
Submitted:
22 January 2025
Posted:
24 January 2025
You are already at the latest version
Abstract
Histone deacetylases (HDACs) become one of the main targets in cancer therapy due to their involvement in various biological processes, including gene regulation, cell proliferation and differentiation. On the other hand, microtubules as key elements of the cell cytoskeleton also represent an important therapeutic target in anticancer drugs research. These proteins are implied in diverse cellular functions, especially mitosis, cell signaling and intracellular trafficking. With the emergence of multi-target therapy during the last decades, the association of HDAC and tubulin inhibitors has been envisioned as a practical approach for optimizing the therapeutic efficacy of antitumor molecules. The HDAC/tubulin dual-targeting inhibitors offer the advantages of a synergistic action of both compounds, along with a significant decrease of their respective toxicities and drug resistance. This review will detail the major recent advancements in the development of HDAC/tubulin dual inhibitors over the last decade, with their impact in anticancer drugs discovery.
Keywords:
Histone desacetylase
; tubulin
; dual compounds
1. Introduction
Cancer constitutes one of the main leading causes of death worldwide, with 19.3 million new cases and around 10 million deaths reported in 2020 [1]. This complex disease involves various biological factors, such as epigenetic alterations, which could modulate genes expression. Among these, the significant variation of reversible acetylation-deacetylation of histones represents a key hallmark of carcinogenesis [2].
Histone deacetylases (HDACs) are a class of epigenetic metalloenzymes capable of removing acetyl groups from N-acetylated lysine residues on histone or non-histone proteins. HDACs mainly target histones, for tightly wrapping DNA. They can regulate the gene expression and protein activity by varying the DNA accessibility [3]. In contrast, their dysregulation can lead to the alteration of oncogenes or tumor suppressor genes transcription, inducing carcinogenic events [4]. Hence, HDAC inhibitors (HDACis) have been developed during the last decades as major molecules in cancer treatment [5]. These compounds are able to hamper the proliferation and differentiation of cancer cells and promote their apoptosis. HDACis are promising antitumor agents, with four US FDA-approved HDACis (Vorinostat, Panobinostat, Belinostat and Romidepsin) [6]. Clinical trials have confirmed their high efficiency in solid and liquid tumors [7]. More recently, Tucinidostat was approved as HDACi in China for relapsed and refractory peripheral T-cell lymphoma [8] (Figure 1). Nonetheless, the main drawback of these HDACis is their low HDAC-isoform specificity causing significant side-effects, e. g. fatigue, nausea, thrombocytopenia, cardiotoxicity, or hematological toxicity [9]. Furthermore, drug resistance to HDACis could also be noted, probably due to the activation of signal transduction pathways, namely Akt (protein kinase B) or CDK (cyclin-dependent protein kinase) signaling processes [10].
To circumvent all these issues, research efforts on dual-targeting HDACis has emerged. This strategy consists of associating different pharmacophores in one drug, which could interact with multiple cancer targets. These hybrid molecules present several advantages compared to the drug combinations, such as their more predictable pharmacodynamic and pharmacokinetic properties, lower toxicities and higher efficiency in advanced-stage diseases thanks to their synergistic effects [11,12]. In the frame of this review, we will particularly focus on the dual inhibitors targeting HDACs and tubulin, as potential anti-cancer agents.
Microtubules also represent a major therapeutic target in the development of new anticancer drugs. They are constituted by dynamic cytoskeletal proteins made of α/β-tubulin heterodimers. These structural cell elements play pivotal roles in the maintenance of cell architecture, migration, and proliferation in eukaryotic cells [13]. Tubulin inhibitors, via their interaction with the binding sites (laulimalide, taxane/epothilone, vinca alkaloid, and colchicine sites), would stop the cell cycle and interrupt the mitosis process [14]. Thus, the cell apoptosis pathway could be activated. Through this approach, these tubulin targeting agents demonstrated their ability to kill tumor cells.
In view of the effective properties of HDACis and tubulin inhibitors, combination therapy was envisioned with these compounds. In fact, synergistic effects were observed by associating HDACis with tubulin inhibitors (e. g. paclitaxel, vincristine) [15,16]. Therefore, the construction of molecular hybrids HDAC/tubulin inhibitors is the next logical step in the optimization of new-generation anti-cancer drugs. With all the benefits of the multi-targeting compounds, numerous research groups have studied the HDAC/tubulin dual inhibitors. These dual-targeted molecules would heighten the synergistic antitumor effect observed in the combination therapy, leading to the suppression of tumor cell proliferation and the induction of the apoptosis process in cancer cells.
Given the wide chemical diversity of these biologically active compounds, this review will mainly classify the main dual molecules developed during the last few decades according to the structural scaffold of the tubulin inhibitors.
2. Combretastatin-A4 motif and related structures
Combretastatin-A4 (CA-4) is a natural product isolated from Combretum caffrum, an African willow tree by Pettit et al. [17] (Figure 2). This molecule is identified as a potent anticancer agent, which inhibits the tubulin polymerization by interacting with the colchicine site [18]. In addition to its selective action on tubulin, CA-4 possesses a nanomolar-scale cytotoxicity against a broad range of human cancer cells, including multidrug resistant cells [19]. Despite such promising properties, CA-4 shows high instability of the Z-stilbene structure, which could be isomerized during the metabolization process to the less active E isomer. Thus, the development of CA-4 derivatives and related structural analogs was envisioned during the last decades in various medicinal chemistry research groups [20]. By combining these compounds with HDACis, significant synergistic effects were found for these newly designed antitumor agents.
2.1. Chalcone derivatives
Among the CA-4 related compounds, chalcones constitute one of the main groups. In this manner, Luan et al. described chalcone derivatives as HDAC/tubulin dual-targeting inhibitors. Inspired by the FDA-approved HDACi (see Figure 1) and the CA-4 structure, the researchers designed a chalcone platform functionalized with the key methoxy substituents of CA-4, ensuring the tubulin inhibition. This part was connected to a hydroxamic acid skeleton, providing HDAC inhibitory activity (Scheme 1). These molecules were constructed through a multi-step synthesis, including a condensation reaction and an alkylation step. The HDAC inhibition rates of the prepared hybrid compounds disclosed that compounds 1a and 1b presented the highest inhibitory activities (87% and 92% of inhibition at a concentration of 1 µM respectively). This result was confirmed in HeLa cell nucleus extract with IC50 values ranging from 71.5 to 132.6 nM towards HDACs. A thorough study showed that 1a and 1b had pan-HDAC inhibitory properties, especially with HDAC 1, 6 and 8. After checking the viability of these molecules as HDACis, their cytotoxicity was evaluated for three cancer cell lines (A549, HeLa and SGC-7901). Despite modest HDAC inhibitory activities, molecule 1c was the most active compound with IC50 values up to 550 nM for A549 cells. 1a, 1b and 1c were then submitted to tubulin polymerization inhibition (TPI) assay, with the best inhibition value observed for 1b (IC50 = 5.0 µM). Further explorations of the anti-proliferative properties of 1a, 1b and 1c confirmed their activities against eight human tumor cell lines (LoVo, HCT-116, NCI-H460, NCI-H226, MCF-7, MDA-MB-231, MGC80-3, BGC-823), especially for 1c (IC50 = 2.40-7.54 µM). More precisely by analyzing the A549 cells apoptosis, 1c was able to stop the cell cycle at the G2/M phase at a 10 µM-dose. Finally, 1c also presented an anti-migratory effect on A549 cells at a concentration of 5 or 10 µM via a wound healing assay [21].
Mourad and his colleagues employed analogous scaffolds for their HDAC/tubulin inhibitors hybrids [22]. The group conceived their dual compounds by mixing a chalcone skeleton, which has the same tubulin inhibitory properties as CA-4 [23], with a phthalimide group presenting interesting biological antitumor activities.[24] To ensure the HDACi efficiency, a benzamide group was used as an Entinostat analog (Scheme 2). Their chemical synthetic route consisted of the condensation of phthalide with potassium phthalimide, followed by an amide coupling and an aldolization reaction. The screening of the in vitro cytotoxicity of the prepared molecules revealed 2a, 2b and 2c provided higher efficacy than CA-4, with IC50 ranges from 1.62 to 2.21 µM against MCF-7 and Hep G2 cells. The measurement of TPI capacity of 2a, 2b and 2c shed light on compounds 2b and 2c, which offered the best IC50 of 3.27 and 2.32 µM respectively. It is worth mentioning that these molecules were selected as hits as their TPI values are comparable to the CA-4 one (IC50 = 2.62 µM). Notably, this outcome was in line with the HDAC inhibitory activity of 2b and 2c, which had a significant specificity for HDAC 1 and 2. In view of the probable involvement of 2b and 2c in the cell cycle due to its interaction with tubulin, in vitro DNA flow cytometry was examined. 2b and 2c seemed to trigger apoptosis by stopping cell growth at G2/M phase. Calculations of energy binding scores confirmed the strong interaction between 2b and 2c in the colchicine binding site of tubulin, mainly due to hydrogen bonds. Molecular docking investigations on HDAC active site gave same conclusions noted in the tubulin protein.
2.2. Stilbene derivatives
Inspired by the isoCA-4 structure, a stilbene isomer of CA-4, proposed by the Alami group [26], the Hamze team conceptualized novel dual-targeting inhibitors of tubulin and HDAC by associating isoCA-4 moiety with the pharmacophore of belinostat as chimeric HDAC/tubulin inhibitors (Scheme 3). Their chemical synthesis involved a palladium-catalyzed Barluenga-Valdés cross-coupling, between an aryl halide and the corresponding N-tosylhydrazone. The functionalization with the HDACi pharmacophore (hydroxamic acid) was then realized with different strategies, including alkylation in standard conditions, Sonogashira or Heck couplings. After submission to in vitro antiproliferative assays on HCT-116 cancer cells, the authors validated the importance of the hydroxamic acid group for the cytotoxic activity. Compounds bearing an alkene linker between the stilbene scaffold and the hydroxamic acid skeleton exhibited the best GI50 values (1.5 nM for 3a and 8 nM for 3b). The TPI character of the dual compounds was next determined, with promising IC50 values of 1.6 and 2.1 µM for 3a and 3b respectively. The HDAC inhibitory activity of 3a and 3b was then settled, with a strong selectivity for HDAC 8. The cytotoxicity of these molecules was also verified with nine cancer cell lines (A549, K562, K562R, PC3, U87-MG, MCF7, BXPC3, MiaPaca2, and HT29), with low GI50 values obtained for 3a, ranging from 0.4 to 5.1 nM. Additional studies by incubating 3a at a concentration of 5 or 10 nM with HCT-116, K562 and BL2 cells, demonstrated the ability of 3a to stop the cell cycle at the G2/M phase and induce the cell death. Finally, the viability of 3a was corroborated with its safety profile on quiescent peripheral blood lymphocytes, with an IC50 value of 7 µM [27].
In 2022, the same team proposed a second generation of HDAC/tubulin chimeric inhibitors, by replacing the trimethoxybenzene ring of CA-4 with quinoline or quinazoline cores [28] (Scheme 4). The designed structures were prepared through a Buchwald-Hartwig coupling or nucleophilic substitution (SNAr) reaction under acidic conditions, followed by the connection to the hydroxamic acid motif via alkylation under basic conditions, or through Sonogashira or Heck cross-couplings. The GI50 values in HCT-116 cells of the thirty-one molecules thus synthesized were then determined. The structure activity relationship (SAR) studies confirmed the importance of the hydroxamic acid and the alkene linker for the cytotoxicity. Thus, 4a and 4b were recognized as lead compounds, with GI50 values of 0.5 and 0.6 nM respectively. With these encouraging outcomes, 4a and 4b were tested against nine additional human cancer cell lines (NCI-N87, K562, K562R, MiaPaca2, SKOV3, A549, MCF7, MDA MB231, HT-29). Notably, 4a and 4b remained efficient, with average IC50 of 0.6 and 0.7 nM respectively. The antiproliferative activities of these second generation of HDAC/tubulin chimeric inhibitors were up to more than 60 times greater than those of the previously described dual molecules, CA-4 and isoCA-4. The HDAC inhibitory profiles of 4a and 4b were next assessed, showing complete inhibition of HDAC 8, and partial effects on HDAC 6 and 11 (60 to 90 % inhibition) at 1 µM. The in vitro TPI assay was realized for 4a, with IC50 value of 20 nM. With the crucial implication of microtubules network in the cell division, extra flow cytometry analyses depicted the cell cycle arrest at the G2/M phase at the concentration of 1 nM of 4a and 4b. The authors also demonstrated the involvement of 4a and 4b in the mitochondrial dysfunction at 2 nM. The metabolic stability of 4a and 4b was also explored in rat and human microsomes. 4a exhibited a longer in vitro half-life, likely due to its quinoline skeleton and the hydroxamic acid scaffold.
The applicability of 4a was further validated using an in vivo allograft mouse model. Significant tumor decrease was observed at 0.25 mg/kg and 0.50 mg/kg doses, without any recidivism after 91-days treatment. No toxicities were noticed after intratumoral injection of these doses of 4a.
In 2021, Yao, Yang, Duan and coworkers studied new tubulin/HDAC dual-targeting inhibitors with benzophenones-related and stilbene-derived structures exhibiting antitumor properties [25] (Scheme 5). These compounds were designed through the fusion of CA-4 derivatives (benzophenone or stilbene groups) with HDACi pharmacophores (hydroxamic acid or benzamide moieties). The construction of these dual molecules was based on an alkylation step or a palladium-catalyzed Heck cross-coupling, followed by amide bond formation. The generated products were subjected to cytotoxicity assays with six cancer cell lines (MGC-803, MCF-7, U937, A549, HepG2, Hela). The SAR examination revealed that stilbene-derived compounds had higher antiproliferative activities than benzophenone analogs. The presence of alkene linker and benzamide functional group also favored the cytotoxicity. Compound 5 was hence selected as the lead compound, with IC50 ranging from 16 to 305 nM. The specific activity of 5 towards cancer cell lines was confirmed through testing on non-tumorigenic cells (293T, THP-1-derived macrophages, and L02 cell lines), with non-observed toxicity (IC50 values up to 556 nM). The biological analyses of 5 were pursued with the evaluation of its tubulin polymerization and HDAC inhibitory capacity. Compound 5 was found as a potent tubulin/HDAC inhibitor, with an IC50 value of 1.2 µM for the TPI activity, and a pan-HDAC selectivity, especially for HDAC 3, 4, 5, 7 and 9. The TPI phenomenon was also visualized under confocal microscope by the disruption of the cell microtubule network. To explain the high affinity of 5 for its expected targets, molecular docking was realized, detecting the binding between 5 and the colchicine site of the tubulin via mainly hydrogen bonds. Similar interactions could be seen between 5 and HDAC 7. By deepening the mechanism of action of 5, the research group discovered that 5 induced the cell cycle arrest at G2/M phase, by upregulating Cyclin B1 and diminishing P21 and Pcdc2 expression. 5 also contributed to the cell death process by decreasing the concentration Caspase-3, Caspase-7, Caspase-9 and PARP and increasing the quantity of cleaved corresponding proteins. Alongside this pro-apoptotic pathway, 5 alter mitochondria membrane potential and promoted the generation of reactive oxygen species (ROS). A wound healing test with human umbilical vein endothelial cells (HUVECs) showed that 5 could hamper cell migration, invasion and angiogenesis.
Finally, the effectiveness of 5 was tested in zebrafish embryos, where angiogenesis was impeded at 0.6 µM of 5. Using xenograft zebrafish models, a significant tumor reduction was noted at 100 nM of 5.
In the same trend, the CA-4 derivatives developed by Chen, Xu and their colleagues featured a stilbene skeleton bearing a quinoline or pyridine motif to enhance tubulin polymerization inhibitory activity. To fulfill the dual property of these molecules, the research team incorporated a hydroxamic acid structure into the stilbene scaffold as an HDACi [29] (Scheme 6). The construction of the stilbene moiety relied on a Barluenga-Valdés cross-coupling. The installation of the hydroxamic acid group was realized by alkylation or a Wittig reaction, followed by amide bond formation. By evaluating the antiproliferative activities of the synthesized dual compounds, compound 6 exhibited the best in vitro IC50 value against K562 cells (3 nM). Further investigations confirmed the superior cytotoxicity of 6 for five cancer cell lines (MCF-7, MDA-MB-231, A549, B16F10, A2780), with IC50 values ranging from 5 to 36 nM. In contrast, 6 demonstrated low cytotoxicity for normal human lung cells (IC50 (HFL-1) = 20 nM). The HDAC inhibitory character of 6 was validated, with a pan-HDAC selectivity, especially for HDAC 1, 2 and 6. The TPI effect of 6 was also established in vitro, with a measured IC50 value of 3.84 µM. Competitive inhibition experiments with tritium-labelled colchicine indicated that 6 acted via binding to the colchicine site of the tubulin. The dual characteristic of 6 was confirmed in K562 cells, with a disruption of the microtubule network and the accumulation of acetylated α-tubulin. In cellulo assays showed that 6 was able to stop the cell cycle at G2 phase, and induce the apoptosis of K562 cells, by increasing the concentration of pro-apoptotic proteins Bad and Bax. Antivascular activity was also found for 6, with the suppression of the HUVECs migration at 8 nM. Finally, an in vivo antitumor assay was performed with 6, using a mouse liver cancer allograft model. Similarly to the in vitro results, 6 furnished an excellent efficacy (Tumor growth inhibition (TGI) = 73.12% at 20 mg/kg), without loss in body weight or significant toxicity in vital organs.
2.3. Amine derivatives
In 2022, Song, Zhang and coworkers combined CA-4-derived skeleton with the key motif of Entinostat, an HDACi agent, to construct SY-65, a novel aromatic amide scaffold targeting HDAC1 and tubulin (Scheme 7). The synthesis of SY-65 involved a three-step synthesis, consisting of two amide couplings and a saponification reaction. This compound demonstrated activity against several human cancer cell lines (MGC-803, HGC-27, SGC-7901, HCT-116 and KYSE450), with IC50 values ranging from 39 to 254 nM. With these promising results, SY-65 was then studied as a tubulin polymerization inhibitor in MGC-803 and HGC-27 cells, showing an IC50 of 3.64 µM. Complementary EBI competition and cellular thermal shift assays, confirmed that SY-65 bind to the colchicine site on β-tubulin. By evaluating the activities of SY-65 against HDACs, the authors reported an inhibition of HDAC1 (IC50 = 525 nM). This effect was also observed in cellulo with MGC-803 and HGC-27 cell lines. Besides, SY-65 exhibited apoptotic properties in MGC-803 and HGC-27, including the reduction of Bcl-xL, Bcl-2 and c-IAP1 rates. The compound also arrested cell proliferation at the G2/M phase in these cell lines, as evidenced by the downregulation of cell cycle-related proteins Wee1, cdc2, and phosphorylated cdc2. An in vivo nude mouse xenograft model bearing MGC-803 cells confirmed all the biological features of SY-65, especially the inhibition of the tumor growth after 14- or 21-day treatment [30].
2.4. Oxazole derivatives
In 2019, Höpner et al. created novel HDAC/tubulin inhibitors by attaching hydroxamic acids to oxazole-bridged CA-4 derivatives (Scheme 8). These bent structures were prepared via a three-step synthesis, with a Von Leusen reaction in basic conditions as the key step. The biological evaluation of these hybrid compounds unveiled that bromo derivatives 7a, 7b and 7c exhibited the highest average anti-proliferative activities for six cancer cell lines (518A2, 518A2, HT-29, DLD-1, HCT-116, KB-V1Vbl, MCF-7Topo) with IC50 values ranging from 0.11 to 14.2 µM. It should be noted that 7a-c were also active against Ea.Hy926 cells (endothelial hybrid cells), indicating their potential antivascular capacity as CA-4. Besides, 7a-c showed strong selectivity towards cancer cells, contrasting with their lack of major cytotoxicity in non-malignant HDFa dermal fibroblasts.
Tubulin polymerization assays were then achieved with 7a-c, revealing 7a as the most effective tubulin polymerization inhibitor at the concentration of 10 µM. Similar results were observed in 518A2 melanoma cells, with a disruption of the microtubule network at 0.5 µM of 7a. The HDAC inhibitory properties of 7a-c were next studied. Although 7a was the most effective compound against cancer cells and microtubules, only moderate HDAC 1 and HDAC 6 inhibition were noted. On the contrary, 7c possesses a high specificity for HDAC 1 and HDAC 6, with IC50 values of 0.49 and 0.32 µM respectively. These data were validated by the molecular docking with stronger affinities for HDAC 1 and HDAC 6 for molecules bearing long alkyl linkers (7c vs 7a) Western-blot analyses highlighted that 7c could also impede the acetylation of tubulin, without disturbing the microtubule skeleton, due to its concomitant HDAC 6 inhibitory activity. The FACS data on 518A2 melanoma cells corroborated the effect of 7a-c on cell cycle: 7a and 7b led to the interruption of the cell growth on the G2/M phase, while 7c caused arrest at G1 phase. The proof of concept was then concretized with 7a in in vivo nude mice model. The treatment was well-tolerated at high doses (up to 200 mg/kg orally), without causing weight loss [31].
2.5. 1,4-Diarylazetidin-2-one derivatives
Based on the chiral 1,4-diarylazetidin-2-one structure, an constrained analog of CA-4, capable of inhibit tubulin by binding the colchicine site [32], and the Vorinostat skeleton, Ding, Wang and coworkers designed an original skeleton as chimeric inhibitors [33] (Scheme 9). The synthesis of these molecules started with a Michael addition. The alcohol intermediate was subsequently condensed with the corresponding carboxylic acid to afford the desired products. In vitro cytotoxicity studies against four tumor cell lines (BE-(2)-C, A549, U87MG and HCT-116), revealed that molecules 8a and 8b exhibited the best antiproliferative activities, with IC50 ranging from 16 to 56 nM. Furhter exploration of the HDAC isoform inhibitory capacity demonstrated a high selectivity towards HDAC 1 and HDAC 8 for 8a and 8b. It should be noted that 8b depicted better IC50 values against HDAC 1 and HDAC 8 compared to 8a. The HDAC inhibitory property of 8b was also confirmed in cellulo through western blot analyses, by the detection of non-deacylated substrates of HDAC 1 and HDAC 8. Compound 8b also acted as a proven TPI, with an IC50 value of 5.4 µM on the in vitro tubulin polymerization inhibition. Molecular docking supported these results with a tight fixation of 8b via hydrophobic interactions between methoxyphenyl groups and the apolar binding site of tubulin. The interaction between 8b and HDAC 8 was ascertained by the chelation of the hydroxamic moiety with the Zn2+ localized in the active pocket and the hydrogen bonds with key amino acids (Tyr341, His141 and His142). Additionally, 8b could inhibit the cancer cell colony formation at a concentration of 100 nM. A thorough inspection of 8b’s biological pathway suggested that 8b induced G2/M mitosis arrest and caused the cell apoptosis by activating the cleavage of proapoptotic proteins PARP-1 and Bax. The microsomal stability of 8b was also evaluated, revealing a long half-life (t1/2 = 68.8 min) and an intrinsic clearance of 18.1 mL/min/kg was detected in human liver microsomes. The antitumor activities of 8b were endorsed in human neuroblastoma xenograft models, with the significant decrease of the tumor growth at a dose of 25 mg/kg with a TGI of 67%. Notably, 8b demonstrated synergistic efficiency with better TGI values than those observed with paclitaxel and Vorinostat used alone. The in vivo low toxicity of 8b was verified in nude mice, with minor weight loss or adverse effects noted.
2.6. Arylpyridine derivatives
In 2022, Ding, Wang and their colleagues proposed a new dual antitumoral agent presenting a diarylpyridine scaffold (Scheme 10).[34] The rational design of these structures relied on the association between pyridine-bridged analogs of CA-4, which exhibited comparable biological properties as CA-4 [35], with hydroxamic acid as HDACi motif. The synthesis of these frameworks involved the use of commercially available 2,6-dibromopyridine, which underwent two Suzuki-Miyaura cross-couplings to form the corresponding diarylpyridine. The branching of the HDACi pharmacophore was established via an alkylation step or a Heck coupling, followed by amide condensation. The resulting molecules were then submitted to diverse in vitro biological assays. After a screening of the antiproliferative activities against four cancer cell lines (BE-(2)–C, A549, U87MG and HCT-116), compound 9 was selected as the hit compound with IC50 values ranging from 17 to 90 nM. The dual property of 9 was then scrutinized. On one hand, 9 presented a high specificity for HDAC 8 (IC50 = 117 nM). On the other hand, 9 significantly inhibited tubulin polymerization with a IC50 value of 2.4 µM. In cellulo experiments showed a disruption of the microtubule network at a concentration of 100 nM of 9. By affecting the microtubule structure, 9 could also arrest the cell mitosis at G2/M phase, by upregulating the expression of Cyclin-1 and promote the hyper-phosphorylation of key mitotic proteins (Bubr-1 and Histone 3). 9 could also participate in the apoptosis process, by inducing the formation of cleaved proapoptotic markers (PARP-1 and Bax). The evaluation of the metabolic stability in liver microsome of 9 showed a better half-life and clearance values that CA-4 (54.8 min and 22.8 mL/ min/kg for 9 vs 13.6 min and 89.5 mL/min/kg for CA-4). Finally, compound 9 was tested in vivo on human neuroblastoma xenograft mouse model. At a dose of 50 mg/kg, 9 had a TGI of 61%, which was higher than those observed for paclitaxel or Vorinostat employed alone (N. B.: TGI = 30% at 5 mg/kg of paclitaxel and TGI = 19% at 50 mg/kg of Vorinostat). In comparison with the combination of paclitaxel/SAHA, the TGI of 9 remains higher (61% vs 44%), demonstrating the synergistic effect of the dual compound 9.
More recently, Abdelhafez et al. constructed a library of dual inhibitors combining key motifs of CA-4 with the pharmacophore of vorinostat. The linkage between the main functional groups was ensured by a central cyanopyridine core [36] (Scheme 11). The chemical approach for the preparation of these scaffolds consisted of a multicomponent reaction between 4-methoxyacetophenone, 3,4,5-trimethoxybenzaldehyde and ethyl cyanoacetate. The cyanopyridine derivatives were then submitted to an alkylation reaction and an amide coupling. The antiproliferative assays allowed the identification of the hit compound 10a, with the best IC50 values for UO-31 (IC50 = 3.52 µM) and T47D cell lines (IC50 = 5.50 µM). The HDAC inhibitory activity of the hybrid compounds was then quantified, highlighting that 10b was more efficient than 10a, especially for HDAC 1 and 6. The TPI assay emphasized that 10a has an IC50 value comparable with the one obtained with vinblastine, an anticancer agent with potent TPI capacity (IC50 values of 3.73 and 2.86 µM respectively). The cytotoxicity of 10a and 10b on WI-38 normal cells was also assessed, showing only minor effects (IC50 ~ 30 µM). With satisfactory results with 10a, its mechanism of action was elucidated with complementary biological studies. Thus, the authors pointed out that 10a favored the cell cycle arrest at the G2/M phase, leading to the apoptosis process. The western blotting of the cells stopped at the G2/M phase by 10a demonstrated the diminution of HDAC and tubulin. This outcome showed the clear influence of 10a on cell mitosis arrest via a biological pathway involving HDAC and tubulin.
3. Colchicine derivatives
Colchicine, a natural product extracted from the corms of Colchicum autumnale L., is one of the first potent tubulin destabilizer. Alongside its therapeutic use in familial mediterranean fever and gout, this compound is also an efficient anti-cancer agent by inhibiting mitosis in diseased cells. Although the colchicine efficacy was well-demonstrated in tumor cell lines, its therapeutic index remains narrow. This major drawback results in significant side effects, such as anemia, neutropenia or bone marrow damage [37].
To overcome these issues, intensive research around the colchicine related molecules has grown during the last decade. In this context, tubulin/HDAC dual-targeting inhibitors have been developed.
In 2013, Lu, Chen and coworkers reported the design and synthesis of tubulin/HDAC inhibitors, based on the colchicine and hydroxamic acid scaffolds (Scheme 12). The synthetic route of these dual molecules relied on a three-step synthesis, including two amidation reactions mediated by HATU. The HDAC inhibitory properties were studied for the five elaborated compounds, showing a pan-HDAC selectivity for HDAC 1, 3 and 6, especially for compound 11a, with IC50 values from 0.44 to 0.83 µM. By treating BEL-7402 cells with all these tubulin/HDAC inhibitors, the authors noticed that 11b significantly promoted the cell cycle arrest at G2/M phase at a concentration of 1 µM. The viability of the hybrid molecules was validated by exploring the antiproliferative activities towards five cancer cell lines (A431, A549, HCT-116, MCF-7 and PC-3). 11b was thus detected as the most active compound with IC50 ranging from 0.242 to 4.672 µM [38].
Among the colchicine derivatives employed as HDAC/tubulin inhibitors, the hybrid compounds developed by Fang, Lu and coworkers merged a colchicine core with TPI activity and a benzamide motif for the HDACi property (Scheme 13). The construction of these hybrid structures was based on an amide coupling between the two main chemical entities, using HATU as the coupling agent. The in vitro HDAC inhibitory activity was evaluated for all the synthesized compounds, emphasizing that compound 12a was the most active molecule against HDAC 1, 2 and 3 with IC50 ranging from 0.19 to 1.50 µM. The TPI effect was confirmed with all the prepared hybrid inhibitors, namely 12a and 12b possessing comparable activities with free colchicine at a concentration of 10 µM. These outcomes corroborated with the cell cycle analyses, where 12a and 12b were found to block mitosis at the G2/M phase. The cytotoxicity of 12a and 12b was also explored. 12b exhibited higher antiproliferative activities towards twelve human cancer cell lines (A549, HCT-116, SW620, Hep3B, HeoG2, MHCC97H, SNU-5, SNU-16, MKN-45, PANC-1 and SJSA-1) compared to 12a, with IC50 values from 2 to 106 nM [39].
4. Aminobenzamide core
Based on their previous outcomes in tubulin inhibitors [40], the Xu group reported in 2022 the elaboration of dual molecules displaying an aminobenzamide core capable of inhibiting the microtubule polymerization combined with a hydroxamic acid core as an HDACi (Scheme 14). The in vitro antiproliferative assays showed that compound 13 exhibited significant IC50 values (18 to 30 nM) for six cancer cell lines (K562, HepG2, HCT-116, MDA-MB-231, H22 and MCF-7 cells). Satisfactory results were obtained for the in vitro inhibitory potencies against HDACs of compound 13, with an inhibition rate of 93% at 10 µM. Further investigations around the HDAC subtype validated the pan-HDAC inhibiting property of molecule 13, with a selectivity toward HDAC 1, 2, 3 and 6. Moreover, compound 13 presented a TPI activity with a IC50 value of 4.06 µM, particularly in the colchicine binding site. The concomitant role of compound 13 was validated in HepG2 cells, where the HDAC/tubulin inhibitory effects were identified by western blot analysis. Mechanistic insights revealed that molecule 13 could use several pathways for the cell death, such as its abilities to stop the cell cycle to the G2/M phases, to activate the expression of pro-apoptotic Bad and Bax proteins, to induce the mitochondria membrane potential depolarization or increase ROS levels. Notably, analogously to its TPI precursor, compound 13 featured an important antivascular activity, demonstrated in HUVECs. Finally, the in vivo antitumoral properties of 13 were evaluated in a liver cancer allograft mouse model. A major reduction of the tumor weights by 82% (at an intravenous dose of 20 mg/kg per day) was observed in the presence of 13, corroborating all the in vitro efficacies [41].
5. Amide derivatives
Zaki and coworkers created a novel series of HDAC/tubulin inhibitors integrating amide functional groups [42] (Scheme 15). The synthesis of these compounds relied on an oxazole ring-opening with the corresponding aniline or hydrazide. The conceived products were then biologically evaluated, with the best IC50 values against HepG2 obsereved for 14a and 14b (0.65 and 0.92 µM respectively). In contrast, low toxicities were reported for 14a and 14b normal liver HL-7022 cell lines (IC50 values of 9.62 and 11.09 µM). 14a provided higher HDAC inhibitory activity than 14b, with IC50 values ranging from 47 to 86 nM for HDAC 1 and HDAC 2. A TPI assay was then performed for 14a, affording an IC50 value of 270 nM. This TPI effect was validated in cellulo, with the cell division arrest at the G2/M phase. This phenomenon could lead to HepG2 cell apoptosis after incubating with 14a at a concentration of 0.65 µM for 48 h. This apoptosis pathway was activated through the upregulation of caspases 3 and 7. As caspases 3 and 7 are key markers of apoptosis in mitochondria, the analysis of mitochondrial events was conducted. It was noticed that 14a was able to activate caspases 3 and 7, resulting in mitochondrial apoptosis.
6. Quinolone derivatives
In 2018, Wang et al. conceived a novel series of dual compounds using a quinolone derivative as TPI attached to a hydroxamic acid side chain as HDACi, through a triazole linkage (Scheme 16). More precisely, the group designed the TPI pharmacophore based on the interesting properties of levofloxacin derivatives as antitumor agents [43]. The HDAC inhibition activity was first explored. The prepared dual conjugates presented high affinity for HDAC 1, 2 and 6, especially the molecule 15 with IC50 values ranging from 21 to 41 nM. The TPI evaluation for the hybrid conjugates showed that all the synthesized compounds were more effective than levofloxacin. As previously, compound 15 exhibited the highest efficacy with an IC50 value of 1.79 µM. After the investigation of the in vitro properties of the dual molecules, the whole cell antiproliferative activity was examined. Again, 15 confirmed its antitumor capacity against five cancer cell lines (A549, HepG2, MCF-7, PC-3 and HeLa), with IC50 values ranging from 0.3 to 4.9 µM. Finally, the viability of the HDAC/tubulin inhibitors was established in the healthy epithelial cell line MCF-10A, where negligible toxicity was found for all these compounds [44].
7. Benzofuran scaffold
Based on their previous works on benzofuran derivatives as inhibitors of tubulin polymerization [45], the Romagnoli group envisaged the development of dual molecules using their lead compound TR187, binding the tubulin colchicine site and a hydroxamic acid as an HDACi pharmacophore [46] (Scheme 17). These compounds were synthesized from diversely substituted α-bromo acetophenones, which underwent a cyclization reaction under basic conditions to afford the benzofuran core. The connection with the hydroxamic group was carried out via Heck or Sonogashira couplings. The molecular hybrids thus prepared were engaged in different biological tests. The cytotoxicity studies led to the identification of 16a and 16b as hit molecules exhibiting promising IC50 ranges from 0.4 to 23.5 nM against HeLa, MDA-MB-231, A549, HT-29, and MCF-7 cells. Despite their interesting results, the TPI assay demonstrated that 16c and 16d had the best tubulin inhibitory potencies, with an IC50 value of 0.42 μM. Although 16a and 16b were less effective (IC50 values of 0.58 and 0.49 µM respectively), these compounds remained more potent than CA-4 (IC50 = 0.75 µM). The ability of these molecules to inhibit HDACs revealed only moderate activities towards HDAC 1, 6, 8 and 10. 16a exhibited a pan-HDAC specificity with HDAC 1, 6, and 10 whereas 16b was more selective for HDAC 1 and 16c had a strong affinity for HDAC 6. In contrast, negligible HDAC inhibitory capacity was found for 16d. In view of these outcomes, the authors suggested that the antiproliferative activities noted for these compounds might mainly come from the TPI effect.
8. 2-Benzylideneindanone scaffold
With their long-lasting interest in indanone and benzylideneindanone derivatives as potential anticancer agents [47,48,49], Negi et al. reported a hybrid inhibitor bearing a hydroxamic acid skeleton as an HDACi joined to an indanone core for the TPI activity (Scheme 18). The installation of the benzylideneindanone moiety involved a condensation reaction under basic conditions between 3,4,5-trimethoxybenzaldehyde and trimethoxyacetophenone, followed by a Nazarov cyclization. The linkage with the hydroxamic acid core was provided by a condensation, a saponification, an amidation and a final deprotection step. The synthesized benzylideneindanone derivatives were then submitted to antiproliferative studies against three human cancer cell lines (MCF-7, MDAMB-231 and K562). Compounds 17a and 17b were thus selected for further biological assays, with their satisfactory IC50 values (IC50 = 0.36-49.67 µM). Their low cytotoxicity towards normal Vero cells (IC50 = 100.32 µM and IC50 = 47.23 µM for 17a and 17b respectively) validated their selectivity for malignant cell lines. The cell cycle analyses in the presence of 17b proved that 17b could promote apoptosis by blocking the G2/M and the S phases. Further mechanistic insights indicated a major stabilization effect of tubulin by 17a and 17b at 5 µM. This phenomenon was also noticed in confocal microscopy as a disorganization of the cytoskeletal tubulin network. Finally, the authors described a pan HDAC inhibitory activity for 17a and 17b, with a high specificity of 17b for HDAC 6 at 20 µM, attesting the dual character of these compounds.
Besides, 17a and 17b presented a potential anti-inflammatory property in macrophage cells with a significant reduction at 10 µM in the rates of TNF-α and IL-6 (12 to 29%), indicators of cancer inflammation. Complementary safety aspects were explored for 17a in mice at various concentrations (5-1000 mg/kg), identifying 17a as a tolerable and quite safe agent [50].
9. Aminothiazoles
Building on their recent discoveries in methoxybenzoyl-aryl-thiazole (SMART) derivatives as TPI agents [51], Chen et al. prepared in 2021 a new class of dual HDAC/tubulin inhibitors (Scheme 19). By associating the SMART core with a benzamide scaffold as the HDACi pharmacophore through a multi-step synthesis, a wide range of dual compounds were elaborated and employed in various biological tests. First, the cytotoxic activities of these molecules were evaluated, leading to the identification of compound 18, which was highly potent against HCT-116, B16-F10, Jurkat and A549 cancer cells (IC50 = 30-140 nM). 18 validated its cytotoxicity with an HDAC inhibitor-resistant cancer cell line (YCC3/7) with an IC50 value of 560 nM. The analysis of the inhibition of HDAC isoforms demonstrated a selectivity of 18 towards HDAC 3 (IC50 = 30 nM). This aspect was reinforced in cellular assays, by treating B16-F10 cancer cell line with 18. The increasing rate of the acetylated histone H3 by enhancing the concentration of 18 proved the HDAC inhibitory activity. The in vitro tubulin polymerization assay was then examined for 18. The TPI property of 18 was thus confirmed with an IC50 value of 12.2 µM. This characteristic was also noted by incubating 18 in B16-F10 cells, where 18 was capable of deeply disturbing the microtubule network. 18 was also involved in the inhibition of cancer cell migration, more especially at a concentration of 100 nM. Moreover, 18 could interfere with the cell cycle, by stopping the mitosis at the G2/M phase, thereby inducing apoptosis. In vivo investigations of 18 using a mouse model revealed a significant reduction in tumor growth (TGI = 70%). Furthermore, no notable changes in body weight were observed throughout the treatment period. Further safety assessments indicated that 18 had no significant effects on major organ tissues, including the heart, liver, and kidney.[52].
In 2023, the Schiedel group imagined an original dual molecule with two specific targets: tubulin deacetylases sirtuin 2 (Sirt2) and HDAC 6. As the disruption of the two enzymes activities could promote cancer processes [53], the team conceived a hybrid structure inspired by the Sirt2 and HDAC6 respective selective inhibitors (Scheme 20). Inspired by the Sirt2 inhibitors previously developed in the laboratory [54], Schiedel and his coworkers installed the aminothiazole core by a diazotation step followed by a Meerwein reaction. This motif was then attached to a hydroxamic acid skeleton, the HDACi pharmacophore, via a copper-catalyzed Huisgen cycloaddition. The in vitro biological efficiency of these dual molecules was then analyzed. Among the dozen synthesized compounds, 19 exhibited the highest selectivity and activity towards Sirt2 and HDAC6 with IC50 values of 320 nM and 43 nM respectively. It should be highlighted that the co-crystallization of 19 and its analogs was achieved with the two respective targets, indicating a strong hydrophobic interaction between Sirt2 and the corresponding pharmacophore, but also the coordination to the hydroxamic acid with the Zn atom. In cellulo assays corroborated the in vitro results with a high level of acetylated α-tubulin observed by incubating 20 µM of 19 in PC-3M-cell line. To prove the synergistic effect of the hybrid molecule 19, cell viability assays were performed on cancer cells sensitive to Sirt2 and HDAC 6 inhibition. 19 could decrease the cell viability towards HGC27, W1, MCF-7, and PC-3M-luc cells, with EC50 ranging from 12.9 to 30.1 µM [55].
10. 2-Methoxyestradiol core
2-Methoxyestradiol, a natural metabolite of estradiol, and its derivatives were known as remarkable tubulin polymerization inhibitors and disruptors of the microtubule skeleton [56]. In this frame, Yao et al. described a dual-targeting inhibitor exhibiting a 2-methoxyestradiol scaffold as TPI pharmacophore linked to a 2-aminobenzamide motif as HDACi. After a thorough examination of the SAR for forty-seven synthesized hybrid molecules, 20 was identified as the compound presenting the best cytotoxicities on six cancer cell lines (MCF-7, MGC-803, HeLa, A549, HepG2 and U937), with IC50 ranges from 0.371 to 4.840 µM. The inhibitory activities of 20 towards HDAC isoforms showed higher selectivities for HDAC 2 and 6, with IC50 values of 60 and 120 nM respectively (Scheme 21). With the in vitro immunofluorescence assays, compound 20 proved its ability to disorganize the microtubule network. Moreover, by incubating 20 at the concentration for 4 µM with purified porcin brain tubulin, the tubulin polymerization was significantly stopped. Similarly to the tubulin polymerization inhibitors it was noted that 20 could efficiently halt the cell cycle at the G2/M phase, induce the apoptosis via a mitochondrial membrane potential change, an increase of the ROS rate and the upregulation of pro-apoptotic proteins (cleaved forms of caspase 3, 7, 9 and PARP). Besides, 20 could also hamper the proliferation, migration, and invasion of tumor cells. All these antitumor activities were validated in the in vivo zebrafish xenograft tumor model [57].
11. Millepachine core
More recently, Yin, Kong and their colleagues developed a new family of dual HDAC/tubulin inhibitors, featuring the millepachine, a natural chalcone with TPI activity, branched to a hydroxamic acid structure as HDACi (Scheme 22). After a large molecule-screening against five cancer cell lines (MDA-MB-231, A549, PC-3, U251 and MCF-7), the compound 21 was determined as the best molecule with highest cytotoxicities, with a notable IC50 value of 16 nM for the human prostate cancer cell line PC-3. The HDAC inhibition activities were then inspected, showing that 21 possessed 67% inhibition of HDACs at a concentration of 1 µM. Furthermore, 21 has a pan-HDAC inhibitory activity towards HDAC 1, 2 and 6. The TPI properties of 21 was validated with purified tubulin protein, with an IC50 value of 4.82 µM. This experiment was also confirmed by immunofluorescence assay, where the intracellular microtubules were disassembled by incubation with 21. The biological 21 action was then clarified by flow cytometry assay and Western blot analysis. The authors demonstrated that 21 could interrupt the cell cycle at the G2/M phase, by enhancing the cleaved PARP and Caspase 3 levels and diminishing Bim and Bcl-2 rates. 21 could also inhibit the cell migration of tumor cells and could be involved in the mitochondrial apoptosis process, by reducing the mitochondrial membrane potential and increasing the ROS levels. Besides, an anti-angiogenesis effect was detected with 21, since the decrease of the HUVECs capillary-like tubular network was observed in the presence of 21. Finally, all the in vitro antitumor aspects of 21 were ascertained in the in vivo PC-3 mice xenografts, with a TGI of 90% in a 21 dosage of 20 mg/kg [58].
12. Deoxypodophyllotoxin derivatives
In 2014, Chen, Li, Lu and coworkers proposed a dual HDAC/tubulin inhibitor, integrating a podophyllotoxin (PPT) analog platform. Based on the design of their topoisomerase II/HDAC hybrid inhibitors [59], the team elaborated an analogous approach for the construction of the novel molecules. With the well-known TPI properties of deoxypodophyllotoxin (DPT) [60,61] combining with the HDACi activities of benzamides, a novel family of molecular hybrids could be generated (Scheme 23). The DPT core was synthesized from PPT, in three steps under standard conditions [62]. The benzamide structure was then linked to the DPT group via an amidation using HATU as the coupling agent. The in vitro HDAC inhibition of the established molecules disclosed a pan inhibitory activity for 22a and 22b towards HDAC 1, 2 and 3 (IC50 = 0.75-11.09 µM). By treating the HCT-116 cells with the dual HDAC/tubulin inhibitors at 40 or 80 nM for 24 h, the authors remarked an accumulation of cells in the G2/M phase, hinting the cell cycle blocking by a TPI effect. The cytotoxicity of the prepared structures validated the biological activities of 22a and 22b in A549 and HCT-116 cancer cell lines, with IC50 values ranging from 36 to 40 nM [63].
13. Paclitaxel scaffold
In view of the high efficiency of paclitaxel as FDA-approved anticancer drug and its activity as tubulin polymerization inhibitor [64], it would seem obvious to combine this molecule with a FDA-approved HDACi, such as vorinostat. This idea came up in the Chen and Lu groups, with the conjugation of paclitaxel with vorinostat through a glycine or a succinic acid linker [65] (Scheme 24). The cytotoxicity of synthesized dual inhibitors were determined. Surprisingly, compound 23 bearing the glycine motif possessed higher antiproliferative activities against drug-resistant cancer cell line MCF-7/ADR from compared to paclitaxel used alone, with IC50 value of 1384 nM. This promising result had prompted the research teams to explore the synergistic effect of 23 in the cellular level. 23 was able to step up the G2/M cell cycle arrest compared to the standard influence of paclitaxel. Western-blot analyses indicated that 23 induced a hyperacetylation of tubulin in HCT-116, MCF-7 and MCF-7/ADR cells at a concentration of 10 nM.
14. Sulfonamide scaffold
The sulfonamide skeleton could be found in the structure of E-7010, an orally active antitumor compound developed by Eisai company. This molecule provided promising biological results, namely its tubulin polymerization inhibition by binding in the colchicine binding site [66]. Based on these, Chen, Liou and coworkers envisioned the construction of hybrid agents, including a sulfonamide group for the TPI activity and a benzamide function as HDACi (Scheme 25). The connection between the two chemical entities was ensured by a stilbene link. As this motif is not a substrate of membrane-bound P-glycoprotein (P-gp), the stilbene could bypass the P-gp-mediated multidrug resistance pathway. With this hypothesis in hand, twenty-two dual molecules were prepared and biologically evaluated. The antiproliferative activities revealed that compound 24 was the most cytotoxic agent against KB cell lines, with a GI50 value of 12 nM. It should be highlighted that the same results were noticed in drug-resistant KB cancer cell lines. The HDAC isoform inhibition showed a strong selectivity of 24 for HDAC 1 and 2, with IC50 values of 1.07 and 1.47 µM respectively. Mechanistic insights disclosed that 24 could activate pro-apoptotic markers, such as activated forms of PARP, γH2AX, caspase 3, 8 and 9 in KB cell lines. With the in vitro complementary analyses in flow cytometry and fluorescence microscopy, the authors found that 24 blocked the cell cycle at the G2/M phase, with a microtubule disassembly [67].
15. Indoline/indole-sulfonamide scaffold
With the promising biological properties of indoline-sulfonamide derivatives as tubulin inhibitors [68], Liou, Chang and coworkers imagined a hybrid dual molecule incorporating this scaffold and a benzamide group as an efficient HDAC pharmacophore (Scheme 26). The two functional motifs were associated with a benzyl linker, installed through a reductive amination conducted under standard conditions. Twenty-one dual compounds were thus synthesized and submitted to biological assays. The antiproliferative activities were determined, with compound 25 as the most cytotoxic compound against KB, A549 and MKN45 cancer cell lines (IC50 = 49-79 nM). The cell-killing capacity of 25 was attested in oral epidermoid carcinoma drug-resistant cell lines (KBVIN10, KB-S15 and KB-7D), with IC50 ranges from 44 to 65 nM. The TPI ability of the prepared hydrid molecules was also measured. Again, 25 was identified as the best tubulin inhibitor with an IC50 value of 1 µM. Further examinations showed that 25 had a better affinity in the colchicine site compared to the colchicine itself, at the concentration of 1 or 5 µM. HDAC isoforms selectivity was detected for 25, especially with a significant inhibition of HDAC 1, 2 and 6. Moreover, the HDACi efficiency of 25 was verified in A549 cells, with the accumulation of acetylated α-tubulin in the presence of 25. Finally, the dual inhibitory feature of 25 was established in an in vivo A549 xenograft mouse model, with a remarkable reduction of the tumor (TGI = 62.9% with a 25 dosage of 50 mg/kg). Similar results were also found with the B-cell lymphoma xenograft tumor mouse model [69].
The same group envisioned a library of dual active compounds fusing a potent tubulin assembly inhibitor currently in phase II clinical trials [70] with a HDACi bearing a indoline-sulfonamide structure [71] (Scheme 27). The construction of the hybrid molecules was based on a three-step synthesis featuring an alkylation step, a Heck coupling and an amide condensation [72]. The developed structures demonstrated an antitumor activity against A549, HCT-116 and PC-3 cancer cell lines (IC50 values as low as 33 nM) for compound 26a. In contrast, despite remarkable cytotoxic properties, the tubulin polymerization inhibitory property of 26a remained less important than 26b. Besides, 26a and 26b displayed a high HDAC 6 specificity, with IC50 values of 275 and 64.5 nM respectively. The in vivo experiments on PC-3 xenograft mice models validated the efficacy of 26b, with the suppression of tumor growth at doses of 100 and 200 mg/kg (TGI of 24.8% and 68.5% respectively). Similar outcomes were obtained with multiple myeloma RPMI-8226 xenografts (TGI values of 35.8% and 58.2% for doses of 50 and 100 mg/kg daily, respectively).
Emulated by the previous research of the Liou team, Fu et al. deepened the investigation of the properties of a chimeric indole-sulfonamide 26a [73] (Scheme 27). 26a also showed a significant antiproliferative activity against liquid tumor cell lines such as HL-60, with IC50 value of 42 nM. Molecular docking validated the dual targeting of 26a, with a partial overlap with colchicine in the tubulin binding site, and analogous interactions with Vorinostat in HDAC active site. Further biological analyses indicated that this molecule was able to stop the mitosis process at G2/M phase, by varying significantly the G2/M transition proteins rates. Along with this aspect, 26a could induce cell apoptosis, through an upregulation of proapoptotic proteins (cleaved caspases 3, 7, 8, 9 and PARP). Mouse xenograft models confirmed the antitumor effects of 26a, with TGI values of 31.1 and 40.9% in PC-3 and HL-60 grafted mice respectively.
In parallel with the conventional molecular hydrid inhibitors connecting TPI and HDACi pharmacophores, some original dual molecules have been recently conceived according to their HDACi chemical structure.
16. Dual HDAC/tubulin inhibitors inspired by the HDACis
Among the wide diversity of the developed dual HDAC/tubulin inhibitors, the ones originated from HDACis constitute an important molecule family. In addition to their main function on histones, HDACs could also react with non-histone proteins, namely α-tubulin. For example, HDAC 6 could use tubulin as a substrate, thus regulating the stability of the microtubule network [74,75,76]. Therefore, the construction of HDACis with a dual HDAC/tubulin action has emerged. In this frame, the medicinal chemists mainly based their compounds design on HDACis developed in the literature. The tubulin inhibitory activity of the synthesized molecules was systematically studied alongside with diverse biological assays.
16.1. Pyrrolo[2,3-d]pyrimidine skeleton
Emulated by the structure of Tucidinostat and their recent advances in the development of HDACis [77], Lee, Liou and their colleagues envisaged at the first glance an ameliorated version of their HDACis (Scheme 28). In this prospect, the team elaborated a rigid and bulky pyrrolo[2,3-d]pyrimidine structure at the cap part of the HDACis, improving the anticancer properties of the molecules by blocking efficiently the active site entrance [77]. As some reported HDACis also targeted tubulin [78], the group considered their newly designed HDACis as potential dual hybrid HDAC/tubulin inhibitors. The in vitro cytotoxicities of these molecules were then studied. Among the nine synthesized compounds, the molecules 27a and 27b disclosed the highest antiproliferative activities towards eight solid tumor cell lines (MDA-MB-231, MDA-MB-468, HeLa, DLD-1, HCT-116, H661, H1299 and A549), with IC50 values from 50 to 1150 nM. It should be noteworthy that these cytotoxicity properties were higher than the Tucidinostat ones. Likewise, 27a and 27b confirmed their antitumor effects in leukemia cell lines (HH, HuT78, HL60 and KG-1), with IC50 values ranging from 80 to 150 nM. Moreover, 27a exhibited an important cell-killing efficacy in multi drug-resistant cell line MES-SA/Dx5, with a IC50 value of 9.54 µM, three times higher than the vinorelbine one.
The analysis of HDAC isoforms inhibitory activities highlighted that 27a had a strong selectivity for HDAC 1 and 2, whereas 27b showed a wider pan-HDAC inhibitory efficiency for HDAC 1, 2, 3 and 8. Mechanistic studies through flow cytometry suggested that 27a and 27b could promote the tumor cells apoptosis, by inducing the production of active forms of caspases-3 and -9 and reducing the formation of anti-apoptotic agents MCL-1 and Bcl-xL. The TPI of 27a and 27b was also scrutinized, with the in vitro tubulin polymerization assay. By incubating tubulin proteins with 10 µM of 27a and 27b, the authors noted that these compounds were able to jeopardize the microtubule assembly. It should be noted that no in vitro inhibition was found for HDAC 6 for 27a and 27b, implying an independent TPI pathway for the two dual compounds [79].
16.2. Benzamide scaffold
In 2021, He, Chen and coworkers described LT-548-133-1, a tucidinostat analog possessing an equivalent HDAC inhibitory activity [80] (Scheme 29). By incubating LT-548-133-1 with MCF-7 cells for 48 h, an IC50 value of 2.1 mM was found. Further investigations indicated that LT-548-133-1 could enhance the rates of acetylated histone H3, probably due to its HDAC inhibitory property. Moreover, LT-548-133-1 promoted the cell cycle arrest at G2/M phase, contrary to tucidinostat which induced G0/G1 cell mitosis arrest. This result indicated that LT-548-133-1 might follow a different mechanism of action compared to tucidinostat. Complementary western-blot analyses specified that the G2/M cell cycle arrest should be induced by the increase of the CyclinB1 protein expression. Besides, LT-548-133-1 could also lead to abnormal cell mitosis and apoptosis in MCF-7 cell lines. With all these effects on the mitosis process, the authors suggested that LT-548-133-1 could interfere with the microtubule network. Putting LT-548-133-1 with MCF-7 cells revealed by immunofluorescence the destructuration of the microtubules, proving the TPI effect of LT-548-133-1.
16.3. Quinazoline scaffold
By scrutinizing the biological pathway of their previously developed SKLB-23bb [81], an HDAC 6 selective inhibitor, the Chen group shed light that this molecule could have a dual-targeting activity [82].
SKLB-23bb integrates in its structure a 2-methylquinazoline core, known key bioactive skeleton,[83,84] linked to a hydroxamic acid group through an alkyl linker (Scheme 30). SKLB-23bb was afforded through a SNAr reaction between a highly functionalized aniline, followed by an amide condensation.[81] This molecule featured a high cytotoxic potential, with nanomolar-range IC50 values (36.68 to 116.56 nM) for a wide panel of liquid and solid cancer cell lines. In addition to its HDAC 6 inhibitory property, SKLB-23bb also acted via another biological pathway to hamper the cancer cells. In fact, the cytotoxic effects of SKLB-23bb remained similar in HDAC 6 knockout tumor cell lines compared to the wild type cells. To clarify the exact mechanism of action of this molecule, the incubation of SKLB-23bb in fluorescent-stained A2780s cell line was carried out. Major morphologic modifications in the microtubule network were noted, and this microtubule dysfunction was similarly observed by adding colchicine to the A2780s cells. This result could suggest that this molecule could also bind the colchicine site of tubulin to block the polymerization process. The validation of this hypothesis was seen in the N,N’-ethylene-bis(iodoacetamide) (EBI) competition assay. Complementary TPI test demonstrated that SKLB-23bb could inhibit tubulin polymerization at a concentration of 10 µM. As a result of this biological characteristic, SKLB-23bb could stop the cell cycle at G2/M phase and trigger the apoptosis by promoting the upregulation of pro-apoptotic proteins, such as Bax.
The effectiveness of SKLB-23bb was preserved in vivo on xenograft mice imitating B-lymphoma model, with a tumor growth inhibition (58.22% tumor-inhibitory rate) at a dose of 40 mg/kg. Analogous results were afforded with solid tumor models (HCT-116, A2780, and MCF-7 xenografts).
16.4. Imidazolyl motif
In 2021, Taylor, Tillekeratne et al. built a novel class of heterocyclic compounds with a double action on HDACs and tubulin. Inspired by the imidazole derivatives and the FDA-approved hydroxamic acids as HDACi [85,86,87], the group introduced an imidazolyl skeleton with potential HDAC inhibitory activity. To strengthen the dual property of the newly conceived compounds, a chalcone scaffold was added to the molecule’s structure for the antimitotic activity (Scheme 31) [88]. The synthesis of these dual inhibitors was realized via a multi-step synthesis, including a Heck cross-coupling reaction, an olefin oxidative cleavage and an aldol condensation as key steps. A first preliminary biology study on HCT-116 cell line was conducted with the prepared compounds, confirming their cytotoxicity at a micromolar range (IC50 = 5.14-6.95 µM). The HDAC isoforms inhibitory effects were then evaluated. The in vitro results highlighted that compounds 28a and 28c had a pan-HDAC inhibitory character towards HDAC 1, 2, 3, 6 and 8. In contrast, the molecule 28b was a selective HDACi for HDAC 8. A similar analysis was carried out by incubating the synthesized compounds with HCT-116 cells. Unexpectedly, no significant variation in the histone or tubulin acetylation was noticed, implying that these molecules did not mainly target HDACs. Despite these outcomes, complementary assays were performed to evaluate their cytotoxicity against a wide range of cancer cell lines. The compounds demonstrated their effectiveness in HeLa and NCI-60 cancer cell lines, with micromolar GI50 values. The examination of the antimitotic activity of 28a in HeLa cells unveiled that 28a was able to arrest the mitosis process, by destabilizing microtubules. Molecular docking studies indicated a probable interaction between 28a and tubulin in the colchicine binding site [89].
17. Conclusions
The rise of the dual-targeting drugs during the last decades represents a promising alternative to conventional chemotherapeutics in cancer treatment. By associating two pharmacophores in one anticancer agent, medicinal chemists aim to target “two birds in one stone”, thereby overcoming the limitations of the traditional chemotherapy, such as excessive toxicity, the diminution of immunity and the development of drug resistance.
As highlighted in this review, tubulin and HDACs proved to be highly effective and complementary targets for the development of dual active agents. The remarkable efficacy of the described dual molecules is evident from their outstanding in vitro antiproliferative activities, with nanomolar IC50 values across various human cancer cell lines. These compounds provided a pan-HDAC inhibitory property, notably for HDAC 1, 2 and 6, and effectively inhibited tubulin polymerization, which further led to the arrest of the cell cycle at the G2 phase. Some of the HDAC/tubulin inhibitors, such as CA-4 derivatives, also display potent anti-angiogenic properties, contributing to their antitumor characteristics. In addition to their synergistic effects towards HDACs and tubulin, these inhibitors showed minimal toxicity at both the cellular and in vivo levels, highlighting their potential for therapeutic application. However, despite their promising preclinical results, no dual HDAC/tubulin inhibitors have entered clinical phase trials yet, since their pharmacokinetics are still under investigation. Hence, the conception of dual-targeting drug candidates remains more than ever a real challenge in cancer therapy. The elaboration of such molecules would require substantial efforts to synthesize the lead compounds, along with their thorough pharmacomodulation to reach the pharmacokinetics and toxicity criteria.
References
- Sung, H.; Ferlay, J.; Siegel, R. L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209-249. [CrossRef]
- Fraga, M. F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; Iyer, N. G.; Pérez-Rosado, A.; Calvo, E.; Lopez, J. A.; Cano, A.; Calasanz, M. J.; Colomer, D.; Piris, M. A.; Ahn, N.; Imhof, A.; Caldas, C.; Jenuwein, T.; Esteller, M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391-400. [CrossRef]
- Huang, M.; Huang, J.; Zheng, Y.; Sun, Q. Histone acetyltransferase inhibitors: An overview in synthesis, structure-activity relationship and molecular mechanism. Eur. J. Med. Chem. 2019, 178, 259-286. [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8. [CrossRef]
- He, X.; Hui, Z.; Xu, L.; Bai, R.; Gao, Y.; Wang, Z.; Xie, T.; Ye, X.-Y. Medicinal chemistry updates of novel HDACs inhibitors (2020 to present). Eur. J. Med. Chem. 2022, 227, 113946. [CrossRef]
- Squarzoni, A.; Scuteri, A.; Cavaletti, G. HDACi: The Columbus’ Egg in Improving Cancer Treatment and Reducing Neurotoxicity? Cancers 2022, 14, 5251. [CrossRef]
- Yoon, S.; Eom, G. H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1-11. [CrossRef]
- Rai, S.; Kim, W. S.; Ando, K.; Choi, I.; Izutsu, K.; Tsukamoto, N.; Yokoyama, M.; Tsukasaki, K.; Kuroda, J.; Ando, J.; Hidaka, M.; Koh, Y.; Shibayama, H.; Uchida, T.; Yang, D. H.; Ishitsuka, K.; Ishizawa, K.; Kim, J. S.; Lee, H. G.; Minami, H.; Eom, H. S.; Kurosawa, M.; Lee, J. H.; Lee, J. S.; Lee, W. S.; Nagai, H.; Shindo, T.; Yoon, D. H.; Yoshida, S.; Gillings, M.; Onogi, H.; Tobinai, K. Oral HDAC inhibitor tucidinostat in patients with relapsed or refractory peripheral T-cell lymphoma: phase IIb results. Haematologica 2023, 108, 811-821. [CrossRef]
- Zhang, X.-H.; Qin, M.; Wu, H.-P.; Khamis, M. Y.; Li, Y.-H.; Ma, L.-Y.; Liu, H.-M. A Review of Progress in Histone Deacetylase 6 Inhibitors Research: Structural Specificity and Functional Diversity. J. Med. Chem. 2021, 64, 1362-1391. [CrossRef]
- Peng, X.; Sun, Z.; Kuang, P.; Chen, J. Recent progress on HDAC inhibitors with dual targeting capabilities for cancer treatment. Eur. J. Med. Chem. 2020, 208, 112831. [CrossRef]
- de Lera, A. R.; Ganesan, A. Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin. Epigenet. 2016, 8, 105. [CrossRef]
- Soltan, O. M.; Shoman, M. E.; Abdel-Aziz, S. A.; Narumi, A.; Konno, H.; Abdel-Aziz, M. Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy. Eur. J. Med. Chem. 2021, 225, 113768. [CrossRef]
- Dumontet, C.; Jordan, M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790-803. [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D. D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943-2971. [CrossRef]
- Zuco, V.; De Cesare, M.; Cincinelli, R.; Nannei, R.; Pisano, C.; Zaffaroni, N.; Zunino, F. Synergistic Antitumor Effects of Novel HDAC Inhibitors and Paclitaxel In Vitro and In Vivo. PLOS ONE 2011, 6, e29085. [CrossRef]
- Chao, M.-W.; Lai, M.-J.; Liou, J.-P.; Chang, Y.-L.; Wang, J.-C.; Pan, S.-L.; Teng, C.-M. The synergic effect of vincristine and vorinostat in leukemia in vitro and in vivo. J. Hematol. Oncol. 2015, 8, 82. [CrossRef]
- Pettit, G. R.; Singh, S. B.; Hamel, E.; Lin, C. M.; Alberts, D. S.; Garcia-Kendal, D. Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 1989, 45, 209-211. [CrossRef]
- Lin, C. M.; Ho, H. H.; Pettit, G. R.; Hamel, E. Antimitotic natural products combretastatin A-4 and combretastatin A-2: studies on the mechanism of their inhibition of the binding of colchicine to tubulin. Biochemistry 1989, 28, 6984-6991. [CrossRef]
- McGown, A. T.; Fox, B. W. Differential cytotoxicity of combretastatins A1 and A4 in two daunorubicin-resistant P388 cell lines. Cancer Chemother. Pharmacol. 1990, 26, 79-81. [CrossRef]
- Tseng, C.-H.; Li, C.-Y.; Chiu, C.-C.; Hu, H.-T.; Han, C.-H.; Chen, Y.-L.; Tzeng, C.-C. Combretastatin A-4 derivatives: synthesis and evaluation of 2,4,5-triaryl-1H-imidazoles as potential agents against H1299 (non-small cell lung cancer cell). Mol. Divers. 2012, 16, 697-709. [CrossRef]
- Wang, B.; Chen, X.; Gao, J.; Su, L.; Zhang, L.; Xu, H.; Luan, Y. Anti-tumor activity evaluation of novel tubulin and HDAC dual-targeting inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 2638-2645. [CrossRef]
- Mourad, A. A. E.; Mourad, M. A. E.; Jones, P. G. Novel HDAC/Tubulin Dual Inhibitor: Design, Synthesis and Docking Studies of α-Phthalimido-Chalcone Hybrids as Potential Anticancer Agents with Apoptosis-Inducing Activity. Drug Des. Devel. Ther. 2020, 14, 3111-3130. [CrossRef]
- Sylvie, D. Antimitotic Chalcones and Related Compounds as Inhibitors of Tubulin Assembly. Anticancer Agents Med. Chem. 2009, 9, 336-347.
- Belluti, S.; Orteca, G.; Semeghini, V.; Rigillo, G.; Parenti, F.; Ferrari, E.; Imbriano, C. Potent Anti-Cancer Properties of Phthalimide-Based Curcumin Derivatives on Prostate Tumor Cells. Int. J. Mol. Sci. 2019, 20, 28. [CrossRef]
- Wang, Y.; Sun, M.; Wang, Y.; Qin, J.; Zhang, Y.; Pang, Y.; Yao, Y.; Yang, H.; Duan, Y. Discovery of novel tubulin/HDAC dual-targeting inhibitors with strong antitumor and antiangiogenic potency. Eur. J. Med. Chem. 2021, 225, 113790. [CrossRef]
- Khelifi, I.; Naret, T.; Renko, D.; Hamze, A.; Bernadat, G.; Bignon, J.; Lenoir, C.; Dubois, J.; Brion, J.-D.; Provot, O.; Alami, M. Design, synthesis and anticancer properties of IsoCombretaQuinolines as potent tubulin assembly inhibitors. Eur. J. Med. Chem. 2017, 127, 1025-1034. [CrossRef]
- Lamaa, D.; Lin, H.-P.; Zig, L.; Bauvais, C.; Bollot, G.; Bignon, J.; Levaique, H.; Pamlard, O.; Dubois, J.; Ouaissi, M.; Souce, M.; Kasselouri, A.; Saller, F.; Borgel, D.; Jayat-Vignoles, C.; Al-Mouhammad, H.; Feuillard, J.; Benihoud, K.; Alami, M.; Hamze, A. Design and Synthesis of Tubulin and Histone Deacetylase Inhibitor Based on iso-Combretastatin A-4. J. Med. Chem. 2018, 61, 6574-6591. [CrossRef]
- Hauguel, C.; Ducellier, S.; Provot, O.; Ibrahim, N.; Lamaa, D.; Balcerowiak, C.; Letribot, B.; Nascimento, M.; Blanchard, V.; Askenatzis, L.; Levaique, H.; Bignon, J.; Baschieri, F.; Bauvais, C.; Bollot, G.; Renko, D.; Deroussent, A.; Prost, B.; Laisne, M.-C.; Michallet, S.; Lafanechère, L.; Papot, S.; Montagnac, G.; Tran, C.; Alami, M.; Apcher, S.; Hamze, A. Design, synthesis and biological evaluation of quinoline-2-carbonitrile-based hydroxamic acids as dual tubulin polymerization and histone deacetylases inhibitors. Eur. J. Med. Chem. 2022, 240, 114573. [CrossRef]
- Zhu, H.; Zhu, W.; Liu, Y.; Gao, T.; Zhu, J.; Tan, Y.; Hu, H.; Liang, W.; Zhao, L.; Chen, J.; Zhu, Z.; Chen, J.; Xu, J.; Xu, S. Synthesis and bioevaluation of novel stilbene-based derivatives as tubulin/HDAC dual-target inhibitors with potent antitumor activities in vitro and in vivo. Eur. J. Med. Chem. 2023, 257, 115529. [CrossRef]
- Li, Y.-R.; Liu, F.-F.; Liu, W.-B.; Zhang, Y.-F.; Tian, X.-Y.; Fu, X.-J.; Xu, Y.; Song, J.; Zhang, S.-Y. A novel aromatic amide derivative SY-65 co-targeted tubulin and histone deacetylase 1 with potent anticancer activity in vitro and in vivo. Biochem. Pharmacol. 2022, 201, 115070. [CrossRef]
- Schmitt, F.; Gosch, L. C.; Dittmer, A.; Rothemund, M.; Mueller, T.; Schobert, R.; Biersack, B.; Volkamer, A.; Höpfner, M. Oxazole-Bridged Combretastatin A-4 Derivatives with Tethered Hydroxamic Acids: Structure–Activity Relations of New Inhibitors of HDAC and/or Tubulin Function. Int. J. Mol. Sci. 2019, 20, 383. [CrossRef]
- Zhou, P.; Liang, Y.; Zhang, H.; Jiang, H.; Feng, K.; Xu, P.; Wang, J.; Wang, X.; Ding, K.; Luo, C.; Liu, M.; Wang, Y. Design, synthesis, biological evaluation and cocrystal structures with tubulin of chiral β-lactam bridged combretastatin A-4 analogues as potent antitumor agents. Eur. J. Med. Chem. 2018, 144, 817-842. [CrossRef]
- Tang, H.; Liang, Y.; Yu, M.; Cai, S.; Ding, K.; Wang, Y. Discovery of chiral 1,4-diarylazetidin-2-one-based hydroxamic acid derivatives as novel tubulin polymerization inhibitors with histone deacetylase inhibitory activity. Bioorg. Med. Chem. 2023, 92, 117437. [CrossRef]
- Tang, H.; Liang, Y.; Shen, H.; Cai, S.; Yu, M.; Fan, H.; Ding, K.; Wang, Y. Discovery of a 2,6-diarylpyridine-based hydroxamic acid derivative as novel histone deacetylase 8 and tubulin dual inhibitor for the treatment of neuroblastoma. Bioorg. Chem. 2022, 128, 106112. [CrossRef]
- Zheng, S.; Zhong, Q.; Mottamal, M.; Zhang, Q.; Zhang, C.; LeMelle, E.; McFerrin, H.; Wang, G. Design, Synthesis, and Biological Evaluation of Novel Pyridine-Bridged Analogues of Combretastatin-A4 as Anticancer Agents. J. Med. Chem. 2014, 57, 3369-3381. [CrossRef]
- El-Zoghbi, M. S.; Bass, A. K. A.; A Abuo-Rahma, G. E.-D.; Mohamed, M. F. A.; Badr, M.; Al-Ghulikah, H. A.; Abdelhafez, E.-S. M. N. Design, Synthesis and Mechanistic Study of New Dual Targeting HDAC/Tubulin Inhibitors. Future Med. Chem. 2024, 16, 601-622. [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D. D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943-2971. [CrossRef]
- Zhang, X.; Zhang, J.; Tong, L.; Luo, Y.; Su, M.; Zang, Y.; Li, J.; Lu, W.; Chen, Y. The discovery of colchicine-SAHA hybrids as a new class of antitumor agents. Bioorg. Med. Chem. 2013, 21, 3240-3244. [CrossRef]
- Zhang, X.; Kong, Y.; Zhang, J.; Su, M.; Zhou, Y.; Zang, Y.; Li, J.; Chen, Y.; Fang, Y.; Zhang, X.; Lu, W. Design, synthesis and biological evaluation of colchicine derivatives as novel tubulin and histone deacetylase dual inhibitors. Eur. J. Med. Chem. 2015, 95, 127-135. [CrossRef]
- Zhu, H.; Li, W.; Shuai, W.; Liu, Y.; Yang, L.; Tan, Y.; Zheng, T.; Yao, H.; Xu, J.; Zhu, Z.; Yang, D.-H.; Chen, Z.-S.; Xu, S. Discovery of novel N-benzylbenzamide derivatives as tubulin polymerization inhibitors with potent antitumor activities. Eur. J. Med. Chem. 2021, 216, 113316. [CrossRef]
- Zhu, H.; Tan, Y.; He, C.; Liu, Y.; Duan, Y.; Zhu, W.; Zheng, T.; Li, D.; Xu, J.; Yang, D.-H.; Chen, Z.-S.; Xu, S. Discovery of a Novel Vascular Disrupting Agent Inhibiting Tubulin Polymerization and HDACs with Potent Antitumor Effects. J. Med. Chem. 2022, 65, 11187-11213. [CrossRef]
- Al-Warhi, T.; Aldhahrani, A.; Althobaiti, F.; Fayad, E.; Abu Ali, O. A.; Albogami, S.; Abu Almaaty, A. H.; Khedr, A. I. M.; Bukhari, S. N. A.; Zaki, I. Design, Synthesis and Cytotoxic Activity Evaluation of Newly Synthesized Amides-Based TMP Moiety as Potential Anticancer Agents over HepG2 Cells. Molecules 2022, 27, 3960.
- Korolyov, A.; Dorbes, S.; Azéma, J.; Guidetti, B.; Danel, M.; Lamoral-Theys, D.; Gras, T.; Dubois, J.; Kiss, R.; Martino, R.; Malet-Martino, M. Novel lipophilic 7H-pyrido[1,2,3-de]-1,4-benzoxazine-6-carboxylic acid derivatives as potential antitumor agents: improved synthesis and in vitro evaluation. Bioorg. Med. Chem. 2010, 18, 8537-8548. [CrossRef]
- Wang, X.; Jiang, X.; Sun, S.; Liu, Y. Synthesis and biological evaluation of novel quinolone derivatives dual targeting histone deacetylase and tubulin polymerization as antiproliferative agents. RSC Adv. 2018, 8, 16494-16502. [CrossRef]
- Romagnoli, R.; Baraldi, P. G.; Carrion, M. D.; Cara, C. L.; Cruz-Lopez, O.; Tolomeo, M.; Grimaudo, S.; Cristina, A. D.; Pipitone, M. R.; Balzarini, J.; Zonta, N.; Brancale, A.; Hamel, E. Design, synthesis and structure–activity relationship of 2-(3′,4′,5′-trimethoxybenzoyl)-benzo[b]furan derivatives as a novel class of inhibitors of tubulin polymerization. Bioorg. Med. Chem. 2009, 17, 6862-6871. [CrossRef]
- Mariotto, E.; Canton, M.; Marchioro, C.; Brancale, A.; Hamel, E.; Varani, K.; Vincenzi, F.; De Ventura, T.; Padroni, C.; Viola, G.; Romagnoli, R. Synthesis and Biological Evaluation of Novel 2-Aroyl Benzofuran-Based Hydroxamic Acids as Antimicrotubule Agents. Int. J. Mol. Sci. 2024, 25, 7519. [CrossRef]
- Singh, A.; Fatima, K.; Singh, A.; Behl, A.; Mintoo, M. J.; Hasanain, M.; Ashraf, R.; Luqman, S.; Shanker, K.; Mondhe, D. M.; Sarkar, J.; Chanda, D.; Negi, A. S. Anticancer activity and toxicity profiles of 2-benzylidene indanone lead molecule. Eur. J. Pharm. Sci. 2015, 76, 57-67. [CrossRef]
- Saxena, H. O.; Faridi, U.; Srivastava, S.; Kumar, J. K.; Darokar, M. P.; Luqman, S.; Chanotiya, C. S.; Krishna, V.; Negi, A. S.; Khanuja, S. P. S. Gallic acid-based indanone derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2008, 18, 3914-3918. [CrossRef]
- Negi, A. S., Prakasham, A.P., Saxena, A.K., Luqman, S., Chanda, D., Kaur, T., Gupta, A. Anticancer and Tubulin Polymerisation Activity of Benzylidene Indanones and the Process of Preparing the Same. US8633242 B2, 2014.
- Kumar, K.; Das, R.; Thapa, B.; Rakhecha, B.; Srivastava, S.; Savita, K.; Israr, M.; Chanda, D.; Banerjee, D.; Shanker, K.; Bawankule, D. U.; Santini, B.; Di Paolo, M. L.; Via, L. D.; Passarella, D.; Negi, A. S. Dual targeted 2-Benzylideneindanone pendant hydroxamic acid group exhibits selective HDAC6 inhibition along with tubulin stabilization effect. Bioorg. Med. Chem. 2023, 86, 117300. [CrossRef]
- Li, L.; Quan, D.; Chen, J.; Ding, J.; Zhao, J.; Lv, L.; Chen, J. Design, synthesis, and biological evaluation of 1-substituted -2-aryl imidazoles targeting tubulin polymerization as potential anticancer agents. Eur. J. Med. Chem. 2019, 184, 111732. [CrossRef]
- Peng, X.; Chen, J.; Li, L.; Sun, Z.; Liu, J.; Ren, Y.; Huang, J.; Chen, J. Efficient Synthesis and Bioevaluation of Novel Dual Tubulin/Histone Deacetylase 3 Inhibitors as Potential Anticancer Agents. J. Med. Chem. 2021, 64, 8447-8473. [CrossRef]
- Yang, M. H.; Laurent, G.; Bause, A. S.; Spang, R.; German, N.; Haigis, M. C.; Haigis, K. M. HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS. Mol. Cancer Res. 2013, 11, 1072-1077. [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Oláh, J.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Aminothiazoles as Potent and Selective Sirt2 Inhibitors: A Structure–Activity Relationship Study. J. Med. Chem. 2016, 59, 1599-1612. [CrossRef]
- Sinatra, L.; Vogelmann, A.; Friedrich, F.; Tararina, M. A.; Neuwirt, E.; Colcerasa, A.; König, P.; Toy, L.; Yesiloglu, T. Z.; Hilscher, S.; Gaitzsch, L.; Papenkordt, N.; Zhai, S.; Zhang, L.; Romier, C.; Einsle, O.; Sippl, W.; Schutkowski, M.; Gross, O.; Bendas, G.; Christianson, D. W.; Hansen, F. K.; Jung, M.; Schiedel, M. Development of First-in-Class Dual Sirt2/HDAC6 Inhibitors as Molecular Tools for Dual Inhibition of Tubulin Deacetylation. J. Med. Chem. 2023, 66, 14787-14814. [CrossRef]
- Sun, M.; Zhang, Y.; Qin, J.; Ba, M.; Yao, Y.; Duan, Y.; Liu, H.; Yu, D. Synthesis and biological evaluation of new 2-methoxyestradiol derivatives: Potent inhibitors of angiogenesis and tubulin polymerization. Bioorg. Chem. 2021, 113, 104988. [CrossRef]
- Sun, M.; Qin, J.; Kang, Y.; Zhang, Y.; Ba, M.; Yang, H.; Duan, Y.; Yao, Y. 2-Methoxydiol derivatives as new tubulin and HDAC dual-targeting inhibitors, displaying antitumor and antiangiogenic response. Bioorg. Chem. 2022, 120, 105625. [CrossRef]
- Xie, S.; Leng, J.; Zhao, S.; Zhu, L.; Zhang, M.; Ning, M.; Zhao, B.; Kong, L.; Yin, Y. Design and biological evaluation of dual tubulin/HDAC inhibitors based on millepachine for treatment of prostate cancer. Eur. J. Med. Chem. 2024, 268, 116301. [CrossRef]
- Zhang, X.; Bao, B.; Yu, X.; Tong, L.; Luo, Y.; Huang, Q.; Su, M.; Sheng, L.; Li, J.; Zhu, H.; Yang, B.; Zhang, X.; Chen, Y.; Lu, W. The discovery and optimization of novel dual inhibitors of topoisomerase II and histone deacetylase. Bioorg. Med. Chem. 2013, 21, 6981-6995. [CrossRef]
- Yong, Y.; Shin, S. Y.; Lee, Y. H.; Lim, Y. Antitumor activity of deoxypodophyllotoxin isolated from Anthriscus sylvestris: Induction of G2/M cell cycle arrest and caspase-dependent apoptosis. Bioorg. Med. Chem. Lett. 2009, 19, 4367-4371. [CrossRef]
- Shin, S. Y.; Yong, Y.; Kim, C. G.; Lee, Y. H.; Lim, Y. Deoxypodophyllotoxin induces G2/M cell cycle arrest and apoptosis in HeLa cells. Cancer Lett. 2010, 287, 231-239. [CrossRef]
- Chen, S.-W.; Gao, Y.-Y.; Zhou, N.-N.; Liu, J.; Huang, W.-T.; Hui, L.; Jin, Y.; Jin, Y.-X. Carbamates of 4′-demethyl-4-deoxypodophyllotoxin: Synthesis, cytotoxicity and cell cycle effects. Bioorg. Med. Chem. Lett. 2011, 21, 7355-7358. [CrossRef]
- Zhang, X.; Zhang, J.; Su, M.; Zhou, Y.; Chen, Y.; Li, J.; Lu, W. Design, synthesis and biological evaluation of 4′-demethyl-4-deoxypodophyllotoxin derivatives as novel tubulin and histone deacetylase dual inhibitors. RSC Adv. 2014, 4, 40444-40448. [CrossRef]
- Schiff, P. B.; Fant, J.; Horwitz, S. B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665-667. [CrossRef]
- Liu, S.; Zhang, K.; Zhu, Q.; Shen, Q.; Zhang, Q.; Yu, J.; Chen, Y.; Lu, W. Synthesis and biological evaluation of paclitaxel and vorinostat co-prodrugs for overcoming drug resistance in cancer therapy in vitro. Bioorg. Med. Chem. 2019, 27, 1405-1413. [CrossRef]
- Yoshimatsu, K.; Yamaguchi, A.; Yoshino, H.; Koyanagi, N.; Kitoh, K. Mechanism of Action of E7010, an Orally Active Sulfonamide Antitumor Agent: Inhibition of Mitosis by Binding to the Colchicine Site of Tubulin. Cancer Res. 1997, 57, 3208-3213.
- Wu, W.-C.; Liu, Y.-M.; Lin, M.-H.; Liao, Y.-H.; Lai, M.-J.; Chuang, H.-Y.; Hung, T.-Y.; Chen, C.-H.; Liou, J.-P. Design, synthesis, and evaluation of N-phenyl-4-(2-phenylsulfonamido)-benzamides as microtubule-targeting agents in drug-resistant cancer cells, displaying HDAC inhibitory response. Eur. J. Med. Chem. 2020, 192, 112158. [CrossRef]
- Chang, J.-Y.; Hsieh, H.-P.; Chang, C.-Y.; Hsu, K.-S.; Chiang, Y.-F.; Chen, C.-M.; Kuo, C.-C.; Liou, J.-P. 7-Aroyl-aminoindoline-1-sulfonamides as a Novel Class of Potent Antitubulin Agents. J. Med. Chem. 2006, 49, 6656-6659. [CrossRef]
- Lai, M.-J.; Ojha, R.; Lin, M.-H.; Liu, Y.-M.; Lee, H.-Y.; Lin, T. E.; Hsu, K.-C.; Chang, C.-Y.; Chen, M.-C.; Nepali, K.; Chang, J.-Y.; Liou, J.-P. 1-Arylsulfonyl indoline-benzamides as a new antitubulin agents, with inhibition of histone deacetylase. Eur. J. Med. Chem. 2019, 162, 612-630. [CrossRef]
- Kuo, C.-C.; Hsieh, H.-P.; Pan, W.-Y.; Chen, C.-P.; Liou, J.-P.; Lee, S.-J.; Chang, Y.-L.; Chen, L.-T.; Chen, C.-T.; Chang, J.-Y. BPR0L075, a Novel Synthetic Indole Compound with Antimitotic Activity in Human Cancer Cells, Exerts Effective Antitumoral Activity in Vivo. Cancer Res. 2004, 64, 4621-4628. [CrossRef]
- Lee, H.-Y.; Tsai, A.-C.; Chen, M.-C.; Shen, P.-J.; Cheng, Y.-C.; Kuo, C.-C.; Pan, S.-L.; Liu, Y.-M.; Liu, J.-F.; Yeh, T.-K.; Wang, J.-C.; Chang, C.-Y.; Chang, J.-Y.; Liou, J.-P. Azaindolylsulfonamides, with a More Selective Inhibitory Effect on Histone Deacetylase 6 Activity, Exhibit Antitumor Activity in Colorectal Cancer HCT116 Cells. J. Med. Chem. 2014, 57, 4009-4022. [CrossRef]
- Lee, H.-Y.; Lee, J.-F.; Kumar, S.; Wu, Y.-W.; HuangFu, W.-C.; Lai, M.-J.; Li, Y.-H.; Huang, H.-L.; Kuo, F.-C.; Hsiao, C.-J.; Cheng, C.-C.; Yang, C.-R.; Liou, J.-P. 3-Aroylindoles display antitumor activity in vitro and in vivo: Effects of N1-substituents on biological activity. Eur. J. Med. Chem. 2017, 125, 1268-1278. [CrossRef]
- Wu, Y.-W.; Hsu, K.-C.; Lee, H.-Y.; Huang, T.-C.; Lin, T. E.; Chen, Y.-L.; Sung, T.-Y.; Liou, J.-P.; Hwang-Verslues, W. W.; Pan, S.-L.; HuangFu, W.-C. A Novel Dual HDAC6 and Tubulin Inhibitor, MPT0B451, Displays Anti-tumor Ability in Human Cancer Cells in Vitro and in Vivo. Front. Pharmacol. 2018, 9, 205. [CrossRef]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; Horinouchi, S.; Yoshida, M. In vivodestabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820-6831. [CrossRef]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.-F.; Yao, T.-P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455-458. [CrossRef]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168-79. [CrossRef]
- Nepali, K.; Chang, T.-Y.; Lai, M.-J.; Hsu, K.-C.; Yen, Y.; Lin, T. E.; Lee, S.-B.; Liou, J.-P. Purine/purine isoster based scaffolds as new derivatives of benzamide class of HDAC inhibitors. Eur. J. Med. Chem. 2020, 196, 112291. [CrossRef]
- Schemies, J.; Sippl, W.; Jung, M. Histone deacetylase inhibitors that target tubulin. Cancer Lett. 2009, 280, 222-232. [CrossRef]
- Singh, A.; Chang, T.-Y.; Kaur, N.; Hsu, K.-C.; Yen, Y.; Lin, T. E.; Lai, M.-J.; Lee, S.-B.; Liou, J.-P. CAP rigidification of MS-275 and chidamide leads to enhanced antiproliferative effects mediated through HDAC1, 2 and tubulin polymerization inhibition. Eur. J. Med. Chem. 2021, 215, 113169. [CrossRef]
- Xue, J.; Wu, G.; Ejaz, U.; Akhtar, F.; Wan, X.; Zhu, Y.; Geng, A.; Chen, Y.; He, S. A novel histone deacetylase inhibitor LT-548-133-1 induces apoptosis by inhibiting HDAC and interfering with microtubule assembly in MCF-7 cells. Invest. New Drugs 2021, 39, 1222-1231. [CrossRef]
- Yang, Z.; Wang, T.; Wang, F.; Niu, T.; Liu, Z.; Chen, X.; Long, C.; Tang, M.; Cao, D.; Wang, X.; Xiang, W.; Yi, Y.; Ma, L.; You, J.; Chen, L. Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer. J. Med. Chem. 2016, 59, 1455-1470. [CrossRef]
- Wang, F.; Zheng, L.; Yi, Y.; Yang, Z.; Qiu, Q.; Wang, X.; Yan, W.; Bai, P.; Yang, J.; Li, D.; Pei, H.; Niu, T.; Ye, H.; Nie, C.; Hu, Y.; Yang, S.; Wei, Y.; Chen, L. SKLB-23bb, A HDAC6-Selective Inhibitor, Exhibits Superior and Broad-Spectrum Antitumor Activity via Additionally Targeting Microtubules. Mol. Cancer Ther. 2018, 17, 763-775. [CrossRef]
- Cai, X.; Zhai, H.-X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.-J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-Ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a Potent Multi-Acting HDAC, EGFR, and HER2 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 2000-2009. [CrossRef]
- Wang, X.-F.; Guan, F.; Ohkoshi, E.; Guo, W.; Wang, L.; Zhu, D.-Q.; Wang, S.-B.; Wang, L.-T.; Hamel, E.; Yang, D.; Li, L.; Qian, K.; Morris-Natschke, S. L.; Yuan, S.; Lee, K.-H.; Xie, L. Optimization of 4-(N-Cycloamino)phenylquinazolines as a Novel Class of Tubulin-Polymerization Inhibitors Targeting the Colchicine Site. J. Med. Chem. 2014, 57, 1390-1402. [CrossRef]
- Gryder, B. E.; Sodji, Q. H.; Oyelere, A. K. Targeted Cancer Therapy: Giving Histone Deacetylase Inhibitors All They Need To Succeed. Future Med. Chem. 2012, 4, 505-524. [CrossRef]
- Ning, Z.-Q.; Li, Z.-B.; Newman, M. J.; Shan, S.; Wang, X.-H.; Pan, D.-S.; Zhang, J.; Dong, M.; Du, X.; Lu, X.-P. Chidamide (CS055/HBI-8000): a new histone deacetylase inhibitor of the benzamide class with antitumor activity and the ability to enhance immune cell-mediated tumor cell cytotoxicity. Cancer Chemother. Pharmacol. 2012, 69, 901-909. [CrossRef]
- Vasudevan, A.; Ji, Z.; Frey, R. R.; Wada, C. K.; Steinman, D.; Heyman, H. R.; Guo, Y.; Curtin, M. L.; Guo, J.; Li, J.; Pease, L.; Glaser, K. B.; Marcotte, P. A.; Bouska, J. J.; Davidsen, S. K.; Michaelides, M. R. Heterocyclic ketones as inhibitors of histone deacetylase. Bioorg. Med. Chem. Lett. 2003, 13, 3909-3913. [CrossRef]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762-7810. [CrossRef]
- Al-Hamashi, A. A.; Koranne, R.; Dlamini, S.; Alqahtani, A.; Karaj, E.; Rashid, M. S.; Knoff, J. R.; Dunworth, M.; Pflum, M. K. H.; Casero, R. A.; Perera, L.; Taylor, W. R.; Tillekeratne, L. M. V. A new class of cytotoxic agents targets tubulin and disrupts microtubule dynamics. Bioorg. Chem. 2021, 116, 105297. [CrossRef]
Figure 1.
Approved HDACis.
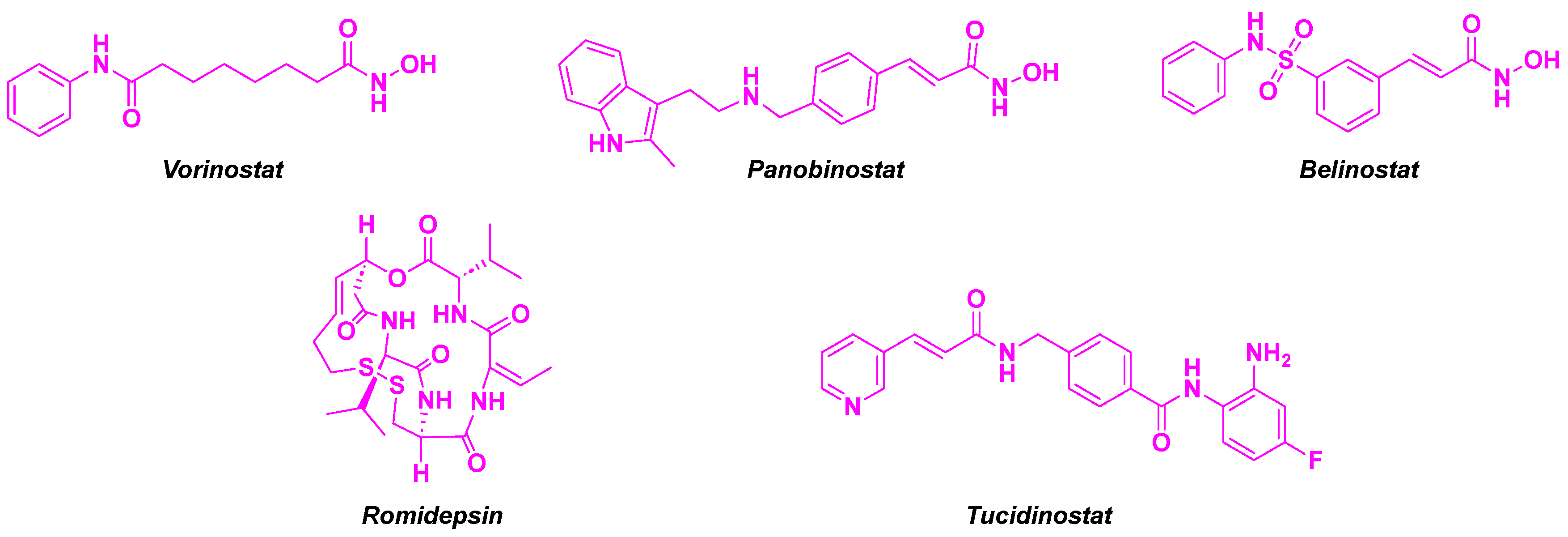
Figure 2.
Structure of CA-4.
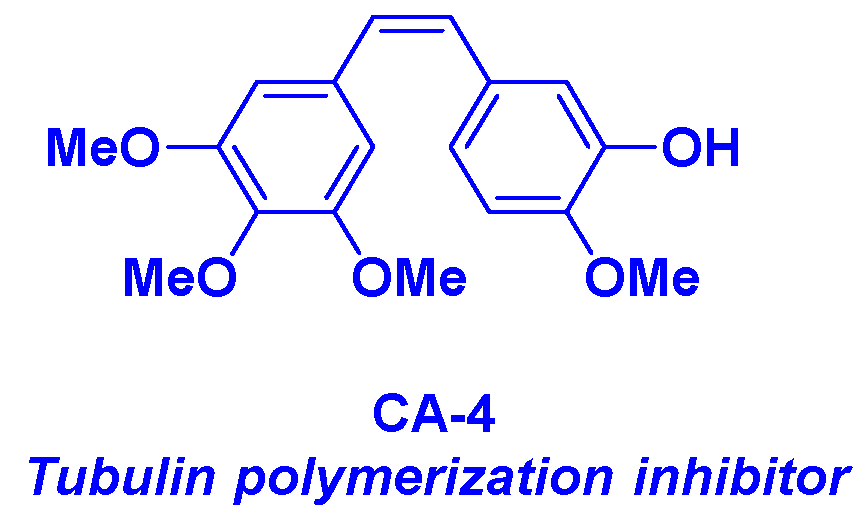
Scheme 1.
HDAC/tubulin inhibitors designed by Luan et al.
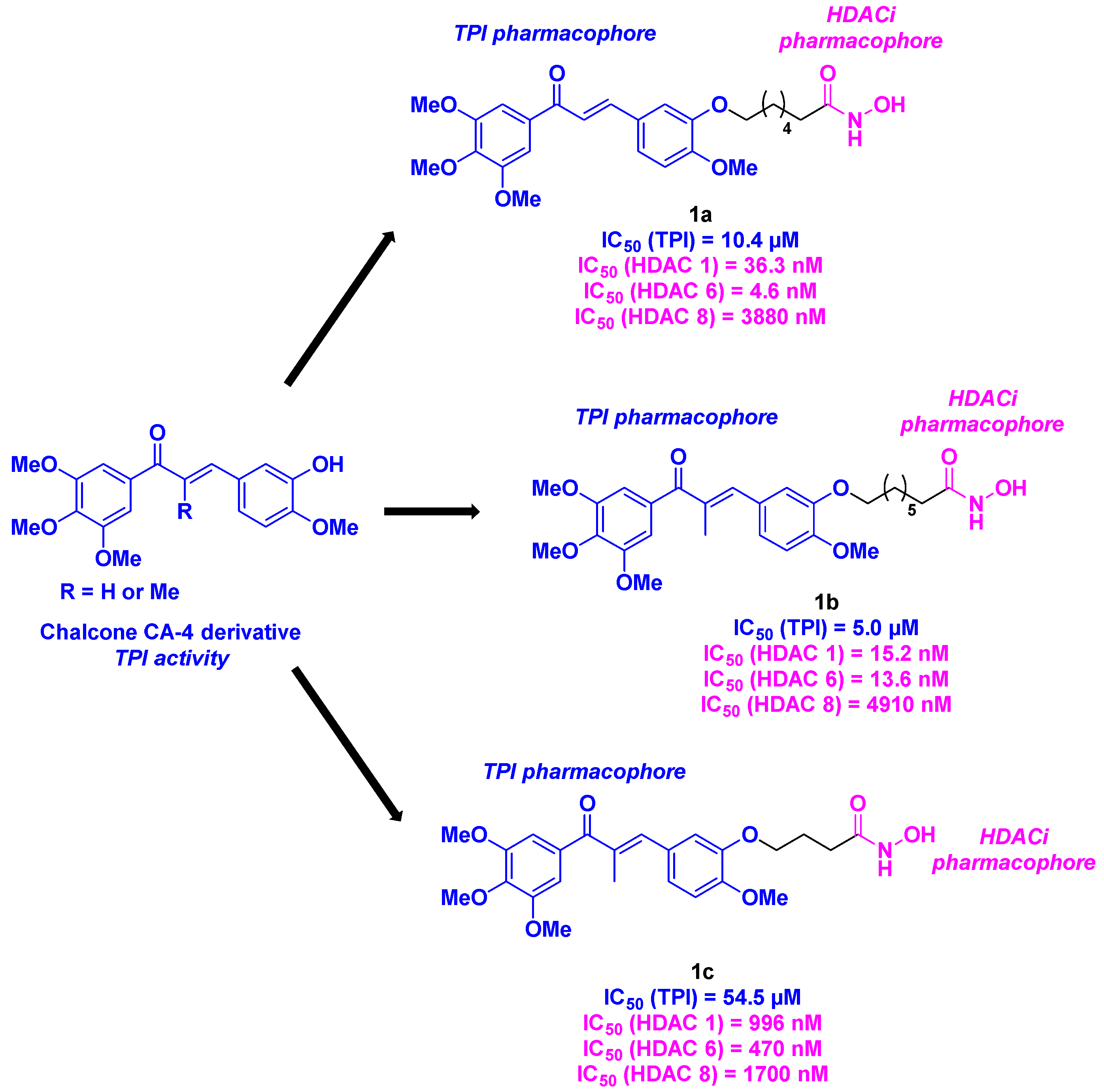
Scheme 2.
HDAC/tubulin inhibitors conceived by Mourad and his colleagues.
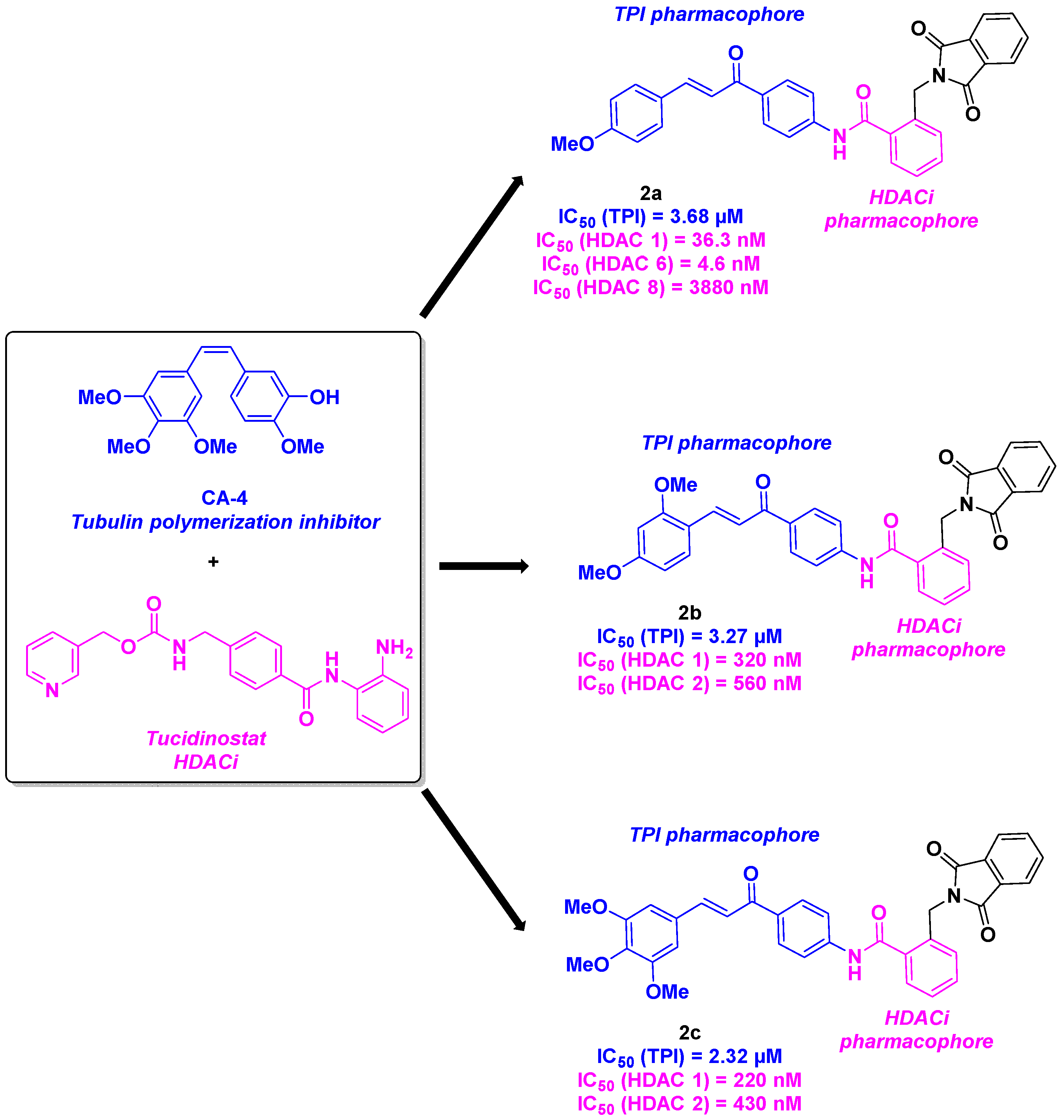
Scheme 3.
HDAC/tubulin dual inhibitors elaborated by the Hamze group.
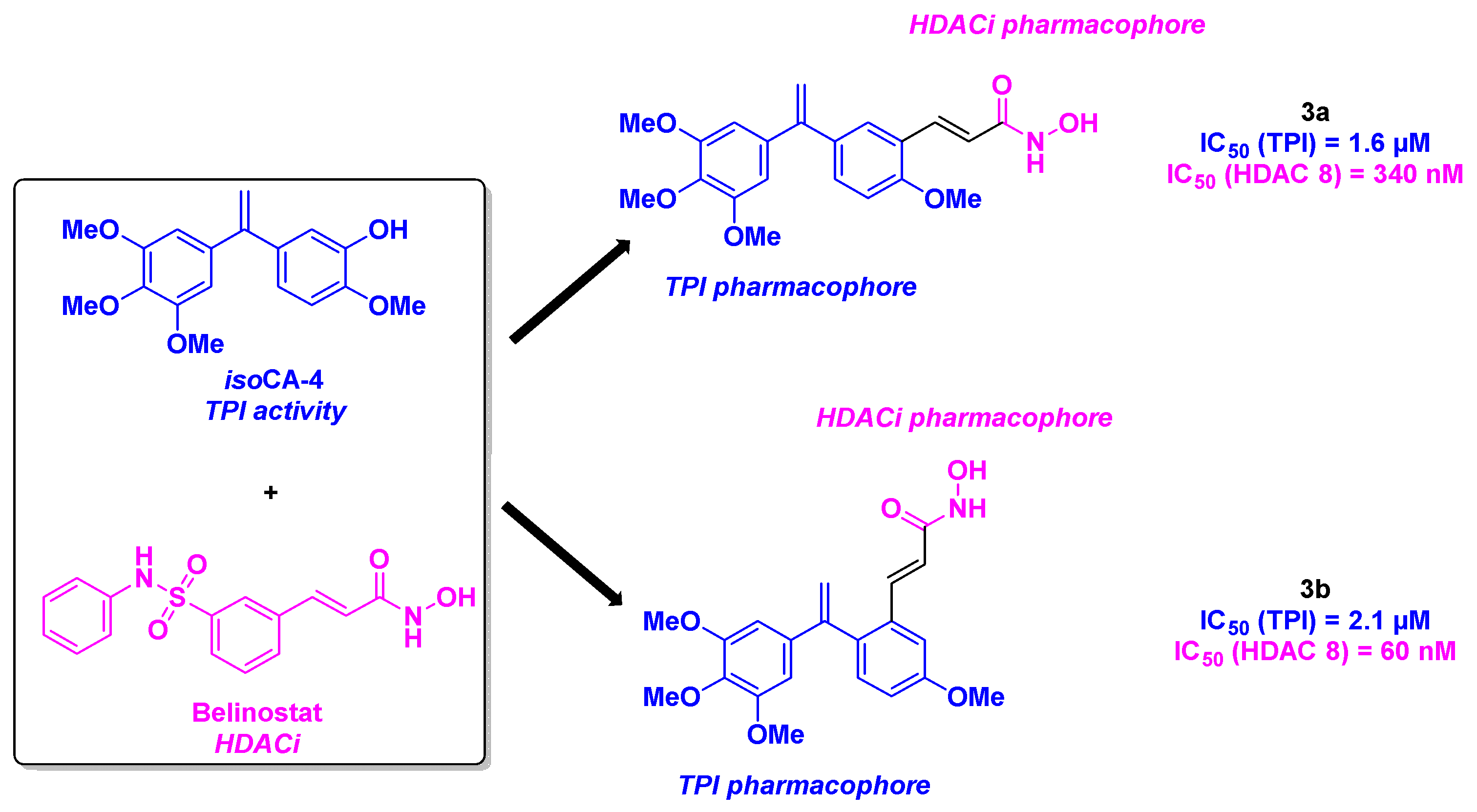
Scheme 4.
Second generation of HDAC/tubulin dual inhibitors elaborated by the Hamze group.
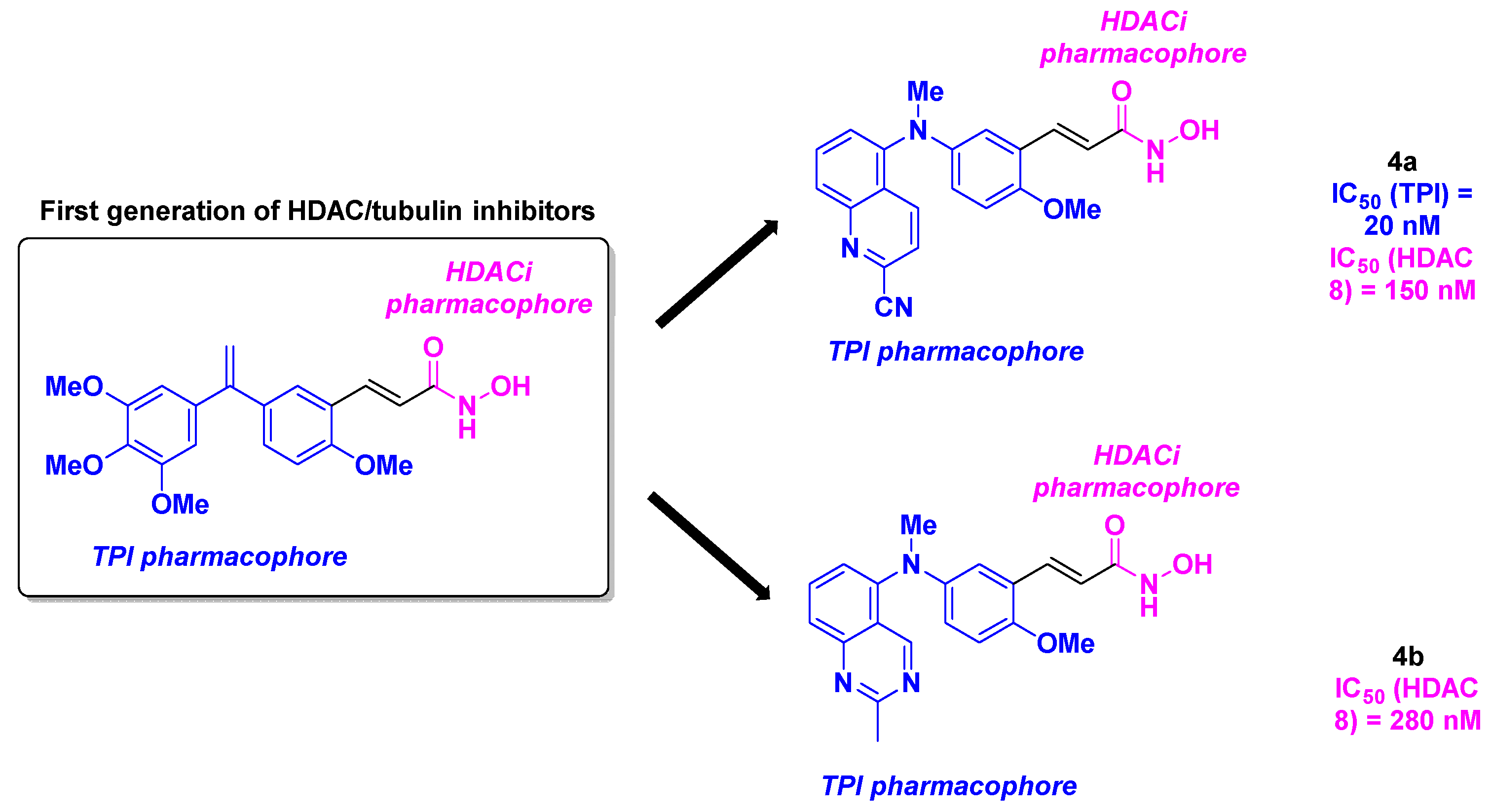
Scheme 5.
HDAC/tubulin dual inhibitor conceived by Yao, Yang, Duan and coworkers.
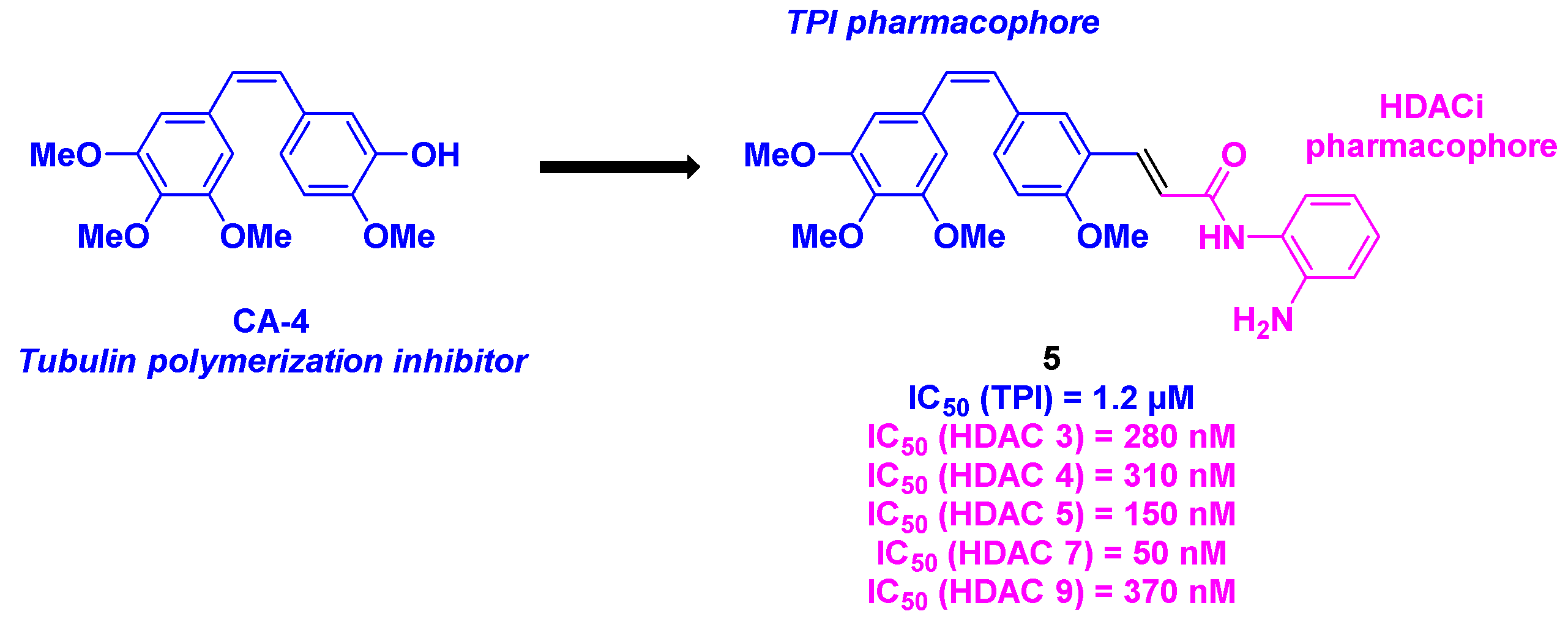
Scheme 6.
HDAC/tubulin dual inhibitor proposed by Chen, Xu and their colleagues.
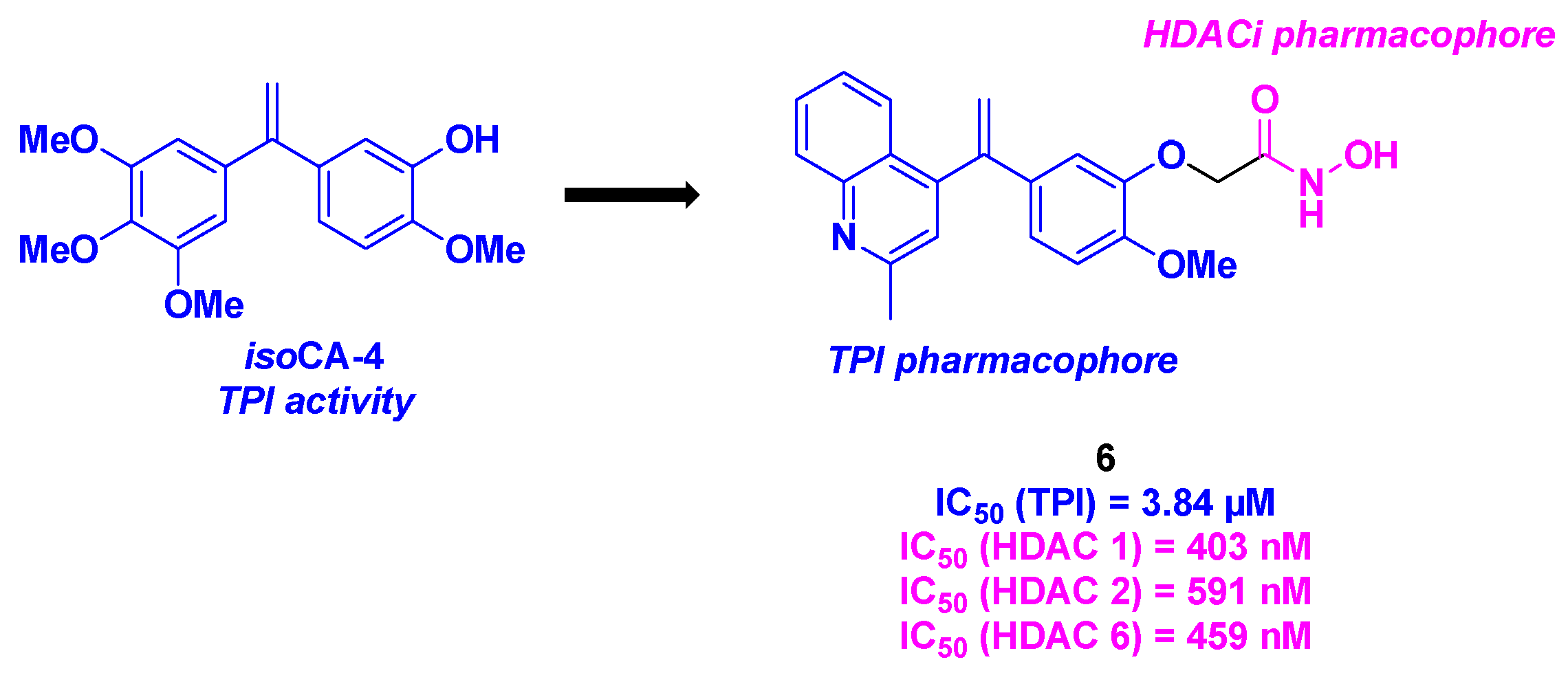
Scheme 7.
HDAC/tubulin inhibitor SY-65 prepared by Song, Zhang and coworkers.
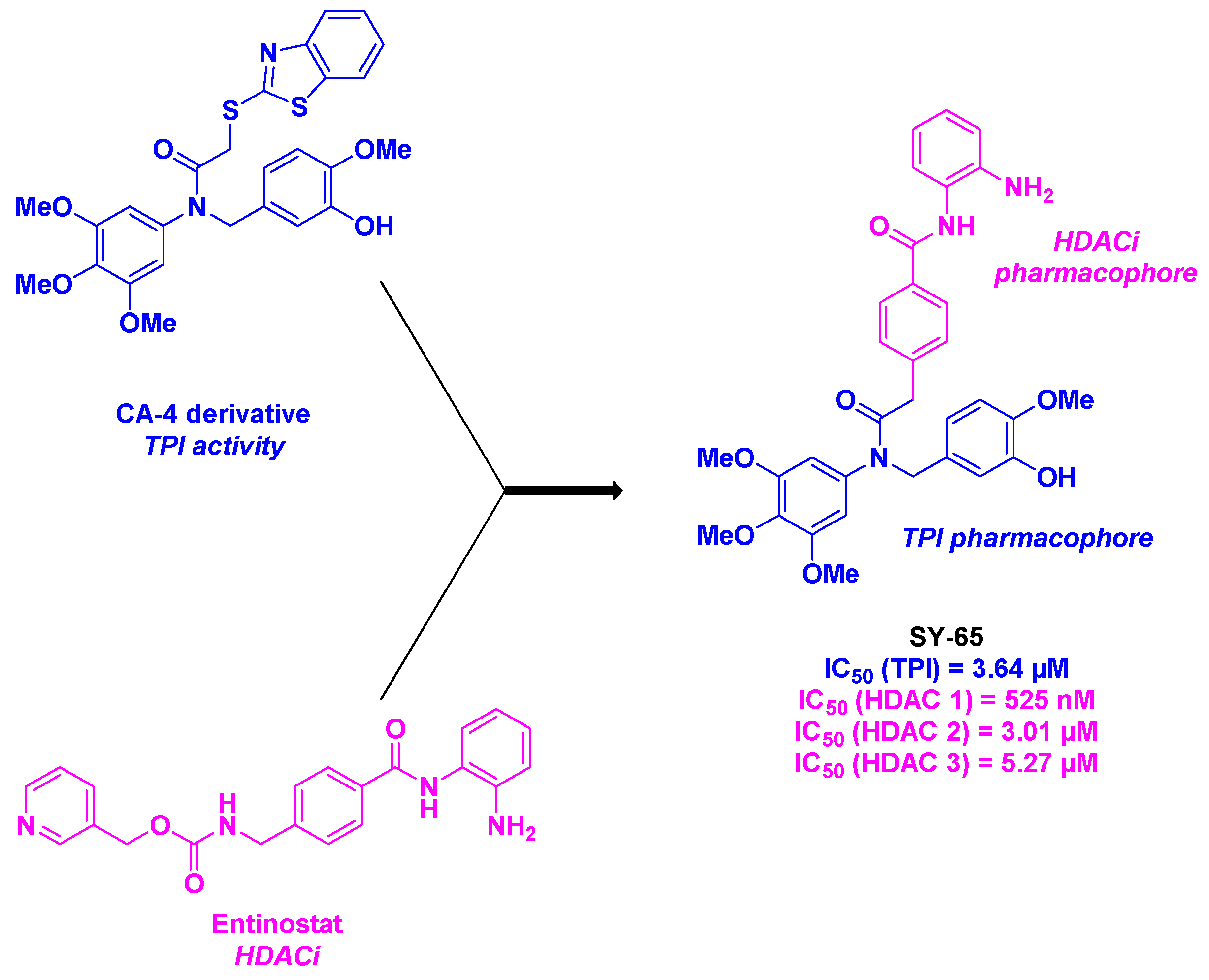
Scheme 8.
HDAC/tubulin inhibitors prepared by Höpner et al.
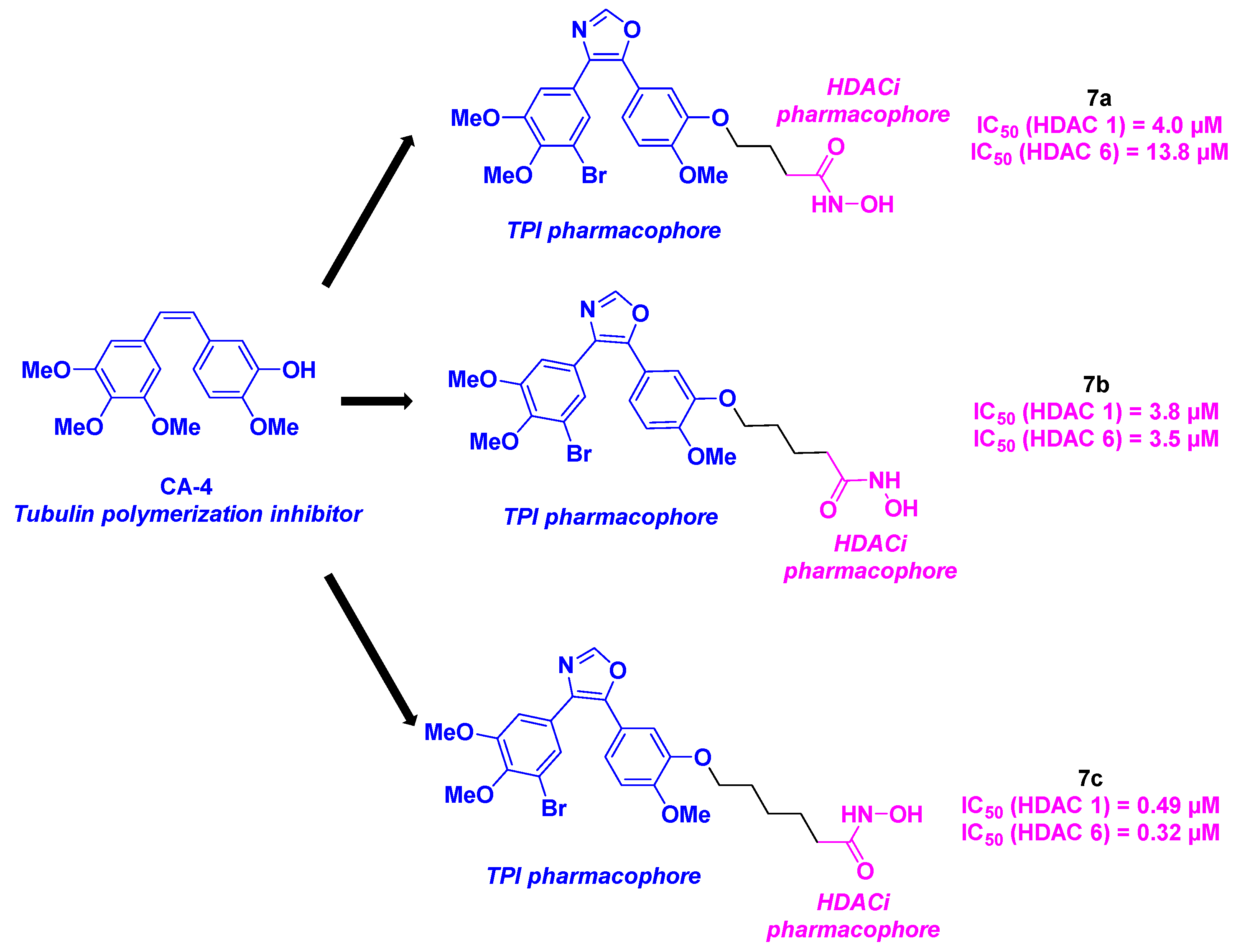
Scheme 9.
HDAC/tubulin inhibitors designed by Ding, Wang and coworkers.
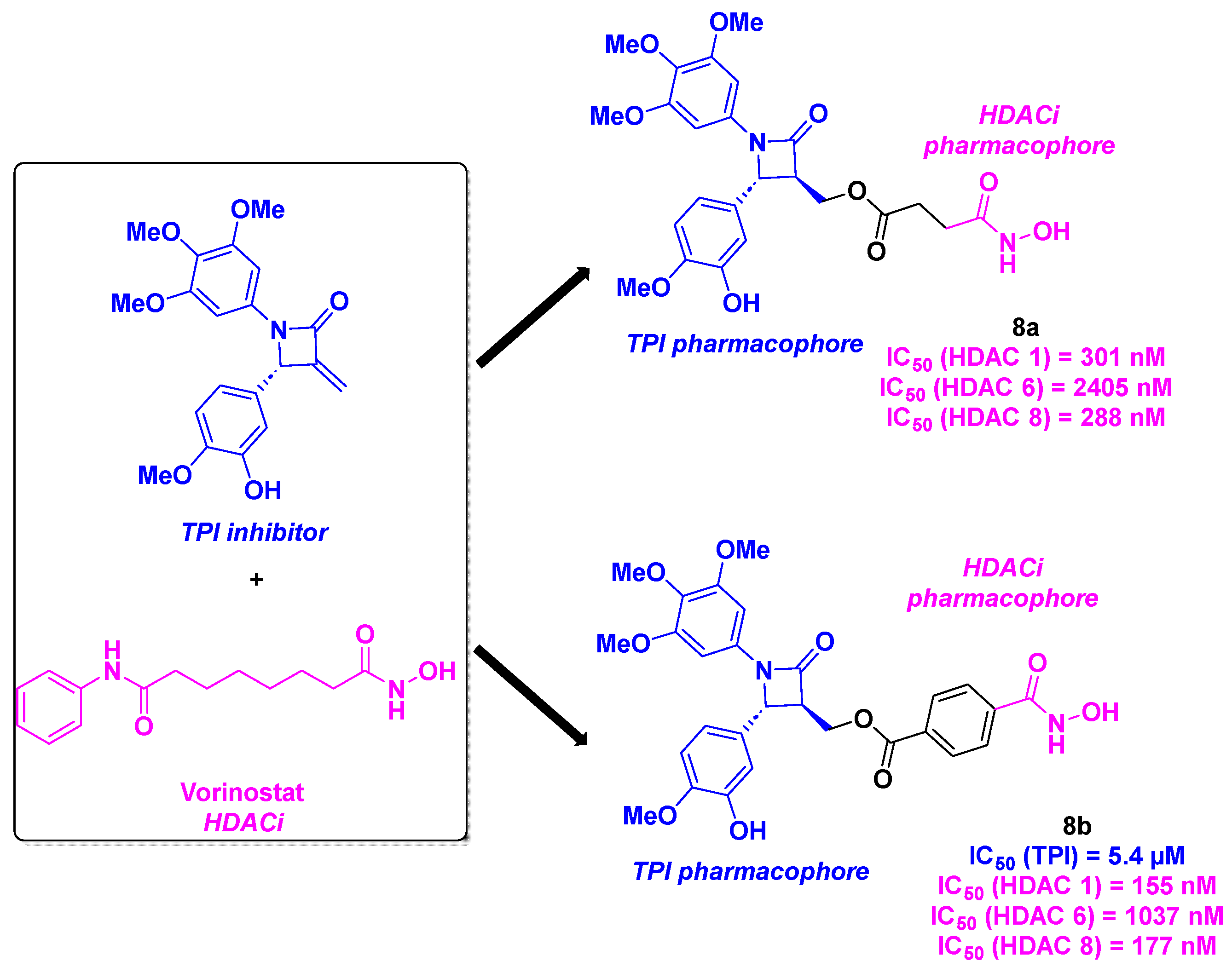
Scheme 10.
HDAC/tubulin inhibitor designed by Ding, Wang and coworkers.
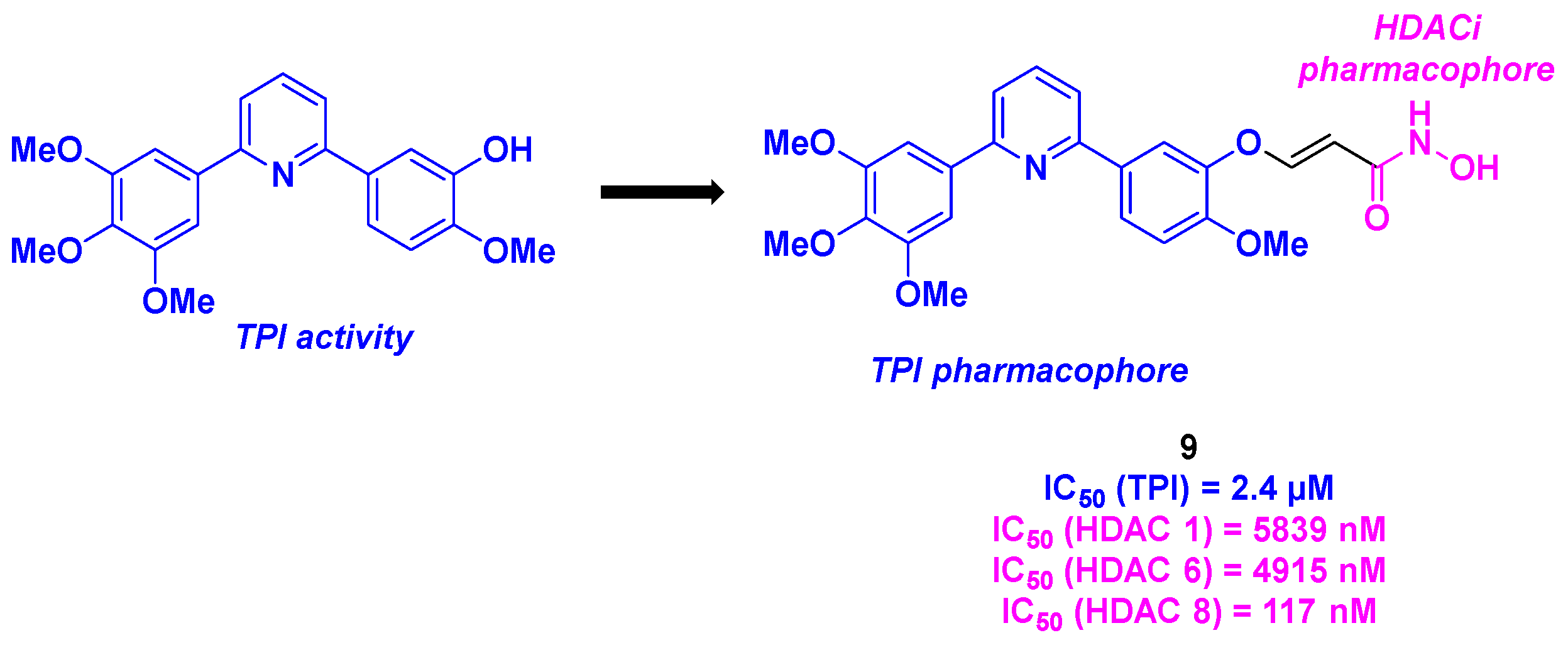
Scheme 11.
HDAC/tubulin inhibitors synthesized by Abdelhafez et al.
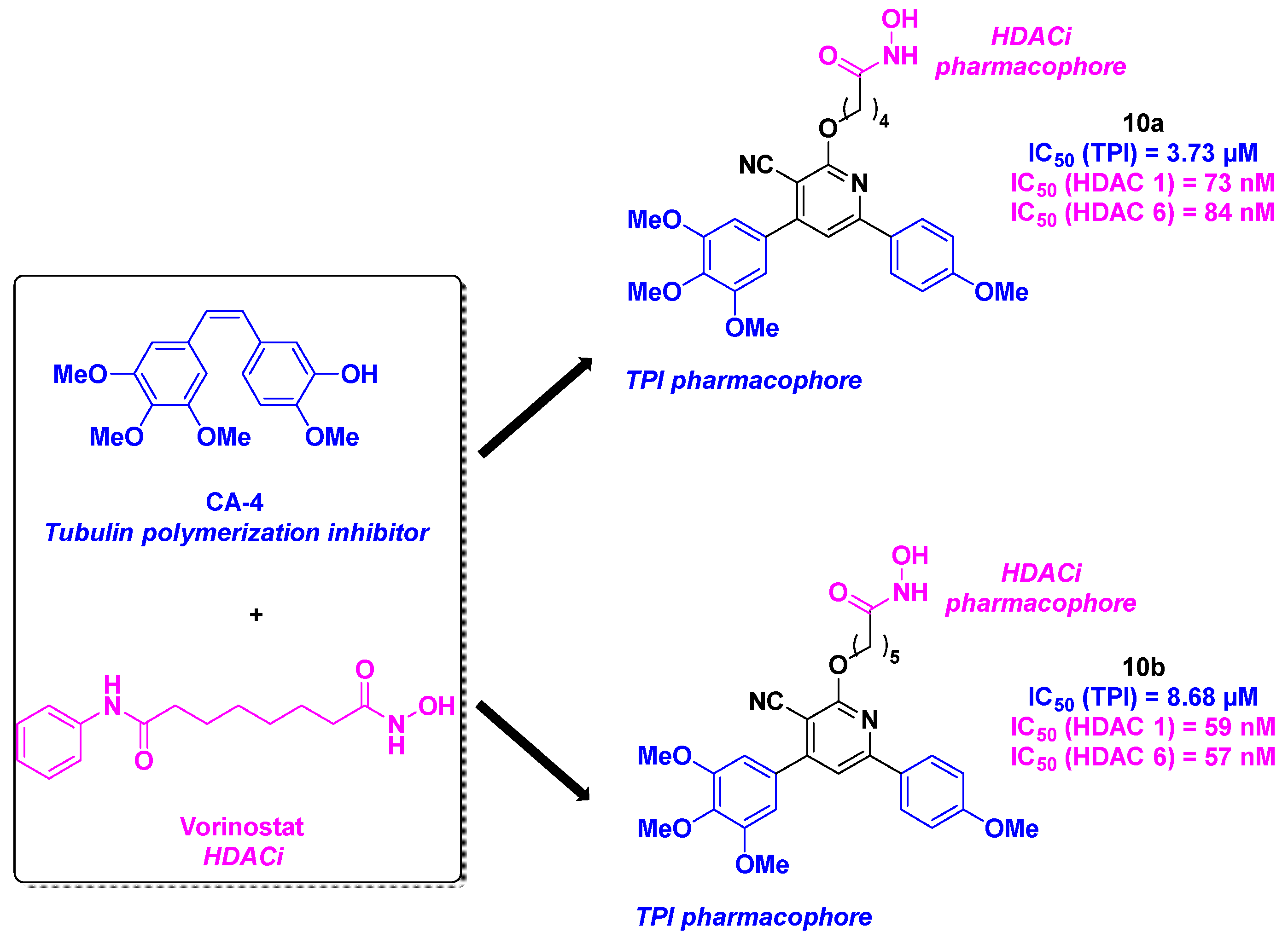
Scheme 12.
HDAC/tubulin inhibitors designed by Lu, Chen and coworkers.
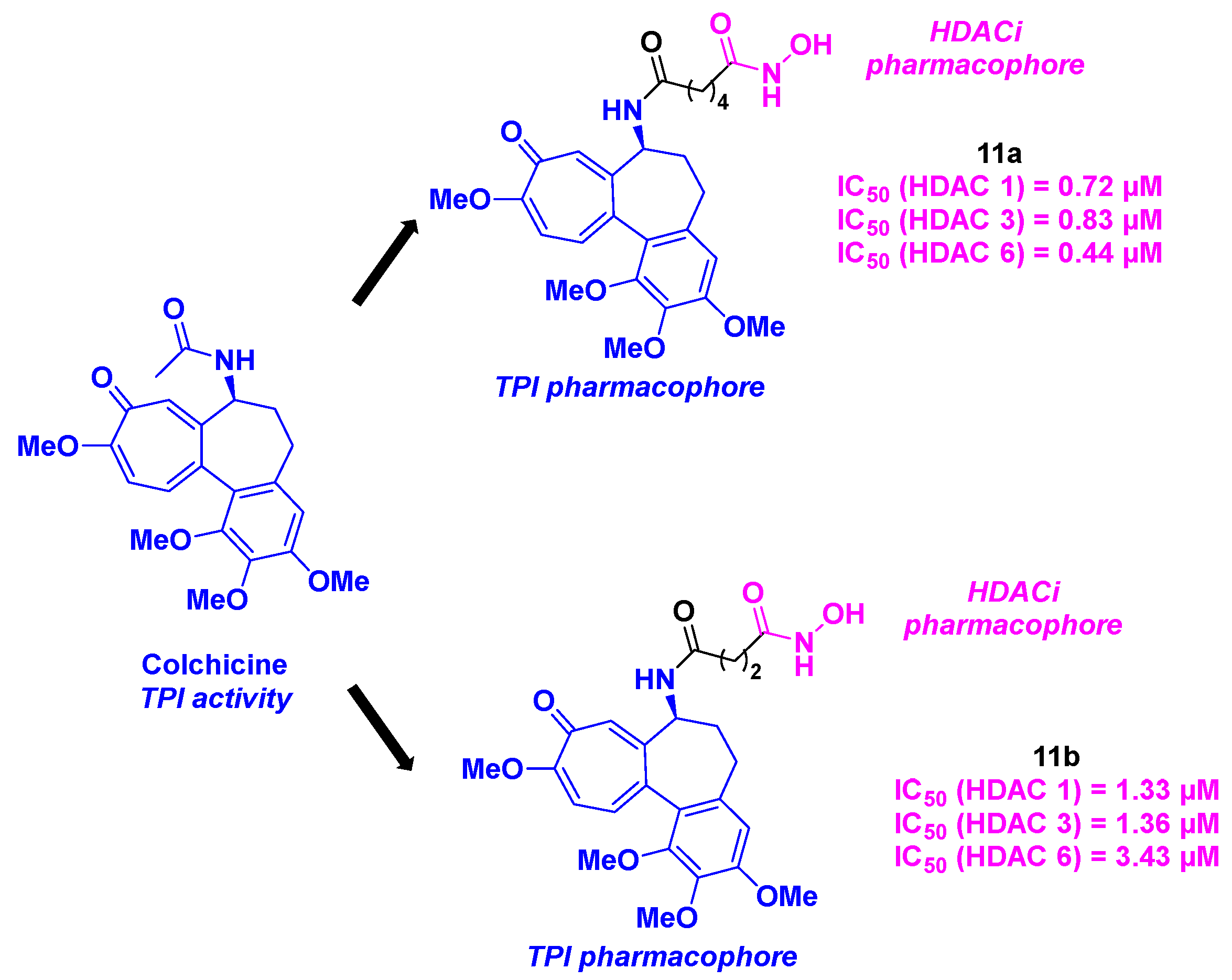
Scheme 13.
HDAC/tubulin inhibitors described by Fang, Lu and coworkers.
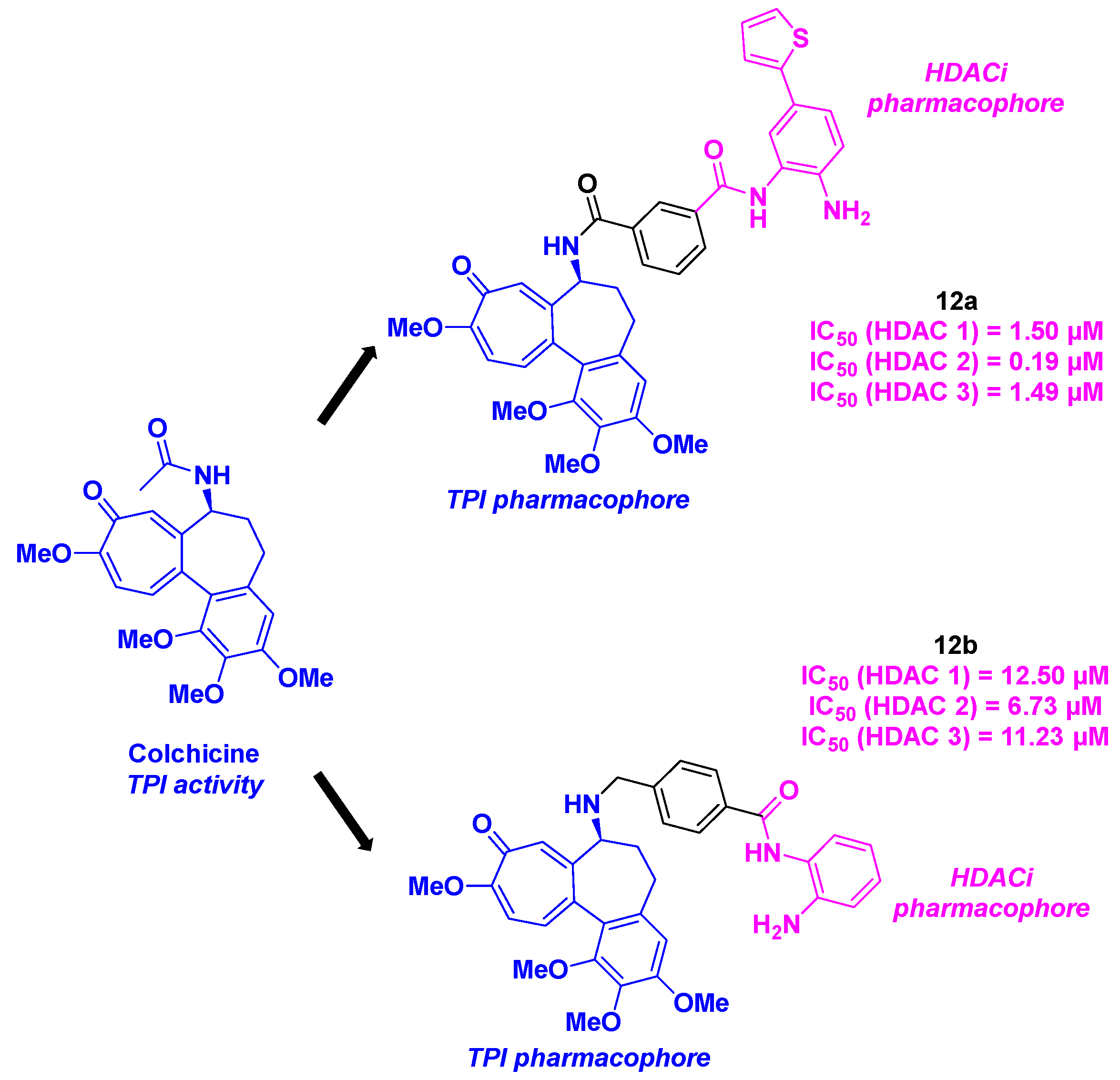
Scheme 14.
HDAC/tubulin hybrid inhibitor bearing an aminobenzamide core developed by the Xu group.
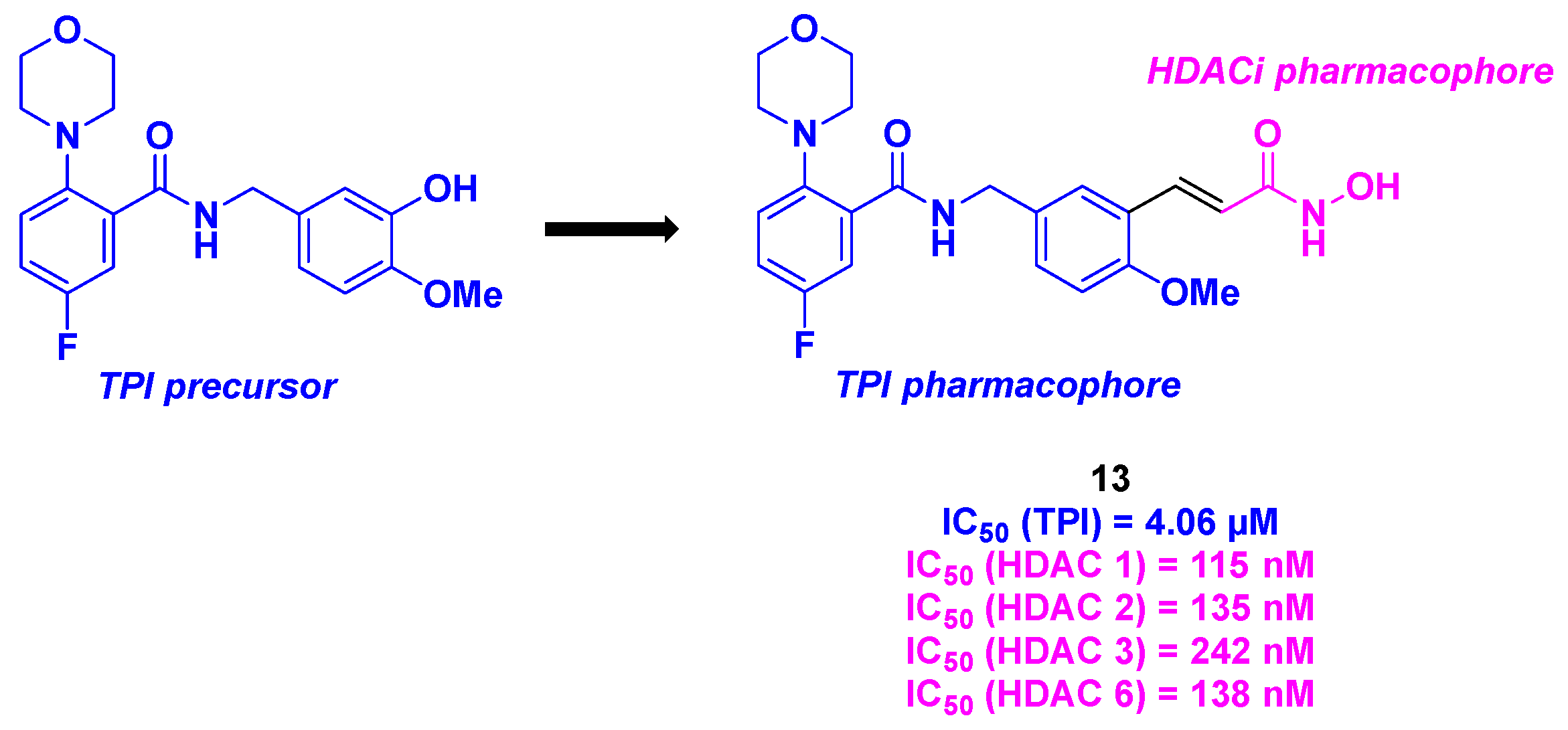
Scheme 15.
HDAC/tubulin hybrid inhibitors created by Zaki and coworkers.
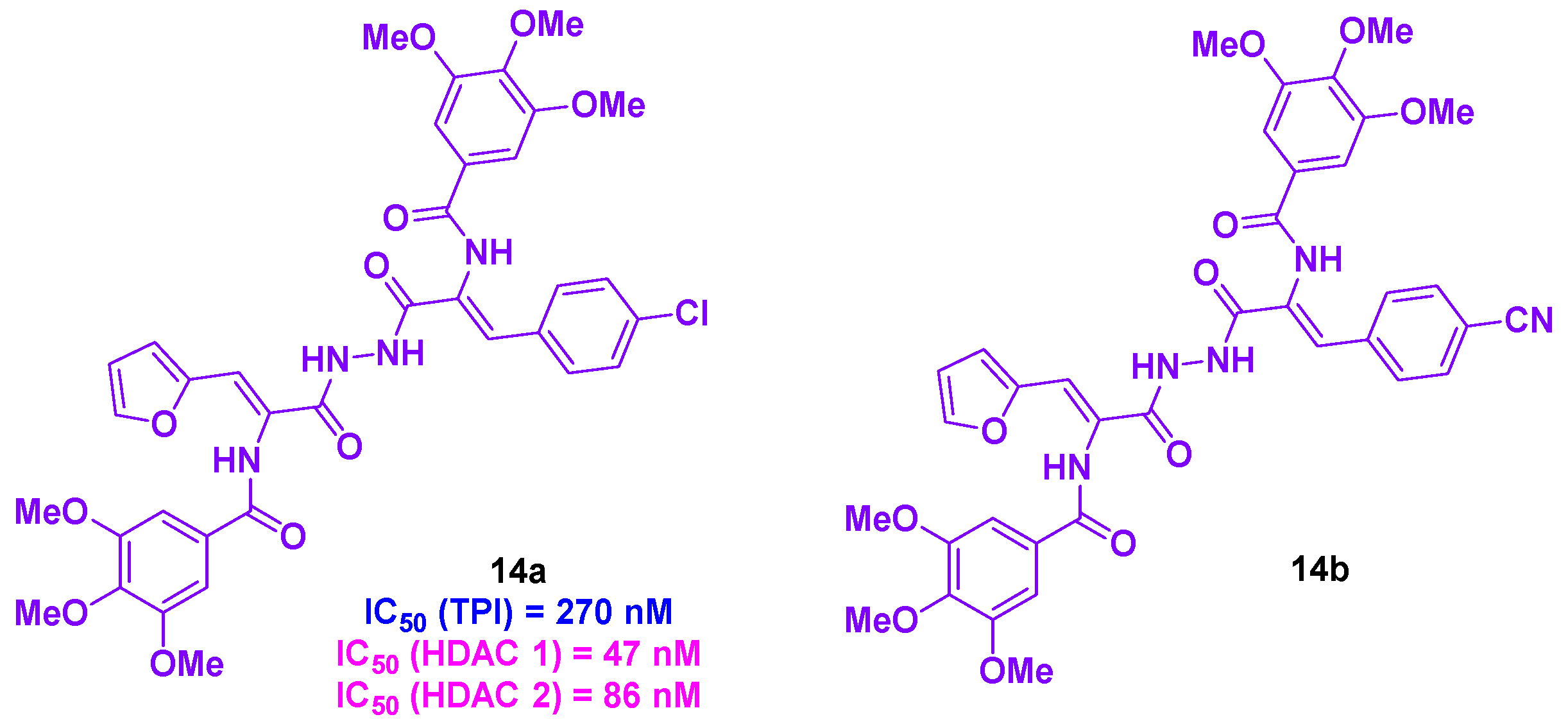
Scheme 16.
Dual HDAC/tubulin inhibitor conceived by Wang et al.
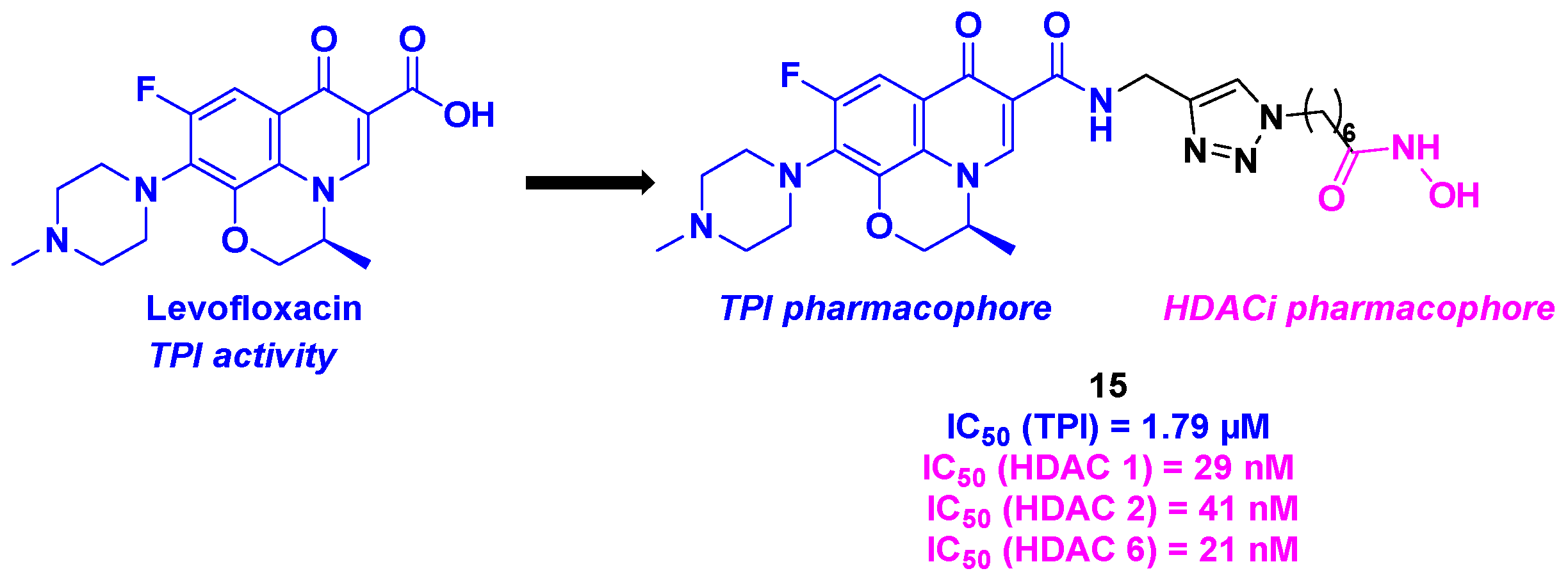
Scheme 17.
Dual HDAC/tubulin inhibitors developed by the Romagnoli group.
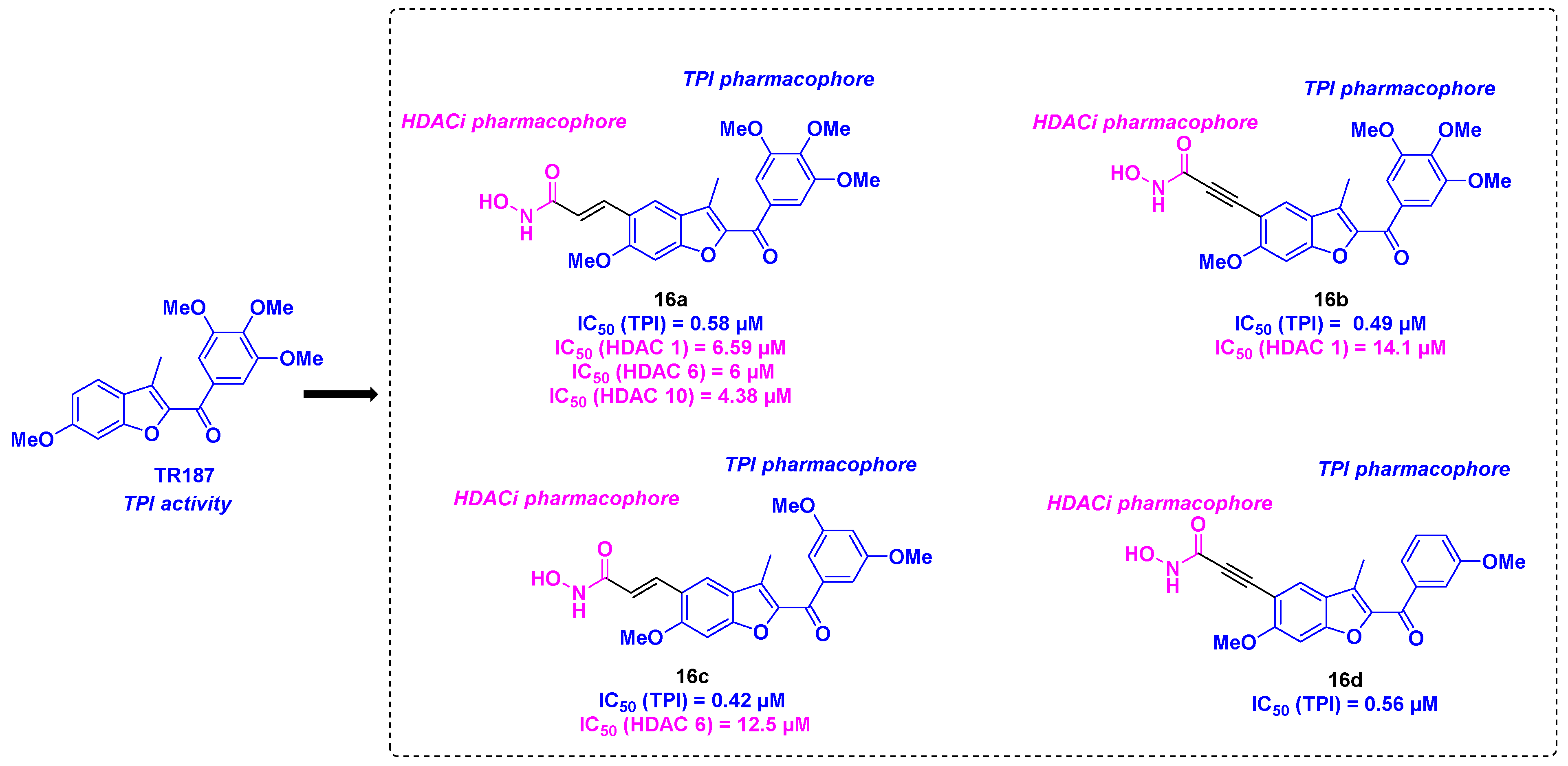
Scheme 18.
Dual HDAC/tubulin inhibitors described by Negi et al.
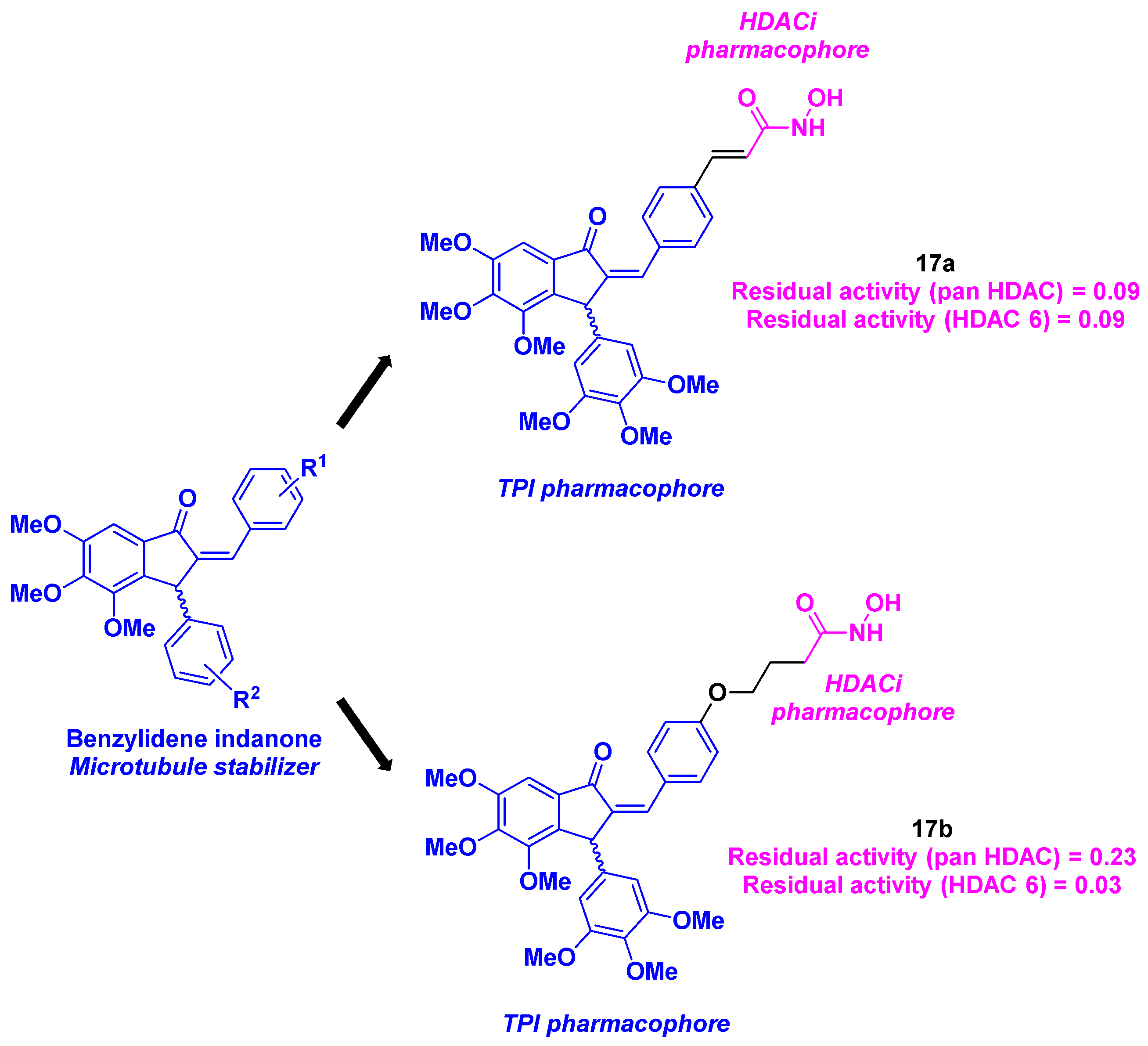
Scheme 19.
Dual HDAC/tubulin inhibitor prepared by Chen et al.
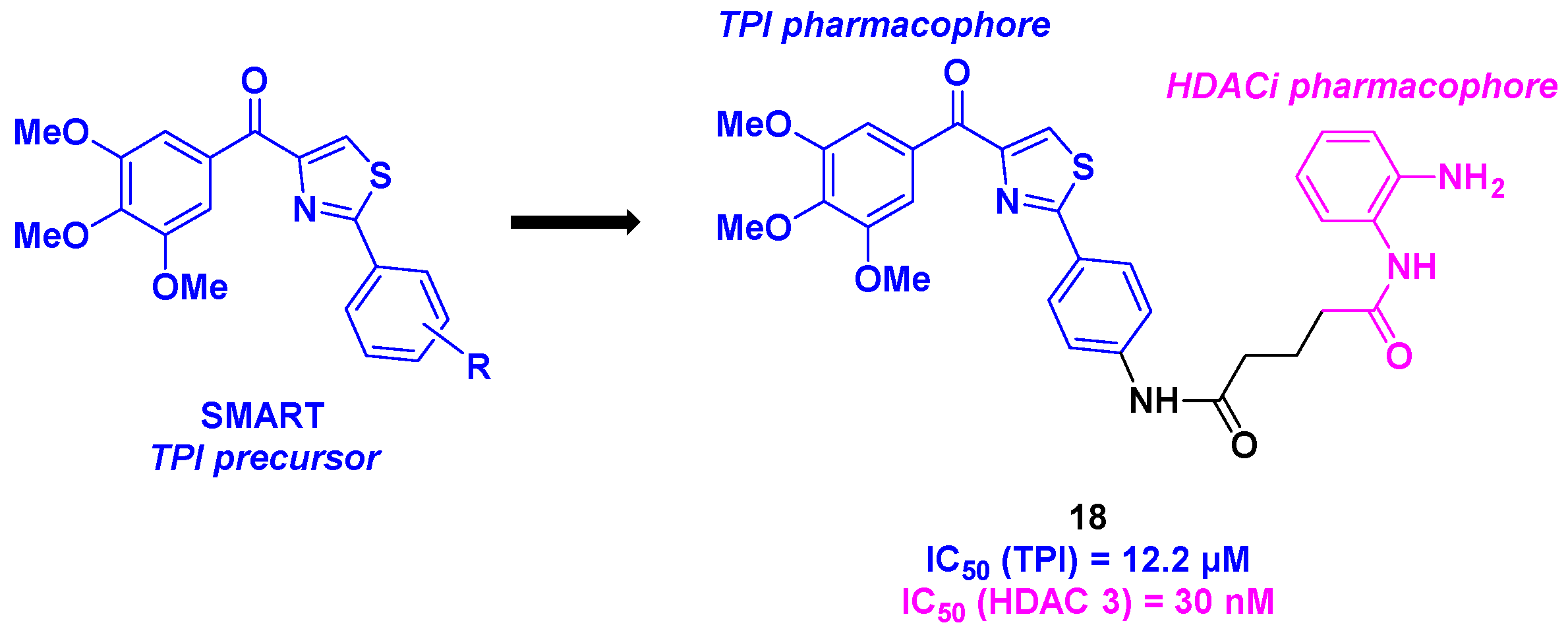
Scheme 20.
Dual Sirt2/HDAC 6 inhibitor developed by the Schiedel group.
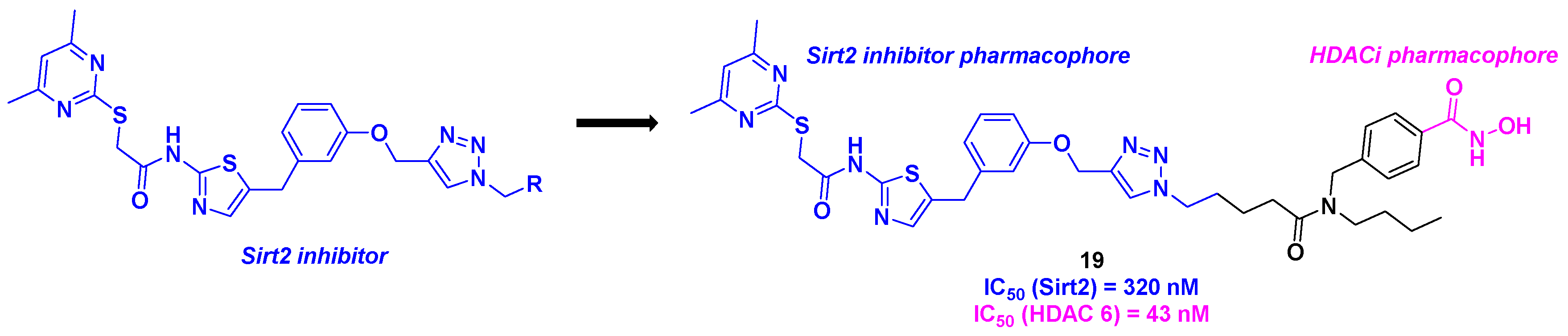
Scheme 21.
HDAC/tubulin dual inhibitor developed by Yao et al.
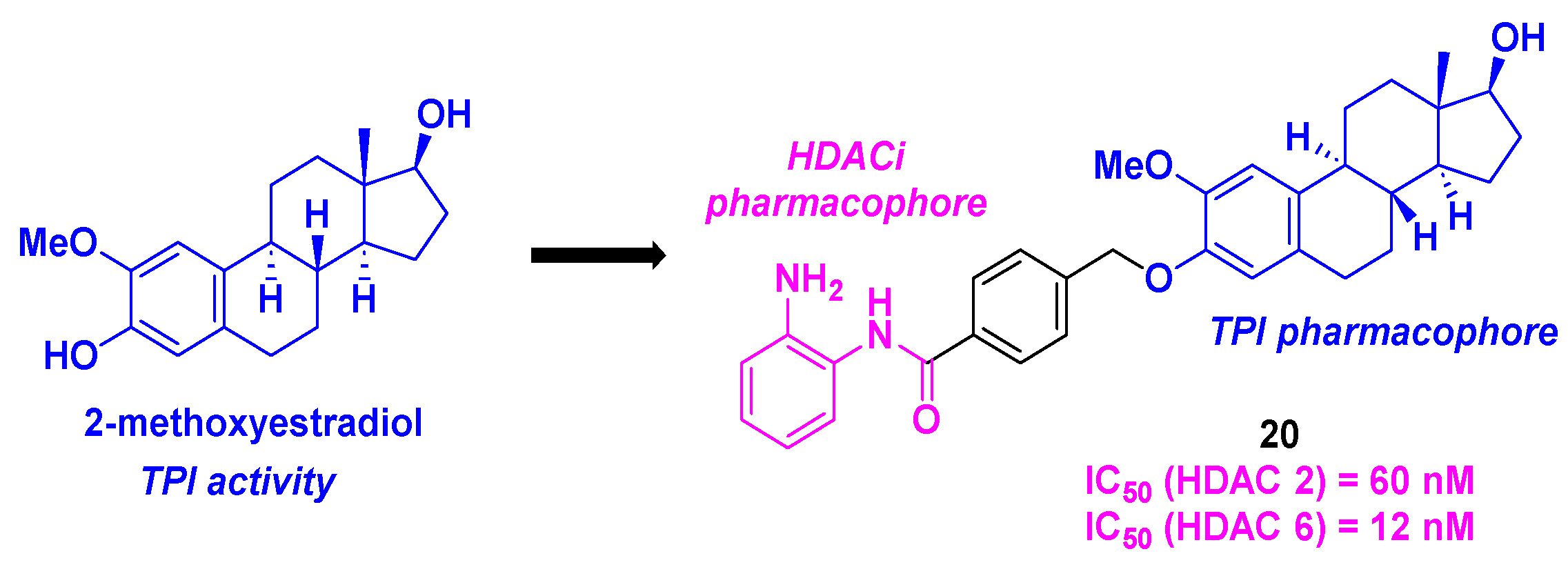
Scheme 22.
Dual HDAC/tubulin inhibitor developed by Yin, Kong and their colleagues.
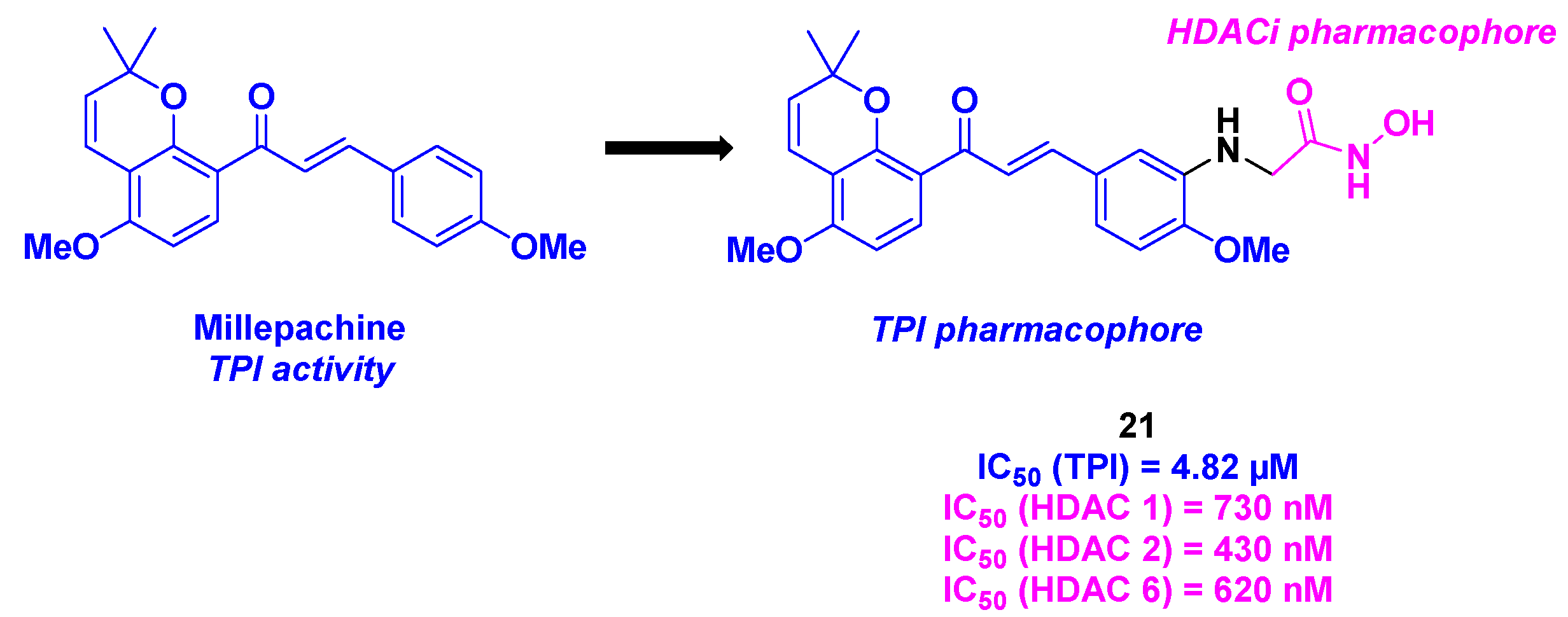
Scheme 23.
Dual HDAC/tubulin inhibitors elaborated by Chen, Li, Lu and coworkers.
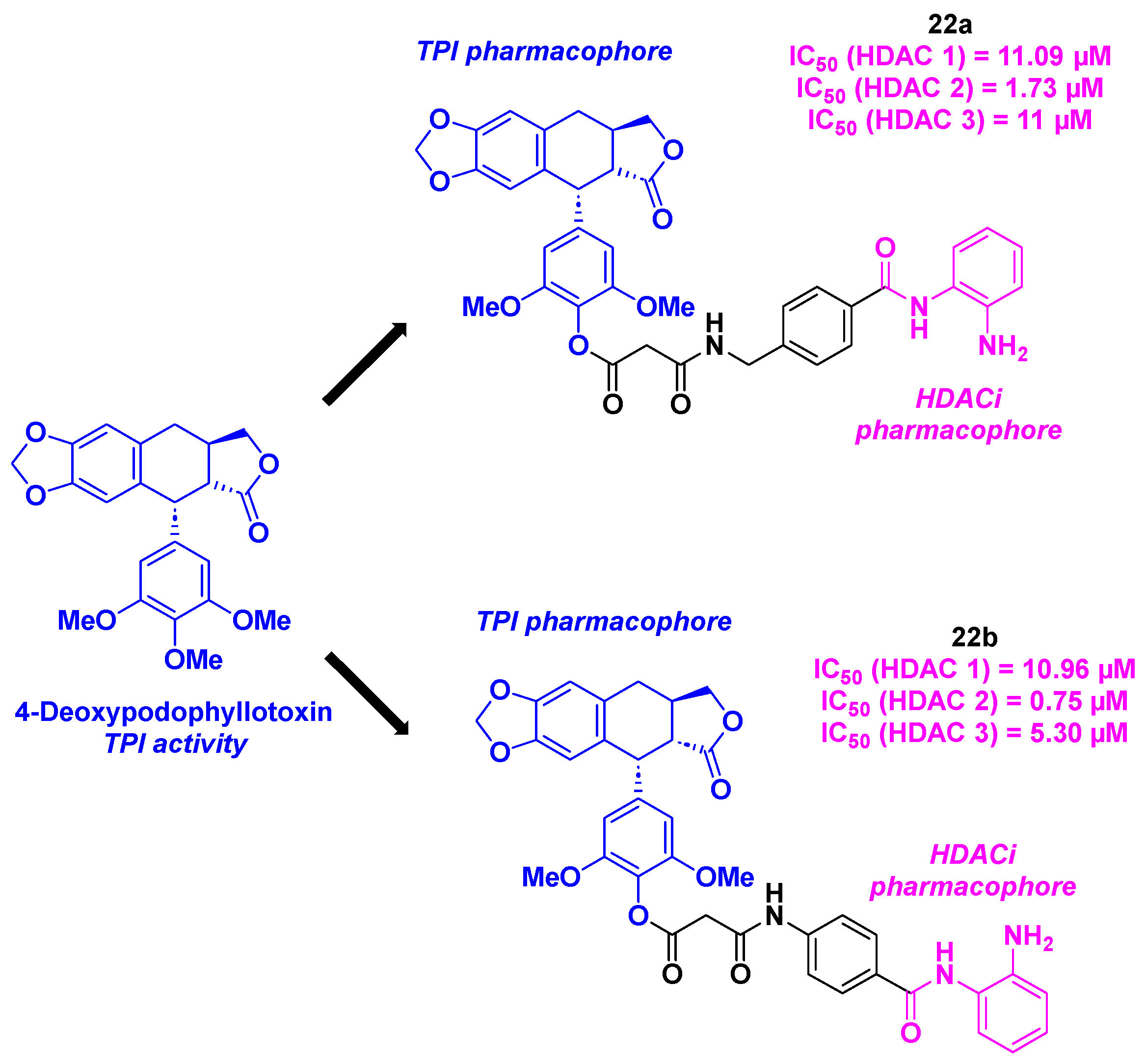
Scheme 24.
Dual HDAC/tubulin inhibitor conceived by the Chen and Lu groups.
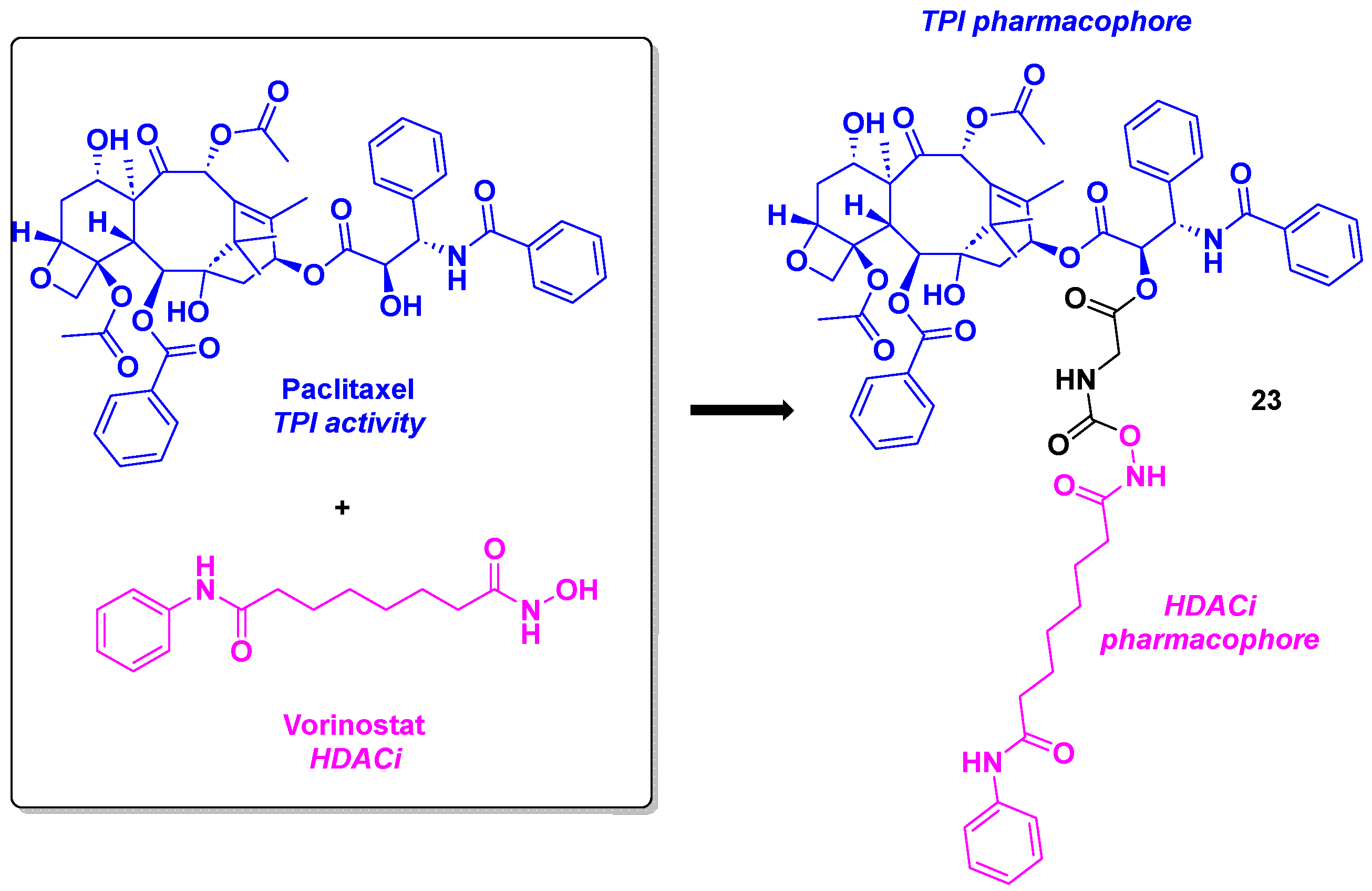
Scheme 25.
Molecular hybrids HDAC/tubulin inhibitor developed by Chen, Liou and coworkers.
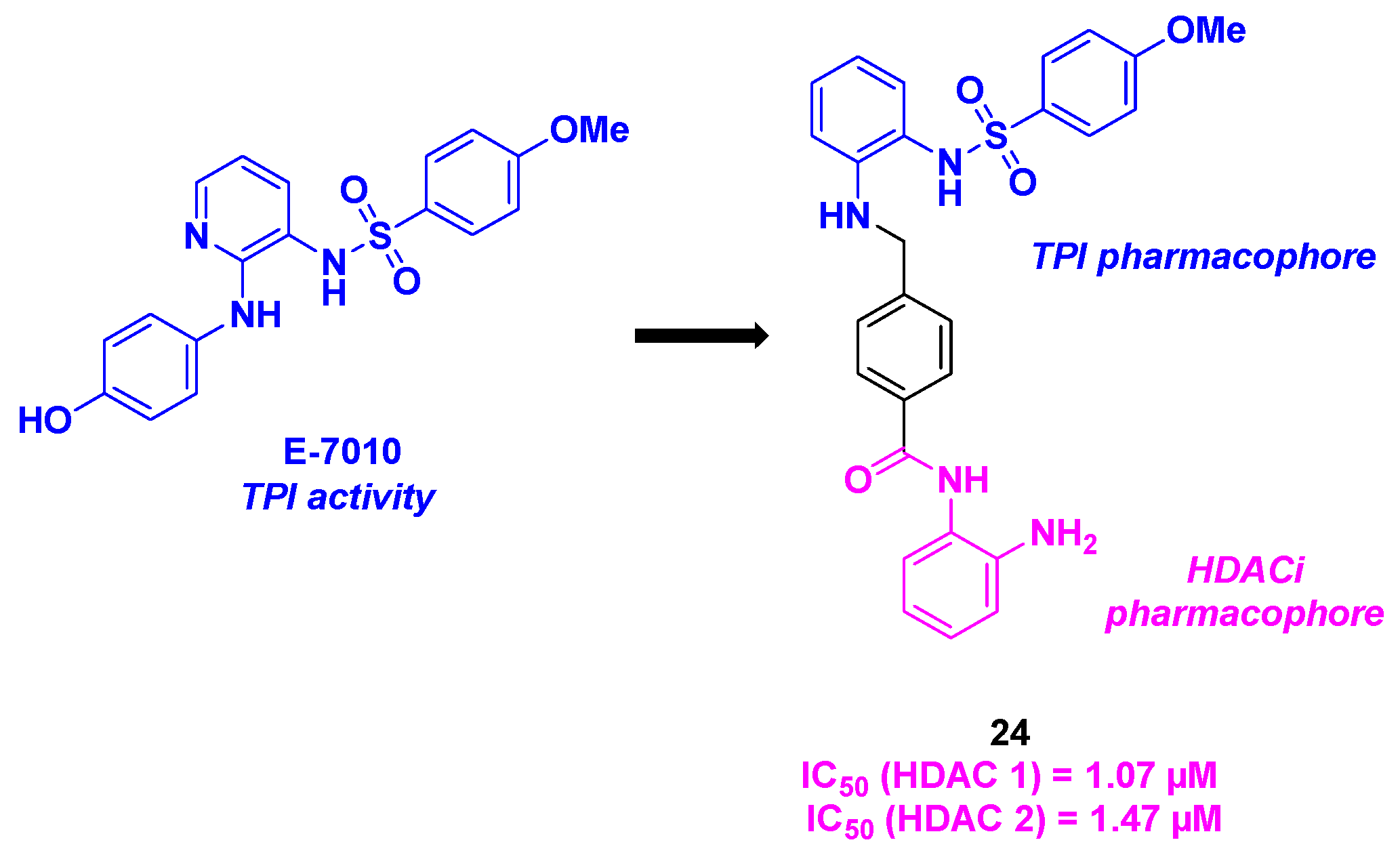
Scheme 26.
Hybrids HDAC/tubulin inhibitors developed by Chen, Liou and their colleagues.
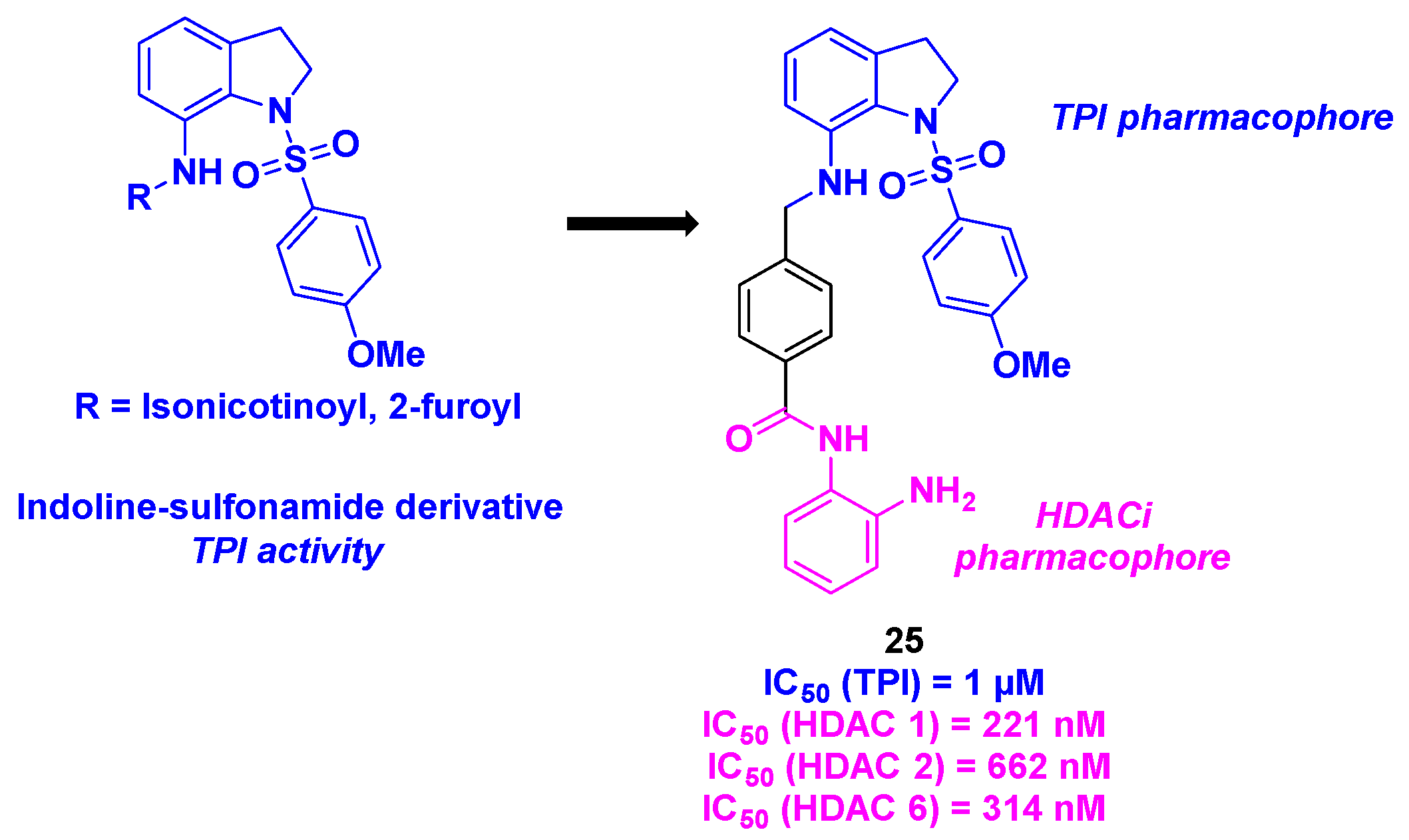
Scheme 27.
Hybrid HDAC/tubulin inhibitors conceived by Yang, Liou and coworkers.
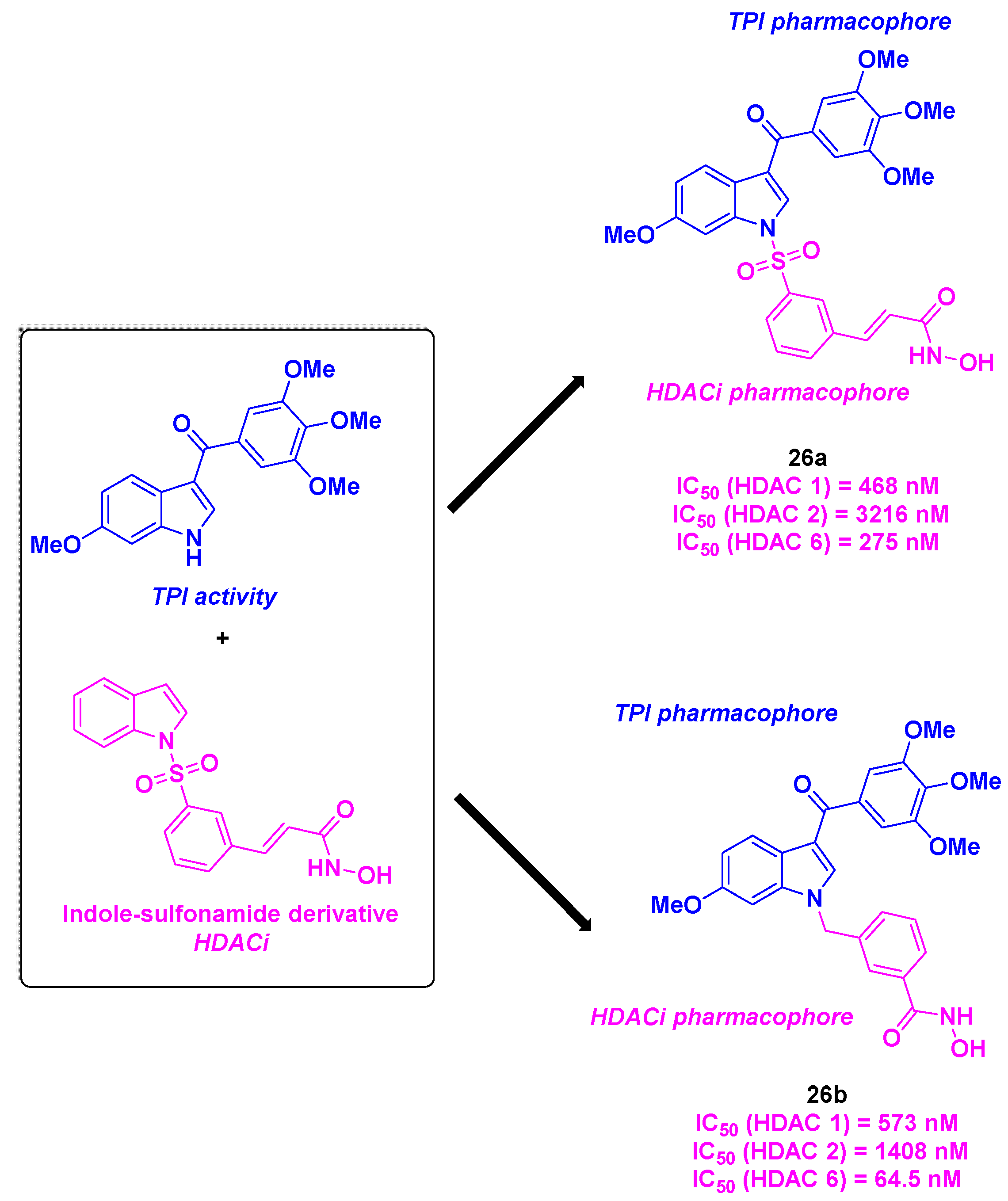
Scheme 28.
Dual HDAC/tubulin inhibitor designed by Lee, Liou and their colleagues.
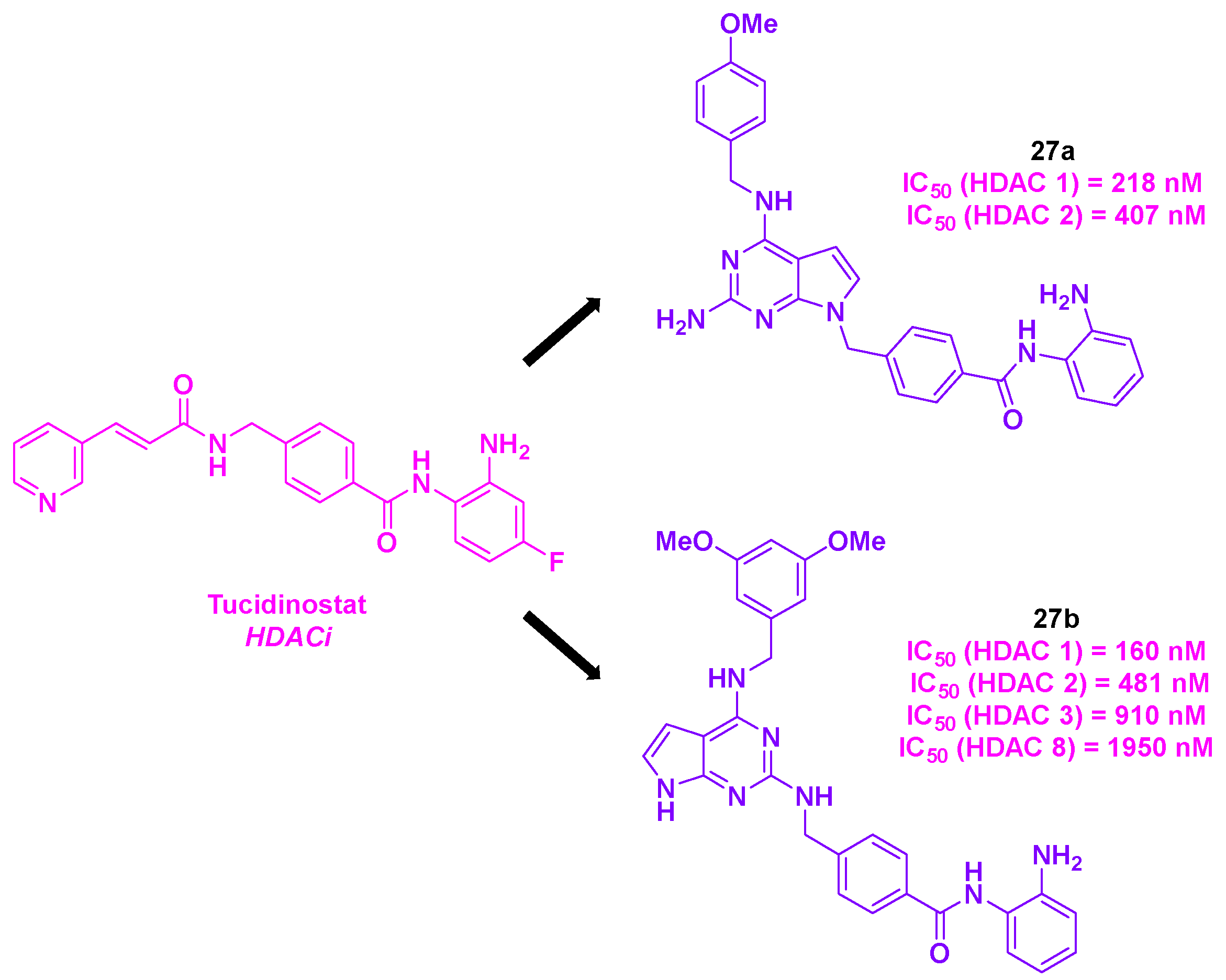
Scheme 29.
Dual HDAC/tubulin inhibitor LT-548-133-1 reported by He, Chen and coworkers.
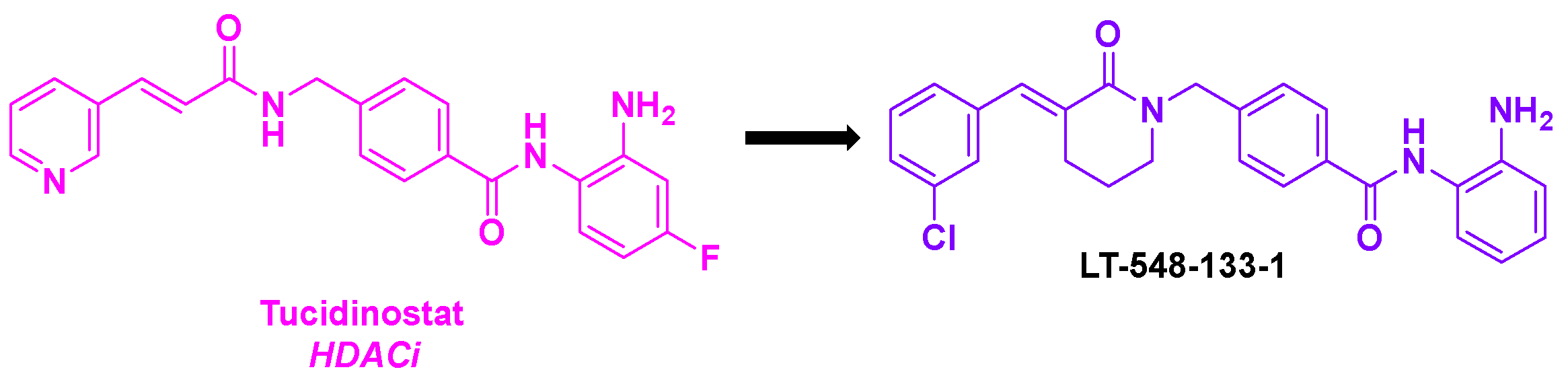
Scheme 30.
Dual HDAC/tubulin inhibitor SKLB-23bb developed by the Chen group.
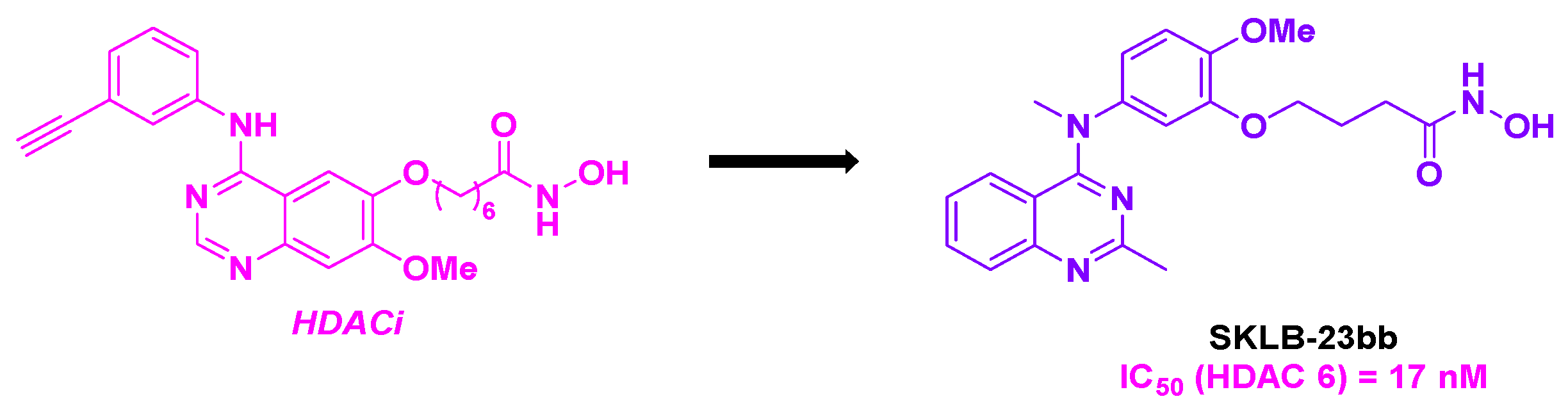
Scheme 31.
Dual HDAC/tubulin inhibitor conceived by Taylor, Tillekeratne et al.
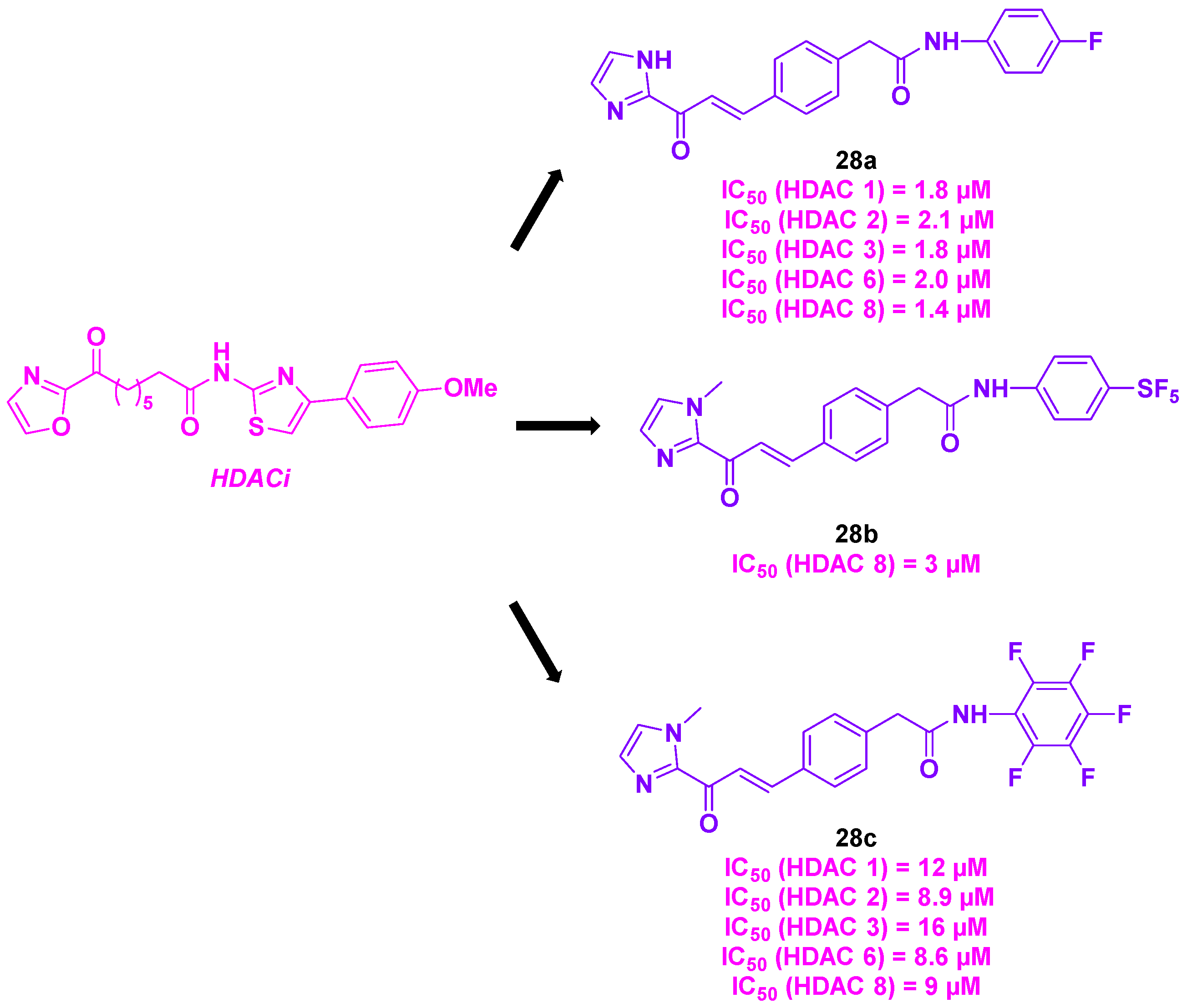
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



