Submitted:
30 January 2025
Posted:
31 January 2025
You are already at the latest version
Abstract
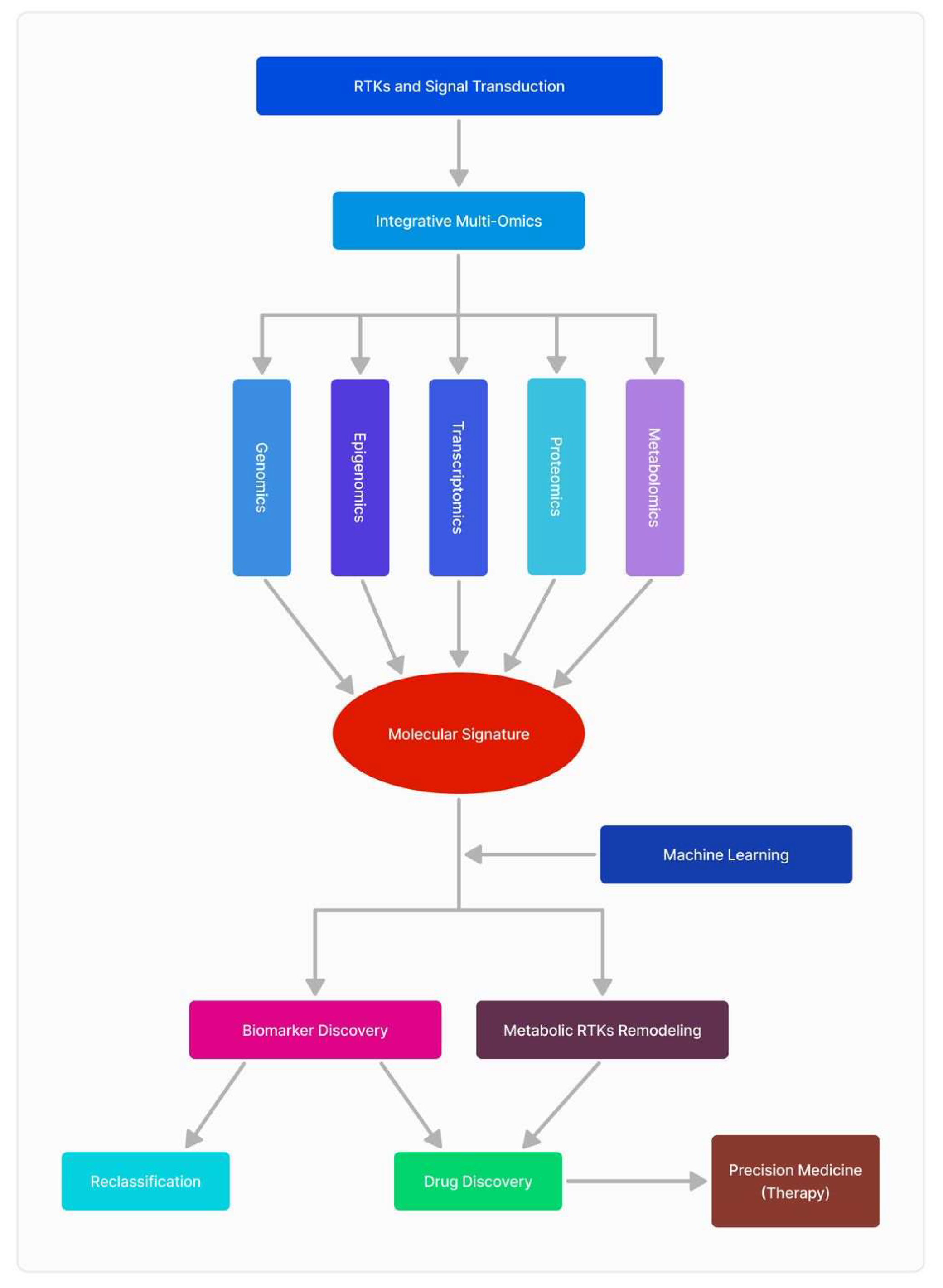
Glioblastoma (GBM) is an aggressive brain tumor characterized by its molecular complexity and resistance to conventional treatments, including surgery, radiation, and chemotherapy. Despite these challenges, advancements in receptor tyrosine kinase (RTK) research, combined with multi-omics approaches, hold promise for improving patient outcomes and survivability. RTKs, are central to GBM progression, influencing cell proliferation, survival, and angiogenesis. However, the complexity of RTK signaling necessitates a broader, integrative perspective, which has been enabled by the emergence of -omics sciences. Multi-omics technologies—including genomics, transcriptomics, proteomics, and metabolomics—offer unprecedented insights into the molecular landscape of GBM and its RTK-driven pathways. Genomic studies reveal mutations and amplifications in RTK-related genes, while transcriptomics uncovers alterations in gene expression patterns, providing a clearer picture of how these aberrations drive tumor behavior. Proteomics further delineates changes in protein expression and post-translational modifications linked to RTK signaling, highlighting novel therapeutic vulnerabilities. Metabolomics complements these findings by identifying RTK-associated metabolic reprogramming, such as shifts in glycolysis and lipid metabolism, which sustain tumor growth and therapy resistance. The integration of these multi-omics layers enables a comprehensive understanding of RTK biology in GBM. For example, studies have linked metabolic alterations with RTK activity, offering new biomarkers for tumor classification and therapeutic targeting. Additionally, single-cell transcriptomics has unveiled intratumoral heterogeneity, a critical factor in therapy resistance. This article highlights the transformative potential of multi-omics in unraveling the complexity of RTK signaling in GBM. By combining these approaches, researchers are paving the way for precision medicine strategies that may significantly enhance diagnostic accuracy and treatment efficacy, providing new hope for patients facing this devastating disease.
Keywords:
1. Introduction
2. RTK Signaling Pathways in GBM
2.1. Epithelial Growth Factor Receptor (EGFR)
2.2. Platelet Derived Growth Factor Receptor (PDGFR)
2.3. Vascular Endothelial Growth Factor Receptor (VEGFR)
2.4. c-MET and Hepatocyte Growth Factor (HGF) Pathway
2.5. AXL Receptor
2.6. RTK’s Downstream Signaling Pathways
2.6.1. RAS/MAPK/ERK Pathway
2.6.2. JAK/STAT Pathway
2.6.3. PI3K/AKT Pathway
2.6.4. PLC/PKC Pathway

| RTK | Genomics | Transcriptomics | Proteomics | Metabolomics |
| EGFR | EGFR amplifications and EGFRvIII mutations drive tumor aggressiveness. PIK3CA mutations cause disruption in the PI3K pathway, contributing to recurrence. | EGFR activation induces significant transcriptomic changes that promote tumor proliferation and resistance mechanisms. Increased expression of PTK2 enhances cell survival. | EGFR overexpression and PTEN downregulation promote tumor growth and resistance. Phosphorylation (Y1068, Y1173) and PI3K/AKT signaling enhance cell survival and migration. | Activation of EGFR leads to reprogramming of lipid metabolism and glycolysis, enhancing energy production and tumor survival. Studies show elevated glycerophospholipids (PC ae C42:4). |
| VEGFR | VEGFR alterations and the VEGF-HIF1α axis drive tumor angiogenesis. Gene amplifications and mutations contribute to GBM growth and progression. | VEGFR expression is significantly upregulated in hypoxic regions, promoting angiogenesis and tumor survival through enhanced RTK signaling. | VEGFR phosphorylation at key sites (Y951, Y1175) activates angiogenesis and cell survival pathways. Interactions with neuropilin enhance signaling. | VEGFR signaling promotes glycolysis, fatty acid oxidation, and mitochondrial biogenesis, supporting tumor survival under low-oxygen conditions. |
| PDGFR | PDGFR amplifications and mutations in the proneural subtype drive tumor progression by altering extracellular matrix (ECM) remodeling and promoting invasion. | PDGFR is enriched in the proneural subtype of GBM, affecting migration, adhesion, and immune evasion. Altered transcriptional networks support these processes. | PDGFR phosphorylation (Y751, Y1021) regulates cell migration and immune checkpoint interactions. Modifications in ECM support tumor progression. | Metabolic coupling between tumor and stromal cells promotes lactate production and aerobic glycolysis, supporting tumor invasiveness. |
| MET | MET amplifications, exon 14 skipping, and gene fusions (e.g., TPR-MET, PTPRZ1-MET) lead to persistent kinase activity and poor prognosis in GBM. | MET upregulation in invasive subpopulations enhances tumor migration and invasiveness, supported by transcriptomic alterations in invasive genes. | MET phosphorylation (Y1234, Y1235) promotes invasive signaling and MAPK pathway activation. MET fusions result in persistent oncogenic signaling. | NADPH production and redox homeostasis are key to maintaining oxidative stress tolerance and cell survival, aiding invasive tumor growth. |
| AXL | AXL overexpression is associated with epithelial-mesenchymal transition (EMT), enhancing immune evasion and metastasis in GBM. | AXL transcriptional upregulation by HIF2α and TWIST1 promotes EMT, immune evasion, and tumor progression. | AXL signaling bypasses PI3K/AKT and NF-κB pathways to promote cell survival and metastasis in GBM. | Fatty acid uptake/storage increases under nutrient-limited conditions, supporting cell survival and growth under metabolic stress. |
| HER2 | HER2 overexpression is linked to therapy resistance and aggressive GBM phenotypes, contributing to tumor progression and poor prognosis. | HER2 upregulation in therapy-resistant GBM phenotypes is correlated with transcriptional changes promoting tumor growth and resistance. | HER2 signaling modulates protein pathways that affect apoptosis resistance, involving ubiquitin-proteasome system dysregulation and PTK2 phosphorylation. | Glutamine dependency is a key feature of HER2-overexpressing GBM, aiding proliferation and survival in resistant phenotypes. |
3. Recent Advances in RTKs-Omics Approaches and Their Impact on Diagnosis and Therapeutic Targets in GBM
3.1. Genomics
3.2. Transcriptomics
3.3. Proteomics
3.4. Metabolomics
3.5. The Inter-Play of Multi-Omic Sciences and Clinical Data
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grochans, S. et al., Epidemiology of glioblastoma multiforme–literature review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Vigneswaran, K. , S. Neill, and C.G. Hadjipanayis, Beyond the World Health Organization grading of infiltrating gliomas: advances in the molecular genetics of glioma classification. Annals of translational medicine 2015, 3. [Google Scholar]
- Sevastre, A.-S. , et al., Glioblastoma pharmacotherapy: A multifaceted perspective of conventional and emerging treatments. Experimental and Therapeutic Medicine 2021, 22, 1–18. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B. , et al., Glioblastoma stem cells—useful tools in the battle against cancer. International Journal of Molecular Sciences 2022, 23, 4602. [Google Scholar] [CrossRef]
- Koukourakis, G.V. , et al., Temozolomide with radiation therapy in high grade brain gliomas: pharmaceuticals considerations and efficacy; a review article. Molecules 2009, 14, 1561–1577. [Google Scholar] [CrossRef]
- Daianu, O. , et al., Temozolomide and targeted therapy against epidermal growth factor receptor in glioma. 2016, 9, 15249–15261. [Google Scholar]
- Kubben, P.L. et al., Intraoperative MRI-guided resection of glioblastoma multiforme: a systematic review. The lancet oncology 2011, 12, 1062–1070. [Google Scholar] [CrossRef]
- Alexandru, O. , et al., Platelet-derived growth factor receptor and ionizing radiation in high grade glioma cell lines. International Journal of Molecular Sciences 2019, 20, 4663. [Google Scholar] [CrossRef]
- Alexandru, O. , et al., The influence of EGFR inactivation on the radiation response in high grade glioma. International Journal of Molecular Sciences 2018, 19, 229. [Google Scholar] [CrossRef]
- Fisher, J.P. and D.C. Adamson, Current FDA-approved therapies for high-grade malignant gliomas. Biomedicines 2021, 9, 324.
- Artene, S.-A. , et al., Dendritic cell immunotherapy versus bevacizumab plus irinotecan in recurrent malignant glioma patients: a survival gain analysis. OncoTargets and therapy 2016, 6669–6677. [Google Scholar] [CrossRef]
- Brada, M. , et al., Temozolomide versus procarbazine, lomustine, and vincristine in recurrent high-grade glioma. Journal of clinical oncology 2010, 28, 4601–4608. [Google Scholar] [CrossRef]
- Agosti, E. , et al., Glioblastoma immunotherapy: A systematic review of the present strategies and prospects for advancements. International Journal of Molecular Sciences 2023, 24, 15037. [Google Scholar] [CrossRef]
- Zeng, J. , et al., Oncolytic viro-immunotherapy: an emerging option in the treatment of gliomas. Frontiers in immunology 2021, 12, 721830. [Google Scholar] [CrossRef]
- Tilak, M. , et al., Receptor tyrosine kinase signaling and targeting in glioblastoma multiforme. International Journal of Molecular Sciences 2021, 22, 1831. [Google Scholar] [CrossRef]
- Carapancea, M. , et al., Growth factor receptors signaling in glioblastoma cells: therapeutic implications. Journal of neuro-oncology 2009, 92, 137–147. [Google Scholar] [CrossRef]
- Rodriguez, S.M.B. , et al., An overview of EGFR mechanisms and their implications in targeted therapies for glioblastoma. International Journal of Molecular Sciences 2023, 24, 11110. [Google Scholar] [CrossRef]
- Serban, F. , et al., Epidermal growth factor, latrophilin, and seven transmembrane domain-containing protein 1 marker, a novel angiogenesis marker. OncoTargets and therapy 2015, 3767–3774. [Google Scholar]
- Sevastre, A.-S. , et al., ELTD1—An emerging silent actor in cancer drama play. International Journal of Molecular Sciences 2021, 22, 5151. [Google Scholar] [CrossRef]
- Onciul, R. , et al., Deciphering Glioblastoma: Fundamental and Novel Insights into the Biology and Therapeutic Strategies of Gliomas. Current Issues in Molecular Biology 2024, 46, 2402–2443. [Google Scholar] [CrossRef]
- Deleanu, R. L.C. Ceafalan, and A. Dricu, Transcriptomic crosstalk between gliomas and telencephalic neural stem and progenitor cells for defining heterogeneity and targeted signaling pathways. International Journal of Molecular Sciences 2021, 22, 13211. [Google Scholar] [CrossRef]
- Carrasco-García, E. M. Saceda, and I. Martínez-Lacaci, Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells 2014, 3, 199–235. [Google Scholar] [CrossRef]
- Nazarenko, I. et al., PDGF and PDGF receptors in glioma. Upsala journal of medical sciences 2012, 117, 99–112. [Google Scholar] [CrossRef]
- Cenciarelli, C. , et al., PDGFRα depletion attenuates glioblastoma stem cells features by modulation of STAT3, RB1 and multiple oncogenic signals. Oncotarget 2016, 7, 53047. [Google Scholar] [CrossRef]
- Heldin, C.-H. , Targeting the PDGF signaling pathway in tumor treatment. Cell Communication and Signaling 2013, 11, 1–18. [Google Scholar] [CrossRef]
- Liu, T. et al., PDGF-mediated mesenchymal transformation renders endothelial resistance to anti-VEGF treatment in glioblastoma. Nature communications 2018, 9, 3439. [Google Scholar] [CrossRef]
- Caporarello, N. , et al., Pericytes in microvessels: from “mural” function to brain and retina regeneration. International journal of molecular sciences 2019, 20, 6351. [Google Scholar] [CrossRef]
- Andersson, P. , Mechanisms of Tumor Microenvironment in Promoting Metastasis. 2016, Karolinska Institutet (Sweden).
- Balaziova, E. , et al., Fibroblast activation protein expressing mesenchymal cells promote glioblastoma angiogenesis. Cancers 2021, 13, 3304. [Google Scholar] [CrossRef]
- di Tomaso, E. , et al., PDGF-C induces maturation of blood vessels in a model of glioblastoma and attenuates the response to anti-VEGF treatment. PloS one 2009, 4, e5123. [Google Scholar] [CrossRef]
- Ivy, S.P. J.Y. Wick, and B.M. Kaufman, An overview of small-molecule inhibitors of VEGFR signaling. Nature reviews Clinical oncology 2009, 6, 569–579. [Google Scholar] [CrossRef]
- Reardon, D.A. , et al., A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. Journal of the National Comprehensive Cancer Network 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Barzegar Behrooz, A. , et al., Wnt and PI3K/Akt/mTOR survival pathways as therapeutic targets in glioblastoma. International journal of molecular sciences 2022, 23, 1353. [Google Scholar] [CrossRef] [PubMed]
- Ceci, C. , et al., Role of VEGFs/VEGFR-1 signaling and its inhibition in modulating tumor invasion: experimental evidence in different metastatic cancer models. International journal of molecular sciences 2020, 21, 1388. [Google Scholar] [CrossRef] [PubMed]
- Jenny, B. , et al., Expression and localization of VEGF-C and VEGFR-3 in glioblastomas and haemangioblastomas. The Journal of Pathology: A Journal of the Pathological Society of Great Britain and Ireland 2006, 209, 34–43. [Google Scholar] [CrossRef]
- Treps, L. , et al., Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. Journal of extracellular vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef]
- Gong, J. , et al., Interplay of VEGFa and MMP2 regulates invasion of glioblastoma. Tumor Biology 2014, 35, 11879–11885. [Google Scholar] [CrossRef]
- Lamszus, K. , et al., Levels of soluble vascular endothelial growth factor (VEGF) receptor 1 in astrocytic tumors and its relation to malignancy, vascularity, and VEGF-A. Clinical cancer research 2003, 9, 1399–1405. [Google Scholar]
- Cheng, F. and D. Guo, MET in glioma: signaling pathways and targeted therapies. Journal of Experimental & Clinical Cancer Research 2019, 38, 1–13. [Google Scholar]
- Kahlert, U.D. J.V. Joseph, and F.A. Kruyt, EMT-and MET-related processes in nonepithelial tumors: importance for disease progression, prognosis, and therapeutic opportunities. Molecular oncology 2017, 11, 860–877. [Google Scholar] [CrossRef]
- Burel-Vandenbos, F. , et al., MET immunolabelling is a useful predictive tool for MET gene amplification in glioblastoma. Neuropathology and Applied Neurobiology 2017, 43, 252–266. [Google Scholar] [CrossRef]
- Cruickshanks, N. , et al., Role and therapeutic targeting of the HGF/MET pathway in glioblastoma. Cancers 2017, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, M. , et al., Axl and growth arrest–specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clinical Cancer Research 2008, 14, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Repici, A. , et al., Signaling Pathways of AXL Receptor Tyrosine Kinase Contribute to the Pathogenetic Mechanisms of Glioblastoma. Cells 2024, 13, 361. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I. , et al., Quercetin induces apoptosis in glioblastoma cells by suppressing Axl/IL-6/STAT3 signaling pathway. The American Journal of Chinese Medicine 2021, 49, 767–784. [Google Scholar] [CrossRef]
- Oprita, A. , et al., Updated insights on EGFR signaling pathways in glioma. International Journal of Molecular Sciences 2021, 22, 587. [Google Scholar] [CrossRef]
- Allahverdi, A. , et al., Involvement of EGFR, ERK-1, 2 and AKT-1, 2 activity on human glioma cell growth. Asian Pacific journal of cancer prevention: APJCP 2020, 21, 3469. [Google Scholar] [CrossRef]
- Aldape, K. , et al., Glioblastoma: pathology, molecular mechanisms and markers. Acta neuropathologica 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Lo, H.-W. , Targeting Ras-RAF-ERK and its interactive pathways as a novel therapy for malignant gliomas. Current cancer drug targets 2010, 10, 840–848. [Google Scholar] [CrossRef]
- Ou, A. , et al., The role and therapeutic targeting of JAK/STAT signaling in glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef]
- Luwor, R.B. S.S. Stylli, and A.H. Kaye, The role of Stat3 in glioblastoma multiforme. Journal of clinical neuroscience 2013, 20, 907–911. [Google Scholar] [CrossRef]
- Li, X. , et al., PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440. [Google Scholar] [CrossRef] [PubMed]
- Margolis, B. , et al., EGF induces tyrosine phosphorylation of phospholipase C-II: a potential mechanism for EGF receptor signaling. Cell 1989, 57, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Reyland, M.E. , Protein kinase C isoforms: Multi-functional regulators of cell life and death. Frontiers in bioscience (Landmark edition) 2009, 14, 2386. [Google Scholar] [CrossRef] [PubMed]
- An, Z. , et al., Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Ding, J. , et al., EGFR suppresses p53 function by promoting p53 binding to DNA-PKcs: a noncanonical regulatory axis between EGFR and wild-type p53 in glioblastoma. Neuro-oncology 2022, 24, 1712–1725. [Google Scholar] [CrossRef]
- Al-Ghabkari, A. B. Huang, and M. Park, Aberrant MET receptor tyrosine kinase signaling in glioblastoma: targeted therapy and future directions. Cells 2024, 13, 218. [Google Scholar] [CrossRef]
- Ghanem, P. et al., Druggable genomic landscapes of high-grade gliomas. Frontiers in Medicine 2023, 10, 1254955. [Google Scholar] [CrossRef]
- Fan, F. , et al., A comprehensive prognostic signature for glioblastoma patients based on transcriptomics and single cell sequencing. Cellular Oncology 2021, 44, 917–935. [Google Scholar] [CrossRef]
- Vastrad, B. , et al., Molecular mechanisms underlying gliomas and glioblastoma pathogenesis revealed by bioinformatics analysis of microarray data. Medical Oncology 2017, 34, 1–30. [Google Scholar] [CrossRef]
- Akiyama, Y. , et al., YKL-40 downregulation is a key factor to overcome temozolomide resistance in a glioblastoma cell line. Oncology Reports 2014, 32, 159–166. [Google Scholar] [CrossRef]
- Mansouri, S. , et al., Sox2: regulation of expression and contribution to brain tumors. CNS Oncol 2016, 5, 159–73. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. , et al., TIMP1/CHI3L1 facilitates glioma progression and immunosuppression via NF-κB activation. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2024, 1870, 167041. [Google Scholar]
- Shapovalov, V. , et al., Transcriptomics-based phenotypic screening supports drug discovery in human glioblastoma cells. Cancers 2021, 13, 3780. [Google Scholar] [CrossRef]
- Atanaki, F.F. , et al., Integrative analysis of single-cell transcriptomic and multilayer signaling networks in glioma reveal tumor progression stage. Frontiers in Genetics 2024, 15, 1446903. [Google Scholar] [CrossRef]
- Mueller, C. , et al., Glioblastoma cell enrichment is critical for analysis of phosphorylated drug targets and proteomic–genomic correlations. Cancer research 2014, 74, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.-H. , et al., Novel sphingomyelin biomarkers for brain glioma and associated regulation research on the PI3K/Akt signaling pathway. Oncology Letters 2019, 18, 6207–6213. [Google Scholar]
- Pooladi, M. S. Abad, and M. Hashemi, Proteomics analysis of human brain glial cell proteome by 2D gel. Indian journal of cancer 2014, 51, 159–162. [Google Scholar]
- Jayaram, S. , et al., Multi-omics data integration and mapping of altered kinases to pathways reveal gonadotropin hormone signaling in glioblastoma. Omics: a journal of integrative biology 2016, 20, 736–746. [Google Scholar] [CrossRef]
- Jaroch, K. P. Modrakowska, and B. Bojko, Glioblastoma metabolomics—in vitro studies. Metabolites 2021, 11, 315. [Google Scholar] [CrossRef]
- Zhou, Y. , et al., Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. Journal of Biological Chemistry 2011, 286, 32843–32853. [Google Scholar] [CrossRef]
- Hesse, F. , et al., Imaging glioblastoma response to radiotherapy using 2H magnetic resonance spectroscopy measurements of fumarate metabolism. Cancer research 2022, 82, 3622–3633. [Google Scholar] [CrossRef] [PubMed]
- SongTao, Q. , et al., IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer science 2012, 103, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Kallenberg, K. , et al., Untreated glioblastoma multiforme: increased myo-inositol and glutamine levels in the contralateral cerebral hemisphere at proton MR spectroscopy. Radiology 2009, 253, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Fontanilles, M. , et al., Metabolic remodeling in glioblastoma: a longitudinal multi-omics study. Acta Neuropathologica Communications 2024, 12, 1–13. [Google Scholar] [CrossRef]
- Neftel, C. , et al., An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 2019, 178, 835–849. e21. [Google Scholar] [CrossRef]
- Seifert, C. , et al., PIM1 inhibition affects glioblastoma stem cell behavior and kills glioblastoma stem-like cells. International journal of molecular sciences 2021, 22, 11126. [Google Scholar] [CrossRef]
- Martínez, A.H. , et al., Unravelling glioblastoma heterogeneity by means of single-cell RNA sequencing. Cancer letters 2022, 527, 66–79. [Google Scholar] [CrossRef]
- Chakraborty, S. , et al., Multi-OMICS approaches in cancer biology: New era in cancer therapy. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 2024, 1870, 167120. [Google Scholar] [CrossRef]
- Liu, J. , et al., Multi-scale signaling and tumor evolution in high-grade gliomas. Cancer cell 2024, 42, 1217–1238. e19. [Google Scholar] [CrossRef]
- Alom, M.W. , et al., Integrated Gene Expression Data-Driven Identification of Molecular Signatures, Prognostic Biomarkers, and Drug Targets for Glioblastoma. BioMed Research International 2024, 2024, 2024, 6810200. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



