
Submitted:
09 February 2025
Posted:
11 February 2025
You are already at the latest version
Abstract
Acute myeloid leukemia (AML) is characterized by clonal expansion of myeloid progenitors blocked at various stages of their differentiation process and drugs that bypass this differentiation block are therapeutically efficient, as supported by retinoic acid and arsenic trioxide in acute promyelocytic leukemia. However, the successful application of differentiation therapy to APL was not translated into clinical benefit for other non-APL subtypes of AML, in which intensive chemotherapy regimens represent the standard of care. However, the development of molecular studies has led to the identification of therapeutic targets (such as mutated proteins, deregulated pathways) have led to the generation of a new category of specific pharmacologic agents. Some of these agents, such as inhibitors of mutant isocitrate dehydrogenase (IDH1 and IDH2), Lysine-specific demethylase-1 (LSD1) and Menin have shown the capacity of inducing leukemic cell differentiation and a significant therapeutic efficacy.
Keywords:
Leukemia
; Acute Myeloid Leukemia
; Differentiation
; Targeted Therapy
; Genetic Abnormalities
1. Introduction
A hallmark of acute myeloid leukemia (AML) is represented by a differentiation block of myeloid progenitors/precursors that accumulate in bone marrow and in blood.
One AML subtype, acute promyelocytic leukemia (APL) displayed a unique sensitivity to retinoic acid promoting the differentiation of leukemic promyelocytes to mature neutrophils [1]. This discovery provided the rationale for initial clinical studies based on the use of all-trans retinoic acid (ATRA) in APL, which supported the capacity of this drug to promote in vivo differentiation of leukemic cells [2-4]. Subsequent studies have clarified the molecular basis of the peculiar sensitivity of APL to ATRA; in fact, APL is characterized by a 15;17 chromosome translocation with breakpoints within the retinoic acid receptor α (RARA) gene on chromosome 17 and the PML gene, which encodes a transcription factor on chromosome 15: a PML-RARA fusion protein is formed because of the translocation. The specific targeting of this protein represents the molecular basis of the peculiar sensitivity of APL to ATRA [5]. The PML-RARA protein is responsible for all the peculiar features of APL cells, such as sensitivity to ATRA, block of cell differentiation at a promyelocyte stage and increased proliferation due to diminished apoptotic cell death [6]. This peculiar molecular vulnerability of APL was exploited and allowed the development of a curative dual differentiation therapy based on the association of ATRA and arsenic trioxide (ATO), leading to more than 90% of patients being cured from this leukemia without the use of cytotoxic chemotherapy [7].
Although differentiation therapy has completely revolutionized the therapy of APL, changing the outcome of this leukemic subtype once highly fatal to a curable disease, great challenges were observed attempting to develop differentiation-based treatments in other AML subtypes. However, some recent developments of targeted therapies have led to the identification of newer targets for differentiation therapy. Particularly, in this context, the targeting of some molecules in non-APL AMLs, such as isocitrate dehydrogenase 1 and 2 (ODH1 and IDH2), lysine-specific demethylase 1 (LSD1) and Menin was associated with induction of leukemic cell differentiation and significant therapeutic effects.
This review will analyze recent developments in the therapy of AML based on IDH, LSD1 and Menin inhibitors.
2. IDH Inhibitors
Isocitrate dehydrogenase (IDH) is an enzyme that catalyzes the oxidative decarboxylation of isocitrate, producing α-ketoglutarate and CO2. In humans, three isoforms of IDH exist: IDH3 catalyzes the third step of the citric cycle, while converting NAD to NADH in the mitochondria; the isoforms 1 and 2 (IDH1 and IDH2) catalyze th same reaction outside of the citric acid cycle and use NADP as cofactor.
Mutations of IDH are frequently observed in AML and their prevalence increases with age. While WT-IDH1 and IDH2 catalyze the decarboxylation of isocitrate to α-ketoglutarate, mutant IDH1 and IDH2 enzymes catalyze the conversion of α-KG to 2-hydroxyglutarate (2-HG), an oncometabolite responsible for oncogenic activity of IDH1 and IDH2 mutations [8]. Cancer-associated IDH mutations occur at the level of distinct arginine residues in the enzyme active sites.
Inhibitors of IDH are small molecules that bind within the IDH enzymatic active site, blocking aberrant 2-HG production and inducing myeloid differentiation.
The largest study of molecular characterization of 1023 older AML patients reported a frequency of 9.7% IDH1MUT, 18.9% IDH2MUT, including 1% of double-mutant IDH1/IDH2; IDH1MUT significantly co-occurred with DNMT3AMUT (42.4%), while IDH2MUT was associated with DNMT3AMUT (35.8%), NPM1MUT (31.1%), SRFSF2MUT (38.3%) [9]. IDH1MUT was less frequently associated with TET2MUT (6.3%) and TP53MUT (9.3%) [9]. Normal karyotype was more frequently observed in IDH1MUT and IDH2MUT, while complex karyotype was less frequent in IDH1MUT and IDH2MUT AMLs [9]. IDH2MUT AMLs exhibited an improved survival when treated with low-intensity treatments compared to IDHWT AMLs.
The co-mutation profile significantly differed in IDH1R132C and IDH1R132H AMLs in that in the former ones there is a significantly higher frequency of SRSF2, ASXL1 and RUNX1 mutations and lower frequency of NPM1 mutations than in the latter ones [10].
The frequency of both IDH1 and IDH2 mutations in AML is strongly influenced by age. IDH1+IDH2 mutations increased from 3.4%, 11.3%, 17.7% up to 21% in groups pediatric (0-12 yrs), 13-39 yrs, 40-59 yrs and >60 yrs of patients, respectively [11]. ASXL1 and RUNX1 mutations markedly increased in older AML patients [11].
IDH Mutations and Cell Differentiation
Experimental studies have in part elucidated the mechanisms through which IDH mutations exert a leukemogenic effect. Studies in mouse models have shown that IDH mutations alone cannot induce in vivo the development of a full leukemic process but need to cooperate with additional genetic lesions to initiate leukemia [12, 13]. More recently, transgenic IDH2MUT zebrafish models have confirmed these findings and have reported in transgenic embryos co-expressing IDH2 mutations and FLT3ITD the development of a leukemic process recapitulating features of human IDHMUT AML [14]. Single-cell transcriptomic analysis showed increased myeloid skewing, differentiation blockade and leukemia-associated gene signatures [14].
Figure 1.
Various steps involved in the development of IDH-mutant AMLs, in relation to the pathogenetic contribution of IDH1 and IDH2 mutations.
Figure 1.
Various steps involved in the development of IDH-mutant AMLs, in relation to the pathogenetic contribution of IDH1 and IDH2 mutations.

In vitro models of IDH mutations have shown a consistent pattern of DNA hypermethylation, similar to that observed in primary IDHMUT AML cells [15]. Conditional knock-in experiments have allowed to explore the effects of DNA hypermethylation in hematopoietic progenitor cells (HPCs): using this approach, IDH1 mutation (IDH1R132H) was inserted into the endogenous murine IDH1 locus and was expressed in all hematopoietic cells or specifically in cells of myeloid cell lineage [15]. These mutant mice showed increased numbers of HPCs and self-renewal capacity [12]. Expansion of HPC and increased self-renewal were observed also in a model of transgenic mice expressing IDH2R140Q in the hematopoietic system; in addition, it was observed also a block of cell differentiation [13].
In addition to hematopoietic cells, IDH mutations impair histone demethylation and inhibit cell differentiation in normal astrocytes [16].
Additional studies have shown that IDH1R132H mutant promotes cytokine independence and blocks differentiation in hematopoietic cells and these effects can be recapitulated by 2-HG [17].
The key role of IDH mutations in blocking HPC differentiation was further and strongly supported by the observation that a small molecule acting as an inhibitor of mutant IDH2 induced cell differentiation of IDH2MUT leukemic cells [18].
A recent study showed that the expression of IDH1R132H and IDHR140Q mutants into human CD34+ cells via lentiviral transduction markedly impaired the colony forming capacity along erythroid, monocytic and granulocytic lineages [19]. In line with this observation, CD34+ cells isolated from a IDH2MUT AML patient undergoing treatment with Enasidenib showed in the time a progressive improvement in their differentiation capacity, as supported by the in vitro generation of colonies of mature hematopoietic cells [19].
Finally, a recent study additionally supported the role of IDH mutations in blocking hematopoietic differentiation. Thus, Landberg et al. have generated CD34+ cells edited by the CRISPR/Cas9 and AAV6-mediated homology-directed repair to express IDH1R132H or IDHR140Q mutations: mutant CD34+ cells displayed a pronounced decrease in their colony and differentiation capacity; the block of cell differentiation of IDH1R132H – edited cells was rescued by Ivosidenib [20-21].
All these observations support a key role of IDH mutations in blocking cell differentiation of IDHMUT AMLs.
It is important to underline that the effects of IDH mutations on cell differentiation are largely dependent on the methylation changes induced by these mutations. IDH1 and IDH2 mutations in AML are associated with increased DNA methylation. The effects of these DNA methylation changes are not widespread but occur at the level thousands of focal regions that are specifically hypermethylated compared to normal CD34+ cells and AMLs without IDH mutations [22]. The methylation profile observed at these focal regions is different compared to that observed at the level of loci commonly hypermethylated in AML [22]. The profile of hypermethylated loci is highly comparable in IDH1 and IDH2-mutant AMLs, but DNA hypermethylation is more pronounced in IDH1MUT AMLs than in IDH2MUT AMLs [22]. AMLs with biallelic inactivating TET2 mutations had less dramatic methylation phenotype, but many of the hypermethylated regions correspond to those observed in IDHMUT AMLs [22]. 5 hydroxy methyl cytosine (5hmC) levels in hypermethylated DNA regions are significantly lower in IDHMUT or TET2MUT AMLs, thus providing evidence that both the mutations determine increased DNA methylation by blocking TET2-mediated demethylation (TET2 promotes DNA demethylation through hydroxylation of 5mC) [22]. DNA hypermethylation in IDHMUT AML cells requires the activity of the DNMT3A metyltransferase. Importantly, IDHMUT-specific DNA hypermethylated regions are enriched at the level of enhancers involved in the interaction with gened involved in normal hematopoiesis and AML [22].
IDH1 Inhibitors
Ivosidenib (AG-120) is a first-in-class inhibitor of IDH1MUT that exhibits marked 2-HG lowering in IDH1MUT AMLs and induction of cell differentiation of these leukemic blasts [23]. A pivotal study of Di Nardo and coworkers reported a rate of complete remission (CR) or complete remission with partial hematological recovery (CRi) of 30.4%, with an overall response rate (ORR) of 41.6% and a median duration of response (mDR) of 8.2 months [23]. Bone marrow studies showed that Ivosidenib induced myeloid differentiation and hematopoietic recovery without an intervening period of bone marrow aplasia, a finding consistent with a mechanism of action involving induction of myeloid cell differentiation [24]. Differentiation syndrome was observed in 11% of treated patients and all these patients showed different degrees of response to treatment [24].
17 patients enrolled in this study responded to Ivosidenib treatment and subsequently underwent allogeneic HSCT [25]. 12% of these patients had relapsed following prior transplant before starting Ivosidenib therapy, and 76% were refractory to initial therapy; most common baseline co-mutations were NPM1 (29%), DNMT3A (24%) and SRFSF2 (18%) [24]. Prior response to Ivosidenib before allo-HSCT were CR in 10/17, CRi in 4/7 and MLFS (morphologic leukemia-free state) 3/17) [25]. Post-HSCT mOS was 7.7 months, survival rates of 76.5% and 47.1% at 6 and 12 months, respectively; mRFS was 7.3 months, RFS rates of 58.8% and 47.1% at 6 and 12 months [23]. Of all patients whose best OR was CR following Ivosidenib therapy and who underwent HSCT, median OS was not reached [25].
In newly diagnosed IDH1MUT AML patients ineligible to standard chemotherapy induction, Ivosidenib treatment achieved a CR/CRi rate of 42.4%; 18% of treated patients developed a DS [26].
The phase III trial AG120-C-009 evaluated newly diagnosed IDH1MUT AML patients ineligible for intensive chemotherapy to receive Ivosidenib and Azacitidine (IA) or Placebo and Azacitidine (PA): event-free survival was significantly longer in IA than in PB; the median OS was 24 months with IA and 7.9 months with PA [27]. DS was observed in 14% of patients receiving IA and 8% in those treated with PA [27]. The addition of Venetoclax to the Ivosidenib and Azacitidine doublet was safe and increased the rate of response s and improved long-term survival [28].
A phase I clinical study evaluated safety and efficacy of Ivosidenib in association with induction chemotherapy in newly diagnosed IDH1MUT AML patients, resulting in a CR+CRi rate of 72% [29]. In this clinical setting the frequency of DS was low [29]. A more careful examination of these patients at the level of BM morphology and flow cytometric phenotype at day 14 and 21 showed three different patterns of response: (i) aplasia pattern, <10% cellular, <5% blasts; (ii) >10% cellular, <5% blasts, with morphologic and flow cytometric evidence of blast cell differentiation; (iii) persistence of leukemic blasts [30]. The differentiation response was observed in 20% of patients treated with Ivosidenib and chemotherapy [30].
Fathi et al. reported the results of a multicenter phase I trial involving treatment post-HSCT with Ivosidenib for IDH1-mutated AML [31]. 16 patients were enrolled and 8 discontinued maintenance with Ivosidenib. Two-year PFS was 81% and two-year OS was 88% [31].
The analysis of the primary response and relapse after Ivosidenib treatment allowed to define the mechanisms of resistance and relapse in R/R IDH1MUT AML patients. Clinical response to Ivosidenib was not predicted by the position of IDH1MUT within the conal hierarchy: IDH1MUT was subclonal in 28% of cases and clonal in 72% of cases and there was no association between IDH1MUT subclonal or clonal status and response to Ivosidenib [32]. Baseline RTK mutations (NRAS, KRAS, FLT3-ITD, FLT3-TKD) are associated with primary treatment resistance [31]. Relapse is characterized by multiple mechanisms, including emergence of RTK pathway mutations and IDH mutations conferring resistance to Ivosidenib [32].
The TARGET-seq+ method was used for single-cell genotyping to explore sequential bone marrow samples from 8 patients (6 relapsed and 2 in sustained remission), 6 treated with Ivosidenib/Venetoclax and 2 with Ivosidenib/Venetoclax/Azacitidine [32]. Relapse was associated with either genetic clonal evolution or impaired differentiation of pre-existing clones fully differentiated into mature myeloid cells prior to treatment, within HSPC/precursor cell compartments known to demonstrate LSC potential [33]. In the 2 patients remained in sustained remission, therapy eradicated all leukemic clones within 3 cycles of treatment [33].
Olutasidenib is an allosteric IDH1MUT inhibitor that binds at the level of hydrophobic pocket located near the IDH1 heterodimer interfaces and determines the stabilization of the mutant IDH1 molecule in an inactive conformation, thus blocking its neomorphic enzymatic activity. At structural level, Olutasidenib is a quinoline derivative [34]. Given these inhibitory properties, Olutasidenib inhibits 2-HG production and promotes granulo-monocytic differentiation in primary IDH1MUT AML cells and inhibits the growth of IDH1MUT AML xenograft models [35]. A phase I/II clinical study evaluated the safety and the efficacy of Olutasidenib in IDH1MUT AML and MDS patients. The phase I of the study evaluated the safety and efficacy of Olutasidenib as a single agent and in combination with azacitidine in patients with IDH1MUT AML and MDS; Olutasidenib monotherapy was evaluated in R/R patients and Olutasidenib as monotherapy or in combination with Azacitidine. Grade 3-4 hematologic adverse events were more common with combination therapy than with monotherapy; differentiation syndrome was observed in both monotherapy and combination treatment [36]. ORR was 41% in patients receiving monotherapy and 46% in patients treated with combination therapy; for treatment-naïve AML patients, ORR was 25% in monotherapy and 77% in combination therapy [36]. For R/R IDH1MUT AML patients, the median OS was 8.7 months for monotherapy and 12.1 months for combination therapy [36].
The phase II study involving monotherapy treatment enrolled 153 R/R IDH1MUT AML patients, with a median age fo 71 years and a median 2 prior therapy regimens; the CR+CRi was 35% and ORR was 48%; the median duration of CR+CRi was 25.9 months; the median OS was 11.6 months; response rates were similar in patients who received or not prior Venetoclax treatment [37]. OS occurred in 22% of patients and in 9% of patients was grade ≥3, with one fatal case [37]. The final analysis 5-year follow-up showed a CR+CRi rate of 35% with a median duration of CR+CRi of 25.3 months; ORR was 48%, with median duration of 15.5 months; mOS was 11.6 months and in patients R/R to prior Venetoclax mOS was 16.2 months [38]. Transfusion independence from red blood cells and platelets was achieved in 39% and 41% of patients, respectively [38]. The analysis restricted to elderly patients (≥75 years) showed that Olutasedinib was well tolerated and 31% of these patients achieved a CR+CRi, with a median time to CR+CRi of 15 months, and with a median duration of CR of 25.9 months [39].
The phase II of the combination of Olutasidenib and Azacitidine was based on a study involving 4 cohorts of patients: treatment-naïve patients with AML; R/R AML and MDS patients with no prior exposure to HMA or IDH1 inhibitors; R/R to HMA; patients with prior exposure to IDH1 inhibitors [40]. In an initial report, 72 patients with AML/MDS were reported (20 R/R without prior HMA/HD1 inhibitor therapy; 21 R/R with prior HMA therapy; 20 R/R with prior HD1 therapy; 11 treatment-naïve AML patients) [40]. In treatment-naïve AML patients, the CR+CRi rates were 45%; in R/R setting, CR+CRi rates were 47% in those without prior HMA/HD1 inhibitor therapy, 38% with prior HMA/HD1 inhibitor therapy, and 30% with prior IDH1 inhibitor therapy [40]. The duration of CR+CRi was longest among treatment-naïve patients [40].
More recently, it was presented a pooled analysis of R/R IDH1-mutant AML patients included in this trial: 43% were refractory and 57% relapsed after a prior treatment; 83% of these patients had at least 2 prior treatment regimens, including HMA (40%), IDH1 inhibitor (31%), HSCT (10%) CR+CRi was observed in 31% of patients, with a median duration of 15 months; CR was achieved in 27% of patients with a median duration of 20 months; ORR was 51%; median OS was 13 months in all patients, 24 months in overall responders, 30.6 months in patients with CR+CRi and not reached in patients with CR [41].
A multicenter, investigator-initiated phase I/II study will evaluate the safety and the efficacy of the triplet regimen based on Olutasidenib in combination with Decitabine and Venetoclax [42].
The comparison of outcomes of Olutasidenib-treated from the 2102-HEM-101 single-arm trial were compared to a real-world external central arm of Ivosidenib suggestive of favorable effectiveness of Olutasidenib for patients with IDH1-mutant AML who are R/R to a Venetoclax-based regimen [43].
A recent study reported a comparative analysis of Olutasidenib and Ivosidenib. Both these drugs have been approved by FDA for treatment of IDH1-mutant AML. In the absence of head-to-head studies, this analysis compared the outcomes observed in studies carried out in R/R AML patients: the rates of CR+CRi observed with Olutasidenib and ivosidenib were similar; however, the duration of response observed in patients treated with Olutasidenib was longer than that observed in patients treated with Ivosidenib (25.9 months vs 8.2 months, respectively). Another notable difference is that Olutasidenib is structurally smaller with a lower molecular weight than Ivosidenib thus occupying less space in the binding pocket of IDH1 dimers, making it resistant to displacement by IDH1 second-mutation [44]. In biochemical studies, Olutasidenib was found to inhibit mutant IDH1 but not wild-type IDH1 [44].
IDH2 Inhibitors
Enasidenib (AG-221) is a clinically useful IDH2-mutant inhibitor developed by Agios Pharmaceuticals/Celgene, reducing 2HG levels and promoting cell differentiation [45]. A pivotal study by Stein and coworkers in relapsed or refractory IDH2 mutant AML patients reported an overall response rate of 40.3%, with a median OS of 9.3 months (19.7 months in patients who attained a CR); responses were associated with cellular differentiation of leukemic bast and not cytotoxicity was the main driver of anti-leukemic activity of Enasidenib [46]. Enasidenib was explored also in older AML patients who are unfit for intensive chemotherapy, showing an ORR of 30.8%, with 18% of CR and with a mOS of 11.3 months [47]. The analysis of R/R AML patients treated with Enasidenib showed that: Enasidenib induced a marked decrease of 2-HG levels, preceding the clinical response; 2-HG suppression by Enasidenib did not predict response since most non-responding patients also exhibited 2-HG suppression; complete remission and normalization of stem and progenitor compartments were associated with emergence of functional neutrophils bearing IDH2 mutations; in a subset of responding patients, IDH2MUT burden decreased and remained undetectable during response; co-mutations in NRAS and MAPK pathway effectors are enriched in patients resistant to treatment [48].
A randomized phase 3 clinical trial explored Enasidenib vs conventional care in a population of 319 older IDHMUT R/R AML patients who received 2 to 3 prior AML-directed therapies [48]. Enasidenib improved EFS and hematological parameters but failed to improve OS compared to standard treatment; OS could be confounded by early dropout and use of subsequent AML therapies. [49].
In a phase Ib/II clinical trial, Enasidenib was evaluated in 101 newly diagnosed IDH2-mutated AML patients not eligible for standard induction chemotherapy, the treatment based on Enasidenib plus Azacitidine (2/3 of patients) or Azacitidine alone (1/3 of patients) [50]. Significant differences were observed between the two groups of patients concerning ORR and CRR, but not in median OS [50]. About one-third of patients in the group Azacitidine alone received Enasidenib at progression as a post-protocol therapy and this may have confounded survival analysis [50].
Explorative correlative studies performed in patients treated with Enasidenib and Azacitidine showed that responding patients displayed a decrease of balst cell population markers and an increase of differentiated myeloid markers, such as CD11b and these changes were paralleled by a decrease of VAF of IDH2 mutations or NPM1 mutations up to low/very low levels, no patients with NRAS, KRAS or PTPN11 mutations achieved a CR [51].
A recent study explored Enasidenib combined with Venetoclax in a population of patients with R/R AML or other myeloid malignancies, showing that this drug combination induced an ORR higher than that observed with Enasidenib alone: an ORR was significantly higher in IDH2R172 than IDH2R140-mutant patients (83% vs 55%, respectively) [52]. In the whole treated population, ORR was 70%, 57% CRR; the median duration of response was 16.6 months, and 3 patients proceeded to HSCT following CR [52].
The combination of Enasidenib with intensive induction and consolidation chemotherapy was explored in 93 fit IDH2MUT AML patients, showing that it was well tolerated and was associated with a CR+CRi rate of 77%, with 23% of patients achieving IDH2 mutation clearance [29].
A recent pilot clinical trial (NCT 03728335) assessed the use of Enasidenib as a post-HSCT maintenance therapy fro 15 patients with IDH2-mutated AML: the patients received 24 cycles of Enasidenib therapy with leukemia-free survival rates of 100% and 93% of chromic GVHD-free survival and 87% relapse-free survival [53]. At the safety level, the treatment was well tolerated, with adverse events easily managed [53]. The results were considered highly promising and the enrollment of 20 additional patients was scheduled [53].
Concurrent RAS-signaling mutations are a great challenge in the treatment of IDH-mutant AML patients in that they infer resistance to IDH inhibitors. Thus, as supported by preclinical studies, it was recently proposed a phase Ib study based on the administration of Enasidenib and MEK inhibitor Cobimetinib in R/R AML patients who have co-occurring IDH2 and RAS signaling mutations [54].
Differentiation Syndrome in Patients Treated with IDH Inhibitors
Treatment with IDH inhibitors was associated with the development of differentiation syndrome (DS), a potentially lethal adverse reaction triggered by agents that induce myeloid differentiation.
DS was initially described in acute promyelocytic leukemia (APL) treated with agents inducing myeloid differentiation, such as all-trans retinoic acid (ATRA) and arsenic trioxide (ATO) [56]. DS is a relatively common and potentially severe complication observed in APL patients treated with ATRA and/or ATO; this syndrome is characterized by the association of a number of symptoms, including unexplained fever, weight gain, dyspnea with pulmonary infiltrates, pleuropericardial effusion, hypertension, and renal failure [56-57]. This syndrome may occur in a mild and in a severe form. The mechanisms responsible for the development of DS remain not fully understood but seem to be related to the induction of differentiation of leukemic blasts, resulting in the massive release of cytokines (“cytokine storm”) and chemokines, and subsequent systemic inflammatory response and increased expression of adhesion molecules on the surface of differentiated leukemic cells, mediating their adhesion to vascular endothelium and then their migration into tissues [58].
The prevalence, risk factors and clinical outcomes of IDH-mutant AML patients treated with IDH inhibitors were not prospectively evaluated. A retrospective analysis performed by an independent differentiation review committee, composed by investigators who have participated to the development of Enasidenib showed that DS was observed in 11.7% of R/R AML patients treated with Enasidenib and that most frequent symptoms were dyspnea, fever, pulmonary infiltrates, and hypoxia; in 395 of cases DS was associated with concomitant leukocytosis [59].
The Food and Drug Administration performed a systematic analysis of DS observed in 393 AML patients treated with Ivosidenib or Enasidenib [60]. According to this analysis, DS was identified in 19% of patients; the predominant symptoms observed in these patients are pulmonary (dyspnea, pulmonary infiltrates and effusions) [60]. In these patients, the onset of symptoms occurred at a median of 19-20 days after treatment [61]. Predictors of DS were represented by higher leukemic burden in the bone marrow (≥48% of blasts) and in the blood (≥15%-25% blasts); furthermore, concurrent mutations in TET2 and SRFSF2 were associated with a higher risk of developing DS, CR+CR rates were lower in patients with versus those without DS (Ivosidenib 18% vs 36%; Enasidenib 18% vs 25%) [60].
Montesinos and coworkers have performed a pooled analysis from four clinical trials involving Enasidenib in AML patients, either in monotherapy or in combination with Azacitidine or with chemotherapy [58]. Highest incidence of DS was observed in patients who received Enasidenib plus Azacitidine (17.6%) and lowest incidence in patients who received Enasidenib plus chemotherapy (2.2%) [61]. The most common symptoms were dyspnea/hypoxia (80.6%) and pulmonary infiltrate (73.1%) [61]. Baseline risk factors for developing DS were represented by higher levels of bone marrow blasts and lactate dehydrogenase [61].
Inhibitors of Lysine-Specific Demethylase 1 (LSD1 or KMD1A)
LSD1 is a member of the flavin adenine dinucleotide-dependent (FAD-dependent) amine oxidase family of demethylase, participating to the formation of various chromatin multiprotein complexes involved in gene regulation, such as rest co-repressor (CoREST) and nucleosome and remodeling and deacetylase (NuRD) [62-63]. LSD1 acts demethylating lysine 4 and 9 on histone 3 and also on non-histone substrates, such as p53 and, through these effects, it represses gene expression [62]. LSD1 is essential for the function of HSCs and its knockout results in a reduction of granulopoiesis, thrombocytopoiesis and erythropoiesis, associated with an expansion of granulo-monocytic, megakaryocytic and erythroid progenitors, thus supporting a physiological role for LSD1 in myeloid differentiation and maturation [64]. LSD1 is overexpressed in many cancer types, including AML.
A number of compounds with amine oxidase inhibitory activity (trancylcypromine, paragyline and phenelzine) and SP-2509 and GSK-LSD1 have been utilized in initial studies aiming to explore their anticancer activity. New LDS1 inhibitors suitable for clinical studies, such as ORY-1001 (Iadademstat) SP-2577 (Seclidemstat), IMG-7289 (Bodemstat) and CC-90011 (Pulodenstat) have been evaluated in clinical studies [62-63]. These compounds act as irreversible or reversible LSD1 inhbitors, with various degrees of specificity and display in vitro efficacy in AML with inhibition of cell proliferation and induction of cell differentiation [62-63].
Experimental studies in suitable models supported the clinical use of LSD1 inhibitors as antileukemic drugs. A pivotal study by Lynch and coworkers provided evidence that pharmacological inhibitors of LSD1 promote differentiation of myeloid leukemic cells through a mechanism independent of inhibition of LSD1 enzymatic activity (histone demethylation) but dependent of the interaction between LSD1 and transcription factor GFI1, essential to maintain the differentiation block in AML [65]. Particularly, LSD1 inhibitors disrupt the interaction of LSD1 and RCOR1 with the transcription repressor GFI1, which is bound to a set of enhancers located close to transcription factor genes involved in the regulation of myeloid cell differentiation; the inactivation of GFI1 leads to increased enhancer histone acetylation and consequent gene activation [66]. LSD1 inhibitors interfere with GFI-mediated repression of PU.1 and C/EBPα target genes and induce differentiation of AML cells [67-68]. The stabilization of the binding of LSD1 on chromatin at GFI1 binding sites requires the interaction of the HMG-box protein HMG 20B with LSD1 [69].
Figure 2.
Molecular mechanisms underlying induction of cell differentiation by LSD1 inhibitors. LSD1 inhibition drives expression of genes involved in myeloid cell differentiation through disruption of transcriptional repression mediated by GFI1. Top Panel: when LSD1 interacts with the transcriptional repressor GFI1 and with HMG20B, it allows the recruitment of repressors to chromatin and catalyzes H3K4 demethylation and histone deacetylation though HDAC activity, resulting in transcriptional repression. Bottom Panel: LSD1 inhibitors block the interaction between LSD1 and GFi1, thus destabilizing the whole LSD1 repressor complex and leading to activation of myeloid enhancer elements with consequent transcriptional activation of master transcription factors of myeloid differentiation, such as PU.1.
Figure 2.
Molecular mechanisms underlying induction of cell differentiation by LSD1 inhibitors. LSD1 inhibition drives expression of genes involved in myeloid cell differentiation through disruption of transcriptional repression mediated by GFI1. Top Panel: when LSD1 interacts with the transcriptional repressor GFI1 and with HMG20B, it allows the recruitment of repressors to chromatin and catalyzes H3K4 demethylation and histone deacetylation though HDAC activity, resulting in transcriptional repression. Bottom Panel: LSD1 inhibitors block the interaction between LSD1 and GFi1, thus destabilizing the whole LSD1 repressor complex and leading to activation of myeloid enhancer elements with consequent transcriptional activation of master transcription factors of myeloid differentiation, such as PU.1.
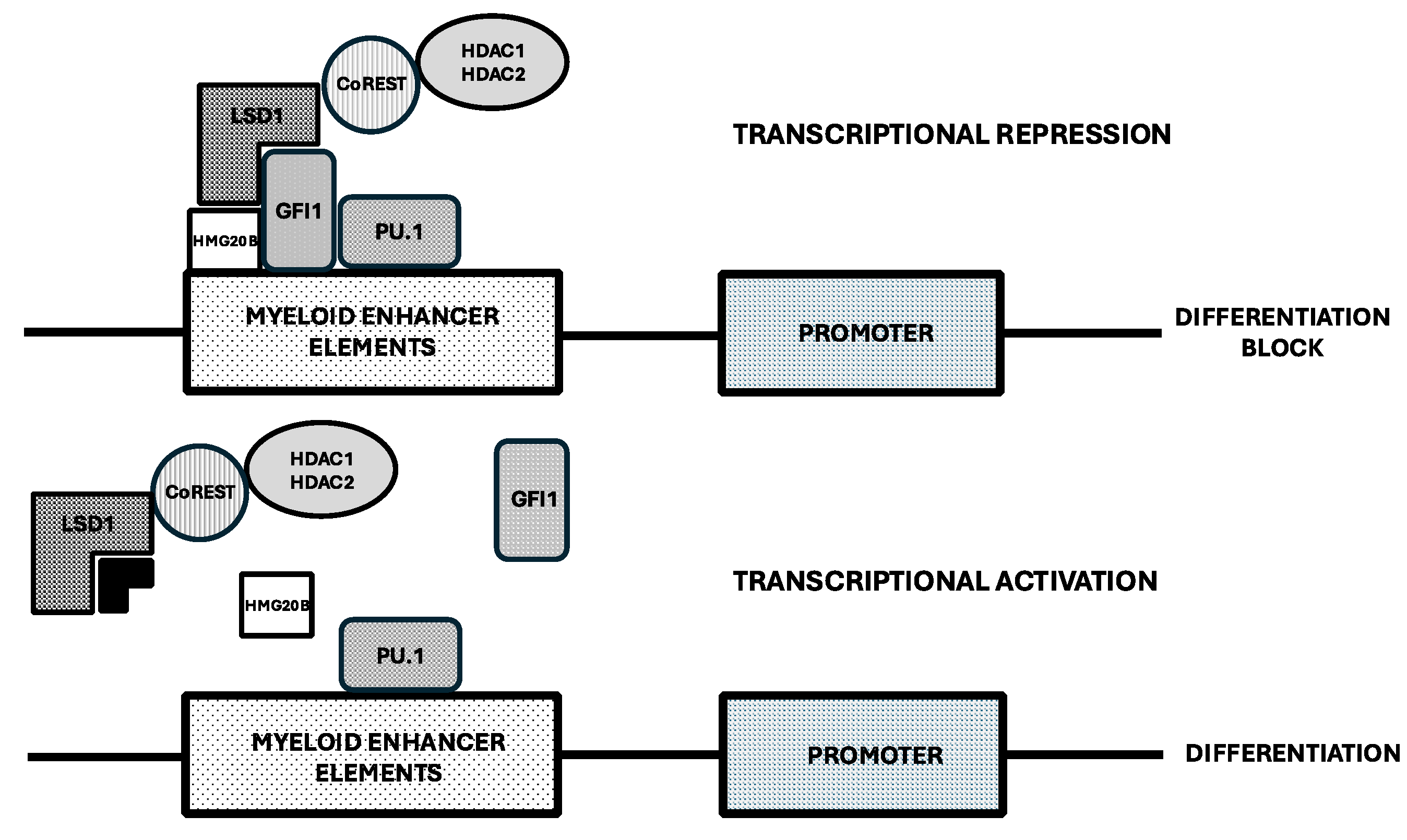
The LSD1/CoREST/HDCA2 complex is recruited by the SNAG domain of GFI1 [66]. In addition to GFI1, GFI1B is another repressor containing a SNAG domain and mediating the recruitment of LSD1/CoREST/HDCA2 complexes [70]. These molecular complexes involving LSD1 act as epigenetic regulators of hematopoietic differentiation [70]. GFI1 plays an important role in the control of granulo-monocytic differentiation, while GFI1B plays a role in erythroid and megakaryocytic differentiation [71].
The N-terminal intrinsically disordered region (IDR) is required for the regulation of LSD1-transcription factors interactions, controlling enhancer activation that is necessary for AML cell differentiation [72].
PRY-1001 is a potent and selective LSD1 inhibitor that induces H3K4me2 accumulation on LSD1 target genes, differentiation of leukemic cells and inhibition of leukemic cells and inhibition of leukemic stem cell activity in AML models [73]. ORY-1001 synergizes with other anti-leukemic drugs currently used in the treatment of AML and in a patient-derived xenograft model of T-ALL significantly reduces leukemia development and improves survival [73]. These observations have supported the clinical evaluation of ORY-1001 (Iadademstat) in AML patients.
A phase I clinical study evaluated Iadademstat in relapsed or refractory AML patients: the dose-escalation section of the study carried out in 27 patients showed the recommended dose of Iadademstat for an extension cohort was 140 μg/m2 administered as single agent from day 1 to 5 of 28-day cycles; in the extension cohort 14 additional patients were treated, including 5 patients with MLL/KMT2A-rearranged AMLs [74]. Reduction of blood and bone marrow percentages of blasts were observed, with one patient achieving CR; inhibition of blast cell differentiation was frequently observed, particularly in patients with MLL translocations: cell differentiation was observed in 80% of these patients and was particularly pronounced in two patients, with one of these patients developing a rapid and fulminant differentiation syndrome [74].Preclinical studies have shown a synergism between Azacitidine and Iadademstat, thus supporting the clinical evaluation of this drug combination. Thus, the phase II ALICE clinical study evaluated 36 newly diagnosed AML patients with intermediate or advanced-risk disease, with a median follow-up of 22 months; dose-response studies showed that optimal dose of Iadademstat in combination with Azacitidine was 90 μg/m2 [75]. In 27 patients, the ORR was 22727 with 14/27 CR+CRi (11/27 MRD-) [75]. 3 patients displayed DS and one patient had fatal grade 5 intracranial hemorrhage.
Iadademstat is also being evaluated in combination with Venetoclax and Azacitidine in newly diagnosed AML patients in an investigator-initiated phase I clinical trial at Oregon Health & Science University Knight Cancer Institute (NCT 06357182) and in company-sponsored phase Ib clinical trial in combination with Gilterinib in patients with R/R AML patients harboring FLT3 mutations (NC 05546580).
Particularly, concerning the association of Iadademstat with Gilertinib, he FRIDA study aims to establish he safety, tolerability and the recommended phase II dose in R/R FLT3-mutant AML patients [76]. This trial was supported by preclinical studies showing a strong synergy of Iadademstat with FLT3 inhibitors and particularly with Gilterinib in FLT3-mutant AML cells [76]. The FRIDA sudy was based on classical design for phase I/II studies, with an escalation phase (from 75 to 150 μg/orally of Iadademstat) and an expansion phase at the selected safe and pharmacologically active dose [76]. Preliminary results on the FRIDA trial were presented at the EHA 2024 Meeting on the first 13 patients: the combination of Iadademstat and Gliteritinib appeared to be safe and well tolerated, with no dose limiting toxicities at initial dose (75 μg) and DL1 (100 μg) of Iadademstat; encouraging antileukemic activity was shown, with 5 out of 13 patients (38%) achieving CR+CRi and 9 out of 13 patients (69%) achieving bone marrow blast clearance in the first cycle of treatment [77].
Two other LSD1 inhibitors, SP-2577 (Seclidemstat) and CC-90011 (Purodemstat) are under evaluation in patients with hematological malignancies but their evaluation in AML patients is at the moment very limited.
Menin Inhibitors in the Treatment of AML
Menin protein is encoded by the MEN 1 gene, whose germline mutations are the causation of sporadic or autosomal dominant hereditary cancer syndromes affecting the endocrine system; menin plays a key role as epigenetic regulator of gene expression for its role as scaffold protein able to interact with various partners; thus, Menin acts as an adaptor protein between H3K4 (histone 3 protein with the lysine amino acid al position 4) methyltransferase KMT2A (also known as MLL) and LEDGF (lens epithelium derived growth factor). Menin interacts with both the wild-type and rearranged KMT2A, regardless of the fusion partner. Menin was found to be crucial for KMT2A activity and the maintenance of HOXA expression but not essential for normal hematopoiesis. The normal regulation of the KMT2A activity is required for the maintenance of expression of HOX family cluster genes in tissues.
Upregulated HOXA/MEIS1 expression is observed in AMLs characterized by the presence of NPM1 mutations or by rearrangements of the MLL gene [78-80]. In NPM1-mutant AMLs (20-30% of total AMLs) the nucleolar protein is mutated and is usually delocalized in the cytoplasm; however, a part of the mutated NPM1 protein resides in the nucleus where it interacts and is co-localized with KMT2A at the level of the HOXA locus [78-80]. The mutant NPM1c protein amplifies the function of KMT2A [79] and inhibits also the activity of histone deacetylases [80] to maintain active transcription of HOXA/B cluster genes and MEIS1. Mutant NPM1 upregulates the expression of HOXA/B gene clusters also through another mechanism dependent upon the interaction between mutant NPM1c protein and FOXM1 protein with its consequent inactivation and delocalization in the cytoplasm: the transcriptional inactivation of FOXM1 by cytoplasmic sequestration induces de-repression of HOX A/B cluster genes [81].
KMT2A-rearranged AMLs represent 5-10% of adult AMLs, but are much more frequent in younger patients, while they are rare in older patients; in these leukemias the rearrangement of KMT2A gene upregulates HOX-MEIS1 expression [82]. KMT2A protein directly binds to and uses its SET domain H3 (Lys4) methyltransferase to regulate HOX gene promoters. A third AML subset associated with increased HOX A/B expression is represented by partial tandem duplication (PTD) of KMT2A gene, occurring in 3-11% of adult de novo AML and associated with adverse outcome. At the level of gene expression, KMT2A-PTD are characterized by a peculiar profile, showing increased expression of several HOX genes, including HOX-B5, HOX-B7, HOX-B8 and HOX-B9. The comparative analysis of the profile of HOX-A/B genes deregulated in these AMLs overexpressing HOX genes showed a different profile in KMT2A-PTD and KMT2A-rearranged AMLs and a similar profile in KMT2A-PTD and NPM1-mutant AMLs [83]. The analysis of the whole gene expression profile allowed to define three gene signatures along the HOX-targeted gene axis (HOX primitive, HOX transient and HOX committed profiles); KMT2A-rearranged AM Ls were mostly classified as HOX-committed, while KMT2A-PTD as HOX-primitive/transient and NPM1-mutant AMLs were distributed along the three HOX profiles [83].
A recent study provided an analysis of pediatric AMLs subdivided according to HOX family gene expression. No expression or very low expression of HOXA and HOXB genes was observed in normal bone marrow cells, while 25% of pediatric AML had significant upregulation of HOX A/B gene expression. The cluster of AMLs with highest HOX A expression contained 88% of KMT2A-rearranged AMLs and these patients displayed a significantly worse outcome compared to those with low/moderate HOX expression; patients without KMT2A rearrangements that clustered within the group at highest HOX A expression had a dismal outcome [84]. Patients with high HOXB expression mainly pertain to patients with FLT3-ITD or NUP98-NSD1 gene alterations [84]. AMLs with HOX-A expression display a clear co-expression of MEIS-1 [84].
Figure 3.
Role of Menin in the regulation of HOX a/B gene expression and cell differentiation in KMT2A-rearranged AMLs. Top Panel: KMT2A fusion protein binding to its cofactor Menin acts as a stimulator of HOX A7B gene expression, with consequent effects resulting in a block of cell differentiation. Bottom Panel: Menin inhibitors bind to Menin and impair its interaction with HOX A/B genes, inhibit HOX A/B gene expression, with consequent induction of cell differentiation.
Figure 3.
Role of Menin in the regulation of HOX a/B gene expression and cell differentiation in KMT2A-rearranged AMLs. Top Panel: KMT2A fusion protein binding to its cofactor Menin acts as a stimulator of HOX A7B gene expression, with consequent effects resulting in a block of cell differentiation. Bottom Panel: Menin inhibitors bind to Menin and impair its interaction with HOX A/B genes, inhibit HOX A/B gene expression, with consequent induction of cell differentiation.
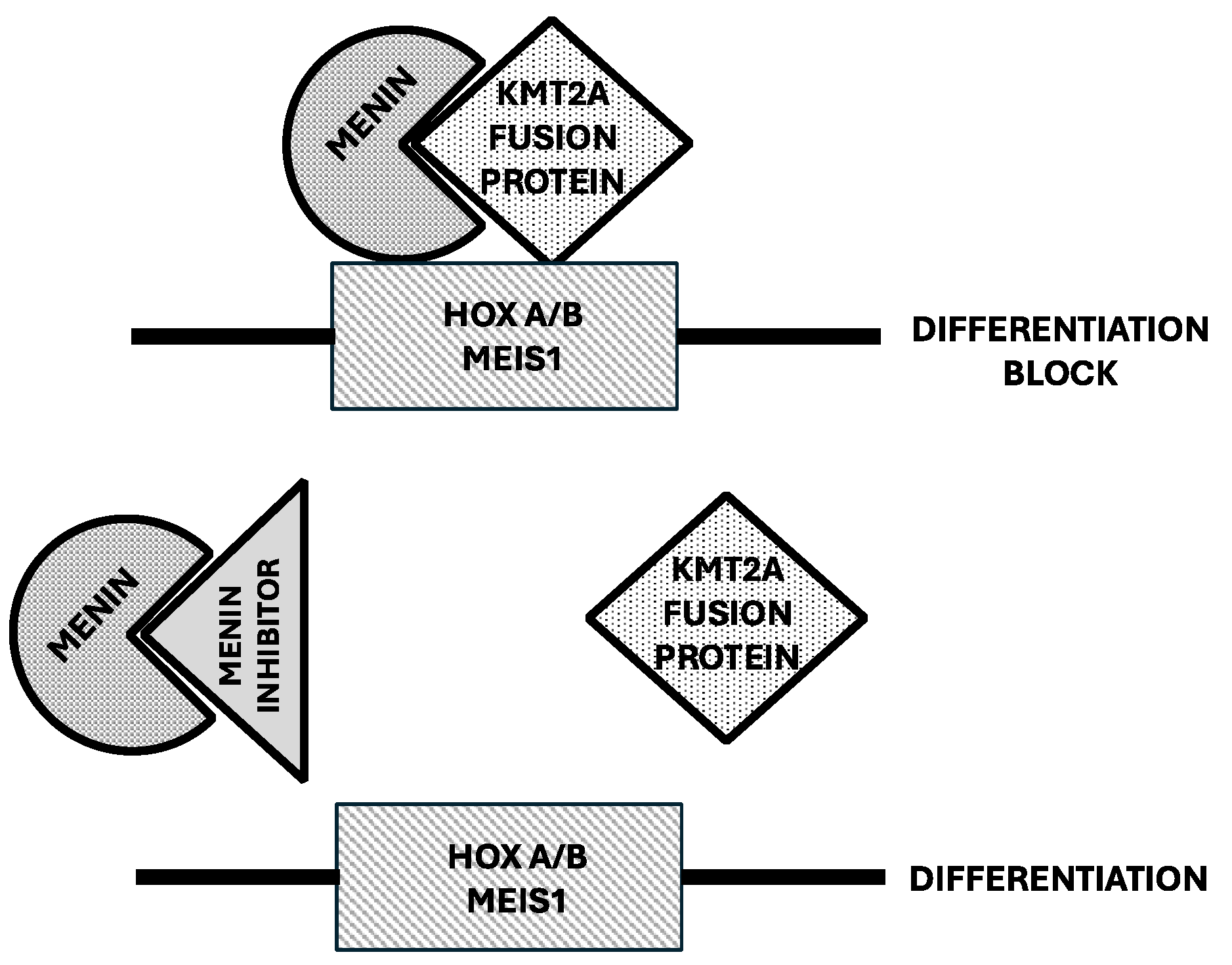
Efficient drug discovery techniques have led to the development of potent and selective Menin inhibitors that block the Menin-KMT2A interaction through a hydrophobic pocket on surface of Menin protein. Preclinical studies have supported the efficacy of Menin inhibitors in MLL-rearranged and NPM1-mutated models of leukemia. Thus, Krivstov et al showed that the highly selective Menin inhibitor VTP50469 displaced Menin from protein complexes and inhibited chromatin occupancy by MLL of selected genes in leukemic cells bearing MLL rearrangements resulting in induction of cell differentiation and apoptosis [81]. Patient-derived xenograft models derived from MLL-rearranged AML cells showed dramatic reduction of leukemia when treated with VTP50469 [85]. Klossowski et al. confirmed that the menin-specific inhibitor MI-3454 inhibited cell proliferation and induced differentiation in primary patient samples with MLL translocations or NPM1 mutations and induced remission or regression of leukemia in mouse models of MLL-rearranged or NPM1-mutated leukemia [85]. Menin inhibitors silence both a canonical HOX and MEIS-1-dependent oncogenic gene expression program and a noncanonical transcriptional program involving tumor suppressor genes and both these events are required to achieve a good therapeutic response [86].
In vitro studies in NPM1-mutant or MLL-rearranged leukemic cell lines or primary AML cells using the Menin inhibitor DS-1594b showed that the primary effect was induction of cell differentiation and not of apoptosis; when the Menin inhibitor was associated to the BCL-2 inhibitor Venetoclax the apoptotic effect was predominant [87].

Table 1.
A-rerrangend and NPM1-mutant AML patients treated with Menin inhibitors in monotherapy.
| Compound | Revumenib | Bleximenib | Enzomenib | Ziftomenib |
|---|---|---|---|---|
| Trial | AUGMENT-101 Phase I/II |
CAMELOT-1 Phase I/II |
DSP-5336-101 Phase I/II |
KOMET-001Phase I/II |
| Number of patients | 161 | 21 | 40 | 58 |
| ORR | KMT2Ar 64% NPM1m 47% |
KMT2Ar 30% NPM1m 50% |
KMT2Ar 59% NPM1m 54% |
KMT2Ar 17% NPM1m 42% |
| CR+CRi | KMT2Ar 23% NPM1m 23% |
KMT2Ar 33% NPM1m 33% |
KMT2Ar 30% NPM1m 47% |
KMT2Ar 17% NPM1m 35% |
| MRD negativity(in CR+CRi) | KMT2Ar 58% NPM1m 64% |
NR | NR | KMT2Ar 100% NPM1m 63% |
| HSCT amongresponders | KMT2Ar 36% NPM1m 17% |
NR | NR | KMT2Ar 33% NPM1m 33% |
| DifferentiationSyndrome (%) | 22% | 19% | 11% | 11% |
Revumenib
Revumenib ia a potent oral small molecule inhibitor of the Menin-KMT2A interaction. This compound was evaluated in monotherapy in R/R AML patients with KMT2A rearrangements or NPM1 mutations. The AUGMENT-101 trial (NCT 04065399) is a phase I clinical stud evaluating the safety and the efficacy of Revumenib in patients with R/R heavily pretreated NPM1-mutant or KMT2A-rearranged AMLs [88]. Since Revumenib is a substrate of CP3A4, the study was subdivided into two cohorts: a cohort A of patients receiving a CP3A4 inhibitor and a cohort B of patients not receiving a CP3A4 inhibitor [85]. A first evaluation of this study was based on 60 patients (46 with KMT2A-rearranged AMLs and 14 with NPM1-mutant AMLs); the ORR was 59% in KMT2A-rearranged AMLs and 36% in NPM1-mutant AMLs; the rate of CR+CRi was 30%; with a median follow-up of 11.9 months in patients who achieved morphologic CR+CRi the median duration of response was 9.1 months; mOS was 14.3 months; 56% of the responder patients achieved MRD negativity and 38% underwent allogeneic hematopoietic stem cell transplantation [88]. Interestingly, in many patients with KMT2A rearrangements achieving morphological remission after one cycle of treatment, there was continued evidence of KMT2A fusions, a phenomenon seemingly related to the induction of leukemic blast differentiation elicited by Revumenib. Differentiation syndrome was observed in 16% of patients, with all cases classified as grade 2 [88]. Additional 8 AML patients NPM1-WT and KMT2A-WT did not respond to Revumenib [88]. Analysis of the pharmacodynamic effects of Revumenib assessed through transcriptional analysis of RNA expression of bone marrow cells showed that Menin inhibition resulted in downregulation of HOXA9 and MEIS1, associated with an increase in the expression of genes related to differentiation, such as CD11b and CD14 [88]. Single cell studies carried out on 4 patients treated with Revumenib showed a differentiation continuum that starts with immature AML blasts (CD34+/c-kit+), progresses through intermediate blast cells (CD68+, CD11b-, CD14-) and ends with more differentiated monocytic cells (CD68+, CD11b+, CD14+); intermediate AML blasts and monocytic cells were enriched in post-treatment samples [89].
A second study reported the results of phase II carried out in 94 R/R KMT2A-rearranged acute leukemia (78 with AML and 14 with ALL); 13% of patients discontinued treatment for adverse events; DS was observed in 26% of patients (14.9% had grade 3 and 1% grade4) [90]. CR+CRi rate was 22.8% with an ORR of 63.2%; the median duration of response was 6.4 months; mOS was 8.0 months; among responding patients, 38.9% received allo-HSCT [90]. Transcriptional changes in bone marrow cells showed a decrease of MEIS1 and HOXA expression and an increase of differentiation-related genes CD11b and CD14 [90]. An updated evaluation of KMT2A-rearranged AML patients enrolled in AUGMENT-001 trial extended to a total of 116 patients showed 23% of patients achieving CR+CRi, with an ORR of 64% and with 5.8% of MRD negativity among patients achieving CR+CRi; 34% of patients who achieved ORR proceeded to HSCT [91].
Recently, the Syndex Pharmaceuticals announced the results of the phase II of the AUGMENT-001 study evaluating the efficacy of Revumenib in 64 R/R heavily pretreayted (75% relapsing after Ventoclax therapy) NPM1-mutated AML patients; 23% of patients achieved a CR+CRi; ORR was 47%; MRD negativity was 64% among patients who achieved CR+CRi; 17% of the responding patients undergo HSCT [92].
ALLG AMLM26 INTERCEPT multiarm study was design to obtain proof-of-concept for novel theraoies targeting MRD or early relapse in AML [93]. In the context of this study, preliminary analysis on 9 NPM1-mutant AML patients with MRD-positivity showed a a NPM1 mutant ≥1 log10 MRD reduction in 62.5 of patients, including 37.5 of patients achieving MRD negativity [93].
A phase I/II study evaluated combination of Revumenib with decitabine/cedazuridine and Venetoclax in R/R AML patients (NPM1-mutant, KMT2A-rearranged and NUP98-rearranged), reporting an ORR of 88%, with a CR+CRi of 58% and with a MRD negativity by flow cytometry of 93% among patients with CR+CRi [94].
In pediatric patients, KMT2A rearrangements are frequently associated with RAS pathway mutations (51% of cases) and with a worse prognosis [95]. A recent preclinical study showed that the combination of a Menin inhibitor (VTP-50469, an analog of Revumenib) and Selutenib (a MEK 1-2 inhibitor) exerted a synergistic antitumor effect in vitro and in PDX models of KMT2A-rearranged AML cells bearing RAS pathway mutations [95].
About 40% of patients treated with Revumenib developed MEN1 mutations and some acquired resistance to Menin inhibitors without MEN1 mutations. Particularly, Perner et al. identified Menin gene mutations that were not present at diagnosis and developed on Revumenib treatment; clonal expansion of these mutations was observed in about 39% of evaluable patients who have performed at least two cycles of treatment [96]. These mutations were found at residues M327, G331 or T349 which do not impact the interaction between KMT2A and Menin or its oncogenic properties but decrease binding affinity of Revumenib to KMT2A and mediate therapeutic resistance [96-97]. A recent study characterized in detail the consequences of Menin mutations in the interaction with Menin inihbitors [93]. The crystal structure of Menin mutants T349M, M327I, G331R and G331D and the N-terminal of MLL1 showed that: drug-resistant mutations in Mnwin occur at a site located in proximity of the MLL1 binding site, but do not affect t MLL1 binding to Menin; all these point mutations in Menin generate a steric collision with Mwnin inhibitors; the G331D mutant show a particularly slow dissociation of MLL1 from Menin and thus seems to be particularly difficult to be inhibited with small molecule inhibitory drugs [98].
The identification of MEN1 mutations in AML patients undergoing treatment with Revumenib is important because may offer opportunities for patients with some MEN1 mutations to derive benefit from treatment with some of new Menin inhibitors.
Bleximenib
JNJ-75276617 (Bleximenib) is a novel potent inhibitor of the protein-protein interaction between Menin and KMT2A; in KMT2A-rearranged and NPM1-mutant leukemia cells, this compound inhibited the interaction of the Menin-KMT2A complex with chromatin at the level of target genes resulting in reduced expression of several target genes such as MEIS1 and FLT3 and exerted a potent antiproliferative activity [94]. Bleximenib displayed a synergistic antileukemic activity with Gilteritinib, Venetoclax and Azacitidine [93]. Interestingly, Bleximenib displayed a strong antiproliferative activity in leukemic cells bearing mutations (MEN1M371 or MEN1T349M) observed in patients refractory to Revumenib [99]. In these mutant AMLs, Bleximenib was still able to displace KMT2A and prevent its interaction with Menin despite the presence of MEN1 mutations that block the acivity of other Menin inhibitors.
A first-in-human phase I clinical study evaluated the safety and preliminary the efficacy of Bleximenib in 58 R/R acute leukemia patients (56 AML and 2 ALL) showing an acceptable safety profile and preliminary evidence of antileukemic efficacy and biologic activity [100].
A phase I clinical study (NCT 04811560) carried out in 121 R/R acute leukemias (mostly AML, 108) patients identified the optimal dose of Bleximenib (100 mg) in monotherapy to be used in phase II studies; at this dose, the ORR was 50% and the CR+CRi was 40% [101]. The enrolled patients had either KMT2A rearrangements or NPM1 mutations. Another recent phase I clinical study explored Bleximenib in association with standard chemotherapy in a group of 22 newly diagnosed AML patients (11 KMT2A-rearranged and 11 NPM1-mutated) showing an acceptable safety profile with no DS or dose-limiting toxicities [102]. The ORR was 93% (83% in KMT2A-rearranged AML and 100% in NPM1-mutated AML); 6 patients proceeded to HSCT [102]. A phase Ib (NCT 05453903) clinical study explored the safety and the efficacy of Bleximenib in combination with Venetoclax and Azacitidine in R/R AML patients with KMT2A rearrangements or NPM1 mutations [98]. In the safety dataset, 45 patients received the triplet combination treatment, with a median of 2 prior lines of treatment, including prior Venetoclax treatment in 56% of cases and allo-HSCT in 27% of cases [102]. The safety profile was acceptable and no patient developed DS or tumor lysis syndrome [103]. In the efficacy dataset, ORR was was 86%, CR+CRi was 48%; for patients with prior Venetoclax exposure, ORR was 82% and CR+CRi was 36% [103]. 9 responder patients discontinued treatment and proceeded to allo-HSCT [103].
Hogeling et al. recently reported a large in vitro careening of primary AML cells incubated in the presence of Bleximenib [104]. This study showed that AML cells bearing NPM1 mutations or KMT2A rearrangements are sensitive to the antiproliferative and differentiation-inducing effects of Bleximenib [104]. Concerning NPM1-mutant AMLs, those bearing both NPM1 and DNMT3A mutations are those markedly sensitive to the induction of differentiation [104]. In addition to these AML subtypes, also AML characterized b a granulo-monocyte progenitor (GMP)-like phenotype display sensitivity to Bleximenib: these cases include leukemias bearing CEBPA mutations [104]. Gene expression studies showed that AMLs responding to Bleximenib display some epigenetic alterations inducing a striking upregulation of HLA class I and class II expression through a mechanism independent of MEIS 1 loss [99]. These epigenetic changes resulted in an enhanced sensitivity of leukemic cells to T cell-mediated cytotoxicity in allogeneic and autologous settings [104].
In line with this observation, previous studies have shown that: CEBPA is an essential collaborator in HOXA9/MEIS 1-mediated leukemogenesis [105]; CEBPA and KMT2A are co-localized on chromatin and CEBPA-mutated HPCs are hypersensitive to pharmacologic targeting of KMT2A complex using Menin inhibitors [106].
DSPP-5336 (Enzomenib)
Enzomenib is a potent, orally bioavailable inhibitor of menin-MLL interaction exhibiting antiproliferative activity in vitro in cell lines, primary leukemic blasts and patient-derived xenograft mouse models of KMT2A-rearranged or NPM1-mutant AMLs [102]. Enzomenib directly binds to Menin (Kd 6.0 nM) and inhibits Menin-KMT2A interaction (IC50 1.4 nM) [107].
A phase I/II clinical study evaluated 81 patients in Arm A (without CYYP3A4 inhibitor ) or in Arm B (with CYP3A4 inhibitor) for the safety and efficacy of a treatment based on Enzamenib monotherapy [108]. In the phase I portion of the stud it was evaluated that the active doses of Enzomenib correspond to ≥140 mg [109]. 35 patients were treated at the active Enzomenib dose: 22 KMT2A-rearranged (20 AML and 2 ALL) with an ORR of 59% and a CR+CRi of 22.7%; 13 NPM1-mutant AML patients with an ORR of 53.8% and CR+CRi of 23% [109]. Pharmacodynamic changes supported induction of cell differentiation with decrease in stemness markers (HOXA9, MEIS1) and increase in differentiation markers (CD11b) [104]. DS was observed in 11% of treated patients.
KO-539 (Ziftomenib)
Ziftomenib is an oral menin inhibitor targeting the Menin-KMT2A protein-protein interaction; Ziftomenib is metabolized to two metabolites with comparable activity to Ziftomenib itself. Preclinical studies have supported the antileukemic activity of Ziftomenib in models of NPM1-mutant and KMT2A-rearranged AMLs. Particularly, Ziftomenib induced Menin protein degradation through the ubiquitin-proteasome pathway, reduces MEIS1, FLT3, CDK6 and BCL2 protein levels, in association with induction of differentiation and induction of cell apoptosis of leukemic cells harboring MLL-rearrangements or NPM1 mutations [110]. Furthermore, co-treatment with Ziftomenib with BCL-2,CDK6 or BET inhibitors induced synergistic lethality in MLL-rearranged or NPM1-mutant leukemic cells [110]. These findings were confirmed and extended in a second study showing that Ziftomenib had a marked anti-proliferative activity in combination with drugs from various classes, including those targeting chromatin regulation and DNA damage as well as apoptosis and cell cycle block [111]. Particularly pronounced was the synergistic interaction between Ziftomenib and Venetoclax resulting in a pronounced anti-proliferative activity in MLL-rearranged and NPM1-mutant leukemic cells [111].
The KOMET-001 multicentre, open-label, multicohort, phase I/II clinical trial evaluated Ziftomenib in adult R/R AML patients; the study was subdivided into two phases: a phase Ia (dose-response) in which the patients received Ziftomenib (from 50 to 1000 mg) orally once daily in 28 day-cycles; a phase Ib in which patients with KMT2A rearrangements or NPM1 mutations were randomly assigned to two parallel dose cohorts (200 and 600 mg Ziftomenib) [112]. In phase Ib no responses were observed in patients treated at 200 mg of Ziftomenib; at the recommended dose for phase II (600 mg) the rate of CR+CRi was 12.5% (2/16) in KMT2A-rearranged and 35% (7/20) in NPM1-mutant AML patients; the ORR in KMT2A-rearranged was 17% and 45% in NPM1-mutant AMLs; the median OS was 6.0 and 5.6 months, respectively in KMT2A-rearranged and NPM1-mutant AMLs [112].
The KOMET-007 phase I clinical study evaluated the safety and the efficacy of Ziftomenib combined with standard chemotherapy in 34 newly diagnosed AML patients with KMT2A rearrangements or NPM1 mutations; the patients were treated either with 200 or 400 mg of Ziftomenib [113]. For NPM1-mutant AML patients, the CR+CRi rate was 100% at 200 mg and 86% at 400 mg, with MRD negativity among responders of 100% and 80%, respectively; for KMT2A-rearranged patients, CR+CRi rates were 90% at 200 mg and 63% at 400 mg, with MRD negativity among responders of 83% and 100%, respectively [113].
The ongoing KOMET-008 trial is an open-label, dose-escalation and expansion study aiming to determine the safety, tolerability, and preliminary efficacy of Ziftomenib in association with standard of care regimens for treatment of either R/R NPM1-mutant AML (arm A) or KMT2A-rearranged AML (arm B). The arm A is subdivided into 3 cohorts: cohort A-1 (Ziftomeniob+FLAG-IDA), cohort A-2 (Ziftomenib plus LDAC) and cohort A-3 (Ziftomenib plus Gilteritinib in NPM1/FLT3 double mutant AML). The arm B is subdivided in two cohorts: cohort B-1 (Ziftomenib plus FLAG-IDA), cohort B-2 (Ziftomenib plus LDAC) [114].
BMF-219 (Icovamenib)
BMF-219 is the only covalent Menin inhibitor in clinical development under evaluation in multiple hematologic malignancies, solid tumors and diabetes mellitus, Preclinical studies have shown that BMF-219 show sustained and marked inhibition of Menin-dependent oncogenic signaling.
The phase I COVALENT-101 phase I dose-escalation and dose-expansion study evaluated BMF-219 in R/R acute leukemia (cohort 1), DLBCL (cohort 2), multiple myeloma (Cohort 3) and CLL (cohort 4) [115] A recent report analyzed the results observed in 26 R/R AL patients (24 AML and 2 ALL) enrolled in two parallel arms, with or without a CYPP314 inhibitor [115]. BMF-219 was usually well tolerated, with no dose-limiting toxicities; DS was observed in 13% of cases [115]. 5 patients were evaluable for response and 2 achieved a CR (1 CR and 1CRi) [115].
Recurrent chromosomal rearrangements involving the Nucleoporin 98 (NUP98) gene, detected in 5-10% of pediatric AML cases and in 2-4% of adult AMLs, are classified as high-risk AMLs [116]. In NUP98 rearrangements the NUP98 gene is fused with various partners, the most frequent being NSD1 and KDM5A [116]. NUP98 fusions elicit leukemogenesis through interaction with histone modifying complexes: particularly, NUP98 fusion proteins interact with KMT2A complexes and are co-localized with KMT2A at the level of HOX A/B genes [117]. NUP98-rearranged AMLs are Menin-dependent as supported by the observation that the Menin inhibitor VTP50469 induced antiproliferative effects and prolongation of survival in mouse AML models driven NUP98 fusion proteins [118].
Another study showed that the Menin inhibitor Revumenib inhibited proliferation and survival of primary NUP08 fusion protein-positive AML cells and inhibited numerous NUP fusion protein target genes, such as MEIS1 and CDK6 [114].
In order to exert their leukemogenetic activity NUP98 fusion proteins and KMT2A-Menin antagonize the non-canonical polycomb repressive complex 1.1 (PRC1.1) [115].
Interestingly, Carraway et al. reported the case of a patient with NUP98-fusion relapsed after prior treatments, including allo-HSCT; the patient was treated with BMF-219 and achieved a CR after few cycles of treatment; unfortunately, after 5 months of treatment, the patient relapsed with NUP98-NSD1-positive AML [115].
The findings of this case report suggest that Menin inhibitor monotherapy is not sufficient to obtain a complete eradication of leukemic clones in NUP98 fusion-positive AML patients and that the cooperation between Menin inhibition and kinase inhibitors targeting either CDK6 or nFLT3 strongly cooperate in NUP98 rearranged primary AML cells and in PDX models [116].
3. Conclusions
In AML, different genetic events determine a block in the differentiation of leukemic cells at various stages of the differentiation/maturation process. Targeted therapy developed in the late twentieth century have introduced a new therapeutic approach based on the targeting of a specific abnormality present in a subset of AML cells. One of the aims of target therapy consists in forcing leukemic cells to differentiate. The prototype of successful differentiation therapy was represented by the treatment of APL using differentiation-inducing agents ATRA and ATO that have completely revolutionized the therapy of this AML subtype, transforming the outcome of this leukemic subtype from the deadliest to the most curable.
However, ATRA and ATO resulted to be not effective in AML outside APL, thus providing evidence that differentiation block observed in various AML subtypes does not have a uniform underlying mechanism causing differentiation impairment. Thus, it emerged the idea that the strategy to induce differentiation in other AML subtypes could be induced through the targeting of specific alterations during the leukemic process. This view was strongly supported by recent studies showing that IDH inhibitors, LSD1 inhibitors and Menin inhibitors induce the differentiation of some AML subtypes. Although these agents have shown a marked capacity to induce leukemic cell differentiation associated with antileukemic effects, the therapeutic success was clearly lower than that observed using ATRA+ATO in APL. This lower efficacy of differentiation therapy based on IDH, LSD1 and Menin inhibitors compared to ATRA+ATO in APL is seemingly related to a higher genetic complexity and heterogeneity of IDH-mutant AMLs, KMT2A-rearranged and NPM1-mutant AMLs compared to APL cells in which all the pathogenic events are driven by PML-RARA fusion protein.
However, in spite these difficulties, these agents represent an important therapeutic tool for improving the outcomes of IDH-mutant, IDH-mutant and KMT2A-rearranged AML patients and possibly also of other AML subtypes. Optimal responses induced by IDH, LSD1 and Menin inhibitors may be significantly improved by strategic partnering with other therapies: ongoing and future clinical studies will clarify the impact of these association therapies, appropriately extended to AML patients at various disease stages. In this context, promising is the introduction of some of these agents into first-line treatment of elderly non-fit patients using triplet drug combinations.
Future studies will be required to evaluate the impact of these agent on overall survival in the context of various therapeutic settings involving R/R and first-line patients. Future studies will assess also the potential therapeutic impact of these drugs as maintenance therapy or treatment for eradication of MRD.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
All authors equally contributed to the conceptualization and writing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Breitman, T.R.; Colling, S.J.; Keene, B.R. Terminal differentiation of human promyelocytic leukemic cells in primary culture in response to retinoic acid. Blood 1981, 57, 100–1004. [Google Scholar] [CrossRef]
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.R.; Lu, J.X.; Zhou, L.; Gu, L.J.; Wang, Z.Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Degos, L.; Chomienne, C.; Daniel, M.T.; Berger, R.; Dombret, H.; Fenaux, P.; Castaigne, S. Treatment of rist relapse in acute promyelocytic leukemia with all-trans retinoic acid. Lancet 1990, 336, 1440–1441. [Google Scholar] [CrossRef] [PubMed]
- Castaigne, S.; Chomienne, C.; Daniel, M.T.; Ballerini, P.; Berger, R.; Fenaux, P.; Degos, L. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia, I: clinical results. Blood 1990, 76, 1704–1709. [Google Scholar] [CrossRef]
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejan, A. The t(15;17) translocation of acute promyelocytic leukemia fuses the retinoic acid receptor alpha geneto a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef]
- Grignani, F.; Ferrucci, P.F.; Testa, U.; Talamo, G.; Fagioli, M.; Alacalay, M.; Mencarelli, A.; Grignani, F.; Peschle, C.; Nicoletti, I.; et al. The acute promyelocytic leukemia-specific PML-RARα fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell 1993, 74, 423–431. [Google Scholar] [CrossRef]
- Lo-Coco, F.; Avvisati, G.; Vignetti, M.; Thiede, C.; Orlando, S.M.; Iacobelli, S.; Ferrara, F.; fazzi, P.; Cicconi, L.; Di Bona, E.; et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med 2013, 369, 111–121. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Isocitrate dehydrogenase mutations in myelodysplastic syndromes and in acute myeloid leukemia. Cancers 2020, 12, 2427. [Google Scholar] [CrossRef]
- Hoff, F.W.
- Falini, B.; Spinelli, O.; Meggendorfer, M.; Martelli, M.P.; Bigerna, B.; Ascani, S.; Stein, H.; Rambaldi, A.; Haferlach, T. IDH1-R132 changes vary according to NPM1 and other mutations status in AML. Leukemia 2019, 33, 1043–1047. [Google Scholar] [CrossRef]
- Zamegar-Lumley, S.; Alonzo, T.A.; Gerbing, R.B.; Othus, M.; Sun, Z.; Reis, R.E.; Wang, J.; Leonti, A.; Kutny, M.A.; et al. Characteristics and prognostic impact of IDH mutations in AML: a COG, SWOG and ECOG analysis. Blood Adv 2023, 7, 5941–5953. [Google Scholar] [CrossRef]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brustle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Heigth, J.; et al. IDH1(R132H) mutation increase murine hematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Buegess, K.; Bhagarva, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant Idh2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef]
- Wang, D.; Zheng, L.; Cheng, B.Y.L.; Sin, C.F.; Li, R.; Tsui, S.P.; Yi, X.; Ma, A.C.H.; he, B.L.; Leung, A.Y.H.; et al. Transgenic IDH2R172K and IDH2R140Q zebrafish model recapitulated features of human acute myeloid leukemia. Oncogene 2023, 42, 1272–1281. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagvat, N. , Vasannthakumar, A.; Fernandez, H.F.; et al. Leukemic Idh1 and Idh2 mutations result in a hypermethylation phenotype, disrupt Tet2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohe, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; Wellen, K.E. IDH mutation impais histohe demethylation and results in a block of cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, P.; Schneider, R.K.; McMahon, R.K.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G. 8R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Trafins, J.; DeLa Barre, B.; Penard-Lacronique, V.; Schalm, V.; Hansen, E.; Starley, K.; Kewrnytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant Idh2 in leukemnia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Pierangeli, S.; Donnini, S.; Ciaurro, V.; Milano, F.; Cardinali, V.; Sciabolacci, S.; Cimino, G.; Gianfriddo, I.; Ranieri, R.; Cipriani, S.; et al. The leukemic isocitrate dehydrogenase (IDH) 1 / 2 mutations impair myeloid and erythroid cell differentiation of primary human hematopoietic stem and progenitor cells (HSPCs). Cancers 2024, 16, 2675. [Google Scholar] [CrossRef]
- Landberg, N.; Koehnke, T.; Nakauchi, Y.; Fan, A.; Linde, M.H.; Karigane, D.; Thomas, D.; Majeti, R. Targeting Idh1-mutated pre-leukemic hematopoietic stem cells in myeloid disease, including CCUS and AML. Blood 2022, 140, 2234–2235. [Google Scholar] [CrossRef]
- Landberg, N.; Koehnke, T.; Feng, Y.; Nakauchi, Y.; Fan, A.; Karigane, D.; Lim, K.; Sinha, R.; Malcovati, L.; et al. Idh1-mutant preleukemic hematopoietic stem cells can be eliminated by inhibition of oxidative phosphorylation. Blood Cancer Discov 2024, 5, 114–131. [Google Scholar] [CrossRef]
- Wilson, E.R.; Helton, E.M.; Heath, S.E.; Fulton, S.R.; Payton, J.E.; Welch, J.S.; Walter, M.J.; Westervelt, P.; DiPersio, J.F.; Link, D.C.; et al. Focal disruption of DNA methylation dynamics at enhancers in IDH-Mutant AML cells. Leukemia 2022, 36, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Di Nardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, J.K.; Altman, A.S.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N. , Pollyea, D.A.; et al. Durable remissions with ivodisenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Di Nardo, C.D.; Stein, E.M.; Pigneux, A.; Altman, J.K.; Collins, R.; Erba, E.P.; Watts, J.M.; Uy, G.L.; Winkler, T.; Wang, H.; et al. Outcomes of patients with IDH1-mutant relapsed or refractory acute myeloid leukemia receiving ivosidenib who proceeded to hematopoietic stem cell transplant. Leukemia 2021, 35, 3278–3281. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Mims, A.S.; Prince, G.T.; Altman, J.K.; Arellano, M.L.; Donnellan, W.; Erba, H.P.; et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1-mutant acute myeloid leukemia. Blood 2020, 135, 463–471. [Google Scholar] [CrossRef]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycha, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.P.; et al. Ivosidenib and azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med 2022, 386, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Marvin-Peek, J.; Garcia, J.S.; Borthakur, G.; Garcia-Manero, G.; Short, N.J.; Kadia, T.M.; Loghavi, S.; Masarova, L.; Daver, N. A phase Ib/II study of Ivosidenib ± Azacitidine in IDH1-mutated hematologic malignancies: a 2024 update. Blood 2024, 144 (suppl.1), 219–221. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Fathi, A.T.; Mims, A.S.; Pratz, K.W.; Savona, M.R.; Stein, A.S.; Stone, R.M.; Winer, E.S.; et al. Ivosidenib or enasidenib combined with intensive chemotherapy in patients with newly diagnosed AML: a phase I study. Blood 2021, 137, 1792–1803. [Google Scholar] [CrossRef]
- Mason, E.F.; Pozdynakova, O.; Roshal, M.; Fathi, A.T.; Stein, E.M.; Ferrell, P.B.; Shaver, A.C.; Frattini, M.; Wang, H.; Hua, L.; et al. A novel differentiation response with combination IDH inhibitor and intensive induction therapy for AML. Blood Adv 2021, 5, 2279–2283. [Google Scholar] [CrossRef]
- Fathi, A.T.; Kim, H.T.; Soiffer, R.J. ; Levis. M.J.; Li, S.; Kim, A.S; DeFilipp, Z.; El-Jewari, A.; MCAfee, S; Brunner, AM; et al. Multicenter phase I trial of ivosidenib as maintenance treatment following as maintenance treatment following allogeneic hematopoietic cell transplantation for IDH1-mutated acute myeloid leukemia. Clin Cancer Res 2023, 29, 2034–2042. [Google Scholar]
- Choe, S.; Wang, H.; Di Nardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following Ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv 2020, 4, 1894–1905. [Google Scholar] [CrossRef] [PubMed]
- Turkalj, S.; Stoilova, B.; Groom, A.J.; Radtke, F.E.; Mecklenbrauck, R.; Jakobsen, N.A.; Lachowiez, C.A.; Metzner, M.; Usukhbayar, B.; Salazar, M.A; et al. Clonal basis of resistance and response to ivosidenib combination therapies is established early during treatment in IDH1-mutated myeloid malignancies. Blood 2024, 144, 642–644. [Google Scholar] [CrossRef]
- Caravella, J.A.; Lin, j.; Diebold, R.B.; Campbell, A.M.; Ericsson, A.; Gustafsson, G.; Wang, Z.; Castro, J.; Clarke, A.; Gofur, D.; et al. Structure-based design and identification of FT-2102 (Olutasidenib), a potent mutant-selective IDH1 inhibitor. J Med Chem 2020, 63, 1612–1623. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lu, W.; Caravella, J.A.; Campbell, A.M.; Diebold, FR.B.; Ericsson, A.; Fritzen, E.; Gustafson, G.R.; Lancia, D.R.; Shelekhin, T. Discovery and optimization of quinolone derivatives as potent, selective, and orally bioavailable mutant ioscitreate dehydrogenase 1 (mIDH1) inhibitors. J Med Chem 2019, 62, 6575–6592. [Google Scholar] [CrossRef]
- Watts, J.M.; Baer, M.R.; Yang, J.; Prebet, T.; Lee, S.; Schiller, G.J.; Dinner, S.N.; Pigneux, A.; Montesinos, P.; Wang, A.S.; et al. Olutasedinib alone or with azicitidine in IDH1-mutated acute myeloid leukemia and myelodysplastic syndrome: phase 1 results fo a phase 1-2 trial. Lancet Haematol 2023, 10, e46–e58. [Google Scholar] [CrossRef]
- De Botton, S.; Fenaux, P.; Yee, K.; Récher, C.; Wei, A.H.; Montesinos, P.; Taussig, D.C.; Pigneux, A.; Braun, T.; Curti, A.; et al. Olutasedinib (FT-2102) induces durable complete remissions in patients with relapsed or refractory IDH1-muatted AML. Blood Adv 2023, 7, 3117–3127. [Google Scholar] [CrossRef]
- Cortes, J.E.; Jonas, B.A.; Watts, J.M.; Chao, M.M.; De Botton, S. Olutasidenib for mutated IDH1 acute myeloid leukemia: final five-year results from the phase 2 pivotal cohort. J Clin Oncol 2024, suppl 16, 6528. [Google Scholar] [CrossRef]
- De Botton, S.; Jonas, B.A.; Ferrell, B.; Choa, M.M.; Mims, A.S. Safety and efficacy of olutasidenib treatment in elderly patients with relapsed/refractory mIDH1 acute myeloid leukemia. J Clin Oncol 2024, 42, suppl–16. [Google Scholar] [CrossRef]
- Cortes, J.E.; Esteve, J.; Bajel, A.; Yee, K.; Braun, T.; De Botton, S.; Peterlin, P.; Recher, C.; Thomas, X.; Watts, J.; et al. Olutasidenib (FT-2102) in combination with azacitidine induces durable complete remissions in patients with mIDH1 acute myeloid leukemia. Blood 2021, 138, 698–701. [Google Scholar] [CrossRef]
- Cortes, J.E.; Roboz, G.J.; Watts, J.; Baer, M.R.; Jonas, B.A.; Schiller, G.J.; Yee, K.; Ferrell, B.; Yang, J.; Wang, E.S.; et al. Combination of olutasidenib and azacitidine induces durable complete remissions in mIDH1 acute myeloid leukemia: a multicohort open-label phase ½ trial. Blood 2024, 144 (suppl.1), 2886. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Chien, K.S.; Mullin, J.; Hammond, D.; Ramdial, J.; Kadia, T.M.; Haddad, F.G.; Yilmaz, M.; Sasaki, K.; Issa, G.C.; et al. Phase ½ study of decitabine and venetoclax in combination with the targeted mutant IDH1 inhibitor olutasidenib for patients with relapsed/refractory AML, high risk MDS, or newly diagnosed AML not eligible for chemotherapy with an IDH1 mutation. Blood 2024, 144 (suppl.1), 617. [Google Scholar] [CrossRef]
- Lai, C.E.; Leahy, T.P.; Turner, A.; Thomassen, A.; Wang, L.; Sheppard, A.; Cortes, J.E. Effectiveness of olutasidenib versus ivosidenib in patients with mutated isocitrate dehydrogenase 1 acute myeloid leukemia who are relapsed or refractory to venetoclax: the 2102-HEM-101 trial versus a US electronic health record-based external control arm. Blood 2024. [Google Scholar] [CrossRef]
- Watts, J.M.; Shaw, S.J.; Jona, B.A. Looking beyond the surface: olutasidenib and ivosidenib for treatment of mIDH1 acute myeloid leukemia. Curr Treat Options Oncol 2024, 25, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Travens, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLa Barre, B.; Tobin, E.; et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discover. 2017, 7, 478–493. [Google Scholar] [CrossRef]
- Stein, E.M.; Di Nardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; De Angelo, D.J.; Levine, R.L.; Finn, J.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Tallman, M.S.; De Botton, S.; Komborjian, A.M.; Collins, R.; Stein, A.S.; Frattini, M.G.; Xu, Q.; Tosolini, A.; See, W.L.; et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia 2019, 33, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Amatangelo, M.D.; Quek, L.; Shih, A.; Stein, E.M.; Roshal, M.; David, M.D.; Marteyn, B.; Farnoud, N.R.; de Botton, S.; Bernard, O.A.; et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood 2017, 130, 732–741. [Google Scholar] [CrossRef]
- De Botton, S.; Montesinos, P.; Schuh, A.C.; Papayannidis, C.; Vyes, P.; Wei, A.H.; Ommen, H.; Semochkin, S.; Kim, H.J.; Lrsom, R.A.; et al. Enasidenib versus conventional care in older patients with late-stage mutant IDH2-relapsed/refractory AML: a randomized phase 3 trial. Blood 2023, 141, 156–164. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Schuh, A.C.; Stein, E.M.; Montesinos, P.; Wei, A.H.; de Botton, S.; Ziedan, A.M.; Fathi, A.T.; Kantarjian, H.M.; Bennett, J.M.; et al. Enasidenib plus azacitidine versus azacitidine alone in patients with newly diagnosed, mutant-IDH2 acute myeloid leukemia (AG221-AML-005): a single-arm, phase 1b and randomized, phase 2 trial. Lancet Oncol 2021, 22, 1597–1608. [Google Scholar] [CrossRef]
- Cai, S.F.; Huang, Y.; Lance, J.R.; Mao, H.C.; Dunbar, A.J.; McNulty, S.N.; Druley, T.; Li, Y.; Baer, M.R.; Stock, W.; et al. A study to assess the efficacy of enasidenib and risk-adapted addition of azacitidine in newly diagnosed IDH2-mutant AML. Blood Adv 2024, 8, 429–440. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Gupta, G.; Cameron, C.; Chatelin, S.; Bankar, A.; Davidson, M.B.; Gupta, V.; Maze, D.C.; Minden, M.D.; et al. Final results of the phase Ib/II study evaluating enasidenib in combination with venetoclax in patients with IDH2-mutated relapsed/refractory myeloid malignancies. Blood 2023, 142 (suppl.1), 159–161. [Google Scholar] [CrossRef]
- Salhotra, A.; Bejanyan, N.; Yang, D. Multicenter pilot clinical triasl of enasidenib as maintenance therapy after allogeneic hematopoietic cell transplantation (allo-HCT) in patienys with acute myeloid leukemia (AML) carrying IDH2 mutations. Transplantation and Cellular Therapies Meeting 2024, Sant Antonio, Texas, USA, abst 10.
- Ball, B.J.; Zhang, J.; Afkhami, M.; Robbins, M.; Chang, L.; Humpal, S.; Porras, V.; Liu, Y.; Synold, T.; Farzinkhou, S.; et al. Study of IDH inhibition with Enasidenib and MEK inhibition with Cobimetinib in patients with relapsed or refractory acute myeloid leukemia who have co-occurring IDH2 and RAS signling gene mutations. Blood 2024, 144 (suppl.1), 6043–6044. [Google Scholar] [CrossRef]
- Sebert, M.; Chevrer, S.; Dimicoli-Salazar, S.; Cluzeau, T.; Rauzy, O.; Bastard, A.S.; Lribi, K.; Fosard, G.; Thépot, S.; Gloaguen, S.; et al. Enasidenib (ENA) monotherapy in patients with IDH2 muatted myelodysplastic syndrome (MDS), the ideal phase 2 study by the GFM and Emsco groups. Blood 2024, 144, 1839–1840. [Google Scholar] [CrossRef]
- Montesinos, P.; Bergua, J.M.; Vellenga, E.; Rayon, C.; Parody, R.; de la Serna, J.; Leon, A.; Esteve, J.; Milone, G.; Debén, G.; et al. Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors. Blood 2009, 113, 775–783. [Google Scholar] [CrossRef]
- Luesink, M.; Pennings, J.; Wissink, W.; Linssen, P.; Muus, P.; mPfundt, R.; de Witte, T.; van der Reijden, B.; Jansen, J.H. Chemokine induction by all-trans retinoic acid and arsenic trioxide in acute promyelocytic leukemia: triggering the differentiation syndrome. Blood 2009, 114, 5512–5521. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Montesinos, P. How we prevent and treat differentiation syndrome in patients with acute promyelocytic leukemia. Blood 2014, 123, 2777–2782. [Google Scholar] [CrossRef]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; AG221-C-001 Study Investigators. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1-2 study. JAMA Oncol 2018, 4, 1106–1110. [Google Scholar] [CrossRef]
- Nosworthy, K.J.; Mulkey, F.; Scott, E.C.; Ward, A.F.; Przepiorka, D.; Charlab, R.; Dorff, S.E.; Deisseroth, A.; Kazandjian, D.; Sridhara, R.; et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. food and drug administration systematic analysis. S. food and drug administration systematic analysis. Clin Cancer Res 2020, 26, 4280–4288. [Google Scholar]
- Montesinos, P.; Fathi, A.T.; de Botton, S.; Stein, E.M.; Zeidan, A.M.; Zhu, Y.; Prtebet, T.; Vigil, C.E.; Bluemmert, I.; Yu, X.; et al. Differentiation syndrome associated with treatment with IDH2 inhibitor enasidenib: pooled analysis from clinical trials. Blood Adv 2024, 6, 2509–2520. [Google Scholar] [CrossRef]
- Sacilotto, N.; Dessanti, P.; Lufino, M.M.P.; Ortega, A.; Rodriguiez-Gimeno, A.; Salas, J.; Maes, T.; Bues, C.; Mascarò, C.; Soliva, R. Comprehensive in vitro characterization of the LSD1 small molecule inhibitor class in oncology. ACS Pharmacol Transl Sci 2021, 4, 1818–1834. [Google Scholar] [CrossRef]
- Baby, S.; Shinde, S.D.; Kulkarni, N.; Sahu, B. Lysine-specific demethylase 1 (LSD1) inhibitors: peptides as emerging class of therapeutics. ACS Chem Biol 2023, 18, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Sprussel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Handschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; Konig, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef]
- Lynch, J.T.; Spence, G.J.; Harris, W.J.; Maiques-Diaz, A.; Ciceri, F.; Huang, X.; Somervaille, T. ; Pharmacological inhibitors of LSD1 promote differentiation of myeloid leukemia cells through a mechanism independent of histone demethylation. Blood Adv 2014, 124 (suppl.1), 267. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Spencer, G.J.; Lynch, J.T.; Ciceri, F.; Williams, E.L.; Amaral, F.; Wiseman, D.H.; Harris, W.J.; Li, Y.; Sahoo, S.; et al. Enhancer activation by pharmacologic displacement of LSD1 from GFI1 induces differentiation in acute myeloid leukemia. Cell Rep 2018, 22, 3641–3659. [Google Scholar] [CrossRef]
- Barth, J.; Abou-El-Ardat, K.; Dalic, D.; Kurrle, N.; Maier, A.M.; Mohr, S.; Schutte, J.; Vassen, L.; Greve, G.; Schulz-Fincke, J.; et al. Lsd1 inhibition by tranylcypromine derivatives interferes with GFI1.mediated repression of PU.1 target genes and induces differentiation in AML. mediated repression of PU.1 target genes and induces differentiation in AML. Leukemia 2019, 33, 1411–1426. [Google Scholar]
- Cusan, M.; Cai, S.F.; Mohammad, H.P.; Krivstov, A.; Chramiec, A.; Loizou, E. LSD1 inhibition exerts in antileukemic effect by recommissioning PU.1 and C/EBPα-dependent enhancers in AML. Blood 2018, 131, 1730–1742. [Google Scholar] [CrossRef]
- Maiques-Diaz, A.; Nicosia, L.; Bosma, N.J.; Romero-Camarero, I.; Camera, F.; Spencer, G.J.; Amad, F.; Sineom, F.; Wingelhafer, B.; Williamson, A.; et al. HMG20B stabilizes association of LSD1 with GFI1 on chromatin to confer transcription repression and leukemia differentiation block. Oncogene 2022, 44, 4841–4854. [Google Scholar] [CrossRef] [PubMed]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic regulation of hematopoietic differentiation by GFI1 and GFI1B is mediated by the cofactors CoREST and LSD1. Mol Cell 2007, 27, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Moroy, T.; Vassen, L. , Wilkes, B.; Khandanpour, C. From cytopenia to leukemia. The role of GFI1 and GFI1B in blood formation. Blood 2015, 126, 2561–2569. [Google Scholar] [CrossRef]
- Waterbury, A.L.; Kwok, H.S.; Lee, C.; Narducci, D.N.; Freedy, A.M.; Su, C.; Raval, S.; Reiter, A.H.; Hawkins, W.; Lee, K. An autoinhibitory switch of the LSD1 disordered region controls enhancer silencing. Mol Cell 2024, 84, 2238–2254. [Google Scholar] [CrossRef]
- Maes, T.; Mascarò, C.; Tirapu, I.; Estiarte, A.; Ciceri, F.; Lunardi, S.; Guibourt, N.; Perdone, A.; Lufino, M.M.; Somervaille, T.; et al. ORY-1001, a potent and selective covalent KMD1A inhibitor, for the treatment of acute leukemia. Cancer Cell 2018, 33, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Salamero, O.; Montesinos, P.; Willekens, C.; Perez-Simon, J.A.; Pigneux, A.; Rècher, C.; Popat, R.; Carpo, C.; Molinero, C.; Mascaro, C.; et al. First-in-human phase I study of Iadademstat (ORY-1001): a first-in-class lysine-specific histone demethylase 1A inhibitor, in relapsed or refractory acute myeloid leukemia. J Clin Oncol 2020, 38, 4260–4273. [Google Scholar] [CrossRef] [PubMed]
- Salamero, O.; Molero, A.; Perez-Simon, J.A.; Arnan, M.; Cokli, R.; Garcia-Avila, S.; Acuna-Criz, E.; Cano, I.; Somervaille, T.; Gutierrez, S.; et al. Iadademstat in combination with azacitidine in patients with newly diagnosed acute myeloid leukemia (ALICE): an open-label, phase 2a dose-finding study. Lancet Hematol 2024, 2024. 11, e487–e498. [Google Scholar] [CrossRef]
- Fathi, A.; Braun, T.P.; Abinder, A.J.; Borthakur, G.; Redner, R.L.; Arevalo, M.; Gutierrez, S.; Limon, A.; Faller, D.V. Iadademstat and gilteritinib for the treatment of FLT3-mutated relapsed/refractory acute myeloid leukemia: the Frida study. Blood 2023, 142 (suppl.1), 5974–5975. [Google Scholar] [CrossRef]
- Fathi, A.; Braun, T.P.; Abinder, A.J.; Palmisano, N.; Khurana, S.; Strickland, S.; Murthy, G.; Venugopal, S.; Feld, J.; Sanchez, E.; et al. Preliminary results of the FRIDA study: iadademstat and gilteritinib in FLT3-mutated R/R AML. EHA 2024.
- Brunett, L.; Gundry, M.C.; Sorcini, D.; Guzman, A.G.; Huang, Y.H.; Ramabradan, R.; et al. Mutant NPM1 maintains the leukemic state through HOX expression. Cancer Cell 2018, 34, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Uckelmann, H.J.; Haaer, E.L.; Takeda, R.; Wong, E.M.; Hatton, C.; Marinaccio, C.; et al. Mutant NPM1 directly regulates oncogenic transcription in acute myeloid leukemia. Cancer Discov 2023, 13, 746–765. [Google Scholar] [CrossRef]
- Wang, X.Q.D.; Fan, D.; Han, Q.; Liu, Y.; Miao, H.; Wang, X.; et al. Mutant NPM1 hiajcacks transcriptional hubs to maintain pathogenic gene programs in acute myeloid leukemia. Cancer Discov 2023, 13, 724–745. [Google Scholar] [CrossRef]
- Khan, I.; Amin, M.A.; Eklund, E.A.; Gartel, A.L. Regulation of HOX gene expression in AML. Blood Cancer J 2024, 14, 42. [Google Scholar] [CrossRef]
- Kubota, Y.; Reynold, M.; Williams, N.D.; Kawashima, N.; Bravo-Perez, C.; Guarnera, L.; Haddad, C.; Mandala, A.; Guarnari, C.; Durmaz, A.; et al. Genomic analyses unveil the pahtogenesis and inform on therapeutic targeting in KMT2A-PTD AML. Blood 2023, 142 (suppl.1), 5696. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.P.; Smith, K.S.; et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Wallace, L.K.; Peplinski, J.H.; Ries, R.E.; Kirkey, D.C.; Meshinchi, S. The AML HOX-ome: the landscape of developmental transcription factors across pediatric acute myeloid leukemia. Blood 2024, 144 (suppl.1), 618. [Google Scholar] [CrossRef]
- Klossowski, S.; Miao, H.; Kempinska, K.; Wu, T.; Purohit, T.; Kim, E.; Linhares, B.; Chen, D.; Jih, G.; Perkey, E.; Huang, H.; et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J Clin Invest 2020, 130, 981–997. [Google Scholar] [CrossRef] [PubMed]
- Soto-Feliciano, Y.; Sanchez-Rovera, F.; Perner, F.; Barrows, D.; Kastenhuber, E.; Ho, Y.J.; Carool, T.; Xiong, Y.; Aand, D.; Soshnev, A.A.; et al. A molecular switch between mammalian MLL complexes dictates response to menin-MLL inhibition. Cancer Discov 2023, 13, 146–169. [Google Scholar] [CrossRef] [PubMed]
- Ciaurro, V.; Skwarska, A.; Daver, N.; Konopleva, M. .; Menin inhibtor DS-1594b drives differentiation and induces synergistic lethality in combination with venetoclax in AML cells with MLL-rearranged and NPM1 mutation. Blood 2022, 140 (suppl.1), 3082–3083. [Google Scholar] [CrossRef]
- Issa,.C.; Aldoss, I.; DiPersio, J.; Cuglievan, B.; Stone, R.; Arellano, M.; Thirman, M.J.; Patel, M.R.; Dickens, D.S.; Shenoy, S.; t al. The menin inhibitor revumenib in KMT2A-rearraned or NPM1-mutant leukemia. Nature 2023, 615, 920–926.
- Muftuoglu, M.; Basyal, M. ; Ayoub,nE.; Lv, J.; Bedoy, A.; Patsilevas, T.; Bidikian, A., Issa, G.S.; Andreef, M. Single-cell proteome analysis reveal menin inhibition-induced proteomic alterations in AML patients treated with Revumenib. Blood 2023, 142, 2933. [Google Scholar] [CrossRef]
- Issa, G.C.; Aldoss, I.; Thirman, M.J.; DiPersio, J.; Arellano, M.; Blachly, J.S.; Mannis, G.N.; Perl, A.; Dickens, D.S.; McMahon, C.M.; et al. Menin inhibition with Revumenib for KMT2A-rearranged relapsed or refractory acue leukemia (AUGMANY-101). J Clin Oncol 2024, 43, 75–84. [Google Scholar] [CrossRef]
- Aldoss, I.; Issa, G.C.; Blachly, J.; Thirman, M.J.; Mannis, G.N.; Arellano, M.; DiPersio, J.; Traer, E. , Zwaan, M.; Shukla, N.; et al. updated results and longer follow-up from AUGMENT-101 phase 2 study of remuvenib in all patients with relapsed or refractor (R/R) KMT2A-Ar acute leukemia. Blood 2024, 144, 211. [Google Scholar] [CrossRef]
- Syndax Pharmaceuticals announced pivotal topline results from relapsed or refractory AML cohort in AUGMAENT-101 trial of revumenib. Nes release. Syndax Pharmaceuticals. November 12, 2024.
- Loo, S.; Iland, H.; Tiong, I.S.; Westerman, D.; Othman, J.; Masrlton, P.; Chua, C.C.; Putill, D.; Rose, H.; Fleming, S.; et al. Revumenib as pre-emptive therapy for measurable residual disease in NPM1 mutated or KMT2A-rearranged acute myeloid leukemia: a domain of the multi-arm ALLG AMLM26 intercept platform trial. Blood 2024, 144, 223–225. [Google Scholar] [CrossRef]
- Issa, G.C.; Cuglievan, B.; Daver, N.; DiNardo, C.; Farhat, A.; Short, N.J.; McCall, D.; Pike, A.; Tan, S.; Kamerer, B.; et al. Phase I/II stud of the all-oral combination of remivenib (SNDX-5613) with decitabine/cedazuridine (ASTX727) and venetoclax (SAVE) in R/R AML. Blood 2024, 144. [Google Scholar] [CrossRef]
- Scheidegger, N.; Alexa, G.; Khalid, D.; Ries, R.E.; Wang, J.; Alonzo, T.A.; Perr, J.; Armstrong, S.A.; Meshinchi, S.; Pikman, Y.; et al. Combining menin and MEK inhibition to target poor prognostic KMT”A-rearranged RAS pathway-mutant acute leukemia. Blood 2023, 142 (suppl.1), 166–167. [Google Scholar] [CrossRef]
- Perner, F.; Stein, E.M.; Wenge, D.V.; Singh, S.; Kim, J.; Apazidis, A.; Rahnamoun, H.; Anand, D.; Marinaccio, C.; Hatton, C.; et al. MEN1 mutations mediate clinical resistance to menin inhibition, Nature 2023, 615, 913-919.
- Perner, F.; Cai, S.F. .; Wenge, D.V.; Kim, J.; Cutler, J.; Nowak, R.; Cassel, J.; Singh, S.; Bijpuria, S.; Miller, W.H.; et al. Characterization of acquired resistance mutations to Menin inhibitors. Cancer Res 2023, 83 (suppl. 7), 3457. [Google Scholar] [CrossRef]
- Ray, J.; Clegg, B.; Grembecka, J.; Cierpicki, T. Drug-resistant menin variants retain high binding affinity and interactions with MLL1. J Biol Chem 2024, 300, 107777. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.C.; Thuring, J.W.; Querolle, O.; Dai, X.; Verhulst, T.; Pande, V.; Marien, A.; Goffin, D.; Wenge, D.V.; Yue, H.; et al. Preclinical efficacy of the potent, selective menin-KMT2A inhibitor JNJ-75276617 (bleximenib) in KMT2A- and NPM1-altered leukemias. Blood 2024, 144, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, E.; Searle, E.; Abdul-Hay, M.; Abedin, S.; Aldoss, I.; Pierola, A.A.; Alonso-Dominguez, j.M.; Chevallier, P.; Cost, C.; Diskalakis, N.; et al. A first-in-human phase 1 stud of the menin-KMT2A (MLL-1) inhibitor JNJ-75276617 in adult patients relapsed/refractory acute leukemia harboring KMT2A or NPM1 alterations. Blood 2023, 142 (suppl.1), 57–60. [Google Scholar] [CrossRef]
- Searle, E.; Recher, C. ; Abdul, Hay, M.; Abedin, S.; Aldoss, I.; Pierole, A.; Aolnso-Dominguiez, J.; Chevallier, P.; Cost, C.; Daskalakis, N.; et al. Bleximenib dose optimization and determination of RP2D from a phase 1 stud in relapsed/refractor acute leukemia patients with KMt2A and NPM1 alterations. Blood 2024, 144, 212. [Google Scholar]
- Recher, C.; O’Nions, J.; Aldoss, I.; Pierola, A.A.; Allred, A.; Alonso-Dominguez, J.M.; Barreyro, L.; Bories, P.; Curtis, M.; Daskalakis, N.; et al. Phase Ib study of menin-KMT2A inhibitor Blixemenib in combination with intensive chemotherapy in newly diagnosed acute myeloid leukemia with KMT2A or NPM1 alterations. Blood 2024, 144 (suppl.1). [Google Scholar] [CrossRef]
- Wei, A.H.; Searle, E.; Aldoss, I.; Alfonso-Pierole, A.; Alonso-Dominguez, J.M.; Curtis, M.; Dsakalakis, N.; Della Porta, M.; Dohner, H.; D’Souza, A.; Dugan, J.P.; et al. A phase 1B study of the menin-KMT2A inhibitor JNJ-75276617 in combination with venetoclax and azacytidine in relapsed/refractory acute myeloid leukemic with alterations in KMT2A or NPM1. EHA Meeting 2024, abst S133.
- Hogeling, S.M.; Le, D.M.; La Rose, N.; Kwon, M.C.; Wierenga, A.; van den Heuvei, F.; van den Boom, V.; Kucknio, A.; Philippar, U.; Huls, G.; et al. Bleximinab, the novel menin-KMT2A inhibitor JNJ-75276617, impairs long-term proliferation and immune evasion in acute myeloid leukemia. Haematologica, 2025, in press.
- Collins, C.; Wang, J.; Miao, H.; Bronstein, J.; Nawer, H.; Xu, T.; Figueroa, M.; Muntean, A.; Hess, J.L. C/ERBPα is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci USA 2014, 111, 9899–9904. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Heyes, E.; Scheiblecker, L; Eder, T.; Volpe, G.; Frampton, J.; Nerlov, C.; Valent, P.; Germbecka, J.; Grebien, F. Leukemia 2019, 33, 1608-1619.
- Eguchi, K.; Shimizu, T.; Kato, D. ; Furuta,.; Kamioka, S.; Ban, H.; Ymamoto, S.; Yokoyama, A.; Kitabaayshi, I. Preclinical evaluation of a novel orally bioavailable menin-MLL interaction inhibitor, DSP-5336, for the treatment of acute leukemia patients with MLL-rearrangement of NPM1 mutation. Blood 2021, 138, 3339. [Google Scholar]
- Daver, N.; Zeidner, J.F.; Yuda, J.; Watts, J.M.; Levis, M.J.; Fukushima, K.; Ikezoe, T.; Ogawa, .; Brandwein, J.; Wang, E.S.; et al. phase 1-2 first-in-human study of the menin-MLL inhibitor DSP-5336 in patients with relapsed or refractory acue leukemia. Blood 2023, 142 (suppl.1), 2911. [CrossRef]
- Zeidner, J.F.; Yuda, J.; Watts, J.M. , Levis, M.J.; Erba, H.P.; Fukushima, K.; Shaima, T.; Palmisano, N.D.; Wang, E.S.; borate, U-; et al. Phase 1 results: first-in-human phase 1-2 study of the menin-MLL inhibitor enzomenib (DSP-5336) in patients with relapsed or refractor acute leukemia. Blood 2024, 144, 213. [Google Scholar] [CrossRef]
- Fisku, W.; Daver, N.; Boettcher, S.; Mill, C.P.; Sasaki, K.; Bindwell, C.E.; Davis, J.A.; Das, K. , Takashi, K.; Kadia, T.M.; et al. Activity of menin inhibitor zitfomenib (KO-539) as monotherapy or in combinations against AML cells with MLL1 rearrangement or mutant NPM1. Leukemia 2022, 36, 2729–2733. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.; Dzama, M.M. ; Dolgikh, N:; Stiller, H.; Bohl, S.; Lahrmann, C.; Kunz, K.; Kessler, L.; Echannoni, H.; Chei, C.W.; et al. Menin inhibitor ziftomeninb (KO-539) synergizes with drugs targeting chromatin regulation or apoptosis and sensitizes acute myeloid leukemia with MLL rearrangement or NPM1 mutation to venetoclax. Haematologica 2023, 108, 2837–2847. [Google Scholar]
- Wang, E.S.; Issa, G.C.; Erba, H.P.; Altman, J.K.; Montesinos, P.; DeBotton, S.; Walter, R.B.; Pettit, K.; Savona, M.R.; Shah, M.V.; et al. Ziftomenib in relapsed or refractory acute myeloid leukemia (KOPMET-001): a multicenter, open-label, multi-cohort, phase I trial. Lancet Oncol 2024, 25, 1310–1324. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Wang, E.S.; Issa, G.C.; Altman, J.; Balasubramianan, S.K.; Strickland, S.A.; Roboz, G.J.; Schiller, G.J.; McMahon, C.M.; Palmisano, N.D.; et al. Ziftomenib combined with intensive induction (7+3) in newly diagnosed NPM1-m or KMTY2A-r acute myeloid leukemia: interim phase 1a results from KOMET-007. Blood 2024, 144 (suppl.1), 214. [Google Scholar] [CrossRef]
- Golobberg, A.D.; Corun, D.; Ahsan, J.; Nie, K.; Koziek, T.; Leoni, M.; Dale, S. Kamet-008: a phase 1 study to determine the safety and tolerability of Ziftomernib combinations for the treatment of KMT2A-rearranged or NPM1-mutant relapsed/refractory acute myeloid leukemia. Blood 2023, 142 (suppl.1), 1553. [Google Scholar] [CrossRef]
- Lancet, J.; Ravandi, F.; Montesinos, P.; Barrientos, J.C.; Badar, T.; Alegre, A.; Bashey, A.; Bergua Bugues, J.M.; et al. Covalent menin inhibitor Bmf-219 in patients with relapsed or refractory (R/R) leukemia (AL): preliminary phase 1 data from the Covalent-101 stud. Blood 2023, 142 (suppl.1), 2916–2918. [Google Scholar] [CrossRef]
- Rasouli, M.; Troester, S.; Grebien, F.; Goemans, B.F.; Zwaan, G.M.; Heidenreich, O. NUP98 oncofusions in myeyloid malignancies: an update on molecular mechanisms and therapeutic opportunities. HemaSphere 2024, 8, 70013. [Google Scholar] [CrossRef]
- Xu, H.; Valerio, D.G.; Eisold, M.E.; Sinha, A.; Koche, R.P.; Hu, W.; Chen, C.W.; Chu, S.H.; Brien, G.L.; Park, C.Y.; et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer Cell 2016, 30, 863–878. [Google Scholar] [CrossRef]
- Heikamp, E.B.; Heinroich, J.A.; Perner, F.; Wong, E.M.; Hatton, C.; Wen, Y.; Barwe, S.P.; Golalakrishnapillai, A.; Xu, H.; et al. The menin-MLL1 interaction in a molecular dependency in NUP98-rearranghed AML. Blood 2022, 139, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Rasouli, M.; Blair, H.; Troester, S.; Szoltysek, K.; Cameron, R.; Ashtiani, M.; Krippmner-Heidenreich, A.; Grebien, F.; McGeehan, G.; McGeehanm, G.; et al. The MLL-menin interaction is a therapeutic vulnerability in NUP98-rearranged AML. HemaSphere 2023, 7, e935. [Google Scholar] [CrossRef] [PubMed]
- Heikamp, E.D.; Martucci, C.; Henrich, J.A.; Neel, D.S.; Mahendra-Rajah, S.; Rice, H.; Wenge, D.V.; Perner, F.; Wen, Y.; Hatton, C.; et al. NUP98 fusion proteins and KMT2A-Menin antagonize PRC1.1 to drive gene expression in AML. Cell Rep 2024, 43, 114901. [Google Scholar] [CrossRef] [PubMed]
- Carraway, H.E.; Nakitandwe, J.; Cacovean, A.; Ma, Y.; Mumeke, B.; Waghmare, G.; Mandap, C.; Ahmed, U.; Kawalkezyk, N.; Butler, T.; et al. Complete remission of NUP98 fusion-positive acute myeloid leukemia with the covalent menin inhibitor BMF-219, icovamenib. Haematologica in press. 2025. [Google Scholar] [CrossRef]
- Miao, H.; Cheri, D.; Ropa, J.; Purohit, T.; Kim, E.; Sulis, M.L.; Ferrando, L.; Cierpicki, T.; Grembecka, J.; et al. Combination of menin and kinase inhibitors as an effective treatment for leukemia with NUP98 translocations. Leukemia 2024, 34, 1674–1686. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



