
Submitted:
03 February 2025
Posted:
04 February 2025
You are already at the latest version
Abstract
There are five different types of the hepatotropic hepatitis viruses (HAV, HBV, HCV, HDV, and HEV). Infection with all hepatitis viruses leads to the development of disease, and all of them are capable of co-infection and super-infection; i.e., the presence of more than one type of hepatitis virus in an infected individual. Typically, co-infections cause more severe illness. Although many facets of virus-host interactions are known, important aspects related to the prevalence and functionality of intrinsic disorder in viral interactomes remain mostly unexplored. Even less is known about prevalence and roles of intrinsic disorder in proteins related to the hepatotropic co-infections. The goal of this study is to fill this gap by conducting the bioinformatics analysis of intrinsic disorder in host proteins interacting with hepatotropic viruses, with special focus on host proteins that can interact with more than one type of hepatitis viruses. To this end, a set of computational tools was used to evaluate disorder status of proteins, their predisposition for liquid-liquid phase separation (LLPS), and interactivity. This analysis revealed that some viral proteins were predicted to have high LLPS potential. Host proteins interacting with hepatotropic viruses were characterized by noticeable variation in their intrinsic disorder status and LLPS potential. Although global disorder distribution within the sets of host proteins interacting with hepatitis viruses was not too different from that of the entire human proteome, more host proteins interacting with hepatitis viruses were predicted as moderately disordered in comparison with the entire human proteome. Intrinsic disorder was shown to be commonly present in host proteins shared by several hepatotropic viruses, where it is used for various functional properties.
Keywords:
HAV
; HBV
; HCV
; HDV
; HEV
; viral co-infection
; intrinsically disordered proteins
; liquid-liquid phase separation
; protein-protein interactions
; PPI network
1. Introduction
Among various diseases affecting liver, a very significant place is hold by viral hepatitis caused by a viral infection leading to liver inflammation and damage, which, being untreated is often associated with serious health issues, such as liver scarring or cancer. Although there are five different types of the hepatotropic hepatitis viruses, A, B, C, D, and E (HAV, HBV, HCV, HDV, and HEV) the most common types of viral hepatitis are hepatitis A, hepatitis B, and hepatitis C. In fact, it is estimated that there are more than 100 million HAV infections (∼1.5 million of HAV clinical cases occurring globally every year) [1], with hepatitis A caused by HAV being globally the most common form of the acute viral hepatitis [2]; 400 million people worldwide are chronically infected with HBV [3,4]; whereas the estimated global prevalence of HCV infection mounts to 2.2%, which corresponds to ~130 million HCV-positive persons worldwide [5], with the highest prevalence of HCV infection (15%-20%) being reported from Egypt [6,7,8]. However, the most severe form of viral hepatitis is associated with the HBV-HDV co-infection, which is observed in ~20 million people [9,10,11]. Furthermore, it was estimated that ~ 939 million (i.e., 1 in 8 individuals globally) have ever experienced HEV infection, with 15-110 million individuals having recent or ongoing HEV infection [12].
Although infection with all hepatitis viruses leads to the development of disease, and although they are not easily distinguishable clinically from each other, not all types of hepatitis are equally serious, being manifested with different characteristics. Hepatitis A, being transmitted mainly by direct contact with patients who have been infected or by ingesting contaminated water or food, is very common in n low-income countries but typically does not lead to serious complications or long-lasting illness and does not cause chronic infections [13]. Hepatitis B, also known as post-transfusion hepatitis, being transmitted through contact with infected blood or body fluids (i.e., injection drug use, unscreened blood transfusions, unsafe health care, unsafe injection practice, birth, sexual practices that lead to exposure to blood, tattoos and body piercings) [14], can be very serious if the initial viral infection progresses to a chronic infection, which is a common cause of liver cirrhosis and hepatocellular carcinoma (HCC) worldwide [15]. While no initial symptoms are typically caused by HCV infection, which is most efficiently transmitted through large or repeated direct percutaneous exposures to blood (e.g., transfusion or transplantation from infectious donors, injecting drug use) [16], it is estimated that a lifelong, chronic infection can be developed in 60-80% of patients, and can be associated with 27% of cirrhosis and 25% of HCC worldwide [8]. Being a sub-viral agent, whose genome codes for the only virus protein, the Delta antigen, for its replication and spread, HDV depends on the presence of HBV in the infected cells [11,17,18]. Similar to HBV, HDV is transmitted through contact with infected blood or blood products or broken skin (via injection, tattooing etc.), or through personal contacts [19]. Severity of hepatitis dramatically increases as a result of the HDV-HBV co-infection, as in HDV/HBV carriers, HDV causes threefold and twofold increase of the HCC and mortality risks, respectively, in comparison with the chronic carriers of HBV alone [11,20,21]. Finally, an emerging zoonotic pathogen HEV, being known worldwide as a leading cause of acute viral hepatitis, typically results in the asymptomatic or self-limiting infection in the general population but might show a high risk of developing chronic infection in immunocompromised patients or severe clinical outcomes in pregnant women [12,22].
An important feature of hepatitis viruses is their propensity for co-infection and super-infection. Both of these phenomena reflect the presence of more than one type of hepatitis virus in an individual, but they are based on different mechanisms, where co-infection corresponds to a situation, where a person is infected with multiple types of viruses simultaneously, whereas super-infection refers to a case, where primary infection with one type of hepatitis virus is followed by a secondary infection with a different hepatitis virus type. Since often it is difficult to unequivocally determine how hepatitis viruses of different types ended up in one individual, we will not differentiate between these mechanisms. For HCV, co-infections were found with HAV [23,24,25,26], HBV [27,28], HBV-HDV [29,30,31,32,33,34,35,36,37], and HEV [38,39,40,41]. Co-infections typically cause more severe illness, as evidenced by increased morbidity and mortality in chronic HCV patients co-infected with HAV and HBV [24] and already emphasized dramatically increased severity of hepatitis in the HDV-HBV co-infection [11,20,21].
It is known that intrinsic disorder is commonly present in viral proteins, where it plays a number of crucial functional roles [42,43,44,45,46,47,48,49]. It was pointed out that intrinsic disorder acts both as an armor and weapon of viruses [45,47]. On the other hand, intrinsic disorder in host proteins is crucial for the appropriate response to infection and also mediates interactions between virus and host cell proteins [50,51,52]. Generally, the protein-protein interactions (PPIs) between hosts and viruses are known to play important roles in host immune responses and define the peculiarities of viral infections [53]. It is not surprising therefore that virus-host interactomes have being established and intensively analyzed for many viruses, including hepatotropic viruses, such as the HBV [54,55,56,57,58], HCV [59,60,61,62,63,64,65,66,67], and HEV [68,69,70]. Although many facets of virus-host interactions are known, important aspects related to the prevalence and functionality of intrinsic disorder in viral interactomes remain mostly unexplored. In fact, there are only a few studies looking at the roles of intrinsic disorder in interactions between the host proteins and hepatotropic viruses [50,62]. Even less is known about prevalence and roles of intrinsic disorder in proteins related to the hepatotropic co-infections. The goal of this study is to fill this gap by conducting the bioinformatics analysis of intrinsic disorder in human proteins interacting with hepatotropic viruses, with special focus on host proteins that can interact with more than one type of hepatitis viruses.
2. Materials and Methods
2.1. Protein Datasets
Information about proteomes of human hepatitis viruses A, B, C, D, and E was retrieved from UniProt [197]. For HAV, we analyzed genome polyprotein from human hepatitis A virus genotype IB (isolate HM175) (HHAV) (human hepatitis A virus (isolate Human/Australia/HM175/1976) (UniProt ID: P08617). For HBV, we used hepatitis B virus genotype D subtype ayw (isolate France/Tiollais/1979) (HBV-D) (Proteome ID: UP000007930). For HCV, genome polyprotein from Hepatitis C virus genotype 1a (isolate H77) (HCV) (UniProt ID: P27958) was used. For HDV, we analyzed S-HDAg (UniProt ID: P0C6L3) and L-HDAg (UniProt ID: P29996) from hepatitis delta virus genotype I (isolate D380). Hepatitis E virus genotype 1 (isolate Human/China/HeBei/1987) was selected to analyze its non-structural poly-protein ORF1 (UniProt ID: Q81862), pro-secreted protein ORF2 (UniProt ID: Q81871), and protein ORF3 (UniProt ID: Q81870).
To get host proteins interacting with hepatotropic viruses, we used the STRING_Viruses platform (http://viruses.string-db.org/). Resulting HAV-host network included 7 HAV proteins and 486 Pan troglodytes proteins. Note that with the settings used in this study, instead of human proteins, STRING_Viruses pulled out Pan troglodytes proteins for the HAV-host interactome. Since the euchromatic regions of chimpanzee (Pan troglodytes) genome share ~98% sequence similarity with the human (Homo sapiens) [86], the derived set of the host proteins interacting with HAV can be used for the purposes of this study. PPI network linking HBV proteins with human proteins contained 5 HBV proteins (proteins P, X, external core antigen, middle envelope protein, and capsid) and 78 human proteins. Human interactome of HCV proteins included 8 viral and 202 human proteins. In HDV-human PPI network, there are 2 HDV and 11 human proteins. In the PPI network between the HEV and human proteins there were 9 human proteins. All retrieved host proteins interacting with hepatotropic viruses were used in global disorder analysis.
Assuming that in co-infection (or super-infection), different hepatotropic viruses infecting the same cell can be involved in interaction with same set of host proteins, we looked for host proteins shared by different hepatitis viruses within the corresponding virus-host interactome. This analysis retrieved 33 such proteins. Their IDs and major properties are listed in Table 1.
The interactability of these proteins within the set of shared host proteins and within individual PPI networks was also evaluated in terms of internal and global node degree, respectively. Next, we selected a subset of 11 host proteins interacting with at least three different hepatitis viruses and conducted more focused functional disorder analysis.
2.2. Computational Analyses of Viral and Host Proteins
The sequence-based and structure-based analyses of proteins in this study were conducted using the web-based computational tools, such as UniProt [197] and AlphaFold [198]. Disorder-based analysis was done using the RIDAO platform [199]. For functional disorder analysis we used the D2P2 database [200], whereas liquid-liquid phase separation potential of query proteins was evaluated by the FuzDrop platform [147,148,201]. The STRING database [202] in multi-protein and single-protein modes was used for evaluation of the protein-protein interactions (PPIs) and analysis of functional enrichment in the resulting PPI networks and interactomes.
3. Results and Discussion
3.1. Intrinsic Disorder in Proteomes of Human Hepatitis Viruses
We started with the evaluation of the intrinsic disorder status of the proteomes of five human hepatitis viruses.
3.1.1. Hepatitis A Virus
HAV is a non-enveloped picornavirus containing a positive-sense, single-stranded RNA genome, which is packaged in a protein shell [71] and encodes a single genome polyprotein of 2,227 residues. At maturation, polyprotein is cleaved into functional viral proteins, such as capsid proteins VP1 (residues 492-765), VP2 (residues 24-245), and VP3 (residues 246-491) that form an icosahedral capsid enclosing the viral genome [72], capsid protein VP0 precursor that serve as a component of immature procapsid (residues 1-245) [72], capsid protein VP4 (residues 1-23) that participates in the assembly of an icosahedral capsid [73], and a capsid protein VP1-2A (residues 492-836) containing an assembly signal (residues 766-836) [73]. In addition to capsid proteins, genome polyprotein includes protein 2B (residues 837-1,087) acting as a viroporin among other functions [74], protein 2BC (residues 837-1,422) affecting membrane integrity and increasing the membrane permeability [74,75], protein 2C (residues 1,088-1,422) that has RNA-binding activity [76] and causes structural rearrangements of intracellular membranes [75], precursor protein 3ABCD (residues 1,423-2,227), precursor protein 3ABC (residues 1,423-1,738) that is targeted to the mitochondrial membrane where the viral protease 3C cleaves and inhibits the host antiviral protein MAVS [77] and also can efficiently cleave the 2BC precursor [78], protein 3AB (residues 1,423- 1,519) interacts with the 3CD precursor and shows RNA binding activity [79], protein 3A (residues 1,423-1,496) that anchors the 3AB and 3ABC precursors to the membrane via its hydrophobic domain [80] and shows RNA binding activity [79], viral protein genome-linked (residues 1,497-1,519) acting as a primer for the viral RNA replication [81], serine protease 3C (residues 1,520-1,738) that is responsible for generation of the mature viral proteins from the precursor polyprotein [82], protein 3CD (residues 1,520-2,227) interacting with the 3AB precursor and RNA [79], and RNA-directed RNA polymerase 3D-POL (residues 1,739-2,227) that replicates genomic and antigenomic RNA by recognizing replications specific signals.
Figure 1 represents intrinsic disorder profile generated for genome polyprotein from human hepatitis A virus genotype IB (isolate HM175) (HHAV) (human hepatitis A virus (isolate Human/Australia/HM175/1976) (UniProt ID: P08617) and shows that this genome polyprotein is moderately disordered. This is based on the accepted classification of proteins based on their percent of predicted intrinsically disordered residues (PPIDR) values as highly ordered, moderately disordered, and highly disordered, if their corresponding PPIDR values are below 10%, between 10% and 30%, and above 30%, respectively [83,84]. Additional classification mode is given by the analysis of the protein average disorder score (ADS) values, as proteins ADS < 0.15, 0.15 ≤ ADS < 0.5, and ADS ≥ 0.5 are considered as highly ordered, moderately disordered/flexible, or highly disordered, respectively. Since polyprotein has PPIDR PONDR VSL2 of 16.3% and ADS PONDR VSL2 of 0.306, it is classified as moderately disordered. Importantly, Figure 1 shows that most of the cleavage site are located within the intrinsically disordered regions (IDRs, i.e., regions with the disorder scores above 0.5 threshold). Furthermore, according to this analysis, mature HAV proteins show different levels of intrinsic disorder and based on their ADSPONDR VSL2 values, can be grouped as follows: VP4 (0.891±0.025) > VP0 (0.692±0.094) > VP2 (0.671±0.072) > VP3 (0.501±0.032) > VP1 (0.409±0.025) > 2A (0.397±0.032) > 2B (0.304±0.020) > 2C (0.239±0.022) > 3A (0.192±0.003) > 3B (0.184±0.001) > 3C (0.161±0.014) > 3D (0.087±0.033). Therefore, VP0, VP2, VP3, and VP4 are classified as highly disordered proteins. Remaining HAV proteins, with the only exception for the RNA-directed RNA polymerase 3D-POL, are moderately disordered. Detailed discussion of the functional roles of intrinsic disorder in individual HAV proteins and analysis of the variability of their disorder predispositions in different human HAV genotypes and subtypes are outside the scopes of this work and will be a subject of dedicated study.
Next, we looked at the HAV-host interactome via the STRING_Viruses platform (http://viruses.string-db.org/) using low confidence 0.15 for minimum required interaction score (to ensure maximal inclusion of HAV proteins in the corresponding virus-host PPI network). Based on these settings, the platform generated a dense network containing almost 500 host proteins (see Figure 2).
FuzDrop analysis indicated that although neither the whole-length genome polyprotein nor mature HAV proteins can undergo spontaneous liquid-liquid phase separation (they are characterized by the probability of spontaneous liquid-liquid phase separation, pLLPS, below the 0.6 threshold), capsid proteins VP0 (pLLPS = 0.2160), VP1 (pLLPS = 0.3729), and VP2 (pLLPS = 0.1640), can act as droplet-client proteins as they contain droplet-promoting regions, which can induce their partitioning into condensates. These observations suggest that HAV can somehow affect cellular LLPS processes, and corresponding distortions can be related to the pathogenic mechanisms of this virus. This is an intriguing hypothesis in lights of the recently recognized importance of such capability for many viruses [87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107], including HBV (see below) [108,109].
3.1.2. Hepatitis B Virus
HBV is a partially double-stranded DNA virus of the genus Orthohepadnavirus and a member of the Hepadnaviridae family of viruses. HBV genome that replicates through an RNA intermediate, encodes seven proteins. Based on the antigenic epitopes found on its envelope proteins, HBV can be divided into four major serotypes (adr, adw, ayr, and ayw), which, based on the overall nucleotide sequence variation of the genome, are further divided into ten genotypes (A–J) and forty subgenotypes [110]. Since the evaluation of the related variability in the intrinsic disorder predispositions of the HBV proteins is outside the copes of this study, we present below results of the intrinsic disorder analysis of proteins in just one variant, namely hepatitis B virus genotype D subtype ayw (isolate France/Tiollais/1979) (HBV-D) (Proteome ID: UP000007930). The large envelope protein encoded by the gene S is a transmembrane protein that exists in two topological conformations, external and internal, Le-HBsAg and Li-HBsAg, respectively, with Le-HBsAg being responsible for initiating infection via attachment to the cell receptors, and with Li-HBsAg being involved in virion morphogenesis and acting as a matrix protein by mediating the contact with the nucleocapsid. Three isoforms are produced by alternative splicing and alternative initiation, a 389-residue-long canonical form known as large envelope protein (L, LHB, and L-HBsAg), a 281-residue-long middle envelope protein (M, MHB, or M-HBsAg), which is different from L by missing residues 1-108, and a 226-residue-long small envelope protein (S, SHB, or S-HBsAg), which is different from L by missing residues 1-163. Capsid protein (183-residue-long) and external core antigen (212-residue-long) are both encoded by the same gene C via the alternative initiation. Capsid protein is responsible for formation of an icosahedral capsid, whereas external core antigen can regulate the immune response to the intracellular capsid [111]. A 153-residue-long protein X is a multifunctional protein with roles in promoting viral transcription, genome replication, and silencing host antiviral defenses [112]. Protein P is an 832-residue-long multifunctional enzyme responsible for the conversion of the viral RNA genome into dsDNA in viral cytoplasmic capsids, which also possesses DNA polymerase activity, being capable of copying either DNA or RNA templates, and shows a ribonuclease H (RNase H) activity utilized for cleavage of the RNA strand of RNA-DNA heteroduplexes [111]. HBV genome also encodes two putative uncharacterized proteins of 10.4 and 15.3 kDa. Figure 3 represents results of the intrinsic disorder analysis of these proteins and shows that all of them contain noticeable levels of disorder. Based on their ADSPONDR VSL2 values, these proteins can be grouped as follows: putative uncharacterized 10.4 kDa protein (0.88±0.13) > protein X (0.48±0.28) > capsid protein (0.44±0.34) > putative uncharacterized 15.3 kDa protein (0.43±0.25) > large envelope protein (0.42±0.31) > protein P (0.38±0.27) > external core antigen (0.37±0.34) > middle envelope protein (0.31±0.28) > small envelope protein (0.25±0.28).
Figure 4 represents the PPI network linking HBV proteins with human proteins. This network, which was generated by STRING_Viruses platform (http://viruses.string-db.org/) using medium confidence of 0.4 for the minimum required interaction score, contains 5 HBV proteins (proteins P, X, external core antigen, middle envelope protein, and capsid) and 78 human proteins.
Analysis of the LLPS predisposition of HBV proteins indicated that all of them are somehow related phase separation. In fact, two HBV proteins, large envelope protein (pLLPS = 0.9812, 5 DPRs, residues 1-23, 44-108, 124-153, 201-230, and 265-285) and putative uncharacterized 10.4 kDa protein (pLLPS = 0.9938, one DPR, residues 1-74) are expected to serve as droplet drivers, since they have high probability of spontaneous liquid-liquid phase separation. Furthermore, all the remaining HBV proteins (protein P (pLLPS = 0.4030, one DPR, residues 162-313); capsid protein (pLLPS = 0.3354, one DPR, residues 130-183); protein X (pLLPS = 0.2173, one DPR, residues 18-56); external core antigen (pLLPS = 0.2376, one DPR, residues 159-212); and putative uncharacterized 15.3 kDa protein (pLLPS = 0.3429, one DPR, residues 1-29)) are potential droplet-clients, since although their pLLPS values are below the 0.6 threshold, all of them contain droplet-promoting regions. Importantly, the role of LLPS in HBV infection and development of HBV-driven hepatocellular carcinoma (HCC) were shown to be dependent on LLPS. In fact, it was shown that protein X binds to CREB1 and increases the cellular prion protein (PrPC) expression promoting PrPC phase separation and thereby activating the NF-κB signaling pathway and promoting tumor progression [109]. Another side of the HBV-specific LLPS mechanism is given by the roles G-quadruplexes of the HBV covalently closed circular DNA (cccDNA) in virus replication via phase separation [108].
3.1.3. Hepatitis C Virus
HCV, with at least 6 genotypes and numerous subtypes, is a member of the hepacivirus genus from the Flaviviridae family. This enveloped virus has a positive-strand single-stranded RNA genome encoding a genome polyprotein of ~3,000 residues, which is cleaved at maturation to 10 functional proteins (core protein C, and glycoproteins E1 and E2, integral membrane protein p7, and nonstructural proteins NS2, NS3, NS4A, NS4B, NS5A, and NS5B). Prevalence and functionality of intrinsic disorder in 32 HCV polyproteins across the 18 genotypes with 8 isolates for genotype 1b, 2 isolates for genotypes 1a, 1c, 2a, 2b, 3a, 5a, 6a, and 1 isolate for genotypes 2c, 2k, 3b, 3k, 4a, 6b, 6d, 6g, 6g, and 6k were analyzed in a previous study [43]. Using a wide spectrum of bioinformatics techniques it was shown that HCV proteins contain noticeable levels of intrinsic disorder, and that the associated structural flexibility is crucial for many of their functions [43]. In line with those earlier observations, Figure 5 represents the RIDAO-generated per-residues disorder profile of genome polyprotein from Hepatitis C virus genotype 1a (isolate H77) (HCV) and shows that the entire polyprotein can be classified as moderately disordered. In fact, it is characterized by PPIDR PONDR VSL2 of 19.5% and ADS PONDR VSL2 of 0.32±0.24. Based on their disorder status measured in terms of the ADSPONDR VSL2 values, HCV proteins can be arranged as follows: C (0.60±0.30) > NS5A (0.53±0.31) > NS3 (0.33±0.19) > NS5B (0.31±0.17) > E2 (0.25±0.15) > NS4B (0.24±0.13) > E1 (0.19±0.12) > NS4A (0.17±0.16) > NS2 (0.13±0.09) > p7 (0.10±0.08). When using PPIDR-based classification, these proteins for the following order: C (65.9%) > NS5A (46.7%) > NS3 (20.3%) > NS5B (14.6%) > NS4A (7.4%) > NS4B (4.6%) > E2 (2.3%) > E1 (1.6%) > NS2 (0.0%) > p7 (0.5%).
Human interactome of HCV proteins is shown in Figure 6. It includes 8 viral and 202 human proteins. FuzDrop analysis revealed that the HCV genome polyprotein has a noticeable LLPS potential, possessing the pLLPS of 0.7034 and containing 7 droplet-promoting regions (residues 1-97, 96-21, 1206-1217, 1485-1495, 2191-2212, 2304-2418, and 2682-2693). Mature HCV proteins C (pLLPS = 0.9364, three DPRs, residues 1-28, 36-81, and 96-116) and NS5A (pLLPS = 0.6757, two DPRs, residues 219-240 and 332-444) are predicted as droplet-drivers, proteins E2 (pLLPS = 0.1706, one DPR, residues 1-13), serine protease/helicase NS3 (pLLPS = 0.1873, two DPRs, residues 179-194 and 459-469), and RNA-directed RNA polymerase (pLLPS = 0.2638, one DPR, residues 262-273) are acpected to serve as droplet-clients, whereas proteins E1 (pLLPS = 0.1119, no DPRs), viroporin p7 (pLLPS = 0.1152, no DPRs), protease NS2 (pLLPS = 0.1031, no DPRs), and NS4A (pLLPS = 0.0990, no DPRs), and NS4B (pLLPS = 0.1223, no DPRs) are predicted to be unrelated to the cellular phase separation processes. HS5A of HCV was shown to colocalize with Ras-GTPase-activating protein-binding protein 1 (G3BP1) in the HCV replication complex (RC) [113].
Although HCV RCs, which are virus-induced structures that serve as factories for viral RNA synthesis, are not membrane-less organelles, being instead membrane-associated structures [114], it is tempting to hypothesize that they might represent an example of the 2D LLPS (i.e., LLPS happening in or on the membrane) [115]. This hypothesis is in line with the notion that the viral replication complexes also known as viral inclusion bodies can be considered as biomolecular condensates formed through phase separation [116]. Both C and NS5B were shown to be associated with the processing body (P-body) biogenesis by relocalizing the P-body-specific protein Mov10 to circular structures surrounding cytoplasmic lipid droplets with NS5A and C proteins [117]. It was also speculated that NS5A phosphorylation might affect assembly and disassembly of stress granules and other membrane-less organelles [102].
3.1.4. Hepatitis D Virus
HDV, being a sub-viral agent whose replication and spread depends on the presence of HBV in the infected cells [11,17,18], is an enveloped virus with the negative-sense, single-stranded, covalently closed circular RNA genome classified as the genus Deltavirus, within the realm Ribozyviria. Virus is characterized by considerable genetic variability, existing as eight major HDV genotypes [118] showing specific geographic distributions and being linked to distinctive clinical outcomes and disease progression [119]. Genome of this virus encodes the only virus protein, the hepatitis delta antigen (HDAg). However, its envelope containing host phospholipids has three proteins taken from the HBV, L-HBsAg, M-HBsAg, and S-HBsAg. The delta antigen exists in two forms generated from the same viral genome, small (S-HDAg, 195 residue-long) and large (L-HDAg, 213 residue-long) [120], with different functions, with the small delta antigen being crucial for viral RNA replication, and with the large delta antigen acting as a potent trans-dominant inhibitor for HDV replication [121,122] being primarily involved in the assembly of new viral particles [11,123]. The difference between the S-HDAg and L-HDAg is the presence of 19 extra residues added to the C-terminal region of L-HDAg via an RNA editing mechanism taking place during the late stages of the replication cycle and causing conversion of an amber stop codon into a tryptophan codon thereby leading to the addition of 57 nucleotides to the ORF and consequently 19-residue extension to the protein [11,124,125].
Figure 7 represents RIDAO-generated disorder profiles of S-HDAg and L-HDAg and shows that both forms of delta antigen are highly disordered. The proteins are predicted to have several disorder-based binding sites, molecular recognition features, MoRFs, which are disordered segments that become ordered at interaction with binding partners. In S-HDAg, these MoRFs are regions comprising residues 40-62, 67-96, 106-123, and 143-195. In L-HDAg, C-terminal MoRF is extended to cover residues 143-210. Therefore, despite of (or due to) their highly disordered status, both proteins contain important functional regions, such as the dimerization coiled-coil region (residues 12-60), nuclear localization signal (residues 66-75), two RNA-binding regions (residues 97-107 and 136-146), and, in S-HDAg, region involved in interaction with host RNA polymerase II complex (residues 130-195). C-terminal region of L-HDAg contains a prenylation site required for viral particle assembly with HBsAg [126,127].
Both proteins are predicted to be characterized very high probability of spontaneous liquid-liquid phase separation, pLLPS of 0.9914 and 0.9927 for S-HDAg and L-HDAg, respectively. Therefore, it is tempting to hypothesize that virus-driven LLPS could be related to the mechanism of the HDV pathogenicity. In line with this hypothesis, it was reported that HDAg and viral RNA accumulate in the nucleus of infected cell in the form of massive RNA-protein hubs that often occupy the majority of the nuclei [128]. It was emphasized that the HDV genome is exclusively enriched in G-quadruplex forming sequences, with density of these sequences being more than four times greater than that of the human genome [129]. Since in HBV, the G-quadruplex forming sequences were linked to the efficient liquid-liquid phase separation associated with virus replication [108], it is likely that similar LLPS mechanism can also be applicable to HDV as well.
Figure 8 shows that S-HDAg has more host protein partners than L-HDAg does (10 vs. 3). Overall, there are 11 human proteins that can interact by the HDV proteins.
3.1.5. Hepatitis E Virus
The HEV belongs to the genus Orthohepevirus from the Hepeviridae family. Its genome is a single-stranded, positive-sense RNA that contains three open reading frames (ORFs), ORF1, ORF2, and ORF3 [130,131]. Of seven HEV genotypes infecting various animal species, HEV-1, HEV-2, HEV-3, HEV-4 [132], and HEV-7 [133] are able to infect humans. ORF1 encodes a non-structural polyprotein pORF1, which is a 1,693-residue-long multifunctional protein containing a methyltransferase domain (residues 1-240), Y-domain (residues 241-439), which is involved in gene regulation and membrane binding in intracellular replication complexes [134], a papain-like cysteine protease (residues 442-509), zinc-binding region (residues 510-691), a proline-rich disordered hypervariable hinge region (residues 712-770), a macro domain also known as X-domain (residues 775-921), a +RNA virus helicase ATP-binding domain (residues 934-1082), an NTPase/helicase domain (residues 960-1204), a C-terminal +RNA virus helicase domain (residues 1083-1216), and an RNA-directed RNA polymerase (residues 1207-1693) containing RdRp catalytic domain (residues 1454-1565) [135,136]. ORF2 encodes a pro-secreted protein pORF2, which is considered as a canonical pORG2 form containing a signal peptide (residues 1-19) and a pro-peptide (residues 20-33), removal of which generate secreted protein ORF2s (ORF2g). Because of alternative initiation, a capsid protein (ORF2c or ORF2i) is synthesized as well, which is different from the canonical form by missing residues 1-15. pORG2 contains an Arginine-Rich Motif (ARM, residues 28-33) acting as a nuclear localization signal, a region responsible for particle formation (residues 368-394) and an oligomerization region (residues 585-610). The major role of secreted protein ORF2s is the inhibition of the host antibody-mediated neutralization, whereas capsid protein ORFc is responsible for the formation of an icosahedral capsid. ORF3 encodes the ORF3 multifunctional phosphoprotein also known as Vp13 with crucial roles in counteracting host innate immunity, as well as virion morphogenesis and egress [137,138,139,140]. Despite its small size (114 residues), pORF3 contains a multitude of functional regions requires for membrane association (residues 1-28), induction of host signal regulator protein alpha (SIRPA) expression required for the type I interferon down-regulation [141], interaction with host HPX (residues 28-68), interaction with the the ORP2c (residues 48-72), homodimerization and interaction with host AMBP/bikunin (residues 72-114), as well as interaction with host FYN, GRB2, HCK, PIK3R3, and SRC (residues 95-104), and PSAP/PTAP motif required for the virion release from infected cells (residues 96-99) [139].
Figure 9 illustrate the intrinsic disorder status of these proteins and shows that all of them are expected to contain noticeable levels of disorder, being classified as moderately or highly disordered. In fact, pORF1 is characterized by ADSPONDR VSL2 of 0.30±0.19 and PPIDRPONDR VSL2 of 14.9%, being, therefore, moderately disordered, whereas both pORF2 and pORF3 are highly disordered, as their ADSPONDR VSL2 values are 0.47±0.25 and 0.74±0.18, and PPIDRPONDR VSL2 values are 36.5% and 87.7%, respectively.
An important feature of pORF2 and pORF3 is that they are encoded by the bicistronic mRNA, where ORF3 substantially overlaps the 5′ coding sequence of ORF2 in a separate reading frame [131]. Therefore, it is not surprising that both of these proteins are highly disordered, as this is in line with well-established fact that intrinsic disorder is commonly found in viral proteins produced from overlapping reading frames [45,48,142,143,144].
An obvious consequence of high intrinsic disorder status of HEV proteins is their aforementioned multifunctionality and binding promiscuity. This is illustrated by Figure 10 representing PPI network of HEV and human proteins and showing that the most disordered pORF3 is highly connected.
FuzDrop-based analysis of the LLPS predisposition of HEV proteins revealed that the pro-secreted protein pORF2 can act as droplet driver (pLLPS = 0.7344, three DPRs, residues 1-53, 57-126, and 444-465), whereas both pORF1 (pLLPS = 0.2675, four DPRs, residues 488-501, 633-643, 672-694, and 711-773) and pORF3 (pLLPS = 0.4066, one DPR, residues 85-114) are expected to be droplet clients. Although involvement of LLPS in the HEV life cycle was not discussed in the literature, it is very likely that similar to other hepatotropic viruses (as well as viruses in general), HEV might use phase separation either for replication or for fighting against host immune response. Clearly, further analysis of these possibilities is needed.
3.2. Intrinsic Disorder Status of Host Proteins Interacting with Hepatotropic Viruses
It is known that host cells use numerous advantages of IDPs and IDRs to fight “flexible” invaders, viruses, and to successfully overcome the viral invasion [52]. Therefore, we decided to check the intrinsic disorder status of human proteins involved in interaction with different hepatitis viruses. To be consistent with selection of human proteins for such analysis, we used datasets generated by STRING_Virus. The corresponding PPI networks were shown and briefly described in the corresponding sections above. We present results of the analysis of global intrinsic disorder predisposition of host proteins interacting with different hepatotropic viruses below.
Figure 11 and Figure 12 show results of the evaluation of global intrinsic disorder propensity of host proteins in all analyzed datasets; i.e., 486, 78, 202, 11, and 9 host proteins interacting with HAV, HBV, HCV, HDV, and HEV proteins, respectively. As a reminder, STRING_Virus generated networks for human proteins interacting with HBV, HCV, HDV, and HEV, whereas HAV-host interactome represents proteins from Pan troglodytes, despite our request to show HAV-human interactome. As we already indicated, since chimpanzee (Pan troglodytes) and human (Homo sapiens) genomes share ~98% sequence similarity [86], the derived set of the chimpanzee proteins interacting with HAV can be used for the purposes of this study. Figure 11A represents the outputs of this analysis as the PONDR® VSL2 Score vs. PONDR® VSL2 (%) plot, which can be used for the classification of proteins based on their Average Disorder Score (ADS) and Percentage of Predicted Disordered Residues (PPDR) values. Based on the outputs of these analysis, the proteins were classified as highly ordered if they had a PPDR of less than 10% and/or an ADS of less than 0.15. Proteins with 10% ≤ PPIDR < 30% and/or 0.15 ≤ ADS < 0.5 were considered moderately disordered. Proteins with PPDR ≥ 30% and ADS of ≥ 0.5 were defined as highly disordered. This classification is in line with the practice accepted in the field [83]. To make results of this analysis more visual, the ADS vs. PPIDR plot has differently colored blocks, where regions containing mostly ordered, moderately disordered (), or mostly disordered proteins are shown by blue/light blue, pink/light pink, and color, respectively. If the two parameters agree, the corresponding part of the background is dark (blue or pink), whereas light blue and light pink reflect regions, where the ADS and PPIDR disagree with each other. Figure 11A shows that host proteins interacting with hepatotropic viruses are characterized by noticeable variation in their intrinsic disorder status.
Another accepted way of global classification of protein intrinsic disorder utilizes position of query proteins within the CH-CDF phase space [145,146] originating from the combined consideration of two binary predictors, the charge-hydropathy (CH) plot and cumulative distribution function (CDF) analysis. CH-plot classifies proteins based on their absolute mean charge and mean normalized hydrophobicity, as highly disordered proteins are often characterized by low hydropobicity and high net charge [147,148]. The CDF describes the cumulative frequency of disordered residues along the length of a given protein, and position of the resulting CDF curve relative to the order-disorder boundary is used for protein classification [147]. Combination of the outputs of these binary predictors produces CH-CDF classifier that allows more detailed characterization of the disorder status of query proteins based on the agreement between the predictors generating four quadrants [145,146]. In the resulting ΔCH-ΔCDF plot, Quadrant 1 contains proteins that are likely to be structured (both predictors agree, CHordered and CDFordered). Quadrant 2 includes proteins that are either molten globular or hybrid; i.e., proteins that are compact yet lack a distinctive 3D structure or contain noticeable levels of ordered and disordered residues (predictors disagree, CDFdisordered but CHordered). Quadrant 3 contains highly disordered proteins (both predictors agree, CHdisordered and CDFdisordered), whereas Quadrant 4 contains proteins that are predicted to be disordered according to the CH-plot yet ordered according to the CDF-plot [145,146]. Figure 11B represents the results of global disorder analysis of the host proteins interacting with different hepatitis viruses in the form of the ∆CH-∆CDF plot and shows that these proteins are spread between all four quadrants, further reflecting variability of their global disorderedness.
Quantification of distribution of host proteins within the different areas the ADSPONDR VSL2 vs. PPIDRPONDR VSL2 plot and the ΔCH-ΔCDF plot is illustrated by Figure 12 showing contents of different areas of these plots. Figure 11A and Figure 12A show that the vast majority of host proteins are predicted as moderately or highly disordered (none of them is found in the blue area, and less than 10% of them are located within the cyan area). Most of the host proteins are within the pink or light pink areas, being therefore predicted as moderately disordered. At least 30% of host proteins interacting with hepatitis viruses are expected to be highly disordered, as they are located within the red area of the ADSPONDR VSL2 vs. PPIDRPONDR VSL2 plot. In comparison with the entire human proteome, more host proteins interacting with hepatitis viruses were predicted as moderately disordered.
Figure 12.
Quantification of host proteins interacting with various hepatitis viruses based on their positions within the ADSPONDR VSL2 vs. PPIDRPONDR VSL2 plot (A) and ΔCH-ΔCDF plot (B). Analogous data for the entire human proteome are shown for comparison as well (black bars). Corresponding data are taken from [149].
Figure 12.
Quantification of host proteins interacting with various hepatitis viruses based on their positions within the ADSPONDR VSL2 vs. PPIDRPONDR VSL2 plot (A) and ΔCH-ΔCDF plot (B). Analogous data for the entire human proteome are shown for comparison as well (black bars). Corresponding data are taken from [149].
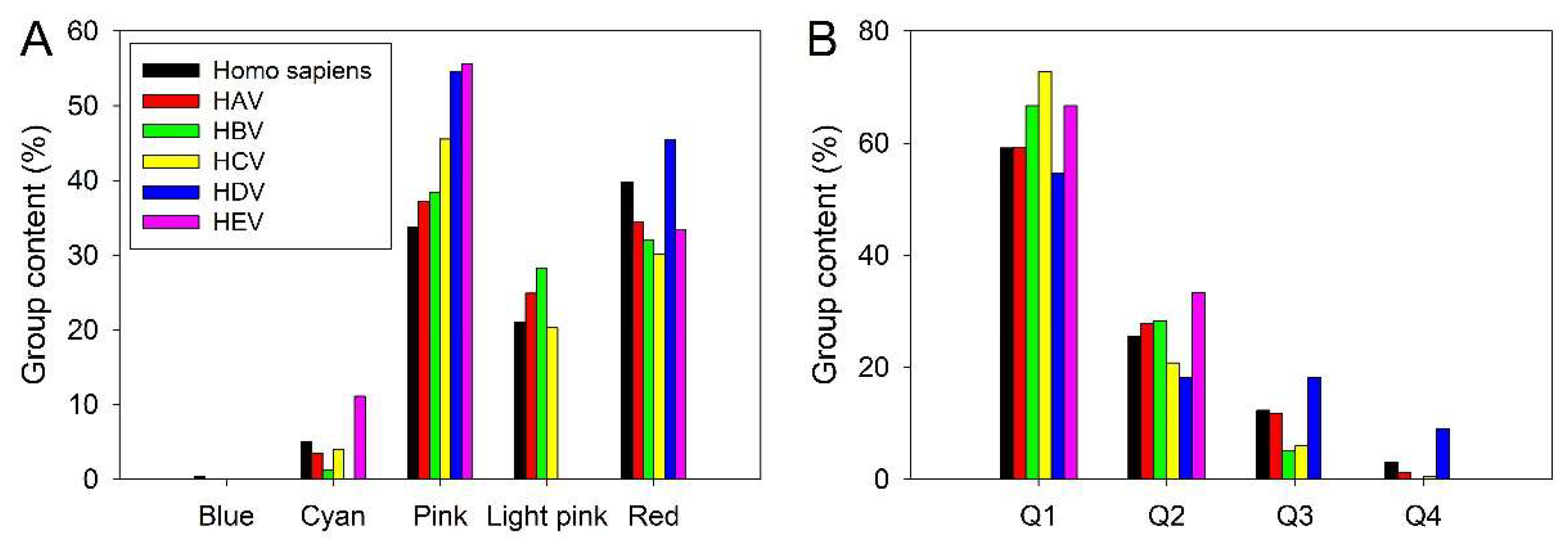
Figure 11B and Figure 12B show that many host proteins interacting with various hepatitis viruses are expected to be mostly ordered (~60% are located within the Q1). All HEV-interacting proteins are predicted as either ordered/compact or molten globule/hybrids. Altogether, their distribution within the ΔCH-ΔCDF plot is not too different from the distribution of entire human proteome.
3.3. Intrinsic Disorder Status of Host Proteins Shared by Different Hepatotropic Viruses
Since co-infection and super-infection reflects simultaneous presence of more than one type of virus in the infected cells, it is logical to assume that the co-infecting viruses might interact with some common host proteins. Analysis of the host interactomes of HAV, HBV, HCV, HDV, and HEV revealed the presence of 33 shared proteins. Interestingly, 32 of these shared host proteins are found in the HBV and HCV interactomes, whereas interactomes of HAV, HDV, and HEV contains 11, 5, and 1 shared host proteins, respectively. These host proteins are listed in Table 1, which also contains information on their physico-chemical properties, disorder and LLPS propensities.
We also looked if these shared host proteins are involved in interaction with each other using the STRING platform. Figure 13A summarize the results of this analysis and shows that most of these proteins do form a dense PPI network. In this network, 28 human proteins are connected by 214 interactions. Four proteins, GDI1, CDSN, TBXT, and FUT1 are loners (i.e., do not interact with other members of the network), whereas STRING does not have information for VEGFA. The average node degree of this network is 15.3, indicating, than on average, each member of network interacts with 15 partners. The average local clustering coefficient of this network, which is a measure of the degree to which nodes in a network tend to cluster together, is 0.859, indicating that the network has densely connected clusters (note that value of 1 represents the highest possible level of clustering within a network, and if every node has a clustering coefficient of 1, then immediate neighbors of each node are completely connected to one another, resembling a clique). PPI enrichment p-value of PPI network connecting shared host proteins is 1.54×10-13, indicating that this network has significantly more interactions among themselves than what would be expected for a set of proteins of the same size and degree distribution randomly drawn from the genome, and thereby suggesting that the proteins in this network can be biologically connected, as a group.
We also used STRING-embedded routing to visualize the functional enrichment of this network based lowest values of false discovery rate (which is the measure describing the significance of the enrichment using p-values corrected for multiple testing within each category using the Benjamini–Hochberg procedure [150]. Results of this analysis are shown in Figure 13B-G as plots representing enriched GO biological processes (Figure 13B), GO molecular functions (Figure 13C), GO cellular components (Figure 13D), disease-gene associations (Figure 13E), KEGG pathways (Figure 13F), and reactome pathways (Figure 13G). From the viewpoint of biological process enrichment, the most members of this group of proteins are involved in cell surface receptor signaling pathway, animal organ development, cellular response to chemical stimulus, positive regulation of cell population proliferation, regulation of cell population proliferation, and regulation of cell death (Figure 13B). Their most common molecular function is protein binding (Figure 13C). Most often they are found in protein-containing complexes, endomembrane system, and intracellular organelle lumen (Figure 13D). They are most commonly associated with liver diseases, gastrointestinal system diseases, gastrointestinal system cancer, intestinal diseases, disease of cellular proliferation, organ system cancer, and respiratory system diseases (Figure 13E).
Among most enriched KEGG pathways, in which members of this network can be found, are hepatitis B, pathways in cancer, AGE-RAGE signaling pathway in diabetic complications, Epstein-Barr virus infection, measles, human T-cell leukemia virus 1 infection, hepatitis C, and Kaposi sarcoma-associated herpesvirus infection (Figure 13F). Finally, many members of this network are significantly enriched in signaling by interleukins, cytokine signaling in immune system, immune system, signal transduction, and developmental biology (Figure 13G).
Since binding promiscuity and multifunctionality of proteins are frequently associated with intrinsic disorder, at the next stage, we analyzed intrinsic disorder predisposition of shared host proteins. Figure 14A represents results of this analysis and shows none of the shared host proteins was predicted as mostly ordered, indicating that all these proteins were moderately or highly disordered. In fact, among 33 shared host proteins, 12 (36.4%) were characterized by 10% ≤ PPIDRPONDR VSL2 < 30% and 0.15 ≤ ADS PONDR VSL2 < 0.5, 11 (33/3%) had PPIDRPONDR VSL2 ≥ 30% and 0.15 ≤ ADSPONDR VSL2 < 0.5, and 10 (30.3%) possessed PPDRPONDR VSL2 ≥ 30% and ADSPONDR VSL2 ≥ 0.5 (see Figure 14A).
Figure 14B represents an interesting observation on the correlation between the internal node degree (i.e., connectivity of shared host proteins within the PPI network they form according to STRING (see Figure 13A)) and the global node degree of individual proteins (i.e., their interactability in the STRING-generated networks centered at each of these proteins individually). Figure 14B shows that the global interactivity of the shared host proteins measured as their node degree within their individual PPI networks ranged 42 to 469 partners. Furthermore, this global node degree seems to increase exponentially with the increase in the internal node degree, being virtually independent on the internal node degree ranging from 0 to 15 but experiencing dramatic increase at higher internal node degree values.
Figure 14C indicates that both global and internal interactivity of shared host proteins show weak positive correlation with the intrinsic disorder propensity of these proteins. This behavior is not surprising, since all these proteins contain noticeable levels of disorder. Curiously, although the probability of host proteins to be shared by two or three different types of hepatitis viruses is almost independent on the host protein disorder status, two of the three proteins shared by four viruses (CDSN and TBXT) are highly disordered (see Table 1), with CDSN being the most disordered protein in the set. On the other hand, FUT1, which is also shared by for different hepatitis viruses is the most ordered member of the set of shared proteins.
We also used FuzDrop [151] to analyze the predisposition of shared host proteins to undergo spontaneous LLPS and found that 8 of these proteins (CDSN, CREB1, JUN, MYC, VEGFA, TP53, PRDM10, and TBXT) can serve as droplet drivers. There were also six proteins (CD4, SMYD3, HBE1, AFP, PSMB7, and GDI1) that were not related to cellular LLPS processes being characterized by low pLLPS value and not possessing DPRs. Remaining 19 shared proteins were predicted to act as droplet-clients (see Table 1). Generally, a significant part of cellular processes is known to be determined by the functioning of liquid-droplet-like condensates – membrane-less organelles (MLOs), which are very diverse and commonly found in cytoplasm, nucleus, and mitochondria of various eukaryotic cells [152,153]. Figure 14D shows that there is a strong positive correlation between PPIDRPONDR VSL2 and pLLPS, and that all 8 proteins predicted as droplet drivers (i.e., proteins characterized by pLLPS ≥ 0.60) are also predicted to be highly disordered. The intracellular LLPS processes, also known as liquid-liquid demixing phase separation, were recognized as important biogenesis drivers of various MLOs [154,155]. These processes are strongly dependent on protein intrinsic disorder [156,157]. In fact, many of the MLO resident proteins are IDPs or contain IDRs, and the formation of all the MLOs analyzed so far relies on IDPs/IDRs, indicating that intrinsic disorder is important for MLO biogenesis [154]. Furthermore, the fact that many shared host proteins potentially associated with various hepatitis co-infections can participate in LLPS is in line with a recent observation that the liquid-liquid phase separation process is emerging as an important cellular mechanism in liver innate immunity [158].
3.4. Functional Intrinsic Disorder in Most Shared Host Proteins
Next, we looked at functionality of intrinsic disorder in 11 proteins shared by four or three different hepatitis viruses. The results of these analyses are presented below, with corresponding sections being ranged based on the intrinsic disorder status of the discussed proteins.
3.4.1. Corneodesmosin (CDSN, UniProt ID: Q15517; PPIDR: 94.52%) Shared by HAV, HBV, HCV, and HDV
Corneodesmosin, being important for the epidermal barrier integrity, is one of the crucial extracellular constituents of corneodesmosomes, which are the major intercellular adhesion structures in the outermost layer of the skin known as stratum corneum and which help maintain the protective barrier of the skin [159]. The stratum corneum is characterized by relative impermeability to water and water soluble substances. These properties are defined by the unique structure of the stratum corneum, which is composed of corneocytes, “mummified” dead and flattened cells containing a cornified envelope that provides extreme individual resistance [160]. In humans, corneodesmosomes are located in the epidermis, the hard palate epithelium, and the inner root sheath of the hair follicles [161]. The crucial role of corneodesmosomes and corneodesmosin in cell adhesion is illustrated by the facts that a complete loss of the CDSN expression is associated with the generalized peeling skin disease, which is an autosomal-recessive ichthyosiform erythroderma characterized by lifelong patchy peeling of the skin [162] and that the mutations the CDSN gene located in the major psoriasis-susceptibility locus (PSORS1) may be responsible for the susceptibility to psoriasis [163]. This is also in line with the observations that the Cdsn gene ablation in mice induced neonatal death as a result of epidermal tearing upon minor mechanical stress and hair-follicle degeneration related to the desmosome dysfunction [164] and that grafting of the Cdsn-deficient skin onto immunodeficient mice resulted in rapid hair loss together with epidermal abnormalities resembling psoriasis [163].
Potential link of corneodesmosin to hepatitits is determined by the fact that there is a potential link between hepatitis and psoriasis. In fact, these two conditions could be linked interacting with each other and exacerbating each other’s symptoms. For example, it was noted that adults with psoriasis have higher rates of the HCV infection compared to those who do not have psoriasis, and HCV-infected patients with moderate-severe psoriasis are characterized by a higher rate of hepatic decompensation [165]. Curiously, a patient with chronic HCV and psoriasis showed remission of psoriasis after receiving antiviral treatment for the HCV infection [166]. Furthermore, hepatitis is known to be manifested by some extrahepatic simproms, such as mixed cryoglobulinemia, lichen planus, and porphyria cutanea tarda reflecting skin involvement [167]. The CDSN interactors from HAV proteome are P2C, P3C, P3D-POL, VP3, and VP2. In HBV-host PPI interactome, CDSN interacts with proteins X and capsid. In the case of HCV, CDSN interacts with core p19, envelope glycoprotein E2, NS4A, NS4B, NS5A, NS5B/RNA-directed RNA polymerase, and serine protease NS3. It also binds both S-HDAh and L-HDAg of HDV.
Despite obvious importance of CDSN for healthy skin, not much is known about structure of this 529-residue-long protein. It is discussed that CDSN is a 52-56 kDa basic phosphoprotein characterized by very high serine and glycine content (27.5% and 16%, respectively), especially within its N- and C-termini of the protein (residues 43-222 and 385-470) [160,164]. Capability of CDSN to act as an adhesive molecule is attributed to the presence of “glycine loops” in this protein, which are formed as a result of the association of interspersed aromatic or aliphatic residues [168]. The presence of such “glycine loops” mediates intermolecular adhesion via the Velcro-type action [168]. The progressive proteolysis of CDSN in the stratum corneum represents another important feature of this protein [160].
Figure 15 represents the results of functional disorder analysis of human CDSN. It is clear that this protein is highly disordered (Figure 15A–C). CDSN utilizes its disordered segments for protein-protein interactions, as evidenced by the presence of 12 molecular recognition features (MoRFs), which are spread throughout its entire sequence (see Figure 15B). The CDSN-centered PPI network includes 113 members linked by 1549 interactions. This protein is characterized by the pLLPS of 1.00 (see Table 1), and therefore is predicted as droplet-driver, which is a protein that can spontaneously undergo liquid-liquid phase separation.
3.4.2. Myc Proto-Oncogene Protein (MYC, UniProt ID: P01106; PPIDR: 79.74%) Shared by HAV (H2QWQ2), HBV and HCV
Neither MYC (also known as proto-oncogene c-Myc and class E basic helix-loop-helix protein 39 (bHLHe39)) nor its disorder status require special introduction. In fact, since MYC drives growth and proliferation of cells, this transcription factor is considered as critical oncogene involved in the majority of human tumors [169]. The link of MYC to hepatitis is manifested by significantly elevated levels of this protein in liver cell infected with hepatitis viruses [170,171,172,173,174,175]. It was also reported that HCV may accelerate breast cancer progression by increasing circulating levels of c-Myc oncoprotein [176]. MYC is one of the proteins which are crucially related to the transition from cirrhosis to hepatocellular carcinoma in HCV-infected patients [177]. The intrinsic disorder nature of this important transcription factor was discussed in several dedicated studies (e.g., see [169,178,179,180,181,182,183,184,185]).
Figure 16 shows that this 454-residue-long protein is characterized by high levels of intrinsic disorder. It is predicted to have three MoRFs (residues 208-214, 229-355, and 385-394) and pLLPS of 0.9563. The capability of MYC to undergo LLPS and be involved in formation of biomolecular condensates was reported in several recent studies, e.g. it was found in nuclear puncta induced by the Epstein-Barr virus (EBV) proteins EBNA2 and EBNALP [186], was included to a LLPS-related gene-based risk index associated with HCC [187], as well as to the list of LLPS-associated proteins in rheumatoid arthritis based on the single-cell sequencing and transcriptome analyses [188]. Furthermore, it was pointed out that LLPS of MYC, which is related to the cell fate regulation, its dependent on the disordered domains of this protein [189].
Human MYC forms a very dense PPI network containing 349 members connected by 3,103 interactions (see Figure 16C). Among most enriched biological processes associated with the members of this network are regulation of gene expression, regulation of primary metabolic process, regulation of macromolecule metabolic process, and regulation of cellular metabolic process. Their most enriched molecular functions are binding, protein binding, DNA binding, heterocyclic compound binding, nucleic acid binding, organic cyclic compound binding, transcription factor binding, and transcription regulator activity. They are mostly enriched in the following cellular components: nucleoplasm, nuclear lumen, intracellular organelle lumen, nucleus, intracellular membrane-bounded organelle, chromosome, chromatin, protein-containing complex, intracellular non-membrane-bounded organelle, and transcription regulator complex. The most enriched disease-gene associations are cancer, disease of cellular proliferation, organ system cancer, cell type cancer, carcinoma, gastrointestinal system cancer, nervous system cancer, hepatobiliary system cancer, and hepatobiliary disease. Most enriched KEGG pathways associated with the members of the MYC-centered PPI network are pathways in cancer, micrornas in cancer, prostate cancer, viral carcinogenesis, cell cycle, human t-cell leukemia virus 1 infection, cellular senescence, human papillomavirus infection, FoxO signaling pathway, and hepatitis. Among the most enriched reactome pathways associated with these proteins are generic transcription pathway, RNA polymerase II transcription, gene expression (Transcription), signal transduction, disease, ESR-mediated signaling, signaling by nuclear receptors, estrogen-dependent gene expression, cellular responses to stress, and cellular Senescence.
3.4.3. Cellular Tumor Antigen p53 (TP53, UniProt ID: P04637; PPIDR: 68.11%) Shared by HAV, HBV and HCV
Being one of the most well-studied proteins (e.g., as of January 29, 2025, more 123,500 papers talking about different aspects of p53 were reported in PubMed), cellular tumor antigen p53 (TP53) requires even less introduction than MYC. In fact, this protein is broadly known as a guardian of the genome, mutations in which are involved in ~60% of human cancers [190]. Among functions ascribed to p53 are negative regulator of cell cycle progression via growth arrest [191] and apoptosis [192], as well as a tumor suppressor, a regulator of senescence and differentiation [193], and a guardian of the genome responsible for guiding DNA repair [194]. TP53 gene is expressed as multiple isoforms (with sometimes antagonistic functions) due to alternative splicing, alternative promoter usage and alternative initiation of translation [195]. In one of the comprehensive reviews dedicated to p53 and published in 2016 [196], it was pointed out: “…with its countless biological functions and well-known capability to interact with a myriad of unrelated binding partners, p53 acts as a polyfunctional multibinder, whose functional molecular mechanisms are clearly opposed to the lock-and-key-like functionality described for many globular proteins. This rich functional spectrum of p53 is a reflection of richness and complexity of its structure with multiple proteoforms generated due to the presence of intrinsically disordered regions, numerous posttranslational modifications, and multiple isoforms created by alternative splicing, alternative promoter usage, or alternative initiation of translation, and the ability of p53 to homotetramerize. All these factors play a role in defining the biological multifariousity of this protein. Therefore, for understanding its multifunctionality and roles in carcinogenesis, p53 should be considered through the prism of the proteoform-based protein structure-function continuum, and not in the line of the classical lock-and-key model” [196].
There is a strong link between p53 and pathogenesis of viral hepatitis. For example, p53 is frequently mutated in HCC, which is associated with the HCV and HBV infection [197,198,199,200,201,202,203]. In addition to point mutations, allelic deletions of p53 have been commonly found in human HCCs triggered by hepadnavirus infection [204]. It was also shown that the HBV X protein disrupts the normal p53 function, thereby enhancing the HBV-related carcinogenesis [205,206,207,208,209,210,211,212]. In HCV as well, the core, NS5A, and NS3 proteins were shown to interact with p53 and prevent its correct function [213,214,215,216,217].
In the realm of IDPs, p53 serves as an illustrative example of the importance of intrinsic disorder example for protein functionality and dysfunction [196,218,219]. Figure 16 demonstrate this notion by showing that human TP53 is characterized by highly disordered N- and C-terminal domains, contains multiple MoRFs, numerous PTMs and forms very dense PPI network that includes 470 proteins linked by 5,562 interactions. Most enriched biological processes associated with the members of this network are positive regulation of macromolecule metabolic processes, cellular response to stress, and negative regulation of cellular process. Their most enriched molecular functions are protein binding, transcription factor binding, heterocyclic compound binding, DNA binding, and nucleic acid binding. They are most enriched components of nucleoplasm, nuclear lumen, intracellular organelle lumen, nucleus, intracellular membrane-bounded, organelle, intracellular non-membrane-bounded organelle, chromosome, protein-containing complex, and chromatin. Most enriched disease-gene associations are cancer and disease of cellular proliferation. Most enriched KEGG pathways are pathways in cancer, microRNAs in cancer, p53 signaling pathway, FoxO signaling pathway, cellular senescence, hepatitis B, apoptosis, human T-cell leukemia virus 1 infection, Epstein-Barr virus infection, and viral carcinogenesis.
The predicted high potential of p53 to undergo spontaneous LLPS (pLLPS = 0.9848) is supported by numerous experimental observations reporting engagement of this protein in formation of nuclear biomolecular condensates [220,221,222,223,224,225,226,227,228,229,230,231]. In the norm, such liquid-like condensates can bind to DNA and perform transcriptional activity, whereas cancer-associated p53 mutants form condensates with increased rigidity and impaired DNA binding ability [221].
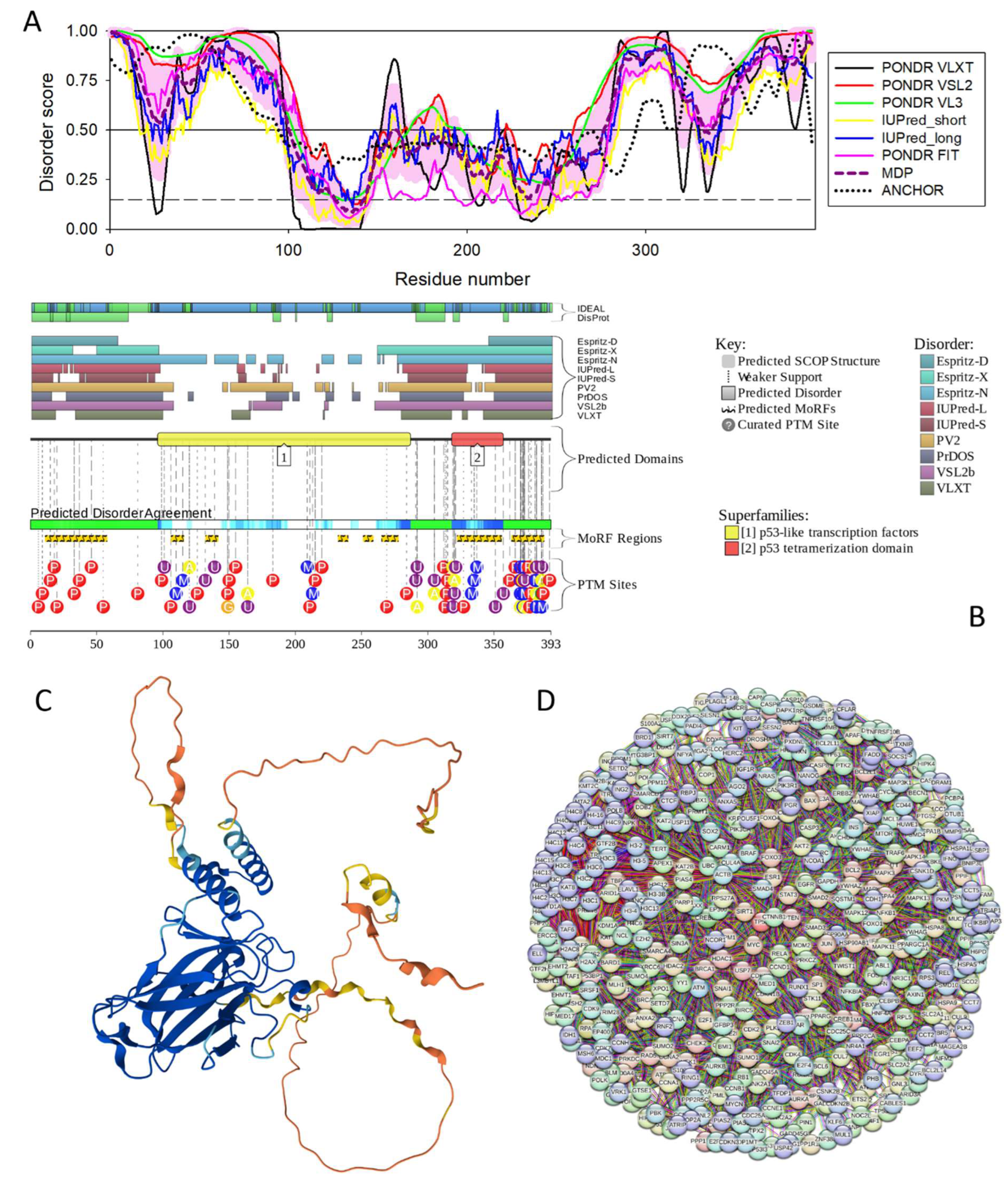
Figure 17.
Functional disorder analysis of human Cellular tumor antigen p53 (TP53, UniProt ID: P04637). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. Here, the IDR localization predicted by IUPred, PONDR® VLXT, PONDR® VSL2, PrDOS, PV2, and ESpritz are shown by 9 differently colored bars on the top of the plot, whereas the blue-green-white bar in the middle of the plot shows the agreement between the outputs of these disorder predictors, with disordered regions by consensus being shown by blue and green. The two lines with colored and numbered bars above the disorder consensus bar show the positions of functional SCOP domains [232,233] predicted using the SUPERFAMILY predictor [234]. Positions of the predicted disorder-based binding sites (MoRF regions) identified by the ANCHOR algorithm are shown by yellow zigzagged bars [235]. Locations of the sites of different posttranslational modifications (PTMs) identified by the PhosphoSitePlus platform [236] are shown at the bottom of the plot by the differently colored circles. C. 3D structural model generated by AlphaFold. D. TP53-centered PPI network generated by STRING using confidence level of 0.815 for minimum required interaction score (permalink https://version-12-0.string-db.org/cgi/network?networkId=b8c6hliT45pI).
Figure 17.
Functional disorder analysis of human Cellular tumor antigen p53 (TP53, UniProt ID: P04637). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. Here, the IDR localization predicted by IUPred, PONDR® VLXT, PONDR® VSL2, PrDOS, PV2, and ESpritz are shown by 9 differently colored bars on the top of the plot, whereas the blue-green-white bar in the middle of the plot shows the agreement between the outputs of these disorder predictors, with disordered regions by consensus being shown by blue and green. The two lines with colored and numbered bars above the disorder consensus bar show the positions of functional SCOP domains [232,233] predicted using the SUPERFAMILY predictor [234]. Positions of the predicted disorder-based binding sites (MoRF regions) identified by the ANCHOR algorithm are shown by yellow zigzagged bars [235]. Locations of the sites of different posttranslational modifications (PTMs) identified by the PhosphoSitePlus platform [236] are shown at the bottom of the plot by the differently colored circles. C. 3D structural model generated by AlphaFold. D. TP53-centered PPI network generated by STRING using confidence level of 0.815 for minimum required interaction score (permalink https://version-12-0.string-db.org/cgi/network?networkId=b8c6hliT45pI).
3.4.4. PR Domain Zinc Finger Protein 10 (PRDM10, UniProt ID: Q9NQV6; PPIDR: 64.34%) Shared by HAV, HBV and HCV
PR domain zinc finger protein 10 (PRDM10, also known as tristanin) is a 1,147-residue long transcriptional activator belonging to a small group of zinc-finger transcription factors known as positive regulatory domain members (PRDM). Members of this family serve as important transcriptional regulators controlling cell fate specification in various developmental contexts. They are responsible for the transduction of signals that control cell proliferation and differentiation and consequently neoplastic transformation [237]. PRDMs are characterized by the presence of the PR/SET domain (PRDI-BF1 and RIZ1 homology domain closely related to the lysine methyltransferase SET (Suppressor of variegation 3–9, Enhancer of zeste and Trithorax) domain) followed by a set of C2H2-type zinc finger repeats mediating the sequence-specific DNA binding, as well as interactions with other protens and RNA [238,239]. Although in humans, there are 17 PRDM genes [240], because of alterative splicing or utilization of alternative promoters, members of the PRDM family are expressed in multiple molecular forms [237,239,240,241]. Often, PRDM members are expressed as PR-plus and PR-minus forms, which are different by the presence or absence of the PR domain and typically have opposite functional roles [242].
Similar to the other PRDM family members, PRDM10 was shown to be mutated in multiple different cancers [242]. Furthermore, PRDM10 mutations are associated with Birt-Hogg-Dube syndrome 2, an autosomal dominant disease characterized by fibrofolliculomas, pulmonary cysts, pneumothoraces, and renal cell carcinomas [243]. Association of PRDM10 with hepatitis is based on the analysis of the aberrant DNA methylation profile associated with HBV infection, where this protein was shown to be important transformation from the normal to chronic infection [244].
In comparison with other members of the PRDM family, this protein remains rather understudied. Human PRDM10 protein contains a degenerated PR/SET domain (residues 208-326), which possibly has lost the methyltransferase activity, followed by 10 C2H2-type zinc finger domains (residues 355-377, 530-552, 560-582, 588-610, 616-639, 644-666, 672-695, 727-750, 772-795, and 834-857). Figure 18A–C show that human PRDM is predicted to contain high level of disorder. Since it has ten C2H2-type zinc finger domains, its cysteine content (28) is high (2.44% of its sequence are cysteine residues, which is almost 2-fold higher than the average cysteine content in UniProt (1.37%)). Since cysteine is considered as one of the strongest order-promoting residues [245,246,247,248], and since its oxidative state can contribute to protein structural stability, we also checked PRDM10 for the presence of redox-sensitive disordered regions (i.e., regions that undergo order-disorder transition at reduction of disulfide bonds). Figure 18B shows that PRDM contains 3 such regions. Although human PRDM10 is predicted to be characterized by high LLPS potential (pLLPS = 0.9912), current literature does not have data demonstrating involvement of this protein (and as a matter of fact of any human PRDM family member) in cellular phase separation processes. However, it was reported that Caenorhabditis elegans protein SET-17 with a PRDM9/7-like SET domain localizes to chromatin-associated foci and promotes spermatocyte gene expression, sperm production and fertility [249]. It is tempting to speculate that the LLPS of PRMD10 can be related to its physiological and pathological functions.
Figure 18C shows that PRDM10 forms a network containing 43 members linked by 117 interactions. Members of this network are involved in histone lysine methylation, peptidyl-lysine modification, histone modification, methylation, peptidyl-lysine trimethylation, rhythmic process. Their most enriched molecular functions are histone-lysine N-methyltransferase activity, methyltransferase activity, ion binding, and metal ion binding. The most enriched disease-gene association of these proteins are urinary bladder cancer, hepatocellular carcinoma, hepatobiliary system cancer, urinary system disease, and endocrine gland cancer. They are most enriched in the following KEGG pathways: mitophagy, ErbB signaling pathway, thyroid hormone signaling pathway, hepatitis b, viral carcinogenesis, kaposi sarcoma-associated herpesvirus infection, lysine degradation, platinum drug resistance, EGFR tyrosine kinase inhibitor resistance, and axon guidance.
3.4.5. T-Box Transcription Factor T (TBXT, UniProt ID: O15178; PPIDR: 60.00%) Shared by HAV, HBV, HCV, and HDV
T-box transcription factor T (TBXT, also known as T or brachyury) is a 435-residue-long transcription factor regulating genes required for mesoderm formation and differentiation. TBXT activates gene transcription when bound to a palindromic T site 5’-TTCACACCTAGGTGTGAA-3’ DNA sequence. TBXT plays crucial role at early stages of embryonic development, being exquisitely orchestrated and controlled to transform the nascent dorsal mesoderm into the notochord, the rudimentary axial skeleton [250,251]. TBXT is believed to define the midline of a bilaterian organism and the establishment of the anterior-posterior axis [252]. In the human fetus, TBXT is silenced at ~ 12 weeks of development [253], and in adults is minimally detected in the pituitary gland, thyroid, and testes [254]. However, TBXT became expressed in several cancers of epithelial origin (lung, prostate and breast cancer), promoting tumor progression and epithelial-mesenchymal transition [255]. Furthermore, TBXT is specifically expressed in chordomas [256,257], which are rare malignant tumors originating from the notochordal remnants in the axial bones [258]. Therefore, it is used as one of the most important markers in the diagnosis of chordoma [257]. Mutations in TBXT are associated with neural tube defects, which are congenital malformations of the central nervous system and adjacent structures related to defective neural tube closure during the first trimester of pregnancy [259], as well as with sacral agenesis with vertebral anomalies (SAVA) [260]. In the interactomes of different hepatitis viruses with the host, TBXT/brachyury interacts with P2C, P3C, P3D-POL, VP2, and VP3 of HAV; HBeAG, HBsAg, capsid, and protein X of HBV; core p19, envelope glycoprotein E2, NS4A, NS4B, NS5A, NS5B/RNA-directed RNA polymerase, and serine protease NS3 of HCV; and S-HDAh and L-HDAg of HDV.
TBXT/brachyury contains DNA-binding domain (residues 42-219), structure of which was resolved (e.g., PDB ID: 8FMU, [261]). Figure 19A–C show that both N- and C-terminal regions of this protein are predicted as disordered. There are 9 MoRFs in human TBXT/brachyury, which also has several phosphorylation sites. In line with the predicted high LLPS potential of this protein (pLLPS = 0.9862), brachyury was shown to form liquid-like transcriptional condensates in nucleus [254].
Figure 19D represents the STRING-generated PPI network centered at human TBXT. This network includes 46 proteins connected by 415 interactions. Among the enriched biological processes associated with the members of this network are regionalization, anterior/posterior pattern specification, embryo development, embryonic morphogenesis, anatomical structure formation involved in morphogenesis, positive regulation of transcription by RNA polymerase II, circulatory system development, gastrulation, cell fate commitment, and embryonic organ development. Their enriched molecular functions are cis-regulatory region sequence-specific DNA binding, RNA polymerase II cis-regulatory region sequence-specific DNA binding, DNA-binding transcription factor, activity, RNA polymerase II-specific, transcription regulator activity, DNA binding, transcription factor binding, RNA polymerase II-specific DNA-binding transcription factor binding, DNA-binding transcription factor binding, DNA-binding transcription activator activity, and RNA polymerase II-specific co-SMAD binding. The enriched cellular components associated with this network are chromosome, chromatin, intracellular non-membrane-bounded organelle, nucleus, transcription regulator complex, RNA polymerase II transcription regulator complex, nucleoplasm, intracellular organelle lumen, activin responsive factor complex, and heteromeric SMAD protein complex. The most enriched disease-gene associations are endocrine system disease, microphthalmia, coloboma, right atrial isomerism, holoprosencephaly, embryonal carcinoma, teratoma, carcinoma, cell type cancer, and syndromic microphthalmia. The most enriched KEGG pathways are signaling pathways regulating pluripotency of stem cells, Hippo signaling pathway, gastric cancer, adherens junction, pathways in cancer, colorectal cancer, hepatocellular carcinoma, TGF-beta signaling pathway, Wnt signaling pathway, and Basal cell carcinoma.
3.4.6. Human Serum Albumin (ALB; UniProt ID: P02768; PPIDR: 32.02%) Shared by HAV, HBV and HCV
Being one of the most studied proteins, human serum albumin (HSA) is the most abundant protein in plasma. It is synthesized in the liver and exported as a single non-glycosylated chain, reaching a blood concentration of about 7×10-4 M. It is a monomeric multi-domain macromolecule, and the main plasma component determining plasma oncotic pressure (also known as colloid osmotic pressure) [262]. Albumin is also the main regulator of fluid distribution between compartments of the body [263,264,265]. It shows a very high ligand binding ability, serving as a depot and carrier for a wide variety of compounds that may be available in quantities well beyond their solubility in plasma. In fact, the physiological importance of HSA stems from the fact that it is involved in bioregulatory and transport phenomena. It binds various metal ions: Ca2+ [266], Cu2+ and Ni2+ [267,268], Zn2+ [269], Mn2+, Mg2+, Co2+, and Cd2+ [270], and many others [262]. HSA takes part in transport and storage of different fatty acids [271,272,273]. It also binds bilirubin [274], steroids [275,276], amino acids [277], and many other ligands, usually with hydrophobic moieties [262,278]. Curiously, in human plasma, about 90% of β-amyloid peptide is carried by HSA [279]. This unique property enables HSA to fulfil a fundamental biological role as a universal carrier and reservoir in blood plasma, tissues, and secretions throughout the mammalian body [280]. Being the main carrier for fatty acids, serum albumin is known to affect pharmacokinetics of numerous drugs [281] and serves as a regulator of the metabolic modification of some ligands. It is a valuable biomarker of many diseases, including cancer, rheumatoid arthritis, and ischemia [282,283,284,285].
In relation to subject of this study, low albumin levels serve as a strong predictor of acute liver failure in patients with acute HAV [286], can be a sign of cirrhosis in HCV patients [287,288], and serve as indicator of higher hepatocellular carcinoma recurrence in HBV patients [289]. Albumin can be incorporated into the HBV coat, likely via the disulfide bonding with the hepatitis B surface antigen [290]. Polymerized HSA may play a role in the infection of hepatocytes by hepatitis B virus, as HBsAg particles contain specific binding sites for transglutaminase-cross-linked HSA, suggesting that the albumin polymers may play a role in the attachment of HBV to hepatocytes [291].
Although HSA contains disordered or structurally flexible regions (see Figure 20A,B), which are heavily decorated by various PTMs (Figure 20B), it has a well-defined structure (see Figure 20C). Predicted levels of intrinsic disorder in this protein (30.2%) correlate well with the content of random coil structure in this protein (29.1%) evaluated by Fourier-transform infrared (FTIR) spectroscopy [292]. Prevalence and functionality of intrinsically disordered regions in serum albumins was a subject of a dedicated study [293]. Among various other observations, it was indicated that structural flexibility is required for interaction of serum albumins with their partners [293]. Although HSA cannot undergo LLPS spontaneously (its pLLPS of 0.1403 is too low), it still can act as a droplet client, since it contains one disorder-promoting region (DRP, residues 210-220).
In line with these considerations, Figure 20D shows that human serum albumin forms a densely connected PPI network, where 241 partners are linked by 3,009 edges. Multifunctionality of the members of this network is reflected in the fact that their most enriched molecular functions include signaling receptor binding, molecular function regulator activity, signaling receptor regulator activity, signaling receptor activator activity, receptor ligand activity, growth factor activity, protein-containing complex binding, protein binding, glycosaminoglycan binding, and growth factor receptor binding. Their most enriched biological processes are response to stimulus, response to stress, regulation of multicellular organismal process, cellular response to chemical stimulus, response to chemical, response to organic substance, biological process involved in interspecies interaction between organisms, regulation of cell population proliferation, defense response, and positive regulation of protein phosphorylation. Among enriched cellular components of these proteins are extracellular space, extracellular region, blood microparticle, extracellular vesicle, extracellular exosome, vesicle, cell surface, secretory granule, platelet alpha granule, and collagen-containing extracellular matrix. The most enriched disease-gene associations of these proteins are disease of anatomical entity, vascular disease, cardiovascular system disease, hematopoietic system disease, respiratory system disease, artery disease, disease by infectious agent, immune system disease and urinary system disease. The most enriched KEGG pathways linked to these proteins are pathways in cancer, PI3K-Akt signaling pathway, Rap1 signaling pathway, melanoma, gastric cancer, tuberculosis, complement and coagulation cascades, MAPK signaling pathway, RAS signaling pathway, and malaria.
3.4.7. T-Cell Surface Glycoprotein CD8 Alpha Chain (CD8A; UniProt ID: P01732; PPIDR: 26.38%) Shared by HAV, HBV and HCV
T-cell surface glycoprotein CD8 (cluster of differentiation 8) alpha chain (CD8A also known as T-lymphocyte differentiation antigen T8/Leu-2) is a 235-residue-long transmembrane protein on the surface of T cells with a crucial role in the immune response by helping T cells to response to pathogens [294]. CD8 acts as a co-receptor for the T-cell receptor. CD8 exists as a homo- or heterodimer of CD8 alpha and beta chains (CD8A and CDB, respectively) encoded by different genes, with three different dimers, CD8AA, CD8AB and CD8BB being reported [295]. CD8A, being primarily expressed on the surface of cytotoxic T cells (in fact, CD8 is a marker for cytotoxic T cell population), can also be found on cortical thymocytes, dendritic cells, and natural killer cells. This protein is responsible for the cell-mediated immune defense and T-cell development [296,297], also acts as a co-receptor for MHC class I molecule:peptide complex [298]. CD8A can serve as a diagnostic and prognosis marker for several diseases, such as inflammatory disorders and tumors, including chronic rhinosinusitis [299], rheumatoid arthritis [300], lung adenocarcinoma [301], and was proposed as a biomarker associated with immunocytes infiltration in hyperoxia-induced bronchopulmonary dysplasia [302]. Furthermore, the CD8A expression levels are positively correlated with the degree of liver inflammation and fibrosis [303], and CD8A was proposed to be used as a potential diagnostic biomarker for HBV-induced liver fibrosis in chronic hepatitis B patients [304].
Being a type I membrane protein, CD8A has four major modules, an N-terminal extracellular immunoglobulin variable (IgV)-like domain (residues 22–135), a highly dynamic proline-rich stalk region known as a hinge region (residues 136-182), a single transmembrane helix (residues 183–203), and a small cytoplasmic region (residues 204–235). IgV-like domain and hinge region together constitute an ectodomain. Structurally, the extracellular IgV-like domain is characterized by a typical IgV fold containing two β-sheets (e.g., PDB ID: 8EW6, [305]). Figure 21A–D show that the extracellular IgV-like domain and the transmembrane region of human CD8A are predicted as mostly ordered, whereas its hinge/stalk region and cytoplasmic C-tail are both expected to be disordered. In line with these observations, based on the results of NMR analysis it was reported that the hinge/stalk of human CD8A represents an intrinsically disordered region characterized by a dynamic exchange that includes proline cis-trans isomerization [306]. Furthermore, CD8A contains several important cysteine residues playing essential structural and functional roles. Since this protein contains 9 cysteine residues, its cysteine content of 3.83% almost 3-fold exceed that of UniProt (1.37%). Therefore, it is expected that it might possess some redox-sensitive disordered regions. Validity of this assumption is illustrated by Figure 21B showing that the C-tail of human CD8A indeed represents a redox-sensitive region. Curiously, this tail also contain phosphorylation sites. Furthermore, a flexible region located within the IgV-like domain (residues 74-88, see Figure 21A–C) corresponds to the functionally important CDR2 loop, which is responsible for interaction with MHC-I, and whose structure was not resolved (it is a region with missing electron density in PDB ID 8EW6 [305]). With pLLPS of 0.2472 and with presence of one DPR (residues 135-155) that is included into the hinge/stalk region, human CD8A can serve as a droplet-driver.
Figure 21E represents the STRING-generated PPI network centered at human CD8A that includes 263 partners connected by 5,362 interactions. Among the most enriched biological processes of the members of this network are immune system process, immune response, regulation of immune system process, positive regulation of immune system process, leukocyte activation, cell activation, regulation of leukocyte activation, regulation of cell activation, regulation of lymphocyte activation and defense response. Some of their most enriched molecular functions are signaling receptor binding, signaling receptor activity, cytokine receptor binding, cytokine activity, immune receptor activity, protein binding, transmembrane signaling receptor activity, cytokine receptor activity, antigen binding, and cytokine binding.
These proteins are most enriched in the following cellular components: cell surface, external side of plasma membrane, side of membrane, plasma membrane, cell periphery, intrinsic component of membrane, integral component of membrane, intrinsic component of plasma membrane, integral component of plasma membrane, and extracellular region. Their most enriched disease-gene associations are immune system disease, primary immunodeficiency disease, autoimmune disease, autoimmune disease of musculoskeletal system, lymphatic system disease, hematopoietic system disease, disease of anatomical entity, hematologic cancer, disease by infectious agent, and organ system cancer. Most enriched KEGG pathways associated with these proteins are cytokine-cytokine receptor interaction, viral protein interaction with cytokine and cytokine receptor, hematopoietic cell lineage, natural killer cell mediated cytotoxicity, cell adhesion molecules, inflammatory bowel disease, JAK-STAT signaling pathway, Epstein-Barr virus infection, Th17 cell differentiation, and measles.
3.4.8. α-Fetoprotein (AFP; UniProt ID: P02771; PPIDR: 20.53%) Shared by HAV, HBV and HCV
α-Fetoprotein (AFP), a 590-residue-long glycoprotein (contains 4.5% carbohydrates), being a major protein of the embryonic plasma [307,308,309] produced by the yolk sac and the fetal liver during fetal development and showing high sequence and structural similarity to HSA [310,311], is commonly considered as an embryonic albumin or embryonic α-globulin. In fact, similar to HSA, AFP serves as a major carrier of bilirubin, dioxin, dyes, fatty acids, flavonoids, heavy metals, phytoestrogens, retinoids, steroids, and various drugs [310,311,312,313]. Although in fetus, AFP is present at a high concentration of 1-10 mg/ml, its content decreases abruptly soon after the birth, as only a trace amount of this protein (typically <10 ng/mL) can be detected by the end of second month postpartum, and AFP is replaced by HSA as the major serum protein [307,309,310,314]. However, being an oncofetal protein, AFP reemerges under several pathological conditions, and increased levels of this protein in the plasma of adults is the hallmark of the development of hepatocellular carcinoma (HCC) and other malignant tumors [315,316,317,318] including germ cell tumors of the testis [319], acinar cell carcinoma of the pancreas [320], intracranial yolk sac tumor [321], and many others. Furthermore, variation in the AFP levels during pregnancy are related to various fetal abnormalities, such as Down’s syndrome, as well as open neural tube defects, such as spina bifida [310,322,323,324,325]. Decrease in serum AFP levels serves as a predictor of poor prognosis of acute hepatic failure in patients with chronic hepatitis B [326]. Importantly, changes in the AFP levels have intricate connections to HCV and HBV infections as well. In fact, AFP levels were shown to be elevated in some patients with chronic viral hepatitis and cirrhosis who do not have HCC [327,328,329,330,331].
Figure 22A–C show that human AFP is a mostly ordered protein that possesses several disordered/flexible regions. This protein was shown to be characterized by the extremely high conformational stability, which was attributed to the large number of disulfide bonds (this protein, being 590 amino acid residues long, contains 15 S–S bridges) [311]. Therefore, it is not surprising that it has 4 redox-sensitive disordered regions (see Figure 22B). AFP has several PTM sites. In addition to shown in Figure 22C three phosphorylation and acetylation sites, AFP can be phosphorylated at Ser111, Ser115, Ser117, Ser344, Ser444, and Ser445 by FAM20C, which is a kinase that phosphorylates S-x-E/pS motifs in the majority of secreted proteins [332]. Furthermore, AFP is glycosylated at Asn251 [333,334], and is characterized by molecular microheterogeneity determined by differences in the attached carbohydrate moiety [335,336]
Despite its obvious functional and diagnostic importance and its remarkable similarity to HSA, no structural information about AFP was available until quite recently. In fact, the first 3D structure of this protein resolved by cryo-electron microscopy (cryo-EM) was reported in 2023 (PDB ID: 7YIM, [337]) (see Figure 22D). AFP is characterized by the V-like (or a heart-like) shape with three well-defined domains (domains I, II, and III), with each domain consisting of ~195 residues with 4, 5, and 6 disulfide bonds being positioned in domains I, II and II, respectively [338]. Since human AFP does not have DPRs and shows low pLLPS of 0.1510, this protein is not related to the cellular LLPS processes.
Besides being a carrier, AFP has several other functions, possessing immunosuppressive activity and playing a role in regulation of cell proliferation [313], and, therefore, can interact with multiple partner proteins. This is illustrated by Figure 22E presenting a PPI network centered at AFP containing 239 proteins linked by 6,092 interactions. This network is characterized by the average node degree of 51. Members of this network are engaged in various biological processes, such as response to stimulus, response to organic substance, cellular response to chemical stimulus, response to chemical, regulation of multicellular organismal process, animal organ development, negative regulation of biological process, multicellular organismal process, positive regulation of metabolic process, and positive regulation of macromolecule metabolic process. Their most enriched molecular functions are protein binding, signaling receptor binding, identical protein binding, binding, signaling receptor regulator activity, signaling receptor activator activity, receptor ligand activity, growth factor activity, molecular function regulator activity, and protein-containing complex binding. They are enriched within the following cellular components: extracellular region, extracellular space, blood microparticle, extracellular exosome, cell surface, vesicle, platelet alpha granule lumen, platelet alpha granule, endoplasmic reticulum lumen, and vesicle lumen. Among the most enriched disease-gene associations of the members of this network are disease of cellular proliferation, cancer, organ system cancer, cell type cancer, hepatobiliary disease, gastrointestinal system disease, disease of anatomical entity, carcinoma, reproductive system disease, gastrointestinal system cancer, reproductive organ cancer, endocrine gland cancer, female reproductive system disease, and liver disease. These proteins are most enriched in the following KEGG pathways: pathways in cancer, PI3K-Akt signaling pathway, proteoglycans in cancer, gastric cancer, melanoma, breast cancer, MAPK signaling pathway, colorectal cancer, FoxO signaling pathway, maturity onset diabetes of the young, focal adhesion, hepatitis B, EGFR tyrosine kinase inhibitor resistance, AGE-RAGE signaling pathway in diabetic complications, and toxoplasmosis. The most enriched Reactome pathways associated with these proteins are regulation of insulin-like growth factor (IGF) transport and uptake by insulin-like growth factor binding proteins (IFGBPs), post-translational protein phosphorylation, hemostasis, platelet degranulation, platelet activation, signaling and aggregation, interleukin-4 and Interleukin-13 signaling, cell surface interactions at the vascular wall, immune system, metabolism of proteins, signaling by receptor tyrosine kinases, developmental biology, diseases of signal transduction by growth factor receptors and second messengers, cytokine signaling in immune system, negative regulation of the PI3K/AKT network, and extracellular matrix organization.
3.4.9. Proteasome Subunit Beta Type-7 (PSMB7; UniProt ID: Q99436; PPIDR: 17.33%) Shared by HAV, HBV and HCV
Proteasome subunit beta type-7 (PSMB7, also known as macropain chain Z, multicatalytic endopeptidase complex chain Z, proteasome subunit Z, and proteasome subunit beta-2) is a 277-residue-long component of the 20S core proteasome complex possessing a trypsin-like activity. PSMB7, being a member of the proteasome B-type family, also known as the T1B family, is one the essential subunits of the 20S core proteasome, which is composed of four rings of 28 non-identical subunits, with two rings being composed of seven alpha subunits (alpha-1 to -7) and two rings being composed of seven beta subunits (beta-1 to -7). It was reported that PSMB7 can be phosphorylated, and phosphorylation/dephosphorylation of this subunit can be related to the regulation of the adjacent beta-1 subunit [339], which interacts with PSMB7 and has the peptidylglutamyl-peptide hydrolyzing (PGPH) activity [340]. It was reported also that the levels of the phosphorylated PSMB7 were noticeable reduced in the proteasomes isolated from tumor cells as compared to normal cells lines [339]. PSMB7 is overexpressed in colorectal carcinomas [341], with elevated levels of both PSMB7 protein as well as PSMB7 mRNA showing associations with lower overall survival [342]. In breast cancer, overexpression of the PSMB7 gene was shown to be associated with poor prognostic outcome and with anthracycline resistance [343]. PSMB7 was one of the differently expressed proteins in an experimental mesangial proliferative nephritis model [344]. In multiple myeloma, PSMB7 was one of the strongest bortezomib sensitizers; i.e., genes, silencing of which is not directly cytotoxic but synergistically potentiate the growth inhibitory effects of bortezomib [345]. Furthermore, PSMB7 was recognized as a key gene involved in the development of multiple myeloma and resistance to bortezomib [346]. In HCC, PSMB7 was found as one of the proteins involved in 5-fluorouracil resistance [347]. Based on the transcriptomic analyses of thousands of samples from The Cancer Genome Atlas it was concluded that the PSMB7 expression is increased in most cancer types [348]. In Alzheimer’s disease (AD), expression of PSMB7 was shown to be significantly decreased compared to the control group, with more noticeable decrease being associated with the high Braak stages [349]. Inherited and/or de novo loss-of-function mutations in the PSMB7 gene are linked to proteasome-associated autoinflammatory syndromes (PRAAS) [350].
Analysis of the mechanisms behind the interferon lambda (IFN-λ)-induced inhibition of the HBV replication in HepG2.2.15 cells revealed that PSMB7 was one of the proteins up-regulated by IFN-λ3 treatment [351]. Furthermore, hepatitis B virus X protein was shown to interact with PSMB7, with this interaction being functionally important in the pleiotropic effect of this viral protein [352,353,354], and affecting HBV replication through the proteasome-dependent pathway [355]. PSMB7 is one of the proteins involved in interaction with the HCV proteins [356].
Figure 23 shows that although human PSMB7 is expected to be mostly ordered, it has several disordered or flexible regions, including N- and C-terminal regions. In agreement with these observations, in a cryo-EM resolved structures of 26S proteasome (e.g., PDB ID: 5LN3 [357]; 5L4G [358]) two regions of PSMB7 corresponding to its N- and C-termini (residues 1-43 and 264-277) were unmodeled, indicating their highly flexible nature. Human PSMB7 has multiple phosphorylation and ubiquitination sites (Figure 23B). With low pLLPS of 0.1497 and lack of DPRs, human PSMB7 is not expected to be related to the cellular LLPS processes.
Figure 23D shows that human PSMB7 forms a very dense PPI network with average node degree of 97.1 that contains 295 proteins connected via 14,323 interactions. Members of this network are most enriched in biological processes, such as proteolysis involved in protein catabolic process, protein catabolic process, modification-dependent protein catabolic process, proteasomal protein catabolic process, ubiquitin-dependent protein catabolic process, proteasome-mediated ubiquitin-dependent protein catabolic process, macromolecule catabolic process, cellular macromolecule catabolic process, proteolysis, and cellular macromolecule metabolic process. Their most enriched molecular functions are ubiquitin-like protein ligase binding, ubiquitin protein ligase binding, enzyme binding, protein binding, structural constituent of ribosome, proteasome binding, kinase binding, protein kinase binding, threonine-type endopeptidase activity, and metal-dependent deubiquitinase activity. Most enriched disease-gene associations of these proteins are nervous system cancer, retinal cancer, sensory system cancer, cancer, infratentorial cancer, medulloblastoma, brain cancer, central nervous system cancer, basal cell carcinoma, and organ system cancer. These proteins are most enriched in the following KEGG pathways: proteasome, ubiquitin mediated proteolysis, Epstein-Barr virus infection, Parkinson disease, spinocerebellar ataxia, prion disease, amyotrophic lateral sclerosis, Alzheimer disease, cell cycle, and pathways in cancer.
3.4.10. RAB GDP Dissociation Inhibitor Alpha (GDI1; UniProt ID: P31150; PPIDR: 17.23%) Shared by HAV, HBV, HCV, and HDV
Guanosine diphosphate dissociation inhibitor 1 (GDI1, also known as RAB GDP dissociation inhibitor alpha, RAB GDI alpha, oligophrenin-2, and protein XAP-4) is a 447-residue-long regulator of GDP/GTP exchange if most RAS-related proteins in brain (RAB proteins). RAB proteins constitute a family of small GTPases (G proteins) with a number of crucial roles in regulating intracellular membrane trafficking in eukaryotic cells [359]. GD11 inhibits the GDP dissociation from and GTP binding to RAB proteins. It can promote dissociation of RAB1A, RAB3A, RAB5A, and RAB10 from membranes [360]. GDI1 is one of the proteins involved in nonspecific X-linked mental retardation [361]. Mutations in GDI1 can disturb vesicle trafficking and thereby act as one of the causes of the synaptic vesicle cycling (SVC) disorders [362]. Other pathological conditions associated with distorted GDI1 include Alzheimer’s disease [363], colorectal cancer [364], isolated adrenocorticotropin deficiency (IAD) [365], and oligodendrogliomas [366]. In HCC, loss of GDI1 was shown to serve as a prognostic factor, being closely related to long-term survival of patients with HCC [367,368]. In patients with chronic HCV infection, GDI1 was one of the proteins whose levels were increased in the circulating extracellular vesicles (EV) in comparison to the EVs from healthy donors [369].
No actual structural information is currently available for human GDI1. Figure 24A–C show that it is predicted as mostly ordered protein with several disordered segments. Despite its mostly ordered nature, GDI1 has multiple PTM sites (see Figure 24B). This protein is not related to cellular LLPS processes, since it does not have DPRs and shows pLLPS of 0.1538. In GDI1-centered PPI network, there are 163 protein connected by 1,736 interactions. Members of this network are entiched in various biological processes, such as vesicle-mediated transport, cellular localization, intracellular transport, establishment of localization in cell, establishment of localization, transport, localization, protein transport, establishment of protein localization, and nitrogen compound transport. Their most enriched molecular function are GTPase activity, GTP binding, nucleoside-triphosphatase activity, purine ribonucleoside triphosphate binding, purine ribonucleotide binding, nucleotide binding, guanyl-nucleotide exchange factor activity, GTPase regulator activity, GDP binding, and small GTPase binding. The most enriched cellular components associated with these proteins are cytoplasmic vesicle, vesicle, endosome, cytosol, Golgi apparatus, endomembrane system, endocytic vesicle, bounding membrane of organelle, cytoplasmic vesicle membrane, and TRAPP complex. Endocytosis, salmonella infection, vasopressin-regulated water reabsorption, bacterial invasion of epithelial cells, synaptic vesicle cycle, AMPK signaling pathway, tuberculosis, and autophagy are among the most enriched KEGG pathways associated with these proteins. Some of the most enriched reactome pathways linked to these proteins are RAB regulation of trafficking, RAB GEFs exchange GTP for GDP on RABs, membrane trafficking, RAB geranylgeranylation, post-translational protein modification, TBC/RABGAPs, COPII-mediated vesicle transport, intra-Golgi and retrograde Golgi-to-ER traffic, retrograde transport at the trans-Golgi-network, and intra-Golgi traffic. Some of the most enriched disease-gene associations are X-linked monogenic disease, intellectual disability, developmental disorder of mental health, disease of mental health, non-syndromic X-linked intellectual disability, Warburg micro syndrome 1, Warburg micro syndrome 2, Warburg micro syndrome 3, Martsolf syndrome, and Rett syndrome.
3.4.11. Galactoside Alpha-(1,2)-Fucosyltransferase 1 (FUT1; UniProt ID: P19526; PPIDR: 10.14%) Shared by HAV, HBV, HCV, and HDV
Galactoside alpha-(1,2)-fucosyltransferase 1 (FUT1, also known as alpha(1,2)FT 1, blood group H alpha 2-fucosyltransferase, fucosyltransferase 1, GDP-L-fucose:beta-D-galactoside 2-alpha-L-fucosyltransferase 1, and type 1 galactoside alpha-(1,2)-fucosyltransferase FUT1) is a 465-residue-long enzyme catalyzing the L-fucose transfer from a guanosine diphosphate-beta-L-fucose to the terminal galactose residue of glycoconjugates through an alpha(1,2) linkage [370]. This results in the synthesis of the H antigen, a precursor to the A and B antigens, which determine a person’s ABO blood type [370]. Several mutations in FUT1 are linked to para-Bombay phenotype, which is a rare blood type that occurs when red blood cells lack the H antigen [371,372].
Changes in FUT expression were shown to have a number of significant roles in cancer development and malignancy [373], with FUT1 being highly expressed in various types of cancers [374]. For example, in ovarian carcinoma cells, overexpression of FUT1 positively regulates 215 genes [375]. In breast cancer, FUT1-driven fucosylation of the lysosome-associated membrane proteins 1 and 2 (LAMP-1 and LAMP-2) leads to the formation of the alpha1,2-fucosylated Lewis Y (LeY) antigens, whose levels were substantial in less invasive ER+/PR+/HER- breast cancer cells (MCF-7 and T47D) but negligible in highly invasive triple-negative MDA-MB-231 cells [374]. FUT1 downregulation resulted in the perinuclear localization of LAMP-1 and LAMP-2 and correlated with the increased rate of autophagic flux by decreasing mTOR signaling and increasing autolysosome formation [374].
High FUT1 expression was significantly associated with HCC advanced stages and poor outcome [376]. FUT1, being highly upregulated in acute HCV infection, was shown to regulate and facilitate HCV replication in hepatocytes [377]. HCV infection increased the expression of fucosylated epitopes, resulting in the reorganization of the glycocalyx (which is a thin, carbohydrate-rich layer covering the surface of epithelial and endothelial cells) that favors the spread of viral progeny and alters various biological functions mediated by glycans, including recognition by the adaptive immune system [378]. In HBV-associated HCC, upregulation of FUT1, 4, and 8 resulted in elevated levels of fucosylated N-glycans as evidenced by integrating transcriptomics, glycomics, and glycoproteomics [379].
Despite its physiological and pathological importance, no structural information is currently available for human FUT1. Figure 25 shows that this protein is predicted to be mostly ordered with several disordered/flexible regions. Being a single-pass type II membrane protein, FUT1 has a transmembrane helical region (residues 9-25), which, together with the most N-terminal positively charged residues forms a signal-anchor domain. Based on the evaluation of its LLPS potential, FUT1 can serve as a droplet-client, since with low pLLPS of 0.2215, it possess one DPR (residues 58-69).
FUT1-centered PPI network contains 53 members connected by 491 interactions. Most enriched biological processes associated with the members of this network are protein glycosylation, carbohydrate derivative biosynthetic process, organonitrogen compound biosynthetic process, cellular biosynthetic process, organic substance biosynthetic process, protein O-linked glycosylation, carbohydrate metabolic process, oligosaccharide biosynthetic process, carbohydrate biosynthetic process, and fucosylation. Their most enriched molecular functions are glycosyltransferase activity, hexosyltransferase activity, transferase activity, and catalytic activity. The cellular components most enriched in these proteins are Golgi membrane, Golgi apparatus, organelle membrane, endomembrane system, integral component of membrane, cytoplasm, Golgi cisterna membrane, Golgi cisterna, Golgi apparatus subcompartment, and organelle subcompartment. Among the most enriched reactome pathways associated with these proteins are blood group systems biosynthesis, Lewis blood group biosynthesis, metabolism of carbohydrates, metabolism, O-linked glycosylation of mucins, O-linked glycosylation, keratan sulfate biosynthesis, post-translational protein modification, asparagine N-linked glycosylation, and N-glycan antennae elongation in the medial/trans-Golgi.
4. Conclusions
The bioinformatics analysis performed on the proteins of different hepatitis viruses and on the host proteins interacting with different hepatitis viruses provided important observations regarding the roles of intrinsic disorder in the life cycle of hepatitis viruses. Many of the viral and host proteins were predicted to be related to cellular LLPS processes, serving either as droplet drivers or droplet clients. These observations suggest that phase separation and formation of biomolecular condensates are important for the viral replication and fight with host immunity on one side and for the host response to the viral infection on the other side.
Host proteins interacting with hepatotropic viruses were shown to be characterized by noticeable variation in their intrinsic disorder status. Although global disorder distribution within the sets of host proteins interacting with hepatitis viruses was not too different from that of the entire human proteome, more host proteins interacting with hepatitis viruses were predicted as moderately disordered in comparison with the entire human proteome.
Analysis of the host interactomes of HAV, HBV, HCV, HDV, and HEV revealed the presence of 33 shared proteins. Interestingly, 32 of these shared host proteins are found in the HBV and HCV interactomes, whereas interactomes of HAV, HDV, and HEV contains 11, 5, and 1 shared host proteins, respectively. All the host proteins shared by several hepatotropic viruses were predicted as noticeably disordered.
Detailed bioinformatics analysis of the 11 host proteins shared by four or three different hepatitis viruses indicated that intrinsic disorder can play several important roles in functionality of these proteins. It is also very likely that intrinsic disorder of the host proteins shared by different hepatotropic viruses can be related to the hepatitis co-infection, joint pathogenicity of co-infecting viruses, modulating their mechanisms of immune evasion, promoting the development of antiviral drug resistance and, thereby, contributing to the virus adaptability and evolution.
Author Contributions
Conceptualization, V.N.U.; methodology, V.N.U.; validation, E.M.R., A.A.A., V.N.U.; formal analysis, V.N.U.; investigation, V.N.U.; data curation, E.M.R., A.A.A., V.N.U.; writing—original draft preparation, V.N.U.; writing—review and editing, E.M.R., A.A.A., V.N.U.; visualization, V.N.U.; project administration, E.M.R., A.A.A., V.N.U.; funding acquisition, E.M.R., A.A.A., V.N.U. All authors have read and agreed to the published version of the manuscript.
Funding
The authors extend their appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia, for funding this research work through project number 838.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Cao, G.; Jing, W.; Liu, J.; Liu, M. The global trends and regional differences in incidence and mortality of hepatitis A from 1990 to 2019 and implications for its prevention. Hepatol Int 2021, 15, 1068-1082. [CrossRef]
- Linder, K.A.; Malani, P.N. Hepatitis A. JAMA 2017, 318, 2393. [CrossRef]
- Gish, R.G. Current treatment and future directions in the management of chronic hepatitis B viral infection. Clin Liver Dis 2005, 9, 541-565, v. [CrossRef]
- Akbar, F.; Yoshida, O.; Abe, M.; Hiasa, Y.; Onji, M. Engineering immune therapy against hepatitis B virus. Hepatol Res 2007, 37 Suppl 3, S351-356. [CrossRef]
- Global Burden Of Hepatitis, C.W.G. Global burden of disease (GBD) for hepatitis C. J Clin Pharmacol 2004, 44, 20-29. [CrossRef]
- Shepard, C.W.; Finelli, L.; Fiore, A.E.; Bell, B.P. Epidemiology of hepatitis B and hepatitis B virus infection in United States children. Pediatr Infect Dis J 2005, 24, 755-760. [CrossRef]
- Frank, C.; Mohamed, M.K.; Strickland, G.T.; Lavanchy, D.; Arthur, R.R.; Magder, L.S.; El Khoby, T.; Abdel-Wahab, Y.; Aly Ohn, E.S.; Anwar, W.; et al. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet 2000, 355, 887-891. [CrossRef]
- Alter, M.J. Epidemiology of hepatitis C virus infection. World J Gastroenterol 2007, 13, 2436-2441. [CrossRef]
- Heidrich, B.; Manns, M.P.; Wedemeyer, H. Treatment options for hepatitis delta virus infection. Curr Infect Dis Rep 2013, 15, 31-38. [CrossRef]
- Reinheimer, C.; Doerr, H.W.; Berger, A. Hepatitis delta: on soft paws across Germany. Infection 2012, 40, 621-625. [CrossRef]
- Cunha, C.; Tavanez, J.P.; Gudima, S. Hepatitis delta virus: A fascinating and neglected pathogen. World J Virol 2015, 4, 313-322. [CrossRef]
- Li, P.; Liu, J.; Li, Y.; Su, J.; Ma, Z.; Bramer, W.M.; Cao, W.; de Man, R.A.; Peppelenbosch, M.P.; Pan, Q. The global epidemiology of hepatitis E virus infection: A systematic review and meta-analysis. Liver Int 2020, 40, 1516-1528. [CrossRef]
- Migueres, M.; Lhomme, S.; Izopet, J. Hepatitis A: Epidemiology, High-Risk Groups, Prevention and Research on Antiviral Treatment. Viruses 2021, 13. [CrossRef]
- Villena, E.Z. Module lTransmission routes of hepatitis C virus infection. Annals of Hepatology 2006, 5, 12-14. [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479-480, 672-686. [CrossRef]
- Recommendations for prevention and control of hepatitis C virus (HCV) infection and HCV-related chronic disease. Centers for Disease Control and Prevention. MMWR Recomm Rep 1998, 47, 1-39.
- Rizzetto, M.; Hoyer, B.; Canese, M.G.; Shih, J.W.; Purcell, R.H.; Gerin, J.L. delta Agent: association of delta antigen with hepatitis B surface antigen and RNA in serum of delta-infected chimpanzees. Proc Natl Acad Sci U S A 1980, 77, 6124-6128. [CrossRef]
- Ponzetto, A.; Negro, F.; Popper, H.; Bonino, F.; Engle, R.; Rizzetto, M.; Purcell, R.H.; Gerin, J.L. Serial passage of hepatitis delta virus in chronic hepatitis B virus carrier chimpanzees. Hepatology 1988, 8, 1655-1661. [CrossRef]
- Niro, G.A.; Casey, J.L.; Gravinese, E.; Garrubba, M.; Conoscitore, P.; Sagnelli, E.; Durazzo, M.; Caporaso, N.; Perri, F.; Leandro, G.; et al. Intrafamilial transmission of hepatitis delta virus: molecular evidence. J Hepatol 1999, 30, 564-569. [CrossRef]
- Rizzetto, M.; Verme, G.; Recchia, S.; Bonino, F.; Farci, P.; Arico, S.; Calzia, R.; Picciotto, A.; Colombo, M.; Popper, H. Chronic hepatitis in carriers of hepatitis B surface antigen, with intrahepatic expression of the delta antigen. An active and progressive disease unresponsive to immunosuppressive treatment. Ann Intern Med 1983, 98, 437-441. [CrossRef]
- Fattovich, G.; Giustina, G.; Christensen, E.; Pantalena, M.; Zagni, I.; Realdi, G.; Schalm, S.W. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep). Gut 2000, 46, 420-426. [CrossRef]
- Khuroo, M.S.; Kamili, S. Aetiology, clinical course and outcome of sporadic acute viral hepatitis in pregnancy. J Viral Hepat 2003, 10, 61-69. [CrossRef]
- Koff, R.S. Risks associated with hepatitis A and hepatitis B in patients with hepatitis C. J Clin Gastroenterol 2001, 33, 20-26. [CrossRef]
- Kramer, J.R.; Hachem, C.Y.; Kanwal, F.; Mei, M.; El-Serag, H.B. Meeting vaccination quality measures for hepatitis A and B virus in patients with chronic hepatitis C infection. Hepatology 2011, 53, 42-52. [CrossRef]
- Syed, N.A.; Hearing, S.D.; Shaw, I.S.; Probert, C.S.; Brooklyn, T.N.; Caul, E.O.; Barry, R.E.; Sarangi, J. Outbreak of hepatitis A in the injecting drug user and homeless populations in Bristol: control by a targeted vaccination programme and possible parenteral transmission. Eur J Gastroenterol Hepatol 2003, 15, 901-906. [CrossRef]
- Vento, S. Fulminant hepatitis associated with hepatitis A virus superinfection in patients with chronic hepatitis C. J Viral Hepat 2000, 7 Suppl 1, 7-8. [CrossRef]
- Mavilia, M.G.; Wu, G.Y. HBV-HCV Coinfection: Viral Interactions, Management, and Viral Reactivation. J Clin Transl Hepatol 2018, 6, 296-305. [CrossRef]
- Maqsood, Q.; Sumrin, A.; Iqbal, M.; Younas, S.; Hussain, N.; Mahnoor, M.; Wajid, A. Hepatitis C virus/Hepatitis B virus coinfection: Current prospectives. Antivir Ther 2023, 28, 13596535231189643. [CrossRef]
- Weakley, T.; Rajender Reddy, K. Hepatitis B. Curr Treat Options Gastroenterol 1999, 2, 463-472. [CrossRef]
- Weltman, M.D.; Brotodihardjo, A.; Crewe, E.B.; Farrell, G.C.; Bilous, M.; Grierson, J.M.; Liddle, C. Coinfection with hepatitis B and C or B, C and delta viruses results in severe chronic liver disease and responds poorly to interferon-alpha treatment. J Viral Hepat 1995, 2, 39-45. [CrossRef]
- Jardi, R.; Rodriguez, F.; Buti, M.; Costa, X.; Cotrina, M.; Galimany, R.; Esteban, R.; Guardia, J. Role of hepatitis B, C, and D viruses in dual and triple infection: influence of viral genotypes and hepatitis B precore and basal core promoter mutations on viral replicative interference. Hepatology 2001, 34, 404-410. [CrossRef]
- Bumbea, H.; Vladareanu, A.M.; Vintilescu, A.; Radesi, S.; Ciufu, C.; Onisai, M.; Baluta, C.; Begu, M.; Dobrea, C.; Arama, V.; et al. The lymphocyte immunophenotypical pattern in chronic lymphocytic leukemia associated with hepatitis viral infections. J Med Life 2011, 4, 256-263.
- Gozlan, J.; Lacombe, K.; Gault, E.; Raguin, G.; Girard, P.M. Complete cure of HBV-HDV co-infection after 24weeks of combination therapy with pegylated interferon and ribavirin in a patient co-infected with HBV/HCV/HDV/HIV. J Hepatol 2009, 50, 432-434. [CrossRef]
- Grabowski, J.; Wedemeyer, H. Hepatitis delta: immunopathogenesis and clinical challenges. Dig Dis 2010, 28, 133-138. [CrossRef]
- Lacombe, K.; Boyd, A.; Desvarieux, M.; Serfaty, L.; Bonnord, P.; Gozlan, J.; Molina, J.M.; Miailhes, P.; Lascoux-Combe, C.; Gault, E.; et al. Impact of chronic hepatitis C and/or D on liver fibrosis severity in patients co-infected with HIV and hepatitis B virus. AIDS 2007, 21, 2546-2549. [CrossRef]
- Lorenc, B.; Sikorska, K.; Stalke, P.; Bielawski, K.; Zietkowski, D. Hepatitis D, B and C virus (HDV/HBV/HCV) coinfection as a diagnostic problem and therapeutic challenge. Clin Exp Hepatol 2017, 3, 23-27. [CrossRef]
- Shukla, N.B.; Poles, M.A. Hepatitis B virus infection: co-infection with hepatitis C virus, hepatitis D virus, and human immunodeficiency virus. Clin Liver Dis 2004, 8, 445-460, viii. [CrossRef]
- Burkard, T.; Proske, N.; Resner, K.; Collignon, L.; Knegendorf, L.; Friesland, M.; Verhoye, L.; Sayed, I.M.; Bruggemann, Y.; Nocke, M.K.; et al. Viral Interference of Hepatitis C and E Virus Replication in Novel Experimental Co-Infection Systems. Cells 2022, 11. [CrossRef]
- Elhendawy, M.; Abo-Ali, L.; Abd-Elsalam, S.; Hagras, M.M.; Kabbash, I.; Mansour, L.; Atia, S.; Esmat, G.; Abo-ElAzm, A.R.; El-Kalla, F.; et al. HCV and HEV: two players in an Egyptian village, a study of prevalence, incidence, and co-infection. Environ Sci Pollut Res Int 2020, 27, 33659-33667. [CrossRef]
- Oluremi, A.S.; Ajadi, T.A.; Opaleye, O.O.; Alli, O.A.T.; Ogbolu, D.O.; Enitan, S.S.; Alaka, O.O.; Adelakun, A.A.; Adediji, I.O.; Ogunleke, A.O.; et al. High seroprevalence of viral hepatitis among animal handlers in Abeokuta, Ogun State, Nigeria. J Immunoassay Immunochem 2021, 42, 34-47. [CrossRef]
- Salu, O.B.; Akinbamiro, T.F.; Orenolu, R.M.; Ishaya, O.D.; Anyanwu, R.A.; Vitowanu, O.R.; Abdullah, M.A.; Olowoyeye, A.H.; Tijani, S.O.; Oyedeji, K.S.; et al. Detection of hepatitis viruses in suspected cases of Viral Haemorrhagic Fevers in Nigeria. PLoS One 2024, 19, e0305521. [CrossRef]
- Uversky, V.N.; Roman, A.; Oldfield, C.J.; Dunker, A.K. Protein intrinsic disorder and human papillomaviruses: increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J Proteome Res 2006, 5, 1829-1842. [CrossRef]
- Fan, X.; Xue, B.; Dolan, P.T.; LaCount, D.J.; Kurgan, L.; Uversky, V.N. The intrinsic disorder status of the human hepatitis C virus proteome. Mol Biosyst 2014, 10, 1345-1363. [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci 2015, 72, 137-151. [CrossRef]
- Xue, B.; Blocquel, D.; Habchi, J.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. Structural disorder in viral proteins. Chem Rev 2014, 114, 6880-6911. [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: disorder in 3500 proteomes from viruses and the three domains of life. J Biomol Struct Dyn 2012, 30, 137-149. [CrossRef]
- Xue, B.; Mizianty, M.J.; Kurgan, L.; Uversky, V.N. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell Mol Life Sci 2012, 69, 1211-1259. [CrossRef]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Goh, G.K.; Dunker, A.K.; Uversky, V.N. Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept Lett 2010, 17, 932-951. [CrossRef]
- Mishra, P.M.; Verma, N.C.; Rao, C.; Uversky, V.N.; Nandi, C.K. Intrinsically disordered proteins of viruses: Involvement in the mechanism of cell regulation and pathogenesis. Prog Mol Biol Transl Sci 2020, 174, 1-78. [CrossRef]
- Dolan, P.T.; Roth, A.P.; Xue, B.; Sun, R.; Dunker, A.K.; Uversky, V.N.; LaCount, D.J. Intrinsic disorder mediates hepatitis C virus core-host cell protein interactions. Protein Sci 2015, 24, 221-235. [CrossRef]
- Xue, B.; Ganti, K.; Rabionet, A.; Banks, L.; Uversky, V.N. Disordered interactome of human papillomavirus. Curr Pharm Des 2014, 20, 1274-1292. [CrossRef]
- Xue, B.; Uversky, V.N. Intrinsic disorder in proteins involved in the innate antiviral immunity: another flexible side of a molecular arms race. J Mol Biol 2014, 426, 1322-1350. [CrossRef]
- Li, S.; Zhou, W.; Li, D.; Pan, T.; Guo, J.; Zou, H.; Tian, Z.; Li, K.; Xu, J.; Li, X.; et al. Comprehensive characterization of human-virus protein-protein interactions reveals disease comorbidities and potential antiviral drugs. Comput Struct Biotechnol J 2022, 20, 1244-1253. [CrossRef]
- Chabrolles, H.; Auclair, H.; Vegna, S.; Lahlali, T.; Pons, C.; Michelet, M.; Coute, Y.; Belmudes, L.; Chadeuf, G.; Kim, Y.; et al. Hepatitis B virus Core protein nuclear interactome identifies SRSF10 as a host RNA-binding protein restricting HBV RNA production. PLoS Pathog 2020, 16, e1008593. [CrossRef]
- Kar, A.; Samanta, A.; Mukherjee, S.; Barik, S.; Biswas, A. The HBV web: An insight into molecular interactomes between the hepatitis B virus and its host en route to hepatocellular carcinoma. J Med Virol 2023, 95, e28436. [CrossRef]
- Nakai, Y.; Miyakawa, K.; Yamaoka, Y.; Hatayama, Y.; Nishi, M.; Suzuki, H.; Kimura, H.; Takahashi, H.; Kimura, Y.; Ryo, A. Generation and Utilization of a Monoclonal Antibody against Hepatitis B Virus Core Protein for a Comprehensive Interactome Analysis. Microorganisms 2022, 10. [CrossRef]
- Van Damme, E.; Vanhove, J.; Severyn, B.; Verschueren, L.; Pauwels, F. The Hepatitis B Virus Interactome: A Comprehensive Overview. Front Microbiol 2021, 12, 724877. [CrossRef]
- Whitworth, I.T.; Romero, S.; Kissi-Twum, A.; Knoener, R.; Scalf, M.; Sherer, N.M.; Smith, L.M. Identification of Host Proteins Involved in Hepatitis B Virus Genome Packaging. J Proteome Res 2024, 23, 4128-4138. [CrossRef]
- Budzko, L.; Marcinkowska-Swojak, M.; Jackowiak, P.; Kozlowski, P.; Figlerowicz, M. Copy number variation of genes involved in the hepatitis C virus-human interactome. Sci Rep 2016, 6, 31340. [CrossRef]
- Colpitts, C.C.; El-Saghire, H.; Pochet, N.; Schuster, C.; Baumert, T.F. High-throughput approaches to unravel hepatitis C virus-host interactions. Virus Res 2016, 218, 18-24. [CrossRef]
- de Chassey, B.; Navratil, V.; Tafforeau, L.; Hiet, M.S.; Aublin-Gex, A.; Agaugue, S.; Meiffren, G.; Pradezynski, F.; Faria, B.F.; Chantier, T.; et al. Hepatitis C virus infection protein network. Mol Syst Biol 2008, 4, 230. [CrossRef]
- Dolan, P.T.; Zhang, C.; Khadka, S.; Arumugaswami, V.; Vangeloff, A.D.; Heaton, N.S.; Sahasrabudhe, S.; Randall, G.; Sun, R.; LaCount, D.J. Identification and comparative analysis of hepatitis C virus-host cell protein interactions. Mol Biosyst 2013, 9, 3199-3209. [CrossRef]
- Germain, M.A.; Chatel-Chaix, L.; Gagne, B.; Bonneil, E.; Thibault, P.; Pradezynski, F.; de Chassey, B.; Meyniel-Schicklin, L.; Lotteau, V.; Baril, M.; et al. Elucidating novel hepatitis C virus-host interactions using combined mass spectrometry and functional genomics approaches. Mol Cell Proteomics 2014, 13, 184-203. [CrossRef]
- Matthaei, A.; Joecks, S.; Frauenstein, A.; Bruening, J.; Bankwitz, D.; Friesland, M.; Gerold, G.; Vieyres, G.; Kaderali, L.; Meissner, F.; et al. Landscape of protein-protein interactions during hepatitis C virus assembly and release. Microbiol Spectr 2024, 12, e0256222. [CrossRef]
- Mosca, E.; Alfieri, R.; Milanesi, L. Diffusion of information throughout the host interactome reveals gene expression variations in network proximity to target proteins of hepatitis C virus. PLoS One 2014, 9, e113660. [CrossRef]
- Mukhopadhyay, A.; Maulik, U. Network-based study reveals potential infection pathways of hepatitis-C leading to various diseases. PLoS One 2014, 9, e94029. [CrossRef]
- Saik, O.V.; Ivanisenko, T.V.; Demenkov, P.S.; Ivanisenko, V.A. Interactome of the hepatitis C virus: Literature mining with ANDSystem. Virus Res 2016, 218, 40-48. [CrossRef]
- Martino, C.; Di Luca, A.; Bennato, F.; Ianni, A.; Passamonti, F.; Rampacci, E.; Henry, M.; Meleady, P.; Martino, G. Label-Free Quantitative Analysis of Pig Liver Proteome after Hepatitis E Virus Infection. Viruses 2024, 16. [CrossRef]
- Ojha, N.K.; Lole, K.S. Hepatitis E virus ORF1 encoded non structural protein-host protein interaction network. Virus Res 2016, 213, 195-204. [CrossRef]
- Tian, Y.; Huang, W.; Yang, J.; Wen, Z.; Geng, Y.; Zhao, C.; Zhang, H.; Wang, Y. Systematic identification of hepatitis E virus ORF2 interactome reveals that TMEM134 engages in ORF2-mediated NF-kappaB pathway. Virus Res 2017, 228, 102-108. [CrossRef]
- Cristina, J.; Costa-Mattioli, M. Genetic variability and molecular evolution of hepatitis A virus. Virus Res 2007, 127, 151-157. [CrossRef]
- Wang, X.; Ren, J.; Gao, Q.; Hu, Z.; Sun, Y.; Li, X.; Rowlands, D.J.; Yin, W.; Wang, J.; Stuart, D.I.; et al. Hepatitis A virus and the origins of picornaviruses. Nature 2015, 517, 85-88. [CrossRef]
- Probst, C.; Jecht, M.; Gauss-Muller, V. Intrinsic signals for the assembly of hepatitis A virus particles. Role of structural proteins VP4 and 2A. J Biol Chem 1999, 274, 4527-4531. [CrossRef]
- Jecht, M.; Probst, C.; Gauss-Muller, V. Membrane permeability induced by hepatitis A virus proteins 2B and 2BC and proteolytic processing of HAV 2BC. Virology 1998, 252, 218-227. [CrossRef]
- Teterina, N.L.; Bienz, K.; Egger, D.; Gorbalenya, A.E.; Ehrenfeld, E. Induction of intracellular membrane rearrangements by HAV proteins 2C and 2BC. Virology 1997, 237, 66-77. [CrossRef]
- Kusov, Y.Y.; Probst, C.; Jecht, M.; Jost, P.D.; Gauss-Muller, V. Membrane association and RNA binding of recombinant hepatitis A virus protein 2C. Arch Virol 1998, 143, 931-944. [CrossRef]
- Yang, Y.; Liang, Y.; Qu, L.; Chen, Z.; Yi, M.; Li, K.; Lemon, S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A 2007, 104, 7253-7258. [CrossRef]
- Kusov, Y.; Gauss-Muller, V. Improving proteolytic cleavage at the 3A/3B site of the hepatitis A virus polyprotein impairs processing and particle formation, and the impairment can be complemented in trans by 3AB and 3ABC. J Virol 1999, 73, 9867-9878. [CrossRef]
- Beneduce, F.; Ciervo, A.; Kusov, Y.; Gauss-Muller, V.; Morace, G. Mapping of protein domains of hepatitis A virus 3AB essential for interaction with 3CD and viral RNA. Virology 1999, 264, 410-421. [CrossRef]
- Ciervo, A.; Beneduce, F.; Morace, G. Polypeptide 3AB of hepatitis A virus is a transmembrane protein. Biochem Biophys Res Commun 1998, 249, 266-274. [CrossRef]
- Sun, Y.; Guo, Y.; Lou, Z. Formation and working mechanism of the picornavirus VPg uridylylation complex. Curr Opin Virol 2014, 9, 24-30. [CrossRef]
- Jia, X.Y.; Ehrenfeld, E.; Summers, D.F. Proteolytic activity of hepatitis A virus 3C protein. J Virol 1991, 65, 2595-2600. [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J Cell Biochem 2011, 112, 3256-3267. [CrossRef]
- Uversky, V.N. Analyzing IDPs in interactomes. In Intrinsically Disordered Proteins, Kragelund, B.B., Skriver, K., Eds.; Humana New York, NY, 2020; Volume Methods in Molecular Biology, pp. 895-945.
- Lanford, R.E.; Walker, C.M.; Lemon, S.M. The Chimpanzee Model of Viral Hepatitis: Advances in Understanding the Immune Response and Treatment of Viral Hepatitis. ILAR J 2017, 58, 172-189. [CrossRef]
- Luke, S.; Verma, R.S. Human (Homo sapiens) and chimpanzee (Pan troglodytes) share similar ancestral centromeric alpha satellite DNA sequences but other fractions of heterochromatin differ considerably. Am J Phys Anthropol 1995, 96, 63-71. [CrossRef]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid-Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus-Host Interactions. Int J Mol Sci 2020, 21. [CrossRef]
- Brodrick, A.J.; Broadbent, A.J. The Formation and Function of Birnaviridae Virus Factories. Int J Mol Sci 2023, 24. [CrossRef]
- Cao, Z.; Yang, Y.; Zhang, S.; Zhang, T.; Lu, P.; Chen, K. Liquid-liquid phase separation in viral infection: From the occurrence and function to treatment potentials. Colloids Surf B Biointerfaces 2025, 246, 114385. [CrossRef]
- Caragliano, E.; Brune, W.; Bosse, J.B. Herpesvirus Replication Compartments: Dynamic Biomolecular Condensates? Viruses 2022, 14. [CrossRef]
- Chau, B.A.; Chen, V.; Cochrane, A.W.; Parent, L.J.; Mouland, A.J. Liquid-liquid phase separation of nucleocapsid proteins during SARS-CoV-2 and HIV-1 replication. Cell Rep 2023, 42, 111968. [CrossRef]
- Di Nunzio, F.; Uversky, V.N.; Mouland, A.J. Biomolecular condensates: insights into early and late steps of the HIV-1 replication cycle. Retrovirology 2023, 20, 4. [CrossRef]
- Dolnik, O.; Gerresheim, G.K.; Biedenkopf, N. New Perspectives on the Biogenesis of Viral Inclusion Bodies in Negative-Sense RNA Virus Infections. Cells 2021, 10. [CrossRef]
- Eltayeb, A.; Al-Sarraj, F.; Alharbi, M.; Albiheyri, R.; Mattar, E.H.; Abu Zeid, I.M.; Bouback, T.A.; Bamagoos, A.; Uversky, V.N.; Rubio-Casillas, A.; et al. Intrinsic factors behind long COVID: IV. Hypothetical roles of the SARS-CoV-2 nucleocapsid protein and its liquid-liquid phase separation. J Cell Biochem 2024, 125, e30530. [CrossRef]
- Etibor, T.A.; Yamauchi, Y.; Amorim, M.J. Liquid Biomolecular Condensates and Viral Lifecycles: Review and Perspectives. Viruses 2021, 13. [CrossRef]
- Glon, D.; Leonardon, B.; Guillemot, A.; Albertini, A.; Lagaudriere-Gesbert, C.; Gaudin, Y. Biomolecular condensates with liquid properties formed during viral infections. Microbes Infect 2024, 26, 105402. [CrossRef]
- Li, H.; Ernst, C.; Kolonko-Adamska, M.; Greb-Markiewicz, B.; Man, J.; Parissi, V.; Ng, B.W. Phase separation in viral infections. Trends Microbiol 2022, 30, 1217-1231. [CrossRef]
- Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S.S.; Visentin, A.; Peralta-Martinez, R.; Loureiro, M.E.; de Prat-Gay, G. Deconstructing virus condensation. PLoS Pathog 2021, 17, e1009926. [CrossRef]
- Mouland, A.J.; Chau, B.A.; Uversky, V.N. Methodological approaches to studying phase separation and HIV-1 replication: Current and future perspectives. Methods 2024, 229, 147-155. [CrossRef]
- Nevers, Q.; Albertini, A.A.; Lagaudriere-Gesbert, C.; Gaudin, Y. Negri bodies and other virus membrane-less replication compartments. Biochim Biophys Acta Mol Cell Res 2020, 1867, 118831. [CrossRef]
- Papa, G.; Borodavka, A.; Desselberger, U. Viroplasms: Assembly and Functions of Rotavirus Replication Factories. Viruses 2021, 13. [CrossRef]
- Saito, A.; Shofa, M.; Ode, H.; Yumiya, M.; Hirano, J.; Okamoto, T.; Yoshimura, S.H. How Do Flaviviruses Hijack Host Cell Functions by Phase Separation? Viruses 2021, 13. [CrossRef]
- Scoca, V.; Di Nunzio, F. Membraneless organelles restructured and built by pandemic viruses: HIV-1 and SARS-CoV-2. J Mol Cell Biol 2021, 13, 259-268. [CrossRef]
- Su, J.M.; Wilson, M.Z.; Samuel, C.E.; Ma, D. Formation and Function of Liquid-Like Viral Factories in Negative-Sense Single-Stranded RNA Virus Infections. Viruses 2021, 13. [CrossRef]
- Wei, W.; Bai, L.; Yan, B.; Meng, W.; Wang, H.; Zhai, J.; Si, F.; Zheng, C. When liquid-liquid phase separation meets viral infections. Front Immunol 2022, 13, 985622. [CrossRef]
- Zhang, Q.; Ye, H.; Liu, C.; Zhou, H.; He, M.; Liang, X.; Zhou, Y.; Wang, K.; Qin, Y.; Li, Z.; et al. PABP-driven secondary condensed phase within RSV inclusion bodies activates viral mRNAs for ribosomal recruitment. Virol Sin 2024, 39, 235-250. [CrossRef]
- Zhang, X.; Zheng, R.; Li, Z.; Ma, J. Liquid-liquid Phase Separation in Viral Function. J Mol Biol 2023, 435, 167955. [CrossRef]
- Giraud, G.; Roda, M.; Huchon, P.; Michelet, M.; Maadadi, S.; Jutzi, D.; Montserret, R.; Ruepp, M.D.; Parent, R.; Combet, C.; et al. G-quadruplexes control hepatitis B virus replication by promoting cccDNA transcription and phase separation in hepatocytes. Nucleic Acids Res 2024, 52, 2290-2305. [CrossRef]
- Liu, Y.; Zhang, J.; Zhai, Z.; Liu, C.; Yang, S.; Zhou, Y.; Zeng, X.; Liu, J.; Zhang, X.; Nie, X.; et al. Upregulated PrP(C) by HBx enhances NF-kappaB signal via liquid-liquid phase separation to advance liver cancer. NPJ Precis Oncol 2024, 8, 211. [CrossRef]
- Hundie, G.B.; Stalin Raj, V.; Gebre Michael, D.; Pas, S.D.; Koopmans, M.P.; Osterhaus, A.D.; Smits, S.L.; Haagmans, B.L. A novel hepatitis B virus subgenotype D10 circulating in Ethiopia. J Viral Hepat 2017, 24, 163-173. [CrossRef]
- Zhao, F.; Xie, X.; Tan, X.; Yu, H.; Tian, M.; Lv, H.; Qin, C.; Qi, J.; Zhu, Q. The Functions of Hepatitis B Virus Encoding Proteins: Viral Persistence and Liver Pathogenesis. Front Immunol 2021, 12, 691766. [CrossRef]
- Leupin, O.; Bontron, S.; Schaeffer, C.; Strubin, M. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J Virol 2005, 79, 4238-4245. [CrossRef]
- Yi, Z.; Pan, T.; Wu, X.; Song, W.; Wang, S.; Xu, Y.; Rice, C.M.; Macdonald, M.R.; Yuan, Z. Hepatitis C virus co-opts Ras-GTPase-activating protein-binding protein 1 for its genome replication. J Virol 2011, 85, 6996-7004. [CrossRef]
- Wolk, B.; Buchele, B.; Moradpour, D.; Rice, C.M. A dynamic view of hepatitis C virus replication complexes. J Virol 2008, 82, 10519-10531. [CrossRef]
- Nesterov, S.V.; Ilyinsky, N.S.; Uversky, V.N. Liquid-liquid phase separation as a common organizing principle of intracellular space and biomembranes providing dynamic adaptive responses. Biochim Biophys Acta Mol Cell Res 2021, 1868, 119102. [CrossRef]
- Wu, C.; Holehouse, A.S.; Leung, D.W.; Amarasinghe, G.K.; Dutch, R.E. Liquid Phase Partitioning in Virus Replication: Observations and Opportunities. Annu Rev Virol 2022, 9, 285-306. [CrossRef]
- Liu, D.; Ndongwe, T.P.; Puray-Chavez, M.; Casey, M.C.; Izumi, T.; Pathak, V.K.; Tedbury, P.R.; Sarafianos, S.G. Effect of P-body component Mov10 on HCV virus production and infectivity. FASEB J 2020, 34, 9433-9449. [CrossRef]
- Mello, F.C.A.; Barros, T.M.; Angelice, G.P.; Costa, V.D.; Mello, V.M.; Pardini, M.; Lampe, E.; Lago, B.V.; Villar, L.M. Circulation of HDV Genotypes in Brazil: Identification of a Putative Novel HDV-8 Subgenotype. Microbiol Spectr 2023, 11, e0396522. [CrossRef]
- Sunbul, M. Hepatitis B virus genotypes: global distribution and clinical importance. World J Gastroenterol 2014, 20, 5427-5434. [CrossRef]
- Weiner, A.J.; Choo, Q.L.; Wang, K.S.; Govindarajan, S.; Redeker, A.G.; Gerin, J.L.; Houghton, M. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta antigen polypeptides p24 delta and p27 delta. J Virol 1988, 62, 594-599. [CrossRef]
- Chao, M.; Hsieh, S.Y.; Taylor, J. Role of two forms of hepatitis delta virus antigen: evidence for a mechanism of self-limiting genome replication. J Virol 1990, 64, 5066-5069. [CrossRef]
- Glenn, J.S.; White, J.M. trans-dominant inhibition of human hepatitis delta virus genome replication. J Virol 1991, 65, 2357-2361. [CrossRef]
- Huang, W.H.; Yung, B.Y.; Syu, W.J.; Lee, Y.H. The nucleolar phosphoprotein B23 interacts with hepatitis delta antigens and modulates the hepatitis delta virus RNA replication. J Biol Chem 2001, 276, 25166-25175. [CrossRef]
- Polson, A.G.; Bass, B.L.; Casey, J.L. RNA editing of hepatitis delta virus antigenome by dsRNA-adenosine deaminase. Nature 1996, 380, 454-456. [CrossRef]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc Natl Acad Sci U S A 2002, 99, 15118-15123. [CrossRef]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a prenylation site in delta virus large antigen. Science 1992, 256, 1331-1333. [CrossRef]
- Hwang, S.B.; Lai, M.M. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J Virol 1993, 67, 7659-7662. [CrossRef]
- Khalfi, P.; Denis, Z.; McKellar, J.; Merolla, G.; Chavey, C.; Ursic-Bedoya, J.; Soppa, L.; Szirovicza, L.; Hetzel, U.; Dufourt, J.; et al. Comparative analysis of human, rodent and snake deltavirus replication. PLoS Pathog 2024, 20, e1012060. [CrossRef]
- Brazda, V.; Valkova, N.; Dobrovolna, M.; Mergny, J.L. Abundance of G-Quadruplex Forming Sequences in the Hepatitis Delta Virus Genomes. ACS Omega 2024, 9, 4096-4101. [CrossRef]
- Zhang, M.; Purcell, R.H.; Emerson, S.U. Identification of the 5’ terminal sequence of the SAR-55 and MEX-14 strains of hepatitis E virus and confirmation that the genome is capped. J Med Virol 2001, 65, 293-295. [CrossRef]
- Kenney, S.P.; Meng, X.J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb Perspect Med 2019, 9. [CrossRef]
- Smith, D.B.; Simmonds, P.; Members Of The International Committee On The Taxonomy Of Viruses Study, G.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J Gen Virol 2014, 95, 2223-2232. [CrossRef]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355-357 e353. [CrossRef]
- Parvez, M.K. Mutational analysis of hepatitis E virus ORF1 “Y-domain”: Effects on RNA replication and virion infectivity. World J Gastroenterol 2017, 23, 590-602. [CrossRef]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: delineation of an additional group of positive-strand RNA plant and animal viruses. Proc Natl Acad Sci U S A 1992, 89, 8259-8263. [CrossRef]
- Purdy, M.A. Evolution of the hepatitis E virus polyproline region: order from disorder. J Virol 2012, 86, 10186-10193. [CrossRef]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc Natl Acad Sci U S A 2017, 114, 1147-1152. [CrossRef]
- Gouttenoire, J.; Pollan, A.; Abrami, L.; Oechslin, N.; Mauron, J.; Matter, M.; Oppliger, J.; Szkolnicka, D.; Dao Thi, V.L.; van der Goot, F.G.; et al. Palmitoylation mediates membrane association of hepatitis E virus ORF3 protein and is required for infectious particle secretion. PLoS Pathog 2018, 14, e1007471. [CrossRef]
- Nagashima, S.; Takahashi, M.; Jirintai; Tanaka, T.; Yamada, K.; Nishizawa, T.; Okamoto, H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J Gen Virol 2011, 92, 269-278. [CrossRef]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Nagashima, S.; Tanaka, T.; Okamoto, H. ORF3 protein of hepatitis E virus is essential for virion release from infected cells. J Gen Virol 2009, 90, 1880-1891. [CrossRef]
- Huang, F.; Yang, C.; Yu, W.; Bi, Y.; Long, F.; Wang, J.; Li, Y.; Jing, S. Hepatitis E virus infection activates signal regulator protein alpha to down-regulate type I interferon. Immunol Res 2016, 64, 115-122. [CrossRef]
- Rancurel, C.; Khosravi, M.; Dunker, A.K.; Romero, P.R.; Karlin, D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol 2009, 83, 10719-10736. [CrossRef]
- Willis, S.; Masel, J. Gene Birth Contributes to Structural Disorder Encoded by Overlapping Genes. Genetics 2018, 210, 303-313. [CrossRef]
- Tokuriki, N.; Oldfield, C.J.; Uversky, V.N.; Berezovsky, I.N.; Tawfik, D.S. Do viral proteins possess unique biophysical features? Trends Biochem Sci 2009, 34, 53-59. [CrossRef]
- Mohan, A.; Sullivan, W.J., Jr.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst 2008, 4, 328-340. [CrossRef]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac Symp Biocomput 2012, 128-139.
- Oldfield, C.J.; Cheng, Y.; Cortese, M.S.; Brown, C.J.; Uversky, V.N.; Dunker, A.K. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989-2000. [CrossRef]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415-427. [CrossRef]
- Mohammed, A.S.; Uversky, V.N. Intrinsic Disorder as a Natural Preservative: High Levels of Intrinsic Disorder in Proteins Found in the 2600-Year-Old Human Brain. Biology (Basel) 2022, 11. [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal statistical society: series B (Methodological) 1995, 57, 289-300. [CrossRef]
- Hardenberg, M.; Horvath, A.; Ambrus, V.; Fuxreiter, M.; Vendruscolo, M. Widespread occurrence of the droplet state of proteins in the human proteome. Proc Natl Acad Sci U S A 2020, 117, 33254-33262. [CrossRef]
- Turoverov, K.K.; Kuznetsova, I.M.; Fonin, A.V.; Darling, A.L.; Zaslavsky, B.Y.; Uversky, V.N. Stochasticity of Biological Soft Matter: Emerging Concepts in Intrinsically Disordered Proteins and Biological Phase Separation. Trends Biochem Sci 2019, 44, 716-728. [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357. [CrossRef]
- Uversky, V.N.; Kuznetsova, I.M.; Turoverov, K.K.; Zaslavsky, B. Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates. FEBS Lett 2015, 589, 15-22. [CrossRef]
- Brangwynne, C.P. Phase transitions and size scaling of membrane-less organelles. The Journal of cell biology 2013, 203, 875-881. [CrossRef]
- Uversky, V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Current opinion in structural biology 2016, 44, 18-30. [CrossRef]
- Brangwynne, Clifford P.; Tompa, P.; Pappu, Rohit V. Polymer physics of intracellular phase transitions. Nat Phys 2015, 11, 899-904. [CrossRef]
- Zhang, X.; Yang, Z.; Fu, C.; Yao, R.; Li, H.; Peng, F.; Li, N. Emerging roles of liquid-liquid phase separation in liver innate immunity. Cell Commun Signal 2024, 22, 430. [CrossRef]
- Ishida-Yamamoto, A.; Igawa, S. The biology and regulation of corneodesmosomes. Cell Tissue Res 2015, 360, 477-482. [CrossRef]
- Jonca, N.; Caubet, C.; Guerrin, M.; Simon, M.; Serre, G. Corneodesmosin: structure, function and involvement in pathophysiology. 2010.
- Jonca, N.; Leclerc, E.A.; Caubet, C.; Simon, M.; Guerrin, M.; Serre, G. Corneodesmosomes and corneodesmosin: from the stratum corneum cohesion to the pathophysiology of genodermatoses. Eur J Dermatol 2011, 21 Suppl 2, 35-42. [CrossRef]
- Oji, V.; Eckl, K.M.; Aufenvenne, K.; Natebus, M.; Tarinski, T.; Ackermann, K.; Seller, N.; Metze, D.; Nurnberg, G.; Folster-Holst, R.; et al. Loss of corneodesmosin leads to severe skin barrier defect, pruritus, and atopy: unraveling the peeling skin disease. Am J Hum Genet 2010, 87, 274-281. [CrossRef]
- Matsumoto, M.; Zhou, Y.; Matsuo, S.; Nakanishi, H.; Hirose, K.; Oura, H.; Arase, S.; Ishida-Yamamoto, A.; Bando, Y.; Izumi, K.; et al. Targeted deletion of the murine corneodesmosin gene delineates its essential role in skin and hair physiology. Proc Natl Acad Sci U S A 2008, 105, 6720-6724. [CrossRef]
- Leclerc, E.A.; Huchenq, A.; Mattiuzzo, N.R.; Metzger, D.; Chambon, P.; Ghyselinck, N.B.; Serre, G.; Jonca, N.; Guerrin, M. Corneodesmosin gene ablation induces lethal skin-barrier disruption and hair-follicle degeneration related to desmosome dysfunction. J Cell Sci 2009, 122, 2699-2709. [CrossRef]
- Noe, M.H.; Grewal, S.K.; Shin, D.B.; Ogdie, A.; Takeshita, J.; Gelfand, J.M. Increased prevalence of HCV and hepatic decompensation in adults with psoriasis: a population-based study in the United Kingdom. J Eur Acad Dermatol Venereol 2017, 31, 1674-1680. [CrossRef]
- Enomoto, M.; Tateishi, C.; Tsuruta, D.; Tamori, A.; Kawada, N. Remission of Psoriasis After Treatment of Chronic Hepatitis C Virus Infection With Direct-Acting Antivirals. Ann Intern Med 2018, 168, 678-680. [CrossRef]
- Sayiner, M.; Golabi, P.; Farhat, F.; Younossi, Z.M. Dermatologic Manifestations of Chronic Hepatitis C Infection. Clin Liver Dis 2017, 21, 555-564. [CrossRef]
- Steinert, P.M.; Mack, J.W.; Korge, B.P.; Gan, S.Q.; Haynes, S.R.; Steven, A.C. Glycine loops in proteins: their occurrence in certain intermediate filament chains, loricrins and single-stranded RNA binding proteins. Int J Biol Macromol 1991, 13, 130-139. [CrossRef]
- Beaulieu, M.E.; Castillo, F.; Soucek, L. Structural and Biophysical Insights into the Function of the Intrinsically Disordered Myc Oncoprotein. Cells 2020, 9. [CrossRef]
- Higgs, M.R.; Lerat, H.; Pawlotsky, J.M. Hepatitis C virus-induced activation of beta-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene 2013, 32, 4683-4693. [CrossRef]
- Sun, Y.; Yu, M.; Qu, M.; Ma, Y.; Zheng, D.; Yue, Y.; Guo, S.; Tang, L.; Li, G.; Zheng, W.; et al. Hepatitis B virus-triggered PTEN/beta-catenin/c-Myc signaling enhances PD-L1 expression to promote immune evasion. Am J Physiol Gastrointest Liver Physiol 2020, 318, G162-G173. [CrossRef]
- Tappero, G.; Natoli, G.; Anfossi, G.; Rosina, F.; Negro, F.; Smedile, A.; Bonino, F.; Angeli, A.; Purcell, R.H.; Rizzetto, M.; et al. Expression of the c-myc protooncogene product in cells infected with the hepatitis delta virus. Hepatology 1994, 20, 1109-1114.
- Terradillos, O.; Billet, O.; Renard, C.A.; Levy, R.; Molina, T.; Briand, P.; Buendia, M.A. The hepatitis B virus X gene potentiates c-myc-induced liver oncogenesis in transgenic mice. Oncogene 1997, 14, 395-404. [CrossRef]
- Li, W.; Miao, X.; Qi, Z.; Zeng, W.; Liang, J.; Liang, Z. Hepatitis B virus X protein upregulates HSP90alpha expression via activation of c-Myc in human hepatocarcinoma cell line, HepG2. Virol J 2010, 7, 45. [CrossRef]
- Wu, W.B.; Shao, S.W.; Zhao, L.J.; Luan, J.; Cao, J.; Gao, J.; Zhu, S.Y.; Qi, Z.T. Hepatitis C virus F protein up-regulates c-myc and down-regulates p53 in human hepatoma HepG2 cells. Intervirology 2007, 50, 341-346. [CrossRef]
- Fathy, A.; Abdelrazek, M.A.; Attallah, A.M.; Abouzid, A.; El-Far, M. Hepatitis C virus may accelerate breast cancer progression by increasing mutant p53 and c-Myc oncoproteins circulating levels. Breast Cancer 2024, 31, 116-123. [CrossRef]
- Wang, Y.; Li, J.; Chen, J.; Liu, L.; Peng, Z.; Ding, J.; Ding, K. From cirrhosis to hepatocellular carcinoma in HCV-infected patients: genes involved in tumor progression. Eur Rev Med Pharmacol Sci 2012, 16, 995-1000.
- Bayliss, R.; Burgess, S.G.; Leen, E.; Richards, M.W. A moving target: structure and disorder in pursuit of Myc inhibitors. Biochem Soc Trans 2017, 45, 709-717. [CrossRef]
- Das, S.K.; Lewis, B.A.; Levens, D. MYC: a complex problem. Trends Cell Biol 2023, 33, 235-246. [CrossRef]
- Khaitin, A.M.; Guzenko, V.V.; Bachurin, S.S.; Demyanenko, S.V. c-Myc and FOXO3a-The Everlasting Decision Between Neural Regeneration and Degeneration. Int J Mol Sci 2024, 25. [CrossRef]
- Metallo, S.J. Intrinsically disordered proteins are potential drug targets. Curr Opin Chem Biol 2010, 14, 481-488. [CrossRef]
- Ross, J.; Miron, C.E.; Plescia, J.; Laplante, P.; McBride, K.; Moitessier, N.; Moroy, T. Targeting MYC: From understanding its biology to drug discovery. Eur J Med Chem 2021, 213, 113137. [CrossRef]
- Sammak, S.; Zinzalla, G. Targeting protein-protein interactions (PPIs) of transcription factors: Challenges of intrinsically disordered proteins (IDPs) and regions (IDRs). Prog Biophys Mol Biol 2015, 119, 41-46. [CrossRef]
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J Mol Biol 2018, 430, 2321-2341. [CrossRef]
- Feris, E.J.; Hinds, J.W.; Cole, M.D. Formation of a structurally-stable conformation by the intrinsically disordered MYC:TRRAP complex. PLoS One 2019, 14, e0225784. [CrossRef]
- Peng, Q.; Wang, L.; Qin, Z.; Wang, J.; Zheng, X.; Wei, L.; Zhang, X.; Zhang, X.; Liu, C.; Li, Z.; et al. Phase Separation of Epstein-Barr Virus EBNA2 and Its Coactivator EBNALP Controls Gene Expression. J Virol 2020, 94. [CrossRef]
- Fang, Z.S.; Zhang, Z.; Liang, Z.J.; Long, Z.R.; Xiao, Y.; Liang, Z.Y.; Sun, X.; Li, H.M.; Huang, H. Liquid-Liquid Phase Separation-Related Genes Associated with Tumor Grade and Prognosis in Hepatocellular Carcinoma: A Bioinformatic Study. Int J Gen Med 2021, 14, 9671-9679. [CrossRef]
- Tai, J.; Wang, L.; Yan, Z.; Liu, J. Single-cell sequencing and transcriptome analyses in the construction of a liquid-liquid phase separation-associated gene model for rheumatoid arthritis. Front Genet 2023, 14, 1210722. [CrossRef]
- Pei, X.; Chen, Y.; Liu, L.; Meng, L.; Zhang, J.; Liu, Y.; Chen, L. E242-E261 region of MYC regulates liquid-liquid phase separation and tumor growth by providing negative charges. J Biol Chem 2024, 300, 107836. [CrossRef]
- Soussi, T. The p53 tumor suppressor gene: from molecular biology to clinical investigation. Ann N Y Acad Sci 2000, 910, 121-137; discussion 137-129. [CrossRef]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817-825. [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293-299.
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413-431. [CrossRef]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15-16. [CrossRef]
- Bourdon, J.C. p53 Family isoforms. Current pharmaceutical biotechnology 2007, 8, 332-336. [CrossRef]
- Uversky, V.N. p53 Proteoforms and Intrinsic Disorder: An Illustration of the Protein Structure-Function Continuum Concept. Int J Mol Sci 2016, 17. [CrossRef]
- Hirohashi, S. Pathology and molecular mechanisms of multistage human hepatocarcinogenesis. Princess Takamatsu Symp 1991, 22, 87-93.
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49-53. [CrossRef]
- Gallinger, S.; Langer, B. Primary and secondary hepatic malignancies. Curr Opin Gen Surg 1993, 257-264.
- Tabor, E. Tumor suppressor genes, growth factor genes, and oncogenes in hepatitis B virus-associated hepatocellular carcinoma. J Med Virol 1994, 42, 357-365. [CrossRef]
- Tabor, E. Hepatocarcinogenesis: hepatitis viruses and altered tumor suppressor gene function. Princess Takamatsu Symp 1995, 25, 151-161.
- Tornesello, M.L.; Buonaguro, L.; Tatangelo, F.; Botti, G.; Izzo, F.; Buonaguro, F.M. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics 2013, 102, 74-83. [CrossRef]
- Eskander, E.F.; Abd-Rabou, A.A.; Yahya, S.M.; El Sherbini, A.; Mohamed, M.S.; Shaker, O.G. “P53 codon 72 single base substitution in viral hepatitis C and hepatocarcinoma incidences”. Indian J Clin Biochem 2014, 29, 3-7. [CrossRef]
- Robinson, W.S. Molecular events in the pathogenesis of hepadnavirus-associated hepatocellular carcinoma. Annu Rev Med 1994, 45, 297-323. [CrossRef]
- Koike, K.; Takada, S. Biochemistry and functions of hepatitis B virus X protein. Intervirology 1995, 38, 89-99. [CrossRef]
- Cromlish, J.A. Hepatitis B virus-induced hepatocellular carcinoma: possible roles for HBx. Trends Microbiol 1996, 4, 270-274. [CrossRef]
- Feitelson, M.A. Hepatitis B x antigen and p53 in the development of hepatocellular carcinoma. J Hepatobiliary Pancreat Surg 1998, 5, 367-374. [CrossRef]
- Feitelson, M.A.; Duan, L.X. Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am J Pathol 1997, 150, 1141-1157.
- Kew, M.C. Increasing evidence that hepatitis B virus X gene protein and p53 protein may interact in the pathogenesis of hepatocellular carcinoma. Hepatology 1997, 25, 1037-1038. [CrossRef]
- Henkler, F.F.; Koshy, R. Hepatitis B virus transcriptional activators: mechanisms and possible role in oncogenesis. J Viral Hepat 1996, 3, 109-121. [CrossRef]
- Dewantoro, O.; Gani, R.A.; Akbar, N. Hepatocarcinogenesis in viral Hepatitis B infection: the role of HBx and p53. Acta Med Indones 2006, 38, 154-159.
- Kew, M.C. Hepatitis B virus x protein in the pathogenesis of hepatitis B virus-induced hepatocellular carcinoma. J Gastroenterol Hepatol 2011, 26 Suppl 1, 144-152. [CrossRef]
- Anzola, M.; Burgos, J.J. Hepatocellular carcinoma: molecular interactions between hepatitis C virus and p53 in hepatocarcinogenesis. Expert Rev Mol Med 2003, 5, 1-16. [CrossRef]
- McGivern, D.R.; Lemon, S.M. Tumor suppressors, chromosomal instability, and hepatitis C virus-associated liver cancer. Annu Rev Pathol 2009, 4, 399-415. [CrossRef]
- Mitchell, J.K.; McGivern, D.R. Mechanisms of hepatocarcinogenesis in chronic hepatitis C. Hepat Oncol 2014, 1, 293-307. [CrossRef]
- Tornesello, M.L.; Annunziata, C.; Tornesello, A.L.; Buonaguro, L.; Buonaguro, F.M. Human Oncoviruses and p53 Tumor Suppressor Pathway Deregulation at the Origin of Human Cancers. Cancers (Basel) 2018, 10. [CrossRef]
- Brychtova, V.; Hrabal, V.; Vojtesek, B. Oncogenic Viral Protein Interactions with p53 Family Proteins. Klin Onkol 2019, 32, 72-77. [CrossRef]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 2008, 9 Suppl 1, S1. [CrossRef]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit 2005, 18, 343-384. [CrossRef]
- Chen, C.; Fu, G.; Guo, Q.; Xue, S.; Luo, S.Z. Phase separation of p53 induced by its unstructured basic region and prevented by oncogenic mutations in tetramerization domain. Int J Biol Macromol 2022, 222, 207-216. [CrossRef]
- Datta, D.; Navalkar, A.; Sakunthala, A.; Paul, A.; Patel, K.; Masurkar, S.; Gadhe, L.; Manna, S.; Bhattacharyya, A.; Sengupta, S.; et al. Nucleo-cytoplasmic environment modulates spatiotemporal p53 phase separation. Sci Adv 2024, 10, eads0427. [CrossRef]
- de Oliveira, G.A.P.; Cordeiro, Y.; Silva, J.L.; Vieira, T. Liquid-liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases. Adv Protein Chem Struct Biol 2019, 118, 289-331. [CrossRef]
- Garg, A.; Kumar, G.; Singh, V.; Sinha, S. Doxorubicin catalyses self-assembly of p53 by phase separation. Curr Res Struct Biol 2024, 7, 100133. [CrossRef]
- Kamagata, K.; Kanbayashi, S.; Honda, M.; Itoh, Y.; Takahashi, H.; Kameda, T.; Nagatsugi, F.; Takahashi, S. Liquid-like droplet formation by tumor suppressor p53 induced by multivalent electrostatic interactions between two disordered domains. Sci Rep 2020, 10, 580. [CrossRef]
- Kilic, S.; Lezaja, A.; Gatti, M.; Bianco, E.; Michelena, J.; Imhof, R.; Altmeyer, M. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J 2019, 38, e101379. [CrossRef]
- Liebl, M.C.; Hofmann, T.G. Regulating the p53 Tumor Suppressor Network at PML Biomolecular Condensates. Cancers (Basel) 2022, 14. [CrossRef]
- Marques, M.A.; de Oliveira, G.A.P.; Silva, J.L. The chameleonic behavior of p53 in health and disease: the transition from a client to an aberrant condensate scaffold in cancer. Essays Biochem 2022, 66, 1023-1033. [CrossRef]
- Silonov, S.A.; Mokin, Y.I.; Nedelyaev, E.M.; Smirnov, E.Y.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N.; Fonin, A.V. On the Prevalence and Roles of Proteins Undergoing Liquid-Liquid Phase Separation in the Biogenesis of PML-Bodies. Biomolecules 2023, 13. [CrossRef]
- Usluer, S.; Spreitzer, E.; Bourgeois, B.; Madl, T. p53 Transactivation Domain Mediates Binding and Phase Separation with Poly-PR/GR. Int J Mol Sci 2021, 22. [CrossRef]
- Yu, Y.; Liu, Q.; Zeng, J.; Tan, Y.; Tang, Y.; Wei, G. Multiscale simulations reveal the driving forces of p53C phase separation accelerated by oncogenic mutations. Chem Sci 2024, 15, 12806-12818. [CrossRef]
- Zong, Z.; Xie, F.; Wang, S.; Wu, X.; Zhang, Z.; Yang, B.; Zhou, F. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell 2024, 187, 2375-2392 e2333. [CrossRef]
- Andreeva, A.; Howorth, D.; Brenner, S.E.; Hubbard, T.J.; Chothia, C.; Murzin, A.G. SCOP database in 2004: refinements integrate structure and sequence family data. Nucleic Acids Res 2004, 32, D226-229. [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol 1995, 247, 536-540. [CrossRef]
- de Lima Morais, D.A.; Fang, H.; Rackham, O.J.; Wilson, D.; Pethica, R.; Chothia, C.; Gough, J. SUPERFAMILY 1.75 including a domain-centric gene ontology method. Nucleic Acids Res 2011, 39, D427-434. [CrossRef]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol 2009, 5, e1000376. [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 2012, 40, D261-270. [CrossRef]
- Fog, C.K.; Galli, G.G.; Lund, A.H. PRDM proteins: important players in differentiation and disease. Bioessays 2012, 34, 50-60. [CrossRef]
- Han, B.Y.; Seah, M.K.Y.; Brooks, I.R.; Quek, D.H.P.; Huxley, D.R.; Foo, C.S.; Lee, L.T.; Wollmann, H.; Guo, H.; Messerschmidt, D.M.; et al. Global translation during early development depends on the essential transcription factor PRDM10. Nat Commun 2020, 11, 3603. [CrossRef]
- Di Zazzo, E.; De Rosa, C.; Abbondanza, C.; Moncharmont, B. PRDM Proteins: Molecular Mechanisms in Signal Transduction and Transcriptional Regulation. Biology (Basel) 2013, 2, 107-141. [CrossRef]
- Fumasoni, I.; Meani, N.; Rambaldi, D.; Scafetta, G.; Alcalay, M.; Ciccarelli, F.D. Family expansion and gene rearrangements contributed to the functional specialization of PRDM genes in vertebrates. BMC Evol Biol 2007, 7, 187. [CrossRef]
- Hohenauer, T.; Moore, A.W. The Prdm family: expanding roles in stem cells and development. Development 2012, 139, 2267-2282. [CrossRef]
- Sorrentino, A.; Federico, A.; Rienzo, M.; Gazzerro, P.; Bifulco, M.; Ciccodicola, A.; Casamassimi, A.; Abbondanza, C. PR/SET Domain Family and Cancer: Novel Insights from the Cancer Genome Atlas. Int J Mol Sci 2018, 19. [CrossRef]
- van de Beek, I.; Glykofridis, I.E.; Oosterwijk, J.C.; van den Akker, P.C.; Diercks, G.F.H.; Bolling, M.C.; Waisfisz, Q.; Mensenkamp, A.R.; Balk, J.A.; Zwart, R.; et al. PRDM10 directs FLCN expression in a novel disorder overlapping with Birt-Hogg-Dube syndrome and familial lipomatosis. Hum Mol Genet 2023, 32, 1223-1235. [CrossRef]
- Jin, J.; Xu, H.; Wu, R.; Niu, J.; Li, S. Aberrant DNA methylation profile of hepatitis B virus infection. J Med Virol 2019, 91, 81-92. [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J Mol Graph Model 2001, 19, 26-59. [CrossRef]
- Williams, R.M.; Obradovic, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: amino acid determinants of intrinsic order and disorder. Pac Symp Biocomput 2001, 89-100.
- Radivojac, P.; Iakoucheva, L.M.; Oldfield, C.J.; Obradovic, Z.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder and functional proteomics. Biophys J 2007, 92, 1439-1456. [CrossRef]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinformatics 2007, 8, 211. [CrossRef]
- Engert, C.G.; Droste, R.; van Oudenaarden, A.; Horvitz, H.R. A Caenorhabditis elegans protein with a PRDM9-like SET domain localizes to chromatin-associated foci and promotes spermatocyte gene expression, sperm production and fertility. PLoS Genet 2018, 14, e1007295. [CrossRef]
- Cerdan, C.; McIntyre, B.A.; Mechael, R.; Levadoux-Martin, M.; Yang, J.; Lee, J.B.; Bhatia, M. Activin A promotes hematopoietic fated mesoderm development through upregulation of brachyury in human embryonic stem cells. Stem Cells Dev 2012, 21, 2866-2877. [CrossRef]
- Wilkinson, D.G.; Bhatt, S.; Herrmann, B.G. Expression pattern of the mouse T gene and its role in mesoderm formation. Nature 1990, 343, 657-659. [CrossRef]
- Le Gouar, M.; Guillou, A.; Vervoort, M. Expression of a SoxB and a Wnt2/13 gene during the development of the mollusc Patella vulgata. Dev Genes Evol 2004, 214, 250-256. [CrossRef]
- Risbud, M.V.; Shapiro, I.M. Notochordal cells in the adult intervertebral disc: new perspective on an old question. Crit Rev Eukaryot Gene Expr 2011, 21, 29-41. [CrossRef]
- Sheppard, H.E.; Dall’Agnese, A.; Park, W.D.; Shamim, M.H.; Dubrulle, J.; Johnson, H.L.; Stossi, F.; Cogswell, P.; Sommer, J.; Levy, J.; et al. Targeted brachyury degradation disrupts a highly specific autoregulatory program controlling chordoma cell identity. Cell Rep Med 2021, 2, 100188. [CrossRef]
- Hamilton, D.H.; David, J.M.; Dominguez, C.; Palena, C. Development of Cancer Vaccines Targeting Brachyury, a Transcription Factor Associated with Tumor Epithelial-Mesenchymal Transition. Cells Tissues Organs 2017, 203, 128-138. [CrossRef]
- Robinson, H.; McFarlane, R.J.; Wakeman, J.A. Brachyury: Strategies for Drugging an Intractable Cancer Therapeutic Target. Trends Cancer 2020, 6, 271-273. [CrossRef]
- Ma, T.; Bai, J.; Zhang, Y. Current understanding of brachyury in chordoma. Biochim Biophys Acta Rev Cancer 2023, 189010. [CrossRef]
- Walcott, B.P.; Nahed, B.V.; Mohyeldin, A.; Coumans, J.V.; Kahle, K.T.; Ferreira, M.J. Chordoma: current concepts, management, and future directions. Lancet Oncol 2012, 13, e69-76. [CrossRef]
- Jensen, L.E.; Barbaux, S.; Hoess, K.; Fraterman, S.; Whitehead, A.S.; Mitchell, L.E. The human T locus and spina bifida risk. Hum Genet 2004, 115, 475-482. [CrossRef]
- Postma, A.V.; Alders, M.; Sylva, M.; Bilardo, C.M.; Pajkrt, E.; van Rijn, R.R.; Schulte-Merker, S.; Bulk, S.; Stefanovic, S.; Ilgun, A.; et al. Mutations in the T (brachyury) gene cause a novel syndrome consisting of sacral agenesis, abnormal ossification of the vertebral bodies and a persistent notochordal canal. J Med Genet 2014, 51, 90-97. [CrossRef]
- Chase, D.H.; Bebenek, A.M.; Nie, P.; Jaime-Figueroa, S.; Butrin, A.; Castro, D.A.; Hines, J.; Linhares, B.M.; Crews, C.M. Development of a Small Molecule Downmodulator for the Transcription Factor Brachyury. Angew Chem Int Ed Engl 2024, 63, e202316496. [CrossRef]
- Peters, T., Jr. Serum albumin. Adv Protein Chem 1985, 37, 161-245. [CrossRef]
- Peters, T. All About Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: San Diego, CA, 1996.
- Evans, T.W. Review article: albumin as a drug--biological effects of albumin unrelated to oncotic pressure. Aliment Pharmacol Ther 2002, 16 Suppl 5, 6-11. [CrossRef]
- Mendez, C.M.; McClain, C.J.; Marsano, L.S. Albumin therapy in clinical practice. Nutr Clin Pract 2005, 20, 314-320. [CrossRef]
- Pedersen, K.O. An analysis of measured and calculated calcium quantities in serum. Scand J Clin Lab Invest 1978, 38, 659-667. [CrossRef]
- Bradshaw, R.A.; Shearer, W.T.; Gurd, F.R. Sites of binding of copper (II) ion by peptide (1-24) of bovine serum albumin. J Biol Chem 1968, 243, 3817-3825. [CrossRef]
- Sarkar, B. Albumin as the major plasma protein transporting metals. Life Chem. Reports 1983, 1, 165-209.
- Gurd, F.R.; Wilcox, P.E. Complex formation between metallic cations and proteins, peptides and amino acids. Adv Protein Chem 1956, 11, 311-427. [CrossRef]
- Nandedkar, A.K.; Nurse, C.E.; Friedberg, F. Mn++ binding by plasma proteins. Int J Pept Protein Res 1973, 5, 279-281. [CrossRef]
- Klopfenstein, W.E. Thermodynamics of binding lysolecithin to serum albumin. Biochimica et Biophysica Acta (BBA)-Lipids and Lipid Metabolism 1969, 187, 272-274. [CrossRef]
- Roda, A.; Cappelleri, G.; Aldini, R.; Roda, E.; Barbara, L. Quantitative aspects of the interaction of bile acids with human serum albumin. Journal of lipid research 1982, 23, 490-495. [CrossRef]
- Savu, L.; Benassayag, C.; Vallette, G.; Christeff, N.; Nunez, E. Mouse alpha 1-fetoprotein and albumin. A comparison of their binding properties with estrogen and fatty acid ligands. J Biol Chem 1981, 256, 9414-9418. [CrossRef]
- Jacobsen, J. Studies of the affinity of human serum albumin for binding of bilirubin at different temperatures and ionic strength. Int J Pept Protein Res 1977, 9, 235-239. [CrossRef]
- Pearlman, W.H.; Crepy, O. Steroid-protein interaction with particular reference to testosterone binding by human serum. J Biol Chem 1967, 242, 182-189. [CrossRef]
- Unger, W. Binding of prostaglandin to human serum albumin. Journal of Pharmacy and Pharmacology 1972, 24, 470-477. [CrossRef]
- Sollenne, N.P.; Wu, H.L.; Means, G.E. Disruption of the tryptophan binding site in the human serum albumin dimer. Arch Biochem Biophys 1981, 207, 264-269. [CrossRef]
- Kragh-Hansen, U. Molecular aspects of ligand binding to serum albumin. Pharmacol Rev 1981, 33, 17-53. [CrossRef]
- Biere, A.L.; Ostaszewski, B.; Stimson, E.R.; Hyman, B.T.; Maggio, J.E.; Selkoe, D.J. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem 1996, 271, 32916-32922. [CrossRef]
- He, X.M.; Carter, D.C. Atomic structure and chemistry of human serum albumin. Nature 1992, 358, 209-215. [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: from bench to bedside. Mol Aspects Med 2012, 33, 209-290. [CrossRef]
- Gupta, D.; Lis, C.G. Pretreatment serum albumin as a predictor of cancer survival: a systematic review of the epidemiological literature. Nutr J 2010, 9, 69. [CrossRef]
- Koga, M.; Kasayama, S. Clinical impact of glycated albumin as another glycemic control marker. Endocr J 2010, 57, 751-762. [CrossRef]
- Sbarouni, E.; Georgiadou, P.; Voudris, V. Ischemia modified albumin changes - review and clinical implications. Clin Chem Lab Med 2011, 49, 177-184. [CrossRef]
- Shevtsova, A.; Gordiienko, I.; Tkachenko, V.; Ushakova, G. Ischemia-Modified Albumin: Origins and Clinical Implications. Dis Markers 2021, 2021, 9945424. [CrossRef]
- Jiang, A.A.; Greenwald, H.S.; Sheikh, L.; Wooten, D.A.; Malhotra, A.; Schooley, R.T.; Sweeney, D.A. Predictors of Acute Liver Failure in Patients With Acute Hepatitis A: An Analysis of the 2016-2018 San Diego County Hepatitis A Outbreak. Open Forum Infect Dis 2019, 6, ofz467. [CrossRef]
- Tillmann, H.L.; Manns, M.P.; Rudolph, K.L. Merging models of hepatitis C virus pathogenesis. Semin Liver Dis 2005, 25, 84-92. [CrossRef]
- Chitturi, S.; George, J. Predictors of liver-related complications in patients with chronic hepatitis C. Ann Med 2000, 32, 588-591. [CrossRef]
- Jeng, L.B.; Li, T.C.; Hsu, S.C.; Chan, W.L.; Teng, C.F. Association of Low Serum Albumin Level with Higher Hepatocellular Carcinoma Recurrence in Patients with Hepatitis B Virus Pre-S2 Mutant after Curative Surgical Resection. J Clin Med 2021, 10. [CrossRef]
- Michalak, T.; Krawczynski, K.; Ostrowski, J.; Nowoslawski, A. Hepatitis B surface antigen and albumin in human hepatocytes. An immunofluorescent and immunoelectron microscopic study. Gastroenterology 1980, 79, 1151-1158. [CrossRef]
- Thung, S.N.; Wang, D.F.; Fasy, T.M.; Hood, A.; Gerber, M.A. Hepatitis B surface antigen binds to human serum albumin cross-linked by transglutaminase. Hepatology 1989, 9, 726-730. [CrossRef]
- Halder, K.; Sengupta, P.; Chaki, S.; Saha, R.; Dasgupta, S. Understanding Conformational Changes in Human Serum Albumin and Its Interactions with Gold Nanorods: Do Flexible Regions Play a Role in Corona Formation? Langmuir 2023, 39, 1651-1664. [CrossRef]
- Litus, E.A.; Permyakov, S.E.; Uversky, V.N.; Permyakov, E.A. Intrinsically disordered regions in serum albumin: what are they for? Cell Biochemistry and Biophysics 2018, 76, 39-57. [CrossRef]
- Zhang, N.; Bevan, M.J. CD8(+) T cells: foot soldiers of the immune system. Immunity 2011, 35, 161-168. [CrossRef]
- Srinivasan, S.; Zhu, C.; McShan, A.C. Structure, function, and immunomodulation of the CD8 co-receptor. Front Immunol 2024, 15, 1412513. [CrossRef]
- Xu, Q.; Chen, Y.; Zhao, W.M.; Huang, Z.Y.; Zhang, Y.; Li, X.; Tong, Y.Y.; Chang, G.B.; Duan, X.J.; Chen, G.H. DNA methylation and regulation of the CD8A after duck hepatitis virus type 1 infection. PLoS One 2014, 9, e88023. [CrossRef]
- Tregaskes, C.; Kong, F.; Paramithiotis, E.; Chen, C.; Ratcliffe, M.; Davison, T.; Young, J. Identification and analysis of the expression of CD8 alpha beta and CD8 alpha alpha isoforms in chickens reveals a major TCR-gamma delta CD8 alpha beta subset of intestinal intraepithelial lymphocytes. Journal of immunology (Baltimore, Md.: 1950) 1995, 154, 4485-4494. [CrossRef]
- Gao, G.F.; Jakobsen, B.K. Molecular interactions of coreceptor CD8 and MHC class I: the molecular basis for functional coordination with the T-cell receptor. Immunol Today 2000, 21, 630-636. [CrossRef]
- Alromaih, S.; Mfuna-Endam, L.; Bosse, Y.; Filali-Mouhim, A.; Desrosiers, M. CD8A gene polymorphisms predict severity factors in chronic rhinosinusitis. Int Forum Allergy Rhinol 2013, 3, 605-611. [CrossRef]
- Zhou, S.; Lu, H.; Xiong, M. Identifying Immune Cell Infiltration and Effective Diagnostic Biomarkers in Rheumatoid Arthritis by Bioinformatics Analysis. Front Immunol 2021, 12, 726747. [CrossRef]
- Ma, K.; Qiao, Y.; Wang, H.; Wang, S. Comparative expression analysis of PD-1, PD-L1, and CD8A in lung adenocarcinoma. Ann Transl Med 2020, 8, 1478. [CrossRef]
- Du, Y.; Zuo, L.; Xiong, Y.; Wang, X.; Zou, J.; Xu, H. CD8A is a Promising Biomarker Associated with Immunocytes Infiltration in Hyperoxia-Induced Bronchopulmonary Dysplasia. J Inflamm Res 2023, 16, 1653-1669. [CrossRef]
- Bai, Y.M.; Liang, S.; Zhou, B. Revealing immune infiltrate characteristics and potential immune-related genes in hepatic fibrosis: based on bioinformatics, transcriptomics and q-PCR experiments. Front Immunol 2023, 14, 1133543. [CrossRef]
- Song, J.; Liu, L.; Wang, Z.; Xie, D.; Azami, N.L.B.; Lu, L.; Huang, Y.; Ye, W.; Zhang, Q.; Sun, M. CCL20 and CD8A as potential diagnostic biomarkers for HBV-induced liver fibrosis in chronic hepatitis B. Heliyon 2024, 10, e28329. [CrossRef]
- Sriraman, S.K.; Davies, C.W.; Gill, H.; Kiefer, J.R.; Yin, J.; Ogasawara, A.; Urrutia, A.; Javinal, V.; Lin, Z.; Seshasayee, D.; et al. Development of an (18)F-labeled anti-human CD8 VHH for same-day immunoPET imaging. Eur J Nucl Med Mol Imaging 2023, 50, 679-691. [CrossRef]
- Chen, X.; Mirazee, J.M.; Skorupka, K.A.; Matsuo, H.; Youkharibache, P.; Taylor, N.; Walters, K.J. The CD8alpha hinge is intrinsically disordered with a dynamic exchange that includes proline cis-trans isomerization. J Magn Reson 2022, 340, 107234. [CrossRef]
- Bergstrand, C.G.; Czar, B. Demonstration of a new protein fraction in serum from the human fetus. Scand J Clin Lab Invest 1956, 8, 174. [CrossRef]
- Galdo, A.; Casado, J.P.; Talavera, R. [Demonstration of a new protein fraction in the serum of the human fetus by means of paper electrophoresis]. Arch Fr Pediatr 1959, 16, 954-962.
- Ruoslahti, E.; Seppala, M. alpha-Foetoprotein in normal human serum. Nature 1972, 235, 161-162. [CrossRef]
- Deutsch, H.F. Chemistry and biology of alpha-fetoprotein. Adv Cancer Res 1991, 56, 253-312. [CrossRef]
- Gillespie, J.R.; Uversky, V.N. Structure and function of alpha-fetoprotein: a biophysical overview. Biochim Biophys Acta 2000, 1480, 41-56. [CrossRef]
- Mizejewski, G.J. Alpha-fetoprotein structure and function: relevance to isoforms, epitopes, and conformational variants. Exp Biol Med (Maywood) 2001, 226, 377-408. [CrossRef]
- Terentiev, A.A.; Moldogazieva, N.T. Structural and functional mapping of alpha-fetoprotein. Biochemistry (Mosc) 2006, 71, 120-132. [CrossRef]
- Tatarinov, J. Content of embryo-specific alpha-globulin in the blood serum of human fetus, newborn and adult man in primary cancer. Vopr. Med. Khim. 1964, 10, 90-91.
- Abelev, G. Alpha-fetoprotein in ontogenesis and its association with malignant tumors. Advances in cancer research 1971, 14, 295-358.
- Tatarinov, Y.S. Detection of embryospecific alpha-globulin in serum of patients with primary liver cancer. In Proceedings of the 1st All-Union Biochem Congress Abstract Book. Moscow–Leningrad, 1963.
- Okuda, K.; Kotoda, K.; Obata, H.; Hayashi, N.; Hisamitsu, T. Clinical observations during a relatively early stage of hepatocellular carcinoma, with special reference to serum alpha-fetoprotein levels. Gastroenterology 1975, 69, 226-234. [CrossRef]
- Terentiev, A.A.; Moldogazieva, N.T. Alpha-fetoprotein: a renaissance. Tumour Biol 2013, 34, 2075-2091. [CrossRef]
- Nochomovitz, L.E.; DeLa Torre, F.E.; Rosai, J. Pathology of germ cell tumors of the testis. Urol Clin North Am 1977, 4, 359-378. [CrossRef]
- Itoh, T.; Kishi, K.; Tojo, M.; Kitajima, N.; Kinoshita, Y.; Inatome, T.; Fukuzaki, H.; Nishiyama, N.; Tachibana, H.; Takahashi, H.; et al. Acinar cell carcinoma of the pancreas with elevated serum alpha-fetoprotein levels: a case report and a review of 28 cases reported in Japan. Gastroenterol Jpn 1992, 27, 785-791. [CrossRef]
- Yoshiki, T.; Itoh, T.; Shirai, T.; Noro, T.; Tomino, Y.; Hama Jima, T.I. Primary intracranial yolk sac tumor: immunofluorescent demonstration of alpha-fetoprotein synthesis. Cancer 1976, 37, 2343-2348. [CrossRef]
- Seppala, M.; Ruoslahti, E. Alpha fetoprotein: Physiology and pathology during pregnancy and application to antenatal diagnosis. J Perinat Med 1973, 1, 104-113. [CrossRef]
- Brock, D.J.; Scrimgeour, J.B.; Nelson, M.M. Amniotic fluid alphafetoprotein measurements in the early prenatal diagnosis of central nervous system disorders. Clin Genet 1975, 7, 163-169. [CrossRef]
- Seppala, M. Fetal pathophysiology of human alpha-fetoprotein. Ann N Y Acad Sci 1975, 259, 59-73. [CrossRef]
- Ruoslahti, E.; Seppala, M. alpha-Fetoprotein in cancer and fetal development. Adv Cancer Res 1979, 29, 275-346. [CrossRef]
- Yang, S.S.; Cheng, K.S.; Lai, Y.C.; Wu, C.H.; Chen, T.K.; Lee, C.L.; Chen, D.S. Decreasing serum alpha-fetoprotein levels in predicting poor prognosis of acute hepatic failure in patients with chronic hepatitis B. J Gastroenterol 2002, 37, 626-632. [CrossRef]
- Di Bisceglie, A.M.; Hoofnagle, J.H. Elevations in serum alpha-fetoprotein levels in patients with chronic hepatitis B. Cancer 1989, 64, 2117-2120. [CrossRef]
- Liaw, Y.F.; Tai, D.I.; Chen, T.J.; Chu, C.M.; Huang, M.J. Alpha-fetoprotein changes in the course of chronic hepatitis: relation to bridging hepatic necrosis and hepatocellular carcinoma. Liver 1986, 6, 133-137. [CrossRef]
- Bayati, N.; Silverman, A.L.; Gordon, S.C. Serum alpha-fetoprotein levels and liver histology in patients with chronic hepatitis C. Am J Gastroenterol 1998, 93, 2452-2456. [CrossRef]
- Goldstein, N.S.; Blue, D.E.; Hankin, R.; Hunter, S.; Bayati, N.; Silverman, A.L.; Gordon, S.C. Serum alpha-fetoprotein levels in patients with chronic hepatitis C. Relationships with serum alanine aminotransferase values, histologic activity index, and hepatocyte MIB-1 scores. Am J Clin Pathol 1999, 111, 811-816. [CrossRef]
- Chu, C.W.; Hwang, S.J.; Luo, J.C.; Lai, C.R.; Tsay, S.H.; Li, C.P.; Wu, J.C.; Chang, F.Y.; Lee, S.D. Clinical, virologic, and pathologic significance of elevated serum alpha-fetoprotein levels in patients with chronic hepatitis C. J Clin Gastroenterol 2001, 32, 240-244. [CrossRef]
- Tagliabracci, V.S.; Wiley, S.E.; Guo, X.; Kinch, L.N.; Durrant, E.; Wen, J.; Xiao, J.; Cui, J.; Nguyen, K.B.; Engel, J.L.; et al. A Single Kinase Generates the Majority of the Secreted Phosphoproteome. Cell 2015, 161, 1619-1632. [CrossRef]
- Yoshima, H.; Mizuochi, T.; Ishii, M.; Kobata, A. Structure of the asparagine-linked sugar chains of alpha-fetoprotein purified from human ascites fluid. Cancer Res 1980, 40, 4276-4281.
- Mizejewski, G. α-fetoprotein as a biologic response modifier: relevance to domain and subdomain structure. Proceedings of the Society for Experimental Biology and Medicine 1997, 215, 333-362. [CrossRef]
- Breborowicz, J. Microheterogeneity of human alphafetoprotein. Tumour Biol 1988, 9, 3-14. [CrossRef]
- Lester, E.P.; Miller, J.B.; Yachnin, S. Human alpha-fetoprotein: immunosuppressive activity and microheterogeneity. Immunol Commun 1978, 7, 137-161. [CrossRef]
- Zheng, L.; Liu, N.; Gao, X.; Zhu, W.; Liu, K.; Wu, C.; Yan, R.; Zhang, J.; Gao, X.; Yao, Y.; et al. Uniform thin ice on ultraflat graphene for high-resolution cryo-EM. Nat Methods 2023, 20, 123-130. [CrossRef]
- Morinaga, T.; Sakai, M.; Wegmann, T.G.; Tamaoki, T. Primary structures of human alpha-fetoprotein and its mRNA. Proc Natl Acad Sci U S A 1983, 80, 4604-4608. [CrossRef]
- Eang, R.; Girbal-Neuhauser, E.; Xu, B.; Gairin, J.E. Characterization and differential expression of a newly identified phosphorylated isoform of the human 20S proteasome beta7 subunit in tumor vs. normal cell lines. Fundam Clin Pharmacol 2009, 23, 215-224. [CrossRef]
- Jayarapu, K.; Griffin, T.A. Protein–protein interactions among human 20S proteasome subunits and proteassemblin. Biochemical and biophysical research communications 2004, 314, 523-528. [CrossRef]
- Rho, J.H.; Qin, S.; Wang, J.Y.; Roehrl, M.H. Proteomic expression analysis of surgical human colorectal cancer tissues: up-regulation of PSB7, PRDX1, and SRP9 and hypoxic adaptation in cancer. J Proteome Res 2008, 7, 2959-2972. [CrossRef]
- Yoon, J.Y.; Wang, J.Y.; Roehrl, M.H.A. An Investigation Into the Prognostic Significance of High Proteasome PSB7 Protein Expression in Colorectal Cancer. Front Med (Lausanne) 2020, 7, 401. [CrossRef]
- Munkacsy, G.; Abdul-Ghani, R.; Mihaly, Z.; Tegze, B.; Tchernitsa, O.; Surowiak, P.; Schafer, R.; Gyorffy, B. PSMB7 is associated with anthracycline resistance and is a prognostic biomarker in breast cancer. Br J Cancer 2010, 102, 361-368. [CrossRef]
- Lu, Y.; Liu, X.; Shi, S.; Su, H.; Bai, X.; Cai, G.; Yang, F.; Xie, Z.; Zhu, Y.; Zhang, Y.; et al. Bioinformatics analysis of proteomic profiles during the process of anti-Thy1 nephritis. Mol Cell Proteomics 2012, 11, M111 008755. [CrossRef]
- Zhu, Y.X.; Tiedemann, R.; Shi, C.X.; Yin, H.; Schmidt, J.E.; Bruins, L.A.; Keats, J.J.; Braggio, E.; Sereduk, C.; Mousses, S.; et al. RNAi screen of the druggable genome identifies modulators of proteasome inhibitor sensitivity in myeloma including CDK5. Blood 2011, 117, 3847-3857. [CrossRef]
- Wu, D.; Miao, J.; Hu, J.; Li, F.; Gao, D.; Chen, H.; Feng, Y.; Shen, Y.; He, A. PSMB7 Is a Key Gene Involved in the Development of Multiple Myeloma and Resistance to Bortezomib. Front Oncol 2021, 11, 684232. [CrossRef]
- Tan, Y.; Qin, S.; Hou, X.; Qian, X.; Xia, J.; Li, Y.; Wang, R.; Chen, C.; Yang, Q.; Miele, L.; et al. Proteomic-based analysis for identification of proteins involved in 5-fluorouracil resistance in hepatocellular carcinoma. Curr Pharm Des 2014, 20, 81-87. [CrossRef]
- Rouette, A.; Trofimov, A.; Haberl, D.; Boucher, G.; Lavallee, V.P.; D’Angelo, G.; Hebert, J.; Sauvageau, G.; Lemieux, S.; Perreault, C. Expression of immunoproteasome genes is regulated by cell-intrinsic and -extrinsic factors in human cancers. Sci Rep 2016, 6, 34019. [CrossRef]
- Zhang, Y.; Wu, J.; You, G.; Guo, W.; Wang, Y.; Yu, Z.; Geng, Y.; Zhong, Q.; Zan, J.; Zheng, L. Bioinformatic Analysis and Experimental Validation of Ubiquitin-Proteasomal System-Related Hub Genes as Novel Biomarkers for Alzheimer’s Disease. J Integr Neurosci 2023, 22, 138. [CrossRef]
- Ebstein, F.; Poli Harlowe, M.C.; Studencka-Turski, M.; Kruger, E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front Immunol 2019, 10, 2756. [CrossRef]
- Makjaroen, J.; Somparn, P.; Hodge, K.; Poomipak, W.; Hirankarn, N.; Pisitkun, T. Comprehensive Proteomics Identification of IFN-lambda3-regulated Antiviral Proteins in HBV-transfected Cells. Mol Cell Proteomics 2018, 17, 2197-2215. [CrossRef]
- Huang, J.; Kwong, J.; Sun, E.; Liang, T.J. Proteasome complex as a potential cellular target of hepatitis B virus X protein. Journal of virology 1996, 70, 5582-5591. [CrossRef]
- Zhang, Z.; Torii, N.; Furusaka, A.; Malayaman, N.; Hu, Z.; Liang, T.J. Structural and functional characterization of interaction between hepatitis B virus X protein and the proteasome complex. J Biol Chem 2000, 275, 15157-15165. [CrossRef]
- Hu, Z.; Zhang, Z.; Doo, E.; Coux, O.; Goldberg, A.L.; Liang, T.J. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. Journal of virology 1999, 73, 7231-7240. [CrossRef]
- Zhang, Z.; Protzer, U.; Hu, Z.; Jacob, J.; Liang, T.J. Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. Journal of virology 2004, 78, 4566-4572. [CrossRef]
- James, S.A.; Ong, H.S.; Hari, R.; Khan, A.M. A systematic bioinformatics approach for large-scale identification and characterization of host-pathogen shared sequences. BMC genomics 2021, 22, 1-18. [CrossRef]
- Wang, X.; Cimermancic, P.; Yu, C.; Schweitzer, A.; Chopra, N.; Engel, J.L.; Greenberg, C.; Huszagh, A.S.; Beck, F.; Sakata, E.; et al. Molecular Details Underlying Dynamic Structures and Regulation of the Human 26S Proteasome. Mol Cell Proteomics 2017, 16, 840-854. [CrossRef]
- Schweitzer, A.; Aufderheide, A.; Rudack, T.; Beck, F.; Pfeifer, G.; Plitzko, J.M.; Sakata, E.; Schulten, K.; Forster, F.; Baumeister, W. Structure of the human 26S proteasome at a resolution of 3.9 A. Proc Natl Acad Sci U S A 2016, 113, 7816-7821. [CrossRef]
- Stenmark, H.; Olkkonen, V.M. The Rab GTPase family. Genome Biol 2001, 2, REVIEWS3007. [CrossRef]
- Kirsten, M.L.; Baron, R.A.; Seabra, M.C.; Ces, O. Rab1a and Rab5a preferentially bind to binary lipid compositions with higher stored curvature elastic energy. Mol Membr Biol 2013, 30, 303-314. [CrossRef]
- Castellvi-Bel, S.; Mila, M. Genes responsible for nonspecific mental retardation. Mol Genet Metab 2001, 72, 104-108. [CrossRef]
- John, A.; Ng-Cordell, E.; Hanna, N.; Brkic, D.; Baker, K. The neurodevelopmental spectrum of synaptic vesicle cycling disorders. J Neurochem 2021, 157, 208-228. [CrossRef]
- Liu, L.; Liu, L.; Lu, Y.; Zhang, T.; Zhao, W. GDP dissociation inhibitor 1 (GDI1) attenuates beta-amyloid-induced neurotoxicity in Alzheimer’s diseases. Neurosci Lett 2024, 818, 137564. [CrossRef]
- Xie, X.; Lin, H.; Zhang, X.; Song, P.; He, X.; Zhong, J.; Shi, J. Overexpression of GDP dissociation inhibitor 1 gene associates with the invasiveness and poor outcomes of colorectal cancer. Bioengineered 2021, 12, 5595-5606. [CrossRef]
- Kiyota, A.; Iwama, S.; Sugimura, Y.; Takeuchi, S.; Takagi, H.; Iwata, N.; Nakashima, K.; Suzuki, H.; Nishioka, T.; Kato, T.; et al. Identification of the novel autoantigen candidate Rab GDP dissociation inhibitor alpha in isolated adrenocorticotropin deficiency. Endocr J 2015, 62, 153-160. [CrossRef]
- Erdem-Eraslan, L.; Heijsman, D.; de Wit, M.; Kremer, A.; Sacchetti, A.; van der Spek, P.J.; Sillevis Smitt, P.A.; French, P.J. Tumor-specific mutations in low-frequency genes affect their functional properties. J Neurooncol 2015, 122, 461-470. [CrossRef]
- Li, W.; Wang, H.; Jin, X.; Zhao, L. Loss of RhoGDI is a novel independent prognostic factor in hepatocellular carcinoma. Int J Clin Exp Pathol 2013, 6, 2535-2541.
- Lai, M.C.; Zhu, Q.Q.; Owusu-Ansah, K.G.; Zhu, Y.B.; Yang, Z.; Xie, H.Y.; Zhou, L.; Wu, L.M.; Zheng, S.S. Prognostic value of Rho GDP dissociation inhibitors in patients with hepatocellular carcinoma following liver transplantation. Oncol Lett 2017, 14, 1395-1402. [CrossRef]
- Montaldo, C.; Terri, M.; Riccioni, V.; Battistelli, C.; Bordoni, V.; D’Offizi, G.; Prado, M.G.; Trionfetti, F.; Vescovo, T.; Tartaglia, E.; et al. Fibrogenic signals persist in DAA-treated HCV patients after sustained virological response. J Hepatol 2021, 75, 1301-1311. [CrossRef]
- Larsen, R.D.; Ernst, L.K.; Nair, R.P.; Lowe, J.B. Molecular cloning, sequence, and expression of a human GDP-L-fucose:beta-D-galactoside 2-alpha-L-fucosyltransferase cDNA that can form the H blood group antigen. Proc Natl Acad Sci U S A 1990, 87, 6674-6678. [CrossRef]
- Intharanut, K.; Khumsuk, P.; Chidtrakoon, S.; Glab-Ampai, K.; Nathalang, O. Identification of two novel variants, c.-35A>T and c.[-35A>T, 725T>G], in the FUT1 gene in a patient exhibiting the para-Bombay phenotype. Transfusion 2024, 64, 2252-2259. [CrossRef]
- Scharberg, E.A.; Olsen, C.; Bugert, P. The H blood group system. Immunohematology 2016, 32, 112-118. [CrossRef]
- Vasconcelos, A.O.; Vieira, L.M.; Rocha, C.R.C.; Beltrao, E.I.C. The contribution of fucosyltransferases to cancer biology. Braz J Biol 2024, 84, e278681. [CrossRef]
- Tan, K.P.; Ho, M.Y.; Cho, H.C.; Yu, J.; Hung, J.T.; Yu, A.L. Fucosylation of LAMP-1 and LAMP-2 by FUT1 correlates with lysosomal positioning and autophagic flux of breast cancer cells. Cell Death Dis 2016, 7, e2347. [CrossRef]
- Gao, S.; Zhu, L.; Feng, H.; Hu, Z.; Jin, S.; Song, Z.; Liu, D.; Liu, J.; Hao, Y.; Li, X.; et al. Gene expression profile analysis in response to alpha1,2-fucosyl transferase (FUT1) gene transfection in epithelial ovarian carcinoma cells. Tumour Biol 2016, 37, 12251-12262. [CrossRef]
- Kuo, H.H.; Lin, R.J.; Hung, J.T.; Hsieh, C.B.; Hung, T.H.; Lo, F.Y.; Ho, M.Y.; Yeh, C.T.; Huang, Y.L.; Yu, J.; et al. High expression FUT1 and B3GALT5 is an independent predictor of postoperative recurrence and survival in hepatocellular carcinoma. Sci Rep 2017, 7, 10750. [CrossRef]
- Papic, N.; Maxwell, C.I.; Delker, D.A.; Liu, S.; Heale, B.S.; Hagedorn, C.H. RNA-sequencing analysis of 5’ capped RNAs identifies many new differentially expressed genes in acute hepatitis C virus infection. Viruses 2012, 4, 581-612. [CrossRef]
- Gutierrez-Huante, K.; Salinas-Marin, R.; Mora-Montes, H.M.; Gonzalez, R.A.; Martinez-Duncker, I. Human adenovirus type 5 increases host cell fucosylation and modifies Ley antigen expression. Glycobiology 2019, 29, 469-478. [CrossRef]
- Li, Z.; Zhang, N.; Dong, Z.; Wang, X.; Zhou, J.; Gao, J.; Yang, Y.; Li, J.; Guan, F.; Zhou, Y.; et al. Integrating transcriptomics, glycomics and glycoproteomics to characterize hepatitis B virus-associated hepatocellular carcinoma. Cell Commun Signal 2024, 22, 200. [CrossRef]
Figure 1.
Per-residue intrinsic disorder profile generated for genome polyprotein from human hepatitis A virus genotype IB (isolate HM175) (HHAV) (human hepatitis A virus (isolate Human/Australia/HM175/1976) (UniProt ID: P08617) by RIDAO. The outputs of PONDR® VLXT, PONDR® VSL2, PONDR® VL3, PONDR® FIT, IUPred long, and IUPred short are shown by black, red, green, pink, blue, and yellow lines, respectively. Mean disorder profile (or mean disorder prediction, MDP) calculated as an average of outputs of these six predictors is shown by dashed dark pink line, whereas error distribution are shown as light pink shadow. In this per-residue disorder analysis, a disorder score was assigned to each residue. A residue with disorder score equal to or above 0.5 is considered as disordered and a residue with disorder score below 0.5 is predicted as ordered. Residues/regions with disorder scores between 0.15 and 0.5 were considered as ordered but flexible. The corresponding thresholds are shown by solid (0.5) and long-dashed lines (0.15). Positions of cleavage sites leading to the release of mature viral proteins are indicated by verical dark red bars.
Figure 1.
Per-residue intrinsic disorder profile generated for genome polyprotein from human hepatitis A virus genotype IB (isolate HM175) (HHAV) (human hepatitis A virus (isolate Human/Australia/HM175/1976) (UniProt ID: P08617) by RIDAO. The outputs of PONDR® VLXT, PONDR® VSL2, PONDR® VL3, PONDR® FIT, IUPred long, and IUPred short are shown by black, red, green, pink, blue, and yellow lines, respectively. Mean disorder profile (or mean disorder prediction, MDP) calculated as an average of outputs of these six predictors is shown by dashed dark pink line, whereas error distribution are shown as light pink shadow. In this per-residue disorder analysis, a disorder score was assigned to each residue. A residue with disorder score equal to or above 0.5 is considered as disordered and a residue with disorder score below 0.5 is predicted as ordered. Residues/regions with disorder scores between 0.15 and 0.5 were considered as ordered but flexible. The corresponding thresholds are shown by solid (0.5) and long-dashed lines (0.15). Positions of cleavage sites leading to the release of mature viral proteins are indicated by verical dark red bars.
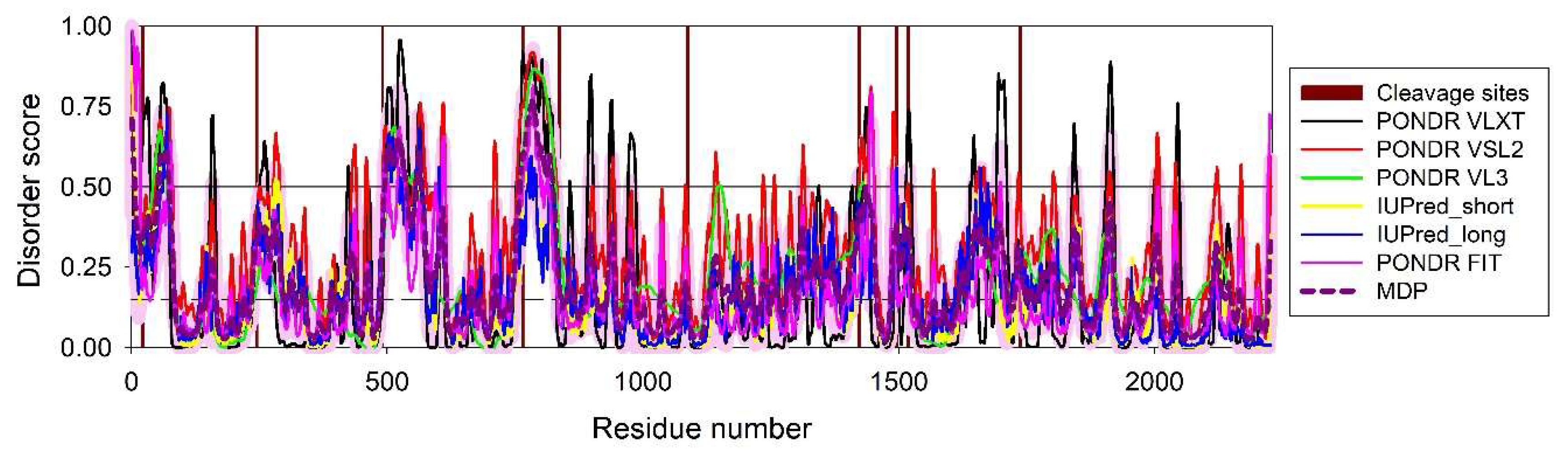
Figure 2.
PPI network between the HAV and Pan troglodytes proteins generated using the STRING_Viruses platform. This network includes 7 HAV proteins and 486 Pan troglodytes proteins. Note that with these settings, instead of human proteins, STRING_Viruses pulled out Pan troglodytes proteins. This is no surprising as chimpanzee is considered as the most appropriate animal model for the analysis if HAV infection [85]. Since the euchromatic regions of chimpanzee (Pan troglodytes) genome share ~98% sequence similarity with the human (Homo sapiens) [86], the derived set of the host proteins interacting with HAV can be used for the purposes of this study.
Figure 2.
PPI network between the HAV and Pan troglodytes proteins generated using the STRING_Viruses platform. This network includes 7 HAV proteins and 486 Pan troglodytes proteins. Note that with these settings, instead of human proteins, STRING_Viruses pulled out Pan troglodytes proteins. This is no surprising as chimpanzee is considered as the most appropriate animal model for the analysis if HAV infection [85]. Since the euchromatic regions of chimpanzee (Pan troglodytes) genome share ~98% sequence similarity with the human (Homo sapiens) [86], the derived set of the host proteins interacting with HAV can be used for the purposes of this study.
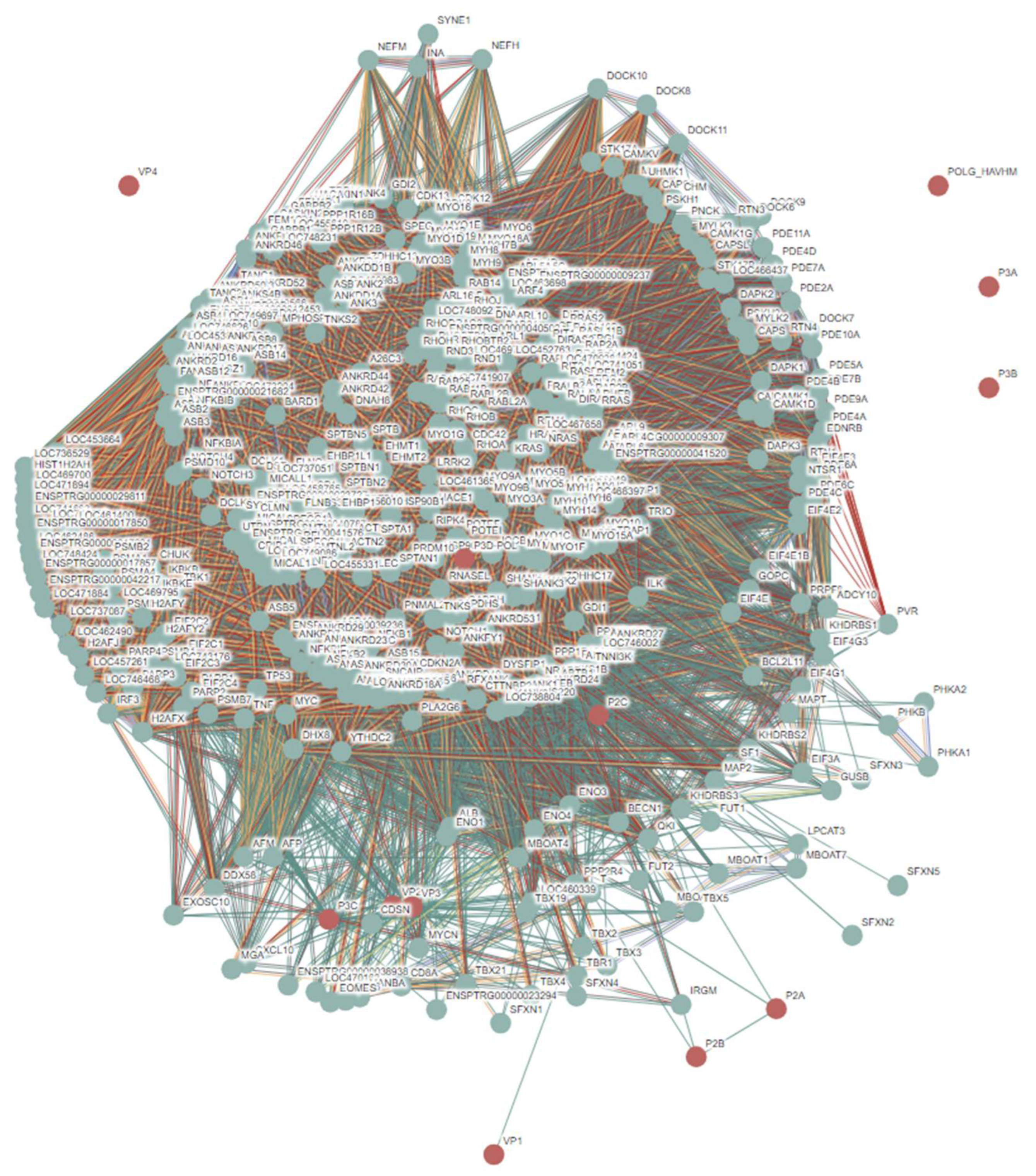
Figure 3.
Evaluation of intrinsic disorder propensity of proteins in hepatitis B virus genotype D subtype ayw (isolate France/Tiollais/1979) (HBV-D). A. Protein P (UniProt ID: P03156); B. Large envelope protein (UniProt ID: P03138); C. Capsid protein (UniProt ID: P03146); D. Protein X (UniProt ID: P03165); E. External core antigen (UniProt ID P0C573); F. Putative uncharacterized 15.3 kDa protein (UniProt ID: P03164); G. Putative uncharacterized 10.4 kDa protein (UniProt ID: P03163).
Figure 3.
Evaluation of intrinsic disorder propensity of proteins in hepatitis B virus genotype D subtype ayw (isolate France/Tiollais/1979) (HBV-D). A. Protein P (UniProt ID: P03156); B. Large envelope protein (UniProt ID: P03138); C. Capsid protein (UniProt ID: P03146); D. Protein X (UniProt ID: P03165); E. External core antigen (UniProt ID P0C573); F. Putative uncharacterized 15.3 kDa protein (UniProt ID: P03164); G. Putative uncharacterized 10.4 kDa protein (UniProt ID: P03163).
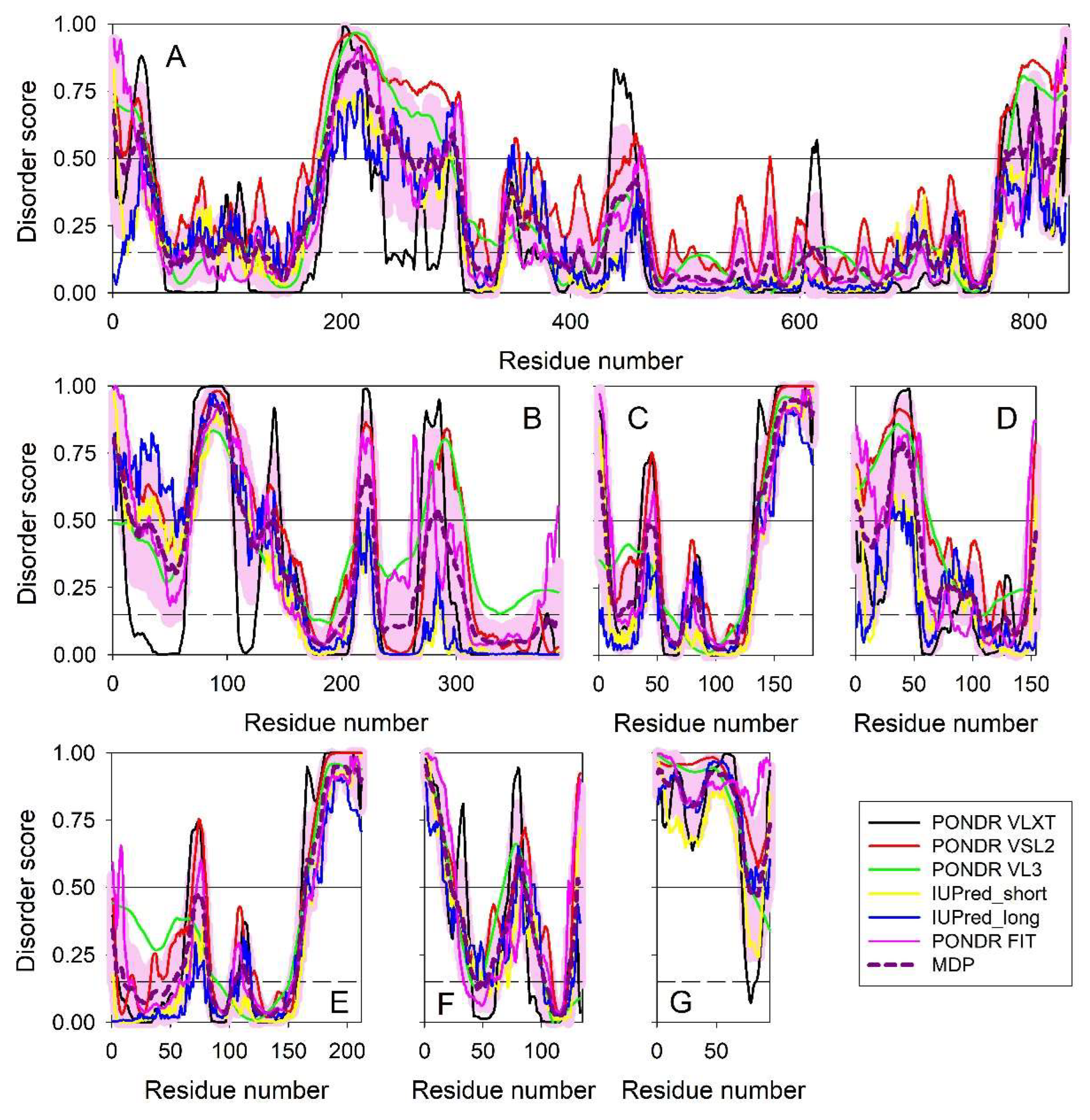
Figure 4.
PPI network between the HBV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.
Figure 4.
PPI network between the HBV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.
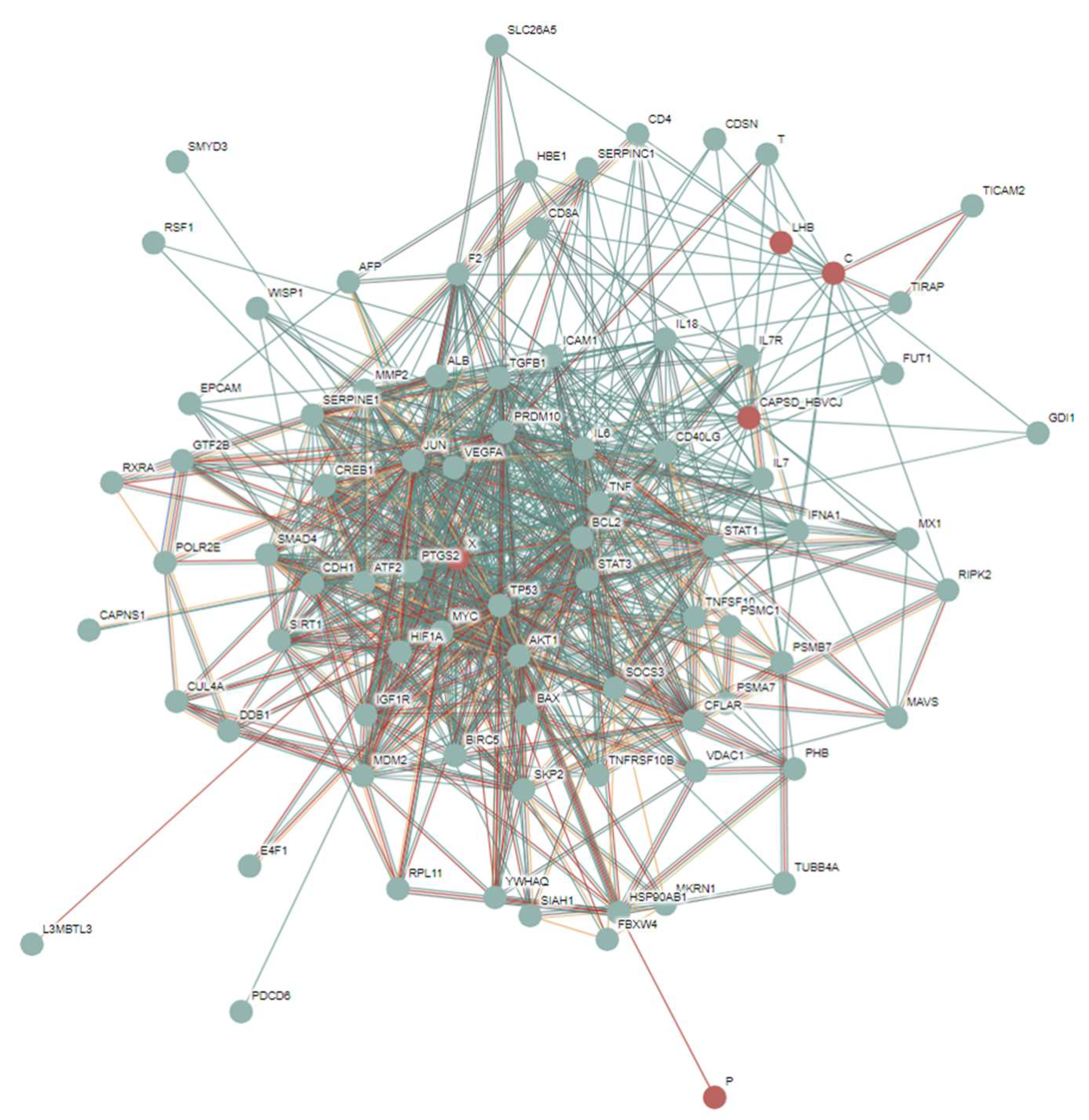
Figure 5.
Per-residue intrinsic disorder profile generated for genome polyprotein from Hepatitis C virus genotype 1a (isolate H77) (HCV) (UniProt ID: P27958) by RIDAO.
Figure 5.
Per-residue intrinsic disorder profile generated for genome polyprotein from Hepatitis C virus genotype 1a (isolate H77) (HCV) (UniProt ID: P27958) by RIDAO.
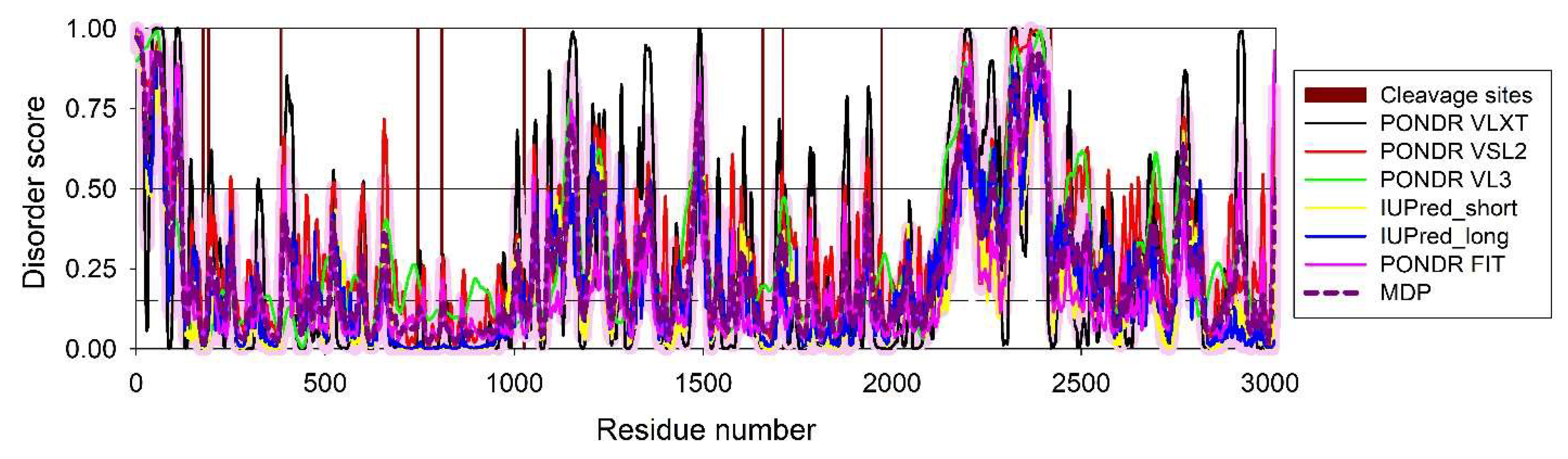
Figure 6.
PPI network between the HCV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.
Figure 6.
PPI network between the HCV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.

Figure 7.
Per-residue intrinsic disorder profiles generated for S-HDAg (UniProt ID: P0C6L3; A) and L-HDAg (UniProt ID: P29996; B) from hepatitis delta virus genotype I (isolate D380).
Figure 7.
Per-residue intrinsic disorder profiles generated for S-HDAg (UniProt ID: P0C6L3; A) and L-HDAg (UniProt ID: P29996; B) from hepatitis delta virus genotype I (isolate D380).
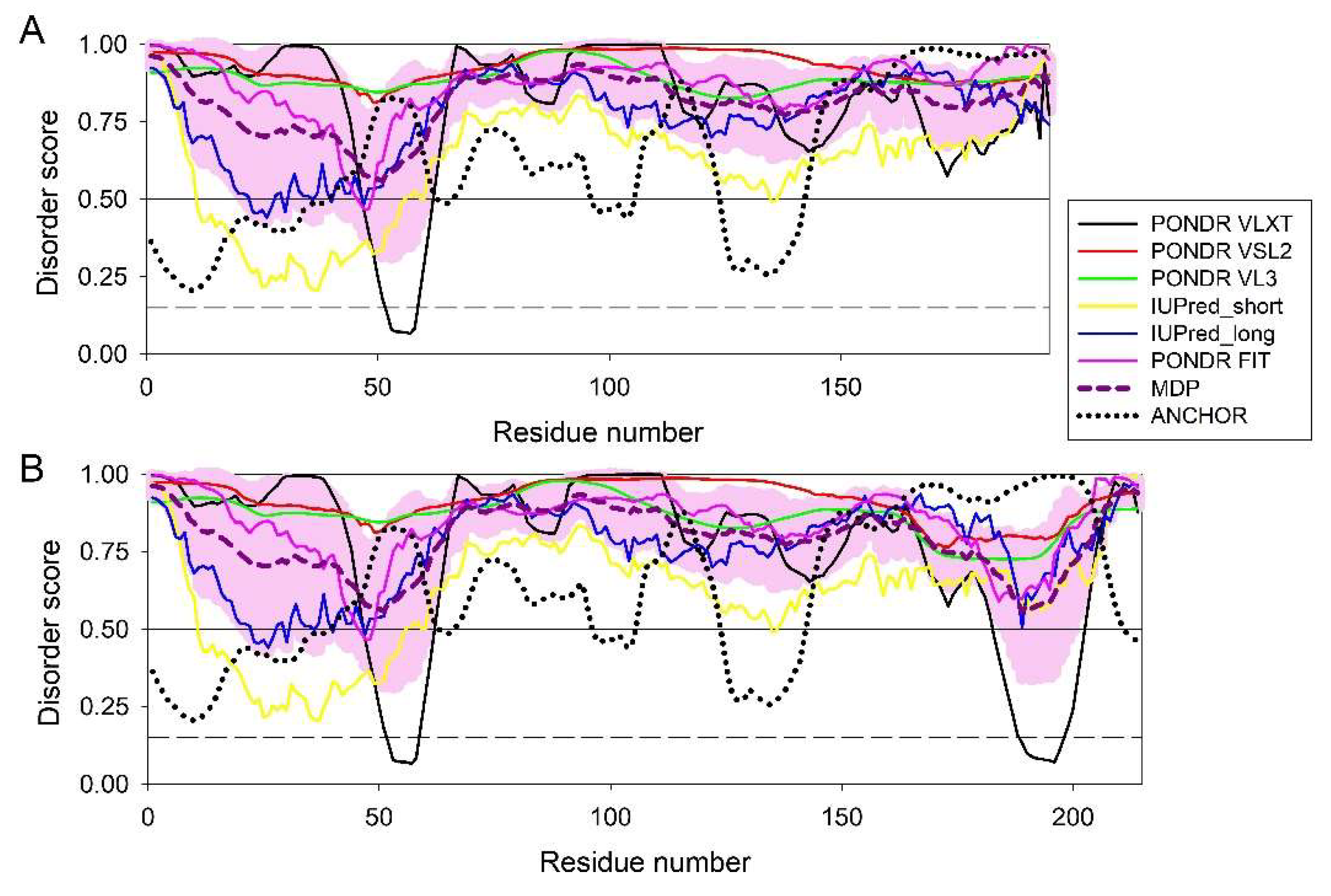
Figure 8.
PPI network between the HDV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.
Figure 8.
PPI network between the HDV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.
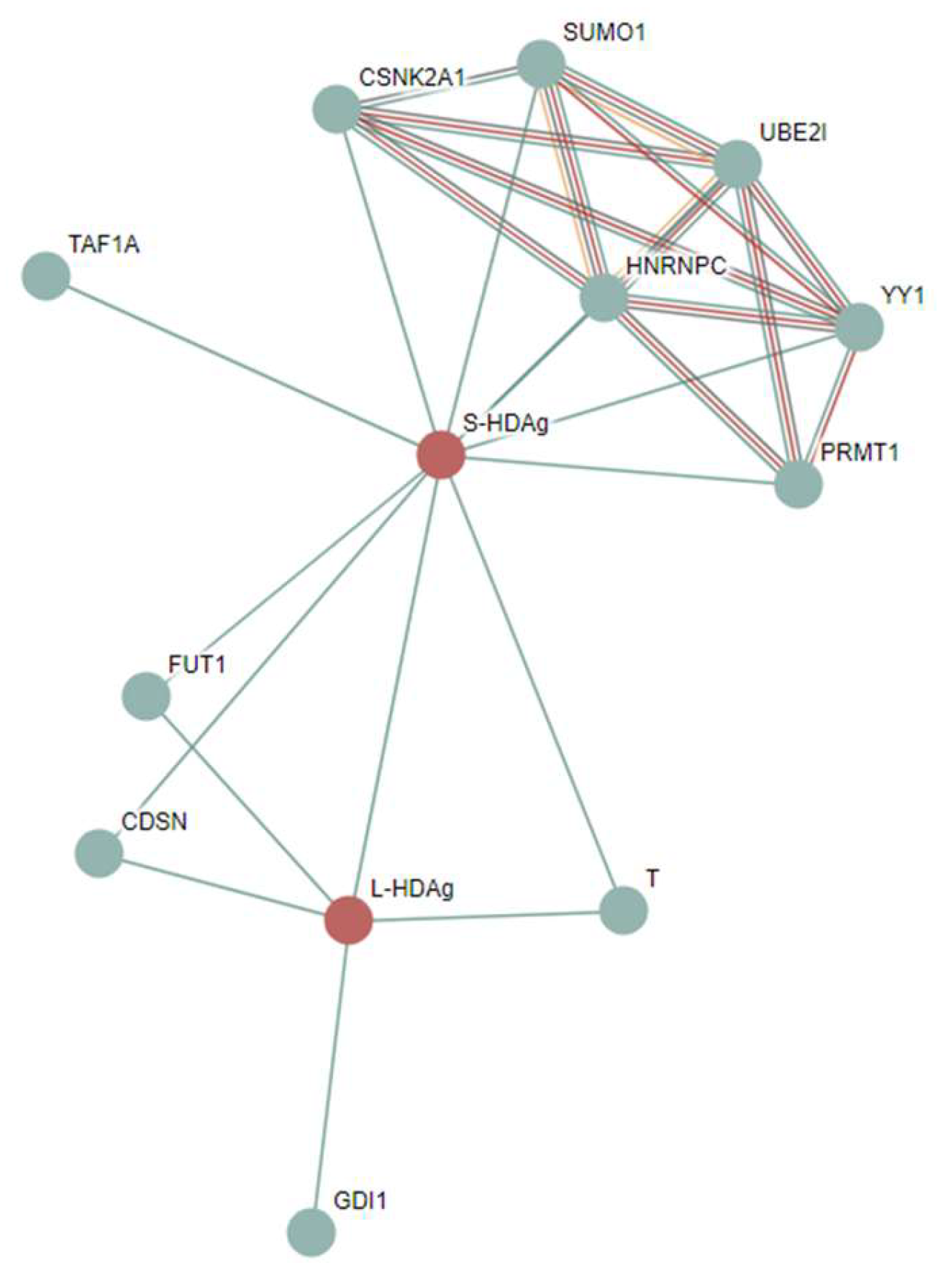
Figure 9.
RIDAO-based analysis of the intrinsic disorder propensity of proteins from the hepatitis E virus genotype 1 (isolate Human/China/HeBei/1987) (HEV). A. Non-structural polyprotein ORF1 (UniProt ID: Q81862); B. Pro-secreted protein ORF2 (UniProt ID: Q81871); C. Protein ORF3 (UniProt ID: Q81870).
Figure 9.
RIDAO-based analysis of the intrinsic disorder propensity of proteins from the hepatitis E virus genotype 1 (isolate Human/China/HeBei/1987) (HEV). A. Non-structural polyprotein ORF1 (UniProt ID: Q81862); B. Pro-secreted protein ORF2 (UniProt ID: Q81871); C. Protein ORF3 (UniProt ID: Q81870).
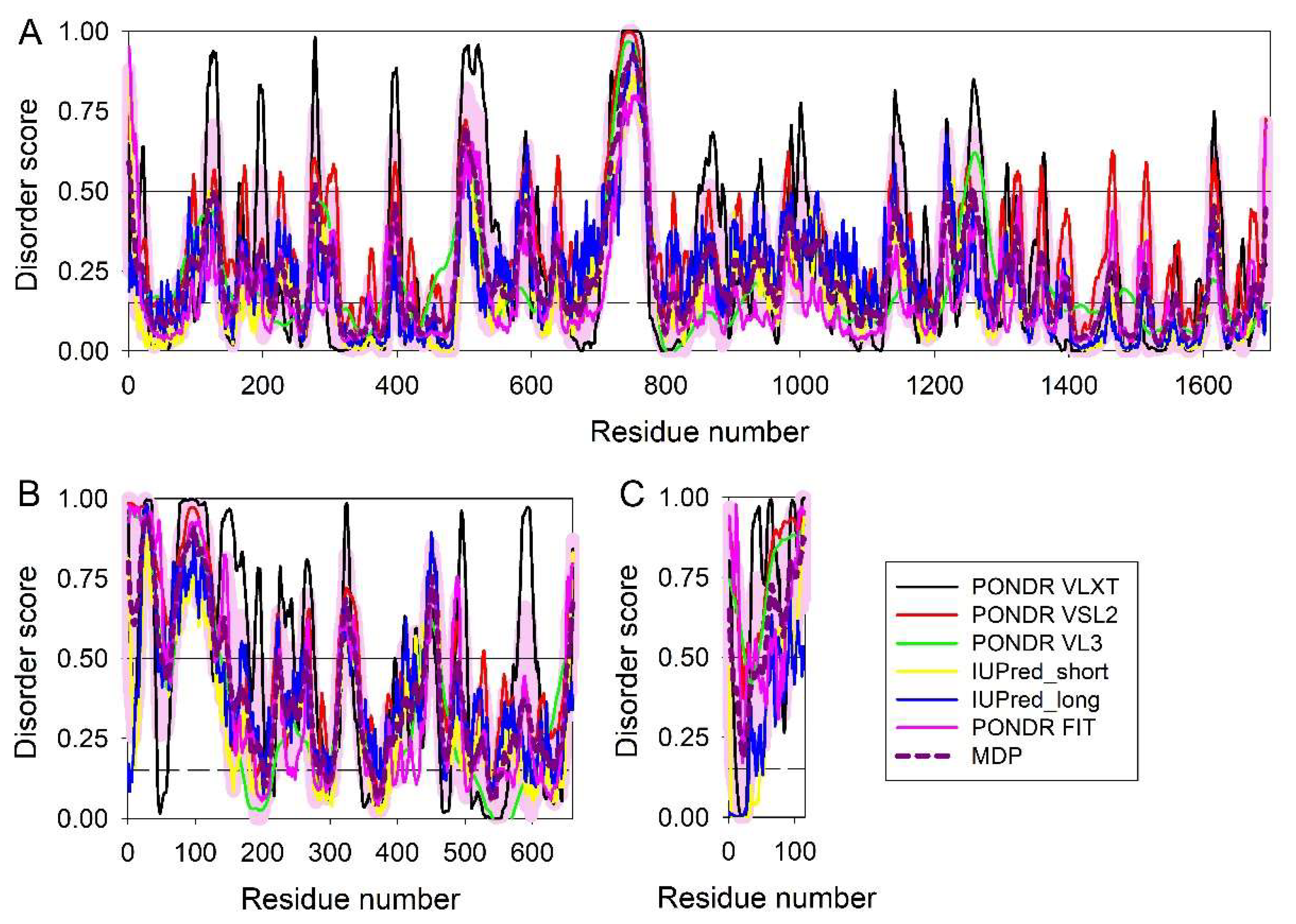
Figure 10.
PPI network between the HEV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.
Figure 10.
PPI network between the HEV and human proteins generated by the STRING_Viruses platform using medium confidence of 0.4 for the minimum required interaction score.
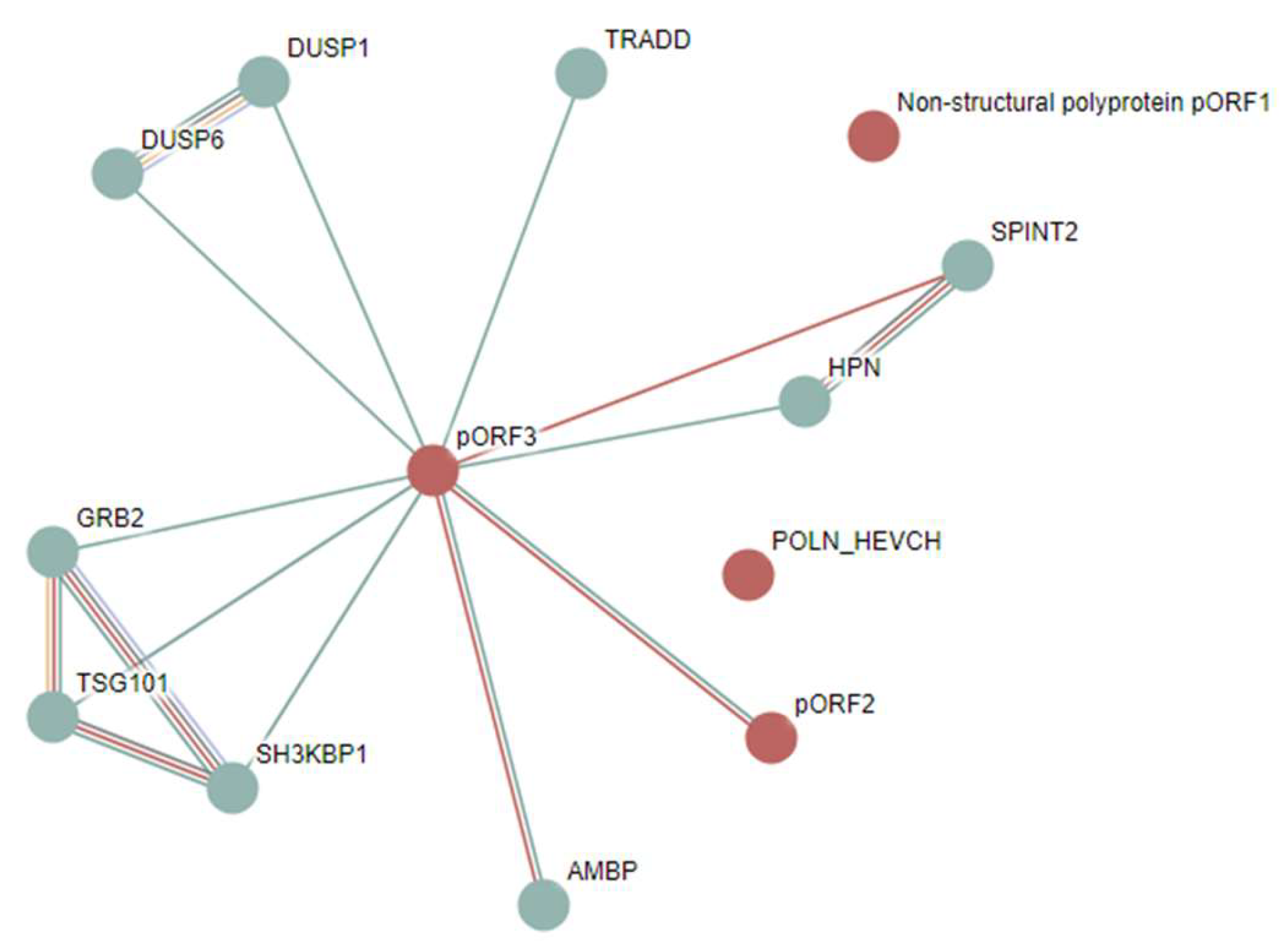
Figure 11.
Global disorder analysis of host proteins interacting with various hepatitis viruses: HAV (open circles), HBV (yellow circles), HCV (gray circles), HDV (cyan circles), and HEV (green circles). A. PONDR® VSL2 score vs. PONDR® VSL2 (%) plot. PONDR® VSL2 (%) is a percent of predicted disordered residues (PPDR), i.e., residues with disorder scores above 0.5. PONDR® VSL2 score is the average disorder score (ADS) for a protein. The boundaries of the colored regions represent arbitrary and accepted cutoffs for ADS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). B. Charge-hydropathy (CH) and Cumulative Distribution Function (CDF) analysis (ΔCH-ΔCDF plot) of human proteins interacting with hepatitis viruses. The Y-axis (ΔCH) represents the protein’s distance from the CH boundary, indicating the balance between charge and hydrophobicity, while the X-axis (ΔCDF) represents the deviation of a protein’s disorder frequency from the CDF boundary. Proteins are found in four quadrants: Quadrant 1 (bottom right, blue) contains proteins likely to be structured; Quadrant 2 (bottom left, pink) includes proteins that may be in a molten globule state or hybrid proteins containing ordered domains and disordered regions; Quadrant 3 (top left, red) consists of proteins predicted to be highly disordered; Quadrant 4 (top right, violet) contains proteins with a mixed prediction of being disordered according to CH but ordered according to CDF.
Figure 11.
Global disorder analysis of host proteins interacting with various hepatitis viruses: HAV (open circles), HBV (yellow circles), HCV (gray circles), HDV (cyan circles), and HEV (green circles). A. PONDR® VSL2 score vs. PONDR® VSL2 (%) plot. PONDR® VSL2 (%) is a percent of predicted disordered residues (PPDR), i.e., residues with disorder scores above 0.5. PONDR® VSL2 score is the average disorder score (ADS) for a protein. The boundaries of the colored regions represent arbitrary and accepted cutoffs for ADS (y-axis) and the percentage of predicted disordered residues (PPDR; x-axis). B. Charge-hydropathy (CH) and Cumulative Distribution Function (CDF) analysis (ΔCH-ΔCDF plot) of human proteins interacting with hepatitis viruses. The Y-axis (ΔCH) represents the protein’s distance from the CH boundary, indicating the balance between charge and hydrophobicity, while the X-axis (ΔCDF) represents the deviation of a protein’s disorder frequency from the CDF boundary. Proteins are found in four quadrants: Quadrant 1 (bottom right, blue) contains proteins likely to be structured; Quadrant 2 (bottom left, pink) includes proteins that may be in a molten globule state or hybrid proteins containing ordered domains and disordered regions; Quadrant 3 (top left, red) consists of proteins predicted to be highly disordered; Quadrant 4 (top right, violet) contains proteins with a mixed prediction of being disordered according to CH but ordered according to CDF.
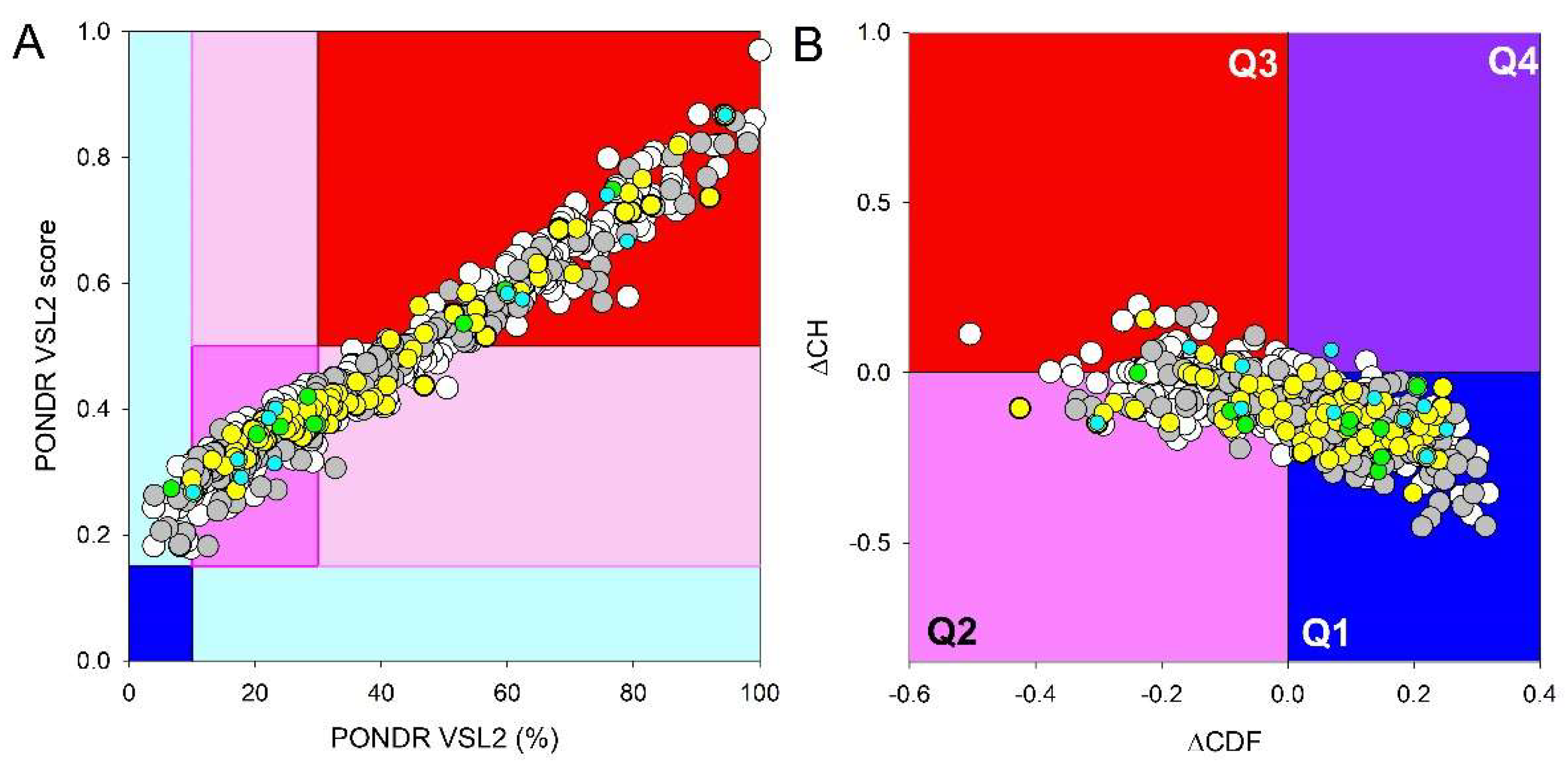
Figure 13.
STRING-based connectivity analysis of human proteins shared by hepatitis viruses. A. PPI network generated using medium confidence of 0.4 for the minimum required interaction score. Functional enrichment of this network is shown in terms of enriched GO biological proceses (B), GO molecular function (C), GO cellular component (D), disease-gene associations (E), KEGG pathways (F), and reactome pathways (G).
Figure 13.
STRING-based connectivity analysis of human proteins shared by hepatitis viruses. A. PPI network generated using medium confidence of 0.4 for the minimum required interaction score. Functional enrichment of this network is shown in terms of enriched GO biological proceses (B), GO molecular function (C), GO cellular component (D), disease-gene associations (E), KEGG pathways (F), and reactome pathways (G).
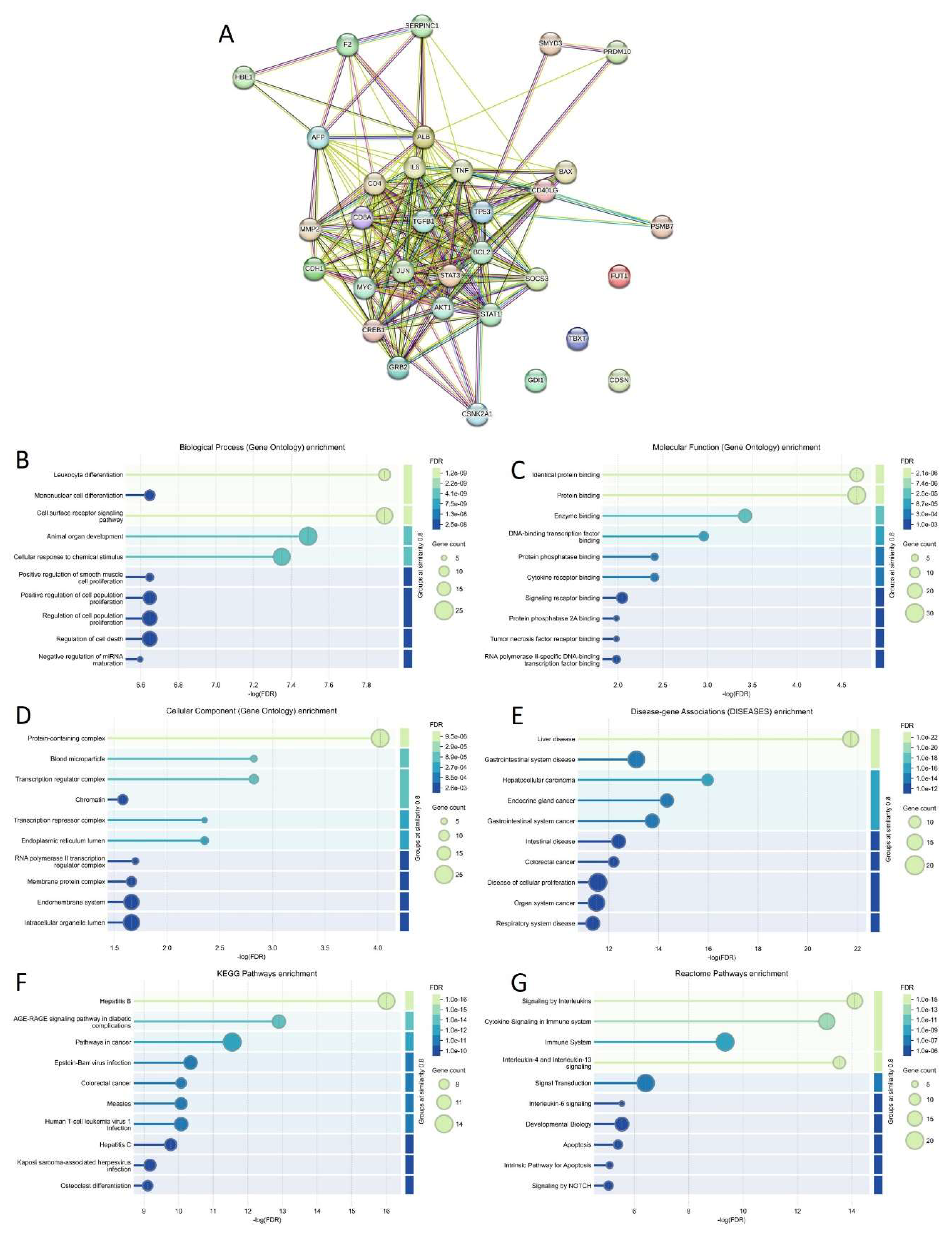
Figure 14.
Disorder, functionality, and LLPS predisposition analysis of of host proteins shared by hepatitis viruses. A. PONDR® VSL2 score vs. PONDR® VSL2 (%) (ADS vs. PPIDR) plot. B. Global node degree versus internal node degree plot. C. Dependencies of the global node degree (large symbols) and internal node degree (smaller symbols) on VSL2 PONDR® (%). D. Correlation between the predisposition to be involved in liquid-liquid phase separation (LLPS), pLLPS, and intrinsic disorder levels in these host proteins. Solid black lines in all the plots show fits of the reported data, whereas blue and red dashed lines show 95% confidence and prediction bands of corresponding fits, respectively. Dashed black lines represent boundaries between different disorder catego-ries (A) and LLPS promoters and other proteins (D).
Figure 14.
Disorder, functionality, and LLPS predisposition analysis of of host proteins shared by hepatitis viruses. A. PONDR® VSL2 score vs. PONDR® VSL2 (%) (ADS vs. PPIDR) plot. B. Global node degree versus internal node degree plot. C. Dependencies of the global node degree (large symbols) and internal node degree (smaller symbols) on VSL2 PONDR® (%). D. Correlation between the predisposition to be involved in liquid-liquid phase separation (LLPS), pLLPS, and intrinsic disorder levels in these host proteins. Solid black lines in all the plots show fits of the reported data, whereas blue and red dashed lines show 95% confidence and prediction bands of corresponding fits, respectively. Dashed black lines represent boundaries between different disorder catego-ries (A) and LLPS promoters and other proteins (D).
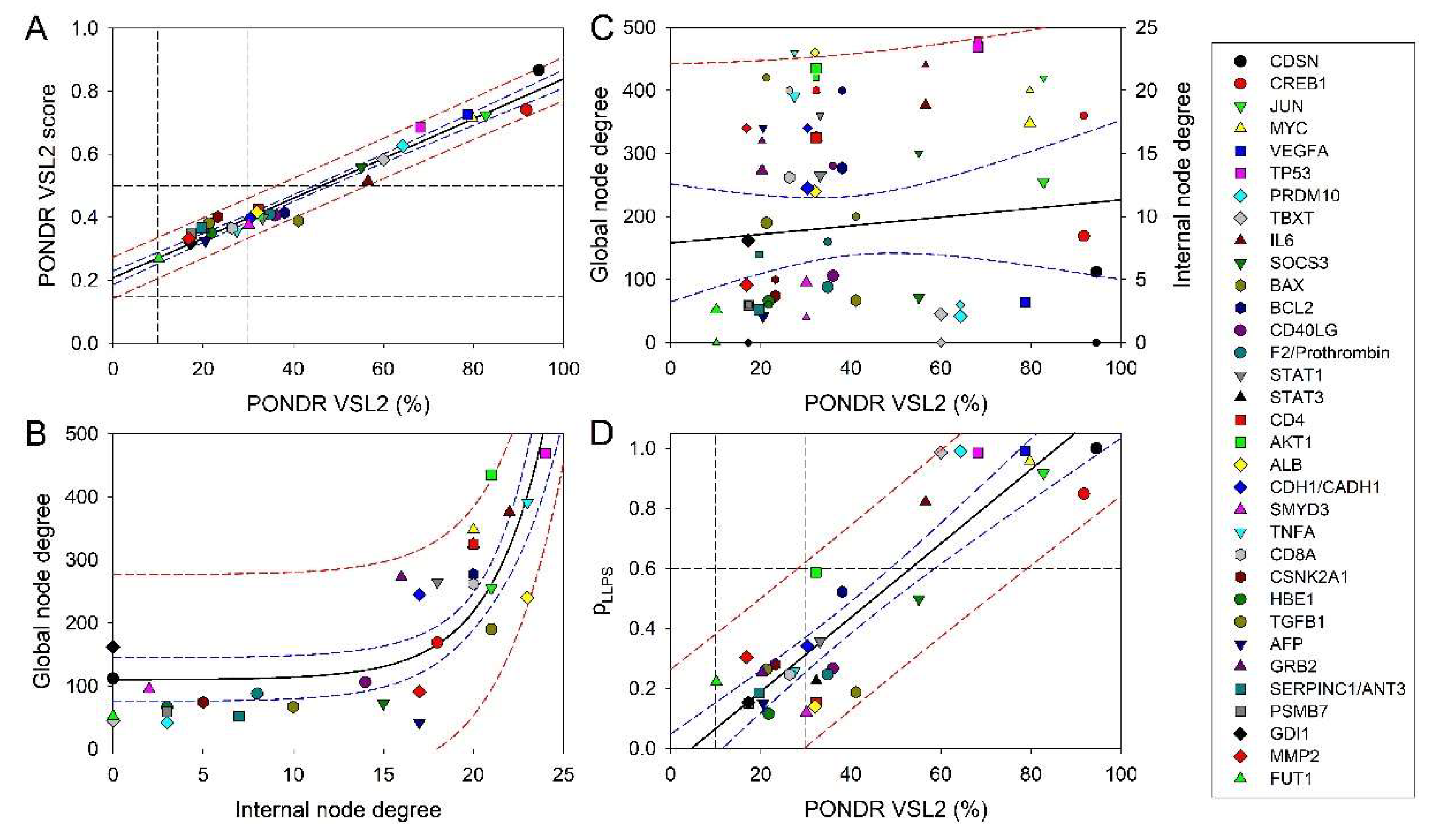
Figure 15.
Functional disorder analysis of human Corneodesmosin (CDSN, UniProt ID: Q15517). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2 containing the outputs of several disorder predictors such as VLXT, VSL2b, PrDOS, IUPred and Espritz. Positions of molecular recognition feature (MoRF) regions, unique disordered binding sites that become ordered at interaction with specific partners, found through the ANCHOR algorithm, are shown by yellow zigzagged bars. C. 3D structural model generated by AlphaFold. The structure is colored according to the per-residue model confidence score (pLDDT), where very high confidence (pLDDT > 90), high confidence (70 < pLDDT < 90), low confidence (50 < pLDDT < 70), and very low confidence (pLDDT < 50) are shown by blue, cyan, yellow, and orange colors, respectively. D. CDSN-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bV6EsisP3Znr).
Figure 15.
Functional disorder analysis of human Corneodesmosin (CDSN, UniProt ID: Q15517). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2 containing the outputs of several disorder predictors such as VLXT, VSL2b, PrDOS, IUPred and Espritz. Positions of molecular recognition feature (MoRF) regions, unique disordered binding sites that become ordered at interaction with specific partners, found through the ANCHOR algorithm, are shown by yellow zigzagged bars. C. 3D structural model generated by AlphaFold. The structure is colored according to the per-residue model confidence score (pLDDT), where very high confidence (pLDDT > 90), high confidence (70 < pLDDT < 90), low confidence (50 < pLDDT < 70), and very low confidence (pLDDT < 50) are shown by blue, cyan, yellow, and orange colors, respectively. D. CDSN-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bV6EsisP3Znr).
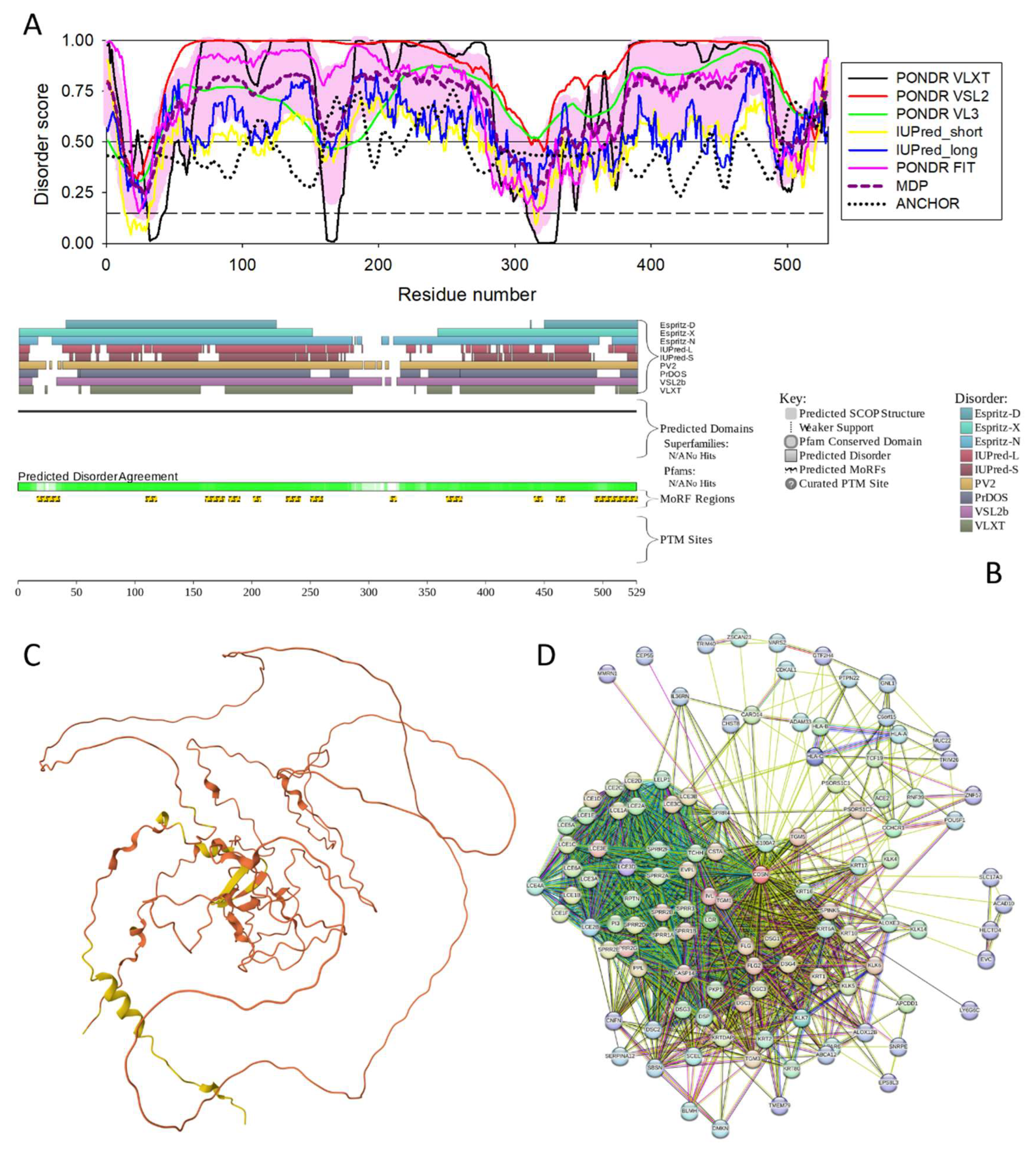
Figure 16.
Functional disorder analysis of human Myc proto-oncogene protein (MYC, UniProt ID: P01106). A. Per-residue disorder profile generated by RIDAO. B. 3D structural model generated by AlphaFold. C. MYC-centered PPI network generated by STRING using high confidence of 0.7 for minimum required interaction score B (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bpBTApPOOZcr).
Figure 16.
Functional disorder analysis of human Myc proto-oncogene protein (MYC, UniProt ID: P01106). A. Per-residue disorder profile generated by RIDAO. B. 3D structural model generated by AlphaFold. C. MYC-centered PPI network generated by STRING using high confidence of 0.7 for minimum required interaction score B (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bpBTApPOOZcr).
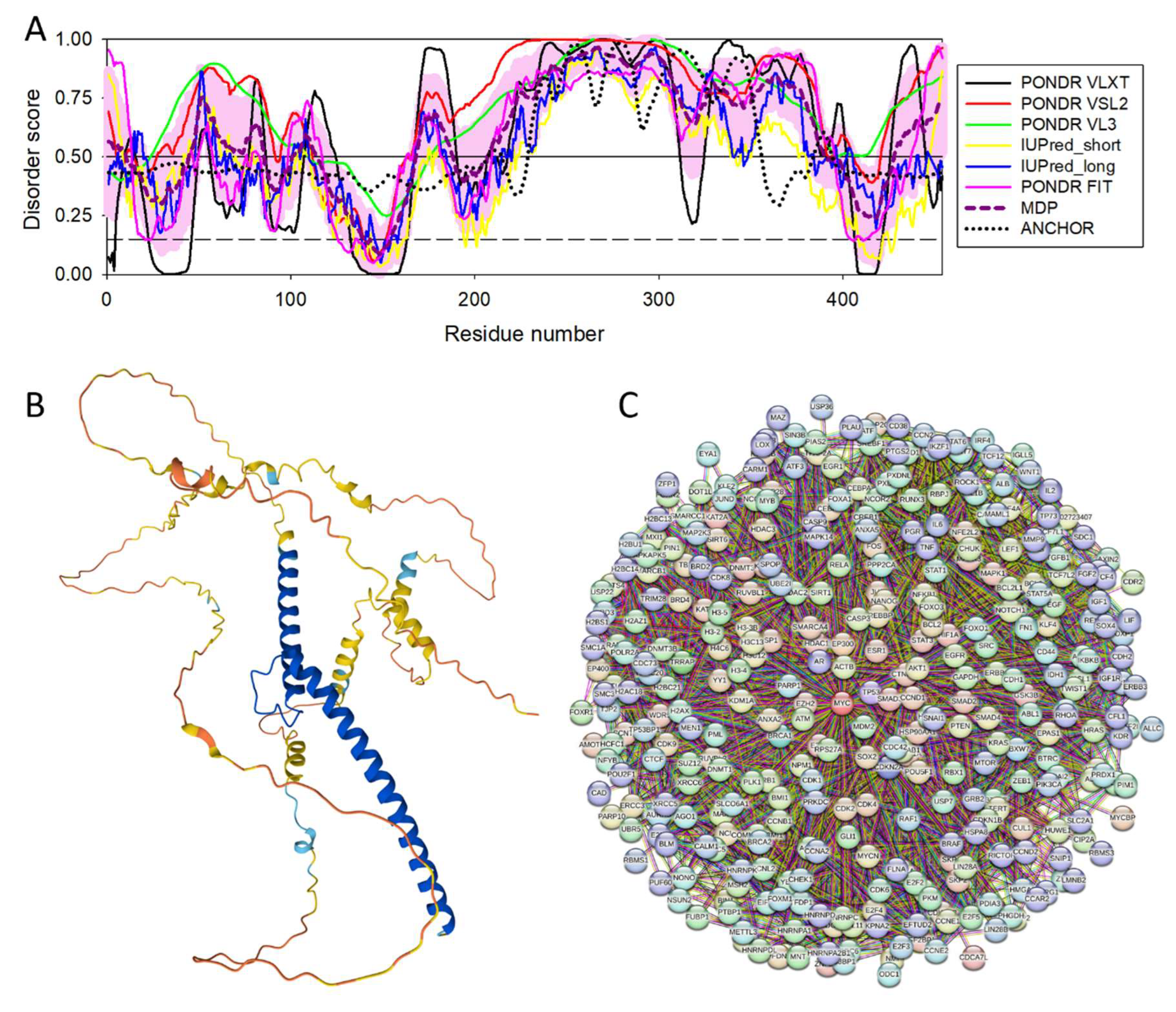
Figure 18.
Functional disorder analysis of human PR domain zinc finger protein 10 (PRDM10, UniProt ID: Q9NQV6). A. Per-residue disorder profile generated by RIDAO. B. Evaluation of the redox sensitive disorder propensity of human PRDM10 by IUPred2A. Redox-sensitive disordered regions are shown as shaded purple areas. C. 3D structural model generated by AlphaFold. D. PRDM10-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bsPRlymzQpJR).
Figure 18.
Functional disorder analysis of human PR domain zinc finger protein 10 (PRDM10, UniProt ID: Q9NQV6). A. Per-residue disorder profile generated by RIDAO. B. Evaluation of the redox sensitive disorder propensity of human PRDM10 by IUPred2A. Redox-sensitive disordered regions are shown as shaded purple areas. C. 3D structural model generated by AlphaFold. D. PRDM10-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bsPRlymzQpJR).

Figure 19.
Functional disorder analysis of human T-box transcription factor T (TBXT, UniProt ID: O15178). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. TBXT-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=b6lU22eVsjXg).
Figure 19.
Functional disorder analysis of human T-box transcription factor T (TBXT, UniProt ID: O15178). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. TBXT-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=b6lU22eVsjXg).
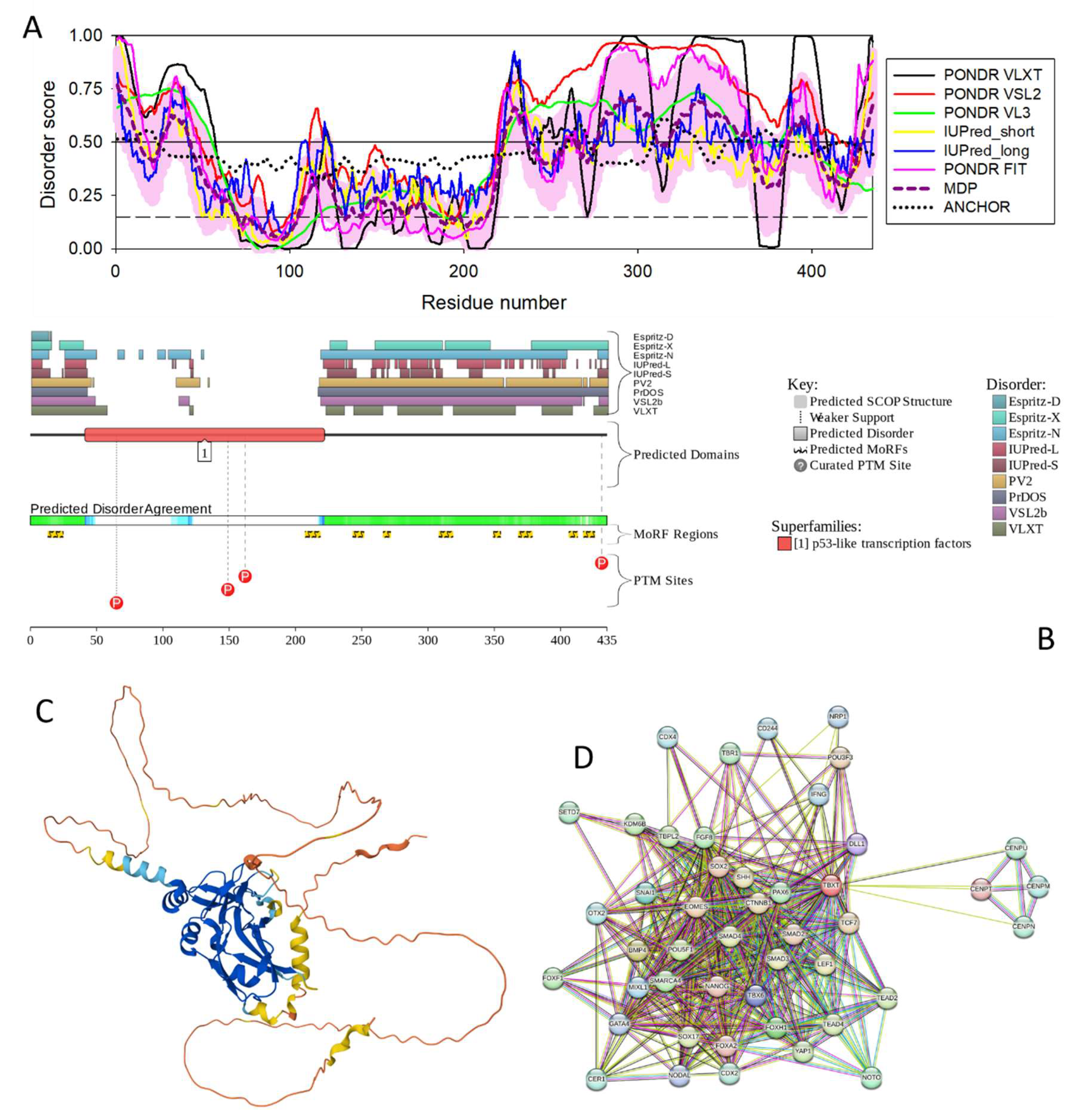
Figure 20.
Functional disorder analysis of human serum albumin (ALB; UniProt ID: P02768). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. ALB-centered PPI network generated by STRING using high confidence of 0.7 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=buFq1uwRvjnM).
Figure 20.
Functional disorder analysis of human serum albumin (ALB; UniProt ID: P02768). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. ALB-centered PPI network generated by STRING using high confidence of 0.7 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=buFq1uwRvjnM).
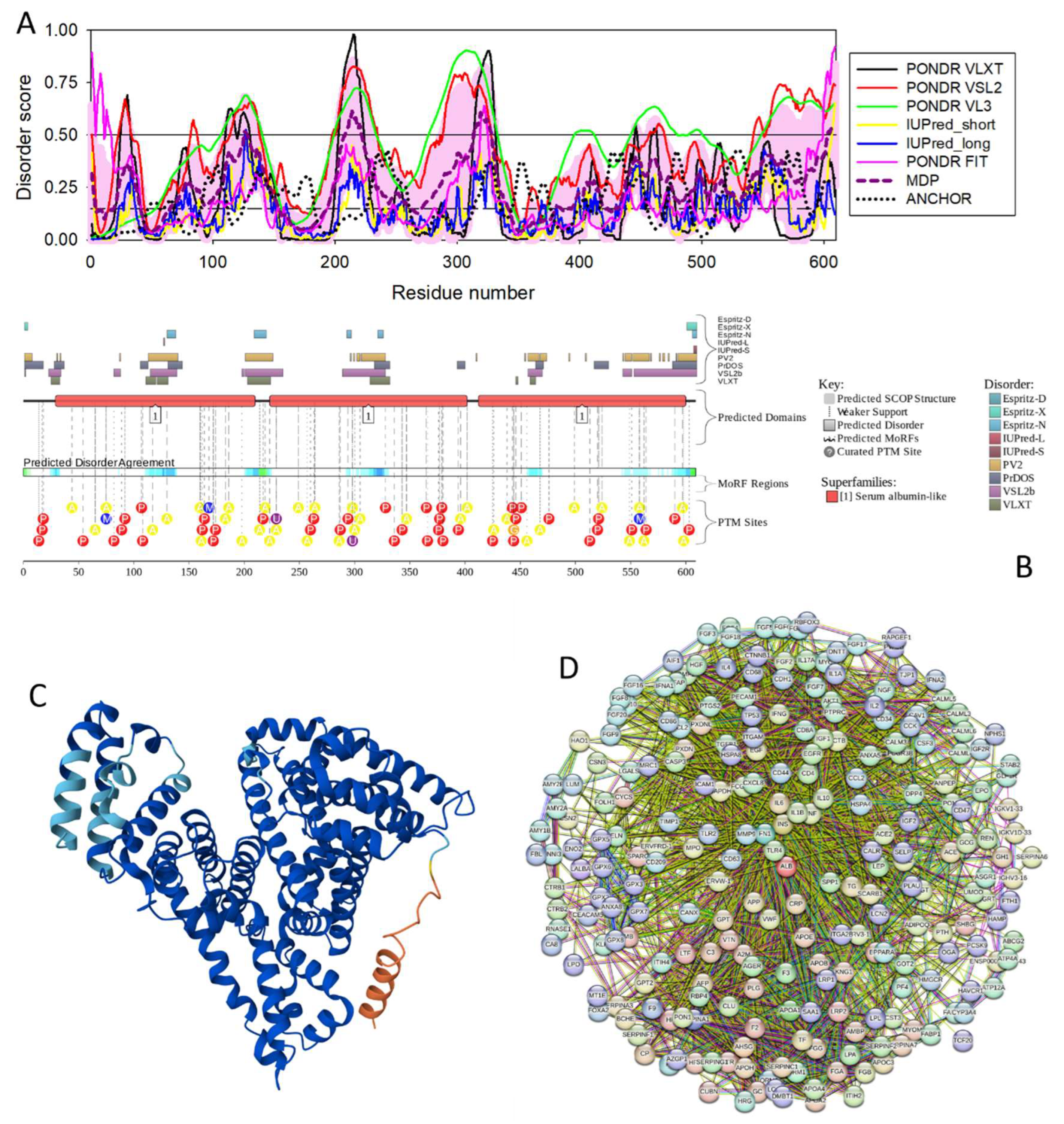
Figure 21.
Functional disorder analysis of human T-cell surface glycoprotein CD8 alpha chain (CD8A; UniProt ID: P01732). A. Per-residue disorder profile generated by RIDAO. B. Evaluation of the redox sensitive disorder propensity of human CD8A by IUPred2A. Redox-sensitive disordered region is shown as shaded purple area. C. Functional disorder profile generated by D2P2. D. 3D structural model generated by AlphaFold. E. CD8A-centered PPI network generated by STRING using high confidence of 0.7 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bBj2ohdw8iVA).
Figure 21.
Functional disorder analysis of human T-cell surface glycoprotein CD8 alpha chain (CD8A; UniProt ID: P01732). A. Per-residue disorder profile generated by RIDAO. B. Evaluation of the redox sensitive disorder propensity of human CD8A by IUPred2A. Redox-sensitive disordered region is shown as shaded purple area. C. Functional disorder profile generated by D2P2. D. 3D structural model generated by AlphaFold. E. CD8A-centered PPI network generated by STRING using high confidence of 0.7 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bBj2ohdw8iVA).
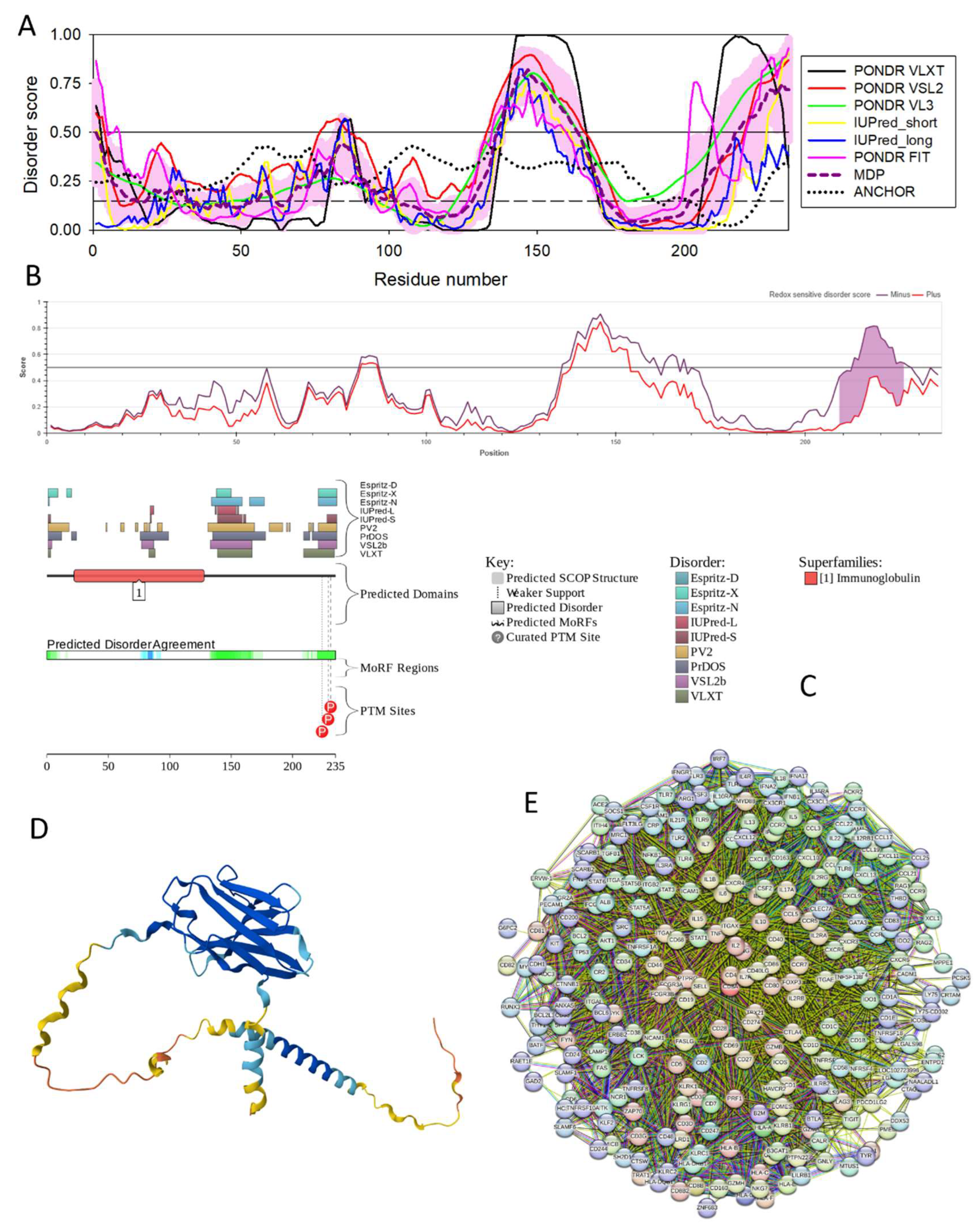
Figure 22.
Functional disorder analysis of human α-fetoprotein (AFP; UniProt ID: P02771). A. Per-residue disorder profile generated by RIDAO. B. Evaluation of the redox sensitive disorder propensity of human AFP by IUPred2A. Redox-sensitive disordered regions are shown as shaded purple areas. C. Functional disorder profile generated by D2P2. D. 3D structural model generated by AlphaFold. E. AFP-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bXOriTks553m).
Figure 22.
Functional disorder analysis of human α-fetoprotein (AFP; UniProt ID: P02771). A. Per-residue disorder profile generated by RIDAO. B. Evaluation of the redox sensitive disorder propensity of human AFP by IUPred2A. Redox-sensitive disordered regions are shown as shaded purple areas. C. Functional disorder profile generated by D2P2. D. 3D structural model generated by AlphaFold. E. AFP-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bXOriTks553m).
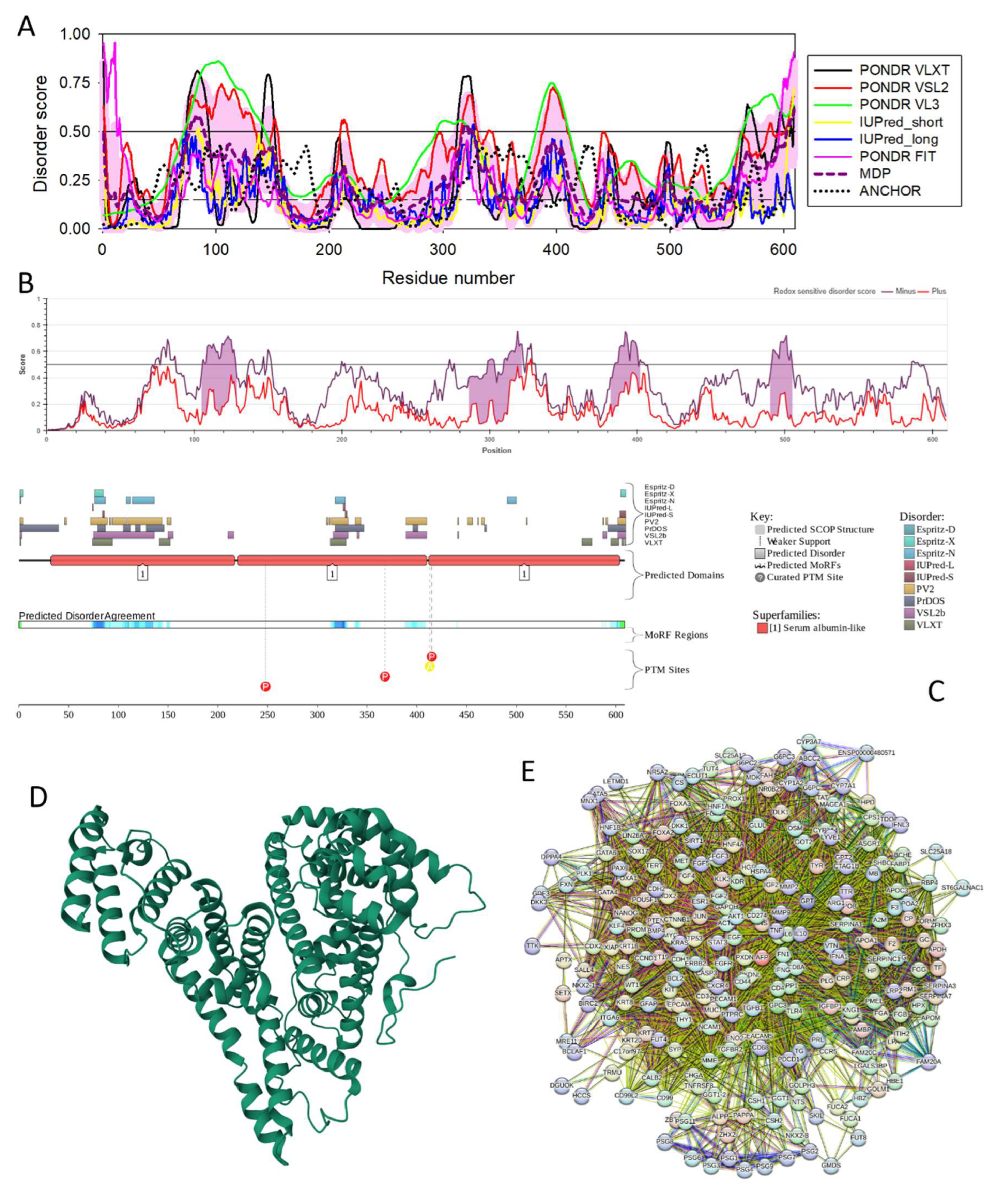
Figure 23.
Functional disorder analysis of human proteasome subunit beta type-7 (PSMB7; UniProt ID: Q99436). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. PSMB7-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bBRDCNM4HXzZ).
Figure 23.
Functional disorder analysis of human proteasome subunit beta type-7 (PSMB7; UniProt ID: Q99436). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. PSMB7-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bBRDCNM4HXzZ).
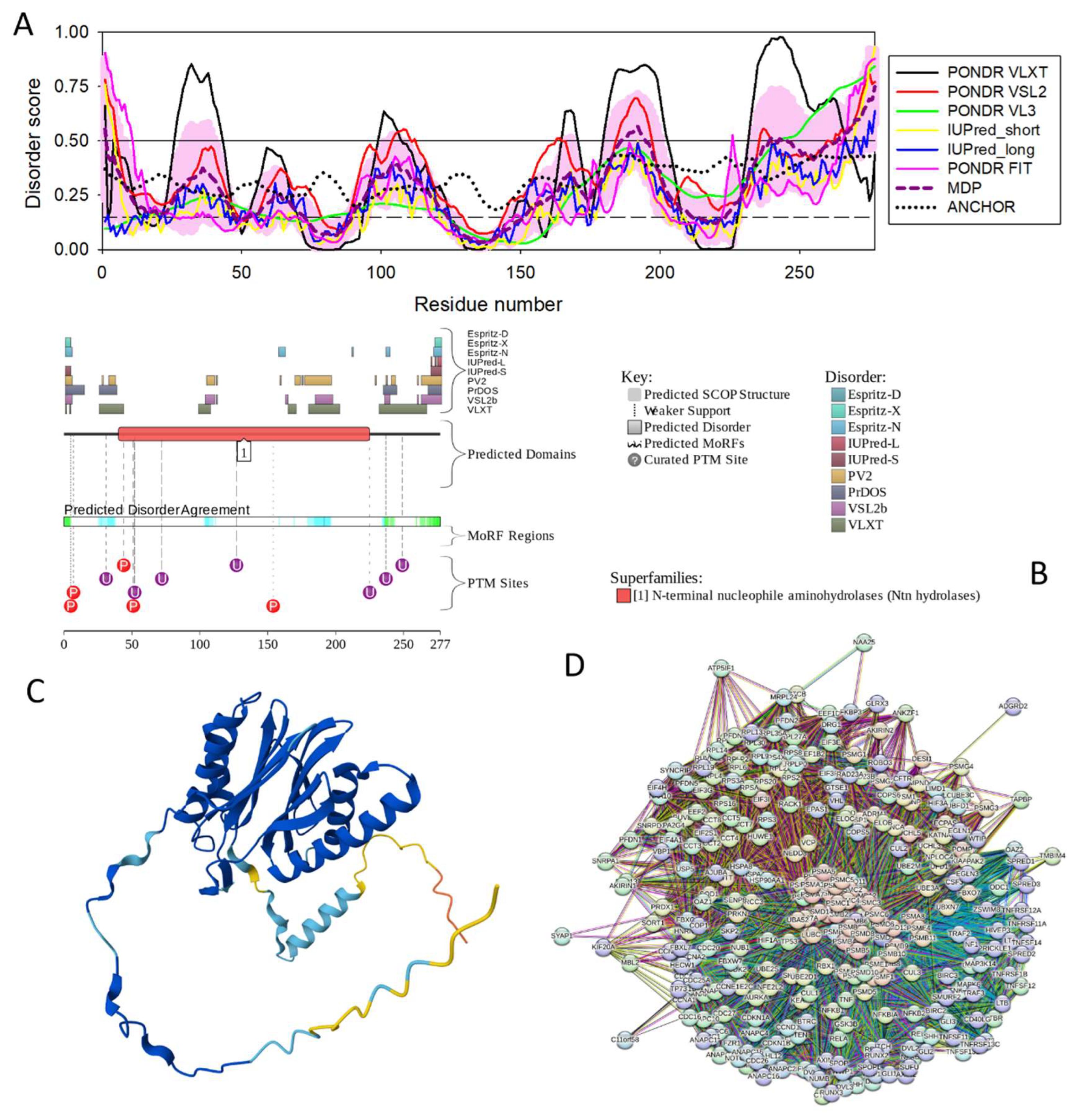
Figure 24.
Functional disorder analysis of human RAB GDP dissociation inhibitor alpha (GDI1; UniProt ID: P31150). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. GDI1-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bVNyFB0A32QY).
Figure 24.
Functional disorder analysis of human RAB GDP dissociation inhibitor alpha (GDI1; UniProt ID: P31150). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. GDI1-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=bVNyFB0A32QY).
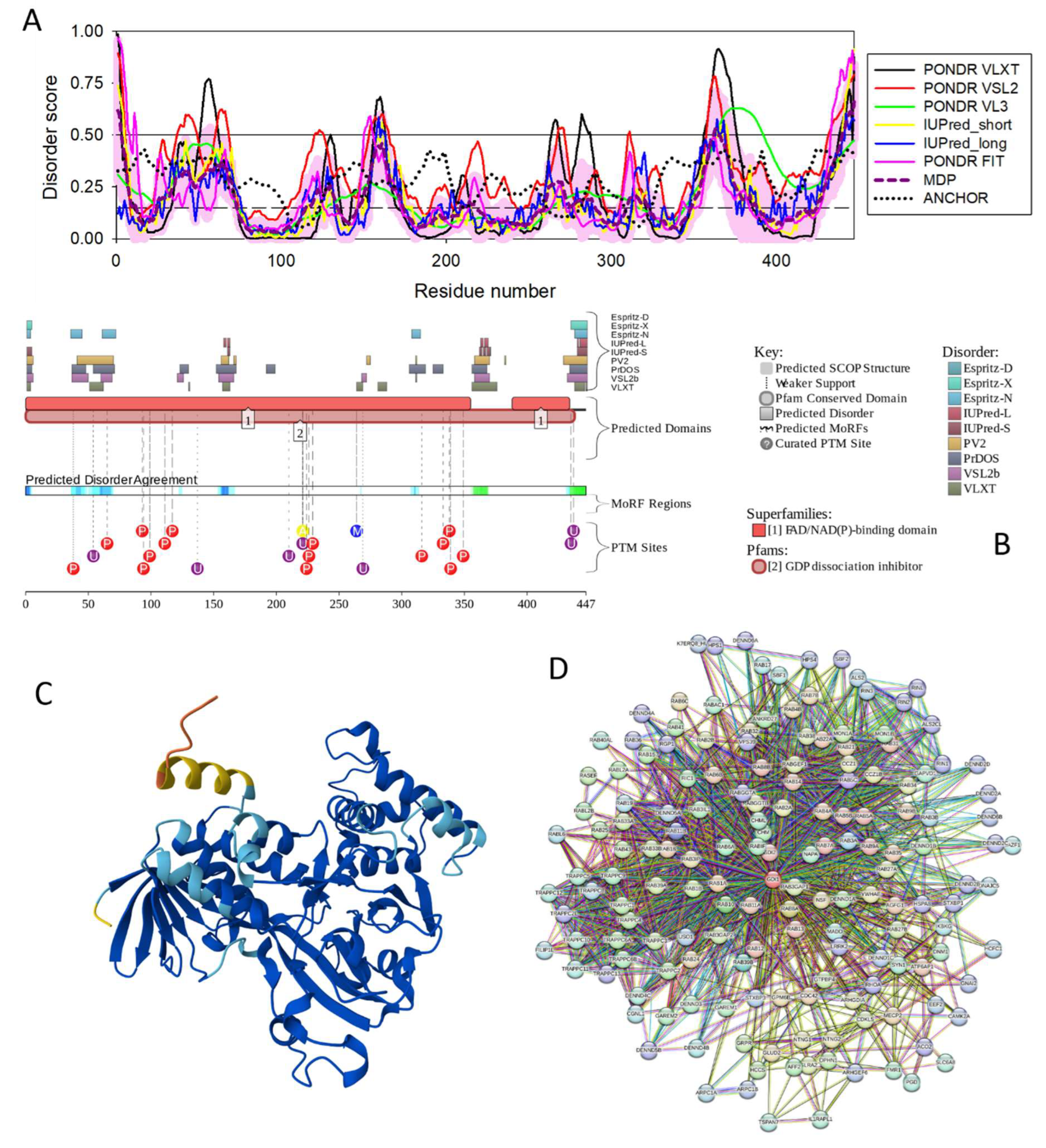
Figure 25.
Functional disorder analysis of human galactoside alpha-(1,2)-fucosyltransferase 1 (FUT1; UniProt ID: P19526). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. GDI1-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=b1kXOX9Lrx6K).
Figure 25.
Functional disorder analysis of human galactoside alpha-(1,2)-fucosyltransferase 1 (FUT1; UniProt ID: P19526). A. Per-residue disorder profile generated by RIDAO. B. Functional disorder profile generated by D2P2. C. 3D structural model generated by AlphaFold. D. GDI1-centered PPI network generated by STRING using medium confidence of 0.4 for the minimum required interaction score (permalink: https://version-12-0.string-db.org/cgi/network?networkId=b1kXOX9Lrx6K).
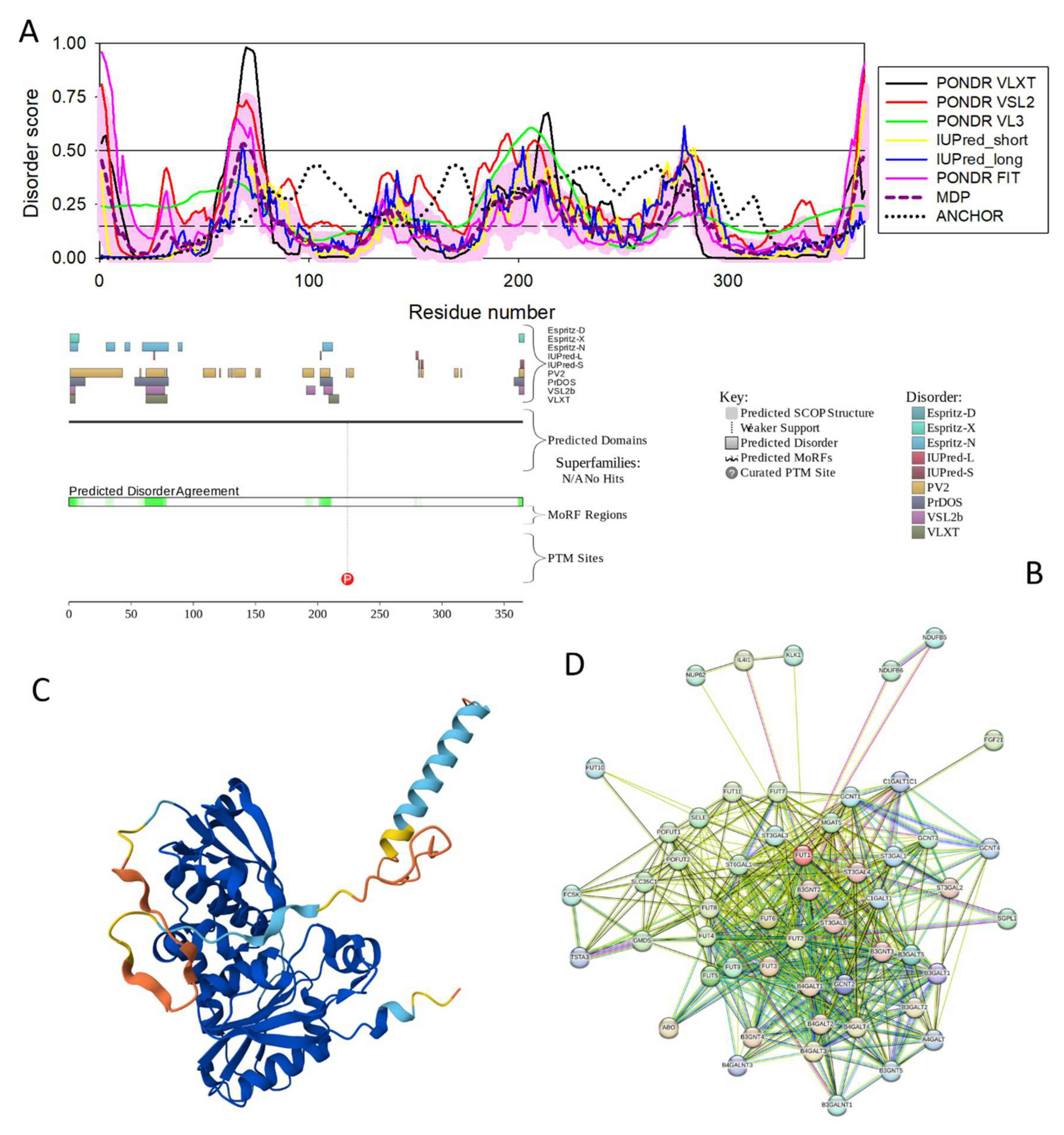

Table 1.
Characteristics of host proteins shared by different hepatotropic viruses. Proteins are ordered by the decrease in their intrinsic disorder status as evidenced by the PPIDR values.
Table 1.
Characteristics of host proteins shared by different hepatotropic viruses. Proteins are ordered by the decrease in their intrinsic disorder status as evidenced by the PPIDR values.
| Name | UniProt ID | Shared by | Number of residues | PPIDR | ADS | pLLPS | Number of DPRs | Internal node degree | Global node degree |
| CDSN | Q15517 | HAV,a HBV, HCV, and HDV | 500 | 94.52 | 0.8669 | 1.00 | 5 | 0 | 28m/112n |
| CREB1 | P16220 | HBV and HCV | 300 | 91.74 | 0.7414 | 0.85 | 3 | 18 | 169 |
| JUN | P05412 | HBV and HCV | 274 | 82.78 | 0.7235 | 0.92 | 2 | 21 | 255 |
| MYC | P01106 | HAV,b HBV, and HCV | 454 | 79.74 | 0.7152 | 0.96 | 4 | 20 | 348 |
| VEGFA | P15692 | HBV and HCV | 395 | 78.73 | 0.7263 | 0.99 | 4 | N.D. | 64 |
| TP53 | P04637 | HAV,c HBV, and HCV | 393 | 68.19 | 0.6849 | 0.98 | 4 | 24 | 469 |
| PRDM10 | Q9NQV6 | HAV,d HBV, and HCV | 1147 | 64.34 | 0.6271 | 0.99 | 9 | 3 | 0m/42n |
| TBXT | O15178 | HAV,e HBV, HCV, and HDV | 425 | 60.00 | 0.5834 | 0.99 | 4 | 0 | 12m/45n |
| IL6 | P05231 | HBV and HCV | 212 | 56.60 | 0.5152 | 0.82 | 4 | 22 | 376 |
| SOCS3 | O14543 | HBV and HCV | 225 | 55.11 | 0.5583 | 0.50 | 3 | 15 | 72m/359n |
| BAX | Q07812 | HBV and HCV | 192 | 41.15 | 0.3883 | 0.19 | 2 | 10 | 67m/172n |
| BCL2 | P10415 | HBV and HCV | 239 | 38.08 | 0.4142 | 0.52 | 2 | 20 | 277 |
| CD40LG | P29965 | HBV and HCV | 261 | 36.02 | 0.4052 | 0.27 | 1 | 14 | 106m/434n |
| F2/prothrombin | P00734 | HBV and HCV | 622 | 34.89 | 0.4094 | 0.25 | 2 | 8 | 88m/284n |
| STAT1 | P42224 | HBV and HCV | 750 | 33.20 | 0.3970 | 0.36 | 4 | 18 | 265 |
| STAT3 | P40763 | HBV and HCV | 770 | 32.34 | 0.4026 | 0.22 | 3 | 20 | 325 |
| CD4 | P01730 | HBV and HCV | 458 | 32.31 | 0.4247 | 0.15 | 0 | 20 | 325 |
| AKT1 | P31749 | HBV and HCV | 480 | 32.29 | 0.4069 | 0.59 | 3 | 21 | 435 |
| ALB | P02768 | HAV,f HBV, and HCV | 609 | 32.02 | 0.4168 | 0.14 | 1 | 23 | 240 |
| CDH1 | P12830 | HBV and HCV | 882 | 30.39 | 0.3957 | 0.34 | 4 | 17 | 245 |
| SMYD3 | Q9H7B4 | HBV and HCV | 428 | 30.14 | 0.3760 | 0.12 | 0 | 2 | 21m/95n |
| TNF | P01375 | HBV and HCV | 233 | 27.47 | 0.3559 | 0.26 | 2 | 23 | 391 |
| CD8A | P01732 | HAV,g HBV, and HCV | 235 | 26.38 | 0.3652 | 0.25 | 1 | 20 | 262 |
| CSNK2A1 | P68400 | HCV and HDV | 391 | 23.27 | 0.4007 | 0.28 | 1 | 5 | 74m/377n |
| HBE1 | P02100 | HBV and HCV | 147 | 21.77 | 0.3503 | 0.12 | 0 | 3 | 16m/66n |
| TGFB1 | P01137 | HBV and HCV | 390 | 21.28 | 0.3814 | 0.26 | 2 | 21 | 190 |
| AFP | P02771 | HAV,h HBV, and HCV | 609 | 20.53 | 0.3250 | 0.15 | 0 | 17 | 42m/239n |
| GRB2 | P62993 | HBV and HEV | 217 | 20.28 | 0.3598 | 0.25 | 1 | 16 | 273 |
| SERPINC1 | P01008 | HBV and HCV | 464 | 19.61 | 0.3664 | 0.19 | 1 | 7 | 52m/193n |
| PSMB7 | Q99436 | HAV,i HBV, and HCV | 277 | 17.33 | 0.3476 | 0.15 | 0 | 3 | 59m/294n |
| GDI1 | P31150 | HAV,j HBV, and HCV | 447 | 17.23 | 0.3195 | 0.15 | 0 | 0 | 33m/162n |
| MMP2 | P08253 | HBV and HCV | 660 | 16.82 | 0.3324 | 0.30 | 2 | 17 | 91m/465n |
| FUT1 | P19526 | HAV,k HBV, HCV, and HDV | 365 | 10.14 | 0.2677 | 0.22 | 1 | 0 | 25m/52n |
a Chimpanzee homolog of human corneodesmosin (CDSN) has a UniProt ID Q7YR44 and is 99.4% identical and 99.8% similar to human protein. b Chimpanzee homolog of human Myc proto-oncogene protein (MYC) has a UniProt ID H2QWQ2 and is 95.8% identical and 96.0% similar to human protein. c Chimpanzee homolog of human cellular tumor antigen p53 (TP53) has a UniProt ID H2QC53 and is 100.0% identical to human protein. d Chimpanzee homolog of human PR domain zinc finger protein 10 (PRDM10) has a UniProt ID H2Q545 and is 87.6% identical and 88.2% similar to human protein. e Chimpanzee homolog of human T-box transcription factor T (TBXT) has a UniProt ID H2QU10 and is 99.5% identical and 99.5% similar to human protein. f Chimpanzee homolog of human serum albumin (ALB) has a UniProt ID H2RBT1 and is 98.9% identical and 99.7% similar to human protein. g Chimpanzee homolog of human T-cell surface glycoprotein CD8 alpha chain (CD8A) has a UniProt ID H2QC53 and is 83.4% identical and 83.4% similar to human protein. h Chimpanzee homolog of human alpha-fetoprotein (AFP) has a UniProt ID H2QPM9 and is 95.5% identical and 96.5% similar to human protein. i Chimpanzee homolog of human proteasome subunit beta (PSMB7) has a UniProt ID H2QXV4 and is 99.6% identical and 99.6% similar to human protein. j Chimpanzee homolog of human Rab GDP dissociation inhibitor alpha (GDI1) has a UniProt ID P60028 and is 100.0% identical to human protein. k Chimpanzee homolog of human L-fucosyltransferase (FUT1) has a UniProt ID Q9TUD6 and is 98.4% identical and 98.4% similar to human protein. l N.D., not determined. m Evaluated using high confidence of 0.7 for minimum required interaction score. n Evaluated using medium confidence of 0.4 for minimum required interaction score.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



