
Submitted:
22 January 2025
Posted:
23 January 2025
You are already at the latest version
Abstract
Neurological involvement in the course of Systemic Lupus Erythematosus (SLE) is a challenging for the large number of patients it affects. It has a profound impact on morbidity, mortality, and quality of life. This review provides an update on current concepts regarding pathogenesis, clini-cal presentation, diagnosis issues, and management strategies for neurological SLE. The patho-physiology is complex and involves immune-mediated and vascular mechanisms of neurological involvement in SLE, including autoantibodies, neuroinflammation, dysregulation of complement, and genetic predisposition. Neuropsychiatric systemic lupus erythematosus (NPSLE) can present with a broad spectrum of central and peripheral nervous system manifestations, ranging from neuropsychiatric SLE, one of the most challenging manifestations, such as cognitive dysfunction, mood disorders, and psychosis, to cerebrovascular disease with an increased incidence of stroke and transient ischemic attacks; demyelinating syndromes mimicking multiple sclerosis; and neuropathies of sensory, motor, or autonomic function. The diagnosis of neurological SLE is very challenging due to subtle, nonspecific, and fluctuating symptoms. A meticulous neurological ex-amination along with neuroimaging techniques, autoantibody profiling, and cerebrospinal fluid analysis is needed. Current treatment strategies will be discussed with corticosteroids, conven-tional immunosuppressants, and emerging biological agents targeting specific immune pathways. It is worth mentioning the complexities of neuropsychiatric symptom management, seizure man-agement, and neuropathic pain.
Keywords:
systemic lupus erythematosus
; collagen vascular disease
; neuropsychiatric lupus
; NPSLE
; central nervous system syndromes
; neurolupus
; neurologic disease
; cognitive dysfunction
; peripheral neuropathy
; acute ischemic stroke
1. Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder characterized by inflammation involving connective tissues. SLE typically tends to impact all organs and tissues in the body, and the specific symptoms experienced by patients might vary significantly. The exact cause of SLE is not fully understood. However, it is thought to arise from the complex interaction of genetic and hormonal factors and environmental exposures, which determine the occurrence of antibodies to nuclear and cytoplasmic antigens [1]. In addition, patients with SLE may have other autoantibodies, including anti-Scl-70 antibodies (found in systemic sclerosis), anti-La and anti-Ro antibodies (found in Sjogren's disease), anticardiolipin antibodies, and antiphospholipid antibodies, suggesting a broad connection between SLE and other autoimmune disorders [2].
The worldwide incidence of SLE varies between 1.5 [3] and 11 [4] cases per 100,000 person-years, whereas the prevalence ranges from 13 to 7,713.5 cases per 100,000 individuals [5]. The female gender predominance in SLE has been extensively documented, with a male-to-female patient ratio of 1:9. The occurrence and frequency of SLE in females frequently peak between the ages of 15 and 44 and 45 and 64, respectively [6]. SLE has variability across all age groups, but it is more prevalent among those aged 15 to 45 years [7]. Research indicates that between 10% and 20% of individuals diagnosed with SLE develop the disease during childhood. Interestingly, patients with childhood-onset SLE have a higher prevalence of renal, neuropsychiatric, and cardiovascular complications than other groups [8,9,10].
In North America, the incidence of SLE was estimated to be 23.2 per 100,000 person-years (95% CI: 23.4, 24.0), while the prevalence was estimated to be 241 per 100,000 individuals (95% CI: 130, 352). Africa and Ukraine had the lowest rates of SLE, with an incidence of 0.3 occurrences per 100,000 person-years. Northern Australia had the lowest prevalence, with no recorded cases among a sample of 847 subjects. Individuals of black ethnicity had the highest incidence and prevalence of SLE, while those of white ethnicity displayed the lowest incidence and prevalence. Over time, an increase in the prevalence of SLE was observed [11]. African American populations have the highest SLE incidence and mortality, followed by Hispanic and Asian populations, while Caucasian populations have the lowest rates of the disease [12].
SLE is a prominent contributor to mortality among young women [13]. A meta-analysis of over 26,000 female patients with SLE in the United States (US) found that the overall mortality rate was 2.6 times higher compared to the general population. The standardized mortality ratio (SMR) was more than 2 for cardiovascular disease and over 5 for infection and renal disease [14].
A notable manifestation of nervous system in SLE is known as neuropsychiatric systemic lupus erythematosus (NPSLE). The documented frequency of neurological symptoms associated with SLE varies from 14% to 95%, with a higher occurrence in children compared to adults [15,16]. It can potentially impact the central and peripheral nervous systems (PNS) in various ways. The total prevalence rate was 56.3%, with a higher incidence in the central nervous system (CNS) at 93.1% compared to the PNS at 6.9% [17,18].
In 1999, the American College of Rheumatology (ACR) published a collection of NPSLE case definitions, which included 12 manifestations related to the CNS and seven manifestations associated with the PNS (Table 1) [19]. The CNS manifestations can be categorized into four psychiatric syndromes and eight neurological syndromes. They can also be classified as either focal (presenting as focal neurologic deficits) or diffuse (including cognitive disorder, mood disorder, psychosis, acute confusional state, and anxiety disorder) [19]. However, a study of 128 participants uncovered a significant occurrence of NPSLE in this group of individuals with SLE who were not specifically chosen. The most prevalent NPSLE manifestations seen were headaches, cognitive impairment, and mental illnesses [20].
Physicians face difficulties in diagnosing NPSLE due to the absence of specific and sensitive laboratory serum or CSF biomarkers, radiographic imaging abnormalities, and formal criteria for establishing the diagnosis and guiding therapy and management decisions in NPSLE [21]. NPSLE diagnosis is facilitated by many clinical, serological, immunological, electrophysiological, and neuroimaging tests [18]. Regrettably, even when healthcare personnel can recognize neurological or psychiatric disorders in patients with SLE, these patients are rarely subjected to thorough evaluation, resulting in a lack of diagnosis and treatment. This is because the required tests are demanding in terms of resources and time [22]. Magro-Checa et al. introduced a diagnostic method for patients with NPSLE that relies on the clinical symptoms observed in patients with SLE [23]. Both patients with significant NPSLE and patients with moderate or non-NPSLE exhibited a higher rate of death compared to the general population [24]. A study revealed that people with NPSLE have a death rate that is 11 to 14 times higher than that of the general population. Interestingly, acute confusional state was the most important indicator for predicting a negative outcome [24].
SLE, a complex autoimmune disease, can profoundly affect the nervous system, leading to a wide range of debilitating conditions that significantly affect patient outcomes. This narrative review presents the current information regarding NPSLE, focusing on the clinical symptoms, pathophysiological pathways that play a role in the condition, diagnostic challenges, and the current strategy for management and treatment.
2. Neurological Involvement In SLE: Proposed Mechanisms
In patients with SLE, approximately one-third of neuropsychiatric events can be directly attributed to SLE, and they occur in 21% of SLE patients within the first 6.6 years of their illness [25]. CNS involvement is observed in 90% of the cases, while the PNS is only responsible for 10% [25]. Noteworthy, there is extensive research on CNS disease in people with SLE, but the impact on the PNS is less well-established (Table 2). Such studies have provided a valuable understanding of the range of CNS symptoms as well as the connection between certain CNS disorders and autoantibodies, immunologic markers, and elements of SLE activity [25,26]. However, only a few studies have been conducted on the relationship between peripheral neuropathies and autoantibody patterns and the characteristics of SLE activity [27].
The CNS is affected by SLE and presents a wide range of neuropsychiatric symptoms. These symptoms include aseptic meningitis, cerebrovascular disease, demyelinating syndrome, various types of headaches (such as migraine and benign intracranial hypertension), movement disorders, myelopathy, seizure disorders, acute confusion, anxiety disorders, and psychosis [19]. Although less common than CNS involvement, PNS involvement can still significantly impact patients. The signs include Guillain-Barré syndrome, autonomic dysfunction, mononeuropathy, myasthenia gravis, cranial neuropathy, plexopathy, and polyneuropathy [19].
The autoimmune/inflammatory route involves activating pro-inflammatory substances or producing autoantibodies that target neuronal cells. This leads to damage by forming intrathecal immune complexes and disrupting the blood-brain barrier (BBB). Prior research has commonly maintained that the BBB's permeability significantly contributes to the development of NPSLE [33].
The complement system plays a crucial role in the inflammatory pathway, particularly in disrupting the integrity of the BBB, mainly through the action of complement C5a/C5aR. In vitro experiments have shown that C5a triggers death in endothelial cells, most likely via binding to the C5aR1 receptor. It has been observed that microglia have a high level of C5aR1 receptor mRNA [34]. Endothelial cells are stimulated by immune complexes to produce pro-inflammatory cytokines and cellular adhesion molecules by activating nuclear factor-kappa B (NF-κB) signaling. Jacob et al. investigated the impact of C5a/cluster of differentiation 88 (CD88) signaling on the integrity of the BBB in SLE throughout the NF-κB pathway. The authors revealed an increase in NF-κB translocation. A following study by the same group revealed that the levels of zona occludens have decreased, but the expression of microtubule-associated protein (MAP) levels has increased, which may contribute to an increase in the permeability of the blood-brain barrier [35]. Therefore, elevated systemic signaling, mediated by cytokines and complement, is the primary factor for triggering apoptosis in CNS endothelial cells, resulting in BBB damage and increased permeability. Of particular significance is the involvement of the tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK)/fibroblast growth factor-inducible 14 (Fn14) pathway [34]. Moreover, diffuse NPSLE was already associated with anti-C1q, C3/AP50, and focal NPSLE with C4 levels. This suggests that the disease activity marked by the complement pathway activation may play a role in neurocognitive dysfunction in NPSLE [23].
Inflammatory mediators can disrupt the BBB. At the same time, neuronal or glial cells can also generate these cytokines and chemokines within the intrathecal space, thereby enhancing the BBB's permeability. Both processes could facilitate circulating autoantibodies and leukocytes infiltration into the CNS.
The presence of inflammatory cytokines, including TNF-α, TWEAK, interferon-γ (IFN-γ), interleukin-6 (IL-6), interleukin-8 (IL-8), and B-cell activating factor (BAFF), has been observed in the cerebrospinal fluid of patients with NPSLE. This suggests that the acute inflammatory response plays a significant role in the development of NPSLE [36]. Yoshio et al. discovered that the levels of certain cytokines (IL-6, IL-8, MCP-1/CCL2, IP-10/CXCL10, G-CSF, and GM-CSF) in the spinal fluid were not affected by the levels of these cytokines in the blood of patients with central NPSLE. This suggests that these cytokines likely produce chemokines in the CNS, which is apparently needed to develop central NPSLE [37]. Other interleukins involved in the development of NPSLE are interleukin-1 (IL-1), interleukin-2 (IL-2), interleukin-10 (IL-10), a proliferation-inducing ligand (APRIL), and interferon-⍺ (IFN-⍺) [38]. Also, the chemokine-ligand 5 (CCL-5) was already associated with NPSLE [38].
Previous research has shown strong evidence that IFN-α can lead to neuropsychiatric symptoms in both human and mouse models. Santer et al. propose that the serum and cerebrospinal fluid (CSF) of individuals with NPSLE may have abnormally high levels of IFN-α activity [39]. In addition, inhibiting IFN-α signaling decreases the synaptic loss associated with microglia and improves anxiety-like behavior and cognitive impairments in 564Igi Lupus-prone mice [40].
Among the cytokines that have been documented, IL-6 is considered to have a strong positive relationship with NPSLE activity. Also, IL-6 in the CSF is a reliable indicator for diagnosing central NPSLE, with high sensitivity and specificity. A recent study has identified a more detailed categorization of NPSLE. It has been observed that levels of blood IL-6 and CSF IL-6 are significantly higher in cases of acute confusion states (ACS) compared to non-ACS diffuse NPSLE, which includes conditions such as anxiety disorders, cognitive impairment, mood disorders, and psychosis, as well as localized NPSLE. Furthermore, the Q albumin (CSF/serum albumin quotient) was significantly elevated in ACS compared to the other two categories of NPSLE. Interestingly, Q albumin correlates more strongly with serum IL-6 than CSF IL-6 in patients with diffuse NPSLE, including both ACS and non-ACS cases [41].
An overactive response of microglia in the brains of mice with SLE, known as MRL/MpJ-Faslpr/J mice (MRL-lpr; Strain 000485), was observed, indicating the presence of brain innate immunity. In NPSLE, the interaction between CD40 and hippocampus microglia played a role in the progression of cognitive dysfunction. In mice, inhibiting the activation of microglia can decrease neuropsychiatric symptoms [42].
2.1. Autoantibodies in NPSLE
A prominent characteristic of SLE is the generation of autoantibodies, and many antibodies have been discovered to be linked to NPSLE symptoms. A total of 116 antibodies have been identified in SLE thus far. Among them, at least 20 antibodies have been linked to NPSLE, including 11 that specifically target the brain and nine that affect the entire system [43].
2.1.1. Antiphospholipid Antibodies Include β2-Glycoprotein 1, Cardiolipin Anticardiolipin (Anti-CL), and Lupus Anticoagulant (LA)
The aPL antibodies specifically bind to anionic phospholipids, such as β2GPI, instead of being directed against these phospholipids, as their name might imply. These phospholipids are found in the plasma membrane and have a role in regulating the blood clotting cascade [44]. Following this, procoagulant activation leads to thrombosis and cerebral infarction [45]. aPL antibodies have been found to be associated with focal and diffuse NPSLE symptoms, including cognitive dysfunction [46], seizures [47], and myelopathy [29].
2.1.2. Anti-Ribosomal P protein Antibodies (anti-RP Ab)
Anti-ribosomal P (anti-RP Ab) antibodies are serological indicators that are frequently seen in patients with SLE psychosis and might be associated with peripheral nervous system complications [48]. Anti-ribosomal-P antibodies are found near the carboxy-terminal end of the 60S subunit of ribosomes. They specifically bind to three phosphorylated proteins, namely P0, P1, and P2 [49]. Anti-ribosomal P-antibodies are thought to cross the BBB, enter neuronal cells, and hinder protein synthesis [50].
2.1.3. Antibodies Against the N-Methyl-D-Aspartate Receptor (anti-NMDA)
The NMDA receptor is a type of glutamate receptor found in the CNS that plays a crucial role in synaptic plasticity and memory formation [51]. The NR2A and NR2B subunits are present in the hippocampus, amygdala, and hypothalamus [52]. NMDA receptors consist of tetramers that are made up of NR1 subunits and two of the four NR2 (A–D) subunits [53]. Anti-NMDAR encephalitis is a neurological disorder caused by the immune system attacking the NMDAR receptors. There are reports of anti-NMDAR encephalitis in patients with SLE, but the exact mechanisms behind its development is not completely understood [54].
2.1.4. Antibodies Against Aquaporin 4 (NMO-IgG/AQP4-Ab)
Aquaporin 4 (AQP4) is the primary protein responsible for facilitating the movement of water in the central nervous system. It is predominantly found at the extremities of astrocytes and is positioned at both the BBB and the brain-cerebrospinal fluid barrier. AQP4 plays a crucial role in regulating the flow of water within cells [55]. Anti-AQP4 antibodies induce damage to astroglial cells by triggering an inflammatory immune response. This response activates complement-dependent cytotoxicity, which in turn disrupts the BBB and leads to the infiltration of white blood cells and the release of cytokines. Consequently, this process causes damage to oligodendrocytes, myelin, and neurons [56]. Myelitis and antiphospholipid syndrome and AQP4-IgG positivity were observed in 6.7% of the patients with SLE [57].
2.1.5. Structural Endothelial Proteins
Endothelial cells (ECs) are located on the inner linings of blood vessels and constitute a layer of cells known as the endothelium. Endothelial cells have not been previously recognized as constituents of the immune system. Endothelial cells (ECs) have a crucial role in maintaining blood pressure and are involved in various physiological and pathological processes such as coagulation, fibrinolysis, angiogenesis, and immune cell activation [58]. In autoimmune disorders, the regulation of endothelial cells by the adaptive and innate immune systems is critical, as endothelial cells contribute to persistent inflammation through angiogenesis, immune cell recruitment, and antigen presentation [59]. The EC dysfunction may lead to microvascular changes in the white matter of patients with SLE leading to vascular dementia features. Also, microvascular abnormalities in the peripheral nerves may be one of the causes of neuropathy in SLE [60].
Anti-suprabasin antibody (Anti-SBSN Ab) are induced by astrocytes exposure to antibodies that have altered senescence and autophagy pathways [61]. It can serve as a useful indicator to distinguish NPSLE from SLE without neuropsychiatric symptoms, as anti-SBSN antibodies and their related immune complex were found solely in the CSF of NPSLE [62].
2.1.6. Anti-Endothelial Cell Antibodies (AECAb)
2.1.7. Anti-Ubiquitin Carboxyl Hydrolase L1 Antibodies (Anti-UCH-L1 Ab)
Li et al. revealed that antibodies against UCH-L1 can be a dependable biomarker in the CSF to diagnose NPSLE. Additionally, the concentrations of UCH-L1 in the CSF may serve as an indicator of the intensity of NPSLE [65]. Furthermore, this marker was linked to increased severity of the disease and overall disease activity [66]. Similarly, a recent study on autoantibodies found that the autoantibody UCH58-69, which targets amino acids 58–69 of UCH-L1, has a high level of specificity and diagnostic significance in distinguishing NPSLE patients from SLE patients without neuropsychiatric symptoms. The study found that NPSLE patients had significantly elevated levels of anti-UCH58-69 in their serum compared to SLE patients without neuropsychiatric symptoms. Furthermore, these levels were correlated with the severity of the condition [63].
2.1.8. Antibodies Against Glyceraldehyde 3-Phosphate Dehydrogenase (GAPDH)
GAPDH has been identified as a new autoantigen linked to neuropsychiatric diseases. Delunardo et al. revealed a strong positive relationship between levels of anti-GAPDH antibodies in the blood and detrimental cognitive and mood conditions (such as schizophrenia and major depression) in patients with SLE. The levels of anti-GAPDH antibodies were elevated in SLE patients who had psychotic symptoms compared to SLE patients who did not have psychotic symptoms [32]. Anti-GAPDH Ab can induce neurite interaction and impairment of neuronal plasticity by blocking and binding of synaptic molecules. These antibodies level are increased in NPSLE and they are associated with cognitive dysfunction and psychiatric manifestations [67].
2.1.9. Triosephosphate Isomerase (Anti-TPI Antibody)
Triosephosphate isomerase (TPI) is an enzyme involved in the conversion of dihydroxyacetone phosphate (DHAP) to glyceraldehyde-3-phosphate (G3P). It is present in neurons and red blood cells [68]. Anti-TPI antibodies have been linked to NPSLE, mainly showing a greater occurrence of aseptic meningitis and higher levels of serum IgG in NPSLE patients who test positive for anti-TPI compared to those who test negative for anti-TPI [69].
2.1.10. Microtubule-Associated Protein (Anti-MAP-2 Ab)
MAP-2 is a cytoskeletal protein found in neuronal cells that plays a crucial role in initiating and stabilizing microtubules, regulating the movement of organelles and protein kinases involved in signal transduction [70]. Anti-MAP-2 antibodies are linked to damage and death of neurons and are notably increased in the CSF of individuals with NPSLE [30]. Neuropsychiatric symptoms, such as psychosis, schizophrenia, bipolar disorder, and major depression, have been linked to the presence of anti-MAP-2 antibodies [71].
2.1.11. U1 Ribonucleoprotein (Anti-U1RNP Ab)
Autoimmune disorders such as mixed connective tissue disease (MCTD), systemic sclerosis (SSc), and SLE are associated with the presence of anti-UIRNP antibodies [72]. SnRNPs, or small nuclear ribonucleoproteins, are abundant RNA-protein complexes located in the nucleus. They play a crucial role in the processing of pre-mRNA and other proteins that make up the spliceosome [73]. Anti-U1RNP antibodies are associated with a increased risk of NPSLE, mainly when detected in the CSF [74].
2.1.12. Others
Brain cytoplasmic ribonucleic acid (BC RNA) refers to a type of non-coding RNA molecule, specifically BC200 RNA (also known as brain cytoplasmic RNA 1 or BCYRN1), which is primarily found in the cytoplasm of neurons within the brain and plays a role in regulating protein translation by inhibiting translation initiation factors; essentially controlling gene expression at the post-transcriptional level. Anti-BC RNA induction decreases the delivery of BC RNA to synaptodendritic sites in the brain. The decrease of BC RNA in CNS was already associated with some types of NPSLE, mainly involving cognitive impairment [75].
2.2. The Cerebrovascular Pathway
Cerebrovascular events frequently manifest in the presence of increased disease activity and evidence of illness-related tissue injury [76]. In NPSLE, the most widely accepted cause of cerebrovascular disease is thrombosis induced by antiphospholipid (aPL) antibodies (APLAs). Prominent risk factors for this condition include chronic and high disease activity, a high cumulative corticosteroid dosage, persistent positivity for aPL antibodies at moderate-to-high levels, heart valve disease, and systemic hypertension [77]. The prevalence of SLE infections ranges from 3% to 20%, with a potential contribution of up to 15% to overall mortality [78,79].
Antiphospholipid syndrome (APS) is an autoimmune disorder distinguished by the presence of defined APLAs, namely anti-cardiolipin IgM and IgG, anti-beta-2 glycoprotein IgM and IgG, and lupus anticoagulant. These APLAs induce a condition of hypercoagulability, resulting in the formation of venous and arterial thrombi. The presence of APS can manifest as a primary disease. Nevertheless, it is worth noting that approximately 20–30% of patients with SLE test positive for APLAs [80]. Moreover, when APLA levels reach moderate-to-high levels, patients are at an elevated risk of thrombosis, even in the absence of any prior thrombotic episodes. APLAs have been identified as agents involved in the vascular pathway of NPSLE pathogenesis, leading to various adverse outcomes such as strokes, venous thromboembolism, cerebral venous sinus thrombosis, cognitive impairment, peripheral neuropathy, and chorea. APS has been associated with many clinical manifestations, such as maternal health complications and livedoid vasculopathy [81]. The CNS exhibits a higher vulnerability to thrombus formation than other tissues in APS, which is believed to be associated with some specific receptors in the brain vasculature [82].
Vasculitis is a group of illnesses characterized by inflammation in blood vessels of varying sizes, occasionally accompanied by fibrinoid necrosis and subsequent vessel wall loss. It is an infrequent manifestation in SLE, with a prevalence of less than 7% in patients with NPSLE [83]. Interestingly, CNS SLE vasculopathy is usually a non-inflammatory condition that impacts small arterioles and capillaries, forming micro-infarcts and hemorrhages [84]. Immune complexes can induce endothelial dysfunction by stimulating endothelial cells to express vascular cell adhesion molecule 1, hence facilitating the recruitment of monocytes into the arterial wall [85]. Anti-endothelial cell antibodies, generated in the context of SLE, elicit the production of proinflammatory factors and foster the adherence of monocytes to endothelial cells, resulting in the subsequent inflammation of vessel walls. The presence of anti-endothelial cell antibodies leads to an increased release of cytokines, including interleukin 1, interleukin 6, and interleukin 8. This physiological response ultimately results in endothelial death [85]. Anti-endothelial cell antibodies and complement significantly contribute to vasculitis development in medium-sized arteries. This condition can lead to various complications, including luminal stenosis, small-vessel noninflammatory vasculopathy, microvessel occlusion, multifocal microinfarcts, intracranial embolism, and microhemorrhages [17].
Hypertension, diabetes mellitus, cigarette smoking, dyslipidemia, and a sedentary lifestyle are considered to be both traditional and modifiable risk factors for stroke and cardiovascular diseases. Hypertension is more common in SLE and APS than in the general population, and it can be explained by chronic inflammation [86]. Also, systemic inflammatory processes have been identified as the underlying cause of early atherosclerosis, which is also known as accelerated atherosclerosis [87]. Other factors influencing atherosclerosis are lupus nephritis, steroid therapy, and vitamin D deficiency.
SLE patients who have previously experienced a transient ischemic attack (TIA) have a 57% chance of developing an ischemic stroke [88]. The incidence of stroke in SLE ranges from 2% to 19% [89,90]. The incidence of ischemic stroke is twice as high in patients with SLE than in the general population and encompasses elements beyond the conventional Framingham risk factors. Additional factors that contribute to the higher risk of an ischemic cerebrovascular event include the presence of vasculitis, associated APS and positive aPL autoantibodies (found in 9.4% of cases), endocarditis, Libman-Sacks endocarditis, hyperviscosity (increased homocysteine levels), genetic polymorphism (the presence of allele GT20), and hypertension [76]. Consistent with this observation, a research study revealed that males with β2GPI-dependent immunoglobulin (Ig) G anticardiolipin antibodies had a 1.5-fold increased risk of stroke and acute myocardial infarction (AMI) compared to those without such antibodies [20]. Additionally, another study indicated that elevated levels of anticardiolipin antibodies in the bloodstream, irrespective of other cardiovascular risk factors, were a significant predictor of future stroke and TIA in females but not in males [91].
Individuals with autoimmune disorders have a twice-fold increased risk of experiencing an ischemic stroke within the first year of hospitalization compared to those without hospitalization. Interestingly, the risk of having a stroke remains elevated for at least ten years after the first hospitalization [92]. Also, after the first ischemic stroke, the risk of having another stroke in ten years is as high as 35% [93].
2.3. Genetic And Environmental Factors
Numerous studies have proven the link between genetic factors and the development of SLE. Nevertheless, investigating the genes responsible for humans' susceptibility to SLE has proven to be a formidable task, mainly due to limited epistasis, genetic heterogeneity, ethnic heterogeneity, and environmental factors. Determining the specific genotypes that contribute to NPSLE pathophysiology remains elusive. A meta-analysis found that the Fc³RIIIa, Fc³RIIIb, and ITGAM genotypes have been identified as probable susceptibility genes for NPSLE following a comprehensive examination of genetic variants associated with this disorder [94].
Specifically, it has been observed that patients with NPSLE have mutations in TREX1, a gene responsible for encoding three-prime repair exonuclease 1 (DNase III) [95]. Furthermore, variations in this gene have been linked to the presentation of CNS involvement, including seizures [96]. There is evidence suggesting that the HLA-DRB1*04/*13 genotype and the SNP rs10181656 in STAT4, which encodes the signal transducer and activator of transcription 4, are linked to strokes in individuals with SLE [97]. This association remains significant even after controlling for aPL antibody status and conventional cardiovascular risk factors [98,99]. A comprehensive assessment has been conducted on the collective impact of single nucleotide polymorphisms (SNPs) in multiple genes linked to SLE, namely HLADRB1, IRF5, STAT4, BLK, TNFAIP3, TNIP1, FCGR2B, and TNFSF13, in Japanese individuals diagnosed with SLE. The presence of ten or more SNPs doubled the likelihood of neurological symptoms in these patients [100].
The induction or exacerbation of autoimmune diseases, such as SLE, by infection can be attributed to various mechanisms, including molecular mimicry, epitope spreading, polyclonal activation of B cells, bystander expansion of auto-reactive T cells, and viral and bacterial super-antigens [101]. The clinical presentations observed in NPSLE are a consequence of immunological dysregulation, characterized by the development of autoantibodies or vaso-occlusive events associated with the presence of APLAs. Disease-associated immunological dysfunction and the immunosuppressive effects of treatment contribute to an increased vulnerability to infection [102]. Furthermore, the bimodal survival pattern in SLE has been reevaluated, revealing that death resulting from infections persisted throughout the progression of the disease [103].
The prevalence of CNS involvement in patients with SLE is estimated to range from 18% to 67% [104]. In SLE, infections contribute to around 20–55% of morbidity and mortality [105]. The CNS is accountable for 3% of these infections [105]. However, a significant proportion of NPSLE patients, up to 81%, do not exhibit any accompanying systemic SLE activity [104]. The predominant pathogens are Mycobacterium tuberculosis and Cryptococcus neoformans [106]. Notwithstanding their infrequent occurrence, CNS infections result in significant morbidity and mortality [107]. The results of the correlation study revealed a subtle positive relationship between rubella IgM antibody titers and the presence of psychosis in individuals with NPSLE [108].
The ultraviolet (UV) radiation emitted by sunlight is typically categorized into three broad bands: UV-C (200–290 nm: distant UV, germicidal UV), UV-B (290–320 nm: midrange UV, sunburn radiation), and UV-A (320–400 nm: near UV, black light). It is observed that the stratospheric ozone layer fully absorbs UV-C light, but UV-B radiation is only partially absorbed. The penetration of UV-B radiation is limited to the epidermis of window glass. Still, UV-A radiation can penetrate both ordinary and colored glass and the epidermis [109]. UV-B radiation, when administered in low concentrations, has been found to induce DNA damage, lymphocyte apoptosis [110], and the production of pro-inflammatory cytokines, adhesion molecules [111], and nitric oxide synthase in keratinocytes [112]. This might potentially have systemic ramifications. The transformation of trans- to cis-urocanic acid produced by UV-B radiation has been observed to inhibit cell-mediated immunity. Furthermore, UV-B radiation reduces Langerhans cells' ability to stimulate CD4+ Th1 cells. It activates CD4+CD45RA+ suppressor-inducer T cells, thereby altering the immunological landscape in favor of B-cell activation [111,113]. Ultraviolet A (UV-A) radiation has the potential to induce systemic flares by directly infiltrating the subcutaneous vasculature. Conversely, it has been suggested that UV-A1 light with a longer wavelength range of 340–400 nm may possess protective properties against flare-ups in SLE [114]. It found instances of organ involvement, including lupus nephritis, in individuals diagnosed with SLE due to exposure to sunlight [115]. Interestingly, there are anecdotal reports of NPSLE after sunlight in individuals with NPSLE [116].
Pharmaceutical compounds linked to drug-induced lupus erythematosus (DILE) have diverse chemical compositions, including aromatic amines, hydrazine, and sulfhydryl groups. This suggests that DILE does not possess a singular unifying chemical configuration [117]. Pharmacological agents implicated in DILE can be classified into four distinct categories, which include drugs that are definitive, likely, potentially, and recently documented to induce DILE [118]. The medications that are most frequently associated with DILE include hydralazine, procainamide, isoniazid, and TNF-α inhibitors [119]. Hydrochlorothiazide, calcium channel blockers, and angiotensin-converting enzyme inhibitors [120] are among the drugs that are most prone to inducing subacute cutaneous lupus erythematosus (SCLE). Other medications like proton-pump inhibitors (PPIs) [121], terbinafine [122], immunomodulators (leflunomide [123], TNF-κ inhibitors [124], and chemotherapeutic agents [125], have the potential to elicit SCLE. Grönhagen et al. found higher odds ratio (OR) for terbinafine (OR 52.9), TNF-κ inhibitors (OR 8.0), antiepileptics (OR 3.4), and PPIs (OR 2.9) and SCLE [126]. Fluorouracil substances or their contemporary counterparts, such as capecitabine, have traditionally been identified as the primary triggers of chronic cutaneous DILE [127].
3. Spectrum of Neurological Manifestations in SLE
The American College of Rheumatology developed a set of criteria for NPSLE. These include 19 NPSLEs. They are classified into 12 central and seven peripheral nervous system manifestations [19]. NPSLE could develop as a presenting symptom or throughout the disease course [19]. Neuropsychiatric syndromes occur in 56.3% of SLE patients [18]. Furthermore, their prevalence varies according to the age at which the disease begins. Pamuk et al. showed that seizures and psychosis were more common in early-onset SLE patients, while peripheral neuropathy was more prevalent in the late-onset SLE group [128].
3.1. Psychiatric Disorders (Anxiety, Depression, and Psychosis)
Mood disorders are prevalent in around 20% of patients with SLE [18]. The severity of depression and anxiety is associated with SLE activity [129]. Moreover, depression could affect drug adherence [130]. Thus leading to frequent SLE flares and a worse prognosis [131]. Psychosis is considered rare, occurring in 2.5% of patients with SLE [132]. Psychosis is a disorder in the perception of reality; patients with psychosis could experience hallucinations or delusions [19]. In around 5% of patients with SLE, psychosis could be caused by steroid administration [133]. SLE-related psychosis could be managed with steroids and immunosuppressive drugs. This resulted in the full remission of about 66.7% of patients [132]. Meanwhile, rituximab resulted in a significant improvement in the refractory cases [134].
3.2. Cognitive Dysfunction
The ACR defines cognitive dysfunction as impairment in cognitive domains, including attention, memory, reasoning, and language [19]. Cognitive dysfunction is present in 19.7% of SLE patients [18]. However, it is poorly recognized by physicians [135]. Cognitive dysfunction is not correlated with the severity of the disease. However, it is associated with the presence of organ damage. Moreover, anti-dsDNA antibodies are negatively related to cognitive impairment [135]. The ACR has recommended a 1-hour battery test for diagnosing cognitive impairment [19]. The use of psychosocial education helped to improve memory and functioning [136]. Memantine did not significantly improve the cognitive impairment in NPSLE [137].
3.3. Seizures
Seizures affect approximately 9.9% of SLE patients. The generalized tonic-clonic seizure is the most frequent semiology in SLE patients [138]. Females and patients who had SLE at a young age are at a higher risk of developing seizures related to SLE [138]. There is no association between auto-antibodies, including the lupus anticoagulant and anticardiolipin, and the risk of developing seizures [138]. The outcome of seizures related to SLE is favorable. Most seizures are resolved without requiring long-term medications or significantly affecting the quality of life [138].
3.4. Stroke and Transient Ischemic Attacks (TIAs)
Cerebrovascular diseases (CVD) are prevalent among 8% of SLE patients [18]. The typical risk factors for CVD include diabetes, hypertension, and dyslipidemia [139]. Other risk factors include cutaneous vasculitis, anticardiolipin antibodies, and lupus anticoagulant antibodies [140]. Moreover, the antiphospholipid syndrome is an important risk factor as it leads to a hypercoagulable state [139]. This is done by inhibiting protein C, reducing prostacyclin, activating platelets, and activating endothelial cells [141]. TIAs are reversible neurological events. However, they are considered important stroke predictors in SLE patients [139].
3.5. Peripheral Neuropathy
Polyneuropathy occurs in 3% of SLE patients, while mononeuropathy occurs in 1.5% [18]. Jasmin et al. reported that sensory neuropathy was the most prevalent type among SLE patients [142]. Mahran et al. showed that neuromuscular ultrasound could be used in addition to nerve conduction studies in patients with SLE and concerning neuropathy as it provided additional information on the pathophysiology of nerve affection [143]. Guillain-Barré syndrome (GBS) is a rare presenting symptom in around 1.3% of SLE patients [144]. However, its incidence has been increasing. There are 27 cases of GBS associated with SLE that have been reported in the literature [145]. These patients were aged 20–60, and 77% were females. They were managed with various immunosuppressive drugs, including steroids and cyclophosphamide; 63% of them achieved complete recovery [145]. However, it is essential to differentiate between GBS as a symptom of SLE and pure GBS co-occurring with SLE, as cyclophosphamide is ineffective in patients with pure GBS [146].
3.6. Demyelinating Syndromes
Demyelinating syndromes (DS) are rare, occurring in around 0.3% of SLE patients [18]. The manifestations of DS include vision loss, diplopia, lower limb weakness, cranial nerve palsy, nystagmus, cognitive impairment, and an acute confusional state [147]. Differentiating SLE from multiple sclerosis (MS) can be challenging in clinical practice. However, the presence of certain manifestations could guide the diagnosis of SLE. These include the presence of rash, arthralgia, myalgia, and renal affection [148]. Furthermore, the involvement of the peripheral nervous system could indicate SLE, since MS only affects the CNS [149].
The pathology of MS is different compared to SLE. Plaques, which are focal areas of demyelination, characterize MS. They are present, along with inflammatory changes and axonal degeneration. Meanwhile, the pathology of SLE-related DS relies on cerebrovascular disease. Therefore, there are multiple areas of infarction, along with atherosclerosis and angiopathy [149]. However, Nikolopoulos et al. reported that patients with SLE demyelinating diseases (SLE-DS) and those with SLE fulfilling the criteria for multiple sclerosis had similar neurological findings on MRI. Moreover, they had similar rates of optic nerve affection. Patients with SLE-DS, however, had low odds of achieving an elevated IgG index. None of the SLE-DS patients had positive type II oligoclonal bands [150].
4. Diagnostic Challenges in Neurological SLE
Neurological and psychiatric symptoms are known as NPSLE. The reported prevalence of NPSLE ranges between 14 to 95% and is more common in children than adults [15,16]. Diagnosing neurological SLE is challenging due to the complexity and variability of its manifestations. There are no specific criteria to diagnose NPSLE; based on the diagnosis of exclusion, less than 40 % of neuropsychiatric symptoms will be attributed to SLE-induced nervous system damage. In the remaining cases, other causes (e.g., drug-induced, primary disorders) better explain these symptoms. Thus, the physician has to exclude other causes and assess neurological and psychiatric symptoms. Further evaluation includes assessing general SLE activity, cardiovascular risk factors, atherosclerotic disease, and thrombotic events [18,76].
Neurological assessment should be focused on headaches, signs of seizures, and motor and sensory deficits, while psychiatric evaluation should assess behavior, cognition, perception, thinking, mood, and affect. The diagnostic evaluation of patients with neurological manifestations in SLE should involve all the appropriate investigations that would typically be conducted for non-SLE patients presenting with similar symptoms and signs [76]. For example, a patient with stroke symptoms should undergo a screening echocardiogram and vessel imaging of carotids and vertebral arteries, in addition to testing for SLE-specific causes such as APLAs [151]. Another example is when a patient presents with a psychiatric disorder, such as confusion. The physician should identify the cause, whether it is metabolic abnormalities, infection, or psychoactive drug use.
Several studies have investigated a correlation between disease activity and neuropsychiatric events in SLE patients. Some studies have demonstrated a connection between heightened overall SLE disease activity and neuropsychiatric manifestations attributed to the disease. This correlation is stronger for diffuse rather than focal events [27]. Neuroimaging and laboratory tests are integral to diagnosing neurological SLE, particularly distinguishing its manifestations from other neurological conditions. The table below will provide examples of investigations used for diagnosis (Table 3).
5. Management and Treatment Strategies for Neurological SLE
The primary goal in managing neurological SLE is to control SLE disease activity, minimize neurological symptoms, and improve overall function and quality of life. Due to the diverse clinical presentations and difficulties in diagnosis, managing patients with SLE who experience neuropsychiatric symptoms is most effective when approached by a multidisciplinary team coordinated by a rheumatologist. Identifying and treating non-SLE-related factors in all cases is essential, even when SLE is the primary contributor. For instance, infections or metabolic issues must be managed in patients presenting with acute neuropsychiatric symptoms, while those with vascular-related neuropsychiatric events should be evaluated for cardiovascular risk factors. Specific therapies for primary SLE manifestations are chosen based on the predominant immunopathogenic mechanism in neuropsychiatric SLE, whether the injury is inflammatory-mediated or vascular-mediated.
When the etiology is thought to be inflammatory/neurotoxic (especially aseptic meningitis, optic neuritis, transverse myelitis, peripheral neuropathy, refractory seizures, psychosis, and acute confusional state) and in the presence of lupus activity, management includes glucocorticoids alone or in combination with other immunosuppressants such as azathioprine or cyclophosphamide [163]. In cases of severe symptoms refractory to standard therapy, plasma exchange [164], intravenous immunoglobulin [165], and rituximab (an anti-CD20 monoclonal antibody) have been used [166]. A randomized controlled trial by Barile-Fabris in 32 patients with acute, severe neuropsychiatric SLE reported a significantly better response to therapy with intermittent intravenous cyclophosphamide than with intravenous methylprednisolone (95% versus 54%, P <0.03) [167]. In patients with APS, anticoagulation therapy is more effective than antiplatelet therapy. For secondary prevention of arterial events such as stroke and TIA [168]. In patients with recurrent thrombosis while on warfarin, adjuvant therapies are added to their treatment plans, such as antiplatelet agents, antimalarial agents, and statins [169].
Antidepressants, anxiolytics, and antipsychotic agents are prescribed according to their standard indications in psychiatric disorders. At the same time, antiseizure therapy is initiated when high-risk features are present, such as serious brain injury, brain structural abnormalities (MRI), focal neurological signs, and epileptiform discharges [170].
Antimalarial drugs (hydroxychloroquine and chloroquine) are used in the primary prevention of SLE symptoms, especially for cutaneous and musculoskeletal involvement [76] and reduction in mortality [171]. Although no studies specifically address its effect on neuropsychiatric SLE symptoms, a preventive role of these drugs in CNS lupus has been suggested, especially in cerebrovascular disease [172]. As shown in Table 4, immunosuppressants and biological therapy are the main lines of treatment for NPSLE.
As shown in the previous table, corticosteroids are first-line for acute symptoms, especially during flares, but they are associated with significant long-term risks. Cyclophosphamide is preferred in severe cases due to its efficacy despite its side effects. Azathioprine and Mycophenolate Mofetil are used for maintenance therapy, though their role in neuropsychiatric symptoms is not well studied. Rituximab offers the potential for refractory cases.
In the case of ischemic NPSLE, the main line of treatments are anticoagulants and antiplatelets. Platelet activation is increased in SLE patients, and the presence of antiphospholipid antibodies add additional risk of thrombosis [188].
It’s recommended to use low-dose acetylsalicylic acid as primary thromboprophylaxis for SLE in patients who have positive lupus anticoagulant (LAC) or persistently elevated anticardiolipin antibodies (aCL) at medium to high levels [189].
For secondary prevention for patients with antiphospholipid antibodies, acetylsalicylic acid monotherapy, clopidogrel 75 mg monotherapy, and the combination of acetylsalicylic acid and extended-release dipyridamole are acceptable options for initial treatment [190,191]. On the other hand, anticoagulation therapy is preferred for secondary prevention in high-risk patients. It is more effective at preventing stroke recurrence. However, the bleeding risk is higher, necessitating close monitoring with INR to ensure that the benefits of preventing thrombosis outweigh the risk of hemorrhagic complications. Sometimes, a combination of antiplatelet and anticoagulation therapies is used [192]. The following chart summarizes the management of neuropsychiatric SLE according to its etiology (Figure 1).
6. Prognosis And Long-Term Outcomes In NPSLE
NPSLE is associated with a negative impact on quality of life and severe fatigue, regardless of the level of disease activity [193]. Moreover, patients with SLE are at increased risk of work incapacity compared to normal individuals [194]. Up to 34% of patients with SLE are unable to work. This is attributed to many factors, including socioeconomic status, disease activity, pain, fatigue, anxiety, and neurocognitive disorders [195]. Furthermore, cognitive impairment negatively impacts quality of life and participation in social activities [196].
Patients with NPSLE have an increased risk of mortality and morbidity compared to the general population. Li et al. reported an 11-fold increase in the risk of death [62]. The leading causes of death were related to NPLSE and involved increased intracranial pressure, cerebrovascular disease, and motor neuron disorders [62]. On the other hand, a focal CNS lesion is associated with a 7-fold increase in mortality [197]. Furthermore, the state of acute confusion, the level of C-reactive protein, and the increased intracranial pressure are associated with mortality [62]. Also, receiving the appropriate treatment could affect the prognosis. Patients with neuropsychiatric events attributed to SLE who received steroids and immunosuppressive drugs had improved outcomes [198].
Many biomarkers have the potential to predict the prognosis of patients with SLE. Kostopoulou et al. showed that low levels of complement C3 and high levels of anti-dsDNA were associated with SLE flares [199]. Moreover, the presence of certain biomarkers could predict certain neuropsychiatric manifestations. Jiang et al. reported an association between psychosis and anti-β2GPI antibodies; polyneuropathy and anti-Scl70 antibodies; and demyelinating syndrome and anti-cardiolipin antibodies. Meanwhile, anti-Sjogren syndrome antigen B antibodies and high complement C3 were associated with a low risk of NPSLE [200]. Additionally, the prognosis of NPSLE could be predicted. Low C4 levels, lupus anticoagulant, and anti-cardiolipin antibodies were associated with severe neuropsychiatric manifestations [201,202]. Moreover, the treatment response could also be predicted. Zheng et al. showed that patients with low C3 or C4 levels or positive anti-dsDNA had better outcomes following belimumab administration [203].
For patients with SLE, there is a need for regular follow-up to monitor disease activity and treatment effects. Hsu et al. reported a case of GBS that was regularly followed up for a year after complete recovery [204]. This is critical because patients may not respond to treatment or experience a relapse. For example, in the case of demyelinating diseases, approximately 42% of patients with SLE and demyelinating syndrome experienced a relapse [150]. In GBS associated with SLE, only 63% of patients experienced complete recovery [145]. Meanwhile, refractory symptoms could be managed with other approaches. Tokunaga et al. reported that patients with refractory NPSLE significantly improved following cyclophosphamide administration [134]. Moreover, SLE activity should be monitored as it could be associated with the severity of NPSLE [129].
7. Neurological SLE Research's Future Directions
In recent years, numerous multinational initiatives have recorded advancements in systemic lupus erythematosus clinical management. The 2019 classification by the European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) established positive antinuclear antibodies (ANA) as a mandatory entry criterion, organized items into weighted organ domains, and substituted individual exclusion criteria with a singular attribution rule, stipulating that items should only be considered if no more plausible explanation than SLE exists. The revised EULAR recommendations explicitly outline significant advancements in SLE therapy, emphasizing the administration of hydroxychloroquine to all SLE patients without contraindications, the necessity of risk factor modification, treatment aimed at specific targets, and the reduction of glucocorticoid exposure. The Latin American Grupo Latino Americano de Estudio del Lupus (GLADEL) recommendations conveyed the same perspectives concerning antimalarials for all SLE patients and advocated for maintaining modest glucocorticoid dosages.
SLE is a frequently observed disease in clinical practice, but the neurological symptoms related to this rheumatologic disorder are still poorly understood (Figure 2). There is an urgent need for studies involving this specific group of patients because they have a significantly worsened quality of life when compared to individuals without neuropsychiatric manifestations. Therefore, the development of biomarkers specific to NPSLE is mandatory. Also, the current biomarkers should be evaluated with meta-analysis regarding their benefit in diagnosing and assessing disease activity. As a general statement, a prompt diagnosis and treatment of NPSLE will likely result in a better prognosis.
8. Conclusions
NPSLE is a broad term for a long list of neurological manifestations associated with SLE, which can significantly impact a patient's quality of life and may lead to long-term disability. The pathophysiology is complex and involves autoantibodies, neuroinflammation, dysregulation of complement, and genetic predisposition. Although challenging, early recognition and diagnosis of NPSLE are essential for initiating appropriate treatment and preventing irreversible neurological damage.
Author Contributions
J.P.R., R.S., I.K., K.I., M.A., A.F.G., and A.L.F.C. conceived and designed the methodology of the literature review. J.P.R., R.S., I.K., K.I., M.A., and A.F.G. extracted and collected the relevant information and drafted the manuscript. A.L.F.C. supervised the article selection and reviewed and edited the manuscript. J.P.R. and A.L.F.C. reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Karrar, S.; Cunninghame Graham, D.S. Abnormal B Cell Development in Systemic Lupus Erythematosus: What the Genetics Tell Us. Arthritis Rheumatol. 2018, 70, 496–507.
- Didier, K.; Bolko, L.; Giusti, D.; Toquet, S.; Robbins, A.; Antonicelli, F.; Servettaz, A. Autoantibodies Associated With Connective Tissue Diseases: What Meaning for Clinicians? Front. Immunol. 2018, 9, 541.
- Magro, R.; Borg, A.A. Characterisation of Patients with Systemic Lupus Erythematosus in Malta: A Population Based Cohort Cross-Sectional Study. Biomed. Res. Int. 2018, 2018, 2385386.
- Anstey, N.M.; Bastian, I.; Dunckley, H.; Currie, B.J. Systemic Lupus Erythematosus in Australian Aborigines: High Prevalence, Morbidity and Mortality. Aust. N. Z. J. Med. 1993, 23, 646–651.
- Grennan, D.M.; Bossingham, D. Systemic Lupus Erythematosus (SLE): Different Prevalences in Different Populations of Australian Aboriginals. Aust. N. Z. J. Med. 1995, 25, 182–183.
- Danchenko, N.; Satia, J.A.; Anthony, M.S. Epidemiology of Systemic Lupus Erythematosus: A Comparison of Worldwide Disease Burden. Lupus 2006, 15, 308–318.
- McMurray, R.W.; May, W. Sex Hormones and Systemic Lupus Erythematosus: Review and Meta-Analysis. Arthritis Rheum. 2003, 48, 2100–2110.
- Dave, M.; Rankin, J.; Pearce, M.; Foster, H.E. Global Prevalence Estimates of Three Chronic Musculoskeletal Conditions: Club Foot, Juvenile Idiopathic Arthritis and Juvenile Systemic Lupus Erythematosus. Pediatr. Rheumatol. Online J. 2020, 18, 49.
- Lewandowski, L.B.; Schanberg, L.E.; Thielman, N.; Phuti, A.; Kalla, A.A.; Okpechi, I.; Nourse, P.; Gajjar, P.; Faller, G.; Ambaram, P.; et al. Severe Disease Presentation and Poor Outcomes among Pediatric Systemic Lupus Erythematosus Patients in South Africa. Lupus 2017, 26, 186–194.
- Aggarwal, A.; Phatak, S.; Srivastava, P.; Lawrence, A.; Agarwal, V.; Misra, R. Outcomes in Juvenile Onset Lupus: Single Center Cohort from a Developing Country. Lupus 2018, 27, 1867–1875.
- Rees, F.; Doherty, M.; Grainge, M.J.; Lanyon, P.; Zhang, W. The Worldwide Incidence and Prevalence of Systemic Lupus Erythematosus: A Systematic Review of Epidemiological Studies. Rheumatology (Oxford) 2017, 56, 1945–1961.
- Stojan, G.; Petri, M. Epidemiology of Systemic Lupus Erythematosus: An Update. Curr. Opin. Rheumatol. 2018, 30, 144–150.
- Yen, E.Y.; Singh, R.R. Brief Report: Lupus-An Unrecognized Leading Cause of Death in Young Females: A Population-Based Study Using Nationwide Death Certificates, 2000-Arthritis Rheumatol. 2018, 70, 1251–1255.
- Lee, Y.H.; Choi, S.J.; Ji, J.D.; Song, G.G. Overall and Cause-Specific Mortality in Systemic Lupus Erythematosus: An Updated Meta-Analysis. Lupus 2016, 25, 727–734.
- Muscal, E.; Brey, R.L. Neurologic Manifestations of Systemic Lupus Erythematosus in Children and Adults. Neurol. Clin. 2010, 28, 61–73.
- Sibbitt, W.L.J.; Brandt, J.R.; Johnson, C.R.; Maldonado, M.E.; Patel, S.R.; Ford, C.C.; Bankhurst, A.D.; Brooks, W.M. The Incidence and Prevalence of Neuropsychiatric Syndromes in Pediatric Onset Systemic Lupus Erythematosus. J. Rheumatol. 2002, 29, 1536–1542.
- Govoni, M.; Bortoluzzi, A.; Padovan, M.; Silvagni, E.; Borrelli, M.; Donelli, F.; Ceruti, S.; Trotta, F. The Diagnosis and Clinical Management of the Neuropsychiatric Manifestations of Lupus. J. Autoimmun. 2016, 74, 41–72.
- Unterman, A.; Nolte, J.E.S.; Boaz, M.; Abady, M.; Shoenfeld, Y.; Zandman-Goddard, G. Neuropsychiatric Syndromes in Systemic Lupus Erythematosus: A Meta-Analysis. Semin Arthritis Rheum. 2011, 41, 1–11.
- The American College of Rheumatology Nomenclature and Case Definitions for Neuropsychiatric Lupus Syndromes. Arthritis Rheum. 1999, 42, 599–608.
- Brey, R.L.; Holliday, S.L.; Saklad, A.R.; Navarrete, M.G.; Hermosillo-Romo, D.; Stallworth, C.L.; Valdez, C.R.; Escalante, A.; del Rincón, I.; Gronseth, G.; et al. Neuropsychiatric Syndromes in Lupus: Prevalence Using Standardized Definitions. Neurology 2002, 58, 1214–1220.
- Zardi, E.M.; Giorgi, C.; Zardi, D.M. Diagnostic Approach to Neuropsychiatric Lupus Erythematosus: What Should We Do? Postgrad. Med. 2018, 130, 536–547.
- Meier, A.L.; Bodmer, N.S.; Wirth, C.; Bachmann, L.M.; Ribi, C.; Pröbstel, A.-K.; Waeber, D.; Jelcic, I.; Steiner, U.C. Neuro-Psychiatric Manifestations in Patients with Systemic Lupus Erythematosus: A Systematic Review and Results from the Swiss Lupus Cohort Study. Lupus 2021, 30, 1565–1576.
- Magro-Checa, C.; Zirkzee, E.J.; Huizinga, T.W.; Steup-Beekman, G.M. Management of Neuropsychiatric Systemic Lupus Erythematosus: Current Approaches and Future Perspectives. Drugs 2016, 76, 459–483.
- Monahan, R.C.; Fronczek, R.; Eikenboom, J.; Middelkoop, H.A.M.; Beaart-van de Voorde, L.J.J.; Terwindt, G.M.; van der Wee, N.J.A.; Rosendaal, F.R.; Huizinga, T.W.J.; Kloppenburg, M.; et al. Mortality in Patients with Systemic Lupus Erythematosus and Neuropsychiatric Involvement: A Retrospective Analysis from a Tertiary Referral Center in the Netherlands. Lupus 2020, 29, 1892–1901.
- Hanly, J.G.; Li, Q.; Su, L.; Urowitz, M.B.; Gordon, C.; Bae, S.-C.; Romero-Diaz, J.; Sanchez-Guerrero, J.; Bernatsky, S.; Clarke, A.E.; et al. Cerebrovascular Events in Systemic Lupus Erythematosus: Results From an International Inception Cohort Study. Arthritis Care Res. (Hoboken) 2018, 70, 1478–1487.
- Abdel-Nasser, A.M.; Ghaleb, R.M.; Mahmoud, J.A.; Khairy, W.; Mahmoud, R.M. Association of Anti-Ribosomal P Protein Antibodies with Neuropsychiatric and Other Manifestations of Systemic Lupus Erythematosus. Clin. Rheumatol. 2008, 27, 1377–1385.
- Florica, B.; Aghdassi, E.; Su, J.; Gladman, D.D.; Urowitz, M.B.; Fortin, P.R. Peripheral Neuropathy in Patients with Systemic Lupus Erythematosus. Semin. Arthritis Rheum. 2011, 41, 203–211.
- Bonfa, E.; Golombek, S.J.; Kaufman, L.D.; Skelly, S.; Weissbach, H.; Brot, N.; Elkon, K.B. Association between Lupus Psychosis and Anti-Ribosomal P Protein Antibodies. N. Engl. J. Med. 1987, 317, 265–271.
- Sanna, G.; Bertolaccini, M.L.; Cuadrado, M.J.; Laing, H.; Khamashta, M.A.; Mathieu, A.; Hughes, G.R.V. Neuropsychiatric Manifestations in Systemic Lupus Erythematosus: Prevalence and Association with Antiphospholipid Antibodies. J. Rheumatol. 2003, 30, 985–992.
- Williams, R.C.J.; Sugiura, K.; Tan, E.M. Antibodies to Microtubule-Associated Protein 2 in Patients with Neuropsychiatric Systemic Lupus Erythematosus. Arthritis Rheum. 2004, 50, 1239–1247.
- Conti, F.; Alessandri, C.; Bompane, D.; Bombardieri, M.; Spinelli, F.R.; Rusconi, A.C.; Valesini, G. Autoantibody Profile in Systemic Lupus Erythematosus with Psychiatric Manifestations: A Role for Anti-Endothelial-Cell Antibodies. Arthritis Res. Ther. 2004, 6, R366-372.
- Delunardo, F.; Soldati, D.; Bellisario, V.; Berry, A.; Camerini, S.; Crescenzi, M.; Alessandri, C.; Conti, F.; Ceccarelli, F.; Francia, A.; et al. Anti-GAPDH Autoantibodies as a Pathogenic Determinant and Potential Biomarker of Neuropsychiatric Diseases. Arthritis Rheumatol. 2016, 68, 2708–2716.
- Stock, A.D.; Gelb, S.; Pasternak, O.; Ben-Zvi, A.; Putterman, C. The Blood Brain Barrier and Neuropsychiatric Lupus: New Perspectives in Light of Advances in Understanding the Neuroimmune Interface. Autoimmun. Rev. 2017, 16, 612–619.
- Jacob, A.; Hack, B.; Chiang, E.; Garcia, J.G.N.; Quigg, R.J.; Alexander, J.J. C5a Alters Blood-Brain Barrier Integrity in Experimental Lupus. FASEB J. 2010, 24, 1682–1688.
- Jacob, A.; Hack, B.; Chen, P.; Quigg, R.J.; Alexander, J.J. C5a/CD88 Signaling Alters Blood-Brain Barrier Integrity in Lupus through Nuclear Factor-κB. J. Neurochem. 2011, 119, 1041–1051.
- Kivity, S.; Agmon-Levin, N.; Zandman-Goddard, G.; Chapman, J.; Shoenfeld, Y. Neuropsychiatric Lupus: A Mosaic of Clinical Presentations. BMC Med. 2015, 13, 43.
- Yoshio, T.; Okamoto, H.; Kurasawa, K.; Dei, Y.; Hirohata, S.; Minota, S. IL-6, IL-8, IP-10, MCP-1 and G-CSF Are Significantly Increased in Cerebrospinal Fluid but Not in Sera of Patients with Central Neuropsychiatric Lupus Erythematosus. Lupus 2016, 25, 997–1003.
- Duan, L.; Yao, Y.; Kong, H.; Zhou, Y.; Cui, D. Chemokines and Chemokine Receptors: Potential Therapeutic Targets in Systemic Lupus Erythematosus. Cytokine 2024, 184, 156770.
- Santer, D.M.; Yoshio, T.; Minota, S.; Möller, T.; Elkon, K.B. Potent Induction of IFN-Alpha and Chemokines by Autoantibodies in the Cerebrospinal Fluid of Patients with Neuropsychiatric Lupus. J. Immunol. 2009, 182, 1192–1201.
- Bialas, A.R.; Presumey, J.; Das, A.; van der Poel, C.E.; Lapchak, P.H.; Mesin, L.; Victora, G.; Tsokos, G.C.; Mawrin, C.; Herbst, R.; et al. Retraction Note: Microglia-Dependent Synapse Loss in Type I Interferon-Mediated Lupus. Nature 2020, 578, 177.
- Hirohata, S.; Kikuchi, H. Role of Serum IL-6 in Neuropsychiatric Systemic Lupus Erythematosus. ACR Open Rheumatol. 2021, 3, 42–49.
- Qiao, X.; Wang, H.; Lu, L.; Chen, J.; Cheng, Q.; Guo, M.; Hou, Y.; Dou, H. Hippocampal Microglia CD40 Mediates NPSLE Cognitive Dysfunction in Mice. J. Neuroimmunol. 2021, 357, 577620.
- Sciascia, S.; Bertolaccini, M.L.; Roccatello, D.; Khamashta, M.A.; Sanna, G. Autoantibodies Involved in Neuropsychiatric Manifestations Associated with Systemic Lupus Erythematosus: A Systematic Review. J. Neurol. 2014, 261, 1706–1714.
- Salmon, J.E.; de Groot, P.G. Pathogenic Role of Antiphospholipid Antibodies. Lupus 2008, 17, 405–411.
- Harper, B.E.; Wills, R.; Pierangeli, S.S. Pathophysiological Mechanisms in Antiphospholipid Syndrome. Int. J. Clin. Rheumtol. 2011, 6, 157–171.
- Katzav, A.; Ben-Ziv, T.; Blank, M.; Pick, C.G.; Shoenfeld, Y.; Chapman, J. Antibody-Specific Behavioral Effects: Intracerebroventricular Injection of Antiphospholipid Antibodies Induces Hyperactive Behavior While Anti-Ribosomal-P Antibodies Induces Depression and Smell Deficits in Mice. J. Neuroimmunol. 2014, 272, 10–15.
- de Carvalho, J.F.; Pasoto, S.G.; Appenzeller, S. Seizures in Primary Antiphospholipid Syndrome: The Relevance of Smoking to Stroke. Clin. Dev. Immunol. 2012, 2012, 981519.
- Shi, Z.-R.; Han, Y.-F.; Yin, J.; Zhang, Y.-P.; Jiang, Z.-X.; Zheng, L.; Tan, G.-Z.; Wang, L. The Diagnostic Benefit of Antibodies against Ribosomal Proteins in Systemic Lupus Erythematosus. Adv. Rheumatol. 2020, 60, 45.
- Caponi, L.; Bombardieri, S.; Migliorini, P. Anti-Ribosomal Antibodies Bind the Sm Proteins D and B/B’. Clin. Exp. Immunol. 1998, 112, 139–143.
- Alajangi, H.K.; Kaur, M.; Sharma, A.; Rana, S.; Thakur, S.; Chatterjee, M.; Singla, N.; Jaiswal, P.K.; Singh, G.; Barnwal, R.P. Blood-Brain Barrier: Emerging Trends on Transport Models and New-Age Strategies for Therapeutics Intervention against Neurological Disorders. Mol. Brain. 2022, 15, 49.
- Li, F.; Tsien, J.Z. Memory and the NMDA Receptors. N. Engl. J. Med. 2009, 361, 302–303.
- Levite, M. Glutamate Receptor Antibodies in Neurological Diseases: Anti-AMPA-GluR3 Antibodies, Anti-NMDA-NR1 Antibodies, Anti-NMDA-NR2A/B Antibodies, Anti-mGluR1 Antibodies or Anti-mGluR5 Antibodies Are Present in Subpopulations of Patients with Either: Epilepsy, Encephalitis, Cerebellar Ataxia, Systemic Lupus Erythematosus (SLE) and Neuropsychiatric SLE, Sjogren’s Syndrome, Schizophrenia, Mania or Stroke. These Autoimmune Anti-Glutamate Receptor Antibodies Can Bind Neurons in Few Brain Regions, Activate Glutamate Receptors, Decrease Glutamate Receptor’s Expression, Impair Glutamate-Induced Signaling and Function, Activate Blood Brain Barrier Endothelial Cells, Kill Neurons, Damage the Brain, Induce Behavioral/Psychiatric/Cognitive Abnormalities and Ataxia in Animal Models, and Can Be Removed or Silenced in Some Patients by Immunotherapy. J. Neural Transm. (Vienna) 2014, 121, 1029–1075.
- Schüler, T.; Mesic, I.; Madry, C.; Bartholomäus, I.; Laube, B. Formation of NR1/NR2 and NR1/NR3 Heterodimers Constitutes the Initial Step in N-Methyl-D-Aspartate Receptor Assembly. J. Biol. Chem. 2008, 283, 37–46.
- Zhang, S.; Yang, Y.; Long, T.; Li, Z. Systemic Lupus Erythematosus Associated with Recurrent Anti-NMDA Receptor Encephalitis during Pregnancy. Arch. Womens Ment. Health 2021, 24, 525–528.
- Saikali, P.; Cayrol, R.; Vincent, T. Anti-Aquaporin-4 Auto-Antibodies Orchestrate the Pathogenesis in Neuromyelitis Optica. Autoimmun. Rev. 2009, 9, 132–135.
- Rocca, M.A.; Cacciaguerra, L.; Filippi, M. Moving beyond Anti-Aquaporin-4 Antibodies: Emerging Biomarkers in the Spectrum of Neuromyelitis Optica. Expert Rev. Neurother. 2020, 20, 601–618.
- Asgari, N.; Jarius, S.; Laustrup, H.; Skejoe, H.P.; Lillevang, S.T.; Weinshenker, B.G.; Voss, A. Aquaporin-4-Autoimmunity in Patients with Systemic Lupus Erythematosus: A Predominantly Population-Based Study. Mult. Scler. 2018, 24, 331–339.
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91.
- Bergkamp, S.C.; Wahadat, M.J.; Salah, A.; Kuijpers, T.W.; Smith, V.; Tas, S.W.; van den Berg, J.M.; Kamphuis, S.; Schonenberg-Meinema, D. Dysregulated Endothelial Cell Markers in Systemic Lupus Erythematosus: A Systematic Review and Meta-Analysis. J. Inflamm. (Lond) 2023, 20, 18.
- Moschetti, L.; Piantoni, S.; Vizzardi, E.; Sciatti, E.; Riccardi, M.; Franceschini, F.; Cavazzana, I. Endothelial Dysfunction in Systemic Lupus Erythematosus and Systemic Sclerosis: A Common Trigger for Different Microvascular Diseases. Front. Med. (Lausanne) 2022, 9, 849086.
- Ichinose, K.; Ohyama, K.; Furukawa, K.; Higuchi, O.; Mukaino, A.; Satoh, K.; Nakane, S.; Shimizu, T.; Umeda, M.; Fukui, S.; et al. Novel Anti-Suprabasin Antibodies May Contribute to the Pathogenesis of Neuropsychiatric Systemic Lupus Erythematosus. Clin. Immunol. 2018, 193, 123–130.
- Li, X.; Xiang, X.; Sun, J.; Liu, S.; Liu, Y.; Feng, L.; Li, C.; Li, Z. Prevalence, Outcome and Prognostic Factors of Neuropsychiatric Systemic Lupus Erythematosus: A Real World Single Center Study. Mod. Rheumatol. 2020, 30, 321–326.
- Liu, Y.; Tu, Z.; Zhang, X.; Du, K.; Xie, Z.; Lin, Z. Pathogenesis and Treatment of Neuropsychiatric Systemic Lupus Erythematosus: A Review. Front. Cell. Dev. Biol. 2022, 10, 998328.
- Stock, A.D.; Wen, J.; Putterman, C. Neuropsychiatric Lupus, the Blood Brain Barrier, and the TWEAK/Fn14 Pathway. Front. Immunol. 2013, 4, 484.
- Li, X.; Sun, J.; Mu, R.; Gan, Y.; Wang, G.; He, J.; Yi, L.; Wang, Q.; Sun, X.; Li, Z. The Clinical Significance of Ubiquitin Carboxyl Hydrolase L1 and Its Autoantibody in Neuropsychiatric Systemic Lupus Erythematosus. Clin. Exp. Rheumatol. 2019, 37, 474–480.
- Muslimov, I.A.; Iacoangeli, A.; Eom, T.; Ruiz, A.; Lee, M.; Stephenson, S.; Ginzler, E.M.; Tiedge, H. Neuronal BC RNA Transport Impairments Caused by Systemic Lupus Erythematosus Autoantibodies. J. Neurosci. 2019, 39, 7759–7777.
- Lindblom, J.; Mohan, C.; Parodis, I. Biomarkers in Neuropsychiatric Systemic Lupus Erythematosus: A Systematic Literature Review of the Last Decade. Brain Sci. 2022, 12.
- Myers, T.D.; Palladino, M.J. Newly Discovered Roles of Triosephosphate Isomerase Including Functions within the Nucleus. Mol. Med. 2023, 29, 18.
- Sato, S.; Yashiro, M.; Asano, T.; Kobayashi, H.; Watanabe, H.; Migita, K. Association of Anti-Triosephosphate Isomerase Antibodies with Aseptic Meningitis in Patients with Neuropsychiatric Systemic Lupus Erythematosus. Clin. Rheumatol. 2017, 36, 1655–1659.
- Sánchez, C.; Díaz-Nido, J.; Avila, J. Phosphorylation of Microtubule-Associated Protein 2 (MAP2) and Its Relevance for the Regulation of the Neuronal Cytoskeleton Function. Prog. Neurobiol. 2000, 61, 133–168.
- Kang, H.J.; Voleti, B.; Hajszan, T.; Rajkowska, G.; Stockmeier, C.A.; Licznerski, P.; Lepack, A.; Majik, M.S.; Jeong, L.S.; Banasr, M.; et al. Decreased Expression of Synapse-Related Genes and Loss of Synapses in Major Depressive Disorder. Nat. Med. 2012, 18, 1413–1417.
- Vlachoyiannopoulos, P.G.; Guialis, A.; Tzioufas, G.; Moutsopoulos, H.M. Predominance of IgM Anti-U1RNP Antibodies in Patients with Systemic Lupus Erythematosus. Br. J. Rheumatol. 1996, 35, 534–541.
- Dema, B.; Charles, N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies (Basel) 2016, 5, 2.
- Ota, Y.; Srinivasan, A.; Capizzano, A.A.; Bapuraj, J.R.; Kim, J.; Kurokawa, R.; Baba, A.; Moritani, T. Central Nervous System Systemic Lupus Erythematosus: Pathophysiologic, Clinical, and Imaging Features. Radiographics 2022, 42, 212–232.
- Zhou, Z.; Sun, B.; Huang, S.; Zhao, L. Roles of Circular RNAs in Immune Regulation and Autoimmune Diseases. Cell Death Dis. 2019, 10, 503.
- Bertsias, G.K.; Ioannidis, J.P.A.; Aringer, M.; Bollen, E.; Bombardieri, S.; Bruce, I.N.; Cervera, R.; Dalakas, M.; Doria, A.; Hanly, J.G.; et al. EULAR Recommendations for the Management of Systemic Lupus Erythematosus with Neuropsychiatric Manifestations: Report of a Task Force of the EULAR Standing Committee for Clinical Affairs. Ann. Rheum. Dis. 2010, 69, 2074–2082.
- Murray, S.G.; Yazdany, J.; Kaiser, R.; Criswell, L.A.; Trupin, L.; Yelin, E.H.; Katz, P.P.; Julian, L.J. Cardiovascular Disease and Cognitive Dysfunction in Systemic Lupus Erythematosus. Arthritis Care Res. (Hoboken) 2012, 64, 1328–1333.
- Cervera, R.; Khamashta, M.A.; Font, J.; Sebastiani, G.D.; Gil, A.; Lavilla, P.; Mejía, J.C.; Aydintug, A.O.; Chwalinska-Sadowska, H.; de Ramón, E.; et al. Morbidity and Mortality in Systemic Lupus Erythematosus during a 10-Year Period: A Comparison of Early and Late Manifestations in a Cohort of 1,000 Patients. Medicine (Baltimore) 2003, 82, 299–308.
- Saadatnia, M.; Sayed-Bonakdar, Z.; Mohammad-Sharifi, G.; Sarrami, A.H. The Necessity of Stroke Prevention in Patients with Systemic Lupus Erythematosus. J. Res. Med. Sci. 2012, 17, 894–895.
- Zandman-Goddard, G.; Chapman, J.; Shoenfeld, Y. Autoantibodies Involved in Neuropsychiatric SLE and Antiphospholipid Syndrome. Semin. Arthritis Rheum. 2007, 36, 297–315.
- Barbhaiya, M.; Zuily, S.; Naden, R.; Hendry, A.; Manneville, F.; Amigo, M.-C.; Amoura, Z.; Andrade, D.; Andreoli, L.; Artim-Esen, B.; et al. The 2023 ACR/EULAR Antiphospholipid Syndrome Classification Criteria. Arthritis Rheumatol. 2023, 75, 1687–1702.
- Giannakopoulos, B.; Krilis, S.A. The Pathogenesis of the Antiphospholipid Syndrome. N. Engl. J. Med. 2013, 368, 1033–1044.
- Hanly, J.G.; Walsh, N.M.; Sangalang, V. Brain Pathology in Systemic Lupus Erythematosus. J. Rheumatol. 1992, 19, 732–741.
- Ellis, S.G.; Verity, M.A. Central Nervous System Involvement in Systemic Lupus Erythematosus: A Review of Neuropathologic Findings in 57 Cases, 1955--1977. Semin. Arthritis Rheum. 1979, 8, 212–221.
- Cohen, D.; Rijnink, E.C.; Nabuurs, R.J.A.; Steup-Beekman, G.M.; Versluis, M.J.; Emmer, B.J.; Zandbergen, M.; van Buchem, M.A.; Allaart, C.F.; Wolterbeek, R.; et al. Brain Histopathology in Patients with Systemic Lupus Erythematosus: Identification of Lesions Associated with Clinical Neuropsychiatric Lupus Syndromes and the Role of Complement. Rheumatology (Oxford) 2017, 56, 77–86.
- Bruce, I.N.; Urowitz, M.B.; Gladman, D.D.; Ibañez, D.; Steiner, G. Risk Factors for Coronary Heart Disease in Women with Systemic Lupus Erythematosus: The Toronto Risk Factor Study. Arthritis Rheum. 2003, 48, 3159–3167.
- Holmqvist, M.; Simard, J.F.; Asplund, K.; Arkema, E.V. Stroke in Systemic Lupus Erythematosus: A Meta-Analysis of Population-Based Cohort Studies. RMD Open 2015, 1, e000168.
- Ward, M.M. Premature Morbidity from Cardiovascular and Cerebrovascular Diseases in Women with Systemic Lupus Erythematosus. Arthritis Rheum. 1999, 42, 338–346.
- Mikdashi, J.; Handwerger, B.; Langenberg, P.; Miller, M.; Kittner, S. Baseline Disease Activity, Hyperlipidemia, and Hypertension Are Predictive Factors for Ischemic Stroke and Stroke Severity in Systemic Lupus Erythematosus. Stroke 2007, 38, 281–285.
- Urowitz, M.B.; Gladman, D.; Ibañez, D.; Bae, S.C.; Sanchez-Guerrero, J.; Gordon, C.; Clarke, A.; Bernatsky, S.; Fortin, P.R.; Hanly, J.G.; et al. Atherosclerotic Vascular Events in a Multinational Inception Cohort of Systemic Lupus Erythematosus. Arthritis Care Res. (Hoboken) 2010, 62, 881–887.
- Levine, S.R.; Brey, R.L.; Tilley, B.C.; Thompson, J.L.P.; Sacco, R.L.; Sciacca, R.R.; Murphy, A.; Lu, Y.; Costigan, T.M.; Rhine, C.; et al. Antiphospholipid Antibodies and Subsequent Thrombo-Occlusive Events in Patients with Ischemic Stroke. JAMA 2004, 291, 576–584.
- Fernández-Nebro, A.; Rúa-Figueroa, Í.; López-Longo, F.J.; Galindo-Izquierdo, M.; Calvo-Alén, J.; Olivé-Marqués, A.; Ordóñez-Cañizares, C.; Martín-Martínez, M.A.; Blanco, R.; Melero-González, R.; et al. Cardiovascular Events in Systemic Lupus Erythematosus: A Nationwide Study in Spain From the RELESSER Registry. Medicine (Baltimore) 2015, 94, e1183.
- Saadatnia, M.; Sayed-Bonakdar, Z.; Mohammad-Sharifi, G.; Sarrami, A.H. Prevalence and Prognosis of Cerebrovascular Accidents and Its Subtypes Among Patients with Systemic Lupus Erythematosus in Isfahan, Iran: A Hospital Clinic-Based Study. Int. J. Prev. Med. 2014, 5, 123–126.
- Ho, R.C.; Ong, H.; Thiaghu, C.; Lu, Y.; Ho, C.S.; Zhang, M.W. Genetic Variants That Are Associated with Neuropsychiatric Systemic Lupus Erythematosus. J. Rheumatol. 2016, 43, 541–551.
- de Vries, B.; Steup-Beekman, G.M.; Haan, J.; Bollen, E.L.; Luyendijk, J.; Frants, R.R.; Terwindt, G.M.; van Buchem, M.A.; Huizinga, T.W.J.; van den Maagdenberg, A.M.J.M.; et al. TREX1 Gene Variant in Neuropsychiatric Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2010, 69, 1886–1887.
- Namjou, B.; Kothari, P.H.; Kelly, J.A.; Glenn, S.B.; Ojwang, J.O.; Adler, A.; Alarcón-Riquelme, M.E.; Gallant, C.J.; Boackle, S.A.; Criswell, L.A.; et al. Evaluation of the TREX1 Gene in a Large Multi-Ancestral Lupus Cohort. Genes Immun. 2011, 12, 270–279.
- Karageorgas, T.P.; Tseronis, D.D.; Mavragani, C.P. Activation of Type I Interferon Pathway in Systemic Lupus Erythematosus: Association with Distinct Clinical Phenotypes. J. Biomed. Biotechnol. 2011, 2011, 273907.
- Lundström, E.; Gustafsson, J.T.; Jönsen, A.; Leonard, D.; Zickert, A.; Elvin, K.; Sturfelt, G.; Nordmark, G.; Bengtsson, A.A.; Sundin, U.; et al. HLA-DRB1*04/*13 Alleles Are Associated with Vascular Disease and Antiphospholipid Antibodies in Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2013, 72, 1018–1025.
- Rullo, O.J.; Tsao, B.P. Recent Insights into the Genetic Basis of Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2013, 72 Suppl 2, ii56-61.
- Koga, M.; Kawasaki, A.; Ito, I.; Furuya, T.; Ohashi, J.; Kyogoku, C.; Ito, S.; Hayashi, T.; Matsumoto, I.; Kusaoi, M.; et al. Cumulative Association of Eight Susceptibility Genes with Systemic Lupus Erythematosus in a Japanese Female Population. J. Hum. Genet. 2011, 56, 503–507.
- Vista, E.S.; Farris, A.D.; James, J.A. Chapter 24 - Roles for Infections in Systemic Lupus Erythematosus Pathogenesis. In Systemic Lupus Erythematosus (Fifth Edition); Lahita, R.G., Ed.; Academic Press: San Diego, 2011; pp. 425–435 ISBN 978-0-12-374994-9.
- Iliopoulos, A.G.; Tsokos, G.C. Immunopathogenesis and Spectrum of Infections in Systemic Lupus Erythematosus. Semin Arthritis Rheum. 1996, 25, 318–336.
- Telles, R.W.; Lanna, C.C.D.; Souza, F.L.; Rodrigues, L.A.; Reis, R.C.P.; Ribeiro, A.L. Causes and Predictors of Death in Brazilian Lupus Patients. Rheumatol. Int. 2013, 33, 467–473.
- Warnatz, K.; Peter, H.H.; Schumacher, M.; Wiese, L.; Prasse, A.; Petschner, F.; Vaith, P.; Volk, B.; Weiner, S.M. Infectious CNS Disease as a Differential Diagnosis in Systemic Rheumatic Diseases: Three Case Reports and a Review of the Literature. Ann. Rheum. Dis. 2003, 62, 50–57.
- Zhong, Y.; Li, M.; Liu, J.; Zhang, W.; Peng, F. Cryptococcal Meningitis in Chinese Patients with Systemic Lupus Erythematosus. Clin. Neurol. Neurosurg. 2015, 131, 59–63.
- Yang, C.-D.; Wang, X.-D.; Ye, S.; Gu, Y.-Y.; Bao, C.-D.; Wang, Y.; Chen, S.-L. Clinical Features, Prognostic and Risk Factors of Central Nervous System Infections in Patients with Systemic Lupus Erythematosus. Clin. Rheumatol. 2007, 26, 895–901.
- Lu, X.Y.; Zhu, C.Q.; Qian, J.; Chen, X.X.; Ye, S.; Gu, Y.Y. Intrathecal Cytokine and Chemokine Profiling in Neuropsychiatric Lupus or Lupus Complicated with Central Nervous System Infection. Lupus 2010, 19, 689–695.
- Zandman-Goddard, G.; Berkun, Y.; Barzilai, O.; Boaz, M.; Ram, M.; Anaya, J.M.; Shoenfeld, Y. Neuropsychiatric Lupus and Infectious Triggers. Lupus 2008, 17, 380–384.
- Nived, O.; Johansson, I.; Sturfelt, G. Effects of Ultraviolet Irradiation on Natural Killer Cell Function in Systemic Lupus Erythematosus. Ann. Rheum. Dis. 1992, 51, 726–730.
- Abe, M.; Ishikawa, O.; Miyachi, Y.; Kanai, Y. In Vitro Spontaneous and UVB-Induced Lymphocyte Apoptosis Are Not Specific to SLE. Photodermatol. Photoimmunol. Photomed. 1997, 13, 204–207.
- Cohen, M.R.; Isenberg, D.A. Ultraviolet Irradiation in Systemic Lupus Erythematosus: Friend or Foe? Br. J. Rheumatol. 1996, 35, 1002–1007.
- Kuhn, A.; Fehsel, K.; Lehmann, P.; Krutmann, J.; Ruzicka, T.; Kolb-Bachofen, V. Aberrant Timing in Epidermal Expression of Inducible Nitric Oxide Synthase after UV Irradiation in Cutaneous Lupus Erythematosus. J. Invest. Dermatol. 1998, 111, 149–153.
- Norris, D.A. Pathomechanisms of Photosensitive Lupus Erythematosus. J. Invest. Dermatol. 1993, 100, 58S-68S.
- Molina, J.F.; McGrath, H.J. Longterm Ultraviolet-A1 Irradiation Therapy in Systemic Lupus Erythematosus. J. Rheumatol. 1997, 24, 1072–1074.
- Schmidt, E.; Tony, H.-P.; Bröcker, E.-B.; Kneitz, C. Sun-Induced Life-Threatening Lupus Nephritis. Ann. N. Y. Acad. Sci. 2007, 1108, 35–40.
- Geara, A.S.; Torbey, E.; El-imad, B. Lupus Cerebritis after Visiting a Tanning Salon. Bull. NYU Hosp. Jt. Dis. 2009, 67, 391–393.
- Yung, R.L.; Richardson, B.C. Chapter 22 - Drug-Induced Lupus Mechanisms. In Systemic Lupus Erythematosus (Fifth Edition); Lahita, R.G., Ed.; Academic Press: San Diego, 2011; pp. 385–403 ISBN 978-0-12-374994-9.
- Sarzi-Puttini, P.; Atzeni, F.; Capsoni, F.; Lubrano, E.; Doria, A. Drug-Induced Lupus Erythematosus. Autoimmunity 2005, 38, 507–518.
- Aguirre Zamorano, M.A.; Lopez Pedrera, R.; Cuadrado Lozano, M.J. Drug-Induced Lupus. Medicina Clinica 2010, 135, 124–129.
- Pretel, M.; Marquès, L.; España, A. Lupus Eritematoso Inducido Por Fármacos. Actas Dermo-Sifiliográficas 2014, 105, 18–30.
- Bracke, A.; Nijsten, T.; Vandermaesen, J.; Meuleman, L.; Lambert, J. Lansoprazole-Induced Subacute Cutaneous Lupus Erythematosus: Two Cases. Acta Derm. Venereol. 2005, 85, 353–354.
- McKay, D.A.; Schofield, O.M.V.; Benton, E.C. Terbinafine-Induced Subacute Cutaneous Lupus Erythematosus. Acta Derm. Venereol. 2004, 84, 472–474.
- Suess, A.; Sticherling, M. Leflunomide in Subacute Cutaneous Lupus Erythematosus - Two Sides of a Coin. Int. J. Dermatol. 2008, 47, 83–86.
- Costa, M.F.; Said, N.R.; Zimmermann, B. Drug-Induced Lupus Due to Anti-Tumor Necrosis Factor Alpha Agents. Semin. Arthritis Rheum. 2008, 37, 381–387.
- Guhl, G.; Diaz-Ley, B.; García-García, C.; Fraga, J.; Garcia-Diez, A. Chemotherapy-Induced Subacute Lupus Erythematosus. Lupus 2009, 18, 859–860.
- Grönhagen, C.M.; Fored, C.M.; Linder, M.; Granath, F.; Nyberg, F. Subacute Cutaneous Lupus Erythematosus and Its Association with Drugs: A Population-Based Matched Case-Control Study of 234 Patients in Sweden. Br. J. Dermatol. 2012, 167, 296–305.
- Merlin, F.; Prochilo, T.; Kildani, B.; Lombardi, C.; Pasolini, G.; Bonetti, F.; Beretta, G.D. Discoid Lupus Erythematosus (DLE)-like Lesions Induced by Capecitabine. Int. J. Colorectal Dis. 2008, 23, 715–716.
- Pamuk, O.N.; Raza, A.A.; Hasni, S. Neuropsychiatric Lupus in Late- and Early-Onset Systemic Lupus Erythematosus Patients: A Systematic Review and Meta-Analysis. Rheumatology (Oxford) 2024, 63, 8–15.
- Liao, J.; Kang, J.; Li, F.; Li, Q.; Wang, J.; Tang, Q.; Mao, N.; Li, S.; Xie, X. A Cross-Sectional Study on the Association of Anxiety and Depression with the Disease Activity of Systemic Lupus Erythematosus. BMC Psychiatry 2022, 22, 591.
- Mehat, P.; Atiquzzaman, M.; Esdaile, J.M.; AviÑa-Zubieta, A.; De Vera, M.A. Medication Nonadherence in Systemic Lupus Erythematosus: A Systematic Review. Arthritis Care Res. (Hoboken) 2017, 69, 1706–1713.
- Costedoat-Chalumeau, N.; Pouchot, J.; Guettrot-Imbert, G.; Le Guern, V.; Leroux, G.; Marra, D.; Morel, N.; Piette, J.-C. Adherence to Treatment in Systemic Lupus Erythematosus Patients. Best Pract. Res. Clin. Rheumatol. 2013, 27, 329–340.
- Abrol, E.; Coutinho, E.; Chou, M.; Hart, M.; Vincent, A.; Howard, R.; Zandi, M.S.; Isenberg, D. Psychosis in Systemic Lupus Erythematosus (SLE): 40-Year Experience of a Specialist Centre. Rheumatology (Oxford) 2021, 60, 5620–5629.
- Chau, S.Y.; Mok, C.C. Factors Predictive of Corticosteroid Psychosis in Patients with Systemic Lupus Erythematosus. Neurology 2003, 61, 104–107.
- Tokunaga, M.; Saito, K.; Kawabata, D.; Imura, Y.; Fujii, T.; Nakayamada, S.; Tsujimura, S.; Nawata, M.; Iwata, S.; Azuma, T.; et al. Efficacy of Rituximab (Anti-CD20) for Refractory Systemic Lupus Erythematosus Involving the Central Nervous System. Ann. Rheum. Dis. 2007, 66, 470–475.
- Raghunath, S.; Glikmann-Johnston, Y.; Golder, V.; Kandane-Rathnayake, R.; Morand, E.F.; Stout, J.C.; Hoi, A. Clinical Associations of Cognitive Dysfunction in Systemic Lupus Erythematosus. Lupus Sci. Med. 2023, 10, e000835.
- Harrison, M.J.; Morris, K.A.; Horton, R.; Toglia, J.; Barsky, J.; Chait, S.; Ravdin, L.; Robbins, L. Results of Intervention for Lupus Patients with Self-Perceived Cognitive Difficulties. Neurology 2005, 65, 1325–1327.
- Petri, M.; Naqibuddin, M.; Sampedro, M.; Omdal, R.; Carson, K.A. Memantine in Systemic Lupus Erythematosus: A Randomized, Double-Blind Placebo-Controlled Trial. Semin. Arthritis Rheum. 2011, 41, 194–202.
- Hanly, J.G.; Urowitz, M.B.; Su, L.; Gordon, C.; Bae, S.-C.; Sanchez-Guerrero, J.; Romero-Diaz, J.; Wallace, D.J.; Clarke, A.E.; Ginzler, E.; et al. Seizure Disorders in Systemic Lupus Erythematosus Results from an International, Prospective, Inception Cohort Study. Ann Rheum Dis 2012, 71, 1502–1509.
- de Amorim, L.C.D.; Maia, F.M.; Rodrigues, C.E.M. Stroke in Systemic Lupus Erythematosus and Antiphospholipid Syndrome: Risk Factors, Clinical Manifestations, Neuroimaging, and Treatment. Lupus 2017, 26, 529–536.
- Su, L.; Qi, Z.; Guan, S.; Wei, L.; Zhao, Y. Exploring the Risk Factors for Ischemic Cerebrovascular Disease in Systemic Lupus Erythematosus: A Single-Center Case-Control Study. Front. Immunol. 2022, 13, 978910.
- Harris, E.N.; Pierangeli, S. Antiphospholipid Antibodies and Cerebral Lupus. Ann. N. Y. Acad. Sci. 1997, 823, 270–278.
- Jasmin, R.; Sockalingam, S.; Ramanaidu, L.P.; Goh, K.J. Clinical and Electrophysiological Characteristics of Symmetric Polyneuropathy in a Cohort of Systemic Lupus Erythematosus Patients. Lupus 2015, 24, 248–255.
- Mahran, S.A.; Galluccio, F.; Khedr, T.M.; Elsonbaty, A.; Allam, A.E.-S.; Garcia Martos, A.; Osman, D.M.M.; Matucci-Cerinic, M.; Guiducci, S.; Galal, M.A.A. Peripheral Neuropathy in Systemic Lupus Erythematosus: What Can Neuromuscular Ultrasonography (NMUS) Tell Us? A Cross-Sectional Study. Lupus Sci. Med. 2021, 8, e000521.
- Xianbin, W.; Mingyu, W.; Dong, X.; Huiying, L.; Yan, X.; Fengchun, Z.; Xiaofeng, Z. Peripheral Neuropathies Due to Systemic Lupus Erythematosus in China. Medicine (Baltimore) 2015, 94, e625, doi:10.1097/MD.0000000000000625.
- Bhoi, S.K.; Jha, M.; Jaiswal, B. Guillain-Barre Syndrome as the Initial Presentation of Systemic Lupus Erythematosus: Case Report with a Systematic and Literature Review. Med. J. Armed Forces India 2023, 79, S360–S364.
- Xiong, H.; Tang, F.; Guo, Y.; Xu, R.; Lei, P. Neural Circuit Changes in Neurological Disorders: Evidence from in Vivo Two-Photon Imaging. Ageing Res. Rev. 2023, 87, 101933.
- Chessa, E.; Piga, M.; Floris, A.; Mathieu, A.; Cauli, A. Demyelinating Syndrome in SLE: Review of Different Disease Subtypes and Report of a Case Series. Reumatismo 2017, 69, 175–183.
- Miller, D.H.; Weinshenker, B.G.; Filippi, M.; Banwell, B.L.; Cohen, J.A.; Freedman, M.S.; Galetta, S.L.; Hutchinson, M.; Johnson, R.T.; Kappos, L.; et al. Differential Diagnosis of Suspected Multiple Sclerosis: A Consensus Approach. Mult. Scler. 2008, 14, 1157–1174.
- Magro Checa, C.; Cohen, D.; Bollen, E.L.E.M.; van Buchem, M.A.; Huizinga, T.W.J.; Steup-Beekman, G.M. Demyelinating Disease in SLE: Is It Multiple Sclerosis or Lupus? Best Pract. Res. Clin. Rheumatol. 2013, 27, 405–424.
- Nikolopoulos, D.; Kitsos, D.; Papathanasiou, M.; Kapsala, N.; Garantziotis, P.; Pieta, A.; Gioti, O.; Grivas, A.; Voumvourakis, K.; Boumpas, D.; et al. Demyelinating Syndromes in Systemic Lupus Erythematosus: Data From the “Attikon” Lupus Cohort. Front. Neurol. 2022, 13, 889613.
- Esdaile, J.M.; Abrahamowicz, M.; Grodzicky, T.; Li, Y.; Panaritis, C.; du Berger, R.; Côte, R.; Grover, S.A.; Fortin, P.R.; Clarke, A.E.; et al. Traditional Framingham Risk Factors Fail to Fully Account for Accelerated Atherosclerosis in Systemic Lupus Erythematosus. Arthritis Rheum. 2001, 44, 2331–2337.
- Kozora, E.; West, S.G.; Kotzin, B.L.; Julian, L.; Porter, S.; Bigler, E. Magnetic Resonance Imaging Abnormalities and Cognitive Deficits in Systemic Lupus Erythematosus Patients without Overt Central Nervous System Disease. Arthritis Rheum. 1998, 41, 41–47.
- Nomura, K.; Yamano, S.; Ikeda, Y.; Yamada, H.; Fujimoto, T.; Minami, S.; Fukui, R.; Takaoka, M.; Yamamoto, Y.; Dohi, K. Asymptomatic Cerebrovascular Lesions Detected by Magnetic Resonance Imaging in Patients with Systemic Lupus Erythematosus Lacking a History of Neuropsychiatric Events. Intern. Med. 1999, 38, 785–795.
- Sibbitt, W.L.J.; Sibbitt, R.R.; Brooks, W.M. Neuroimaging in Neuropsychiatric Systemic Lupus Erythematosus. Arthritis Rheum. 1999, 42, 2026–2038.
- Waterloo, K.; Omdal, R.; Sjöholm, H.; Koldingsnes, W.; Jacobsen, E.A.; Sundsfjord, J.A.; Husby, G.; Mellgren, S.I. Neuropsychological Dysfunction in Systemic Lupus Erythematosus Is Not Associated with Changes in Cerebral Blood Flow. J. Neurol. 2001, 248, 595–602.
- Zirkzee, E.J.M.; Magro Checa, C.; Sohrabian, A.; Steup-Beekman, G.M. Cluster Analysis of an Array of Autoantibodies in Neuropsychiatric Systemic Lupus Erythematosus. J. Rheumatol. 2014, 41, 1720–1721.
- Birnbaum, J.; Kerr, D. Devic’s Syndrome in a Woman with Systemic Lupus Erythematosus: Diagnostic and Therapeutic Implications of Testing for the Neuromyelitis Optica IgG Autoantibody. Arthritis Rheum. 2007, 57, 347–351.
- Birnbaum, J.; Petri, M.; Thompson, R.; Izbudak, I.; Kerr, D. Distinct Subtypes of Myelitis in Systemic Lupus Erythematosus. Arthritis Rheum. 2009, 60, 3378–3387.
- González-Duarte, A.; Cantú-Brito, C.G.; Ruano-Calderón, L.; García-Ramos, G. Clinical Description of Seizures in Patients with Systemic Lupus Erythematosus. Eur. Neurol. 2008, 59, 320–323.
- Appenzeller, S.; Cendes, F.; Costallat, L.T.L. Epileptic Seizures in Systemic Lupus Erythematosus. Neurology 2004, 63, 1808–1812.
- Hanly, J.G. Diagnosis and Management of Neuropsychiatric SLE. Nat. Rev. Rheumatol. 2014, 10, 338–347.
- Zirkzee, E.J.M.; Steup-Beekman, G.M.; van der Mast, R.C.; Bollen, E.L.E.M.; van der Wee, N.J.A.; Baptist, E.; Slee, T.M.; Huisman, M.V.; Middelkoop, H.A.M.; Luyendijk, J.; et al. Prospective Study of Clinical Phenotypes in Neuropsychiatric Systemic Lupus Erythematosus; Multidisciplinary Approach to Diagnosis and Therapy. J. Rheumatol. 2012, 39, 2118–2126.
- Stojanovich, L.; Stojanovich, R.; Kostich, V.; Dzjolich, E. Neuropsychiatric Lupus Favourable Response to Low Dose i.v. Cyclophosphamide and Prednisolone (Pilot Study). Lupus 2003, 12, 3–7.
- Neuwelt, C.M. The Role of Plasmapheresis in the Treatment of Severe Central Nervous System Neuropsychiatric Systemic Lupus Erythematosus. Ther. Apher. Dial. 2003, 7, 173–182.
- Milstone, A.M.; Meyers, K.; Elia, J. Treatment of Acute Neuropsychiatric Lupus with Intravenous Immunoglobulin (IVIG): A Case Report and Review of the Literature. Clin. Rheumatol. 2005, 24, 394–397.
- Vina, E.R.; Fang, A.J.; Wallace, D.J.; Weisman, M.H. Chronic Inflammatory Demyelinating Polyneuropathy in Patients with Systemic Lupus Erythematosus: Prognosis and Outcome. Semin. Arthritis Rheum. 2005, 35, 175–184.
- Barile-Fabris, L.; Ariza-Andraca, R.; Olguín-Ortega, L.; Jara, L.J.; Fraga-Mouret, A.; Miranda-Limón, J.M.; Fuentes de la Mata, J.; Clark, P.; Vargas, F.; Alocer-Varela, J. Controlled Clinical Trial of IV Cyclophosphamide versus IV Methylprednisolone in Severe Neurological Manifestations in Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2005, 64, 620–625.
- Muñoz-Rodriguez, F.J.; Font, J.; Cervera, R.; Reverter, J.C.; Tàssies, D.; Espinosa, G.; López-Soto, A.; Carmona, F.; Balasch, J.; Ordinas, A.; et al. Clinical Study and Follow-up of 100 Patients with the Antiphospholipid Syndrome. Semin. Arthritis Rheum. 1999, 29, 182–190.
- Jung, H.; Bobba, R.; Su, J.; Shariati-Sarabi, Z.; Gladman, D.D.; Urowitz, M.; Lou, W.; Fortin, P.R. The Protective Effect of Antimalarial Drugs on Thrombovascular Events in Systemic Lupus Erythematosus. Arthritis Rheum. 2010, 62, 863–868.
- Hermosillo-Romo, D.; Brey, R.L. Diagnosis and Management of Patients with Neuropsychiatric Systemic Lupus Erythematosus (NPSLE). Best Pract. Res. Clin. Rheumatol. 2002, 16, 229–244.
- Fessler, B.J.; Alarcón, G.S.; McGwin, G.J.; Roseman, J.; Bastian, H.M.; Friedman, A.W.; Baethge, B.A.; Vilá, L.; Reveille, J.D. Systemic Lupus Erythematosus in Three Ethnic Groups: XVI. Association of Hydroxychloroquine Use with Reduced Risk of Damage Accrual. Arthritis Rheum. 2005, 52, 1473–1480.
- Petri, M. Use of Hydroxychloroquine to Prevent Thrombosis in Systemic Lupus Erythematosus and in Antiphospholipid Antibody-Positive Patients. Curr. Rheumatol. Rep. 2011, 13, 77–80.
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms Involved in the Side Effects of Glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43.
- Buttgereit, F.; da Silva, J.A.P.; Boers, M.; Burmester, G.-R.; Cutolo, M.; Jacobs, J.; Kirwan, J.; Köhler, L.; Van Riel, P.; Vischer, T.; et al. Standardised Nomenclature for Glucocorticoid Dosages and Glucocorticoid Treatment Regimens: Current Questions and Tentative Answers in Rheumatology. Ann. Rheum. Dis. 2002, 61, 718–722.
- Hejaili, F.F.; Moist, L.M.; Clark, W.F. Treatment of Lupus Nephritis. Drugs 2003, 63, 257–274.
- Houssiau, F.A.; Vasconcelos, C.; D’Cruz, D.; Sebastiani, G.D.; de Ramon Garrido, E.; Danieli, M.G.; Abramovicz, D.; Blockmans, D.; Cauli, A.; Direskeneli, H.; et al. The 10-Year Follow-up Data of the Euro-Lupus Nephritis Trial Comparing Low-Dose and High-Dose Intravenous Cyclophosphamide. Ann. Rheum. Dis. 2010, 69, 61–64.
- Ognenovski, V.M.; Marder, W.; Somers, E.C.; Johnston, C.M.; Farrehi, J.G.; Selvaggi, S.M.; McCune, W.J. Increased Incidence of Cervical Intraepithelial Neoplasia in Women with Systemic Lupus Erythematosus Treated with Intravenous Cyclophosphamide. J. Rheumatol. 2004, 31, 1763–1767.
- DiPiero, J.; Teng, K.; Hicks, J.K. Should Thiopurine Methyltransferase (TPMT) Activity Be Determined before Prescribing Azathioprine, Mercaptopurine, or Thioguanine? Cleve. Clin. J. Med. 2015, 82, 409–413.
- Oelzner, P.; Abendroth, K.; Hein, G.; Stein, G. Predictors of Flares and Long-Term Outcome of Systemic Lupus Erythematosus during Combined Treatment with Azathioprine and Low-Dose Prednisolone. Rheumatol. Int. 1996, 16, 133–139.
- Allison, A.C. Mechanisms of Action of Mycophenolate Mofetil. Lupus 2005, 14, s2-8.
- Pisoni, C.N.; Sanchez, F.J.; Karim, Y.; Cuadrado, M.J.; D’Cruz, D.P.; Abbs, I.C.; Khamasta, M.A.; Hughes, G.R.V. Mycophenolate Mofetil in Systemic Lupus Erythematosus: Efficacy and Tolerability in 86 Patients. J. Rheumatol. 2005, 32, 1047–1052.
- Zhou, H.Q.; Zhang, F.C.; Tian, X.P.; Leng, X.M.; Lu, J.J.; Zhao, Y.; Tang, F.L.; Zhang, X.; Zeng, X.F.; Zhang, Z.L.; et al. Clinical Features and Outcome of Neuropsychiatric Lupus in Chinese: Analysis of 240 Hospitalized Patients. Lupus 2008, 17, 93–99.
- Wang, J.; Zhao, Y.; Zhang, J.; Lei, H.; Zhu, G.; Fu, B. Impact Analysis of Autoantibody Level and NR2 Antibody Level in Neuropsychiatric SLE Treated by Methylprednisolone Combined with MTX and DXM Intrathecal Injection. Cell Biochem. Biophys. 2014, 70, 1005–1009.
- Bambauer, R.; Schwarze, U.; Schiel, R. Cyclosporin A and Therapeutic Plasma Exchange in the Treatment of Severe Systemic Lupus Erythematosus. Artif. Organs. 2000, 24, 852–856.
- Yang, M.; Li, M.; He, W.; Wang, B.; Gu, Y. Calcineurin Inhibitors May Be a Reasonable Alternative to Cyclophosphamide in the Induction Treatment of Active Lupus Nephritis: A Systematic Review and Meta-Analysis. Exp. Ther. Med. 2014, 7, 1663–1670.
- Glennie, M.J.; French, R.R.; Cragg, M.S.; Taylor, R.P. Mechanisms of Killing by Anti-CD20 Monoclonal Antibodies. Mol. Immunol. 2007, 44, 3823–3837.
- Camara, I.; Sciascia, S.; Simoes, J.; Pazzola, G.; Salas, V.; Karim, Y.; Roccatello, D.; Cuadrado, M.J. Treatment with Intravenous Immunoglobulins in Systemic Lupus Erythematosus: A Series of 52 Patients from a Single Centre. Clin Exp Rheumatol 2014, 32, 41–47.
- Baigent, C.; Blackwell, L.; Collins, R.; Emberson, J.; Godwin, J.; Peto, R.; Buring, J.; Hennekens, C.; Kearney, P.; Meade, T.; et al. Aspirin in the Primary and Secondary Prevention of Vascular Disease: Collaborative Meta-Analysis of Individual Participant Data from Randomised Trials. Lancet 2009, 373, 1849–1860.
- Tektonidou, M.G.; Laskari, K.; Panagiotakos, D.B.; Moutsopoulos, H.M. Risk Factors for Thrombosis and Primary Thrombosis Prevention in Patients with Systemic Lupus Erythematosus with or without Antiphospholipid Antibodies. Arthritis Rheum. 2009, 61, 29–36.
- Adams, R.J.; Albers, G.; Alberts, M.J.; Benavente, O.; Furie, K.; Goldstein, L.B.; Gorelick, P.; Halperin, J.; Harbaugh, R.; Johnston, S.C.; et al. Update to the AHA/ASA Recommendations for the Prevention of Stroke in Patients with Stroke and Transient Ischemic Attack. Stroke 2008, 39, 1647–1652.
- Ruiz-Irastorza, G.; Cuadrado, M.J.; Ruiz-Arruza, I.; Brey, R.; Crowther, M.; Derksen, R.; Erkan, D.; Krilis, S.; Machin, S.; Pengo, V.; et al. Evidence-Based Recommendations for the Prevention and Long-Term Management of Thrombosis in Antiphospholipid Antibody-Positive Patients: Report of a Task Force at the 13th International Congress on Antiphospholipid Antibodies. Lupus 2011, 20, 206–218.
- Cuadrado, M.J.; Bertolaccini, M.L.; Seed, P.T.; Tektonidou, M.G.; Aguirre, A.; Mico, L.; Gordon, C.; Ruiz-Irastorza, G.; Egurbide, M.V.; Gil, A.; et al. Low-Dose Aspirin vs Low-Dose Aspirin plus Low-Intensity Warfarin in Thromboprophylaxis: A Prospective, Multicentre, Randomized, Open, Controlled Trial in Patients Positive for Antiphospholipid Antibodies (ALIWAPAS). Rheumatology (Oxford) 2014, 53, 275–284.
- Nikolopoulos, D.; Cetrez, N.; Lindblom, J.; Palazzo, L.; Enman, Y.; Parodis, I. Patients with NPSLE Experience Poorer HRQoL and More Fatigue than SLE Patients with No Neuropsychiatric Involvement, Irrespective of Neuropsychiatric Activity. Rheumatology (Oxford) 2024, 63, 2494–2502.
- Jönsen, A.; Bengtsson, A.A.; Nived, O.; Ryberg, B.; Sturfelt, G. Outcome of Neuropsychiatric Systemic Lupus Erythematosus within a Defined Swedish Population: Increased Morbidity but Low Mortality. Rheumatology (Oxford) 2002, 41, 1308–1312.
- Baker, K.; Pope, J. Employment and Work Disability in Systemic Lupus Erythematosus: A Systematic Review. Rheumatology (Oxford) 2009, 48, 281–284.
- Mendelsohn, S.; Khoja, L.; Alfred, S.; He, J.; Anderson, M.; DuBois, D.; Touma, Z.; Engel, L. Cognitive Impairment in Systemic Lupus Erythematosus Is Negatively Related to Social Role Participation and Quality of Life: A Systematic Review. Lupus 2021, 30, 1617–1630.
- Ahn, G.Y.; Kim, D.; Won, S.; Song, S.T.; Jeong, H.-J.; Sohn, I.-W.; Lee, S.; Joo, Y.B.; Bae, S.-C. Prevalence, Risk Factors, and Impact on Mortality of Neuropsychiatric Lupus: A Prospective, Single-Center Study. Lupus 2018, 27, 1338–1347.
- Bortoluzzi, A.; Fanouriakis, A.; Silvagni, E.; Appenzeller, S.; Carli, L.; Carrara, G.; Cauli, A.; Conti, F.; Costallat, L.T.L.; De Marchi, G.; et al. Therapeutic Strategies and Outcomes in Neuropsychiatric Systemic Lupus Erythematosus: An International Multicentre Retrospective Study. Rheumatology (Oxford) 2024, 63, 2711–2720.
- Kostopoulou, M.; Ugarte-Gil, M.F.; Pons-Estel, B.; van Vollenhoven, R.F.; Bertsias, G. The Association between Lupus Serology and Disease Outcomes: A Systematic Literature Review to Inform the Treat-to-Target Approach in Systemic Lupus Erythematosus. Lupus 2022, 31, 307–318.
- Jiang, Y.; Yuan, F.; Xu, X.; Liu, Y.; Liang, Y.; Zhang, Y.; Lin, Z.; Zhao, C. Correlation between Neuropsychiatric Systemic Lupus Erythematosus and Immunological Markers: A Real-World Retrospective Study. Clin. Rheumatol. 2024, 43, 2833–2842.
- Abdul-Sattar, A.B.; Goda, T.; Negm, M.G. Neuropsychiatric Manifestations in a Consecutive Cohort of Systemic Lupus Erythematosus; a Single Center Study. Int. J. Rheum. Dis. 2013, 16, 715–723.
- Aso, K.; Kono, M.; Kono, M.; Watanabe, T.; Shimizu, Y.; Ogata, Y.; Fujieda, Y.; Kato, M.; Oku, K.; Amengual, O.; et al. Low C4 as a Risk Factor for Severe Neuropsychiatric Flare in Patients with Systemic Lupus Erythematosus. Lupus 2020, 29, 1238–1247.
- Zheng, J.; Gu, J.; Su, Y.; Li, Y.; Li, X.; Xiong, C.; Cao, H.; Quasny, H.; Chu, M.; Curtis, P.; et al. Efficacy of Belimumab in Patients with Systemic Lupus Erythematosus from North East Asia: Results of Exploratory Subgroup Analyses. Mod. Rheumatol. 2023, 33, 751–757.
- Hsu, T.-Y.; Wang, S.-H.; Kuo, C.-F.; Chiu, T.-F.; Chang, Y.-C. Acute Inflammatory Demyelinating Polyneuropathy as the Initial Presentation of Lupus. Am J Emerg Med 2009, 27, 900.e3-5.
Figure 1.
Algorithm for the management of NPSLE (neuropsychiatric systemic lupus erythematosus).
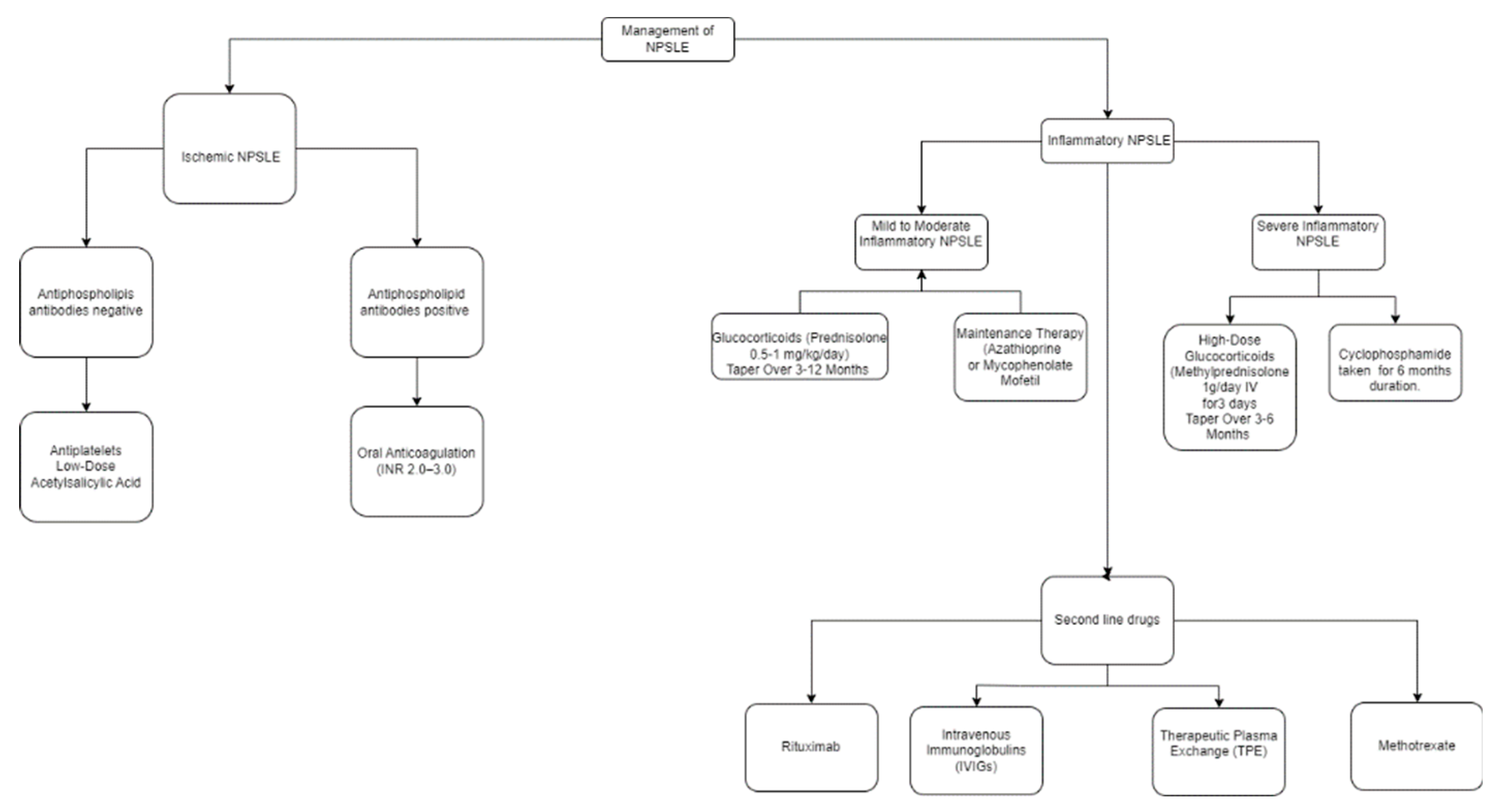
Figure 2.
Pathophysiology, clinical manifestations, and treatment of neuropsychiatric systemic lupus erythematosus.
Figure 2.
Pathophysiology, clinical manifestations, and treatment of neuropsychiatric systemic lupus erythematosus.
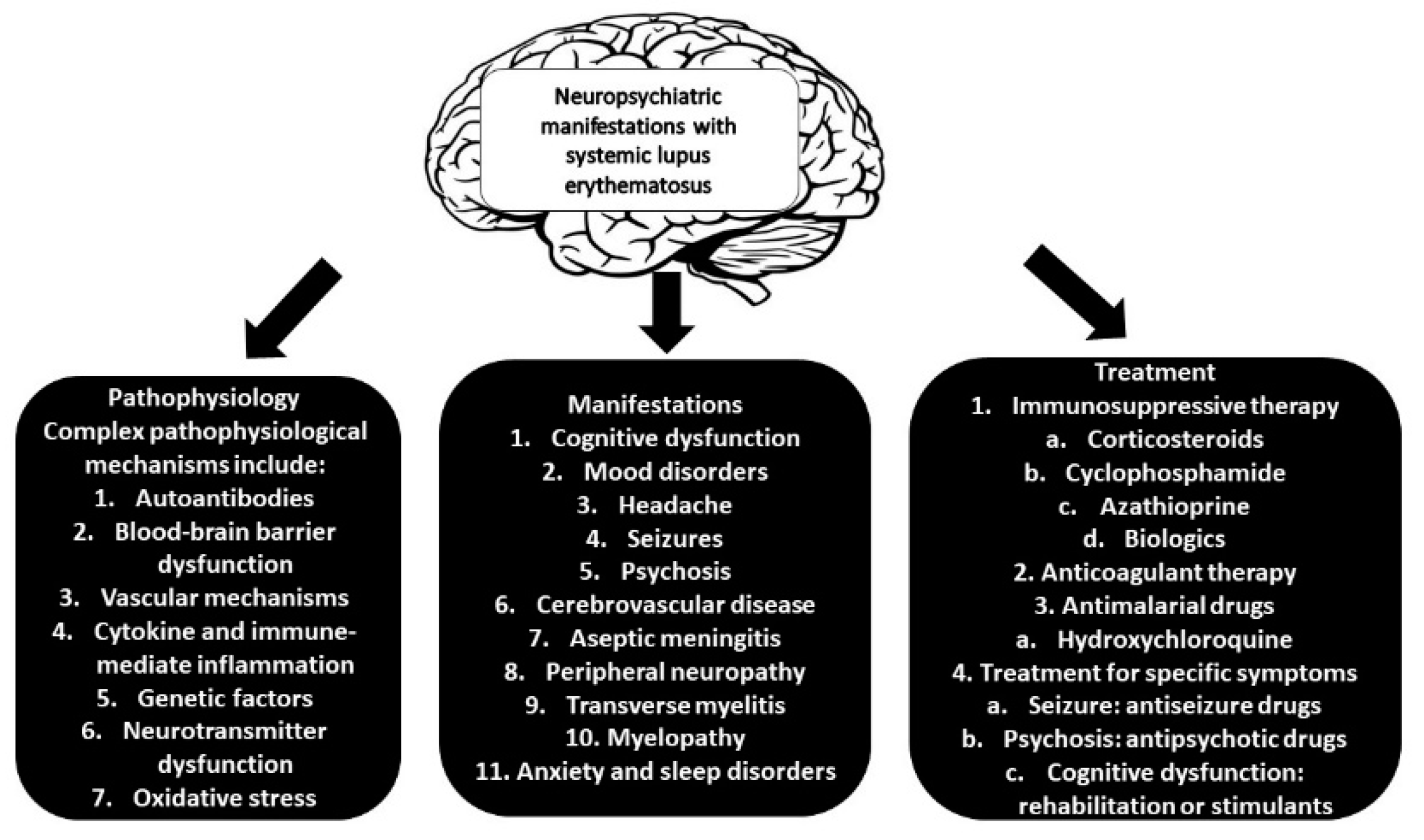

Table 1.
The American College of Rheumatology Nomenclature and Case Definitions for Neuropsychiatric Lupus Syndromes (1999), and their frequency.
Table 1.
The American College of Rheumatology Nomenclature and Case Definitions for Neuropsychiatric Lupus Syndromes (1999), and their frequency.
| Central Nervous system | Peripheral Nervous system |
| Aseptic meningitis (0.3–2.7%) Cerebrovascular disease (8.0–15%) Headache (including migraine and benign intracranial hypertension) (12.2–28.3%) Movement disorder (chorea) (0.9%) Myelopathy (0.9–3.9%) Seizure disorders (7.0–20%) Acute confusional state (0.9–7%) Anxiety disorder (6.4–40%) Cognitive dysfunction (6.6–80%) Mood disorder (7.4–65%) Psychosis (0.6–11%) |
Acute inflammatory demyelinating polyradiculoneuropathy (Guillain-Barre syndrome) (0.08–1.2%) Autonomic disorder (0.08–1.3%) Mononeuropathy, single/multiplex (0.9–6.9%) Myasthenia gravis (0.2%) Neuropathy, cranial (1.0%) Plexopathy (Not available data) Polyneuropathy (1.5–5.4%) |
Table 2.
Immune-mediated mechanisms in neurological SLE.
| Mechanism | Description | Autoantibodies | Manifestations | References |
| The anti-P antiidiotypic antibody population could induce disease by binding to membrane receptors, as has been shown with experimentally induced antibodies to insulin and acetylcholine. | In 18 of 20 patients with psychosis secondary to systemic lupus erythematosus (SLE), autoantibodies to ribosomal P proteins were detected by immunoblotting and measured with a new radioimmunoassay using a synthetic peptide as antigen. | Anti-ribosomal P protein antibodies (Anti-RP Ab) |
Psychosis | Bonfa et al. (1987) [28] |
| The strong association between aPL and NP manifestations in this study supports the theory that an occlusive vasculopathy may be a major mechanism for NPSLE | The study included 323 consecutive SLE patients and investigated the associations between aPL and neuropsychiatric manifestations. | Antiphospholipid antibodies (aPL antibodies) |
Cerebrovascular disease, headache, and seizures | Sanna et al. (2003) [29] |
| MAP-2, have repetitive microtubule-binding motifs, so that they not only control cytoskeletal integrity but also interact with other structural elements of the cell | Sera from 100 patients with SLE, 74 patients with other neurologic disorders and injuries, and 60 normal controls were examined both by enzyme immunoassays and by Western immunoblotting for autoantibodies to MAP-2 | Anti-microtubule-associated protein 2 antibodies (Anti-MAP-2 Ab) |
Psychosis, seizure, neuropathy, and cerebritis | Williams et al. (2004) [30] |
| It has been postulated that AECA reactivity may be due in part to the binding to a complex of β2-GPI with phospholipids on endothelial cells | This study included 51 unselected outpatients with SLE (44 women, 7 men; mean age 36.8 years, range 22–54 years; mean disease duration 9.4 years, range 0.5–26 years) | Anti-endothelial-cell antibodies (AECA) | Psychosis and mood disturbances | Conti et al. (2004) [31] |
| Suggesting that these antibodies, generated in the periphery, penetrate the CNS from the peripheral blood across the altered blood–brain barrier and bind to cell surfaces, possibly interfering with neuronal function | Investigate the potential role of circulating autoantibodies specific to neuronal cell surface antigens in the pathophysiology of neuropsychiatric disorders. | Antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) |
Cognitive dysfunction | Delunardo et al. (2016) [32] |
Table 3.
Diagnostic tests for neurological SLE.
| Test | Utility in Neurological SLE | Reference |
| MRI (Magnetic Resonance Imaging) | The imaging technique of choice is MRI especiallyT2-weighted images. The most frequent pathological pattern is small punctate hyperintense T2-weighted focal lesions in subcortical and periventricular white matter (WM), usually in the frontal-parietal regions. Unfortunately, these MRI lesions are also present in many patients without neuropsychiatric manifestations. MRI can exclude multiple sclerosis, malignancy, infarction, subarachnoid hemorrhage. | Kozora et al. (1998) [152] Nomura et al. (1999) [153] Magro-Checa et al. (2016) [23] |
| Computed tomography | Used to exclude other causes of neurological symptoms such as bleeding, tumors and hemorrhage. | Magro-Checa et al. (2016) [23] |
| PET (Positron Emission Tomography) | SPECT imaging has detected both widespread and localized deficits in patients with SLE, which may be either persistent or reversible. SPECT imaging can be abnormal in up to half of patients with SLE with no clinical manifestations of neuropsychiatric disease. Thus, SPECT findings are not unique to SLE. | Sibbitt et al. (1999) [154] Waterloo et al. (2001) [155] |
| Autoantibody Profiling | Several circulating autoantibodies like apl (antiphospholipid antibodies) and β2-glycoprotein antibodies both correlate with disease activities specially with focal events such as cerebrovascular disease and seizures. Antiribosomal P antibodies found to be specifically related to lupus psychosis. Aquaporin 4 autoantibodies can help in the diagnostic process of a patient presenting with myelopathy and optic neuritis and Neuromyelitis optica (NMO). | Zirkzee et al. (2014) [156] Birnbaum et al. (2007) [157] Birnbaum et al. (2009) [158] |
| Cerebrospinal Fluid (CSF) Analysis | Help to exclude CNS infection in patients with fever or other signs and symptoms suggestive of infection; mild CSF abnormalities are common (40–50%) but are not specific to the neuropsychiatric SLE. It can exclude infection,malignancy,myasthenia gravis and oligoclonal bands. | Kozora et al. (1998) [152] Nomura et al. (1999) [153] 1/22/25 2:18:00 PM |
| Electroencephalogram (EEG) | EEG studies help to diagnose underlying seizure disorder. |
González-Duarte et al. (2008) [159] Appenzeller et al. (2004) [160] |
| Neuropsychological Testing | It’s carried out when suspicion of impaired cognitive abilities is present. Patients with suspected impaired cognitive ability should be referred for full neuropsychological assessment. | Hanly et al. (2014) [161] Zirkzee et al. (2012) [162] |
Table 4.
Management of neuropsychiatric manifestations in SLE.
| Drug name | Mechanism of action | Indication | Administration | Side effects | References |
| Corticosteroids | Modulate immune response and inflammation via glucocorticoid receptors | First line drug and used to control of SLE flares (mild-severe) | IV Methylprednisolone (1g/day x 3 days), oral Prednisolone (1 mg/kg/day) tapering over 3-12 months | Hypertension, dyslipidemia, osteoporosis, diabetes, cataract, glaucoma psychiatric issues (10% risk), infections, and peptic ulcer disease | Schäcke et al. (2002) [173] Bertsias et al. (2010) [76] Buttgereit et al. (2002) [174] |
| Cyclophosphamide | Impairs DNA replication and immune cell proliferation. It is a prodrug that is converted by liver cytochrome 450 enzymes to its metabolite 4-hydroxy cyclophosphamide. | Severe NPSLE manifestations, mainly CNS involvement | Monthly IV regimen (0.75–1.5 g/m²), 500 mg fixed dose in less severe cases | Alopecia, nausea, leukopenia, hemorrhagic cystitis, cardiotoxicity, gonadal failure | Hejaili et al. (2003) [175] Houssiau et al. (2010) [176] Ognenovski et al. (2004) [177] |
| Azathioprine | Inhibits purine synthesis, affects both cellular and humoral immune functions | Used as maintenance therapy. In prevention of flares. It is a first option in mild NPSLE symptoms as a glucocorticoid-sparing agent | Oral (2–3 mg/kg/day) | Bone marrow suppression, hepatotoxicity,increased risk of infection | DiPiero et al. (2015) [178] Oelzner et al. (1996) [179] |
| Mycophenolate mofetil | Inhibits lymphocyte proliferation via inosine-5'-monophosphate dehydrogenase | Maintenance after induction in renal SLE the efficacy of this drug in NPSLE patients is very modest and lacks strong evidence for its efficacy in neuropsychiatric symptoms | Oral (1000–3000 mg/day) | GI intolerance, bone marrow suppression, infections | Allison et al. (2005) [180] Pisoni et al. (2005) [181] |
| Methotrexate | Methotrexate is a folic acid antagonist. It inhibits IL-2 transcription, suppresses T-cell activity | Limited evidence for NPSLE, some reports suggest positive effects with intrathecal administration | Oral, subcutaneous, or intrathecal in CNS involvement | GI symptoms, stomatitis, increased levels of liver enzymes and mild cytopenia. In severe cases liver fibrosis, interstitial pneumonitis, and severe pancytopenia | Zhou et al. (2008) [182] Wang et al. (2014) [183] |
| Cyclosporin A | Inhibits IL-2 transcription, suppresses T-cell activity | Rarely used in NPSLE with Limited evidence, but may aid in organic brain syndrome and psychosis when combined with therapeutic plasma exchange (TPE) | Oral (2.5–3 mg/kg/day), sometimes with plasma exchange | Hypertension, renal dysfunction, hypertrichosis | Bambauer et al. (2000) [184] Yang et al. (2014) [185] |
| Rituximab | Rituximab is a chimeric monoclonal antibody directed against the B-cell-specific antigen CD20. | It is used in severe refractory NPSLE. But, it lacks of long-term data. | IV (1000 mg doses separated by 15 days) | Infusion reactions, infections | Glennie et al. (2007) [186] Tokunaga et al. (2007) [134] |
| IVIG (intravenous immunoglobulin) |
Mixture of natural IgG antibodies derived from the blood of healthy donors. It inhibits the activity of autoreactive B lymphocytes and suppresses typeI interferon-driven differentiation of dendritic cells and reduces nucleosome endocytosis. | Positive effects in small case reports, with limited evidence. But, it is used a adjunctive therapy in refractory cases | IV administration 2g/kg, with divided doses over 2–5 days | Mild and self-limited such as headaches, fever, flushing, chills, arthralgia and myalgia. Infusion reactions, risk of thrombosis | Camara et al. (2014) [187] Milstone et al. (2005) [165] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



