Submitted:
18 February 2025
Posted:
18 February 2025
You are already at the latest version
Abstract
One of the most challenging issues in catalytic water dissociation hydrogen production technology is understanding the activation mechanism of water. To gain insights into this process, a first-principles molecular dynamics method was used to simulate the catalytic dissociation of H2O on 88 alloy catalysts. The study's results revealed that a larger red shift of the center of the v1 and v3 modes of adsorbed H2O corresponds to a lower dissociation temperature. The resonance absorption of heat from water molecules by the frontier electron promotes the dissociation reaction. Comparing the frontier orbitals of precursors and intermediate states shows that the number of involved frontier orbitals significantly influences the catalytic reaction.
Keywords:
Water Dissociation
; First-principles Molecular Dynamics
; Catalysis
; Infrared Spectrum of Vibration
; Frontier Orbitals
; Resonance Absorption of Heat
1. Introduction
Hydrogen, a clean and renewable energy source, has the potential to address global warming, environmental pollution, and the fossil energy crisis [1,2,3]. The overall reaction for hydrogen production by water splitting is an “uphill reaction” with an unfavorable positive free Gibbs energy of ΔGo = 237.13 kJ/mol-1, and it usually cannot occur spontaneously due to a barrier. Catalysts with electronic and optical properties that can be tailored to specific dimensions, shapes, and compositions can reduce the energy barriers associated with chemical reactions, thereby facilitating their occurrence [4]. Since the initial report in 1972 demonstrating photoelectrochemical water splitting under UV irradiation with an n-type TiO2 photoanode and a Pt cathode for H2 evolution (the Honda-Fujishima effect) [5], there has been a vast quantity of research articles and reviews focusing on various materials for photocatalytic water splitting [6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21].
The dissociation of water can be achieved by electronic excitations [22,23] or by vibrational excitation [24,25]. The water molecule exhibits three distinct vibrational normal modes: bending (v2), symmetric (v1), and antisymmetric (v3) stretching modes. The two stretching modes are strongly coupled (Darling–Dennison coupling) due to the close vibrational frequencies and anharmonicity, while the coupling between the bending mode and the stretching motion is weak [26,27]. Polanyi’s rule posits that a “late” barrier is more readily surmounted by vibrational excitation, whereas an “earlier” barrier is more readily overcome by translational energy. This is based on the observation that the “promoting coordinate” coincides with the reaction coordinate near the transition state in each case [28]. The dissociative chemisorption of H2O has a late barrier, where the bond cleavage occurs after the molecule adsorbs on the surface. Consequently, vibrational excitation of H2O may have a significant impact on reactivity. Vibrationally excited molecular hydrogen has been observed in the dense photodominated regions [29]. Experimentalists have previously reported that in the presence of a laser field, vibrational excitation increases the reactivity of unimolecular and bimolecular reactions more than translational and rotational excitation [30,31]. Theoretical studies of water dissociative chemisorption have predicted that water on Cu(111) and Ni(111) exhibits strong mode specificity, bond selectivity, and steric effects [32,33,34,35]. In a pioneering study, Zhang et al. employed full-dimensional quantum dynamics to investigate the dissociative chemisorption of H2O on rigid Cu(111). Their findings revealed that the excitations in vibrational modes of H2O play a more pivotal role than increasing the translational energy in promoting the reaction. Moreover, they demonstrated that the enhancement of the excitation in asymmetric stretching is the most pronounced [36]. The influence of alloying on mode-selectivity in H2O dissociation on Ni(100), Ni(110), Ni(111) and Cu/Ni bimetallic surfaces has been investigated by Tiwari et al. The authors employed a fully quantum approach based on the reaction path Hamiltonian, which revealed that mode specificity of H2O dissociation was observed on all the surfaces. This invariably suggested that the vibrational excitation resulted in a significant enhancement in reactivity for all systems [37,38]. These conclusions are consistent with those of quantum state-resolved molecular beam experiments on the dissociation of heavy water on Ni (111) [39]. The vibrational enhancement of reactivity can be understood by the sudden vector projection (SVP) model, and the enhancement of reactivity by Polanyi’s rule mode reflects its coupling with the reaction coordinate at the transition state [40,41]. Indeed, Kim et al. have successfully manipulated the state-selective dissociation of a single water molecule using inelastic tunneling electrons with a low-temperature scanning tunnelling microscope. This process results in the dissociation of water into hydroxyl by the excitation of the vibrational state of the stretching modes and into atomic oxygen by the excitation of the electronic state [42]. Additionally, vibrationally excited hydrolysis dissociation experiments are an effective means of measuring hydrolysis dissociation thresholds. For instance, Maksyutenko et al. accurately determined the value of the first dissociation threshold of water, D0 = 41145.94 ± 0.15 cm−1, from the onset of the dissociative continuum in triple-resonance spectra [43].
The atomic-scale insights into the interactions of water molecules with catalysts have long been recognized as essential, given the fundamental role of these interactions in the elementary process of hydrogen generation. The theoretical calculations based on density functional theory (DFT) have been employed to elucidate the mechanism of water interaction with metal surfaces at the molecular level [33]. Despite substantial efforts in both experimental and theoretical studies, to date only a few works have been published on the role of chemical shifts in the infrared spectra of water molecules when adsorbed on catalysts in characterizing the reactivity of water molecules. Since vibrational spectroscopy can provide information on the structure and the bonding of the molecules, the correlation between reactivity and chemical shift in the infrared vibrational spectra of H2O on the Cu(10-n)Ptn and Cu(9-n)Pdn alloy catalysts has been studied [44,45]. In these studies, we introduced the concept of the center of the v1 and v3 modes (the center of the v1 and v3 modes = (v1 ×intensity of v1 + v3×intensity of v3)/ (intensity of v1 + intensity of v3)) is used to characterize the reactivity of water molecules due to the strong coupling of the two stretching modes of H2O with the reaction coordinate. The results indicated that the dissociation barrier of H2O is closely dependent on the red shift of the center of the v1 and v3 modes of H2O adsorbed. The larger the red shift of the center of the v1 and v3 modes is, the lower the corresponding barrier is. It is therefore hypothesized that the red shift of the center due to the adsorption of water molecules on the catalyst can be used to characterize the activation properties of the catalysts. This study examines the chemical shifts observed in the infrared vibrational spectra of H2O adsorbed on 88 alloy clusters, with the objective of testing this hypothesis and developing a predictive understanding of the activity of the catalysts.
2. Computational Details
In this study, we optimized the structures of catalysts using the generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) functional [46] and a density functional theory (DFT)-based relativistic semicore pseudo-potential (DSPP) [47]. Subsequently, infrared spectra were calculated. In order to investigate the effect of the heat of adsorption reaction of water molecules, we selected the NVE ensemble and conducted a first-principles molecular dynamics simulation of the catalytic dissociation of H2O molecules at temperatures ranging from 0 to 1500 K, with a temperature step of 100 K. The time step and the total simulation time were 1 fs and 5 ps, respectively. All pertinent details have been provided in the supplementary materials.
3. Results and Discussions
3.1. Identification and Construction of Research Subjects
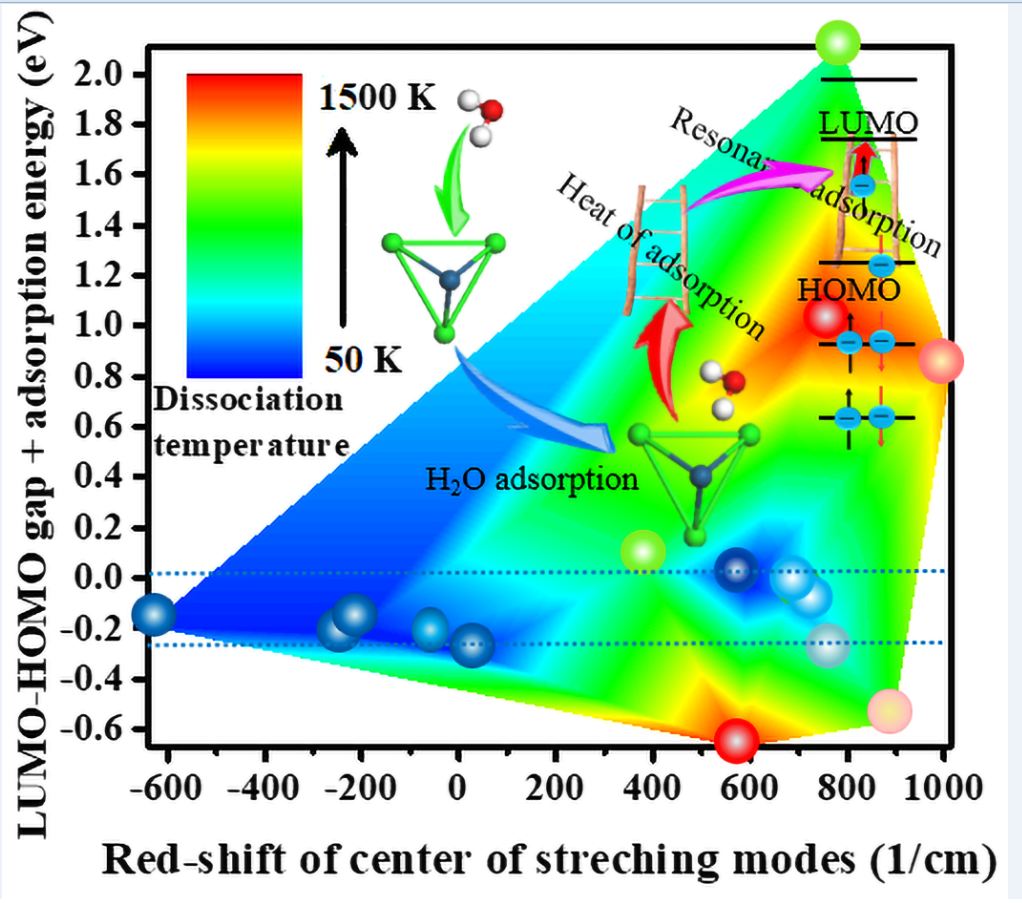
The adsorption behavior of H2O on the first 75 single atoms in the periodic table was initially investigated. Three cases of being positively charged, uncharged, and negatively charged were considered separately, as illustrated in Figure 1. The red shifts of the center of the v1 and v3 modes of H2O adsorbed on single atoms were then plotted in relation to that of the isolated H2O in an electrically neutral state. It is evident that the red shift of the center of the stretching vibration modes of H2O adsorbed on the single atom carrying a negative charge is considerably larger than that of the other two cases. This phenomenon exhibits a certain periodic behavior that is closely dependent on the nature of the elements. The reason why inert elements exhibit high activity in negative charge is that their high stability makes it easier for electrons to transfer to water molecules, thereby leaving the water molecules in a more active state. Consequently, the extent of the red shift of the center of the v1 and v3 modes of H2O adsorbed on inert elements is comparable to that observed when the water molecule carries a direct negative charge.
The catalytic dissociation of water is typically achieved through the use of Cu and Pt metal surfaces [9,38]. The red shift of the center of the stretching (v1 and v3) modes of H2O adsorbed on the negatively charged Cu atom is 615 1/cm, as illustrated by the green dash-dotted line in Figure 1. The construction of 24 diatomic alloy clusters, 30 triatomic alloy clusters, and 34 tetraatomic alloy clusters, based mainly on Pt, was carried out using sodium elements representing alkali metals and elements with redshift scales exceeding that of copper, as shown in Table 1. Subsequently, the catalytic dissociation of H2O molecules over the aforementioned catalysts was simulated within a temperature range of 50–1500 K.
3.2. Dependence of Dissociation Temperature on Infrared Vibrational Spectra
The dependence of the catalytic dissociation temperature of water on the red shift of the center of the v1 and v3 modes of H2O on different clusters is shown in Figure 2. For comparison, the red shift of the center of the v1 and v3 modes of H2O is relative to the negatively charged isolated H2O (∆IR). For catalysts with a water dissociation temperature above 1500K, the dissociation temperatures are all marked as 1500K for ease of plotting. It is evident that clusters with less than 300 1/cm of ∆IR have high catalytic water dissociation activity, corresponding to a dissociation temperature of no more than 200K. For ∆IR is between 300 and 700 1/cm, except for Sr3Pt and CuPt, these clusters still showed high catalytic activity with dissociation temperatures not higher than 600K. For the red shifts in the 700-1016 1/cm range, different catalysts showed significant variations in catalytic activity. Other catalysts corresponding to higher ∆IR showed no visible catalytic activity. The results here show that the red shift of the center of the v1 and v3 modes of H2O, which takes into account the strong coupling of the two stretching modes [26,27], allows a good characterization of the catalytic reactivity of water molecules. Given that the dissociation of water on these catalysts occurs mainly through a late-barrier reaction, as shown in Figure S1, it is reasonable to expect the above results from the Polanyi’s law and the Sudden Variation Vector Projection (SVP) model [39,40] as well. It is also consistent with the results observed by Tiwari’s group in their work on vibrationally excited dissociation of water molecules on Ni surfaces, where the softening of symmetric stretching and bending modes suggests that the excitation of these modes would lead to a significant enhancement in reaction probabilities [37].
As illustrated in Figure 2, while the red shift of the center of the stretching modes is an effective descriptor of catalytic reactivity in water, the origin of some exceptions remains unknown. A multitude of factors, including molecular orbitals in proximity to the Fermi level, tunneling, resonances, the zero-point energy (ZPE) effect, and electron spin polarization, among others, can influence the reactivity of water [36,37,38,48,49,54]. The following section will provide a detailed examination of the mechanism of catalytic water activation.
3.3. Resonant Absorption of Heat of Adsorption Versus Dissociation Temperature
Faradzhev et al. found that water dissociation on Ru(0 0 0 1) can be achieved by thermal or by electronic excitation [50]. Therefore, charge transfer, adsorption energy, bond length, frontier orbitals, and density of states were considered to elucidate the underlying mechanisms of catalytic water activation. In order to examine the relationship between the heat of reaction of adsorption of water molecules and the excitation of the electronic state, we combine the LUMO-HOMO gap (Eg) and the adsorption energy of H2O (Ea). The adsorption energies of the water molecule on catalysts with high catalytic activity and several inactive catalysts, the LUMO-HOMO gaps of the catalysts adsorbing H2O, and the corresponding dissociation temperature are listed in Table 2. From these data, it can be seen that lower dissociation temperatures always tend to correspond to systems where the sum of LUMO-HOMO gap and the adsorption energy of H2O (Eg+Ea) is close to zero or the adsorption energy slightly higher than the LUMO-HOMO gap. If the exceptions are excluded, as in the cases of SrRe3 and FPt, the dissociation temperature and the absolute values of Eg+Ea are strongly correlated in Table 2 with a Pearson correlation coefficient of 0.90. This means that if the heat released by the adsorption of H2O is equal to or slightly greater than the energy required to excite an electron from the HOMO to the LUMO orbital, the catalytic water dissociation reaction will tend to occur. On the contrary, when the absolute value of (Eg+Ea) is greater than 0.25 eV, the reaction is generally difficult to occur. Accordingly, electronic state excitation triggered by resonant absorption of the heat of adsorption reaction is one of the important reasons for promoting the catalytic dissociation process of water molecules. This result is readily understandable when one considers that some scientists have observed that catalytic dissociation of water can be achieved on some metal substrates and oxide monolayers by providing energy corresponding to the lowest unoccupied molecular orbital (LUMO) energy of water [41,51,52,53]. Although the adsorption energy of water on Sr2Re is slightly 0.05 eV lower than that of the LUMO-HOMO gap, the quantum tunneling effect may be contributing factors to the catalytic water dissociation reaction.
For the case of SrRe3, it has the absolute value of (Eg+Ea) of 0.3 eV, but a low dissociation temperature of 100 K, which attributes to the Frontier molecular orbitals and will be discussed in Section 3.4. For FPt, the corresponding (Eg+Ea) value is 2.11 eV, which are considerably far from the resonance region, but with a dissociation temperature of 600 K. This is mainly due to the fact that the products of dissociation of water molecules on FPt are an H adsorbed on the Pt atom and a free HO group, which leads to the entropy from the intermediate state to the product is increased, that is, the entropy-driven effect plays an important role in this reaction. This is opposite to the entropy change in the dissociation reaction of water on other catalysts, as shown in Figure S1.
The correlation between dissociation temperature and both (Eg+Ea) and ∆IR has been illustrated in Figure 3, thereby facilitating a more straightforward comprehension of this relationship. One can see that when the ∆IR of water molecules after adsorption is lower and the value of (Eg+Ea) within the range of 0 to -0.30 eV (between the two blue dashed lines), the corresponding catalytic dissociation temperature of water molecules is usually lower, as shown in Figure 3. It is in alignment with the conclusions presented in both Section 3.2 and Section 3.3.
3.4. The Function of the Frontier Molecular Orbitals (FMOs)
The concept of locality in physical space is of paramount importance in the understanding of the underlying mechanisms of chemical reactivity. Frontier molecular orbitals (FMOs) serve to identify the locality of chemical bonds that are chemically reactive, due to the associated orbital energies, and have thus far proven to be an invaluable tool in the description of chemical reactivity [55,56]. In order to gain a more comprehensive understanding of the reaction kinetics of water dissociation, a detailed analysis was performed of the frontier orbitals of the catalysts with adsorbed H2O molecules and the frontier orbitals of the intermediate state in the catalytic dissociation reaction, as illustrated in Figure S1. By comparing the frontier orbitals of the reaction precursors and the intermediate states, it is possible to identify which orbitals play a role in the dissociation process. The configuration of the frontier orbitals (HOMO-1, HOMO, and LUMO) of precursors is essentially identical to that of the intermediate states in the reaction, which indicates that the frontier orbitals of reaction precursors play a pivotal role in the catalytic reaction process. In a few instances, the order of frontier orbits underwent a slight alteration. For instance, as illustrated in Figure S1, the energy hierarchy of the HOMO and LUMO of SrReH2O (precursor) underwent a reversal, with the energy levels of HOMO-1 and HOMO of Sr2ReH2O (precursor) shifting upwards to become the HOMO and LUMO of the intermediate state, respectively. A comparison of the frontier orbitals (HOMO-1, HOMO, and LUMO) of the precursor and intermediate state in the dissociation process reveals that the number of orbitals involved in the reaction process (orbitals from catalysts with increased overlap with H atoms during H2O dissociation) is inversely proportional to the corresponding H2O dissociation temperature. This is a logical conclusion, as the larger the overlap integral, the stronger the interaction between H and catalytic atoms will be. This phenomenon has been observed by Kawai and colleagues in their study of the role of molecular orbitals near the Fermi level in the excitation of vibrational modes of a single water molecule. In this study, a molecule is chemisorbed onto the metal surface, resulting in the formation of a molecule-metal bond and the rehybridization of the molecule’s orbitals [57]. In Figure 4, we utilize CuPt, SrPt, and Sr2Re as illustrative examples to elucidate the influence of frontier orbitals on catalytic water dissociation reactions. For CuPt, a comparison of the two sets of frontier orbitals on the left in Figure 4 reveals that only the LUMO orbitals are involved in the catalytic separation of H from H2O molecules, without the assistance of resonance absorption. Consequently, this material is unable to effectively catalyze the water dissociation reaction at temperatures below 1500K. From an energetic standpoint, the HOMO-LUMO gap (Eg) and the heat generated by the adsorption of water molecules are nearly identical, which evidently satisfies the prerequisite for the resonance absorption in the case of SrPt. Furthermore, the incorporation of both the HOMO and LUMO orbitals into the process of H separation can be observed in the frontier orbitals of the intermediate state. Both of these processes result in lower dissociation temperatures on SrPt. In the case of Sr2Re, as illustrated in the two sets of images on the right in Figure 4, the HOMO-1 and LUMO orbitals of the intermediate state participate in the catalytic process of H2O decomposition through changing the level order, thus lowering the corresponding dissociation temperature compared to that on CuPt. Ditto for the dissociation of water molecules on SrRe3 as shown in Figure S1(e), where the homo-1, homo and LUMO orbitals are all involved in the catalytic water dissociation reaction, and thus the corresponding dissociation temperatures are as low as 100 K under conditions away from the energy resonance.
4. Conclusions
In conclusion, the redshift of the center of the v1 and v3 modes of H2O (∆IR) can reflect the catalytic activation of water molecules to a certain extent. However, a more profound comprehension of the activation mechanism of water molecules necessitates a comprehensive examination of the degree of involvement of the frontier orbitals and entropy changes. It is our conviction that these findings will stimulate further experimental and theoretical research in this domain.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Materials and Methods, Supplementary Text, Figure S1.
Data Availability Statement
All data are available in the main text or the supplementary materials.
Acknowledgments
The Natural Science Foundation of Jiangsu Province (Grant BK20150118), the Natural Science Foundation of Wuxi Institute of Technology (Grant ZK202104), the National Natural Science Foundation of China (Grant 11905084), the Colleges and Universities in Jiangsu Province Natural Science Research Projects (Grant 20KJB140002) have provided financial support for this research, and Jiangsu University ”Qing Lan Project” have also contributed to this endeavor.
Conflicts of Interest
There are no conflicts of interest to declare.
References
- Li, Y.; et al. Recent Advances on Water-Splitting Electrocatalysis Mediated by Noble-Metal-Based Nanostructured Materials. Adv. Energy Mater. 2020, 10, 1903120. [Google Scholar] [CrossRef]
- Yuan, W.; et al. Visualizing H2O Molecules Reacting at TiO2 Active Sites with Transmission Electron Microscopy. Science 2020, 367, 428–430. [Google Scholar] [CrossRef]
- Nishiyama, H.; et al. Photocatalytic Solar Hydrogen Production from Water on a 100-m2 Scale. Nature 2021, 598, 304–307. [Google Scholar] [CrossRef]
- Kranz, C.; Wächtler, M. Characterizing photocatalysts for water splitting: from atoms to bulk and from slow to ultrafast processes. Chem. Soc. Rev. 2021, 50, 1407–1437. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I.; Takahashi, A. H2O Dissociation at the Perimeter Interface between Gold Nanoparticles and TiO2 Is Crucial for Oxidation of CO. ACS Catal. 2020, 10, 2517–2521. [Google Scholar] [CrossRef]
- Fajin, J.L.C.; Cordeiro, M.N.D.S.; Illas, F.; Gomesc, J.R.B. Influence of step sites in the molecular mechanism of the water gas shift reaction catalyzed by copper. J. Catal. 2009, 268, 131–141. [Google Scholar] [CrossRef]
- Prats, H.; Gamallo, P.; Illas, F.; Sayós, R. Comparing the catalytic activity of the water gas shift reaction on Cu(321) and Cu(111) surfaces: Step sites do not always enhance the overall reactivity. J. Catal. 2016, 342, 75–83. [Google Scholar] [CrossRef]
- Huang, S.C.; Lin, C.H.; Wang, J.H. Trends of Water Gas Shift Reaction on Close-Packed Transition Metal Surfaces. J. Phys. Chem. C 2010, 114, 9826–9834. [Google Scholar] [CrossRef]
- Ahmad, F.; Agusta, M.K.; Maezono, R.; Dipojono, H.K. DFT + U study of H2O adsorption and dissociation on stoichiometric and nonstoichiometric CuO(111) surfaces. J. Phys. Condens. Matter. 2020, 32, 045001. [Google Scholar] [CrossRef]
- Karim, N.A.; Alias, M.S.; Kamarudin, S.K. The Mechanism of the Water Dissociation and Dehydrogenation of Glycerol on Au(111) and PdAu Alloy Catalyst Surfaces. Int. J. Hydrog. Energy 2021, 46, 30937–30947. [Google Scholar] [CrossRef]
- Wang, J.; et al. Amorphization Activated Ruthenium-Tellurium Nanorods for Efficient Water Splitting. Nat. Commun. 2019, 10, 5692. [Google Scholar] [CrossRef]
- Fajin, J.L.C.; Cordeiro, M.N.D.S.; Illas, F.; Gomesc, J.R.B. Descriptors controlling the catalytic activity of metallic surfaces toward water splitting. J. Catal. 2010, 276, 92–100. [Google Scholar] [CrossRef]
- Wu, R.; et al. A Janus Nickel Cobalt Phosphide Catalyst for High-Efficiency Neutral-PH Water Splitting. Angew. Chem. Int. Ed. 2018, 47, 15671–15675. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, Z.; Zhang, S.; Liang, Y.; Wang, R. Engineering MoS2 Basal Planes for Hydrogen Evolution via Synergistic Ruthenium Doping and Nanocarbon Hybridization. Adv. Sci. 2019, 6, 1900090. [Google Scholar] [CrossRef]
- Tang, Y.; et al. d-Band Center Modulating of CoOx/Co9S8 by Oxygen Vacancies for Fast-Kinetics Pathway of Water Oxidation. Chem. Eng. J. 2022, 427, 130915. [Google Scholar] [CrossRef]
- Miao, X.; et al. Quadruple Perovskite Ruthenate as a Highly Efficient Catalyst for Acidic Water Oxidation. Nat. Commun. 2019, 10, 3809. [Google Scholar] [CrossRef]
- Takata, T.; Domen, K. Defect Engineering of Photocatalysts by Doping of Aliovalent Metal Cations for Efficient Water Splitting. J. Phys. Chem. C 2009, 113, 19386–19388. [Google Scholar] [CrossRef]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Domen, K. Photocatalytic Water Splitting with a Quantum Efficiency of Almost Unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef]
- Mu, L.; et al. Enhancing Charge Separation on High Symmetry SrTiO3 Exposed with Anisotropic Facets for Photocatalytic Water Splitting. Energy Environ. Sci. 2016, 9, 2463–2469. [Google Scholar] [CrossRef]
- Zhang, Y.; et al. Single-atom Cu anchored catalysts for photocatalytic renewable H2 production with a quantum efficiency of 56%. Nat. Commun. 2022, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Haxton, D.J.; Rescigno, T.N.; McCurdy, C.W. Dissociative electron attachment to the H2O molecule. II. Nuclear dynamics on coupled electronic surfaces within the local complex potential model. Phys. Rev. A 2007, 75, 012711. [Google Scholar] [CrossRef]
- Adaniya, H.; et al. Imaging the molecular dynamics of dissociative electron attachment to water. Phys. Rev. Lett. 2009, 103, 233201. [Google Scholar] [CrossRef]
- Jiang, B.; Ren, X.; Xie, D.; Guo, H. Enhancing dissociative chemisorption of H2O on Cu(111) via vibrational excitation. PNAS 2012, 109, 10224–10227. [Google Scholar] [CrossRef]
- Persson, B.N.J.; Baratoff, A. Inelastic electron tunneling from a metal tip: the contribution from resonant processes. Phys. Rev. Lett. 1987, 59, 339–342. [Google Scholar] [CrossRef]
- Yu, C.C.; Chiang, K.Y.; Okuno, M.; et al. Vibrational couplings and energy transfer pathways of water’s bending mode. Nat. Commun. 2020, 11, 5977. [Google Scholar] [CrossRef]
- Darling, T.; Dennison, D.M. The Water Vapor Molecule. Phys. Rev. 1940, 57, 128–139. [Google Scholar] [CrossRef]
- Polanyi, J.C. Concepts in reaction dynamics. Acc. Chem. Res. 1972, 5, 161–168. [Google Scholar] [CrossRef]
- Chang, Y.; An, F.; Chen, Z.; et al. Vibrationally excited molecular hydrogen production from the water photochemistry. Nat. Commun. 2021, 12, 6303. [Google Scholar] [CrossRef]
- Crim, F.F. State- and Bond-Selected Unimolecular Reactions. Science 1990, 249, 1387. [Google Scholar] [CrossRef]
- Crim, F.F. Vibrational state control of bimolecular reactions: discovering and directing the chemistry. Acc. Chem. Res. 1999, 32, 877. [Google Scholar] [CrossRef]
- Jiang, B.; Xie, D.; Guo, H. Vibrationally mediated bond selective dissociative chemisorption of HOD on Cu(111). Chem. Sci. 2013, 4, 503–508. [Google Scholar] [CrossRef]
- Mondal, A.; Seenivasan, H.; Tiwari, A.K. Water dissociation on Cu (111): Effects of molecular orientation, rotation, and vibration on reactivity. J. Chem. Phys. 2012, 137, 094708. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Li, J.; Xie, D.; Guo, H. Effects of reactant internal excitation and orientation on dissociative chemisorption of H2O on Cu(111): Quasi-seven-dimensional quantum dynamics on a refined potential energy surface. J. Chem. Phys. 2013, 138, 044704. [Google Scholar] [CrossRef]
- Seenivasan, H.; Tiwari, A.K. Water dissociation on Ni(100) and Ni(111): Effect of surface temperature on reactivity. J. Chem. Phys. 2013, 139, 174707. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, T.; Fu, B.; Yang, X.; Zhang, D.H. First-Principles Quantum Dynamical Theory for the Dissociative Chemisorption of H2O on Rigid Cu(111). Nat. Commun. 2016, 7, 11953. [Google Scholar] [CrossRef] [PubMed]
- Seenivasan, H.; Jackson, B.; Tiwari, A.K. Water dissociation on Ni(100), Ni(110), and Ni(111) surfaces: Reaction path approach to mode selectivity. J. Chem. Phys. 2017, 146, 074705. [Google Scholar] [CrossRef]
- Ghosh, S.; Ray, D.; Tiwari, A.K. Effects of alloying on mode-selectivity in H2O dissociation on Cu/Ni bimetallic surfaces. J. Chem. Phys. 2019, 150, 114702. [Google Scholar] [CrossRef]
- Hundt, P.M.; Jiang, B.; van Reijzen, M.E.; Guo, H.; Beck, R.D. Vibrationally Promoted Dissociation of Water on Ni(111). Science 2014, 344, 504–507. [Google Scholar] [CrossRef]
- Jiang, B.; Guo, H. Control of mode/bond selectivity and product energy disposal by the transition state: X + H2O (X = H, F, O(3P), and Cl) reactions. J. Am. Chem. Soc. 2013, 135, 15251–15256. [Google Scholar] [CrossRef]
- Jiang, B.; Guo, H. Relative efficacy of vibrational vs. translational excitation in promoting atom-diatom reactivity: Rigorous examination of Polanyi’s rules and proposition of sudden vector projection (SVP) model. J. Chem. Phys. 2013, 138, 234104. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Jung, J.; Motobayashi, K.; et al. State-selective dissociation of a single water molecule on an ultrathin MgO film. Nature Mater. 2010, 9, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Pavlo, M.; Maxim, G.; Thomas, R.R.; Oleg, V.B. State-resolved spectroscopy of high vibrational levels of water up to the dissociative continuum. Phil. Trans. R. Soc. A 2012, 370, 2710–2727. [Google Scholar]
- Wang, Y.; Zhang, X.; Wu, G. Shift of Infrared Vibrational Spectra and H2O Activation on PtCu Alloy Clusters. AIP Adv. 2020, 10, 085019. [Google Scholar] [CrossRef]
- Qian, C.; Wang, Y.; Wang, Z. A DFT Study of Enhancement of H2O Activation in Vibrational Excitation. Inorganica Chim. Acta. 2020, 514, 119980. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Sun, T.; Tang, Z.; Zang, W.; et al. Ferromagnetic single-atom spin catalyst for boosting water splitting. Nat. Nanotechnol. 2023, 18, 763–771. [Google Scholar] [CrossRef]
- Mtangi, W.; Kiran, V.; Fontanesi, C.; Naaman, R. Role of the Electron Spin Polarization in Water Splitting. J. Phys. Chem. Lett. 2015, 6, 4916–4922. [Google Scholar] [CrossRef]
- Faradzhev, N.S.; Kostov, K.L.; Feulner, P.; Madey, T.E.; Menzel, D. Stability of water monolayers on Ru(0 0 0 1): Thermal and electronically induced dissociation. Chemical Physics Letters 2005, 415, 165–171. [Google Scholar] [CrossRef]
- Lauhon, L.J.; Ho, W. Inducing and observing the abstraction of a single hydrogen atom in bimolecular reactions with a scanning tunneling microscope. J. Phys. Chem. B 2001, 105, 3987–3992. [Google Scholar] [CrossRef]
- Morgenstern, K.; Rieder, K.H. Dissociation of water molecules with the scanning tunnelling microscope. Chem. Phys. Lett. 2002, 358, 250–256. [Google Scholar] [CrossRef]
- Mugarza, A.; Shimizu, T.K.; Ogletree, D.F.; Salmeron, M. Chemical reaction of water molecules on Ru(0001) induced by selective excitation of vibrational modes. Surf. Sci. 2009, 603, 2030–2036. [Google Scholar] [CrossRef]
- Maksyutenko, P.; Rizzo, T.R.; Boyarkin, O.V. A direct measurement of the dissociation energy of water. J. Chem. Phys. 2006, 125, 181101. [Google Scholar] [CrossRef]
- Yu, J.; Su, N.Q.; Yang, W. Describing Chemical Reactivity with Frontier Molecular Orbitalets. JACS Au 2022, 2, 1383–1394. [Google Scholar] [CrossRef]
- Browna, J.J.; Cockroft, S.L. Aromatic reactivity revealed: beyond resonance theory and frontier orbitals. Chem. Sci. 2013, 4, 1772–1780. [Google Scholar] [CrossRef]
- Ohara, M.; Kim, Y.; Yanagisawa, S.; Morikawa, Y.; Kawai, M. Role of Molecular Orbitals Near the Fermi Level in the Excitation of Vibrational Modes of a Single Molecule at a Scanning Tunneling Microscope Junction. PRL 2008, 100, 136104. [Google Scholar] [CrossRef]
Figure 1.
The red shifts of the centers of v1 and v3 modes of H2O adsorbed on the first 75 single atoms in the periodic table relative to isolated electrically neutral H2O are presented. The black squares, red circles, and blue triangles represent negative charge, no charge, and a positive charge, respectively.
Figure 1.
The red shifts of the centers of v1 and v3 modes of H2O adsorbed on the first 75 single atoms in the periodic table relative to isolated electrically neutral H2O are presented. The black squares, red circles, and blue triangles represent negative charge, no charge, and a positive charge, respectively.
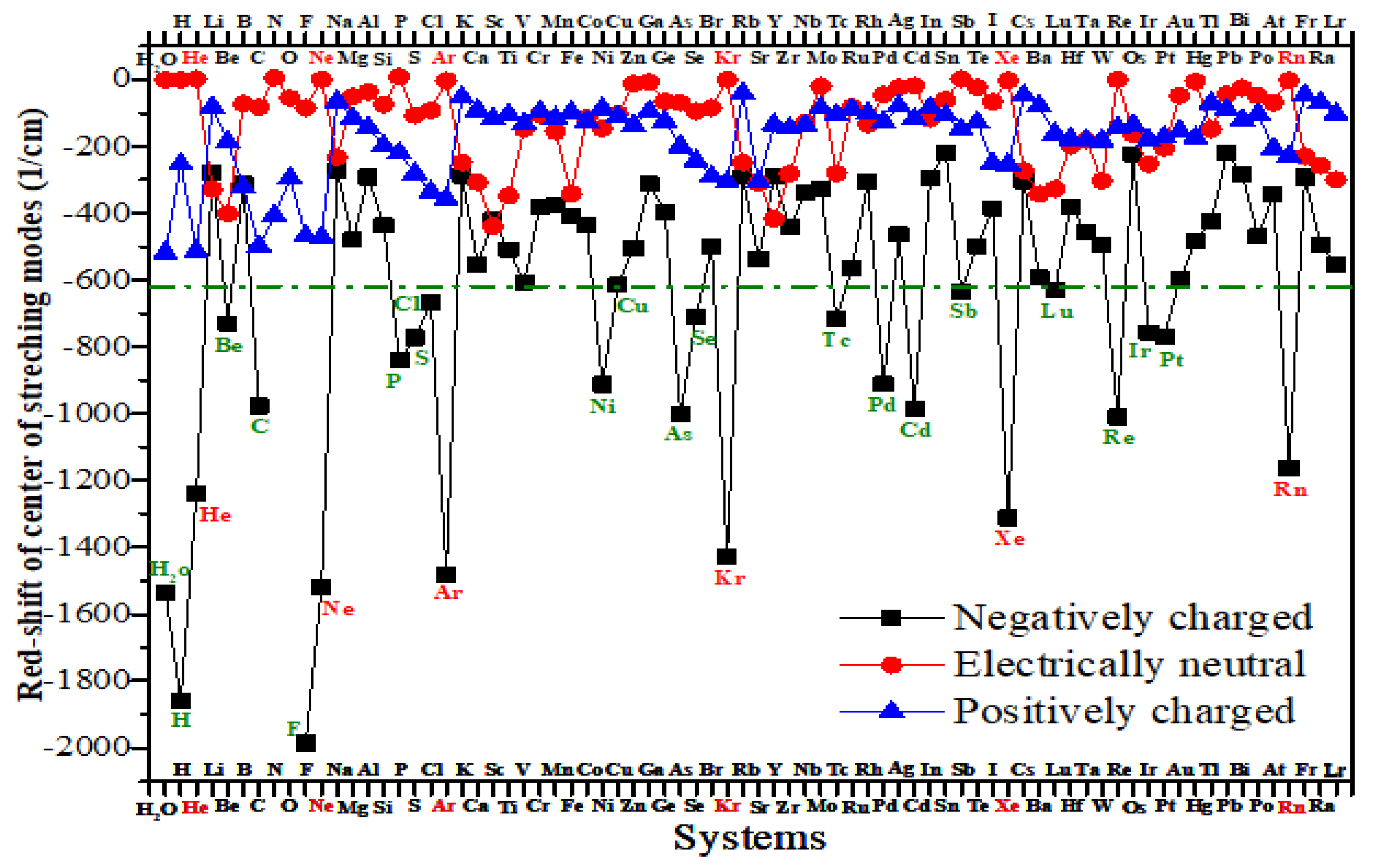
Figure 2.
The dependence of the catalytic dissociation temperature of water on the red shift of the center of the v1 and v3 modes of H2O on various clusters. The red shift of the center of the v1 and v3 modes of H2O is relative to that of the negatively charged isolated H2O. Blue circles represent catalysts.
Figure 2.
The dependence of the catalytic dissociation temperature of water on the red shift of the center of the v1 and v3 modes of H2O on various clusters. The red shift of the center of the v1 and v3 modes of H2O is relative to that of the negatively charged isolated H2O. Blue circles represent catalysts.
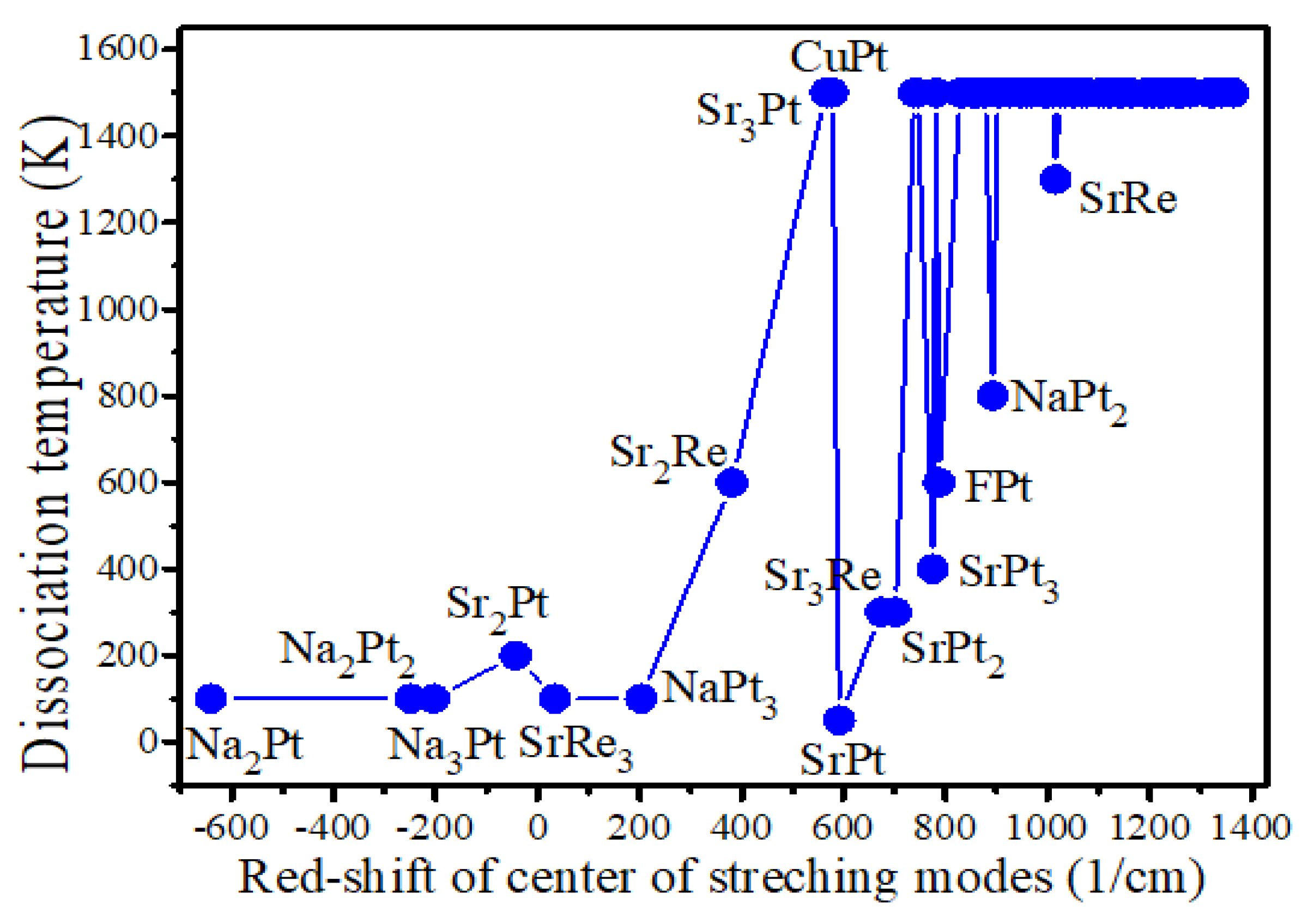
Figure 3.
The dependence of dissociation temperature on (Eg+Ea) and the red shift of the centre of v1 and v3 modes of H2O was plotted. The color of the ball is indicative of the dissociation temperature of H2O on the corresponding catalyst.
Figure 3.
The dependence of dissociation temperature on (Eg+Ea) and the red shift of the centre of v1 and v3 modes of H2O was plotted. The color of the ball is indicative of the dissociation temperature of H2O on the corresponding catalyst.
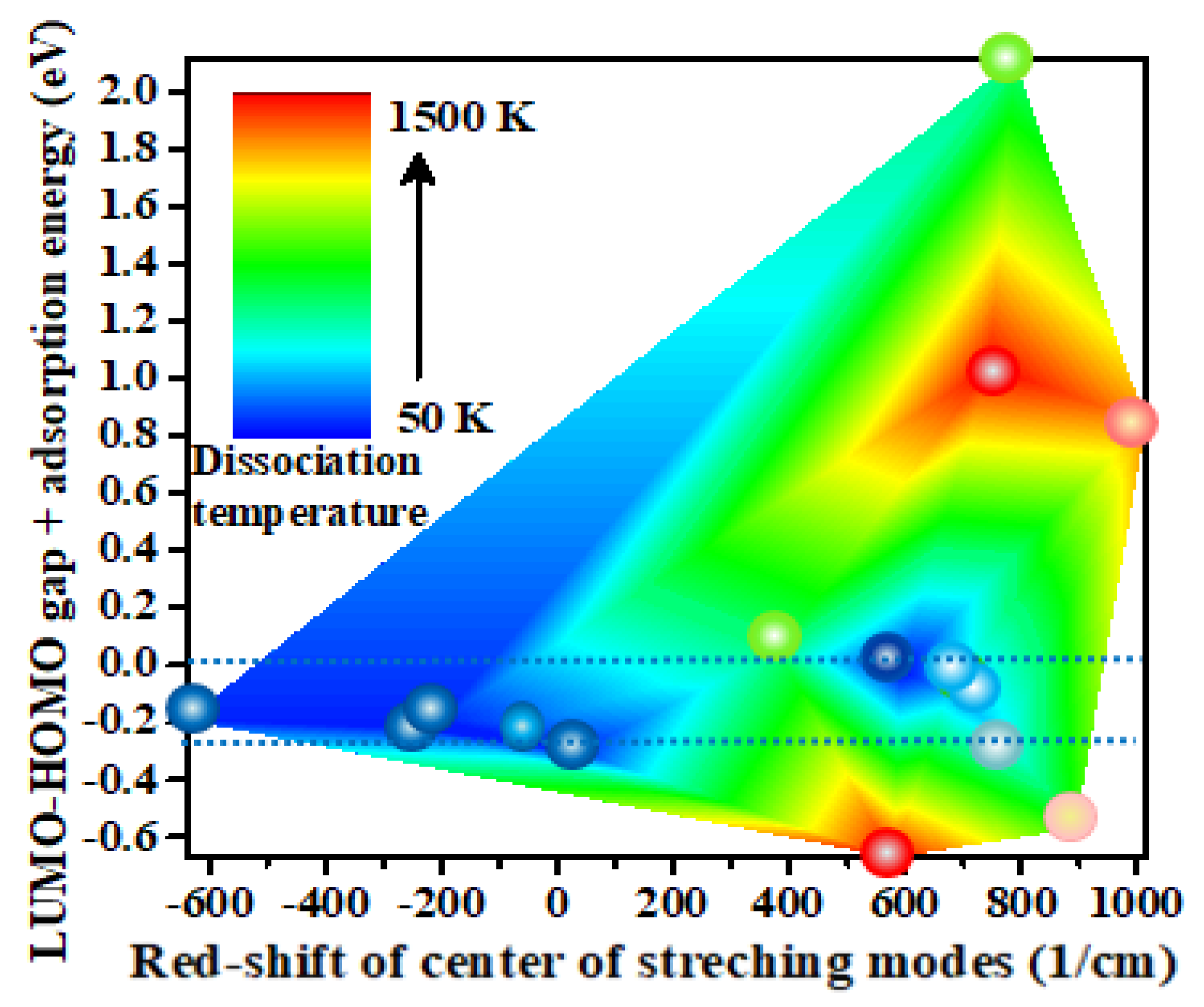
Figure 4.
Frontier orbitals (HOMO-1, HOMO, and LUMO) of the precursors and intermediate states in the dissociation reaction for the examples of CuPt, SrPt, and Sr2Re. Ea stands for adsorption energy of H2O, ∆IR for red-shift of center of v1 and v3 modes of H2O, Td for dissociation temperature.
Figure 4.
Frontier orbitals (HOMO-1, HOMO, and LUMO) of the precursors and intermediate states in the dissociation reaction for the examples of CuPt, SrPt, and Sr2Re. Ea stands for adsorption energy of H2O, ∆IR for red-shift of center of v1 and v3 modes of H2O, Td for dissociation temperature.
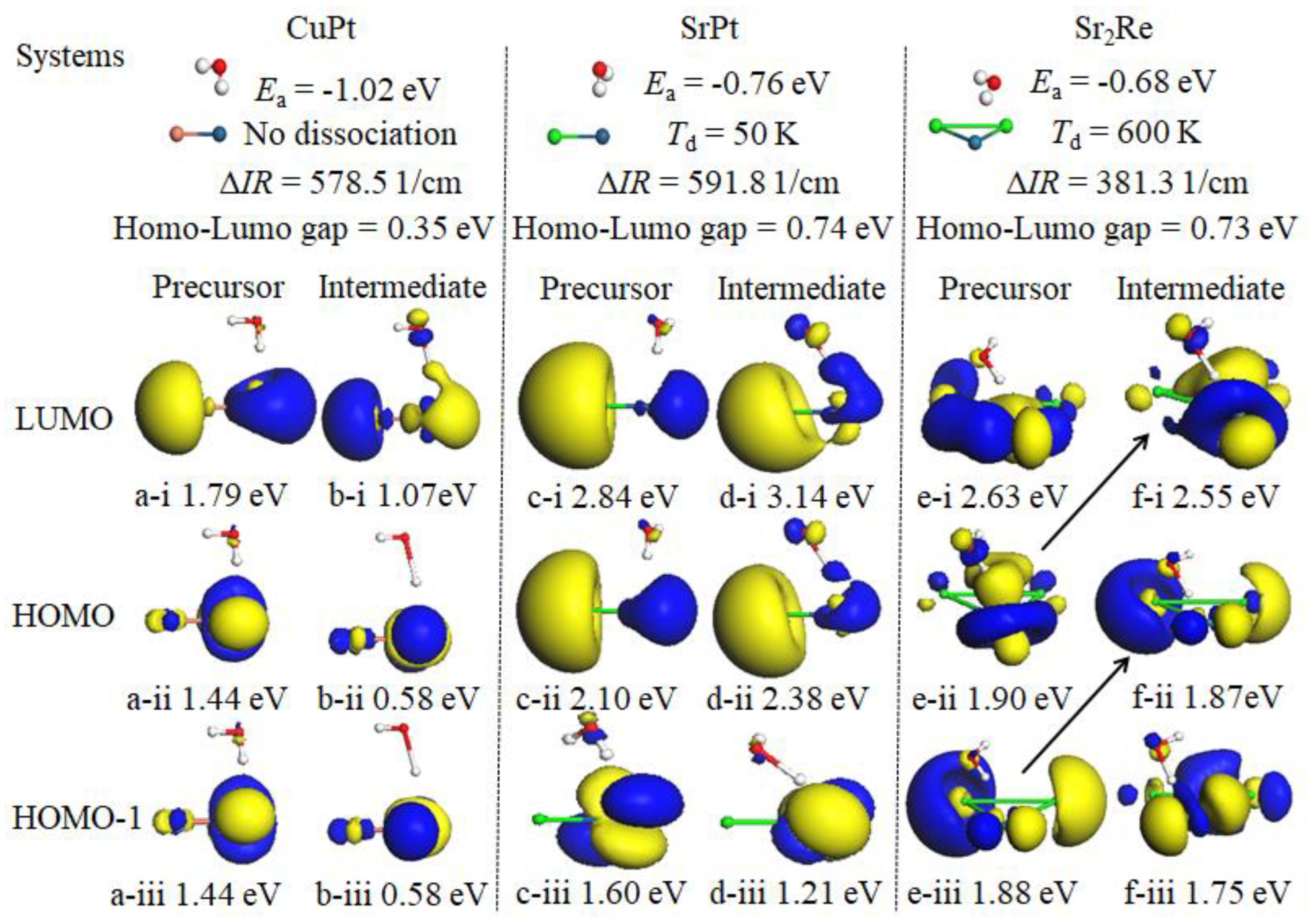

Table 1.
24 diatomic, 30 triatomic, and 34 tetraatomic clusters were constructed and listed below.
|
Diatomic clusters 24 |
BePt | CPt | NPt | FPt | NaPt | PPt |
| NiPt | Cu2 | CuPt | AsPt | SrCd | SrRe | |
| SrPt | TcPt | PdPt | Cd2 | CdRe | CdPt | |
| SbPt | Re2 | RePt | IrPt | Pt2 | PtAu | |
| Triatomic clusters30 | Be2Pt | C2Pt | N2Pt | F2Pt | Na2Pt | NaPt2 |
| P2Pt | Ni2 | Ni2Pt | Cu3 | Cu2Pt | As2Pt | |
| Sr2Cd | Sr2Re | Sr2Pt | SrCd2 | SrRe2 | SrPt2 | |
| Tc2Pt | Pd2Pt | Cd3 | Cd2Re | Cd2Pt | CdRe2 | |
| Sb2Pt | Re3 | Re2Pt | Ir2Pt | Pt3 | PtAu2 | |
| Tetraatomic clusters 34 |
Be3Pt | C3Pt | N3Pt | F3Pt | Na4 | Na3Pt |
| Na2Pt2 | NaPt3 | P3Pt | Ni3Pt | NiPt3 | Cu4 | |
| Cu3Pt | As3Pt | Sr4 | Sr3Cd | Sr3Re | Sr3Pt | |
| Sr2Re2 | Sr2Pt2 | SrCd3 | SrRe3 | SrPt3 | Tc3Pt | |
| Pd3Pt | Cd4 | Cd3Re | Cd3Pt | CdRe3 | Sb3Pt | |
| Re4 | Re3Pt | Ir3Pt | Pt4 |
Table 2.
∆IR represents the redshift of the stretching mode center relative to negatively charged H2O. Eg denotes the LUMO-HOMO gap, Ea is the adsorption energy, and Td is the dissociation temperature.
Table 2.
∆IR represents the redshift of the stretching mode center relative to negatively charged H2O. Eg denotes the LUMO-HOMO gap, Ea is the adsorption energy, and Td is the dissociation temperature.
| Systems | ∆IR (1/cm) | Eg (eV) | Ea (eV) | Ea+Eg (eV) | Td (K) |
| Na2Pt | -640.5 | 0.71 | -0.90 | -0.19 | 100 |
| Na2Pt2 | -249.5 | 0.75 | -0.97 | -0.22 | 100 |
| Na3Pt | -203.1 | 0.67 | -0.86 | -0.19 | 100 |
| Sr2Pt | -42.8 | 0.60 | -0.80 | -0.20 | 200 |
| SrRe3 | 34.1 | 0.28 | -0.58 | -0.30 | 100 |
| Sr2Re | 381.3 | 0.73 | -0.68 | 0.05 | 600 |
| CuPt | 578.5 | 0.35 | -1.02 | -0.67 | 1500 |
| SrPt | 591.8 | 0.74 | -0.76 | -0.02 | 50 |
| Sr3Re | 673.8 | 0.59 | -0.68 | -0.09 | 300 |
| SrPt2 | 702.0 | 0.96 | -1.09 | -0.13 | 300 |
| Sr2Pt2 | 745.3 | 0.45 | 0.59 | 1.04 | 1500 |
| SrPt3 | 774.9 | 0.73 | -0.98 | -0.25 | 400 |
| FPt | 787.4 | 2.95 | -0.84 | 2.11 | 600 |
| NaPt2 | 892.9 | 0.44 | -1.01 | -0.57 | 800 |
| SrRe | 1015.9 | 0.55 | 0.30 | 0.85 | 1300 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



