Submitted:
08 February 2025
Posted:
10 February 2025
You are already at the latest version
Abstract
The Survivin protein has roles in repairing incorrect microtubule-kinetochore attachments at prometaphase, and the faithful execution of cytokinesis, both as part of the chromosomal passenger complex (CPC). In its absence, mitotic errors often occur that lead to chromosome missegregation, a cause of aneuploidy, polyploidy and ultimately cancer. Adding to these well-known roles of Survivin, this paper now shows for the first time that this protein is required for cancer cells to enter mitosis, and that, in its absence, HeLa cells accumulate at early prophase, or prior to prometaphase as reported before. This early prophase blockage is demonstrated by the inability of Survivin-depleted cells to disassemble their nuclear lamina and their low Cdk1 activity. Importantly, escaping the arrest induced by the Survivin double mutant SUR D70A/D71A leads to multiple mitotic defects, or mitotic catastrophe, and eventually cell death. Mechanistically, Cdk1 does not localize at the centrosome in the absence of Survivin, pointing at this latter protein contributing to the activation of the mitotic kinase via Cdc25B. Furthermore, absence of Survivin leads to an inactive cytosolic Cdc25B-Cdk1-Cyclin B1 complex, which seems to indicate a role for Survivin in bridging this complex and its centrosomal activator/s. Interestingly, the drop in Cdc25B activity caused by interference with the Survivin’s function could be rescued when Survivin-depleted HeLa cell lysates were incubated with the recombinant Survivin protein. Also, a role for Survivin in the Cdc25B-mediated activation of Cdk1, and concomitant prophase to prometaphase transition could be confirmed by expression of a gain-of-function Cdc25B mutant, which overrode the G2/prophase blockage caused by Survivin depletion.
Keywords:
Survivin
; Cdk1
; Cdc25B
; mitosis
; kinase
; phosphatase
; G2/M-phase checkpoint
; mitotic catastrophe
; cancer
Introduction
Precise sequential and spatial activation of the Cdk1 protein kinase is essential for correct entry of cells into mitosis [1]. This process involves formation of a complex between Cdk1 and its activator Cyclin B1 [1], and removal of inhibitory phosphates on the kinase residues Threonine 14 and Tyrosine 15 by the phosphatase Cdc25 [1]. In this respect, Cdk1 can be activated by three Cdc25 isoforms [1], being Cdc25B the first one that acts on the kinase at the centrosome [2,3], and the other two the ones that amplify the centrosomal signal in the cytosol and the nucleus [1].
Full activation of Cdk1 in the nucleus at late prophase is considered a point-of-no-return in mitosis [4]. Here, the nuclear lamina is disassembled as a result of Cdk1 phosphorylation of the Lamin B subunits, triggering the nuclear membrane breakdown (NMBD) [5,6,7]. As a result cytosolic microtubules are able to reach the kinetochores on the chromosomes, coinciding with cells entering prometaphase [8,9]. This so-called mitotic commitment, which requires high levels of Cdk1 activity in the nucleus [10,11], when erroneously triggered leads to mitotic catastrophe and cell death [12].
On a related aspect of mitosis, it is well known the role of the Survivin protein in establishing proper microtubule-kinetochore attachments in prometaphase as part of the Chromosome Passenger Complex (CPC) [13]. Here, errors often lead to multiple mitotic defects and sustained spindle checkpoint [14,15,16,17] that when overridden, can cause chromosome mis-segregation and aneuploidy, both typical features of many tumors [13].
Survivin has also been localized at the centrosome [18] but its role at this non-membranous organelle has not been elucidated yet. In this regard, Survivin depletion causes a mini spindle phenotype in Xenopus egg extracts [17] and HeLa cells [16,19], which seems to indicate a failure in centrosome separation, an early mitotic event for which centrosomal Cdk1 activity is required through its effect on Eg5 [20,21]. These data, and the fact that Survivin binds to both Cdk1 [22] and microtubules [17,23], seem to suggest a role of Survivin in the correct operation of the main mitotic kinase at the centrosome, which would place the former protein at the hub where decisions to initiate mitosis are made, however, challenge the current model, which envisions Survivin as only acting in mitosis via the CPC [14,15]. Surprisingly, despite the interest of this theory, the role of Survivin at an earlier time than prometaphase has not been investigated so far. Therefore, with the intention to unravel the function of Survivin at mitotic onset in cancer cells, I decided to revisit the early mitotic events that follow abrogation of the Survivin protein in HeLa cell cultures.
Results
siRNA-Mediated Loss of Survivin Induces an Early Prophase Blockage
To clarify the stage at which cancer cells first arrest at mitosis following Survivin abrogation, I initially repeated the RNAi preliminary experiments carried out by others in HeLa cultures using a fully-characterized Survivin siRNA [16]. As Figure 1A, left indicates, treatment of asynchronous HeLa samples with the Survivin reagent (S4) caused a complete ablation of this protein versus the control (VIII), which correlated with cells piling up at the G2/M-phase checkpoint, and the detection of endoreplication by FACS analysis (Figure 1A, right) as seen before [16].
In order to discern the cell cycle phase at which the above blocked Survivin-depleted samples were at, HeLa cell cultures identically treated as in Figure 1A were analyzed by immunofluorescence microscopy (Figure 1B). As it can be seen, control cells (VIII) had many mitotic figures, which contrasted with the slow progression into mitosis and high degree of endoreplication observed in the Survivin-ablated samples (S4). Also, the cultures lacking Survivin showed a peculiar phenotype, consisting of rounded and often large cells with multiple layers of bright microtubules surrounding the central cellular space in which no noticeable spindle could be observed, and where chromatin was not fully condensed. The immunofluorescence experiment was repeated using a Survivin antibody to make sure that the cells lacking the spindle did not express this protein. As seen in Figure 1C, left all the round control cells formed a spindle and accumulated Survivin, which colocalized with the chromatin. In contrast, in many Survivin-depleted cells, the microtubules formed a ring around the partially condensed chromatin and no spindle was clearly visible. Figure 1C right shows this abnormal phenotype in more detail. See how the control cell assembled a perfect spindle around its condensed chromatin, and how the Survivin-depleted cell just formed a ring of microtubules but not mitotic apparatus, a typical characteristic of prophase. To accurately determine the difference in the number of mitotic cells as a result of the different siRNA treatment, I repeated several times the immunofluorescence experiment described above, and each time counted the number of rounded cells in several microscope fields. As seen in Figure 1D, HeLa cell cultures where Survivin was ablated (VIII) contained 72% less rounded cells than the control ones (S4), suggesting a role for Survivin in mitotic entry. The drop in cells committing to mitosis in cultures where Survivin was absent could also be reproduced in siRNA-transfected HeLa cultures analyzed by immunofluorescence microscopy using an antibody against the bona fide mitotic protein Cyclin B1 (Figure S1). Here, I could see that, following Survivin abrogation (S4), most cells had a dispersed Cyclin B1 protein throughout the cytosol, a typical phenotype of late G2 or early prophase cells [24]. Furthermore, the few Survivin-depleted cells that managed to enter mitosis appeared to be mostly in prophase, as judged by the colocalization of their Cyclin B1 protein and DNA [24], in agreement with the data in Figure 1. In contrast, Cyclin B1 overlapped with the spindle in the control samples (VIII), a typical feature of cells in prometaphase or metaphase [25]. From these observations and Figure 1, it seemed that abrogation of Survivin in HeLa cells led to an impairment in their capacity to enter prometaphase, an event controlled by nuclear Cdk1 activation [8].
In order to obtain more visual data with which to confirm the prophase to prometaphase blockage in the Survivin-depleted samples, siRNA-treated rounded HeLa cells were further analyzed by immunofluorescence microscopy, this time using a Lamin B antibody (Figure 2). Nuclear lamina disassembly is an excellent marker of nuclear Cdk1 activation and subsequent progression into prometaphase [5,6,7], and therefore should clearly indicate the cell cycle phase at which the rounded cells lacking Survivin were at. Figure 2A, Survivin panels again show the round cells without spindle already described in Figure 1. In the bottom Lamin B panels, it can also be observed that while control rounded cells (VIII) generally had their Lamin B protein colocalizing with the spindle (spindle-like structures, SLS), in the Survivin-depleted samples (S4), rounded cells had an intact nuclear lamina (intact lamina, IL) enclosing the chromatin, a typical prophase feature. Individual cells expressing or not the Survivin protein are shown magnified in Figure 2B. Here, it can be seen that in interphase, cells looked the same independently of the siRNA treatment, and had long microtubules and an intact nuclear lamina (arrows) engulfing the chromatin. Moreover, the control rounded mitotic cell (VIII) shown in the picture as an example, assembled a spindle, which embraced the condensed chromatin situated at the spindle equator, and had a disassembled nuclear lamina, a phenotype previously ascribed to full Cdk1 activation [7], where the Lamin B monomers clearly colocalized with the spindle (Figure 2B, Lamin B VIII panel, arrow head), also a typical pro/metaphase characteristic [26]. In contrast, the rounded cell lacking Survivin (S4) (Lamin B S4 panel, arrow) had a partially condensed chromatin surrounded by layers of short microtubules, and an intact lamina, which encircled the DNA. This latter phenotype was described before as a sign of cells in interphase or early prophase, and absence of nuclear Cdk1 activation [5,6,7]. To determine the extent of the abnormal presence of the nuclear lamina in rounded cells lacking Survivin (S4), this phenotype was tallied in a number of control or Survivin siRNA-treated samples from several independent experiments. As Figure 2C shows, while 94% of the rounded cells treated with the control siRNA (VIII) had a Lamin B spindle-like structure, indicative of cells in prometaphase or metaphase, only 10% of the Survivin-depleted rounded cells (S4) had a similar phenotype. Conversely, 90% of the Survivin-depleted rounded mitotic cells had an intact lamina versus 6% in the control. From this data, I strongly supported the idea of Survivin being first required for the prophase to prometaphase transition, or mitotic commitment. The Lamin B experiments were reproduced in immunofluorescence experiments where siRNA-treated HeLa cells were also incubated with a Cyclin B1 antibody (Figure 1SC). Here, it could clearly be seen that in the control cells (VIII), Cyclin B1 colocalized with the spindle as seen before [25], and that this phenotype coincided with Lamin B spindle-like structures (SLS) as those seen in Figure 2B. In contrast, in the Survivin-ablated samples (S4), the nuclear lamina was intact (IL), in otherwise cells that seemed committed to mitosis as judged by their chromatin condensation.
Finally, if a role for Survivin in the activation of Cdk1 at prophase were real, HeLa cells treated with a Cdk1 inhibitor should also show the Lamin B ring detected when Survivin was absent. To check this possibility, HeLa cells were incubated with the Cdk1 inhibitor purvalanol A, and analyzed by immunofluorescence microscopy. As Figure 2D shows the purvalanol A treatment replicated the intact-lamina and partially-condensed-chromatin phenotype seen in Survivin-depleted cells, supporting the role of Survivin in Cdk1 activation and prophase to prometaphase transition.
To obtain direct biochemical evidence on the regulation of the Cdk1 activity by Survivin, I decided to check this kinase’s activity in the presence and absence of the latter protein. To this aim, siRNA-transfected HeLa cultures were lysed, subjected to Cdk1 immunoprecipitation (IP), and their kinase activity was measured by a Histone H1 phosphorylation assay. As Figure 3A, right shows, the activity of the Cdk1 protein was reduced when Survivin was absent (S4) compared to the control (VIII). Also, the same lysates did not show significant differences in the amounts of Cdk1 and Cyclin B1 (Figure 3A, left), demonstrating that the lower Cdk1 activity in the Survivin-depleted samples was not due to lower amounts of the proteins forming the Cdk1-Cyclin B1 complex.
Together, the immunofluorescence and Cdk1 activity data contradicted previous findings, which claimed an accumulation of cells at prometaphase following Survivin abrogation [14,15]. To clearly show the difference between the blockage caused by Survivin depletion, and a real high Cdk1 activity prometaphase arrest [27,28], HeLa cells were either treated with siRNA oligonucleotides, or incubated with the microtubule-depolymerizing agent nocodazole, which causes a potent prometaphase blockage, and their Cdk1 activities were compared by using a Histone H1 phosphorylation assay. As Figure 3B shows, HeLa cells lacking Survivin (S4) had a low Cdk1 activity, which contrasted with the potent Cdk1 activation observed when similar cells were incubated with nocodazole.
The Early Prophase Blockage Caused by Survivin Abrogation in Asynchronous Cultures Can Be Replicated in Synchronized HeLa Cells, Precedes Endoreplication and Can Be Rescued by Exogenous Survivin
It is well known that Survivin and Cdk1 form a complex in vivo [22]. In order to track the assembly of this complex through the cell cycle, untreated HeLa cells were synchronized and upon their release, samples were taken at different times, and either Survivin was immunoprecipitated (IP) (Figure 4A, left) or cells were analyzed by FACS (Figure 4A, right). As seen, Survivin and Cdk1 first formed a clear complex at 8 h, or exactly when cells started to complete mitosis and accumulate in G1. This complex persisted up to 10 h, while cells were still dividing, and started to decline at 12 h, or when all cells had completed mitosis.
Synchronized HeLa cells treated with siRNAs were released in order to follow their cell cycle progression and compare it with the untreated samples. As seen in Figure 4B, bottom, for the first 9 h following the thymidine release, cells, independently of the siRNA treatment, progressed exactly the same while moving towards the G2/M-phase, and only behaved slightly sluggish, probably due to the siRNA transfection. After this initial stage however, cells started to diverge at 14 h depending on the siRNA being used. On one hand, control cells (VIII) started to complete mitosis and accumulate at G1, continuing this trend for the remainder of the experiment. Here, not all the control cells managed to complete mitosis as a result of the short time course (18 h) (Figure 4B, bottom, upper graphs). In contrast, Survivin-depleted cultures (S4) once reached G2, remained stuck at this peak for the remainder of the experiment (Figure 4B, bottom, lower graphs).
According to Figure 4A, the Survivin complex in the control siRNA-treated synchronized HeLa cells should have been formed before the G1 peak was first detected in these samples. This occurred between 9 and 14 h (Figure 4B, bottom, upper graphs). In order to check the activity of this complex in the control (VIII), and compare it with the Survivin-depleted samples (S4), the same experiment shown in Figure 4B was repeated and samples were taken between 12 and 18 h, which spanned mitotic transition (Figure 4C, left). As shown in Figure 4C, right, the Cdk1 activity of control siRNA-transfected (VIII), synchronized HeLa cultures was high as measured by a Histone H1 kinase assay of immunoprecipitated (IP) Cdk1 from all times tested. In comparison, the Survivin-depleted samples (S4) mounted some initial kinase activity but this response could not be sustained during the remainder of the time course, gradually declining as cells remained stuck at the G2/M-phase checkpoint (Figure 4, left).
A Survivin Peptide Spanning Amino Acids Ala55 through Asp70 (SUR A55-D70) Binds the Cdk1 αC/β4 Loop
Next, I wanted to see whether the Survivin and Cdk1 interaction was direct. To this aim, I expressed and purified GST-Survivin and His-Cdk1 from E. coli, and used these recombinant proteins in a GST-pull down experiment. As Figure S2A shows, GST-Survivin but not GST pulled down the His-Cdk1 protein kinase, demonstrating that these two proteins bind directly.
From the above result, I could now address the question of whether Survivin is directly implicated in the in vivo activation of Cdk1 by making a peptide that might interfere with the Survivin-Cdk1 interface. To this end, N- and C-terminal Survivin deletion mutants attached to a GST-tag were made, and used in GST-pull downs with His-Cdk1. Figure S2A, left shows that His-Cdk1 interacted with the Survivin N-terminus (i.e., first 70 amino acids) but not the C-terminus (i.e., amino acids from 71 to 142). To narrow down the Cdk1-binding site in Survivin, more GST-Survivin deletion mutants were generated. As Figure S2A, right shows, GST-pull downs using the new Survivin fragments showed that the minimal region in Survivin that binds to Cdk1 comprises the residues Ala55 (A55) through Asp70 (D70), a region in the BIR domain with an abundant number of acidic amino acids [29] (Figure S3A and Figure S3C).
Next, I was also interested in knowing the region in Cdk1 that binds to Survivin. For this reason, I made several Cdk1 deletion mutants (Figure S2B), which I used in binding assays with Survivin. Figure S2B, left shows GST-Survivin pull downs using these kinase fragments. As seen, only the Cdk1 mutants His-Cdk1 1-183 and His-Cdk1 1-85, which contained the kinase N-terminal region, could bind to GST-Survivin but not GST. Next, I biotinylated the smallest Survivin region that binds to Cdk1 (SUR A55-D70), and used it in streptavidin-binding assays with all the His-Cdk1 fragments (Figure S2B, right). As shown, all the Cdk1 mutants but His-Cdk1 182-297, His-Cdk1 1-56 and His-Cdk1 1-43 bound to SUR A55-D70. From these data, I concluded that the minimal region in Cdk1 that binds to Survivin is located in the kinase N-terminal sequence, and comprises the amino acids from Lys56 (K56) to Met85 (M85), or a sequence that includes the αC/β4 loop of the kinase (Figures S3B and S3D).
The SUR A55-D70 Peptide Targets the Survivin-Cdk1 Complex, Inducing its Disassembly, Loss of Cdk1 Activity, Spindle Abnormalities and Apoptosis
Before using the SUR A55-D70 peptide in vivo, I carried out a few preliminary experiments to assess its potential. First, I wanted to know whether the SUR A55-D70 peptide could pull down the Cdk1 protein from HeLa cell lysates. For this purpose, I incubated streptavidin beads pre-bound to the biotinylated scrambled or SUR A55-D70 peptide with HeLa cell extracts, and observed whether the endogenous Cdk1 protein bound or not to the resin. As Figure S4A shows, endogenous Cdk1 bound to the beads that contained the Survivin peptide but not to the control reagent.
Second, I wanted to know whether the SUR A55-D70 peptide could displace the endogenous Survivin protein from its complex with Cdk1, and, if that was the case, whether this displacement affected the kinase activity. To this aim, I used a cell-free system, consisting of HeLa cell lysates from nocodazole-treated cultures, which should contain a large number of active Survivin-Cdk1 complexes. These cell extracts were supplemented with an ATP-regenerating system to make them biochemically active, and then incubated with the control or Survivin peptide to see their effect. Figure S4B shows that when the SUR A55-D70 peptide was incubated with nocodazole-treated HeLa cell lysates, and then Cdk1 was immunoprecipitated (IP), the kinase did not bind to the endogenous Survivin protein, and the Cdk1 activity was very low. In contrast, no effect on the Survivin binding to Cdk1 or the kinase activity was observed with the control peptide.
Last, before using the SUR A55-D70 peptide to analyze its effect in vivo, I wanted to know whether this reagent could transverse the HeLa cell membrane when attached to an N-terminal HIV tat cell-permeable sequence. Figure S4C shows that both the scrambled and Survivin A55-D70 peptides attached to the HIV sequence, readily accumulated inside the cells following their incubation with HeLa cultures.
Following the above controls, asynchronous HeLa cells were incubated with HIV tat-peptides (50 μM) for 6 h, and samples were analyzed by FACS, or immunofluorescence using an α-Tubulin antibody and DAPI staining. In contrast to the FACS analysis, which did not show major differences (data not shown), mitotic cells treated with the SUR A55-D70 peptide but not with the control reagent, showed several spindle abnormalities (Figure 5A), which are summarized in Figure 5B. Here, it can be noticed that normal spindles were reduced by more than 50% in the mitotic cells treated with the Survivin peptide, as compared to the control. Also, this time I could see the mini spindles reported before [16,17,19]. The mini spindle phenotype was 6 fold higher in the Survivin peptide-treated samples versus the control (28% vs. 5% in the control). Also, to a lesser extent, I could see other abnormal mitotic phenotypes with the SUR A55-D70 peptide, including microtubule-depleted, aberrant or multipolar spindles.
When looking in detail at the DAPI staining of Survivin peptide-incubated cells, some chromosomes appeared to mis-segregate or be fragmented (Figure 5A, white arrows). This phenotype was not detected in the RNAi experiments, and might represent damaged cells earmarked for cell death. To investigate this possibility, I repeated the treatment of HeLa cells with the Survivin or control peptide but this time checked the DNA content by FACS analysis at a later time point. As Figure 6A, top graphs show when HeLa cells were incubated with 50 μM SUR A55-D70 peptide for 24 h, a sub-G1 peak (9%) corresponding to cells with fragmented DNA (<2N) that had not been seen before, could be observed in contrast to almost no effect in the control. Also, looking at these cells under phase contrast (Figure 6A, top panels), they presented evident signs of cell death manifested as cell bodies full of dark material (arrows).
With the intention to enhance the above cell death phenotype, I incubated HeLa cells with higher concentrations of the Survivin peptide, and again measured the amount of DNA by FACS analysis. Figure 6B, bottom graphs show that at 200 μM (the highest peptide concentration that I could use without causing too much toxicity in the control samples), the sub-G1 increased to 22% in the Survivin peptide-treated cells versus 3% in the control. On the other hand, Figure 6B bottom panels show that at this higher concentration of the Survivin peptide, a higher number of dead cells accumulated, as concluded from the black cell bodies (arrows) already seen before (Figure 6A, top panels), and a new abnormal phenotype consisting of bubbles containing some cell material (arrow heads). Also, at this higher Survivin peptide concentration, I could see less cells under the microscope, suggesting a cytostatic or lytic effect of this reagent, which could be deducted from the lower amount of cells that could be counted in the FACS profile (Figure 6B, bottom graphs), and the higher percentage of compromised cells after the Survivin peptide treatment (55%) in comparison with the control (15%), according to the Trypan Blue data (Figure 6B, right).
In order to determine the type of cell death detected in the HeLa cells transfected with the Survivin peptide, I treated HeLa cell cultures with 200 μM peptides, and assayed for caspase activity either by using the fluorescence probe FAM-DEVD-FMK, which labels the active Caspase 3 and 7 enzymes in living cells [30], or incubating the samples with an antibody against Procaspase 3, which cleavage indicates activation of the enzyme by Caspases 8 and 9 [31]. As Figure 6B, left panels show, the same cell death phenotype as in Figure 6A, Survivin peptide panels was observed in the Survivin peptide-treated cells but not in the control samples. Also, when these images were compared to the FAM-DEVD-FMK labeling (Figure 6B, right panels), the same exact cells, which had signs of cell death by phase-contrast microscopy, showed activation of Caspases 3 and 7 that could hardly be seen with the control reagent (Figure 6B, top right panel). Identically treated HeLa cell cultures as in Figure 6B were used to analyze apoptosis using the Procaspase 3 antibody. As Figure 6C shows, following the incubation of HeLa cells with the Survivin peptide for 24 or 48 h, a strong activation of Caspase 3, as demonstrated by the disappearance of the caspase’s proform, was seen in these samples but not in the control.
Since the Survivin peptide was targeting the Survivin-Cdk1 complex at mitosis (Figure S4B), I reasoned that if I enriched for mitotic cells by using synchronous cultures, and then incubated these samples with the Survivin peptide, apoptosis might be enhanced. To this aim, synchronous HeLa cells released for 9 h into fresh media (G2/M-phase) (Figure 6D, top, left graph), were incubated with the peptides for 12 h, and then analyzed by FACS (Figure 6D, top, right graphs). As shown in this specific experiment, not all G2/M-phase cells treated with the control peptide had enough time to exit mitosis following the 12 h treatment (31% G1 population), with some still remaining in G2/M-phase after this time (32%). However, the G2/M-phase to G1 transition in these samples was smooth, as concluded by the almost absence of cell death in these cultures (1%). In contrast, the Survivin peptide-treated cells had a higher sub-G1 population (16%), which seemed to originate from the cell pool that stalled at G2/M-phase in the control, suggesting the better effectiveness of the Survivin peptide at causing cell death in cells transiting through mitosis.
Parallel peptide-treated, synchronous cell cultures entering mitosis as in Figure 4D, top were analyzed by phase-contrast microscopy. As Figure 4D, bottom shows, HeLa cells targeted with the SUR A55-D70 peptide at the G2/M-phase border died more readily than the asynchronous ones, as attested by the massive amount of dead cells seen under these conditions, which interestingly was again higher than that observed in the FACS analysis. As with the asynchronous cultures, a lower number of cells could also be observed in the Survivin peptide-treated samples, reiterating the possibility that this reagent might have cytostatic or lytic properties.
A Survivin Asp70Ala/Asp71Ala Double Mutant (SUR D70A/D71A) Fails to Bind Cdk1, and Causes G2/M-Phase Arrest, Mitotic Abnormalities and Cell Death
The Survivin peptide did not cause a G2/prophase blockage as shown in the RNAi experiments. Interestingly, the former reagent caused spindle abnormalities, which were reminiscent of the prometaphase arrest seen by others [14,15] and apoptosis. In order to clarify which of these two phenotypes was prevalent following interference with the Survivin function, a second molecular antagonist of the Survivin-Cdk1 complex was made, consisting of a Survivin mutant that could not bind to Cdk1. To make this polypeptide, the Survivin region spanning the the Cdk1-binding domain (i.e., amino acids Ala55 through Asp70) was subjected to Alanine Scanning Mutagenesis, and the resulting proteins were screened for binding to recombinant His-Cdk1. As a result of this approach, several mutants were identified that had a reduced kinase binding capacity. These polypeptides included the single mutants SUR E63A, SUR E65A, SUR W67A and SUR D71A, and the Survivin double mutant SUR D70A/D71A (Figure S5). From these binding assays, several things could be concluded. First, all the Survivin mutants, with the exception of SUR W67A, that did not bind to the Cdk1 kinase corresponded to mutations in acidic residues in the SUR A55-D70 region. Second, the best of these mutants was SUR D70A/D71A, and therefore, this was the reagent chosen for the following experiments. Finally, once the Survivin structure was revisited, it was hypothesized that the SUR D70A/D71A double mutant might act as a dominant-negative protein since residues Asp70 and Asp71 appear on the surface of the Survivin dimer, and do not contribute to the monomers’ interface [32].
Next, the SUR D70A/D71A protein was attached to a HA-tag and expressed in asynchronous HeLa cell cultures (Figure 7A, left), which were analyzed by fluorescence microscopy (Figure 7A, right), and by counting cells from several independent experiments according to their phenotype (Figure 7B). As shown in Figure 7A, right and Figure 7B, following the Survivin double mutant transfection, a large population of cells (37% vs. 8% in the control) had a phenotype that resembled the cells in prophase seen before with the Survivin siRNA (Figure 1 and Figure 2). In effect, these cells were larger than normal, and had microtubules that did not form spindles but instead surrounded the chromatin, and which colocalized with the HA-tagged Survivin mutant protein. Also, this time some HA-SUR D70A/D71A-transfected HeLa cells appeared to have mis-segregated or fragmented DNA (Figure 7A, right, cells labeled fragmented chromatin), as some of the cells treated with the Survivin peptide. Coincidentally with the mitotic defects observed with the Survivin double mutant, HeLa cells expressing this protein also had a low Cdk1 activity that corresponded to a large decrease in the binding of the Survivin protein to the Cdk1 kinase (Figure 7C, top), and the same amount of total Survivin in the lysate independently of the treatment (Figure 7C, bottom). Last, when using an antibody against the inactive form of Cdk1, I could also see the inactive Cdk1 protein accumulating in the SUR D70A/D71A-transfected cells but not in the control (Figure 7D).
The above results with the Survivin double mutant pointed at this protein being necessary for both Cdk1 activation at prophase and spindle assembly at pro/metaphase. In order to clearly determine which abnormality is preponderant following Survivin malfunction, cell cycle progression of individual synchronized HeLa cells transfected either with the GFP-tagged wild type Survivin protein (GFP-SUR WT) or SUR D70A/D71A (GFP-SUR D70A/D71A) 12 h prior to their release into fresh media was monitored. This approach allowed for a full expression of the recombinant protein prior to cells entering mitosis. Specifically, two said experiments were carried out from which 7 control and 30 Survivin double mutant-transfected HeLa cells (green cells) were studied by time-lapse video microscopy (Figure 7E,F).
As shown in Figure 7E, top graph, synchronized control HeLa cells rounded around 9.3 h, which correlated with them reaching the G2/M-phase, as previously shown in the FACS analysis (see Figure 4B, bottom, VIII graphs). These control cells remained rounded for an average of 61 min before initiating telophase/cytokinesis, a stage at which they stayed for an additional 119 min, as average, before spreading and entering G1.
Following a very different behavior (Figure 7E, bottom graph), 13 out of the initial 30 GFP-SUR D70A/D71A-expressing HeLa cells (43%) remained flat or rounded without progressing to telophase (G2/M-phase blocked) for the full length of the time course. Of the other 17 green cells, 7 (23% of the initial count) died either early after their release into fresh media (3 cells in between 40-180 min), or later during the G2/M-phase blockage (4 cells in between 540-1,400 min or 9-24 h). The remaining 10 green cells, or 33% of the initial monitored population, rounded at an average of 11.5 h, or approximately 2.5 h later than their control counterparts, and then entered telophase. During telophase, 4 out of these 10 cells died (40%) (2 of them collapsing after 700 min or 11.7 h), and the remaining 6 cells managed to divide (half of them after more than 200 min). Finally, once mitosis was completed, 2 of the 6 cells that managed to enter G1 died (33%), 1 cell aborted cytokinesis, and only 3 cells managed to survive, or 10% of the initial 30 cells monitored in the 2 time-lapse experiments. From these results, I concluded that cells lacking a functional Survivin protein first stably arrest in G2/M-phase with those overriding the mitotic checkpoint most likely dying in the successive mitotic phases. This result was in agreement with the asynchronous experiments (Figure 7B) where 37% of the HA-SUR D70A/D71A positive cells were in prophase vs. 8% in the control.
A detailed frame-by-frame analysis of the HeLa cells expressing the Survivin double mutant provided further interesting information (Figure 7F). Here, 3 cells labeled with a magenta, yellow or light blue arrow are shown (Figure 7F, bottom panels) that are representative of a population of cells in the pool (remember that some of these cells never rounded up and looked like they were in S-phase or G2). First focussing on the cell labeled with the yellow arrow, which corresponds to the yellow column shown in Figure 7E, bottom graph, this cell appeared round at 16.3 h (978 min), flat at 22.3 h (1,338 min) and died around 24 h. This behavior was typical of some of the cells expressing the Survivin double mutant, and could be explained either as the cell exiting mitosis without completing cytokinesis and then dying, or alternatively, returning to G2, after entering mitosis, and then dying trying to re-enter prophase. Since, according to Figure 7E, bottom graph, very few cells expressing the Survivin double mutant reached G1, I found the second explanation more plausible. Regarding the cells in Figure 7F, bottom panels labeled with the magenta and light blue arrows, they also rounded very late, and remained in this fashion for the rest of the time course. More interestingly, these cells had an elongated shape and were larger than normal, phenotypes already seen in the RNAi experiments (Figure 1 and Figure 2), which have been connected to deregulation of the Cdc25-Cdk1 axis [33].
Survivin Is Required for Recruitment and Activity of Cdk1 at the Centrosome
The results so far pointed at a role of Survivin in the early activation of Cdk1. This kinase activity is first detected at the centrosome [2,3], and interestingly, I could see a fast-migrating Cdk1 band previously ascribed to the active kinase [27] bound to Survivin in crude centrosomal preparations from untreated synchronized HeLa cells (Figure 8A). The putatively active centrosomal Cdk1 isoform first accumulated at 7 h, preceding the FACS time point at which untreated HeLa cells started to come out of mitosis (8 h) (Figure 4A, right). Furthermore, the Survivin-Cdk1 complex continued to be visible until 11 h, or right before all the synchronized HeLa cells reached G1 (12 h) (Figure 4A, right).
To answer the question of whether Survivin is required for localization and/or activation of Cdk1 at the centrosome, I treated asynchronous HeLa cell cultures with siRNA oligonucleotides, and prepared centrosomes, which were used to analyze their Cdk1 content by Western blotting. Figure 8B shows that Survivin ablation (S4) caused a large decrease in the levels of the centrosomal Cdk1 protein, and a partial drop in γ-Tubulin, which coincided with a constant level of the Ran protein, a subpopulation of which, also associates with this organelle. Also, a decrease in the amount of the Caspase 3 proform could be seen in the centrosomes of Survivin-depleted cells (Figure S7A). To see if this reduction in centrosomal Cdk1 could be reproduced in synchronous cultures, HeLa cells were transfected with siRNA oligonucleotides, synchronized, and aliquots were collected 12 to 18 h following their release, coinciding with mitosis exit in the control cells (Figure 8C, bottom, upper graphs). These samples were then used to prepare centrosomes from which Cdk1 was immunoprecipitated (IP), and the kinase activity measured by a Histone H1 assay. Figure 8C, IP Cdk1, left panels show that a Survivin-Cdk1 complex accumulated in the centrosome of control cells (VIII), which coincided with a strong Cdk1 activity at this location. In contrast, much less amount of Cdk1 was found and hardly any kinase activity was detected in the centrosomes of synchronized HeLa cells depleted of Survivin (S4) (Figure 8C, IP Cdk1, right panels and 8C, bottom, lower graphs), despite the constant levels of the mitotic kinase in the lysate (Figure 8C, lysate).
Centrosome dynamics is a well known process in eukaryotic cells [34]. Briefly, in a G2 cell, the centrosome remains intact, although with duplicated centrioles. Following centrosomal Cdk1 activation at early prophase, the centrosome splits into two, and the daughter centrosomes gradually separate. Finally, as cells approach metaphase, the two spindle poles (i.e., daughter centrosomes) are fully separated, which coincides with the chromatin being totally condensed at the spindle equator. Interestingly, when I started using my Cdk1 antibody in immunofluorescence microscopy experiments, I could identify one or two dots in mitotic HeLa cells, which seemed to mimic the above described centrosome dynamics during mitosis. This observation, I believed, could be used to identify the mitotic phase at which cells treated with the different siRNAs were at. To this aim, I treated HeLa cells with siRNA oligonucleotides as before, and looked at them under the fluorescence microscope, following their labeling with the Cdk1 antibody (Figure 8D, top). Here, I classified control cells (VIII) transiting through mitosis in 3 categories (Figure 8D, top, left panels): a, b, cells that contained a single Cdk1 dot and partially condensed chromatin, which I assigned to early prophase, c, d, cells with a splitted Cdk1 signal, and uncompleted condensed chromatin, which I placed in prometaphase, and e, f, cells with two well separated Cdk1 dots, some spindle-localized Cdk1, and fully condensed chromatin, which I believed were at metaphase. In contrast to the control cells, a majority of the Survivin-depleted samples (S4) (Figure 8D, top, right panels) appeared to be in S-phase or G2 (g, h), or early prophase (i-l), as indicated by their unseparated Cdk1 dot, and low chromatin condensation. To statistically determine the relevance of the unsplitted Cdk1 signal in cells depleted of Survivin, I counted this phenotype in several independent experiments. As shown in Figure 8D, bottom, lack of Survivin caused a reduction of more than 2 fold (38% vs. 15%) in the number of cells with a splitted Cdk1 signal, supporting the role of Survivin in centrosomal Cdk1 activation and dynamics.
Recombinant Survivin Induces Cdc25 Activity In Vitro
The centrosomal data showed that Cdk1 does not localize at this location following Survivin abrogation. Since Cdk1 is first activated by Cdc25B at this organelle [2,3], it was tempting to speculate that Survivin might be bridging these two proteins at this place, thus contributing to the initial Cdk1 activity. Mislocalization and accumulation of Cdk1 at the cytosol in the absence of Survivin would tip the balance towards the inactive kinase, which is phosphorylated at residues Thr 14 and Tyr 15 (PT14/PY15-Cdk1 protein) [27,35] by the action of its negative regulators Wee1 and Myt1 [36,37], and indeed, this was already detected in the cells transfected with the Survivin double mutant (Figure 7D). To confirm this result using a different Survivin antagonist, asynchronous HeLa cultures were treated with siRNA oligonucleotides, and lysates were prepared from shaken-off cells (i.e., samples enriched in mitotic cells), which were analyzed by Western blotting using an antibody against the inactive Cdk1 form. As Figure 9A, left shows, Survivin siRNA (S4) but not control (VIII) treated cells showed the inactive Cdk1 band. Here, as a control, a Histone H1 phosphorylation assay of Cdk1 immunoprecipitated (IP) from the same samples was also performed to confirm that the PT14/PY15-Cdk1 band correlated with low Cdk1 activity (Figure 9A, right).
As already introduced in the centrosomal studies, another way to differentiate between the inactive and active Cdk1 forms is by running proteins longer during SDS-PAGE [27,35]. To see whether I could use this approach to easily follow the state of Cdk1 activation in cells subjected to RNAi, I used siRNA-treated synchronized cells that were released for 14 h (i.e., control cells transiting through mitosis, see FACS analysis in Figure 4B, bottom and kinase assay in Figure 4C, right), and lysates were prepared, which were subjected to a long SDS-PAGE run followed by Western blotting analysis using a Cdk1 antibody. As seen in Figure 9B, a strong Cdk1 band was detected in the control cells (VIII). In contrast, in the Survivin siRNA-treated samples (S4), I could see a doublet, consisting of a faster migrating band, similar to the one in the control, but fainter, and a slow-running Cdk1 form that should correspond to the phosphorylated inactive kinase [27,35].
In a reverse experiment to the one described above, if Survivin had a role in the Cdc25B-mediated activation of Cdk1, the former protein should induce the kinase activity when added back to cell lysates priorly depleted of it. To check this possibility, lysates from untreated synchronized HeLa cells entering mitosis (7-9 h) were prepared, which were subjected to Survivin depletion by incubating them with a Survivin antibody. The Survivin-immunodepleted supernatants (Figure 9C, left) were then supplemented with an ATP-generating system to make them biochemically active, and incubated with either recombinant GST or GST-Survivin protein to see whether Survivin could induce the accumulation of the active Cdk1 form. As Figure 9C, right shows, when Survivin-depleted HeLa cell lysates were incubated with GST, and Cdk1 was immunoprecipitated (IP), a small amount of the fast-migrating Cdk1 band could be seen that slightly increased from 7 to 9 h, as should be expected in cells approaching mitosis. In contrast, the same Survivin-depleted HeLa lysates supplemented with GST-Survivin clearly showed a higher amount of the immunoprecipitated fast-migrating Cdk1 protein at all times versus the control, which also significantly increased throughout the time course. Interestingly, in this latter case, a band corresponding to the recombinant GST-Survivin protein could be seen bound to the kinase at all time points in the coomassie blue-stained PVDF membrane (*) (Figure 9C, lower right panel).
Next, I performed a titration experiment to see whether the accumulation of the fast-migrating Cdk1 band was dose dependent. This time, I used S-phase extracts, which should have almost no active Cdk1, and therefore provide a cleaner background. Here, it was also interesting to test whether Survivin could induce the activation of Cdk1 in interphase. This approach however, had a caveat, which was that, under these conditions, Cdk1 might not be activated at all due to the predicted low upstream positive signaling in interphase samples. To check these possibilities, Survivin-depleted HeLa cell lysates (Figure 9D, top left) prepared from synchronized cultures collected 2 h post thymidine release (Figure 9D, bottom left) were incubated with increasing amounts of GST-Survivin or GST, and accumulation of the fast-migrating Cdk1 band was monitored. As seen in Figure 9D, right, increasing amounts of GST-Survivin but not GST induced the accumulation of the fast-migrating Cdk1 form in a dose-dependent manner. Finally, to see whether this S-phase, fast-migrating Cdk1 protein was active, a Histone H1 phosphorylation assay was carried out. As seen in Figure 9E, even in interphase, where Cdk1 and Cdc25 activities should be low, recombinant Survivin could induce some activation of the mitotic kinase. From all these in vitro experiments, I concluded that Survivin contributes to Cdk1 activity through Cdc25 possibly through its B isoform.
Survivin is Involved in the Activation of the Cdc25B Phosphatase Isoform
As I mentioned above, an explanation for the Survivin-mediated activation of Cdk1 in early prophase might be its mediation in the formation of the Cdc25B-Cdk1 complex at the centrosome. Accordingly, and since earlier in this paper, the direct interaction between Survivin and Cdk1 [22] was confirmed (Figure S2A), I also wanted to know whether Survivin can directly bind to Cdc25B. Figure 10A shows that when GST-Survivin was incubated with recombinant His-Cdc25B, these two proteins bound to each other. From this result, I was confident that I would find an in vivo Cdc25B-Cdk1 complex in the cells expressing Survivin. To my surprise, however, when I immunoprecipitated (IP) the Cdk1-Cyclin B1 complex from siRNA-transfected HeLa cells using a Cyclin B1 antibody, I could not see Cdc25B attached to it in the control samples (VIII) but counterintuitively, only in the cells that lacked Survivin (S4) (Figure 10B). The Cdc25B-Cdk1-Cyclin B1 complex correlated with a faint, fast-migrating Cdk1 band, or active kinase, and the slow-moving Cdk1 form previously ascribed to the inactive protein [27,35]. This Cdc25B-Cdk1-Cyclin B1 complex had to be cytosolic, as almost no Cdk1 protein was found in the centrosomes of the Survivin-depleted samples (Figure 8). The difference in the amount of Cdc25B bound to the Cdk1-Cyclin B1 complex in the siRNA-treated samples was replicated in cell lysates (Figure 10C, top), and probably due to the different cell cycle phase, which these two samples were at (i.e., G1 in VIII vs. G2/M in S4, see Figure 1A, right). Indeed, when aliquots from synchronized oligonucleotide-treated HeLa cultures were analyzed by Western blotting, control cells showed an increasing amount of Cdc25B that peaked at 9 h (G2/M-phase, see Figure 4B, bottom, upper graphs) and later declined (Figure 10C, bottom), which coincided with a similar amount of this protein in the Survivin-depleted samples (S4) also at 9 h (Figure 10C, bottom). Curiously, in these latter samples this amount of the Cdc25B protein was sustained for the remainder of the time course (Figure 10C, bottom).
The Cdc25B-Cdk1-Cyclin B1 complex detected in Figure 10B, and specifically the Cdc25B protein bound to it, seemed to be inactive, and indeed had to be in order to agree with the Histone H1 phosphorylation assays (Figure 3A and Figure 9A). To check whether this was true, I decided to immunoprecipitate (IP) Cdc25B from siRNA-treated synchronous cells entering mitosis (Figure 10D), or the moment when this phosphatase is activated, and measure this protein’s activity by employing the artificial substrate 3-O-methylfluorescein phosphate (OMFP) [38]. As seen, the Cdc25B activity of Survivin-depleted cells (S4) at G2/M-phase was residual (0.18 a.u.) in comparison with the control (VIII) (1.2 a.u.), representing a 6.7 fold difference. This experiment was repeated four times and the results were very reproducible as shown in Figure 10E.
To find out when the Cdc25B-Cdk1-Cyclin B1 complex first forms and activates, siRNA-treated synchronous samples were prepared (Figure 10F, bottom), and subjected to immunoprecipitation (IP) using a Cyclin B1 antibody (Figure 10F, top left). Alternatively, Cdc25B was immunoprecipitated, and used to measure its activity by OMFP hydrolysis (Figure 10F, right). As Figure 10F, top left shows, Cdc25B formed a transient complex with Cdk1 and Cyclin B1 in the control HeLa cells (VIII), which peaked at 14 h, and correlated with maximal phosphatase activity (Figure 10F, right) and transition through mitosis (Figure 10F, bottom). In contrast, in the absence of Survivin (S4), the Cdc25B-Cdk1-Cyclin B1 complex appeared earlier and continued being visible until the end of the time course (Figure 10F, top left). These latter samples remained stuck at the G2/M-phase checkpoint (Figure 10F, bottom), and did not show any phosphatase activity (Figure 10F, right).
A Gain-of-Function Cdc25B Mutant Can Bypass the Early Prophase Blockage Caused by Survivin Abrogation
To confirm the effect of Survivin abrogation on Cdc25B activity, the potent capacity of overexpressing the phosphatase Cdc25B to induce mitotic entry when transfected into HeLa cultures [39] could be used. In effect, if Survivin had a role in Cdc25B activation, overexpression of the phosphatase in Survivin-depleted cells would not induce entry into mitosis. To this end, the FACS profiles of siRNA-treated, synchronized HeLa cells transfected with a Cdc25B construct (Figure 11A, left) were analyzed as they transited through mitosis (Figure 11A, right). Figure 11A, right, upper graphs show that Cdc25B overexpression in control samples (VIII) resulted in an increase in the number of cells that progressed through mitosis (28% vs. 19%, or a 47% increase), as indicated by their larger G1 peak in comparison with the control (empty vector). In contrast, the Survivin-ablated, synchronized HeLa cells transfected with the Cdc25B plasmid remained stuck at the G2/M-phase, as attested by the negligible change in the G1 peak when comparing these cells to the empty vector-transfected samples (16% vs. 15%, or a 6.7% difference).
Because Survivin appeared to be indispensable to activate the Cdc25B phosphatase, and subsequently Cdk1, I predicted that a constitutively-active Cdc25B mutant being expressed in the Survivin-ablated cells would bypass the G2/M-phase blockage. For this purpose, a construct encoding a truncated Cdc25B protein, containing its catalytic domain but not its N-terminal regulatory region (∆NCdc25B) was generated as before [40] and transfected together with a GFP-monitoring plasmid in siRNA-treated synchronous cells (a full-length Cdc25B construct was also used as a control) (Figure 11B, right). Simultaneously, I checked that the recombinant proteins were correctly expressed. As Figure 11B, top left shows, lysates of siRNA-treated HeLa cells transfected with the pCdc25B or p∆NCdc25B constructs showed accumulation of these proteins. Here, an interesting pattern could be noticed, which was that both the exogenously-expressed full-length and truncated Cdc25B phosphatases accumulated to a large extent in the cells lacking Survivin, mimicking the behavior of the cells only expressing the endogenous phosphatase. Individual synchronized HeLa cells transfected with the control (VIII) or Survivin (S4) oligonucleotide, and expressing the recombinant Cdc25B proteins as they approached the G2/M-phase are shown in Figure 11B, right. Here, it can be seen that control siRNA-treated HeLa cells (VIII) that were transfected with pCdc25B showed more mitotic cells than cultures, which were transfected with pGFP alone, in agreement with the data in Figure 11A, right. In contrast, Survivin-knocked down cells (S4) transfected with the same pCdc25B construct did not enter mitosis when compared to the control but, on the contrary, died more readily. On the other hand, both synchronous HeLa cells treated with the control (VIII) or Survivin (S4) siRNA, followed by transfection of the constitutively-active Cdc25B mutant, clearly increased their mitotic entry when compared to their respective controls. The results from three different experiments of this type appear quantified in Figure 11B, bottom left. Here, it can be seen that ∆Ncdc25B was effective in bypassing the G2/M-phase blockage associated with Survivin abrogation, and concomitant low Cdk1 activity reported in these studies.
Discussion
The current model of Survivin function assigns two roles to this protein, namely: i. Its mediation in the correction of faulty microtubule-kinetochore attachments, and ii. the process of cytokinesis, both as part of the CPC [13]. In agreement with this, it is firmly believed that the first abnormal phenotype, following the interference with Survivin protein, is a prometaphase blockage that responds to errors in the repair of incorrect microtubule-kinetochore connections [14,15]. In this regard, before this project started, I was aware of experiments carried out with Xenopus egg extracts [17] and HeLa cells [16,19], which revealed that, following Survivin abrogation, a peculiar phenotype ensues, consisting of spindles with a very short pole-to-pole distance, also called mini spindles, which pointed out to a failure in centrosome separation. Here, I believed that these results could poorly be explained by the sole role of the Survivin protein in the CPC since this complex had not been reported to have a function so early in mitosis [13]. A more plausible explanation here might be a role of Survivin in this organelle’s splitting at early mitosis, an event for which Cdk1 is necessary [20,21]. In support of this model, it has been reported that interference with Survivin leads to centrosomal abnormalities, and the disruption of a Survivin-Caspase-3-p21 complex at this location [18]. In this respect, if Survivin had a role in centrosomal Cdk1 activation, this would place this protein at the hub where decisions to enter mitosis are made, prior to its CPC function, explain why cancer cells show preference for taking over this pathway, and might also provide the explanation for the connection between Survivin and cell death, as a default centrosomal-based apoptotic route [18] triggered when mitotic onset misfires. This possible new mitotic role of Survivin, however interesting, had never been pursued before, and therefore, I decided to make it the center of my investigation.
Survivin Is Required to Activate Cdk1 in Early Prophase and for Cancer Cells to Commit to Mitosis
To start this project, I first wanted to clarify when cells that lack Survivin first arrest in mitosis. For this purpose, I repeated the RNAi experiments carried out by others in HeLa cells, which led them to conclude that, in the absence of Survivin, a prometaphase blockage ensues [14,15]. To my surprise, when I replicated this work, I not only could see less cells entering mitosis (Figure 1D) but those, which managed to progress into this stage, being blocked at early prophase instead of prometaphase. The evidence supporting this finding in the Survivin-depleted samples was ample, and encompassed: i. The presence of a microtubule ring surrounding their chromatin (Figure 1B,C, and Figure 2A,B), ii. the absence of a spindle or the presence of a very faint one (Figure 1C, Figure 2A and Figure S1C), iii. the detection of an intact nuclear lamina (Figure 2A,B), iv. the partially condensed chromatin (Figure 1C and Figure 2A,B), v. the lower amount of Cdk1 activity, and vi. the detection of the microtubule ring and intact nuclear lamina also in cells incubated with the Cdk1 inhibitor Purvalanol (Figure 2D). From this data, I concluded that Survivin has a role upstream of its CPC function at the level of early Cdk1 activation, an event which occurs in early prophase at the centrosome [2,3]. This conclusion was further supported by comparing the activity of Cdk1 in the absence of Survivin, and that of cells incubated with a real prometaphase blocker such as nocodazole [28] (Figure 3B).
The results obtained with asynchronous HeLa cultures could be reproduced with synchronous cells. Here, a much more robust mitotic blockage, and declining Cdk1 activity was observed in the cells depleted of Survivin (Figure 4B,C). From this data, I could easily infer that the mitotic cells that accumulated in the G2/M-phase peak in the absence of Survivin had to be in either G2 or early prophase (i.e., stable checkpoint), as their asynchronous counterparts, since if these cells had committed to mitosis (i.e., prometaphase) in the absence of Cdk1 activity (Figure 4C), they would have averted the spindle checkpoint due to their low Cdk1 activity, and progressed into a new round of DNA synthesis, as HeLa cells possess very low levels of the p53 tumor suppressor [41], and therefore lack an effective G1 checkpoint [42].
The use of the Survivin double mutant in these studies provided a more precise description of the mitotic anomalies that occur when the Survivin’s function is impaired in cells about to enter mitosis. First, these results showed that Survivin is crucial to commit to mitosis (i.e., transition from prophase to prometaphase), as demonstrated by the accumulation of flat or large rounded cells when the SUR D70A/D71A mutant was transfected (Figure 7A,E,F). Interestingly, these large rounded mitotic cells were already observed in the siRNA studies, and assigned to early prophase (see above explanation). Since in these two cases, this phenotype coincided with a low Cdk1 activity (Figure 3A and Figure 7C), and it is well know the need of Cdk1 activation for cells to commit to mitosis, it was only logical to conclude that the impairment in entering mitosis when using either of the above mentioned Survivin antagonists was probably due to the impossibility of the transfected cells to mount a high enough Cdk1 activity with which to transverse the G2/M-phase checkpoint. Second, the Survivin double mutant experiments also showed that in the absence of a functional Survivin protein, and despite their low Cdk1 activity, some cells can still transverse the G2/M-phase checkpoint (33%, see Figure 7E, bottom), however, in most cases, they stall or die at this stage. Similar results were also obtained by other researchers in human cells when interfering with their Cdk1 levels via RNAi [43]. In this case, 20% of cells still could enter mitosis, however, these Cdk1-attenuated cells were impaired in their ability to phosphorylate their mitotic targets. At this point, it is impossible not to think about the targets that Survivin has in cells entering prometaphase and proceeding into cytokinesis [13], and conclude that all the abnormalities due to the Survivin double mutant seen at this late mitotic stages should be attributed to the role of this protein at the CPC. Focussing on prometaphase however, we should not forget about the extended regulation exerted by Cdk1 on the CPC at this phase [13,44,45,46], and conclude that maybe, we should start considering the abnormal phenotype caused by Survivin impairment at prometaphase more as a combined outcome due to both CPC malfunction (i.e., Survivin’s traditional role) and Cdk1 misactivation (i.e., new role of Survivin proposed in this paper). Briefly, INCENP is phosphorylated by Cdk1, which regulates the localization and kinase activity of Aurora-B [44]. Second, Plk1 binds to Haspin in a Cdk1-dependent manner, and reducing Plk1 activity inhibits Haspin phosphorylation of Histone H3 [45]. Third, Survivin binds to phosphorylated Histone H3, and this step is necessary for CPC accumulation at the centromere [46]. Interestingly, in this same work, an identical mutant as the one I used in my studies, SUR D70A/D71A, showed no binding to a phosphorylated Histone H3 peptide in vitro, implicating the same two residues, Asp70 and Asp71, in both Cdk1 activation and Survivin centromeric localization, and providing a clever mechanism for Survivin to exert its two functions in a sequential accurate manner. Finally, Borealin, a Survivin-binding partner at the centromere, is phosphorylated by Cdk1, which allows it to bind to Shughosin, and subsequently phosphorylate Histone H2A [13]. From all the above evidence and the data shown in this paper, it seems fair to conclude that Survivin appears to have an undiscovered role on the CPC via its regulation of the Cdk1 kinase.
Centrosomal Cdk1 Does Not Accumulate in the Absence of Survivin, and it May Cause a Short-Circuit between this Kinase and its Activators
Cdk1 is activated by the dual-specificity phosphatase Cdc25 [1], with isoform B being the first one that acts on Cdk1 at the centrosome during early prophase [2,3], and isoforms A and C being subsequently active at the cytosol and nucleus in a process that amplifies the initial kinase activity [1], and commits cells to mitosis [10,11]. The data in this article now shows that when the Survivin function was compromised in HeLa cells that had not entered mitosis yet, these samples arrested at early prophase (see above discussion), pointing out to a short-circuit in the centrosomal Cdc25B-Cdk1 axis according to the literature. In agreement with this model, the Cdk1 amount and activity at this organelle were greatly reduced in the Survivin-depleted samples (Figure 8B,C). Interestingly, still a low quantity and activity of centrosomal Cdk1 protein could be seen at early time points that correlated with an also unexpected Cdk1 activity at 12 h in cytosolic extracts from Survivin siRNA-treated samples. This early kinase activity might be due to the initial Cdk1-Cyclin A complex, which escapes Wee1/Myt1-inhibitory phosphorylation, and is required to activate Aurora A [47], itself the activator of Cdc25B at the centrosome [48], before Cdk1-Cyclin B1 activity can be triggered in this organelle. The biochemical centrosomal data could be confirmed by immunofluorescence experiments, which also showed a reduced amount of Cdk1 at this location and an unsplitted kinase signal (Figure 8D), which is indicative of lack of Cdk1 activity at this organelle [20,21]. As a conclusion, from all the centrosomal results, I initially envisioned Survivin probably acting as some kind of bridge that could facilitate the activation of Cdk1 via Cdc25B at the centrosome, being that role the reason why, in its absence, cells stalled at early prophase.
Survivin Activates the Cdk1 Kinase via Phosphatase Cdc25B
Apart from the centrosomal Cdk1 only being active in the presence of Survivin, I could detect a fast migrating Cdk1 band, which was previously assigned to the active kinase [27], being bound to Survivin in the same location (Figure 8A). This result pointed at Cdc25B being responsible for this effect, and could also be replicated in cytosolic extracts, which also contained centrosomes, of HeLa cells treated with the control siRNA (Figure 9B). In contrast, similar lysates of Survivin-depleted cells had a much lower amount of the active Cdk1 form and more of the inactive isoform in comparison with the control. The Cdk1 protein in the Survivin-depleted samples had to be cytosolic as hardly any kinase was found in the centrosomes of cells depleted of Survivin (Figure 8B,C). Interestingly, the putatively Cdc25B-induced shift between the inactive and active Cdk1 forms could be induced in a cell-free assay where Survivin was depleted from mitotic lysates, and then added back (Figure 9C). Here, I could see that this shift correlated with the recombinant Survivin protein being bound to Cdk1. The accumulation of the faster migrating active Cdk1 protein could also be reproduced in a dose-dependent manner using interphase extracts (Figure 9D) that phosphorylated Histone H1 (Figure 9E), a result that might indicate an unknown function of Survivin at interphase that could be behind the cell death observed in cells expressing the Survivin double mutant while progressing towards the G2/M-phase checkpoint (Figure 7E, bottom).
Although, I initially supported a role for Survivin as a bridge between Cdc25B and Cdk1 at the centrosome, I discarded this possibility when I could only detect a Cdc25B-Cdk1-Cyclin B1 complex in lysates of HeLa cells treated with the Survivin siRNA (Figure 10B). As already mentioned above, this complex could only be cytosolic. Also, according to the Histone H1 phosphorylation data (Figure 3A and Figure 9A), the Cdc25B bound to Cdk1 in the cytosolic complex had to be inactive. Indeed, when I looked at this immunoprecipitate in detail, I could see the accumulation of the slow-migrating Cdk1 band previously ascribed to the inactive kinase [27,35]. The conclusion of Cdc25B being inactive in the absence of Survivin was corroborated by directly measuring the Cdc25B activity in samples where this protein was ablated (Figure 10D–F). As a consequence of these results, I opted for a new model where Survivin might act as some kind of centrosomal anchor or scaffold for the inactive Cdc25B-Cdk1-Cyclin B1 complex, which by this means, could come close to its centrosomal activator/s.
In the absence of Survivin, Cdc25B somehow accumulated (Figure 10B,C,F and Figure 11B), and remained in complex with the Cdk1 protein for a longer period of time (Figure 10F). This result was very different to what was reported before about this protein’s short stability [49]. Moreover, Cdc25B normally accumulates at G2, and its activity precedes Cdk1 activity, followed by both events rapidly declining after G2/M-phase [49,50,51] (a result also reproduced in my control experiment (Figure 10F)), as a consequence of Cdk1-dependent proteasomal degradation [52].
Overexpression of Cdc25B normally induces mitotic entry [39,53]. However, when the recombinant Cdc25B phosphatase was expressed in the Survivin-depleted G2/M-phase blocked HeLa cells, this protein could not rescue the blockage (Figure 11A, right), confirming that Survivin is required to activate Cdc25B. In agreement with this theory, when a gain-of-function Cdc25B mutant was expressed in G2/M-phase cells lacking Survivin, they managed to enter mitosis (Figure 11B), supporting a role for Survivin in signaling through the Cdc25B-Cdk1 axis.
Early Prophase Physiognomy and Reversibility
Following the accumulation of data in this project, it was clear that the Survivin protein is necessary to assemble a functional Cdk1 complex in early prophase. This was a valid conclusion except for the fact that a lower than expected number of rounded cells was observed following the treatment with the different Survivin antagonists. Searching through the literature to find an explanation for this observation, I discovered that early prophase cells do not always look round, but on the contrary, many times remain spread, resembling the physiognomy of interphase or G2 cells [10,11]. The choice between these two different appearances seems to rely on the amount of Cdk1 activity in the nucleus. Accordingly, it was quite possible that many of the cells that looked like being in G2, or interphase, following interference with the Survivin function, were actually in early prophase.
Early prophase is a reversible phase, and this reversibility can be triggered by multiple insults. Antephase is the stage to which cells resort following the activation of a G2/M-phase checkpoint branch that responds to stress insults, and is mediated by p38 [54]. This checkpoint is different to the one triggered by DNA damage, which depends on ATM and/or ATR activity [55]. Therefore, I would also speculate here that many of the cells that entered mitosis with a low Cdk1 activity as a result of Survivin interference, might have activated their antephase rather than their DNA damage checkpoint, and as a consequence returned to G2, hoping to re-enter mitosis at a better time. This was precisely what could be seen with some of the Survivin double mutant- transfected cells (Figure 7F). The antephase checkpoint only works while the activity of Cdk1 is not very high, or before nucleolar breakdown [54]. This would explain why cells that were treated with the Survivin peptide could not return to G2, and continued into the apoptotic program (see below).
Interference with Committed Mitotic Cells via a Cdk1-Binding Survivin Peptide Results in Apoptosis
The Survivin peptide was made with the intention to interfere with the assembly of the Survivin-Cdk1 complex. As expected, incubation of HeLa cell lysates from nocodazole-treated HeLa cells (i.e., prometaphase) with this reagent caused complex disassembly, and loss of Cdk1 activity (Figure S4B), pointing at Survivin binding being necessary for Cdk1 not only to activate but to remain active.
Interestingly, the Survivin peptide did not cause a G2/prophase blockage, as seen with the other two antagonists of this protein used in this work (i.e., Survivin siRNA and double mutant), and this might have been due to the short half-life of peptides in general, which would have allowed cells approaching mitotic entry in the presence of this reagent to briefly halt at the G2/M-phase checkpoint until the peptide cleared up. A brief G2/M-phase blockage would be difficult to detect by FACS analysis, and this is probably why I could not observe this abnormal phenotype under these conditions. In contrast, once transitioning into prometaphase, a stage at which the Cdk1 kinase is fully active [10,11], the presence of the Survivin peptide in cells should have been very troublesome, since at this stage and moving forward both Cdk1 and Survivin are required for events that normally occur fairly rapidly such as spindle formation (17, 20, 21, 56, 57), CPC functioning [13,44,45,46] and decisions on cell fate [12,18]. Here, a short-life potent antagonist of Survivin and Cdk1 function should indeed have a strong effect, and these precisely were the places at which anomalies were detected when committed mitotic HeLa cells were incubated with the Survivin peptide.
Specifically on the cell death caused by the Survivin peptide, it should be noticed that this phenotype was not or minimally observed when the other two Survivin interfering reagents were used in this study. The reason for this might have been the possibility for cells to find refuge in the G2/M-phase checkpoint when subjected to slow-accumulating Survivin antagonists. In support of this theory and the vulnerability of tumoral cells committed to mitosis (i.e., nuclear Cdk1 activation and progression into prometaphase), which are then targeted at their Cdk1 function, cancer cells and tumors in a cancer xenograft model were very sensitive to a combination of taxol and purvalanol A (i.e prometaphase blockage followed by Cdk1 targeting) but not to the reverse order of the treatment (i.e., Cdk1 inhibition leading to G2/M-phase blockage, and therefore lack of effectiveness of a prometaphase drug such as taxol) [58], indicating that when challenged cancer cells are allowed to rest at the G2/M-phase checkpoint, they can avoid cell death.
More specifically on the cell death observed when HeLa cells were treated with the Survivin peptide, it has been reported that Cdk1-Cyclin B1 activity is required to phosphorylate Procaspase 8 [59] and Procaspase 9 [60], and subsequently block the extrinsic [59] and intrinsic [60] apoptotic pathways in cancer cell lines. Here, blockage of Cdk1 activity by Cyclin B1 RNAi or the Cdk1 inhibitor RO-3306 [59], or short-circuit of the kinase by the expression of non-phosphorylatable caspase proforms [59,60], led to apoptosis. In agreement with these results, I could also detect Caspase 8 and 9 activity being triggered in HeLa cells after treatment with the Survivin peptide (see Figure 6C,D). From these results, the Cdk1 kinase rises as some kind of mitotic shield but also, as a factor, which might contribute to tumorigenesis if deregulated. Traditionally, these have been roles assigned to the Survivin protein, and here, a partner in causing these phenotypes may have been found.
There has been a lot of controversy regarding the two seemingly roles of Survivin in cancer regulation, namely: the one in mitosis and the one in apoptosis (for reviews on these two different views see 61 and 62). In this respect, I propose here a model, which attempts to integrate these two areas of research. Herein, Survivin’s initial function would be to initiate mitotic onset by contributing to the activation of Cdk1 at the centrosome. At this stage, interference with Survivin would not lead to fatal consequences, as cells would be able to wait at the G2/M-phase checkpoint until being fit to move on. In contrast, sustained Survivin malfunction in arrested cells resulting in bypass of this checkpoint, or interference with this protein in committed mitotic cells (i.e., prometaphase) that require high Cdk1 activity and a functional CPC, both functions regulated by Survivin, would lead to a myriad of mitotic defects and ultimately cell death.
Survivin Binds to the αC/β4 Loop in Cdk1, a Region Involved in Protein-Protein Interactions, Binding of the Hsp90-Cdc37 Complex and Regulation of Kinases’ Activity
The Survivin peptide that binds to Cdk1 (Figure S3A and Figure S3C) comprises several negatively-charged residues that reside on the Survivin dimer’s acidic surface [32]. In fact, even though I used the SUR D70A/D71A double mutant as a Survivin antagonist, both the GST-SUR 71-142 truncated protein, which could not bind the kinase (Figure S2B), and the alanine point mutant SUR D71, which slightly did (Figure S5, bottom), suggested that only the Asp70 residue is crucial for binding to Cdk1. From these observations, I predicted that Survivin should probably recognize a region in the Cdk1 kinase with a net positive charge, and indeed, I was able to narrow down the Survivin-binding sequence in the Cdk1 kinase to amino acids Lys56 through Met85, a region that includes the kinase’s αC/β4 loop, which contains several basic residues (Figure S3B). Residues 66 through 85 were also part of the original Cdk1 sequence recognized by Survivin, however this kinase region harbors numerous hydrophobic amino acids, and therefore I found it unlikely to be part of the Survivin-binding domain.
The αC/β4 loop is a conserved structural motif present in all eukaryotic protein kinases, which bridges the αC helix in the N lobe and regions at the top of the C lobe [63]. In most kinases, this loop spans 8 amino acids, which follow the consensus sequence: L-x-H-P-N-T-V-x, where x represents any amino acid [64]. Functionally, it has been hypothesized that the αC-β4 loop may act as a molecular brake for protein kinases by maintaining auto-inhibitory interactions via hydrogen bonding with the hinge region in the C lobe [65,66,67]. In support of this theory, many mutations have been found at this location, which confer constitutive kinase activity and/or drug resistance, and play a direct role in cancer progression [64]. The αC-β4 loop is also a site for protein–protein interactions, and as such, it recognizes the molecular chaperone Hsp90 and its co-chaperone Cdc37 [68,69], proteins that together promote the proper folding of 60% of the human kinome [69,70]. By cryoEM structure of an Hsp90-Cdc37-Cdk4 complex, a general mechanism of Hsp90-Cdc37 action has been postulated [71], where a conserved H-P-N motif in Cdc37 would mimic the turn within the kinase αC-β4 loop, and push against the kinase αE helix in the C-lobe, preventing the contact between the kinase N and C lobes. As a result of this intermolecular interaction, the kinase would be initially stabilized, and would expose other motifs to which Hsp90 could bind. From all this information, it is tempting to speculate that Survivin might also have a role in the regulation of Cdk1 inter-lobe movement and its activation. In support of this view, Survivin recapitulates some of Cdc37’s features. In effect, apart from binding to the αC-β4 loop region as already stated, Survivin, like Cdc37, binds to the N-terminus of the Hsp90 chaperone [72,73]. Also, I could detect a complex between Survivin, Hsp90 and the active Cdk1 protein form at mitosis (Figure S6). For these reasons, it is plausible to believe that coinciding with mitotic onset, Survivin might function as some kind of Hsp90 co-chaperone. Alternatively, Survivin might bind the chaperone machinery, once the Cdk1 complex has been assembled, and bring it close to its activator/s at the centrosome. Still, a third possibility might be that Survivin unloads the Hsp90-assembled Cdk1 complexes, and delivers them to its centrosomal activator/s. In this regard, this work showed that when Survivin was absent, an inactive cytosolic Cdc25B-Cdk1-Cyclin B1 complex accumulated, and here, it would be interesting to find out whether this complex was bound to Hsp90. A function of Survivin in mediating Cdk1 through Hsp90 is however a bit controversial, as it encounters those who claim that Cdk1 is not a Hsp90 client [70], and others that support the opposite view [69,74,75].
Conclusions
Survivin has been called a cancer gene [76]. This paper now shows that this title may have in part been earned due to an up-to-date unreported role of Survivin in the activation of the Cdc25B-Cdk1 complex. In this regard, there is plenty of research showing a correlation between Cdc25B signaling and tumorigenesis, and it would make perfect sense that cancer cells hijack this pathway in order to control mitotic entry. The connection between Cdc25B and cancer has led to an overwhelming effort trying to develop efficient Cdc25B inhibitors [77]. These inhibitors however most of the time target the Cdc25B catalytic domain, and here, an exciting alternative might be finding small molecules that disrupt the Survivin-Cdc25B interaction.
In this work, I have also shown that the absence of Survivin leads to an early prophase blockage due to low Cdk1 activity, and propose that this phenotype is very sensitive to events, which override the G2/M-phase checkpoint. Therefore, a combination of drugs that promote mitotic entry, and then inhibit Cdk1 activity might be a very effective way to combat cancer.
Allosteric inhibitors that bind to the αC-β4 loop in Cdks are also on the way [78], and this paper reinforces the importance of this site by proving that Survivin is another protein that binds to this epitope. From different studies, the αC-β4 loop is now rising as some sort of a hub through which kinases communicate with other proteins, and signals can be sent through a myriad of pathways. Therefore, elucidating the exact function of this region in kinases, and the role of the chaperone machinery at this location, may help refine the design of new drugs against cancer.
To end, I would like to reiterate the connection between Cdk1 activity and CPC’s regulation, and the possible role of Survivin at multitasking between kinases. In this context, Survivin might operate as a coordinator of upstream and downstream mitotic events, which would lead to smooth progression through mitosis.
Materials And Methods
Plasmids, Cloning, Recombinant Proteins and Peptides
Full-length human cDNAs encoding Cdk1 (BC014563) and Cdc25B (BC051711) were obtained from Open Biosystems. Cdk1 and Cdc25B cDNAs were subcloned in frame with the N-terminal His-tag into pRSETa (Novagen, Merck Biosciences). Full-length Cdc25B cDNA or a construct encoding a truncated Cdc25B protein, containing its catalytic domain but not its N-terminal regulatory region (∆NCdc25B) [40] were subcloned into pcDNA3. pGST-Survivin (plasmid containing a Survivin sequence cloned in frame with the N-terminal GST-tag into pGEX (Pharmacia)) and pGFP-Survivin (plasmid made in pcDNA3 including an N-terminal GFP sequence (pGFP)) were available in the laboratory. Deletion mutants for His-tagged Cdk1 (residues 1-43, 1-56, 1-85, 1-183, 9-85, 36-85 and 182-297), Cdc25B (residues 391-580 (ΔNCdc25B)) and GST-Survivin (residues 1-70, 15-142, 38-142, 55-142, 71-142 and 81-142) were generated by PCR. Alanine mutagenesis of the Survivin sequence (pGST-Survivin) was carried out using the QuikChangeR Site-Directed Mutagenesis Kit (Stratagene). Plasmid encoding GFP-Survivin D70A/D71A (pGFP-Survivin D70A/D71A) was made by cloning the Survivin D70A/D71A sequence obtained by alanine mutagenesis into pGFP. All constructs were confirmed by DNA sequencing. His-tagged and GST-fusion proteins were expressed in BL21 E. Coli strain, and isolated by affinity chromatography on Ni2+-charged agarose (Novagen, Merck Biosciences), or glutathione agarose (Sigma-Aldrich). A peptide comprising residues Ala55 throughout Asp70 in Survivin (AQCFFCFKELEGWEPD), and a scrambled version of the same peptide (EPCWDECFAEKFQGFL), both containing a Biotin moiety and a HIV tat cell-permeable sequence at the N-terminus, were synthesized using f-BOC chemistry, purified by HPLC and analyzed by mass spectrometry (Peptron, Inc., Daejeon, South Korea or W. M. Keck Biotechnology Research Center, Yale University, School of Medicine).
Cell Culture, Synchronization and Transfections
Cervical carcinoma HeLa cells (ATCC, Manassas, VA) were maintained in culture as described [16]. Cells were synchronized by incubation with 2 mM thymidine (G1/S-phase) or 10 μM nocodazole (M-phase). In some experiments, cells were treated with the Cdk1 inhibitor purvalanol A (10-20 μM) for 16 h. On other occasions, mitotic cells were prepared by shaking off the plates, or collecting the samples 9 to 14 h post thymidine release. Gene targeting by small interfering RNA (siRNA) was carried out with a control or a Survivin-siRNA oligonucleotide characterized in earlier studies [16]. When using synchronized cultures, cells were first treated with the appropriate siRNAs, and then incubated with thymidine for 48 h. Cells were then released into fresh medium, and samples taken at increasing time intervals. For plasmid transfection, cells (50-70% confluency) were incubated with 1-2 μg of plasmid in 1 mL Optimem-I and 4 μL lipofectamine/well for 4 h. In the case of transfections with siRNAs and Cdc25B plasmids, cells remained in thymidine for 32 h after the siRNA treatment, plasmids were transfected in the presence of thymidine for 4 h, and cells were finally released 12 h post-transfection and harvested as indicated.
Preparation of Cell Lysates and Isolation of Centrosomes
Cells were lysed in 1 mM HEPES, pH 7.2, 0.5% IGEPAL CA-630, 0.5 mM MgCl2, 0.1% β-mercaptoethanol, 50 mM NaF, 1 mM Na3VO4 plus protease inhibitors for 30 min on ice. The cell lysate was pipetted up and down five times, centrifuged at 2,500 g for 10 min, and supernatant was adjusted to 10 mM HEPES, pH 7.2 final concentration. For isolation of crude centrosomes, 1 μg/mL DNAse I was added to the lysate, and incubation proceeded for 30 min on ice. The mixture was then overlaid on a 60% sucrose cushion, containing 10 mM HEPES, pH 7.2, 0.1% Triton-X100 and 0.1% β-mercaptoethanol, and centrifuged at 10,000 g for 30 min. The interface of the sucrose cushion containing enriched centrosomes was collected for the various experiments. Protein concentration was estimated by the BCA assay (Pierce).
GST-Pull Downs, Immunoprecipitations and Antibodies
Pull-down experiments with His-tagged proteins mixed with GST, GST-Survivin or GST-Survivin mutants were carried out as described [78]. For streptavidin-binding experiments, 50 μg of biotinylated peptides were bound to 25-50 μl of a 50:50 slurry of streptavidin agarose (Sigma-Aldrich), mixed with recombinant proteins or cell lysates, and analyzed by Western blotting. Immunoprecipitations using primary antibodies (10 μg/mL), and 200-400 μg of total or fractionated cell extracts were carried out as described [79]. The following antibodies to Survivin (NOVUS Biologicals), Cdk1, Lamin B, 14-3-3 β (all from Santa Cruz Biotechnology), PT14/PY15-Cdk1, pro-Caspase 3, GFP, HA-Tag, RCC1, (all from Fisher Scientific), Cdc25B, HSP90, γ-Tubulin, p21, β-Actin, Ran (all from Cell Signaling), Cyclin B1 (PharMingen), α-Tubulin (Sigma-Aldrich) and His-tag (Invitrogen) were used. Protein bands were visualized by enhanced chemiluminescence (Amersham Biosciences).
Analysis of Cdk1 Activity
Cell lysates in 10 mM HEPES, pH 7.2, 0.5% IGEPAL CA-630, 0.5 mM MgCl2, 0.1% β-mercaptoethanol, 50 mM NaF, 1 mM Na3VO4 plus protease inhibitors were used to immunoprecipitate Cdk1. Kinase activity was assayed as described [22]. For the in vitro Cdk1 activation experiments, synchronized HeLa cell lysates were prepared by incubation for 30 min on ice in 10 μM HEPES, pH 7.4, 10 μM KCl, 1.5 mM MgCl2 and 2 μM DTT plus protease inhibitors, and passing the cells ten times through a 27 ½ G needle. Lysates were centrifuged at 2,500 g for 10 min, and supernatants were adjusted to 10 mM HEPES, pH 7.4, 100 μM KCl and 1 mM DTT. Survivin was depleted in the lysates by rotation with 25-50 μl of a 50:50 slurry of protein A agarose (Boehringer Ingelheim) bound to Survivin antibody overnight at 4°C. Survivin-depleted supernatants were supplemented with an ATP-regenerating system (2 mM ATP, 10 mM phosphocreatine and 3.5 U creatine kinase per 100 μL lysate), and incubated with 0.5-3 μM GST or GST-Survivin for 1 h at 30°C. For the peptide interference of Cdk1 activity in vitro, lysates were prepared from nocodazole-treated cells as described for the Cdk1 activation experiments but this time the endogenous Survivin protein was not depleted, and samples were incubated with 1-5 μM peptides for 1 h at 30°C. All reactions were terminated by a 10-fold dilution in a buffer containing 50 mM NaF and 1 mM Na3VO4. Phosphorylated Cdk1 proteins were immunoprecipitated, resolved by SDS-PAGE and identified by Western blotting. Cdk1 activity was assayed as described [22].
Cdc25B Activity Assay
Cdc25B protein was immunoprecipitated from cell lysates as described [79], and phosphatase activity was assayed by 3-O-methyl fluorescein phosphate (OMFP) hydrolysis at 30°C in assay buffer (100 mM Tris, pH 8.2, 40 mM NaCl, 1 mM DTT and 20% glycerol) [38]. Fluorometric detection was carried out at 485 nm excitation and 530 nm emission wavelengths (Photon Technology International, Inc.). Fluorescence slopes were calculated and normalized against values obtained with control, non-binding IgG.
FACS Analysis
DNA content in asynchronous or thymidine-synchronized HeLa cell cultures was determined by FACS analysis (propidium iodide staining and flow cytometry) as described [22]. Data were quantified using the FlowJo cell cycle analysis software.
Analysis of Apoptosis
DNA integrity was analyzed by scoring the sub-G1 population (<2N) in FACS analysis. Caspase activity was detected by incubating siRNA transfected HeLa cells with a fluorescein-conjugated caspase inhibitor (FAM-DEVD-FMK). Alternatively, HeLa cell lysates were analyzed by Western blotting using an antibody to procaspase 3.
Fluorescence Microscopy
Transfected HeLa cells seeded onto optical grade glass coverslips were fixed in ice-cold 4% paraformaldehyde/PBS, pH 7.2 for 1 h, washed and permeabilized in 0.2% Triton-X100/PBS, pH 7.2 for 30 min at 22°C, except when they were transfected with GFP constructs, in which case, cells were directly analyzed under the microscope. Coverslips were blocked in 3% BSA/0.1 % Tween-20/PBS, pH 7.2 for 30 min at 22°C, and incubated with primary antibodies appropriately diluted in blocking buffer for 1 h at 22°C. Cells were incubated with Alexa Fluor (Invitrogen) secondary antibodies for 1 h at 22°C in the dark. When cells were transfected with biotinylated peptides, they were directly incubated with Texas red-streptavidin, following fixation and permeabilization. DNA was stained with 10 μg/mL DAPI (Sigma-Aldrich). Coverslips were analyzed using an inverted fluorescence microscope (Olympus IX71). Pictures were taken using a microscope-attached camera (Nikon), and data was computer-analyzed by the IPLab software.
Time-Lapse Video Microscopy Analysis
Thymidine synchronized HeLa cells seeded onto Lab-Tek two-chambered borosilicate coverglass slides (Nalge Nunc International) were transfected with the appropriate GFP plasmid, released into fresh medium, and imaged using an inverted microscope (Zeiss) with a 20X numerical aperture 0.5 objective lens, a spinning-disk confocal scan head (Perkin Elmer Life and Analytical Sciences) and an Orca-ER cooled CCD camera (Hamamatsu) under conditions of CO2 exchange (0.5 L/min) at 37ºC. Time-lapse imaging was conducted using a multidimensional image software (Metamorph 6.3r6) to acquire images every 10 min for 24 h, observe multiple x,y-coordinates at every time point and acquire six optical sections over a 10 μm z-series (2 μm step size) at each coordinate. A z-stack of GFP images was taken at the start and end of each time-lapse experiment.
Supplementary Materials
The following supporting information can be downloaded at: preprints.org.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Acknowledgments
The author wants to thank Dr. Stephen J. Doxsey for his help in the time-lapse video microscopy studies, and his laboratory colleagues for their input. This work was supported by NIH grants CA78810, CA90917 and HL54131.
Conflicts of Interest
The author declares no competing interests.
References
- Crncec, A. and Hochegger, H. (2019). Triggering mitosis. FEBS Lett. 593, 2868-2888.
- Krämer, A., Mailand, N., Lukas, C., Syljuåsen, R. G., Wilkinson, C. J., Nigg, E. A., Bartek, J. and Lukas, J. (2004). Centrosome-associated Chk1 prevents premature activation of cyclin-B-Cdk1 kinase. Nat. Cell Biol. 6, 884-891. [CrossRef]
- Lindqvist, A., Källström, H., Lundgren, A., Barsoum, E. and Rosenthal, C. K. (2005). Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J. Cell Biol. 171, 35-45. [CrossRef]
- Potapova, T. A., Sivakumar, S., Flynn, J. N., Li, R. and Gorbsky, G. J. (2011). Mitotic progression becomes irreversible in prometaphase and collapses when Wee1 and Cdc25 are inhibited. Mol. Biol. Cell 22, 1191–1206. [CrossRef]
- Peter, M., Nakagawa, J., Dorée, M., Labbé, J. C., Nigg, E. A. (1990). In vitro disassembly of the nuclear lamina and M phase-specific phosphorylation of lamins by cdc2 kinase. Cell 61, 591-602. [CrossRef]
- Miake-Lye, R. and Kirschner, M. W. (1985). Induction of early mitotic events in a cell-free system. Cell 41, 165-75. [CrossRef]
- Dessev, G., Iovcheva-Dessev, C., Bischoff, J. R., Beach, D. and Goldman, R. (1991). A complex containing p34cdc2 and cyclin B phosphorylates the nuclear lamin and disassembles nuclei of clam oocytes in vitro. J. Cell Biol. 112, 523-533. [CrossRef]
- Georgatos, S. D., Pyrpasopoulou, A. and Theodoropoulos, P. A. (1997). Nuclear envelope breakdown in mammalian cells involves stepwise lamina disassembly and microtubule-drive deformation of the nuclear membrane. J. Cell Sci. 110, 2129- 2140.
- Beaudouin, J., Gerlich, D., Daigle, N., Eils, R. and Ellenberg. J. (2002). Nuclear envelope breakdown proceeds by microtubule-induced tearing of the lamina. Cell 108, 83-96. [CrossRef]
- Gavet, O. and Pines, J. (2010). Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J. Cell Biol. 189, 247-259. [CrossRef]
- Gavet, O. and Pines, J. (2010). Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533-543. [CrossRef]
- McCloy, R.A., Rogers, S., Caldon, C. E., Lorca, T., Castro, A. and Burgess, A. (2014). Partial inhibition of Cdk1 in G2 phase overrides the SAC and decouples mitotic events. Cell Cycle 13, 1400-1412. [CrossRef]
- Van der Horst, A. and Lens, S. M. A. (2014). Cell division: control of the chromosomal passenger complex in time and space. Chromosoma 123, 25-42.
- Lens, S. M. A., Wolthuis, R. M. F., Klompmaker, R., Kauw, J., Agami, R., Brummelkamp, T., Kops, G. and Medema, R. H. (2003). Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J. 22, 2934-2947. [CrossRef]
- Carvalho, A., Carmena, M., Sambade, C., Earnshaw, W. C. and Wheatley, S. P. (2003). Survivin is required for stable checkpoint activation in taxol-treated HeLa cells. J. Cell Sci. 116, 2987-2998. [CrossRef]
- Beltrami, E., Plescia, J., Wilkinson, J. C., Duckett, C. S. and Altieri, D. C. (2004). Acute ablation of survivin uncovers p53-dependent mitotic checkpoint functions and control of mitochondrial apoptosis. J. Biol. Chem. 279, 2077-2084. [CrossRef]
- Cánovas, P. M. and Guadagno, T. M. (2007). Functional analysis of Survivin in spindle assembly in Xenopus egg extracts. J. Cell. Biochem. 100, 217-229.
- Li, F., Ackermann, E. J., Bennett, C. F., Rothermel, A. L., Plescia, J., Tognin, S., Villa, A., Marchisio, P. C. and Altieri, D. C. (1999). Pleiotropic cell-division defects and apoptosis induced by interference with survivin function. Nat. Cell Biol. 1, 461-466. [CrossRef]
- Giodini, A., Kallio, M. J., Wall, N. R., Gorbsky, G. J., Tognin, S., Marchisio, P. C., Symons, M. and Altieri, D. C. (2002). Regulation of microtubule stability and mitotic progression by survivin. Cancer Res. 62, 2462-2467.
- Cahu, J., Olichon, A., Hentrich, C., Schek, H., Drinjakovic, J., Zhang, C., Doherty-Kirby, A., Lajoie, G. and Surrey, T. (2008). Phosphorylation by Cdk1 increases the binding of Eg5 to microtubules in vitro and in Xenopus egg extract spindles. PLoS One 3, e3936. [CrossRef]
- Smith, E., Hégarat, N., Vesely, C., Roseboom, I., Larch, C., Streicher, H., Straatman, K., Flynn, H., Skehel, M., Hirota, T., Kuriyama, R. and Hochegger, H. (2011). Differential control of Eg5-dependent centrosome separation by Plk1 and Cdk1. EMBO J. 30, 2233-45. [CrossRef]
- O’Connor, D. S., Grossman, D., Plescia, J., Li, F., Zhang, H., Villa, A., Tognin, S., Marchisio, P. C. and Altieri, D. C. (2000). Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc. Natl. Acad. Sci. U. S. A. 97, 13103-13107. [CrossRef]
- Li, F., Ambrosini, G., Chu, E. Y., Plescia, J., Tognin, S., Marchisio, P. C. and Altieri, D. C. (1998). Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396, 580-584. [CrossRef]
- Santos, S. D. M., Wollman, R., Meyer, T. and Ferrell Jr., J. E. (2012). Spatial positive feedback at the onset of mitosis. Cell 149, 1500-1513.
- Pines, J. and Hunter, J. (1991). Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J. Cell Biol. 115, 1-17.
- Tsai, M.-Y., Wang, S., Heidinger, J. M., Shumaker, D. K., Adam, S. A., Goldman, R. D. and Zheng, Y. (2006). A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science 31, 1887-1893. [CrossRef]
- Borgne, A. and Meijer, L. (1996). Sequential dephosphorylation of p34(cdc2) on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem. 271, 27847-27854.
- Brizuela, L, Draetta, G. and Beach, D. (1989). Activation of human CDC2 protein as a histone H1 kinase is associated with complex formation with the p62 subunit. Proc. Natl. Acad. Sci. U. S. A. 86, 4362-4366. [CrossRef]
- Muchmore, S. W., Chen, J., Jakob, C., Zakula, D., Matayoshi, E. D., Wu, W., Zhang, H., Li, F., Ng, S. C. and Altieri, D. C. (2000). Crystal structure and mutagenic analysis of the inhibitor-of-apoptosis protein survivin. Mol. Cell 6, 173-182. [CrossRef]
- Grabarek, J., Amstad, P. and Darzynkiewicz, Z. (2002). Use of fluorescently labeled caspase inhibitors as affinity labels to detect activated caspases. Hum. Cell 15, 1-12. [CrossRef]
- Mazumder, S., Plesca, D. and Almasan, A. (2008). Caspase-3 activation is a critical determinant of genotoxic stress-induced apoptosis. Methods Mol. Biol. 414, 13-21.
- Chantalat, L., Skoufias, D. A., Kleman, J. P., Jung, B., Dideberg, O. and Margolis, R. L. (2000). Crystal structure of human survivin reveals a bow tie-shaped dimer with two unusual alpha-helical extensions. Mol. Cell 6, 183-189. [CrossRef]
- Keifenheim, D., Sun, X.-M., D’Souza, E., Ohira, M. J., Magner, M., Mayhew, M. B., Marguerat, S., and Rhind, N. (2017). Size-Dependent Expression of the Mitotic Activator Cdc25 Suggests a Mechanism of Size Control in Fission Yeast. Curr. Biol. 27, 1491-1497. [CrossRef]
- Fu, J., Hagan, I. M. and Glover, D. M. (2015). The centrosome and its duplication cycle. Cold Spring Harb. Perspect. Biol. 7, a015800. [CrossRef]
- Norbury, C., Blow, J. and Nurse, P. (1991). Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 10, 3321-3329. [CrossRef]
- Heald, R., McLoughlin, M. and McKeon, F. (1993). Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated Cdc2 kinase. Cell 74, 463-474. [CrossRef]
- Liu, F., Stanton, J. J., Wu, Z., Piwnica-Worms, H. (1997). The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol. Cell Biol. 17, 571-583. [CrossRef]
- Gottlin, E. B., Xu, X., Epstein, D. M., Burke, S. P., Eckstein, J. W., Ballou, D. P. and Dixon, J. E. (1996). Kinetic analysis of the catalytic domain of human cdc25B. J. Biol. Chem. 271, 27445-27449. [CrossRef]
- Karlsson, C., Katich, S., Hagting, A., Hoffmann, I. and Pines, J. (1999). Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J. Cell. Biol. 146, 573-584. [CrossRef]
- Varmeh-Ziaie, S. and Manfredi, J. J. (2007). The dual specificity phosphatase Cdc25B, but not the closely related Cdc25C, is capable of inhibiting cellular proliferation in a manner dependent upon its catalytic activity. J. Biol. Chem. 282, 24633-24641. [CrossRef]
- Tommasino, M., Accardi, R., Caldeira, S., Dong, W., Malanchi, I., Smet, A. and Zehbe, I. (2003). The role of TP53 in Cervical carcinogenesis. Hum. Mutat. 21, 307-312. [CrossRef]
- Hyun, S.-Y., Rosen, E. M. and Jang, Y.-J. (2012). Novel DNA damage checkpoint in mitosis: Mitotic DNA damage induces re-replication without cell division in various cancer cells. Biochem. Biophys. Res. Commun. 423, 593-5999. [CrossRef]
- Lindqvist, A., Van Zon, W., Rosenthal, C. K. and Wolthuis, R. M. F. (2007). Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol. 5, e123. [CrossRef]
- Goto, H., Kiyono, T., Tomono, Y., Kawajiri, A., Urano, T., Furukawa, K., Nigg, E. A. and Inagaki, M. (2006). Complex formation of Plk1 and INCENP required for metaphase-anaphase transition. Nat. Cell Biol. 8, 180-187. [CrossRef]
- Zhou, L., Tian, X., Zhu, C., Wang, F. and Higgins, J. M. G. (2014). Polo-like kinase-1 triggers histone phosphorylation by Haspin in mitosis. EMBO Rep. 15, 273–281.
- Wang, F., Dai, J., Daum, J. R., Niedzialkowska, E., Banerjee, B., Stukenberg P. T., Gorbsky, G. J. and Higgins, J. M. (2010). Histone H3 Thr-3 phosphorylation by Haspin positions Aurora B at centromeres in mitosis. Science 303, 231–235. [CrossRef]
- Vigneron, S., Sundermann, L., Labbé, J.-C., Pintard, L., Radulescu, O., Castro, A. and Lorca, T. (2018). Cyclin A-cdk1-Dependent Phosphorylation of Bora Is the Triggering Factor Promoting Mitotic Entry. Dev. Cell 45, 637-650. [CrossRef]
- Dutertre, S., Cazales, M., Quaranta, M., Froment, C., Trabut, V., Dozier, C., Mirey, G., Bouché, J. P., Theis-Febvre, N., Schmitt, E., Monsarrat, B., Prigent, C. and Ducommun, B. (2004). Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J. Cell Sci. 117, 2523-2531. [CrossRef]
- Nishijima, H., Nishitani, H., Seki, T. and Nishimoto, T. (1997). A dual-specificity phosphatase Cdc25B is an unstable protein and triggers p34(cdc2)/cyclin B activation in hamster BHK21 cells arrested with hydroxyurea. J. Cell Biol. 138, 1105-1116.
- Gabrielli, B. G., De Souza, C. P., Tonks, I. D., Clark, J. M., Hayward, N. K. and Ellem, K. A. (1996). Cytoplasmic accumulation of cdc25B phosphatase in mitosis triggers centrosomal microtubule nucleation in HeLa cells. J. Cell Sci. 109, 1081-1093. [CrossRef]
- Lammer, C., Wagerer, S., Saffrich, R., Mertens, D., Ansorge, W. and Hoffmann, I. (1998). The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J. Cell Sci. 111, 2445-2453.
- Baldin, V., Cans, C., Knibiehler, M. and Ducommun, B. (1997). Phosphorylation of human CDC25B phosphatase by CDK1-cyclin A triggers its proteasome-dependent degradation. J. Biol. Chem. 272, 32731-32734. [CrossRef]
- Russell, P. and Nurse, P. (1986). cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell 45, 145-153. [CrossRef]
- Matsusaka, T. and Pines, J. (2004). Chfr acts with the p38 stress kinases to block entry to mitosis in mammalian cells. J. Cell Biol. 166, 507-516. [CrossRef]
- Rieder, C. L. and Cole, R. W. (2000). Microtubule disassembly delays the G2-M transition in vertebrates. Curr. Biol. 10, 1067–1070. [CrossRef]
- Wu, Z., Jiang, Q., Clarke, P. R. and Zhang, C. (2013). Phosphorylation of Crm1 by CDK1-cyclin-B promotes Ran-dependent mitotic spindle assembly. J. Cell Sci. 126, 3417-3428. [CrossRef]
- Guo, Li, Mohd, K. S., Ren, H., Xin, G., Jiang, Q., Clarke, P. R. and Zhang, C. (2019). Phosphorylation of importin-α1 by CDK1-cyclin B1 controls mitotic spindle assembly. J. Cell Sci. 132, 232314. [CrossRef]
- O’Connor, D. S., Wall, N. R., Porter, A. C. G. and Altieri, D. C. A p34(cdc2) survival checkpoint in cancer. Cancer Cell 2, 43-54. [CrossRef]
- Matthess, Y., Raab, M., Sanhaji, M., Lavrik, I. N. and Strebhardt, K. (2010). Cdk1/cyclin B1 controls Fas-mediated apoptosis by regulating caspase-8 activity. Mol. Cell Biol. 30, 5726-5740.
- Allan, L. A. and Clarke, P. R. (2007). Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol. Cell 26, 301-310.
- Lens, S. M. A., Vader, G. and Medema, R. H. (2006). The case for Survivin as mitotic regulator. Curr. Opin. Cell Biol. 18, 616-622.
- Altieri, D. C. (2006). The case for survivin as a regulator of microtubule dynamics and cell-death decisions. Curr. Opin. Cell Biol. 18, 609-615.
- Kannan, N. and Neuwald, A. F. (2005). Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J. Mol. Biol. 351, 956-972.
- Yeung, W., Ruan, Z. and Kannan, N. (2020). Emerging roles of the αC-β4 loop in protein kinase structure, function, evolution, and disease. IUBMB Life 72, 1189-1202.
- Chen, H., Ma, J., Li, W., Eliseenkova, A. V., Xu, C., Neubert, T. A., Miller, W. T. and Mohammadi, M. (2007). A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol. Cell. 27, 717–730. [CrossRef]
- Ruan, Z. and Kannan, N. (2015). Mechanistic Insights into R776H Mediated Activation of Epidermal Growth Factor Receptor Kinase. Biochemistry 54, 4216-4225. [CrossRef]
- Ruan, Z. and Kannan, N. (2018). Altered conformational landscape and dimerization dependency underpins the activation of EGFR by αC-β4 loop insertion mutations. Proc. Natl. Acad. Sci. U. S. A. 115, E8162-E8171.
- Xu, W., Yuan, X., Xiang, Z., Mimnaugh, E., Marcu, M. and Neckers, L. (2005). Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat. Struct. Mol. Biol. 12, 120-126. [CrossRef]
- Citri, A., Harari, D., Shohat, G., Ramakrishnan, P., Gan, J., Lavi, S., Eisenstein, M., Kimchi, A., Wallach, D., Pietrokovski, S. and Yarden, Y. (2006). Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 281, 14361-14369.36. [CrossRef]
- Taipale, M., Krykbaeva, I., Koeva, M., Kayatekin, C., Westover, K. D., Karras, G. I. and Lindquist, S. (2012). Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987-1001. [CrossRef]
- Verba, K. A., Wang, R. Y.-R., Arakawa, A., Liu, Y., Shirouzu, M., Yokoyama, S. and Agard, D. A. (2016). Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547. [CrossRef]
- Plescia, J., Whitney, S., Xia, F., Pennati, M., Zaffaroni, N., Daidone, M. G., Meli, M., Dohi, T., Fortugno, P., Nefedova, Y., Gabrilovich, D. I., Colombo, G. and Altieri, D. C. (2005). Rational design of shepherdin, a novel anticancer agent. Cancer Cell 7, 457-468. [CrossRef]
- Roe, S. M., Ali, M. M. U., Meyer, P., Vaughan, C. K., Panaretou, B., Piper, P. W., Prodromou, C. and Pearl, L. H. (2004). The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116, 87-98. [CrossRef]
- Turnbull, E. L., Martin, I. V. and Fantes, P. A. (2006). Activity of Cdc2 and its interaction with the cyclin Cdc13 depend on the molecular chaperone Cdc37 in Schizosaccharomyces pombe. J. Cell Sci. 119, 292-302. [CrossRef]
- García-Morales, P., Carrasco-García, E., Ruiz-Rico, P., Martínez-Mira, R., Menéndez-Gutiérrez, M. P., Ferragut, J. A., Saceda, M. and Martínez-Lacaci, I. (2007). Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene 26, 7185-7193.
- Altieri, D. C. (2003). Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer 3, 46-54. [CrossRef]
- Abdelwahab, A. B., El-Sawy, E. R., Hanna, A. G., Bagrel, D. and Kirsch, G. (2022). A Comprehensive Overview of the Developments of Cdc25 Phosphatase Inhibitors. Molecules 27, 2389. [CrossRef]
- Faber, E. B., Sun, L., Tang, J., Roberts, E., Ganeshkumar, S., Wang, N., Rasmussen, D., Majumdar A., Hirsch, L. E., John, K., Yang, A., Khalid, H., Hawkinson, J. E., Levinson, N. M., Chennathukuzhi, V., Harki, D. A., Schönbrunn, E. and Georg, G. I. (2023). Development of allosteric and selective CDK2 inhibitors for contraception with negative cooperativity to cyclin binding. Nat. Commun. 14, 3213. [CrossRef]
- Fortugno, P., Beltrami, E., Plescia, J., Fontana, J., Pradhan, D., Marchisio, P. C., Sessa, W. C. and Altieri, D. C. (2003). Regulation of survivin function by Hsp90. Proc. Natl. Acad. Sci. U. S. A. 100, 13791-13796. [CrossRef]
Figure 1.
siRNA-mediated loss of Survivin induces an early prophase blockage in asynchronous HeLa cell cultures. A, Survivin knockdown. Cells transfected with control (VIII) or Survivin (S4) siRNA were analyzed by Western blotting (left) or FACS analysis (right). B, Survivin knockdown cells. Cells transfected with the indicated siRNA were analyzed by Western blotting (left) or fluorescence microscopy using an antibody to α-Tubulin (right). DNA was stained with DAPI. C, Absence of spindle in knockdown cells. Cells transfected with the indicated siRNA were analyzed by fluorescence microscopy (left). Cells were incubated with antibodies against α-Tubulin (top) and Survivin (middle). DNA was stained with DAPI. Right, One cell transfected control (VIII) or Survivin (S4) siRNA at higher magnification. D, Bar graph shows the percentage of mitotic cells in siRNA transfected cultures (n=4).
Figure 1.
siRNA-mediated loss of Survivin induces an early prophase blockage in asynchronous HeLa cell cultures. A, Survivin knockdown. Cells transfected with control (VIII) or Survivin (S4) siRNA were analyzed by Western blotting (left) or FACS analysis (right). B, Survivin knockdown cells. Cells transfected with the indicated siRNA were analyzed by Western blotting (left) or fluorescence microscopy using an antibody to α-Tubulin (right). DNA was stained with DAPI. C, Absence of spindle in knockdown cells. Cells transfected with the indicated siRNA were analyzed by fluorescence microscopy (left). Cells were incubated with antibodies against α-Tubulin (top) and Survivin (middle). DNA was stained with DAPI. Right, One cell transfected control (VIII) or Survivin (S4) siRNA at higher magnification. D, Bar graph shows the percentage of mitotic cells in siRNA transfected cultures (n=4).
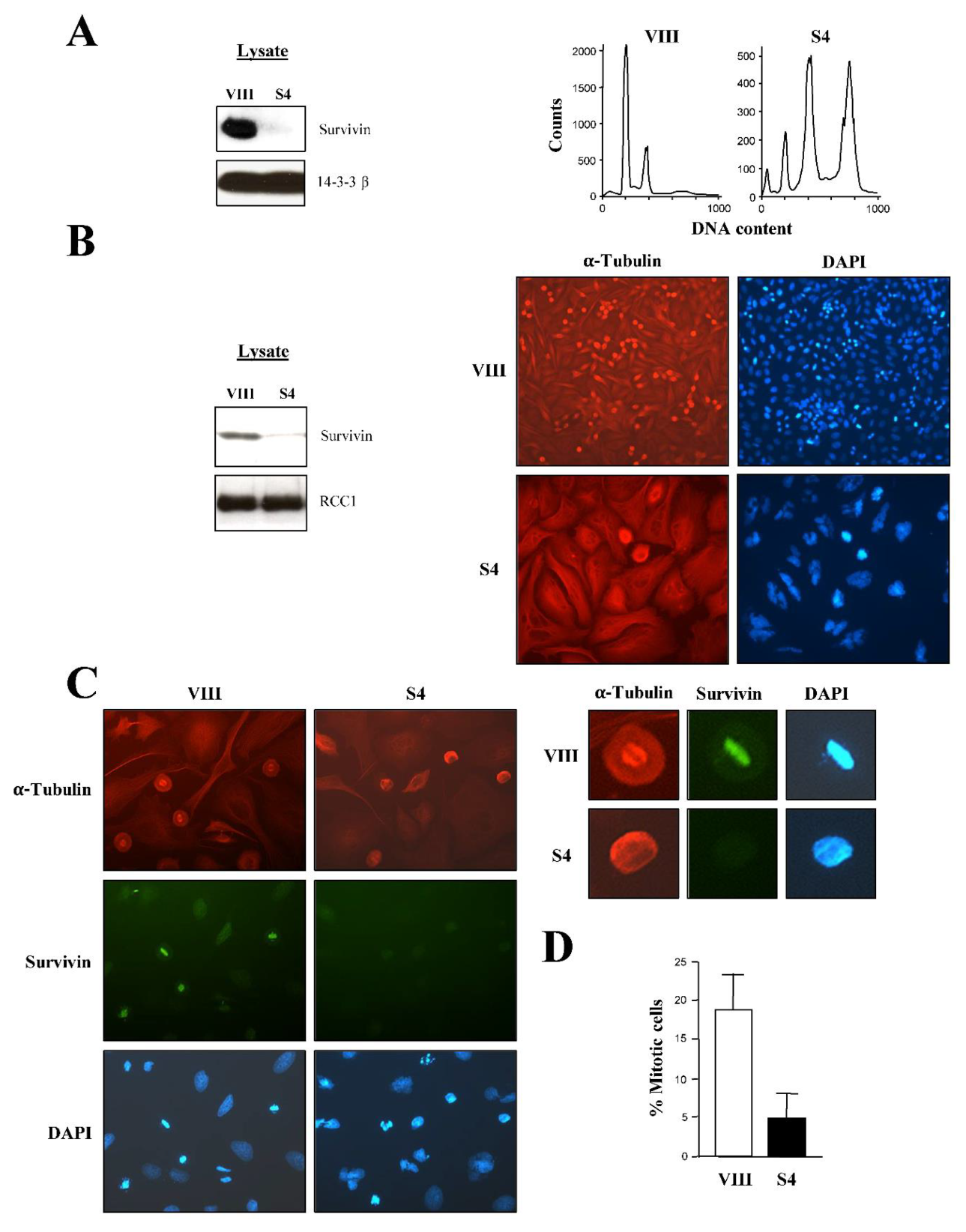
Figure 2.
Nuclear lamina stays assembled in Survivin-depleted HeLa cells. A, Nuclear lamina integrity in siRNA-transfected cells. Cells transfected with the indicated siRNA were analyzed by fluorescence microscopy using an antibody to α-Tubulin (left), and Survivin (top middle) or Lamin B (bottom middle). DNA was stained with DAPI. Several cells are labeled with arrow heads (S: Interphase, P: Prophase, PM: Prometaphase, M: Metaphase, T: Telophase, MN: Multinucleated, IL: Intact lamina and SLS: Spindle-like structure). B, Nuclear lamina in interphase or mitotic cells. Cells were transfected with the indicated siRNA, stained with an antibody to Lamin B and α-Tubulin, and analyzed by fluorescence microscopy. DNA was stained with DAPI. Cells are shown at the same scale. Arrows show intact nuclear lamina, and arrow head points out at Lamin B colocalization with the mitotic spindle. C, Percentage of rounded mitotic cells with intact or disassembled nuclear lamina. Rounded mitotic cells transfected and stained as in A were tallied for the presence or absence of an intact lamina (n=3). D, Purvalanol A treatment of cells. Cells were treated with the Cdk1 inhibitor purvalanol A (20 μM), stained with an antibody to Lamin B and α-Tubulin, and analyzed by fluorescence microscopy. DNA was stained with DAPI.
Figure 2.
Nuclear lamina stays assembled in Survivin-depleted HeLa cells. A, Nuclear lamina integrity in siRNA-transfected cells. Cells transfected with the indicated siRNA were analyzed by fluorescence microscopy using an antibody to α-Tubulin (left), and Survivin (top middle) or Lamin B (bottom middle). DNA was stained with DAPI. Several cells are labeled with arrow heads (S: Interphase, P: Prophase, PM: Prometaphase, M: Metaphase, T: Telophase, MN: Multinucleated, IL: Intact lamina and SLS: Spindle-like structure). B, Nuclear lamina in interphase or mitotic cells. Cells were transfected with the indicated siRNA, stained with an antibody to Lamin B and α-Tubulin, and analyzed by fluorescence microscopy. DNA was stained with DAPI. Cells are shown at the same scale. Arrows show intact nuclear lamina, and arrow head points out at Lamin B colocalization with the mitotic spindle. C, Percentage of rounded mitotic cells with intact or disassembled nuclear lamina. Rounded mitotic cells transfected and stained as in A were tallied for the presence or absence of an intact lamina (n=3). D, Purvalanol A treatment of cells. Cells were treated with the Cdk1 inhibitor purvalanol A (20 μM), stained with an antibody to Lamin B and α-Tubulin, and analyzed by fluorescence microscopy. DNA was stained with DAPI.
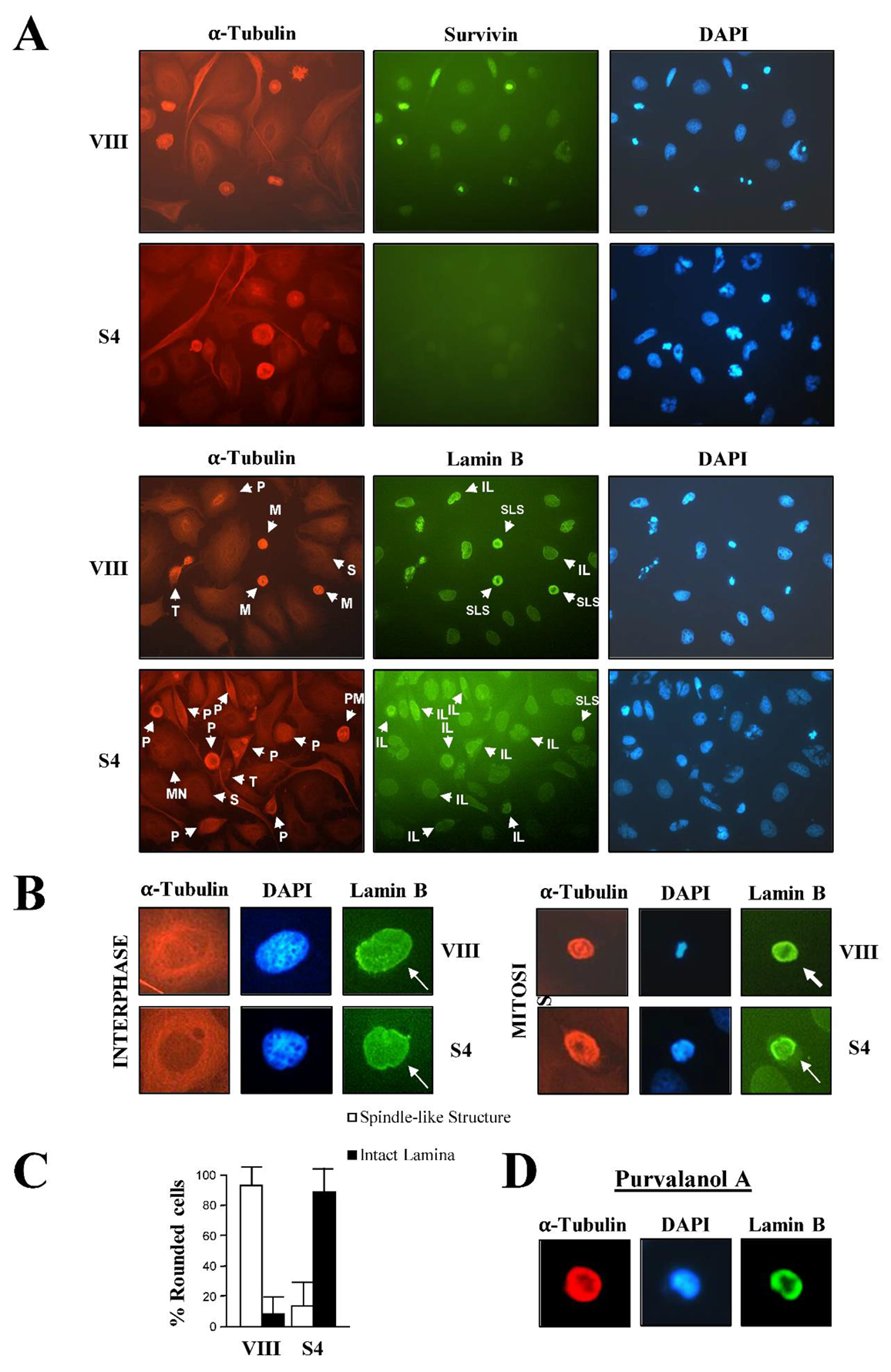
Figure 3.
Cdk1 activity is lower in Survivin-depleted HeLa cells. A, Cdk1 kinase assay. siRNA-transfected asynchronous Cell lysates were used to immunoprecipitate (IP) Cdk1, and the kinase activity was analyzed by a Histone H1 phosphorylation assay (right). Amounts of Cyclin B1, Cdk1 and Survivin were analyzed by Western blotting as a control (left). B, Survivin ablation vs. prometaphase blockage. Cells were treated with 10 μM nocodazole, and their Cdk1 activity was analyzed by an IP and a Histone H1 phosphorylation assay. Survivin siRNA-transfected HeLa cells were used as a control.
Figure 3.
Cdk1 activity is lower in Survivin-depleted HeLa cells. A, Cdk1 kinase assay. siRNA-transfected asynchronous Cell lysates were used to immunoprecipitate (IP) Cdk1, and the kinase activity was analyzed by a Histone H1 phosphorylation assay (right). Amounts of Cyclin B1, Cdk1 and Survivin were analyzed by Western blotting as a control (left). B, Survivin ablation vs. prometaphase blockage. Cells were treated with 10 μM nocodazole, and their Cdk1 activity was analyzed by an IP and a Histone H1 phosphorylation assay. Survivin siRNA-transfected HeLa cells were used as a control.
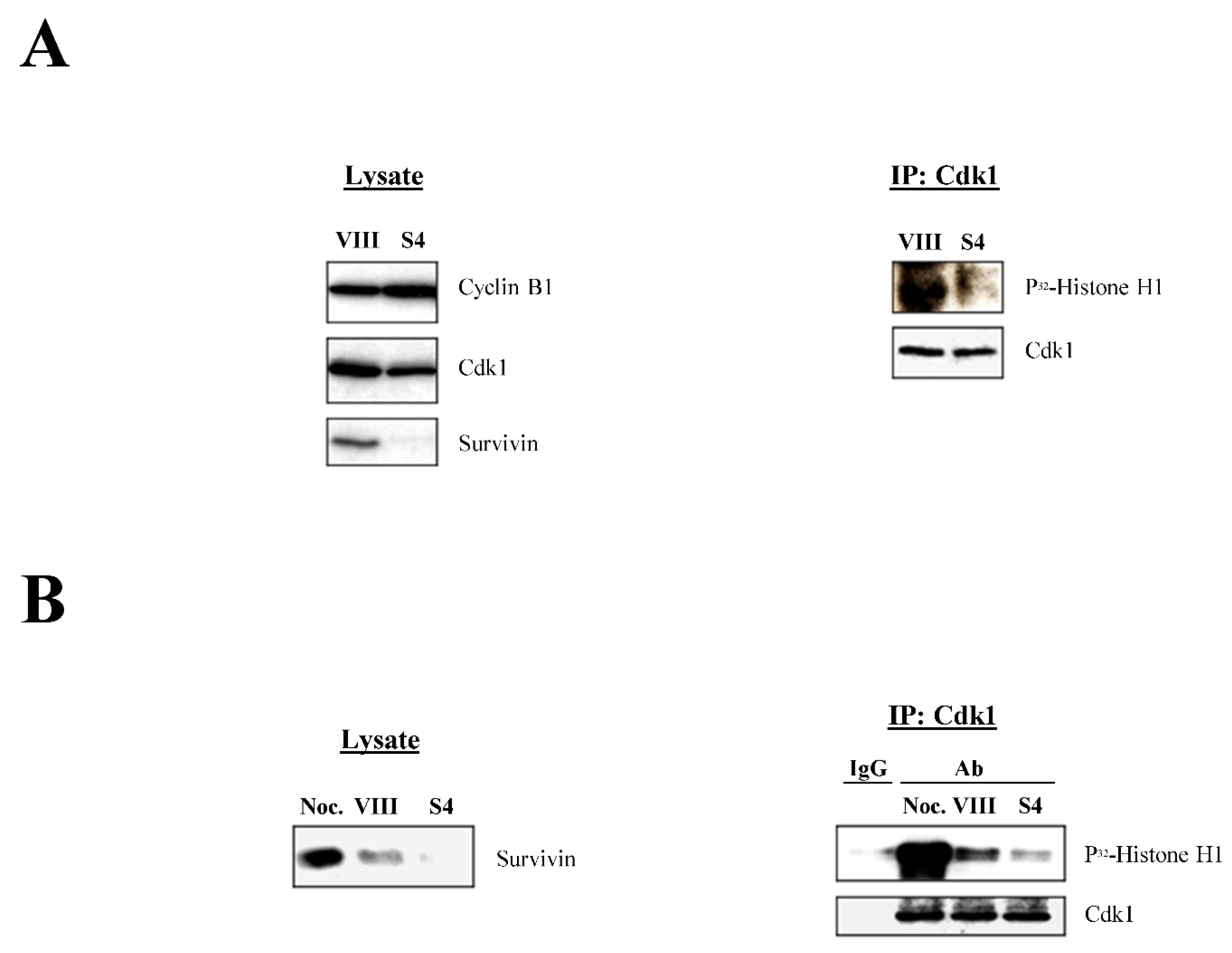
Figure 4.
Survivin abrogation in synchronous HeLa cells causes a sustained G2/M-phase blockage and diminished Cdk1 activity. A, Cell cycle analysis of untreated synchronous cells. Cells were synchronized with 2 mM thymidine for 48 h, released into fresh medium, and harvested at the indicated time intervals. Collected samples were subjected to an IP using an antibody to Survivin, and analyzed by Western blotting (left) or FACS analysis (right). B, Cell cycle analysis of synchronous cells depleted of Survivin. Cells transfected with control (VIII) or Survivin (S4) siRNA were synchronized as in A, released into fresh medium, and harvested at the indicated time intervals. Collected samples were subjected to Western blotting using a Survivin or Cdk1 antibody (top), or FACS analysis (bottom). C, Cdk1 activity during mitotic transition. Synchronized cells, previously transfected with the indicated siRNA, were harvested when they transitioned through mitosis, and analyzed by FACS (left). Lysates were prepared and used to IP Cdk1, and the immune complexes were analyzed in a Histone H1 phosphorylation assay (top right). The amount of Survivin in the lysates is shown as a control (bottom right).
Figure 4.
Survivin abrogation in synchronous HeLa cells causes a sustained G2/M-phase blockage and diminished Cdk1 activity. A, Cell cycle analysis of untreated synchronous cells. Cells were synchronized with 2 mM thymidine for 48 h, released into fresh medium, and harvested at the indicated time intervals. Collected samples were subjected to an IP using an antibody to Survivin, and analyzed by Western blotting (left) or FACS analysis (right). B, Cell cycle analysis of synchronous cells depleted of Survivin. Cells transfected with control (VIII) or Survivin (S4) siRNA were synchronized as in A, released into fresh medium, and harvested at the indicated time intervals. Collected samples were subjected to Western blotting using a Survivin or Cdk1 antibody (top), or FACS analysis (bottom). C, Cdk1 activity during mitotic transition. Synchronized cells, previously transfected with the indicated siRNA, were harvested when they transitioned through mitosis, and analyzed by FACS (left). Lysates were prepared and used to IP Cdk1, and the immune complexes were analyzed in a Histone H1 phosphorylation assay (top right). The amount of Survivin in the lysates is shown as a control (bottom right).
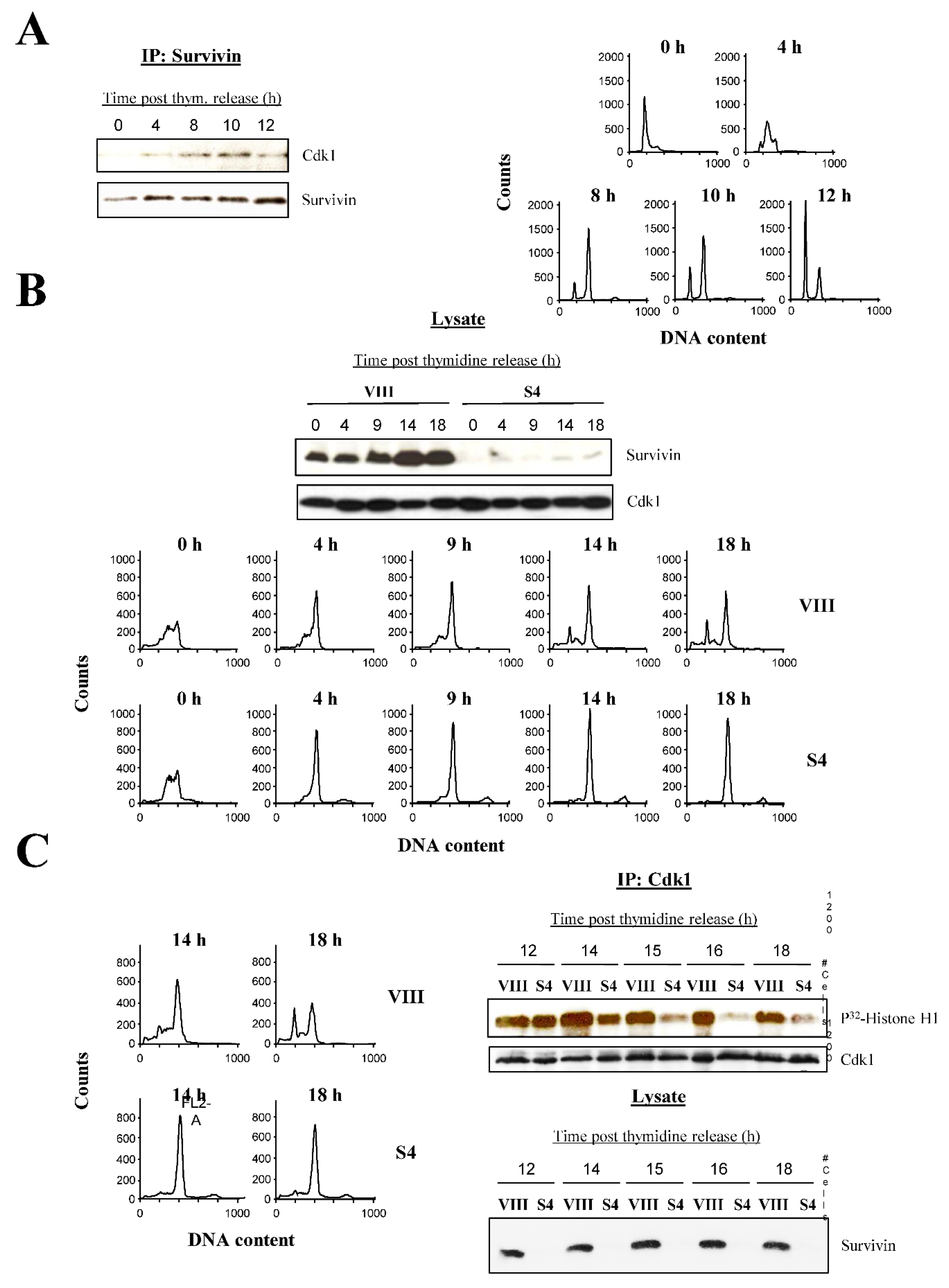
Figure 5.
Treatment of HeLa cells with the SUR A55-D70 peptide causes spindle abnormalities. A, Spindle abnormalities caused by the SUR A55-D70 peptide. Asynchronous cell cultures were transfected with 50 μM scrambled or SUR A55-D70 peptide for 6 h, and cells were stained with an antibody to α-Tubulin. DNA was stained with DAPI. B, Mitotic phenotypes were quantified (n=2).
Figure 5.
Treatment of HeLa cells with the SUR A55-D70 peptide causes spindle abnormalities. A, Spindle abnormalities caused by the SUR A55-D70 peptide. Asynchronous cell cultures were transfected with 50 μM scrambled or SUR A55-D70 peptide for 6 h, and cells were stained with an antibody to α-Tubulin. DNA was stained with DAPI. B, Mitotic phenotypes were quantified (n=2).
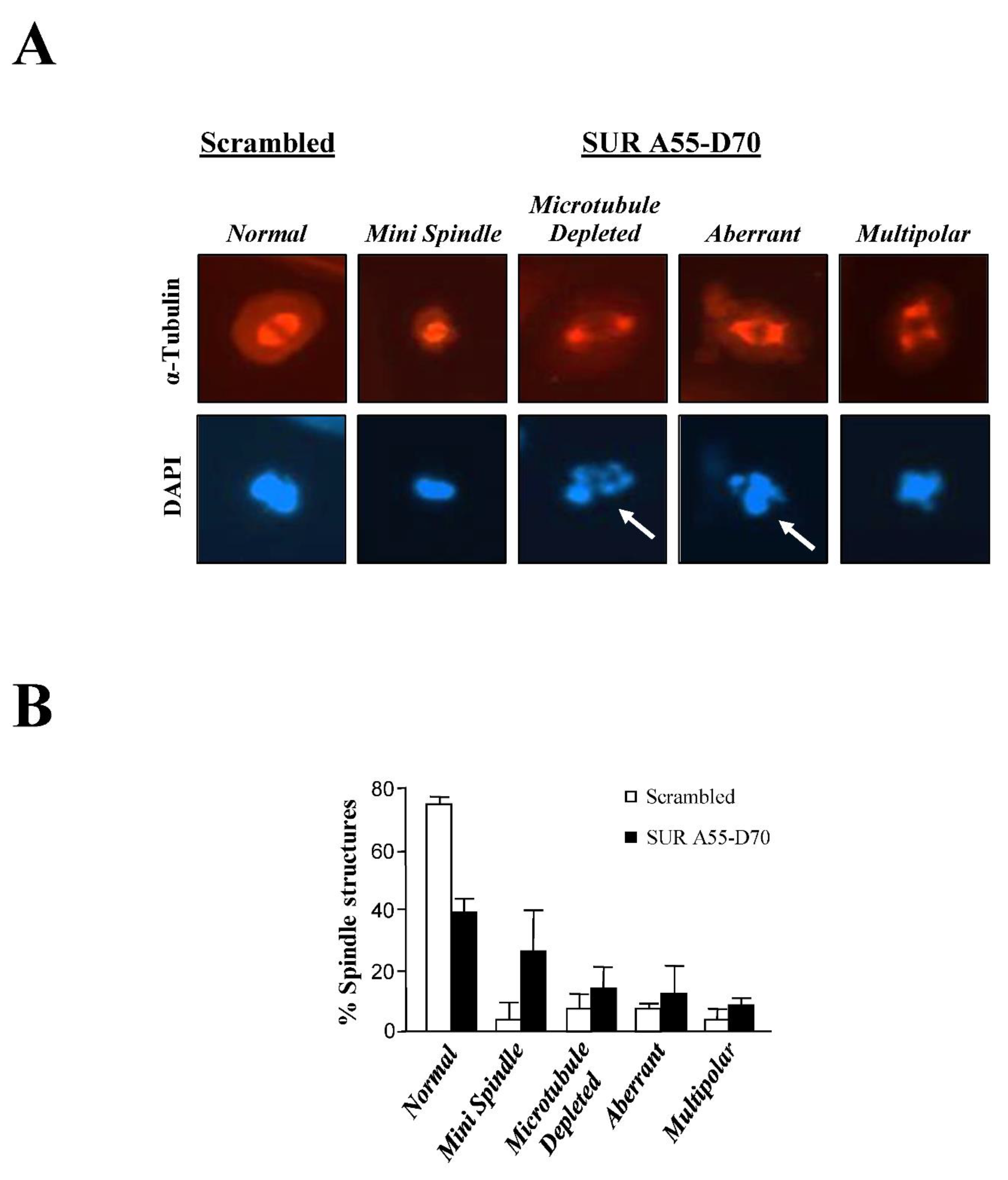
Figure 6.
Prolonged incubation of HeLa cells with the SUR A55-D70 peptide causes apoptosis. A, SUR A55-D70 peptide-induced cell death. Asynchronous cell cultures were treated with 50 μM scrambled or SUR A55-D70 peptides for 24 h, and cells were analyzed by FACS (top), or phase-contrast microscopy (bottom). Images represent one of several experiments (n=4). The percentage of apoptotic (<2N), G1 (2N) and G2/M-phase (4N) cells is indicated at top. Dead cells showing some kind of dark material inside are indicated by arrows. B, SUR A55-D70 peptide dose response of transfected HeLa cells. Asynchronous cell cultures were treated with 200 μM scrambled or SUR A55-D70 peptides for 24 h, and cells were analyzed by FACS (left), phase-contrast microscopy (middle) or Trypan Blue staining (right). Images represent one of several experiments (n=4). The percentage of apoptotic (<2N), G1 (2N) and G2/M-phase (4N) cells is indicated on left. Dead cells showing some kind of dark material inside are indicated by arrows, and dead cells looking like bubbles are indicated by arrow heads. C, SUR A55-D70 peptide-induced apoptosis. Asynchronous cell cultures were transfected with 200 μM scrambled or SUR A55-D70 peptides, and cells were analyzed by phase-contrast microscopy (left) and FAM-DEVD-FMK fluorescence microscopy (right) after 24 h. Images represent one of several experiments (n=3). D, SUR A55-D70 peptide activation of caspase 3. Cells treated as in C for 24-48 h were incubated with an antibody to the caspase 3 proform. E, SUR A55-D70 peptide-induced apoptosis during G2/M-phase. G2/M-phase synchronized cells were transfected with 200 μM scrambled or SUR A55-D70 peptides for 12 h, and analyzed by FACS (top), or phase-contrast microscopy (bottom). The percentage of apoptotic (<2N), G1 (2N) and G2/M-phase (4N) cells is indicated.
Figure 6.
Prolonged incubation of HeLa cells with the SUR A55-D70 peptide causes apoptosis. A, SUR A55-D70 peptide-induced cell death. Asynchronous cell cultures were treated with 50 μM scrambled or SUR A55-D70 peptides for 24 h, and cells were analyzed by FACS (top), or phase-contrast microscopy (bottom). Images represent one of several experiments (n=4). The percentage of apoptotic (<2N), G1 (2N) and G2/M-phase (4N) cells is indicated at top. Dead cells showing some kind of dark material inside are indicated by arrows. B, SUR A55-D70 peptide dose response of transfected HeLa cells. Asynchronous cell cultures were treated with 200 μM scrambled or SUR A55-D70 peptides for 24 h, and cells were analyzed by FACS (left), phase-contrast microscopy (middle) or Trypan Blue staining (right). Images represent one of several experiments (n=4). The percentage of apoptotic (<2N), G1 (2N) and G2/M-phase (4N) cells is indicated on left. Dead cells showing some kind of dark material inside are indicated by arrows, and dead cells looking like bubbles are indicated by arrow heads. C, SUR A55-D70 peptide-induced apoptosis. Asynchronous cell cultures were transfected with 200 μM scrambled or SUR A55-D70 peptides, and cells were analyzed by phase-contrast microscopy (left) and FAM-DEVD-FMK fluorescence microscopy (right) after 24 h. Images represent one of several experiments (n=3). D, SUR A55-D70 peptide activation of caspase 3. Cells treated as in C for 24-48 h were incubated with an antibody to the caspase 3 proform. E, SUR A55-D70 peptide-induced apoptosis during G2/M-phase. G2/M-phase synchronized cells were transfected with 200 μM scrambled or SUR A55-D70 peptides for 12 h, and analyzed by FACS (top), or phase-contrast microscopy (bottom). The percentage of apoptotic (<2N), G1 (2N) and G2/M-phase (4N) cells is indicated.
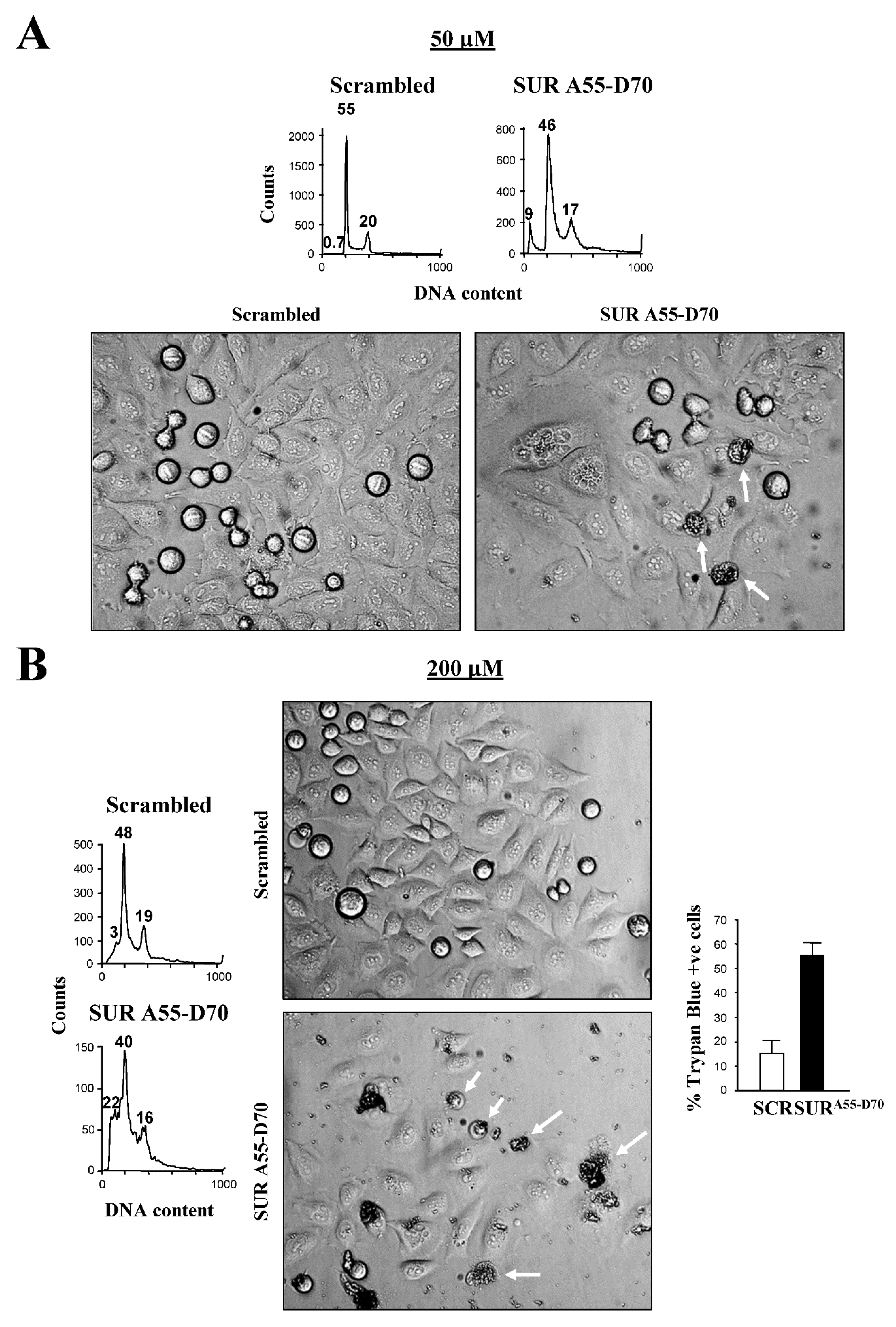
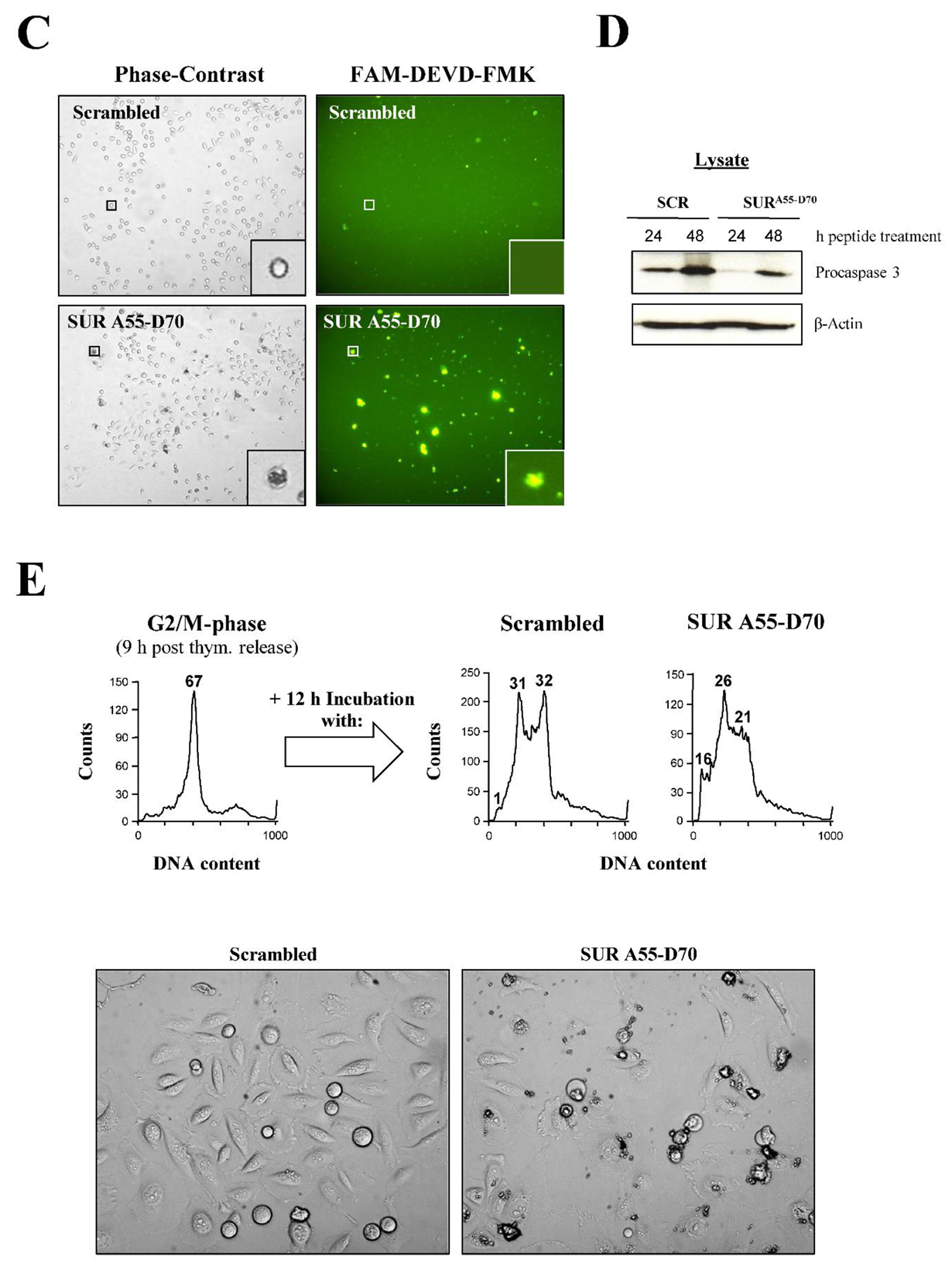
Figure 7.
The Survivin Asp70Ala/Asp71Ala double mutant (SUR D70A/D71A) causes G2/M-phase arrest, mitotic abnormalities and cell death in HeLa cells. A, SUR D70A/D71A causes mitotic abnormalities. Asynchronous cell cultures were transfected with constructs expressing HA-tagged wild type Survivin (HA-SUR WT) or SUR D70A/D71A (HA-SUR D70A/D71A), and subjected to Western blotting using antibodies to HA-Tag, Survivin or β-Actin (left), or analyzed by fluorescence microscopy using antibodies to HA-Tag or α-Tubulin (right). DNA was stained with DAPI. Cells are labeled according to their mitotic phase or chromatin fragmentation. B, Phenotypes observed in SUR D70A/D71A-expressing cells. Graph shows the averages of two independent experiments. C, Cdk1 activity in SUR D70A/D71A-expressing cells. HA-SUR WT- or HA-SUR D70A/D71A-transfected cell lysates were used to IP Cdk1, and pellets were analyzed in a Histone H1 phosphorylation assay (up), or by Western blotting (bottom). D, Detection of phosphorylated inactive Cdk1 form in cells expressing SUR D70A/D71A. Parallel samples as in C were used to determine the level of inactive Cdk1 by Western blotting. E, F, Time-lapse video microscopy of SUR D70A/D71A-expressing cells. Synchronous cell cultures were transfected with GFP-SUR WT or GFP-SUR D70A/D71A, released into fresh medium, and imaged every 10 min continuously for 24 h. Images were taken at the indicated time points. E, Individual cells (bars), transfected with GFP-SUR WT (top) or GFP-SUR D70/D71A mutant (bottom) were quantified for time spent at each indicated mitotic transition and assigned specific phenotypes (see legend). Bottom F, Selected cells undergoing sustained mitotic arrest (magenta and light blue arrows), or cell death after mitosis re-entry (yellow arrow) are shown. Top F, White arrow shows a control-transfected cell that progressed normally through mitosis.
Figure 7.
The Survivin Asp70Ala/Asp71Ala double mutant (SUR D70A/D71A) causes G2/M-phase arrest, mitotic abnormalities and cell death in HeLa cells. A, SUR D70A/D71A causes mitotic abnormalities. Asynchronous cell cultures were transfected with constructs expressing HA-tagged wild type Survivin (HA-SUR WT) or SUR D70A/D71A (HA-SUR D70A/D71A), and subjected to Western blotting using antibodies to HA-Tag, Survivin or β-Actin (left), or analyzed by fluorescence microscopy using antibodies to HA-Tag or α-Tubulin (right). DNA was stained with DAPI. Cells are labeled according to their mitotic phase or chromatin fragmentation. B, Phenotypes observed in SUR D70A/D71A-expressing cells. Graph shows the averages of two independent experiments. C, Cdk1 activity in SUR D70A/D71A-expressing cells. HA-SUR WT- or HA-SUR D70A/D71A-transfected cell lysates were used to IP Cdk1, and pellets were analyzed in a Histone H1 phosphorylation assay (up), or by Western blotting (bottom). D, Detection of phosphorylated inactive Cdk1 form in cells expressing SUR D70A/D71A. Parallel samples as in C were used to determine the level of inactive Cdk1 by Western blotting. E, F, Time-lapse video microscopy of SUR D70A/D71A-expressing cells. Synchronous cell cultures were transfected with GFP-SUR WT or GFP-SUR D70A/D71A, released into fresh medium, and imaged every 10 min continuously for 24 h. Images were taken at the indicated time points. E, Individual cells (bars), transfected with GFP-SUR WT (top) or GFP-SUR D70/D71A mutant (bottom) were quantified for time spent at each indicated mitotic transition and assigned specific phenotypes (see legend). Bottom F, Selected cells undergoing sustained mitotic arrest (magenta and light blue arrows), or cell death after mitosis re-entry (yellow arrow) are shown. Top F, White arrow shows a control-transfected cell that progressed normally through mitosis.
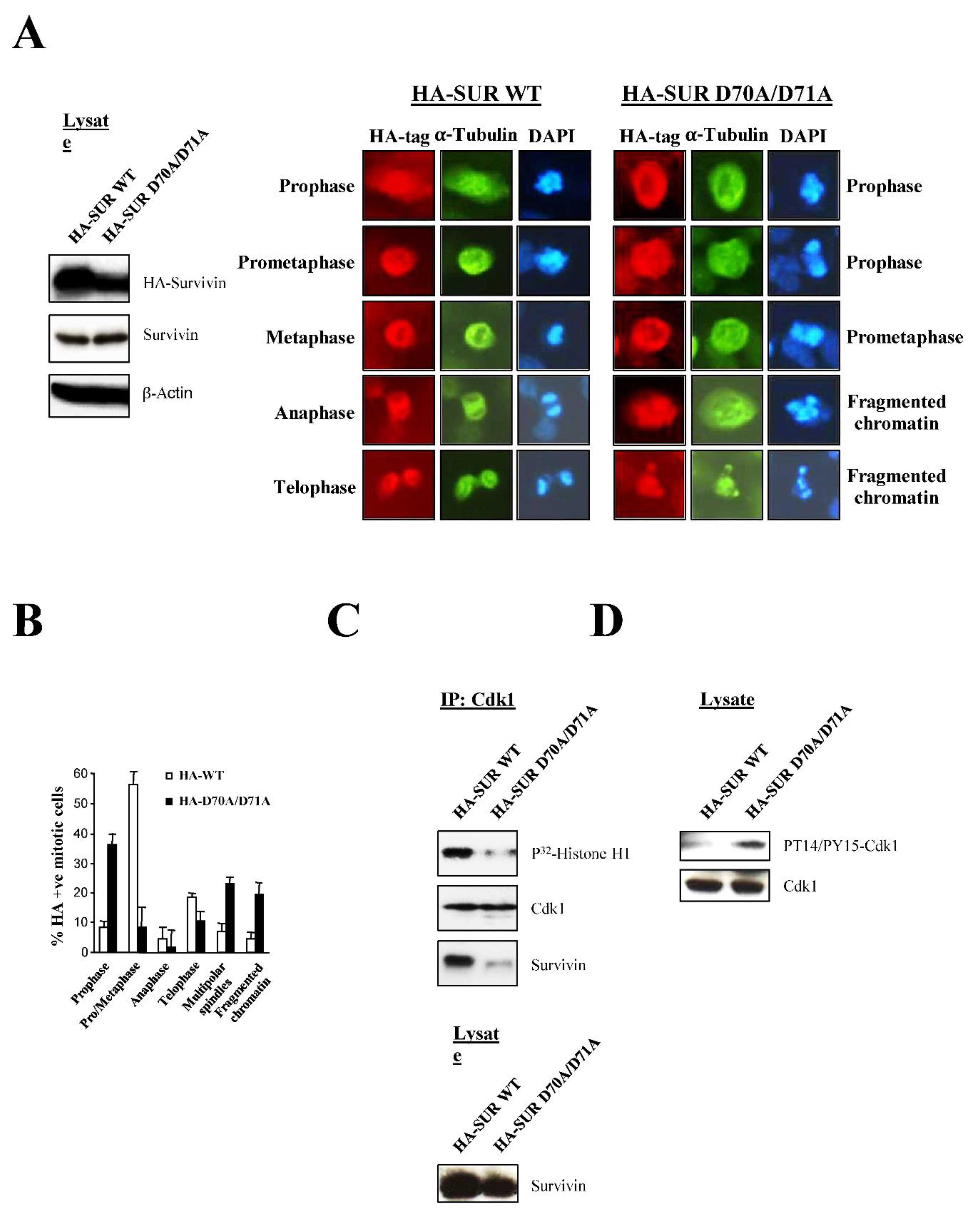
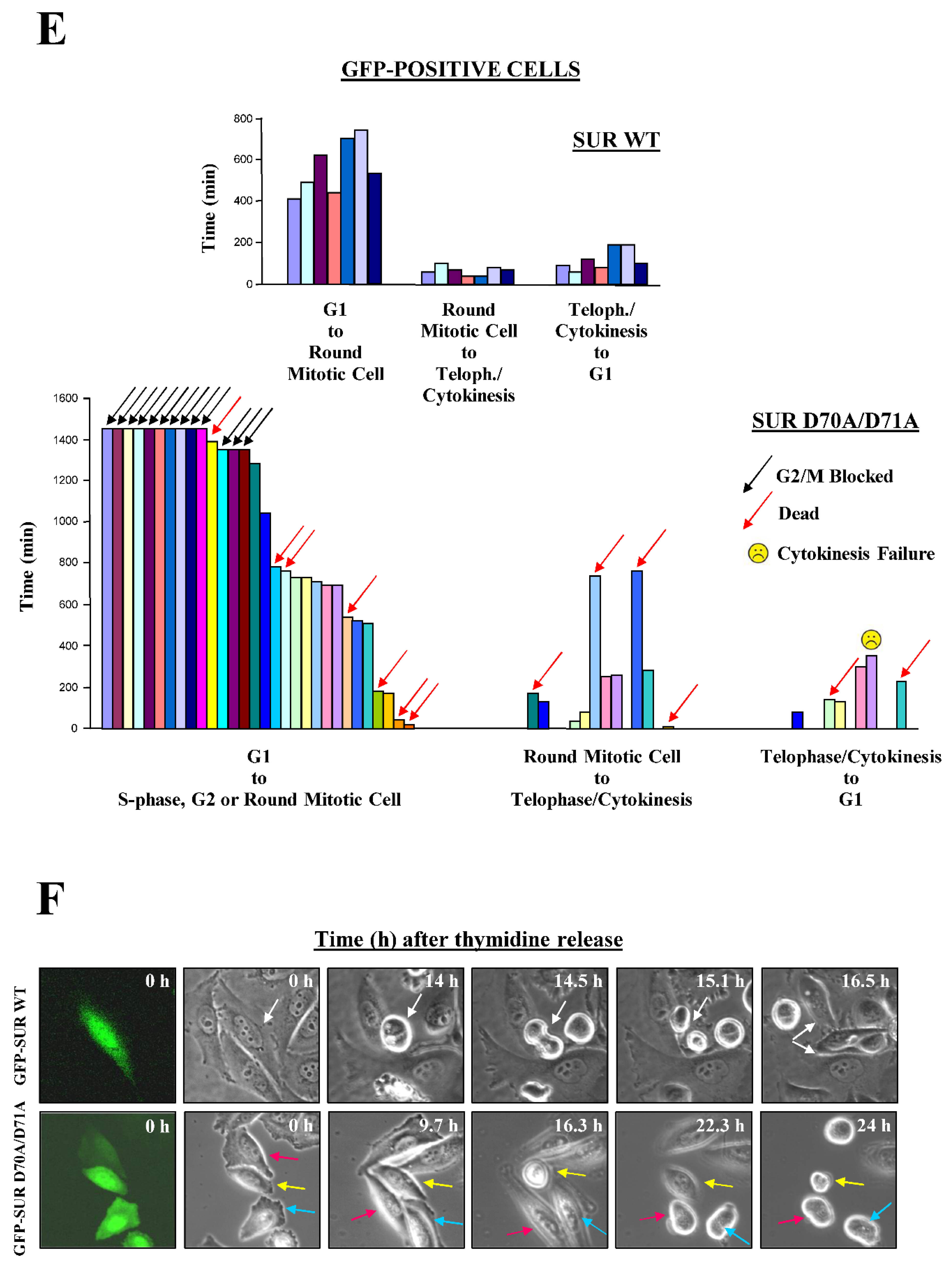
Figure 8.
Survivin is needed to recruit Cdk1 to Hela cell centrosomes. A, Fast-migrating centrosomal Survivin-Cdk1 complex at mitosis. Centrosomes were isolated from lysates of untreated synchronous HeLa cell cultures, subjected to Survivin IP, and analyzed by Western blotting using antibodies to Cdk1 or Survivin (left). B, Centrosomal Cdk1 levels in cells depleted of Survivin. Centrosomes were isolated from asynchronous HeLa cells transfected with control (VIII) or Survivin (S4) siRNA, and analyzed by Western blotting using several antibodies (see original data in Figure S7A). C, Centrosomal Cdk1 levels and activity in siRNA-treated synchronous HeLa cultures. Centrosomal preparations from synchronized HeLa cells transfected with the indicated siRNA that were collected after release into fresh media at the mentioned times, were analyzed by FACS (bottom) or IP with an antibody to Cdk1, and pellets were analyzed by Western blotting or a Histone H1 phosphorylation assay (*: 16 h time point was omitted in both treatments due to a loading error (see original data in Figure S7B). Right panel shows the levels of Cyclin B1, Cdk1 and Survivin in the lysate as a control. D, Centrosomal Cdk1 signal in Survivin-depleted cells. siRNA-treated cells were analyzed by fluorescence microscopy using an antibody to Cdk1 (top). DNA was stained with DAPI. Control cells (VIII) were either in early prophase (a, b), prometaphase (c, d) or metaphase (e, f), and Survivin-depleted cells (S4) were in S-phase/G2 (g, h) or early prophase (i-l). Percentage of splitted Cdk1 signal was scored (bottom) (n=3).
Figure 8.
Survivin is needed to recruit Cdk1 to Hela cell centrosomes. A, Fast-migrating centrosomal Survivin-Cdk1 complex at mitosis. Centrosomes were isolated from lysates of untreated synchronous HeLa cell cultures, subjected to Survivin IP, and analyzed by Western blotting using antibodies to Cdk1 or Survivin (left). B, Centrosomal Cdk1 levels in cells depleted of Survivin. Centrosomes were isolated from asynchronous HeLa cells transfected with control (VIII) or Survivin (S4) siRNA, and analyzed by Western blotting using several antibodies (see original data in Figure S7A). C, Centrosomal Cdk1 levels and activity in siRNA-treated synchronous HeLa cultures. Centrosomal preparations from synchronized HeLa cells transfected with the indicated siRNA that were collected after release into fresh media at the mentioned times, were analyzed by FACS (bottom) or IP with an antibody to Cdk1, and pellets were analyzed by Western blotting or a Histone H1 phosphorylation assay (*: 16 h time point was omitted in both treatments due to a loading error (see original data in Figure S7B). Right panel shows the levels of Cyclin B1, Cdk1 and Survivin in the lysate as a control. D, Centrosomal Cdk1 signal in Survivin-depleted cells. siRNA-treated cells were analyzed by fluorescence microscopy using an antibody to Cdk1 (top). DNA was stained with DAPI. Control cells (VIII) were either in early prophase (a, b), prometaphase (c, d) or metaphase (e, f), and Survivin-depleted cells (S4) were in S-phase/G2 (g, h) or early prophase (i-l). Percentage of splitted Cdk1 signal was scored (bottom) (n=3).
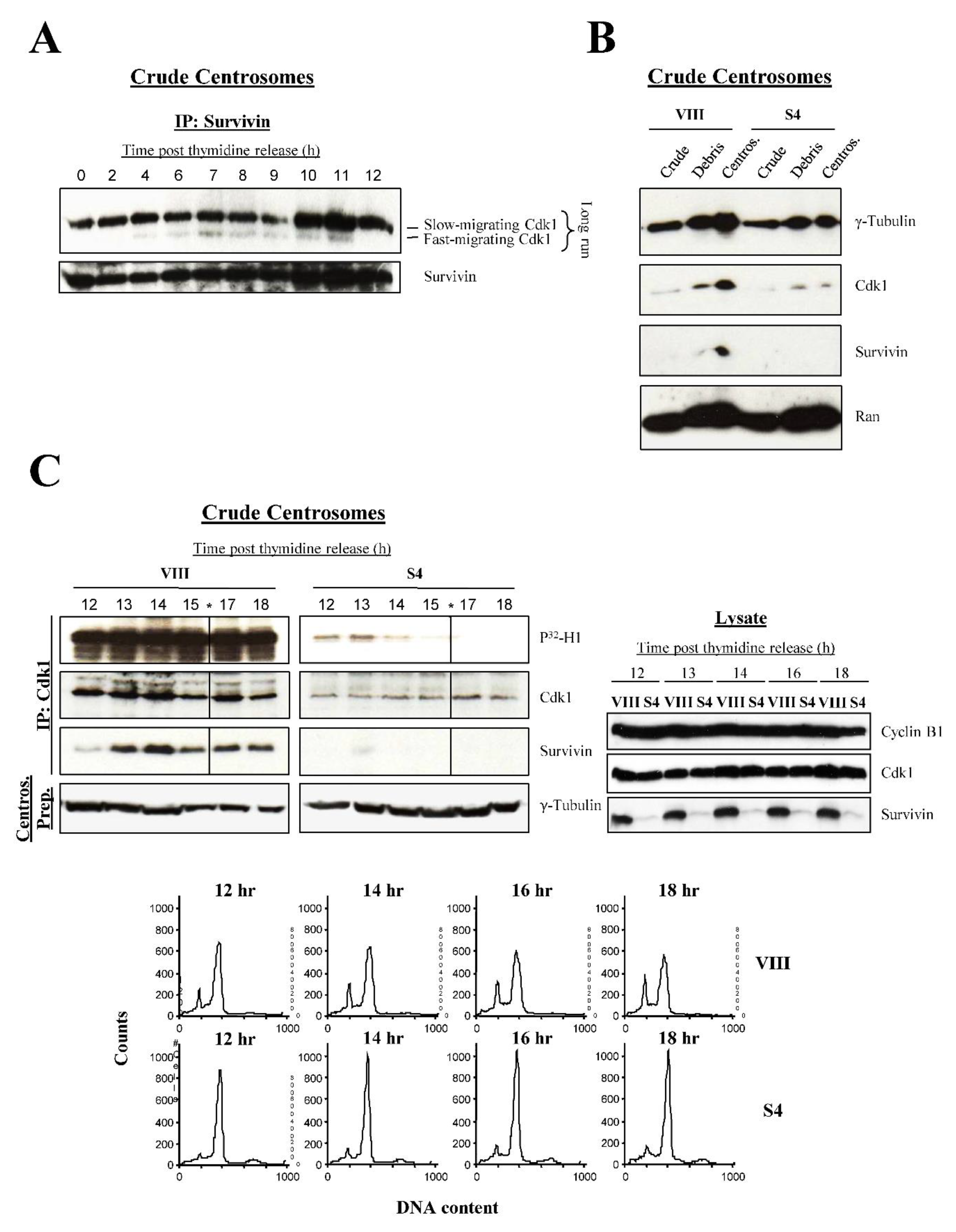
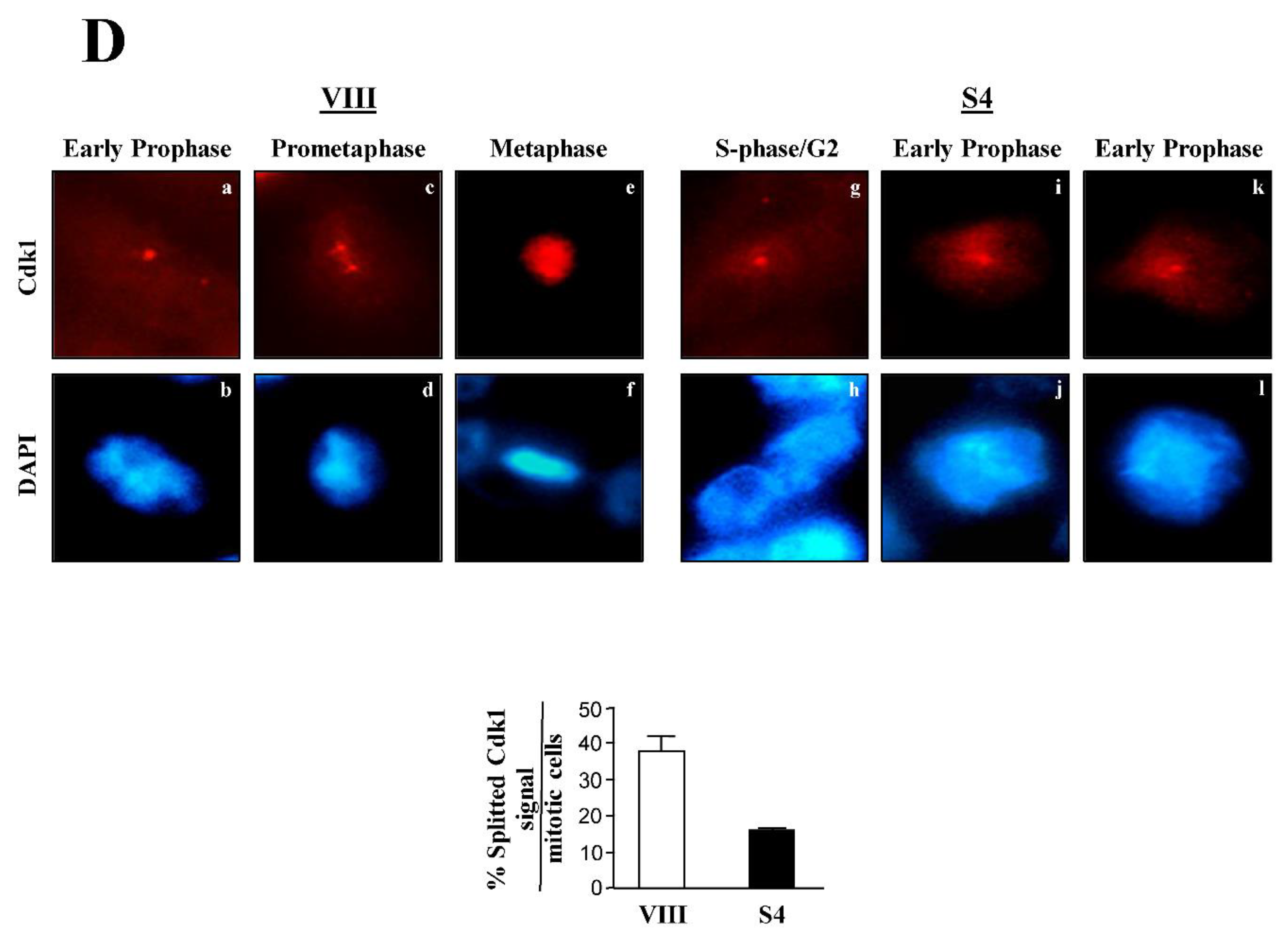
Figure 9.
Cdc25 activity is induced by recombinant Survivin in vitro. A, Phosphorylated inactive Cdk1 isoform in Survivin-depleted Hela cells. Shaken-off cells transfected with control (VIII) or Survivin (S4) siRNA were analyzed by Western blotting using several antibodies (left) or a Histone H1 phosphorylation assay (right). B, Impaired Cdc25 activity in Survivin-knocked down mitotic HeLa cells. Synchronized mitotic cells (14 h) transfected with the indicated siRNA were collected, and analyzed by Western blotting. C, In vitro induction of Cdc25 activity by recombinant Survivin. G2/M-phase HeLa cell lysates were immunodepleted of Survivin (left), supplemented with an ATP-regenerating system, incubated with GST or GST-Survivin, used to IP Cdk1, and analyzed by Western blotting (right up). Binding of the recombinant GST-Survivin protein (*) to the immunoprecipitated Cdk1 was checked by staining the IP membrane with Coomassie Blue. U indicates a nonspecific protein band. D, E, Cdk1 activation by recombinant Survivin in interphase. D, Interphase HeLa cell lysates (top left), depleted of Survivin (bottom left), and supplemented with an ATP-regenerating system, were incubated with different concentrations of GST or GST-Survivin, and analyzed by Western blotting (top right), or a Histone H1 phosphorylation assay (E).
Figure 9.
Cdc25 activity is induced by recombinant Survivin in vitro. A, Phosphorylated inactive Cdk1 isoform in Survivin-depleted Hela cells. Shaken-off cells transfected with control (VIII) or Survivin (S4) siRNA were analyzed by Western blotting using several antibodies (left) or a Histone H1 phosphorylation assay (right). B, Impaired Cdc25 activity in Survivin-knocked down mitotic HeLa cells. Synchronized mitotic cells (14 h) transfected with the indicated siRNA were collected, and analyzed by Western blotting. C, In vitro induction of Cdc25 activity by recombinant Survivin. G2/M-phase HeLa cell lysates were immunodepleted of Survivin (left), supplemented with an ATP-regenerating system, incubated with GST or GST-Survivin, used to IP Cdk1, and analyzed by Western blotting (right up). Binding of the recombinant GST-Survivin protein (*) to the immunoprecipitated Cdk1 was checked by staining the IP membrane with Coomassie Blue. U indicates a nonspecific protein band. D, E, Cdk1 activation by recombinant Survivin in interphase. D, Interphase HeLa cell lysates (top left), depleted of Survivin (bottom left), and supplemented with an ATP-regenerating system, were incubated with different concentrations of GST or GST-Survivin, and analyzed by Western blotting (top right), or a Histone H1 phosphorylation assay (E).
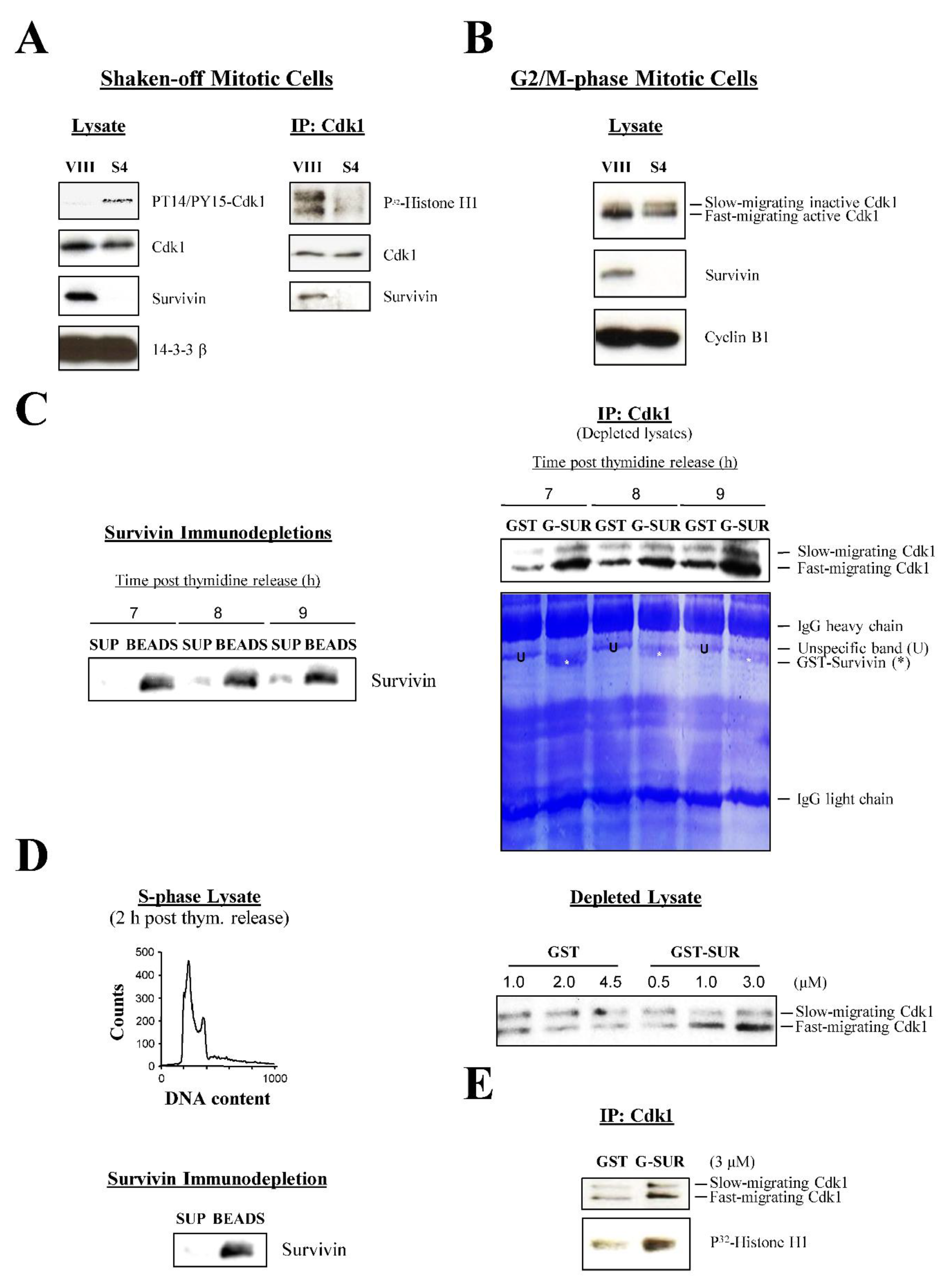
Figure 10.
Survivin regulation of Cdc25B phosphatase activity in HeLa cells. A, Survivin binds directly to Cdc25B. GST or GST-Survivin were mixed with His-Cdc25B, and analyzed by Western blotting. B, Accumulation of a cytosolic Cdc25B-Cdk1-Cyclin B1 complex in the absence of Survivin. Asynchronized cells transfected with control (VIII) or Survivin (S4) siRNA were collected, lysates were used to IP Cyclin B1, and pellets were analyzed by Western blotting. C, Cdc25B accumulation in Survivin-depleted cell cultures. Asynchronous (top) or synchronous (bottom) cells treated with the indicated siRNA were analyzed by Western blotting. D, Cdc25B activity at mitosis onset in siRNA-treated cells. Synchronized HeLa cells transfected with the indicated siRNA were collected at G2/M-phase (top left), analyzed for Survivin expression (bottom left), and used to IP Cdc25B (bottom right). Cdc25B phosphatase activity in the immune complexes was measured by the rate of OMFP hydrolysis (top right). E, Quantification of several experiments as in D (n=4) is shown. F, Cdc25B activity during the cell cycle in siRNA-treated cells. Synchronized cells transfected with the indicated siRNA were collected at the indicated times following thymidine release, analyzed by FACS (bottom), and used to IP Cyclin B1. Immune complexes were analyzed by Western blotting (top left). Cdc25B was IP from the same samples and its activity measured by the rate of OMFP hydrolysis (right).
Figure 10.
Survivin regulation of Cdc25B phosphatase activity in HeLa cells. A, Survivin binds directly to Cdc25B. GST or GST-Survivin were mixed with His-Cdc25B, and analyzed by Western blotting. B, Accumulation of a cytosolic Cdc25B-Cdk1-Cyclin B1 complex in the absence of Survivin. Asynchronized cells transfected with control (VIII) or Survivin (S4) siRNA were collected, lysates were used to IP Cyclin B1, and pellets were analyzed by Western blotting. C, Cdc25B accumulation in Survivin-depleted cell cultures. Asynchronous (top) or synchronous (bottom) cells treated with the indicated siRNA were analyzed by Western blotting. D, Cdc25B activity at mitosis onset in siRNA-treated cells. Synchronized HeLa cells transfected with the indicated siRNA were collected at G2/M-phase (top left), analyzed for Survivin expression (bottom left), and used to IP Cdc25B (bottom right). Cdc25B phosphatase activity in the immune complexes was measured by the rate of OMFP hydrolysis (top right). E, Quantification of several experiments as in D (n=4) is shown. F, Cdc25B activity during the cell cycle in siRNA-treated cells. Synchronized cells transfected with the indicated siRNA were collected at the indicated times following thymidine release, analyzed by FACS (bottom), and used to IP Cyclin B1. Immune complexes were analyzed by Western blotting (top left). Cdc25B was IP from the same samples and its activity measured by the rate of OMFP hydrolysis (right).
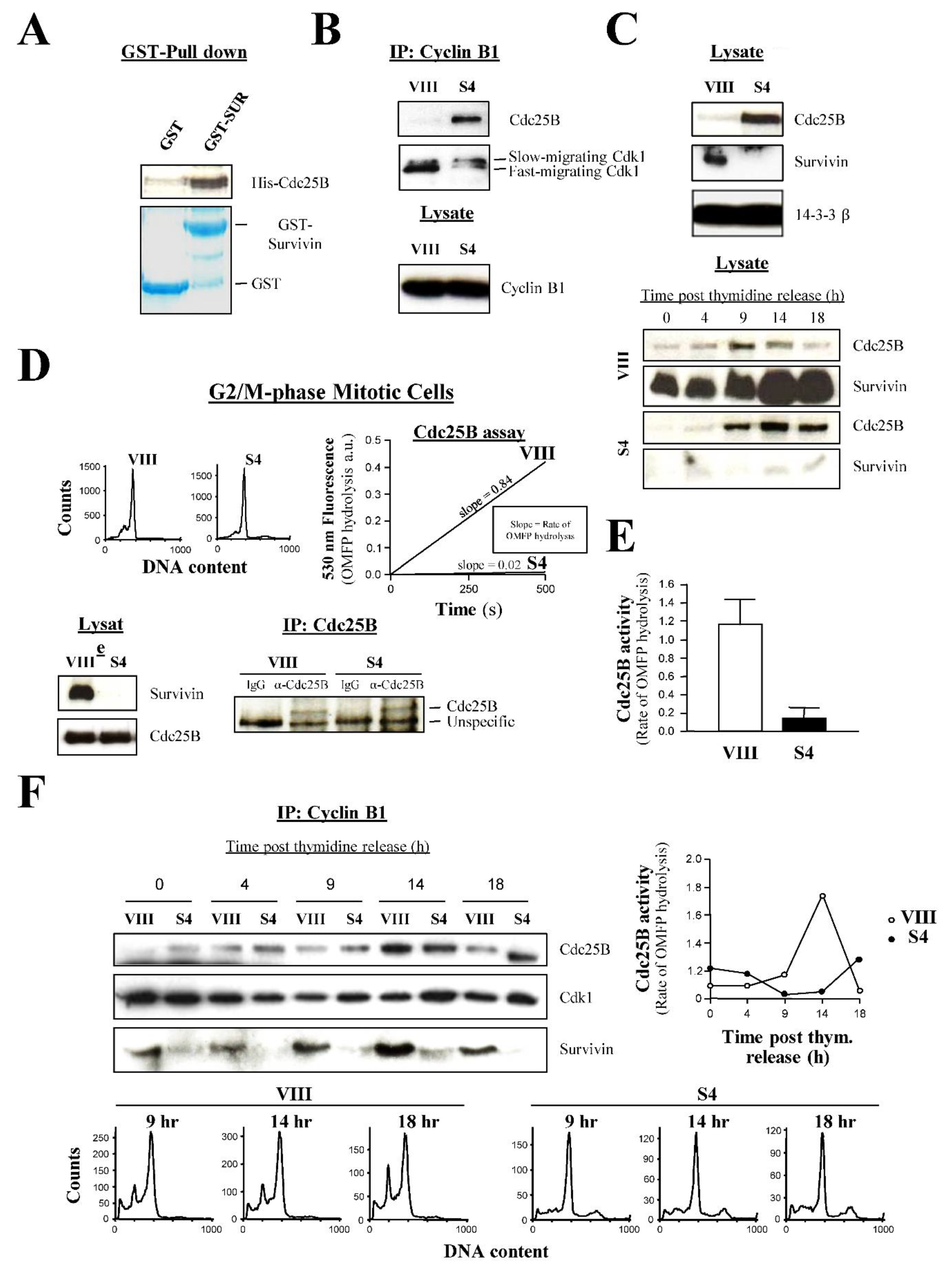
Figure 11.
A gain-of-function Cdc25B mutant can override the blockage induced by Survivin abrogation in HeLa cells. A, Cdc25B-mediated mitotic entry. Cells transfected with control (VIII) or Survivin (S4) siRNA were synchronized for 40 h. Cells were then transfected with a pcDNA3 or pCdc25B plasmid, and placed back in thymidine for another 8 h. Upon release, cells were collected at 16 h, as they transited through G2/M-phase. Cells were analyzed by Western blotting (left) and FACS analysis (right). The percentage of cells in apoptosis (<2N), G1 (2N), G2/M-phase (4N) and polyploid (>4N) is indicated. Arrows and percentages indicate changes in cell populations. B, Gain-of-function ∆NCdc25B mutant overrides blockage induced by Survivin abrogation. siRNA-treated synchronized cells were transfected with a pGFP (control), pCdc25B (wild type Cdc25B) or p∆NCdc25B (constitutively active Cdc25B mutant) plasmid (the last 2 were also transfected with the GFP monitoring plasmid), released into fresh medium, and analyzed by Western blotting (left), or fluorescence and phase-contrast microscopy (right) after 12 h (cells entering mitosis). Averages of 3 independent experiments are shown in bottom left.
Figure 11.
A gain-of-function Cdc25B mutant can override the blockage induced by Survivin abrogation in HeLa cells. A, Cdc25B-mediated mitotic entry. Cells transfected with control (VIII) or Survivin (S4) siRNA were synchronized for 40 h. Cells were then transfected with a pcDNA3 or pCdc25B plasmid, and placed back in thymidine for another 8 h. Upon release, cells were collected at 16 h, as they transited through G2/M-phase. Cells were analyzed by Western blotting (left) and FACS analysis (right). The percentage of cells in apoptosis (<2N), G1 (2N), G2/M-phase (4N) and polyploid (>4N) is indicated. Arrows and percentages indicate changes in cell populations. B, Gain-of-function ∆NCdc25B mutant overrides blockage induced by Survivin abrogation. siRNA-treated synchronized cells were transfected with a pGFP (control), pCdc25B (wild type Cdc25B) or p∆NCdc25B (constitutively active Cdc25B mutant) plasmid (the last 2 were also transfected with the GFP monitoring plasmid), released into fresh medium, and analyzed by Western blotting (left), or fluorescence and phase-contrast microscopy (right) after 12 h (cells entering mitosis). Averages of 3 independent experiments are shown in bottom left.
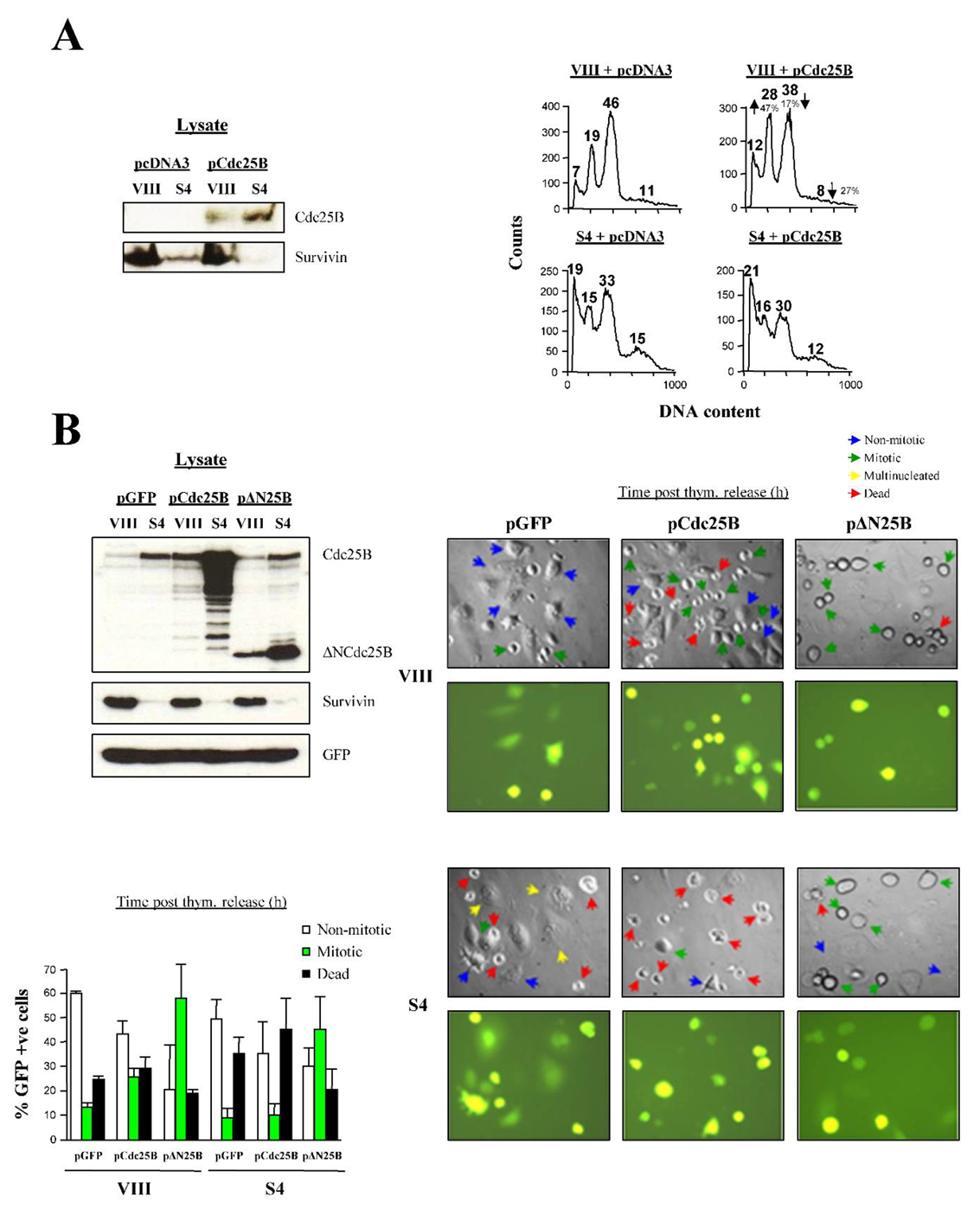
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



