
Submitted:
27 January 2025
Posted:
29 January 2025
You are already at the latest version
Abstract
Physiologic aging and insults from the environment lead to DNA damage. In response, cells in any organ will undergo senescence-induced growth arrest to prevent damaged cells from further propagation. This review focuses on senescence pharmacology. First, we describe senescence induction mechanisms and a unique feature of senescent cells, the SASP (senescence-associated secretory phenotype). Signaling pathways that control and respond to the SASP provide the framework for a better understanding of senescence pharmacology. We describe how several commonly used drugs can induce cellular senescence and how that impacts their efficacy and produces unexpected effects. Thereafter, we discuss the potential and challenges of senolytic drugs that eliminate senescent cells, and we describe targeting of components of the SASP as well as pathways that control expression of genes contributing to the SASP. Lastly, we discuss studies that have exemplified the significant impact of senescence-targeted therapy in various disease states.
Keywords:
cellular senescence
; senolytics
; SASP
1. Senescence. History and Mechanisms
Cellular senescence is a programmed state of cell cycle arrest that is accompanied by complex phenotypes. Senescent cells play a role in physiological processes such as tumor suppression, wound healing and embryonic development, whilst paradoxically also contributing to cancer progression as well as aging and age-related disease [1]. As such, cellular senescence has broad, multidisciplinary reach.
Cellular senescence occurs when cells are not able to progress normally through the cell cycle [2]. Senescence in cells was first described by Weismann in 1881 when evaluating the principles that govern the duration of life and was subsequently re-discovered by Hayflick and Moorhead in the early 1960s as reviewed by Kirkwood and Cremer in 1982 [3]. Hayflick and Moorhead observed that normal human fibroblasts entered an irreversible growth arrested state through continuous states of passaging in vitro [4,5,6]. The growth arrest occurred at the G1 phase of the cell cycle. Though senescent cells do not have the ability to undergo mitosis, they still show high metabolic activity [2]. Once they are arrested, senescent cells will not replicate their DNA even if subjected to conditions that are suitable for growth and proliferation [2]. This contrasts with cellular quiescence, which has been established as a reversible state that can be exited if a cell is given a strong enough stimulus. Also, quiescent cells show reduced metabolic activity. In general, the senescent cell state is believed to be permanent. Nonetheless, some studies have observed that senescence is not as irreversible as once believed. Such instances of senescent cells re-entering the cell cycle have been found when proteins activated during senescence such as p16 or p38MAPK are inhibited [7].
To understand the complex network of cellular senescence first requires understanding its drivers. Senescence can be induced in multiple ways as illustrated in Figure 1. Specific examples of senescence induction include activation of oncogenes (RAS, BRAF), inhibition of tumor suppressors (PTEN, PTTG1, CSNK1A1), DNA damage through chemical (e.g., oxidative stress, alkylating drugs) or physical insults (e.g., radiotherapy and environmental radiation or excitotoxic injury in the CNS) as well as dysfunctional and shortened telomeres [8,9]. With every cycle of cell division, DNA damage occurs at the telomere ends. After many cell divisions, the telomere ends will enter a short enough stage where repair can no longer occur with fidelity and DNA breaks will accumulate leading to activation of the tumor suppressor protein p53. Along with another tumor suppressor, retinoblastoma protein (Rb), p53 and Rb activity are controlled by the INK4a/ARF or Cyclin Dependent Kinase inhibitor 2a (CDKN2a) locus [4]. CDKN2a encodes the two isoforms p16ink4a and p14ARF. While p16ink4a inhibits cyclin-dependent kinase 4 and 6 (CDK4/6), p14ARF inhibits Mdm2 which prevents the degradation of p53 [10]. Thus, when the CDKN2a locus is activated, a cell cycle arrest is induced [4]. In general, activation of CDK4/6 will lead to the hyper-phosphorylation of Rb, the dissociation of the Rb-E2 transcription factor (E2F) complex, transcription of S phase genes and progression of the cell cycle [11,12]. However, if CDK4/6 is inhibited by expression of p16, this will cause hypo-phosphorylation of Rb allowing the Rb-E2F complex to stay intact and inhibiting S phase gene transcription and leading to senescence [11,13]. In addition, CDKN1a which encodes p21Cip1, another CDK inhibitor, impacts the cell cycle through downstream interactions with p53 [8]. Thus, p16ink4a establishes the Rb regulated growth arrest, while p21Cip1 maintains the p53 mediated growth arrest [8]. The proteins and pathways involved are shown in Figure 2.
The molecular and cellular characteristics that identify senescent cells are numerous and equally complex as the pathways leading to senescence. Multiple markers have been established as identifiers of senescent cells, yet there is no single, universal, biomarker specific only to senescent cells. In cultured cells, morphological changes (i.e., a large and flattened morphology) is characteristic of senescence. One of the most widely used biomarkers used for the identification of senescent cells has been Senescence-Associated Beta-galactosidase (SA-β-gal) described in 1995 by the Campisi laboratory [14]. This beta-galactosidase activity is detectable in senescent cells at pH 6.0 in contrast to the optimum pH of 4.0 for this enzyme [2,14,15]. It is believed that increased lysosomal biogenesis present in senescent cells is linked to the SA-β-gal staining [2,15]. Most interestingly, recent studies have identified GLB1 or galactosidase Beta 1 as the specific enzyme responsible for SA-Beta-Gal activity [16]. This is an important finding as GLB1 expression is detectable in formalin-fixed paraffin-embedded tissues, while measurements of SA-β-Gal activity measurements are typically limited to viable cells, e. g. in frozen tissue or cultured cells [16]. Also, p16ink4a, p14ARF, and p21Cip1, cyclin-dependent kinase inhibitors of the cell cycle discussed above have been used as indicators of senescence although p16ink4a can be increased irrespective of senescence [2,17]. Lastly, other markers used for senescent cell identification are shown in Figure 3 and include less specific chromatin alterations (lamin B1 deficiency and heterochromatin foci) and DNA damage markers (γH2AX foci) as well as the senescence-associated secretory phenotype (SASP) a striking feature of senescent cells discussed in more detail in the next section [17].
1.1. SASP: Senescence Associated Secretory Phenotype
The SASP consists of proteins and other components shed by cells with complex systemic and local microenvironment effects depending on the senescence inducers, the type of cell secreting the SASP and the duration of senescence. Acute induction of the SASP provides for optimal fitness of the organism and such scenarios occur during wound healing, tumor growth suppression, and somewhat counterintuitively also during embryonic development [9,18]. In contrast, chronic senescence and thus prolonged SASP exposure have been shown to promote detrimental effects including tumorigenesis, tissue dysfunction, and immunosuppression [19,20,21]. The SASP consists of various cytokines, chemokines, and interleukins including IL-6, 8, 10, 13, MCP-2, IFNγ, TNFα, CXCLs, TGFb, proteases such as MMPs and MIP-3a, growth factors such as FGF2, HGF, and IGFBPs. Shed cell surface molecules include ICAMs, uPAR, and TNF receptors[22,23]. These factors signal in both autocrine and paracrine fashions, impacting both the senescent cell and the tissue microenvironment. Unique effects due to the distinct and tissue-specific composition of the SASP, and its associated secretomes, range from tissue repair to tumor growth promotion and priming of the immune system to clear out senescent cells [8]. Emerging studies show that the immune system itself is also subject to regulation by senescence with an impact on its functionality that range from the response to immunization and the efficacy of anti-cancer treatments [24].
It is interesting to note that while the induction of SA-β-galactosidase activity and change in cellular morphology to a large and flattened phenotype occur quickly, the induction of the SASP occurs at a slower rate, with changes seen only detectable after several days of exposure to genotoxic stress [8,22]. The SASP in cells exposed to genotoxic stress is pro-inflammatory and characterized by IL-6 and -8 as the major components. Combined actions of IL-6/8 enable the clearance of the damaged cells but also tumorigenesis, epithelial-mesenchymal transition (EMT) and induction of invasiveness of premalignant lesions [22,25]. IL-6 and -8 expression and secretion are controlled by IL-1α. IL-1α acts through an autocrine loop signaling through the IL-1R on senescent cells with IRAK4 and 1 (Interleukin-1-receptor kinase) as well as MyD88 (myeloid differentiation primary-response protein 88) binding to the IL-1/IL-1R complex. IRAK1 is then phosphorylated by IRAK4 ultimately leading to NF-κB translocation to the nucleus and subsequent transcriptional activation of the genes encoding IL-6 and -8 [25]. IL-1α plays a central role in the SASP network, specifically by regulating IL-6 and -8 expression and promoting its own secretion in a positive feedback loop [17,25]. IL-1α signaling is thus a possible target of senolytics although other components of the SASP are induced in parallel and independent of this axis. Senescence induction in endothelia and hyperinflammation was also observed after COVID-19 infection and thought to cause acute and chronic pathologies that may be mitigated by anti-inflammatory agents, e.g., dexamethasone or senolytics [26].
Another factor in the SASP is CXCL1 (C-X-C motif ligand 1), a cytokine previously called Gro-1 (growth regulated oncogene 1), that promotes tumor growth, invasion, and metastasis through its interactions with stromal and epithelial cells within the tumor microenvironment. It is secreted by RAS-transformed cells, binds to its receptor (CXCR2) on fibroblasts and can induce senescence. In turn, these senescent fibroblasts secrete SASP factors that lead to a pro-tumorigenic environment [27].
A functionally distinct component of the SASP are matrix-metalloproteinases (MMPs). DNA damaging agents can lead to senescence of fibroblasts and these senescent cells can cause fluid accumulation within the tissues through actions by the MMPs secreted as part of the SASP [28]. During the physiologic repair of tissue injury, the MMPs lead to microvascular permeability and extravasation of plasma components which allows for new blood vessel ingrowth, healing and restoration of functions of the injured tissue. In contrast to this physiologic setting, MMPs released from senescent cells within a malignant lesion as part of the SASP can promote tumor growth and angiogenesis [28].
The SASP components discussed above highlight several of the detrimental effects on the host organism. It is noteworthy that transient (acute) exposure to the SASP leads to more restorative effects that include cell growth, stem cell marker expression and increased regeneration capacity [29]. However, continuous exposure to the SASP can lead to cell cycle arrest and is a possible mechanism to prevent the over-proliferation of cells with enhanced plasticity or progression of pre-malignant lesions [29].
In conclusion, the SASP has varying impacts dependent on the senescent cell type as well as the inducer of senescence and the microenvironment. Depending on the timing, secretions of the SASP can be either beneficial or detrimental to the organism. Thus, it will be important to gain further understanding of the various components of the SASP as this will allow insight on potential therapeutic targets.
1.2. Senescence Signaling Networks
DNA double strand breaks (DSBs) can induce cellular senescence due to DNA damage response (DDR) signaling and activation of p53. ATM, NBS1, and CHK2 are important components of the DDR and in turn regulate the secretion of IL-6 that occurs in response to DNA damage. Senescence-driven IL-6 secretion and its ability to cause cancer invasiveness is decreased if ATM of the DDR is depleted. However, IL-6 and IL-8 are not the only components of the SASP that are regulated by the DDR [30]. Furthermore, only certain senescent phenotypes are present at the time the DDR is activated, while detection of SASP occurs at much later time points. For example, beta-galactosidase positive staining and the DDR occur at similar time points after the insulting damage [30,31]. On the other hand, SASP expression is apparent after a few days which suggests that the induction of SASP is dependent on the DDR [30,31]. This would suggest that other proteins and/or pathways bridge the “slow” and “fast” phases of senescence responses. A protein believed to play such an essential role is p38MAPK. P38MAPK interacts with NF-kB and through this combined signaling leads to expression of the SASP, specifically pro-inflammatory cytokines [31]. Furthermore, p38MAPK activation and signaling occurs slowly after DDR and mirrors SASP expression kinetics [31]. As one consequence of this mechanism, a transient DDR that fully resolves DSBs does not promote SASP expression [30]. The delay of p38MAPK signaling is brought about by p53 suppressing p38MAPK allowing for repairable DNA damage to be completed but initiating SASP expression upon failed repair. The SASP components under control of p38MAPK consist of chemokines, cytokines and growth factors. In contrast, MMP secretions are not p38MAPK dependent. Notably, depleting the RelA subunit of NF-KB decreases SASP secretion regulated by p38MAPK indicating the contribution of NF-KB [31].
Another pathway that plays a unique role in the function of the SASP and the composition of its secretome is the NOTCH pathway. At the early stages of senescence, NOTCH1 is highly expressed on the cell surface but decreased in later stages. Activated NOTCH1 leads to a decreased expression of pro-inflammatory SASP secretions, while it promotes increased TGF-beta expression [32,33]. The decreased expression of pro-inflammatory cytokines is likely caused by repression of the C/EBPB transcription factor, which under normal circumstances promotes the expression of the cytokines in cooperation with NF-kB. Interestingly, the NOTCH1-promoted TGF-beta secretion initiates senescence in adjacent cells [32,33]. On the other hand, NOTCH1 inhibition allows for expression of the pro-inflammatory cytokines which leads to lymphocyte recruitment and senescence surveillance. It is noteworthy that two distinct senescence secretomes are controlled by the NOTCH1 signaling pathway [32,33].
A central pathway regulating the SASP is the mTOR pathway. mTOR activity controls and senses changes in the levels of growth and nutrient signals in a cell. Generally speaking, highly active mTOR leads to somatic growth and decreased lifespan whilst suppression of mTOR is associated with an increase in lifespan [34]. Rapamycin (sirolimus), an inhibitor of mTOR used clinically, inhibits expression of several SASP components by impacting the ability of Elf4A1 helicases to unwind secondary mRNA structures [34]. Failure to unwind reduces the translation of mRNAs coding for SASP pro-inflammatory cytokines. As such, rapamycin has been shown to decrease IL-1a levels, which also control IL-6 via NF-kB [34]. This reflects the potential of Rapamycin as a modulator of SASP expression and secretion in addition to its control of cellular metabolism [34]. Finally, by inhibiting IL-1a expression, Rapamycin prevents senescent cells from promoting tumor growth [34].
1.3. Therapeutic Targeting of Senescence
Directly targeting senescent cells or modulating the SASP has been increasingly explored to reverse the functional deterioration of healthy tissues. In the context of cancer treatment, this may enhance the efficacy of cancer treatment or reverse treatment resistance. Given that senescent cells can promote either an anti- or pro-tumorigenic environment depending on the cellular context, it would be of great benefit to be able to enhance the former over the latter in the setting of cancer therapeutics. The following sections will evaluate senescence pharmacology with highlights of proteins that either lead to senescence or can be targeted for elimination of senescent cells. Figure 4 provides an overview and is discussed further below.
1.4. Impact of Senescence on Drug Action
As stated earlier, one of the many roles of the SASP is in immune system activation to clear senescent cells and result in an anti-tumor response [18,35]. As such, a goal is to isolate the anti-tumor SASP effects and in doing so provide a synergistic effect with other well-established cancer therapies. However, implementing this concept may not be as straightforward as there is also the possibility that senescence induction could reduce the efficacy of chemotherapeutics as the SASP secreted by the senescent cells can promote immunosuppression counteracting the tumor killing effects. This conundrum was assessed by Toso et al., 2014 and reviewed recently [1,36]. The study utilized the Pten-null prostate cancer mouse model. By seven weeks of age, mice express senescent cells and their SASP was shown to promote an immunosuppressive microenvironment. CD11b+ myeloid derived suppressor cells (MDSCs) were highly prevalent in the Pten-null tumors and their presence inhibited the activity and proliferation of CD4+ and CD8+ T cells and natural killer cells. The SASP of the Pten-null senescent tumor cells included the cytokines CXCL1, CXCL2, IL-6 and IL-10. The expression of these immunosuppressive cytokines was controlled by the transcription factor Stat3 [36,37]. Inactivating Stat3 led to changes in the senescent phenotype and the SASP composition but no change in the expression of p53, NF-kB, or Senescence-associated Beta galactosidase staining, but a significant reduction in IL-10, IL-13, and GM-CSF [36]. In contrast, factors such as CXCL10 which lead to B cell, T cell, and monocyte recruitment were increased. Furthermore, inactivating Stat3 led to p16ink4a expression reduction, suggesting that targeting Stat3 could be used to clear senescent tumor cells. Thus, this study highlighted that basic senescence characteristics were still intact after inactivation of Stat3, but found that the composition of the SASP can be changed from a pro-tumor to an anti-tumor phenotype. Knowing that the SASP of Pten-null senescent cancer cells can be modulated is of interest for its impact on chemotherapy efficacy. One such drug assessed was docetaxel, an inhibitor of microtubule depolymerization, that can induce senescence in stromal cells and thus impact tumor growth [38]. The anti-tumor effect of docetaxel was lost in Pten-null mice and restored if Jak2, the upstream activator of Stat3 and one of the main modulators of the SASP secretion in mice, is inhibited [39].
Lastly, the SASP composition will differ based on senescence inducers and cell type. For example, the SASP composition in Pten-null versus mutant KRAS oncogene-induced senescent tumor cells is different [18]. Immunosuppressive cytokines and Stat3 levels are higher in Pten-null versus KRAS oncogene induced senescent cells. Furthermore, efficient senescence surveillance occurs in KRAS mutant mice as the oncogene-induced senescent cells are removed, while in the Pten null mice senescent cells are not cleared out[18,39,40]. Thus, the variation of the composition of the SASP due to different oncogenic drivers in subtypes of cancers can impact the efficacy of treatments due to distinct host responses to the SASP signaling.
1.5. Therapy Induced Senescence
In addition to the impact of senescence on drug efficacy, the mechanisms of action of some well-established treatments are directly linked to the induction of cellular senescence. An example of such a drug is the a CDK4/6 inhibitor palbociclib, which keeps Rb hypophorylated, preventing mitosis [41] (Figure 4). Interestingly, the long-term impact of palbociclib exposure can induce fibroblast senescence via Mdm2 degradation which allows for maintained expression of p21 [41,42]. Furthermore, palbociclib-induced senescent fibroblasts secrete more pro-inflammatory SASP than fibroblasts that may undergo senescence through ultraviolet radiation or mitomycin C exposure [41]. SASP components released from the senescent fibroblast cells also produce a highly immunosuppressive environment by recruiting Gr1+ immunosuppressive myeloid-derived suppressor cells (MDSCs)[41]. We have found that this extends to endothelial cell senescence induced by palbociclib exposure and primes the lung metastatic niche resulting in increased metastatic seeding of breast cancer cells in a syngeneic mouse model [43].
Another notable example of a chemotherapeutic drug that induces senescence is doxorubicin. Doxorubicin intercalates with DNA and forms complexes with topoisomerase II that lead to a lack of relegation of cleaved DNA strands by topoisomerase II [44,45]. As a consequence, the DNA damage response is activated resulting in cellular senescence as discussed above [46]. The SASP secreted by doxorubicin-induced senescent cells includes high levels of pro-inflammatory cytokines and chemokines that have negative impacts on cardiac function and bone marrow proliferation. Similar to palbociclib, a significant decrease in tumor growth is observed when doxorubicin is used early during treatment. However, similar to palbociclib, long term doxorubicin exposure can cause tumors to relapse and an increase in metastatic foci [47]. These studies highlight that both short- and long-term exposure induce senescence though with uniquely different outcomes in terms of tumor growth and immune infiltration.
2. Senotherapy
The next section will discuss targeted elimination of senescent cells also illustrated in Figure 4 and summarized in Table 1 further below. Senotherapy represents a balancing act between beneficial aspects of senescence for proper wound healing and some anti-cancer therapies and detrimental effects of senescence with respect to tumor growth and metastasis. The last decade has seen a rapid increase in studies demonstrating beneficial effects of senotherapies in restoring normal function. The senotherapy discussed below includes senolytics (direct elimination of senescent cells), SASP neutralization and targeting of senescence-related proteins.
2.1. Senolytics
Elimination of senescent cells, “senolysis”, is based on studies in the Kirkland lab, who observed that within the SASP, senescent cells can also secrete pro-apoptotic factors. To prevent self-inflicted, autocrine-induced apoptosis, senescent cells increase activation of anti-apoptotic pathways [48,49,50]. These “senescent cell anti-apoptotic pathways” (SCAPs) include proteins of the BCL2 family, phosphoinositide 3 kinase (PI3K), p53, p21, and ephrin receptor tyrosine kinases. They have been studied as targets for the elimination of senescent cells with the most commonly used drugs being navitoclax (ABT-263), dasatinib and quercetin [49,50].
2.1.1. Navitoclax
Navitoclax (ABT-263) is a BH3 mimetic that can induce apoptosis by binding to the BH3 domain in BCL2 and preventing its interaction with Bax. Chang et al. reported in 2016 that ABT-263 lead to stem cell regeneration and an increase in health span. Within this study, decreased p16ink4a, TNFa, and chemokines (e. g. CCL5) were observed after ABT-263 [51]. Furthermore, targeted elimination of the senescent cells through ABT-263 also led to increased hematopoietic stem cell proliferation to replace the senescent cells that were eliminated. Though navitoclax (ABT-263) shows the desired effects on senescent cells, it also caused transient thrombocytopenia and neutropenia which limit its potential use in age-related diseases with possible immunosuppression [52]. Dasatinib, an inhibitor of different kinases including abl, src and c-kit is another potential senolytic drug. Dasatinib is used to treat patients with chronic myelogenous leukemia (CML), Philadelphia Chromsome+ acute lymphoblastic leukemia and chronic lymphoblastic leukemia as a second line drug after imatinib [53]. Adverse effects are overall mild with GI symptoms and fluid retention the most frequent. Myelosuppression, hepato- and cardiotoxicity are possible but relatively infrequent.
2.1.2. Fisetin & Quercetin
Fisetin & Quercetin are some of the most prevalent plant flavonoids that are reportedly present in many fruits and vegetables such as apples and onions. The bioactive potential of fisetin and quercetin has been established, especially in the modulation of a range of cancer signaling pathways. The anti-cancer, anti-inflammatory, and antioxidant roles exhibited by these flavonols have been reportedly found to be associated with their ability of apoptotic activation, cell cycle arrest, regulating ECM remodeling and inhibiting EMT. Many studies of fisetin and quercetin found a modulatory potential in different cancer-related signaling pathways and growth factors including Akt, JNK, p38MAPK, NF-κB, and VEGF, cytokines and chemokines indicating a broad range of mechanisms of action of these flavonols [54,55].
2.1.3. Quercetin
Quercetinhas been used synergistically with dasatinib and promoted as a potential senolytic. Quercetin functions as an anti-oxidant that reduces inflammation by inhibiting the PI3K pathway. Studies utilizing both dasatinib and quercetin have shown elimination of senescent cells and decreased production of SASP pro-inflammatory cytokines in human adipose tissue grafts [56]. A more recent study revealed that senescent cells may play a significant role in the neuropathophysiology related to Trisomy 21 (T21) resulting in Down Syndrome. In neural progenitor cells with T21, treatment with dasatinib and quercetin significantly alleviated the senescence-related impact at the genome architecture, transcriptional and cellular level [57].
2.1.4. Salidroside
Salidroside is similar to quercetin, is found in Chinese herbal roots and showed an effect on endothelial cellular senescence [58]. Senescent endothelial cells have been found to occur in atherosclerosis and their presence leads to a pro-inflammatory environment [59]. Hyperhomocysteinemia (HHC) mice were used as a model to study the effects of atherosclerosis and exhibit aortic medial thickness, increased collagen deposition and increased macrophage infiltration [58,60]. The HHC mice also have low levels of SIRT3, a deacetylase that eliminates reactive oxygen species (ROS) in the mitochondria one of the main damage inducers in the vasculature. When HHC mice were administered salidroside, the vasculature exhibited decreased collage deposition and aortic intima hyperplasia as well as an increased SIRT3. Finally, salidroside treatment decreased expression of p16 and p21 in addition to reduced SASP which links its mechanism of action to senescence.
2.1.5. SSK1
SSK1 is a prodrug of gemcitabine, a cytotoxic difluoro-analogue of deoxycytidine that causes cellular apoptosis and is approved and used for chemotherapy of a range of different cancers causing inhibition of DNA synthesis amongst other mechanisms [45]. The elegantly designed inactive SSK1 prodrug contains a site cleavable by SA-beta-galactosidase thus releasing cytotoxically active gemcitabine in senescent cells [61]. The prodrug did not show cytotoxic activity in non-senescent cells and provides a very promising therapeutic window.
2.1.6. Conclusions and Outlook.
There is a great potential for the use of senolytics that target specific rate-limiting proteins in subpopulations of senescent cells [7,62]. Adaptation of treatment schedules could avoid eliminating beneficial senescent cells induced earlier and temporarily while targeting long-lived senescent cells [63]. Furthermore, short treatment cycles would also decrease the chances of adverse effects. One of the advantages of senolytics is that senescent cells do not replicate and the potential for selection of mutated, resistant subpopulations in later cell generations is thus negligible [49]. However, there is significant need for further drug development including the recent concept underlying the inactive prodrug SSK1 (see above [64]) with the potential of exploiting a senescence mechanism-based activation of the cytotoxic effect of the prodrug. A limitation of most of the above senolytic compounds is relatively broad targets, which may or may not be selective for senescent cells. While effective at clearing senescent cells, the potential for off target impacts producing disease modifying effects remains. Parallel studies using highly specific genetic strategies to ablate senescent cells (e.g., targeted apoptosis of p16 expressing cells), however, provides strong validation that the therapeutic effects observed in a range of disease states with pharmacological senotherapies are due to senolysis and due to off target effects. The biggest challenge is certainly the limited understanding of the complexity of senescence induction and the responses in different cell types to produce a synergistic effect with established therapies be that an improvement in efficacy or a reduction of adverse effects.
2.2. Reducing SASP effects
Along with the direct senolytic approach that targets senescent cells, there are also indirect ways to impact cellular senescence. One of the potential targets that was touched upon earlier is the SASP, a defining feature of senescent cells. The SASP impacts the microenvironment in unique ways and inhibition of selective components could have unique potential. One of the approaches is to target proteases that are important for the maturation of many of the SASP components including IL-1alpha [7,65]. Furthermore, MMPs secreted with the SASP promote extracellular matrix degradation and their inhibition could prevent an environment conducive for invasion and metastasis.
A second approach to blocking the effects of the SASP is to target ligands or receptors exemplified by IL-6: siltuximab (anti-IL-6) or tocilizumb (anti-IL-6R) [7]. These blocking antibodies are approved for the treatment of rheumatoid arthritis and Castleman’s disease [7,66,67].
A third avenue for targeting the SASP is to inhibit signaling pathways. Potential targeted therapies can be split into NF- κB dependent and NF- κB independent due the central role NF-κB plays in the expression and function of numerous SASP inflammatory cytokines [68]. Glucocorticoids are potential candidates as they possess anti-inflammatory effects and have been shown to cause decreased NF- κB-based transcription of SASP genes and thus reducing expression of SASP related inflammatory factors. However, longer use of glucocorticoids at effective doses induces significant endocrine adverse effects [34,69]. Similarly, metformin, a well-established drug in the treatment of patients with type 2 diabetes mellitus prevents NF-κB translocation to the nucleus [70]. Interestingly, metformin also decreased the expression of SASP cytokines that led to reduction in the growth of prostate cancer cells [71]. In addition, other proteins and cytokines that are part of the NF-κB pathway could be targeted. Anakinra inhibits the binding of IL-1alpha to its receptor and prevents IL-1alpha / NF- κB signaling and induction of expression of IL-6 and IL-8 [25]. Lastly, rapamycin, the mTOR inhibitior can also regulate IL-1alpha expression and signaling [34].
On the other hand, there is also the potential of targeting NF-κB independent pathways. One such example discussed earlier is the JAK/STAT pathway [68]. By binding to its receptor, IL-6 leads to JAK/Stat signaling which in turn activates C/EBPB, the transcription factor important for SASP cytokine expression [72]. A study by Toso et al. in 2015 illustrated that the JAK2 inhibitor ruxolitinib decreased inflammatory cytokines in the SASP [36].
2.3. Targeting Senescence-related Proteins
As discussed above, senescent cells can secrete factors that induce apoptosis while at the same time secreting anti-apoptotic factors that counteract those effects. Thus, understanding the balance between senescence and apoptosis is a challenge when targeting senescent cells. Baar et al. discovered that senescent IMR90 fibroblasts secrete pro-apoptotic molecules but also increase expression of FOXO4 preventing apoptosis [77]. Upon shRNA-mediated depletion of FOXO4 from the senescent fibroblasts, mitochondrial cytochrome C was released and BAX/BAK-dependent caspase 3 cleavage induced apoptosis [77]. Furthermore, during senescence, PML bodies and 53BP1 foci in the nucleus fuse with DNA segments with chromatin alterations reinforcing senescence (DNA-SCARS) [35]. The complex of DNA-SCARS/PML bodies and p53 work to regulate the expression of the SASP [35,77]. In addition, FOXO4 interacts with p53 and the PML bodies in the nucleus[77]. With FOXO4, p53 complexes with p21 and leads to senescence induction [78]. Baar et al. used a peptide, FOXO4-DRI which interrupts the FOXO4-p53 interaction and by doing so saw a reduction in complex formation between FOXO4 and the PML bodies. Subsequently, p53 did not localize to the nucleus leading to decreased levels of p21 [77]. Instead, the cytostolic p53 translocated to the mitochondria inducing mitochondrial-based apoptosis [77]. Most interestingly, the anti-senescence effects of FOXO4-DRI were shown to have an impact on therapy-induced senescence by using the transgenic p16-3MR model. In this model developed in the Campisi laboratory, p16 positive cells can be selectively eliminated through induction of cell death by p16-dependent expression of a ganciclovir-sensitive cassette. Doxorubicin administration to mice induced numerous senescence related effects, including FOXO4 localization with PML/DNA-SCARS bodies, increased IL-6 and decreased body weight [77]. However, if these mice were administered FOXO4-DRI, these senescence effects were drastically reduced[77]. Within the same study, Baar et al. also studied the effect of FOXO4-DRI on a premature aging mouse model (XPD-TTD) which models trichothiodystrophy [79]. They observed that characteristics of TTD, which included renal function loss, loss of hair, increased abdominal temperatures, were reversed by FOXO4-DRI [77]. Similar to SSK1 discussed above this study provides another ground-breaking mechanistic concept whereby FOXO4-DRI selectively targets and eliminates senescent cells without obvious adverse effects.
3. Concluding Remarks—Promises and Limitations
There are numerous potential targets for both senescence induction and eliminating senescent cells (Figure 4 and Table 1). The most challenging question is when either of these approaches should be prioritized. With senescent cells, knowing when they have a beneficial tumor targeting effect or a detrimental tumor promoting impact is key to establishing treatment protocols. In addition, numerous studies have been carried out to see if inducing senescence in certain cancers promotes or negates the efficacy of well-known cancer therapies [80]. In order to be used optimally, not only do senolytics have to be carefully assessed for off-target effects but also for the best time point and duration they should be administered for relative to established treatment regimen including chemotherapy, pathway-targeted therapies, radiation treatment, immune-modulatory and combination of different treatment modalities.
Author Contributions
Conceptualization, draft and final editing I.S.K. and A.W.; review and editing, B.N.F., P.A.F. and M.O.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by grants from different Institutes at the National Institutes of Health, USA. F30 CA243357-01 and T32CA009686 to I.S.K.; R21NS125552 to P.A.F.; T32GM142520 to P.A.F. and A.W.; R01CA231291 and P30CA51008 to A.W.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Huang, W.; Hickson, L.J.; Eirin, A.; Kirkland, J.L.; Lerman, L.O. Cellular Senescence: The Good, the Bad and the Unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Campisi, J.; Fagagna, F. d’Adda di Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Kirkwood, T.B.L.; Cremer, T. Cytogerontology since 1881: A Reappraisal of August Weismann and a Review of Modern Progress. Hum. Genet. 1982, 60, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular Senescence in Cancer and Aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Hayflick, His Limit, and Cellular Ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Deursen, J.M. van Senescent Cells: An Emerging Target for Diseases of Ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.-Y.; Campisi, J. Tumor Suppressor and Aging Biomarker P16INK4a Induces Cellular Senescence without the Associated Inflammatory Secretory Phenotype*. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and P16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and Functions of Cellular Senescence. J. Clin. Investig. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Khan, I.; Schmidt, M.O.; Kallakury, B.; Jain, S.; Mehdikhani, S.; Levi, M.; Mendonca, M.; Welch, W.; Riegel, A.T.; Wilcox, C.S.; et al. Low Dose Chronic Angiotensin II Induces Selective Senescence of Kidney Endothelial Cells. Front. Cell Dev. Biol. 2021, 9, 782841. [Google Scholar] [CrossRef]
- Zhang, X.; Wharton, W.; Donovan, M.; Coppola, D.; Croxton, R.; Cress, W.D.; Pledger, W.J. Density-Dependent Growth Inhibition of Fibroblasts Ectopically Expressing P27kip1. Mol. Biol. Cell 2000, 11, 2117–2130. [Google Scholar] [CrossRef]
- Fischer, M.; Müller, G.A. Cell Cycle Transcription Control: DREAM/MuvB and RB-E2F Complexes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 638–662. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated Β-galactosidase Is Lysosomal Β-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Damaschke, N.; Yang, B.; Truong, M.; Guenther, C.; McCormick, J.; Huang, W.; Jarrard, D. Overexpression of the Novel Senescence Marker β-Galactosidase (GLB1) in Prostate Cancer Predicts Reduced PSA Recurrence. PLoS ONE 2015, 10, e0124366. [Google Scholar] [CrossRef] [PubMed]
- Sieben, C.J.; Sturmlechner, I.; Sluis, B. van de; Deursen, J.M. van Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- Kang, T.-W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence Surveillance of Pre-Malignant Hepatocytes Limits Liver Cancer Development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Choi, Y.W.; Lee, J.; Soh, E.Y.; Kim, J.-H.; Park, T.J. Senescent Tumor Cells Lead the Collective Invasion in Thyroid Cancer. Nat. Commun. 2017, 8, 15208. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; Doorn, R. van; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal Senescence Establishes an Immunosuppressive Microenvironment That Drives Tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Pathol.: Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.S.; Sojo, G.B.; Sun, H.; Friedland, B.N.; McNamara, M.E.; Schmidt, M.O.; Wellstein, A. The Role of Aging and Senescence in Immune Checkpoint Inhibitor Response and Toxicity. Int. J. Mol. Sci. 2024, 25, 7013. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell Surface-Bound IL-1α Is an Upstream Regulator of the Senescence-Associated IL-6/IL-8 Cytokine Network. Proc. Natl. Acad. Sci. 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Tchkonia, T.; Niedernhofer, L.J.; Robbins, P.D.; Kirkland, J.L.; Lee, S. COVID-19 and Cellular Senescence. Nat Rev Immunol 2023, 23, 251–263. [Google Scholar] [CrossRef]
- Yang, G.; Rosen, D.G.; Zhang, Z.; Bast, R.C.; Mills, G.B.; Colacino, J.A.; Mercado-Uribe, I.; Liu, J. The Chemokine Growth-Regulated Oncogene 1 (Gro-1) Links RAS Signaling to the Senescence of Stromal Fibroblasts and Ovarian Tumorigenesis. Proc. Natl. Acad. Sci. 2006, 103, 16472–16477. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hornsby, P.J. Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. P38MAPK Is a Novel DNA Damage Response-independent Regulator of the Senescence-associated Secretory Phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Ito, Y.; Kang, T.-W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 Mediates a Switch between Two Distinct Secretomes during Senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Müller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell Rep. 2019, 27, 997–1007.e5. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Muñoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppé, J.-P.; Campeau, E.; Beauséjour, C.M.; Kim, S.-H.; et al. DNA-SCARS: Distinct Nuclear Structures That Sustain Damage-Induced Senescence Growth Arrest and Inflammatory Cytokine Secretion. J. Cell Sci. 2010, 124, 68–81. [Google Scholar] [CrossRef]
- Toso, A.; Mitri, D.D.; Alimonti, A. Enhancing Chemotherapy Efficacy by Reprogramming the Senescence-Associated Secretory Phenotype of Prostate Tumors. OncoImmunology 2015, 4, e994380. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, S.R.; Fu, V.X.; Desotelle, J.A.; Kenowski, M.L.; Jarrard, D.F. The Identification of Senescence-Specific Genes during the Induction of Senescence in Prostate Cancer Cells. Neoplasia 2005, 7, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing Chemotherapy Efficacy in Pten-Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Lee, K.E.; Bar-Sagi, D. Oncogenic KRas Suppresses Inflammation-Associated Senescence of Pancreatic Ductal Cells. Cancer Cell 2010, 18, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; LaPak, K.M.; Hennessey, R.C.; Yu, C.Y.; Shakya, R.; Zhang, J.; Burd, C.E. Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol. Cancer Res. 2017, 15, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK Inhibitors (P16/P19/P21) Induce Senescence and Autophagy in Cancer-Associated Fibroblasts, “Fueling” Tumor Growth via Paracrine Interactions, without an Increase in Neo-Angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef]
- Gallanis, G.T.; Sharif, G.M.; Schmidt, M.O.; Friedland, B.N.; Battina, R.; Rahhal, R.; Davis, J.E.; Khan, I.S.; Wellstein, A.; Riegel, A.T. Stromal Senescence Following Treatment with the CDK4/6 Inhibitor Palbociclib Alters the Lung Metastatic Niche and Increases Metastasis of Drug-Resistant Mammary Cancer Cells. Cancers 2023, 15, 1908. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Wellstein, A.; Sausville, E.A. Cytotoxics and Antimetabolites. In Goodman and Gilman’s The Pharmacological Basis of Therapeutics.; Brunton, L., Knollmann, B., Eds.; 2023; pp. 1340–1380.
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Chaib, S.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and Senolytics: The Path to the Clinic. Nat. Med. 2022, 28, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Garon, E.B.; Oliveira, M.R. de; Bonomi, P.D.; Camidge, D.R.; Chu, Q.; Giaccone, G.; Khaira, D.; Ramalingam, S.S.; et al. Phase II Study of Single-Agent Navitoclax (ABT-263) and Biomarker Correlates in Patients with Relapsed Small Cell Lung Cancer. Clin. Cancer Res. 2012, 18, 3163–3169. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Jimenez, C.A.; Mauro, M.J.; Geyer, A.; Pinilla-Ibarz, J.; Smith, B.D. Pleural Effusion in Dasatinib-Treated Patients With Chronic Myeloid Leukemia in Chronic Phase: Identification and Management. Clin. Lymphoma Myeloma Leuk. 2017, 17, 78–82. [Google Scholar] [CrossRef]
- Kashyap, D.; Garg, V.K.; Tuli, H.S.; Yerer, M.B.; Sak, K.; Sharma, A.K.; Kumar, M.; Aggarwal, V.; Sandhu, S.S. Fisetin and Quercetin: Promising Flavonoids with Chemopreventive Potential. Biomolecules 2019, 9, 174. [Google Scholar] [CrossRef]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New Agents That Target Senescent Cells: The Flavone, Fisetin, and the BCL-XL Inhibitors, A1331852 and A1155463. Aging (Albany NY) 2017, 9, 955–963. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Meharena, H.S.; Marco, A.; Dileep, V.; Lockshin, E.R.; Akatsu, G.Y.; Mullahoo, J.; Watson, L.A.; Ko, T.; Guerin, L.N.; Abdurrob, F.; et al. Down-Syndrome-Induced Senescence Disrupts the Nuclear Architecture of Neural Progenitors. Cell Stem Cell 2022, 29, 116–130.e7. [Google Scholar] [CrossRef]
- Xing, S.-S.; Li, J.; Chen, L.; Yang, Y.-F.; He, P.-L.; Li, J.; Yang, J. Salidroside Attenuates Endothelial Cellular Senescence via Decreasing the Expression of Inflammatory Cytokines and Increasing the Expression of SIRT3. Mech. Ageing Dev. 2018, 175, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and Molecular Biology of Aging Endothelial Cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent Developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of Senescent Cells by β-Galactosidase-Targeted Prodrug Attenuates Inflammation and Restores Physical Function in Aged Mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Gasek, N.S.; Kuchel, G.A.; Kirkland, J.L.; Xu, M. Strategies for Targeting Senescent Cells in Human Disease. Nat. Aging 2021, 1, 870–879. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally Occurring P16Ink4a-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of Senescent Cells by β-Galactosidase-Targeted Prodrug Attenuates Inflammation and Restores Physical Function in Aged Mice. Cell Res 2020, 30, 1–16. [Google Scholar] [CrossRef]
- Rovillain, E.; Mansfield, L.; Caetano, C.; Alvarez-Fernandez, M.; Caballero, O.L.; Medema, R.H.; Hummerich, H.; Jat, P.S. Activation of Nuclear Factor-Kappa B Signalling Promotes Cellular Senescence. Oncogene 2011, 30, 2356–2366. [Google Scholar] [CrossRef]
- Emery, P.; Keystone, E.; Tony, H.P.; Cantagrel, A.; Vollenhoven, R. van; Sanchez, A.; Alecock, E.; Lee, J.; Kremer, J. IL-6 Receptor Inhibition with Tocilizumab Improves Treatment Outcomes in Patients with Rheumatoid Arthritis Refractory to Anti-Tumour Necrosis Factor Biologicals: Results from a 24-Week Multicentre Randomised Placebo-Controlled Trial. Ann. Rheum. Dis. 2008, 67, 1516. [Google Scholar] [CrossRef] [PubMed]
- Rhee, F. van; Wong, R.S.; Munshi, N.; Rossi, J.-F.; Ke, X.-Y.; Fosså, A.; Simpson, D.; Capra, M.; Liu, T.; Hsieh, R.K.; et al. Siltuximab for Multicentric Castleman’s Disease: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol. 2014, 15, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Demaria, M. Therapeutic Interventions for Aging: The Case of Cellular Senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P.; Kino, T. Glucocorticoid Signaling in the Cell. Ann. N. York Acad. Sci. 2009, 1179, 153–166. [Google Scholar] [CrossRef]
- Stavri, S.; Trusca, V.G.; Simionescu, M.; Gafencu, A.V. Metformin Reduces the Endotoxin-Induced down-Regulation of Apolipoprotein E Gene Expression in Macrophages. Biochem. Biophys. Res. Commun. 2015, 461, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin Inhibits the Senescence-associated Secretory Phenotype by Interfering with IKK/NF-κB Activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Niehof, M.; Streetz, K.; Rakemann, T.; Bischoff, S.C.; Manns, M.P.; Horn, F.; Trautwein, C. Interleukin-6-Induced Tethering of STAT3 to the LAP/C/EBPβ Promoter Suggests a New Mechanism of Transcriptional Regulation by STAT3*. J. Biol. Chem. 2001, 276, 9016–9027. [Google Scholar] [CrossRef] [PubMed]
- Palsuledesai, C.C.; Distefano, M.D. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol. 2015, 10, 51–62. [Google Scholar] [CrossRef]
- Liu, S.; Uppal, H.; Demaria, M.; Desprez, P.-Y.; Campisi, J.; Kapahi, P. Simvastatin Suppresses Breast Cancer Cell Proliferation Induced by Senescent Cells. Sci. Rep. 2015, 5, 17895. [Google Scholar] [CrossRef]
- Rezaie-Majd, A.; Maca, T.; Bucek, R.A.; Valent, P.; Müller, M.R.; Husslein, P.; Kashanipour, A.; Minar, E.; Baghestanian, M. Simvastatin Reduces Expression of Cytokines Interleukin-6, Interleukin-8, and Monocyte Chemoattractant Protein-1 in Circulating Monocytes From Hypercholesterolemic Patients. Arter., Thromb., Vasc. Biol. 2002, 22, 1194–1199. [Google Scholar] [CrossRef]
- Sakoda, K.; Yamamoto, M.; Negishi, Y.; Liao, J.K.; Node, K.; Izumi, Y. Simvastatin Decreases IL-6 and IL-8 Production in Epithelial Cells. J. Dent. Res. 2006, 85, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; Willigenburg, H. van; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [PubMed]
- Keizer, P.L.J. de; Packer, L.M.; Szypowska, A.A.; Riedl-Polderman, P.E.; Broek, N.J.F. van den; Bruin, A. de; Dansen, T.B.; Marais, R.; Brenkman, A.B.; Burgering, B.M.T. Activation of Forkhead Box O Transcription Factors by Oncogenic BRAF Promotes P21cip1-Dependent Senescence. Cancer Res. 2010, 70, 8526–8536. [Google Scholar] [CrossRef] [PubMed]
- Boer, J. de; Donker, I.; Wit, J. de; Hoeijmakers, J.H.; Weeda, G. Disruption of the Mouse Xeroderma Pigmentosum Group D DNA Repair/Basal Transcription Gene Results in Preimplantation Lethality. Cancer Res. 1998, 58, 89–94. [Google Scholar]
- Guarente, L.; Sinclair, D.A.; Kroemer, G. Human Trials Exploring Anti-Aging Medicines. Cell Metab. 2024, 36, 354–376. [Google Scholar] [CrossRef]
- Khan, T.; Hussain, A.I.; Casilli, T.P.; Frayser, L.; Cho, M.; Williams, G.; McFall, D.; Forcelli, P.A. Prophylactic Senolytic Treatment in Aged Mice Reduces Seizure Severity and Improves Survival from Status Epilepticus. Aging Cell 2024, 23, e14239. [Google Scholar] [CrossRef]
- Guarente, L.; Sinclair, D.A.; Kroemer, G. Human Trials Exploring Anti-Aging Medicines. Cell Metab. 2024, 36, 354–376. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Inducers of Senescence. There are numerous types of senescence classifications with different inducers: Replicative Senescence can occur as cells continue to proliferate and create DNA sequence errors. DNA Damage-Induced Senescence is due to the activation of the DNA Damage Response (DDR) by DNA breaks (e. g. shortened telomeres) or excitotoxic injury in the CNS [81]. Tumor Suppressor Induced Senescence and Oncogene Induced Senescence occur when proteins like PTEN or KRAS are either lost or activated, respectively through mutations. Therapy-Induced Senescence is due to drug treatment with senescence-inducing drugs such as some anti-cancer drugs. Lastly, Developmental-Induced Senescence only occurs during embryogenesis and in certain regions of the growing embryo.
Figure 1.
Inducers of Senescence. There are numerous types of senescence classifications with different inducers: Replicative Senescence can occur as cells continue to proliferate and create DNA sequence errors. DNA Damage-Induced Senescence is due to the activation of the DNA Damage Response (DDR) by DNA breaks (e. g. shortened telomeres) or excitotoxic injury in the CNS [81]. Tumor Suppressor Induced Senescence and Oncogene Induced Senescence occur when proteins like PTEN or KRAS are either lost or activated, respectively through mutations. Therapy-Induced Senescence is due to drug treatment with senescence-inducing drugs such as some anti-cancer drugs. Lastly, Developmental-Induced Senescence only occurs during embryogenesis and in certain regions of the growing embryo.
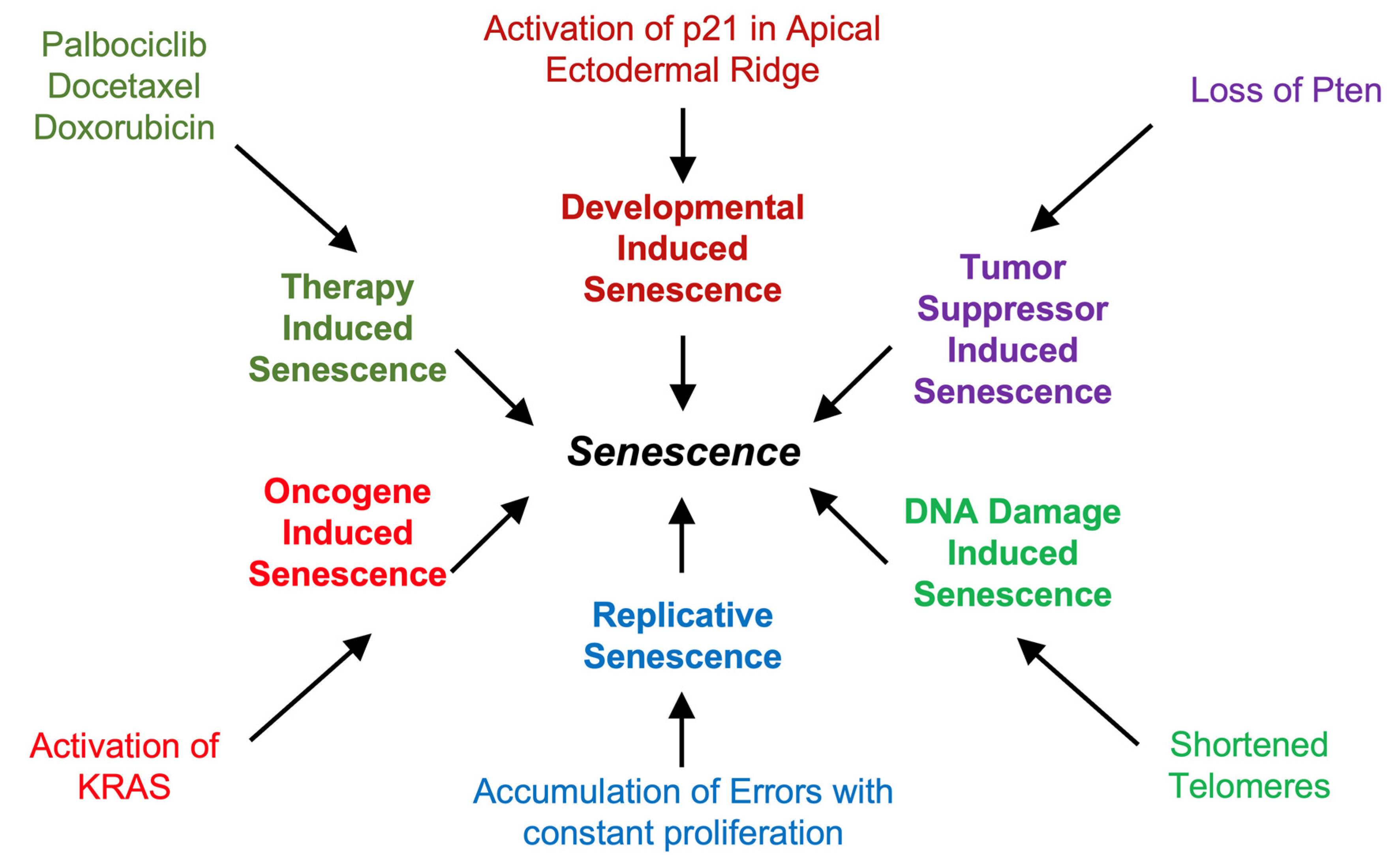
Figure 2.
Pathways and Proteins Controlling Senescence. The DDR promotes activation of p53 and p21Cip1 while activation of the INK4A/ARF locus leads to expression of p16ink4a and p14ARF which inhibit Cyclin kinases and Mdm2 respectively.
Figure 2.
Pathways and Proteins Controlling Senescence. The DDR promotes activation of p53 and p21Cip1 while activation of the INK4A/ARF locus leads to expression of p16ink4a and p14ARF which inhibit Cyclin kinases and Mdm2 respectively.
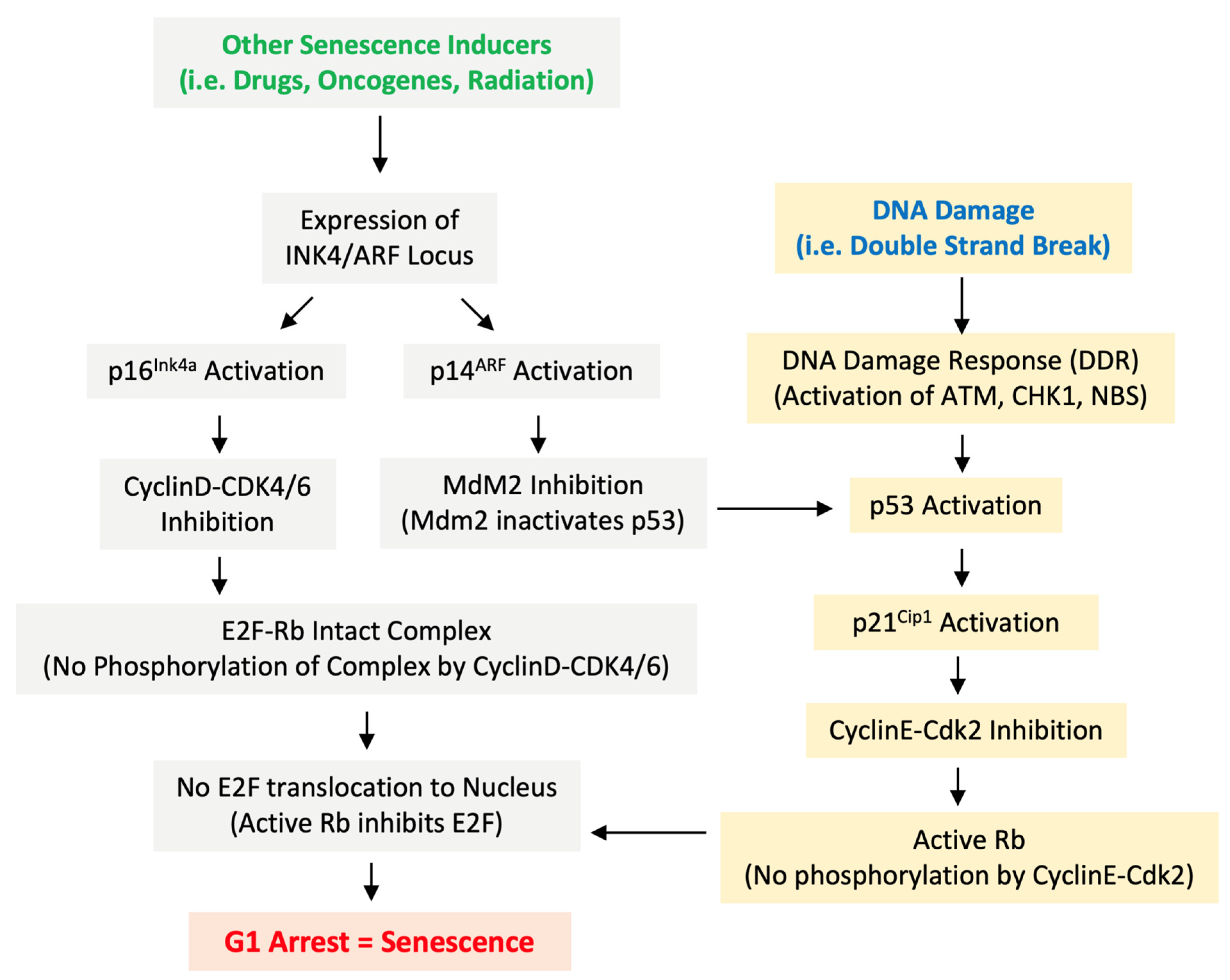
Figure 3.
Cellular changes during senescence. Recognizable features of senescent cells include SA-b-gal, chromatin changes (SAHF), nuclear lamina and envelope defects (Lamin B1 deficiency), secretion of SASP, DNA Damage markers (gamma-H2Ax) with an overall flattened and enlarged morphology.
Figure 3.
Cellular changes during senescence. Recognizable features of senescent cells include SA-b-gal, chromatin changes (SAHF), nuclear lamina and envelope defects (Lamin B1 deficiency), secretion of SASP, DNA Damage markers (gamma-H2Ax) with an overall flattened and enlarged morphology.
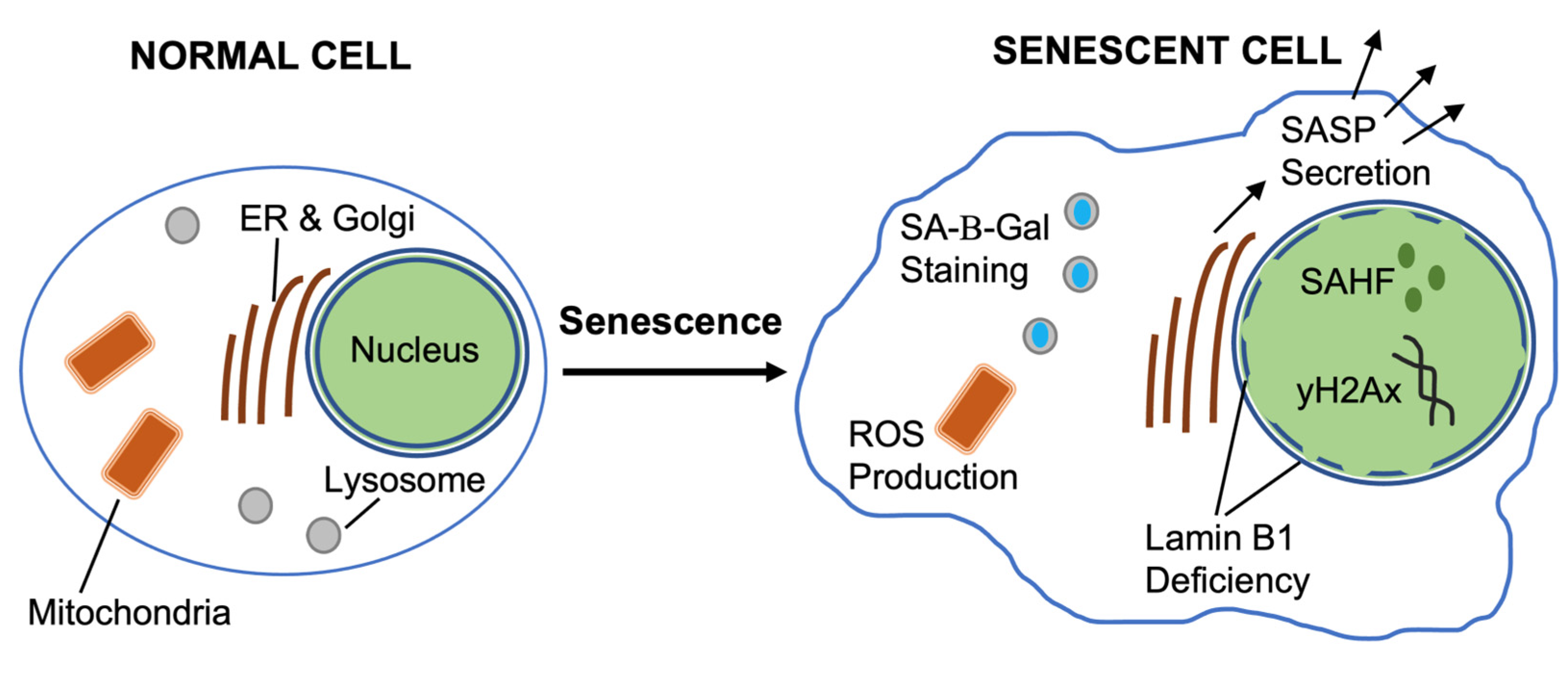
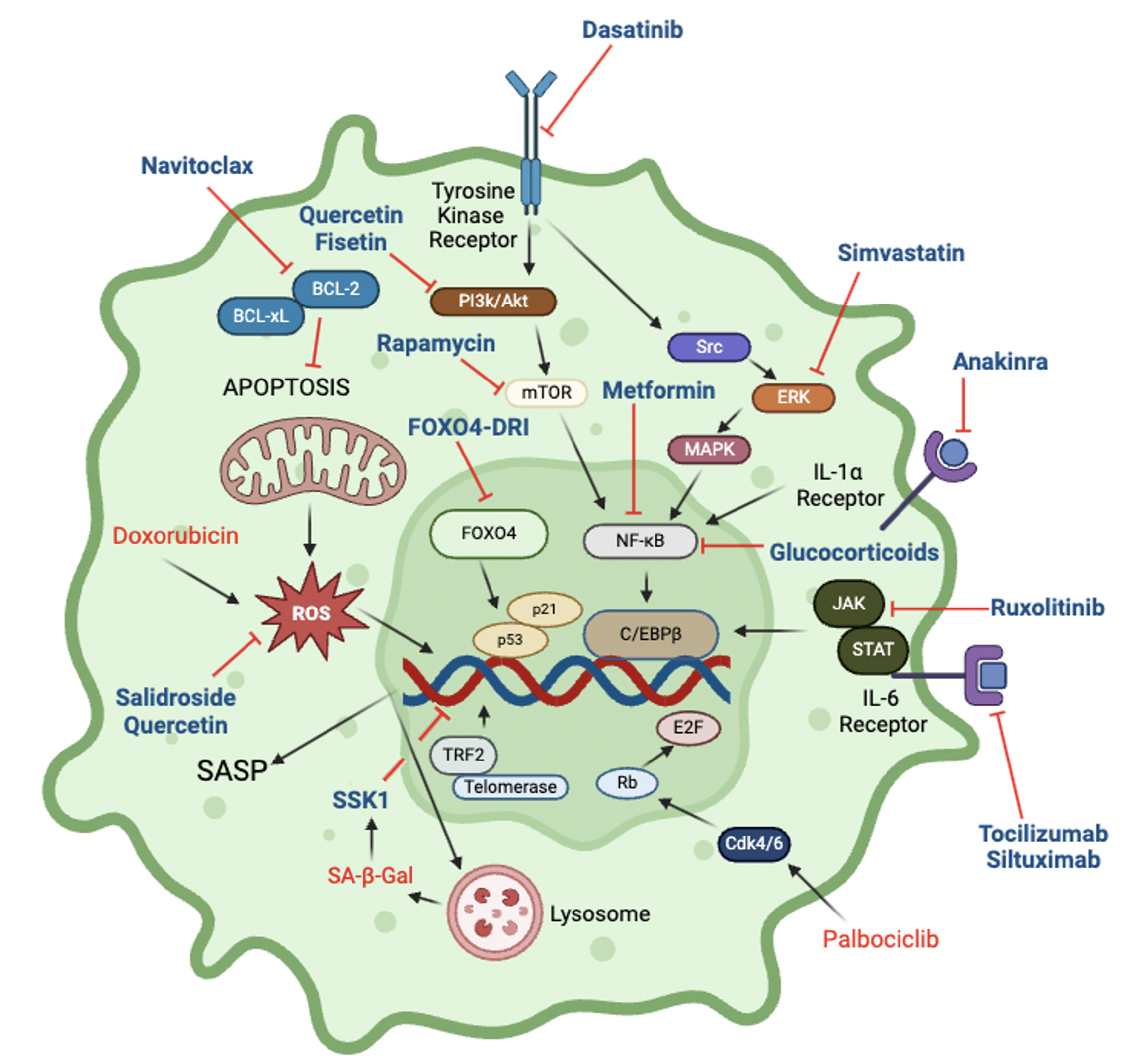
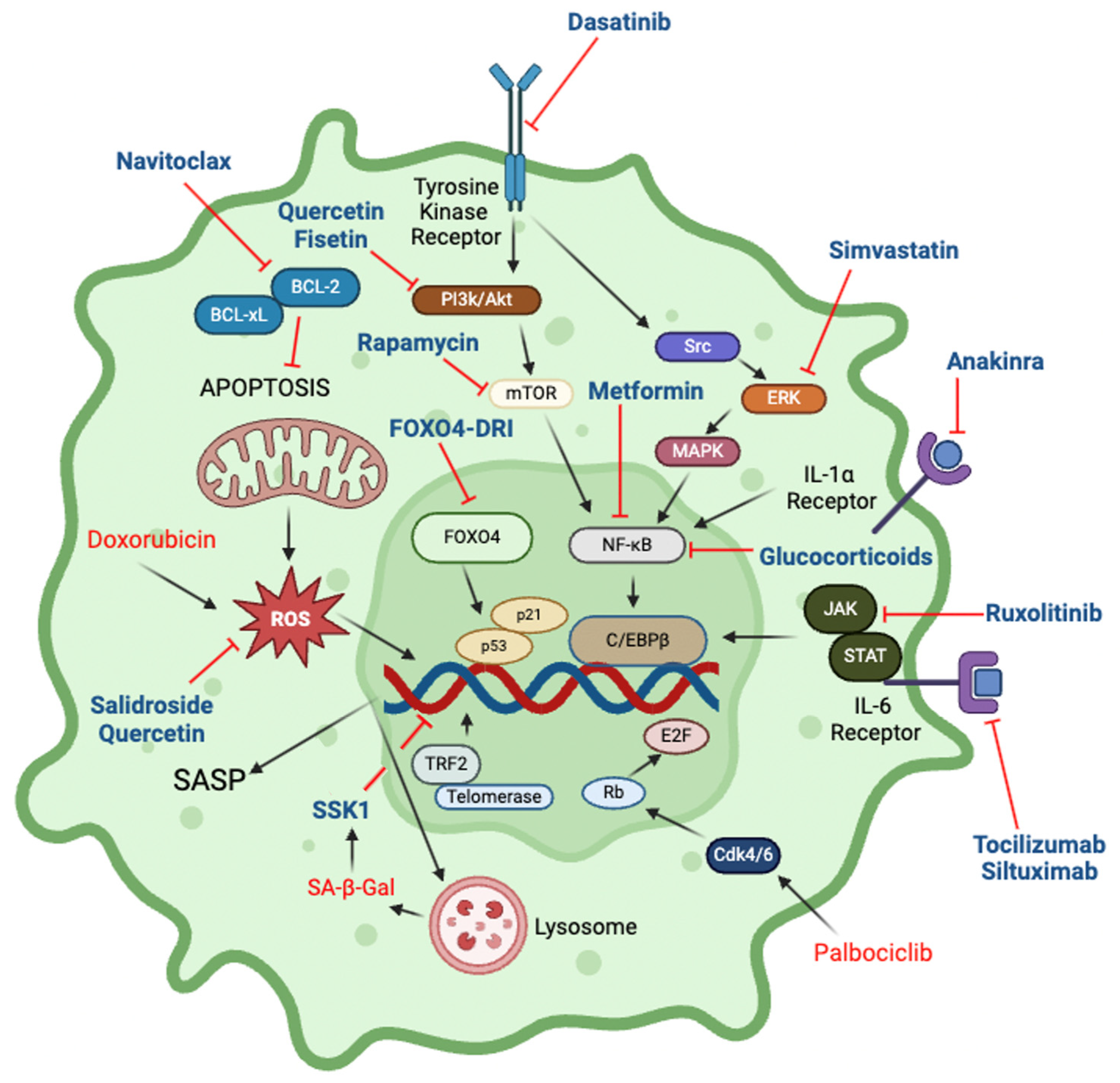
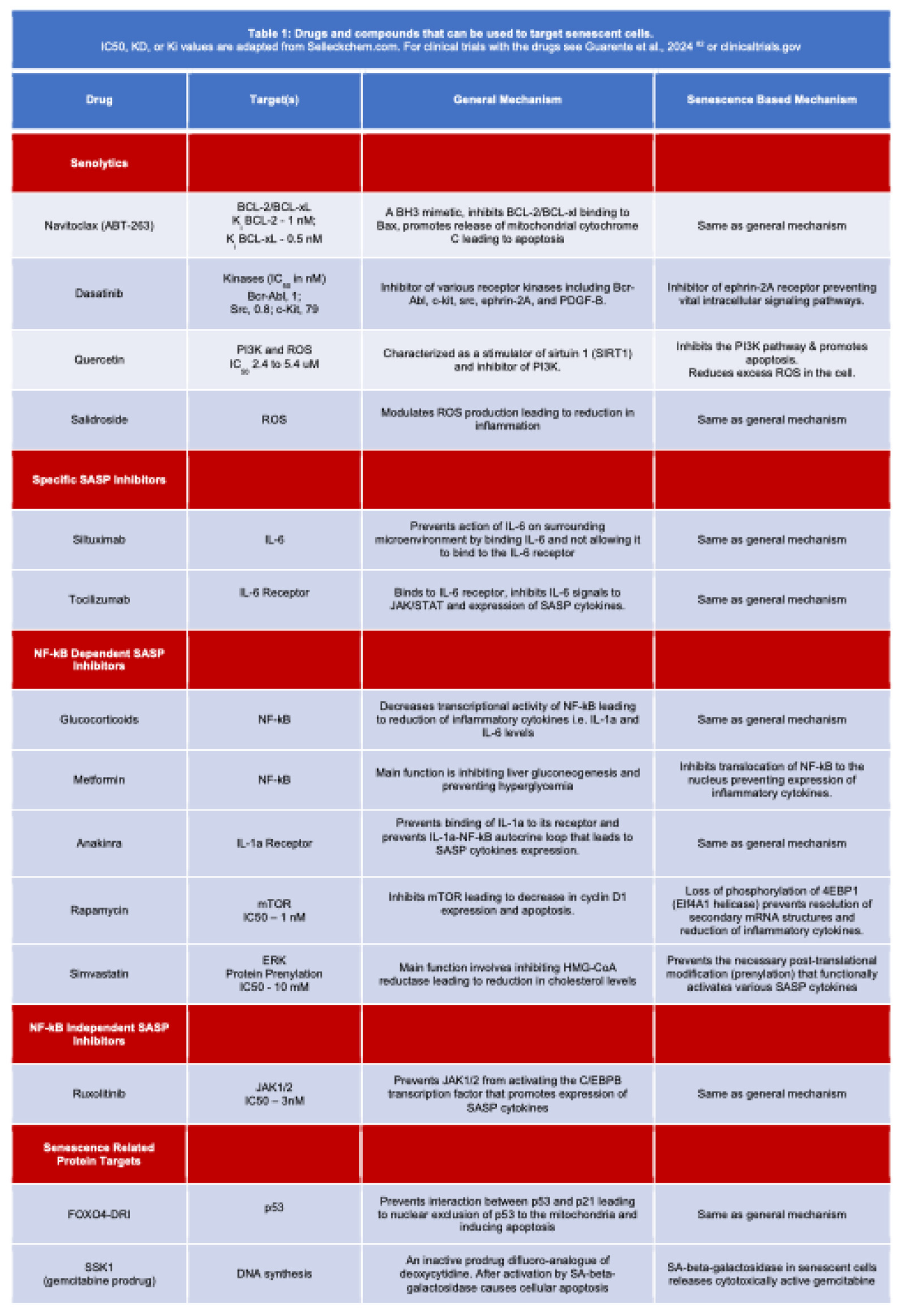
Figure 4.
Cellular pathways and drugs that impact senescence. Inhibitors are shown in blue font, inducers in red font. Further details are in Table 1.
Figure 4.
Cellular pathways and drugs that impact senescence. Inhibitors are shown in blue font, inducers in red font. Further details are in Table 1.

 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



