
Submitted:
22 February 2025
Posted:
25 February 2025
You are already at the latest version
Abstract
Soil health is a critical determinant of agricultural productivity and environmental sustainability. Traditional assessment methods often fail to provide a comprehensive understanding of soil microbial communities and their functions. This study addresses this challenge by employing metagenomic techniques to assess the functionality of soil microbiomes in Russian black soil, renowned for its high fertility. We utilized shotgun metagenomic sequencing to analyze soil samples from Western Siberia subjected to different degrees of agro-soil disturbance. We identified functional genes involved in carbon (accA, argG, acsA, mphE, miaB), phosphorus (phoB, ppa, pstB, pnp, phnJ), and nitrogen (queC, amiF, pyrG, guaA, guaB, napA) metabolic pathways and associated with changes in microbial diversity in general and higher representation of certain bacterial species - Bradyrhizobium spp. Results demonstrated significant differences in microbial composition and functional potential between tillage treatments. No-Till technology and conventional tillage practices promoted beneficial microbial communities and enhanced soil health compared to long-term fallow soil. This work underscores the potential of metagenomic analysis in providing a comprehensive understanding of soil health, marking a significant advancement in the field.
Keywords:
soil metagenome
; Russian black soil
; shotgun sequencing
; agricultural productivity
; No-Till technology
1. Introduction
Soil health is a critical component of agricultural productivity, environmental sustainability, and overall ecosystem function. Healthy soils support plant growth by providing essential nutrients, water, and a stable structure for roots, while harboring beneficial microorganisms that enhance nutrient availability and protect plants from diseases. Soil health is linked to environmental sustainability through its role in carbon sequestration, water infiltration, and erosion control, which contribute to climate change mitigation and water quality protection [1,2]. Despite the importance of soil health, traditional assessment methods often fail to provide a comprehensive understanding of soil microbial communities and their functions [3]. Metagenomic analysis has emerged as a powerful tool for investigating soil health by characterizing microbial communities and their functional potential. This approach involves sequencing the collective genomes of microorganisms present in a soil sample, offering insights into the diversity, structure, and functional capabilities of the soil microbiome [4]. However, the high microbial diversity and variable evenness in soil pose challenges for metagenomic assembly, making it difficult to reconstruct microbial genomes from soil samples [5].
Recent advances in metagenomic methods have allowed the characterisation of microbial indicators of soil health as influenced by different types of tillage. These techniques can assess both compositional and functional changes in microbial communities, providing valuable insights into soil health [6]. Field-scale studies investigating microbial taxa from agricultural experiments are crucial for understanding the long-term effects of crop rotation and tillage on microbial indicator species [7] and to identify bioindicators of soil health by characterizing the changes caused by tillage [8].
Our research centers on evaluating soil health through metagenomic analysis of the microbiomes found in Russian black soil. Renowned for its exceptional fertility and high organic matter content [9], Russian black soil provides a distinctive setting for investigating soil microbial communities. Nevertheless, the impact of various tillage practices on the microbial diversity and functional potential of this soil type has not been thoroughly explored. This study will enable the examination of soil samples from fields that have undergone different tillage methods: Long-term fallow (for 16 years) and two types of grain‒fallow crop rotation: conventional tillage and No-Till technology [10].
We applied metagenomic sequencing using Illumina technology to assess the taxonomic diversity of soil microbiomes and to identify genes related to the cycles of phosphorus, nitrogen and carbon accumulation in soil. Our results showed significant differences in microbial composition between different tillage practices. No-Till technology and convetional tillage practices promoted beneficial microbial communities and soil health by improving nutrient cycling and organic matter decomposition. This paper describes a process for assessing soil health using metagenomic analysis of Russian chernozem microbiomes, demonstrating the potential of metagenomic methods in providing a comprehensive understanding of soil microbial communities and their functions. Our study highlights the significant impact of different tillage practices on soil health and the need for sustainable soil management practices to ensure long-term soil fertility and productivity.
2. Materials and Methods
1. Study area and collection of soil samples
Soil samples were collected in August 2023 from medium-humus, medium-loamy black soil (Luvic Chernozem) in the forest-steppe region of the Ob area (coordinates: 54° 53' 13.5" N, 82° 59' 36.7" E). The size of the experimental plots occupied by wheat was 15m x 18m. Soil samples were collected at the 0-10 and 10-20 cm soil layers. The sample for analysis was an average sample of 5 individual ones, collected at each spatial replication of the experiment. On the long term fallow, soil sampling was carried out according to the same scheme. Thus, 18 samples were obtained: 3 variants, 2 layers, 3 replicates. The soil samples were delivered to the laboratory, passed through a sieve with a cell diameter of 1 mm under sterile conditions, placed in sterile containers and stored at a temperature of minus 80 degrees until analysis.
2. Analysis of physical and chemical properties of soil samples
The total organic carbon (TOC) content was determined via the dichromate oxidation method [11]. The mortmass was separated by decanting the soil with water on a sieve with a cell diameter of 0.25 mm. The biomass was dried to an absolutely dry state. The carbon content was determined by thermal analysis on a Vario EL Cube elemental analyzer (Elementar Analysensysteme GmbH, Germany) according to manufacturer’s protocols. The amount of microbial biomass was determined using a substrate-induced respiration method [12]. N-NO3 was determined by the ionometric method. Extractant 1 N K2SO4 solution at a soil:solution ratio of 1:2. Тhe determination of nitrates was carried out using ion-selective electrodes [13]. Phosphatase activity in soil samples was determined using sodium phenolphthalein phosphate as a substrate (pH 6.5), incubation at 30 oC for 60 min [14].
3. DNA extraction:
DNA was extracted using the Quick-DNA Fungal/Bacterial Microprep Kit/Quick-DNA Fecal/Soil Microbe Microprep kit (Zymo Research) following the manufacturer’s protocol. The extracted DNA was quantified using a Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and quality was assessed by electrophoresis in 1% agarose gel. Additionally, DNA quality was assessed according to the manufacturer's instructions from Agilent Technologies using the 2100 Bioanalyzer.
4. ‘Shotgun’ sequencing using Illumina technology.
DNA samples were used to create libraries using Illumina DNA Prep, (M) Tagmentation (100 ng). Shotgun sequencing using Illumina Novaseq technology was performed using the manufacturer's standard techniques. Q30 value: ≥93.00 % (Percentage of sequenced bases with a Phred score ≥30. This value is based on the entire sequencing run). Read length: 2x101 bp. The data was uploaded to the NCBI under BioProject: PRJNA1226397.
5. Primary bioinformatic data processing.
Demultiplexing of sequencing reads was performed using Illumina bcl2fastq (2.20). Adapters were trimmed using Skewer software (version 0.2.2) [15]. The quality of FASTQ files was analysed using FastQC software (version 0.11.5-cegat).
6. Bioinformatic analyses of taxonomic diversity
Taxonomic analysis was conducted using Kraken2 (v.2.1.13) [16] and relative abundance was calculated using Bracken (v.2.9) at the ‘genus’ and ‘species’ levels[17]. Alpha diversity was conducted using Shannon index calculating to establish the level of taxons’ diversity within groups. Beta diversity was calculated using Bray-Curtis distance matrix and PERMANOVA analysis to identify taxons’ differences between groups. The vegan, adonis and edgeR packages were used [18,19]. To establish differences in relative abundance among different sample groups of the soil microbiome, the U-criterion (Mann-Whitney test) and false positive rate (FDR) by the Benjamini–Hochberg method were calculated. The stats, matplotlib, numpy and pandas libraries were used (Available online: https://matplotlib.org/stable/index.html, https://numpy.org/doc/, https://api.semanticscholar.org/CorpusID:61539023).
7. Functional analysis
To perform a functional analysis, it was necessary to collect the catalog of target amino acid sequences. To do this, we have compiled the list of target carbon, nitrogen, and phosphorus exchange genes and taxa that we obtained at the stage of taxonomic analysis. Using this data, we conducted multiple searches in the UniProt [20] and KEGG [21] database. The catalog was compiled from the detected sequences. All sequences satisfy the following requirements: i) the amino acid sequence belongs to the carbon, phosphorus or nitrogen metabolic pathway; ii) the amino acid sequence belongs to an organism that was obtained in the results of the taxonomic analysis (up to class level) iii) the sequence must have at least 2 annotations in the UniProt database. Full catalog information can be viewed in Table S1.
Target genes from the compiled catalog were identified within the sequencing reads using blastx Diamond (v.2.1.11) [22]. To interpret the results, we conducted the following requirements: i) the alignment must have at least 90% homology; ii) the alignment length corresponding to the length of the read. The readcounts were calculated for the detected coding sequences of genes. The calculation was performed using our own code. Then readcounts were normalized using the Trimmed Mean of M-values (TMM) normalization method. To identify significant differences in gene abundance between the groups, the Mann-Whitney test and FDR by the Benjamini–Hochberg method were calculated. If FDR < 0.05, we assume that the abundance of genes in the groups is statistically significantly different in comparing groups.
The results of the taxonomic and functional analysis were compared into a signature matrix consisting of pairs (taxon; gene). Matrix data was processed with TMM normalization, the U-criterion and FDR using the Benjamini–Hochberg method.
3. Results
3.1. Assessment of Soil Physico-Chemical Parameters
The central forest-steppe of the Novosibirsk Priobye, where soil samples were taken, belongs to the eastern edge of the Priob plateau and is located at the junction of two major geomorphological provinces of the West Siberian Plain and the mountain systems of the Kuznetsk Ala-Tau, Altai and Sayan. Absolute heights of the Priobie region are 200-250 metres above sea level. According to the agroclimatic zoning of the Novosibirsk Oblast, the central forest-steppe of the Priobie region belongs to the moderately warm, moderately humidified agroclimatic sub-area (Agroclimatic Resources of the Novosibirsk Oblast, 1971). The difference between the average monthly temperatures of the hottest month and the coldest month is 38°C. Winter is cold and long. Stable snow cover is retained for 157-162 days. Soils freeze up to 1.8-2 meters.
Leached black earths (Luvic Chernozem) are the most fertile soils of Western Siberia. The main crops of grain crops are located on these soils. Since the mid-1980s of the 20th century, large-scale research has been carried out to develop soil-protective farming in the forest-steppe zone of Western Siberia [23]. In particular, it is shown that redistribution of C in mortmass in the profile of root-bearing layer at minimization of mechanical tillage is the main reason of change of its living phase [24].
Long term permanent fallows are an informative tool for studying the problems of soil system stability under conditions of agrogenic degradation [25,26]. In our experiment, the soil was without plants for 16 years, annual plowing was carried out in the spring and during the growing season weeds were removed with a cultivator. Conventional moldboard plowing to a depth of 25 cm is a generally accepted technology for cultivating soil for grain crops in Siberia. No-Till technology is currently being implemented in Siberia
The selected series of options represents 3 degrees of disturbance of the agro-soil. Long-term fallow is a model of extreme degradation of agro-soil, in conditions of No-Till technology, when the intensity of mechanical impact is reduced, soil properties change towards enrichment of mobile fractions of organic matter due to redistribution and some conservation of plant residues, while conventional tillage is a kind of standard to average soil samples found in a given region. Previous research has demonstrated that various tillage methods significantly impact soil quality, carbon and nitrogen stocks, and soil aggregates [27,28].
Our physico-chemical analysis of the soil samples focused on phosphorus, carbon, and nitrogen content. The granulometric composition of the soil is medium loamy, coarse silt-sandy, the content of physical clay (particles <0.01 mm) is 38.3%, coarse silt (0.05-0.01 mm) - 25.0%, fine sand (0.25-0.05 mm) - 36.2%. All samples have pH=7. The main physical and chemical properties of the soil are presented in Table 1
3.2.2. Taxonomy Analysis
The alpha diversity analysis revealed a high level of species diversity, with no statistically significant differences observed among subgroups subjected to varying tillage practices. The Shannon diversity index, averaging approximately 7.7 across all groups, indicates a notably high level of species richness and evenness [29]. This suggests a substantial heterogeneity in species composition within each soil subgroup. Such elevated biodiversity is essential for sustaining soil health and fertility, as it plays a pivotal role in regulating nutrient cycling and the decomposition of organic matter (Figure 1).
The beta diversity analysis, conducted using the Bray-Curtis dissimilarity matrix and PERMANOVA, revealed statistically significant differences among the subgroups. The NMDS graph (Figure 2) further corroborates these findings, demonstrating clear clustering of samples into three distinct subgroups corresponding to the tillage practices. At the same time, the data exhibited considerable dispersion at the species level. The PERMANOVA results indicated that approximately 40% of the observed variability in the data set can be attributed to the classification of soil samples into the three tillage practice groups.
The relative representation of taxa in groups was also compared using the Wilcoxon-Mann-Whitney test (U-test) and FDR. The graph (Figure 3) shows statistically significant results, with median values for each group.
At the species level, the most obvious difference is in the representation of Streptomyces mobaraensis, Capillimicrobium parvum, Conexibacter woesei, Bradyrhizobium sp. 170, Bradyrhizobium sp. 200, Bradyrhizobium sp. CCBAU 051011, Bradyrhizobium sp. S12-14-2. The differences in the representation of these species are more than 0.5%.
The abundance of species Streptomyces mobaraensis, Capillimicrobium parvum and Conexibacter woesei is higher for soil samples with Long-term fallow in comparison with the conventional tillage and No-Till technology groups.
The species Bradyrhizobium sp. 170 is the most represented in subgroup of No-Till technology and the least represented in the Long-term fallow group, as well as the species Bradyrhizobium sp. 200, Bradyrhizobium sp. CCBAU 051011, Bradyrhizobium sp. S12-14-2, and Bradyrhizobium license.
3.3. Analysis of Genes
The genes of carbon, nitrogen and phosphorus metabolism were searched using the catalog of homologues and reads. Relative abundances for the putative genes encoding enzymes were counted and compared, as described in the ‘Materials and Methods’ section. The genes with statistically significant difference in relative abundance between soil subgroups are presented in Figure 4.
The relative abundance of the genes from Figure 4 is presented as median relative abundance of the gene in soil samples subgroups (Appendix A, Table A1). The annotation (function and belonging to the metabolic pathway) to these genes is performed in Table 2.
The relative abundance of genes differs significantly for soil subgroup ‘Long-term fallow’ and ‘Conventional tillage’, ‘Long-term fallow’ and ‘No-Till technology’. The difference between ‘Conventional tillage’ and ‘No-Till technology’ is not statistically significant. In all cases, the relative abundance of genes for ‘Long-term fallow’ is significantly lower than for the other subgroups. Genes acsA, accA, pstB are the most represented (much more than 106). Genes amiF and pnp are not represented in ‘Long-term fallow’ (the median value of the readcounts is zero). Genes mphE, phnJ and queC are presented in ‘Long-term fallow’ at a significantly lower level compared to other subgroups and genes (lower than 104).
3.3. Signature Analysis
To provide the results, all pairs containing the species discussed in the ‘Taxonomy analysis’ section (the species with the largest difference in representation between the subgroups) were selected. These species are Streptomyces mobaraensis, Capillimicrobium parvum, Conexibacter woesei, Bradyrhizobium sp. 170, Bradyrhizobium sp. 200, Bradyrhizobium sp. CCBAU 051011, Bradyrhizobium sp. S12-14-2. The Mann-Whitney test and FDR by the Benjamini–Hochberg method was calculated for the pairs. Then a heatmap was built to visualize the differences in their median relative abundance (Figure 5).
The heatmap shows that the signature pair ‘Bradyrhizobium sp. CCBAU 051011|acsA’ has the highest abundance for ‘No-Till technology’ (over 1000 readcounts in median) and ‘Conventional tillage’ (about 1000 readcounts in median) in comparison with ‘Long-term fallow’. The signature pair ‘Bradyrhizobium sp. 200|pstB’ has much more high abundance (about 700 readcounts) in tilled groups compared with ‘Long-term fallow’ subgroup. These results support the results of taxonomic and functional analysis.
4. Discussion
Modern agriculture faces a number of challenges such as soil degradation, loss of fertility and the need for sustainable agricultural practices. Integrated soil health assessment methods, particularly through metagenomic analysis, can help to address these challenges. Our study contributes to this growing body of knowledge by employing metagenomic tools to evaluate the microbial communities and functional potential of Russian black soil (chernozem), a soil type renowned for its high fertility and agricultural importance [9]. Despite its inherent fertility, chernozem is not immune to degradation, and its sustainable management is critical to preserving its productivity [30]. By employing metagenomic techniques, we have been able to provide a comprehensive understanding of the microbial communities inhabiting this soil type and their functional potential. Using advanced metagenomic methods, we have gained insight into microbial diversity and genes that may be critical for maintaining soil health and productivity.
Our key findings highlight significant differences in microbial community composition nder different tillage regimes. No-Till technology and conventional tillage practices both promoted beneficial microbial communities (Figure 1, Figure 2 and Figure 3) that potentially enhanced soil health by improving nutrient cycling and organic matter decomposition comparing to long-term bare fallow soil. These findings correspond to the increase in total carbon in microbial biomass (Table 1) and are consistent with previous studies demonstrating the positive impact of permanent tillage on soil microbial diversity and community stability [31]. At the same time, No-Till technology, which is considered a reduced tillage practice [32], was associated with a more heterogeneous soil environment than in case of conventional tillage, providing diverse ecological niches that support a wider range of microbial lifestyles. This diversity is crucial for maintaining the resilience of soil ecosystems and their ability to provide essential services, such as nutrient availability and disease suppression, to plants and their environment [33]. The increased microbial diversity under reduced tillage conditions is likely due to the preservation of soil structure and organic matter, which provides a stable habitat for microbial communities. In contrast, conventional tillage disrupts soil aggregates, leading to the loss of organic matter resulted in less extensive microbial diversity [34]. In general, both tillage methods positively influenced soil microbial diversity, with reduced tillage No-Till technology demonstrating greater effectiveness.
The relative representation of the major soil species, Streptomyces mobaraensis, Capillimicrobium parvum and Conexibacter woesei, inversely correlates with the increase in total microbial diversity (Figure 3). S. mobaraensis and C. woesei are known to form extended hyphal networks, and their abundance is associated with stable, structured soils where they thrive in protective microhabitats within intact soil aggregates [35,36]. C. parvum, a small-sized actinobacterium, is likely sensitive to rapid changes in soil moisture and organic matter distribution, which occur while applying regular tillage procedures [37]. Both tillage practices that break up soil aggregates can expose the described major bacterial species to harsher conditions—reducing their capacity to colonize and persist in the soil matrix, but at the same time, enabling the growth and development of other bacterial species.
The results of the study indicate that both treatment technologies led to a significant increase in the abundance of a certain microbial taxa: the members of the genus Bradyrhizobium. Bradyrhizobium spp. are well-known symbiotic nitrogen-fixing bacteria that form nodules on the roots of leguminous plants [38] and are responsible for heavy metal resistance and bioremediation in a long-term heavy metal-contaminated ecosystem[39]. Bradyrhizobium spp. have been shown to stimulate plant growth and improve plant resistance to biotic and abiotic stresses, further highlighting their importance as indicators of soil health and plant vitality [40,41]. Extended presence of Bradyrhizobium spp. under reduced tillage conditions (No-Till technology) suggests that these practice not only enhances soil fertility but also contribute to the resilience of soil ecosystems in the face of environmental stressors.
The functional analysis (Figure 4) revealed that genes associated with carbon metabolism, such as accA, argG, acsA, mphE, and miaB, were more abundant in soils subjected to No-Till technology and conventional tillage practices. Among them, accA (acetyl-coenzyme A carboxylase) and acsA (acetyl-coenzyme A synthetase) are especially crucial for carbon fixation and the synthesis of acetyl-CoA, a key intermediate in the carbon cycle [42]. The increased abundance of these genes due to the application of tillage suggests enhanced carbon sequestration and organic matter decomposition, which are critical for maintaining soil fertility [34]. This assumption is supported by the fact that both methods of soil treatment led to significantly higher carbon content in microbial biomass and the mortmass, as well as increased СО2 emission, comparing to an untreated soil (Table 1). Signature analysis (FIgure 5) identified Bradyrhizobium sp. CCBAU 051011 as a crucial bacterial species associated with the increased abundance of acsA in tilled soils, highlighting its significant role in carbon fixation processes.
The optimization of carbon and nitrogen metabolism in soils under agricultural practices has been extensively studied in the literature. However, significantly less attention has been given to the dynamics of phosphorus metabolism [43]. In particular, the influence of soil cultivation methods on phosphorus metabolism remains underexplored. Nonetheless, a global meta-analysis of 5876 observations demonstrated that soil conservation practices significantly enhance soil phosphatase activity [44]. Our findings provide additional support for this conclusion through the lens of soil metagenomic analysis. Phosphorus metabolism was significantly affected by tillage practices, with genes such as phoB, ppa, pstB, pnp and phnJ exhibiting higher abundance in soils subjected to treatment. These genes play essential roles in phosphorus uptake and regulation, processes vital for sustaining soil phosphorus availability and supporting plant nutrition [45]. Specifically, phoB, ppa, phnJ and pstB encode key proteins involved in solubilizing soil phosphate, thereby enhancing its accessibility to plants [46]. For instance, phoB is crucial for the regulation of phosphate uptake and solubilization as well as P-starvation response, since it controls the expression of phosphatases and phosphate transporters [47]. In tilled soils, the higher abundance of phoB suggests an adaptive response to phosphorus scarcity, promoting the solubilization of inorganic phosphate and enhancing its availability to plants. The increase of phoB abundance was 3 times on plowing and 7 times on No-Till compared to fallow, which may be a sign of optimization of phosphorus metabolism conditions under soil conservation technology. Higher presence of ppa, encoding an inorganic pyrophosphatase, corresponds to elevated phosphatase activity (Table 1) in tilled soils that is potentially associated with enhanced mineralization of organic phosphorus compounds, releasing orthophosphate for plant uptake [48]. The abundance of phnJ, a gene associated with the mineralization of organic phosphorus compounds [49], increased by two orders of magnitude compared to fallow conditions. The abundance of pstB gene, encoding a phosphate transporter protein involved in phosphorus uptake and transport, was 2.5 times higher in tilled soils than on fallow. Signature analysis (Figure 5) further highlighted that the elevated presence of pstB in tilled soils is linked to the increased abundance of Bradyrhizobium sp. 200, suggesting this species plays a significant role in phosphorus solubilization.
In the context of nitrogen metabolism, genes such as queC, amiF, pyrG, guaA, guaB, napA were more abundant in soils subjected to tillage practices. These genes are essential for nitrogen fixation and nitrate reduction, processes that are critical for soil fertility and plant growth. The increased abundance of these genes under tillage conditions is consistent with the higher levels of microbial biomass and the significant reduction in nitrate-nitrogen (N-NO3) soil content (Table 1). N-NO3 reduction is likely due to the enhanced activity of microbial communities carrying nitrogen metabolism genes, which promotes the immobilization of nitrogen in organic forms and reduces nitrate leaching [50]. Nitrate leaching is a major environmental concern, as it can lead to the contamination of groundwater and surface water bodies, contributing to eutrophication and other ecological imbalances [51]. By minimizing nitrate losses, tillage practices not only improve soil health but also contribute to broader environmental sustainability. High presence of nitrogen metabolism genes in tilled soils comes to an agreement with increased abundance of Bradyrhizobium spp., renowned for their ability to fix free nitrogen through symbiotic relationships with legumes and reported to carry the genes of this cluster [52].
5. Conclusions
This study demonstrates that different tillage practices significantly influence the functional genes and microbial taxa associated with key biogeochemical cycles in Russian black soil. No-tillage technology and conventional tillage practices promote beneficial microbial communities and enhance soil health by improving nutrient cycling and organic matter decomposition. The increased abundance of genes involved in carbon, nitrogen, and phosphorus metabolism, as well as the microbial taxa responsible for these processes, underscores the importance of adopting sustainable tillage practices to maintain soil fertility and productivity. The use of metagenomic analysis provides a comprehensive understanding of soil microbial communities and their functions, demonstrating high correlation with traditional widely-used physical and chemical methods evaluating soil quality/fertility and offering valuable insights for the development of sustainable agricultural practices.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Catalog information;
Author Contributions
Conceptualization, O.O.G., N.A.M., V.V.P., D.A.T. and A.A.V.; methodology, A.A.D., T.V.G. and M.I.M.; software, O.O.G.; formal analysis, O.O.G.; investigation, K.S.G.; resources, A.M.Z.; writing—original draft preparation, O.O.G., N.A.M. and A.A.V..; writing—review and editing, O.O.G., N.A.M. and A.A.V..; visualization, O.O.G., N.A.M. and A.A.V..; supervision,K.S.G.; project administration, K.S.G.; funding acquisition, A.A.V. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Olesya O. Galanova1, Nikita A. Mitkin2, Albina A. Danilova3, Vsevolod V. Pavshintsev2, Denis A. Tsybizov2, Alexander M. Zakharenko3, Kirill S. Golokhvast3,4, Tatiana V Grigoryeva5, Maria I Markelova5, Aleksey A. Vatlin
Funding
This work was supported by the RUDN University Scientific Projects Grant System, project № 202760-2-000
Acknowledgments
We express gratitude to the Association of Specialists in the Field of Molecular, Cellular and Synthetic Biology (Russia) for their efforts in uniting specialists involved in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Appendix A

Table A1.
Median relative abundances for genes with statistically significant differences.
| Gene | Long-term fallow | Conventional tillage (median) | No-Till technology (median) |
|---|---|---|---|
| accA | 1.53·106 | 15.15·106 | 12.48·106 |
| acsA | 2.55·108 | 8.09·108 | 8.08·108 |
| amiF | 0 | 5.38·103 | 9.43·103 |
| argG | 108.81·103 | 624.34·103 | 578.59·103 |
| guaA | 1.44·106 | 3.54·106 | 3.41·106 |
| guaB | 1.73·106 | 4.79·106 | 4.72·106 |
| miaB | 53.03·103 | 441.45·103 | 281.52·103 |
| mphE | 1.27·103 | 10.87·103 | 14.29·103 |
| napA | 106.36·103 | 3.67·106 | 3.42·106 |
| phnJ | 0.94·103 | 14.27·103 | 20.07·103 |
| phoB | 121.40·103 | 343.04·103 | 913.79·103 |
| pnp | 0 | 2.46·106 | 1.22·106 |
| ppa | 236.70·103 | 910.72·103 | 772.52·103 |
| pstB | 1.49·108 | 3.68·108 | 3.72·108 |
| pyrG | 2.22·106 | 5.18·106 | 4.96·106 |
| queC | 1.32·103 | 6.84·103 | 9.72·103 |
References
- Wilhelm, R.C.; Amsili, J.P.; Kurtz, K.S.M.; van Es, H.M.; Buckley, D.H. Ecological Insights into Soil Health According to the Genomic Traits and Environment-Wide Associations of Bacteria in Agricultural Soils. ISME Communications 2023, 3, 1. [Google Scholar] [CrossRef]
- Zapata, J.D.D.; Florez, J.E.M.; Alvarez, D.L. Metagenomics Approaches to Understanding Soil Health in Environmental Research - a Review. Soil Sci. Ann. 2023, 74, 1–11. [Google Scholar] [CrossRef]
- Chang, T.; Feng, G.; Paul, V.; Adeli, A.; Brooks, J. Soil Health Assessment Methods: Progress, Applications and Comparison. In Advances in Agronomy; Academic Press, 2022; Vol. 172, pp. 129–210. [Google Scholar]
- Prosser, J.I. Dispersing Misconceptions and Identifying Opportunities for the Use of “omics” in Soil Microbial Ecology. Nat Rev Microbiol 2015, 13, 439–446. [Google Scholar] [CrossRef] [PubMed]
- White, R.A.; Bottos, E.M.; Roy Chowdhury, T.; Zucker, J.D.; Brislawn, C.J.; Nicora, C.D.; Fansler, S.J.; Glaesemann, K.R.; Glass, K.; Jansson, J.K. Moleculo Long-Read Sequencing Facilitates Assembly and Genomic Binning from Complex Soil Metagenomes. mSystems 2016, 1. [Google Scholar] [CrossRef]
- Song, W.; Wang, Y.; Peng, B.; Yang, L.; Gao, J.; Xiao, C. Structure and Function of Microbiomes in the Rhizosphere and Endosphere Response to Temperature and Precipitation Variation in Inner Mongolia Steppes. Front. Plant Sci. 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Behnke, G.D.; Kim, N.; Zabaloy, M.C.; Riggins, C.W.; Rodriguez-Zas, S.; Villamil, M.B. Soil Microbial Indicators within Rotations and Tillage Systems. Microorganisms 2021, 9, 1244. [Google Scholar] [CrossRef]
- Kim, N.; Zabaloy, M.C.; Riggins, C.W.; Rodríguez-Zas, S.; Villamil, M.B. Microbial Shifts Following Five Years of Cover Cropping and Tillage Practices in Fertile Agroecosystems. Microorganisms 2020, 8, 1773. [Google Scholar] [CrossRef]
- Sorokin, A.; Owens, P.; Lang, V.; Jiang, Z.; Micheli, E.; Krasilnikov, P. “Black Soils” in the Russian Soil Classification System, the US Soil Taxonomy and the WRB: Quantitative Correlation and Implications for Pedodiversity Assessment. CATENA 2021, 196, 104824. [Google Scholar] [CrossRef]
- Coughenour, C.M. Innovating Conservation Agriculture: The Case of No-Till Cropping. 2009, 68, 178–304. [Google Scholar] [CrossRef]
- Heanes, D.L. Determination of Total organic-C in Soils by an Improved Chromic Acid Digestion and Spectrophotometric Procedure. Communications in Soil Science and Plant Analysis 1984. [Google Scholar] [CrossRef]
- Anderson, J.P.E.; Domsch, K.H. A Physiological Method for the Quantitative Measurement of Microbial Biomass in Soils. Soil Biology and Biochemistry 1978, 10, 215–221. [Google Scholar] [CrossRef]
- Milham, P.J.; Awad, A.S.; Paull, R.E.; Bull, J.H. Analysis of Plants, Soils and Waters for Nitrate by Using an Ion-Selective Electrode. Analyst 1970, 95, 751–757. [Google Scholar] [CrossRef]
- Khaziev, F. Enzymatic Activity of Soils. Moscow: Nauka Publ. 1976, 180. [Google Scholar]
- Jiang, H.; Lei, R.; Ding, S.-W.; Zhu, S. Skewer: A Fast and Accurate Adapter Trimmer for next-Generation Sequencing Paired-End Reads. BMC Bioinformatics 2014, 15, 182. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biology 2019, 20, 257. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome Analysis Using the Kraken Software Suite. Nat Protoc 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. Journal of Vegetation Science 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the Manually Annotated Section of the UniProt KnowledgeBase: How to Use the Entry View. In Plant Bioinformatics: Methods and Protocols; Edwards, D., Ed.; Springer: New York, NY, 2016; ISBN 978-1-4939-3167-5. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Research 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kiryushin, V.K.; Vlasenko, A.N.; Kalichkin, V.K. Adaptive Landscape Farming Systems of the Novosibirsk Region. Novosibirsk, Izdatel’stvo SO RASKhN = The Publishing House of the Siberian Branch of the Russian Academy of Sciences 2002, 387. [Google Scholar]
- Danilova, A.A. Biodynamics of Arable Soil at Different Content of Organic Matter. Novosibirsk: SFNCA RAS 2018, 156. [Google Scholar]
- Neal, A.L.; Bacq-Labreuil, A.; Zhang, X.; Clark, I.M.; Coleman, K.; Mooney, S.J.; Ritz, K.; Crawford, J.W. Soil as an Extended Composite Phenotype of the Microbial Metagenome. Sci Rep 2020, 10, 10649. [Google Scholar] [CrossRef]
- Wu, Y.; Kemmitt, S.; White, R.P.; Xu, J.; Brookes, P.C. Carbon Dynamics in a 60 Year Fallowed Loamy-Sand Soil Compared to That in a 60 Year Permanent Arable or Permanent Grassland UK Soil. Plant Soil 2012, 352, 51–63. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, W.; Zheng, J.; Luo, Y.; Li, R.; Wang, H.; Qi, H. Effect of Long-Term Tillage on Soil Aggregates and Aggregate-Associated Carbon in Black Soil of Northeast China. PLOS ONE 2018, 13, e0199523. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qi, S.; Gao, W.; Luo, Y.; Hou, Y.; Liang, Y.; Zheng, H.; Zhang, S.; Li, R.; Wang, M.; et al. Eight-Year Tillage in Black Soil, Effects on Soil Aggregates, and Carbon and Nitrogen Stock. Sci Rep 2023, 13, 8332. [Google Scholar] [CrossRef]
- Chao, A.; Shen, T.-J. Nonparametric Estimation of Shannon’s Index of Diversity When There Are Unseen Species in Sample. Environmental and Ecological Statistics 2003, 10, 429–443. [Google Scholar] [CrossRef]
- Chendev, Y.G.; Sauer, T.J.; Ramirez, G.H.; Burras, C.L. History of East European Chernozem Soil Degradation; Protection and Restoration by Tree Windbreaks in the Russian Steppe. Sustainability 2015, 7, 705–724. [Google Scholar] [CrossRef]
- Sergaki, C.; Lagunas, B.; Lidbury, I.; Gifford, M.L.; Schäfer, P. Challenges and Approaches in Microbiome Research: From Fundamental to Applied. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Skaalsveen, K.; Ingram, J.; Clarke, L. The Effect of No-till Farming on the Soil Functions of Water Purification and Retention in North-Western Europe: A Literature Review. Soil and Tillage Research 2019, 189, 98–109. [Google Scholar] [CrossRef]
- Degrune, F.; Theodorakopoulos, N.; Colinet, G.; Hiel, M.-P.; Bodson, B.; Taminiau, B.; Daube, G.; Vandenbol, M.; Hartmann, M. Temporal Dynamics of Soil Microbial Communities below the Seedbed under Two Contrasting Tillage Regimes. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Six, J.; Elliott, E.T.; Paustian, K. Soil Macroaggregate Turnover and Microaggregate Formation: A Mechanism for C Sequestration under No-Tillage Agriculture. Soil Biology and Biochemistry 2000, 32, 2099–2103. [Google Scholar] [CrossRef]
- Marcel, G.A.; Richard, D.B.; Nico, M. van S. The Unseen Majority: Soil Microbes as Drivers of Plant Diversity and Productivity in Terrestrial Ecosystems.
- Monciardini, P.; Cavaletti, L.; Schumann, P.; Rohde, M.; Donadio, S. Conexibacter Woesei Gen. Nov., Sp. Nov., a Novel Representative of a Deep Evolutionary Line of Descent within the Class Actinobacteria. International Journal of Systematic and Evolutionary Microbiology 2003, 53, 569–576. [Google Scholar] [CrossRef]
- Vieira, S.; Huber, K.J.; Geppert, A.; Wolf, J.; Neumann-Schaal, M.; Luckner, M.; Wanner, G.; Müsken, M.; Overmann, J. Capillimicrobium Parvum Gen. Nov., Sp. Nov., a Novel Representative of Capillimicrobiaceae Fam. Nov. within the Order Solirubrobacterales, Isolated from a Grassland Soil. International Journal of Systematic and Evolutionary Microbiology 2022, 72, 005508. [Google Scholar] [CrossRef]
- Hara, S.; Morikawa, T.; Wasai, S.; Kasahara, Y.; Koshiba, T.; Yamazaki, K.; Fujiwara, T.; Tokunaga, T.; Minamisawa, K. Identification of Nitrogen-Fixing Bradyrhizobium Associated With Roots of Field-Grown Sorghum by Metagenome and Proteome Analyses. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Agashe, R.; George, J.; Pathak, A.; Fasakin, O.; Seaman, J.; Chauhan, A. Shotgun Metagenomics Analysis Indicates Bradyrhizobium Spp. as the Predominant Genera for Heavy Metal Resistance and Bioremediation in a Long-Term Heavy Metal-Contaminated Ecosystem. Microbiology Resource Announcements 2024, 13, e00245–24. [Google Scholar] [CrossRef]
- Chaddad, Z.; Lamrabet, M.; Bennis, M.; Kaddouri, K.; Alami, S.; Bouhnik, O.; El Idrissi, M.M. Nitrogen-Fixing Bradyrhizobium Spp. as Plant Growth-Promoting Bacteria to Improve Soil Quality and Plant Tolerance to Biotic and Abiotic Stresses. In Soil Bacteria: Biofertilization and Soil Health; Dheeman, S., Islam, M.T., Egamberdieva, D., Siddiqui, Md.N., Eds.; Springer Nature: Singapore, 2024; ISBN 978-981-9734-73-3. [Google Scholar]
- Jones, F.P.; Clark, I.M.; King, R.; Shaw, L.J.; Woodward, M.J.; Hirsch, P.R. Novel European Free-Living, Non-Diazotrophic Bradyrhizobium Isolates from Contrasting Soils That Lack Nodulation and Nitrogen Fixation Genes – a Genome Comparison. Sci Rep 2016, 6, 25858. [Google Scholar] [CrossRef]
- Li, Y.; Xiong, L.; Zeng, K.; Wei, Y.; Li, H.; Ji, X. Microbial-Driven Carbon Fixation in Natural Wetland. Journal of Basic Microbiology 2023, 63, 1115–1127. [Google Scholar] [CrossRef]
- Liu, S.; Li, H.; Xie, X.; Chen, Y.; Lang, M.; Chen, X. Long-Term Moderate Fertilization Increases the Complexity of Soil Microbial Community and Promotes Regulation of Phosphorus Cycling Genes to Improve the Availability of Phosphorus in Acid Soil. Applied Soil Ecology 2024, 194, 105178. [Google Scholar] [CrossRef]
- Rocabruna, P.; Domene, X.; Preece, C.; Fernández-Martínez, M.; Maspons, J.; Penuelas, J. Effect of Climate, Crop, and Management on Soil Phosphatase Activity in Croplands: A Global Investigation and Relationships with Crop Yield. European Journal of Agronomy 2024, 161, 127358. [Google Scholar] [CrossRef]
- Richardson, A.E.; Barea, J.-M.; McNeill, A.M.; Prigent-Combaret, C. Acquisition of Phosphorus and Nitrogen in the Rhizosphere and Plant Growth Promotion by Microorganisms. Plant Soil 2009, 321, 305–339. [Google Scholar] [CrossRef]
- Wu, X.; Rensing, C.; Han, D.; Xiao, K.-Q.; Dai, Y.; Tang, Z.; Liesack, W.; Peng, J.; Cui, Z.; Zhang, F. Genome-Resolved Metagenomics Reveals Distinct Phosphorus Acquisition Strategies between Soil Microbiomes. mSystems 2022, 7, e01107–21. [Google Scholar] [CrossRef]
- Qin, L.; Xiao, Z.; Ming, A.; Teng, J.; Zhu, H.; Qin, J.; Liang, Z. Soil Phosphorus Cycling Microbial Functional Genes of Monoculture and Mixed Plantations of Native Tree Species in Subtropical China. Front. Microbiol. 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Tanuwidjaja, I.; Vogel, C.; Pronk, G.J.; Schöler, A.; Kublik, S.; Vestergaard, G.; Kögel-Knabner, I.; Mrkonjic Fuka, M.; Schloter, M.; Schulz, S. Microbial Key Players Involved in P Turnover Differ in Artificial Soil Mixtures Depending on Clay Mineral Composition. Microb Ecol 2021, 81, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.-L.; Liu, J.; Jia, P.; Yang, T.; Zeng, Q.; Zhang, S.; Liao, B.; Shu, W.; Li, J. Novel Phosphate-Solubilizing Bacteria Enhance Soil Phosphorus Cycling Following Ecological Restoration of Land Degraded by Mining. The ISME Journal 2020, 14, 1600–1613. [Google Scholar] [CrossRef]
- Wang, J.; Chen, Z.; Xu, C.; Elrys, A.; Shen, F.; Cheng, Y.; Chang, S. Organic Amendment Enhanced Microbial Nitrate Immobilization with Negligible Denitrification Nitrogen Loss in an Upland Soil. Environmental Pollution 2021, 288, 117721. [Google Scholar] [CrossRef]
- Padilla, F.; Gallardo, M.; Manzano-Agugliaro, F. Global Trends in Nitrate Leaching Research in the 1960–2017 Period. Science of The Total Environment 2018, 643, 400–413. [Google Scholar] [CrossRef]
- Zhong, C.; Hu, G.; Hu, C.; Xu, C.; Zhang, Z.; Ning, K. Comparative Genomics Analysis Reveals Genetic Characteristics and Nitrogen Fixation Profile of Bradyrhizobium. iScience 2024, 27. [Google Scholar] [CrossRef]
Figure 1.
Alpha diversity of the taxonomic composition of Leached black earths. The values of the Shannon index are located on the Y-axis. On the X-axis, there are subgroups of soil samples. The shape of the graph represents the distribution density of the Shannon index. The boxplot shows the median, quartiles, and spread of the Shannon index among the data.
Figure 1.
Alpha diversity of the taxonomic composition of Leached black earths. The values of the Shannon index are located on the Y-axis. On the X-axis, there are subgroups of soil samples. The shape of the graph represents the distribution density of the Shannon index. The boxplot shows the median, quartiles, and spread of the Shannon index among the data.
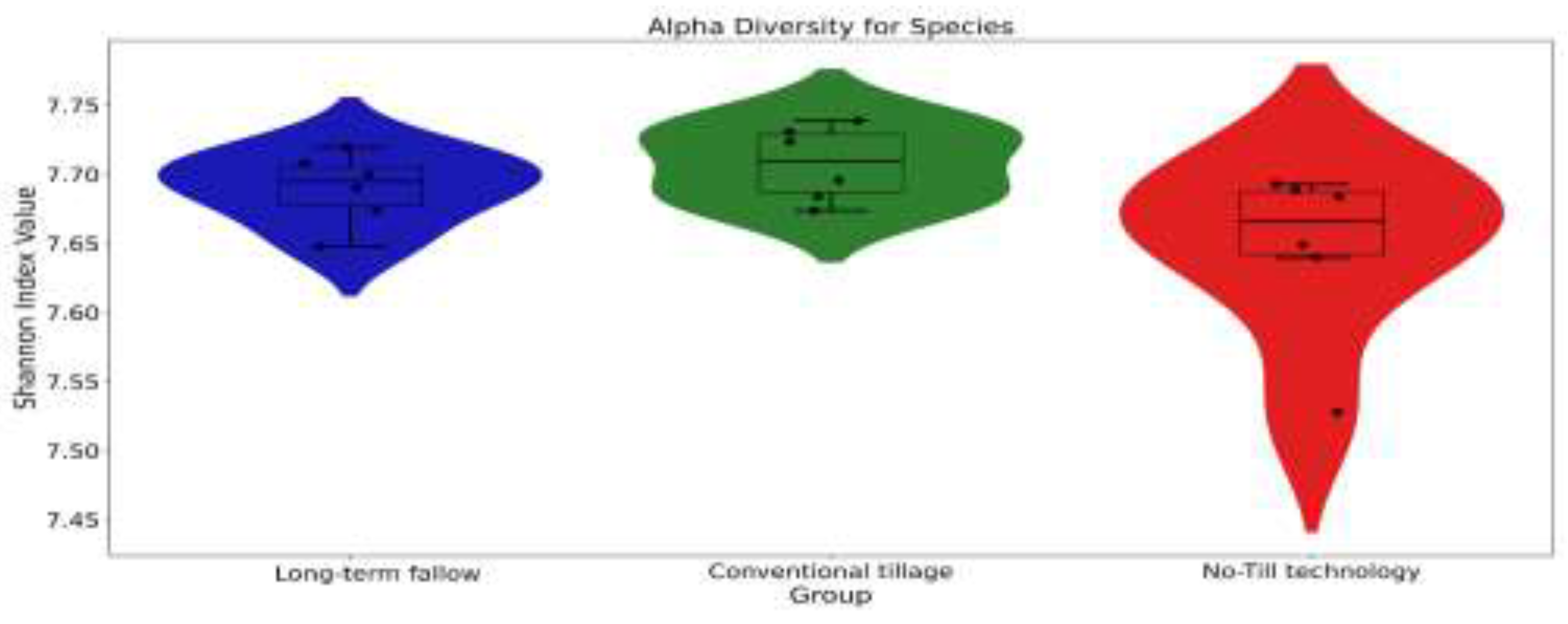
Figure 2.
Beta diversity of the taxonomic composition of Leached black earths at the level of species. The NMDS graph shows the clustering of soil samples (dots on the figure). In our case, the samples are clearly divided into 3 subgroups, the results of the PERMANOVA test indicate that this result is statistically significant (p-value = 0.01). The blue dots represent soil samples from samples subgroup with long-term fallow, green dots for soil samples with conventional tillage and red dots for soil samples with No-Till technology.
Figure 2.
Beta diversity of the taxonomic composition of Leached black earths at the level of species. The NMDS graph shows the clustering of soil samples (dots on the figure). In our case, the samples are clearly divided into 3 subgroups, the results of the PERMANOVA test indicate that this result is statistically significant (p-value = 0.01). The blue dots represent soil samples from samples subgroup with long-term fallow, green dots for soil samples with conventional tillage and red dots for soil samples with No-Till technology.
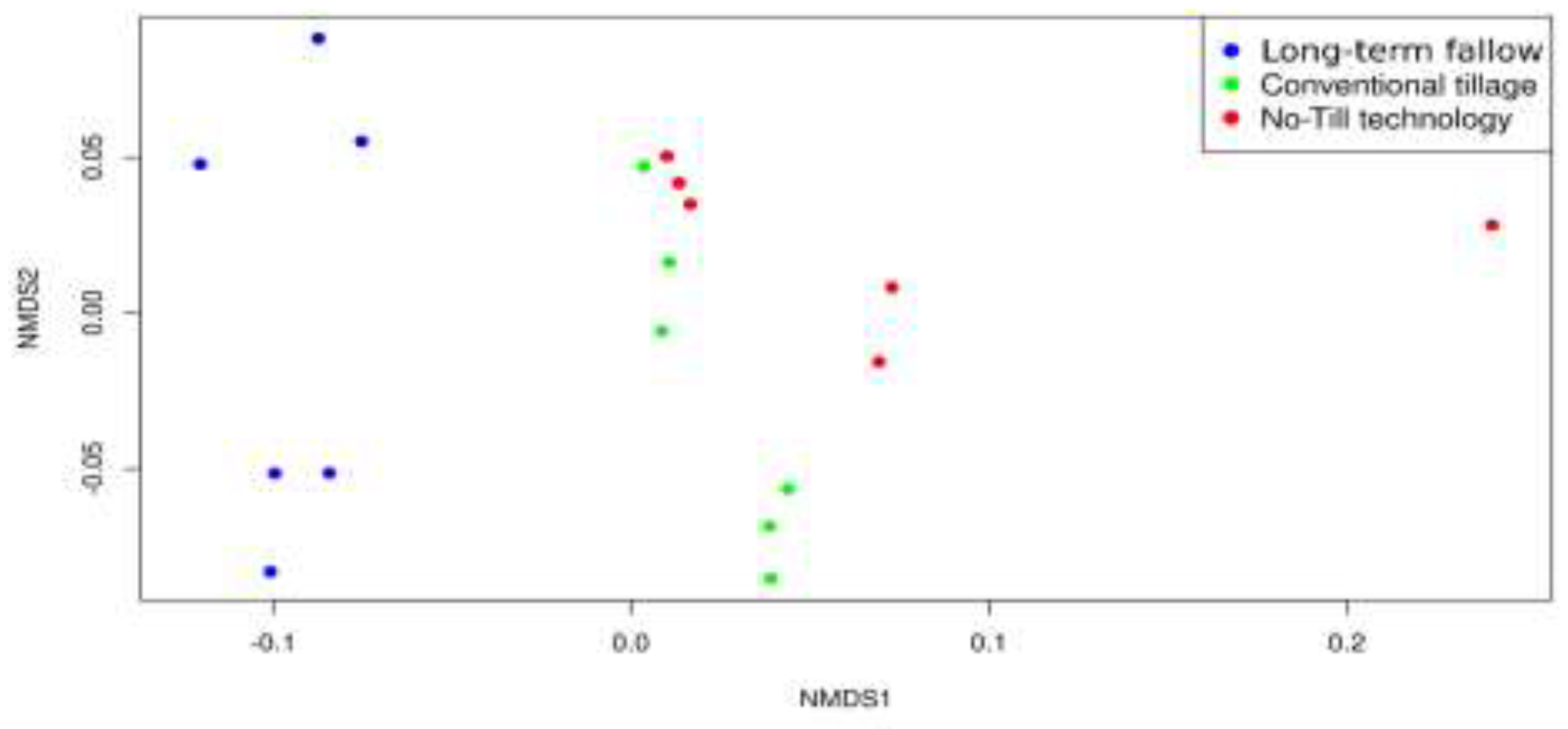
Figure 3.
Comparison of median values of relative representation of species for groups of leached black earths. The relative representation of species in % is located on the Y-axis. On the X-axis, there are subgroups of soil samples.
Figure 3.
Comparison of median values of relative representation of species for groups of leached black earths. The relative representation of species in % is located on the Y-axis. On the X-axis, there are subgroups of soil samples.
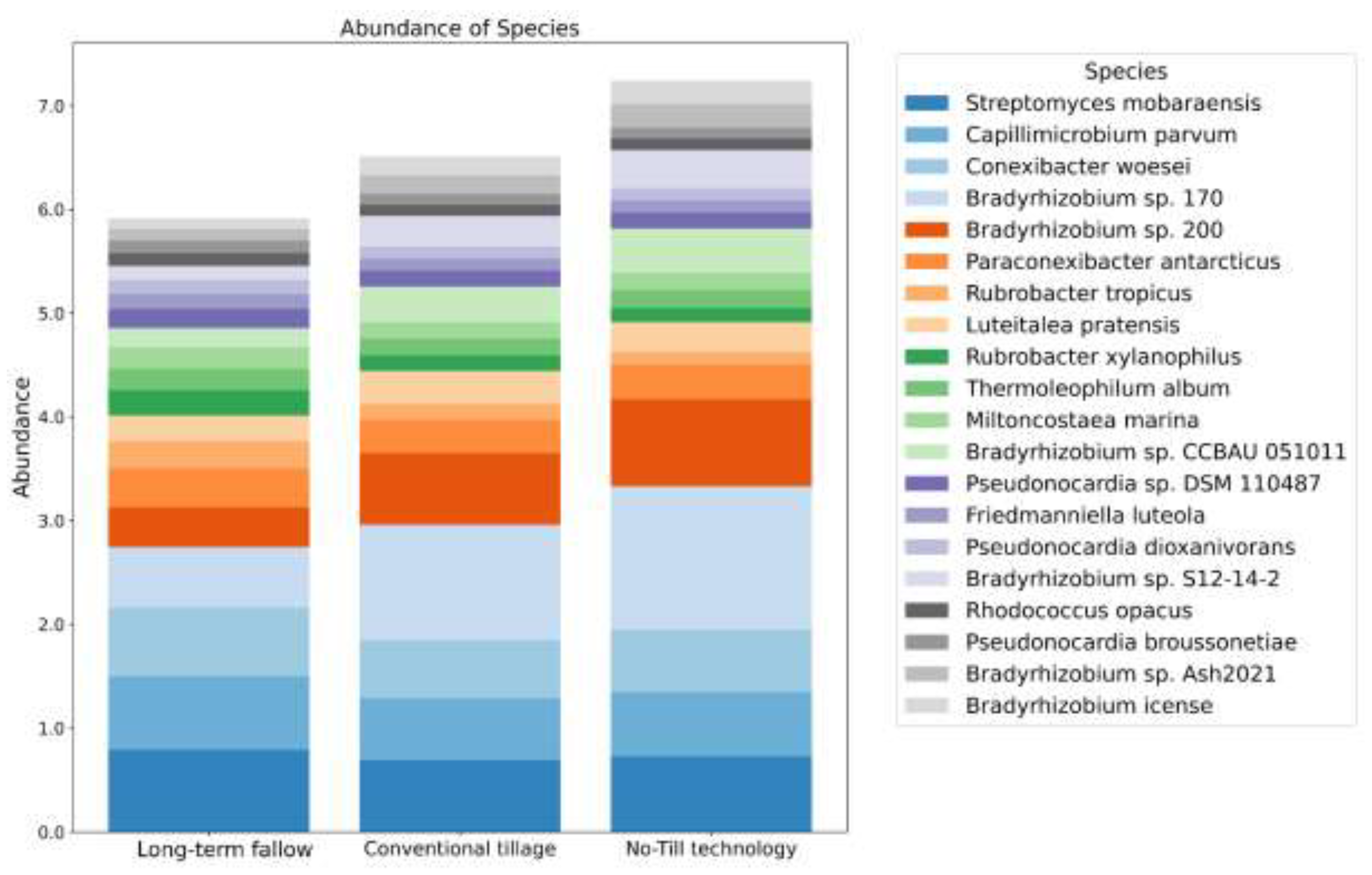
Figure 4.
Comparison of median values of relative representation of species for groups of leached black earths. The relative representation of species in % is located on the Y-axis. On the X-axis, there are subgroups of soil samples.
Figure 4.
Comparison of median values of relative representation of species for groups of leached black earths. The relative representation of species in % is located on the Y-axis. On the X-axis, there are subgroups of soil samples.
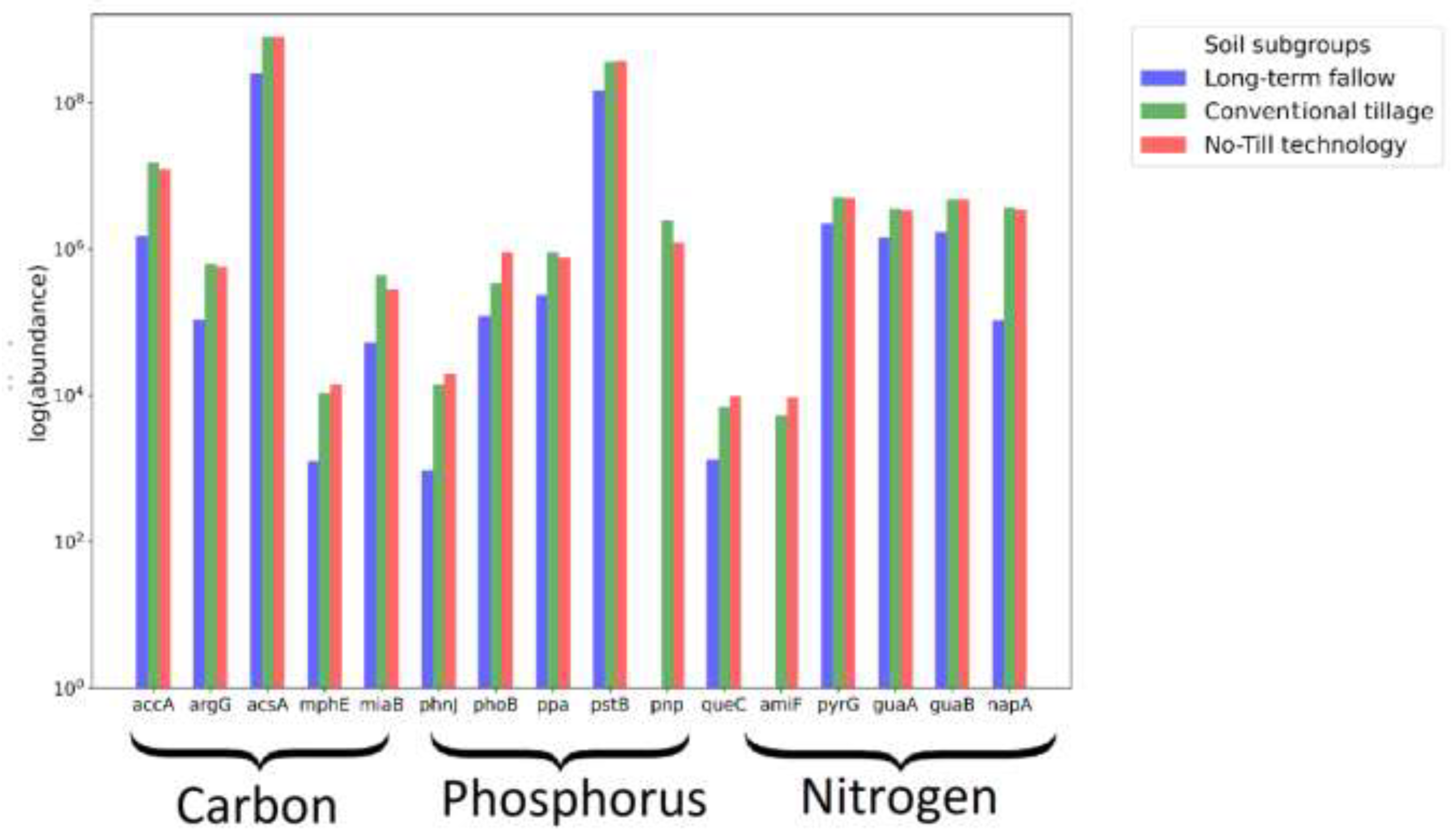
Figure 5.
The heatmap for signature pairs, compiled according to their median relative abundance (FDR < 0.05). The center of the scale is set at 500 readcounts. Here we can see, that the highest abundance is for ‘Bradyrhizobium sp. CCBAU 051011|acsA’ and ‘Bradyrhizobium sp. 200|pstB’
Figure 5.
The heatmap for signature pairs, compiled according to their median relative abundance (FDR < 0.05). The center of the scale is set at 500 readcounts. Here we can see, that the highest abundance is for ‘Bradyrhizobium sp. CCBAU 051011|acsA’ and ‘Bradyrhizobium sp. 200|pstB’
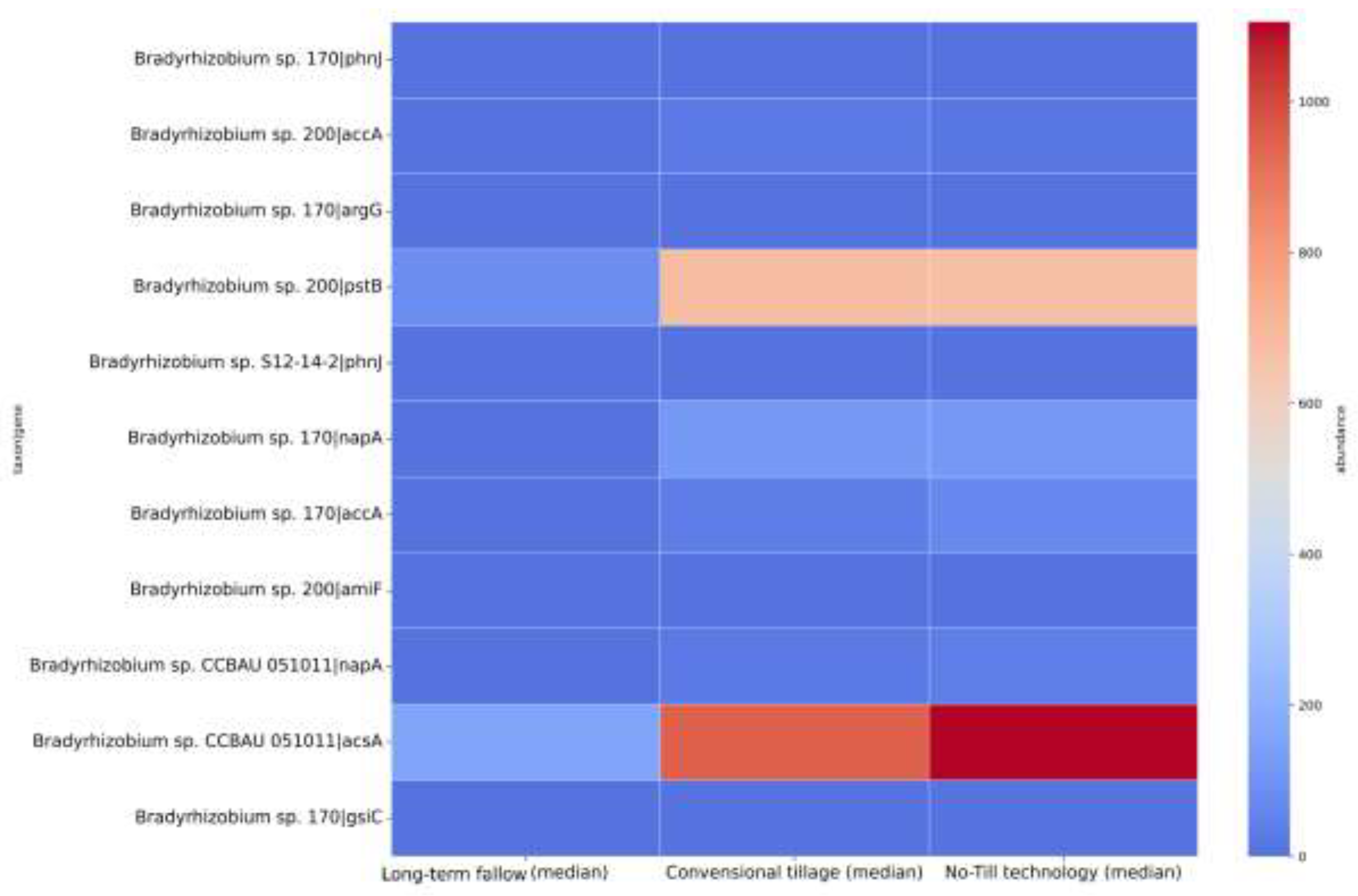
Table 1.
Properties of leached medium-humus, medium-loamy chernozem (Luvic Chernozem) of the Ob forest-steppe of different types of treatment.
Table 1.
Properties of leached medium-humus, medium-loamy chernozem (Luvic Chernozem) of the Ob forest-steppe of different types of treatment.
| Soil indicator | Long-term fallow | Conventional tillage | No-Till technology |
|---|---|---|---|
| TOC % | 4.1 | 4.2 | |
| C in mortmass, mg/kg | 67 | 817 | 1133 |
| C in microbial biomass, μg/g | 30 | 100 | 120 |
| N-NO3, mg/kg | 78.6 | 19.8 | 4.4 |
| Phosphatase activity, μg, Р2О5/g per hour | 13.8 | 14.6 | 26.2 |
Table 2.
Annotation for genes with statistically significant differences in relative abundance about their function and belonging to the metabolic pathway (carbon, nitrogen, phosphorus).
Table 2.
Annotation for genes with statistically significant differences in relative abundance about their function and belonging to the metabolic pathway (carbon, nitrogen, phosphorus).
| Gene name | Enzyme name | KEGG enzyme entry | Metabolic pathway |
| accA | Acetyl-coenzyme A carboxylase carboxyl transferase subunit alpha | 2.1.3.15 | carbon |
| argG | Argininosuccinate synthase (Forming carbon-nitrogen bonds) | 6.3.4.5 | |
| acsA | Acetyl-coenzyme A synthetase | 6.2.1.1 | |
| mphE | 4-hydroxy-2-oxovalerate aldolase | 4.1.3.39 | |
| miaB | tRNA-2-methylthio-N(6)-dimethylallyladenosine synthase (catalyzes methylation) | 2.8.4.3 | |
| phnJ | Alpha-D-ribose 1-methylphosphonate 5-phosphate C-P lyase | 4.7.1.1 | phosphorus |
| phoB | Phosphate regulon transcriptional regulatory protein | 3.6.1.11 | |
| ppa | Inorganic pyrophosphatase | 3.6.1.1 | |
| pstB | Phosphate import ATP-binding protein | 7.3.2.1 | |
| pnp | Polyribonucleotide nucleotidyltransferase (catalyzes the phosphorolysis) | 2.7.7.8 | |
| queC | 7-cyano-7-deazaguanine synthase (Forming carbon-nitrogen bonds) | 6.3.4.20 | nitrogen |
| amiF | Formamidase (Acting on carbon-nitrogen bonds) | 3.5.1.49 | |
| pyrG | CTP synthase (glutamine hydrolysing) (Forming carbon-nitrogen bonds) | 6.3.4.2 | |
| guaA | GMP synthase [glutamine-hydrolyzing] (Forming carbon-nitrogen bonds) | 6.3.5.2 | |
| guaB | Inosine-5'-monophosphate dehydrogenase (Acting on the CH-OH group of donors) | 1.1.1.205 | |
| napA | Periplasmic nitrate reductase | 1.9.6.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



