
Submitted:
17 February 2025
Posted:
18 February 2025
You are already at the latest version
Abstract
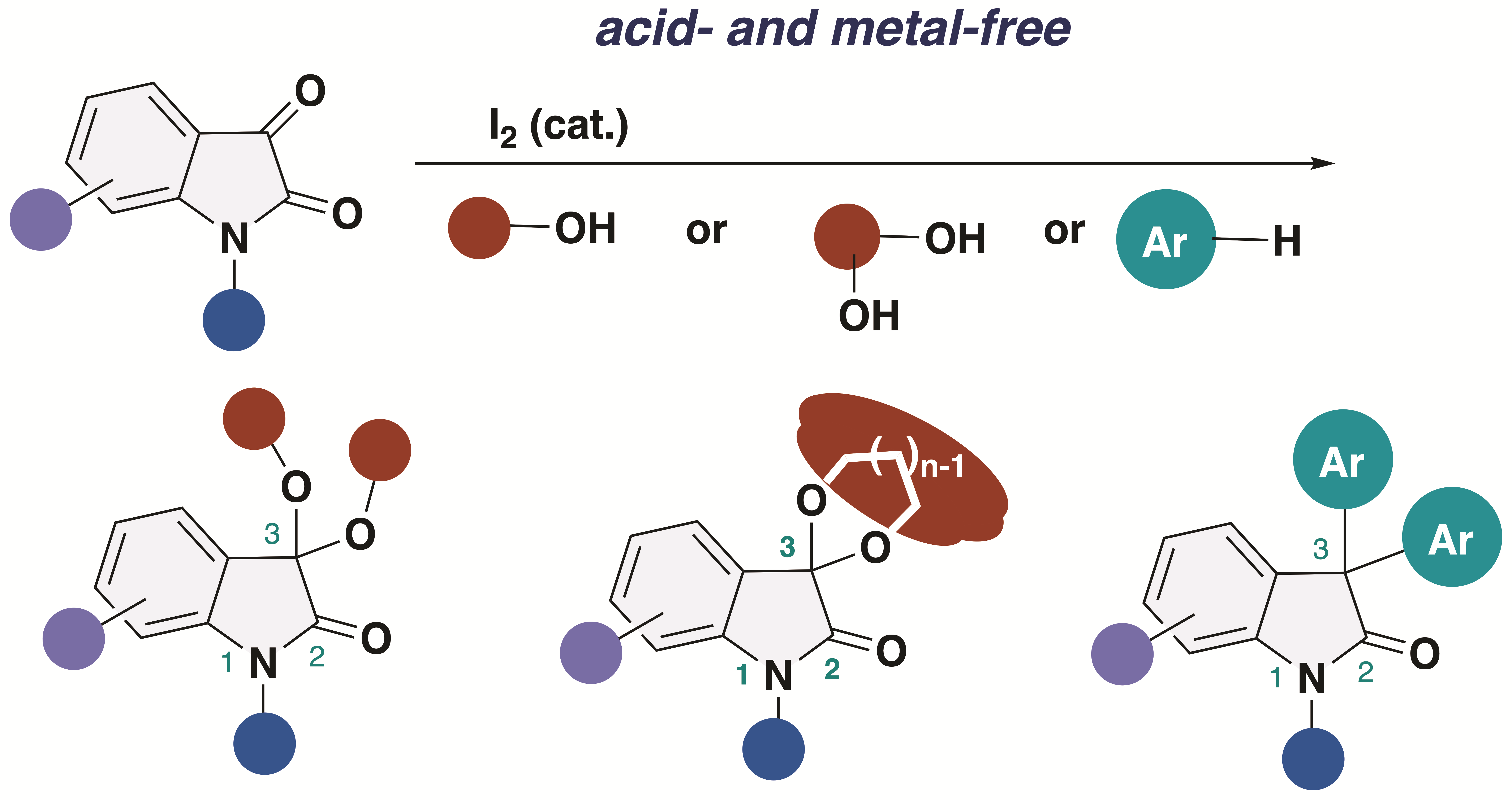
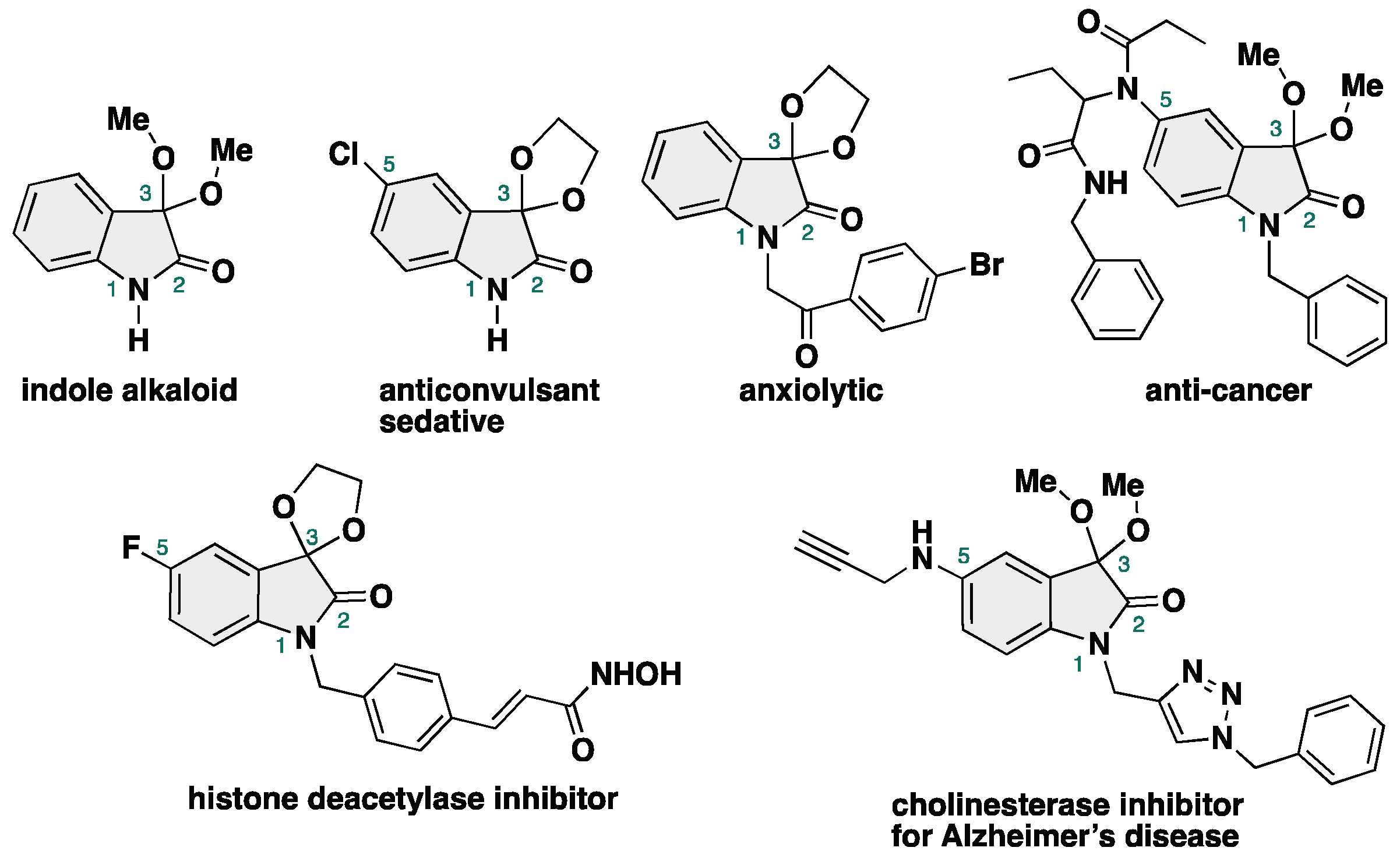
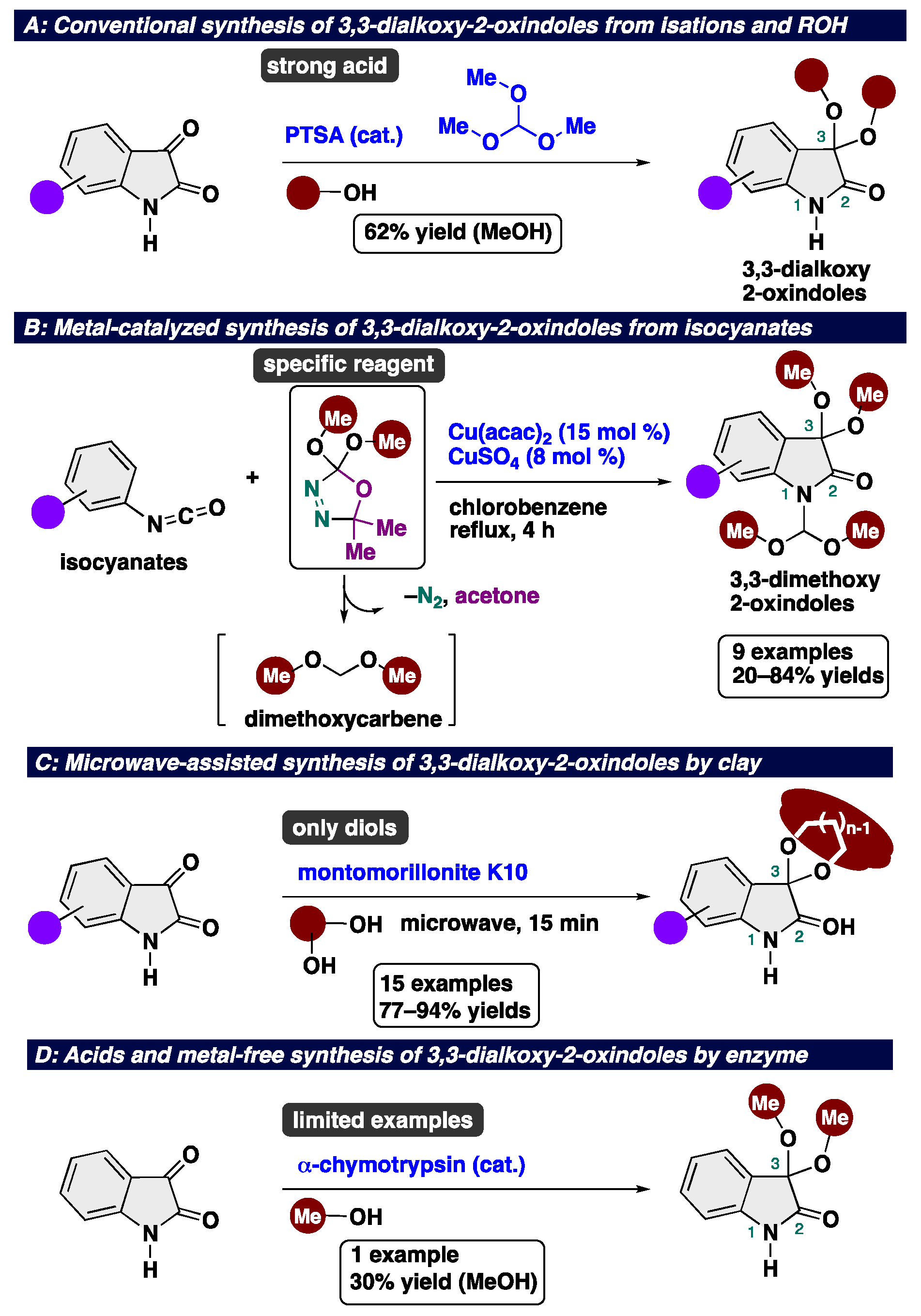
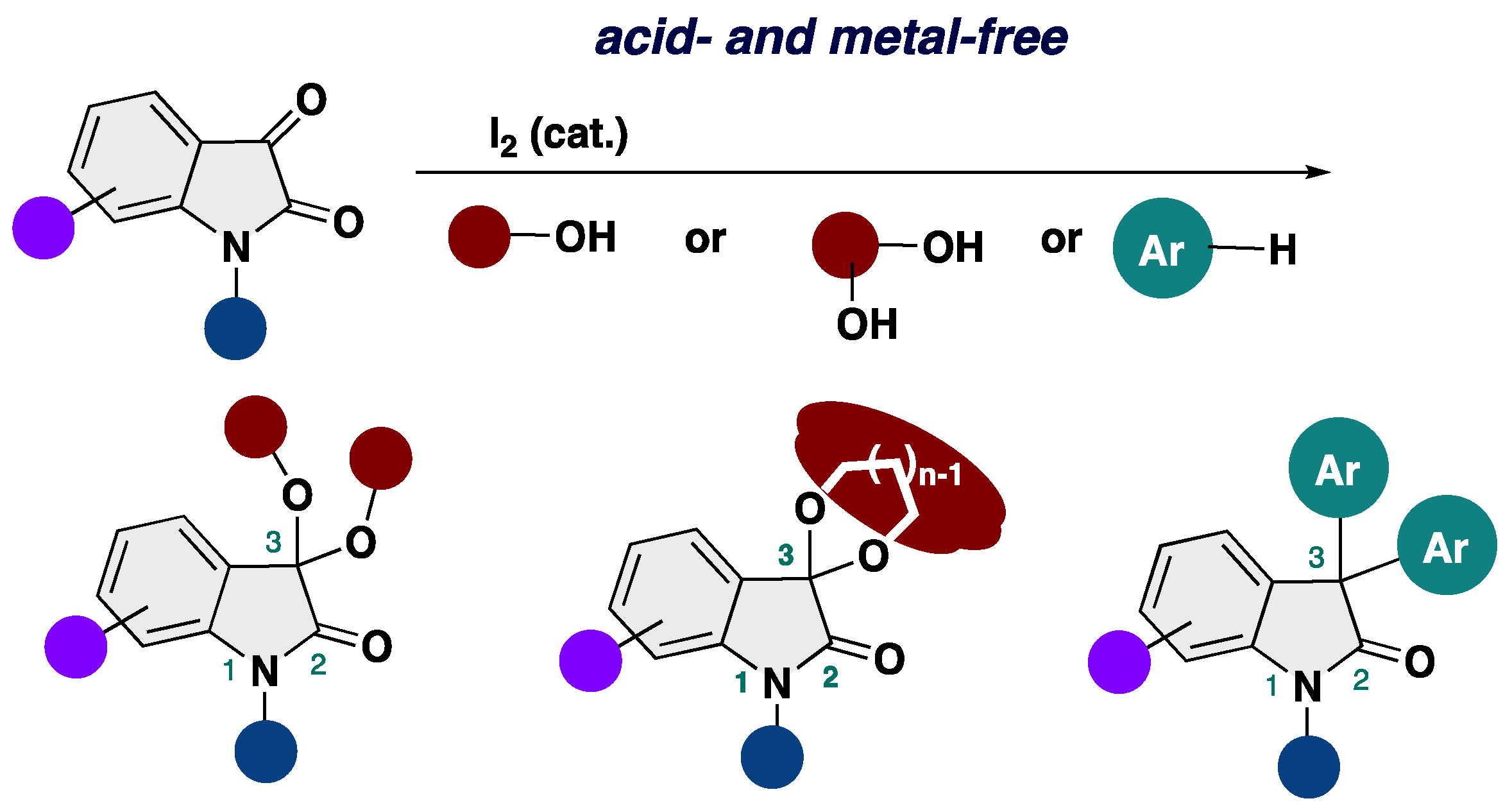
3,3-Dialkoxy-2-oxindoles are prevent in natural product family and exhibit unique biological activities. Among them, acyclic alkoxy analogues shows instability toward acidic conditions, making the access to acyclic isatin ketals highly challenging. Conventional methods for the synthesis of 3,3-dialkoxy-2-oxindoles usually require strong acid and harsh reaction conditions, resulting in low overall efficiency. Herein, we report the acid- and metal-free protocol for the synthesis of 3,3-dialkoxy-2-oxindoles from isatins through an iodine-catalyzed ketalization. This protocol does not require the use of any specific reagents including metal-catalysts. Furthermore, the total synthesis of the unprecedented 2-oxindole alkaloid bearing 3,3-dimethoxy moiety has been achieved.
Keywords:
1. Introduction
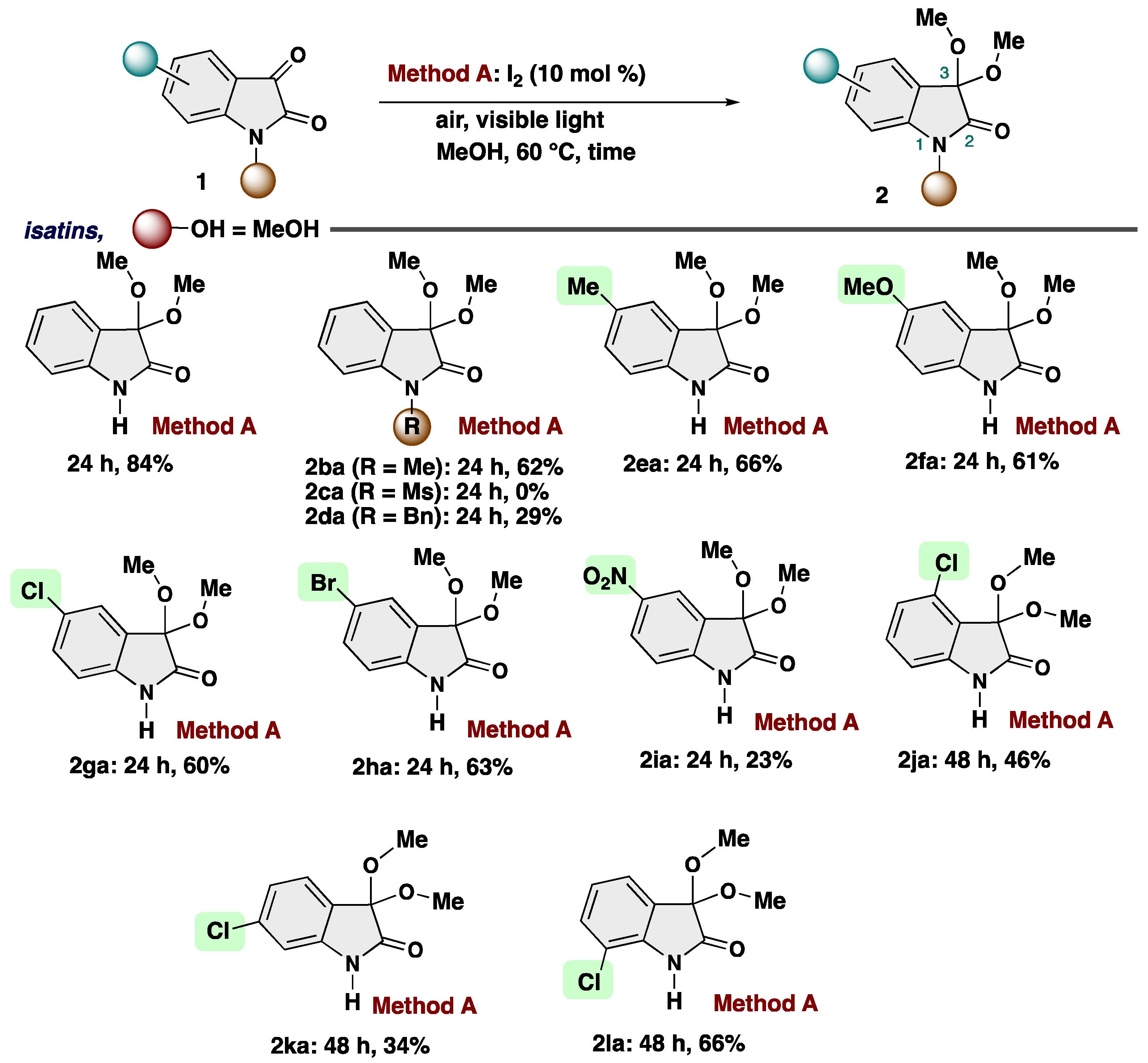
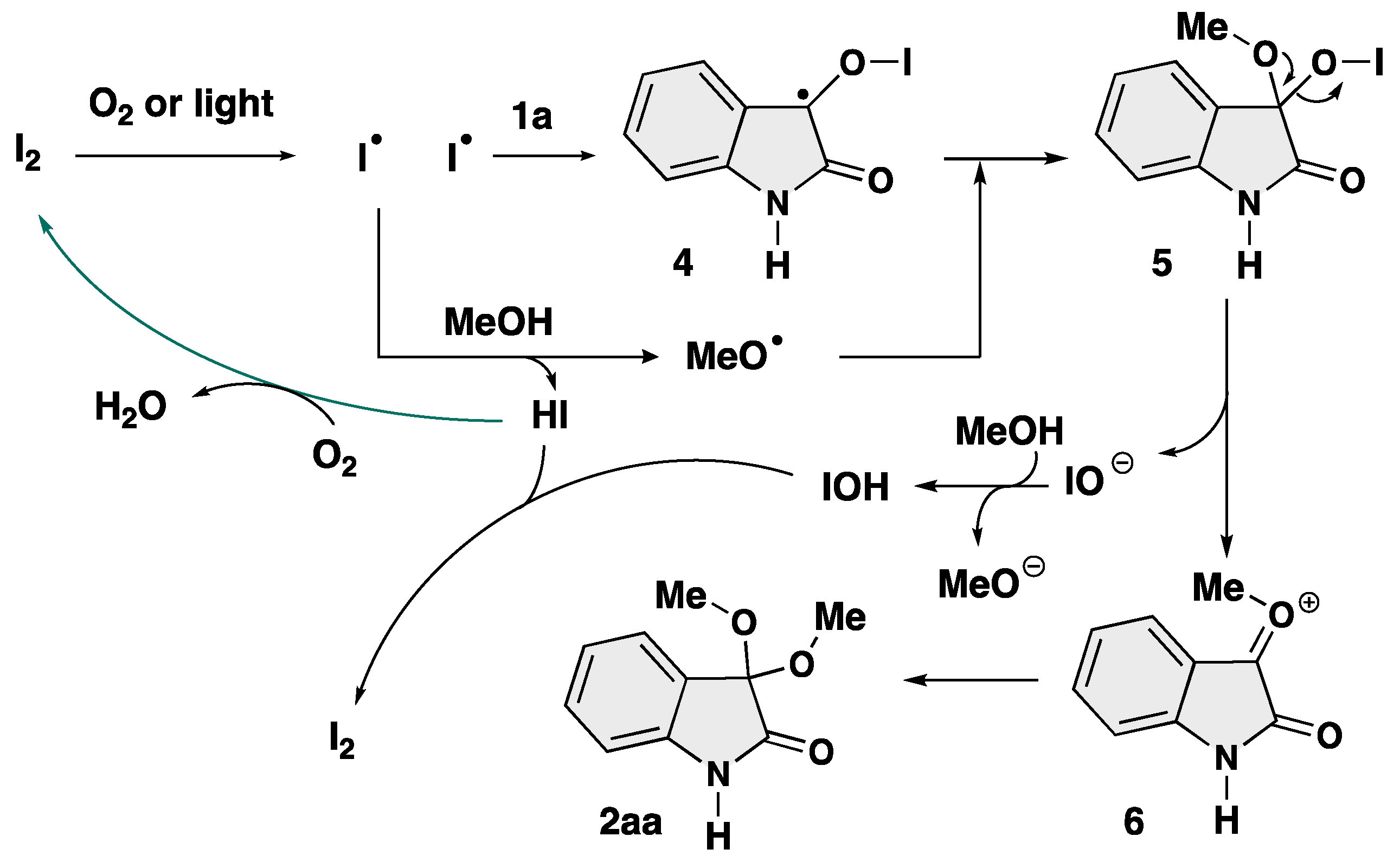
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4-1. Synthesis of N-substituted isatins (1) [69-70]: N-Substituted isatins (1c and 1d) were prepared by reported method. [69-70] Analytical data is in accordance with the literature values. (1c [69], 1d [70])
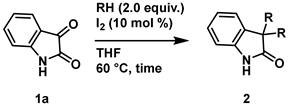
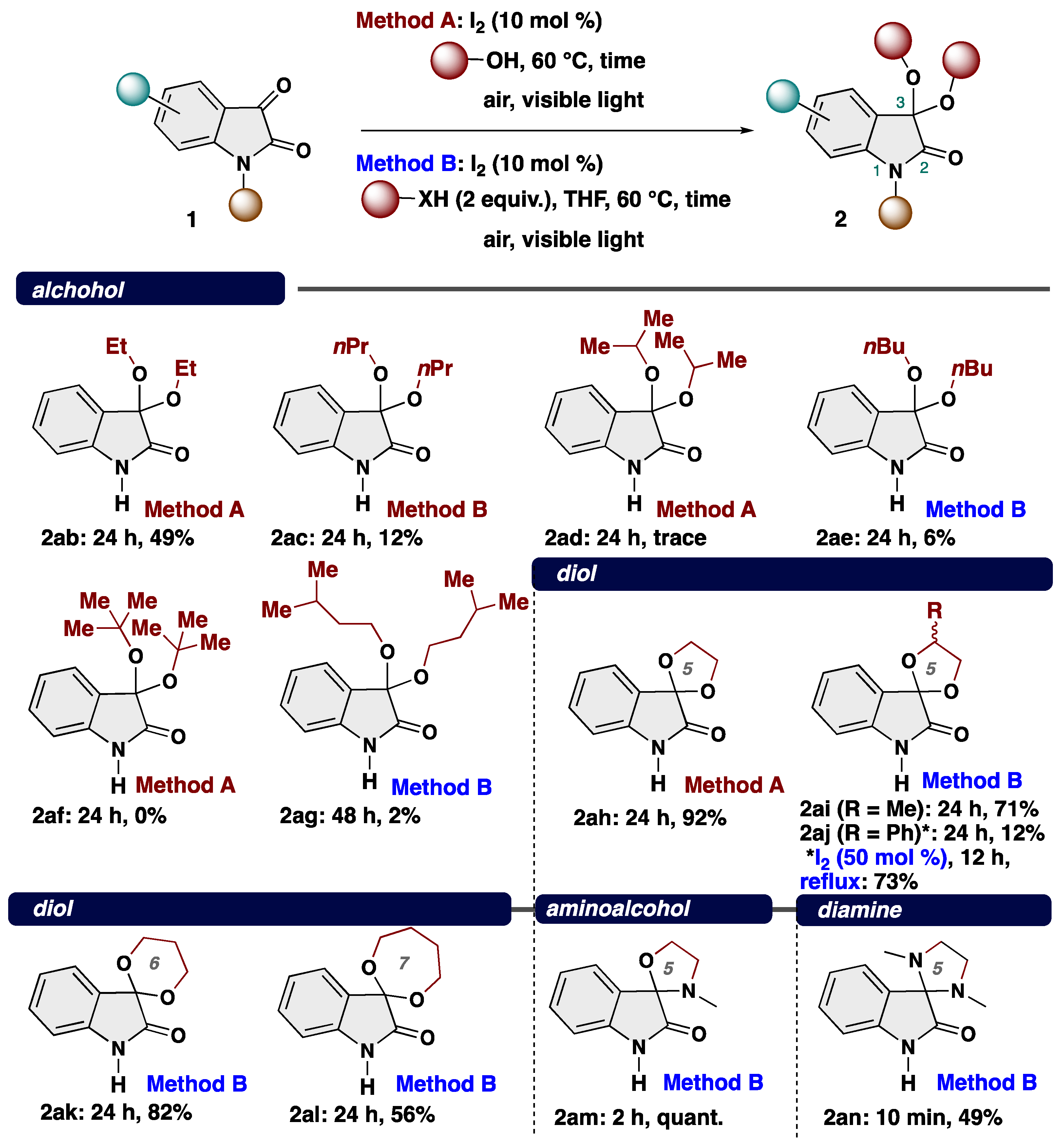
4-2. General procedure for Synthesis of 3,3-dialcohoxyindolin-2-one (2)
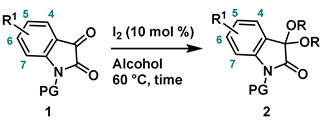 .
.
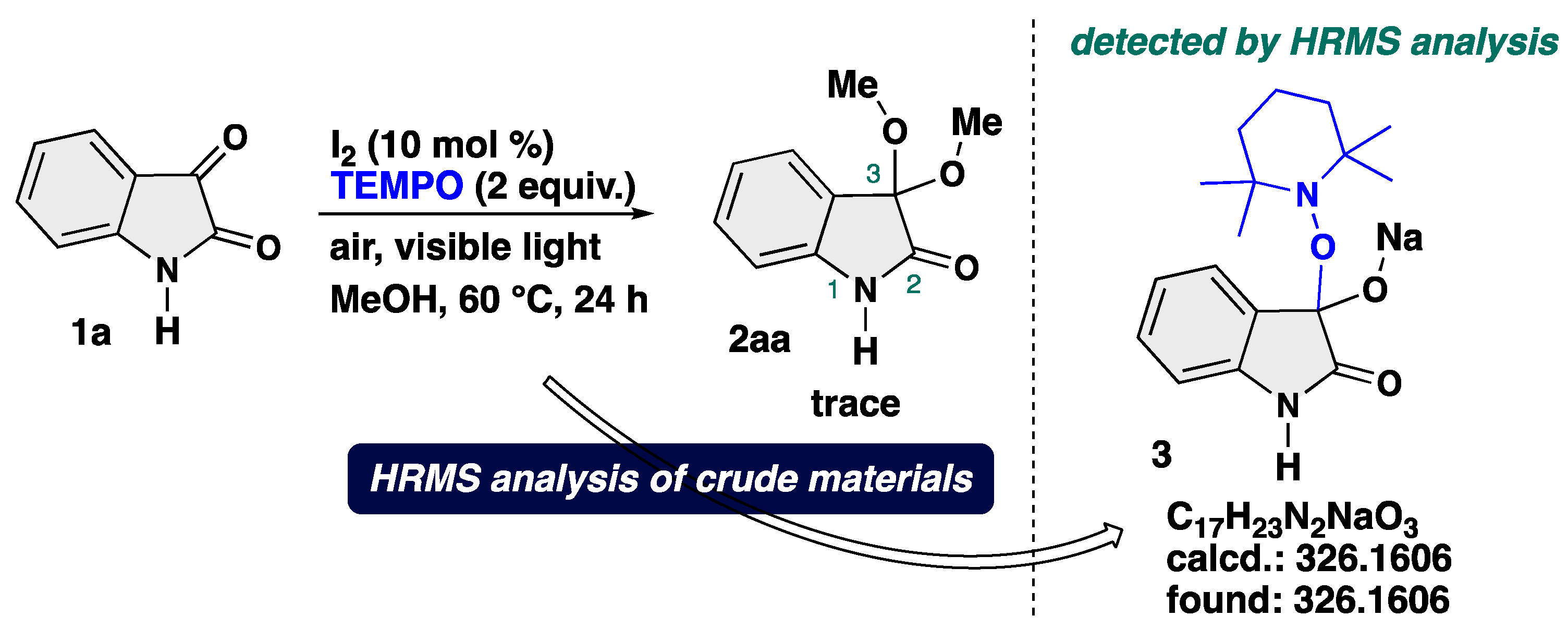
4-3-1. Synthesis of 3,3-Dimethoxyindolin-2-one (2aa) [17] 
4-3-2. Synthesis of 3,3-Dimethoxy-1-methylindolin-2-one (2ba) [71] 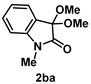
4-3-3. Synthesis of 1-Benzyl-3,3-dimethoxyindolin-2-one (2da) [13] 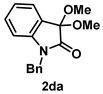
4-3-4. Synthesis of 3,3-Dimethoxy-5-methylindolin-2-one (2ea) 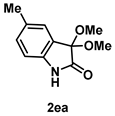
4-3-5. Synthesis of 3,3-Dimethoxy-5-methoxylindolin-2-one (2fa) 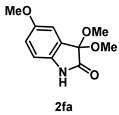
4-3-6. Synthesis of 5-Chloro-3,3-dimethoxyindolin-2-one (2ga) [72] 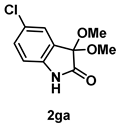
4-3-7. Synthesis of 5-Bromo-3,3-dimethoxyindolin-2-one (2ha) 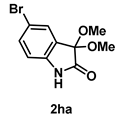
4-3-8. Synthesis of 5-Nitro-3,3-dimethoxyindolin-2-one (2ia) 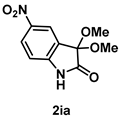
4-3-9. Synthesis of 4-Chloro-3,3-dimethoxyindolin-2-one (2ja) 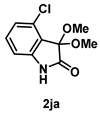
4-3-10. Synthesis of 6-Chloro-3,3-dimethoxyindolin-2-one (2ka)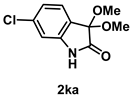
4-3-11. Synthesis of 7-Chloro-3,3-dimethoxyindolin-2-one (2la) 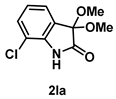
4-3-12. Synthesis of 3,3-Diethoxyindolin-2-one (2ab) [17] 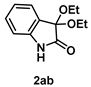
4-3-13. Synthesis of 3,3-Dipropoxyindolin-2-one (2ac) [73] 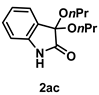
4-3-14. Synthesis of 3,3-Dibutoxyindolin-2-one (2ae) 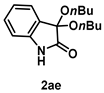
4-3-15. Synthesis of 3,3-Bis(isopentyloxy)indolin-2-one (2ag) 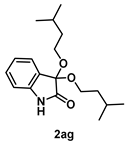
4-4-1. Synthesis of Spiro[1,3-dioxolane-2,3’-indolin]-2’-one (2ah) [17] 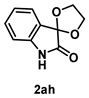
4-4-2. Synthesis of 4'-Methylspiro[indoline-3,2'-[1,3]dioxolan]-2-one (2ai) (dr = 62:38) 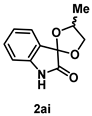
4-4-3. Synthesis of 4'-Phenylspiro[indoline-3,2'-[1,3]dioxolan]-2-one (2aj) (dr = 61:39) 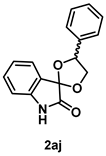
4-4-4. Synthesis of Spiro[1,3-dioxane-2,3’-indolin]-2’-one (2ak) [30] 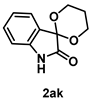
4-4-5. Synthesis of Spiro[1,3-dioxepene-2,3’-indolin]-2’-one (2al) 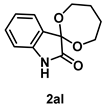
4-4-6. Synthesis of 3'-Methylspiro[indoline-3,2'-oxazolidin]-2-one (2am) 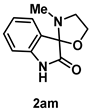
4-4-7. Synthesis of 1,3-Dimethylspiro[imidazolidine-2,3'-indolin]-2'-one (2an) [74] 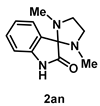
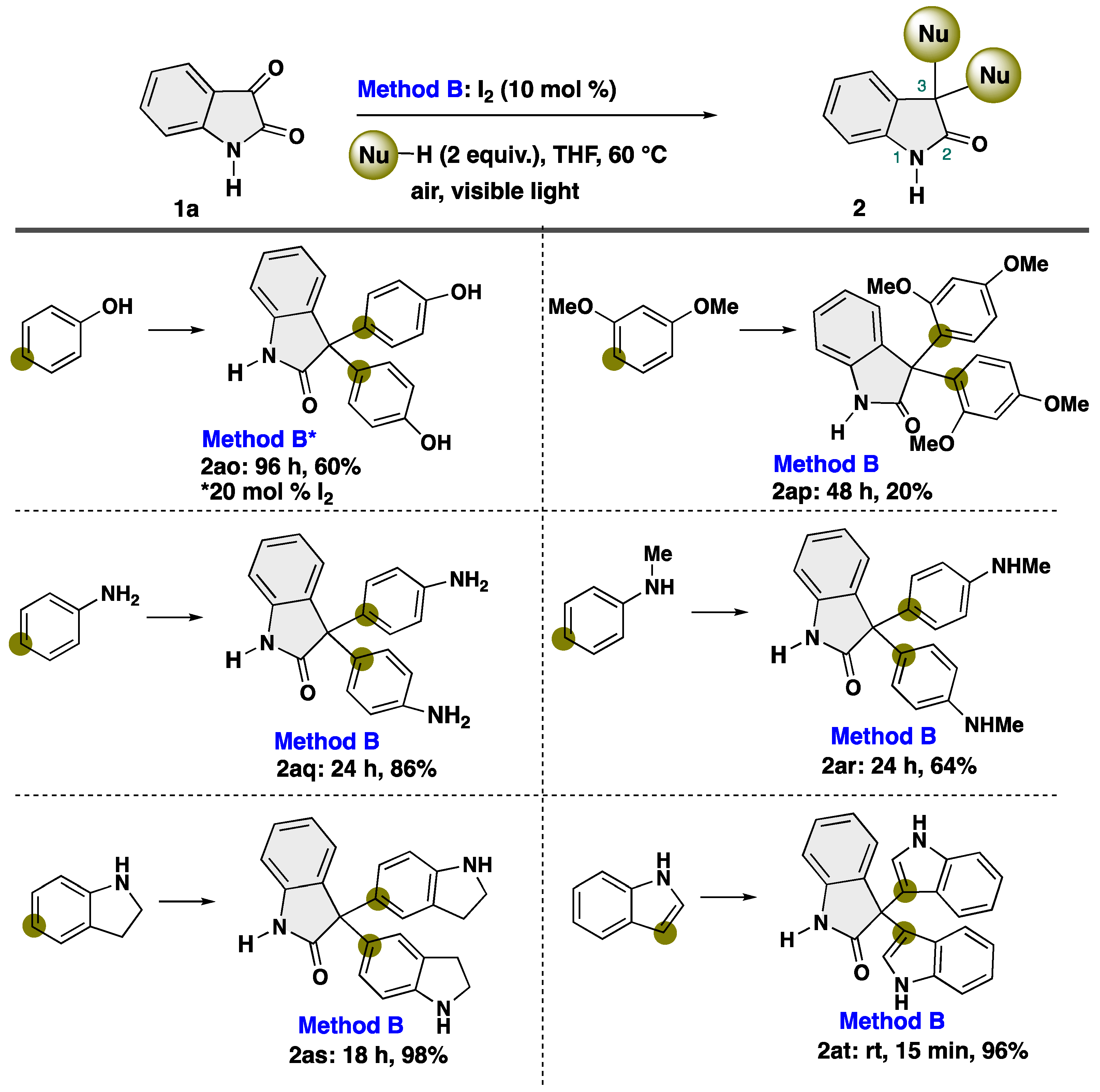
4-5-1. Synthesis of 3,3-Bis(4-hydroxyphenyl)indolin-2-one (2ao) [55] 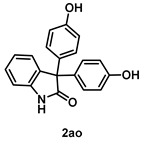
4-5-2. Synthesis of 3,3-Bis(2,4-dimethoxyphenyl)indolin-2-one (2ap) [55] 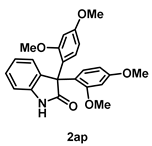
4-5-3. Synthesis of 3,3-Bis(4-aminophenyl)indolin-2-one (2aq) 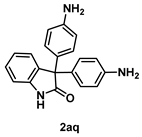
4-5-4. Synthesis of 3,3-Bis(4-(methylamino)phenyl)indolin-2-one (2ar) [55] 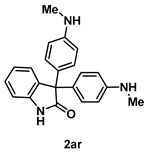
4-5-5. Synthesis of 3,3-Bis[1H-indolin-5-yl]indolin-2-one (2as) 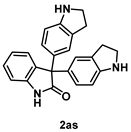
4-5-6. Synthesis of 3,3-Bis[1H-indol-3-yl]indolin-2-one (2at) [55] 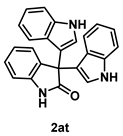
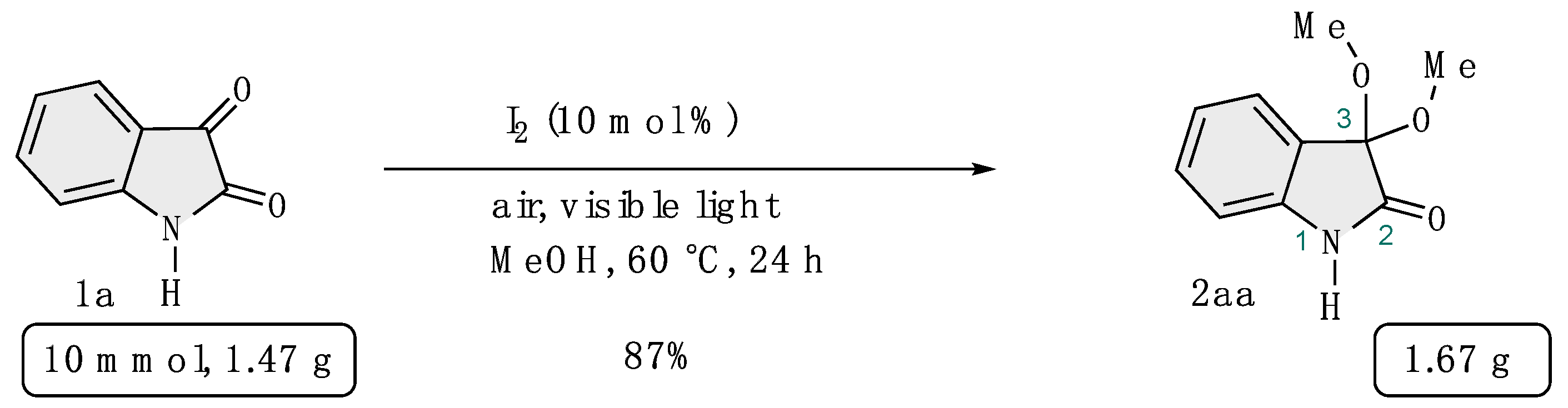
4-6-1: Gram scale synthesis of 3,3-dimethoxyisatin (Scheme 6) 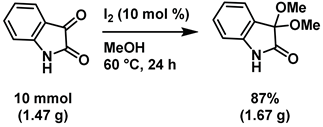
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jao, C.-W.; Hung, T.-H.; Chang, C.-F.; Chuang, T.-H. Chemical Constituents of Phaius mishmensis. Molecules 2016, 21, 1605.
- Ketal.
- Rajopadhye, M.; Popp, F. D. Potential Anticonvulsants. 11. Synthesis and Anticonvulsant Activity of Spiro[1,3-dioxolane-2,3’-indolin]-2’-ones and Structural Analogues. J. Med. Chem. 1988, 31, 1001–1005. [CrossRef]
- Zapata-Sudo, G.; Pontes, L. B.; Gabriel, D.; Mendes, T. C. F.; Ribeiro, N. M.; Pinto, A. C.; Trachez, M. M.; Sudo, R. T. Sedative-hypnotic profile of novel isatin ketals. Pharmaco. Biochem. Behav. 2007, 86, 678–685. [CrossRef]
- Geronikaki, A.; Babaev, E.; Dearden, J.; Dehaen, W.; Filimonov, D.; Galaeva, I.; Krajneva, V.; Lagunin, A.; Macaev, F.; Molodavkin, G.; Poroikov, V.; Pogrebnoi, S.; Saloutin, V.; Stepanchikova, A.; Stingaci, E.; Tkach, N.; Vlad, L.; Voronina, T. Design, synthesis, computational and biological evaluation of new anxiolytics. Bioorg. Med. Chem. 2004, 12, 6559–6568. [CrossRef]
- Marques, C. S.; González-Bakkker, Padrón, J. M.; Burke, A. J. Easy access to Ugi-derived isatin-peptoids and their potential as small-molecule anticancer reagents. New. J. Chem. 2023, 47, 743–750.
- Dung, D. T. M.; Dung, P. T. P.; Oanh, D. T. K.; Vu, T. K.; Hahn, H.; Han, B. W.; Pyo, M.; Kim, Y. G.; Han, S.-B.; Nam, N.-H. Exploration of novel 5’(7’)-substituted-2’-oxospiro[1,3]dioxolane-2,3’-indoline-based N-hydroxypropenamides as histone deacetylase inhibitors and antitumor agents. Arab. J. Chem. 2017, 10, 465–472.
- Marques, C. S.; López, O.; Leitzbach, L.; Fernández-Bolaños, J. G.; Stark, H.; Burke, A. J. Survey of New, Small-Molecule Isatin-Based Oxindole Hybrids as Multi-Targeted Drugs for the Treatment of Alzheimer’s Disease. Synthesis 2022, 54, 4304–4319.
- Singh, G. S.; Desta, Z. Y. Isatins As Privileged Molecules in Design and Synthesis of Spiro-Fused Cyclic Frameworks. Chem. Rev., 2012, 112, 6104–6155. [CrossRef]
- Pinder, J. L.; Weinreb, S. M. Preliminary feasibility studies on total synthesis of the unusual marine bryozoan alkaloids chartellamide A and B. Tetrahedron Lett. 2003, 44, 4141–4143.
- Castaldi, M. P.; Troast, D. M.; Porco Jr., J. A. Stereoselective Synthesis of Spirocyclic Oxindoles via Prins Cyclization. Org. Lett. 2009, 11, 3362–3365.
- Zhang, Y.; Panek, J. S. Stereocontrolled Synthesis of Spirooxindoles through Lewis Acid-Promoted [5 + 2]-Annulation of Chiral Silyl Alcohols. Org. Lett. 2009, 11, 3366–3369. [CrossRef]
- Wang, J.; Crane, E. A.; Scheidt, K. A. Highly Stereoselective Brønsted Acid Catalyzed Synthesis of Spirooxindole Pyrans. Org. Lett. 2011, 13, 3086–3089.
- @Synth. Commun.
- Li, H.; Bonderoff, S. A.; Cheng, B.; Padwa, A. Model Studies Directed toward the Alkaloid Mersicarpine Utilizing a Rh(II)-Catalyzed Insertion/Cycloaddition Sequence. J. Org. Chem. 2014, 79, 392–400.
- Marques, C. S.; González-Bakker, A.; Padrón, J. M. The Ugi4CR as effective tool to access promising anticancer isatin-based α-acetamide carboxamide oxindole hybrids. Beilstein J. Org. Chem. 2024, 20, 1213–1220. [CrossRef]
- Dou, X.; Yao, W.; Jiang, C.; Lu, Y. Enantioselective N-alkylation of isatins and synthesis of chiral N-alkylated indoles. Chem. Commun. 2014, 50, 11354–11357.
- Marques, C. S.; McArdle, P.; Erxleben, A.; Burke, A. J. Accessing New 5-a-(3,3-Disubstituted Oxindole)-Benzylamine Derivatives from Isatin: Stereoselective Organocatalytic Three Component Petasis Reaction. Eur. J. Org. Chem. 2020, 2020, 3622–3634.
- Vine, K. L.; Locke, J. M.; Ranson, M.; Pyne, S. G.; Bremner, J. B. In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorg. Med. Chem. 2007, 15, 931–938.
- Dong, J.-L.; Yu, L.-S.-H.; Xie, J.-W. A Simple and Versatile Method for the Formation of Acetals/Ketals Using Trace Conventional Acids. ACS Omega 2018, 3, 4975–4984.
- Azzena, U.; Carraro, M.; Corrias, M.; Crisafulli, R.; de Luca, L.; Gaspa, S.; Nuvoli, L.; Pintus, S.; Polese, R.; Sanna, M.; Satta, G.; Senes, N.; Urtis, L.; Garroni, S. Ammonium Salts Catalyzed Acetalization Reactions in Green Ethereal Solvents. Catalysts 2020, 10, 1108. [CrossRef]
- Banik, B. K.; Chapa, M.; Marquez, J.; Cardona, M. A remarkable iodine-catalyzed protection of carbonyl compounds. Tetrahedron Lett. 2005, 46, 2341–2343.
- Khan, M. A.; Teixeira, I. F.; Li, M. J.; Koito, Y.; Tsang, S. C. E. Graphitic carbon nitride catalysed photoacetalization of aldehydes/ketones under ambient conditions. Chem. Commun. 2016, 52, 2772–2775.
- Lee, B. W.; Jeong, K.; Seo, J. Y.; Baek, K.-Y. Facile Synthesis of Biomass-Derived Degradable Poly(ketal-ester) Elastomers Using Dual-Acidic Catalysts. ACS Appl. Polym. Mater. 2024, 6, 3507–3516. [CrossRef]
- Putro, W. S.; Iijima, S.; Matsumoto, S.; Hamura, S.; Yabushita, M.; Tomishige, K.; Fukaya, N.; Choi, J.-C. Sustainable synthesis of diethyl carbonate from carbon dioxide and ethanol featuring acetals as regenerable dehydrating agents. RSC Sustain. 2024, 2, 1613–1620.
- Steuernagel, D.; Wagenknecht, H.-A. Photocatalytic Synthesis of Acetals and Ketals from Aldehydes and Silylenolethers without the use of Acids. Chem. Eur. J. 2023, 29, e202203767. [CrossRef]
- Alzard, R. H.; Alsaedi, S.; Alseiari, S.; Aljasmi, S.; El-Maghraby, H. F.; Poulose, V.; Hassan, A.; Kamel, M.; Ali, A.; Abdel-Hafiez, M.; Abdellah, M. Heterogeneous Acetalization of Benzaldehyde over Lanthanoide Oxalate Metal-Organic Frameworks. ACS Omega 2024, s, 37386–37395.
- Design, Synthesis, and Evaluation of Nonpeptidic Inhibitors of Human Rhinovirus 3C Protease. Webber, S. E.; Tikhe, J.; Worland, S. T.; Fuhrman, S. A.; Hendrickson, T. F.; Matthews, D. A.; Love, R. A.; Patick, A. K.; Meador, J. W.; Ferre, R. A.; Brown, E. L.; DeLisle, D. M.; Ford, C. E.; Binford, S. L. J. Med. Chem. 1996, 39, 5072–5082.
- Rigby, J. H.; Brouet, S. A. Metal-mediated reactions of aryl isocyanates with dimethoxycarbene to form isatin derivatives. Tetrahedron Lett., 2013, 54, 2542–2545. [CrossRef]
- Ribeiro, N. M.; da Chunha Pinto, A.; da Silva, B. V.; de Almeida Violante, F.; Dias, M. O. A fast, efficient and eco-friendly procedure to prepare isatin ketals. Cat. Commun. 2007, 8, 2130–2136.
- Shimizu, K.; Higuchi, T.; Takasugi, E.; Hatamachi, T.; Kodama, T.; Satsuma, A. Characterization of Lewis acidity of cation-exchanged montmorillonite K-10 clay as effective heterogeneous catalyst for acetylation of alcohol. J. Mol. Cat. A. Chem. 2008, 284, 89–96.
- Lan, J.; Jiang, G.; Yang, J.; Zhu, H.; Le, Z.; Xie, Z. α-Chymotrypsin-Induced Acetalization of Aldehydes and Ketones with Alcohols. Synthesis 2020, 52, 2121–2126.
- Parvatkar, P. T.; Parameswaran, P. S.; Tilve, S. G. Recent Developments in the Synthesis of Five- and Six-Membered Heterocycles Using Molecular Iodine. Chem. Eur. J. 2012, 18, 5460–5489. [CrossRef]
- Ren, Y.-M.; Cai, C.; Yang, R.-C. Molecular iodine-catalyzed multicomponent reactions: an efficient catalyst for organic synthesis. RSC Adv. 2013, 3, 7182–7204.
- Von der Heiden, D.; Bozkus, S.; Klussmann, M.; Breugst, M. Reaction Mechanism of Iodine-Catalyzed Michael Additions. J. Org. Chem. 2017, 82, 4037–4043.
- Breugst, M.; Von der Heiden, D. Mechanism in Iodine Catalysis. Chem. Eur. J. 2018, 24, 9187–9199.
- Yusubov, M. S.; Zhdankin, V. V. Iodine catalysis: A green alternative to transition metals in organic chemistry and technology. Resour.-Effic. Technol. 2015, 1, 49–67.
- Wang, J.-Q.; Zuo, Z.-Y.; He, W. Recent Advances of Green Catalytic System I2/DMSO in C–C and C–Heteroatom Bonds Formation. Catalysts 2022, 12, 821.
- Finkbeiner, P.; Nachtsheim, B. J. Iodine in Modern Oxidation Catalysis. Synthesis, 2013, 45, 979–999. [CrossRef]
- Jereb, M.; Vrazic, D.; Zupan, M. Iodine-catalyzed transformation of molecules containing oxygen functional groups. Tetrahedron 2011, 67, 1355–1387.
- Das, S.; Borah, R.; Devi, R. R.; Thakur, A. J. Molecular iodine in Protection and Deprotection Chemistry. Synlett 2008, 2008, 2741–2762.
- Togo, H.; Iida, S. Synthetic Use of Molecular Iodine for Organic Synthesis. Synlett 2006, 2006, 2159–2175. [CrossRef]
- Yamashiro, T.; Yamada, K.; Yoshida, H.; Tomisaka, Y.; Nishi, T.; Abe, T. Silver-Mediated Intramolecular Friedel–Crafts-Type Cyclizations of 2-Benzyloxy-3-bromoindolines: Synthesis of Isochromeno[3,4-b]indolines and 3-Arylindoles. Synlett 2019, 30, 2247–2252.
- Abe, T.; Kosaka, Y.; Asano, M.; Harasawa, N.; Mishina, A.; Nagasue, M.; Sugimoto, Y.; Katakawa, K.; Sueki, S.; Anada, M.; Yamada, K. Direct C4-Benzylation of Indoles via Tandem Benzyl Claisen/Cope Rearrangements. Org. Lett. 2019, 21, 826–829.
- Yamashiro, T.; Abe, T.; Tanioka, M.; Kamino, S.; Sawada, D. cis-3-Azido-2-methoxyindolines as safe and stable precursors to overcome the instability of fleeting 3-azidoindoles. Chem. Commun., 2021, 57, 13381–13384. [CrossRef]
- Yamashiro, T.; Abe, T.; Sawada, D. Synthesis of 2-monosubstituted indolin-3-ones by cine-substitution of 3-azido-2-methoxyindolines. Org. Chem. Front., 2022, 9, 1897–1903.
- Abe, T.; Kosaka, Y.; Kawasaki, T.; Ohata, Y.; Yamashiro, T.; Yamada, K. Revisiting 2-Alkoxy-3-bromoindolines: Control C-2 vs. C-3 Elimination for Regioselective Synthesis of Alkoxyindoles. Chem. Pharm. Bull. 2020, 68, 555–558.
- Sugitate, K.; Yamashiro, T.; Takahashi, I.; Yamada, K.; Abe, T. Oxytrofalcatin Puzzle: Total Synthesis and Structural Revision of Oxytrofalcatins B and C. J. Org. Chem. 2023, 88, 9920–9926. [CrossRef]
- Kimata, M.; Abe, T. Total Synthesis of the Proposed Structure of Indolyl 1,2-Propanediol Alkaloids, 1-(1H-Indol-3-yloxy)propan-2-ol. Chemistry 2023, 5, 2772–2784.
- Hirao, S.; Yamashiro, T.; Kohira, K.; Mishima, N.; Abe, T. 2,3-Dimethoxyindolines: a latent electrophile for SNAr reactions triggered by indium catalysts. Chem. Commun. 2020, 56, 5139–5142.
- Tokushige, K.; Yamashiro, T.; Hirao, S.; Abe, T. Aluminum-Catalyzed Cross Selective C3–N1’ Coupling Reactions of N-Methoxyindoles with Indoles. Chemistry 2023, 5, 452–462.
- Abe, T.; Hirao, S. Rapid access to indole-fused bicyclo[2.2.2]octanones by merging the umpolung strategy and molecular iodine as a green catalyst. Org. Biomol. Chem. 2020, 18, 4193–4197. [CrossRef]
- Pitzer, L.; Schäfers, F.; Glorius, F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem. Int. Ed. 2019, 58, 8572–8576.
- Wu, P.-L.; Hsu, Y.-L.; Jao, C.-W. Indole Alkaloids from Cephalanceropsis gracilis. J. Nat. Prod. 2006, 69, 1467–1470.
- Khan, J.; Tyagi, A.; Yadav, N.; Mahato, R.; Hazra, C. K. Lambert Salt-Initiated Development of Friedel–Crafts Reaction on Isatin to Access Distinct Derivatives of Oxindoles. J. Org. Chem. 2021, 86, 17833–17847.
- Garrido, F.; Ibanez, J.; Gonalons, E.; Giraldez, A. Synthesis and laxative properties of some derivative esters of 3,3-bis-(4-hydroxyphenyl)-2-indolinone. Eur. J. Med. Chem. 1975, 10, 143–146. [CrossRef]
- Song, H. M.; Lee, H. J.; Kim, H. R.; Ryu, K.; Kim, J. N. Friedel–Crafts Type Reactions of Some Activated Cyclic Ketones with Phenol Derivatives. Synth. Commun. 1999, 29, 3303–3311.
- Paira, P.; Hazra, A.; Kumar, S.; Paira, R.; Sahu, K. B.; Naskar, S.; Saha, P.; Mondal, S.; Maity, A.; Banerjee, S.; Mondal, N. B. Efficient synthesis of 3,3-diheteroaromatic oxindole analogues and their in vitro evaluation for spermicidal potential. Bioorg. Med. Chem. Lett. 2009, 19, 4786–4789.
- Uddin, M. K.; Reignier, S. G.; Coulter, T.; Montalbetti, C.; Grånäs, C.; Butcher, S.; Krog-Jensen, C.; Felding, J. Syntheses and antiproliferative evaluation of oxyphenisatin derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 2854–2857. [CrossRef]
- Karu, S. K.; Pilli, N.; Malapaka, C. An Efficient Multi-Component Double Friedel–Crafts Alkylation of Isatin: Access to Unsymmetrical and Symmetrical 3,3-Diaryl oxindoles. ChestrySelects 2024, 9, e202403328.
- Kobayashi, M.; Aoki, S.; Gato, K.; Matsunami, K.; Kurosu, M.; Kitagawa, I. Marine Natural Products. XXXIV. Trisindoline, a New Antibiotic Indole Trimer, Produced by a Bacterium of Vibrio sp. Separated from the Marine Sponge Hyrtios altum. Chem. Pharm. Bull. 1994, 12, 2449–2451.
- Abe, T.; Nakamura, S.; Yanada, R.; Choshi, T.; Hibino, S.; Ishikura, M. One-Pot Construction of 3,3’-Bisindolylmethanes through Bartoli Indole Synthesis. Org. Lett. 2013, 15, 3622–3625.
- Reddy, B. V. S.; Rajeswari, N.; Sarangapani, M.; Prashanthi, Y.; Ganji, R. J. Iodine-catalyzed condensation of isatin with indoles: A facile synthesis of di(indolyl)indolin-2-ones and evaluation of their cytotoxicity. Bioorg. Med. Chem. Lett., 2012, 22, 2460–2463.
- Duhamel, T.; Stein, C. J.; Martínez, C.; Reiher, M.; Muñiz, K. Engineering Molecular Iodine Catalysis for AlkylNitrogen Bond Formation. ACS Catal., 2018, 8, 3918–3925.
- Lu, Y.; Chen, C.; Zhu, H.; Luo, Z.; Zhang, Y. Highly efficient and fast synthesis of di-iodinated succinimide derivatives from 1,6-enyne and I2 under air at room temperature. Green Chem., 2022, 24, 8021–8028.
- Meng, Z.; Shi, M.; Wie, Y. Iodine radical mediated cascade [3 + 2] carbocyclization of ene-vinylidecyclopropanes with thiols and selenols via photoredox catalysis. Org. Chem. Front., 2024, 11, 1395–1403.
- Huang, H.-Y.; Wu, H.-R.; Wei, F.; Wang, D.; Liu, L. Iodine-Catalyzed Direct Olefination of 2-Oxindoles and Alkenes via Cross-Dehydrogenative Coupling (CDC) in Air. Org. Lett., 2015, 17, 3702–3705. [CrossRef]
- Huang, H.-M.; Li, Y.-J.; Ye, Q.; Han, L.; Jia, J.-H.; Gao, J.-R. Iodine-Catalyzed 1,3-Dipolar Cycloaddition/Oxidation/Aromatization Cascade with Hydrogen Peroxide as the Terminal Oxidant: General Route to Pyrrolo[2,1-a]isoquinolines. J. Org. Chem., 2014, 79, 1084–1092.
- Sohail, M.; Tanaka, F. Angew. Chem. Int. Ed. 2021, 60, 21256–21260.
- Wee, X. K.; Yeo, W. K.; Zhang, B.; Tan, V. B. C.; Lim, K. M.; Tay, T. E.; Go, M.-L. Bioorg. Med. Chem. 2009, 17, 7562–7571.
- Muschalek, B.; Weidner, I.; Butenschön, H. J. Organomet. Chem. 2007, 692, 2415–2424.
- Hans, R. H.; Gut, J.; Rosenthal, P. J.; Chibale, K. Bioorg. Med. Chem. Lett. 2010, 20, 2234–2237.
- Mohanta, N.; Chaudhari, M. B.; Digrawal, N. K.; Gnanaprakasam, B. Org. Pross. Res. Dev. 2019, 23, 1034–1045.
- Bergman, J.; Stålhandske, C.; Vallberg, H. Acta Chem. Scand. 1997, 51, 753–759.

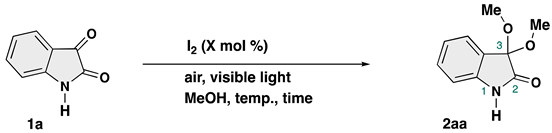 | |||||
| Runa | Catalyst (mol %) | Additive | Temp. (°C) | Time (h) | Yield (%) of 2aab |
| 1 | PhI(OAc)2 (10) | --- | 60 | 24 | trace |
| 2 | PhI(OCOCF3)2 (10) | --- | 60 | 24 | 52 |
| 3 | KI (10) | --- | 60 | 24 | 0 |
| 4 | KIO3 (10) | --- | 60 | 24 | 0 |
| 5 | I2 (10) | --- | 60 | 24 | 84 |
| 6 | CuI (10) | --- | 60 | 24 | 0 |
| 7 | ZnI2 (10) | --- | 60 | 24 | 0 |
| 8 | I2 (5) | --- | reflux | 24 | 68 |
| 9 | I2 (10) | --- | reflux | 24 | 81 |
| 10 | I2 (50) | --- | 60 | 24 | 80 |
| 11 | I2 (50) | MgSO4 | 60 | 24 | 63 |
| 12 | I2 (50) | Na2SO4 | 60 | 24 | 75 |
| 13c | I2 (10) | --- | 60 | 24 | 83 |
| 14d | I2 (50) | --- | 60 | 24 | 54 |
| 15d | I2 (10) | --- | 60 | 24 | 53 |
| 16e | I2 (10) | --- | 60 | 24 | 65 |
| 17f | I2 (10) | --- | 60 | 24 | 42 |
| 18 | I2 (10) | --- | 60 | 12 | 60 |
| 19 | I2 (10) | --- | rt | 24 | 21 |
| 20 | I2 (10) | --- | 0 | 24 | 0 |
| 21g | I2 (10) | --- | 60 | 24 | 68 |
| 22h | I2 (10) | --- | 60 | 24 | 17 |
| 23 | --- | --- | 60 | 24 | nr |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



