
Submitted:
26 January 2025
Posted:
27 January 2025
You are already at the latest version
Abstract
A non-linear (non-additive) increase in stability of hexamers follows an increase in the total number of (i) aad (a double proton acceptor) plus add (a double proton donor) waters commonly linked with anticooperativity and (ii) the total number of intermolecularly delocalized electrons (intermolNdeloc) in the 3D space occupied by a hexamer. Subsequently, we obtained nearly a perfect linear correlation between increase in the cluster stability and intermolNdeloc. Individual water molecules that act as either aad or add: (i) delocalize the largest number of electrons throughout a cluster; (ii) are involved in the strongest attractive, hence energy-stabilizing intermolecular interaction with the remaining five waters; (iii) have the most significant quantum component of the intermolecular interaction energy and (iv) relative to six non-interacting water molecules, stabilize a hexamer the most, as quantified by a purposely derived mol-FAMSEC energy term. Clearly, the all-body approach used in the unified, molecular-wide and electron density (MOWeD)-based concept of chemical bonding contradicts the commonly accepted view that aad and add water molecules are involved in anticooperativity in 3D water hexamers. Consequently, we propose here a general definition of cooperativity that should be applicable to any n-membered molecular clusters, namely: the quantifiable, physics- and quantum-based cooperativity phenomenon is synonymous with the intermolecular all-body delocalization of electrons leading to the increase in stability of individual molecules on an n-membered cluster formation.
Keywords:
cooperativity
; anticooperativity
; water clusters
; molecular-wide and electron density (MOWeD)-based approach
; the Fragment
; Atomic
; Localized
; Delocalized and Interatomic (FALDI)-based electron density decomposition scheme
; the Fragment Attributed Molecular System Energy Change (FAMSEC)-based method
1. Introduction
Water clusters and, in particular, non-additivity in strength and other properties of intermolecular H-bonds have been studied for many decades by exploring underlying processes coined as cooperativity [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25] and anticooperativity [26,27,28,29,30,31,32,33,34,35] between water molecules. Actually, interest in properties of water clusters continues to date due to importance of water in, e.g., supporting life, and a few very informative reviews were published on cooperativity in water clusters [9,10,17,20,36,37,38]. Considering anticooperativity, this concept was introduced well after cooperativity and it is always investigated and discussed in conjunction with cooperativity. Although it is not a subject of this contribution, it is important to stress that the concepts of cooperativity/anticooperativity are also explored in many different kinds of molecular clusters where individual molecules are involved in intermolecular H-bonding [29,39,40,41,42,43,44,45,46].
When water chains or cyclic homodromic structures are formed then all water molecules are of ad character, i.e., being arranged in the (proton acceptor, a)-(proton donor, d) configuration, ⋅⋅⋅O–H⋅⋅⋅O–H⋅⋅⋅O–H⋅⋅⋅. For the ad configurations of water molecules in clusters, non-additivity presents itself as mutual enhancement of H-bonds leading to the binding energy being greater than that computed for a dimer. The process of the enhancement of bonding (binding energy) was named ‘cooperativity’ between water molecules [1].
Ojamäe and Hermansson [20] have arranged four water molecules in the form of tetrahedron using dimers as building blocks. This resulted in the central water molecule acting as a double proton acceptor (aa) and a double proton donor (dd) meaning that the central water molecule in the tetrahedron had aadd functionality. Using OH frequencies computed for tetrahedron, they have concluded that the aadd configuration works in the opposite direction when compared with a single acceptor-single donor ad waters in chains or homodromic pentamer. Such non-additivity that diminishes bonding typically is described as “anticooperativity” but it is clear that they were not in favor of this nomenclature. Instead, they have proposed a concept of ‘strict cooperativity’ that, according to them, is indisputably observed for the energies and the frequencies in chains and homodromic water pentamer (in general, in cyclic homodromic water clusters). Finally, they have concluded that ‘Water molecules in a tetrahedral coordination display cooperativity, but not strict cooperativity’ [20]. Steiner stressed that, because hydrogen bonds may not only enhance, but also reduce the strengths of each other, a change of orientation of H-bonds from homodromic to antidromic arrangement leads to local anticooperativity [36].
Intuitively, the concept of cooperativity among water molecules can be understood as contributions made by 2-, 3-, (many-) body energy terms that enhance the strength of H-bonds in water clusters. Realizing that the same Physics and Quantum Chemical rules apply to all possible sizes and shapes of water clusters, it is difficult to explain the concept of anticooperativity. To this effect, how water molecules ‘decide’ on the mode of action, either enhancing or diminishing bonding using the same 2-, 3-, n-body energy terms? One must realize that neither cooperativity nor anticooperativity are well-defined quantum properties but rather are used to loosely describe ‘positive’ or ‘negative’ additivity of energy terms on a cluster formation.
Recently [47], using the all-body and molecular-wide and electron density (MOWeD)-based approach [48], we proposed a new and quantifiable concept of cooperativity. According to our recent interpretation, the origin of all-body cooperativity is rooted in physics- and quantum-based processes of electron (e) delocalization between water molecules. In other words, cooperativity is the intermolecular all-body e-delocalization leading to the non-additive increase in stability of a water molecule in clusters with an increase in their size.
Cooperativity is defined in Oxford dictionary as “involving doing something together or working together with others toward a shared aim. Cooperative activity is essential to effective community work.” Remarkably, the Oxford definition of cooperativity agrees very well with our definition of cooperativity among molecules, namely:
- (a)
- Intermolecular e-delocalization by water molecules in a cluster can be interpreted as equivalent to doing something together or working together with others from Oxford dictionary definition of cooperativity, and
- (b)
- Reaching the lowest e-energy of a water cluster through intermolecular e-delocalization (from our definition) compares well with Cooperative activity is essential to effective community work to achieve a shared aim as per Oxford dictionary definition.
On the other hand, Oxford dictionary does not provide a definition of anticooperativity. In other words, neither anti-cooperativity nor anticooperativity exist in this dictionary. This might imply that people and, more relevant to this work water molecules, cannot be involved in activity (a physical process) called anticooperativity. This supposition can be written as the following hypothesis that constitutes the focus of the present contribution.
Hypoyhesis-1.
From the fact that:
- 1. 3D 6-water clusters discussed in this work are more stable than 2D cyclic homodromic water hexamer and the latter is restricted to ad configurations showing only positive, non-additivity in the strength of H-bonds, i.e., classical cooperativity, and
- 2. Prism, being the most stable 3D 6-water hexamer, does not have water molecules of ad configurations (there are 3 aad and 3 add waters)
it would fallow that the double-acceptor (aad) and double-donor (add) water molecules must contribute to cluster stability more than ad configuration.
If the above hypothesis holds, then the anticooperativity cannot be supported and hence must be rejected. 3D 6-water clusters (bag, book, cage, and prism) are used here as model systems as they all:
- i.
- Are more stable than the 6-water cyclic hexamer; notably, they are the smallest 3D clusters that are more stable than their cyclic counterparts, and
- i.
- ii. Have water molecules with aad and add configurations and these motifs are observed in each of the ice polymorphs [2].
One must stress that the all-body MOWeD-based protocol treats all atoms as a constellation of nuclei that spontaneously drive electron density (ED) to the global minimum of electronic energy of a molecular system. This approach is in accord with Bader’s view that there are ‘only two forces operative in chemistry, the Feynman force exerted on the nuclei and the Ehrenfest force exerted on the electrons’ [49]
To probe the above hypothesis, we will make use of recently developed tools incorporated in the all-body MOWeD-based approach that will provide qualitative and quantitative interpretation of cooperativity/anticooperativity, namely:
- i.
- i.
- ii. The Fragment Attributed Molecular System Energy Change (FAMSEC) family of methods [53,54] is used to identify molecular fragments that either drive or obstruct a chemical change the most. FAMSEC also meets the all-body requirements as the quantified energy contributions made by fragments are harvested from entire space occupied by a system.
2. Computational Methods
All calculations were performed in Gaussian 09 Rev. D.01 [55] in the gas phase with a keyword ‘opt=verytight’ at the B3LYP level of theory with Grimme’s [56] empirical correction for dispersion using a keyword ‘empiricaldispersion=GD3’. The Dunning triple zeta basis set, aug-cc-pVTZ, which is augmented by diffuse functions, was used throughout. Coordinates of all optimized structures are given in the Supplementary Materials. Frequency calculations were performed for the optimized structures to verify that no imaginary frequency was present. Topological, QTAIM [57] molecular graphs, atomic overlap matrices and IQA [58,59] calculations were performed in AIMAll [60] using B3LYP-generated wavefunctions. The IQA energy terms, and interaction energies in particular, were found to be highly comparable to those obtained at the CCSD/BBC1 level [61]. FAMSEC and FALDI data were calculated using in-house software, and FALDI isosurfaces were visualized using VMD [62]. FALDI codes were incorporated in MOWeD-LAC (molecular-wide electron delocalization and localization atomic counts) and MOWeD-LFC (molecular-wide electron delocalization and localization fragment counts) applications; these two applications are made freely available [63].
3. Theoretical Background
3.1. The FALDI Density Decomposition Scheme
FALDI is a unique electron density decomposition scheme developed recently [50,51,52]. Usefulness and power of FALDI was already demonstrated in the study of numerous bonding and non-bonding interactions in a number of systems [48,50,51,52,64,65,66,67]. It is unique because one can compute, besides classical electron populations, the exact localization and exact delocalization electron counts that are quite different from localization and delocalization indices generated within the QTAIM formalism [57]. Another unique and very instructive feature of FALDI is its ability to generate 3D distributions of localization and delocalization patterns that can be visualized in real-space.
In this contribution, we will focus on useful aspects of FALDI that are directly applicable in the study of cooperativity and anticooperativity in 3D water clusters. Readers interested in full theoretical description of FALDI are referred to our recently published work [47] and references therein.
FALDI-based electron density (ED) decomposition scheme provides (i) atom–ED distribution, (ii) the contribution made by electrons localized to a selected atomic basin ΩA, i.e. loc–ED distribution, and (iii) the contribution made by electrons delocalized between all unique basin-pairs (ΩA,ΩB), i.e., deloc–ED distribution. All these distributions are additive and can be conveniently combined to form fragment F distributions by accounting for selected atomic basins’ contributions. From this follows that the total electron population of a k-atom fragment, e.g., 3-atom fragment of a molecular system, such as a water molecule W in a cluster, Ntotal(W), can be decomposed as:
where the fragment’s electron population is made of two components, namely electrons that can be found only in the space occupied by a water molecule W - this count is called a ‘self’-fragment electron population, Nself(W), and electrons delocalized with the remaining five water molecules in a cluster treated as a fragment R, Ndeloc(W,R). It is important to realize that in the case of multi-atom fragments, such as water molecule, the ‘self’ fragment electron population Nself(W) is made of two kinds of electrons: a) electrons localized to each atom A, Nloc(A,) of a water molecule treated as a fragment W. Hence, for all atom-localized electrons in a water molecule we can write Nloc(W) = ∑Nloc(A) where A∈W, and b) electrons delocalized amongst atoms of the fragment W due to intra-fragment delocalization processes taking place, Ndeloc(W,W). Notably, the two delocalization terms, Ndeloc(W,W) and Ndeloc(W,R), are fundamentally different as the latter represents inter-fragment electron delocalization and it counts electrons that can be found in both fragments, W and R. The term Ndeloc(W,R), is calculated by summing up only the contributions made by atoms within the k-atom (here 3-atom) fragment W to diatomic DIs involving all other atoms in the molecular system (here water cluster):
Ntotal(W) = Nself(W) + Ndeloc(W,R)
3D placement of water molecules in each cluster is unique meaning that each water molecule W founds itself in a different molecular environment. This, in turn, must have an impact on the number of electrons each W is delocalizing to the remaining waters in a cluster. Due to additivity of FALDI-computed terms, one can compute the total number of delocalized electrons in each cluster by summing up the Ndeloc(W,R) terms obtained for individual water molecules.
It might be very useful and informative to investigate electron delocalization patterns throughout a molecular system by computing delocalized electrons counts between molecular fragments. As an example, for the k-atom fragment F and l-atom fragment H we can write
where contributions made by individual atoms are accounted for, as DI(A,B) = ADI(A,B) + BDI(A,B). Notably, DI(F,H) = Ndeloc(F,H) + Ndeloc(H,F) and, in most cases, . This means that measures the degree to which electrons from the k-atom fragment are delocalized within the l-atom fragment . By analogy, similar applies to the Ndeloc(H,F) term. On the other hand, DI(F,H) measures the total electron count due to delocalization of electrons between atoms of both fragments, i.e., the count of electrons shared by the two fragments. Naturally, Eq. 3 is useful to compute the total number of electrons shared by any water-pair in a cluster.
3.2. A Fragment Attributed Molecular System Energy Change (FAMSEC) Protocol
Many molecules, e.g., polyamines, show an incredible flexibility and are able to re-arrange their constellations of nuclei and change the associated electron density distribution in 3D space spontaneously leading to a large number of structural conformers of the same molecule. This can be seen as an intramolecular event. Moreover, many molecules can re-arrange their relative placement in 3D space leading to a countless number of molecular clusters, such as water clusters. This can be seen as intermolecular event. However, what drives a change and what is the energy contribution made by specific atoms, molecular fragments, etc. when a molecular system undergoes a change from its initial (init) to a final (fin) state? From a need of understanding and quantifying changes taking place throughout a molecular system on a chemical event, the concept of FAMSEC [53] was born.
Complex chemical events take place along the reaction energy profile (REP). A dedicated REP-FAMSEC protocol was designed [54] to explore a reaction mechanism leading to new chemical products. The FAMSEC method [53] and its ‘derivatives’, namely REP-FAMSEC [54,68,69] and π-FARMS (Preorganized-Interacting (π) Fragments Attributed to Relative Molecular Stability [70,71]), pinpoint n-atom molecular fragments G (a molecule might also be treated as G) that drive/obstruct a chemical change the most and were also employed to investigate non-covalent interactions in a number of systems [72,73].
Many energy terms can be defined within the FAMSEC method and its derivatives that make use of the exhaustive energy partitioning schemes implemented in QTAIM [57] and IQA [58,59] where entire molecular space is occupied by atoms without voids between them. This assures that harvesting of data from all corners of a molecular system, as required by the MOWeD-based approach, can be met. Making use of the IQA-defined principle energy components of a molecular system, the mol-FAMSEC term was designed to quantify energy contribution made by any n-atom fragment G on a chemical event. To account for molecular-wide contributions made by all atoms of a molecular system, the mol-FAMSEC term is defined as:
The first two terms in Eq. (4) constitute the loc-FAMSEC = + term that quantifies energy change that is localized (loc) to the 3D space occupied by n-atoms of a selected molecular fragment G. This involves a) a change in self-atomic energies of atoms constituting G () and b) the change in all unique intra-fragment diatomic interaction energies, i.e. the term, on the transformation of a molecular system from the initial (init) to final (fin) state. The last term in Eq. (4), , accounts for changes in the strength of all diatomic interactions between atoms of G and all remaining atoms treated as a molecular fragment R. Finally, it is important to stress that all terms in Eq. (4) quantify a specific energy contribution to the electronic energy of a molecular system on the init → fin chemical event.
4. Results and Discussion
Water clusters used to study cooperativity/anticooperativity are shown in Figure 1. Each water molecule is considered as a 3-atom molecular fragment of a 6-membered water cluster named as Wn-mode where ‘mode’ stands for either ad, aad or add mode of action (functionality) a water molecule is involved in. Moreover, each 3-atom fragment representing a classical intermolecular H-bond is denoted as HB-n. All energy terms were computed using non-interacting separate water molecules that served as a reference state throughout the entire investigation.
4.1. Validation of B3LYP-Computed Relative Stabilities of Water Hexamers
3D density distribution varies with the level of theory to some degree; hence, somewhat different electronic energies (e-energies) are expected for each cluster. Our focus, however, is on trends in values of relative electronic energies of hexamers under investigation. As exemplified by the data in Table 1, the relative energies (ΔE = E(hexamer) – E(prism)) computed at a very high level of theory, namely CCSD(T)/CBS [74], compare very well with data obtained at somewhat lower levels, e.g., MP2(full)/aug-cc-pVTZ [75] and CCSD(T)/aug-cc-pVTZ [76].
Most importantly, however, ΔE values computed at the significantly lower level used in our studies, i.e., at the B3LYP/aug-cc-pVTZ level with the empirical GD3 Grimme’s dispersion correction, follow exactly the same trend as obtained at the higher levels – Figure 2.
One can also see that our ΔE values differ from those obtained at the higher levels by a small fraction of kcal/mol – see Table 1 and Figure 2. From that, we conclude that the FALDI- and FAMSEC-based analyses performed on optimized in this work hexamers should provide scientifically sound and highly reliable pictures and conclusions.
4.2. A Cooperativity-Driven Decrease in E(hexamer) Relative to E(6H2O)
Regardless of a chemical system considered, classical interactions and quantum effects involving all atoms govern a relative 3D placement of atoms. This means that the universal laws of Classical and Quantum Physics/Chemistry will spontaneously drive the placement of nuclei and associated distribution of ED that is characteristic for local/global electronic energy minima. Clearly, these are all-body processes leading to a unique constellations of atoms constituting a molecular system at each energy minimum on the potential energy hypersurface. Each constellation of nuclei generates a specific, like a fingerprint, 3D ED distribution from which all the properties of a molecular system can be derived as stressed already over a half of century ago by Hohenberg and Kohn [77], more recently by Bader [57] and in the very recent review by Koch et al [78].
From the above it follows that the electronic energy E of a molecular system and its fragments, like water molecules constituting a water cluster, as well as some components of E can be used to explore physical/quantum properties leading to or linked with cooperativity or anticooperativity using the MOWeD-based approach. Recently [47], we have postulated that a non-linear increase in the stability of a water molecule, i.e., a non-linear decrease in E of a statistical water molecule with an incremental increase in the number
of water molecules in homodromic rings can be used to quantify cooperativity phenomenon taking place in cyclic water clusters. It is evident that our definition of cooperativity phenomenon [47], i.e., ‘the intermolecular all-body e-delocalization leading to the non-additive increase in stability of a water molecule in clusters with an increase in their size’ cannot be applied directly to clusters studied here because all of them are 6-membered 3D structures. However, it can be re-written to a more general form, namely: the cooperativity phenomenon is synonymous with the intermolecular all-body e-delocalization leading to the increase in stability of water molecules on a cluster formation.
From the above it follows that an n-membered water cluster should have lower electronic energy when compared with separate (non-interacting) n water molecules. A trend in Figure 3 shows that (i) all hexamers have much lower energy than six non-interacting water molecules and (ii) all 3D hexamers are more stable than the cyclic homodromic ring. Moreover, a non-linear decrease in electronic energy reaches a minimum value for the prism and this strongly suggests that the cooperativity phenomenon is strongest in this hexamer.
It is accepted for decades that the double functionality of water molecules (aad and add) implies that they are involved in anticooperativity. A change in the electronic energy of a 6-membered water cluster with the total number of double acceptors and donors is shown in Figure 4. Remarkably, the trend seen in Figure 4 is a ‘copy’ of that seen in Figure 3 and it strongly suggests that the larger number of water molecules acting as double donor/double acceptor the more significant cooperativity among water molecules. The combined trends in Figure 3 and Figure 4 provide the first (i) support for our hypothesis-1 stated in the Introduction and (ii) evidence against the concept of anticooperativity as all water molecules in prism act as either double acceptor or double donor.
4.3. Quantifying Cooperativity
Considering our definition of cooperativity and looking at the data presented in Figure 3 and Figure 4 leads us to the second hypothesis that, if confirmed, would not only support our initial hypothesis but also would provide quantified explanation on how double acceptor/donor molecules stabilize clusters more than water molecules with ad functionality.
Hypothesis-2
Water molecules acting as double-donor (add) and double-acceptor (aad) must delocalize a larger number of electrons throughout a cluster than ad water molecules and this makes the 6-membered 3D clusters more stable than the homodromic cyclic hexamer.
To test our hypothesis-2, we make use of the FALDI-defined electron delocalization indices. To gain a full description without any presumptions made, all-body contributions made to the total final property of a system by all water molecules and each atom are accounted for. Recall that FALDI expands the concept of ED sharing to entire 3D space occupied by a molecular system, such as a water cluster, and delocalization indices can be computed for all atom-pairs, atoms as well as any molecular fragment.
It is important to realize that, due to very different distribution of water molecules in 3D hexamers, each water molecule finds itself in a unique environment that must have an impact on the number of delocalized electrons. To this effect, the bag and book clusters have one aad, one add and four ad water molecules. Even though the number of water molecules with a specific functionality is the same, we noted that the number of intermolecularly delocalized electrons intermolNdeloc computed for aad (2.188e) and add (2.075e) waters in bag is slightly larger than in the book hexamer (2.100 and 2.048e, respectively). Also, the number of delocalized electrons by aad configuration appeared to be slightly larger than that by the add configuration in the Cage hexamer.
To account for the impact of the molecular environment and realizing that we are dealing with rather a small sample of hexamers (a total number of 80 conformers was reported by Xantheas [19]), we decided to compute the average number of intermolNdeloc for each ad, aad and add functionality observed in 3D hexamers studied here. We found that, on average, ad, aad and add water molecules in the 3D hexamers delocalize 1.54±0.12, 2.00±0.18 and 1.98±0.12e, respectively. These results show that statistically:
- (i)
- The number of delocalized electrons by ad waters in 3D hexamers examined is the same as found for cyclic hexamer for which intermolNdeloc = 1.557±0.001e [47],
- (ii)
- The difference in intermolNdeloc between aad and add waters in 3D hexamers is insignificant and, the most importantly,
- (iii)
- There is a very significant difference in the number of delocalized electrons between ad and double-acceptor aad and double-donor add waters with ad delocalizing about 0.45e less.
Data seen in Figure 5 shows a non-linear, asymptotic increase in the total number of delocalized by all six water molecules electrons throughout a cluster, from the smallest value found in the cyclic 2D homodromic structure where only ad water molecules are present to the largest value computed for prism where there are no ad molecules. The impact of double functionality (aad/add) is instantly noticeable as a large increase, by over 1e, is observed already in bag and book where only two ad waters are replaced by a pair of aad and add molecules.
There is a striking ‘similarity’ between the trends seen in Figure 4 and Figure 5, where (i) in Figure 4 an asymptotic decrease in electronic energy of hexamers with the number of water molecules having aad plus add functionality is presented, and (ii) in Figure 5 where asymptotic increase in the number of the total intermolNdeloc with the number of waters having double functionality is seen. These asymptotic trends, having similar shape but going in the opposite directions, suggests that there should be a direct link between clusters’ stability and the total number of delocalized electrons throughout the cluster. Indeed, an excellent linear correlation is observed in Figure 6 showing that the least stable cyclic structure has the smallest number of e-delocalized and just opposite is observed for the lowest energy hexamer, prism. Clearly, aad and add waters cooperate more (delocalize more electrons) and hence stabilize clusters more (decrease their electronic energy more) and this is in a strong support of our two hypotheses.
We would like to conclude this section by stating that, from the MOWeD-based perspective, our findings contradict decades-old concept of the aad and add functionality being involved in the anticooperativity when electronic stabilization of entire water clusters goes. It is then reasonable to consider a significant increase in the total number of electrons delocalized by all water molecule throughout the space occupied by 3D clusters as a principal driver and quantifiable measure of cooperativity as we already stipulated for the 2D cyclic clusters [47].
Finally, one might argue that the total number of delocalized electrons does not provide an insight on individual water contributions with different functionalities. One might ask, for instance, if it is possible that, in a specific cluster, ad water molecule is delocalizing more electrons than either aad or add waters believed to be involved in anticooperativity? To address this, relevant data is presented in Table 2 where the number of electrons intermolecularly delocalized by each individual water molecule to the remaining five waters is presented – for brevity it is shown as Ndeloc in Table 2 where, for convenience, functionality of each water and its number (as in Figure 1) are also included.
The impact of an immediate environment on intermolNdeloc is immediately seen in Table 2. For instance, intermolNdeloc varies between 2.11 and 1.78e in reasonably symmetrical structure of prism where, in addition, there are no ad waters. A large spread of delocalized electrons by ad molecules is also seen for book (from 1.65 to 1.41e) and bag (from 1.72 to 1.41e) structures. Fundamentally important, however, is the finding that the aad and add water molecules in either cage, book, or bag delocalize significantly more electrons than ad molecules of these hexamers and this is in direct contrast to the common view of these waters being involved in anticooperativity and provide a quantified support for our hypothesis-2.
4.4. Quantifying Individual Water’s Contribution to the Cluster’s Stability
Classically, delocalizing electrons between two atoms is known to be the measure of covalent character of chemical bonding as defined by Lewis, e.g., two electrons shared (delocalized) between two neighboring atoms represents a single covalent bond. Because in Lewis interpretation of bonding one considers shared electrons between next to each other atoms, it implies that the process of covalent bonding is an intramolecular event. Actually, according to a classic dogma, atoms are bonded only when they are connected through covalent bonds (purely ionic interactions excluding) and, in addition, atoms constituting a molecule (they are covalently bonded) might be involved in either bonding or non-bonding interactions with atoms of the same molecule (intermolecular interactions) or atoms of another molecule (intermolecular interactions).
Our all-body MOWeD-based data presented here and previously for cyclic structures [47] reveal that a large number of electrons is delocalized among water molecules in clusters – see Figure 5 showing that over 11 electrons are delocalized intermolecularly in prism and this is about 2e more than we found in the cyclic hexamer. Furthermore, all the above results clearly demonstrate that gaining stability is synonymous with delocalizing electrons throughout a cluster. The intermolecular e-delocalization can then be seen as intermolecular bonding of a covalent character when Lewis concept is expended to entire molecular system.
We have estimated [47] that the energetic effect of intermolecular e-delocalization on the stability of homodromic cyclic water clusters, where ad functionality is exclusively observed, is about an order of magnitude smaller when compared with intramolecular e-delocalization on a classical covalent single bond formation. Considering the theme of this contribution, it is of upmost interest and importance to quantify the energy contributions made to molecular system’s stability by different functionality of water molecules. To achieve that, we made a use of the dedicated for that purpose FAMSEC protocol that is incorporated in the molecular-wide and electron density-(MOWeD)-based all-body approach. We have computed a number of energy terms describing contributions made by a water molecule toward all remaining five water molecules of the cluster. As the aad and add functionalities were always linked with anticooperativity (or negative cooperativity, or not strict cooperativity) data obtained for bag and cage hexamers will be discussed in some detail as they have waters with ad, aad and add functionalities. Recall that bag and book hexamers have exactly the same number of ad, aad and add water molecules (descriptors computed for bag and book are highly comparable) whereas prism does not have ad water molecules.
Data pertaining to bag hexamer is included in Table 3. Trends seen in Figures that follow were computed for cage hexamer as highly comparable indices were obtained for all 3D hexamers; relevant data obtained for book, cage and prism are placed in Table S1 in the Supplementary Materials. To ease interpretation, water molecules in Table 3 follow the order of their interaction energy with the remaining five waters, Eint(W,R), from the strongest attractive (most negative) to the least attractive. It is immediately seen in Table 3 that water molecules with aad and add functionalities are involved in the strongest interactions with remaining five waters in a cluster. Remarkably and regardless of functionality of a water molecule, intermolecular interactions are governed predominantly by the exchange-correlation (quantum) component VXC(W,R) that constitutes, on average, about 71.5±0.5% for 3D structures (69% for a cyclic hexamer) of the total intermolecular interaction energy, Eint(W,R).
By combining intermolecularly delocalized electrons intermolNdeloc (shown as Ndeloc in Table 2) with the VXC(W,R) component of the interaction energy included in Table 3 one obtains a reasonable correlation – an example generated for cage is shown in Figure 7. It shows that an increase in the number of delocalized electrons throughout a cluster is followed by more significant contribution of the XC-term (quantum component) to the interaction energy Eint(W,R). Furthermore and importantly, ad water molecules contribute the least in terms of delocalized electrons as well as contribution to stability of a cluster as measured by the always stabilizing in nature the VXC(W,R) component. This finding is in direct conflict with a commonly accepted and followed for decades concept of anticooperativity attributed to aad and add functionality of molecules.
The above has demonstrated importance of the VXC-term in understanding water functionality and its cooperativity. There are, however, other energy components impacting (in)stability of a system. The strongest evidence regarding individual water’s contribution to cluster’s (in)stability, i.e. (anti)cooperativity, should come from the purposely-derived energy term mol-FAMSEC. We found highly comparable trends between intermolNdeloc and VXC(W,R) as well as between intermolNdeloc and mol-FAMSEC - see Figure S1 in the Supplementary Materials prepared for cage as an example. This is an important discovery as it illustrates that there is a strong link between the total number of electrons delocalized by a water molecule throughout the space occupied by a 3D cluster (intermolNdeloc) and quantifiable measure of energy contribution to stability of a cluster made by a water molecule (mol-FAMSEC). Recall that the intermolNdeloc term is interpreted here as a principal driver and quantifiable measure of cooperativity. Furthermore, data seen in Figure S1 in the Supplementary Materials suggests that there should be another important and nearly linear correlation between the quantum component of the interaction energy a water molecule is involved in with remaining waters of a cluster VXC(W,R) and mol-FAMSEC. Figure S2 in the Supplementary Materials shows that indeed such a linear correlation exists in the cage hexamer. Actually, we have established that high quality linear correlation between VXC(W,R) and mol-FAMSEC exist for all 3D hexamers discussed in this work In each case, the most significant mol-FAMSEC term is observed for water molecules with aad and add functionality in a specific cluster and this strongly suggests that they are not involved in anticooperativity.
Data presented in Figure 8, where nearly perfect correlation between mol-FAMSEC and the interaction energy Eint(W,R) computed for individual water molecules is plotted for cage (red circles) and bag (violet crosses), is also observed for remaining 3D water hexamers discussed in this work (recall that R stands for the remaining water molecules in a cluster). This means that the combined data from all 3D hexamers follow a single linear trend that might be used as a predictive tool of either mol-FAMSEC or Eint(W,R) – see Figure S3 in the Supplementary Materials.
To fully understand the trend seen in Figure 8, it is fundamentally important to link it with trends and expressions discussed above. According to our definition of cooperativity, this is the Physics and Quantum based quantifiable intermolecular electron delocalization that drives the formation of water clusters (clusters of molecules in general) and it leads to the non-additive decrease in their electronic energy on a n-membered cluster formation. Data in Figure 7 shows a good non-linear correlation between the driving force of cooperativity, i.e., the number of intermolecularly delocalized electrons and quantum contribution to the interaction energy VXC(W,R). In the IQA-defined energy partitioning scheme the interaction energy between any pair of atoms A and B is made of two principle components, namely the classic Coulomb term, Vcl(A,B) and the XC-term (commonly interpreted as covalent or quantum contribution) VXC(A,B). As stated above, the contribution of the VXC-term to the interaction energy is nearly constant (about 71%) meaning that the %-fraction of the classic Coulomb contribution to the interaction energy made by individual water molecule is also constant and much smaller. According to classical orbital-based interpretations the intermolecular interactions, such as e.g., intermolecular H-bonds, are dominated by a classical term. One must stress here that the individual diatomic classic Coulomb intermolecular interaction energy terms Vcl(A,B) in water clusters vary between large positive (repulsive) and large negative (attractive) values. However, their opposite in sign contributions largely cancel each other whereas always negative VXC(A,B) terms sum up to a significant total value of VXC(W,R).
As shown in section 3.2., the mol-FAMSEC term is made of two major contributions, namely loc-FAMSEC and the interaction energy of a selected fragment with all the remaining atoms of a molecular system, which is equivalent, in our case, to the Eint(W,R) term. From this and the relationship seen in Figure 8 it follows that the loc-FAMSEC term contributes nearly constant %-fraction to the mol-FAMSEC energy term computed for individual water molecules in all 3D water clusters. This fully explains the linear relationship observed in Figure 8 and Figure S2 in the Supplementary Materials.
The above undisputedly illustrates that indeed this is not the ad mode of action of water molecules but rather aad and add that stabilize a cluster most. Actually, we have shown that regardless of the physical/quantum property or energy component considered, regardless of the number of ad, aad and add waters in each cluster, the aad and add functionality always provides the most significant contribution within a specific cluster. This does not mean, however, that ad waters always provide smaller contributions relative to that made by aad and add molecules. This is clearly seen in Figure 8 where waters W2 and W5 (both ad) of bag hexamer provide more significant contribution to bag’s stability than energy contributions made to cage by its waters W4-add and W6-aad. This nicely illustrates the overall complexity of clusters’ formation and the impact made by the immediate as well distant neighbors, the relative orientation of water molecules, distances between O-atoms and many more cooperativity-induced effects, such as VXC(W,R), Eint(W,R) and mol-FAMSEC energy terms on the role played by a single water molecule.
4.5. Quantifying Individual Intermolecular H-bonds’ Contribution to the Cluster’s Stability
Considering all the above, it should be then easy to comprehend that three-way interacting aad and add molecules will show a significant spread in the computed descriptors of any kind along each H-bonded link. From the all-body MOWeD approach we conclude that the differences in values observed for the computed indices along each H-bond have nothing to do with anticooperativity as universal and quantifiable quantum and physics-based processes apply throughout all the 3D space occupied by a cluster. Clearly, local environment determines, to some degree, the effectiveness of cooperativity between all water molecules along all possible H-bonded links that provide a unique, ‘privileged’ and the most effective mode of transport for electrons delocalized predominantly by O-atoms throughout a molecular system as documented previously for cyclic structures [47] and here for 3D hexamers discussed – example for bag is shown in Figure S4 in the Supplementary Materials.
Notwithstanding the above and since it is accepted for decades that hydrogen bonds may mutually either enhance their strength when they are involved in cooperativity or reduce their strengths when involved in anticooperativity, we decided to analyze the intermolecular H-bonds in all clusters examined. We decided to make use of energy contributions made to a cluster by H-bonds using a dedicated for this purpose the mol-FAMSEC energy term. One must recall that the stronger a H-bond is the more significant energy-stabilizing contribution to a molecular system it provides. When making use of a classical approach that is focused on individual H-bonds, it is apparent that the values of mol-FAMSEC seen in Table 4 do not follow an obvious pattern that might be used to support a concept of anticooperativity.
A large spread in values of the mol-FAMSEC term is seen in Table 4, e.g. between about –110 and –168 kcal/mol in book. As one would expect, most values computed for the more stable 3D clusters are more significant than that of –115 kcal/mol obtained for a homodromic cyclic hexamer. However, the mol-FAMSEC terms obtained for HB-7 in cage and book are less significant.
Focusing on three H-bonds of the most comparable W1 water molecule in all 3D clusters (it has the same aad functionality throughout), they show different distribution of mol-FAMSEC values in each cluster. This is due to, in our view, different overall molecular environment and we see in Table 4 that mol-FAMSEC: (i) gradually becomes more negative in prism and cage, (ii) has much more negative value for HB-2 in book hexamer whereas comparable and less significant mol-FAMSEC terms are seen for HB-1 and HB-3, and (iii) highly comparable values for all three H-bonds we found in bag cluster. Variation in mol-FAMSEC values seen in Table 4 for W1 water cannot be linked with anticooperativity and any selection made would be highly arbitrary and would have to be supported
A very different and consistent picture immerged when the impact of molecular environment was minimized by making use of the averaged values of the mol-FAMSEC terms seen in Table 4. Based on chemists’ accumulated experience and knowledge, the larger number of intermolecular hydrogen bonds the more significant contribution to the water cluster’s stability is expected and this is exactly what one can see in Figure 9. Relative to non-interacting six water molecules, prism is the most stable and it forms the largest number (nine) of intermolecular hydrogen bonds. Furthermore and importantly, on average, the stabilizing contribution made by a H-bond (as measured by the mol-FAMSEC term) increases from cyclic to prism structure.
Finally, recall that according to our understanding and definition of the cooperativity phenomenon, it is synonymous with the intermolecular all-body e-delocalization leading to the increase in stability of water molecules on a cluster formation. Analysis of data in Figure 9 leads to another supporting observation, namely the largest number of intermolecularly delocalized electrons found for prism (see Figure 6) can be attributed to the largest number of density bridges (DBs) linking water molecules due to the formation of classical intermolecular H-bonds. It becomes then clear that this is not the single DB but the network of DBs that facilitates most efficient e-delocalization that, in turn, determines stability of a cluster.
5. Conclusions
The proposed here general definition of the cooperativity phenomenon is a straightforward extension of our definition proposed for homodromic cyclic clusters of water [47]. The expanded definition should be applicable to any molecular cluster as it states that ‘the quantifiable, physics- and quantum-based cooperativity phenomenon is synonymous with the intermolecular all-body delocalization of electrons leading to the increase in stability of individual molecules on an n-membered cluster formation. From this follows that non-additive decrease in electronic energy of clusters, from the least stable homodromic cyclic hexamer to most stable prism is a direct result of a non-linear increase in the total number of intermolecularly delocalized electrons throughout a 3D space occupied by a hexamer, intermolNdeloc. Water molecules in 3D hexamers studied in this work can exhibit three functionalities, namely they can act as a proton acceptor and proton donor (ad), a double proton acceptor and a proton donor (aad) and a single proton acceptor and a double proton donor (add). Waters with a double functionality (i.e., aad and add) are typically associated with anticooperativity (or negative cooperativity, or not strict cooperativity). Remarkably, prism that is the most stable hexamer among all possible 3D configurations of six water molecules (about 80) has all six water molecules involved in anticooperativity if one accepts the decades long interpretations. So how is that possible that prism is the most stable among them all? Our definition of cooperativity fully explains these two contradicting observations. We showed that all aad and add waters outperform ad molecules when the number of intermolecularly delocalized electrons is accounted for. This means that in all 3D hexamers studied, individual ‘unticooperators’, aad and add water molecules, delocalize significantly more electrons than always ‘cooperating’ ad waters. Hence, one is left with only one possible conclusion that the aad and add water molecules in 3D hexamers are the best ‘cooperators’ and this excludes them as being involved in anticooperativity, a term not defined in proper English (Oxford dictionary). It would be of interest and paramount importance to establish if our finding is equally applicable to all remaining 70+ 3D hexamers. This would be a mammoth job but, due to its fundamental importance, we intend to embark on that challenge and hope to report our results soon.
In our opinion, the intermolNdeloc term is a universal quantifiable measure of cooperativity phenomenon. It can be applied to entire cluster (then it stands for the total number of electrons delocalized by all molecules of a cluster), or individual molecules within a cluster (both these options were explored in this work), or even to examine cooperativity/anticooperativity of a k-membered molecular fragment within a n-membered cluster (k < n).
One must stress that the physics- and quantum-based processes of e-delocalization lead to changes in properties of all imaginable properties of a molecular system, such as a water cluster. It is apparent that some of cooperativity-induced changes in local properties of a cluster were interpreted as anticooperativity. We investigated a few cooperativity-induced properties but from the molecular-wide and electron density (MOWeD) perspective we established that the changes in these properties do not corroborate with commonly accepted concept of anticooperativity. Our main focus was on contributions made to cooperativity-induced properties by individual water molecules within a 3D clusters examined. Contrary to common interpretations, the aad and add molecules have not shown any signature of anticooperativity as they always contributed the most to a cluster stability regardless of a descriptor investigated. Actually, we discovered an excellent linear relationship between mol-FAMSEC (a dedicated energy term to quantify the energy contribution made by a selected molecular fragment to the stability of a molecular system) and the intermolecular interaction energy a fragment (here a water molecule) is involved in with the remaining atoms of a system (here a cluster). Remarkably and considering each cluster separately, all water molecules with the aad and add functionalities outperformed the classically accepted ‘cooperators’, i.e., the ad water molecules, by stabilizing a cluster the most due to most significant mol-FAMSEC and other terms computed.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. The Supporting Information is available free of charge as Cartesian XYZ coordinates of 3D water clusters; energies of 3D clusters; the total interaction energy and its exchange-correlation term between a water molecule and remaining waters in clusters and mol-FAMSEC energy term computed for each water molecule in book, cage and prism; Figures showing trends between selected descriptors computed for individual water molecules in clusters and visual presentation of FALDI 3D density distributions.
Author Contributions
Conceptualization, I.C.; Methodology, I.C. and S.Z.; Software, S.H.; Validation, S.Z. and I.C.; Formal analysis, S.Z. and I.C.; Investigation, S.Z., S.H. and I.C.; Resources, I.C.; Data curation, I.C.; Writing—original draft preparation, I.C.; Writing—review and editing, S.Z. and I.C.; Visualization, I.C. and S.Z.; Supervision, I.C.; Project administration, I.C.; Funding acquisition, I.C. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
On request, computational data are available from I.C.
Acknowledgments
The authors gratefully acknowledge the Centre for High Performance Computing (CHPC), South Africa, for providing computational resources to this research project.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- H. S. Frank, W-Y. Wen, III. Ion-Solvent Interaction. Structural Aspects of Ion-Solvent Interaction in Aaqueous Solutions: A Suggested Picture of Water Structure. Discuss. Faraday Soc. 1957, 24, 133–140. [Google Scholar] [CrossRef]
- D. Hankins, J. W. Moskowitz, F. H. Stillinger, Water Molecule Interactions. J. Chem. Phys. 1970, 53, 4544–4554. [Google Scholar] [CrossRef]
- J. C. White, E. R. Davidson, An Analysis of the Hydrogen Bond in Ice. J. Chem. Phys. 1990, 93, 8029–8035. [Google Scholar] [CrossRef]
- S. S. Xantheas, T. H. Dunning, Jr. The Structure of the Water Trimer From ab Initio Calculations. J. Chem. Phys. 1993, 98, 8037–8040. [Google Scholar] [CrossRef]
- S. S. Xantheas, T. H. Dunning, Jr. Ab Initio Studies of Cyclic Water Clusters (H2O)n, n = 1–6. I. Optimal Structures and Vibrational Spectra. J. Chem. Phys. 1993, 99, 8774–8792. [Google Scholar] [CrossRef]
- S. S. Xantheas Ab Initio Studies of Cyclic Water Clusters (H2O)n, n = 1–6. II. Analysis of Many-Body Interactions. J. Chem. Phys. 1994, 100, 7523–7534. [Google Scholar] [CrossRef]
- W. A.P. Luck, The importance of cooperativity for the properties of liquid water. J. Mol. Struct. 1998, 448, 131–14. [Google Scholar] [CrossRef]
- J. M. Ugalde, I. Alkorta and J. Elguero, Water Clusters: Towards an Understanding Based on First Principles of Their Static and Dynamic Properties. Angew. Chem. Int. Ed., 2000, 39, 717–721. [Google Scholar] [CrossRef]
- R. Ludwig, Water: From Clusters to the Bulk. Angew. Chem. Int. Ed. 2001, 40, 1808–1827. [Google Scholar] [CrossRef]
- F. N. Keutsch, J. D. Cruzan and R. J. Saykally, The Water Trimer. Chem. Rev., 2003, 103, 2533–2577. [Google Scholar]
- K. Ohno, M. Okimura, N. Akaib, Y. Katsumotoa, The effect of cooperative hydrogen bonding on the OH stretching-band shift for water clusters studied by matrix-isolation infrared spectroscopy and density functional theory. Phys. Chem. Chem. Phys. 2005, 7, 3005–3014. [Google Scholar] [CrossRef] [PubMed]
- Y. I. Neela, A. S. Y. I. Neela, A. S. Mahadevi, G. N. Sastry, Hydrogen Bonding in Water Clusters and Their Ionized Counterparts. J. Phys. Chem. B 2010, 114, 17162–17171. [Google Scholar] [CrossRef] [PubMed]
- C. Pérez, M. T. Muckle, D. P. Zaleski, N. A. Seifert, B. Temelso, G. C. Shields, Z. Kisiel, B. H. Pate, Structures of Cage, Prism, and Book Isomers of Water Hexamer from Broadband Rotational Spectroscopy. Science, 2012, 336, 897–901. [Google Scholar] [CrossRef] [PubMed]
- L. Albrecht and R. J. Boyd, Visualizing Internal Stabilization in Weakly Bound Systems Using Atomic Energies: Hydrogen Bonding in Small Water Clusters. J. Phys. Chem. A, 2012, 116, 3946–3951. [Google Scholar] [CrossRef]
- J. M. Guevara-Vela, R. Chávez-Calvillo, M. García-Revilla, J. Hernández-Trujillo, O. Christiansen, E. Francisco, Á. Martín Pendás, and T. Rocha-Rinza, Hydrogen-Bond Cooperative Effects in Small Cyclic Water Clusters as Revealed by the Interacting Quantum Atoms Approach. Chem. Eur. J., 2013, 19, 14304–14315. [Google Scholar]
- L. Albrecht, S. Chowdhury, R. J. Boyd, Hydrogen Bond Cooperativity in Water Hexamers: Atomic Energy Perspective of Local Stabilities. J. Phys. Chem. A, 2013, 117, 10790–10799. [Google Scholar] [CrossRef] [PubMed]
- J. C. Howard and G. S. Tschumper, WIREs Comput. Mol. Sci. Wavefunction Methods for the Accurate Characterization of Water Clusters. 2014, 4, 199–224.
- B. D. Marshall, A Second Order Thermodynamic Perturbation Theory for Hydrogen Bond Cooperativity in Water. J. Chem. Phys. 2017, 146, 174104. [Google Scholar] [CrossRef]
- A. Rakshit, P. Bandyopadhyay, J. P. Heindel and S. S. Xantheas, Atlas of Putative Minima and Low-Lying Energy Networks of Water Clusters n = 3–25. J. Chem. Phys. 2019, 151, 214307. [Google Scholar] [CrossRef] [PubMed]
- Simon, M. Rapacioli, E. Michoulier, L. Zheng, K. Korchagina and J. Cuny, Contribution of the Density-Functional-Based Tight-Binding Scheme to the Description of Water Clusters: Methods, Applications and Extension to Bulk Systems. Mol. Sim. 2019, 45, 249–268. [Google Scholar] [CrossRef]
- M. B. Ahirwar, S. R. Gadre and M. M. Deshmukh, Direct and Reliable Method for Estimating the Hydrogen Bond Energies and Cooperativity in Water Clusters, Wn, n = 3 to 8. J. Phys. Chem. A, 2020, 124, 6699–6706. [Google Scholar] [CrossRef] [PubMed]
- J. A. Bilbrey, J. P. Heindel, M. Schram, P. Bandyopadhyay, S. S. Xantheas, S. Choudhury, A Look Inside the Black Box: Using Graph Theoretical Descriptors to Interpret a Continuous-Filter Convolutional Neural Network (CF-CNN) Trained on the Global and Local Minimum Energy Structures of Neutral Water Clusters. J. Chem. Phys. 2020, 153, 024302. [Google Scholar]
- D. Ben-Amotz, Electric Buzz in a Glass of Pure Water. Hydrogen Bond Charge Transfer in Water May Have Far-Reaching Chemical Iimplications. Science, 2022, 376, 800–801. [Google Scholar]
- L. E. Seijas, C. H. Zambrano, R. Almeida, J. Alí-Torres, L. Rincón, F. J. Torres, Exploring the Non-Covalent Bonding in Water Clusters. Int. J. Mol. Sci. 2023, 24, 5271. [Google Scholar] [CrossRef] [PubMed]
- G. D. Santis, K. M. Herman, J. P. Heindel, S. S. Xantheas, Descriptors of Water Aggregation. J. Chem. Phys., 2024, 160, 054306. [Google Scholar]
- L. Ojamäe, K. Hermansson, Ab Initio Study of Cooperativity in Water Chains: Binding Energies and Anharmonic Frequencies. J. Phys. Chem. 1994, 98, 4271–4282. [Google Scholar] [CrossRef]
- W. A. P. Luck, D. Klein, K. Rangsriwatananon, Anti-Cooperativity of the Two Water OH Groups. J. Mol. Struct. 1997, 416, 287–296. [Google Scholar] [CrossRef]
- M. Huš, T. Urbic, Strength of Hydrogen Bonds of Water Depends on Local Environment. J. Chem. Phys. 2012, 136, 144305. [Google Scholar] [CrossRef]
- L. Albrecht, R. J. Boyd, Atomic energy analysis of cooperativity, anti-cooperativity, and non-cooperativity in small clusters of methanol, water, and formaldehyde. Comput. Theoret. Chem. 2015, 1053, 328–336. [Google Scholar] [CrossRef]
- S. Saha, G. N. Sastry, Quantifying cooperativity in water clusters: an attempt towards obtaining a generalised equation. Mol. Phys. 2015, 113, 3031–3041. [Google Scholar] [CrossRef]
- J.M. Guevara-Vela, E. Romero-Montalvo, V. A. M. Gómez, R. Chávez-Calvillo, M. García-Revilla, E. Francisco, Á. Martín Pendás, and T. Rocha-Rinza, Hydrogen Bond Cooperativity and Anticooperativity Within the Water Hexamer. Phys. Chem. Chem. Phys. 2016, 18, 19557–19566.
- A. S. Mahadevi and G. N. Sastry, Cooperativity in Noncovalent Interactions. Chem. Rev., 2016, 116, 2775–2825. [Google Scholar] [CrossRef]
- P. L. Silvestrelli, Hydrogen bonding characterization in water and small molecules. J. Chem. Phys. 2017, 146, 244315. [Google Scholar] [CrossRef]
- V. M. Castor-Villegas, J. M. Guevara-Vela, W. E. Vallejo Narváez, Á. Martín Pendás, T. Rocha-Rinza, A. Fernández-Alarcón, On the Strength of Hydrogen Bonding Within Water Clusters on the Coordination Limit. J. Comput. Chem. 2020, 41, 2266–2277. [Google Scholar]
- A. Sauza-de la Vega, T. Rocha-Rinza, J. M. Guevara-Vela, Cooperativity and Anticooperativity in Ion-Water Interactions: Implications for the Aqueous Solvation of Ions. Chem. Phys. Chem. 2021, 22, 1269–1285. [Google Scholar] [CrossRef] [PubMed]
- T. Steiner, The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- M. Hakala, K. Nygård, S. Manninen, L. G. M. Pettersson, K. Hämäläinen, Intra- and Intermolecular Effects in the Compton Profile of Water. Phys. Rev. B. 2006, 73, 035432. [CrossRef]
- Ignatov, O. Mosin, Mathematical Models Describing Water Clusters as Interaction Among Water Molecules. Distributions of Energies of Hydrogen Bonds. J. Med. Physiol. Biophys. 2014, 3, 48–70. [Google Scholar]
- M. Weimann, M. Fárník, M. A. Suhm, M. E. Alikhani, J. Sadlej, Cooperative and Anticooperative Mixed Trimers of HCl and Methanol. J Mol. Struct. 2006, 790, 18–26. [Google Scholar] [CrossRef]
- A. Zabardasti, A. Kakanejadi, F. Ghenaatian, Z. Bigleri, A Theoretical Study of Cooperative and Anticooperative Effects on Hydrogen-Bonded Clusters of Water and the Cyanuric Acid. Mol. Sim. 2010, 36, 960–968. [Google Scholar] [CrossRef]
- Zabardasti, A. Kakanejadi, S. Moosavi, Z. Bigleri, M. Solimannejad, Anticooperativity in Dihydrogen Bonded Clusters of Ammonia and BeH42–. J. Mol. Struct: THEOCHEM 2010, 945, 97–100. [Google Scholar] [CrossRef]
- G. Han, Y. Ding, P. Qian, C. Zhang, and W. Song, Theoretical Investigation of Gas Phase Ethanol–(Water)n (n = 1–5) Clusters and Comparison with Gas Phase Pure Water Clusters (Water)n (n = 2–6). Int. J. Quantum Chem., 2013, 113, 1511–1521. [Google Scholar] [CrossRef]
- E. Romero-Montalvo, J. M. Guevara-Vela, A. Costales, Á. Martín Pendás and T. Rocha-Rinza, Cooperative and Aticooperative Effects in Resonance Assisted Hydrogen Bonds in Merged Structures of Malondialdehyde. Phys. Chem. Chem. Phys., 2017, 19, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Monu, B. K. Oram, B. Bandyopadhyay, Simultaneous Exhibition of Positive and Negative Cooperativity by Purely C H⋅⋅⋅O H-bonded (1,3-cyclohexanedione)n (n = 2–6) Clusters: A Density Functional Theoretical Investigation. Int. J. Quantum Chem. 2021, 121, 26581. [Google Scholar] [CrossRef]
- S. Guo, C. Zhu, G, Chen, J. Gu, C. Ma, H. Gao, L. Li, Y. Zhang,·X. Li, Z. Wang, Y. Wei, G. Wang, J. Shen, A Theoretical Study on Intermolecular Hydrogen Bonds of Isopropanol-Water Clusters. Theoret. Chem. Acc. 2022, 141, 6. [Google Scholar]
- D. Patkar, M. B. Ahirwar, M. M. Deshmukh, Energetic Ordering of Hydrogen Bond Strengths in Methanol-Water Clusters: Insights via Molecular Tailoring Approach. Chem. Phys. Chem. 2022, 23, 202200143. [Google Scholar] [CrossRef] [PubMed]
- Cukrowski, S. Zaaiman, S. Hussain, J. H. de Lange, All-Body Concept and Quantified Limits of Cooperativity and Related Effects in Homodromic Cyclic Water Clusters From a Molecular-Wide and Electron Density (MOWeD)-Based Approach. J. Comput. Chem. 2024, 45, 2812–2824. [Google Scholar] [CrossRef] [PubMed]
- Cukrowski, A Unified Molecular-Wide and Electron Density Based Concept of Chemical Bonding. WIREs Comput Mol Sci. 2022, 12, 1579. [CrossRef]
- R. F. W. Bader, Bond Paths are not Chemical Bonds. J. Phys. Chem. A, 2009, 113, 10391–10396. [Google Scholar] [CrossRef]
- H. de Lange, I. Cukrowski, Toward Deformation Densities for Intramolecular Interactions without Radical Reference States Using the Fragment, Atom, Localized, Delocalized, and Interatomic (FALDI) Charge Density Decomposition Scheme. J. Comput. Chem. 2017, 38, 981–997. [CrossRef]
- H. de Lange, D. M. van Niekerk, I. Cukrowski, FALDI-Based Decomposition of an Atomic Interaction Line Leads to 3D Representation of the Multicenter Nature of Interactions. J. Comput. Chem. 2018, 39, 973–985. [Google Scholar] [CrossRef] [PubMed]
- H. de Lange, I. Cukrowski, Exact and Exclusive Electron Localization Indices Within QTAIM Atomic Basins. J. Comput. Chem. 2018, 39, 1517–1530. [Google Scholar] [CrossRef] [PubMed]
- Cukrowski, IQA-Embedded Fragment Attributed Molecular System Energy Change in Exploring Intramolecular Interactions. Comput. Theoret. Chem. 2015, 1066, 62–75. [CrossRef]
- Cukrowski, G. Dhimba and D. L. Riley, A Reaction Energy Profile and Fragment Attributed Molecular System Energy Change (FAMSEC)-Based Protocol Designed to Uncover Reaction Mechanisms: a Case Study of the Proline-Catalysed Aldol Reaction. Phys. Chem. Chem. Phys., 2019, 21, 16694–16705. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Clarendon Press, Oxford, 1990.
- A. Blanco, Á. Martín Pendás, E. Francisco, Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. [Google Scholar] [CrossRef]
- E. Francisco, Á. Martín Pendás, M. A. Blanco, A Molecular Energy Decomposition Scheme for Atoms in Molecules. J. Chem. Theory Comput. 2006, 2, 90–102. [Google Scholar] [CrossRef]
- T. A. Keith, AIMAll (Version 19.02.13); TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Cukrowski, Reliability of HF/IQA, B3LYP/IQA, and MP2/IQA data in interpreting the nature and strength of interactions. Phys. Chem. Chem. Phys., 2019, 21, 10244–10260. [CrossRef]
- W. Humphrey, A. W. Humphrey, A. Dalke and K. Schulten, VMD: Visual molecular dynamics. J. Molec. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Cukrowski, J. H. de Lange and S. Hussain, MOWeD-LAC (Molecular-wide electron (de)localization atomic counts) and MOWeD-LFC (Molecular-wide electron (de)localization fragment counts) software applications, 2023. Available at https://bit.ly/link-to-mowed-software.
- H. de Lange, D. M. E. van Niekerk, I. Cukrowski, FALDI-Based Criterion for and the Origin of an Electron Density Bridge with an Associated (3,–1) Critical Point on Bader’s Molecular Graph. J. Comput. Chem. 2018, 39, 2283–2299. [Google Scholar] [CrossRef]
- S. de Beer, I. Cukrowski, J. H. de Lange, Characterization of bonding modes in metal complexes through electron density cross-sections. J Comput Chem. 2020, 41, 2695–2706. [Google Scholar] [CrossRef] [PubMed]
- D. L. Cooper, J. H. de Lange, R. Ponec, Comparison of DAFH and FALDI-like approaches. Theoret. Chem. Acc. 2020, 139, 179. [Google Scholar] [CrossRef]
- T. G. Bates, J. H. de Lange, I. Cukrowski, The CH⋅⋅⋅HC interaction in biphenyl is a delocalized, molecular-wide and entirely non-classical interaction: Results from FALDI analysis. J Comput Chem. 2021, 42, 706–718. [Google Scholar] [CrossRef] [PubMed]
- K. Mdhluli, W. Nxumalo, I. Cukrowski, A REP-FAMSEC Method as a Tool in Explaining Reaction Mechanisms: A Nucleophilic Substitution of 2-Phenylquinoxaline as a DFT Case Study. Molecules, 2021, 26, 1570. [Google Scholar] [CrossRef]
- Cukrowski, G. Dhimba, D. L. Riley, A Molecular-Wide and Electron Density-Based Approach in Exploring Chemical Reactivity and Explicit Dimethyl Sulfoxide (DMSO) Solvent Molecule Effects in the Proline Catalyzed Aldol Reaction. Molecules 2022, 27, 962. [Google Scholar] [CrossRef] [PubMed]
- Cukrowski, P. Mangondo, Interacting Quantum Fragments-Rooted Preorganized-Interacting Fragments Attributed Relative Molecular Stability of the BeII Complexes of Nitrilotriacetic Acid and Nitrilotri-3-Propionic Acid. J Comput Chem. 2016, 37, 1373–1387. [Google Scholar] [CrossRef]
- P. Mangondo, I. Cukrowski, On the Origin of the Relative Stability of ZnIINTA and ZnIINTPA Metal Complexes. An Insight From the IQA, IQF, and -FARMS methods. Int. J. Quantum Chem. 2017, 117, 25321. [Google Scholar] [CrossRef]
- Cukrowski, F. Sagan and M. P. Mitoraj, On the Stability of Cis- and Trans-2-Butene Isomers. An Insight Based on the FAMSEC, IQA, and ETS-NOCV Schemes. J. Comput. Chem. 2016, 37, 2783–2798. [Google Scholar] [CrossRef]
- P. Mitoraj, F. Sagan, D. W. Szczepanik, J. H. de Lange, A. L. Ptaszek, D. M. E. van Niekerk and I. Cukrowski, Origin of Hydrocarbons Stability from a Computational Perspective: A Case Study of Ortho-Xylene Isomers. Chem. Phys. Chem, 2020, 21, 494–502. [Google Scholar] [CrossRef] [PubMed]
- M. Bates, G. S. Tschumper, CCSD(T) Complete Basis Set Limit Relative Energies for Low-Lying Water Hexamer Structures. J. Phys. Chem. A, 2009, 113, 3555–3559. [Google Scholar] [CrossRef] [PubMed]
- S. Kryachko, Ab Initio Studies of the Conformations of Water Hexamer: Modelling the Penta-Coordinated Hydrogen-Bonded Pattern in liquid water. Chem. Phys. Lett. 1999, 314, 353–363. [CrossRef]
- R. M. Olson, J. L. Bentz, R. A. Kendall, M. W. Schmidt, M. S. Gordon, A Novel Approach to Parallel Coupled Cluster Calculations: Combining Distributed and Shared Memory Techniques for Modern Cluster Based Systems. J. Chem. Theory Comput., 2007, 3, 1312–1328. [Google Scholar] [CrossRef]
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [CrossRef]
- Koch, M. Pavanello, X. Shao, M. Ihara, P. W. Ayers, C. F. Matta, S. Jenkins, S. Manzhos, The Analysis of Electron Densities: From Basics to Emergent Applications. Chem. Rev. 2024, 124, 12661–12737. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Molecular graphs of water hexamers investigated in this work showing atoms’ numbering, numbering of water molecules (Wn) and numbering of hydrogen bonds (HB-n). Functionality of water molecules, in terms of accepting (a) or donating (d) a proton, is also indicated.
Figure 1.
Molecular graphs of water hexamers investigated in this work showing atoms’ numbering, numbering of water molecules (Wn) and numbering of hydrogen bonds (HB-n). Functionality of water molecules, in terms of accepting (a) or donating (d) a proton, is also indicated.
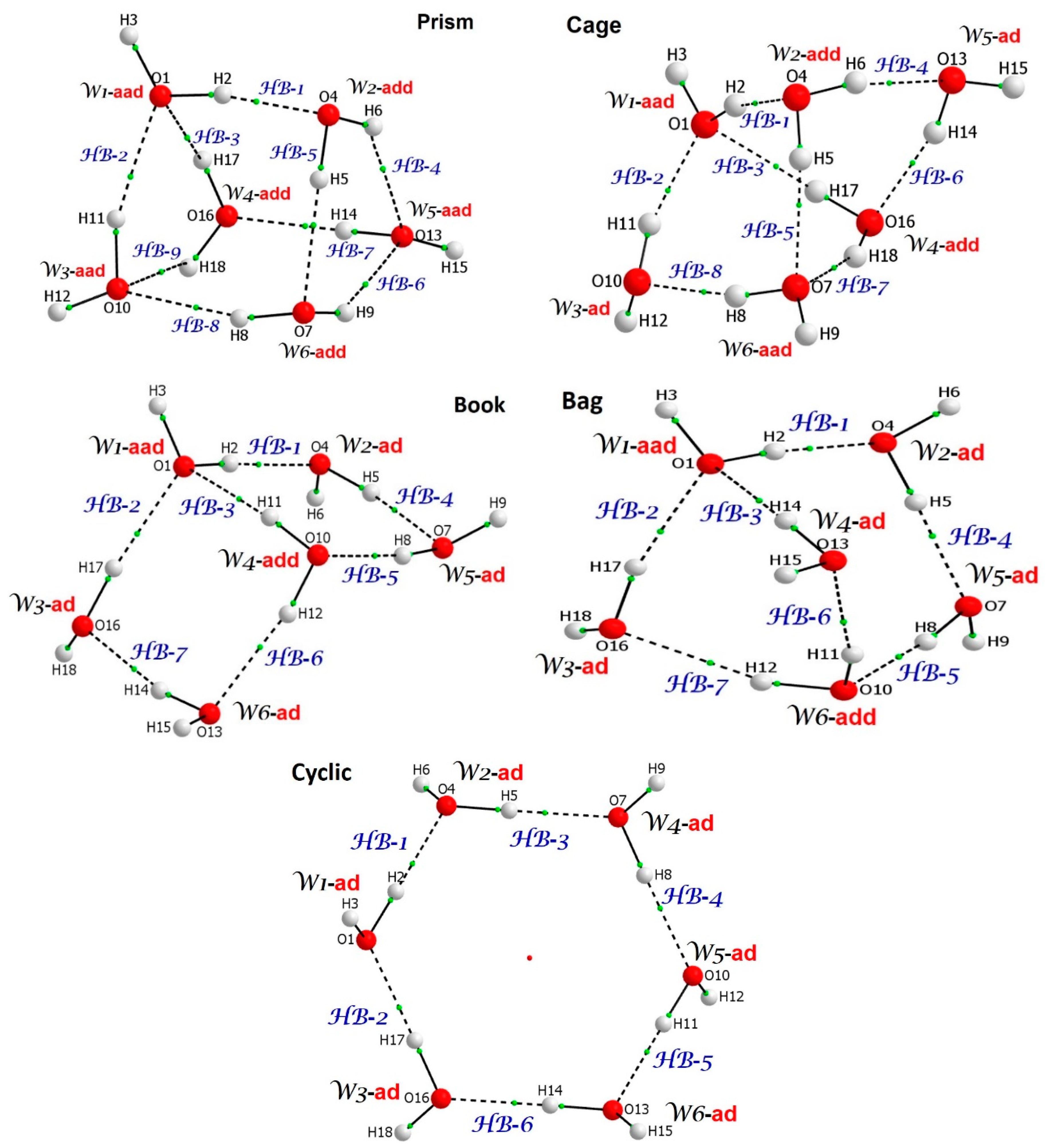
Figure 2.
Trends in relative stability of hexamers obtained at the B3LYP/aug-cc-pVTZ/GD3 level of theory employed in this work (circles with the solid trend line) and best available data (triangles with the dashed trend line) obtained at the CCSD(T)/CBS level. ΔE = E(hexamer) – E(prism).
Figure 2.
Trends in relative stability of hexamers obtained at the B3LYP/aug-cc-pVTZ/GD3 level of theory employed in this work (circles with the solid trend line) and best available data (triangles with the dashed trend line) obtained at the CCSD(T)/CBS level. ΔE = E(hexamer) – E(prism).
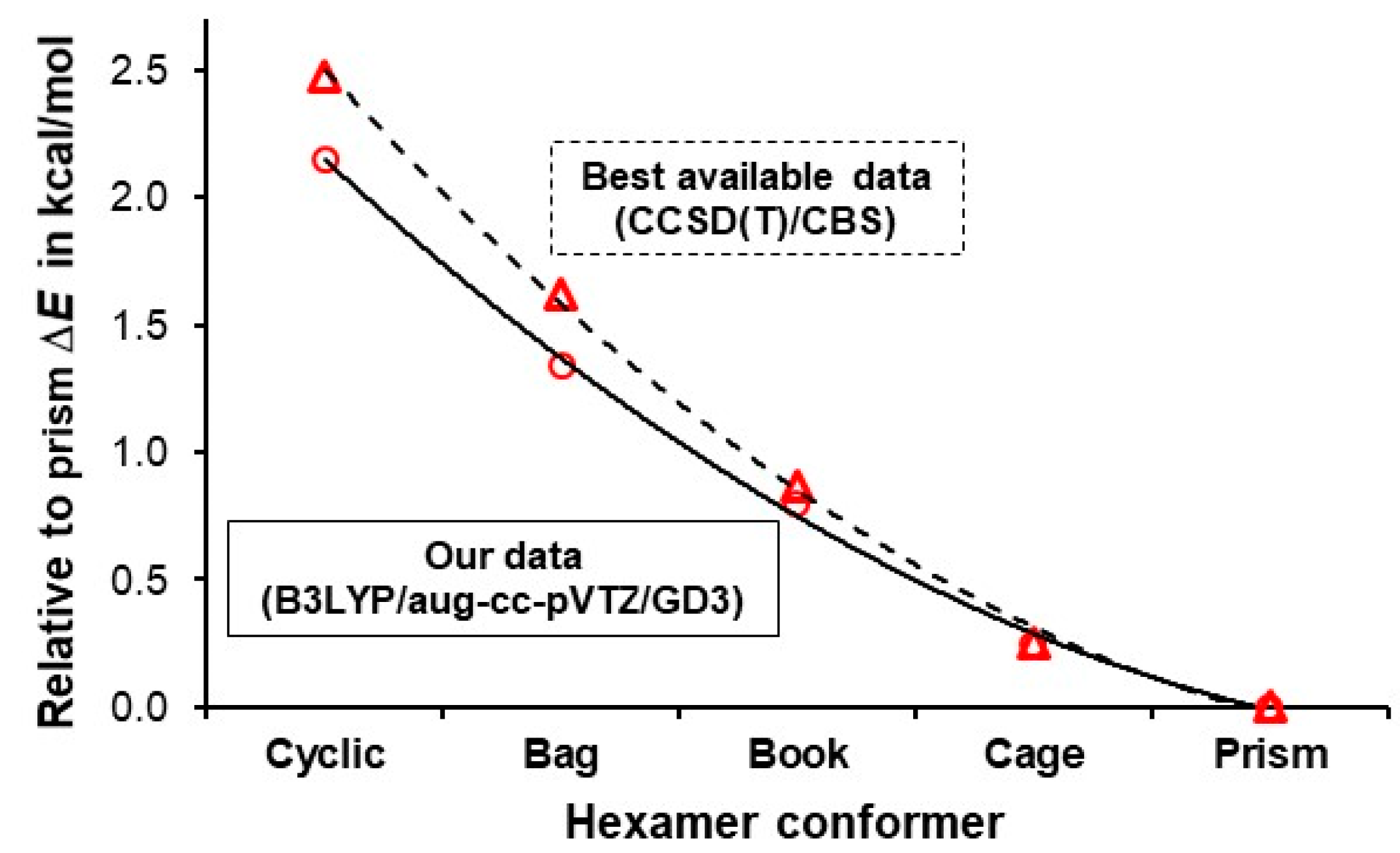
Figure 3.
Relative to six free, non-interacting water molecule, an increase in the stability of indicated water hexamers. ΔE = E(hexamer) – E(6H2O).
Figure 3.
Relative to six free, non-interacting water molecule, an increase in the stability of indicated water hexamers. ΔE = E(hexamer) – E(6H2O).
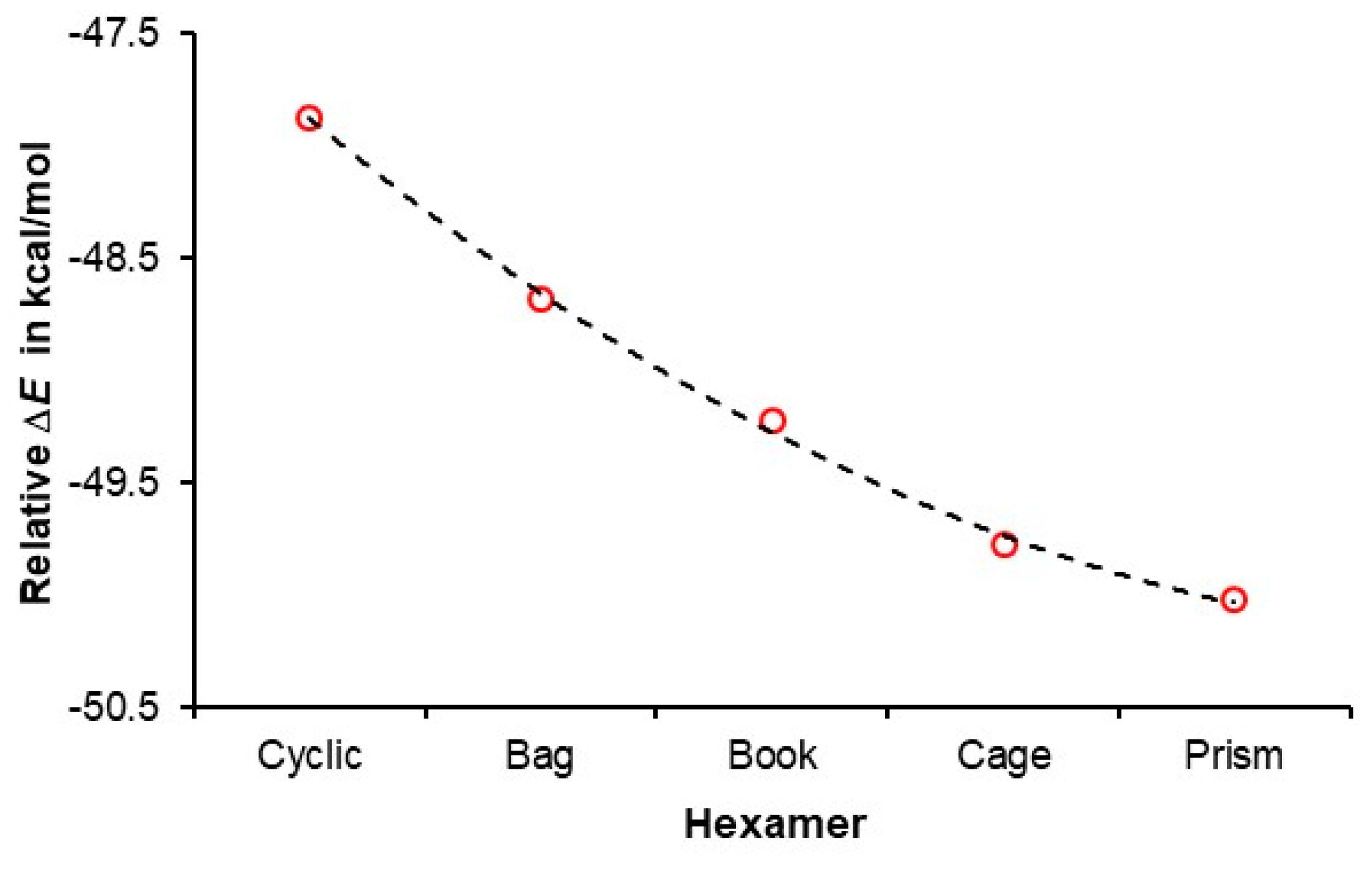
Figure 4.
Relative to six non-interacting water molecules, an increase in the stability of indicated hexamers with an increase in the number of double-acceptor and double-donor water molecules in a cluster. ΔE = E(hexamer) – E(6H2O).
Figure 4.
Relative to six non-interacting water molecules, an increase in the stability of indicated hexamers with an increase in the number of double-acceptor and double-donor water molecules in a cluster. ΔE = E(hexamer) – E(6H2O).
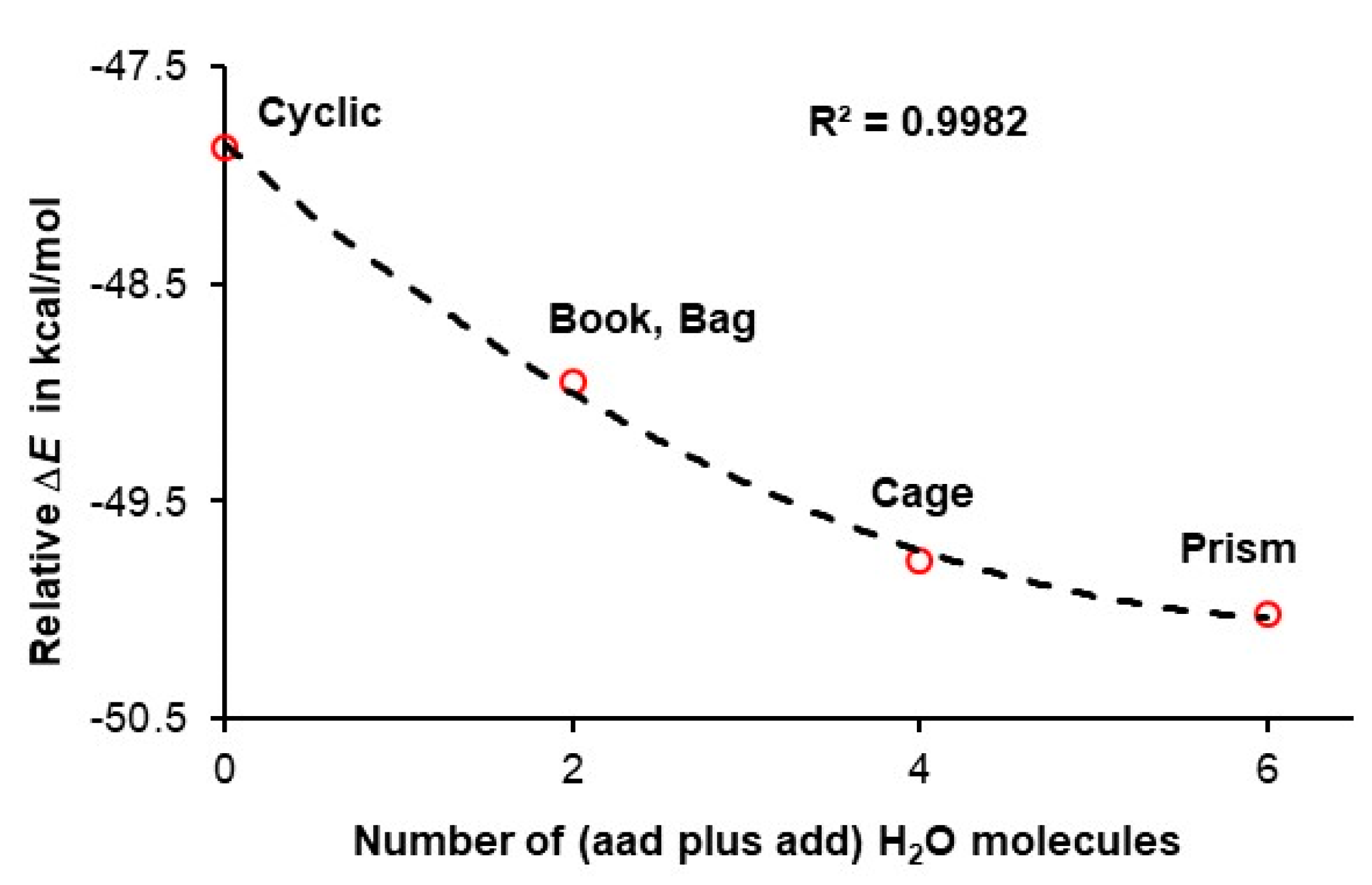
Figure 5.
The trend in the total number of electrons intermolecularly delocalized by all water molecules in a cluster and the number of aad plus add water molecules in a cluster. Regression for the fitted data (dashed line) is also indicated.
Figure 5.
The trend in the total number of electrons intermolecularly delocalized by all water molecules in a cluster and the number of aad plus add water molecules in a cluster. Regression for the fitted data (dashed line) is also indicated.
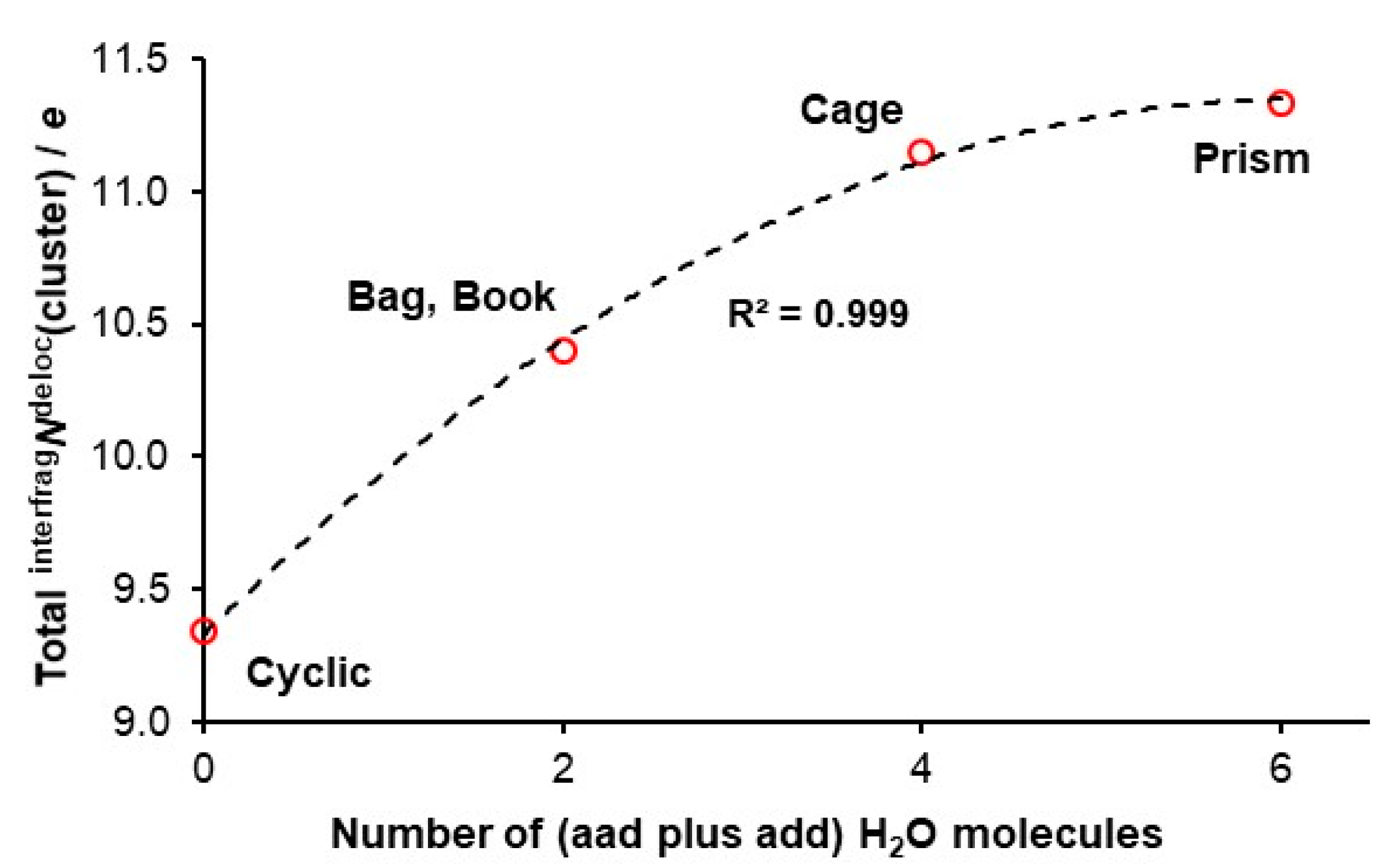
Figure 6.
Relative to E of six non-interacting water molecules, the trend in the decrease in E(cluster) plotted vs. the total number of electrons intermolecularly delocalized by all water molecules in the indicated hexamers. Regression for the fitted data (dashed line) is also indicated.
Figure 6.
Relative to E of six non-interacting water molecules, the trend in the decrease in E(cluster) plotted vs. the total number of electrons intermolecularly delocalized by all water molecules in the indicated hexamers. Regression for the fitted data (dashed line) is also indicated.
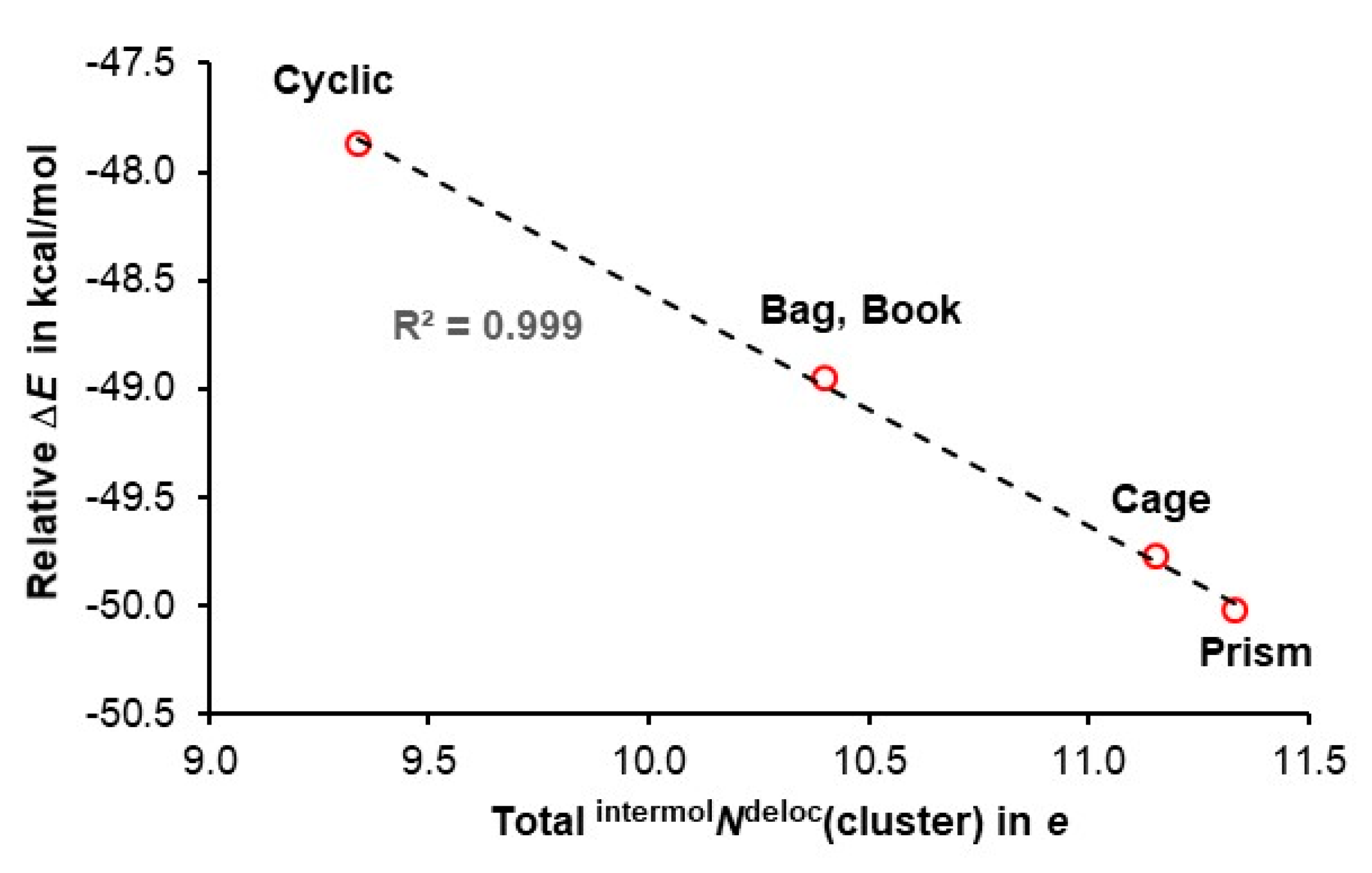
Figure 7.
A trend between intermolecularly delocalized electrons (intermolNdeloc) and the XC-term of a total interaction energy between a water molecule and the remaining five waters VXC(W,R) computed for each individual molecule in the cage hexamer.
Figure 7.
A trend between intermolecularly delocalized electrons (intermolNdeloc) and the XC-term of a total interaction energy between a water molecule and the remaining five waters VXC(W,R) computed for each individual molecule in the cage hexamer.
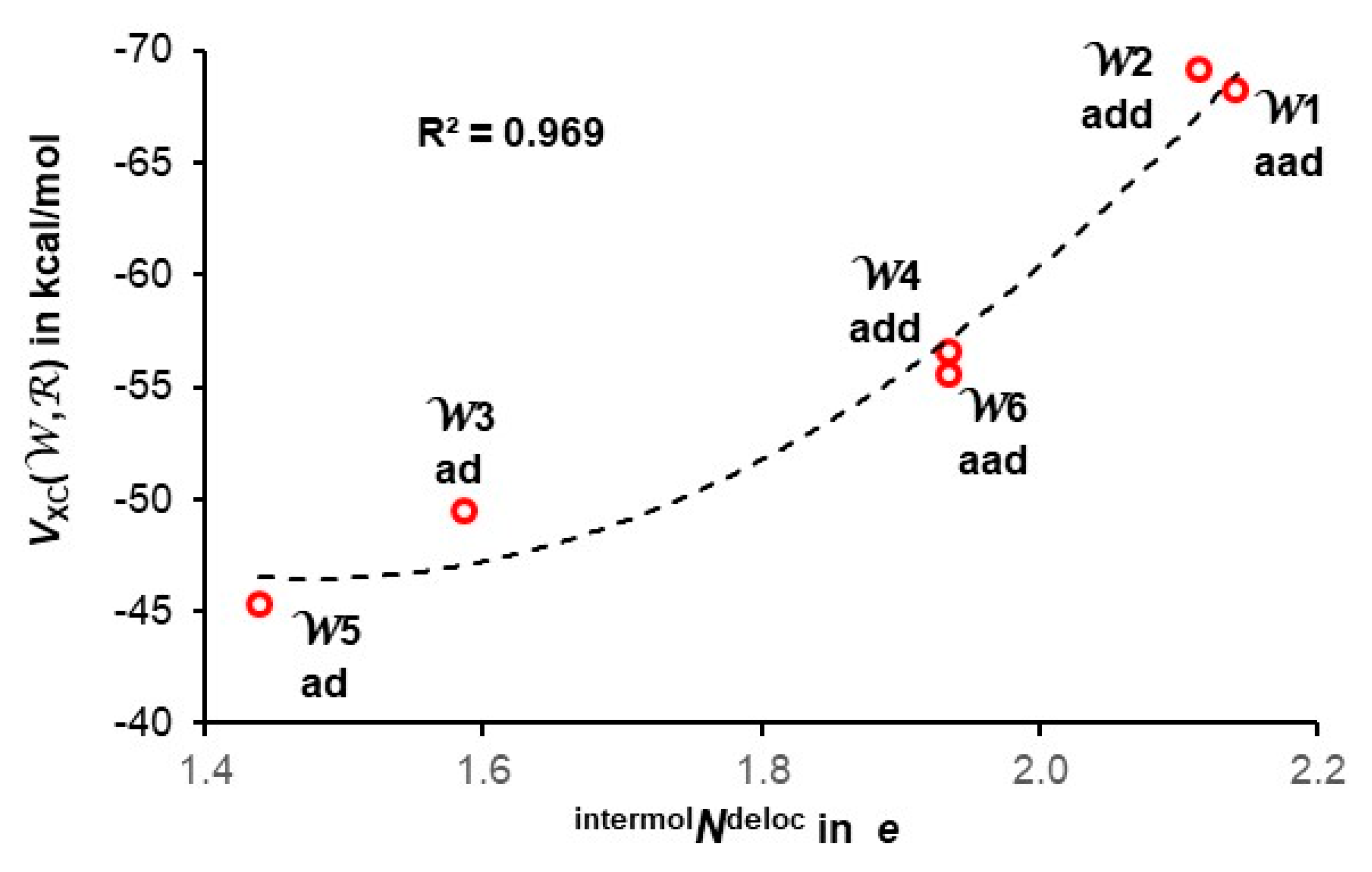
Figure 8.
A correlation between the interaction energy Eint(W,R) and the mol-FAMSEC energy term computed for each individual molecule in the cage hexamer (red color) and bag hexamer (violet color). Black dashed and dotted violet lines are the trend lines for cage and bag, respectively.
Figure 8.
A correlation between the interaction energy Eint(W,R) and the mol-FAMSEC energy term computed for each individual molecule in the cage hexamer (red color) and bag hexamer (violet color). Black dashed and dotted violet lines are the trend lines for cage and bag, respectively.
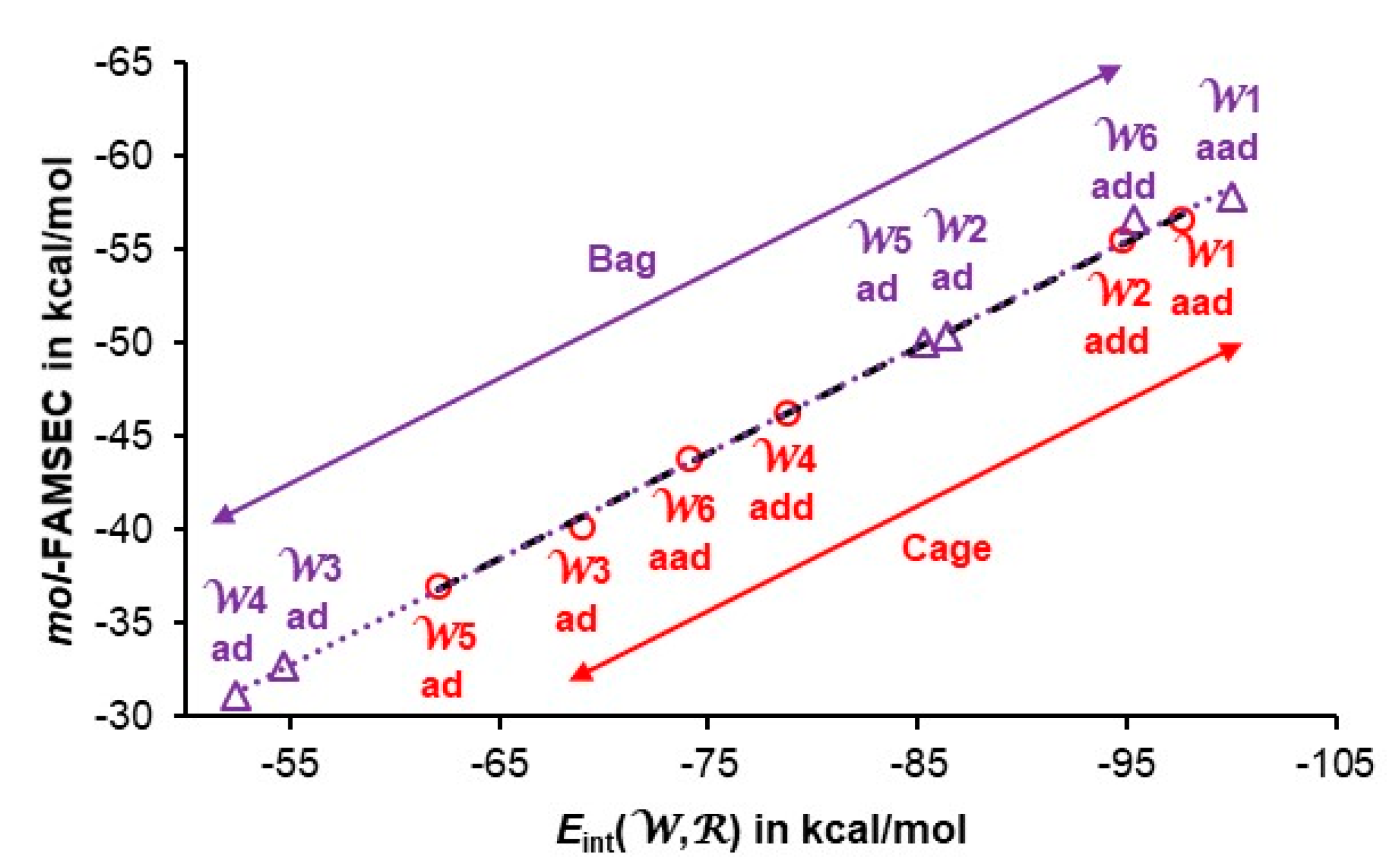
Figure 9.
A trend between average mol-FAMSEC computed for intermolecular H-bond formed in each hexamer and, relative to non-interacting 6 water molecules, an increase in hexamer’s stability.
Figure 9.
A trend between average mol-FAMSEC computed for intermolecular H-bond formed in each hexamer and, relative to non-interacting 6 water molecules, an increase in hexamer’s stability.
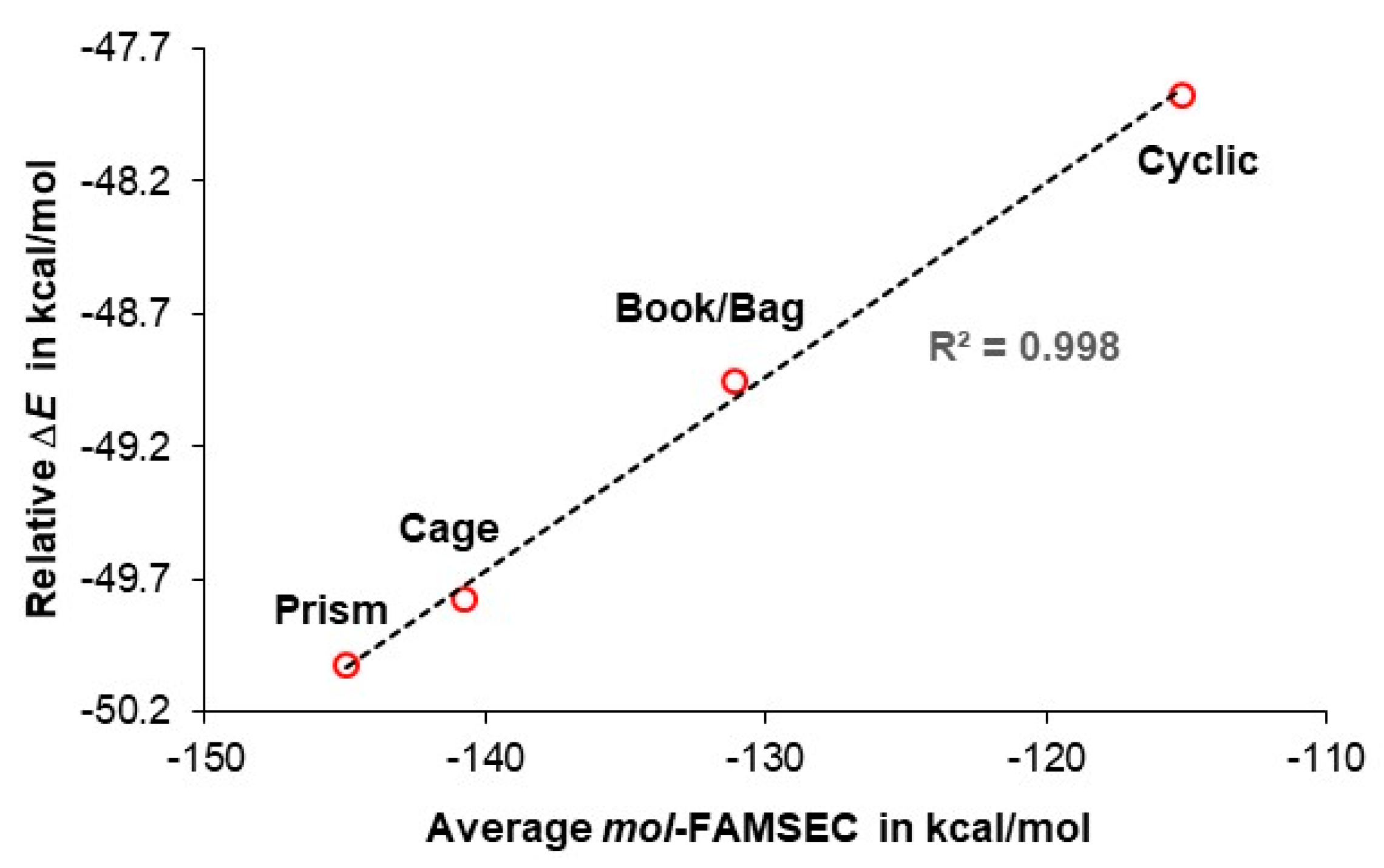

Table 1.
Comparison of the relative energies, as ΔE = E(hexamer) – E(prism) values in kcal/mol, obtained at different levels of theory for the indicated water hexamers.
Table 1.
Comparison of the relative energies, as ΔE = E(hexamer) – E(prism) values in kcal/mol, obtained at different levels of theory for the indicated water hexamers.
| ΔE = E(hexamer) – E(prism) in kcal/mol | ||||||
| Source | Level of theory | Cyclic | Bag | Book | Cage | Prism |
| Our data | B3LYP/aug-cc-pVTZ/GD3 | 2.15 | 1.34 | 0.80 | 0.25 | 0 |
| Bates et al [74] | CCSD(T)/CBS | 2.48 | 1.62 | 0.87 | 0.25 | 0 |
| Kryachko [75] | MP2(full)/aug-cc-pVDZ | 2.06 | N/A | 1.16 | 0.25 | 0 |
| Olson et al [76] | CCSD(T)/aug-cc-pVTZ | 2.10 | N/A | 1.20 | 0.30 | 0 |
Table 2.
The number of electrons delocalized by an indicated water molecule to remaining five water molecules of 3D hexamers. The functionality of water molecules and their numbering is consistent with that seen in Figure 1.
Table 2.
The number of electrons delocalized by an indicated water molecule to remaining five water molecules of 3D hexamers. The functionality of water molecules and their numbering is consistent with that seen in Figure 1.
| Prism | Cage | Book | Bag | ||||
| Water | Ndeloc | Water | Ndeloc | Water | Ndeloc | Water | Ndeloc |
| aad W1 | 2.112 | aad W1 | 2.142 | aad W1 | 2.100 | aad W1 | 2.188 |
| add W2 | 2.027 | add W2 | 2.116 | add W4 | 2.048 | add W6 | 2.074 |
| aad W3 | 1.771 | add W4 | 1.935 | ad W2 | 1.647 | ad W2 | 1.719 |
| add W4 | 1.861 | aad W6 | 1.935 | ad W3 | 1.484 | ad W3 | 1.433 |
| aad W5 | 1.778 | ad W3 | 1.587 | ad W5 | 1.603 | ad W4 | 1.411 |
| add W6 | 1.786 | ad W5 | 1.439 | ad W6 | 1.412 | ad W5 | 1.677 |
Table 3.
Energy terms computed for individual water molecules in bag hexamer. Eint(W,R) and VXC(W,R) stand for the interaction energy and its exchange-correlation (covalent or quantum) component, respectively, computed between an indicated water molecule and remaining waters. mol-FAMSEC quantifies energy contribution made by a water molecule to the stability of bag hexamer.
Table 3.
Energy terms computed for individual water molecules in bag hexamer. Eint(W,R) and VXC(W,R) stand for the interaction energy and its exchange-correlation (covalent or quantum) component, respectively, computed between an indicated water molecule and remaining waters. mol-FAMSEC quantifies energy contribution made by a water molecule to the stability of bag hexamer.
| Energy terms in kcal/mol | |||
| Water | Eint(W,R) | VXC(W,R) | mol-FAMSEC |
| aad W1 | -100.03 | -70.22 | -57.81 |
| add W6 | -95.36 | -69.20 | -56.58 |
| ad W2 | -86.34 | -59.52 | -50.39 |
| ad W5 | -85.31 | -59.23 | -50.07 |
| ad W3 | -54.68 | -40.56 | -32.69 |
| ad W4 | -52.42 | -39.20 | -31.12 |
| Average: | -79.0 | -56.3 | -46.4 |
| St. Dev.: | 20.5 | 13.6 | 11.7 |
Table 4.
mol-FAMSEC-quantified contributions to cluster’s stability made by the indicated classical intermolecular H-bonds.
Table 4.
mol-FAMSEC-quantified contributions to cluster’s stability made by the indicated classical intermolecular H-bonds.
| Water cluster | |||||
| Prism | Cage | Book | Bag | Cyclic | |
| H-bond | mol-FAMSEC in kcal/mol | ||||
| HB-1 | –141.0 | –139.3 | –139.5 | –142.1 | –115.2 |
| HB-2 | –157.3 | –145.1 | –167.7 | –142.7 | |
| HB-3 | –163.0 | –161.0 | –143.4 | –143.7 | |
| HB-4 | –126.4 | –133.0 | –119.4 | –121.8 | |
| HB-5 | –156.9 | –161.5 | –118.8 | –119.2 | |
| HB-6 | –139.1 | –150.1 | –119.4 | –122.4 | |
| HB-7 | –124.4 | –109.8 | –109.2 | –125.5 | – |
| HB-8 | –138.1 | –126.3 | – | – | – |
| HB-9 | –158.5 | – | – | – | – |
| Average: | –145.0 | –140.8 | –131.1 | –131.1 | –115.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



