Submitted:
26 February 2025
Posted:
27 February 2025
You are already at the latest version
Abstract
Aging is the result of various compounding stresses that gradually overcome the homeostatic regulation of the cell, resulting in irreversible damage. This manifests as many acute and chronic conditions, the most common of which are neurodegeneration and dementia. Epidemiological studies have shown significant, strong correlations between viral infection and neurodegenerative diseases. This review overlays the characteristics of viral pathogenesis with the hallmarks of aging to discuss how active and latent viruses contribute to aging. Through our contextualization of myriad basic science papers, we offer explanations for premature aging via viral induction of common stress response pathways. Viruses induce many stresses: dysregulated homeostasis by exogenous viral proteins and overwhelmed protein quality control mechanisms, DNA damage through direct integration and epigenetic manipulation, immune-mediated oxidative stress and immune exhaustion, and general energy theft that is amplified in an aging system. Overall, this highlights the long-term importance of vaccines and antivirals in addition to their acute benefits.
Keywords:
neurotropic viruses
; aging
; neurodegeneration
; dementia
; proteinopathy
; genome instability
; senescence
; herpesviruses
; polyomaviruses
; coronaviruses
1. Introduction
“Aging” describes the gradual damage cells acquire over time, culminating in permanently impaired function and, ultimately, death [1]. In the central nervous system, neurotropic viruses are a major stressor and, therefore, a major driver of aging. Epidemiological studies of multiple large biobanks have shown patients with a history of neurological viral infection are thirty times more likely to develop a neurodegenerative disease [2]. In addition to the pressures of viral proteins during active infection, acute and chronic viral infections disrupt the homeostasis of the cell [3]. This occurs throughout life, with acute causes of neurodegeneration (e.g., hemorrhagic or ischemic stroke, traumatic brain injury) and chronic conditions (e.g., viral latency, metabolic disease) compounding, often synergistically [4]. Here, we define aging, outline the impact viruses have on the brain, and identify the overlapping pathways of viral pathogenesis and age-related neurodegeneration. Previously proposed “Hallmarks of Aging” range in number but can be generally described by three categories: altered proteostasis, genomic compromise, and senescence (Figure 1) [1]. Neurotropic viruses manipulate each of these categories, driving rapid neurodegenerative diseases like Amyotrophic Lateral Sclerosis (ALS) and Parkinson’s Disease (PD) and more progressive neurodegenerative conditions like Alzheimer’s Disease (AD) and Frontotemporal Dementia (FTD) [5].
Although not the sole cause of aging, this review highlights the many ways in which insidious viral infections extend beyond transient colds into lasting, premature aging of the brain [6]. It is important to acknowledge that aging throughout the body has implications on the brain [7,8,9,10,11], however, this review focuses on the direct stressors enacted by non-fatal, neurotropic viruses (Table 1). This includes linear positive sense ssRNA coronaviruses, like novel coronavirus SARS-CoV-2; linear positive sense ssRNA enteroviruses coxsackievirus (CV), echovirus, and poliovirus; linear positive sense ssRNA flaviviruses Dengue, Japanese encephalitis virus (JEV), and West Nile virus (WNV); linear dsDNA herpesviruses herpes simplex 1 (HSV1), varicella zoster (VSV), Epstein Barr (EBV), and cytomegalovirus (CMV); circular dsDNA JC polyomavirus (JCV); and positive sense ssRNA retrovirus human immunodeficiency virus (HIV).
2. Altered Proteostasis
Protein Quality Control (PQC) describes the interconnected pathways of a cell that identify misfolded proteins and direct 1) refolding in ideal conditions, 2) degradation by proteolysis/autophagy for molecular recycling, or 3) controlled apoptosis in high-stress, low-nutrient states [12]. Due to its regulatory role, PQC is interconnected with nutrient sensing, mitochondrial stability, and cell cycle regulation. The most common age-related brain disorders are proteinopathies: tau and amyloid-β (Aβ) aggregation define AD, α-synuclein aggregates characterize both PD and Lewy Body dementia (LBD), and TDP-43 aggregates drive pathogenesis of ALD and FTD [13,14,15]. Evidence discussed here indicates that viral infection not only drives aggregation of proteins, but manipulates the PQC capacity of a cell, preventing the cell from reinstituting proteostasis (Table 2).

Table 1.
Neurotropic viruses implicated in aging.
| Genus | Genome | Capsid | Virus | Associated Diseases |
| Coronavirus | Linear +ssRNA |
Enveloped Icosahedral |
SARS-CoV-2 | COVID19 |
| Enteroviruses | Linear +ssRNA |
Non-enveloped Icosahedral |
Coxsackievirus | Hand-Foot-and-Mouth, Viral Meningitis |
| Echovirus | Viral Meningitis | |||
| Poliovirus | Paralytic Poliomyelitis | |||
| Flaviviruses | Linear +ssRNA |
Enveloped Dimeric αhelix |
Dengue | Breakbone Fever |
| Japanese Encephalitis | Viral Encephalitis | |||
| West Nile | Viral Encephalitis | |||
| Herpesviruses | Linear dsDNA |
Enveloped Icosahedral |
Herpes Simplex 1 | Cold Sores, Viral Encephalitis |
| Varicella Zoster | Chicken Pox, Shingles |
|||
| Epstein Barr | Cancer (Lymphoma, Leukemia, Nasopharyngeal Carcinoma), Infectious Mononucleosis, Multiple Sclerosis |
|||
| Cytomegalovirus | Congenital Birth Defects, Viral Encephalitis |
|||
| Polyomaviruses | Circular dsDNA |
Non-enveloped Icosahedral |
JC | Progressive Multifocal Leukoencephalopathy, Cancer (Glioblastoma, Colorectal Carcinoma) |
| Lentiviruses | Linear +ssRNA |
Enveloped Cone-shaped |
HIV | Acquired Immunodeficiency Syndrome, HIV-associated Neurocognitive Disorder |
2.1. Viral aggregation of intracellular Tau and extracellular Amyloid-β
Extracellular Aβ plaques and intracellular tau tangles are pathognomonic for AD [13]. In a healthy system, both Aβ and tau function as antiviral mediators [16,17]. Prior to entering the host cell, viral capsid and envelope proteins accrue an obstructive “corona” of host proteins, including Aβ, trapping the virus in the extracellular matrix (Figure 2a). This physical obstruction prevents capsid interactions with host cell membranes, impairing cell entry (Figure 2b). This antimicrobial role of Aβ is supported by an increased susceptibility to infection observed following pharmacologic reduction of Aβ [18]. In excess, however, the physical accumulation of antiviral Aβ on compatible viral proteins becomes a nidus for amyloid aggregation (Figure 2c) [19,20]; HSV1 gD, for example, is an envelope glycoprotein that directly agglutinates Aβ via intermolecular hydrogen bonding and van der Waals forces [21]. The heparin binding region of S-protein, the eponymous spike protein of SARS-CoV-2, is exceptionally aggregation prone, physically binding aggregates of Aβ in the extracellular matrix as well as tau, TDP43, and α-synuclein within the cytoplasm [22]. In fact, S-protein has such exceptional proficiency for amyloidosis, it has been shown to produce toxic aggregates within vasculature and other organs, as well as brain tissue. Another herpesvirus, CMV, utilizes this proteinopathy to its advantage, with viral protein M45 independently assembling into amyloid fibrils, impairing host access to virions and preventing virus-induced necroptosis of the host [23]. Similarly, HIV TAT – a secreted viral transcription factor – not only binds extracellular Aβ, it also synergizes with it, increasing neurotoxicity compared to Aβ alone [24,25].
When the virus achieves cell entry, host cell processing of viral DNA and proteins activates the cGAS-STING pathway, an innate antiviral response (Figure 2d) [26,27]. cGAS directly identifies abnormal DNA or proteins in the cytoplasm, triggering STING to activate NFκB and IRF3 (Figure 2e). Downstream phosphorylation of NFκB, IRF3, and tau by TBK1 enables cGAS-STING-dependent degradation of viral proteins, and cytokine activation of the immune response. This was directly evidenced with the ICP27 protein of HSV1, which triggered cGAS-STING antiviral activity as well as tauopathy in neurons and microglia of AD patients [28]. Independent of HSV1, activation of cGAS-STING, alone, is sufficient to induce tau-phosphorylation, improving neuron survivability. This suggests that phosphorylation of tau is an antiviral response requisite for the prevention of acute neurodegeneration. In an abnormal PQC state, however, chronic hyperphosphorylation of tau leads to aggregation, proteinopathy and neurodegeneration (Figure 2f). The viral nucleic acids of CMV, HIV, and SARS-CoV-2 also trigger the cGAS pathway [29,30,31]. The antiviral effect of the cGAS is underscored by the evolution of anti-cGAS viral proteins, that directly or indirectly impair its triggering of the cascade (Figure 2g). This includes CMV tegument proteins pUL31 and pUL83; Dengue NS2B, NS3, NS2B3; HIV capsid protein GAG; HSV1 tegument protein VP22; and VZV tegument protein ORF9 [32,33,34,35,36,37,38,39].
Table 2.
Mechanisms by which neurotropic viruses trigger proteinopathy .
| Disease | Proteinopathy | Pathway | Virus | Protein |
| Alzheimer’s Dementia |
Extracellular Aβ Plaques |
Amyloidogenesis | CMV | M45 |
| HIV | TAT | |||
| HSVI | gD | |||
| SARS-CoV-2 | S-protein | |||
| Intracellular Tau Tangles |
cGAS-STING | CMV | pUL31* pUL83* |
|
| HIV | GAG* | |||
| HSV1 | ICP27 VP11* |
|||
| SARS-CoV-2 | S-protein | |||
| VZV | ORF9* | |||
| Amyotrophic Lateral Sclerosis and Frontotemporal Dementia |
Cytosolic TDP43 Aggregates | RNA Translocation | CV | 2A, 2C |
| Echovirus | 2A, 2C | |||
| Poliovirus | 2A, 2C | |||
| HIV | GAG VIF |
|||
| HERV-K | ASRGL1* | |||
| SARS-CoV-2 | S-protein | |||
| Parkinson’s Disease and Lewy Body Dementia |
Lewy Body Aggregates |
Endoplasmic Reticulum Sequestration |
CMV | Envelope |
| EBV | ||||
| Dengue | ||||
| HIV | ||||
| JEV | ||||
| WNV | ||||
| SARS-CoV-2 | N-protein S-protein |
* These proteins physically obstruct cGAS access to viral nucleic acids, preventing appropriate activation of the antiviral cascade.
2.2. Viral aggregation of TDP43
TDP43 aggregates are implicated in motor neuron degeneration in ALS, as well as cortex neuron degeneration in FTD [15]. Pro-aggregatory genetic mutants of TDP43 are implicated in familial ALS and FTD, but aggregates of wildtype TDP43 have also been shown to form following viral infection including neurotropic HIV, HERV-K, SARS-Cov-2 [22,40,41]. The most established TDP43 translocating viruses are the positive-sense RNA enteroviruses including coxsackievirus, echovirus, and poliovirus [42,43,44]. Normally, transactive response DNA-binding protein 43 is a general regulator of host RNA processing in the nucleus [45]. During infection, TDP43 interaction with viral RNA results in translocation to the cytoplasm where toxic aggregates form (Figure 2j and Figure 2l) [46]. As with hyperphosphorylated tau tangles and production of Aβ fibrils, the translocation of TDP43 has been posited as an antiviral response, with aggregates obstructing viral entry into the nucleus (Figure 2k) [47]. Not only does this cytoplasmic aggregation drive specific neurodegenerative conditions ALS and FTD, but evidence also suggests TDP43 translocation can impair general memory through its proteinopathy [48]. This disruption produces an IFN response independent from cGAS-STING activation, indicating a separate proinflammatory proteinopathy from the intracellular tauopathies discussed.
2.3. Viral aggregation of α-synuclein
Aggregation of α-synuclein is the root proteinopathy of both LBD and PD [49]. Whereas LBD exhibits diffuse cytotoxic “Lewy Bodies” within cortical neurons and first present with cognitive impairments, Lewy Body localization within dopaminergic neurons of the substantia nigra presents with the rigid motor dysfunction characteristic of PD [14]. In a healthy system, α-synuclein functions to traffic synaptic vesicles for neurotransmitter release. As with Aβ, tau, and TDP43, evidence suggests α-synuclein also has an antiviral function: redirecting virion-containing endosomes to the endoplasmic reticulum (ER) (Figure 2h). In addition to genetic predisposition, viral infection has been shown to increase α-synuclein expression and aggregation (Figure 2i) [50]. Knockout models of α-synuclein produced a five-fold increase in the viral load of West Nile Virus encephalitis, whereas a healthy model exhibited a virus-stimulated increase in α-synuclein expression and accompanying sequestration of virions to the endoplasmic reticulum in a Rab1-dependent manner [51]. In addition to impairing the production of viral progeny, this enables α-synuclein modification of the PQC mechanism of the cell, preventing premature triggering of the caspase 3 dependent, intrinsic apoptotic cascade. Because the antiviral role of α-synuclein is unique to neurons, its function – and dysfunction – has been correlated with additional neurotropic viruses including other flaviviruses (i.e., dengue, JEV), herpesviruses (CMV, EBV). HIV, and SARS-CoV-2 [50,52,53,54,55]. Concordantly, treatment with antivirals has been correlated clinically with a reduced risk of Parkinsonism [56].
2.4. Viral aberration of Protein Quality Control
Presuming the tau, amyloid, TDP43, and α-synuclein responses defend against viruses under normal conditions, disruption of the PQC regulatory system must occur to shift the system from protective to pathogenic. In identifying foreign proteins, PQC redirects viral peptides toward degradation by proteolysis or autophagy [57,58]. CMV, EBV, HIV, HSV1 and SARS-CoV-2 express deubiquitinases that remove ubiquitin from proteins the host cell has marked for degradation by lysosomes or proteosomes [59].In instances where lysosomes or proteosomes do reach viral proteins, “hijacking” of PQC chaperones is a characteristic of many viruses. Neurotropic coronaviruses, enteroviruses, and polyomaviruses have all been shown to redirect lysosomes from their traditional autophagic pathways to deliver virions to the nucleus and viral progeny to the cell membrane [60,61,62]. Coronaviruses have also been shown to raise lysosome pH, impairing enzyme function, preventing degradation following endocytosis-mediated cell entry [63].
When stresses exceed the nutrient source of a cell, healthy PQC enactors shift the “cell fate” toward apoptosis [57]. By eliminating the host machinery necessary for viral gene and protein production, this pro-apoptotic shift is inherently antiviral [64]. It is therefore an evolutionary advantage for viruses to immortalize cells, prolonging access to the transcriptional and translational machinery on which they depend [65]. JCV, for example, inhibits tumor suppressors p53 and pRb through the eponymous transcription factor “large tumor antigen” [66]. This is in direct conflict with the endogenous PQC pathway, which directs cell fate toward apoptosis during the increased stress of active infection [64]. Other anti-apoptotic manipulations of PQC include: CMV inhibition of proapoptotic JNK via viral protein, pUL38; EBV sequestration of stress indicator HSP70 via viral protein, EBNA-LP; HSV1 bypassing of nutrient sensing to ensure protein expression via viral proteins gB and γ34.5; and endogenous retrovirus stimulation of stemness via OCT4 expression [65,67,68,69]. These manipulations allow metabolic stress to exceed the predetermined maximum set by PQC, further advancing aging [70]
In this section, we discuss how endogenous proteins – such as tau, Aβ, TDP43, and α-synuclein – react to viral infection and impair viral pathogenesis. It is essential to reiterate an intact PQC system can suppress acute infection and return the cell to proteostasis. In the cases discussed – chronic viral infection, pro-aggregatory antiviral mediators, or disrupted PQC – stressors can compound, resulting in permanent proteinopathy and irreversible damage. The extremes of these conditions present as common neurodegenerative diseases (e.g., AD, ALS, FTD, PD), but subclinical or undiagnosed produce strain conducive to premature aging. In “microdosing” these pro-degenerative stressors, dysregulation of PQC enables gradual progression toward neurodegeneration.
3. Genomic Compromise
Similar to PQC, the DNA damage response (DDR) relies on myriad sensor and enactor molecules to identify and repair strand breaks and aberrations [71]. At checkpoints of the cell cycle, recognized damage is addressed prior to advancement. At the G1S junction, p53, pRb and p21 tumor suppressors are most active in identify aberrant genes, pausing progression to DNA synthesis until genome stability has been established (Figure 3a-c). At the G2M junction, ATM and ATR identify breaks in the chromosome and direct DDR enactors, including Chk1/2 which halt progression to mitosis (Figure 3g-i). For cells to replicate or function during genomic compromise, the DDR must succeed at repairs or be overridden in spite of damage. Irreversible and/or insurmountable genome compromise is known to increase with natural aging [1]. Viruses contribute to this stress, and by extension promote cell death or senescence; it becomes essential that viruses evolve countermeasures to the DDR in order to maintain a long-term host and source of protein machinery [72]. In this section we discuss how age-associated genomic compromise – DNA damage, telomere attrition, and epigenetic shift – are generated by viral infection.
3.1. Viral damage of host DNA
The most overt damage to the host genome occurs during integration. HIV is perhaps the most defined integrating lentivirus; HIV encodes an integrase protein that cleaves the host DNA, inserts the viral DNA, and directs host DNA repair pathways to seal the exogenous DNA in place rather than recognizing it as foreign [73]. This is critical to HIV pathogenesis, with integrase inhibitors (e.g., cabotegravir) utilized for both prophylactic and therapeutic highly active antiretroviral therapy regimens. In the absence of HAART, however, HIV predominantly integrates into introns of active genes [74]. Similar to its preferential infection of CD4 lymphocytes in the periphery, HIV integrates into microglia of the brain. In HIV neurocognitive disorders, integration into astrocytes is also reported [75]. Although HIV integrates semi-randomly, BACH2 and MKL2 integration sites are known to be pro-proliferative and, therefore, selectively persist following infection [76]. The resultant increase in microglia activation advances aging twofold: through rapid increases in proliferative and inflammatory stress [77].
Integration is critical to retroviruses, such as HIV, but is also reported for dsDNA herpesviruses EBV and CMV [78]. EBV – the causative agent of mononucleosis, multiple sclerosis, and multiple cancers including nasopharyngeal carcinoma, B-cell leukemias, and Burkitt lymphoma – is known to integrate at “fragile points” of the host genome [79,80]. This includes the telomeric repeats, resulting in TERT-dependent upregulation of the cell cycle . Association of viral protein EBNA1 with the host chromosome can induce complete breakage and rearrangement of the host chromosome at site 11q23 [81]. The strain this causes to the host does not stop there. In a healthy system, these double stranded DNA breaks are identified by ATM protein at the G2M checkpoint (Figure 3g). ATM (and single-strand break counterpart ATR) is responsible for recruiting enactors of the DNA repair response (Figure 3h). These proteins activate Chk1/2 to pause the cell cycle until repair needs have been met (Figure 3i).
To avoid recognition as foreign and malignant DNA, viruses promote ATM activity (ensuring DNA repair molecules are recruited to facilitate their integration) while impairing Chk1/2 (ensuring progression through the cell cycle) (Figure 3j-k). There are myriad mechanisms by which viruses achieve this (Table 3). The VPR protein of HIV, for example, activates ATR through direct chromatin binding while simultaneously inactivating Chk1 through phosphorylation [82,83]. Conversely, SARS-CoV-2 produces two separate viral proteins, ORF6 and NSP13, which both drive degradation of Chk1 through the separate pathways of proteolysis and autophagy, respectively [84].
In addition to the direct oxidative stress caused by this DNA manipulation, less drastic integration sites can produce more gradual stress through increased proliferative strain [85]. DNA damage as a hallmark of aging is significantly amplified by viral modifications of the DNA damage response, enabling aberrant DNA to persist and accumulate stress. The tumor suppressor p53 is the paramount regulator of the cell cycle [86]. Most expressed during the G1S junction, p53 activates in response to foreign or abnormal DNA (Figure 3a). It upregulates transcription of tumor suppressor, p21, and triggers disinhibition of tumor suppressor pRb. By halting the cell cycle and directing either DDR or apoptosis, p53 and its downstream mediators prevent persistence or exacerbation of gene instability (Figure 3b). Next, during S phase, p53 promotes telomerase elongation (discussed in detail in the next subsection), ostensibly in anticipation of the stress produced by the DDR and future replication (Figure 3f). EBV and HIV have both been shown to amplify host centrosomes through genome manipulation, generating chromosomal instability and increasing aneuploidy risk [87,88].
Tumor suppressors are balanced by cyclins, which inactivate the tumor suppressors via phosphorylation and advance the cell cycle when checkpoints are satisfied (Figure 3c). For the viral genome to persist in the nucleus, it must evade the p53/p21/pRB cascade and promote cyclin activity (Figure 3d). In fact, p53 was first discovered through its interaction with the polyomavirus transcription factor, LTAg, which binds and inhibits both p53 and pRb [89]. This generated the titular “poly” meaning “many” and “oma” meaning “tumor” for the family of small dsDNA viruses. Convergent evolution of viral oncogenes has occurred for other neurotropic viruses as well (Table 3). CMV kinase pUL97 directly inactivates pRb, whereas tegument protein pp71 triggers pRb ubiquitination and degradation [90,91]. Furthermore, CMV transcription factor IE2 triggers pRb phosphorylation via cyclin-E1 [92]. EBV latent proteins LMP1 and EBNA3a promote cyclinD1 expression, inactivating pRb through phosphorylation [93,94]. EBNA3c also binds pRb directly, inactivating it. Similar to CMV IE2 targeting of tumor suppressor pRb, EBNA3c promotes ubiquitination and degradation of tumor suppressor p53 [95,96].
3.3. Viral attrition of telomeres
The protective TTAGGG repeats that cap chromosomes enable lossless replication by DNA polymerases [97]. With each cell cycle, proliferation shortens telomeres, indicating cell age in a way similar to rings of a tree trunk. The shelterin protein complex prevents the IFI16 sensor of the DNA damage response from targeting the uncapped, hanging ends of telomeres, but exogenous toxins (like viruses) can shorten or aberrate telomeres while avoiding the DNA damage response [98]. Innate immune cells exhibit elongated telomeres, an evolutionary selection believed to cope with the telomere expenditure characteristic of infection [99]. Because viruses depend on host cell proliferation for the production of viral progeny, pro-mitotic manipulation by viruses inherently advances telomere attrition and, therefore, advances the speed of cellular aging. Even during AART treatment, telomere length is significantly reduced by latent HIV [100]. Persistent, subclinical viruses like HSV1 and CMV have also been shown to advance the speed of telomere loss through increasing of the host “pathogen burden” [101,102]. During severe acute infection, as observed with epidemic SARS-CoV-2, severe inflammation has been correlated with rapid, and permanent, telomere attrition [103].
The self-sufficient integration and replication by telomerases mirror viral propagation, suggesting telomeres may have evolved through viral integration into the human genome [104]. Similar to telomerases, DNA viruses avoid recognition by the host DNA-damage response through looping/circularization, terminal capping, and telomerase manipulation [98].
3.4. Viral shift of the epigenome
Beyond the physical compromise of the host genome, epigenetic modifications that affect the three-dimensional accessibility of genes can be altered by viruses [105,106]. Just as with manipulation of the PQC, DDR, and cell cycle, viruses manipulate the host epigenome to facilitate viral replication at the expense of the host [107]. This accelerates aging and advances mortality [108]. Not all epigenetic alterations can be included in this review (i.e., microRNA, RNA splicing, RNA degradation), but have been reviewed extensively in recent textbooks [109,110]. Basic epigenetic modifications discussed here include inactivating DNA methylation, inactivating histone methylation, and activating histone acetylation.
By promoting DNA methylation at CpG islands, viruses increase obstructive methyl groups, preventing host gene expression and increasing resources for viral gene expression. This resource theft is characteristic of herpesviruses. HSV1 capsid protein, VP26, binds DNA methyltransferase (DNMT), promoting inactivating methylation of host genes [111]. This is a critical function; in the absence of the VP26-DNMT interaction, HSV1 viral copies are significantly reduced. EBV proteins EBNA3a and EBNA3c drive inactivating methylation of the tumor suppressor Bim by selectively activation DNMT activity within the Bim promotor [112]. This methylation is further advanced by another EBV protein, LMP1, which impairs re-activating demethylation by TET [113]. The resultant inhibition of Bim acts in concordance with EBV’s other anti-apoptotic mechanisms discussed above, and DNA hypermethylation has been proposed as a biomarker, and therapeutic target, for EBV malignancies [114,115]. Other viral infections have been discussed regarding methylation-dependent therapies. CMV exhibits DNA hypermethylation behavior, with CMV seropositivity correlating clinically with a general increase in DNA methylation, epigenetic age, and mortality [116]. Similar hypermethylation patterns have been correlated with other DNA polyomaviruses (i.e., JCV), as well as RNA enteroviruses (i.e., coxsackievirus), coronaviruses, and lentiviruses (i.e., HIV) [117,118,119]. Effective antiviral treatment has shown this methylation pattern is partially reversible, and correlates with viral load [119,120].
In addition to DNA methylation, viruses alter the acetylation and/or methylation status of histones, inhibiting or promoting coiling into inactive nucleosomes, respectively. The Dengue flavivirus capsid directly binds core histones, disrupting host nucleosomes while facilitating viral gene expression [121]. HSV1 and VZV both interact with host protein HCF-1 to promote histone methylation and inactivation of host genes [122]. VZV protein IE63 and EBV protein EBNA3C are both known to recruit deacetylases to the nucleus, reducing accessibility for host gene expression similar to DNA methylation [123,124]. Conversely, CMV proteins IE1/2 antagonize deacetylases, in order to prevent their silencing of the CMV promoter [125]. Therefore, histone deacetylases have been shown to be antiviral in certain infection conditions, and activating in others, dependent on the virus of interest [126].
4. Senescence
If cancer and apoptosis are considered the spectrum of cell fate, senescence acts as a limbo wherein cells are alive but lack functionality or potential. Most cells exhibit a pre-determined lifespan after which they lose their capacity for proliferation and function [127]. This cycle-dependent decline is termed senescence and varies across cell types based on both intrinsic and extrinsic conditions. Although seemingly in conflict, senescence is a secondary alternative to oncogenesis as it spares the host from lysis, prolonging viral access to the replicative machinery needed for the production of viral progeny. In conjunction with being pro-proliferative, latent viruses exhibit pro-senescence characteristics (Table 4). In addition to inducing senescence of infected cells, viruses are enhanced by systemic senescence – with natural aging of the immune and vascular systems disinhibiting latent or persistent viral infections [128,129].
Immunosenescence is a multifactorial and dynamic phenomenon that affects both innate and acquired immunity during aging [140]. The gradual decline of the immune system is further advanced by chronic diseases. Although the development of this immune phenotype is unclear, it may represent exhaustion of the immune system through several factors including inflammation, reactive oxygen species, genomic damage, mitochondrial failure, and chronic viral infection [141]. Accumulating evidence shows that several DNA and RNA viruses are inducers of immunosenescence. DNA herpesviruses can upregulate p16, p21, and p53 senescence-associated molecules, induce inflammaging, and metabolic reprogramming of infected cells, replicative senescence, and telomere shorting, as well as epigenetic modification of DNA and histones [142,143]. The same is true for RNA viruses, such as HIV [100].
CMV is the most established neurotropic virus to induce the immune dysfunctions observed in immunosenescence [144]. Although CMV was once considered the leading cause of age-related immune changes in the elderly, recent data are quite contradictory [144,145]. The current opinion is that CMV infection is not directly detrimental, but rather a recurrent stimulation that maintains sustained immunological alertness [146]. This favors a more robust immune response, at the risk of immune exhaustion in chronic infection. There is growing evidence and consensus that chronic antigen stimulation is necessary to cause immunosenescence [130,147]. Infection can lead to the accumulation of terminally differentiated T cells, or memory inflation (Table 4). This brings potential for both premature immune system aging and biological aging.
The immune phenotypes of cells of the immune system are directly or indirectly modified by viral infection, which has implications for immune responses. Previous studies have highlighted the significance of infection as immune attenuator via affecting the expression of PD1 and CTLA4 [148]. CD4+ and CD8+ T lymphocytes display hallmarks of senescence defined by CD57 expression that is associated with decreased proliferation capacity and function of these cells [149]. Therefore, it is assumed that the continuous exposure of immune cells to viral antigens during chronic infection drives exhaustion and senescence [150]. This is further supported by CMV-specific T lymphocytes which show higher levels of proinflammatory cytokines coupled with lower proliferative response upon stimulation [151]. Although it is difficult to define a singular molecular mechanism, the cycle of inflammation-driven virus reactivation and virus-induced inflammation is clearly implicated in the senescent immune phenotype that has negative consequences for the host.
5. Conclusions
In this review, we discuss the overlap of viral pathogenesis with age-related conditions. Using neurotropic viruses as an example, we highlight major mechanisms established to have chronic implications in addition to the more obvious, and symptomatic, dangers of acute infection. Although neurodegenerative conditions (e.g., AD, ALS, FTD, PD) reflect extremes of stress accumulation, it is critical to note that a spectrum of stressors exist – often subclinically – producing varying speeds at which the brain ages. This is in concordance with intrinsic (i.e., reactive oxygen species produced by metabolism) and extrinsic (i.e., reactive oxygen species produced by physical trauma) stressors and can be countered by protective behaviors (i.e., healthy diet, exercise) [142,152,153]. As described above, neurotropic viruses interact with other stressors, reactivating during deviations from homeostasis due to physical trauma, immune compromise, or systemic aging [4]. Although non-fatal, and potentially asymptomatic, the many ways in which persistent viruses age the brain underline the importance of antiviral therapies, vaccines, and preventative care health habits that counter the stresses of unavoidable environmental stresses, like viruses.
Author Contributions
Conceptualization, A.R., H.S.W., K.K.; writing—original draft preparation, A.R., H.S.W.; writing—review and editing, A.R., H.S.W.; supervision, K.K. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for A.R. was supported by the National Institute of Mental Health of the National Institutes of Health under Award Number T32MH079785.
Acknowledgments
A.R., H.S.W., and K.K. extend sincere thanks to the many interconnected centers of the Lewis Katz School of Medicine, for support, insight, and continued collaboration regarding the complex intricacies of aging. In particular, the authors thank past and present members of the Center for Neurovirology and Gene Editing. All figures were created using BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest in publishing this review.
Abbreviations
The following abbreviations are used in this manuscript:
| DNA | Deoxyribonucleic Acid |
| PQC | Protein Quality Control |
| ALS | Amyotrophic Lateral Sclerosis |
| PD | Parkinson’s Disease |
| AD | Alzheimer’s Disease |
| FTD | Frontotemporal Dementia |
| RNA | Ribonucleic Acid |
| ssRNA | Single Stranded RNA |
| SARS-CoV-2 | Coronavirus |
| CV | Coxsackie Virus |
| JEV | Japanese Encephalitis Virus |
| WNV | West Nile Virus |
| dsDNA | Double Stranded DNA |
| HSV1 | Herpes Simplex Virus 1 |
| VZV | Varicella Zoster Virus (also human herpesvirus 3) |
| EBV | Epstein Barr Virus (also human herpesvirus 4) |
| CMV | Cytomegalovirus (also human herpesvirus 5) |
| JCV | JC polyomavirus |
| HIV | Human Immunodeficiency Virus |
| Aβ | Amyloid-β |
| LBD | Lewy Body Dementia |
| gD/B | Glycoprotein D/B |
| TDP43 | Transactive Response DNA-binding Protein 43 |
| TAT | Transactivator of Transcription |
| cGAS | Cyclic GMP-AMP synthase |
| STING | Synthase-stimulator of Interferon Genes |
| NFκB | Nuclear Factor ΚB |
| IRF3 | Interferon Regulatory Factor 3 |
| TBK1 | TANK-binding Kinase 1 |
| VP | Viral Protein |
| GAG | Group-associated Antigen |
| ORF | Open Reading Frame |
| VIF | Viral Infectivity Factor |
| HERV-K | Human Endogenous Retrovirus |
| ASRGL1 | Asparaginase And Isoaspartyl Peptidase 1 |
| ER | Endoplasmic Reticulum |
| EBNA | Epstein-Barr Virus Nuclear Antigen |
| DDR | DNA Damage Response |
| TERT | Telomerase Reverse Transcriptase |
| DNMT | DNA Methyltransferase |
| TET | Ten-eleven Translocation Methylcytosine Dioxygenase |
| PD1 | Programmed Death Receptor 1 |
| CTLA4 | Cytotoxic T Lymphocyte-associated Protein 4 |
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of Aging: An Expanding Universe. Cell 2023, 186, 243–278. [CrossRef]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus Exposure and Neurodegenerative Disease Risk across National Biobanks. Neuron 2023, 111, 1086-1093.e2. [CrossRef]
- Gao, J.; Feng, L.; Wu, B.; Xia, W.; Xie, P.; Ma, S.; Liu, H.; Meng, M.; Sun, Y. The Association between Varicella Zoster Virus and Dementia: A Systematic Review and Meta-Analysis of Observational Studies. Neurol Sci 2024, 45, 27–36. [CrossRef]
- Cairns, D.M.; Smiley, B.M.; Smiley, J.A.; Khorsandian, Y.; Kelly, M.; Itzhaki, R.F.; Kaplan, D.L. Repetitive Injury Induces Phenotypes Associated with Alzheimer’s Disease by Reactivating HSV-1 in a Human Brain Tissue Model. Science Signaling 2025, 18, eado6430. [CrossRef]
- Levine, K.S.; Leonard, H.L.; Blauwendraat, C.; Iwaki, H.; Johnson, N.; Bandres-Ciga, S.; Ferrucci, L.; Faghri, F.; Singleton, A.B.; Nalls, M.A. Virus Exposure and Neurodegenerative Disease Risk across National Biobanks. Neuron 2023, 111, 1086-1093.e2. [CrossRef]
- Bruno, F.; Abondio, P.; Bruno, R.; Ceraudo, L.; Paparazzo, E.; Citrigno, L.; Luiselli, D.; Bruni, A.C.; Passarino, G.; Colao, R.; et al. Alzheimer’s Disease as a Viral Disease: Revisiting the Infectious Hypothesis. Ageing Res Rev 2023, 91, 102068. [CrossRef]
- Liang, Z.; Zhao, Y.; Ruan, L.; Zhu, L.; Jin, K.; Zhuge, Q.; Su, D.-M.; Zhao, Y. Impact of Aging Immune System on Neurodegeneration and Potential Immunotherapies. Prog Neurobiol 2017, 157, 2–28. [CrossRef]
- Mou, Y.; Du, Y.; Zhou, L.; Yue, J.; Hu, X.; Liu, Y.; Chen, S.; Lin, X.; Zhang, G.; Xiao, H.; et al. Gut Microbiota Interact With the Brain Through Systemic Chronic Inflammation: Implications on Neuroinflammation, Neurodegeneration, and Aging. Front Immunol 2022, 13, 796288. [CrossRef]
- Blinkouskaya, Y.; Caçoilo, A.; Gollamudi, T.; Jalalian, S.; Weickenmeier, J. Brain Aging Mechanisms with Mechanical Manifestations. Mech Ageing Dev 2021, 200, 111575. [CrossRef]
- Andjelkovic, A.V.; Situ, M.; Citalan-Madrid, A.F.; Stamatovic, S.M.; Xiang, J.; Keep, R.F. Blood-Brain Barrier Dysfunction in Normal Aging and Neurodegeneration: Mechanisms, Impact, and Treatments. Stroke 2023, 54, 661–672. [CrossRef]
- Ungvari, Z.; Tarantini, S.; Sorond, F.; Merkely, B.; Csiszar, A. Mechanisms of Vascular Aging, A Geroscience Perspective: JACC Focus Seminar. J Am Coll Cardiol 2020, 75, 931–941. [CrossRef]
- Rajasekaran, N.; Jung, H.S.; Bae, S.H.; Chelakkot, C.; Hong, S.; Choi, J.-S.; Yim, D.-S.; Oh, Y.-K.; Choi, Y.-L.; Shin, Y.K. Effect of HPV E6/E7 siRNA with Chemotherapeutic Agents on the Regulation of TP53/E2F Dynamic Behavior for Cell Fate Decisions. Neoplasia 2017, 19, 735–749. [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol 2014, 71, 505–508. [CrossRef]
- Park, H.; Kam, T.-I.; Dawson, V.L.; Dawson, T.M. α-Synuclein Pathology as a Target in Neurodegenerative Diseases. Nat Rev Neurol 2025, 21, 32–47. [CrossRef]
- Koike, Y. Molecular Mechanisms Linking Loss of TDP-43 Function to Amyotrophic Lateral Sclerosis/Frontotemporal Dementia-Related Genes. Neurosci Res 2024, 208, 1–7. [CrossRef]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The Viral Protein Corona Directs Viral Pathogenesis and Amyloid Aggregation. Nat Commun 2019, 10, 2331. [CrossRef]
- Michiels, E.; Rousseau, F.; Schymkowitz, J. Mechanisms and Therapeutic Potential of Interactions between Human Amyloids and Viruses. Cell Mol Life Sci 2020, 78, 2485–2501. [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H.; Tarenflurbil Phase 3 Study Group Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients with Mild Alzheimer Disease: A Randomized Controlled Trial. JAMA 2009, 302, 2557–2564. [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The Antimicrobial Protection Hypothesis of Alzheimer’s Disease. Alzheimer’s & Dementia 2018, 14, 1602–1614. [CrossRef]
- Hammarström, P.; Nyström, S. Viruses and Amyloids - a Vicious Liaison. Prion 17, 82–104. [CrossRef]
- Wang, H.-C.; Zhang, Q.-X.; Zhao, J.; Wei, N.-N. Molecular Docking and Molecular Dynamics Simulations Studies on the Protective and Pathogenic Roles of the Amyloid-β Peptide between Herpesvirus Infection and Alzheimer’s Disease. J Mol Graph Model 2022, 113, 108143. [CrossRef]
- Idrees, D.; Kumar, V. SARS-CoV-2 Spike Protein Interactions with Amyloidogenic Proteins: Potential Clues to Neurodegeneration. Biochem Biophys Res Commun 2021, 554, 94–98. [CrossRef]
- Pham, C.L.; Shanmugam, N.; Strange, M.; O’Carroll, A.; Brown, J.W.; Sierecki, E.; Gambin, Y.; Steain, M.; Sunde, M. Viral M45 and Necroptosis-Associated Proteins Form Heteromeric Amyloid Assemblies. EMBO Rep 2019, 20, e46518. [CrossRef]
- Hategan, A.; Bianchet, M.A.; Steiner, J.; Karnaukhova, E.; Masliah, E.; Fields, A.; Lee, M.-H.; Dickens, A.M.; Haughey, N.; Dimitriadis, E.K.; et al. HIV Tat Protein and Amyloid-β Peptide Form Multifibrillar Structures That Cause Neurotoxicity. Nat Struct Mol Biol 2017, 24, 379–386. [CrossRef]
- Feng, Q.; Hong, Y.; Pradeep Nidamanuri, N.; Yang, C.; Li, Q.; Dong, M. Identification and Nanomechanical Characterization of the HIV Tat-Amyloid β Peptide Multifibrillar Structures. Chemistry – A European Journal 2020, 26, 9449–9453. [CrossRef]
- Zhang, K.; Huang, Q.; Li, X.; Zhao, Z.; Hong, C.; Sun, Z.; Deng, B.; Li, C.; Zhang, J.; Wang, S. The cGAS-STING Pathway in Viral Infections: A Promising Link between Inflammation, Oxidative Stress and Autophagy. Front Immunol 2024, 15, 1352479. [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [CrossRef]
- Hyde, V.R.; Zhou, C.; Fernandez, J.R.; Chatterjee, K.; Ramakrishna, P.; Lin, A.; Fisher, G.W.; Çeliker, O.T.; Caldwell, J.; Bender, O.; et al. Anti-Herpetic Tau Preserves Neurons via the cGAS-STING-TBK1 Pathway in Alzheimer’s Disease. Cell Rep 2024, 115109. [CrossRef]
- Lio, C.-W.J.; McDonald, B.; Takahashi, M.; Dhanwani, R.; Sharma, N.; Huang, J.; Pham, E.; Benedict, C.A.; Sharma, S. cGAS-STING Signaling Regulates Initial Innate Control of Cytomegalovirus Infection. J Virol 2016, 90, 7789–7797. [CrossRef]
- Maelfait, J.; Bridgeman, A.; Benlahrech, A.; Cursi, C.; Rehwinkel, J. Restriction by SAMHD1 Limits cGAS/STING-Dependent Innate and Adaptive Immune Responses to HIV-1. Cell Rep 2016, 16, 1492–1501. [CrossRef]
- Han, L.; Zhuang, M.-W.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.-L.; Zhang, X.-J.; Gao, C.; Wang, P.-H. SARS-CoV-2 ORF9b Antagonizes Type I and III Interferons by Targeting Multiple Components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING Signaling Pathways. J Med Virol 2021, 93, 5376–5389. [CrossRef]
- Marques, M.; Ferreira, A.R.; Ribeiro, D. The Interplay between Human Cytomegalovirus and Pathogen Recognition Receptor Signaling. Viruses 2018, 10, 514. [CrossRef]
- Huang, Z.-F.; Zou, H.-M.; Liao, B.-W.; Zhang, H.-Y.; Yang, Y.; Fu, Y.-Z.; Wang, S.-Y.; Luo, M.-H.; Wang, Y.-Y. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe 2018, 24, 69-80.e4. [CrossRef]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. Human Cytomegalovirus Tegument Protein Pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. Journal of Virology 2018, 92, 10.1128/jvi.01774-17. [CrossRef]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J Virol 2018, 92, e00841-18. [CrossRef]
- Sumner, R.P.; Blest, H.; Lin, M.; Maluquer de Motes, C.; Towers, G.J. HIV-1 with Gag Processing Defects Activates cGAS Sensing. Retrovirology 2024, 21, 10. [CrossRef]
- Xu, G.; Liu, C.; Zhou, S.; Li, Q.; Feng, Y.; Sun, P.; Feng, H.; Gao, Y.; Zhu, J.; Luo, X.; et al. Viral Tegument Proteins Restrict cGAS-DNA Phase Separation to Mediate Immune Evasion. Mol Cell 2021, 81, 2823-2837.e9. [CrossRef]
- Jin, X.; Wang, W.; Zhao, X.; Jiang, W.; Shao, Q.; Chen, Z.; Huang, C. The Battle between the Innate Immune cGAS-STING Signaling Pathway and Human Herpesvirus Infection. Front Immunol 2023, 14, 1235590. [CrossRef]
- Aguirre, S.; Fernandez-Sesma, A. Collateral Damage during Dengue Virus Infection: Making Sense of DNA by cGAS. J Virol 2017, 91, e01081-16. [CrossRef]
- Garcia-Montojo, M.; Fathi, S.; Rastegar, C.; Simula, E.R.; Doucet-O’Hare, T.; Cheng, Y.H.H.; Abrams, R.P.M.; Pasternack, N.; Malik, N.; Bachani, M.; et al. TDP-43 Proteinopathy in ALS Is Triggered by Loss of ASRGL1 and Associated with HML-2 Expression. Nat Commun 2024, 15, 4163. [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Lorenzo-Sánchez, I.; Estévez-Herrera, J.; García-Luis, J.; Trujillo-González, R.; Valenzuela-Fernández, A. TDP-43 Controls HIV-1 Viral Production and Virus Infectiveness. Int J Mol Sci 2023, 24, 7658. [CrossRef]
- Fung, G.; Shi, J.; Deng, H.; Hou, J.; Wang, C.; Hong, A.; Zhang, J.; Jia, W.; Luo, H. Cytoplasmic Translocation, Aggregation, and Cleavage of TDP-43 by Enteroviral Proteases Modulate Viral Pathogenesis. Cell Death Differ 2015, 22, 2087–2097. [CrossRef]
- Zhang, L.; Yang, J.; Li, H.; Zhang, Z.; Ji, Z.; Zhao, L.; Wei, W. Enterovirus D68 Infection Induces TDP-43 Cleavage, Aggregation, and Neurotoxicity. J Virol 2023, 97, e0042523. [CrossRef]
- Xue, Y.C.; Liu, H.; Mohamud, Y.; Bahreyni, A.; Zhang, J.; Cashman, N.R.; Luo, H. Sublethal Enteroviral Infection Exacerbates Disease Progression in an ALS Mouse Model. J Neuroinflammation 2022, 19, 16. [CrossRef]
- Rahic, Z.; Buratti, E.; Cappelli, S. Reviewing the Potential Links between Viral Infections and TDP-43 Proteinopathies. Int J Mol Sci 2023, 24, 1581. [CrossRef]
- Correia, A.S.; Patel, P.; Dutta, K.; Julien, J.-P. Inflammation Induces TDP-43 Mislocalization and Aggregation. PLoS One 2015, 10, e0140248. [CrossRef]
- Xue, Y.C.; Feuer, R.; Cashman, N.; Luo, H. Enteroviral Infection: The Forgotten Link to Amyotrophic Lateral Sclerosis? Front Mol Neurosci 2018, 11, 63. [CrossRef]
- Licht-Murava, A.; Meadows, S.M.; Palaguachi, F.; Song, S.C.; Bram, Y.; Zhou, C.; Jackvony, S.; Schwartz, R.E.; Froemke, R.C.; Orr, A.L.; et al. Astrocytic TDP-43 Dysregulation Impairs Memory by Modulating Antiviral Pathways and Interferon-Inducible Chemokines 2022, 2022.08.30.503668.
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc Natl Acad Sci U S A 1998, 95, 6469–6473. [CrossRef]
- Gupta, A.; Bohara, V.S.; Chauhan, A.S.; Mohapatra, A.; Kaur, H.; Sharma, A.; Chaudhary, N.; Kumar, S. Alpha-Synuclein Expression in Neurons Modulates Japanese Encephalitis Virus Infection. Journal of Virology 2024, 98, e00418-24. [CrossRef]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J Virol 2016, 90, 2767–2782. [CrossRef]
- Hopkins, H.K.; Traverse, E.M.; Barr, K.L. Viral Parkinsonism: An Underdiagnosed Neurological Complication of Dengue Virus Infection. PLoS Negl Trop Dis 2022, 16, e0010118. [CrossRef]
- Ma, X.; Liao, Z.; Tan, H.; Wang, K.; Feng, C.; Xing, P.; Zhang, X.; Hua, J.; Jiang, P.; Peng, S.; et al. The Association between Cytomegalovirus Infection and Neurodegenerative Diseases: A Prospective Cohort Using UK Biobank Data. eClinicalMedicine 2024, 74, 102757. [CrossRef]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem Neurosci 2021, 13, 143–150. [CrossRef]
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 Proteins Interact with Alpha Synuclein and Induce Lewy Body-like Pathology In Vitro. Int J Mol Sci 2022, 23, 3394. [CrossRef]
- Marreiros, R.; Müller-Schiffmann, A.; Trossbach, S.V.; Prikulis, I.; Hänsch, S.; Weidtkamp-Peters, S.; Moreira, A.R.; Sahu, S.; Soloviev, I.; Selvarajah, S.; et al. Disruption of Cellular Proteostasis by H1N1 Influenza A Virus Causes α-Synuclein Aggregation. Proceedings of the National Academy of Sciences 2020, 117, 6741–6751. [CrossRef]
- Rajendran, A.; Castañeda, C.A. Protein Quality Control Machinery: Regulators of Condensate Architecture and Functionality. Trends Biochem Sci 2025, S0968-0004(24)00275-5. [CrossRef]
- Almasy, K.M.; Davies, J.P.; Lisy, S.M.; Tirgar, R.; Tran, S.C.; Plate, L. Small-Molecule Endoplasmic Reticulum Proteostasis Regulator Acts as a Broad-Spectrum Inhibitor of Dengue and Zika Virus Infections. Proceedings of the National Academy of Sciences 2021, 118, e2012209118. [CrossRef]
- Proulx, J.; Borgmann, K.; Park, I.-W. Role of Virally-Encoded Deubiquitinating Enzymes in Regulation of the Virus Life Cycle. International Journal of Molecular Sciences 2021, 22, 4438. [CrossRef]
- Gavilán, E.; Medina-Guzman, R.; Bahatyrevich-Kharitonik, B.; Ruano, D. Protein Quality Control Systems and ER Stress as Key Players in SARS-CoV-2-Induced Neurodegeneration. Cells 2024, 13, 123. [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [CrossRef]
- Corona, A.K.; Saulsbery, H.M.; Corona Velazquez, A.F.; Jackson, W.T. Enteroviruses Remodel Autophagic Trafficking through Regulation of Host SNARE Proteins to Promote Virus Replication and Cell Exit. Cell Rep 2018, 22, 3304–3314. [CrossRef]
- Zeltzer, S.; Zeltzer, C.A.; Igarashi, S.; Wilson, J.; Donaldson, J.G.; Goodrum, F. Virus Control of Trafficking from Sorting Endosomes. mBio 2018, 9, e00683-18. [CrossRef]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol 2017, 27, 800–809. [CrossRef]
- Johnston, B.P.; McCormick, C. Herpesviruses and the Unfolded Protein Response. Viruses 2020, 12, 17. [CrossRef]
- Rocchi, A.; Sariyer, I.K.; Berger, J.R. Revisiting JC Virus and Progressive Multifocal Leukoencephalopathy. J Neurovirol 2023, 29, 524–537. [CrossRef]
- Qian, Z.; Xuan, B.; Gualberto, N.; Yu, D. The Human Cytomegalovirus Protein pUL38 Suppresses Endoplasmic Reticulum Stress-Mediated Cell Death Independently of Its Ability To Induce mTORC1 Activation. Journal of Virology 2011, 85, 9103–9113. [CrossRef]
- Szymula, A.; Palermo, R.D.; Bayoumy, A.; Groves, I.J.; Ba Abdullah, M.; Holder, B.; White, R.E. Epstein-Barr Virus Nuclear Antigen EBNA-LP Is Essential for Transforming Naïve B Cells, and Facilitates Recruitment of Transcription Factors to the Viral Genome. PLoS Pathog 2018, 14, e1006890. [CrossRef]
- Li, S.; He, S.; Xue, H.; He, Y. Impact of Endogenous Viral Elements on Glioma Clinical Phenotypes by Inducing OCT4 in the Host. Front Cell Infect Microbiol 2024, 14, 1474492. [CrossRef]
- Pan, Z.; Huang, X.; Liu, M.; Jiang, X.; He, G. Research Advances in Chaperone-Mediated Autophagy (CMA) and CMA-Based Protein Degraders. J Med Chem 2025. [CrossRef]
- Groelly, F.J.; Fawkes, M.; Dagg, R.A.; Blackford, A.N.; Tarsounas, M. Targeting DNA Damage Response Pathways in Cancer. Nat Rev Cancer 2023, 23, 78–94. [CrossRef]
- Studstill, C.J.; Mac, M.; Moody, C.A. Interplay between the DNA Damage Response and the Life Cycle of DNA Tumor Viruses. Tumour Virus Res 2023, 16, 200272. [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9. [CrossRef]
- Plaza-Jennings, A.L.; Valada, A.; O’Shea, C.; Iskhakova, M.; Hu, B.; Javidfar, B.; Ben Hutta, G.; Lambert, T.Y.; Murray, J.; Kassim, B.; et al. HIV Integration in the Human Brain Is Linked to Microglial Activation and 3D Genome Remodeling. Mol Cell 2022, 82, 4647-4663.e8. [CrossRef]
- Li, G.-H.; Maric, D.; Major, E.O.; Nath, A. Productive HIV Infection in Astrocytes Can Be Established via a Non-Classical Mechanism. AIDS 2020, 34, 963–978. [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV Integration Sites Are Linked to Clonal Expansion and Persistence of Infected Cells. Science 2014, 345, 179–183. [CrossRef]
- Wahl, A.; Al-Harthi, L. HIV Infection of Non-Classical Cells in the Brain. Retrovirology 2023, 20, 1. [CrossRef]
- Tang, D.; Li, B.; Xu, T.; Hu, R.; Tan, D.; Song, X.; Jia, P.; Zhao, Z. VISDB: A Manually Curated Database of Viral Integration Sites in the Human Genome. Nucleic Acids Research 2020, 48, D633–D641. [CrossRef]
- Xu, M.; Zhang, W.-L.; Zhu, Q.; Zhang, S.; Yao, Y.; Xiang, T.; Feng, Q.-S.; Zhang, Z.; Peng, R.-J.; Jia, W.-H.; et al. Genome-Wide Profiling of Epstein-Barr Virus Integration by Targeted Sequencing in Epstein-Barr Virus Associated Malignancies. Theranostics 2019, 9, 1115–1124. [CrossRef]
- Janjetovic, S.; Hinke, J.; Balachandran, S.; Akyüz, N.; Behrmann, P.; Bokemeyer, C.; Dierlamm, J.; Murga Penas, E.M. Non-Random Pattern of Integration for Epstein-Barr Virus with Preference for Gene-Poor Genomic Chromosomal Regions into the Genome of Burkitt Lymphoma Cell Lines. Viruses 2022, 14, 86. [CrossRef]
- Li, J.S.Z.; Abbasi, A.; Kim, D.H.; Lippman, S.M.; Alexandrov, L.B.; Cleveland, D.W. Chromosomal Fragile Site Breakage by EBV-Encoded EBNA1 at Clustered Repeats. Nature 2023, 616, 504–509. [CrossRef]
- Lai, M.; Zimmerman, E.S.; Planelles, V.; Chen, J. Activation of the ATR Pathway by Human Immunodeficiency Virus Type 1 Vpr Involves Its Direct Binding to Chromatin In Vivo. J Virol 2005, 79, 15443–15451. [CrossRef]
- González, M.E. The HIV-1 Vpr Protein: A Multifaceted Target for Therapeutic Intervention. Int J Mol Sci 2017, 18, 126. [CrossRef]
- Gioia, U.; Tavella, S.; Martínez-Orellana, P.; Cicio, G.; Colliva, A.; Ceccon, M.; Cabrini, M.; Henriques, A.C.; Fumagalli, V.; Paldino, A.; et al. SARS-CoV-2 Infection Induces DNA Damage, through CHK1 Degradation and Impaired 53BP1 Recruitment, and Cellular Senescence. Nat Cell Biol 2023, 25, 550–564. [CrossRef]
- Li, R.; Liao, G.; Nirujogi, R.S.; Pinto, S.M.; Shaw, P.G.; Huang, T.-C.; Wan, J.; Qian, J.; Gowda, H.; Wu, X.; et al. Phosphoproteomic Profiling Reveals Epstein-Barr Virus Protein Kinase Integration of DNA Damage Response and Mitotic Signaling. PLOS Pathogens 2015, 11, e1005346. [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting P53 Pathways: Mechanisms, Structures and Advances in Therapy. Sig Transduct Target Ther 2023, 8, 1–35. [CrossRef]
- Shumilov, A.; Tsai, M.-H.; Schlosser, Y.T.; Kratz, A.-S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein–Barr Virus Particles Induce Centrosome Amplification and Chromosomal Instability. Nat Commun 2017, 8, 14257. [CrossRef]
- Park, J.-E.; Kim, T.-S.; Zeng, Y.; Mikolaj, M.; Il Ahn, J.; Alam, M.S.; Monnie, C.M.; Shi, V.; Zhou, M.; Chun, T.-W.; et al. Centrosome Amplification and Aneuploidy Driven by the HIV-1-Induced Vpr•VprBP•Plk4 Complex in CD4+ T Cells. Nat Commun 2024, 15, 2017. [CrossRef]
- Zheng, H.-C.; Xue, H.; Zhang, C.-Y. The Oncogenic Roles of JC Polyomavirus in Cancer. Front Oncol 2022, 12, 976577. [CrossRef]
- Prichard, M.N.; Sztul, E.; Daily, S.L.; Perry, A.L.; Frederick, S.L.; Gill, R.B.; Hartline, C.B.; Streblow, D.N.; Varnum, S.M.; Smith, R.D.; et al. Human Cytomegalovirus UL97 Kinase Activity Is Required for the Hyperphosphorylation of Retinoblastoma Protein and Inhibits the Formation of Nuclear Aggresomes. Journal of Virology 2008, 82, 5054–5067. [CrossRef]
- Bogdanow, B.; Phan, Q.V.; Wiebusch, L. Emerging Mechanisms of G1/S Cell Cycle Control by Human and Mouse Cytomegaloviruses. mBio 2021, 12, e02934-21. [CrossRef]
- Wiebusch, L.; Asmar, J.; Uecker, R.; Hagemeier, C. Human Cytomegalovirus Immediate-Early Protein 2 (IE2)-Mediated Activation of Cyclin E Is Cell-Cycle-Independent and Forces S-Phase Entry in IE2-Arrested Cells. J Gen Virol 2003, 84, 51–60. [CrossRef]
- Cheerathodi, M.R.; Meckes, D.G. The Epstein-Barr Virus LMP1 Interactome: Biological Implications and Therapeutic Targets. Future Virol 2018, 13, 863–887. [CrossRef]
- Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus Encoded LMP1 Regulates Cyclin D1 Promoter Activity by Nuclear EGFR and STAT3 in CNE1 Cells. J Exp Clin Cancer Res 2013, 32, 90. [CrossRef]
- Saha, A.; Murakami, M.; Kumar, P.; Bajaj, B.; Sims, K.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Augments Mdm2-Mediated P53 Ubiquitination and Degradation by Deubiquitinating Mdm2. J Virol 2009, 83, 4652–4669. [CrossRef]
- Shukla, S.K.; Jha, H.C.; El-Naccache, D.W.; Robertson, E.S. An EBV Recombinant Deleted for Residues 130-159 in EBNA3C Can Deregulate P53/Mdm2 and Cyclin D1/CDK6 Which Results in Apoptosis and Reduced Cell Proliferation. Oncotarget 2016, 7, 18116–18134. [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat Cell Biol 2022, 24, 135–147. [CrossRef]
- Wang, Z.; Deng, Z.; Tutton, S.; Lieberman, P.M. The Telomeric Response to Viral Infection. Viruses 2017, 9, 218. [CrossRef]
- Nassour, J.; Przetocka, S.; Karlseder, J. Telomeres as Hotspots for Innate Immunity and Inflammation. DNA Repair (Amst) 2024, 133, 103591. [CrossRef]
- Montano, M.; Oursler, K.K.; Xu, K.; Sun, Y.V.; Marconi, V.C. Biological Ageing with HIV Infection: Evaluating the Geroscience Hypothesis. The Lancet Healthy Longevity 2022, 3, e194–e205. [CrossRef]
- Dowd, J.B.; Bosch, J.A.; Steptoe, A.; Jayabalasingham, B.; Lin, J.; Yolken, R.; Aiello, A.E. Persistent Herpesvirus Infections and Telomere Attrition Over 3 Years in the Whitehall II Cohort. J Infect Dis 2017, 216, 565–572. [CrossRef]
- Noppert, G.A.; Feinstein, L.; Dowd, J.B.; Stebbins, R.C.; Zang, E.; Needham, B.L.; Meier, H.C.S.; Simanek, A.; Aiello, A.E. Pathogen Burden and Leukocyte Telomere Length in the United States. Immun Ageing 2020, 17, 36. [CrossRef]
- Virseda-Berdices, A.; Behar-Lagares, R.; Martínez-González, O.; Blancas, R.; Bueno-Bustos, S.; Brochado-Kith, O.; Manteiga, E.; Mallol Poyato, M.J.; López Matamala, B.; Martín Parra, C.; et al. Longer ICU Stay and Invasive Mechanical Ventilation Accelerate Telomere Shortening in COVID-19 Patients 1 Year after Recovery. Crit Care 2024, 28, 267. [CrossRef]
- de Lange, T. A Loopy View of Telomere Evolution. Front Genet 2015, 6, 321. [CrossRef]
- Fischer, N. Infection-Induced Epigenetic Changes and Their Impact on the Pathogenesis of Diseases. Semin Immunopathol 2020, 42, 127–130. [CrossRef]
- Silmon de Monerri, N.C.; Kim, K. Pathogens Hijack the Epigenome: A New Twist on Host-Pathogen Interactions. The American Journal of Pathology 2014, 184, 897–911. [CrossRef]
- Pei, Y.; Robertson, E.S. The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection. Viruses 2020, 12, 1377. [CrossRef]
- Christiansen, L.; Lenart, A.; Tan, Q.; Vaupel, J.W.; Aviv, A.; McGue, M.; Christensen, K. DNA Methylation Age Is Associated with Mortality in a Longitudinal Danish Twin Study. Aging Cell 2016, 15, 149–154. [CrossRef]
- Bose, D.; Robertson, E.S. Chapter 108 - Viruses, Cell Transformation, and Cancer. In Molecular Medical Microbiology (Third Edition); Tang, Y.-W., Hindiyeh, M.Y., Liu, D., Sails, A., Spearman, P., Zhang, J.-R., Eds.; Academic Press, 2024; pp. 2209–2225 ISBN 978-0-12-818619-0.
- Chen, M.; Lejeune, S.; Zhou, X.; Nadeau, K. Chapter 5 - Basic Genetics and Epigenetics for the Immunologist and Allergist. In Allergic and Immunologic Diseases; Chang, C., Ed.; Academic Press, 2022; pp. 119–143 ISBN 978-0-323-95061-9.
- Rowles, D.L.; Tsai, Y.-C.; Greco, T.M.; Lin, A.E.; Li, M.; Yeh, J.; Cristea, I.M. DNA Methyltransferase DNMT3A Associates with Viral Proteins and Impacts HSV-1 Infection. PROTEOMICS 2015, 15, 1968–1982. [CrossRef]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-Barr Virus Latency in B Cells Leads to Epigenetic Repression and CpG Methylation of the Tumour Suppressor Gene Bim. PLOS Pathogens 2009, 5, e1000492. [CrossRef]
- Cao, Y.; Xie, L.; Shi, F.; Tang, M.; Li, Y.; Hu, J.; Zhao, L.; Zhao, L.; Yu, X.; Luo, X.; et al. Targeting the Signaling in Epstein–Barr Virus-Associated Diseases: Mechanism, Regulation, and Clinical Study. Sig Transduct Target Ther 2021, 6, 1–33. [CrossRef]
- Leong, M.M.L.; Lung, M.L. The Impact of Epstein-Barr Virus Infection on Epigenetic Regulation of Host Cell Gene Expression in Epithelial and Lymphocytic Malignancies. Front Oncol 2021, 11, 629780. [CrossRef]
- Nehme, Z.; Pasquereau, S.; Herbein, G. Control of Viral Infections by Epigenetic-Targeted Therapy. Clinical Epigenetics 2019, 11, 55. [CrossRef]
- Kananen, L.; Nevalainen, T.; Jylhävä, J.; Marttila, S.; Hervonen, A.; Jylhä, M.; Hurme, M. Cytomegalovirus Infection Accelerates Epigenetic Aging. Exp Gerontol 2015, 72, 227–229. [CrossRef]
- Zhu, Z.; Li, Y.; Zhou, J.; Zhang, Y.; Cao, R. Acute Enterovirus Infections Significantly Alter Host Cellular DNA Methylation Status. Infection, Genetics and Evolution 2020, 80, 104190. [CrossRef]
- Kuss-Duerkop, S.K.; Westrich, J.A.; Pyeon, D. DNA Tumor Virus Regulation of Host DNA Methylation and Its Implications for Immune Evasion and Oncogenesis. Viruses 2018, 10, 82. [CrossRef]
- Balnis, J.; Madrid, A.; Hogan, K.J.; Drake, L.A.; Adhikari, A.; Vancavage, R.; Singer, H.A.; Alisch, R.S.; Jaitovich, A. Persistent Blood DNA Methylation Changes One Year after SARS-CoV-2 Infection. Clin Epigenetics 2022, 14, 94. [CrossRef]
- Esteban-Cantos, A.; Rodríguez-Centeno, J.; Silla, J.C.; Barruz, P.; Sánchez-Cabo, F.; Saiz-Medrano, G.; Nevado, J.; Mena-Garay, B.; Jiménez-González, M.; Miguel, R. de; et al. Effect of HIV Infection and Antiretroviral Therapy Initiation on Genome-Wide DNA Methylation Patterns. eBioMedicine 2023, 88. [CrossRef]
- Colpitts, T.M.; Barthel, S.; Wang, P.; Fikrig, E. Dengue Virus Capsid Protein Binds Core Histones and Inhibits Nucleosome Formation in Human Liver Cells. PLoS One 2011, 6, e24365. [CrossRef]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the Histone Demethylase LSD1 Blocks Alpha-Herpesvirus Lytic Replication and Reactivation from Latency. Nat Med 2009, 15, 1312–1317. [CrossRef]
- Ambagala, A.P.; Bosma, T.; Ali, M.A.; Poustovoitov, M.; Chen, J.J.; Gershon, M.D.; Adams, P.D.; Cohen, J.I. Varicella-Zoster Virus Immediate-Early 63 Protein Interacts with Human Antisilencing Function 1 Protein and Alters Its Ability to Bind Histones H3.1 and H3.3. J Virol 2009, 83, 200–209. [CrossRef]
- Knight, J.S.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C Recruits Histone Deacetylase Activity and Associates with the Corepressors mSin3A and NCoR in Human B-Cell Lines. J Virol 2003, 77, 4261–4272. [CrossRef]
- Park, J.-J.; Kim, Y.-E.; Pham, H.T.; Kim, E.T.; Chung, Y.-H.; Ahn, J.-H. Functional Interaction of the Human Cytomegalovirus IE2 Protein with Histone Deacetylase 2 in Infected Human Fibroblasts. J Gen Virol 2007, 88, 3214–3223. [CrossRef]
- Hackett, B.A.; Dittmar, M.; Segrist, E.; Pittenger, N.; To, J.; Griesman, T.; Gordesky-Gold, B.; Schultz, D.C.; Cherry, S. Sirtuin Inhibitors Are Broadly Antiviral against Arboviruses. mBio 2019, 10, 10.1128/mbio.01446-19. [CrossRef]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol 2018, 28, 436–453. [CrossRef]
- Ferguson, F.G.; Wikby, A.; Maxson, P.; Olsson, J.; Johansson, B. Immune Parameters in a Longitudinal Study of a Very Old Population of Swedish People: A Comparison between Survivors and Nonsurvivors. J Gerontol A Biol Sci Med Sci 1995, 50, B378-382. [CrossRef]
- Hadrup, S.R.; Strindhall, J.; Køllgaard, T.; Seremet, T.; Johansson, B.; Pawelec, G.; thor Straten, P.; Wikby, A. Longitudinal Studies of Clonally Expanded CD8 T Cells Reveal a Repertoire Shrinkage Predicting Mortality and an Increased Number of Dysfunctional Cytomegalovirus-Specific T Cells in the Very Elderly. J Immunol 2006, 176, 2645–2653. [CrossRef]
- Campos, C.; Pera, A.; Sanchez-Correa, B.; Alonso, C.; Lopez-Fernandez, I.; Morgado, S.; Tarazona, R.; Solana, R. Effect of Age and CMV on NK Cell Subpopulations. Exp Gerontol 2014, 54, 130–137. [CrossRef]
- Pita-Lopez, M.L.; Gayoso, I.; DelaRosa, O.; Casado, J.G.; Alonso, C.; Muñoz-Gomariz, E.; Tarazona, R.; Solana, R. Effect of Ageing on CMV-Specific CD8 T Cells from CMV Seropositive Healthy Donors. Immunity & Ageing 2009, 6, 11. [CrossRef]
- Wertheimer, A.M.; Bennett, M.S.; Park, B.; Uhrlaub, J.L.; Martinez, C.; Pulko, V.; Currier, N.L.; Nikolich-Žugich, D.; Kaye, J.; Nikolich-Žugich, J. Aging and Cytomegalovirus Infection Differentially and Jointly Affect Distinct Circulating T Cell Subsets in Humans. J Immunol 2014, 192, 2143–2155. [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A.H. Cytomegalovirus Seropositivity Drives the CD8 T Cell Repertoire Toward Greater Clonality in Healthy Elderly Individuals1. The Journal of Immunology 2002, 169, 1984–1992. [CrossRef]
- Attaf, M.; Malik, A.; Severinsen, M.C.; Roider, J.; Ogongo, P.; Buus, S.; Ndung’u, T.; Leslie, A.; Kløverpris, H.N.; Matthews, P.C.; et al. Major TCR Repertoire Perturbation by Immunodominant HLA-B*44:03-Restricted CMV-Specific T Cells. Front. Immunol. 2018, 9. [CrossRef]
- Nicoli, F.; Clave, E.; Wanke, K.; von Braun, A.; Bondet, V.; Alanio, C.; Douay, C.; Baque, M.; Lependu, C.; Marconi, P.; et al. Primary Immune Responses Are Negatively Impacted by Persistent Herpesvirus Infections in Older People: Results from an Observational Study on Healthy Subjects and a Vaccination Trial on Subjects Aged More than 70 Years Old. eBioMedicine 2022, 76, 103852. [CrossRef]
- Lanfermeijer, J.; de Greef, P.C.; Hendriks, M.; Vos, M.; van Beek, J.; Borghans, J.A.M.; van Baarle, D. Age and CMV-Infection Jointly Affect the EBV-Specific CD8+ T-Cell Repertoire. Front. Aging 2021, 2. [CrossRef]
- Campbell, T.M.; McSharry, B.P.; Steain, M.; Ashhurst, T.M.; Slobedman, B.; Abendroth, A. Varicella Zoster Virus Productively Infects Human Natural Killer Cells and Manipulates Phenotype. PLoS Pathog 2018, 14, e1006999. [CrossRef]
- Gerada, C.; Campbell, T.M.; Kennedy, J.J.; McSharry, B.P.; Steain, M.; Slobedman, B.; Abendroth, A. Manipulation of the Innate Immune Response by Varicella Zoster Virus. Front. Immunol. 2020, 11. [CrossRef]
- Levin, M.J. Immune Senescence and Vaccines to Prevent Herpes Zoster in Older Persons. Current Opinion in Immunology 2012, 24, 494–500. [CrossRef]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and Inflammaging in the Aging Process: Age-Related Diseases or Longevity? Ageing Res Rev 2021, 71, 101422. [CrossRef]
- Ghamar Talepoor, A.; Doroudchi, M. Immunosenescence in Atherosclerosis: A Role for Chronic Viral Infections. Front Immunol 2022, 13, 945016. [CrossRef]
- Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Aging Hallmarks and the Role of Oxidative Stress. Antioxidants 2023, 12, 651. [CrossRef]
- Bellon, M.; Nicot, C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses 2017, 9, 289. [CrossRef]
- Derhovanessian, E.; Maier, A.B.; Hähnel, K.; Zelba, H.; de Craen, A.J.M.; Roelofs, H.; Slagboom, E.P.; Westendorp, R.G.J.; Pawelec, G. Lower Proportion of Naïve Peripheral CD8+ T Cells and an Unopposed Pro-Inflammatory Response to Human Cytomegalovirus Proteins in Vitro Are Associated with Longer Survival in Very Elderly People. Age (Dordr) 2013, 35, 1387–1399. [CrossRef]
- Pawelec, G.; McElhaney, J.E.; Aiello, A.E.; Derhovanessian, E. The Impact of CMV Infection on Survival in Older Humans. Curr Opin Immunol 2012, 24, 507–511. [CrossRef]
- Pawelec, G.; Goldeck, D.; Derhovanessian, E. Inflammation, Ageing and Chronic Disease. Curr Opin Immunol 2014, 29, 23–28. [CrossRef]
- Smithey, M.J.; Li, G.; Venturi, V.; Davenport, M.P.; Nikolich-Žugich, J. Lifelong Persistent Viral Infection Alters the Naive T Cell Pool, Impairing CD8 T Cell Immunity in Late Life. J Immunol 2012, 189, 5356–5366. [CrossRef]
- Serriari, N.-E.; Gondois-Rey, F.; Guillaume, Y.; Remmerswaal, E.B.M.; Pastor, S.; Messal, N.; Truneh, A.; Hirsch, I.; van Lier, R.A.W.; Olive, D. B and T Lymphocyte Attenuator Is Highly Expressed on CMV-Specific T Cells during Infection and Regulates Their Function. J Immunol 2010, 185, 3140–3148. [CrossRef]
- Lopez-Vergès, S.; Milush, J.M.; Pandey, S.; York, V.A.; Arakawa-Hoyt, J.; Pircher, H.; Norris, P.J.; Nixon, D.F.; Lanier, L.L. CD57 Defines a Functionally Distinct Population of Mature NK Cells in the Human CD56dimCD16+ NK-Cell Subset. Blood 2010, 116, 3865–3874. [CrossRef]
- Effros, R.B.; Boucher, N.; Porter, V.; Zhu, X.; Spaulding, C.; Walford, R.L.; Kronenberg, M.; Cohen, D.; Schächter, F. Decline in CD28+ T Cells in Centenarians and in Long-Term T Cell Cultures: A Possible Cause for Both in Vivo and in Vitro Immunosenescence. Exp Gerontol 1994, 29, 601–609. [CrossRef]
- J Heath, J.; D Grant, M. The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cells 2020, 9, 766. [CrossRef]
- Yoon, M.; Yang, P.-S.; Jin, M.-N.; Yu, H.T.; Kim, T.-H.; Jang, E.; Uhm, J.-S.; Pak, H.-N.; Lee, M.-H.; Joung, B. Association of Physical Activity Level With Risk of Dementia in a Nationwide Cohort in Korea. JAMA Network Open 2021, 4, e2138526. [CrossRef]
- Dong, Y.; Zhang, P.; Zhong, J.; Wang, J.; Xu, Y.; Huang, H.; Liu, X.; Sun, W. Modifiable Lifestyle Factors Influencing Neurological and Psychiatric Disorders Mediated by Structural Brain Reserve: An Observational and Mendelian Randomization Study. J Affect Disord 2025, 372, 440–450. [CrossRef]
Figure 1.
Hallmarks shared by aging and chronic viral infection.
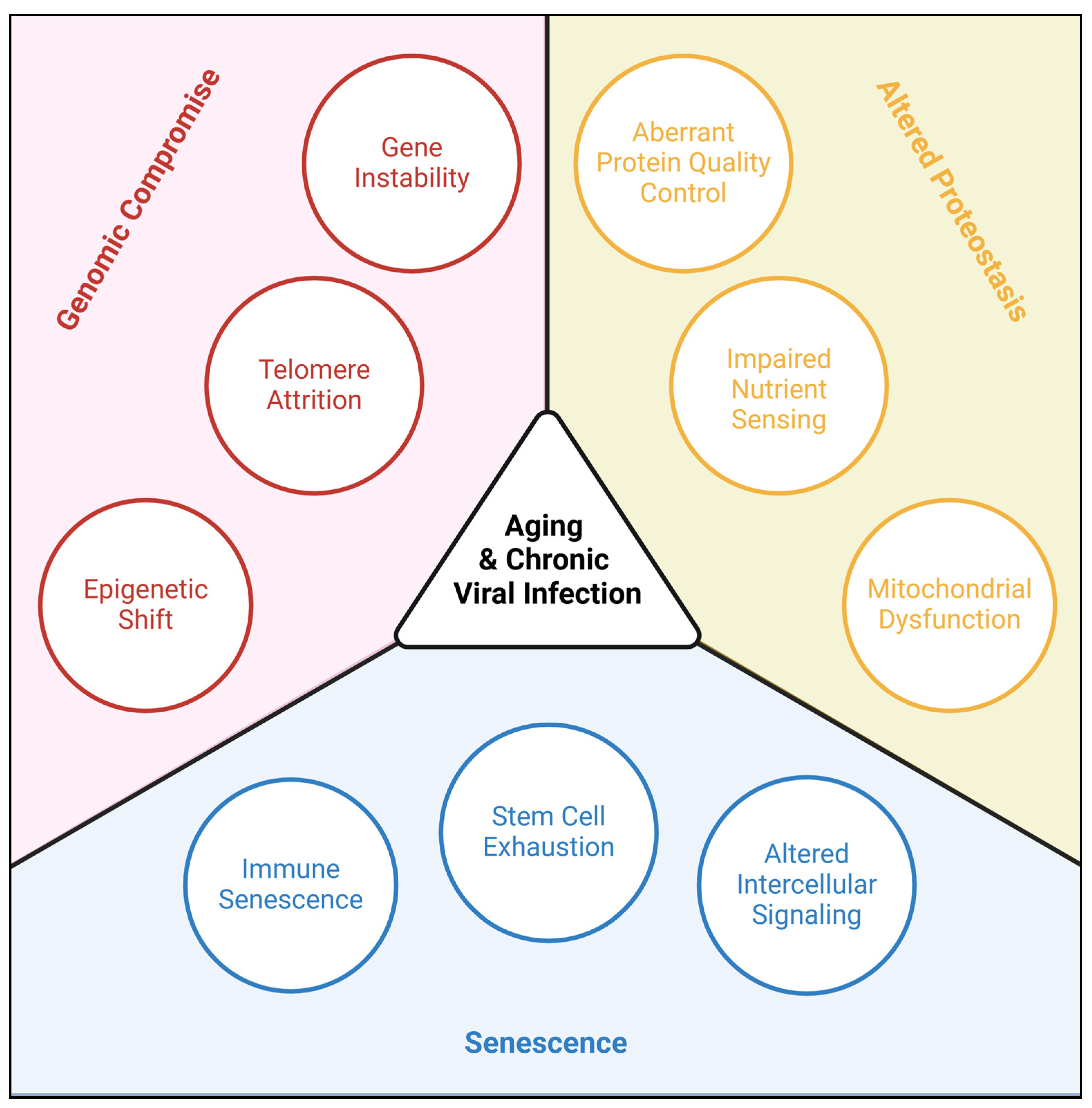
Figure 2.
Viral dysregulation of protein quality control. In the extracellular space, Aβ obstructs viral cell entry (a) by forming an obstructive corona on the viral surface (b). This can lead to amyloid plaque formation (c). Inside the cell, viral molecules are recognized by cGAS (d) triggering the STING pathway (e), but downstream hyperphosphorylation of tau can produce tau tangles (f). Some viruses have evolved anti-cGAS mechanisms, preventing the antiviral cascade (g). Alternative anti-viral mechanisms by the host include α-synuclein sequestration of virions in the ER (h), however, this can lead to formation of Lewy bodies (i). Viral RNA within the nucleus translocate TDP43 to the cytoplasm (j) where it can obstruct delivery of additional virions to the nucleus (k) but can also form obstructive aggregates (l). Ultimately, the virus produces viral progeny using host cell machinery (m) leading to release and spread via cell-sparing exocytosis (n) or fatal lysis (o).
Figure 2.
Viral dysregulation of protein quality control. In the extracellular space, Aβ obstructs viral cell entry (a) by forming an obstructive corona on the viral surface (b). This can lead to amyloid plaque formation (c). Inside the cell, viral molecules are recognized by cGAS (d) triggering the STING pathway (e), but downstream hyperphosphorylation of tau can produce tau tangles (f). Some viruses have evolved anti-cGAS mechanisms, preventing the antiviral cascade (g). Alternative anti-viral mechanisms by the host include α-synuclein sequestration of virions in the ER (h), however, this can lead to formation of Lewy bodies (i). Viral RNA within the nucleus translocate TDP43 to the cytoplasm (j) where it can obstruct delivery of additional virions to the nucleus (k) but can also form obstructive aggregates (l). Ultimately, the virus produces viral progeny using host cell machinery (m) leading to release and spread via cell-sparing exocytosis (n) or fatal lysis (o).
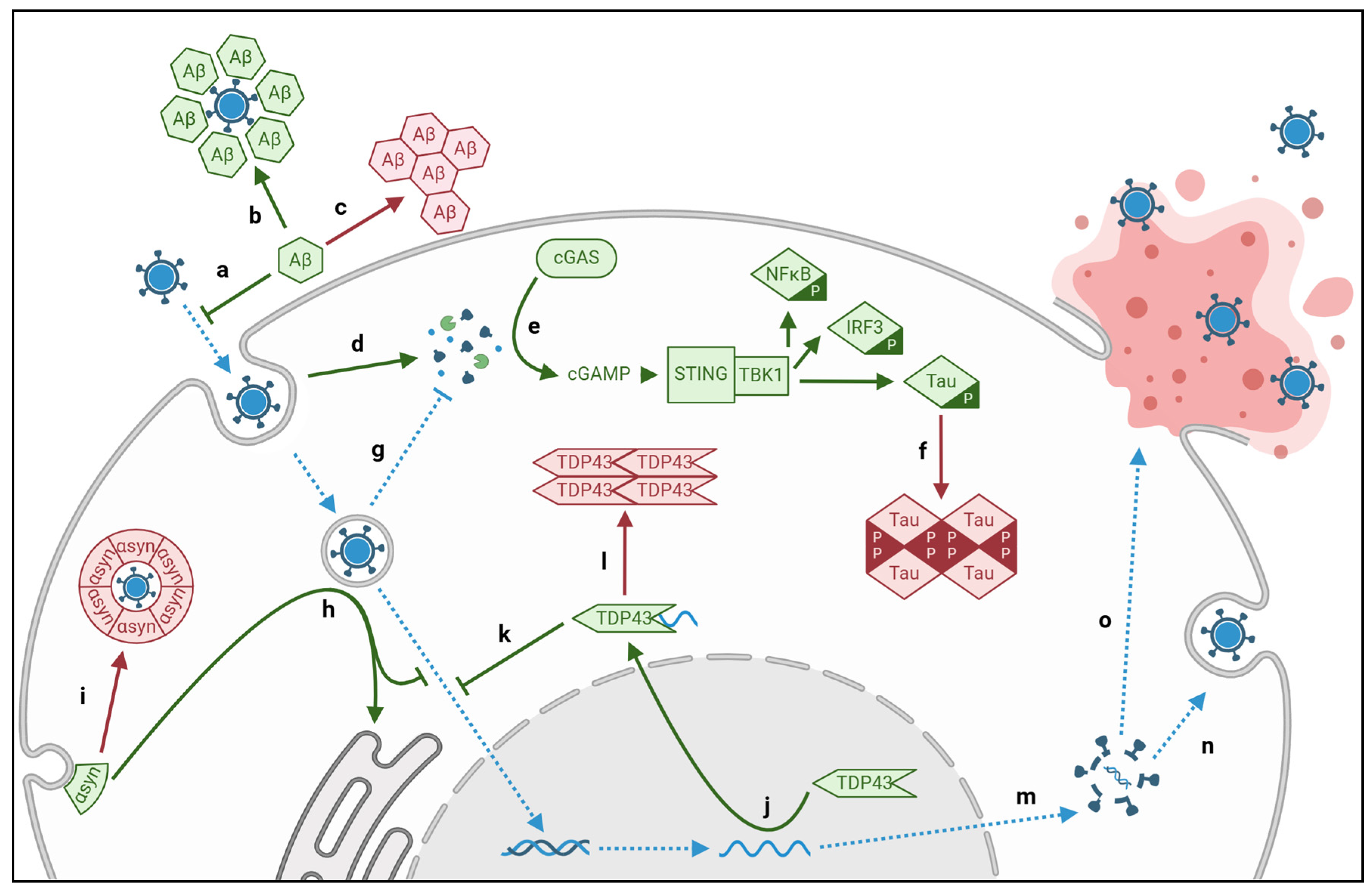
Figure 3.
Viral dysregulation of DNA repair responses. At the G1S junction, DNA damage triggers tumor suppressor p53, p21, and pRb (a) which inhibit progression to the synthesis phase (b). Endogenous cyclins balance this process (c). Viruses directly impair tumor suppressors p53 and p21 and indirectly promote cyclin inhibition of pRb (d), facilitating cell cycle progression regardless of DNA stability. In the synthesis phase, viruses impair telomerases directly (e) and indirectly through inhibition of pro-telomerase p53 (f). At the G2M junction, DNA damage is identified by ATM and ATR proteins (g) activating checkpoint inhibitors Chk1/2 (h) which pause the cycle until DNA is repaired (i). Viruses directly impair Chk1/2, facilitating progression through the cell cycle (j) while also promoting ATM repair of viral genes (k). In addition to the aberrant DNA, viruses duplicate centrosomes leading to asymmetrical division of the already impaired host daughter cells (l).
Figure 3.
Viral dysregulation of DNA repair responses. At the G1S junction, DNA damage triggers tumor suppressor p53, p21, and pRb (a) which inhibit progression to the synthesis phase (b). Endogenous cyclins balance this process (c). Viruses directly impair tumor suppressors p53 and p21 and indirectly promote cyclin inhibition of pRb (d), facilitating cell cycle progression regardless of DNA stability. In the synthesis phase, viruses impair telomerases directly (e) and indirectly through inhibition of pro-telomerase p53 (f). At the G2M junction, DNA damage is identified by ATM and ATR proteins (g) activating checkpoint inhibitors Chk1/2 (h) which pause the cycle until DNA is repaired (i). Viruses directly impair Chk1/2, facilitating progression through the cell cycle (j) while also promoting ATM repair of viral genes (k). In addition to the aberrant DNA, viruses duplicate centrosomes leading to asymmetrical division of the already impaired host daughter cells (l).
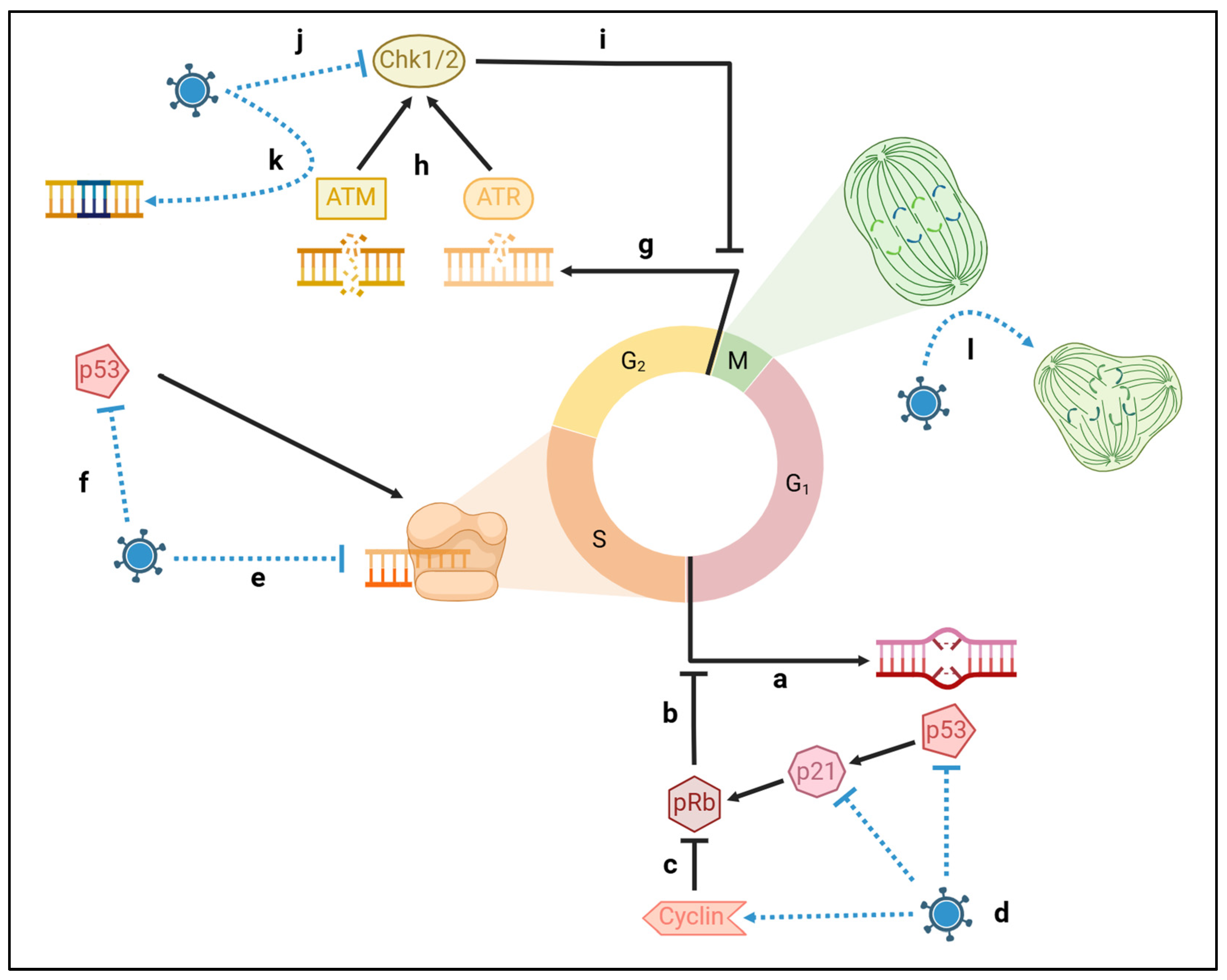
Table 3.
Mechanisms by which neurotropic viruses promote cell cycle progression .
| Target | Virus | Protein | Mechanism |
| ATM/ATR | EBV | EBNA3c | Evasion of ATM via p53 degradation |
| LMP1 | Transcriptional downregulation of ATM | ||
| HIV | VPR | Chromatin binding activates ATR | |
| Chk1/2 | EBV | EBNA3a | Inactivation by direct binding |
| HIV | VPR | Inactivation by phosphorylation | |
| SARS-CoV-2 | ORF6 NSP13 |
Proteolysis Autophagy-mediated degradation |
|
| p53 | EBV | EBNA3c | Ubiquitin-directed degradation |
| JCV | LTAg | Inactivation by direct binding | |
| pRb | CMV | IE2 | Inactivating phosphorylation via Cyclin-E1 |
| pp71 | Ubiquitin-directed degradation | ||
| pUL97 | Inactivation by phosphorylation | ||
| EBV | EBNA3 | Inactivation by direct binding Inactivating phosphorylation via Cyclin-D1 |
|
| LMP1 | Inactivating phosphorylation via Cyclin-D1 | ||
| JCV | LTAg | Inactivation by direct binding |
Table 4.
Adaptive immune changes associated with chronic viral infection. .
| Virus | Immune Changes | Citation |
| CMV | Decrease in percentage of CD16- NK cells | [130] |
| Decrease in percentage of CD16+/CD56bright NK cells | ||
| Increased CD16+/CD56- subset | [131] | |
| Increased CD8+ T-cells with high CD244 expression | [132] | |
| Increased CD4+ and CD8+ effector memory cells | ||
| Exhaustion of peripheral T-cell compartments | ||
| Accumulation of terminally differentiated, apoptosis-resistant, CMV-specific CD8+ lymphocytes |
[133,134] | |
| Reduced diversity of TCR repertoire | [135] | |
| EBV | Increase in differentiated phenotype markers (i.e., KLRG1) | [136] |
| Increase in terminally differentiated T-cells | ||
| Reduced diversity of TCR repertoire | ||
| VZV | Increased population of CD57+, terminally differentiated NK cells | [137] |
| Impaired Type I IFN pathway | [138] | |
| Impaired production of pro-inflammatory cytokines | ||
| Reduced frequency of VZV-specific memory T cells | [139] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



