
Submitted:
29 January 2025
Posted:
03 February 2025
You are already at the latest version
Abstract
In 2022, the World Health Organization reported that tuberculosis (TB) was the second leading cause of death globally from a single infectious agent, following COVID-19. The development of new antitubercular agents with novel mechanisms of action for use in complex TB therapy is considered a key approach to combating TB. In this study, we examined the gene expression profile of M. smegmatis when exposed to a promising antituberculosis agent, quinoxaline 1,4-dioxide (QdNO) 7-chloro-2-(ethoxycarbonyl)-3-methyl-6-(piperazin-1-yl)quinoxaline-1,4-dioxide-1 (LCTA-3368). We investigated how the bacterial response changed with different doses (1/4 × MIC, 1/2 × MIC, and 1 × MIC) and durations (30 minutes and 90 minutes) of treatment with the drug. Our analysis revealed significant upregulation of genes involved in DNA repair and replication processes, as well as changes in the expression of 95 genes encoding proteins with oxidoreductase activity. These findings support the proposed mechanism of antibacterial action of QdNOs, which is associated with the formation of free radicals leading to DNA damage.
Keywords:
M. smegmatis
; QdNOs
; antituberculosis agent
; transcriptome
1. Introduction
According to the World Health Organization (WHO), Mycobacterium tuberculosis and tuberculosis (TB) were the world’s second leading cause of death from a single infectious agent in 2022, after coronavirus disease (COVID-19), and caused nearly twice as many deaths as HIV/AIDS. The global number of people newly diagnosed with TB reached 7.5 million in 2022, the highest number reported since WHO began global TB monitoring in 1995. Globally, an estimated 410,000 people (95% UI: 370,000–450,000) developed multidrug-resistant TB (MDR-TB, defined as TB resistant to rifampicin and isoniazid) or rifampicin-resistant TB (RR-TB) in 2022 [1].
Urgent action is required to end the global TB epidemic by 2030, a goal adopted by all United Nations Member States and the WHO. Given the rising incidence of multidrug-resistant TB, the discovery, development, and rapid adoption of new tools, interventions, and treatment strategies are among the key priorities in TB treatment. Additional priorities include the development of a vaccine to reduce the risk of infection and new drug treatments to combat MDR-TB [1].
Quinoxaline 1,4-dioxides (QdNOs) are promising compounds with a broad spectrum of biological properties, including antitumor, antibacterial, antiparasitic, anti-inflammatory, antioxidant, and herbicidal activities [2]. Recent studies have shown that some QdNO derivatives exhibit excellent inhibitory activity against M. tuberculosis, highlighting the potential of this scaffold for the development of new anti-TB drugs [3,4,5]. The presence of two N-oxide fragments in the quinoxaline ring contributes to their high biological activity by enabling oxidation processes [6]. Additionally, QdNOs can undergo bioreductive activation under the hypoxic conditions present in TB granulomas, where non-replicating persistent forms of M. tuberculosis can survive. This survival mechanism contributes to prolonged treatments and the risk of drug resistance development [7].
In previous work, we identified a lead compound, 7-chloro-2-(ethoxycarbonyl)-3-methyl-6-(piperazin-1-yl)quinoxaline 1,4-dioxide-1 (LCTA-3368), through a screen of a QdNO library using Mycobacterium smegmatis [8], a model organism widely employed for screening anti-TB drug candidates [9,10]. LCTA-3368 demonstrated strong inhibitory activity against both M. smegmatis (4 μg/mL) and M. tuberculosis (1.25 μg/mL), although its cytotoxicity remains to be evaluated. We found that LCTA-3368 induces unique non-synonymous mutations in a variety of genes in spontaneous drug-resistant M. smegmatis mutants, although a clearly defined cellular target has not yet been identified [8]. One proposed mechanism of action for quinoxaline 1,4-dioxide derivatives involves the direct induction of single- and double-stranded DNA breaks in bacteria, which could explain the numerous mutations observed in M. smegmatis strains [11].
Transcriptomic studies, including comprehensive RNA sequencing (RNA-seq) analysis, are increasingly becoming a powerful tool for elucidating additional mechanisms of action of antimicrobials, including anti-tuberculosis drugs, by examining the bacterial transcriptional response to drug exposure [12]. In our previous study, we employed transcriptomic analysis to investigate the impact of imidazo tetrazines on iron metabolism, a set of compounds for which a biotarget could not be identified using the classical approach of generating spontaneous drug-resistant mutants [13].
In this study, we describe the transcriptomic profile of M. smegmatis treated with LCTA-3368 in a time-dependent (30 minutes and 90 minutes) and dose-dependent (1/4 × MIC, 1/2 × MIC, and 1 × MIC) manner to gain insight into the gradual changes in the bacterial response to this drug. We observed significant alterations in the expression of genes involved in DNA repair and replication processes.
2. Materials and Methods
2.1. Microbial Cultures and Growth Conditions
Mycobacterium smegmatis mc² 155 was grown in Middlebrook 7H9 medium (Himedia, India) supplemented with oleic albumin dextrose catalase (OADC, Himedia, India), 0.1% Tween-80 (v/v), and 0.4% glycerol (v/v). Soybean-casein digest agar (M290, Himedia, India) and Middlebrook 7H11 agar (Himedia, India) supplemented with OADC were used as solid media. Liquid cultures were incubated in a Multitron incubator shaker (Infors HT, Bottmingen-Basel, Switzerland) at 37°C and 250 rpm.
For the drug exposure assay and transcriptomic analysis, M. smegmatis mc² 155 was inoculated from agar plates into 7H9 medium and grown until an OD600 of 2.5 (two nights) to obtain a stable liquid culture without clumps. The culture was then diluted 1:200 and grown overnight until reaching an OD600 of 2. Subsequently, it was diluted 1:10 in fresh medium to achieve an approximate OD600 of 0.2. LCTA-3368 100× stock solutions were prepared in DMSO (ACS Grade, Solon, India) and added to the bacterial cultures to final concentrations corresponding to 1/4 × MIC (1 μg/mL), 1/2 × MIC (2 μg/mL), and 1 × MIC (4 μg/mL) in 7H9 OADC medium. The same volume of DMSO (1% v/v) was added to the control samples. Bacterial cultures were incubated for 30 minutes and 90 minutes (1/6 and 1/2 of the cell division time, respectively [14]) at 37°C and 250 rpm, followed by RNA extraction. All experiments were performed in three biological replicates.
2.2. Total RNA Extraction
RNA was extracted using the MagMAX mirVana Total RNA Isolation Kit (Thermo Fisher Scientific, Lithuania) on the KingFisher Flex Purification System (Thermo Fisher Scientific, United States), following the manufacturer’s instructions. The extracted RNA was treated with DNase using the Turbo DNA-Free Kit (Thermo Fisher Scientific) in a 50 µL reaction volume and further purified using Agencourt RNAClean XP (Beckman Coulter, United States) according to the manufacturer’s protocol. The total RNA concentration was measured using the Quant-iT Ribogreen RNA Assay Kit (Thermo Fisher Scientific), and RNA quality was assessed using an Agilent Bioanalyzer with Agilent RNA 6000 Pico Chips (Agilent Technologies, United States).
2.3. Library Preparation and RNA Sequencing
For the preparation of transcriptomic libraries, 250 ng of total RNA was used as input. Ribosomal RNA was selectively removed using the Ribo-Zero Plus rRNA Depletion Kit (Illumina, USA), followed by library preparation with the KAPA RNA Hyper Kit (Roche, Switzerland), according to the manufacturer’s protocol. RNA purification steps were carried out using RNA Clean XP magnetic beads (Beckman Coulter, Brea, USA), and final library purification was performed with Agencourt AMPure XP magnetic beads (Beckman Coulter, Brea, USA). The size distribution and quality of the libraries were evaluated using the Agilent High Sensitivity DNA Kit (Agilent Technologies, USA), while library concentration was quantified using the Quant-iT DNA Assay Kit, High Sensitivity (Thermo Fisher Scientific, USA). Equimolar quantities of all libraries (12 pM) were pooled and sequenced in a high-throughput run on the Illumina HiSeq platform using 2 × 100 bp paired-end reads and a 5% PhiX spike-in control. The RNA-seq read data were deposited in the NCBI Sequence Read Archive under accession number PRJNA1091547.
2.4. Bioinformatics Analysis
Quality control of the raw sequencing data was performed using FastQC (v.0.11.9) [15], and individual reports were merged using MultiQC (v.1.9) [16]. Adapters and low-quality reads were removed with Trimmomatic (v.0.39) [17]. Trimmed reads were mapped to the reference M. smegmatis mc² 155 genome (CP000480.1) using HISAT2 (v.2.2.1) [18]. Mapping quality and gene coverage were assessed with QualiMap (v.2.2.2) [19]. Mapped reads were assigned to genes using featureCounts (v.2.0.1) [20]. Differential gene expression (DGE) analysis was conducted using edgeR (v.3.30.3) [21] in R (v.4.0.2) [22]. Genes with a false discovery rate (FDR) cutoff of 0.05 and a fold change (FC) of log2FC ≤ -1 or log2FC ≥ 1 were considered differentially expressed. Plots were generated in Python 3 (v.3.12.6) [23] using the matplotlib (v.3.9) [24] and numpy (v.2.1.1) [25] packages.
Functional enrichment analysis of Gene Ontology (GO) categories and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways for differentially expressed genes (DEGs) was performed using GOpiscator (v.0.1.5) [26], with categories considered enriched at p ≤ 0.05. Heatmaps were generated in Python 3 (v.3.12.6) using the Seaborn (v.0.13) package. Bubble charts based on GOpiscator (v.0.1.5) [26] results for DEGs were created in Python 3 (v.3.12.6) using the matplotlib (v.3.9) and numpy (v.2.1.1) packages.
3. Results
3.1. Whole-Transcriptome Analysis
A total of 32 RNA-seq libraries were generated to comprehensively characterize the transcriptional profile of M. smegmatis following LCTA-3368 treatment at different concentrations and time points, yielding 167,144,850 raw reads. After the removal of low-quality and adapter sequences, 165,637,398 clean reads were retained, representing an average retention rate of 98.75–99.96% of the raw reads. The average length of bases mapped per library was 256,321,391 bp.
A total of 1,223 differentially expressed genes (DEGs) with at least a two-fold change in expression level were identified (Table S1). Among these, 767 genes were upregulated, with 209 of them consistently expressed across all experimental conditions. Conversely, 456 DEGs were downregulated, with 5 of these consistently expressed at all time points and concentrations.
Figure 1.
Differentially expressed genes (DEGs) in M. smegmatis. Red dots represent positively regulated DEGs (log2FC ≥ 1), while blue dots indicate negatively regulated DEGs (log2FC ≤ -1). Graphs A-C display volcano plots for samples incubated for 30 minutes, whereas graphs D-F correspond to samples incubated for 90 minutes in the presence of LCTA-3368 at concentrations of 1/4 × MIC (A, D), 1/2 × MIC (B, E), and 1 × MIC (C, F).
Figure 1.
Differentially expressed genes (DEGs) in M. smegmatis. Red dots represent positively regulated DEGs (log2FC ≥ 1), while blue dots indicate negatively regulated DEGs (log2FC ≤ -1). Graphs A-C display volcano plots for samples incubated for 30 minutes, whereas graphs D-F correspond to samples incubated for 90 minutes in the presence of LCTA-3368 at concentrations of 1/4 × MIC (A, D), 1/2 × MIC (B, E), and 1 × MIC (C, F).
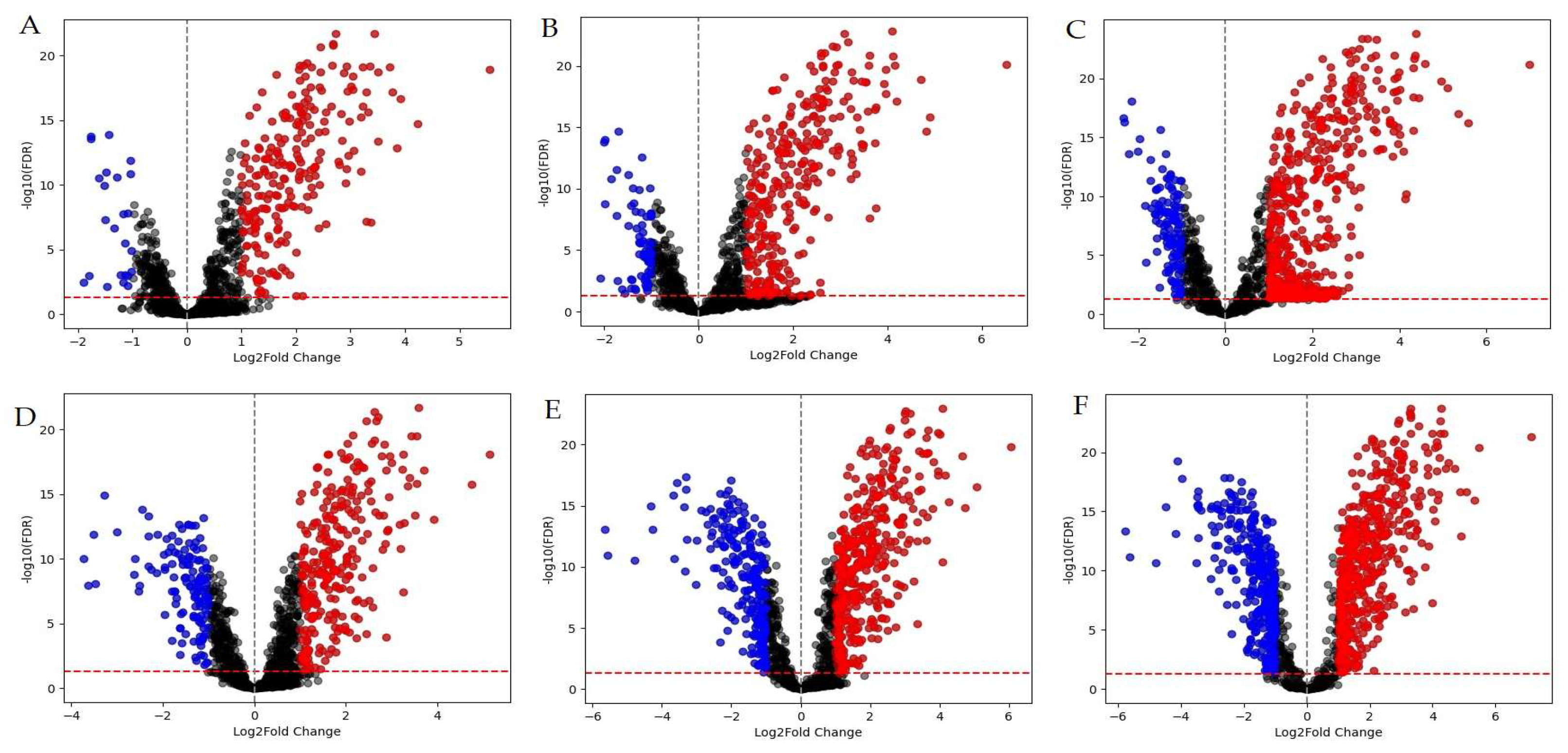
The comparison of different MIC groups reveals that more than half of the differentially expressed genes (DEGs) are represented after 90 minutes of exposure to the test compound, particularly in the 1/2 × MIC and 1 × MIC groups. Regarding the GO analysis (Figure 2), approximately 30% of the DEGs, on average, are associated with the DNA repair pathway (GO:0006281) and the DNA duplex unwinding pathway (GO:0032508) for both 30-minute and 90-minute exposures across all three groups (1/4 × MIC, 1/2 × MIC, 1 × MIC). At the 90-minute time point, negative regulation of DNA-templated transcription (GO:0045892) was observed in the 1/2 × MIC and 1 × MIC groups, but not in the 1/4 × MIC group. A similar pattern was observed for the nucleotide-excision repair (GO:0006289) and cell redox homeostasis (GO:0045454) pathways. Differential expression of genes in the lipid transport pathway (GO:0006869) was detected only in the 30-minute exposure at 1/2 × MIC, while genes related to the DNA unwinding involved in DNA replication pathway (GO:0006268) were identified exclusively in the 30-minute exposure at 1 × MIC.
Since we identified a large number of differentially expressed genes (DEGs), we introduced a dose-dependence criterion to further analyze the results. The primary criterion for dose-dependence is a minimum difference of 0.5 for upregulated genes and -0.5 for downregulated genes in the log2FC values at 1/4 × MIC, 1/2 × MIC, and 1 × MIC, with this difference required at each step. We selected DEGs meeting this criterion at both the 30-minute and 90-minute timepoints, and these will be referred to as "dose-dependent" hereafter. This difference was evaluated separately for the 30-minute and 90-minute timepoints, as well as simultaneously for both timepoints. Thus, DEGs could be dose-dependent at 30 minutes, 90 minutes, or both. An additional criterion is that the log2FC must change in a consistent direction (either increasing or decreasing) with the increasing concentration of LCTA-3368. DEGs satisfying all these conditions are classified as "dose-dependent."
3.2. Functional groups of Differentially Expressed Genes
The analysis identified 42 DEGs involved in DNA repair, 86 DEGs encoding transcriptional regulators, highlighting the robust response to LCTA-3368, 91 DEGs encoding proteins with oxidoreductase functions, 55 DEGs whose products participate in transport processes, 57 DEGs associated with fatty acid metabolism, amino acid metabolism, and the TCA cycle, and 13 DEGs related to transposases (Table S2).
3.2.1. Reparation Genes
The analysis identified 42 DEGs associated with the DNA repair pathway, encompassing all known DNA repair systems in mycobacteria: double-strand break (DSB) repair, homologous recombination (HR), nonhomologous end joining (NHEJ), single-strand annealing (SSA), nucleotide excision repair (NER), and the SOS response system (SOS) (Table S3). All DNA repair-related DEGs were upregulated throughout the experiment, confirming the extensive DNA damage inflicted on bacterial cells by LCTA-3368 [27].
Among the 42 DEGs in Mycobacterium smegmatis, the proteins encoded by 33 DEGs exhibit homology to proteins in Mycobacterium tuberculosis, with sequence similarity ranging from 75% to 98% and differences in amino acid sequence lengths of less than 15% (Table S3). A total of 38 M. smegmatis DEGs have counterparts in the M. tuberculosis genome. However, the amino acid sequences of the primosomal protein PriA (Rv1402 in M. tuberculosis and MSMEG_1238 in M. smegmatis) are not homologous, despite sharing the same function. PriA is involved in replication restart in bacteria by unwinding the lagging strand of stalled DNA replication forks. Similarly, RecF, which participates in homologous recombination, shows low homology between MSMEG_2293 and Rv0003. Notably, M. smegmatis possesses a gene encoding a swt1-family HEPN helicase and an additional gene for a UvrD-like protein, both of which are absent in the M. tuberculosis genome. Importantly, the key genes responsible for the functioning of all DNA repair systems in M. smegmatis have homologs in M. tuberculosis.
Among the DEGs, five genes were upregulated in a dose-dependent manner (Figure 3): MSMEG_1622 (DNA repair) and MSMEG_1633 (SOS) were the most significantly upregulated and dose-dependent at both timepoints. MSMEG_1756 and MSMEG_1757 (NER) exhibited dose-dependent upregulation at 30 minutes, while MSMEG_0211 (NER) showed dose-dependent upregulation at 90 minutes. Interestingly, the upregulation of MSMEG_0211 increased at 90 minutes compared to 30 minutes, whereas the expression of other dose-dependent DEGs either remained stable or slightly decreased at 90 minutes.
3.2.2. Oxidoreductases
The largest group of DEGs (N = 91, comprising 69 upregulated and 22 downregulated DEGs) consisted of genes encoding proteins with predicted oxidoreductase activity. These proteins belong to the following families: SDR family, Nitroreductase superfamily, VOC family, LLM class flavin-dependent oxidoreductases, and FAD-binding oxidoreductases. Previous studies have shown that bacterial oxidoreductases may play a role in the metabolic activation of quinoxalines, generating free radicals that can inhibit DNA synthesis and cause DNA damage [28].
Among the 69 upregulated genes, 8 exhibited dose-dependent regulation (Figure 4). The genes MSMEG_5027 (encoding a VOC family protein) and MSMEG_1623 (encoding an SDR family oxidoreductase) were the most significantly upregulated and the only ones showing dose-dependence at both timepoints. Conversely, the genes MSMEG_3094, MSMEG_5576, and MSMEG_6763 were downregulated throughout the experiment and displayed dose-dependence at the 90-minute timepoint.
3.1.2.3. Other Genes
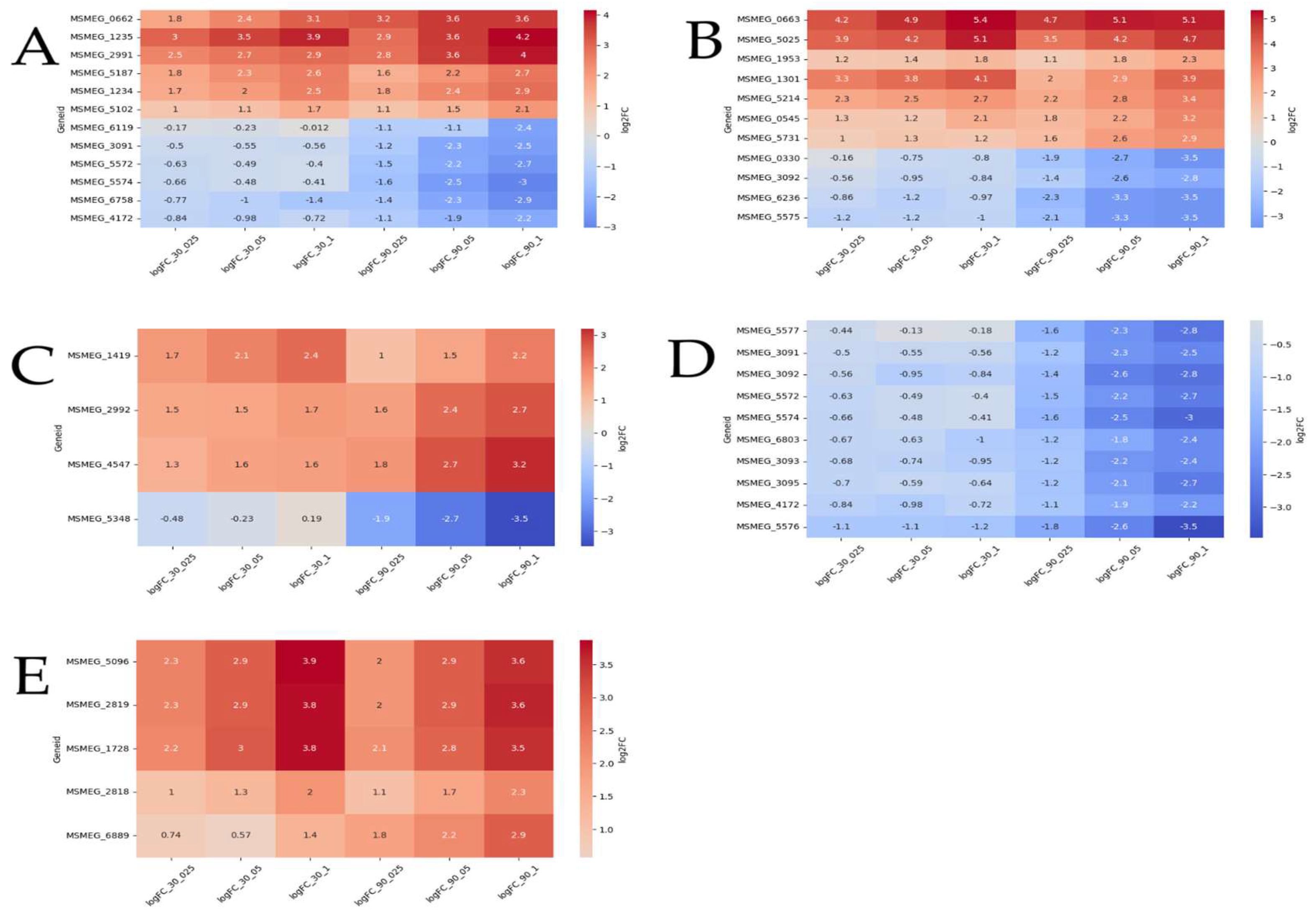
Differential expression analysis identified 55 transport-related genes, with 23 upregulated and 32 downregulated. Among these, 12 exhibited dose-dependent expression patterns (Figure 5A). Specifically, genes MSMEG_2991 and MSMEG_5187 (encoding multidrug efflux MFS transporters), MSMEG_5102, MSMEG_1234, MSMEG_0662 (encoding ABC transporters), and MSMEG_1235 (encoding a SulP transporter) were upregulated in a dose-dependent manner at both timepoints. Additionally, MSMEG_3091, MSMEG_4172, MSMEG_5572, MSMEG_5574 (encoding carbohydrate ABC transporters), MSMEG_6758 (encoding an aquaporin), and MSMEG_6119 (encoding a sulfite exporter) showed dose-dependent upregulation exclusively at the 90-minute timepoint.
Among the 86 differentially expressed genes (DEGs) associated with transcriptional regulators, 12 displayed dose-dependent expression (Figure 5B). Genes such as MSMEG_0663, MSMEG_5025 (TetR family), MSMEG_1953 (WhiB7), MSMEG_1301 (LuxR family), and MSMEG_5214 (sigma-70) were upregulated at both timepoints. MSMEG_0545 (LuxR family) and MSMEG_5731 (GntR family) were upregulated only at 90 minutes. Notably, MSMEG_0663 (TetR family) was the most significantly upregulated and dose-dependent gene at both timepoints. Conversely, four DEGs were downregulated at 90 minutes: MSMEG_0330 (LuxR family), MSMEG_3092 (sugar-binding transcriptional regulator), MSMEG_6236 (MnoR family), and MSMEG_5575 (MarR family).
A total of 18 DEGs were associated with acyl-CoA dehydrogenase/oxidase, 12 with enoyl-CoA hydratase/isomerase (Crotonase), one with phytanoyl-CoA dioxygenase, 11 with AMP-dependent synthetase/ligase, and several with CoA-transferase families I and III, all of which are linked to fatty acid degradation and transport. Among these, three DEGs—MSMEG_2992, MSMEG_4547 (acyl-CoA dehydrogenases), and MSMEG_1419 (phytanoyl-CoA dioxygenase)—were upregulated in a dose-dependent manner at 90 minutes, while MSMEG_5348 (long-chain-fatty-acid-CoA ligase) was downregulated at the same timepoint (Figure 5C).
Of the 56 DEGs associated with carbohydrate metabolism and transport, nine were dose-dependently downregulated at 90 minutes (Figure 5D). These included MSMEG_5572, MSMEG_5574, MSMEG_3091, MSMEG_4172, and MSMEG_6803 (encoding sugar ABC transporters), MSMEG_5577 (encoding fructokinase), MSMEG_3092 (encoding a sugar-binding transcriptional regulator), and MSMEG_3093 and MSMEG_3095 (encoding FGGY carbohydrate kinases). Notably, MSMEG_5576 (encoding D-mannonate oxidoreductase) was the most significantly downregulated gene at both timepoints.
Among the 13 DEGs encoding transposases, four—MSMEG_1728, MSMEG_2819, MSMEG_5096, and MSMEG_2818—were upregulated in a dose-dependent manner at both timepoints, while MSMEG_6889 was upregulated only at 90 minutes.
This comprehensive analysis highlights the dynamic and dose-dependent regulation of genes involved in transport, transcriptional regulation, fatty acid metabolism, carbohydrate metabolism, and transposase activity, providing insights into the molecular responses to the treatment.
4. Discussion
Currently, there is a pressing need to identify new drug targets and elucidate novel mechanisms of action for combating Mycobacterium tuberculosis. Quinoxaline 1,4-di-N-oxides (QdNOs), a class of heterocyclic compounds with N–O groups at the 1- and 4-positions, are known for their reducing potential, which generates free radical intermediates and contributes to their antibacterial activity. QdNOs have demonstrated potent inhibitory activity in vitro against various pathogens, including mycobacteria [3].
In this study, we employed a multi-dose approach with varying levels of inhibition at two timepoints to investigate the early defensive responses of M. smegmatis to the QdNO derivative LCTA-3368 at the transcriptomic level. We hypothesized that genes providing protective effects to the bacterial cell would be upregulated, while those contributing to the mechanism of action of LCTA-3368 would be downregulated.
The antibacterial mechanism of QdNOs is often linked to the generation of free radicals during bioreduction, leading to DNA damage [2, 27, 32, 33]. Both M. smegmatis and M. tuberculosis possess multiple DNA repair pathways, including homologous recombination (HR), nonhomologous end joining (NHEJ), and single-strand annealing (SSA) [34]. In our study, we observed the upregulation of 42 differentially expressed genes (DEGs) involved in DNA repair, with five showing strict dose-dependence.
A hallmark of the bacterial response to antibiotic-induced stress is the activation of the SOS repair system, characterized by the increased expression of error-prone DNA polymerases. In our experiment, DNA polymerase III (MSMEG_1633) was highly upregulated, along with an error-prone DNA polymerase from the Y family (MSMEG_1622), which is known to promote adaptive mutagenesis under stress conditions [35].
Mycobacteria also utilize the nucleotide excision repair (NER) pathway to address DNA damage. This pathway involves the recognition and excision of damaged DNA regions by a complex of UvrA, UvrB, and UvrC proteins, followed by repair synthesis [36, 37]. Additionally, crosslink repair DNA glycosylase (ycaQ) and apurinic/apyrimidinic endonuclease IV play roles in protecting mycobacterial DNA from oxidative stress [38, 39].
Mobile genetic elements (MGEs) further contribute to mutagenesis, with transposases being key drivers of MGE activity. Among the 13 DEGs encoding transposases, five were upregulated in a dose-dependent manner. The increased activity of transposases under drug pressure may accelerate the emergence of drug resistance [40, 41].
The broad upregulation of DNA repair systems in our study supports the proposed mechanism of QdNOs involving DNA damage [2, 32]. Conversely, the dose-dependent upregulation of mutagenic systems, including error-prone polymerases, the NER pathway, and transposases, aligns with the numerous mutations observed in LCTA-3368-resistant mutants [8].
Oxidoreductases are critical for the bioreductive activation of QdNOs [42]. We identified three oxidoreductase genes (MSMEG_6763, MSMEG_5576, and MSMEG_3094) that were downregulated in a dose-dependent manner, suggesting their potential role in LCTA-3368 activation. Conversely, eight oxidoreductase genes were upregulated, with three (MSMEG_1623, MSMEG_4565, and MSMEG_5027) showing the highest levels of differential expression. These oxidoreductases may primarily function to neutralize free radicals generated by LCTA-3368, thereby protecting bacterial cells [43-46].
In previous work, we identified LCTA-3368-resistant strains with multiple mutations, including those in pyruvate synthase (MSMEG_4646), ferredoxin (MSMEG_5122), and the transcriptional repressor MSMEG_1380, which leads to the overexpression of the MmpS5-MmpL5 efflux system [47]. While we did not observe significant changes in the expression of the MmpS5-MmpL5 system, other multidrug resistance (MDR) transporter genes (MSMEG_3815, MSMEG_5187, MSMEG_2991) were overexpressed, indicating their potential role in LCTA-3368 efflux.
Finally, the downregulation of genes involved in lipid and carbohydrate transport, as well as amino acid biosynthesis, suggests a general attenuation of metabolic processes after 90 minutes of LCTA-3368 treatment, ultimately leading to cell death.
While M. smegmatis serves as a useful model, its metabolic differences from M. tuberculosis must be acknowledged. However, most of the dose-dependent DEGs identified in this study have homologs in the M. tuberculosis genome (Table S3), suggesting that our findings may be applicable to the pathogen. Further studies on M. tuberculosis, including experiments with customizable media such as variations of Sauton medium, could provide additional insights into the influence of carbon sources on carbohydrate metabolism and drug efficacy.
5. Conclusions
The QdNO compound LCTA-3368 induces oxidative and/or nitrosative stress conditions, triggering a broad transcriptomic response in mycobacterial cells. The primary mechanisms for damage control in these cells involve DNA repair systems, notably the SOS response and nucleotide excision repair (NER). However, these repair processes can introduce mutations, potentially leading to drug resistance. Additionally, genes encoding proteins with oxidoreductase activity exhibit dose-dependent differential expression. This allows for the classification of oxidoreductases into two groups: those involved in activating LCTA-3368 (downregulated) and those potentially capable of deactivating its active metabolites. Our findings pave the way for the targeted synthesis of more specific quinoxaline 1,4-dioxide (QdNO) derivatives.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
O.B.B. and O.O.G. contributed equally to this work. Conceptualization, O.B.B., A.A.V., D.A.M.; methodology, O.B.B., O.O.G.; investigation, O.B.B, O.O.G., S.G.F., S.V.S., N.I.K., K.M.K., V.A.V., D.A.B.; resources, A.A.V., D.A.M., V.N.D.; data curation, R.A.I., E.A.S; writing—original draft preparation, O.O.G., O.B.B.; writing—review and editing, O.B.B., D.A.M., D.A.R., E.A.S.; visualization, O.O.G.; supervision, O.B.B., D.A.M., V.N.D.; project administration, A.A.V., D.A.M.; funding acquisition, D.A.M., A.A.V. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the RUDN University Scientific Projects Grant System, project № 202760-2-000 and State assignment from the Ministry of Science of the Russian Federation №0092-2022-0003.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Prof. Andrey Shchekotikhin of the Gause Institute of New Antibiotics, for kindly providing LCTA-3368 for the research. Sequenсing was performed using the core facilities of the Lopukhin FRCC PCM “Genomics, proteomics, metabolomics” (http://rcpcm.org/?p=2806). We acknowledge for Government basic research program No. 0088-2024-0009 for supporting Rustem Ilyasov's scientific work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Global Tuberculosis Report 2023. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2023.
- Cheng, G.; Sa, W.; Cao, C.; Guo, L.; Hao, H.; Liu, Z.; Wang, X.; Yuan, Z. Quinoxaline 1,4-Di-N-Oxides: Biological Activities and Mechanisms of Actions. Front Pharmacol 2016, 7, 64. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, S.; Sechi, L.A.; Molicotti, P.; Cannas, S.; Bua, A.; Deriu, A.; Carta, A.; Paglietti, G. In Vitro Activity of New Quinoxalin 1,4-Dioxide Derivatives against Strains of Mycobacterium Tuberculosis and Other Mycobacteria. Int J Antimicrob Agents 2005, 25, 179–181. [Google Scholar] [CrossRef]
- Ortega, M.A.; Montoya, M.E.; Jaso, A.; Zarranz, B.; Tirapu, I.; Aldana, I.; Monge, A. Antimycobacterial Activity of New Quinoxaline-2-Carbonitrile and Quinoxaline-2-Carbonitrile 1,4-Di-N-Oxide Derivatives. Pharmazie 2001, 56, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Villar, R.; Vicente, E.; Solano, B.; Pérez-Silanes, S.; Aldana, I.; Maddry, J.A.; Lenaerts, A.J.; Franzblau, S.G.; Cho, S.-H.; Monge, A.; et al. In Vitro and in Vivo Antimycobacterial Activities of Ketone and Amide Derivatives of Quinoxaline 1,4-Di-N-Oxide. J Antimicrob Chemother 2008, 62, 547–554. [Google Scholar] [CrossRef]
- Crawford, P.W.; Scamehorn, R.G.; Hollstein, U.; Ryan, M.D.; Kovacic, P. Cyclic Voltammetry of Phenazines and Quinoxalines Including Mono- and Di-N-Oxides. Relation to Structure and Antimicrobial Activity. Chem Biol Interact 1986, 60, 67–84. [Google Scholar] [CrossRef]
- Chowdhury, G.; Kotandeniya, D.; Daniels, J.S.; Barnes, C.L.; Gates, K.S. Enzyme-Activated, Hypoxia-Selective DNA Damage by 3-Amino-2-Quinoxalinecarbonitrile 1,4-Di-N-Oxide. Chem Res Toxicol 2004, 17, 1399–1405. [Google Scholar] [CrossRef]
- Frolova, S.G.; Vatlin, A.A.; Maslov, D.A.; Yusuf, B.; Buravchenko, G.I.; Bekker, O.B.; Klimina, K.M.; Smirnova, S.V.; Shnakhova, L.M.; Malyants, I.K.; et al. Novel Derivatives of Quinoxaline-2-Carboxylic Acid 1,4-Dioxides as Antimycobacterial Agents: Mechanistic Studies and Therapeutic Potential. Pharmaceuticals (Basel) 2023, 16, 1565. [Google Scholar] [CrossRef]
- Cooper, C.B. Development of Mycobacterium Tuberculosis Whole Cell Screening Hits as Potential Antituberculosis Agents. J Med Chem 2013, 56, 7755–7760. [Google Scholar] [CrossRef]
- King, G.M. Uptake of Carbon Monoxide and Hydrogen at Environmentally Relevant Concentrations by Mycobacteria†. Appl Environ Microbiol 2003, 69, 7266–7272. [Google Scholar] [CrossRef] [PubMed]
- Junnotula, V.; Sarkar, U.; Sinha, S.; Gates, K.S. Initiation of DNA Strand Cleavage by 1,2,4-Benzotriazine 1,4-Dioxide Antitumor Agents: Mechanistic Insight from Studies of 3-Methyl-1,2,4-Benzotriazine 1,4-Dioxide. J Am Chem Soc 2009, 131, 1015–1024. [Google Scholar] [CrossRef]
- Briffotaux, J.; Liu, S.; Gicquel, B. Genome-Wide Transcriptional Responses of Mycobacterium to Antibiotics. Front Microbiol. 2019, 10, 249. [Google Scholar] [CrossRef] [PubMed]
- Vatlin, A.A.; Shitikov, E.A.; Shahbaaz, M.; Bespiatykh, D.A.; Klimina, K.M.; Christoffels, A.; Danilenko, V.N.; Maslov, D.A. Transcriptomic Profile of Mycobacterium Smegmatis in Response to an Imidazo[1,2-b][1,2,4,5]Tetrazine Reveals Its Possible Impact on Iron Metabolism. Front Microbiol 2021, 12, 724042. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, M.M.; Aldridge, B.B. Stable Regulation of Cell Cycle Events in Mycobacteria: Insights From Inherently Heterogeneous Bacterial Populations. Front Microbiol 2018, 9, 514. [Google Scholar] [CrossRef]
- Babraham Bioinformatics - FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat Biotechnol 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/index.html.
- Python 3.13 Documentation — DevDocs Available online: https://devdocs.io/python/.
- Matplotlib Documentation — Matplotlib 3.9.2 Documentation Available online: https://matplotlib.org/stable/index.html.
- NumPy Documentation Available online: https://numpy.org/doc/.
- GOpiscator 0.1.5 Available online: https://pypi.org/project/gopiscator/.
- Xu, F.; Cheng, G.; Hao, H.; Wang, Y.; Wang, X.; Chen, D.; Peng, D.; Liu, Z.; Yuan, Z.; Dai, M. Mechanisms of Antibacterial Action of Quinoxaline 1,4-di-N-oxides against Clostridium perfringens and Brachyspira hyodysenteriae. Front Microbiol. 2016, 7–1948. [Google Scholar] [CrossRef]
- Suter, W.; Rosselet, A.; Knüsel, F. Mode of Action of Quindoxin and Substituted Quinoxaline-Di-N-Oxides on Escherichia Coli. Antimicrob Agents Chemother 1978, 13, 770–783. [Google Scholar] [CrossRef]
- DuPlessis, E.R.; Pellett, J.; Stankovich, M.T.; Thorpe, C. Oxidase Activity of the Acyl-CoA Dehydrogenases. Biochemistry 1998, 37, 10469–10477. [Google Scholar] [CrossRef] [PubMed]
- Padavattan, S.; Jos, S.; Gogoi, H.; Bagautdinov, B. Crystal Structure of Enoyl-CoA Hydratase from Thermus Thermophilus HB8. Acta Crystallogr F Struct Biol Commun 2021, 77, 148–155. [Google Scholar] [CrossRef]
- Savolainen, K.; Bhaumik, P.; Schmitz, W.; Kotti, T.J.; Conzelmann, E.; Wierenga, R.K.; Hiltunen, J.K. Alpha-Methylacyl-CoA Racemase from Mycobacterium Tuberculosis. Mutational and Structural Characterization of the Active Site and the Fold. J Biol Chem 2005, 280, 12611–12620. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, H.; Huang, L.; Pan, Y.; Li, J.; Chen, D. Deoxidation rates play a critical role in DNA damage mediated by important synthetic drugs, quinoxaline 1,4-dioxides. Chemical Research in Toxicology 2015, 28, 470–481. [Google Scholar] [CrossRef]
- An, H.; Li, Y.; Li, Y.; Gong, S.; Zhu, Y. N.; Li, X.; Wu, Y. Advances in Metabolism and Metabolic Toxicology of Quinoxaline 1,4-Di-N-oxides. Chemical Research in Toxicology 2024, 37, 528–539. [Google Scholar] [CrossRef]
- Gupta, R.; Barkan, D.; Redelman-Sidi, G.; Shuman, S.; Glickman, M.S. Mycobacteria Exploit Three Genetically Distinct DNA Double-Strand Break Repair Pathways. Mol Microbiol 2011, 79, 316–330. [Google Scholar] [CrossRef]
- Sharma, A.; Nair, D.T. Cloning, Expression, Purification, Crystallization and Preliminary Crystallographic Analysis of MsDpo4: A Y-Family DNA Polymerase from Mycobacterium Smegmatis. Acta Crystallogr Sect F Struct Biol Cryst Commun 2011, 67, 812–816. [Google Scholar] [CrossRef]
- Dos Vultos, T.; Mestre, O.; Tonjum, T.; Gicquel, B. DNA Repair in Mycobacterium Tuberculosis Revisited. FEMS Microbiol Rev 2009, 33, 471–487. [Google Scholar] [CrossRef]
- Miggiano, R.; Morrone, C.; Rossi, F.; Rizzi, M. Targeting Genome Integrity in Mycobacterium Tuberculosis: From Nucleotide Synthesis to DNA Replication and Repair. Molecules 2020, 25, 1205. [Google Scholar] [CrossRef]
- Chen, X.; Bradley, N.P.; Lu, W.; Wahl, K.L.; Zhang, M.; Yuan, H.; Hou, X.-F.; Eichman, B.F.; Tang, G.-L. Base Excision Repair System Targeting DNA Adducts of Trioxacarcin/LL-D49194 Antibiotics for Self-Resistance. Nucleic Acids Res 2022, 50, 2417–2430. [Google Scholar] [CrossRef] [PubMed]
- Khanam, T.; Rai, N.; Ramachandran, R. Mycobacterium Tuberculosis Class II Apurinic/Apyrimidinic-Endonuclease/3’-5’ Exonuclease III Exhibits DNA Regulated Modes of Interaction with the Sliding DNA β-Clamp. Mol Microbiol 2015, 98, 46–68. [Google Scholar] [CrossRef] [PubMed]
- Nätt, D.; Thorsell, A. Stress-induced transposon reactivation: a mediator or an estimator of allostatic load? Environ Epigenet 2016, 2. [Google Scholar] [CrossRef]
- Frolova, S.G.; Klimina, K.M.; Kumar, R.; Vatlin, A.A.; Salunke, D.B.; Kendrekar, P.; Danilenko, V.N.; Maslov, D.A. Identification of Mutations Conferring Tryptanthrin Resistance to Mycobacterium Smegmatis. Antibiotics 2021, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Ganley, B.; Chowdhury, G.; Bhansali, J.; Daniels, J. S.; Gates, K. S. Redox-activated, hypoxia-selective DNA cleavage by quinoxaline 1,4-di-N-oxide. Bioorg. Med. Chem 2001, 9, 2395–2401. [Google Scholar] [CrossRef]
- Ashwani, Kumar; Aisha, Farhana; Loni, Guidry; Vikram, Saini; Mary, Hondalus; Adrie, JC. Steyn Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med. 2011, 13. [Google Scholar] [CrossRef]
- Ganley, B.; Chowdhury, G.; Bhansali, J.; Daniels, J. S.; Gates, K. S. . Redox-activated, hypoxia-selective DNA cleavage by quinoxaline 1,4-di-N-oxide. Bioorg. Med. Chem 2001, 9, 2395–2401. [Google Scholar] [CrossRef] [PubMed]
- Gurumurthy, M.; Rao, M.; Mukherjee, T.; Rao, et al. A novel F420-dependent anti-oxidant mechanism protects Mycobacterium tuberculosis against oxidative stress and bactericidal agents. Mol. Microbiol 2013, 87, 744–755. [Google Scholar] [CrossRef]
- Greening, C.; Jirapanjawat, T.; Afroze, S.; Ney, B.; Scott, C.; Pandey, G.; Lee, BM.; Russell, RJ.; Jackson, CJ.; Oakeshott, JG.; Taylor, MC.; Warden, AC. ; Mycobacterial F420H2-Dependent Reductases Promiscuously Reduce Diverse Compounds through a Common Mechanism. Front. Microbiol. 2017, 8, 1000. [Google Scholar] [CrossRef]
- Vatlin, A.A.; Frolova, S.G.; Bekker, O.B.; Danilenko, V.N. Studying the mechanism of action of new derivatives of quinoxalin-1,4-dioxide on the model organism Mycobacterium smegmatis. RUDN Journal of Ecology and Life Safety 2024, 32, 41–50. [Google Scholar] [CrossRef]
Figure 2.
GO analysis of DEGs corresponding to different time points and concentrations of LCTA-3368. The Y-axis represents KEGG pathways, while the X-axis represents fold enrichment (calculated as the ratio of input pathway DEGs to the background gene set). The color of the dots indicates the p-values of enrichment (p < 0.005), the size of the dots corresponds to the number of genes in the pathway, and the shape of the dots represents the treatment time.
Figure 2.
GO analysis of DEGs corresponding to different time points and concentrations of LCTA-3368. The Y-axis represents KEGG pathways, while the X-axis represents fold enrichment (calculated as the ratio of input pathway DEGs to the background gene set). The color of the dots indicates the p-values of enrichment (p < 0.005), the size of the dots corresponds to the number of genes in the pathway, and the shape of the dots represents the treatment time.
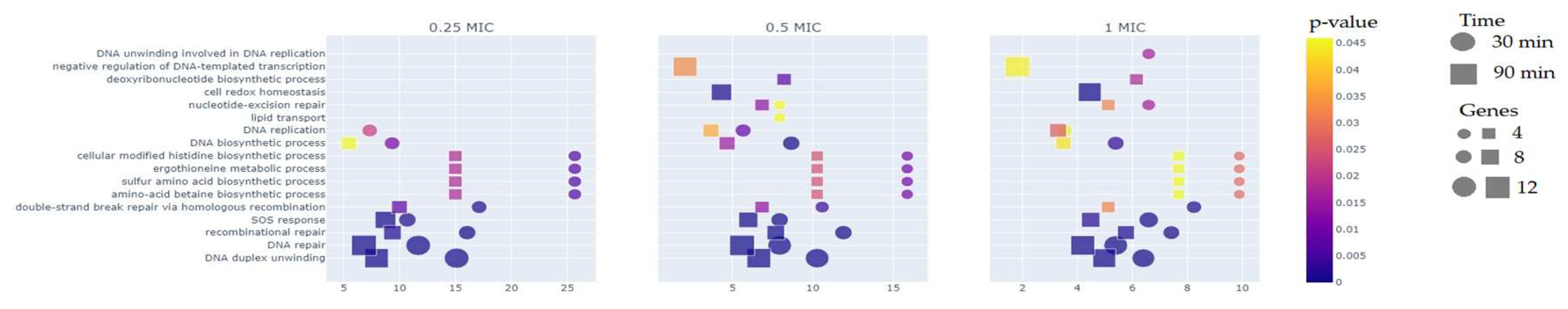
Figure 3.
Differentially expressed genes (DEGs) associated with DNA repair systems exhibiting dose-dependent regulation.
Figure 3.
Differentially expressed genes (DEGs) associated with DNA repair systems exhibiting dose-dependent regulation.
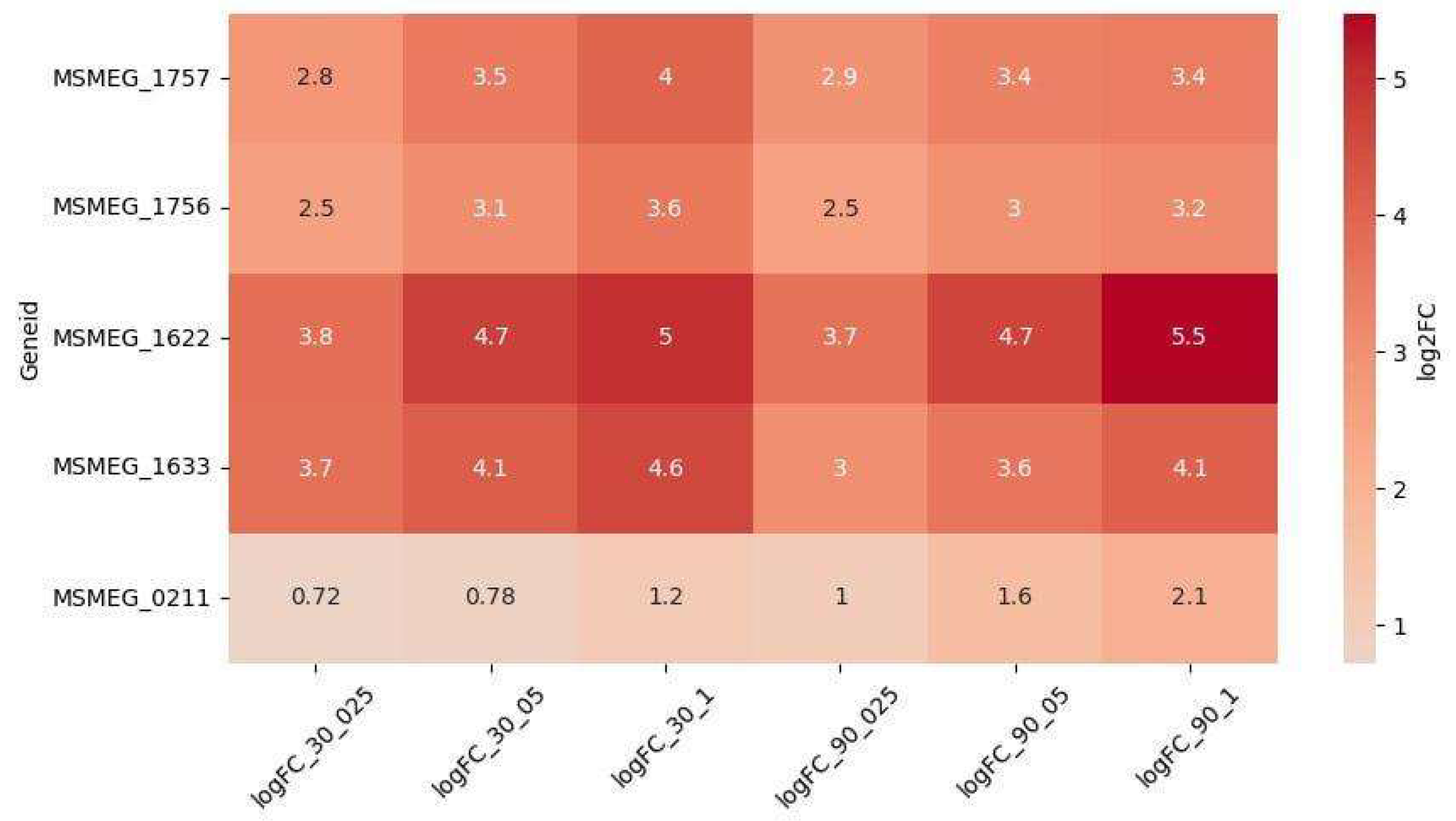
Figure 4.
Dose-dependent expression patterns of oxidoreductase-related differentially expressed genes (DEGs).
Figure 4.
Dose-dependent expression patterns of oxidoreductase-related differentially expressed genes (DEGs).
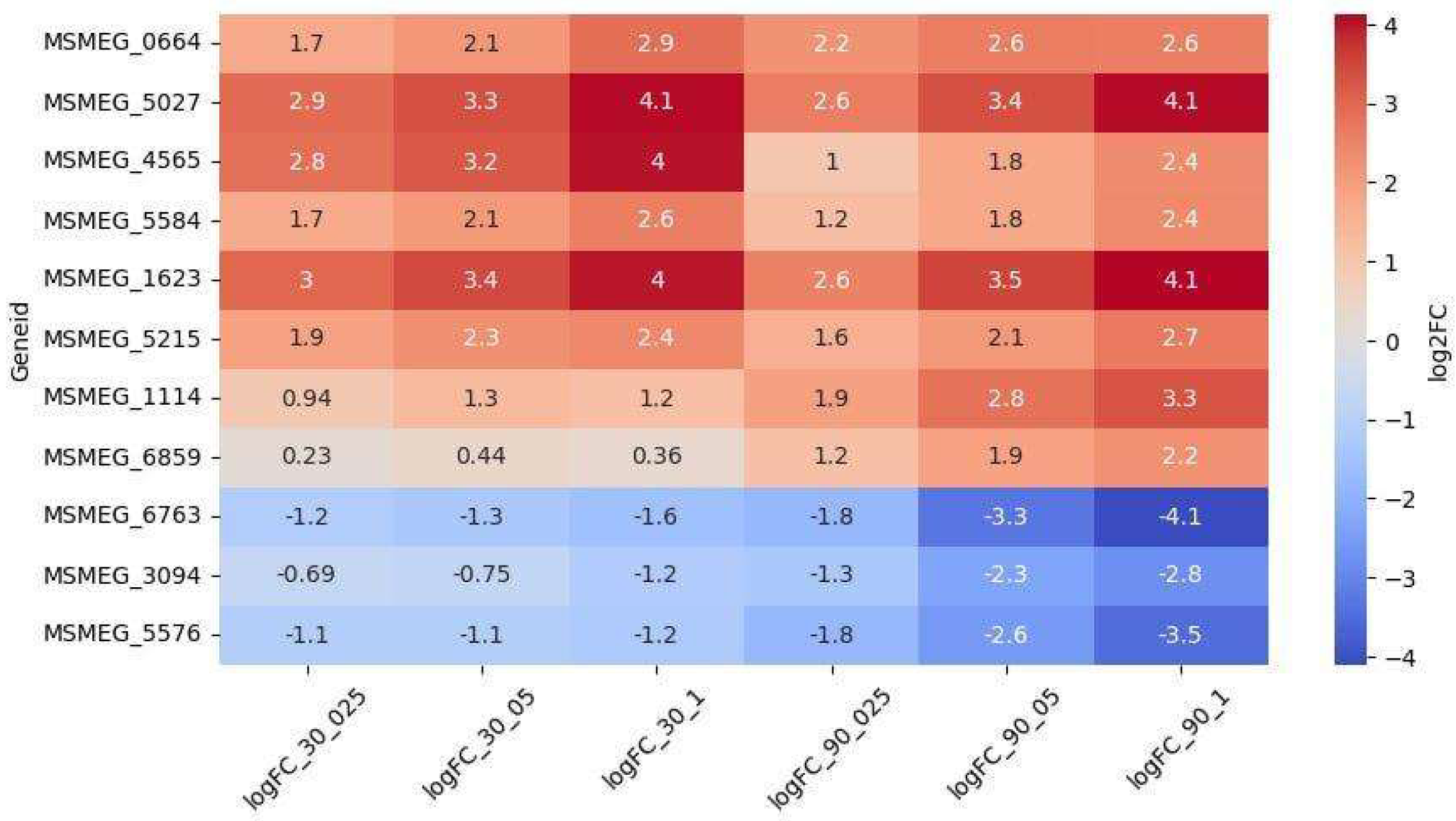
Figure 5.
Heatmap illustrating dose-dependent differentially expressed genes (DEGs). Red indicates upregulated DEGs, while blue represents downregulated DEGs. The graphs depict DEGs associated with: (A) transport activity, (B) transcriptional regulator activity, (C) fatty acid metabolic activity, (D) carbohydrate metabolism and transport, and (E) transposase activity.
Figure 5.
Heatmap illustrating dose-dependent differentially expressed genes (DEGs). Red indicates upregulated DEGs, while blue represents downregulated DEGs. The graphs depict DEGs associated with: (A) transport activity, (B) transcriptional regulator activity, (C) fatty acid metabolic activity, (D) carbohydrate metabolism and transport, and (E) transposase activity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



