
Submitted:
31 January 2025
Posted:
03 February 2025
You are already at the latest version
Abstract
Cancers that are arising from germline mutations of the breast cancer associated gene 1 (BRCA1), which is a crucial player of homologous recombination (HR) DNA repair, are vulnerable to DNA damaging agents such as platinum and PARP inhibitors (PAR-Pi). Increasing evidence suggests that BRCA1 is an essential driver of all phases of the cell cycle, thereby maintaining orderly steps during cell cycle progression. Specifically, loss of BRCA1 activity causes the S-phase, G2/M, spindle checkpoints and centrosome duplication to be dysregulated, thereby blocking cell proliferation and inducing apop-tosis. In vertebrates, loss of HR genes such as BRCA1 and/or BRCA2 is lethal since HR is prerequisite for genome integrity. Thus, cancer cells utilize alternative DNA repair pathways such as non-homologous end joining (NHEJ) to cope with loss of BRCA1 function. In this review, we attempt to update and discuss how these novel compo-nents are crucial for regulating DNA damage repair (DDR) in BRCA1-deficient can-cers.
Keywords:
BRCA1
; PARP1
; TATDN2
; BARD1
; EXO1
; EEPD1
; FANCJ
; BRCA1-deficient cancers
1. Introduction
DNA double-strand breaks (DSBs) are the most consequential DNA lesions threatening genomic integrity [1]. Consequently, failure to repair DBSs has detrimental consequences including oncogenic transformation, tumorigenesis and cell death [1]. Principally, cells use two basic pathways to repair DSBs: (1) homologous recombination (HR) and (2) the classical nonhomologous end-joining pathway (cNHEJ). However, it is worth noting that some recent studies have identified a highly error-prone NHEJ pathway, termed alternative NHEJ (aNHEJ), which operates in both cNHEJ-proficient and cNHEJ-deficient cells [2,3,4,5,6,7]. When the NHEJ pathway is inactivated, DSBs can be repaired by a more error-prone pathway, the microhomology-mediated end joining (MMEJ) [8]. In contrast with cNHEJ typically using microhomologies that have already exposed in single-stranded overhangs on the DBS ends, MMEJ involves alignment of microhomologous sequences internal to the broken ends before joining, therefore it is associated with deletions and insertions of DNA sequence between microhomologies [8].
Breast cancer susceptibility gene 1 (BRCA1) encoding a tumor suppressor BRCA1, also called p220, plays a house-keeping driver of cell cycle checkpoint and DNA repair pathways such as HR, which is a high-fidelity mechanism for DSBs in DNA [9]. So, HR is required for maintaining genetic integrity to ensure the accuracy of DSB repair [10]. Mechanistically, BRCA1 recruits RAB51 and other repair factors to the sites of damage to coat the single-stranded DNA generated at the break site, thereby facilitating the search for a homologous DNA template [11,12,13]. Besides, BRCA1 is a key driver of cell cycle progression though regulating all the phases regulates cell cycle progression, particularly at the G1/S and G2/M checkpoints so as to prevent cells with damaged DNA from proceeding through mitosis [14]. Mutations in BRCA1 gene or loss of BRCA1 function causes a shift away from HR toward NHEJ, which is an error-prone pathway directly ligating the broken DNA ends without the need for a homologous template [15]. Nonetheless, NHEJ frequently results in small deletions or insertions of nucleotides in the damaged loci, leading to chromosomal instability, thereby contributing to the development of various cancers such as breast, ovarian and prostate carcinomas [9,10,11,15,16,17,18].
BRCA1 harbors a highly conserved amino-terminal RING domain located at its N-terminus from residues 24-64, which is a motif found in many E3 ubiquitin ligases [19,20], and BRCA1 C-terminal repeats domain, a motif binding to phosphorylated proteins essential for DNA damage repair [21]. Its RING domain acts as a protein-protein interaction domain, allowing specific proteins to form complexes with other necessary repair factors to facilitate DNA damage repair. The best characterized RING domain (residues 1-109) interaction occurs with BRCA1 associated RING domain protein 1 (BARD1) to form E3 ubiquitin ligase complex. BARD1 is required for HR-mediated DNA repair, replication fork maintenance [22,23].
Etiologically, germline mutations of human BRCA1 gene account for a significantly increased risk of developing several types of cancers including breast, ovarian, prostate and pancreatic cancers. For instance, patients with BRCA1-deficient cancers have a lifetime risk of developing breast cancer as high as 80% as compared to about 12% in the general population [24]. Because BRCA1-deficient breast cancers are often classified as triple-negative breast cancers (TNBCs) that lack estrogen receptors (ERs), progesterone receptors (PRs) and human epidermal growth factor receptor 2 (HER2) amplification, it is challenging to establish the targeted treatments with standard hormone therapies [25,26,27]. In ovarian cancers, BRCA1-deficient tumors are aggressive and often diagnosed at the late stage [28] whereas, in BRCA1-deficient prostate cancers, a higher probability of being an advanced stage and a shorter disease-free survival in patients are reported [29]. Also, although mutations of BRCA1 gene are linked to an increased risk of pancreatic cancer and melanoma, the exact risks are still under investigation [30,31]. Therefore, mutations of BRCA1 gene, mostly those mutating the RING domain, significantly impaired DNA damage repair via HR, forcing them to rely more heavily on the less precise NHEJ pathway for DNA repair, thereby increasing chromosomal abnormalities and cancer risk [32]. Hence, it is mandatory to develop targeted therapies for the treatment of patients with BRCA1-deficient cancers.
One of the most promising strategies is the use of PARP (poly ADP-ribose polymerase) inhibitors, also known as synthetic lethality. Once DNA nicks or breaks (DSBs) occur, PARP, composed of two isoforms PARP1 and PARP2, is activated to generate poly(ADP-ribose) to recruit core DNA repair proteins to the site of damage, including those involved in NHEJ pathway that uses the synaptic activity of PARP and ligation activity of XRCC1-DNA ligase III complex to repair DSBs [33]. In NHEJ, the DSB is first recognized by the Ku70-K80 heterodimer (Ku), which acts as a ‘tool belt’ or loading protein to which other NHEJ proteins can be recruited as needed to promote the joining of DNA ends [34]. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) has a high affinity for Ku-DNA ends and, together with Ku, forms the DNA-PK complex [35]. aNHEJ plays as a secondary DNA repair mechanism to the primary cNHEJ pathway, especially when key proteins such as Ku or other components, which are crucial for cNHEJ, are missing or when PARP1 is competing with Ku to repair DSBs [36]. Interestingly, it was revealed that PARP1 interacted with other core components of cNHEJ pathway such as Ku and the DNA-PKcs; on the contrary, cNHEJ is normally functional in murine PARP1-/- cells [37]. Inhibition of PARP activity causes accumulation of SSBs within DNA, and when the replication fork encounters these unrepaired SSBs, they can be converted into DSBs, resulting in DNA damage during DNA replication [38].
PARP inhibitors including olaparib, rucaparib and niraparib have shown significant efficacy of treatment for patients with BRCA1-deficient breast and/or ovarian cancers [39,40,41]. These drugs are particularly effective in patients with BRCA1 mutation tumors that are highly reliant upon PARP-mediated DNA repair pathway [39,40]. Hence, inhibition of PARP function by these drugs selectively killed BRCA1-deficient cancer cells, but not normal cells. Despite the success of PARP inhibitors in treatment of patients with BRCA1-deficienttumors, resistance to these chemotherapies can develop over time [42]. One mechanism of resistance is the restoration of HR through secondary mutations in BRCA1 gene [43]. Other mechanisms of resistance comprise (1) the upregulation of alterative DNA repair pathways, (2) increased drug efflux, and (3) changes in PARP expression [44]. In addition to PARP inhibitors, other therapeutic strategies such as combinations of PARP inhibitors (PARPis) with chemotherapy, anti-angiogenic agents, immunotherapy, immune checkpoint inhibitors, PI3Kinase inhibitors and other inhibitors of DNA damage repair machinery has been vigorously examined for the treatment of the patients with BRCA1-deficient tumors [45,46,47]. For instance, inhibiting ATR, which controls both BRCA1-independent HR and fork protection by accelerating RAD51 loading to DSB and stalled forks, leads to block BRCA1-independent HR and fork stability, thereby resensitizing resistant cells to PARPis [48]. In addition, immunotherapeutic approaches that target BRCA1-deficient cancers have been under vigorous investigation since immunotherapies using the immune checkpoint inhibitors, are able to strengthen the immune system in order to recognize and attack BRCA1-deficienttumors more efficiently [49].
Recent development of targeted therapies, particularly PARP inhibitors, has revolutionized the treatments for patients with BRCA1-deficient cancers that heavily rely upon aNHEJ pathway. However, resistance to these therapies is an unresolved issue, and ongoing research is aimed at overcoming such resistance and developing new therapeutic strategies, including the use of ATR inhibitors and/or immunotherapy. To resolve this issue, a comprehensive understanding of DNA repair pathways and all the core components of these pathways in BRCA1-deficient cancers is mandatorily required. So, in this review, we have struggled to summarize these components of DNA repair pathways in BRCA1-deficient cancers derived from breast, ovarian and prostate in most recent studies.
2. Novel DNA Repair Components Required for Survival of BRCA1-Deficient, but Not BRCA1-Proficient Cancers
Most recent advancements in the understanding of DNA repair mechanisms in BRCA1-deficient cancers have explored some crucial players such as Twin-Arginine Translocation (TatD) DNase domain containing 2 (TATDN2), Exonuclease/Endonuclease/Phosphatase Domain-1 (EEPD1), E3 ubiquitination ligases (e.g. BARD1 and RAD18), Telomeric Repeat-containing RNA (TERRA), FANCJ and EXO1. These molecules are integral to the DNA repair pathways in BRCA1-deficient cancers, contributing to maintaining genomic stability and precise cell proliferation. Therefore, these molecules are expected to be promising candidates for therapeutic treatments because they have been shown to increase sensitivity in BRCA1-deficient cancers. Here we described and summarized the roles of these molecules in regulating DNA damage repair pathways in BRCA1-deficient cancers.
2.1. Role of TATDN2 in BRCA1-Deficient Cancers
TatD proteins, a large family of 3’-to-5’ exonucleases highly conserved from bacteria to humans, consist of TatA, TatB, TatC, and TatD [50]. TatA, TatB and TatC are membrane-bound proteins, and they form a receptor essential for binding and transporting folded proteins bearing with Mg2+-dependent DNase activity, and the expression of TatD and two TatD homologues, YcfH and YjjV, are not essential for protein export in the Tat pathway in E. coli [50,51,52]. In human, there have been three TatD paralogs identified, TATDN1, TATDN2 and TATDN3. Despite TATDN2, a member of the TATD nuclease family, it is distinct from its relatives, TATDN1 and TATDN3, owing to structural differences in their amino-terminal regions and nuclease activities [50,53,54]. This uniqueness is prerequisite for TATDN2 to resolve R-loops and promote replication fork progression in the presence of excessive R-loops [55]. R-loops, which are DNA-RNA hybrids establishing a three-stranded nucleic acid structure consisting of a DNA:RNA hybrid and a displaced single-stranded DNA (ssDNA), regulate a variety of nuclear events such as DNA replication, chromatin structuring, transcription and genome stability [56]. Principally, two types of R-loops including (1) physiological and (2) pathological have been identified [57]. Of which, whereas physiological R-loops are dependent upon the programmed events requiring specific factors essential for their formation, pathological R-loops occur randomly in a non-programmed manner. DNA-RNA hybrid formation is specifically accelerated at specific regions at which this formation is crucial for regulating physiological function [57]. In contrast, R loops may interfere with DNA replication, repair, and transcription, therefore compromising genome integrity and function [57]. In response to it, cells develop other mechanisms to either inhibit and/or resolve such DNA-RNA hybrids [57]. However, if these mechanisms are dysfunctional, R loops may threaten genome integrity and cell proliferation and differentiation [57]. Increasing evidence suggests that R-loop-mediated replication fork stalling is a major feature of transcription-replication conflicts and R-loop-induced DNA damage [58]. However, it is still poorly understood how cells enable to protect themselves from pathological R-loops and how pathological R-loops contribute to chromatin structure and compromise genome integrity.
Notably, excessive R-loops triggering replication fork stalling and oncogene activation are more abundant in BRCA1-deficient cancers as compared to those in BRCA1-replete cancers. In vivo studies have shown that depleting TATDN2 leads to an increase in R-loops while its overexpression reduces them [55]. The relationship between BRCA1 and TATDN2 is mediated by the RNA-degrading Inositol-Requiring Enzyme type 1 (IRE1) and its target microRNA, miR-4638-p [55]. Of note, elevated expression of IRE1 is observed; therefore, its target, IRE1, is resultantly alleviated in BRCA1-deficient cancer cells [55]. The reduced expression of miR4638p enables TATDN2 to be upregulated, thereby resolving excess R-loops and promoting cell survival [55]. Interestingly, TATDN2 is much less essential for the survival of cognate BRCA1-proficient cancer cells (See Figure 1). As a consequence, loss of IRE1 or overexpression of miR-4638, leading to diminish the expression of TATDN2 which subsequently weakens replication fork progression, could be a promising therapeutic strategy in BRCA1-deficient cancers.
2.2. Role of TERRA in BRCA1-Deficient Cancers
TERRA, a long non-coding RNA (lncRNA), is evolutionally conserved in vertebrates, and it is transcribed from the subtelomeric regions of telomeric DNA sequences that is towards chromosomal ends [59]. In human cells, the huge majority of TERRA contain 7-methylguanosine (m7G) cap structures at its 5’-ends whereas some contain a poly(A) tail at its 3’-end, which is essential for their stability [60]. TERRA is believed to interact with telomere through binding to telomerase core factors such as TERT and TERC (or telomeric repeats) since the 3’ end of TERRA is complementary to the telomerase RNA template region [61] It is worth noting that TERRA transcripts regulate telomerase activity at chromosome ends in vitro [62].
Nevertheless, how TERRA regulates telomerase function remains unclear. For this hypothesis, Emilio et al., has confirm that the telomeric TERRA is involved in the nucleation of telomerase molecules into clusters prior to their recruitment at a short telomere in yeast [63]. Furthermore, telomere shortening promoted TERRA expression and subsequently triggered TERRA to be accumulated into nuclear focus in early S phase [63]. Additionally, TERRA interacts with yeast telomerase RNA TLC1, which is functionally similar to human TERC (hTERC) and forms TERRA-TLC1 complex that co-localizes with the telomere of origin during S phage in vivo. This interaction highlights a regulatory role of TERRA in the spatial organization of telomerase activity in telomeres [63].
In DNA Damage Responses (DDR), TERRA transcripts control the assembly of telomere-binding proteins at chromosome ends (capping), thereby maintaining genomic integrity [59]. In addition, TERRA transcripts serve as epi-genomic modulators in trans and as essential regulators of telomere in cis, thereby controlling DDR pathways indirectly by regulating gene expression [64]. Interestingly, TERRA transcripts are shown to establish DNA-RNA hybrid structure (R-loops) with the telomeric C-rich strand [65]. In human cancers, higher levels of telomeric R-loops are detectable in (Alternative Lengthening of Telomere) ALT-positive cancer cells in comparison with telomerase-positive cancer cells [66].
Formation of R-loop structures at telomeres affects telomere maintenance and genome integrity by accelerating HR between telomeres [67]. TERRA can promote telomeric DNA replication through R-loop formation at telomeres; furthermore, TERRA transcripts are crucial for precise telomere capping, helping to prevent the activation of DDR at chromosome ends [68]. It has been suggested that TERRA plays a role in switching at the ends of chromosomes between the DNA-binding protein RPA (crucial for activating ATR checkpoint) and the shelterin component POT1 (acting as a telomeric suppressor of ATR-mediated DDRs in telomeres) in vitro [69] that R-loops lead to genome instability via interfering with DNA polymerases during DNA replication.
BRCA1 was shown to directly interacts with TERRA via its N-terminal nuclear localization sequence as well as with major members of the telomere-specific shelterin complex in an R-loop-associated manner at promoter and telomeric regions [70]. R-loop-mediated BRCA1 binding to CpG-rich TERRA promoters suppresses TERRA transcription, reducing TERRA-loop-induced DNA damage and enhancing DNA repair responses in a SETX/XRN2-dependent manner [70]. Silencing BRCA1 or BRCA1 mutations within the TERRA-binding region increase TERRA expression, subsequently triggering an excessive formation of TERRA R-loops, telomeric replication stress and telomeric aberration phenomena in BRCA1-deficient cancers [70]. In BRCA1-deficient cancers, TERRA transcription is upregulated as a result of increased binding of transcription factors to TERRA type II promoters (devoid of CpG-islands), which results in exceeding expressions of TERRA R-loops and telomeric abnormalities [71,72]. Excessive TERRA levels together with an accumulation of unresolved TERRA R-loops at telomeric regions lead to replication stress and telomeric aberrancies, thereby resulting in telomere shortening [70] (See Figure 2). Interestingly, tumors harboring BRCA1 mutation is highly vulnerable to trabectedin [73] and lurbinectedin [74], inducing R-loop-mediated damage and causing genomic instability, suggesting that R-loop-targeting compounds in combination with telomerase inhibitor(s) might be a promising BRCA1-deficient tumor therapy [70].
2.3. Role of BARD1 in BRCA1-Deficient Cancers
BRCA1-associated RING domain protein 1 (BARD1), originally identified as tumor suppressor of familial breast cancer, forms heterodimer with BRCA1 to function as an E3 ubiquitin ligase that subsequently interacts with DNA and DNA damage response factors, thereby promoting the HR-mediated repair of DSBs, including at damaged replication forks [22]. BRCA1- BARD1 serves as multidirectional driver of DNA replication and replication, DNA repair responses, and tumor suppression. It is reported that mammary-specific ablation of either BRCA1 or BARD1 in conjugation with p53 deficiency triggers a high frequency of basal-like breast carcinomas with a propensity for being triple-negative tumors (that is, lacking the estrogen, progesterone and HER2 receptors) [75]. The N terminal RING domain of BARD1 binds to the BRCA1 BRCT repeats, which is associated with the phosphorylated isoforms of interacting proteins while the C terminal ankyrin motifs and tandem BRCT repeat binding domains of BARD1 bind to CstF-50 to modulate mRNA processing and RNAP II stability in response to DNA damage [76]. Besides, a previous study showed that BRCA1-BARD1 facilitates the nucleolytic resection of DNA ends to generate a single-stranded DNA (ssDNA) template for the recruitment of another tumor suppressor complex BRCA1-PALB2 and the recombinase RAD51 [77]. Structurally and functionally similar to a vast number of RING domain proteins, BRCA1 and BARD1 enable the interaction with E2 ubiquitin-conjugating enzymes and function as E3 ubiquitin ligases [78].
As mentioned above, BRCA1 interacts with the recombinase Rad51, and their co-localization could be observed in ionizing radiation-induced nuclear foci, suggesting the regulatory role of BRCA1 in HR-mediated DNA repair pathway. Indeed, Brca1-/- mouse and human cells alleviates the formation of DNA damage-induced RAD51 foci and causes defects in DNA end resection, which is a crucial early step in the repair of DSBs by HR repair pathway [79], suggesting a crucial role of BRCA1-BARD1 complex in recruiting RAD51 to DSBs. Also, there is an evidence that BRCA1-BARD1 complex has a role in the nucleolytic resection of DSB ends, as first proposed on the basis of the cell cycle-dependent association of BRCA1 with the MRE11-RAD50-NBS1 nuclease complex and with the end resection factor CtBP-interacting protein (CtIP) [80,81]. Relevance of the BRCA1-BARD1 E3 ligase activity for DSBs repair, tumor suppression, and resistance to PARP inhibitors and platinum-based compounds is still debatable. For example, histone H2A K127/129 ubiquitination is required for HR-mediated DSB repair. Nonetheless, defects in histone H2A ubiquitination by BRCA1-BARD1 complex can be compensated by other ubiquitin E3s such as RNF168 [82,83,84]. A recently published paper has revealed that the BRCA1-BARD1 complex is capable of ubiquitinating Proliferating Cell Nuclear Antigen (PCNA) to accelerate continuous DNA synthesis under normal physiological condition, subsequently preventing the accumulation of ssDNA gaps and protecting replication forks [84]. Thus, the BRCA1-BARD1 complex-mediated ubiquitination of PCNA is defective in BRCA1-deficient cancer cells, which leads to an enhanced accumulation of ssDNA gaps and impaired DNA replication under stress [84] (see Figure 3). In addition to BRCA1-BARD1, RAD18 could also ubiquitinate PCNA independently of BRCA1-BARD1. Besides, the PCNA ubiquitination remain considerably higher than that in BRCA1-proficient cancer cells [85].
2.4. Role of EXO1 in BRCA1-Deficient Cancers
Recent findings have identified the end resection factor EXO1 is crucial for the survival of BRCA1-deficient cells [86,87]. EXO1, which processes dsDNA ends by trimming DNA in a 5’-to-3’ direction, is essential for maintaining genomic stability in the absence of BRCA1.
In BRCA1-proficient cells, the initiation of short-range end resection involves the phosphorylation of CtIP (pCtIP), which then stimulates the nucleolytic function of MRE11 within the MRE11-RAD50-NBS1 (MRN) complex [88]. This complex trims up to approximately 300 nucleotides at the DSB ends, exposing short regions of microhomology that flank the break site [88,89]. This initial resection facilitates the recruitment of factors responsible for long-range resection, particularly EXO1 or DNA2, to further process the DNA ends [90]. It should be worth noting that EXO1 and DNA2 have different cofactors involved in long-range resection. This is particularly important following a DSB, as proper DNA end resection is necessary to initiate HR and prevent repair via NHEJ and SSA, which can lead to genomic instability.
EXO1 specializes in resecting dsDNA, while DNA2 is specifically crucial for ssDNA and is promoted by the BRCA1-BARD1 complex [86,87,90]. After long-range resection by EXO1 and/or DNA2, ssDNA is generated, enabling BRCA1 recruit the core HR protein RAD51 for binding [86]. RAD51 plays a pivotal role by facilitating the search for and alignment with a homologous sequence, which guides accurate repair. Although BRCA1 is essential for recruiting RAD51 and enabling HR, DNA end resection can still occur independently of BRCA1 [91,92]. This BRCA1-independent resection suggests the presence of a compensatory pathway in which EXO1 becomes essential for the survival of BRCA1-deficient cells, underscoring its role in these cells. Notably, EXO1 works alongside the helicase BLM to conduct long-range resection of DSBs [93]. In the absence of RAD51 recruitment, this resection instead facilitates the binding of RAD52, promoting the completion of more error-prone DNA repair pathways, such SSA or aNHEJ [94,95]. Consequently, BRCA1-deficient cells exhibit reduced RAD51 foci (biomarkers for HR) following ionizing radiation-induced DNA damage, indicating a diminished capacity for HR repair and a shift toward these alternative repair pathways.
Studies indicate that BRCA1-deficient cells lacking EXO1 show reduced levels of RAD52 and accumulate replication-associated lesions, particularly ssDNA gaps resulting from unprocessed Okazaki fragments during DNA replication [86,96]. These lesions correlate with increased Poly(ADP-ribosyl)ation (PARylation) by PARP1 during the S phase, which signals replication stress and activates the DDRs [86,96]. As a result, the suspicion is BRCA1-deficient cells preferentially utilize the SSA pathway, which depends on RAD52 rather than RAD51 for repair, suggesting that SSA dictates cells to avoid recruiting RAD51, thereby partially maintaining genomic stability despite compromised HR. Taken together, it indicates that HR is the preferred DNA repair pathway in BRCA1-proficient cells because of DNA2-mediated long-range resection, enabling RAD51 recruitment through BRCA1, whereas, in BRCA1-deficient cells, SSA, which is initiated by EXO1 in RAD52-dependent manner, is preferable. While long-range resection (which can extend over thousands of nucleotides) in HR is still disputable, it obviously serves as a key player of both SSA and aNHEJ, highlighting the complex interplay between these DNA repair pathways. Interestingly, a recent study has revealed has revealed silencing of EXO1 and/or DNA2 significantly downregulate ATR/CHEK1 signaling pathway, which leads unreplicated ssDNA gaps, which are repaired by template switching (TS) pathway, to accumulate, thereby leading to cell death in BRCA1-deficient cells [87]. However, in BRCA1-proficient cells, knockdown of EXO1 and/or DNA2 does not markedly alter cell survival [87] (See Figure 4), suggesting that double knockdown of EXO1 and DNA2 would be a promising therapeutic target for BRCA1-deficient cancers.
3. Other Potential Components
The components outlined above are essential in BRCA1-deficient, but not BRCA1-proficient cancer cells. So, these findings significantly contribute to the therapeutic development of drug program in clinical trials. Besides, we describe the functions of several other proteins on regulating the survival of BRCA1-deficient cancer cells, based on the most recently published papers.
3.1. Role of EEPD1 in BRCA1-Deficient Cancers
Exonuclease/Endonuclease/Phosphatase Domain-1 (EEPD1) plays a critical role in addressing oxidative stress damage, functioning independently of BRCA1 [97,98]. Oxidative stress occurs due to reactive oxygen species (ROS), such as hydrogen peroxide () and superoxide (), which are by-products of the mitochondrial electron transport chain [97,98]. ROS has been linked to inflammation, apoptosis, neurodegenerative diseases, diabetes, carcinogenesis and tumor progression [99]. These issues, at least in part, result from ROS-induced DNA damage, particularly during key cellular processes like replication and transcription.
Unresolved oxidative stress poses a substantial threat during DNA replication or transcription. Elevated ROS levels lead to the formation of DNA lesions, specifically through the conversion of guanine bases into 8-oxo-7,8-dihydroguanine (8-oxoG) [100,101]. This oxidative modification disrupts the structure and function of the replisome-associated ROS sensor, peroxiredoxin 2 (PRDX2), ultimately displacing the TIMELESS-TIPIN complex from replication forks [102]. This displacement impairs replication fork progression, depletes deoxynucleotide triphosphates (dNTPs), and may cause fork arrest [102]. Notably, Naoko and Hisao demonstrated the role of TIMELESS and TIPIN as checkpoint mediators critical to S-phase progression and the DNA replication checkpoint [103]. The frequency of guanine oxidation to 8-oxoG is estimated to occur between 100 and 500 times per cell per day, with a higher occurrence in neuronal cells, linking this damage to neurodegenerative diseases [104]. When an oxidized DNA lesion obstructs a replication fork and remains unresolved, the fork may collapse or fuse, resulting in harmful chromosomal end joining, reaffirming the importance of resolving oxidative DNA stress.
Intriguingly, the human base excision repair (BER) pathway provides a means to repair oxidative DNA damage. In BER, damaged bases are recognized and excised by DNA glycosylases, followed by an endonucleolytic cleavage 5’ of the resulting abasic or apurinic/apyrimidinic (AP) site, primarily via AP-endonuclease 1 (APE1) [105]. APE1 is a multifunctional protein involved in HR, and in the context of HR, it facilitates BRCA1-mediated repair while mitigating oxidative stress [105]. However, BER and HR mechanisms can operate independently of APE1 and BRCA1, respectively, through EEPD1-mediated repair [106].
Research by Robert et al., has shown that EEPD1 is recruited to stalled replication forks when treated with HU, a DNA synthesis inhibitor that mimics replication stress [98]. Upon binding, EEPD1 cleaves the lagging parental strand, enabling long-range resection by EXO1 and, in coordination with BLM and RAD51, initiates HR [107]. In the context of BER, like APE1, EEPD1 exhibits nucleolytic activity, cleaving the 5’ parental strand of a stalled replication fork, thus generating a 3’-OH terminus at the AP site [107]. Since EEPD1 has been shown to be recruited to forks stalled by HU, it is reasonable to propose that EEPD1 may similarly respond to replication forks stalled by ROS-induced damage [107].
A study by Aruna et al., demonstrated that when EEPD1 is significantly depleted in human embryonic kidney (HEK293) cells treated with , there is an increase in stalled replication forks and a decrease in replication fork restart events [97]. Of note, this study shows that EEPD1 not only resolves oxidative damage at replication forks but also protects ongoing replication if repair mechanisms fail, suggesting that if BER is compromised, alternative pathways involving EEPD1 may resolve oxidative insults. While this study did not specifically examine BRCA1's role in BER, previous findings on EEPD1's role in replication fork resolution under stress conditions imply that similar mechanisms may occur in BRCA1-deficient cells experiencing oxidative stress [98].
In summary, EEPD1 functions independently of BRCA1 and is thus a critical factor in DNA repair pathways, potentially offering a therapeutic target for BRCA1-deficient cancers.
3.2. Role of FANCJ in BRCA1-Deficient Cancers
FANCJ, a DNA helicase and interacting partner of the tumor suppressor BRCA1, is vital for the repair of DNA interstrand crosslinks (ICL), a highly toxic lesion triggering chromosomal instability and interrupting normal transcription [108]. In diploid cells, FANCJ is best characterized to be crucial for HR-mediated repair of DSBs; however, its precise role(s) and molecule mechanism(s) have not been fully understood. Interestingly, Sanket Awate et al., screened DDR/DNA repair gene targets in FANCJ-/- cells, and then found that RAP80, a ubiquitin-binding protein, which recruits BRCA1 and other proteins to DSBs for DNA end-processing [109]. This study also showed that loss of FANCJ and RAP80 result in severe defects through weakening the maturation of HR repair intermediates as a consequence of ICL-induced DNA damage [109]. Besides, FANCJ interacts with FANCD2, a core component of Fanconi anemia/BRCA (FA-BRCA) DNA repair pathway. FANCJ is not required for FANCD2 activation, yet it necessitates FANCD2 to thoroughly respond to DNA damage [110]. In addition, loss of FANCJ does not change FANCD2 transcription; nonetheless, it remarkably enhances the proteasome-induced FANCD2 proteolysis, which precisely activates DDR in response to HU-induced replication fork stalling [110]. FANCD2 mono-ubiquitination is associated with BRCA1 in DNA damage-induced nuclear foci [111], and truncating FANCJ mutations were shown to trigger chromosomal instability, genetic disorder Fanconi anemia (FA), and hereditary breast cancer [112], indicating that silencing of FANCJ probably enable cancer cells to significantly sensitive to PARPi. Nevertheless, whether this hypothesis is correct needs to be further investigated.
Ke Cong et al., has shown that FANCJ serves as a key driver of PARP1-induced ssDNA gaps and sensitivity during DNA replication in BRCA1 or FANCJ-deficient breast and ovarian cancer cells [113]. The loss of FANC1 triggers deactivation of PARP1 through modifying the replisome composition and/or DNA secondary structures, thereby causing synthetic lethality in FANC1- or BRCA1-deficient cancer cells [114]. However, PARPi triggered fewer replication gaps compared to those in BRCA1-deficient cells [114]. Although not required for PARP1 chromatin localization, FANCJ is important for maintaining appropriate DNA replication activity of PARP1 and MMP pathway [115]. However, whether loss of FANCJ causes cell death in BRCA1-proficient cells has not yet been examined. Together, these findings apparently provide us some promising clues for the development of targeted therapies and precise medicine approaches for treating the patients with BRCA1-deficient tumors.
3.3. Role of USP1 in BRCA1-Deficient Cancers
As mentioned above, ubiquitination of PCNA by BARD1 in BRCA1-deficient cancer cells is important for accelerating continuous DNA synthesis under normal physiological conditions to protect the replication fork [84]. An additional component in the role of fork protection, which indirectly involves ubiquitinated PCNA (PCNA-ub), is USP1 [116]. Along with USP12 and USP46, USP1 is part of the largest deubiquitinating subfamily, ubiquitin-specific protease (USP) [117]. In general, USPs cleave the isopeptide bond between ubiquitin and the target protein. By default, USP12, USP46, and USP1 are in an inactive state; however, upon binding to the WD40-repeat protein UAF1, USP12, USP46, and USP1 become activated [116,118,119]. Notably, this stimulation by UAF1 is not exclusive, as USP1 in particular can also be stimulated by binding to DNA.
Among the many roles of BRCA1 is the protection of the replication fork from degradation. However, since BRCA1-deficient cells lack this component, USP1 serves as an essential substitute in protection. Research has shown that USP1 is overexpressed in BRCA1-deficient cancer cells compared to other cancer types, suggesting a reliance on USP1 [116]. Thus, the question arises: does USP1 have a direct interaction with DNA, thereby protecting the replication fork? Lim et al., confirmed this by tracking and detecting USP1 localization to the replication fork using iPOND and immunoblotting [116]. Prior research has shown that the USP1-UAF1 complex localizes at the replication fork, and USP1’s deubiquitinating activity, stimulated by binding, contributes to fork protection by deubiquitinating proteins in the vicinity, including PCNA [119].
This contrasts with the claim above that BARD1-mediated ubiquitination of PCNA protects the replication fork. Instead, accumulated mono-ubiquitinated PCNA recruits low-fidelity tran-slesion synthesis (TLS) polymerases, such as POLK and REV1, to the replication fork, which can lead to fork instability [120,121,122,123]. USP1 prevents the accumulation of mono-ubiquitinated PCNA through its deubiquitinating activities. Lim et al., confirmed the toxicity of USP1 knockdown in BRCA1-deficient cells, which resulted in decreased viability due to increased fork instability and degradation from accumulated mono-ubiquitinated PCNA, as shown by colony formation and fork stability assays [116]. In USP1-deficient cells with functional BRCA1, these phenotypes are not observed. Nonetheless, it should be noted that Lim et al., studied only the mono-ubiquitinated form of PCNA, leaving it unclear whether poly-ubiquitination might affect USP1’s efficiency [116]. Therefore, the hierarchy of fork protection in PCNA-ub is uncertain, with both USP1’s deubiquitination activity and BARD1-mediated ubiquitination of PCNA suggesting roles in replication fork protection.
Overall, in both BRCA1-deficient and normal cells with functional USP1, cell survival is maintained. In contrast, in BRCA1-deficient and USP1-deficient cells, cell viability decreases. Replication fork protection is critical in these circumstances: despite the loss of BRCA1, cells with functional USP1 persist, though with reduced replication fork protection. However, the absence of both BRCA1 and USP1 genes results in significant replication fork degradation, leading to lower cell viability. Conversely, in cells with normal BRCA1 expression but lacking USP1, cell viability and replication remain normal.
Lim et al., noted that targeting and inhibiting USP1 with ML323 results in decreased viability of BRCA1-deficient cancer cells [116]. Previously, ML323 was reported to block FANCD2 deubiquitination, increasing sensitivity to cisplatin [124]. Thus, targeting USP1 or potentially any deubiquitinating enzyme involved in replication fork protection could be an effective therapeutic strategy against BRCA1-deficient cancers without affecting normal cells. This approach is innovative, as it allows for specific targeting between cancerous and normal cells.
4. Discussion
BRCA1, accompanied by its interacting partners BRCA2 and BARD1, is widely recognized as an essential component in the maintenance of genome stability and DNA repair. BRCA1 is best characterized to be one of the core components for multiple DNA repair mechanisms, consisting of HR, SSA, BER, and NHEJ [125,126,127,128]. These pathways are essentially required for a range of DNA damage types, from SSBs to complex DSBs. Multifaceted role(s) of BRCA1 in such pathways is crucial for protecting cells genomic instability—a hallmark of many cancers.
The loss of BRCA1 is believed to compromise the efficacy of DNA repair, leading these cells to be more sensitive to DNA damage. However, BRCA1-deficient cells rely upon other pathways to compensate for the defects in BRCA1-mediated DNA repair pathways. These compensatory pathways exert a variety of DNA repair components such as TATDN2, TERRA, BARD1, EXO1, and FANC1 for alternative repair pathways such as NHEJ pathways including classical and alternative. Each of these proteins contributes to different facets of the repair process, from controlling telomeric stability (as seen with TERRA) to processing DNA ends (EXO1) and facilitating chromatin remodeling (FANCJ). The coordinated action of these proteins reflects an intricate web of interactions, redundancy, and co-dependence. These proteins functionally work in a concerted manner, filling gaps left by the primary repair pathway and establishing a compensatory framework to preserve genomic stability to a degree. Depending upon secondary DNA repair components highlights an adaptive evolution under selective pressure. The evolutionary pressure exerted by BRCA1 deficiency leads to an increased reliance on alternative pathways, which become more active and sometimes upregulated in response to the genomic challenges presented by cancer. This adaptation allows cancer cells not only to survive but also to evolve under conditions that would otherwise limit proliferation and survival such as inhibitors and the emergence of proteolysis targeting chimeras (PROTACS).
In summary, loss of BRCA1 does not equate to a straightforward defect in DNA repair pathways; instead, it illustrates a dynamic shift toward a new equilibrium for which alternative repair components and/or pathways compensate. This compensatory mechanism, however, often lacks the precision of BRCA1-mediated repair, potentially leading to an accumulation of mutations that drive further tumorigenic changes. Understanding of these compensatory pathways gives us not only a comprehensive outlook on how BRCA1-deficient cancer cells survive and proliferate, thereby contributing to the establishment of drug development program, which targets the secondary DNA repair pathways as well as development of potential therapeutic treatments in patients with BRCA1-deficient tumors.
Conflicts of Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Author Contributions
N.N.: Conceptualization, investigation, writing, reviewing and editing. M.T.T.: Conceptualization, investigation, writing, reviewing and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data was created or analyzed in this study. Data sharing is not applicable to this article.
References
- Ceccaldi, R.; Rondinelli, B.; D'Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol 2016, 26, 52-64. [CrossRef]
- Yu, W.; Lescale, C.; Babin, L.; Bedora-Faure, M.; Lenden-Hasse, H.; Baron, L.; Demangel, C.; Yelamos, J.; Brunet, E.; Deriano, L. Repair of G1 induced DNA double-strand breaks in S-G2/M by alternative NHEJ. Nature Communications 2020, 11, 5239. [CrossRef]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. Journal of Biological Chemistry 2018, 293, 10512-10523. [CrossRef]
- Hussain, S.S.; Majumdar, R.; Moore, G.M.; Narang, H.; Buechelmaier, Erika S.; Bazil, M.J.; Ravindran, P.T.; Leeman, Jonathan E.; Li, Y.; Jalan, M.; et al. Measuring nonhomologous end-joining, homologous recombination and alternative end-joining simultaneously at an endogenous locus in any transfectable human cell. Nucleic Acids Research 2021, 49, e74-e74. [CrossRef]
- Hanscom, T.; McVey, M. Regulation of Error-Prone DNA Double-Strand Break Repair and Its Impact on Genome Evolution. Cells 2020, 9, 1657.
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Translational Cancer Research 2013, 2, 163-177.
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet 2008, 4, e1000110. [CrossRef]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem Sci 2015, 40, 701-714. [CrossRef]
- Paul, A.; Paul, S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Front Biosci (Landmark Ed) 2014, 19, 605-618. [CrossRef]
- Yamamoto, H.; Hirasawa, A. Homologous Recombination Deficiencies and Hereditary Tumors. Int J Mol Sci 2021, 23. [CrossRef]
- Wang, M.; Chen, S.; Ao, D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. MedComm 2021, 2, 654-691. [CrossRef]
- Nickoloff, J.A.; Jaiswal, A.S.; Sharma, N.; Williamson, E.A.; Tran, M.T.; Arris, D.; Yang, M.; Hromas, R. Cellular Responses to Widespread DNA Replication Stress. International Journal of Molecular Sciences 2023, 24, 16903.
- Huang, R.; Zhou, P.K. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct Target Ther 2021, 6, 254. [CrossRef]
- Deng, C.X. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res 2006, 34, 1416-1426. [CrossRef]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J Cell Physiol 2016, 231, 15-24. [CrossRef]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front Genet 2015, 6, 157. [CrossRef]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers (Basel) 2017, 9. [CrossRef]
- Schiewer, M.J.; Knudsen, K.E. DNA Damage Response in Prostate Cancer. Cold Spring Harb Perspect Med 2019, 9. [CrossRef]
- Wu, W.; Koike, A.; Takeshita, T.; Ohta, T. The ubiquitin E3 ligase activity of BRCA1 and its biological functions. Cell Div 2008, 3, 1. [CrossRef]
- Clark, S.L.; Rodriguez, A.M.; Snyder, R.R.; Hankins, G.D.; Boehning, D. Structure-Function Of The Tumor Suppressor BRCA1. Comput Struct Biotechnol J 2012, 1. [CrossRef]
- Leung, C.C.; Glover, J.N. BRCT domains: easy as one, two, three. Cell Cycle 2011, 10, 2461-2470. [CrossRef]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol 2020, 21, 284-299. [CrossRef]
- Rosen, E.M.; Fan, S.; Ma, Y. BRCA1 regulation of transcription. Cancer Letters 2006, 236, 175-185. [CrossRef]
- Pan, H.; He, Z.; Ling, L.; Ding, Q.; Chen, L.; Zha, X.; Zhou, W.; Liu, X.; Wang, S. Reproductive factors and breast cancer risk among BRCA1 or BRCA2 mutation carriers: Results from ten studies. Cancer Epidemiology 2014, 38, 1-8. [CrossRef]
- Shao, F.; Sun, H.; Deng, C.X. Potential therapeutic targets of triple-negative breast cancer based on its intrinsic subtype. Oncotarget 2017, 8, 73329-73344. [CrossRef]
- Peshkin, B.N.; Alabek, M.L.; Isaacs, C. BRCA1/2 mutations and triple negative breast cancers. Breast Dis 2010, 32, 25-33. [CrossRef]
- Burga, L.N.; Hu, H.; Juvekar, A.; Tung, N.M.; Troyan, S.L.; Hofstatter, E.W.; Wulf, G.M. Loss of BRCA1 leads to an increase in epidermal growth factor receptor expression in mammary epithelial cells, and epidermal growth factor receptor inhibition prevents estrogen receptor-negative cancers in BRCA1-mutant mice. Breast Cancer Research 2011, 13, R30. [CrossRef]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.; Peterson, S.K.; Singletary, C.; Arun, B.K.; Litton, J.K. Cancers associated with 1 and 2 mutations other than breast and ovarian. Cancer 2015, 121, 269-275. [CrossRef]
- Castro, E.; Eeles, R. The role of BRCA1 and BRCA2 in prostate cancer. Asian J Androl 2012, 14, 409-414. [CrossRef]
- Narod, S.A.; Metcalfe, K.; Finch, A.; Chan, A.-W.; Armel, S.R.; Aeilts, A.; Eisen, A.; Karlan, B.; Bordeleau, L.; Tung, N.; et al. The risk of skin cancer in women who carry BRCA1 or BRCA2 mutations. Hereditary Cancer in Clinical Practice 2024, 22, 7. [CrossRef]
- Devico Marciano, N.; Kroening, G.; Dayyani, F.; Zell, J.A.; Lee, F.C.; Cho, M.; Valerin, J.G. BRCA-Mutated Pancreatic Cancer: From Discovery to Novel Treatment Paradigms. Cancers (Basel) 2022, 14. [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer 2011, 12, 68-78. [CrossRef]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A 2011, 108, 3406-3411. [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 2017, 18, 495-506. [CrossRef]
- Liang, S.; Blundell, T.L. Human DNA-dependent protein kinase activation mechanism. Nature Structural & Molecular Biology 2023, 30, 140-147. [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol 2017, 18, 610-621. [CrossRef]
- Couto, C.A.; Wang, H.Y.; Green, J.C.; Kiely, R.; Siddaway, R.; Borer, C.; Pears, C.J.; Lakin, N.D. PARP regulates nonhomologous end joining through retention of Ku at double-strand breaks. J Cell Biol 2011, 194, 367-375. [CrossRef]
- Dale Rein, I.; Solberg Landsverk, K.; Micci, F.; Patzke, S.; Stokke, T. Replication-induced DNA damage after PARP inhibition causes G2 delay, and cell line-dependent apoptosis, necrosis and multinucleation. Cell Cycle 2015, 14, 3248-3260. [CrossRef]
- Wang, S.S.Y.; Jie, Y.E.; Cheng, S.W.; Ling, G.L.; Ming, H.V.Y. PARP Inhibitors in Breast and Ovarian Cancer. Cancers (Basel) 2023, 15. [CrossRef]
- Valabrega, G.; Scotto, G.; Tuninetti, V.; Pani, A.; Scaglione, F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int J Mol Sci 2021, 22. [CrossRef]
- Bhamidipati, D.; Haro-Silerio, J.I.; Yap, T.A.; Ngoi, N. PARP inhibitors: enhancing efficacy through rational combinations. British Journal of Cancer 2023, 129, 904-916. [CrossRef]
- Dilmac, S.; Ozpolat, B. Mechanisms of PARP-Inhibitor-Resistance in BRCA-Mutated Breast Cancer and New Therapeutic Approaches. Cancers (Basel) 2023, 15. [CrossRef]
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci 2011, 102, 663-669. [CrossRef]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers (Basel) 2022, 14. [CrossRef]
- Konstantinopoulos, P.A.; Lheureux, S.; Moore, K.N. PARP Inhibitors for Ovarian Cancer: Current Indications, Future Combinations, and Novel Assets in Development to Target DNA Damage Repair. American Society of Clinical Oncology Educational Book 2020, e116-e131. [CrossRef]
- Revythis, A.; Limbu, A.; Mikropoulos, C.; Ghose, A.; Sanchez, E.; Sheriff, M.; Boussios, S. Recent Insights into PARP and Immuno-Checkpoint Inhibitors in Epithelial Ovarian Cancer. Int J Environ Res Public Health 2022, 19. [CrossRef]
- Veneris, J.T.; Matulonis, U.A.; Liu, J.F.; Konstantinopoulos, P.A. Choosing wisely: Selecting PARP inhibitor combinations to promote anti-tumor immune responses beyond BRCA mutations. Gynecologic Oncology 2020, 156, 488-497. [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 2017, 31, 318-332. [CrossRef]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci Transl Med 2017, 9. [CrossRef]
- Chen, Y.C.; Li, C.L.; Hsiao, Y.Y.; Duh, Y.; Yuan, H.S. Structure and function of TatD exonuclease in DNA repair. Nucleic Acids Res 2014, 42, 10776-10785. [CrossRef]
- Petrů, M.; Wideman, J.; Moore, K.; Alcock, F.; Palmer, T.; Doležal, P. Evolution of mitochondrial TAT translocases illustrates the loss of bacterial protein transport machines in mitochondria. BMC Biology 2018, 16, 141. [CrossRef]
- Kudva, R.; Denks, K.; Kuhn, P.; Vogt, A.; Müller, M.; Koch, H.G. Protein translocation across the inner membrane of Gram-negative bacteria: the Sec and Tat dependent protein transport pathways. Res Microbiol 2013, 164, 505-534. [CrossRef]
- Dorival, J.; Eichman, B.F. Human and bacterial TatD enzymes exhibit apurinic/apyrimidinic (AP) endonuclease activity. Nucleic Acids Res 2023, 51, 2838-2849. [CrossRef]
- Yang, H.; Liu, C.; Jamsen, J.; Wu, Z.; Wang, Y.; Chen, J.; Zheng, L.; Shen, B. The DNase domain-containing protein TATDN1 plays an important role in chromosomal segregation and cell cycle progression during zebrafish eye development. Cell Cycle 2012, 11, 4626-4632. [CrossRef]
- Jaiswal, A.S.; Dutta, A.; Srinivasan, G.; Yuan, Y.; Zhou, D.; Shaheen, M.; Sadideen, D.T.; Kirby, A.; Williamson, E.A.; Gupta, Y.K.; et al. TATDN2 resolution of R-loops is required for survival of BRCA1-mutant cancer cells. Nucleic Acids Res 2023, 51, 12224-12241. [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol Cell 2019, 73, 398-411. [CrossRef]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604-618. [CrossRef]
- Chang, E.Y.; Stirling, P.C. Replication Fork Protection Factors Controlling R-Loop Bypass and Suppression. Genes (Basel) 2017, 8. [CrossRef]
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8. [CrossRef]
- Porro, A.; Feuerhahn, S.; Reichenbach, P.; Lingner, J. Molecular Dissection of Telomeric Repeat-Containing RNA Biogenesis Unveils the Presence of Distinct and Multiple Regulatory Pathways. Molecular and Cellular Biology 2010, 30, 4808-4817. [CrossRef]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Research 2010, 38, 5797-5806. [CrossRef]
- Chebly, A.; Ropio, J.; Baldasseroni, L.; Prochazkova-Carlotti, M.; Idrissi, Y.; Ferrer, J.; Farra, C.; Beylot-Barry, M.; Merlio, J.P.; Chevret, E. Telomeric Repeat-Containing RNA (TERRA): A Review of the Literature and First Assessment in Cutaneous T-Cell Lymphomas. Genes (Basel) 2022, 13. [CrossRef]
- Cusanelli, E.; Romero, C.A.; Chartrand, P. Telomeric noncoding RNA TERRA is induced by telomere shortening to nucleate telomerase molecules at short telomeres. Mol Cell 2013, 51, 780-791. [CrossRef]
- Chu, H.P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.G.; Wei, C.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 2017, 170, 86-101.e116. [CrossRef]
- Rivosecchi, J.; Jurikova, K.; Cusanelli, E. Telomere-specific regulation of TERRA and its impact on telomere stability. Seminars in Cell & Developmental Biology 2024, 157, 3-23. [CrossRef]
- MacKenzie, D., Jr.; Watters, A.K.; To, J.T.; Young, M.W.; Muratori, J.; Wilkoff, M.H.; Abraham, R.G.; Plummer, M.M.; Zhang, D. ALT Positivity in Human Cancers: Prevalence and Clinical Insights. Cancers (Basel) 2021, 13. [CrossRef]
- Gong, Y.; Liu, Y. R-Loops at Chromosome Ends: From Formation, Regulation, and Cellular Consequence. Cancers (Basel) 2023, 15. [CrossRef]
- Al-Turki, T.M.; Maranon, D.G.; Nelson, C.B.; Lewis, A.M.; Luxton, J.J.; Taylor, L.E.; Altina, N.; Wu, F.; Du, H.; Kim, J.; et al. Telomeric RNA (TERRA) increases in response to spaceflight and high-altitude climbing. Communications Biology 2024, 7, 698. [CrossRef]
- Cusanelli, E.; Chartrand, P. Telomeric repeat-containing RNA TERRA: a noncoding RNA connecting telomere biology to genome integrity. Front Genet 2015, 6, 143. [CrossRef]
- Vohhodina, J.; Goehring, L.J.; Liu, B.; Kong, Q.; Botchkarev Jr, V.V.; Huynh, M.; Liu, Z.; Abderazzaq, F.O.; Clark, A.P.; Ficarro, S.B.; et al. BRCA1 binds TERRA RNA and suppresses R-Loop-based telomeric DNA damage. Nature Communications 2021, 12, 3542. [CrossRef]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273-277. [CrossRef]
- Deng, Z.; Wang, Z.; Stong, N.; Plasschaert, R.; Moczan, A.; Chen, H.S.; Hu, S.; Wikramasinghe, P.; Davuluri, R.V.; Bartolomei, M.S.; et al. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. Embo j 2012, 31, 4165-4178. [CrossRef]
- Monk, B.J.; Lorusso, D.; Italiano, A.; Kaye, S.B.; Aracil, M.; Tanović, A.; D'Incalci, M. Trabectedin as a chemotherapy option for patients with BRCA deficiency. Cancer Treat Rev 2016, 50, 175-182. [CrossRef]
- Cruz, C.; Llop-Guevara, A.; Garber, J.E.; Arun, B.K.; Pérez Fidalgo, J.A.; Lluch, A.; Telli, M.L.; Fernández, C.; Kahatt, C.; Galmarini, C.M.; et al. Multicenter Phase II Study of Lurbinectedin in BRCA-Mutated and Unselected Metastatic Advanced Breast Cancer and Biomarker Assessment Substudy. J Clin Oncol 2018, 36, 3134-3143. [CrossRef]
- Shakya, R.; Szabolcs, M.; McCarthy, E.; Ospina, E.; Basso, K.; Nandula, S.; Murty, V.; Baer, R.; Ludwig, T. The basal-like mammary carcinomas induced by Brca1 or Bard1 inactivation implicate the BRCA1/BARD1 heterodimer in tumor suppression. Proc Natl Acad Sci U S A 2008, 105, 7040-7045. [CrossRef]
- Edwards, R.A.; Lee, M.S.; Tsutakawa, S.E.; Williams, R.S.; Nazeer, I.; Kleiman, F.E.; Tainer, J.A.; Glover, J.N. The BARD1 C-terminal domain structure and interactions with polyadenylation factor CstF-50. Biochemistry 2008, 47, 11446-11456. [CrossRef]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Jian Ma, C.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360-365. [CrossRef]
- Mallery, D.L.; Vandenberg, C.J.; Hiom, K. Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. Embo j 2002, 21, 6755-6762. [CrossRef]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1–BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nature Structural & Molecular Biology 2016, 23, 647-655. [CrossRef]
- Zhong, Q.; Chen, C.-F.; Li, S.; Chen, Y.; Wang, C.-C.; Xiao, J.; Chen, P.-L.; Sharp, Z.D.; Lee, W.-H. Association of BRCA1 with the hRad50-hMre11-p95 Complex and the DNA Damage Response. Science 1999, 285, 747-750, doi:doi:10.1126/science.285.5428.747.
- Chen, L.; Nievera, C.J.; Lee, A.Y.-L.; Wu, X. Cell Cycle-dependent Complex Formation of BRCA1·CtIP·MRN Is Important for DNA Double-strand Break Repair*. Journal of Biological Chemistry 2008, 283, 7713-7720. [CrossRef]
- Zong, D.; Adam, S.; Wang, Y.; Sasanuma, H.; Callén, E.; Murga, M.; Day, A.; Kruhlak, M.J.; Wong, N.; Munro, M.; et al. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol Cell 2019, 73, 1267-1281.e1267. [CrossRef]
- Sherker, A.; Chaudhary, N.; Adam, S.; Heijink, A.M.; Noordermeer, S.M.; Fradet-Turcotte, A.; Durocher, D. Two redundant ubiquitin-dependent pathways of BRCA1 localization to DNA damage sites. EMBO reports 2021, 22, e53679. [CrossRef]
- Salas-Lloret, D.; García-Rodríguez, N.; Soto-Hidalgo, E.; González-Vinceiro, L.; Espejo-Serrano, C.; Giebel, L.; Mateos-Martín, M.L.; de Ru, A.H.; van Veelen, P.A.; Huertas, P.; et al. BRCA1/BARD1 ubiquitinates PCNA in unperturbed conditions to promote continuous DNA synthesis. Nat Commun 2024, 15, 4292. [CrossRef]
- Salas-Lloret, D.; García-Rodríguez, N.; Soto-Hidalgo, E.; González-Vinceiro, L.; Espejo-Serrano, C.; Giebel, L.; Mateos-Martín, M.L.; de Ru, A.H.; van Veelen, P.A.; Huertas, P.; et al. BRCA1/BARD1 ubiquitinates PCNA in unperturbed conditions to promote continuous DNA synthesis. Nature Communications 2024, 15, 4292. [CrossRef]
- van de Kooij, B.; Schreuder, A.; Pavani, R.S.; Garzero, V.; Van Hoeck, A.; San Martin Alonso, M.; Koerse, D.; Wendel, T.J.; Callen, E.; Boom, J.; et al. EXO1-mediated DNA repair by single-strand annealing is essential for BRCA1-deficient cells. bioRxiv 2023, 2023.2002.2024.529205. [CrossRef]
- García-Rodríguez, N.; Domínguez-García, I.; Domínguez-Pérez, M.D.C.; Huertas, P. EXO1 and DNA2-mediated ssDNA gap expansion is essential for ATR activation and to maintain viability in BRCA1-deficient cells. Nucleic Acids Res 2024, 52, 6376-6391. [CrossRef]
- Howard, S.M.; Ceppi, I.; Anand, R.; Geiger, R.; Cejka, P. The internal region of CtIP negatively regulates DNA end resection. Nucleic Acids Res 2020, 48, 5485-5498. [CrossRef]
- Patterson-Fortin, J.; D'Andrea, A.D. Exploiting the Microhomology-Mediated End-Joining Pathway in Cancer Therapy. Cancer Res 2020, 80, 4593-4600. [CrossRef]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Experimental & Molecular Medicine 2020, 52, 1705-1714. [CrossRef]
- Halder, S.; Sanchez, A.; Ranjha, L.; Reginato, G.; Ceppi, I.; Acharya, A.; Anand, R.; Cejka, P. Double-stranded DNA binding function of RAD51 in DNA protection and its regulation by BRCA2. Molecular Cell 2022, 82, 3553-3565.e3555. [CrossRef]
- Bunting, S.F.; Callén, E.; Kozak, M.L.; Kim, J.M.; Wong, N.; López-Contreras, A.J.; Ludwig, T.; Baer, R.; Faryabi, R.B.; Malhowski, A.; et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell 2012, 46, 125-135. [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 2011, 25, 350-362. [CrossRef]
- Xue, C.; Greene, E.C. DNA Repair Pathway Choices in CRISPR-Cas9-Mediated Genome Editing. Trends Genet 2021, 37, 639-656. [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet 2016, 32, 566-575. [CrossRef]
- van de Kooij, B.; Schreuder, A.; Pavani, R.; Garzero, V.; Uruci, S.; Wendel, T.J.; van Hoeck, A.; San Martin Alonso, M.; Everts, M.; Koerse, D.; et al. EXO1 protects BRCA1-deficient cells against toxic DNA lesions. Molecular Cell 2024, 84, 659-674.e657. [CrossRef]
- Jaiswal, A.S.; Kim, H.S.; Schärer, O.D.; Sharma, N.; Williamson, E.A.; Srinivasan, G.; Phillips, L.; Kong, K.; Arya, S.; Misra, A.; et al. EEPD1 promotes repair of oxidatively-stressed replication forks. NAR Cancer 2023, 5, zcac044. [CrossRef]
- Wu, Y.; Lee, S.H.; Williamson, E.A.; Reinert, B.L.; Cho, J.H.; Xia, F.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Brantley, A.; et al. EEPD1 Rescues Stressed Replication Forks and Maintains Genome Stability by Promoting End Resection and Homologous Recombination Repair. PLoS Genet 2015, 11, e1005675. [CrossRef]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, II; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int J Mol Sci 2022, 23. [CrossRef]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Burrows, C.J. 8-Oxo-7,8-dihydroguanine in the Context of a Gene Promoter G-Quadruplex Is an On–Off Switch for Transcription. ACS Chemical Biology 2017, 12, 2417-2426. [CrossRef]
- Sugden, K.D.; Martin, B.D. Guanine and 7,8-dihydro-8-oxo-guanine-specific oxidation in DNA by chromium(V). Environ Health Perspect 2002, 110 Suppl 5, 725-728. [CrossRef]
- Andrs, M.; Stoy, H.; Boleslavska, B.; Chappidi, N.; Kanagaraj, R.; Nascakova, Z.; Menon, S.; Rao, S.; Oravetzova, A.; Dobrovolna, J.; et al. Excessive reactive oxygen species induce transcription-dependent replication stress. Nat Commun 2023, 14, 1791. [CrossRef]
- Yoshizawa-Sugata, N.; Masai, H. Human Tim/Timeless-interacting protein, Tipin, is required for efficient progression of S phase and DNA replication checkpoint. J Biol Chem 2007, 282, 2729-2740. [CrossRef]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009, 30, 2-10. [CrossRef]
- Thompson, P.S.; Cortez, D. New insights into abasic site repair and tolerance. DNA Repair (Amst) 2020, 90, 102866. [CrossRef]
- Rajan, A.; Varghese, G.R.; Yadev, I.; Anandan, J.; Latha, N.R.; Patra, D.; Krishnan, N.; Kuppusamy, K.; Warrier, A.V.; Bhushan, S.; et al. Modulation of BRCA1 mediated DNA damage repair by deregulated ER-α signaling in breast cancers. Am J Cancer Res 2022, 12, 17-47.
- Kim, H.S.; Nickoloff, J.A.; Wu, Y.; Williamson, E.A.; Sidhu, G.S.; Reinert, B.L.; Jaiswal, A.S.; Srinivasan, G.; Patel, B.; Kong, K.; et al. Endonuclease EEPD1 Is a Gatekeeper for Repair of Stressed Replication Forks. J Biol Chem 2017, 292, 2795-2804. [CrossRef]
- Awate, S.; Sommers, J.A.; Datta, A.; Nayak, S.; Bellani, M.A.; Yang, O.; Dunn, C.A.; Nicolae, C.M.; Moldovan, G.L.; Seidman, M.M.; et al. FANCJ compensates for RAP80 deficiency and suppresses genomic instability induced by interstrand cross-links. Nucleic Acids Res 2020, 48, 9161-9180. [CrossRef]
- Awate, S.; Sommers, J.A.; Datta, A.; Nayak, S.; Bellani, M.A.; Yang, O.; Dunn, C.A.; Nicolae, C.M.; Moldovan, G.-L.; Seidman, M.M.; et al. FANCJ compensates for RAP80 deficiency and suppresses genomic instability induced by interstrand cross-links. Nucleic Acids Research 2020, 48, 9161-9180. [CrossRef]
- Clark, D.W.; Tripathi, K.; Dorsman, J.C.; Palle, K. FANCJ protein is important for the stability of FANCD2/FANCI proteins and protects them from proteasome and caspase-3 dependent degradation. Oncotarget 2015, 6, 28816-28832. [CrossRef]
- Bogliolo, M.; Surrallés, J. The Fanconi Anemia/BRCA Pathway: FANCD2 at the Crossroad between Repair and Checkpoint Responses to DNA Damage. 2006.
- Fang, C.B.; Wu, H.T.; Zhang, M.L.; Liu, J.; Zhang, G.J. Fanconi Anemia Pathway: Mechanisms of Breast Cancer Predisposition Development and Potential Therapeutic Targets. Front Cell Dev Biol 2020, 8, 160. [CrossRef]
- Cong, K.; MacGilvary, N.; Lee, S.; MacLeod, S.G.; Calvo, J.; Peng, M.; Nedergaard Kousholt, A.; Day, T.A.; Cantor, S.B. FANCJ promotes PARP1 activity during DNA replication that is essential in BRCA1 deficient cells. Nat Commun 2024, 15, 2599. [CrossRef]
- Cong, K.; MacGilvary, N.; Lee, S.; MacLeod, S.G.; Calvo, J.; Peng, M.; Kousholt, A.N.; Day, T.; Cantor, S.B. FANCJ promotes PARP1 activity during DNA replication that is essential in BRCA1 deficient cells. bioRxiv 2024. [CrossRef]
- Yu, W.; Lescale, C.; Babin, L.; Bedora-Faure, M.; Lenden-Hasse, H.; Baron, L.; Demangel, C.; Yelamos, J.; Brunet, E.; Deriano, L. Repair of G1 induced DNA double-strand breaks in S-G2/M by alternative NHEJ. Nat Commun 2020, 11, 5239. [CrossRef]
- Lim, K.S.; Li, H.; Roberts, E.A.; Gaudiano, E.F.; Clairmont, C.; Sambel, L.A.; Ponnienselvan, K.; Liu, J.C.; Yang, C.; Kozono, D.; et al. USP1 Is Required for Replication Fork Protection in BRCA1-Deficient Tumors. Mol Cell 2018, 72, 925-941.e924. [CrossRef]
- García-Santisteban, I.; Peters, G.J.; Giovannetti, E.; Rodríguez, J.A. USP1 deubiquitinase: cellular functions, regulatory mechanisms and emerging potential as target in cancer therapy. Molecular Cancer 2013, 12, 91. [CrossRef]
- Dharadhar, S.; Clerici, M.; van Dijk, W.J.; Fish, A.; Sixma, T.K. A conserved two-step binding for the UAF1 regulator to the USP12 deubiquitinating enzyme. J Struct Biol 2016, 196, 437-447. [CrossRef]
- Murai, J.; Yang, K.; Dejsuphong, D.; Hirota, K.; Takeda, S.; D'Andrea, A.D. The USP1/UAF1 complex promotes double-strand break repair through homologous recombination. Mol Cell Biol 2011, 31, 2462-2469. [CrossRef]
- Edmunds, C.E.; Simpson, L.J.; Sale, J.E. PCNA ubiquitination and REV1 define temporally distinct mechanisms for controlling translesion synthesis in the avian cell line DT40. Mol Cell 2008, 30, 519-529. [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: a double-edged sword guarding the genome. Translational Cancer Research 2013, 2, 107-129.
- Nicolae, C.M.; Aho, E.R.; Vlahos, A.H.S.; Choe, K.N.; De, S.; Karras, G.I.; Moldovan, G.-L. The ADP-ribosyltransferase PARP10/ARTD10 Interacts with Proliferating Cell Nuclear Antigen (PCNA) and Is Required for DNA Damage Tolerance*. Journal of Biological Chemistry 2014, 289, 13627-13637. [CrossRef]
- Venkadakrishnan, J.; Lahane, G.; Dhar, A.; Xiao, W.; Bhat, K.M.; Pandita, T.K.; Bhat, A. Implications of Translesion DNA Synthesis Polymerases on Genomic Stability and Human Health. Mol Cell Biol 2023, 43, 401-425. [CrossRef]
- Dexheimer, T.S.; Rosenthal, A.S.; Liang, Q.; Chen, J.; Villamil, M.A.; Kerns, E.H.; Simeonov, A.; Jadhav, A.; Zhuang, Z.; Maloney, D.J. Discovery of ML323 as a Novel Inhibitor of the USP1/UAF1 Deubiquitinase Complex. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda (MD), 2010.
- Tung, N.M.; Garber, J.E. BRCA1/2 testing: therapeutic implications for breast cancer management. British Journal of Cancer 2018, 119, 141-152. [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nature Reviews Cancer 2012, 12, 68-78. [CrossRef]
- Arun, B.; Couch, F.J.; Abraham, J.; Tung, N.; Fasching, P.A. BRCA-mutated breast cancer: the unmet need, challenges and therapeutic benefits of genetic testing. British Journal of Cancer 2024, 131, 1400-1414. [CrossRef]
- Petrucelli, N.; Daly, M.B.; Pal, T. BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer. In GeneReviews(®), Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle. Copyright © 1993-2024, University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.: Seattle (WA), 1993.
Figure 1.
Graphic abstract for the role of TATDN2 in resolving R-loops in BRCA1-proficient cells and BRCA1-deficient cells. Silencing of TATDN2 expression by either miR 6438-5p or TATDN2-targeting siRNA (siTATDN2) does not alter R-loop resolution activity and cell survival in BRCA1-proficient cells but significantly abolish R-loop resolution activity, thereby leading to cell death in BRCA1-mutant cells.
Figure 1.
Graphic abstract for the role of TATDN2 in resolving R-loops in BRCA1-proficient cells and BRCA1-deficient cells. Silencing of TATDN2 expression by either miR 6438-5p or TATDN2-targeting siRNA (siTATDN2) does not alter R-loop resolution activity and cell survival in BRCA1-proficient cells but significantly abolish R-loop resolution activity, thereby leading to cell death in BRCA1-mutant cells.
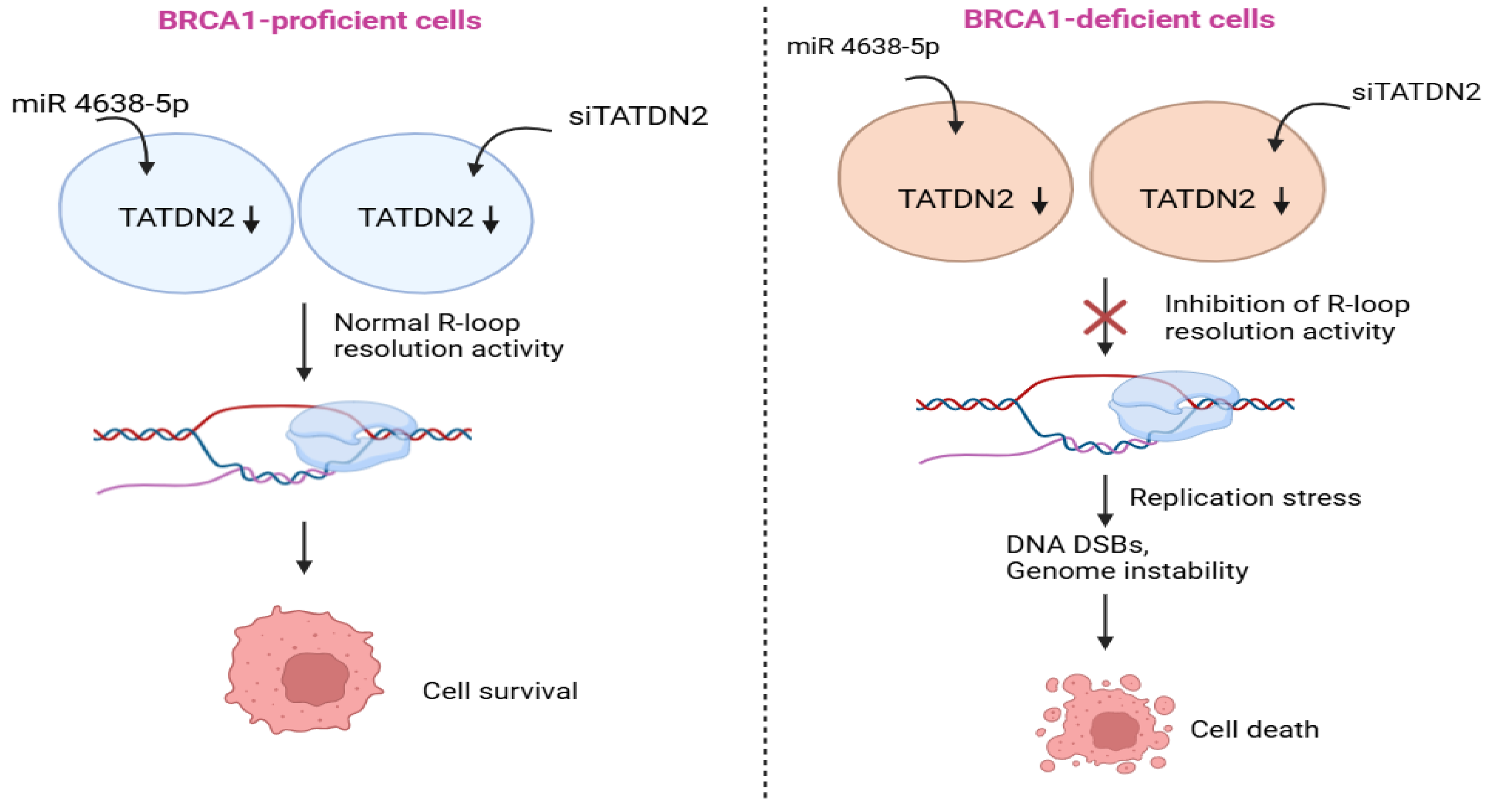
Figure 2.
Graphical abstract for the role of TERRA in resolving R-loops in BRCA1-proficient cells and BRCA1-deficient cells. TERRA transcription and R-loop formation are inhibited in BRCA1-proficient cells, which leads to alleviate telomeric DNA damage, maintain telomere length, and therefore cell survival. Nonetheless, TERRA transcription and R-loop formation are significantly enhanced in BRCA1-deficient cells, which elevates replication stress, telomeric DNA damage, shorten telomere length, and therefore cell death.
Figure 2.
Graphical abstract for the role of TERRA in resolving R-loops in BRCA1-proficient cells and BRCA1-deficient cells. TERRA transcription and R-loop formation are inhibited in BRCA1-proficient cells, which leads to alleviate telomeric DNA damage, maintain telomere length, and therefore cell survival. Nonetheless, TERRA transcription and R-loop formation are significantly enhanced in BRCA1-deficient cells, which elevates replication stress, telomeric DNA damage, shorten telomere length, and therefore cell death.
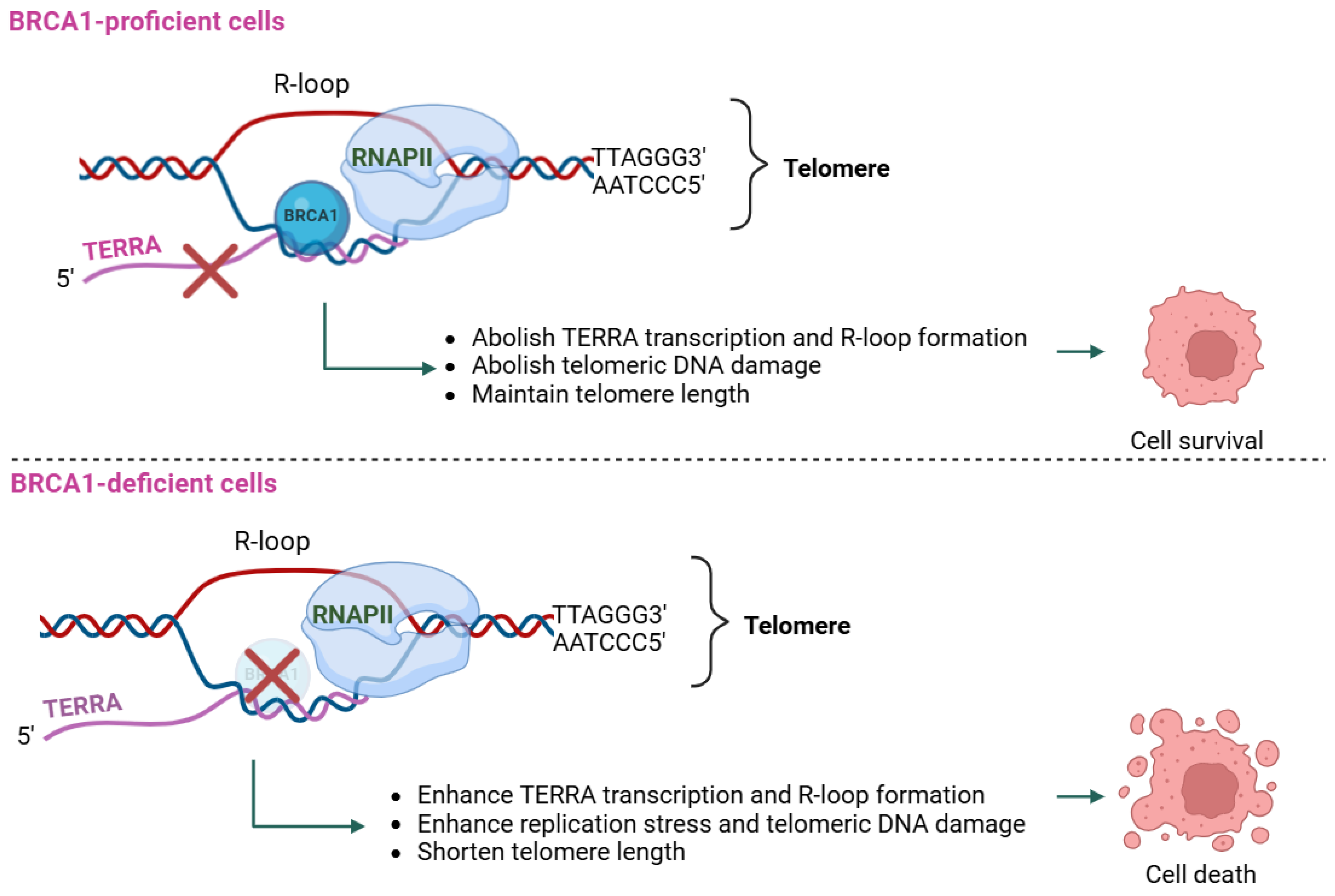
Figure 3.
Graphical abstract for the role of BARD1 in the regulation of DSB repair in BRCA1-proficient cells and BRCA1-deficient cells. In BRCA1-proficient cells, the BRCA1-BARD1 complex is responsible for ubiquitinating PCNA, accelerating DNA synthesis continuously under normal physiological condition, thereby preventing the accumulation of unreplicated ssDNA gaps as well as protecting replication forks. Nonetheless, in BRCA1-deficient cells, PCNA ubiquitination is disturbed, which results in the accumulation of unreplicated ssDNA gaps, impaired DNA replication under stress, thereby leading to the accumulation of unrepaired DSBs and cell death.
Figure 3.
Graphical abstract for the role of BARD1 in the regulation of DSB repair in BRCA1-proficient cells and BRCA1-deficient cells. In BRCA1-proficient cells, the BRCA1-BARD1 complex is responsible for ubiquitinating PCNA, accelerating DNA synthesis continuously under normal physiological condition, thereby preventing the accumulation of unreplicated ssDNA gaps as well as protecting replication forks. Nonetheless, in BRCA1-deficient cells, PCNA ubiquitination is disturbed, which results in the accumulation of unreplicated ssDNA gaps, impaired DNA replication under stress, thereby leading to the accumulation of unrepaired DSBs and cell death.
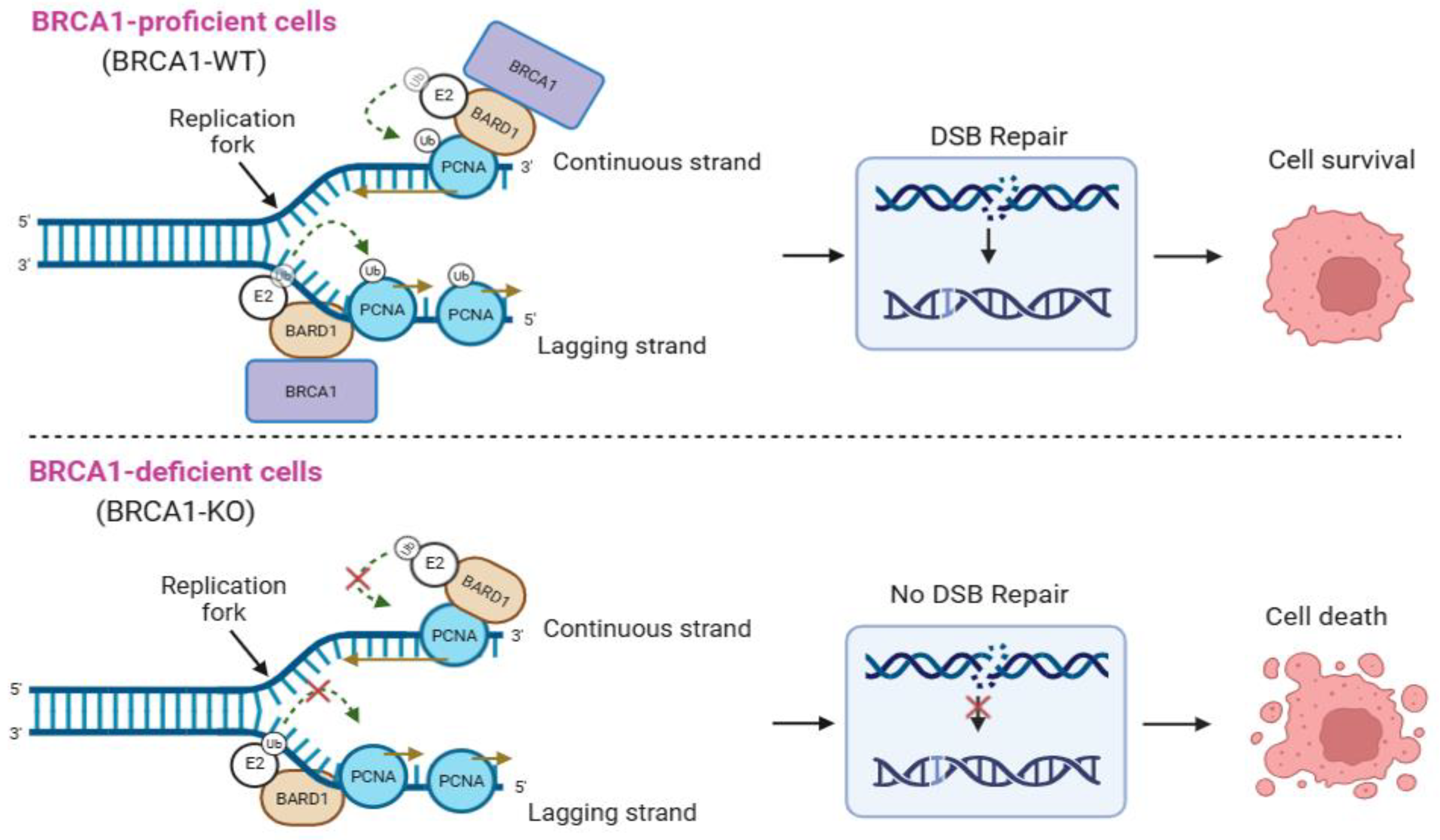
Figure 4.
Graphical abstract for the role of EXO1/DNA2 in the regulation of ssNDA gaps in BRCA1-proficient cells and BRCA1-deficient cells, following replication stress caused by MMS or HU. In BRCA1-proficient cells, EXO1 and/or DNA2 fully activates ATR/CHEK1 signaling pathway, subsequently triggering cell cycle arrest to limit the number of unreplicated ssDNA gaps by TS pathway, thereby maintaining cell survival. Also, in BRCA1-proficient cells with loss of either EXO1 or DNA2, the ATR/CHEK1 signaling pathway is still activated (not full), which promotes ssDNA gap repair and cell survival. However, in BRCA1-deficient cancer cells with silencing of each of EXO1 or DNA2 or both, ATR/CHEK1 signaling pathway is significantly weakened, which results in accumulation of unreplicated ssDNA gaps, thereby leading to cell death.
Figure 4.
Graphical abstract for the role of EXO1/DNA2 in the regulation of ssNDA gaps in BRCA1-proficient cells and BRCA1-deficient cells, following replication stress caused by MMS or HU. In BRCA1-proficient cells, EXO1 and/or DNA2 fully activates ATR/CHEK1 signaling pathway, subsequently triggering cell cycle arrest to limit the number of unreplicated ssDNA gaps by TS pathway, thereby maintaining cell survival. Also, in BRCA1-proficient cells with loss of either EXO1 or DNA2, the ATR/CHEK1 signaling pathway is still activated (not full), which promotes ssDNA gap repair and cell survival. However, in BRCA1-deficient cancer cells with silencing of each of EXO1 or DNA2 or both, ATR/CHEK1 signaling pathway is significantly weakened, which results in accumulation of unreplicated ssDNA gaps, thereby leading to cell death.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



