Submitted:
14 February 2025
Posted:
18 February 2025
You are already at the latest version
Abstract
In the 1980s, the discovery of a 185 KDa transmembrane human receptor protein which is now known as human epidermal growth factor receptor 2 (HER2), paved the way for the development of the most successful and 1st humanized monoclonal antibody, Trastuzumab. The antibody received FDA and EU approval in 1998 and 2000, respectively for the treatment of patients with HER2+ breast cancer (BC). The high affinity of Trastuzumab binding with HER2 having a Kd value of < 1 nM is due to the dual action of two antibody domains. Here, the Fab domain provides selective binding with the HER2 receptor whereas the Fc domain binds with the Fc receptor in natural killer cells and leukocytes to kill the tumour cells by antibody-directed cellular cytotoxicity (ADCC). Trastuzumab is widely used for the treatment of BC where it blocks the over-expressed HER2 receptor-mediated dimerization and consequent intracellular signaling that leads to cancerous growth. The phosphatidylinositol 3′-kinase (PI3K)/protein kinase B (PKB)/mammalian target of rapamycin pathway (mTOR), cross-talk with estrogen receptors, over-expression of Mucin 1 (MUC1) protein, and involvement of tyrosine kinase receptors like insulin-like growth factor I receptor are key pathways involved in trastuzumab resistance (TR). In this review, we have provided a molecular view of how this resistance appears, and what are the possible remedies using BC stem cell (BCSC)-based therapy, PI3K pathway inhibitors, MUC1-directed therapy, etc. We have also elaborated on the claims from different patents and data from clinical trials where overcoming the TR is one of the primary focuses of those studies.
Keywords:
Trastuzumab Resistance
; HER2 receptor
; PI3K/AKT/mTOR pathway
; Mucin1
; Combination therapy
; Patents & Clinical trials
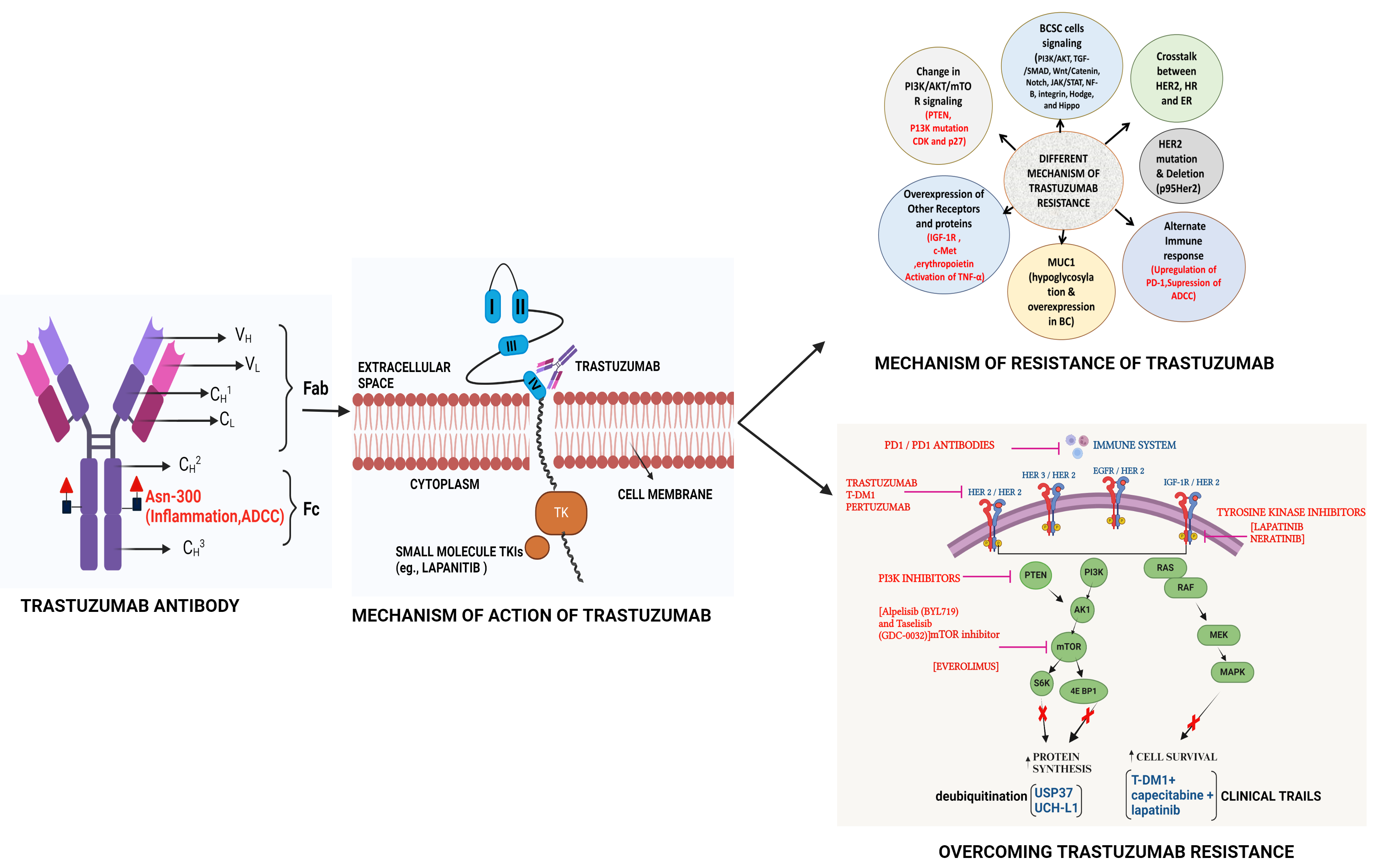
| Graphical Abstract |
 |
The humanized Trastuzumab Monoclonal antibody Targets the domain IV of HER2 receptor to initiate Tyrosine kinase-based signaling to generate BC where activation of different signaling pathways which are involved in resistance can be overcome though use of selective inhibitors of these pathways.
1. Introduction
In 2020, the International Agency for Research on Cancer (IARC), a specialized cancer agency of the World Health Organization (WHO), released a report that showed that BC (BC) has overtaken lung cancer as the most prevalent cancer throughout the globe[1]. BC is also the leading cause of cancer-related deaths in women worldwide [2]. The observed over-expression of HER2 in approximately 20–30% of BC patients has encouraged researchers for years to find a drug/antibody to specifically target HER2 so that HER2-mediated BC can be stopped. In 1984, a gene named ‘neu’ [3]was isolated from rat neuro/glioblastoma encoding a protein with 185 kDa which was found to be similar to a gene located at the human q21 region of chromosome 17[4]. Due to the significant similarities of ‘neu’ gene with the protein sequence of the EGFR (HER1) the ‘neu’ gene was named HER2 which is a kinase receptor at the cell surface. In 1986, it was proven for the first time that the NIH3T3 cells become tumorigenic when transfected with the HER2 gene in vivo[5]. Further, that study also showed that the treatment of the mice with an antibody against the ‘neu’ gene product (named an anti-HER2 antibody) suppresses tumor growth significantly. In 1987, it was established that poor prognosis in BC patients is significantly correlated with the amplification HER2 gene in about 30% of BC tumors[6]. In 1989, a monoclonal antibody (Mab) “4D5” (based on the Hybridoma culture name) was isolated from BALB/c mice, which has a high specificity of binding toward human HER2 and not HER1; thereby selectively suppressing the progression of HER2-positive BC cells [7]. The mouse 4D5 monoclonal antibody was eventually humanized [8] and named Trastuzumab or Herceptin which showed significant clinical efficacy toward patients with HER2-positive BC compared to conventional chemotherapies. The results from two clinical trials demonstrated the efficacy and safety of trastuzumab where it was established that progression-free survival (PFS) was significantly longer due to the addition of trastuzumab to chemotherapy (7.6 vs 4.6 months with chemotherapy alone) as was median overall survival (OS) (25.4 vs 20.3 months) [9,10]. The success of Trastuzumab and its subsequent commercialization emerged from the work of 3 scientists: Axel Ullrich and Michael Shepard of Genentech, and Dennis Slamon of the University of California at Los Angeles (UCLA). Trastuzumab had 2018 worldwide sales of US$7.5 billion. The patents on Trastuzumab expired in the US in June 2019 and Europe in July 2014. There are currently five trastuzumab biosimilars available in the USA. The global trastuzumab biosimilars market is expected to grow from $2.08 billion in 2021 to $2.64 billion in 2022 at a compound annual growth rate (CAGR) of 27.1%. These numbers indicate the importance of Trastuzumab in BC treatment.
The HER2 receptors, which are similar to epithelial growth factor receptor (EGFR, HER1), ErbB3 (HER3), and ErbB4 (HER4) consist of three main domains: (a) the extracellular domain that contains the four subdomains of HER2 receptor allowing ligand-dependent or -independent dimerization, (b) transmembrane domain that connects extracellular and cytoplasmic domains, and (c) the C-terminal cytoplasmic domain that contains tyrosine kinase and regulatory subdomains for activation of downstream signaling pathways [11]. To date, no HER2-specific ligand has been identified indicating that HER2 can go for ligand-independent homo or hetero dimerization to activate the signaling pathway that leads to BC. Therefore, the inhibition of HER2 dimerization, the primary target of trastuzumab, leads to suppression of HER2-mediated cell signaling and tumor growth. One of the major reasons for the highly specific binding of Trastuzumab is that the Mab never has to compete with any intracellular ligand for binding with the HER2 receptor. However, during 2005-2007, it was observed that a fraction (~20%) of early-stage BC patients do not respond to trastuzumab, and ~70% of patients with metastatic disease who receive trastuzumab monotherapy are resistant to treatment within 1 year of treatment [12,13]. This indicates that although the application of anti-HER2-based trastuzumab represents a striking advantage over chemotherapy, resistance to trastuzumab therapy leads to recurrence which limits the survival of HER2-positive BC patients. Therefore, the resistance of trastuzumab has become a global concern for BC treatment considering the early success of the monotherapy of this Mab. This review, therefore, initially focuses on the pathways and biomarkers that are related to TR. In the later stage, we looked at different applications and strategies that dealt with the overcoming of this resistance. We have summarized the claims of different patents &clinical trials targeting the TR with subsequent mechanisms.
2. Different Mechanisms of TR
2.1. HER2 Mutations
The discovery of HER2 receptor and its consequent overexpression in BC cells catalyzed the discovery of 1st humanized monoclonal antibody Trastuzumab, ADC etc. (Figure 1). Trastuzumab cannot bind to the receptor when HER2 is mutated by deletion of its extracellular domain [14].The shortened and N-terminally deleted p95HER2 isoform with constitutive kinase activity is produced by proteolysis or by translation of the HER2 mRNA from internal initiation codons [15,16,17]. This mutant isoform encourages ongoing oncogenic signaling activation while evading the effects of trastuzumab by not allowing any binding sites on the HER2 receptor for the Mab. Those who developed the p95HER2 mutation were less likely to respond to trastuzumab than those who had full-length HER2. This was established in a study of 46 patients with metastatic BC tumor[17].Among 46 patients with metastatic BC only, 11% of patients expressing p95HER2 responded to trastuzumab. But >51% of patients with tumors expressing full-length HER2 achieved either a complete or partial response in the presence of Trastuzumab. However, treatment with lapatinib (a tyrosine kinase inhibitor) shows almost equal response indicating that tyrosine kinase activity is retained in p95HER2-expressing cells. The increased expression of mucin-4 is another factor that masks the trastuzumab-binding on HER2. The presence of mucin-4 (MUC4), a highly O-glycosylated membrane protein that is also thought to be HER2's partner, was associated with a poor prognosis in both BC and other carcinoma types[18]. The study using Trastuzumab-resistant JIMT-1 cell lines showed that MUC4 is overexpressed. Further knockdown of MUC4 at RNA levels increases trastuzumab susceptibility. In another study, TNFα-induced MUC4 expression was proposed as a novel TR mechanism where the administration of TNFα-blocking antibodies downregulated MUC4 and decreased TR by blocking the ADCC[19]. Therefore, steric Inhibition of Antibody-Receptor Interaction in the presence of overexpressed MUC4 is a possible mechanism of TR[20].ABC cell line developed TR by upregulating MUC1*, a cleaved version of the MUC1 protein where treatment with MUC1 antagonists can overcome the resistance[21]. Further, the humanized antibody targeting Mucin1 reduced the growth of HCC1954 xenograft tumors by inhibiting cell proliferation thereby reducing TR[22].
The HER2 protein's inability to bind trastuzumab is caused by several genetic abnormalities, which also contribute to TR. Trastuzumab regimens are now frequently combined with other anti-HER2 medicines that specifically target these variants because of an increased understanding of these polymorphisms [23]. As an illustration, p95HER2, also referred to as the HER2 carboxy-terminal fragment is deficient in the N-terminal extracellular domain required for trastuzumab binding. This fragment is produced either by the metalloprotease ADAM10 shedding the extracellular domain or by the alternate start or translation of the HER2-coding mRNA. ADAM10 are unique metalloproteases that are known as ‘sheddase’ because they cleave off the ectodomain from the membrane proteins also referred to as shedding. Through its capacity to constitutively create homodimers that are stabilized by disulfide linkages, p95HER2 enhances TR[24]. High levels of p95HER2 were found to be associated with shorter PFS and worse OS in metastatic BC patients receiving trastuzumab [25]. Therefore, for a fraction of HER2+ patients with metastatic BC, p95HER2 is an appropriate target.TR has been linked to increased levels of MUC4 in rats also, where it blocks cell-cell and cell-matrix interactions thereby inhibiting binding of trastuzumab [26]. The involvement of MUC4 in cancer pathogenesis is proven by its multifaceted contributions, including attenuation of immune recognition, facilitation of tumor development, suppression of apoptosis, and activation of HER2 signalling[27].
2.2. Mechanism of TR Through Crosstalk Between HER2 and Estrogen Receptors
TR is a significant concern in the treatment of HER2-positive BC, with evidence suggesting a mechanism involving crosstalk between HER2 and Estrogen Receptors (ER)/hormone receptors (HR) [28]. Approximately 50% of HER2-positive breast tumors also express ER, and preclinical models have demonstrated the critical roles played by both HER2 and ER in promoting tumor growth [29]. Notably, therapies targeting HER2 have been shown to increase ER expression, which can enhance cell survival [30]. In 2006, Munzone et al showed for the first time that a patient with HER2+ and ER-negative advanced BC can convert into an ER-positive patient after treatment with trastuzumab and chemotherapy. [31]The data was based on clinical samples with histological assessment for ER markers. More than 50% of all HER2+ BC show the coexistence of HER2 over-expression and ER and/or PR over-expression. It was concluded that inhibition of HER2 by trastuzumab and chemotherapy leads to the activation of ER gene transcription via signaling crosstalk pathways involving more prominently the PI3K/AKT/mTOR pathway. This increased ER signaling pathway acts as a compensatory mechanism for tumor growth and survival under trastuzumab treatment for HER2+ BC.[32] Consequently, there is a growing interest in combination treatments that simultaneously target both HER2 and ER (by endocrine therapy) to effectively combat this resistance mechanism.[33]However, a critical question that remains unanswered is whether patients who have previously received HER2-targeted therapies would benefit from the continuation of endocrine therapy alongside chemotherapy or Antibody-Drug Conjugate (ADC)-based treatments in the future.[34,35] The phase II clinical trial (PERTAIN, NCT01491737) was a randomized, open-label, multicenter-80 sites trial with patients of HER2+ and ER+ BC. In this trial, primary PFS was achieved due to a combination treatment of Trastuzumab and Pertuzumab [35].Therefore it indicates that when ER is overexpressed Trastuzumab alone can’t work and combination therapy is required. In this context, the trastuzumab and tamoxifen combined therapy was found to be effective for HER2+ and ER+ BC. The combination treatment was found to be more growth inhibitory against BT 474 cells than trastuzumab and tamoxifen alone.[36] It was observed that patients with ER+ mBC showed better prognosis after treatment in combination with immunotherapy (pembrolizumab) and antiestrogen agents like letrozole or tamoxifen[37]. All these studies prove that targeting both HER2 and ER is more beneficial than alone HER2-targeted therapy. The mechanism of action of Trastuzuamb with its structure is shown in Figure 2.
One potential avenue of resistance to HER2-directed therapies is the activation of the ER pathway.[38] Several studies have explored the possibility of crosstalk between HER2 and ER.[39,40] This interaction is particularly significant as the upregulation of ER expression has been observed following treatment with lapatinibor aromatase inhibitors which target ER [41]. This phenomenon suggests that the ER may serve as a redundant survival pathway in the context of HER2-targeted therapy. BC progression appears to involve complex interactions between the HER family of receptors and steroid hormone receptors, such as ER and progesterone receptor (PR). Proietti et al. [42] identified the Signal Transducer and Activator of Transcription 3 (Stat3) as a pivotal protein activated by heregulin (HRG). HRG is a soluble and secreted growth factor, which binds and activates HER3 and HER4 transmembrane receptor tyrosine kinases, but not HER2. Stat3 is activated through a co-opted, ligand-independent PR activity, acting as a signaling molecule. In this context, the targeting of Stat3 becomes crucial for impeding the growth of mammary tumors induced by HRG.[38] Their data suggests that HER2+ BC can be blocked by inhibiting the ER/PR signaling pathway. In another study, it was established that the expression of two isoforms of ErbB-2 in the nucleus promotes triple-negative BC (TNBC). These isoforms which act as transcription factors if evicted from the nucleus result in blockage of TNBC and tumor cell growth [43]. The Stat3 was found to be a direct upstream regulator of ErbB-2 and microRNA-21 (miR-21) both of which initiate metastatic BC [44].
These findings underscore the intricate interplay between HER2, ER, and PR in BC growth and the potential for signaling molecules like Stat3 and regulators of PI3K/AKT/mTOR pathways to play key roles in mediating this crosstalk. Understanding these mechanisms is essential for the development of more effective treatment strategies for HER2-positive BC and the prevention of TR.
2.3. HER2/ER Crosstalk in Acquired Endocrine Resistance Models
In the context of acquired endocrine resistance models, recent investigations have uncovered a significant association between enhanced Epidermal Growth Factor Receptor (EGFR) and Human Epidermal Growth Factor Receptor 2 (HER2) signaling and resistance to tamoxifen (an ER modulator) in tumors characterized by initially low levels of EGFR and HER2 expression [45,46]. Nicholson and Gee [47] demonstrated that EGFR/HER2 signaling contributes to the development of acquired tamoxifen resistance in BC cells maintained under long-term culture conditions. These resistant cells exhibit elevated levels of EGFR and HER2 expression, increased activation of EGFR/HER2 heterodimers, and heightened phosphorylation levels of key signaling molecules, including p42/44 Mitogen-Activated Protein Kinase (MAPK), AKT, and nuclear Estrogen Receptor (ER) at serine residues 118 and 167 [48,49]. Notably, the utilization of the EGFR inhibitor gefitinib or the aromatase inhibitor exemestane exerts strong inhibitory effects on the growth of these resistant cells, mirroring the outcomes observed in de novo experimental models of tamoxifen resistance.
Furthermore, increased EGFR and HER2 expression, coupled with the consequent activation of downstream signaling pathways mediated by p42/44 MAPK, has been documented in BC cell models that have developed resistance to estrogen depletion or aromatase inhibitor (AI) therapy over an extended period [50,51,52]. Intriguingly, it has been observed that MCF-7 cells, which have adapted to proliferate in the presence of the potent fulvestrant (a selective ER degrader) exhibit heightened EGFR signaling. Therefore, estrogen deprivation with fulvestrant can effectively inhibit HER-2overexpressing tumors but resistance against tamoxifen develops quickly. This observation implies that growth factor receptors, including EGFR and HER2, play pivotal roles in the development of resistance to endocrine therapies including tamoxifen and beyond [47,53].
These findings underscore the significance of EGFR and HER2 signaling pathways in the context of acquired endocrine resistance, shedding light on potential therapeutic strategies to counteract resistance in BC patients undergoing endocrine therapies [54].
2.4. Failure to Trigger ADCC
The concept of immunomodulatory antibodies came in the limelight due to the discovery of cytotoxic T lymphocyte-associated antigen 4 (CTLA4) and programmed cell death protein 1 (PD1) as immune checkpoint inhibitors. The checkpoint proteins like program death (PD)-1 (present in T cells) bind with PD ligand-1 (PD-L1) which is, generally, overexpressed in most cancer cells. This binding acts as an ‘off-signal’ for immune-mediated signaling so that cancer cells can evade T-cell-mediated death. A very similar mechanism is involved in ADCC where the antibody's constant region (Fc) interacts with Fcγ receptors (FcγRs) which recruits neutrophils and NK cells to the tumor cells which are already bound by the Fab part of the antibody due to receptor-ligand interaction (Figure 3).[55] The NK cells then release perforins and granules to kill the bound tumor cells by apoptosis. "The initial observation of trastuzumab's mechanism of action, involving Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC), was first documented in preclinical animal models. Subsequently, a clinical study confirmed this concept, revealing a significant lymphocytic infiltrate in all patients undergoing neoadjuvant trastuzumab treatment, with a more pronounced effect seen in partial responders.[56] Factors influencing the efficacy of HER2-directed therapy in ADCC induction include Tumor-Infiltrating Lymphocytes (TILs) and Fcγ receptor polymorphisms. The Fcγ receptors are abundant on leukocytes and they recognize IgG-coated immune complexes through the Fc receptor of Antibody. Various strategies have been investigated to enhance ADCC in the context of HER2-targeted therapies. ADCC is governed by three Fcγ receptors expressed on immune cells: CD16A, CD32A, and CD32B [57]. To enhance ADCC, margetuximab (a chimeric Mab targeting HER2+ BC cells) was engineered to preserve the epitope specificity of trastuzumab while introducing five amino acid modifications, increasing its affinity for the activating Fcγ receptor IIIA (CD16A) and reducing its affinity for the inhibitory Fcγ receptor II (CD32B)[58,59]. This antibody is very similar to trastuzumab except that the Fc section is engineered so that it can bind with both alleles of CD16A with improved ADCC. The mutant version of allele CD16A-158V is generated in BC cells due to progressive Trastuzumab treatment. This eventually makes Trastuzumab treatment ineffective for these BC cells due to reduced Fc-FcγRs interaction involved in ADCC [60].In the phase 3 SOPHIA trials, margetuximab+chemotherapy, and trastuzumab+chemotherapy were compared in 536 patients with previously treated HER2+ advanced BC+ metastatic BC (MBC). The PFS was much better for margetuximab+chemotherapy even though Her2+ binding is similar for both margetuximab and Trastuzumab. This indicates better ADCC activity of margetuximab is responsible for BC cure.
The direct involvement of trastuzumab in ADCC and antibody-dependent cell-mediated phagocytosis (ADCP) was observed from a clinical data set where PBMCs from human HER2+ BC patients receiving trastuzumab therapy (both, metastatic and adjuvant) were analyzed for CD14+ (monocytes) and CD56+ (NK) cells. Here, ADCC and ADCP activity was directly correlated with the altered expression of antibody binding (FcγR)I (CD64), FcγRIII (CD16), and FcγRII (CD32) on CD14+ (monocytes), and CD56+ (NK) cells by FACS studies. Further, the expression of CD107a+ (LAMP-1) on NK cells and the total amount of CD4+CD25+FOXP3+ (Treg) cells were measured where ADCC activity was inversely correlated with the expression of LAMP1 on CD56+ cells in adjuvant patients.[61] The study concluded that trastuzumab-mediated ADCC is reduced in BC patients compared to healthy volunteers but efficacy of trastuzumab-specific ADCC and ADCP is not affected by disease progression, treatment duration, or concomitant chemotherapy. Furthermore, results of this work suggested that CD4+CD25+FOXP3+ Treg cells can act as a potential prognostic factor in mBC. In another study it was shown that the extent of phagocytosis and consequent ADCP was influenced by FcγRIIa and FcγRIIIa expression where polymorphism in these receptors (FcγRIIIa 158V and FcγRIIa 131H) may cause significantly altered effect of trastuzumab therapy for different individuals.[62]
The therapeutic landscape for TNBC and various other malignancies has been significantly reshaped by the introduction of immune checkpoint inhibitors (ICIs). [63,64,65] Preclinical studies have demonstrated that ICIs possess the potential to enhance and optimize the therapeutic efficacy of HER2-targeted therapies.[66] However, in a few clinical trials, the combination of immunotherapy with HER2-targeted treatments has not exhibited substantial benefits. In a single-arm, phase 1b-2 PANACEA study, pembrolizumab, an inhibitor of programmed cell death 1 (PD1), was administered in conjunction with trastuzumab for the treatment of advanced BC cases resistant to trastuzumab [67]. Among the forty patients with programmed cell death ligand-1 (PDL1)-positive disease, only six individuals demonstrated an objective response, while resistance was prevalent within the PDL1-positive population. Notably, none of the 12 patients in this subgroup exhibited a clinical response. In total, 58 patients were enrolled in this clinical trial. In the subset of patients with programmed cell death ligand-1-positive (PDL1+) disease, the PFS (progression-free survival) reached 2.7 months with a median follow-up of 13 months, while overall survival (OS) was not reached during this period [67]. In the broader PDL1 population, where the median follow-up was 12 months, the PFS was 2.5 months, and the OS extended to 7 months. 62 The KATE2 study, a phase 2 randomized trial, enrolled 202 patients who had previously received treatment for advanced HER2-positive BC. These patients were randomized to receive T-DM1 either with or without atezolizumab.[68] Although atezolizumab did not significantly improve PFS in the intent-to-treat (ITT) population, a modest enhancement in PFS was observed in the PDL1-positive subset. However, the clinical significance of this improvement remains uncertain, and it is important to note that patients treated with ICIs experienced a higher incidence of toxicities.[68]
The ongoing phase 3 clinical trial, KATE3/MP42319 (NCT04740918), is currently in progress, enrolling patients diagnosed with advanced BC who are both HER2-positive and PD-L1-positive. In this trial, patients are being randomized to receive either Trastuzumab emtansine (T-DM1) as an antibody-drug conjugate (ADC) alone or in combination with atezolizumab as a second-line treatment option.[69] Atezolizumab is a Mab that is commonly used for non-small cell lung cancer (NSCLC), small cell lung cancer (SCLC), hepatocellular carcinoma (HCC) but not used anymore for TNBC. Meanwhile, another clinical trial study (NCT03199885) is focused on assessing the role of atezolizumab in the first-line treatment setting, specifically in a patient population that is not pre-selected based on their PD-L1 status.[69] These investigations aim to elucidate the potential contribution of immune checkpoint inhibitors (ICIs) in the management of advanced HER2-positive BC. Atezolizumab with doxorubicin/cyclophosphamide-paclitaxel and pertuzumab-trastuzumab (PT) for high-risk, HER2-positive early BC did not increase pathologic complete response (pCR) rates versus placebo in the ITT or PD-L1-positive populations. However, this study confirms that PT is better than Atezolizumab with doxorubicin/cyclophosphamide-paclitaxel combination. Additionally, in the neoadjuvant setting, there have been studies exploring the integration of ICIs into standard therapeutic approaches. The ongoing phase 3 clinical trial, KATE3/MP42319 (NCT04740918), is currently in progress and is enrolling patients with advanced BC characterized by HER2 overexpression and PD-L1 positivity. In this trial, patients are randomly assigned to receive T-DM1 either as a standalone treatment or in combination with atezolizumab in a second-line treatment context. In parallel, the study with identifier NCT03199885 is examining the potential of atezolizumab in the first-line treatment setting, specifically in a patient population not pre-selected based on their PDL1 status. These investigations aim to elucidate the potential utility of immune checkpoint inhibitors (ICIs) in managing advanced HER2-positive BC. Furthermore, in the neoadjuvant setting, investigations have been conducted to explore the incorporation of ICIs alongside conventional therapies.[69] In both the intent-to-treat (ITT) and programmed cell death ligand-1-positive (PDL1+) populations, the incorporation of immune checkpoint inhibitors (ICI) did not result in an elevation of pathologic complete response (purr) rates, and the one-year event-free survival rates within these groups remained comparable.
Currently, an ongoing study known as Astefania (NCT04873362) is enrolling patients with high-risk, HER2-positive BC who exhibit residual disease following neoadjuvant therapy. In this study, patients are randomly allocated to receive standard T-DM1 therapy with or without atezolizumab, further examining the potential impact of this treatment approach. These findings indicate that, at present, the use of immune checkpoint inhibitors (ICIs) is not a standard practice in the treatment of HER2-positive BC. Further investigation is imperative to determine whether immunotherapy may offer advantages for a specific subgroup of patients. Like the observations in triple-negative BC,[70] the expression of PDL1 appeared to correlate with treatment response in patients with metastatic disease. However, it's worth noting that these studies were not explicitly designed to assess this patient population, and the utilization of ICIs occurred in later lines of therapy, a factor that has shown modest responses in the context of TNBC.[71,72] Consistent with previous reports on early-stage TNBC, the presence of PDL1 did not emerge as a reliable predictor of treatment response in the IMpassion050 study. Elevated levels of TILs have been associated with favorable prognoses in HER2-positive BC, spanning both early and advanced stages of the disease.[73,74] In the CLEOPATRA phase III clinical study the TILs are isolated and analyzed from 819 patients where early-stage HER2+ BC patients are treated with either pertuzumab or placebo in addition to trastuzumab and docetaxel [74]. It was concluded from the study that higher TIL values are significantly associated with improved OS but not PFS. Nonetheless, it is noteworthy that the potential role of TILs as a predictive biomarker for immune checkpoint inhibitor (ICI) response in HER2-positive BC has yet to be explored mechanistically although the partial role of TILs is evident. In summary, it is evident that ADCC plays a pivotal role in the mechanism of action of trastuzumab, and the concept of immune evasion may function as a defensive strategy against HER2-targeted therapies. Therefore, interventions such as modifying the trastuzumab FcγRs or integrating immune checkpoint inhibitors (ICI) into conventional therapeutic regimens have thus far resulted in only marginal enhancements in patient outcomes.
2.5. Changes in HER2 Downstream Signaling Pathways
2.5.1. Loss of PTEN Expression /Function
Humans have a phosphatase called Phosphatase and Tensin Homolog (PTEN), which is produced by the PTEN gene[75]. The enzyme acts as a tumor suppressor, where it regulates cell division by keeping cells from growing and proliferating too rapidly or in an uncontrolled way through its phosphatase function. Mutations of the PTEN gene are a step in the development of many cancers including BC. PI3K/Akt is constitutively activated as a result of PTEN depletion [76]. Reduced PTEN expression or activity prevented trastuzumab-mediated growth suppression in BC cells overexpressing HER2[77]. Numerous malignancies, including about 50% of BCs, have been shown to have PTEN loss of function brought on by mutation or by transcriptional regulation [38,78]. Trastuzumab treatment outcomes were noticeably lower in patients with PTEN-deficient HER2-overexpressing MBC than in those with normal PTEN tumors. Later, another group showed that tumors from a cohort of 55 patients who were resistant to trastuzumab included 25% PI3K catalytic subunit alpha (PI3KCA) activating mutations and 20–25% PTEN deficiency. The study also showed that HER2-positive BC cell lines with activating PIK3CA mutations (E545K and H1047R) are more trastuzumab-resistant than HER2-positive cell lines without such mutations[79]. The PIK3CA mutation frequency has ranged from 8%-40% in human BCs where the majority of mutations occur at three hotspots: E542K and E545K at exon 9, which encodes the helical domain, and H1047R at exon 20, which encodes the kinase domain.In a retrospective examination of 47 HER2-amplified primary BCs from stage IV patients treated with trastuzumab plus taxane, Nagata and colleagues used immunohistochemistry to correlate PTEN expression with tumor response to treatment [76,78]. It was established Trastuzumab treatment increased PTEN membrane localization with phosphatase activity with reduced PTEN tyrosine phosphorylation via Src inhibition.[80]The observation was further supported by reducing PTEN in BC cells by antisense oligonucleotides conferred TR in vitro and in vivo.
2.5.2. P13K Mutation or Increased Activity
TR has also been linked to oncogenic activating PI3K mutations. Berns and colleagues looked for activating PIK3CA mutations in hotspot areas of 55 HER2-amplified primary tumor samples from trastuzumab-refractory patients. According to earlier investigations on the frequency of mutations in HER2-amplified BC, 25% of tumors had mutations [79,81,82]. In this collection of cancers, PTEN expression was also examined, and in 22% of tumors, decreased PTEN expression was seen. Patients with active PI3K (defined as decreased PTEN expression or PIK3CA mutation) exhibited a substantially lower PFS on trastuzumab-based treatment than patients without evidence of PI3K pathway activation, according to Kaplan-Meier survival curves. An integrative genomic and proteomic analysis that involved mass spectroscopy-based sequencing and reverse-phase protein arrays analyzed 547 human BCs and 41 cell lines to understand the signalling effects of PIK3CA, AKT, and PTEN mutations in HER2+ BC79. The PIK3CA mutations were more common in HR+ (34.5%) and HER2+ (22.7%) compared to basal-like tumors (8.3%). Further, PIK3CA (39%) and PTEN (20%) mutations were more common in cell lines than tumors but PIK3CA mutations did not have a significant effect on clinical efficacy after adjuvant tamoxifen therapy in 157 HR+BC patients. A more recent study that examined PI3K-AKT pathway changes as predictive biomarkers of TR in a larger cohort of tumors from patients with HER2-amplified MBC discovered that the individual biomarkers by themselves were insufficient to predict diminished response to trastuzumab-based therapy[83]. More recent research compared several tumor samples that had been treated with trastuzumab before and after to see if similar alterations in the PI3K pathway were present in the tumors[84]. Here Biopsies and IHC staining were done on 19 patients who are having HER2+ BC under trastuzumab treatment. They observed abnormality in HER3, PI3K, or PTEN implying a critical role for this pathway in mediating resistance to Trastuzumab however abnormalities are not co-existent. For example, loss and PI3K mutation were not seen in the same samples and vice versa. In early reports of this research, tumor samples (n=45) obtained after trastuzumab progression showed that PTEN loss in immunohistochemistry [85] and PIK3CA mutations (in Sequenom assay)[86] were frequently found even in a cohort of patients who had initially benefited from trastuzumab treatment. The PI3K regulatory subunit p85 is encoded by the PIK3R1 gene, which alters its functionality and causes constitutive activation of the PI3K/Akt pathway [87,88]. Additionally, the gene that codes for the catalytic subunit of p100 of PI3K is called PIK3CA (activator of the same pathway and commonly mutated in BC). In human cancer, it is commonly mutated or overexpressed and can give TR in cell culture-based studies [81,89].
2.5.3. Cyclin Dependent Kinases and p27
In vivo, p27 can inhibit the kinase activities of a variety of cyclin/CDK complexes (where N-terminal domain is sufficient to block cyclin/CDK kinase activity) including those containing CDK1, -2, -4, or -6, and cyclins A, E, or D. The p27KIP1 protein downregulation is often correlated with poor prognosis in several types of human cancers where the protein is functionally inactivated by cytoplasmic localization or by phosphorylation. Trastuzumab intracellularly influences cyclin-dependent kinase (CDK)1 inhibitor p27Kip1, the most significant CDK inhibitor, as one of the growth-regulating molecules and
Performs a crucial function in regulating the cell cycle by binding to the cyclin E.CDK2 complex throughout the G1 phase of the cell cycle [90].Proliferating cells are forced out of the cell cycle by an elevation in the protein p27Kip1, while quiescent cells are encouraged to start proliferating again by a reduction in p27Kip1. Several studies have suggested that the p27Kip1 protein plays a role in the development of human cancer because of its crucial role in regulating the cell cycle. A deficiency in the expression of the p27Kip1 protein has been connected to a variety of human malignancies, including BC, where it is linked to increased growth of cells[91]. Low chances of surviving and higher grade neoplasms are strongly correlated with decreased levels of the protein p27Kip1 and also coincide in reverse with HER2 overexpression [92,93]. Maurizio et al found a correlation between Cyclin E amplification/overexpression with TR in a cohort study involving 34 HER2+ BC patients[90]. They have identified genomewide copy-number variation in the trastuzumab-resistant vs. sensitive cells by measuring focal amplification of genomic DNA containing the cyclin E gene. They found that cyclin E amplification/overexpression was associated with a lower PFS (6 mo vs. 14 mo, P < 0.002) and worse clinical benefit (33.3% compared with 87.5%, P < 0.02) compared to non overexpressing cyclin E tumors. In parallel, knockdown of cyclin E expression or treatment with CDK2 inhibitors downregulated cyclin E activity in cyclin E-amplified trastuzumab-resistant clones with concomitant decrease in proliferation and enhanced apoptosis. Their data suggested a direct role for cyclin E overexpression and CDK2 activity in TR and implicate that treatment with CDK2 inhibitors may be a good strategy in patients with BC with HER2 and cyclin E co-amplification/overexpression. During the preclinical study, it was observed that trastuzumab inhibits HER2-overexpressing BC by inducing G1 cell cycle arrest, with an elevation of p27Kip1 and a decrease of CDK2. They further discovered that through a post-translational method,anti-HER2 antibodies control the phosphorylation of the p27Kip1 protein, via which the protein is upregulated. The magnitude of G1 cell cycle arrest induced by trastuzumab or 4D5 is well correlated with the induced level of p27Kip1. Anti-HER2 antibody-induced p27Kip1 protein, G1 arrest, and growth inhibition persist at least 5 days after 14 h of treatment with a single dose of trastuzumab[94].In another study, the magnitude of G1 arrest, the level of p27Kip1 upregulation, and inhibition of CDK2 activity were analyzed by treatment of trastuzumab against BT474 and SKBR3 HER2-overexpressing BC cells [95]. This was also found to be correlated with HER2 receptor expression level. The continuous exposure of cells to Trastuzumab for 3 days results in the downregulation of the cell surface receptor and a concomitant increase in the level of p27Kip1 protein.
2.6. Overxpression of Other Receptors and Proteins in Resistance
2.6.1. Increased Expression of IGF-1R
The insulin-like growth factor 1 receptor (IGF-1R) is another receptor whose abnormal expression may confer TR. According to Lu et al, excessive expression of IGF-1R causes trastuzumab-sensitive SKBR3 cells to become resistant to the therapy. Furthermore, IGF-1R-overexpressing SKBR3 cells have substantially decreased amounts of p27Kip1 and p21Cip1 proteins, and an elevated level of CDK2 kinase function[96]. Jerome et al, proved that IGF1R-overexpressing cells play a role in developing Trastuzumab -resistance by escaping cell cycle arrest caused by trastuzumab. In this study, a human IGF binding protein 3 (rhIGFBP-3) which is an antagonist of IGF-IR signaling was tested in Herceptin-resistant BC cells in vitro and in tumors in vivo. It was observed that rhIGFBP-3 educed IGF-I-induced IGF-IR phosphorylation with a synergistic interaction with Herceptin against cultured HER-2-overexpressing BC cells in vitro. Furthermore, a high degree of IGF-1 receptor (IGF-1R) expression in HER2-amplified cell lines is associated with a reduced response to trastuzumab [97,98] . This is thought to be owing to HER2 and IGF-1R interaction, with IGF-1 activation resulting in HER2 phosphorylation and PI3K stimulation. IGF-1R signaling attenuation stops HER2 phosphorylation and restoration of trastuzumab sensitivity in particular laboratory models[99]. These results provide an opportunity to evaluate the simultaneous blockade of the HER-2 and IGF-IR pathways using a combination therapy which may include rhIGFBP-3 and Herceptin in patients with HER-2-positive BC. Evaluation of primary tumor samples from a neoadjuvant study of vinorelbine and trastuzumab revealed that the existence of membrane expression of IGF-1R was associated with a lower response rate to therapy in contrast with tumors without IGF-1R expression (50 vs 97%; p = 0.001), providing additional evidence that IGF-1R plays a role in mediating TR[96,100,101] .
2.6.2. C-Met Coexpression
The C-MET (mesenchymal-epithelia transition factor) is a receptor tyrosine kinase that binds with its ligand, hepatocyte growth factor (HGF), thereby activating a wide range of different cellular signaling pathways, involved in proliferation, motility, migration, and invasion.[102] C-Met is expressed in epithelial and endothelial cells whereas MET is a membrane receptor involved in embryonic development and wound healing. c-Met, a crucial oncogene, is a 145 KDa tyrosine phosphorylation protein that was found to be the ligand for HGF [103]. Patients with BC who have elevated levels of c-Met are associated with a bad prognosis[104]. When tissue microarray was analyzed with 30-year, disease-specific patient follow-up data, a strong correlation between the expression of HGF, matriptase (an activator of HGF), and Met was confirmed in breast carcinoma. Data from this paper indicated that overexpression of the cytoplasmic tail of Met (c-MET), which can happen due to cleavage or truncating mutation, may play an important role in BC progression. Further, it was discovered that 25% of the tissues from HER2-positive BC patients and HER2-positive cell lines of BC had significantly higher levels of c-Met [100,105,106]. More significantly, it has lately been shown that c-Met physically interacts with HER2 based on receptor tyrosine kinase (TK) capture arrays to identify receptors active in NSCLC cell lines. Overexpression of MET showed physical interaction of c-MET with EGFR and HER2 which implies that c-Met and HER2 signaling may work together to produce TR[107]. This study provides first evidence that the combined use of MET inhibitor with a dual EGFR/Her2 inhibitor provides maximal growth inhibition in cancer. Sweeney's team discovered that trastuzumab quickly increases c-Met expression in vitro and that c-Met deletion increases the cells' sensitivity to the drug [100,108]. Met contribution to TR, was proven as inhibition of Met sensitizes cells to trastuzumab-mediated growth inhibition, whereas Met activation creates by removing the p27 induction. Their study implicated that a certain proportion of Her2 (+) patients may benefit from combined inhibition of MET and HER2. C-met overexpression and specific signal from c-met to HER3 causes developed resistance to the EGFR TKIs gefitinib and erlotinib In non small cell lung cancer[109]. Further, in a pilot phase I trial (NCT03060356) the safety and feasibility for the delivery of intravenous RNA-electroporated chimeric antigen receptor (CAR) T cells targeting the cell-surface antigen cMET was proven [110].
2.6.3. Over Expression of Erythropoietin
Erythropoietin (EPO) is a crucial hematopoietic hormone that controls the differentiation, survival, and proliferation of progenitor cells of erythroid, which are found in the bone marrow[111]. Through contact with particular tyrosine-phosphorylated areas within the activated receptor for EPO(EpoR), EPO-induced cellular signaling activates downstream signaling chains, including the PI3K/Akt and MEK/Erk pathways[112]. These HER2-activated downstream signaling pathways combine with those triggered by EPO via EpoR. They showed that, out of a panel of ten primary cell lines of breast carcinoma, two exhibit relatively high levels of both HER2 and EpoR, and five express the erythropoietin receptor (EpoR)[38].
These findings suggest that the overexpression of, both, HER2 and EpoR occurs in a significant percentage of BCs. It was discovered that recombinant human EPO (rHuEPO) considerably lessens the trastuzumab-induced reduction of cell growth in BC tumors. Here it was observed that rHuEPO antagonizes trastuzumab treatment of BC cells via Jak2-mediated Src activation and PTEN inactivation [23,113,114]. It was hypothesized that since the downstream signaling pathways for EpoR and HER2 are overlapped, partially, therefore EPOR may play a role in the inhibition effect of trastuzumab and its resistance. Interestingly it was observed that EpoR was positive in 42 samples and highly expressed in 24 samples as measured by immunohistochemistry among 55 tested Bc cell lines[112]. In parallel, when the trastuzumab-resistant SKBP3 cell line was generated it showed higher EPO expression with high phosphorylated EPOR level[115]. Therefore, EPO/EpoR expression is directly correlated with tumor progression and proliferation in HER2-positive BC.
2.6.4. Increased Activity of Rac1
The Rho family of GTPases is a family of small (~21 kDa) monomeric G proteins that belong to the Ras superfamily of GTPases. The Ras superfamily of GTPases comprises more than 50 members that share several common features: their molecular weight (18–28 kDa), their C-terminal poly isoprenylation region, and the property to bind to and hydrolyze guanine nucleotides. The Rho GTPases form 8 subfamilies among which one subfamily comprises of Rac1, Rac2, Rac3, and RhoG. Rac1, a tiny GTPase that resembles Ras, has been linked to the regulation of cell development and morphology and is thought to be connected to BC advancement[116,117]. In SKBR3 cells, resistance to trastuzumab was linked to elevated Rac1 activity, which resulted in considerable cytoskeleton disarray. Decreased extracellular signaling regulates kinase function in resistant clones, trastuzumab-facilitated endocytic downregulation of HER2, and low Rac1 activity with a particular inhibitor[118]. Further, it was shown that dysregulation of classical Rho GTPase like Rho, Rac, and Cdc42 can initiate BC and its metastasis [119].
2.6.5. Activation of Tumor Necrosis Factor α
Tumor necrosis factor-α (TNFα) activates c-Src to cause HER2 phosphorylation in BC cells. Additionally, TNFα facilitated the development of the HER2/HER3 heterocomplex, activated AKT, and transcription of NF-κB. TNFα-induced NFkB activation and cell proliferation were inhibited by HER2 knockdown via RNA modification or by the use of AG825, an inhibitor of EGFR/HER2-TK. Trastuzumab, nevertheless, was unable to prevent TNFα from stimulating the growth of BC. It's interesting to note that TNFα can trans-express HER2 and employ it as a downstream signaling molecule that is necessary for the production of mitogenic signaling. Since a considerable percentage of people infiltrating breast tumors have TNFα in their tumor microenvironment, treating HER2-positive with TNF-α as a target may be a potential [120,121]. The blockade of TNF-α was proposed as a therapy to treat BC since TNFα enhances luminal BC cell proliferation by aromatase upregulation. This aromatase enzyme increases estradiol synthesis which after binding to ER promotes luminal cancer cell proliferation[122]. Stable overexpression of TNFα in BT-474 cells, a human HER2-positive BC cell line, led to high activation Akt and NF-κBsignalling pathways. Further, it was proven that TNFα-induced expression of mucin 4 is partially responsible for TR in HER2-positive BC[19]. This was further supported by an experiment where treatment, in combination with trastuzumab and a soluble TNF inhibitor, showed MUC4 downregulation with consequent immunomodulatory effect through M1-like phenotype macrophage polarization and NK cells degranulation [123].
3. Overcoming TR Against HER2-Positive BC
3.1. BC Stem Cell (BCSC)-Specific Therapeutic Approaches forTR
In recent years, multiple studies have found indirect and direct correlations between BCSCs and the development of TR. The aggressive progression and recurrence of BCwas due to the presence of a subset of tumor cells known as BC stem cells (BCSCs). These cells possess the abilities of self-renewal and tumor initiation, which allows them to drive metastases and tumor growth. These cells reside in a microenvironment that is filled with residential inflammatory cells that generate signaling cues for BCSC-mediated self-renewal and survival[124]. Three key proteins were known markers for BCSCs: CD44, CD24, and aldehyde dehydrogenase (ALDH). However, studies have shown that BCSCs contain both CD44 and ALDH, but not CD24 [125]. The role of several signaling pathways, noncoding RNAs, microenvironmental metabolic alterations, and immunological factors that have been shown to generate TR by modulating BCSC biology is discussed there [120]. The behaviour of BCSCs is regulated by several pathways, including PI3K/AKT, TGF-/SMAD, Wnt/Catenin, Notch, JAK/STAT, NF-B, integrin, Hodge, and Hippo signaling pathways; therefore, crosstalk or deregulation of those pathways may be a factor in the growth or transition of BCSCs.
3.1.1. AKT/PI3K Signaling and TGF-β Signalling
Dysregulation of the PI3K/AKT pathway and loss of the tumor suppressor Phosphatase and TENsin homolog (PTEN), a downstream mediator of the PI3K/AKT pathway [80,126] have been linked to adaptation or resistance to trastuzumab and have been found in over 40% of HER2-positive BCs in one study[127]. PTEN loss may cause TR in HER2-positive BC by initiating epithelial-mesenchymal transition (EMT) and switching the CD 44+/CD 24−/low/HER2−/low subtypes. The dimerization of HER2 with its coreceptor HER3 and subsequent direct coupling to the p85 regulatory subunit of PI3K and activation of PI3K-AKT signaling, which drives ALDH+ populations in HER2-overexpressing cells, is another explanation for the emergence of TR[128]. Trastuzumab could decrease the ALDH+ population, but it could not eradicate it [129,130]. As a result, PI3K inhibitors might be able to lower the proportion of BCSCs (including CD44+/CD24/low and ALDH+ cells), hence lowering the HER2-positive BCs susceptibility to trastuzumab [131,132,133,134]. When coupled with trastuzumab, the pan-PI3K inhibitor, XL147 lowered BCSC fractions and slowed the carcinogenesis, possibly by preventing acquired TR[132]. It was discovered that NVP-BKM120, another pan-PI3K inhibitor, effectively silenced the PI3K/AKT signaling pathway, by preventing BCSC growth. In the case of HER2-positive BC, this inhibitor also exhibits a synergistic impact with trastuzumab in the elimination of BCSCs. More importantly, the administration of combinatorial therapy to patients with HER2-positive BC is supported by the fact that this medication combination was well tolerated in xenograft animal models[133]. Additionally, an AKT inhibitor reduced the PI3K/AKT pathway and helped HER2-positive BC patients' refractory BCSCs regain their sensitivity to trastuzumab. Perifosine, an Akt inhibitor, has been shown to drastically lower the ALDH+ population by preventing PI3K/AKT signaling[135]. Disulfiram/copper (DSF/Cu), a substance medication commonly used for overcoming alcohol drinking problems, was discovered to drastically downregulate p185HER2, p95HER2, HER3, and phospho-HER3/This subsequently blocks the PI3K/AKT signaling pathway, which was followed by the eradication of BCSC populations and suppression of BCSC-like characteristics. Particularly, DSF/Cu may inhibit HER2/AKT signaling, cause apoptosis, and abolish BCSC populations, suggesting that DSF may be helpful for the treatment of trastuzumab-refractory HER2-positive malignancies[128]. It was discovered that flubendazole (FLU) plays a similar role to DSF/Cu, which can successfully target BCSC-like traits to increase the effectiveness of trastuzumab in HER2-positive tumors [134]. The mechanism of Trastuzumab resistance involving different signaling pathways are shown in Figure 4.
One study found that by inhibiting TGF- β signaling, which effectively controls the EMT and the maintenance of BCSCs, the resistance to trastuzumab was reduced [136]. According to Chihara et al., TGF- β stimulation results in SMAD3's increased phosphorylation, which causes an increase in the CD44+/CD24/low population, which leads to the development of anti-HER2 medication resistance[137]. It was proposed that A83-01, a different small molecule inhibitor, could alter the TGF- β /SMAD pathway, stop HER2-positive cells from developing a mesenchymal phenotype, and improve the HER2-positive cells' response to trastuzumab therapy[138]. Intriguingly, it was discovered that metformin could reduce the proliferation and capacity for self-renewal of trastuzumab-resistant BCSCs in BC, particularly CD44+/CD 24/low cells[139]. In addition, Cufi and associates showed that metformin might eliminate BCSCs and overcome primary TR in ER+ human BC [140]. These earlier investigations led us to the conclusion that metformin may regulate EMT and suppress TGF- β signaling, as CD44+/CD24/low BCSCs have a strong TGF-β pathway signature which becomes a more epithelial like feature after TGF-β pathway inhibition[141].
3.1.2. Wnt/β-Catenin and Notch Signaling
The Wnt/β -catenin signaling activation has been shown to drive resistance to endocrine treatment, chemotherapy, and radiotherapy in BCSCs[142,143] , Wu et alwere the first to reveal that TR was also accompanied by Wnt/β-catenin signaling. The expression of Wnt3 in trastuzumab-resistant cells is correlated with increased nuclear expression of β-catenin and transactivated the expression of EGFR. The increased Wnt3 in the trastuzumab-resistant cells also promoted a partial EMT-like transition, like increased N-cadherin, Twist, and Slug expression and decreased E-cadherin expression., This was associated with the emergence of TR in HER2-overexpressing BC cells. Conversely, knockdown of Wnt3 by siRNA restored the cytoplasmic expression of β-catenin and decreased EGFR expression in trastuzumab-resistant cells. The Wnt3 transfectants of Trastuzumab-resistant cells, SKBR3/Wnt3-7 and SKBR3/Wnt3-9, showed a significant decrease in E-cadherin and increase in N-cadherin, Twist, and Slug. Another study found that downregulating Wnt3 prevented EMT and cell invasiveness while upregulating Wnt3 promoted EMT in trastuzumab-resistant cells and reduced sensitivity to the drug[143]. Additionally, important relations between the TGF- β and Wnt/β -catenin pathways that induce EMT were found in HER2-overexpressing BCcells.The function of Wnt3 during EMT was found to be regulated by binding to Twist, providing a potential therapeutic target that can inhibit resistance and restore sensitivity to anti-HER2 drugs [144]. Twist served as a linkage between TGF-β and Wnt/β-catenin pathways during TGF-β-induced EMT. Knock-down of Twist by shRNA showed the importance of Twist in response to TGF-β regulating Wnt3 during EMT.
Drug resistance in BCSCs is regulated by mutations in the Notch signaling pathway. Notch receptor inhibitors (DAPT or GSI)and monoclonal antibodies that interrupt receptor-ligand interaction are the two primary forms of Notch signaling inhibitors. The most common GSIs are MK-0752 and PF-03084014. MK-0752 and PF-03084014 have shown excellent efficacy in clinical trials for the treatment of advanced BC. Treatment with GSIs plus docetaxel reduced the frequency of BCSCs, downregulated CD44+/CD24- and ALDH+ biomarkers, and reduced BC volume. OMP-59R5, OMP-21M18, OMP-52M51, and REGN421 are monoclonal antibodies against Notch receptors or ligands, and their specific targets are Notch2/3, DLL-4, Notch1, and DLL-4, respectively [145,146] . The preclinical data with Notch inhibitor + anticancer drug demonstrate that pharmacological inhibition of the Notch pathway can reduce BCSCs in breast tumor graft models[147,148,149,150]. Li et al found that the erythropoietin-producing hepatocellular receptor A5 suppressed BCSC self-renewal via the Notch1 signaling pathway, lowering the probability of TR in HER2-positive BC[151].
3.1.3. JAK/STAT, NF-κB, Integrin and Hodge Signalling Pathways
Although there isn't any concrete evidence to suggest their involvement in the regulation of TR in HER2-positive BC, it's still plausible that altering these pathways through a variety of mediators could lessen TR by changing the phenotype of BCSCs. The mesenchymal CD44+/CD24/low phenotype in HER2-overexpressing BC was strongly related to TR and HER2 overexpression-activated STAT3 might cause CD44 expression to be upregulated. According to reports, the STAT3 inhibitor Stattic kills the CSC phenotype in HER2-positive breast tumors, suggesting that JAK/STAT signaling may be a potential target for trastuzumab-resistant BC[152]. Further, combined treatment of Herceptin and Stattic showed a synergistic effect on the cancer cell growth in vitro. When the STAT3 gene was knocked down, the expression of the stem cell markers (OCT-4, SOX-2, and CD44) was downregulated and tumor formation was abolished. It was concluded from the study that targeting STAT3 may overcome Trastuzumab-induced resistance in HER2-overexpressing breast tumours. Islam and colleagues have shown that, activation of the Hippo transcriptional ROR1(type 1 receptor tyrosine kinase-like orphan receptor) when BC patient tumour cells and BC cell lines were treated with T-DM1. This ROR1 expression could be induced by the coactivator YAP1. They showed that targeting ROR1 and YAP1 may suppress cancer stem cell (CSC) self-renewal efficacy and inhibit tumor progression in BC. Further, it was concluded that treatments that target ROR1 of hippo signaling may overcome the T-DM1 mediated therapeutic trastuzumab resistance and improve clinical outcomes. [153] The Figure 5 represents the proposed inhibitors and their related pathways to overcome Trastuzumab Resistance.
3.1.4. Cytokines and Immunomodulators in BCSC Signaling
Recently, it was discovered that IL-6 and IL-8 levels were considerably increased in BCSCs that were enriched during long-term trastuzumab treatment. Further, it was shown that IL-6 inhibition might eliminate TR. An important approach that could be utilized to combat TR is the inhibition of the IL-6 inflammatory feedback loop and the reduction of the BCSC population, particularly in the CD44+/CD24/low subgroups[154]. When combined with trastuzumab, SCH563705, an inhibitor of the chemokine (C-X-C motif) receptor 1/2 (CXCR1/2), has effectively eliminated BCSC-like cells. These results suggested that HER2-positive BCSCs can be targeted effectively by combining CXCR1/2 signaling with the inhibition of HER2 signalling[154]. PD-L1 is a crucial negative regulator that aids different cancer cell types in evading the immune response. PD-L1 was thought to be related to EMT in earlier investigations. Additionally, it was found that via stimulating the PI3K/AKT signaling pathway, PD-L1 had a significant impact on maintaining the stemness of BC cells. Recent findings indicate that utilizing immunotherapy against PD-L1 may be beneficial for treating HER2-overexpressing human BC cells cocultured with peripheral blood after trastuzumab treatment[155]. It was concluded that Trastuzumab-mediated upregulation of PD-L1 through engagement of immune effector cells may function as a possible mechanism of TR. It indicates that adding anti-PD-1 or anti-PD-L1 therapy to trastuzumab-based treatment may be a good alternative for overcoming TR against HER2+ BCs.
3.1.5. Role of Tumor Micro-Environment and Its Modifications
Strategies that target the BCSC tumour microenvironment (TME) may be effective since, as was already indicated, it plays a role in the emergence of treatment resistance. Collagen type I α1 (COL1A1) is the major component of type I collagen and aberrant expression of COL1A1 and COL1A2 is found in numerous cancers by controlling TME. COL1A1 knockdown decreases cell proliferation and invasion, which results in lower expression of stemness markers that block EMT and stem cell activity, such as sex-determining region Y-box2, octamer-binding transcription factor 4, and CD133[156]. The siRNA-mediated silencing of COL1A1 expression (siCOLIA1)abrogated Slug-dependent EMT and HCC stemness gene-signature, by reducing the expression of stemness markers SOX2, OCT4, and CD133. Although this study was done on HCC it was concluded that the same can be effective against BC. The therapeutic effects may also be hampered by anomalies in the vascular microenvironment. Erlotinib, a specific inhibitor of EGFR, was able to normalize the tumor vascular system, boost the chemotherapeutic effects of nanodrugs, and improve perfusion and oxygenation in a mouse model of BC[157]. Here, when the mouse is pre-treated with 50mg/kg of Erlotinib due to improved tumor vasculature the chemotherapeutic effect of anti-PDL1 antibody was much higher. Tranilast (the TGF-β inhibitor)when combined with Doxil nanomedicine, the TMEin mouse model of TNBC is improved with increased perfusion and oxygenation[158]. This contributes to enhanced anti-tumor immunity with anti-PD-1/anti-CTLA-4 antibodies. BCSCs are more prevalent in hypoxic conditions where an oxygen-deprived environment is crucial to their proliferation, stemness, and self-renewal AzCDF, a small molecule was reported by Kim et al[159] to target BCSCs in a hypoxic environment thereby inhibiting tumor growth and reducing tumorigenesis. By reducing the number of BCSCs and preventing cancer cell metastasis, the suppression of TGF-inducible protein expression decreased hypoxia and tumor angiogenesis[160].
3.1.6. Noncoding RNAs as Target
According to their length, noncoding RNAs can be categorized into siRNAs, piRNAs, microRNAs, and long noncoding RNAs (lncRNAs)[161]. Many miRNAs, lncRNAs, and their "sponges," including Let-7, miR-118, miR-200, miR-221, miR-22, Lnc H19, lncATB, are shown to be connected with EMT and BCSCs progression [162,163,164]. The miR-200c was found to be the most downregulated miRNA in trastuzumab-resistant cells with BCSC-like characteristics. Trastuzumab sensitivity was then restored after inhibiting the miR-200c by ZEB1 or the TGF-signaling transcriptional activator ZNF217. Trastuzumab sensitivity is further increased by miR-200c's ability to suppress ZEB1 or ZNF217 and hence impede TGF- signalling[165]. It was concluded that regulatory circuits of miR-200c/ZEB1 and miR-200c/ZNF217/TGF-β/ZEB1 are involved in synergistically promoting TR. In another study the lnc-RNA activated by TGF-β (lnc-ATB) was found to be the most upregulated lncRNA in trastuzumab-resistant SKBR-3 cells and the tissues of TR BC patients[166]. Their study showed that lnc-ATB could promote TR and invasion-metastasis cascade in BC by competitively biding miR-200c, inducing EMT, and then up-regulating ZEB1 and ZNF-217. Using miRNA microarray, Li et al., identified 13 differentially expressed miRNAs in the serum of HER2+ MBC patients which showed differential response to trastuzumab[167]. Their data showed that miR-940 is released from the tumor cells and other 3 miRNAs (miR-451a, miR-16-5p, and miR-17-3p)are released, mostly, from the immune cells. These 4 miRNAs are concluded as crucial players in regulatingTR[168]. Based on the aforementioned findings, it was concluded that miRNAs and lncRNAs might function independently or cooperatively to promote EMT, and BCSC phenotypes to control TR[169].
4. MUC1 is a Possible Target to Overcome TR
MUC1 is a member of the mucin family and plays oncogenic/mitogenic activities in cancer cells, as well as interacting with multiple other oncogenic receptors and pathways, including HER2, β-catenin, NF-kB, and the estrogen receptor (ER). Furthermore, it has been proven that the MUC1 C-terminal domain (MUC1-C) promotes the development of TR and that silencing the MUC1-C proto-oncogene is related to greater susceptibility of HER2+ cells to trastuzumab-induced inhibitors of growth.
A study found that all BC circulating cells express MUC1 and therefore MUC1 is a possible biomarker for TR. Additionally, MUC1 expression is significantly associated with a poor prognosis in patients with HER2+ and TNBC subtypes of BC. Thus, experts believe MUC1 is a TRmarker. BC patients' peripheral blood contains an increased quantity of soluble MUC1 (sMUC1). CA 15.3 (MUC1 serum marker) can indicate a poor prognosis in ER+ and/or PR+ BC subtypes. MUC1 expression is significantly higher in cancer tissue compared to normal tissue, making it a potential diagnostic for BC diagnosis. Indeed, measuring CA 15.3 can help predict trastuzumab response[170].
4.1. Strategies for Mucin1 Targeting to Overcome TR
Research findings have shown that MUC1 is an excellent antigen for immunotherapy. First, it is found on the surface of a breast tumor. Secondly, malignant MUC1 is hypoglycosylated, indicating that its core antigen is accessible[171] .Third, malignant MUC1 differs from normal MUC1, which is exclusively overexpressed in cancer cells. The fourth important information is that the MUC1 peptide sequence (PDTRP) is one of the most immunogenic MUC1 epitopes, which is recognized by the SM3mAb, which is an antibody against stripped MUC1 core protein.MUC1 is a good target for mAb, vaccines, and inhibitors because of this epitope[172]. The use of anti-MUC1 mAb or siRNA of MUC1 targeting the MUC1 gene would render BC cells vulnerable to trastuzumab-mediated ADCC[173]. The strategies for MUC1 targeting in BC therapy are discussed below. These include both immunotherapeutic and non-immunotherapeutic methods[170].
4.2. MUC1-Based Therapy Using Mab, Vaccines and CAR T Cells
The anti-MUC1 antibody AS1402- is evaluated on patients with metastatic BC in the Phase-I clinical trial. These individuals were previously treated, developed anthracycline or taxol resistance, and were able to tolerate this antibody[174]. Additionally, researchers have created an anti-MUC1 scFv (single chain Fragment variable), also known as nanobodies, that bind to BC cells that express MUC1 and prevent their invasion and survival[175]. It was observed that the cytoplasmic domain of MUC1 (MUC1-C) accelerates the development of resistance to trastuzumab and therefore silencing MUC1-C proto-oncogene is associated with increased sensitivity of HER2+ cells to trastuzumab treatment. In a mouse model of stage IV human BC, a flagella vaccination that targets MUC1 has been tried to decrease metastasis in these animals[176,177]. Additionally, the clinical trial of PANVAC, a MUC1 and carcinoembryonic antigen (CEA)vaccine, on patients with metastatic BC showed promising results[178]. Other MUC1 vaccines have undergone testing in patients with metastatic cancer, demonstrating their safety and ability to promote anti-tumor immunity[179]. In the clinical environment, L-BLP25, a peptide vaccine that targets MUC1 and CEA, is being tested on BC patients[180].After the trial core biopsies of patients were evaluated for MUC1 by immunohistochemistry (IHC; N = 691) and quantitative RT-PCR. High MUC1 protein and mRNA expression were correlated with a lower probability of pathologic complete response and with longer patient survival. Chimeric antigen receptor (CAR) T cells are engineered T cells that have a transmembrane domain, a signaling domain, and scFv from a particular tumor Ag-Antibody[181]. Targeting MUC1 in cancer has also been done using CAR T cell settings. CAR T cells are made using the anti-MUC1 Mab TAB004. Here the antibody TAB004 recognizes the aberrantly glycosylated tumor form of MUC1 (tMUC1). At first, human T cells are transduced with MUC28z, a CAR comprising of the scFv of TAB004 coupled to CD28 and CD3ζ. The MUC28z CAR T cells showed significant target-specific cytotoxicity against human TNBC cells which is evident from the increased production of Granzyme B, IFN-γ and other Th1 type cytokines and chemokines on the TNBC mouse model, Zhou et al. demonstrate that this tactic is effective in generating anti-tumor immunity, with a lethal effect on the TNBC cell line[181].Combination therapies like the COX1,2 inhibitor (indomethacin, celecoxib, etc)along with the MUC1 peptide vaccination dramatically reduce tumor size in BC mouse models[180]. In another work, anti-CTLA4 mAb -9D9 was combined with a DC vaccination fused with MUC1-mRNA to boost anti-tumor immunity in TNBC mouse models[182].In another study, it was shown that the hyperactivated STAT3 signal increases the expression of MUC1 and MUC4, which mediates via masking of trastuzumab binding to HER2 BC cells[183]. A combination of anti-MUC1 antibody (mAb-GP1.4) with an inhibitor of AKT and ERK'1/2 was evaluated in BC cells in one study. This therapy reduced MMP2 and 9 expressions in BC, which triggered G2/M cell cycle arrest and death. The combination of anti-MUC1 mAb -GP1.4- with a berenil complex of platinum (Pt12) causes apoptosis in MCF7 cells[170].
4.3. Other MUC1-Based Therapy
The inhibitors GO-201, 202, and 203 disrupt MUC1-C homodimerization and its associated function by targeting the CQC motif of MUC1-C[184].GO-203 is a cell-penetrating peptide that blocks MUC1-C homodimerization and thereby its oncogenic function. In one study, GO203 reduced HER2 activity[185] and prevented BC cell carcinogenesis[186]. In TNBC cells, GO203 lowers the amounts of BCL2A and MCL protein and mRNA[184,186]. GO-203 was also highly synergistic with doxorubicin in studies of both MCF-7 and ZR-75-1 BC cells. The oligomerization of MUC1 and its transmission to the mitochondria and nucleus is blocked by GO201 and GO202, which inhibit BC proliferation [175,187]. Additionally, GO201 prevents both ER-dependent and independent carcinogenesis and causes cell cycle arrest at the S phase. Additionally, it prevents NF-B activation and NF-B/MUC1 interaction in BC cells[188].In BC cells, siRNA-based silencing of MUC1 increases its sensitivity to oxidative stress[189]. It was proven that the MUC1-C is sufficient to induce FOXO3a activation and its phosphorylation to attenuate oxidative stress. These studies concluded that MUC1 controls the FOXO3a signaling pathway for a survival response to oxidative stress. Another study demonstrated that silencing MUC1-C reverses resistance to trastuzumab where synergistic activity of GO-203 and trastuzumab was observed[190]. Administration of GO-201 to nude mice bearing human breast tumor xenografts showed loss of tumorigenicity and extensive necrosis, which results in prolonged regression of tumor growth[187].
5. Overcoming Trastuzumab or HER2-Mediated Therapy via the PI3K/Akt/mTOR Pathway
Resistance to HER2-targeted therapy may emerge as a result of abnormal activation of signaling pathways downstream of the receptor[79,191,192]. Because HER2 mediates signal transduction via the PI3K/Akt/mTOR pathway[193] inhibiting components of this pathway may be a feasible strategy to overcome resistance and restore sensitivity to HER2-targeted therapy[191,194,195]. The abnormal regulation of PI3K/Akt signaling leads to upregulation of the downstream mTOR pathway, increased mRNA translation, and raised cellular proliferation[196,197], which is mediated by growth factor receptor amplification and loss of the phosphatase and tensin homolog (PTEN) tumor suppressor[198]. BC models with active PI3K/Akt/mTOR pathways demonstrate resistance to HER2-targeted therapies[199] and hyperactivity of the pathway related to BC involves gain-of-function mutations in genes encoding PIK3CA which encodes the catalytic subunit of PI3K, along with mutations in AKT1[200]. PTEN was discovered as the major gene out of 8000 evaluated genes whose suppression resulted in TR75. The study reveals that in a cohort of 55 BC patients, activation of the PI3K pathway, which is based on the presence of oncogenic PIK3CA mutations or low PTEN expression, was associated with poor prognosis after trastuzumab therapy. Trastuzumab in combination with everolimus, an mTOR inhibitor, inhibited the growth of trastuzumab-resistant cells[201]. mTOR is a serine/threonine kinase that controls the signaling pathway of growth factors and hormones, thereby controlling cell growth and angiogenesis. Further, it was observed that combination therapy with an mTOR inhibitor and trastuzumab was more effective in inhibiting tumor growth than monotherapy [202]. Other mTOR, PI3K, and dual PI3K/mTOR inhibitors, including NVP-BKM120, GDC-0941, NVP-BEZ235, andINK-128, have restored sensitivity to HER2-targeted therapy in cell culture-based and in vivo models of BC[203]. The phase 1b dose-escalation study of everolimus in combination with trastuzumab and paclitaxel was done in 33 patients with metastatic HER2+BC who are trastuzumab-resistant. The therapy reported an overall response rate (ORR) of 44%, a disease control rate of 74%, and a median PFS of 34 weeks which is much better than monotherapy of Trastuzumab[204]. The combination of HER2-targeted therapies and BAY 80-6946 (a PI3K inhibitor) inhibited BC growth more effectively than either of the monotherapies. The combination can restore sensitivity to trastuzumab and lapatinib in cells with acquired resistance to either trastuzumab or lapatinib[205].
6. Patents Related to BC Therapy and Associated Trastuzumab Resistanc
6.1. The patent filed by Jiangshu Cancer Prevention Research Institute, China has shown the application of NDUFA4L2 (NADH dehydrogenase ubiquinone 1 alpha subcomplex, 4-like 2)in HER2+ BC resistance. The resistance of HER-2 positive BC towards the trastuzumab can be reversed at the molecular level by NDUFA4L2 through the creation of a treatment-resistant stable cell strain of BC. Here a complete genome study on drug effect on Herceptinsensitivity or resistance on BC cells is performed where the NDUFA4L2 gene was the most highly affected among 453 studied genes. They have claimed that NDUFA 4L2 can play a role in reversing a Herceptin-resistance of a HER2+BC cell line BT474 where the level of NDUFA4L2 is highly overexpressed[206] [CN113308542]. Another application filed by the Jiangshu Cancer Hospital, China the role of UGT1A7 (UDP Glucuronosyltransferase Family 1 Member A7)for HER2-resistant BC. The invention shows that the expression of UGT1A7 is significantly lower in a herceptin-resistant BT474HR cell than in the susceptible parent cell line. UGT1A7 expression is controlled via mitochondria and endoplasmic reticulum (ER) stress after trastuzumab treatment[207] [CN115029435]. Another invention using
CRISPR/Cas9 library it was claimed that inhibition of FGFR4 (fibroblast growth factor receptor 4) enhances the sensitivity of BC in HER2+ therapy. The mechanistic studies show that RNA m6A hypomethylation in BC cells upregulates FGFR4, which phosphorylates GSK-3βto stimulate a beta-catenin/TCF4 signal to drive the drug resistance to HER2 therapy. Furthermore, they have shown synergistic action of anti-HER2 and anti-FGFR4 antibody in HER2+ BC therapy by using the patient derived xenograft and organoid susceptibility testing[208] [CN114736966].
6.2. In another invention (Seagen Inc, France) it was claimed that a mixture of tucatinib, capecitabine, and trastuzumab treatment lessens the consequences of HER2-positive BC. The patients experience at least 7.5 months of progression-free survival after the combination treatment[209] [WO2021080983]. The role of USP37(Ubiquitin specific protease 37), a deubiquitination enzyme, as a drug target in drug-resistant BC was claimed in another invention[210] [CN113640518]. The goal of this discovery is to ascertain whether the USP37 is involved in the progression of BC and whether targeting USP37 in conjunction with chemical-based (cisplatin) or radiation treatments will improve the efficacy of treatment for BC patients. A trinuclear platinum coordinating chemical derived from trimeprazine-based derivatives is disclosed in another invention[211][CN113336798]. Trimeprazine is an antihistamine that is commonly used for allergies. In this case, the trimeprazine-based trinuclear platinum coordination compounds exhibit IC50 of 3.6-18 µM against the BC cells. The invention claims the first trinuclear derivative of cisplatin for the application of BC.
6.3. In another patent the combination therapy of trastuzumab and cold atmospheric plasma (CAP) was claimed as an effective therapy for HER2 positive and negative BC patients with [212][US20200069958]. This is based on previous observations that CAP works very well for wound healing in clinical trials. The US patent WO2019098682 claims the discovery of a new antibody (a humanized Mab with scFv) against HER2 which is more effective against HER2+ BC than trastuzumab because the new antibody targets a new domain on HER2 that is different from trastuzumab [213][WO2019098682].
6.4. The patent of Xiangya Hospital, china claims a new biomarker UCH-L1 (Ubiquitin C-terminal hydrolase L1) which is under-expressed in HER2+ BCs. UCH-L1 is a deubiquitinase that controls the proteasomal degradation of proteins (more commonly for neuronal proteins) by removing Lys48-linked polyubiquitin chains. They claim that UCH-L1 can be used as a drug target alone or in combination with trastuzumab for HER2+ BC since it inhibits the expression of HER2 protein in BC[214][CN112316146]. In another invention, a nanoparticle composed of a peptide, nucleic acid, and doxorubicin (which are formed by non-covalent and anionic interactions) was used for the treatment of a patient who is not responding to trastuzumab. The techniques for treating cancer in patients are described which is targeting the HER3 receptors [215][EP3129066]. Here the peptide NP targets the HER3 receptor, where after endocytosis the doxorubicin is released to treat the BC.
6.5. The potential use of a hormone therapy using melatonin to treat HER2-positive BC is described in an invention. Melatonin is a hormone that is produced by the brain in darkness and controls our circadian rhythm. Studies demonstrate that melatonin can help treat HER2-positive BC by modifying the structural integrity of the HER2 protein. They also demonstrate that melatonin can improve the therapeutic impact of HER2 small-molecule targeted drugs(for example lapatinib and neratinib that is used in this study) on HER2-positive BC that is resistant to these drugs, and that melatonin could be used to stop HER2-positive BC metastasis and reoccurrenc[216][CN112870193]. A therapeutic approach for HER2+ and trastuzumab-resistant BC was claimed by studying the expression level of Mucin 4 protein in those patients. Here a combination therapy including a selective inhibitor of soluble TNF and and trastuzumab is used for those patients where the level of Mucin-4 is greater than a threshold level [217][US20200147175].

Table 1.
Patent applications with major claims to target trastuzumab-resistant HER2+ BC.
| Patent Application No and publication year | .Major claims | Molecular mechanism | Reference No. |
|---|---|---|---|
| CN113308542, 27.08.2021 | 1. A Trastuzumab-resistant cell BT474 HR is generated where protein NDUFA 4L2 is over-expressed significantly among 453 studied genes. 2. NDUFA 4L2 can be new target for studying TR |
In herceptin drug-resistant BT474 HR cells, the expression of a protein, NDUFA 4L2, is significantly increased, and is mainly located in mitochondria of cells. The protein NDUFA 4L2 is identified as a new drug target in HER2+ BC cells. | [206] |
| CN112870193, 01.06.2021 |
1. A new composition for treating HER2-positive BC, composed of melatonin and a Tyrosine kinase inhibitor (TKI) lapatinib or neratinib 2. Both melatonin and Melatonin + TKI reduces the expression of HER2 receptor in sensitive and resistant cells |
Melatonin synergizes the effect of small molecule TKIs in reducing the expression level of HER2 receptor in different HER2 positive and TR BC cells. | [216] |
| CN112316146, 05.02.2021 |
1. The expression of Ubiquitin carboxyl terminal hydrolase-L1 (UCH-L1) through a plasmid based expression in HER2 positive BC cells decreases the expression of HER2. 2. Co treatment of UCH-L1 plasmid + lapatinib kills the BC cells significantly by reducing the HER2 receptor expression |
The expression of UCH-L1, which is a deubiquitinase changes the ubiquitination level of HER2 protein and make them more degradation prone thereby reducing the HER2 protein level in UCH-L1 over-expressing BC cells. | [214] |
| CN115029435 09.09.2022 |
1. UGT1A7 expression can reverse the HER -2 positive BC cells TR 2. UGT1A7 expression is drastically down-regulated in the BT474 resistant cell. |
UGT1A7 expression is controlled via mitochondria and endoplasmic reticulum (ER) stress after trastuzumab treatment | [207] |
| CN114736966 12.07.2022 |
1. Using CRISPR/Cas9 library it was claimed that inhibition of FGFR4 (fibroblast growth factor receptor 4) enhances the sensitivity of BC in HER2+ therapy 2. Synergistic action of anti-HER2 and anti-FGFR4 antibody in HER2+ BC therapy was shown |
RNA m6A hypomethylation in BC cells upregulates FGFR4, which phosphorylates GSK-3β. It stimulate a beta-catenin/TCF4 signal to drive the drug resistance to HER2 therapy. | [208] |
| CN113640518 12.11.2021 | 1. USP37, a new protein, was found to be involved in the progression of BC. 2. Combination of USP37+ cisplatin or radiation treatments will improve the efficacy of treatment for BC patients |
The deubiquitinating enzyme USP37 kills BC more than cisplatin does because the cells become more degradation prone after USP37 expression | [210] |
7. Clinical Trials Targeting HER2+ BC and Associated TR
The phase III SOPHIA trial presented evidence for a novel Mab to HER2, margetuximab (a chimeric Mab of IgG origin), which may outperform trastuzumab's past efficacy. Margetuximabbinds to the same domain of HER2 as trastuzumab but their Fc region provides a better ADCC than currently approved HER2-targeted antibodies, earning the FDA's Fast Track designation for individuals with locally advanced or metastatic HER2+ BC who have previously received anti-HER2+ therapy. The trial found that margetuximab plus chemotherapy had a considerably better PFS than trastuzumab plus chemotherapy (5.8 vs. 4.9 months); a potentially higher overall response rate (ORR) of 22% vs. 16%, and a comparable safety profile. In the trial, 536 patients were recruited and randomly assigned to margetuximab (15 mg/kg intravenously once every 3 weeks; n = 266) plus chemotherapy or trastuzumab (6 mg/kg intravenously once every 3 weeks after an initial dose of 8 mg/kg; n = 270) plus chemotherapy. The trial involves 17 countries with 166 sites. It is worth noting that treatment effects were more significant in patients with CD16A (which was mostly expressed in NK cells) genotypes that contained a lower-affinity 158F allele (PFS 6.9 vs. 5.1 months; P = 0.005), which is present in about 85% of the general population and is known to reduce clinical response to trastuzumab[218]. However, this clinical trial shows that margetuximab + chemotherapy is not significantly cost-effective compared with trastuzumab + chemotherapy[219].
Murthy et al. in a phase II clinical trial reported the efficacy of combination therapy of tucatinib, trastuzumab, and capecitabine for HER2-Positive Metastatic BC (mBC), [220] [NCT02614794]. The primary endpoint was PFS among the first 480 patients who underwent randomization. They found that the tucatinib-combination group had a 1-year PFS of 33.1% compared to 12.3% in the placebo-combination group. Furthermore, the 2-year OS in the tucatinib-combination group was 44.9% vs. 26.6% in the placebo-combination group (P<0.005). Importantly, in a subpopulation of patients with brain metastases, the tucatinib-combination group had a 1-year PFS of 24.9% vs. 0% in the placebo-combination group (P< 0.001). The secondary endpoints were assessed with 612 patients which included OS, PFS among patients with brain metastases, confirmed ORR, and safety. The median duration of PFS was 7.6 months for the tucatinib combination group whereas for the placebo combination group PFS was 4.9 months (P < 0.0001). It was concluded that the tucatinib combination, with additional follow-up, provided a clinically meaningful survival benefit for patients with HER2+ mBC[221].
Table 2.
Important Clinical Trials with study outcomes for treatment of Trastzumab resistant BC and mBC.
Table 2.
Important Clinical Trials with study outcomes for treatment of Trastzumab resistant BC and mBC.
| Sl. No. |
Drugs and Phase Trial | Target | Study design and Outcomes |
ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| 1. | Margetuximab and Phase II | Margetuximab – a chimeric Mab in Phase 2 Study in Individuals with Relapsed or Refractory advanced Breast tumors | 25 participants. 1. Margentuximab is well tolerated. 2. Complete response disappearing of all target lesion within ≥ 4 weeks; whereas ≥ 30% reduction in the target lesions from the beginning, observed at ≥ 4 weeks |
NCT01828021 |
| 2. | Margetuximab and Phase III | In the Treatment of HER2+ mBC (SOPHIA), Margetuximab + Chemotherapy vs. Trastuzumab + Chemotherapy | 624 participants. 1. Better PFS than trastuzumab + chemotherapy (5.8 vs. 4.9 months), a potentially comparable ORR of 22% vs. 16%, and a comparable safety profile. 2. Treatment effects were more significant in patients with CD16A genotypes that contained a lower-affinity 158F allele (PFS 6.9 vs. 5.1 months), which is present in about 85% of the general population and is known to reduce clinical response to trastuzumab. |
NCT02492711 |
| 3. | Trastuzumab deruxtecan (DS-8201) and Phase II | For HER2+ BC patients with two groups – T-DM1 resistant and T-DM1 tolerant | 253 participants. 1. DS-8201 showed durable antitumor activity in a pretreated patient population with HER2-positive MBC. 2. In addition to nausea and myelosuppression, interstitial lung disease was observed in a subgroup of patients which requires attention to pulmonary symptoms. Interstitial lung disease was observed in 13.6% of the patients. |
NCT03248492 |
| 4. | Tucatinib and Phase II | Tucatinib vs. Placebo in the combination with Capecitabine and Trastuzumab in Advanced HER2+ BC Patients (HER2CLIMB) | 612 participants 1. Tucatinib in combination with trastuzumab and capecitabine improved OS (44.9% vs. 26.6% compared to placebo) while reducing the risk of developing new brain lesions. 2. The tucatinib-combination group had a 1-year PFS of 33.1%, compared to 12.3% in the placebo group. 3. A sample of patients with brain metastases showed a 1-year PFS of 24.9% in the tucatinib-combination group compared to 0% in the placebo-combination group. |
NCT02614794 |
| 5. | Tucatinib + T-DM1 and Phase III | Tucatinib vs. Placebo in Combination with Ado-Trastuzumab Emtansine (T-DM1) for Advanced or Metastatic HER2+ Breast carcinoma Patients | 565 participants. 1. Study was done to see if tucatinib with T-DM1 works better than T-DM1 alone. 2. The primary PFS benefit for pre-treated HER2+ mBC patients was 8.2 months, with an ORR of 47% at the highest tolerated dose, where 60% of patients had previously been treated with trastuzumab and had brain metastases. |
NCT03975647 |
| 6. | Pertuzumab and Phase III | Chemotherapy + trastuzumab + pertuzumab vs. chemotherapy + trastuzumab for HER2+ mBC. (CLEOPATRA) |
808 participants. 1. 8-year landmark ORR were 37% in the pertuzumab group and 23% in the other group. 2. The long-term safety and cardiac safety profiles of pertuzumab, trastuzumab, and docetaxel were maintained more compared to other group. |
NCT00567190 |
| 7. | Trastuzumab emtansine (T-DM1) and Phase III | trastuzumab emtansine (T-DM1) vs. capecitabine + lapatinib in participants with HER2+ mBC (EMILIA) |
991 Participants. 1. T-DM1 significantly increased both PFS (10 months vs 6 months, and OS (31 months vs 25 months). 2. T-DM1 improved overall survival in patients with previously treated HER2-positive mBC compared to chemotherapy group. |
NCT00829166 |
| 8. | Neratinib + Capecitabine and Phase III |
A Study of Neratinib+ Capecitabine vs. Lapatinib + Capecitabine in Patients With HER2+ mBC who Have Received HER2Directed treatment (NALA) |
621 participants. 1.After a median follow-up period of 30 months, the PFS rose by two months in the neratinib combination arm (8.8 months vs 6.6 months), but there was no statistical difference in OS. 2. In a subgroup study, patients with non-visceral or hormone-negative illness had superior outcomes. 3. Treatment with neratinib for patients with brain metastases resulted in no difference in PFS when compared to the control group. |
NCT01808573 |
Pyrotinib, an irreversible pan-HER kinase inhibitory substances, was the next new oral drug presented to patients with HER2+ mBC in another clinical trial. Between November 1, 2018, and March 31, 2021, 40 patients were recruited for this study. The study compared pyrotinib plus capecitabine (P+C) to lapatinib plus capecitabine (L+C) in strongly pre-treated patients in a phase II trial. The pyrotinib arm showed a considerably longer PFS (18 vs. 7 months; P<0.0001), an improved ORR (78.5% vs. 57.1%; P < 0.01), and a low toxicity profile at 15 months [222]. The study concludes that pyrotinib plus trastuzumab and chemotherapy offered a better option with a low safety profile for heavily pre-treated HER2-positive mBC, especially for those without liver or/ lung metastases. In 2001, Slamon et al. reported the first big phase III trial confirming the benefits of trastuzumab and chemotherapy in HER2+ mBC which was replicated in many other trials with minor variations. In their research, they noticed longer disease-free survival (DFS, 7.4 vs. 4.6 months; P < 0.001), enhanced ORR (50% vs. 32%; P < 0.001), and longer response duration (9.1 vs. 6.1 months; P < 0.001) for trastuzumab + chemotherapy (anthracycline, doxorubicin or paclitaxel). Further, it showed higher 1-year survival rates (78% vs. 67%; P = 0.008), longer OS (25.1 vs. 20.3 months; P = 0.046), and a 20% risk decrease in death[223]. Another phase III (ALTERNATIVE trial) clinical trial takes advantage of dual pathway inhibition (anti-HER and anti-estrogen) for the treatment of HER2+ mBC. Johnston et al. found that triple therapy (n=120) of lapatinib, trastuzumab, and letrozole was superior in PFS (11 vs. 5.6 months)to dual trastuzumab and letrozole therapy (n=117) in postmenopausal HER2+/HR+ mBC patients who are previously treated with endocrine and trastuzumab therapy[33]. Furthermore, the phase II PERTAIN trial indicated an acceptable safety profile and a considerable improvement in efficacy over dual therapy (PFS of 18.89 vs. 15.80 months; P < 0.007), this time in the first-line setting of metastatic HER2+/HR+ BC33. Here the triple therapy includes pertuzumab + trastuzumab + letrozole/ anastrozole (aromatase inhibitors). The study was done with 129 patients in 80 centers across 8 countries. The study concluded that Pertuzumab plus trastuzumab and an aromatase inhibitor is effective for the treatment of HER2-positive mBC. The FeDriCa clinical trial was a randomized, open-label, international, multicentre(19 countries and 106 sites), phase II study which included 252 patients. The study outlines the efficacy of A subcutaneous formulation of pertuzumab and trastuzumab with recombinant human hyaluronidase in one ready-to-use, fixed-dose combination vial. The vial (pertuzumab, trastuzumab, and hyaluronidase-zzxf) was approved by the US FDA on June 29, 2020, for HER2+ BC[224]. The trial shows that the safety of triple therapy was similar between treatment groups and in line with other pertuzumab, trastuzumab, and chemotherapy trials.
8. Conclusion and Future Directions
It is important to note that in only 20% of BC, the HER2 is overexpressed whereas the other forms of BC can be due to other factors like angiogenesis via VEGF, cytokine imbalance, chronic inflammation, and change in TME or oxidative stress, etc. The plasma and serum levels of VEGF an important biomarkers for BC as it was found that serum and plasma levels of VEGF are more in mBC than in healthy volunteers. [225] The C-reactive protein (CRP) which is considered to be a classic marker for chronic inflammation was identified in nipple aspirate fluid of healthy women and has been positively correlated to BC risk. [226] The crosstalk of inflammatory cytokines in the TME of BC has identified a few stimulatory cytokines (leptin, IL-1B, IL-6, IL-8, etc.) or inhibitory (IL-2, IL-12, and IFN-γ) for BC.[227] Higher levels of 8-hydroxy-2′-deoxyguanosine, a common biomarker for ROS-mediated DNA damage, is always found to be more in BCs than in normal cells.[228] This indicates that higher ROS can cause BC. Although all these factors which are related to HER2-negative BC, it will be interesting to see the fate of these factors when HER2+ BCs are treated with trastuzumab and/or chemotherapy.
Based on the current market sales the top3 Mabs are Humira (adalimumab which targets TNF-α), Keytruda (pembrolizumab which binds PD-1), and Stelara (Ustekinumab which targets IL-12 and IL-23). Among these Humira and its biosimilars are used to treat various inflammatory diseases including juvenile idiopathic arthritis, Crohn’s disease (CD), plaque psoriasis, rheumatoid arthritis (RA), psoriatic arthritis (PsA), etc. This possibly explains its demand and current market sales [65]. Among the top 20 Mabs sales, trastuzumab does not come in the picture and the Mab, Pertuzumab, which acts against HER2+ BC like trastuzumab is in 11th position with market sales of USD4.4B till 2022[207]. But pertuzumab binds to domain II of HER2 whereas trastuzumab binds to domain IV. Further, Pertuzumab efficiently inhibits ligand-induced HER2/HER3 dimerization, whereas trastuzumab's major action is through ADCC. The decrease in sales indicates that resistance against trastuzumab is a serious issue and it is thereby replaced by other antibodies or in combination therapy where trastuzumab is always combined with other Mab, chemotherapy, and/or tyrosine kinase inhibitors. From the information on current patents, the ubiquitin proteosome system was identified as a possible target for HER2+ BC treatment. However, to date no studies were carried out using proteosome inhibitors in combination with trastuzumab for overcoming HER2+ BC in a clinical set up.
Two Mabs that act as immune checkpoint inhibitors (Pembrolizumab and Atezolizumab) are combined with Trastuzumab or T-DM1 (ADC of trastuzumab) in clinical trials to treat the TNBC or HER2+ BC where normal trastuzumab therapy fails[229]. The success of these studies indicates that trastuzumab alone is not recommended for any HER2+ BC therapy because patients generate resistance through different pathways (eg., AKT/PI3K/mTOR, NF-κB, notch signaling, etc.) within 1 year of treatment. To date, most of the combination therapy includes TKIs, among which lapatinib, neratinib, and pyrotinib are extensively used, because of several advantages, including oral administration, reduced cardiotoxicity, and favorable penetration of blood-brain barrier in BC. Apart from combination therapy ADCs like Trastuzumab emtansine (T-DM1) and trastuzumab deruxtecan (T-DXd) are being used for resistance against HER2+ BC but the success is not better than trastuzumab alone [230]. Likely, TR in HER2+ BC overcoming possibly requires a modified Fc-based Trastuzumab with improved ADCC or a better chemotherapeutic agent apart from TKis which can be used in combination with trastuzumab[231].
Author Contributions
Conceptualization -GH, SS, PG and DM. Methodology and collection of information- GH, SS, PG, SMP and DM. Writing and original draft preparation -- GH, SS, PG, SMP, SK and DM. Writing, review and editing – GH, SS, and DM. Supervision- GH, SS, and DM. All authors have read and agreed to the version of manuscript for publication.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors. However, the Post-doctoral fellowship of SMP and Master fellowship of GH, SS and PG are funded by the ministry of chemicals and fertilizers, Department of Pharmaceuticals, Government of India.
Ethics Approval and Consent to Participate
Not Applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Data sharing is not applicable to this article as no datasets were generated oranalysed during the current study.
Acknowledgement
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Abbreviations
| BC | Breast Cancer |
| BCSC | BC stem cell |
| TR | Trastuzumab Resistance |
| ADC | Antibody drug conjugate |
| ADCC | Antibody directed cellular cytotoxicity |
| HER2 | human epidermal growth factor receptor 2 |
| EGFR | Epidermal growth factor receptor |
| ER | Estrogen receptor |
| PR | Progesterone receptor |
| PD-L1 | Programmed death Ligand 1 |
| PD-1 | programmed death 1 |
| PFS | progression free survival |
| OS | overall survival |
| ORR | overall response rate |
| MUC1 | Mucin 1 |
| PI3K | Phosphatidyl Inositol 3 Kinase |
| mTOR | Mammalian target of rapamycin |
| TME | Tumour Micro Environment |
| TKI | Tyrosine kinase inhibitor |
| TNBC | Triple negative BC |
| IGF-1R | Insulin-like Growth Factor 1 receptor |
| MET | Mesenchymal epithelial transition |
| CAR | Chimeric Antigenic Receptor |
| CEA | Carcinoembryonic Antigen |
| STAT3 | Signal transducer and activator of transcription 3 |
| TGF-β | Transforming growth factor β |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians 2021, 71, 209–249. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Gaudet, M.M.; Newman, L.A.; Miller, K.D.; Goding Sauer, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2019. CA: a cancer journal for clinicians 2019, 69, 438–451. [Google Scholar] [CrossRef]
- AL, S. The neu oncogen: an erb-B-releted gene encoding a 185,000-Mr tumour antigen. Nature 1984, 290, 261–264. [Google Scholar]
- Coussens, L.; Yang-Feng, T.L.; Liao, Y.-C.; Chen, E.; Gray, A.; McGrath, J.; Seeburg, P.H.; Libermann, T.A.; Schlessinger, J.; Francke, U. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985, 230, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Drebin, J.A.; Link, V.C.; Weinberg, R.A.; Greene, M.I. Inhibition of tumor growth by a monoclonal antibody reactive with an oncogene-encoded tumor antigen. Proceedings of the National Academy of Sciences 1986, 83, 9129–9133. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. science 1987, 235, 177–182. [Google Scholar] [CrossRef]
- Hudziak, R.M.; Lewis, G.D.; Winget, M.; Fendly, B.M.; Shepard, H.M.; Ullrich, A. p185 HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Molecular and cellular biology 1989, 9, 1165–1172. [Google Scholar]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.; Henner, D.; Wong, W.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proceedings of the National Academy of Sciences 1992, 89, 4285–4289. [Google Scholar] [CrossRef]
- Eiermann, W.; Group, I.H.S. Trastuzumab combined with chemotherapy for the treatment of HER2-positive metastatic breast cancer: pivotal trial data. Annals of oncology 2001, 12, S57–S62. [Google Scholar] [CrossRef]
- Marty, M.; Cognetti, F.; Maraninchi, D.; Snyder, R.; Mauriac, L.; Tubiana-Hulin, M.; Chan, S.; Grimes, D.; Antón, A.; Lluch, A. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2–positive metastatic breast cancer administered as first-line treatment: the M77001 study group. Journal of clinical oncology 2005, 23, 4265–4274. [Google Scholar] [CrossRef]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annual review of biochemistry 2015, 84, 739–764. [Google Scholar] [CrossRef]
- Harris, L.N.; You, F.; Schnitt, S.J.; Witkiewicz, A.; Lu, X.; Sgroi, D.; Ryan, P.D.; Come, S.E.; Burstein, H.J.; Lesnikoski, B.-A. Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clinical Cancer Research 2007, 13, 1198–1207. [Google Scholar] [CrossRef]
- Wolff, A.C.; Somerfield, M.R.; Dowsett, M.; Hammond, M.E.H.; Hayes, D.F.; McShane, L.M.; Saphner, T.J.; Spears, P.A.; Allison, K.H. Human epidermal growth factor receptor 2 testing in breast cancer: ASCO–College of American Pathologists Guideline Update. Journal of Clinical Oncology 2023, 41, 3867–3872. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.; Claret, F.X. Trastuzumab: updated mechanisms of action and resistance in breast cancer. Frontiers in oncology 2012, 2, 62. [Google Scholar] [CrossRef]
- Scott, G.K.; Robles, R.; Park, J.W.; Montgomery, P.A.; Daniel, J.; Holmes, W.E.; Lee, J.; Keller, G.A.; Li, W.-L.; Fendly, B.M. A truncated intracellular HER2/neu receptor produced by alternative RNA processing affects growth of human carcinoma cells. Molecular and cellular biology 1993, 13, 2247–2257. [Google Scholar]
- Christianson, T.A.; Doherty, J.K.; Lin, Y.J.; Ramsey, E.E.; Holmes, R.; Keenan, E.J.; Clinton, G.M. NH2-terminally truncated HER-2/neu protein: relationship with shedding of the extracellular domain and with prognostic factors in breast cancer. Cancer research 1998, 58, 5123–5129. [Google Scholar] [PubMed]
- Scaltriti, M.; Rojo, F.; Ocaña, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. Journal of the National Cancer Institute 2007, 99, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Friedländer, E.; Tanner, M.; Kapanen, A.I.; Carraway, K.L.; Isola, J.; Jovin, T.M. Decreased accessibility and lack of activation of ErbB2 in JIMT-1, a herceptin-resistant, MUC4-expressing breast cancer cell line. Cancer research 2005, 65, 473–482. [Google Scholar] [CrossRef]
- Mercogliano, M.F.; De Martino, M.; Venturutti, L.; Rivas, M.A.; Proietti, C.J.; Inurrigarro, G.; Frahm, I.; Allemand, D.H.; Deza, E.G.; Ares, S. TNFα-induced mucin 4 expression elicits trastuzumab resistance in HER2-positive breast cancer. Clinical Cancer Research 2017, 23, 636–648. [Google Scholar] [CrossRef]
- Wong, A.L.; Lee, S.-C. Mechanisms of resistance to trastuzumab and novel therapeutic strategies in HER2-positive breast cancer. International journal of breast cancer 2012, 2012. [Google Scholar] [CrossRef]
- Fessler, S.P.; Wotkowicz, M.T.; Mahanta, S.K.; Bamdad, C. MUC1* is a determinant of trastuzumab (Herceptin) resistance in breast cancer cells. Breast cancer research and treatment 2009, 118, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Li, L.; Qiu, Y.; Sun, W.; Ren, T.; Lv, Y.; Liu, M.; Wang, X.; Tao, H.; Zhao, L.; et al. A novel humanized MUC1 antibody-drug conjugate for the treatment of trastuzumab-resistant breast cancer. Acta Biochim Biophys Sin (Shanghai) 2021, 53, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandhan, S.; Knutson, K.L. Resistance to trastuzumab. Cancers 2022, 14, 5115. [Google Scholar] [CrossRef]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and breast cancer. Cancer research 2011, 71, 1515–1519. [Google Scholar] [CrossRef]
- Sperinde, J.; Jin, X.; Banerjee, J.; Penuel, E.; Saha, A.; Diedrich, G.; Huang, W.; Leitzel, K.; Weidler, J.; Ali, S.M. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clinical Cancer Research 2010, 16, 4226–4235. [Google Scholar] [CrossRef]
- Price-Schiavi, S.A.; Jepson, S.; Li, P.; Arango, M.; Rudland, P.S.; Yee, L.; Carraway, K.L. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. International journal of cancer 2002, 99, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Carraway, K.L.; Price-Schiavi, S.A.; Komatsu, M.; Jepson, S.; Perez, A.; Carraway, C.A.C. Muc4/sialomucin complex in the mammary gland and breast cancer. Journal of mammary gland biology and neoplasia 2001, 6, 323–337. [Google Scholar] [CrossRef]
- Schlam, I.; Tarantino, P.; Tolaney, S.M. Overcoming resistance to HER2-directed therapies in breast cancer. Cancers 2022, 14, 3996. [Google Scholar] [CrossRef]
- Konecny, G.; Pauletti, G.; Pegram, M.; Untch, M.; Dandekar, S.; Aguilar, Z.; Wilson, C.; Rong, H.-M.; Bauerfeind, I.; Felber, M. Quantitative association between HER-2/neu and steroid hormone receptors in hormone receptor-positive primary breast cancer. Journal of the National Cancer Institute 2003, 95, 142–153. [Google Scholar] [CrossRef]
- Giuliano, M.; Trivedi, M.V.; Schiff, R. Bidirectional crosstalk between the estrogen receptor and human epidermal growth factor receptor 2 signaling pathways in breast cancer: molecular basis and clinical implications. Breast Care 2013, 8, 256–262. [Google Scholar] [CrossRef]
- Munzone, E.; Curigliano, G.; Rocca, A.; Bonizzi, G.; Renne, G.; Goldhirsch, A.; Nolè, F. Reverting estrogen-receptor-negative phenotype in HER-2-overexpressing advanced breast cancer patients exposed to trastuzumab plus chemotherapy. Breast Cancer Res 2006, 8, R4. [Google Scholar] [CrossRef] [PubMed]
- Pegram, M.; Jackisch, C.; Johnston, S.R.D. Estrogen/HER2 receptor crosstalk in breast cancer: combination therapies to improve outcomes for patients with hormone receptor-positive/HER2-positive breast cancer. NPJ Breast Cancer 2023, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.R.D.; Hegg, R.; Im, S.A.; Park, I.H.; Burdaeva, O.; Kurteva, G.; Press, M.F.; Tjulandin, S.; Iwata, H.; Simon, S.D.; et al. Phase III, Randomized Study of Dual Human Epidermal Growth Factor Receptor 2 (HER2) Blockade With Lapatinib Plus Trastuzumab in Combination With an Aromatase Inhibitor in Postmenopausal Women With HER2-Positive, Hormone Receptor-Positive Metastatic Breast Cancer: ALTERNATIVE. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2018, 36, 741–748. [Google Scholar] [CrossRef]
- Llombart-Cussac, A.; Cortés, J.; Paré, L.; Galván, P.; Bermejo, B.; Martínez, N.; Vidal, M.; Pernas, S.; López, R.; Muñoz, M. HER2-enriched subtype as a predictor of pathological complete response following trastuzumab and lapatinib without chemotherapy in early-stage HER2-positive breast cancer (PAMELA): an open-label, single-group, multicentre, phase 2 trial. The lancet oncology 2017, 18, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Rimawi, M.; Ferrero, J.M.; De La Haba-Rodriguez, J.; Poole, C.; De Placido, S.; Osborne, C.K.; Hegg, R.; Easton, V.; Wohlfarth, C.; Arpino, G. First-line trastuzumab plus an aromatase inhibitor, with or without pertuzumab, in human epidermal growth factor receptor 2-positive and hormone receptor-positive metastatic or locally advanced breast cancer (PERTAIN): A randomized, open-label phase II trial. Journal of Clinical Oncology 2018, 36, 2826–2835. [Google Scholar] [CrossRef]
- Ropero, S.; Menéndez, J.A.; Vázquez-Martín, A.; Montero, S.; Cortés-Funes, H.; Colomer, R. Trastuzumab plus tamoxifen: anti-proliferative and molecular interactions in breast carcinoma. Breast Cancer Res Treat 2004, 86, 125–137. [Google Scholar] [CrossRef]
- Wu, D.; Tang, S.; Ye, R.; Li, D.; Gu, D.; Chen, R.; Zhang, H.; Sun, J.; Chen, Z. Case Report: Long-Term Response to Pembrolizumab Combined With Endocrine Therapy in Metastatic Breast Cancer Patients With Hormone Receptor Expression. Front Immunol 2021, 12, 610149. [Google Scholar] [CrossRef]
- Fiszman, G.L.; Jasnis, M.A. Molecular mechanisms of trastuzumab resistance in HER2 overexpressing breast cancer. International journal of breast cancer 2011, 2011. [Google Scholar] [CrossRef]
- Jones, A. Combining trastuzumab (Herceptin®) with hormonal therapy in breast cancer: what can be expected and why? Annals of oncology 2003, 14, 1697–1704. [Google Scholar] [CrossRef]
- Johnston, S.R.; Dowsett, M. Aromatase inhibitors for breast cancer: lessons from the laboratory. Nature Reviews Cancer 2003, 3, 821–831. [Google Scholar] [CrossRef]
- Ocaña, A.; Cruz, J.J.; Pandiella, A. Trastuzumab and antiestrogen therapy: focus on mechanisms of action and resistance. American journal of clinical oncology 2006, 29, 90–95. [Google Scholar] [CrossRef]
- Proietti, C.J.; Rosemblit, C.; Beguelin, W.; Rivas, M.A.; Díaz Flaqué, M.a.C.; Charreau, E.H.; Schillaci, R.; Elizalde, P.V. Activation of Stat3 by heregulin/ErbB-2 through the co-option of progesterone receptor signaling drives breast cancer growth. Molecular and cellular biology 2009, 29, 1249–1265. [Google Scholar] [CrossRef]
- Chervo, M.F.; Cordo Russo, R.I.; Petrillo, E.; Izzo, F.; De Martino, M.; Bellora, N.; Cenciarini, M.E.; Chiauzzi, V.A.; Santa María de la Parra, L.; Pereyra, M.G.; et al. Canonical ErbB-2 isoform and ErbB-2 variant c located in the nucleus drive triple negative breast cancer growth. Oncogene 2020, 39, 6245–6262. [Google Scholar] [CrossRef]
- Venturutti, L.; Romero, L.V.; Urtreger, A.J.; Chervo, M.F.; Cordo Russo, R.I.; Mercogliano, M.F.; Inurrigarro, G.; Pereyra, M.G.; Proietti, C.J.; Izzo, F.; et al. Stat3 regulates ErbB-2 expression and co-opts ErbB-2 nuclear function to induce miR-21 expression, PDCD4 downregulation and breast cancer metastasis. Oncogene 2016, 35, 2208–2222. [Google Scholar] [CrossRef]
- Gutierrez, M.C.; Detre, S.; Johnston, S.; Mohsin, S.K.; Shou, J.; Allred, D.C.; Schiff, R.; Osborne, C.K.; Dowsett, M. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. Journal of clinical oncology 2005, 23, 2469–2476. [Google Scholar] [CrossRef]
- Meng, S.; Tripathy, D.; Shete, S.; Ashfaq, R.; Haley, B.; Perkins, S.; Beitsch, P.; Khan, A.; Euhus, D.; Osborne, C. HER-2 gene amplification can be acquired as breast cancer progresses. Proceedings of the National Academy of Sciences 2004, 101, 9393–9398. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Staka, C.; Boyns, F.; Hutcheson, I.R.; Gee, J.M.W. Growth factor-driven mechanisms associated with resistance to estrogen deprivation in breast cancer: new opportunities for therapy. Endocrine-related cancer 2004, 11, 623–641. [Google Scholar] [CrossRef]
- Jordan, N.J.; Gee, J.M.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Increased constitutive activity of PKB/Akt in tamoxifen resistant breast cancer MCF-7 cells. Breast cancer research and treatment 2004, 87, 167–180. [Google Scholar] [CrossRef]
- Knowlden, J.M.; Hutcheson, I.R.; Jones, H.E.; Madden, T.; Gee, J.M.; Harper, M.E.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 2003, 144, 1032–1044. [Google Scholar] [CrossRef]
- Martin, M.B.; Reiter, R.; Pham, T.; Avellanet, Y.R.; Camara, J.; Lahm, M.; Pentecost, E.; Pratap, K.; Gilmore, B.A.; Divekar, S. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology 2003, 144, 2425–2436. [Google Scholar] [CrossRef]
- Jeng, M.-H.; Yue, W.; Eischeid, A.; Wang, J.-P.; Santen, R.J. Role of MAP kinase in the enhanced cell proliferation of long term estrogen deprived human breast cancer cells. Breast cancer research and treatment 2000, 62, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Brodie, A.; Sabnis, G.; Macedo, L. Xenograft models for aromatase inhibitor studies. The Journal of steroid biochemistry and molecular biology 2007, 106, 119–124. [Google Scholar] [CrossRef]
- Massarweh, S.; Osborne, C.K.; Jiang, S.; Wakeling, A.E.; Rimawi, M.; Mohsin, S.K.; Hilsenbeck, S.; Schiff, R. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor–positive, HER-2/neu-positive breast cancer. Cancer research 2006, 66, 8266–8273. [Google Scholar] [CrossRef]
- Arpino, G.; Wiechmann, L.; Osborne, C.K.; Schiff, R. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: molecular mechanism and clinical implications for endocrine therapy resistance. Endocrine reviews 2008, 29, 217–233. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nature medicine 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Gennari, R.; Menard, S.; Fagnoni, F.; Ponchio, L.; Scelsi, M.; Tagliabue, E.; Castiglioni, F.; Villani, L.; Magalotti, C.; Gibelli, N. Pilot study of the mechanism of action of preoperative trastuzumab in patients with primary operable breast tumors overexpressing HER2. Clinical cancer research 2004, 10, 5650–5655. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ receptors as regulators of immune responses. Nature Reviews Immunology 2008, 8, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, J.L.; Gorlatov, S.; Zhang, W.; Yang, Y.; Huang, L.; Burke, S.; Li, H.; Ciccarone, V.; Zhang, T.; Stavenhagen, J. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Research 2011, 13, 1–14. [Google Scholar] [CrossRef]
- Musolino, A.; Gradishar, W.J.; Rugo, H.S.; Nordstrom, J.L.; Rock, E.P.; Arnaldez, F.; Pegram, M.D. Role of Fcγ receptors in HER2-targeted breast cancer therapy. Journal for immunotherapy of cancer 2022, 10. [Google Scholar] [CrossRef]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. New England Journal of Medicine 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Petricevic, B.; Laengle, J.; Singer, J.; Sachet, M.; Fazekas, J.; Steger, G.; Bartsch, R.; Jensen-Jarolim, E.; Bergmann, M. Trastuzumab mediates antibody-dependent cell-mediated cytotoxicity and phagocytosis to the same extent in both adjuvant and metastatic HER2/neu breast cancer patients. J Transl Med 2013, 11, 307. [Google Scholar] [CrossRef]
- Yin, J.; Albers, A.J.; Smith, T.S.; Riddell, G.T.; Richards, J.O. Differential regulation of human monocytes and NK cells by antibody-opsonized tumors. Cancer Immunol Immunother 2018, 67, 1239–1250. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. The Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.; Hui, R. VP7-2021: KEYNOTE-522: Phase III study of neoadjuvant pembrolizumab+ chemotherapy vs. placebo+ chemotherapy, followed by adjuvant pembrolizumab vs. placebo for early-stage TNBC. Annals of Oncology 2021, 32, 1198–1200. [Google Scholar] [CrossRef]
- Rajana, V.K.; Penumaka, S.M.; Saritha, C.; Ravichandiran, V.; Mandal, D. Yuflyma, A High Concentration and Citrate-free Adalimumab Biosimilar, Received FDA Approval for Treating Different Forms of Inflammato ry Diseases. Antiinflamm Antiallergy Agents Med Chem 2023, 22, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.F.; Duret, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti–ErbB-2 mAb therapy requires type I and II interferons and synergizes with anti–PD-1 or anti-CD137 mAb therapy. Proceedings of the National Academy of Sciences 2011, 108, 7142–7147. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): a single-arm, multicentre, phase 1b–2 trial. The Lancet Oncology 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Emens, L.A.; Esteva, F.J.; Beresford, M.; Saura, C.; De Laurentiis, M.; Kim, S.-B.; Im, S.-A.; Wang, Y.; Salgado, R.; Mani, A. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. The lancet oncology 2020, 21, 1283–1295. [Google Scholar] [CrossRef]
- Huober, J.; Barrios, C.; Niikura, N.; Jarzab, M.; Chang, Y.; Huggins-Puhalla, S.; Graupner, V.; Eiger, D.; Henschel, V.; Gochitashvili, N. VP6-2021: IMpassion050: a phase III study of neoadjuvant atezolizumab+ pertuzumab+ trastuzumab+ chemotherapy (neoadj A+ PH+ CT) in high-risk, HER2-positive early breast cancer (EBC). Annals of Oncology 2021, 32, 1061–1062. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.; Winer, E.; Loirat, D.; Awada, A.; Cescon, D.; Iwata, H.; Campone, M.; Nanda, R. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: cohort A of the phase II KEYNOTE-086 study. Annals of Oncology 2019, 30, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R. Event-free survival with pembrolizumab in early triple-negative breast cancer. New England Journal of Medicine 2022, 386, 556–567. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R. Abstract GS1-01: KEYNOTE-522 study of neoadjuvant pembrolizumab+ chemotherapy vs placebo+ chemotherapy, followed by adjuvant pembrolizumab vs placebo for early-stage TNBC: Event-free survival sensitivity and subgroup analyses. Cancer Research 2022, 82, GS1–01. [Google Scholar] [CrossRef]
- Luen, S.J.; Salgado, R.; Fox, S.; Savas, P.; Eng-Wong, J.; Clark, E.; Kiermaier, A.; Swain, S.M.; Baselga, J.; Michiels, S. Tumour-infiltrating lymphocytes in advanced HER2-positive breast cancer treated with pertuzumab or placebo in addition to trastuzumab and docetaxel: a retrospective analysis of the CLEOPATRA study. The Lancet Oncology 2017, 18, 52–62. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin oncol 2015, 33, 983–991. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Human mutation 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Sansal, I.; Sellers, W.R. The biology and clinical relevance of the PTEN tumor suppressor pathway. Journal of clinical oncology 2004, 22, 2954–2963. [Google Scholar] [CrossRef]
- Nahta, R.; Yu, D.; Hung, M.-C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nature clinical practice Oncology 2006, 3, 269–280. [Google Scholar] [CrossRef]
- Pandolfi, P.P. Breast cancer--loss of PTEN predicts resistance to treatment. N Engl J Med 2004, 351, 2337–2338. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer cell 2007, 12, 395–402. [Google Scholar] [CrossRef]
- Nagata, Y.; Lan, K.-H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer cell 2004, 6, 117–127. [Google Scholar] [CrossRef]
- Saal, L.H.; Holm, K.; Maurer, M.; Memeo, L.; Su, T.; Wang, X.; Yu, J.S.; Malmström, P.-O.; Mansukhani, M.; Enoksson, J. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer research 2005, 65, 2554–2559. [Google Scholar] [CrossRef]
- Stemke-Hale, K.; Gonzalez-Angulo, A.M.; Lluch, A.; Neve, R.M.; Kuo, W.-L.; Davies, M.; Carey, M.; Hu, Z.; Guan, Y.; Sahin, A. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer research 2008, 68, 6084–6091. [Google Scholar] [CrossRef]
- Esteva, F.J.; Guo, H.; Zhang, S.; Santa-Maria, C.; Stone, S.; Lanchbury, J.S.; Sahin, A.A.; Hortobagyi, G.N.; Yu, D. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. The American journal of pathology 2010, 177, 1647–1656. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; King, T.; Sakr, R.; Barbashina, V.; Arroyo, N.; Morrogh, M.; Wynveen, C.; Shanu, M.; Norton, L.; Rosen, N. Hyperactivation of the PI3K-AKT Pathway Commonly Underlies Resistance to Trastuzumab in HER2 Amplified Breast Cancer. Cancer Research 2009, 69, 709–709. [Google Scholar] [CrossRef]
- Sakr, R.A.; Barbashina, V.; Morrogh, M.; Chandarlapaty, S.; Andrade, V.P.; Arroyo, C.D.; Olvera, N.; King, T.A. Protocol for PTEN expression by immunohistochemistry in formalin-fixed paraffin-embedded human breast carcinoma. Applied immunohistochemistry & molecular morphology: AIMM/official publication of the Society for Applied Immunohistochemistry 2010, 18, 371. [Google Scholar]
- Kalinsky, K.; Jacks, L.M.; Heguy, A.; Patil, S.; Drobnjak, M.; Bhanot, U.K.; Hedvat, C.V.; Traina, T.A.; Solit, D.; Gerald, W. PIK3CA mutation associates with improved outcome in breast cancer. Clinical cancer research 2009, 15, 5049–5059. [Google Scholar] [CrossRef]
- Jimenez, C.; Jones, D.R.; Rodríguez-Viciana, P.; Gonzalez-García, A.; Leonardo, E.; Wennström, S.; von Kobbe, C.; Toran, J.L.; Luis, R.; Calvo, V. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. The EMBO journal 1998. [Google Scholar] [CrossRef]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3′-kinase p85α gene is an oncogene in human ovarian and colon tumors. Cancer research 2001, 61, 7426–7429. [Google Scholar]
- McCubrey, J.A.; Abrams, S.L.; Fitzgerald, T.L.; Cocco, L.; Martelli, A.M.; Montalto, G.; Cervello, M.; Scalisi, A.; Candido, S.; Libra, M. Roles of signaling pathways in drug resistance, cancer initiating cells and cancer progression and metastasis. Advances in biological regulation 2015, 57, 75–101. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Philipp-Staheli, J.; Payne, S.R.; Kemp, C.J. p27Kip1: regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Experimental cell research 2001, 264, 148–168. [Google Scholar] [CrossRef]
- Yang, H.-Y.; Zhou, B.P.; Hung, M.-C.; Lee, M.-H. Oncogenic signals of HER-2/neu in regulating the stability of the cyclin-dependent kinase inhibitor p27. Journal of Biological Chemistry 2000, 275, 24735–24739. [Google Scholar] [CrossRef]
- Scaltriti, M.; Eichhorn, P.J.; Cortés, J.; Prudkin, L.; Aura, C.; Jiménez, J.; Chandarlapaty, S.; Serra, V.; Prat, A.; Ibrahim, Y.H. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proceedings of the National Academy of Sciences 2011, 108, 3761–3766. [Google Scholar] [CrossRef]
- Le, X.-F.; Claret, F.-X.; Lammayot, A.; Tian, L.; Deshpande, D.; LaPushin, R.; Tari, A.M.; Bast, R.C. The role of cyclin-dependent kinase inhibitor p27Kip1 in anti-HER2 antibody-induced G1 cell cycle arrest and tumor growth inhibition. Journal of Biological Chemistry 2003, 278, 23441–23450. [Google Scholar] [CrossRef]
- Marches, R.; Uhr, J.W. Enhancement of the p27Kip1-mediated antiproliferative effect of trastuzumab (Herceptin) on HER2-overexpressing tumor cells. International journal of cancer 2004, 112, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zi, X.; Zhao, Y.; Mascarenhas, D.; Pollak, M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). Journal of the National Cancer Institute 2001, 93, 1852–1857. [Google Scholar] [CrossRef]
- Jerome, L.; Alami, N.; Belanger, S.; Page, V.; Yu, Q.; Paterson, J.; Shiry, L.; Pegram, M.; Leyland-Jones, B. Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2–overexpressing breast tumors and potentiates Herceptin activity in vivo. Cancer research 2006, 66, 7245–7252. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fan, Z.; Edgerton, S.M.; Yang, X.; Lind, S.E.; Thor, A.D. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle 2011, 10, 2959–2966. [Google Scholar] [CrossRef]
- Nahta, R.; Yuan, L.X.; Zhang, B.; Kobayashi, R.; Esteva, F.J. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res 2005, 65, 11118–11128. [Google Scholar] [CrossRef] [PubMed]
- Shattuck, D.L.; Miller, J.K.; Carraway III, K.L.; Sweeney, C. Met receptor contributes to trastuzumab resistance of Her2-overexpressing breast cancer cells. Cancer research 2008, 68, 1471–1477. [Google Scholar] [CrossRef]
- Liu, L.; Greger, J.; Shi, H.; Liu, Y.; Greshock, J.; Annan, R.; Halsey, W.; Sathe, G.M.; Martin, A.-M.; Gilmer, T.M. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer research 2009, 69, 6871–6878. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.-L.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef]
- Yen, L.; You, X.-L.; Al Moustafa, A.-E.; Batist, G.; Hynes, N.E.; Mader, S.; Meloche, S.; Alaoui-Jamali, M.A. Heregulin selectively upregulates vascular endothelial growth factor secretion in cancer cells and stimulates angiogenesis. Oncogene 2000, 19, 3460–3469. [Google Scholar] [CrossRef]
- Kang, J.Y.; Dolled-Filhart, M.; Ocal, I.T.; Singh, B.; Lin, C.-Y.; Dickson, R.B.; Rimm, D.L.; Camp, R.L. Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast cancer. Cancer research 2003, 63, 1101–1105. [Google Scholar]
- Yamashita, J.-i.; Ogawa, M.; Yamashita, S.-i.; Nomura, K.; Kuramoto, M.; Saishoji, T.; Shin, S. Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer research 1994, 54, 1630–1633. [Google Scholar]
- Lindemann, K.; Resau, J.; Nährig, J.; Kort, E.; Leeser, B.; Annecke, K.; Welk, A.; Schäfer, J.; Vande Woude, G.; Lengyel, E. Differential expression of c-Met, its ligand HGF/SF and HER2/neu in DCIS and adjacent normal breast tissue. Histopathology 2007, 51, 54–62. [Google Scholar] [CrossRef]
- Agarwal, S.; Zerillo, C.; Kolmakova, J.; Christensen, J.; Harris, L.; Rimm, D.; Digiovanna, M.; Stern, D. Association of constitutively activated hepatocyte growth factor receptor (Met) with resistance to a dual EGFR/Her2 inhibitor in non-small-cell lung cancer cells. British journal of cancer 2009, 100, 941–949. [Google Scholar] [CrossRef]
- Liu, X.; Yao, W.; Newton, R.C.; Scherle, P.A. Targeting the c-MET signaling pathway for cancer therapy. Expert opinion on investigational drugs 2008, 17, 997–1011. [Google Scholar] [CrossRef]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Shah, P.D.; Huang, A.C.; Xu, X.; Orlowski, R.; Amaravadi, R.K.; Schuchter, L.M.; Zhang, P.; Tchou, J.; Matlawski, T.; Cervini, A.; et al. Phase I Trial of Autologous RNA-electroporated cMET-directed CAR T Cells Administered Intravenously in Patients with Melanoma and Breast Carcinoma. Cancer Res Commun 2023, 3, 821–829. [Google Scholar] [CrossRef]
- Jelkmann, W. Erythropoietin: structure, control of production, and function. Physiological reviews 1992, 72, 449–489. [Google Scholar] [CrossRef]
- Damen, J.E.; Liu, L.; Cutler, R.L.; Krystal, G. Erythropoietin stimulates the tyrosine phosphorylation of Shc and its association with Grb2 and a 145-Kd tyrosine phosphorylated protein. 1993. [Google Scholar]
- Liang, K.; Esteva, F.J.; Albarracin, C.; Stemke-Hale, K.; Lu, Y.; Bianchini, G.; Yang, C.-Y.; Li, Y.; Li, X.; Chen, C.-T. Recombinant human erythropoietin antagonizes trastuzumab treatment of breast cancer cells via Jak2-mediated Src activation and PTEN inactivation. Cancer cell 2010, 18, 423–435. [Google Scholar] [CrossRef]
- Puglisi, F.; Minisini, A.M.; De Angelis, C.; Arpino, G. Overcoming treatment resistance in HER2-positive breast cancer: potential strategies. Drugs 2012, 72, 1175–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Duan, X.; Xu, L.; Ye, J.; Zhao, J.; Liu, Y. Erythropoietin receptor expression and its relationship with trastuzumab response and resistance in HER2-positive breast cancer cells. Breast Cancer Res Treat 2012, 136, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Hirsch, D.S.; Shen, Y.; Wu, W.J. Rac1 contributes to trastuzumab resistance of breast cancer cells: Rac1 as a potential therapeutic target for the treatment of trastuzumab-resistant breast cancer. Molecular cancer therapeutics 2009, 8, 1557–1569. [Google Scholar] [CrossRef]
- Schnelzer, A.; Prechtel, D.; Knaus, U.; Dehne, K.; Gerhard, M.; Graeff, H.; Harbeck, N.; Schmitt, M.; Lengyel, E. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000, 19, 3013–3020. [Google Scholar] [CrossRef]
- Cardama, G.; Comin, M.; Alonso, D.; Gómez, D. Rho GTPases as therapeutic targets in cancer and other human diseases. Medicina 2010, 70, 555–564. [Google Scholar]
- Humphries, B.; Wang, Z.; Yang, C. Rho GTPases: Big Players in Breast Cancer Initiation, Metastasis and Therapeutic Responses. Cells 2020, 9. [Google Scholar] [CrossRef]
- Rivas, M.A.; Tkach, M.; Beguelin, W.; Proietti, C.J.; Rosemblit, C.; Charreau, E.H.; Elizalde, P.V.; Schillaci, R. Transactivation of ErbB-2 induced by tumor necrosis factor α promotes NF-κB activation and breast cancer cell proliferation. Breast cancer research and treatment 2010, 122, 111–124. [Google Scholar] [CrossRef]
- Li, Y.; Kong, X.; Xuan, L.; Wang, Z.; Huang, Y.H. Prolactin and endocrine therapy resistance in breast cancer: The next potential hope for breast cancer treatment. Journal of cellular and molecular medicine 2021, 25, 10327–10348. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, M.F.; Bruni, S.; Elizalde, P.V.; Schillaci, R. Tumor Necrosis Factor α Blockade: An Opportunity to Tackle Breast Cancer. Front Oncol 2020, 10, 584. [Google Scholar] [CrossRef] [PubMed]
- Bruni, S.; Mauro, F.L.; Proietti, C.J.; Cordo-Russo, R.I.; Rivas, M.A.; Inurrigarro, G.; Dupont, A.; Rocha, D.; Fernández, E.A.; Deza, E.G.; et al. Blocking soluble TNFα sensitizes HER2-positive breast cancer to trastuzumab through MUC4 downregulation and subverts immunosuppression. J Immunother Cancer 2023, 11. [Google Scholar] [CrossRef]
- Velasco-Velázquez, M.A.; Popov, V.M.; Lisanti, M.P.; Pestell, R.G. The role of breast cancer stem cells in metastasis and therapeutic implications. The American journal of pathology 2011, 179, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Bozorgi, A.; Khazaei, M.; Khazaei, M.R. New findings on breast cancer stem cells: a review. Journal of breast cancer 2015, 18, 303. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh, S.; Yazdanparast, R. Activation of STAT3/HIF-1α/Hes-1 axis promotes trastuzumab resistance in HER2-overexpressing breast cancer cells via down-regulation of PTEN. Biochimica et biophysica acta. General subjects 2017, 1861, 1970–1980. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer cell 2009, 15, 429–440. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cho, Y.; Oh, E.; Lee, N.; An, H.; Sung, D.; Cho, T.M.; Seo, J.H. Disulfiram targets cancer stem-like properties and the HER2/Akt signaling pathway in HER2-positive breast cancer; Elsevier.
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Menard, S.; Tagliabue, E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. In AACRA Magnifico, L Albano, S Campaner, D Delia, F Castiglioni, P Gasparini, G SozziClinical Cancer Research, 2009•AACR; 1078. [Google Scholar] [CrossRef]
- Paik, S.; Kim, C.; Wolmark, N. HER2 Status and Benefit from Adjuvant Trastuzumab in Breast Cancer. New England Journal of Medicine 2008, 358, 1409–1411. [Google Scholar] [CrossRef]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion; nature.comH Korkaya, A Paulson, F Iovino, MS WichaOncogene, 2008•nature.com.
- Chakrabarty, A.; Bhola, N.E.; Sutton, C.; Ghosh, R.; Kuba, M.G.; Dave, B.; Chang, J.C.; Arteaga, C.L. Trastuzumab-resistant cells rely on a HER2-PI3K-FoxO-survivin axis and are sensitive to PI3K inhibitors; AACRA Chakrabarty, NE Bhola, C Sutton, R Ghosh, MG Kuba, B Dave, JC Chang, CL ArteagaCancer research, 2013•AACR.
- Yu, F.; Zhao, J.; Hu, Y.; Zhou, Y.; Guo, R.; Bai, J.; Zhang, S.; Zhang, H.; Zhang, J. The combination of NVP-BKM120 with trastuzumab or RAD001 synergistically inhibits the growth of breast cancer stem cells in vivo; spandidos-publications.comF Yu, J Zhao, Y Hu, Y Zhou, R Guo, J Bai, S Zhang, H Zhang, J ZhangOncology reports, 2016•spandidos-publications.com.
- Kim, Y.J.; Sung, D.; Oh, E.; Cho, Y.; Cho, T.M.; Farrand, L.; Seo, J.H.; Kim, J.Y. Flubendazole overcomes trastuzumab resistance by targeting cancer stem-like properties and HER2 signaling in HER2-positive breast cancer; Elsevier.
- Korkaya, H.; Kim, G.-I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; Angelo, R.D.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. cell.comH Korkaya, G Kim, A Davis, F Malik, NL Henry, S Ithimakin, AA Quraishi, N TawakkolMolecular cell, 2012•cell.com 2012, 47, 570–584. [Google Scholar] [CrossRef]
- Du, L.; Yamamoto, S.; Burnette, B.L.; …, D.H.C.; undefined. Transcriptome profiling reveals novel gene expression signatures and regulating transcription factors of TGFβ-induced epithelial-to-mesenchymal transition. Wiley Online LibraryL Du, S Yamamoto, BL Burnette, D Huang, K Gao, N Jamshidi, MD KuoCancer medicine, 2016•Wiley Online Library 2016, 5, 1962–1972. [Google Scholar] [CrossRef]
- Chihara, Y.; Shimoda, M.; Hori, A.; Ohara, A.; Naoi, Y.; Ikeda, J.i.; Kagara, N.; Tanei, T.; Shimomura, A.; Shimazu, K.; et al. A small-molecule inhibitor of SMAD3 attenuates resistance to anti-HER2 drugs in HER2-positive breast cancer cells. Breast Cancer Research and Treatment 2017, 166, 55–68. [Google Scholar] [CrossRef]
- Wu, Y.; Tran, T.; Dwabe, S.; Sarkissyan, M.; Kim, J.; Nava, M.; Clayton, S.; Pietras, R.; Farias-Eisner, R. A83-01 inhibits TGF-β-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells; SpringerY Wu, T Tran, S Dwabe, M Sarkissyan, J Kim, M Nava, S Clayton, R Pietras, R Farias-EisnerBreast cancer research and treatment, 2017•Springer.
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Barco, S.D.; Martin-Castillo, B.; Menendez, J.A. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. SpringerA Vazquez-Martin, C Oliveras-Ferraros, SD Barco, B Martin-Castillo, JA MenendezBreast cancer research and treatment, 2011•Springer 2010, 126. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, H.E.; Li, H.; Shipitsin, M.; et al. Heterogeneity for stem cell–related markers according to tumor subtype and histologic stage in breast cancer; AACR.
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype; nature.comGB Jang, JY Kim, SD Cho, KS Park, JY Jung, HY Lee, IS Hong, JS NamScientific reports, 2015•nature.com.
- Loh, Y.N.; Hedditch, E.L.; Baker, L.A.; Jary, E.; Ward, R.L.; Ford, C.E. The Wnt signalling pathway is upregulated in an in vitro model of acquired tamoxifen resistant breast cancer. BMC Cancer 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/β-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells; AACRY Wu, C Ginther, J Kim, N Mosher, S Chung, D Slamon, JV VadgamaMolecular Cancer Research, 2012•AACR.
- Wu, Y.; Tran, T.; Dwabe, S.; Sarkissyan, M.; Kim, J.; Nava, M.; Clayton, S.; Pietras, R.; Farias-Eisner, R.; Vadgama, J.V. A83-01 inhibits TGF-β-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells. Breast Cancer Research and Treatment 2017, 163, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clinical Cancer Research 2013, 19, 1512–1524. [Google Scholar] [CrossRef]
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting Notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacology and Therapeutics 2014, 141, 140–149. [Google Scholar] [CrossRef]
- Yen, W.C.; Fischer, M.M.; Axelrod, F.; Bond, C.; Cain, J.; Cancilla, B.; Henner, W.R.; Meisner, R.; Sato, A.; Shah, J.; et al. Targeting notch signaling with a Notch2/Notch3 antagonist (Tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clinical Cancer Research 2015, 21, 2084–2095. [Google Scholar] [CrossRef]
- Chai, H.; Pan, C.; Zhang, M.; Huo, H.; Shan, H.; Wu, J. Histone methyltransferase SETD1A interacts with notch and promotes notch transactivation to augment ovarian cancer development. BMC Cancer 2023, 23. [Google Scholar] [CrossRef]
- McKeage, M.J.; Kotasek, D.; Markman, B.; Hidalgo, M.; Millward, M.J.; Jameson, M.B.; Harris, D.L.; Stagg, R.J.; Kapoun, A.M.; Xu, L.; et al. Phase IB Trial of the Anti-Cancer Stem Cell DLL4-Binding Agent Demcizumab with Pemetrexed and Carboplatin as First-Line Treatment of Metastatic Non-Squamous NSCLC. Targeted Oncology 2018, 13, 89–98. [Google Scholar] [CrossRef]
- Silkenstedt, E.; Arenas, F.; Colom-Sanmartí, B.; Xargay-Torrent, S.; Higashi, M.; Giró, A.; Rodriguez, V.; Fuentes, P.; Aulitzky, W.E.; Van Der Kuip, H.; et al. Notch1 signaling in NOTCH1-mutated mantle cell lymphoma depends on Delta-Like ligand 4 and is a potential target for specific antibody therapy. Journal of experimental & clinical cancer research : CR 2019, 38. [Google Scholar] [CrossRef]
- Li, Y.; Chu, J.; Feng, W.; Yang, M.; Zhang, Y.; Zhang, Y.; Qin, Y.; Xu, J.; Li, J.; Vasilatos, S.N.; et al. EPHA5 mediates trastuzumab resistance in HER2-positive breast cancers through regulating cancer stem cell-like properties. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 2019, 33, 4851–4865. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. International Journal of Oncology 2014, 44, 403–411. [Google Scholar] [CrossRef]
- Islam, S.S.; Uddin, M.; Noman, A.S.M.; Akter, H.; Dity, N.J.; Basiruzzman, M.; Uddin, F.; Ahsan, J.; Annoor, S.; Alaiya, A.A.; et al. Antibody-drug conjugate T-DM1 treatment for HER2+ breast cancer induces ROR1 and confers resistance through activation of Hippo transcriptional coactivator YAP1. EBioMedicine 2019, 43, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Almozyan, S.; Colak, D.; Mansour, F.; Alaiya, A.; Al-Harazi, O.; Qattan, A.; Al-Mohanna, F.; Al-Alwan, M.; Ghebeh, H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. International Journal of Cancer 2017, 141, 1402–1412. [Google Scholar] [CrossRef]
- Chaganty, B.K.R.; Qiu, S.; Gest, A.; Lu, Y.; Ivan, C.; Calin, G.A.; Weiner, L.M.; Fan, Z. Trastuzumab upregulates PD-L1 as a potential mechanism of trastuzumab resistance through engagement of immune effector cells and stimulation of IFNγ secretion. Cancer Letters 2018, 430, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.P.; Chang, H.L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.Y.; Wu, A.T.H.; Yeh, C.T.; Tsai, S.H.; Lee, W.H. Collagen 1A1 (COL1A1) is a reliable biomarker and putative therapeutic target for hepatocellular carcinogenesis and metastasis. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Chen, J.; Yang, Z.; Liang, C.; Yang, Y.; Liu, Z. Tumor vasculature normalization by orally fed erlotinib to modulate the tumor microenvironment for enhanced cancer nanomedicine and immunotherapy. Biomaterials 2017, 148, 69–80. [Google Scholar] [CrossRef]
- Panagi, M.; Voutouri, C.; Mpekris, F.; Papageorgis, P.; Martin, M.R.; Martin, J.D.; Demetriou, P.; Pierides, C.; Polydorou, C.; Stylianou, A.; et al. TGF-β inhibition combined with cytotoxic nanomedicine normalizes triple negative breast cancer microenvironment towards anti-tumor immunity. Theranostics 2020, 10, 1910–1922. [Google Scholar] [CrossRef]
- Kim, J.H.; Verwilst, P.; Won, M.; Lee, J.; Sessler, J.L.; Han, J.; Kim, J.S. A Small Molecule Strategy for Targeting Cancer Stem Cells in Hypoxic Microenvironments and Preventing Tumorigenesis. Journal of the American Chemical Society 2021, 143, 14115–14124. [Google Scholar] [CrossRef]
- Fico, F.; Santamaria-Martínez, A. TGFBI modulates tumour hypoxia and promotes breast cancer metastasis. Molecular Oncology 2020, 14, 3198–3210. [Google Scholar] [CrossRef]
- Kondo, Y.; Shinjo, K.; Katsushima, K. Long non-coding RNAs as an epigenetic regulator in human cancers. Cancer Science 2017, 108, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, Y.; Tsuyada, A.; Ren, X.; Wu, X.; Stubblefield, K.; Rankin-Gee, E.K.; Wang, S.E. Transforming growth factor-Β regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and ATM. Oncogene 2011, 30, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Zare, M.; Bastami, M.; Solali, S.; Alivand, M.R. Aberrant miRNA promoter methylation and EMT-involving miRNAs in breast cancer metastasis: Diagnosis and therapeutic implications. Journal of Cellular Physiology 2018, 233, 3729–3744. [Google Scholar] [CrossRef] [PubMed]
- Tordonato, C.; Di Fiore, P.P.; Nicassio, F. The role of non-coding RNAs in the regulation of stem cells and progenitors in the normal mammary gland and in breast tumors. Frontiers in Genetics 2015, 5. [Google Scholar] [CrossRef]
- Bai, W.D.; Ye, X.M.; Zhang, M.Y.; Zhu, H.Y.; Xi, W.J.; Huang, X.; Zhao, J.; Gu, B.; Zheng, G.X.; Yang, A.G.; et al. MiR-200c suppresses TGF-β signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. International Journal of Cancer 2014, 135, 1356–1368. [Google Scholar] [CrossRef]
- Shi, S.J.; Wang, L.J.; Yu, B.; Li, Y.H.; Jin, Y.; Bai, X.Z. LncRNA-ATB promotes trastuzumab resistance and invasionmetastasis cascade in breast cancer. Oncotarget 2015, 6, 11652–11663. [Google Scholar] [CrossRef]
- Li, H.; Liu, J.; Chen, J.; Wang, H.; Yang, L.; Chen, F.; Fan, S.; Wang, J.; Shao, B.; Yin, D.; et al. A serum microRNA signature predicts trastuzumab benefit in HER2-positive metastatic breast cancer patients. Nature Communications 2018, 9. [Google Scholar] [CrossRef]
- Li, W.; Zhai, L.; Wang, H.; Liu, C.; Zhang, J.; Chen, W.; Wei, Q. Downregulation of LncRNA GAS5 causes trastuzumab resistance in breast cancer. Oncotarget 2016, 7, 27778–27786. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, L.; Liu, H.; Luo, X. Cancer stem cell-targeted therapeutic approaches for overcoming trastuzumabresistance in HER2-positive breast cancer. Stem Cells 2021, 39, 1125–1136. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Merikhian, P.; Naseri, N.; Eisavand, M.R.; Farahmand, L. MUC1 is a potential target to overcome trastuzumab resistance in breast cancer therapy. Cancer cell international 2022, 22. [Google Scholar] [CrossRef]
- Curry, J.M.; Besmer, D.M.; Erick, T.K.; Steuerwald, N.; Roy, L.D.; Grover, P.; Rao, S.; Nath, S.; Ferrier, J.W.; Reid, R.W.; et al. Indomethacin enhances anti-tumor efficacy of a MUC1 peptide vaccine against breast cancer in MUC1 transgenic mice. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, I.A.; Navo, C.D.; Castro-López, J.; Guerreiro, A.; Jiménez-Moreno, E.; Sánchez Fernández, E.M.; García-Martín, F.; Hinou, H.; Nishimura, S.I.; García Fernández, J.M.; et al. Synthesis, conformational analysis and in vivo assays of an anti-cancer vaccine that features an unnatural antigen based on an sp2-iminosugar fragment. Chemical science 2020, 11, 3996–4006. [Google Scholar] [CrossRef] [PubMed]
- Namba, M.; Hattori, N.; Hamada, H.; Yamaguchi, K.; Okamoto, Y.; Nakashima, T.; Masuda, T.; Sakamoto, S.; Horimasu, Y.; Miyamoto, S.; et al. Anti-KL-6/MUC1 monoclonal antibody reverses resistance to trastuzumab-mediated antibody-dependent cell-mediated cytotoxicity by capping MUC1. Cancer letters 2019, 442, 31–39. [Google Scholar] [CrossRef]
- Pegram, M.D.; Borges, V.F.; Ibrahim, N.; Fuloria, J.; Shapiro, C.; Perez, S.; Wang, K.; Schaedli Stark, F.; Courtenay Luck, N. Phase I dose escalation pharmacokinetic assessment of intravenous humanized anti-MUC1 antibody AS1402 in patients with advanced breast cancer. Breast cancer research : BCR 2009, 11. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Choi, J.R.; Tae, N.; Wi, T.M.; Kim, K.M.; Kim, D.H.; Lee, E.S. Novel Antibodies Targeting MUC1-C Showed Anti-Metastasis and Growth-Inhibitory Effects on Human Breast Cancer Cells. International journal of molecular sciences 2020, 21. [Google Scholar] [CrossRef]
- Nathalie, M.; Ruth, A. Therapeutic MUC1-based cancer vaccine expressed in flagella-efficacy in an aggressive model of breast cancer. World Journal of Vaccines 2012, 2012. [Google Scholar]
- Machluf, N.; Arnon, R.; Machluf, N.; Arnon, R. Therapeutic MUC1-Based Cancer Vaccine Expressed in Flagella-Efficacy in an Aggressive Model of Breast Cancer. World Journal of Vaccines 2012, 2, 109–120. [Google Scholar] [CrossRef]
- Mohebtash, M.; Tsang, K.Y.; Madan, R.A.; Huen, N.Y.; Poole, D.J.; Jochems, C.; Jones, J.; Ferrara, T.; Heery, C.R.; Arlen, P.M.; et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clinical Cancer Research 2011, 17, 7164–7173. [Google Scholar] [CrossRef]
- Gulley, J.L.; Arlen, P.M.; Tsang, K.Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2008, 14, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Sinn, B.V.; Von Minckwitz, G.; Denkert, C.; Eidtmann, H.; Darb-Esfahani, S.; Tesch, H.; Kronenwett, R.; Hoffmann, G.; Belau, A.; Thommsen, C.; et al. Evaluation of Mucin-1 protein and mRNA expression as prognostic and predictive markers after neoadjuvant chemotherapy for breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology 2013, 24, 2316–2324. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdanifar, M.; Roy, L.D.; Whilding, L.M.; Gavrill, A.; Maher, J.; Mukherjee, P. CAR T Cells Targeting the Tumor MUC1 Glycoprotein Reduce Triple-Negative Breast Cancer Growth. Frontiers in immunology 2019, 10. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Miao, L.; Liu, Q.; Musetti, S.; Li, J.; Huang, L. Combination Immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade Effectively Inhibits Growth of Triple Negative Breast Cancer. Molecular therapy : the journal of the American Society of Gene Therapy 2018, 26, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhao, L.; Li, W.; Fan, K.; Qian, W.; Hou, S.; Wang, H.; Dai, J.; Wei, H.; Guo, Y. Feedback activation of STAT3 mediates trastuzumab resistance via upregulation of MUC1 and MUC4 expression. Oncotarget 2014, 5, 8317–8329. [Google Scholar] [CrossRef]
- Hiraki, M.; Maeda, T.; Mehrotra, N.; Jin, C.; Alam, M.; Bouillez, A.; Hata, T.; Tagde, A.; Keating, A.; Kharbanda, S.; et al. Targeting MUC1-C suppresses BCL2A1 in triple-negative breast cancer. 2018. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Rajabi, H.; Ahmad, R.; Jin, C.; Kufe, D. Targeting the MUC1-C oncoprotein inhibits self-renewal capacity of breast cancer cells. Oncotarget 2014, 5, 2622–2622. [Google Scholar] [CrossRef]
- Hiraki, M.; Suzuki, Y.; Alam, M.; Hinohara, K.; Hasegawa, M.; Jin, C.; Kharbanda, S.; Kufe, D. MUC1-C Stabilizes MCL-1 in the Oxidative Stress Response of Triple-Negative Breast Cancer Cells to BCL-2 Inhibitors. Scientific Reports 2016 6:1 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Raina, D.; Ahmad, R.; Joshi, M.D.; Yin, L.; Wu, Z.; Kawano, T.; Vasir, B.; Avigan, D.; Kharbanda, S.; Kufe, D. Direct targeting of the mucin 1 oncoprotein blocks survival and tumorigenicity of human breast carcinoma cells. Cancer research 2009, 69, 5133–5141. [Google Scholar] [CrossRef]
- Ahmad, R.; Raina, D.; Joshi, M.D.; Kawano, T.; Ren, J.; Kharbanda, S.; Kufe, D. MUC1-C ONCOPROTEIN FUNCTIONS AS A DIRECT ACTIVATOR OF THE NF-κB p65 TRANSCRIPTION FACTOR. Cancer research 2009, 69, 7013–7013. [Google Scholar] [CrossRef]
- Yin, L.; Huang, L.; Kufe, D. MUC1 oncoprotein activates the FOXO3a transcription factor in a survival response to oxidative stress. The Journal of biological chemistry 2004, 279, 45721–45727. [Google Scholar] [CrossRef]
- Raina, D.; Uchida, Y.; Kharbanda, A.; Rajabi, H.; Panchamoorthy, G.; Jin, C.; Kharbanda, S.; Scaltriti, M.; Baselga, J.; Kufe, D. Targeting the MUC1-C oncoprotein downregulates HER2 activation and abrogates trastuzumab resistance in breast cancer cells. Oncogene 2014, 33, 3422–3431. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Hu, Y.; O'Brien, N.; Finn, R.S. Current approaches and future directions in the treatment of HER2-positive breast cancer. Cancer treatment reviews 2013, 39, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, P.J.A.; Gili, M.; Scaltriti, M.; Serra, V.; Guzman, M.; Nijkamp, W.; Beijersbergen, R.L.; Valero, V.; Seoane, J.; Bernards, R.; et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer research 2008, 68, 9221–9230. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nature reviews. Molecular cell biology 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Nahta, R.; O'Regan, R. Evolving strategies for overcoming resistance to HER2-directed therapy: targeting the PI3K/Akt/mTOR pathway. Clinical breast cancer 2010, 10 Suppl 3. [Google Scholar] [CrossRef]
- Gnant, M. The role of mammalian target of rapamycin (mTOR) inhibition in the treatment of advanced breast cancer. Current oncology reports 2013, 15, 14–23. [Google Scholar] [CrossRef]
- Margariti, N.; Fox, S.B.; Bottini, A.; Generali, D. "Overcoming breast cancer drug resistance with mTOR inhibitors". Could it be a myth or a real possibility in the short-term future? Breast cancer research and treatment 2011, 128, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Kay, A.; Berg, W.J.; Lebwohl, D. Targeting tumorigenesis: development and use of mTOR inhibitors in cancer therapy. Journal of hematology & oncology 2009, 2. [Google Scholar] [CrossRef]
- De Martino, M.C.; Van Koetsveld, P.M.; Pivonello, R.; Hofland, L.J. Role of the mTOR pathway in normal and tumoral adrenal cells. Neuroendocrinology 2010, 92 Suppl 1, 28–34. [Google Scholar] [CrossRef]
- Keck, S.; Glencer, A.C.; Rugo, H.S. Everolimus and its role in hormone-resistant and trastuzumab-resistant metastatic breast cancer. Future oncology (London, England) 2012, 8, 1383–1396. [Google Scholar] [CrossRef]
- Mayer, I. Role of mTOR Inhibition in Preventing Resistance and Restoring Sensitivity to Hormone-Targeted and HER2-Targeted Therapies in Breast Cancer. Clinical advances in hematology & oncology : H&O 2013, 11, 217–217. [Google Scholar]
- Lu, C.-H.; Wyszomierski, S.L.; Tseng, L.-M.; Sun, M.-H.; Lan, K.-H.; Neal, C.L.; Mills, G.B.; Hortobagyi, G.N.; Esteva, F.J.; Yu, D. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clinical Cancer Research 2007, 13, 5883–5888. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Forbes, J.T.; Shah, C.; Wyatt, S.K.; Manning, H.C.; Olivares, M.G.; Sanchez, V.; Dugger, T.C.; de Matos Granja, N.; Narasanna, A. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clinical Cancer Research 2009, 15, 7266–7276. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.-M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Molecular cancer therapeutics 2012, 11, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Campone, M.; O'Regan, R.; Manlius, C.; Massacesi, C.; Sahmoud, T.; Mukhopadhyay, P.; Soria, J.-C.; Naughton, M.; Hurvitz, S.A. Phase I study of everolimus plus weekly paclitaxel and trastuzumab in patients with metastatic breast cancer pretreated with trastuzumab. J Clin Oncol 2009. [Google Scholar] [CrossRef]
- Elster, N.; Cremona, M.; Morgan, C.; Toomey, S.; Carr, A.; O’Grady, A.; Hennessy, B.T.; Eustace, A.J. A preclinical evaluation of the PI3K alpha/delta dominant inhibitor BAY 80-6946 in HER2-positive breast cancer models with acquired resistance to the HER2-targeted therapies trastuzumab and lapatinib. Breast cancer research and treatment 2015, 149, 373–383. [Google Scholar] [CrossRef]
- Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=CN370180537&_cid=P22-LTQ4AC-17139-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=CN375176274&_cid=P22-LTQ49B-16765-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- COMBINED PREPARATION FOR REVERSING DRUG RESISTANCE OF BREAST CANCER AND APPLICATION OF MARKER. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=CN370180537&_cid=P22-LTQ4AC-17139-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- METHODS OF TREATING HER2 POSITIVE BREAST CANCER WITH TUCATINIB IN COMBINATION WITH CAPECITABINE AND TRASTUZUMAB. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021080983&_cid=P22-LTQ4BJ-17652-1#detailMainForm:MyTabViewId:PCTBIBLIO.
- APPLICATION OF DEUBIQUITINATING ENZYME USP37 AS DRUG TARGET IN SCREENING OF DRUGS FOR TREATING DRUG-RESISTANT BREAST CANCER. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=CN343417361&_cid=P22-LTQ4DA-18310-1#detailMainForm:MyTabViewId:NATIONALBIBLIO (accessed on 12.11.2021).
- TRINUCLEAR PLATINUM COORDINATION COMPOUNDBASED ON TRI-HOMOAZINE, PREPARATION METHOD AND APPLICATION THEREOF. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=CN336184951&_cid=P22-LTQ4EX-19092-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- SYSTEM AND METHOD FOR COLD PLASMA THERAPY WITH HER-FAMILY RECEPTORS. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=US289330559&_cid=P22-LTQ4GU-19804-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- ANTI-HER2 ANTIBODY OR ANTIGEN-BINDING FRAGMENT THEREOF, AND CHIMERIC ANTIGEN RECEPTOR COMPRISING SAME. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019098682&_cid=P22-LTQ4HW-20220-1#detailMainForm:MyTabViewId:PCTBIBLIO.
- APPLICATION OF UCH-L1 AS TARGET SITE IN PREPARATION OF HER2 OVEREXPRESSION BREAST CANCER SENSITIZATION TREATMENT DRUGS. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=CN318119386&_cid=P22-LTQ4IY-20615-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- TARGETING TRASTUZUMAB-RESISTANT HER2+BREAST CANCER WITH A HER3-TARGETING NANOPARTICLE.
- APPLICATION OF MELATONIN IN PREPARATION OF DRUG FOR TREATING HER2 POSITIVE BREAST CANCER RESISTANT TO TARGETED DRUG. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=CN326496984&_cid=P22-LTQ4L2-21446-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- METHOD FOR TREATING ANTI-HER2 THERAPY-RESISTANT MUC4+ HER2+ CANCER. Availabe online: https://patentscope.wipo.int/search/en/detail.jsf?docId=US294690999&_cid=P22-LTQ4MA-21848-1#detailMainForm:MyTabViewId:NATIONALBIBLIO.
- Whenham, N.; D'Hondt, V.; Piccart, M.J. HER2-positive breast cancer: from trastuzumab to innovatory anti-HER2 strategies. Clinical breast cancer 2008, 8, 38–49. [Google Scholar] [CrossRef]
- Feng, M.; Yang, Y.; Liao, W.; Li, Q. Margetuximab versus trastuzumab in patients with advanced breast cancer: A cost-effectiveness analysis. Clinical Breast Cancer 2022, 22, e629–e635. [Google Scholar] [CrossRef]
- Murthy, R.K.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.A.; Lin, N.U.; Borges, V.; Abramson, V.; Anders, C. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. New England Journal of Medicine 2020, 382, 597–609. [Google Scholar] [CrossRef]
- Criscitiello, C.; Corti, C.; De Laurentiis, M.; Bianchini, G.; Pistilli, B.; Cinieri, S.; Castellan, L.; Arpino, G.; Conte, P.; Di Meco, F. Tucatinib's journey from clinical development to clinical practice: New horizons for HER2-positive metastatic disease and promising prospects for brain metastatic spread. Cancer Treatment Reviews 2023, 102618. [Google Scholar] [CrossRef]
- Xie, X.F.; Zhang, Q.Y.; Huang, J.Y.; Chen, L.P.; Lan, X.F.; Bai, X.; Song, L.; Xiong, S.L.; Guo, S.J.; Du, C.W. Pyrotinib combined with trastuzumab and chemotherapy for the treatment of human epidermal growth factor receptor 2-positive metastatic breast cancer: a single-arm exploratory phase II trial. Breast Cancer Research and Treatment 2023, 197, 93–93. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England journal of medicine 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Kreutzfeldt, J.; Rozeboom, B.; Dey, N.; De, P. The trastuzumab era: current and upcoming targeted HER2+ breast cancer therapies. American Journal of Cancer Research 2020, 10, 1045–1045. [Google Scholar] [PubMed]
- Adams, J.; et al. Vascular endothelial growth factor (VEGF) in breast cancer: comparison of plasma, serum, and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res. 2000, 60, 2898–2905. [Google Scholar]
- Lithgow, D.; et al. C-reactive protein in nipple aspirate fluid: relation to women's health factors. Nurs Res. 2006, 55, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Habanjar, O.; et al. Crosstalk of Inflammatory Cytokines within the Breast Tumor Microenvironment. Int J Mol Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Tang, Y. Research progress on the role of reactive oxygen species in the initiation, development and treatment of breast cancer. Prog Biophys Mol Biol. 2024, 188, 1–18. [Google Scholar] [CrossRef]
- Duro-Sánchez, S.; Alonso, M.R.; Arribas, J. Immunotherapies against HER2-Positive Breast Cancer. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Zimmerman, B.S.; Esteva, F.J. Next-Generation HER2-Targeted Antibody–Drug Conjugates in Breast Cancer. Cancers 2024, 16, 800. [Google Scholar] [CrossRef]
- Kumbhar, P.R.; Kumar, P.; Lasure, A.; Velayutham, R.; Mandal, D. An updated landscape on nanotechnology-based drug delivery, immunotherapy, vaccinations, imaging, and biomarker detections for cancers: recent trends and future directions with clinical success. Discov Nano 2023, 18, 156. [Google Scholar] [CrossRef]
Figure 1.
Timeline describing the brief History of development of Trastuzumab (1994-2023) from gene discovery to ADC and its use for overcoming resistance.
Figure 1.
Timeline describing the brief History of development of Trastuzumab (1994-2023) from gene discovery to ADC and its use for overcoming resistance.
Figure 2.
Schematic structure of Trastuzumab with Fab and Fc part, where residue important for Inflammation and ADCC is indicated (A). Specific binding domain of Trastuzumab, Pertuzumab and Tyrosine kinase inhibitors on HER-2 is indicated (B).
Figure 2.
Schematic structure of Trastuzumab with Fab and Fc part, where residue important for Inflammation and ADCC is indicated (A). Specific binding domain of Trastuzumab, Pertuzumab and Tyrosine kinase inhibitors on HER-2 is indicated (B).
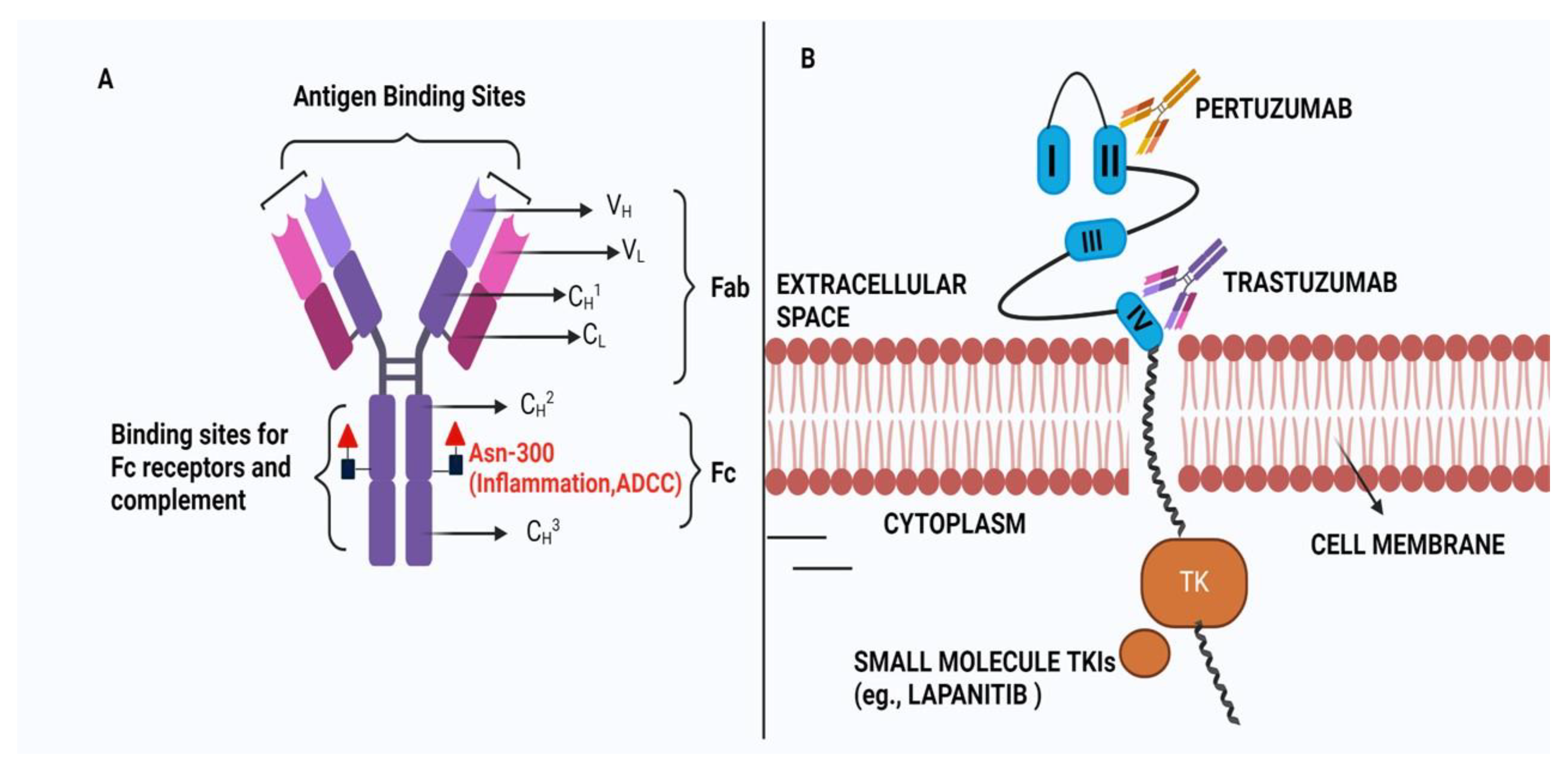
Figure 3.
A schematic diagram of HER2-mediated therapy using Trastuzumab with a specific focus on ADCC.
Figure 3.
A schematic diagram of HER2-mediated therapy using Trastuzumab with a specific focus on ADCC.
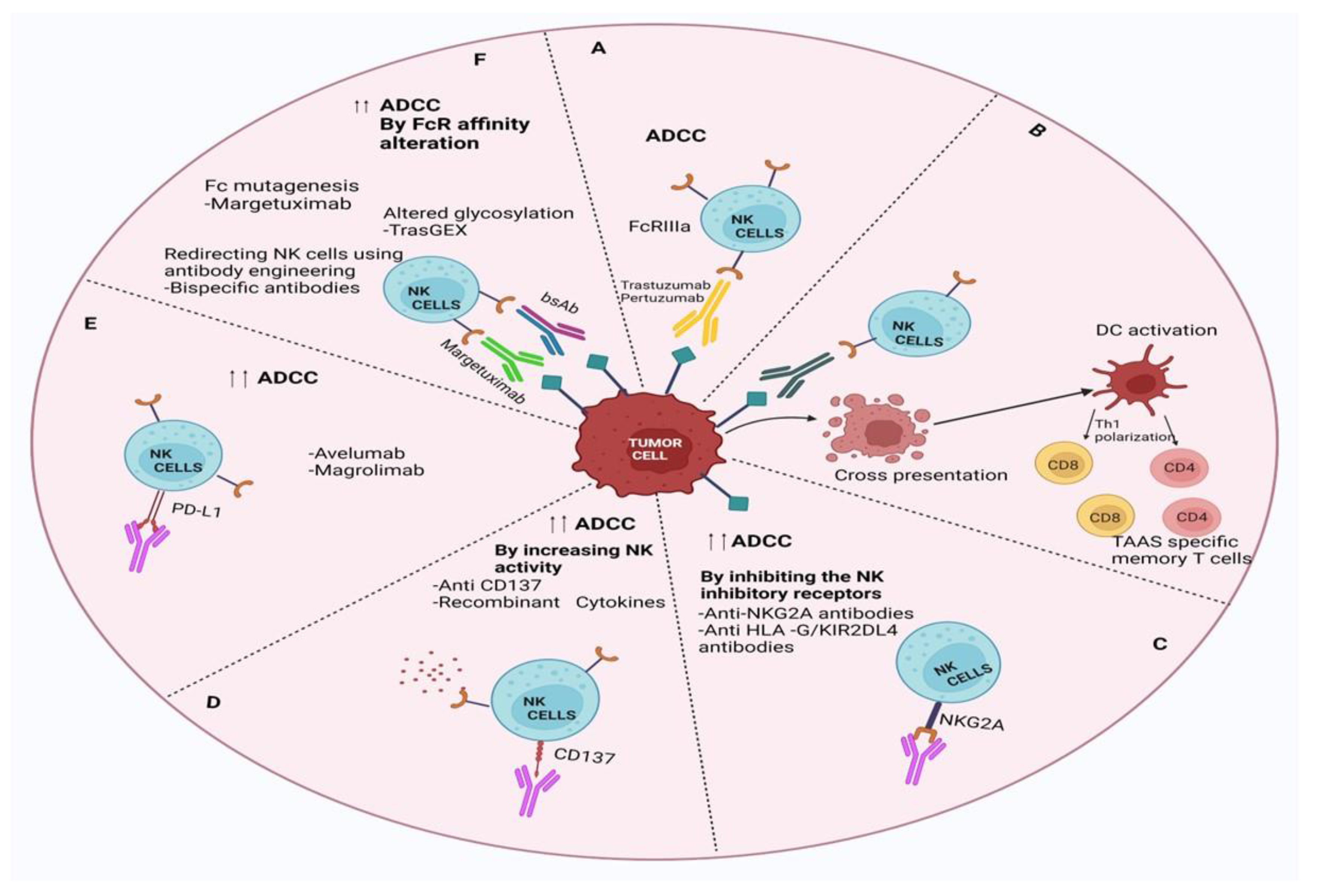
Figure 4.
A schematic presentation of Trastuzumab resistance mechanism involving different signaling pathways where PI3K/AKT/mTOR pathway is highlighted.
Figure 4.
A schematic presentation of Trastuzumab resistance mechanism involving different signaling pathways where PI3K/AKT/mTOR pathway is highlighted.
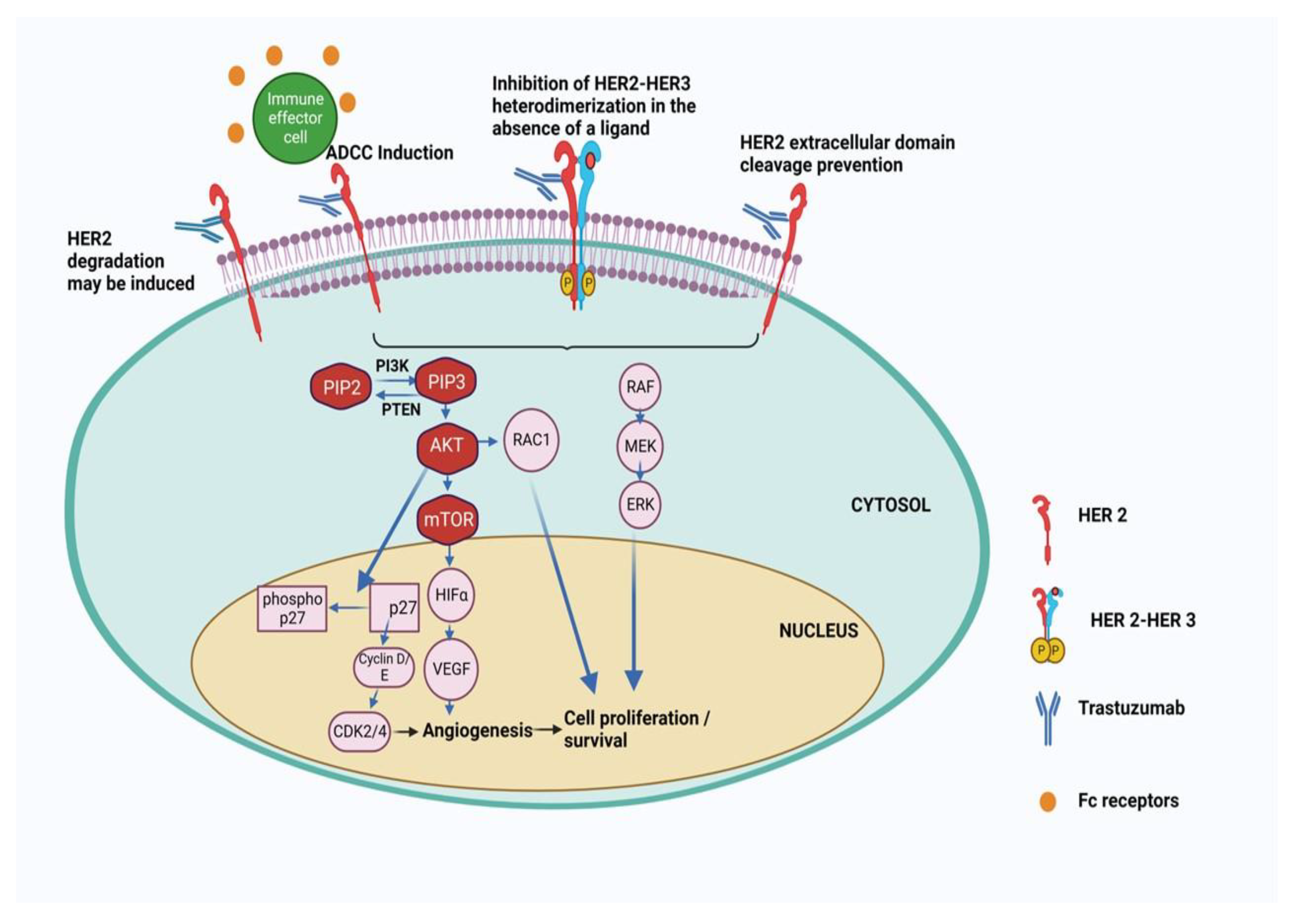
Figure 5.
A timeline of FDA approval of development of small molecule inhibitors for the therapy of BC in combination with Trastuumab and its ADC (A). A schematic presentation of inhibitors which are used to target different pathways to overcome Trastuzumab resistance (B).
Figure 5.
A timeline of FDA approval of development of small molecule inhibitors for the therapy of BC in combination with Trastuumab and its ADC (A). A schematic presentation of inhibitors which are used to target different pathways to overcome Trastuzumab resistance (B).
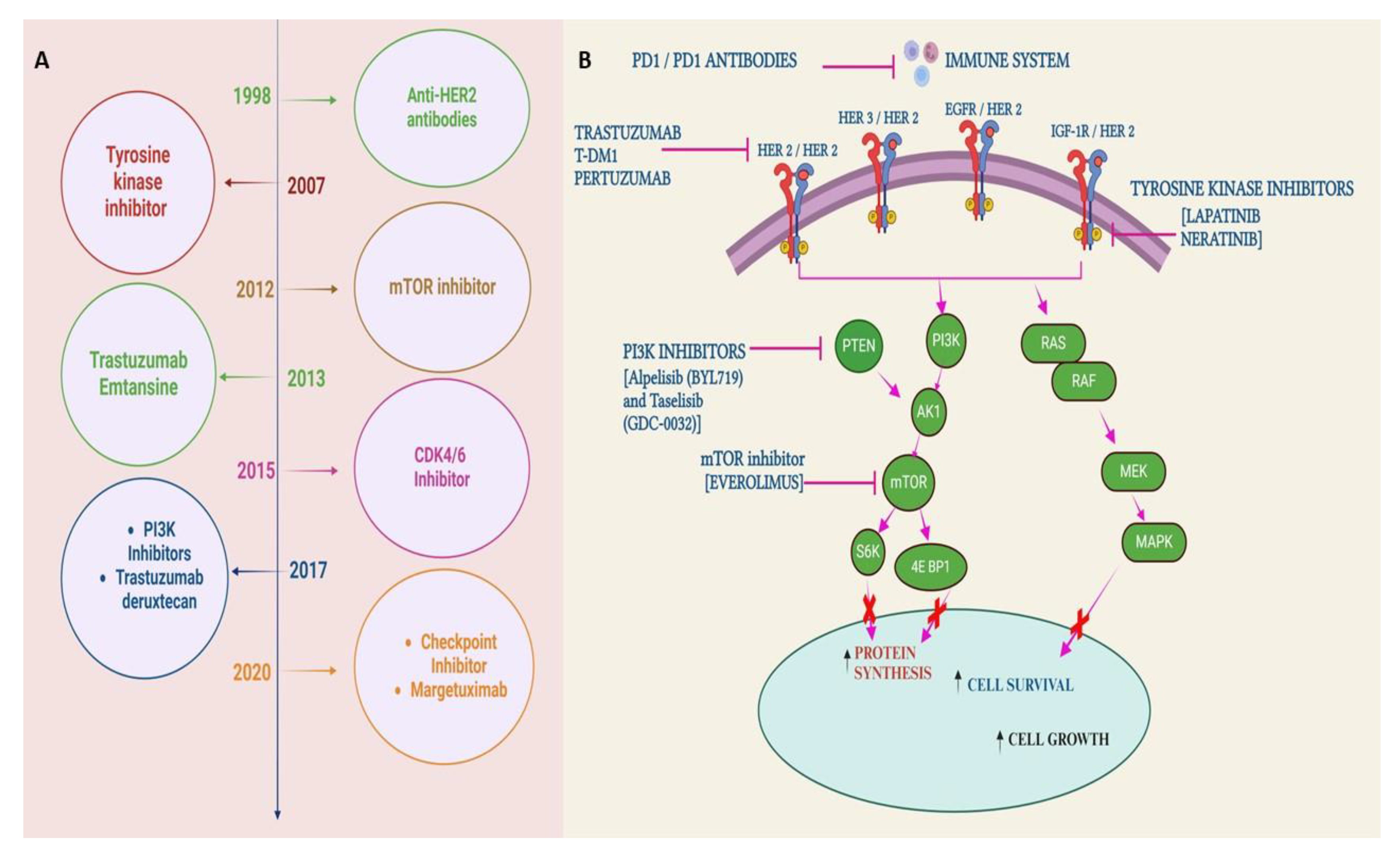
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



