5 protein with and without the critical amino acids at both N and C terminal ends. The mAb reacted GP5 protein bands were marked with red stars underneath.
Figure 5.
Precise identification of linear B cell epitopes recognized by GP5 mAbs (A) Schematic diagram of linear B cell epitope precise mapping by GP5 mAb 3A8. The precise epitope (155WR156) recognized by GP5 mAb was shown. WB detected the reactivity of GP5 mAb 3A8 with GP5 protein with and without the critical amino acids at both N and C terminal ends. (B) Schematic diagram of linear B cell epitope precise mapping by GP5 mAbs 11E6 and 12D1. The precise epitope (196QWGRP200) recognized by both GP5 mAbs was shown. WB detected the reactivity of GP5 mAbs 11E6 and 12D1 with GP5 protein with and without the critical amino acids at both N and C terminal ends. The mAb reacted GP5 protein bands were marked with red stars underneath.
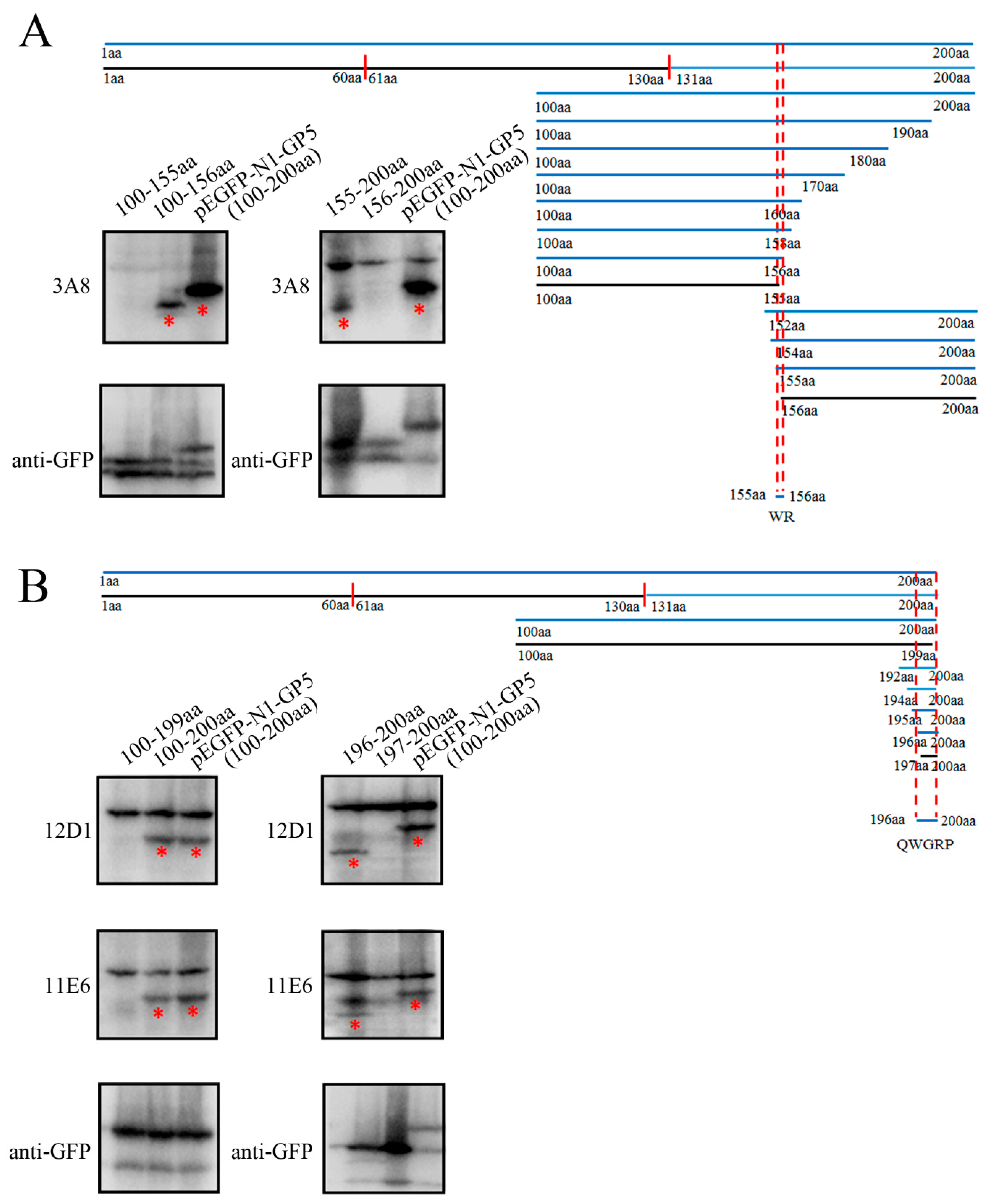
Figure 6.
Conservation analysis of the identified antigenic epitopes across different PRRSV strains. The GP5 protein sequences of PRRSV strains were downloaded from GenBank database, and BioEdit software was used for the alignment analysis as reference to the two identified GP5 epitope sequences. The epitope-containing regions are boxed areas. “-” indicated the identical amino acids. Colored letters indicated the different amino acids. The corresponding mAbs recognizing the epitopes are marked at the bottom. The alignment of representative PRRSV strains was shown, including those of PRRSV-1 subtypes 1-3, and PRRSV-2 lineages 1, 3, 5 and 8, with their corresponding names and GenBank accession numbers. The alignment to complete list of PRRSV GP5 sequences are shown in Table S1.
Figure 6.
Conservation analysis of the identified antigenic epitopes across different PRRSV strains. The GP5 protein sequences of PRRSV strains were downloaded from GenBank database, and BioEdit software was used for the alignment analysis as reference to the two identified GP5 epitope sequences. The epitope-containing regions are boxed areas. “-” indicated the identical amino acids. Colored letters indicated the different amino acids. The corresponding mAbs recognizing the epitopes are marked at the bottom. The alignment of representative PRRSV strains was shown, including those of PRRSV-1 subtypes 1-3, and PRRSV-2 lineages 1, 3, 5 and 8, with their corresponding names and GenBank accession numbers. The alignment to complete list of PRRSV GP5 sequences are shown in Table S1.
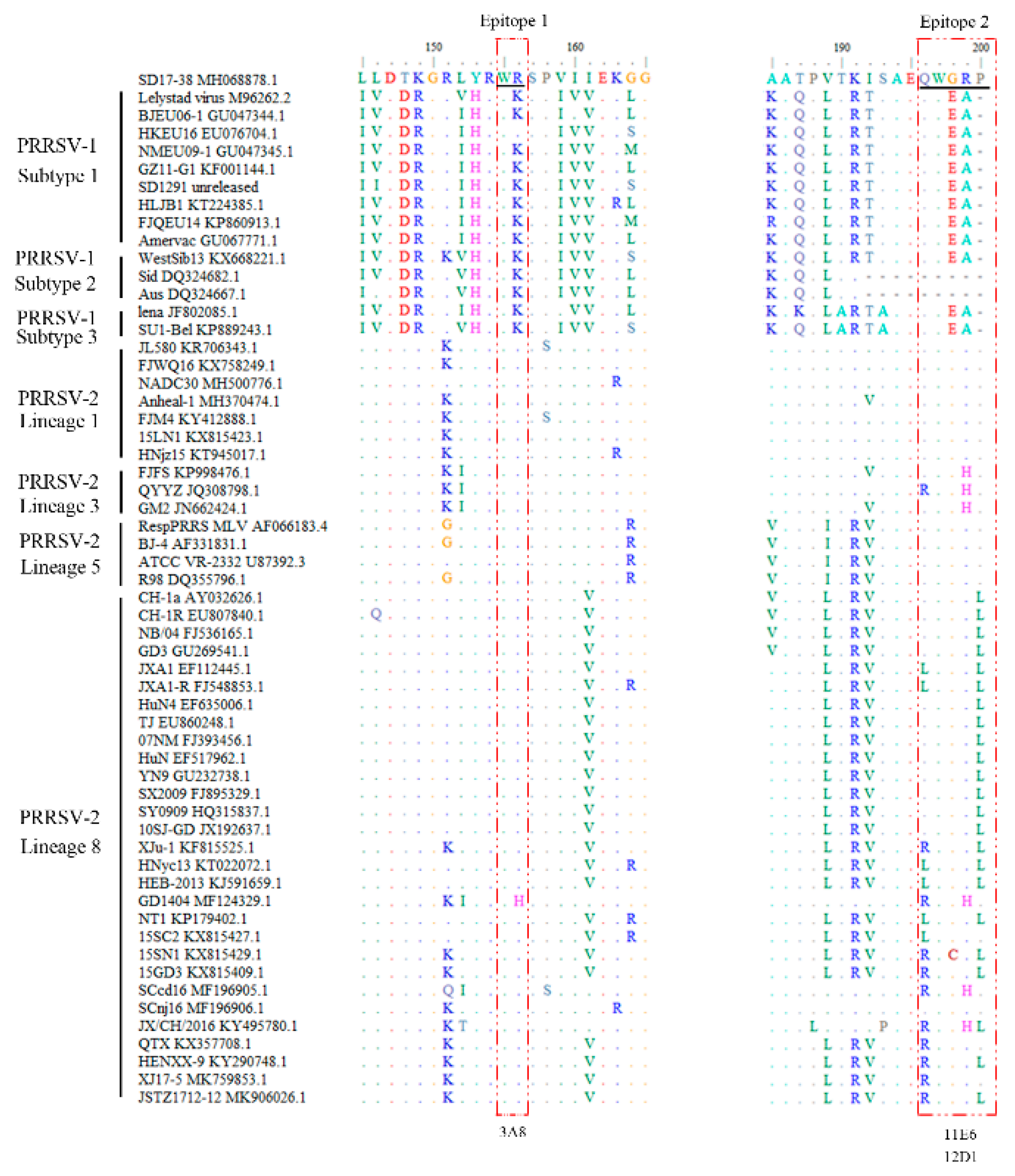
Figure 7.
Reactivity of the GP5 specific mAbs with different PRRSV strains in WB. WB was performed to detect the reactivity of GP5 mAbs 3A8 (A), 11E6 (B), and 12D1 (C) with the PRRSV-1 strains (SD1291, HLJB1), NADC30-like PRRSV-2 strain SD17-38, NADC34-like PRRSV-2 strain Anheal-1, Classic PRRSV-2 VR-2332-like strain R98, classical PRRSV-2 vaccine strain CH-1R, and HP-PRRSV-2 virulent strains XJ17-5 and avirulent JSTZ1712-12. The PRRSV infected cell lysates were used, with mock infected Marc-145 cell lysate as the negative control samples. The GAPDH incubated with the mouse mAb (1:1000, Proteintech, Wuhan, China) was used as the internal controls.
Figure 7.
Reactivity of the GP5 specific mAbs with different PRRSV strains in WB. WB was performed to detect the reactivity of GP5 mAbs 3A8 (A), 11E6 (B), and 12D1 (C) with the PRRSV-1 strains (SD1291, HLJB1), NADC30-like PRRSV-2 strain SD17-38, NADC34-like PRRSV-2 strain Anheal-1, Classic PRRSV-2 VR-2332-like strain R98, classical PRRSV-2 vaccine strain CH-1R, and HP-PRRSV-2 virulent strains XJ17-5 and avirulent JSTZ1712-12. The PRRSV infected cell lysates were used, with mock infected Marc-145 cell lysate as the negative control samples. The GAPDH incubated with the mouse mAb (1:1000, Proteintech, Wuhan, China) was used as the internal controls.
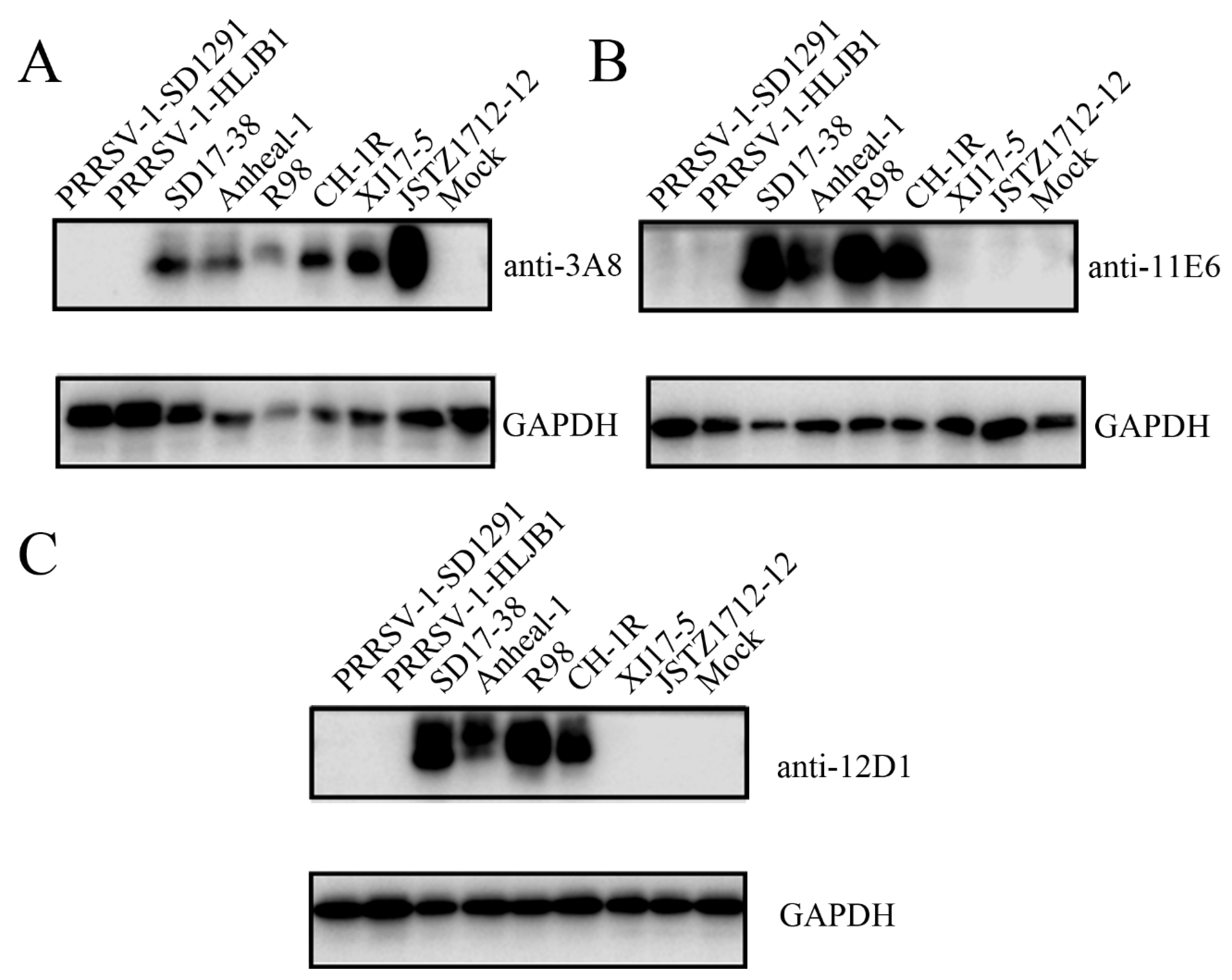
Figure 8.
Neutralizing activity of GP5 mAbs against different PRRSV strains (A) The three GP5 mAbs 3A8, 11E6 and 12D1 were purified and subjected into SDS-PAGE analysis, with the heavy chain and light chain marked, respectively. (B-E) The purified GP5 mAbs together purified N mAbs in our lab were all normalized to 0.1 mg/mL, 10 fold diluted and incubated with different PRRSV strains, SD17-38 (B), Anheal-1 (C), R98 (D), and XJ17-5 (E), followed with infection of 3D4/21-CD163 cells (B and C) and Marc-145 cells (D and E) for 72 h. The medium incubated PRRSVs were used as the controls for infection of 3D4/21-CD163 cells and Marc-145 cells. The infected 3D4/21-CD163 cells were stained with N mAb (6D7) and the goat anti-mouse IgG (H+L) secondary antibody DyLight™ 594 (1:800, Thermo Fisher Scientific) to measure the inhibition of virus infection relative to control infections. The infected Marc-145 cells were observed for CPE to measure the inhibition of the virus infection relative to control infection. The fifty percent end points of neutralization are marked as dotted lines.
Figure 8.
Neutralizing activity of GP5 mAbs against different PRRSV strains (A) The three GP5 mAbs 3A8, 11E6 and 12D1 were purified and subjected into SDS-PAGE analysis, with the heavy chain and light chain marked, respectively. (B-E) The purified GP5 mAbs together purified N mAbs in our lab were all normalized to 0.1 mg/mL, 10 fold diluted and incubated with different PRRSV strains, SD17-38 (B), Anheal-1 (C), R98 (D), and XJ17-5 (E), followed with infection of 3D4/21-CD163 cells (B and C) and Marc-145 cells (D and E) for 72 h. The medium incubated PRRSVs were used as the controls for infection of 3D4/21-CD163 cells and Marc-145 cells. The infected 3D4/21-CD163 cells were stained with N mAb (6D7) and the goat anti-mouse IgG (H+L) secondary antibody DyLight™ 594 (1:800, Thermo Fisher Scientific) to measure the inhibition of virus infection relative to control infections. The infected Marc-145 cells were observed for CPE to measure the inhibition of the virus infection relative to control infection. The fifty percent end points of neutralization are marked as dotted lines.
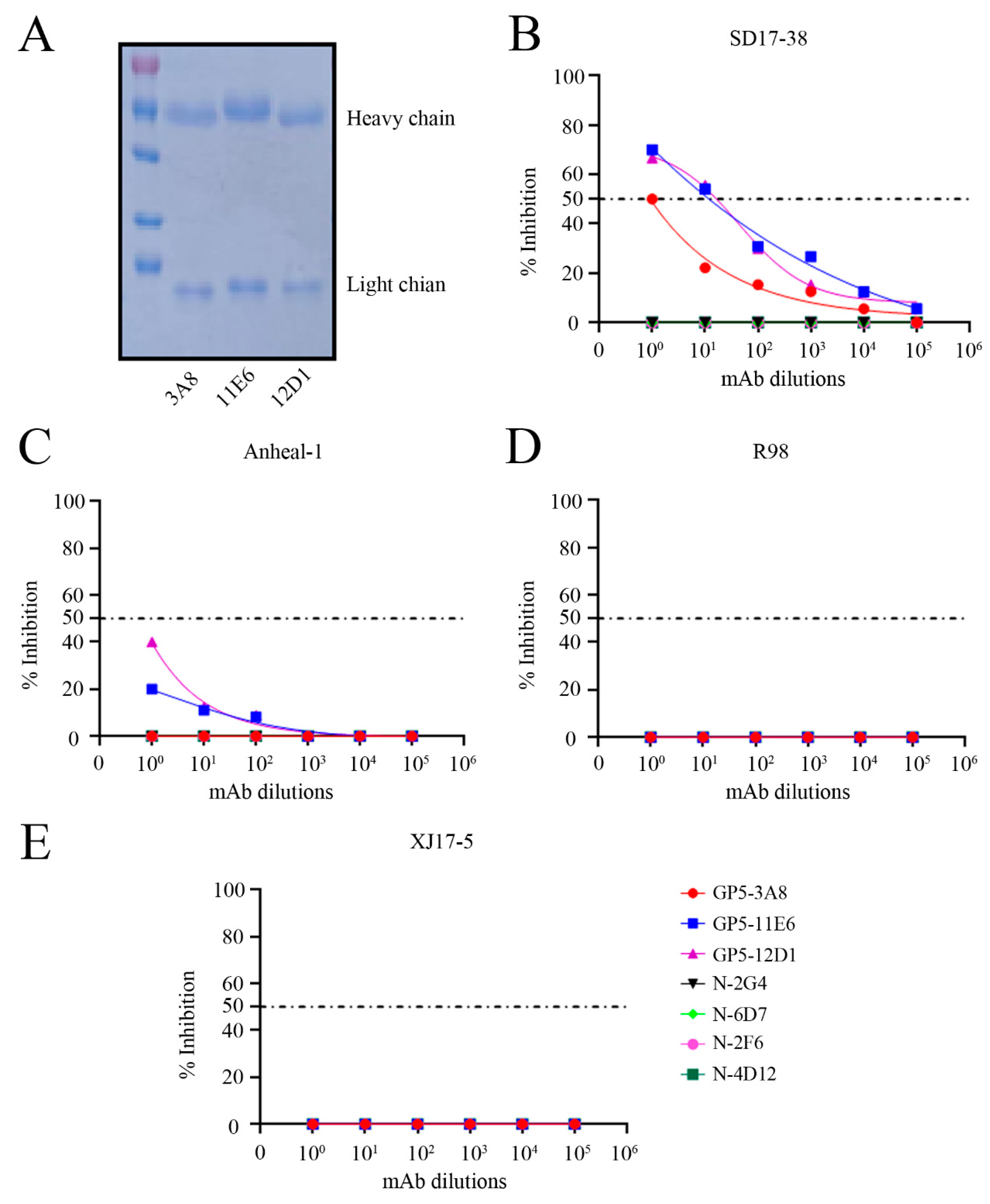
Figure 9.
Development and optimization of the peptide-based ELISA (A) Determination of the optimal ratio of two synthetic peptides P1 (153-158 aa) and P2 (196-200 aa). (B) Checkerboard assay determined the optimal working concentration of synthetic peptides and serum dilution by the peptide-based ELISA. The amounts of synthetic peptides (0.25, 0.5, 1, 2, 4, 8 ng/μL) and diluted serum (1:10, 1:20, 1:40, 1:80, 1:160, 1:320, 1:640, 1:1280) were used. (C) Determination of the optimal coating and blocking conditions. BSA, bovine serum albumin; SMP, skim milk protein. (D-F) Determination of the optimal serum incubation time (D), secondary antibody incubation time (E) and developing time (F).
Figure 9.
Development and optimization of the peptide-based ELISA (A) Determination of the optimal ratio of two synthetic peptides P1 (153-158 aa) and P2 (196-200 aa). (B) Checkerboard assay determined the optimal working concentration of synthetic peptides and serum dilution by the peptide-based ELISA. The amounts of synthetic peptides (0.25, 0.5, 1, 2, 4, 8 ng/μL) and diluted serum (1:10, 1:20, 1:40, 1:80, 1:160, 1:320, 1:640, 1:1280) were used. (C) Determination of the optimal coating and blocking conditions. BSA, bovine serum albumin; SMP, skim milk protein. (D-F) Determination of the optimal serum incubation time (D), secondary antibody incubation time (E) and developing time (F).
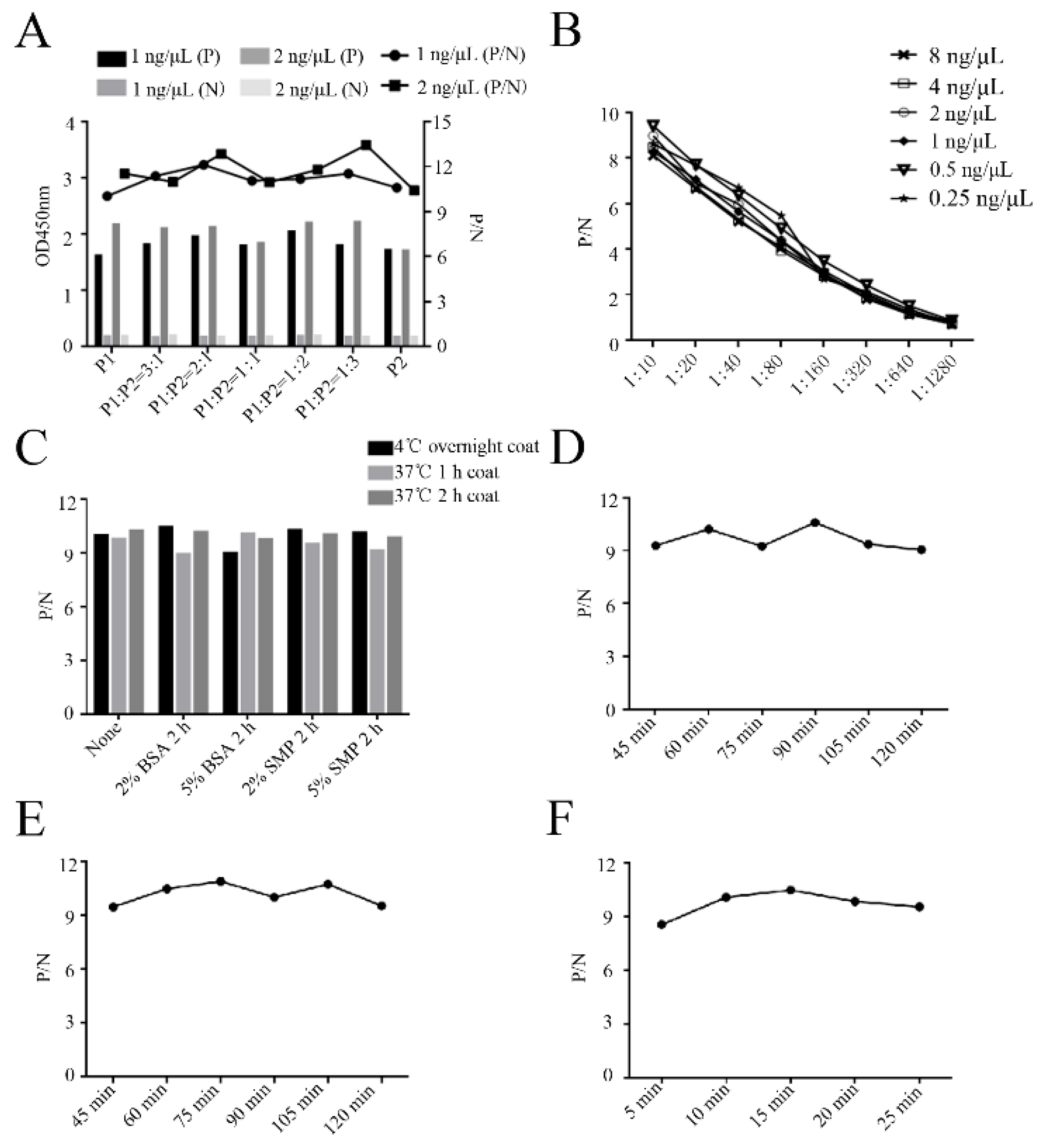
Figure 10.
Specificity, sensitivity and repeatability of the peptide-based ELISA (A) Specificity of the peptide-based ELISA was analyzed using PRRSV, ASFV, SIV, and PEDV positive sera, with normal healthy porcine serum as the negative control. (B) Sensitivity analysis of the peptide-based ELISA was analyzed using two-fold serially diluted PRRSV-positive pig serum. (C and D) Intra-assay (C) and inter-assay (D) repeatability of the peptide-based ELISA (n=3). ***, p < 0.001 vs negative control.
Figure 10.
Specificity, sensitivity and repeatability of the peptide-based ELISA (A) Specificity of the peptide-based ELISA was analyzed using PRRSV, ASFV, SIV, and PEDV positive sera, with normal healthy porcine serum as the negative control. (B) Sensitivity analysis of the peptide-based ELISA was analyzed using two-fold serially diluted PRRSV-positive pig serum. (C and D) Intra-assay (C) and inter-assay (D) repeatability of the peptide-based ELISA (n=3). ***, p < 0.001 vs negative control.
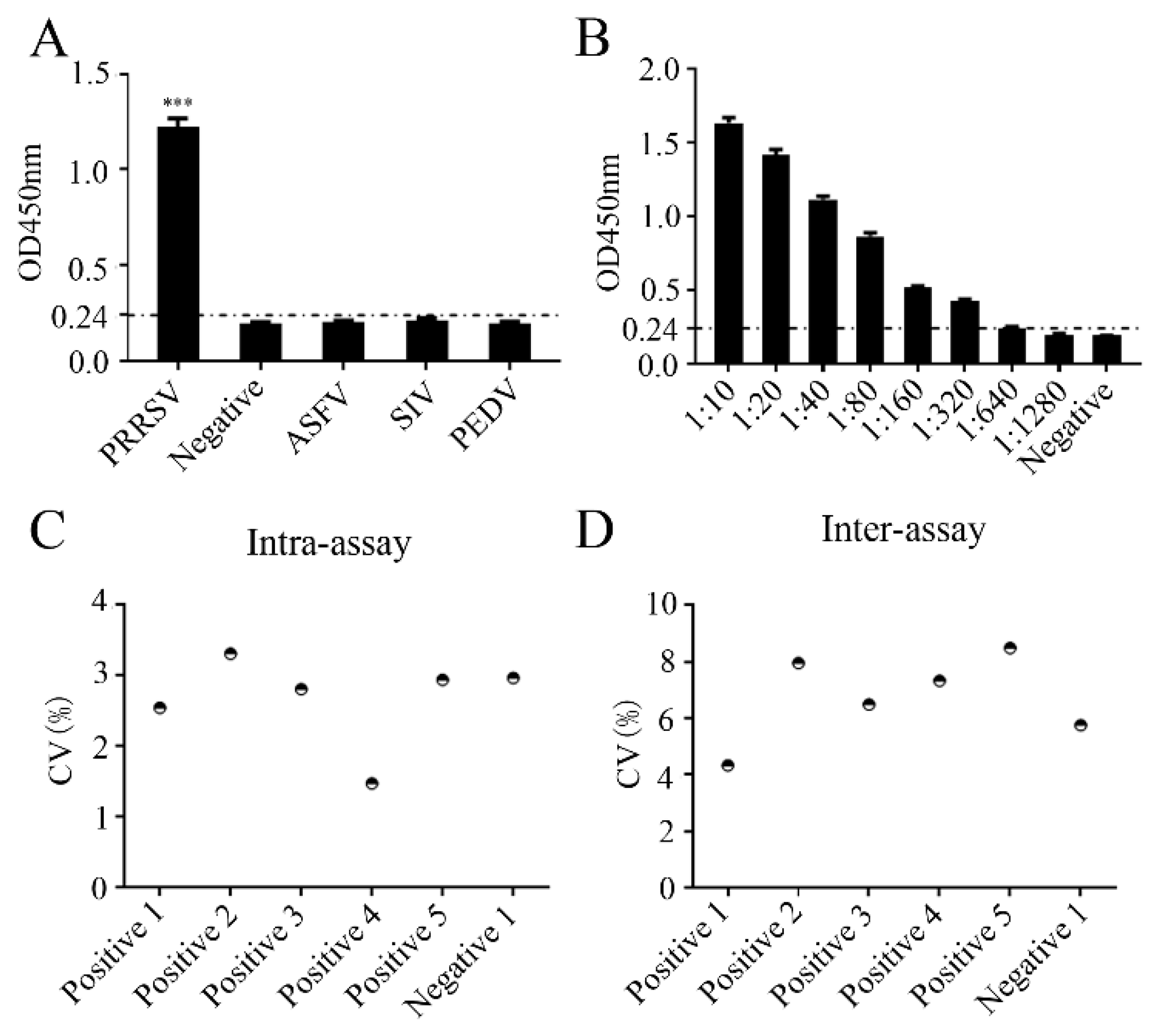

Table 1.
Detection of the serum samples using the peptide-based ELISA together with the IDEXX PRRSV X3 Ab ELISA.
Table 1.
Detection of the serum samples using the peptide-based ELISA together with the IDEXX PRRSV X3 Ab ELISA.
| Samples | Peptide-based ELISA | IDEXX | Coincidence rate (%) | ||||
| Positive | Negative | Positive | Negative | ||||
| Sera (81) | 19 (1*) | 62 | 31 (13#) | 50 | 82.7 | ||
Note: *denotes the number of positive only by peptide-based ELISA, whereas # denotes the number of positive only by IDEXX PRRSV X3 Ab ELISA.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



