Submitted:
06 January 2025
Posted:
07 January 2025
You are already at the latest version
Abstract
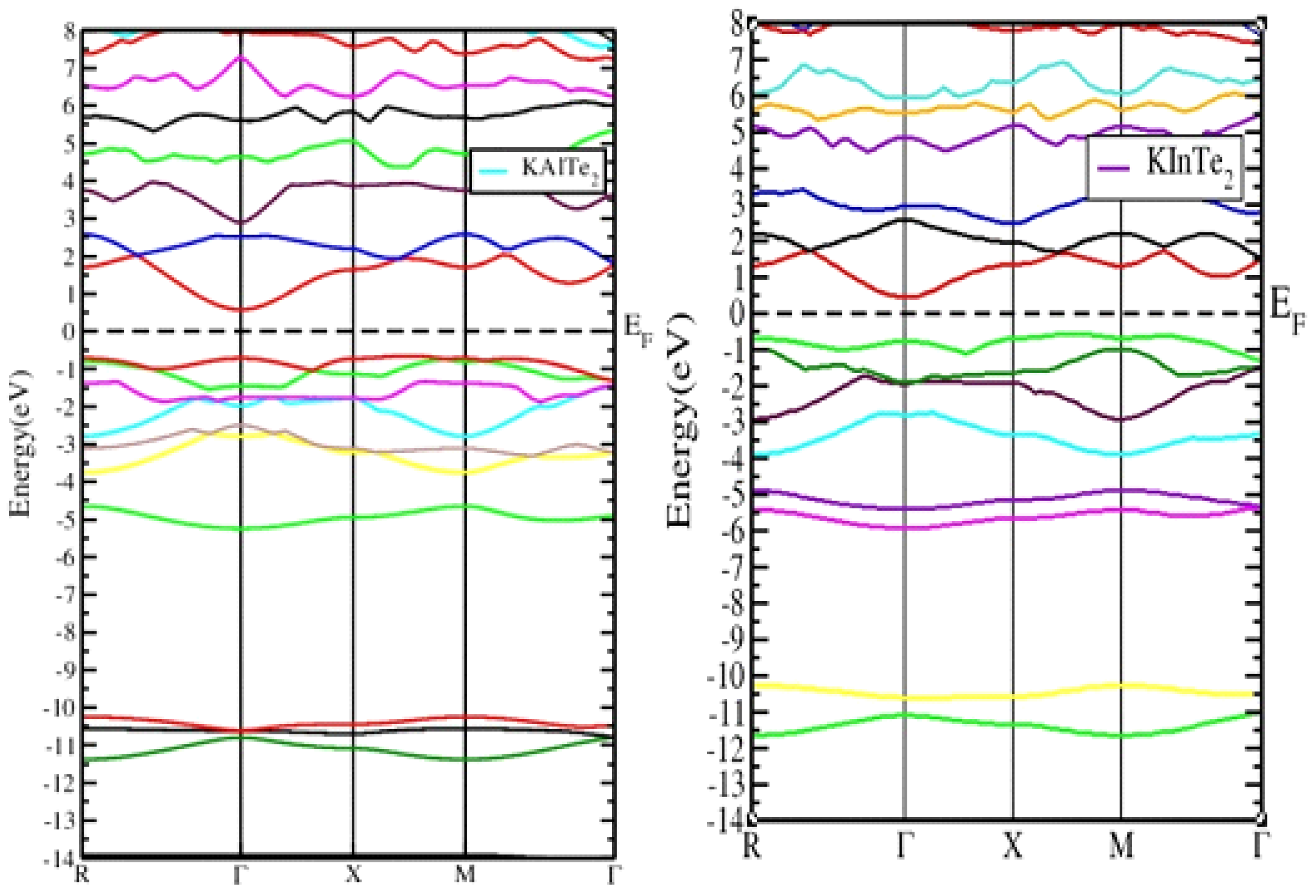
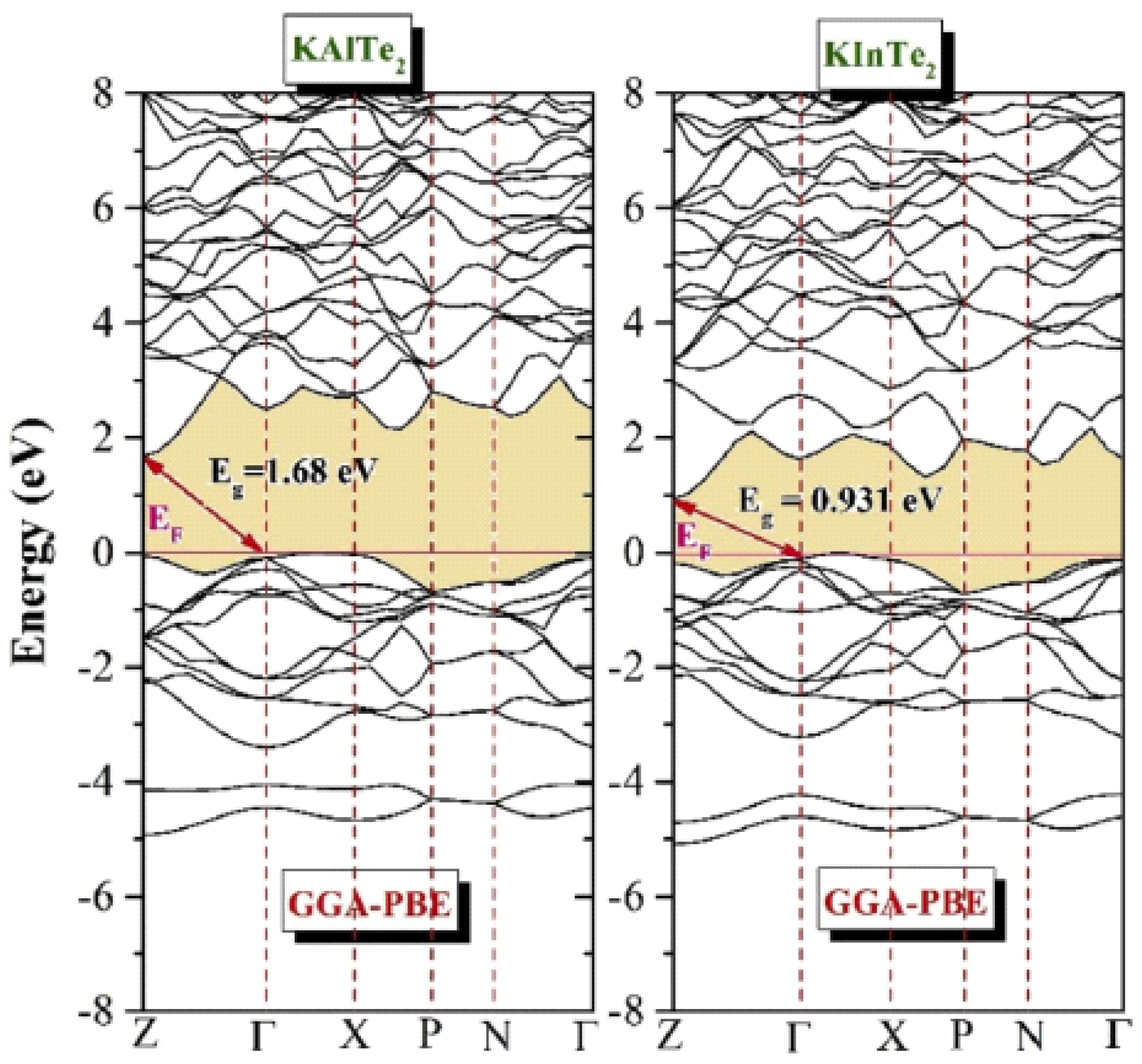
A first-principles study was performed on the anisotropic ternary compounds KAlTe2 and KInTe2 using the WIEN2k and CASTEP codes to optimize their energy to the ground state. The optimized results were then compared with existing theoretical and experimental data for validation. The findings reveal that both KAlTe2 and KInTe2 exhibit both direct and indirect band gaps, depending on the chosen symmetry points during the calculations. The calculated band gaps for KAlTe2 and KInTe2 are found to be 1.68 eV and 0.931 eV, respectively. This dual band-gap behavior indicates that these materials have significant potential for applications in both thermoelectric and optical devices, where direct band gap semiconductors are particularly desirable for efficient energy conversion and light emission. Additionally, the study highlights that the bonding in these materials is characterized by a mixture of covalent and ionic interactions, contributing to their unique electronic properties. The combination of these features makes KAlTe2 and KInTe2 promising candidates for advanced material applications, particularly in fields where the manipulation of band structure and bonding characteristics is crucial for optimizing device performance. This work provides valuable insights into the fundamental properties of these materials and paves the way for further exploration in practical applications.
Keywords:
1. Introduction
2. Method of Calculations
3. Results and Discussion
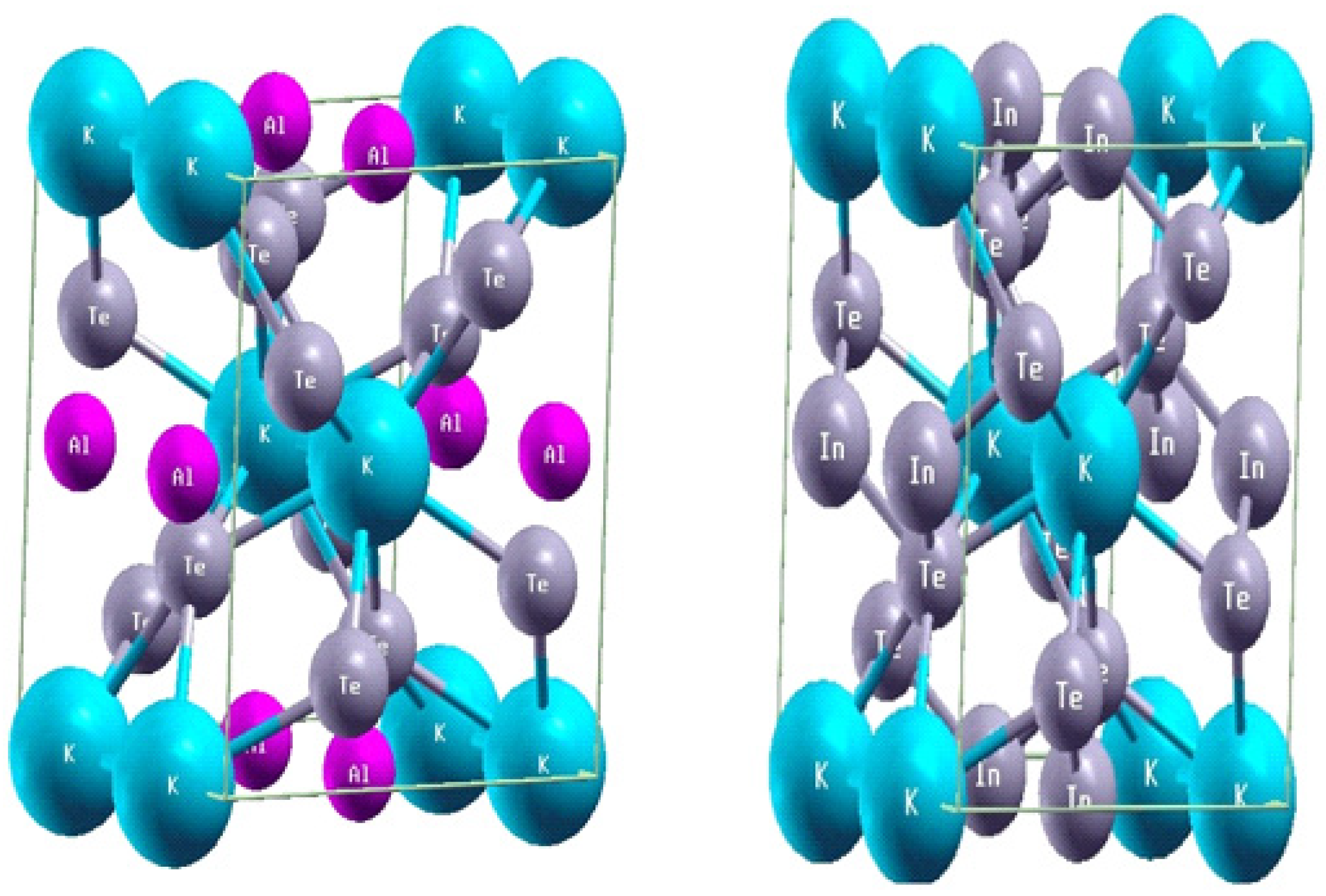
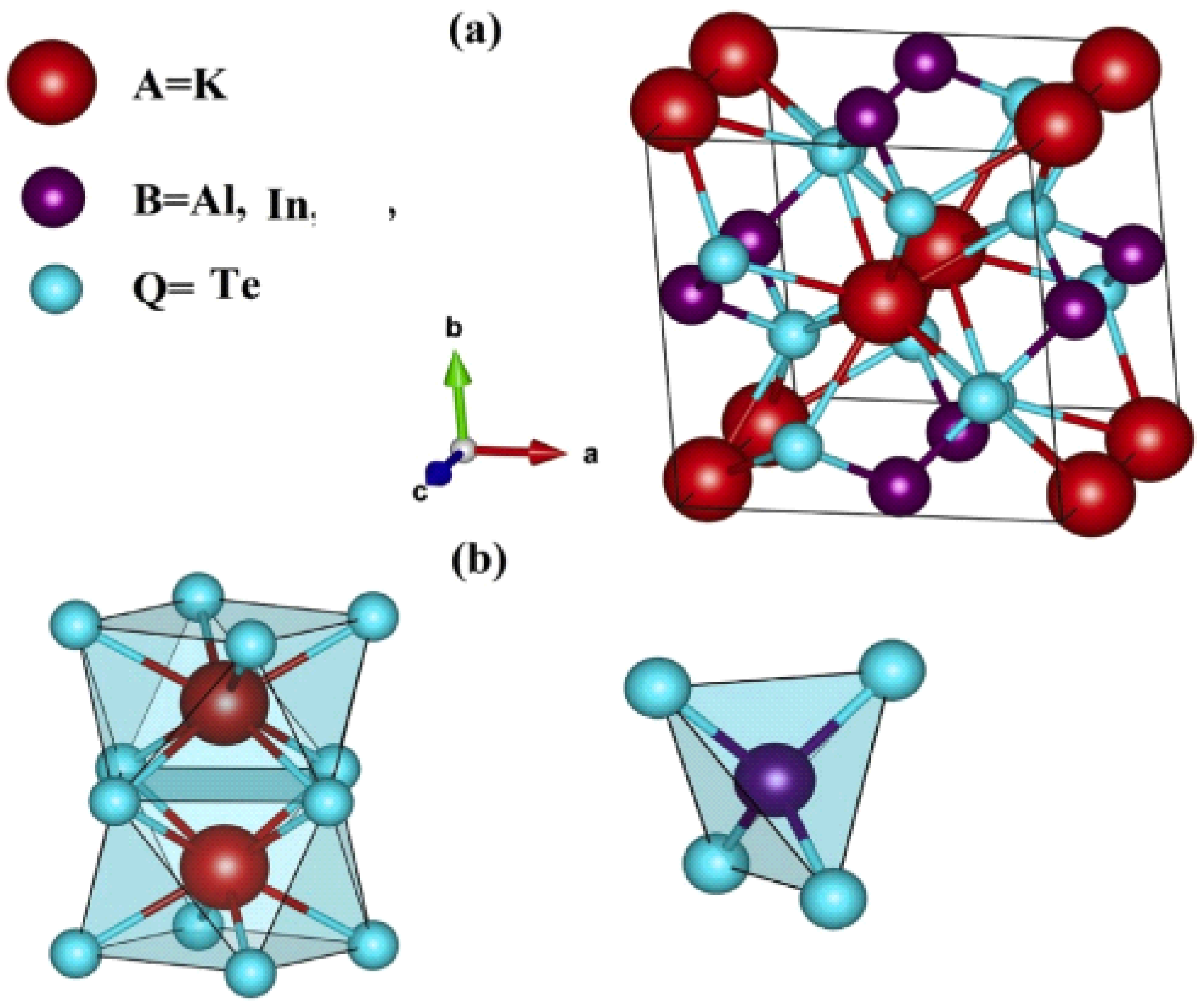
3.1. Structural Properties:
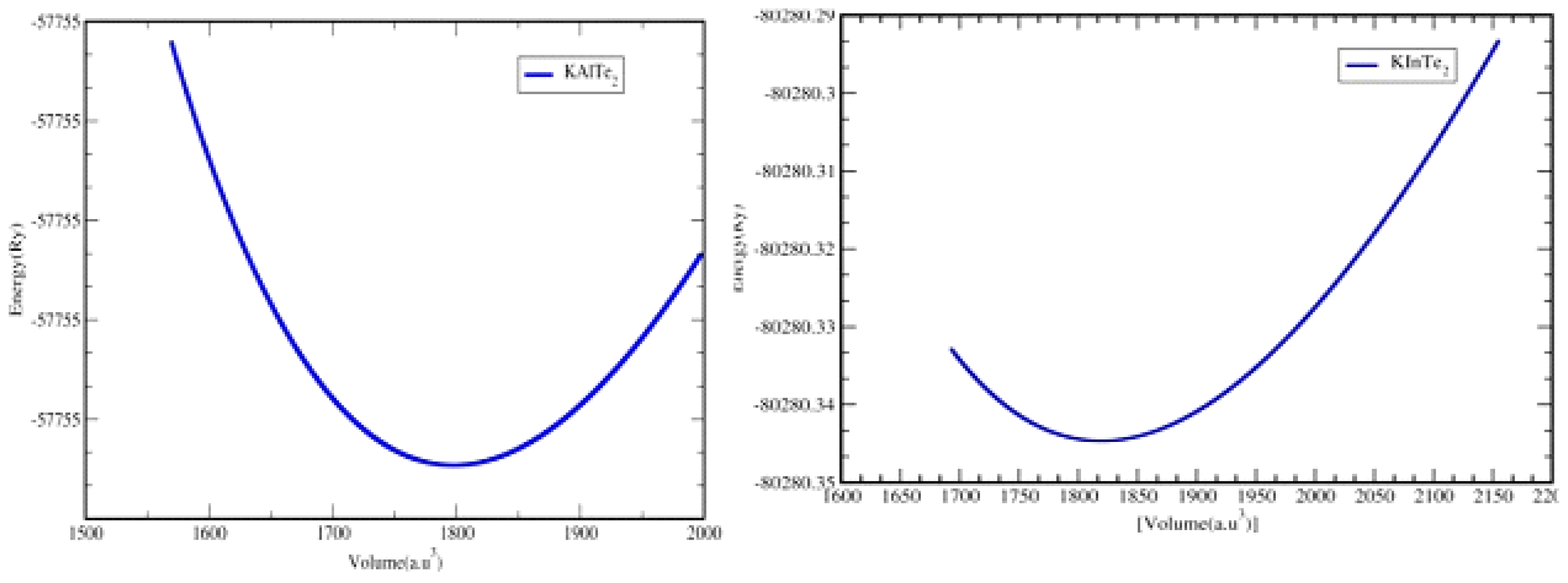
3.2. Optimization Plots:
3.3. Band Structure:
3.4. Bonding Nature/Electron charge density:
3.5. Mechanical Nature:
Conclusion
Funding
References
- Bouchenafa, M.; Benmakhlouf, A.; Sidoumou, M.; Bouhemadou, A.; Maabed, S.; Halit, M.; Al-Douri, Y. Theoretical investigation of the structural, elastic, electronic, and optical properties of the ternary tetragonal tellurides KBTe2 (B= Al, In). Materials Science in Semiconductor Processing 2020, 114, 105085–105088. [Google Scholar] [CrossRef]
- Benmakhlouf, A.; Bentabet, A.; Bouhemadou, A.; Maabed, S.; Khenata, R.; Bin-Omran, S. Structural, elastic, electronic and optical properties of KAlQ2 (Q= Se,Te): A DFT study. Solid State Sciences 2015, 48, 72–81. [Google Scholar] [CrossRef]
- Ullah, Z.; Amir, M.; Bazilla, A.; Ullah, S.; Shahzad, U.; Ullah, N.; Khan, J.; Gul, S. Electronic, Thermoelectric and Magnetic properties of Ternary Telluride KAlTe2 and KInTe2 from Theoretical Perspective. Next Research 2024, 1, 100077. [Google Scholar] [CrossRef]
- Belgoumri, G.; Bentabet, A.; Khenata, R.; Bouhadda, Y.; Benmakhlouf, A.; Rai, D.P.; Bounab, S. Insight into the structural, electronic and elastic properties of AInQ2 (A: K, Rb and Q: S, Se, Te) layered structures from first-principles calculations. Chinese Journal of Physics 2018, 56, 1074–1088. [Google Scholar] [CrossRef]
- Benmakhlouf, A. Structural, elastic, electronic and optical properties of KAlQ2 (Q¼ Se, Te): a DFT study. Solid State Sci 2015, 48, 72–81. [Google Scholar] [CrossRef]
- Feng, K.K. Synthesis structure, physical properties, and electronic structure of KGaSe2. Solid State Sci 2012, 14, 1152–1156. [Google Scholar] [CrossRef]
- Kim, J.; Hughbanks, T. Synthesis and structures of ternary chalcogenides of aluminum and gallium w ith stacking faults: KMQ2 (M= Al, Ga; Q= Se, Te). Journal of Solid-State Chemistry 2000, 149, 242–251. [Google Scholar] [CrossRef]
- Witt, C.W.; Shires, W.B.; Tan, W.C.; Jankowski, J.W.; Pickard, J.C. Random Structure Searching with Orbital-Free Density Functional Theory. The Journal of Physical Chemistry A 2021, 125, 1650–1660. [Google Scholar] [CrossRef]
- Trani, F.; Ninno, D.; Cantele, G.; Iadonisi, G.; Hameeuw, K.; Degoli, E.; Ossicini, S. Screening in semiconductor nanocrystals: Ab initio results and Thomas-Fermi theory. Physical Review B—Condensed Matter and Materials Physics 2006, 73, 245430. [Google Scholar] [CrossRef]
- Xie, Q.X.; Wu, J.; Zhao, Y. Accurate correlation energy functional for uniform electron gas from an interpolation ansatz without fitting parameters. Physical Review B 2021, 103, 045130. [Google Scholar] [CrossRef]
- Claeys, C.; Hsu, P.C.; Mols, Y.; Han, H.; Bender, H.; Seidel, F. . Simoen, E. Electrical Activity of Extended Defects in Relaxed InxGa1− xAs Hetero-Epitaxial Layers. ECS Journal of Solid State Science and Technology 2020, 9, 033001. [Google Scholar] [CrossRef]
- Ikeda, A.; Koibuchi, S.; Kitao, S.; Oudah, M.; Yonezawa, S.; Seto, M.; Maeno, Y. Negative ionic states of tin in the oxide superconductor Sr 3− x SnO revealed by Mössbauer spectroscopy. Physical Review B 2019, 100, 245145. [Google Scholar] [CrossRef]
- Tran, F. (2018). WIEN2k: An Augmented Plane Wave Plus Local Orbitals Program for Calculating Crystal Properties.
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]
- Wu, C.S.; Lee, P.Y.; Chai, J.D. Electronic properties of cyclacenes from TAO-DFT. Scientific reports 2016, 6, 37249. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Francis, S.; Roldan, A. The influence of support materials on the structural and electronic properties of gold nanoparticles–a DFT study. Physical Chemistry Chemical Physics 2019, 21, 19011–19025. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Wang, N.; Li, M.; Xiao, H.; Liu, Z.; Zu, X.; Qiao, L. The thermal and electrical transport properties of layered LaCuOSe under high pressure. Journal of Alloys and Compounds 2021, 861, 157984. [Google Scholar] [CrossRef]
- Wiebeler, H. (2020). A linear scaling DFT-Method and high-throughput calculations for p-type transparent semiconductors (Doctoral dissertation, Universitätsbibliothek).
- Schwarz, K.; Blaha, P. Solid state calculations using WIEN2k. Computational Materials Science 2003, 28, 259–273. [Google Scholar] [CrossRef]
- Schwarz, K. DFT calculations of solids with LAPW and WIEN2k. Journal of Solid State Chemistry 2003, 176, 319–328. [Google Scholar] [CrossRef]
- Labrim, H.; Jabar, A.; Laanab, L.; Jaber, B.; Bahmad, L.; Selmani, Y.; Benyoussef, S. Optoelectronic and thermoelectric properties of the perovskites: NaSnX3 (X= Br or I)—a DFT study. Journal of Inorganic and Organometallic Polymers and Materials 2023, 33, 3049–3059. [Google Scholar] [CrossRef]
- Selmani, Y.; Labrim, H.; Mouatassime, M.; Bahmad, L. Structural, optoelectronic and thermoelectric properties of Cs-based fluoroperovskites CsMF3 (M= Ge, Sn or Pb). Materials Science in Semiconductor Processing 2022, 152, 107053. [Google Scholar] [CrossRef]
- Bouhmaidi, S.; Marjaoui, A.; Talbi, A.; Zanouni, M.; Nouneh, K.; Setti, L. A DFT study of electronic, optical and thermoelectric properties of Ge-halide perovskites CsGeX3 (X= F, Cl and Br). Computational Condensed Matter 2022, 31, e00663. [Google Scholar] [CrossRef]
- Prihadi, H.L.; Zen, F.P.; Ariwahjoedi, S.; Dwiputra, D. (Replica trick calculation for entanglement entropy of static black hole spacetimes. International Journal of Geometric Methods in Modern Physics 2023, 20, 2350132. [Google Scholar] [CrossRef]
- Madsen, H.K.G.; Singh, J.D. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef]
- Kaur, T.; Sinha, M.M. (2021). Probing thermoelectric properties of high potential Ca3PbO: An Ab Initio Study. In IOP Conference Series: Materials Science and Engineering (Vol. 1033, No. 1, p. 012080). IOP Publishing.
- Mahmood, Q.; Hassan, M.; Ahmad SH, A.; Bhamu, K.C.; Mahmood, A.; Ramay, S.M. Study of electronic, magnetic and thermoelectric properties of AV2O4 (A= Zn, Cd, Hg) by using DFT approach. Journal of Physics and Chemistry of Solids 2019, 128, 283–290. [Google Scholar] [CrossRef]
- Feng, S.; Wang, N.; Li, M.; Xiao, H.; Liu, Z.; Zu, X.; Qiao, L. The thermal and electrical transport properties of layered LaCuOSe under high pressure. Journal of Alloys and Materials 2021, 861, 157984–157989. [Google Scholar] [CrossRef]
- Ribeiro, R.A.; Andres, J.; Longo, E.; Lazaro, S.R. Magnetism and multiferroic properties at MnTiO3 surfaces: A DFT study. Applied Surface Science 2018, 452, 463–472. [Google Scholar] [CrossRef]
- Benmekideche, N.; Bentabet, A.; Bouhadda, Y.; Boubatra, D.; Belgoumri, G.; Fetah, S.; Benyelloul, K. DFT study of structural, electronic and elastic properties of two polymorphs of monoclinic CsGaQ2 (Q= S, Se). Chinese Journal of Physics 2019, 56, 1345–1352. [Google Scholar] [CrossRef]


 |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



