Submitted:
05 January 2025
Posted:
06 January 2025
Read the latest preprint version here
Abstract
The connection between acetaminophen use in the pediatric population and the etiology of autism spectrum disorder (ASD) have been a subject of misunderstanding, miscalculation, and misinformation for more than a decade. This narrative review summarizes 27 lines of evidence pointing with no reasonable doubt to the conclusion that exposure of susceptible babies and children to acetaminophen is responsible for many if not most cases of ASD. Susceptibility to acetaminophen-induced injury is imposed by a range of environmental, genetic, and epigenetic factors associated with oxidative stress, and is apparently the greatest immediately after birth. Susceptibility then decreases until about six years of age, which is likely outside of the developmental window in which regression into ASD can occur. Exposure to acetaminophen very early in life can occur for a variety of reasons, including treatment of pain during administration to the mother during labor and delivery, during vaccination, and during circumcision. Although acetaminophen use during vaccination is generally not recommended, the vaccine against meningococcal serogroup B (MenB), administered at 2, 4, and 12 months of life, is now recommended with 3 accompanying doses of acetaminophen in some countries with dramatically rising levels of ASD. Unfortunately, based on current knowledge, it is expected that such exposures to acetaminophen will induce ASD in some susceptible individuals. Evidence therefore strongly and unequivocally indicates that medical recommendations should mandate the MenB vaccine be given separately from other vaccines and without acetaminophen, which some national healthcare services (e.g., Australian and Canadian) have already identified as an acceptable clinical approach.
Keywords:
Acetaminophen
; Autism
; Inflammation
; Paracetamol
; Vaccine
Introduction
Abundant evidence has led us to conclude, without reasonable doubt, that exposure of susceptible babies and children to acetaminophen causes neurodevelopmental injury, leading to many if not most cases of autism spectrum disorder (ASD) [1,2,3,4]. Evidence also demonstrates that a wide range of genetic, epigenetic and environmental factors associated with oxidative stress create susceptibility to acetaminophen-induced injury. Furthermore, the developmental period of greatest susceptibility appears to be at the time of birth, with susceptibility diminishing over time and ending at about six years of age [3]. The most recently published tally listed 22 lines of evidence [3] from clinical observations, pharmacokinetic consideration, and laboratory animal studies. Since that list of evidence was published in 2023, additional lines of evidence were described in the literature in 2024, including the numerous similarities between ASD and fetal alcohol spectrum disorder [4], which demonstrate that a single drug, interacting with environmental and genetic factors, can cause a complex spectrum disorder. As another example, Graeca and Kulesza found that exposure of laboratory rats to acetaminophen in utero leads to problems with auditory processing later in life [5]. As reviewed by the authors [5], some degree of auditory dysfunction is seen in the majority of individuals with ASD. However, it remains unknown whether the acetaminophen-induced auditory dysfunction observed in laboratory rats is related to auditory dysfunction observed in individuals with ASD. Thus, this line of evidence, by itself, does not lead to the conclusion that acetaminophen can act as a trigger for the induction of ASD. Nevertheless, the study by Graeca and Kulesza adds to an already overwhelming body of evidence connecting acetaminophen with developmental problems and the induction of ASD in particular, and therefore should be included in an updated tally of evidence. Other evidence that can be included for consideration are the relationships between arachidonic acid metabolism and acetaminophen [6,7] and between arachidonic acid metabolism and ASD [8]. These studies, taken together, suggest that arachidonic acid could be involved in the induction of ASD and acetaminophen by mechanisms that have yet to be elucidated, and constitute yet another link between acetaminophen and ASD.
A current summary of information that, when considered together, demonstrates the induction of ASD in susceptible individuals by acetaminophen is shown in Table 1. A total of 27 lines of evidence are listed. Despite this overwhelming evidence demonstrating that acetaminophen is a critical component in the etiology of many if not most cases of ASD, the drug continues to be used in the pediatric population. For example, although acetaminophen is not generally recommended for vaccinations, three doses of acetaminophen are now recommended for use with each dose of the vaccine against meningococcal serogroup B (MenB), administered to 2, 4, and 12 months of age (see discussion below). Given the existence of this policy, one obvious question is, do we have enough evidence of harm from acetaminophen to change practice regarding its use?
How Much Evidence Is Enough?
Although randomized, blinded, placebo-controlled experiments to obtain absolute proof that acetaminophen induces ASD in humans might seem worthwhile at first glance, such experiments would be both impractical and unethical. In terms of trial design, acetaminophen effectively treats fevers, so a blinded placebo control could prove difficult if study subjects were able to effectively guess their study group. In addition, even without a blinded placebo control, exposure would need to be controlled from conception to age 5 in thousands of individuals, consuming vast amounts of time and resources for the trial. Even more important is the fact that evidence (Table 1) already conclusively demonstrates that acetaminophen is a developmental neurotoxin. Thus, exposure of any fetus, baby or child to the drug for the sake of research is unethical.
Changes in clinical practice should not require enough evidence to conclude without any reasonable doubt that exposure of susceptible children to acetaminophen causes many if not most cases of ASD. For example, the facts that, (a) children with ASD are deficient in a metabolic pathway (sulfation) that is necessary for safe metabolism of acetaminophen (Table 1, line of evidence #1), (b) relatively low doses acetaminophen in early life cause long-term brain dysfunction in laboratory mice and rats (Table 1, line of evidence #5), and (c) acetaminophen can’t be used in some domestic animals because they are deficient in the same pathway (glucuronidation) that is deficient in all newborn babies (Table 1, line of evidence #22) should have been sufficient to remove the drug entirely from the pediatric market more than a decade ago.
From another perspective, the 2008 study by Schultz and colleagues [9] should have been sufficient to have the safety of acetaminophen for pediatric use completely and immediately reevaluated. That study (see Table 1, lines of evidence #2 and #3) provided an explanation for the repeated observation that many parents believe that a vaccine was involved in the induction of their child’s ASD (Table 1, line of evidence #23). Although the study suggested that the vast majority of all regressive ASD might be induced by acetaminophen, it was largely ignored for reasons that, based on an in-depth analysis [1], are invalid. Thus, the widespread belief that acetaminophen was proven safe for use in babies and children was not disproven until more than a decade later, in 2022 [10] (Table 1, line of evidence #4).
The bottom line is that evidence sufficient to drive regulatory changes and changes in clinical practice has long been ignored. Further, proof without reasonable doubt that exposure of susceptible children to acetaminophen causes ASD exceeds what should be required to remove the drug entirely from the pediatric market. Suspicion of danger should be sufficient. At this point, the evidence (Table 1) leads to a level of certainty that far exceeds suspicion.
Means, Motive and Opportunity
One potentially useful perspective on the link between acetaminophen and ASD involves comparison with establishment of guilt in criminal court. Does acetaminophen have the means, “motive”, and opportunity to induce ASD? Given that the metabolism of acetaminophen yields a toxic metabolite (N-acetyl-p-benzoquinone imine; (NAPQI), a reactive electrophile, similar to that produced by the metabolism of ethanol) especially under conditions associated with ASD, and given that the toxic metabolite affects mitochondrial and neuronal function, acetaminophen has the means to induce ASD (Table 1, lines of evidence #1,#8, #9). The observation that ASD is similar in many regards to fetal alcohol spectrum disorder (Table 1, line of evidence #27) demonstrates that a single chemical does, in fact, have the means to produce a complex developmental spectrum disorder.
While “motive” is not a feature that can be attributed to a chemical compound, the observation that acetaminophen affects social function in adults (Table 1, line of evidence #11) demonstrates that the drug does indeed have a propensity to affect aspects of brain function involved in all ASD phenotypes. This view is corroborated by studies in laboratory animals showing acetaminophen-mediated damage to cortical neurons (Table 1, line of evidence #8), a cell type apparently involved in ASD phenotypes.
Although use of acetaminophen is frequently undocumented [1], evidence suggests that most individuals throughout the US and Europe are exposed to the drug both in utero and during early childhood [1], indicating that acetaminophen does indeed have the opportunity to induce ASD. As we have previously discussed [2], despite low levels (< 10 % of the population) of exposure to acetaminophen are reported in the Danish and Swedish databases, assessments of acetaminophen use in those populations indicate that more than 50 % of those populations are exposed [11,12]. Further, factors associated with oxidative stress, which determine sensitivity to acetaminophen-mediated injury [13], are connected with reasons for using acetaminophen. Such factors include infection, antibiotics and headaches. Thus, exposure to acetaminophen of individuals susceptible to acetaminophen-mediated neurological injury is almost certainly higher than in the population as a whole, increasing risk. Also convincing is the direct measurement of acetaminophen metabolites in humans [14,15], which demonstrates widespread exposure to the drug during neurodevelopment, at least in the populations assessed. More recently, the introduction of the MenB vaccine into pediatric use at 2, 4, and 12 months of age, with mandatory administration of acetaminophen in many countries (see discussion below), will ensure that acetaminophen has the opportunity to induce ASD in many countries. Thus, it would seem that acetaminophen has means, propensity, and opportunity to induce ASD.
Two Lines of Easily Misconstrued Evidence Dominate the Field
Observational studies assessing healthcare databases have dominated the medical literature and public discourse surrounding the link between acetaminophen and ASD. Numerous studies assessing the connection between prenatal acetaminophen use and ASD (Table 1, line of evidence #13), and one study assessing the connection between postnatal acetaminophen use and ASD (Table 1, line of evidence #14) have been published. Such studies are conducted without the need for time-consuming and costly collection of new data, and the associations thus determined provide insight into potential causality. Indeed, if no association exists, causality is not a possibility.
The “raw analysis” from observational studies generally show a strong connection between acetaminophen use and ASD. That is to say, without using statistical methods to adjust for factors that the authors believe might confound the conclusions, an association between acetaminophen and ASD is usually found. For example, in a widely publicized study published by Ahlqvist and colleagues in 2024 [16], the raw data showed a strong connection between acetaminophen use during pregnancy and ASD (Figure 1). The relationship was dose-dependent, with high doses of acetaminophen associated with a hazard ratio of 1.87 (C.I.:1.71-2.06) and statistically significant associations (p < 0.001) at low, medium and high doses of acetaminophen (Figure 1). These associations are particularly concerning given the high prevalence of acetaminophen use in the population. As we have previously pointed out [2], a combination of the hazard ratio associated with a given factor and the prevalence of that factor in the population dictates the potential impact of that factor on human health. Thus, for something as common as acetaminophen use, any statistically significant risk for ASD is probably unacceptable.
However, the conclusion of the widely publicized paper by Ahlqvist noted above [16] was that “Acetaminophen use during pregnancy was not associated with children’s risk of autism…”. One factor potentially affecting the conclusions of the Ahlqvist work is that it was funded to an undisclosed extent by legal experts employed by the drug manufacturer. The results of clinical research studies sponsored by pharmaceutical companies are more likely to yield a favorable outcome for the drug than are studies without pharmaceutical backing [17,18,19]. Further, numerous companies from a variety of industries, including the tobacco industry [20], the food industry [21], and the pharmaceutical industry [22,23] have a known history of trying to influence scientific research in favor of their products [24]. Although it remains unknown whether any industry-related bias affected the outcome of the Ahlqvist study [16], the study was unique among numerous other studies [14,25,26,27,28,29,30,31,32,33,34,35,36,37] in finding no association between heavy acetaminophen use during pregnancy and adverse neurodevelopmental outcomes.
The conclusion reached by Ahlqvist and colleagues [16], that acetaminophen use during pregnancy is not associated with ASD, was based on statistical “correction” for confounding factors that are, in fact, cofactors rather than confounding factors. The effect of correction for confounding factors found in the Ahlqvist study is shown in Figure 1. As we have previously pointed out [2,4], such corrections are not valid or justifiable. Such an error will be evident to trained statisticians, suggesting that experts on the Ahlqvist study team performing statistical tests may have been unaware of the pharmacokinetics of acetaminophen, including interactions of the drug with factors related to inflammation and oxidative stress. In effect, Ahlqvist and colleagues catalogued the conditions under which acetaminophen is dangerous for neurodevelopment. They did not determine that the drug is safe for neurodevelopment.
A formal demonstration of the problem of statistical correction for cofactors (aka, predisposing factor or interacting variables) is shown in Table 2. That table shows the results of an analysis of a virtual (artificially generated) database in which 50 % of all cases of ASD were caused by a combination of oxidative stress and exposure to acetaminophen during an arbitrary time period. In the analysis, the raw (“uncorrected” analysis) shows a hazard ratio for ASD with acetaminophen use of 2.55 (CI 2.41-2.71, p = 2 x 10-16), close to the actual hazard ratio of 2.667 built into the model. However, after correcting for 100 % of the factors that account for all susceptibility to acetaminophen-mediated injury, the calculated hazard ratio for ASD with acetaminophen use is 0.85 (CI 0.80-0.90; p = 2 x 10-7). Thus, when oxidative stress factors are treated as confounding factors, acetaminophen is “shown” to be protective from ASD despite the fact that it actually caused 50 % of all ASD cases in this virtually constructed dataset.
Another problem with analyses of healthcare databases is that such analyses do not take into account that oxidative stress may be associated with genetic, epigenetic, or persistent environmental factors, causing persistence of susceptibility in an individual. The resulting effect is that assessment of acetaminophen exposure versus oxidative stress-related variables at a given time (e.g., during pregnancy or during infancy) may miss important injury-inducing exposure to acetaminophen that happened at a different time (e.g., during labor and delivery). The net result of this situation is that the analysis can identify associations between oxidative-stress related variables but not acetaminophen, even under ideal circumstances. That is to say, even if 100 % of acetaminophen exposures are documented in a given study period, the net effect of limiting the study period (e.g., to prenatal or postnatal exposure) is that only a fraction of potentially important acetaminophen exposures will be documented. The only way to avoid this problem would be to assess all exposures to acetaminophen which might trigger ASD, from conception to age 5. Although such an approach is not feasible, sufficient evidence is already available (Table 1) to draw conclusions for clinical practice. Therefore, the fact that an ideal observational study is not feasible should be of little concern.
Misconstrued analyses of healthcare databases can be influential. An example of placing heavy emphasis on a misconstrued line of evidence is found in Graeca and Kulesza’s groundbreaking work showing acetaminophen-induced developmental problems related to auditory function [5]. The overreliance on observational studies as a basis for understanding the connection between acetaminophen and ASD is evident in the Introduction of that paper:
“Specifically, in utero (but not postnatal) exposure to paracetamol results in a 19% increased risk of ASD (Masarwa et al., 2018, Alemany et al., 2021, Khan et al., 2022)…”
The studies by Masarwa et al. in 2018 [38] and by Khan et al. in 2022 [39] cited by Graeca and Kulesza address only prenatal exposure. Therefore, Graeca and Kulesza base their conclusion that postnatal exposure to acetaminophen (paracetamol) is not associated with ASD solely on the study by Alemany and colleagues in 2021 [30]. However, Alemany and colleagues did not conclude that postnatal acetaminophen use is not associated with ASD. Rather, they concluded that postnatal use of acetaminophen is not associated with “autism spectrum condition symptoms”, which has a prevalence from 6 % to 13 % of the samples they evaluated, much higher than the prevalence of ASD. However, more than 80 % of Alemany’s sample (48,161 out of 58,006 total individuals) came from the Danish National Birth Cohort (DNBC), and ASD, not autism spectrum condition symptoms, was measured in that cohort. In Alemany’s analysis of the connection between postnatal acetaminophen use and ASD in the DNBC, they found a positive association (OR = 1.30, CI 1.02-1.66). Given that the DNBC dramatically underreports acetaminophen use, and given that the authors corrected for oxidative stress-related variables in an invalid fashion (see discussion above), the odds ratio of 1.30 is probably underestimated dramatically. Nevertheless, an odds ratio of 1.30 is profoundly concerning given the high prevalence of acetaminophen exposure. However, when reporting overall results, for example in the abstract of the paper, Alemany and colleagues combined results from analysis of ASD in the DNBC with results from analysis of smaller databases that reported only autism spectrum condition symptoms [30]. In their combined analysis, they counted (weighted) the very concerning results from the DNBC as only 31.84 % of the total, despite the facts that it contained more than 80 % of the total individuals, was the only database to contain measures of ASD, and was the only database which yielded statistically significant results in the analysis. Alemany and colleagues provided no explanation for the weighting scheme or for the lack of emphasis on the very concerning results from their analysis of the DNBC.
In short, analysis of the connection between prenatal acetaminophen use and ASD (Table 1, line of evidence #13) and the connection between postnatal acetaminophen use and ASD (Table 1, line of evidence #14) is informative and useful, but those lines of evidence have been fraught with misinformation, miscalculation, and misinterpretation. As we concluded recently [4]:
“It is concluded that risks of acetaminophen use for neurodevelopment obtained from multivariate analysis of cohort data depend on underlying assumptions in the analyses, and that other evidence, both abundant and robust, demonstrate the critical role of acetaminophen in the etiology of ASD.”
Barriers to Moving Forward
Compelling evidence of the neurodevelopmental toxicity of acetaminophen has continued to mount. Studies in laboratory mice showing long-term, profound (almost complete) loss of important aspects of cognitive function following early life exposure to acetaminophen was published more than decade ago, in 2013 [40]. In that study, two doses of 30 mg/kg acetaminophen were administered in one day. That dose is not exceedingly higher than the oral dosage of acetaminophen in babies and children, who can receive up to 4 doses of 14.7 mg/kg of acetaminophen on multiple, consecutive days. Given these results, acetaminophen could never be approved for pediatric use today, even in clinical trials, if it was evaluated using modern safeguards in place to prevent adverse drug reactions. The drug would not pass preclinical testing.
Shortly after Viberg’s study in laboratory mice in 2013, Frisch and Simonsen, two Danish investigators, found more than double the risk of infantile ASD associated with circumcision when assessing the DNBC [41]. They initiated that investigation in part because Bauer had proposed that acetaminophen exposure during circumcision may induce ASD [42]. Although Frisch and Simonsen could not evaluate acetaminophen use during circumcision, their report did confirm the predictions of Bauer.
Several factors doubtless underly the continued and apparently cavalier use of acetaminophen during neurodevelopment despite overwhelming evidence that the drug is a neurodevelopmental toxin. The dismissal of compelling results is certainly one factor. Dismissal of the report by Frisch and Simonsen [41] is an excellent example. One dismissal, by Morris and colleagues [43], was based on the perplexing assertion that “Sneppen and Thorup, in particular, found ASD prevalence was 7.2 % in uncircumcised Danish boys and suggested Frisch (and Simonsen)'s study suffered from confounding.” [43] However, Sneppen and Thorup [44] explained that their study could not be confounded by ASD induction during circumcision because ASD had already been diagnosed in their cohort prior to circumcision. They never suggested in any way that the initial report by Frisch and Simonsen was confounded [44].
Part of the argument by Morris and colleagues cited above [43] for dismissing the results of Frisch and Simonsen seems to be that the Sneppen and Thorup study found higher rates of ASD than did Frish and Simonsen. However, the children assessed by Sneppen and Thorup were selected because they had been sent to the surgery department as a result of problems with their penis, most (95 %) because of a problem called “phimosis” that is usually treated effectively with anti-inflammatory drugs. More than a quarter of the boys had “severe voiding problems”, which often involve pain when urinating, and more than a quarter of the boys were suspected of having inflammation of their penile tissue. Thus, we expect the boys selected by Sneppen and Thorup to have more ASD than average boys because inflammation and ASD are connected, and because pain management with acetaminophen is connected with ASD. Further, Sneppen and Thorup assessed about 2500-fold less uncircumcised individuals than did Frish and Simonsen (137 versus about 340,000), and did not report or discuss ASD versus circumcision status in their patients. With that in mind, finding a higher prevalence of ASD in a group of 137 uncircumcised boys, most of whom have a painful and/or inflamed penis, does not suggest that a study of all children of certain ages in the entire Danish population could be “confounded”. The view that Frisch and Simonsen’s study could be confounded by unknown factors was apparently not the intent of Sneppen and Thorup’s statements, and it remains a mystery as to how their study could be used as justifiable grounds for dismissal of the Frisch and Simonsen study.
Misinterpretation or misrepresentation of results are not limited to Morris’s analyses of the Frisch and Simonsen study [43] or to high-profile studies of associations between acetaminophen and ASD in healthcare databases [16,30]. Indeed, the initial study by Schultz showing that acetaminophen but not vaccination was associated with ASD [9] has been widely criticized, although a detailed assessment of those criticisms reveals no valid arguments (for review, see Zhao et al. [1]). Further, explanations that attempt to dismiss profound and long-term increases in the prevalence of ASD abound, despite being untenable [1,4].
Misinformation, miscalculation, and misinterpretation plague investigation of the role of acetaminophen in the etiology of ASD and create significant barriers to progress. However, these problems are likely only a symptom of underlying causes. Given the long history of acetaminophen use in the pediatric population and the extensive damage already incurred as a result, most if not all stakeholders are potentially faced with a variety of hurdles (Figure 2), many of which reinforce one another. For example, conflicts of interest and emotional compromises face scientists and clinicians whose careers and reputations may be damaged by acknowledging the role of acetaminophen in the etiology of ASD (Figure 2). The pressure from these factors could lead to continued support of acetaminophen use by authorities and a lack of published work demonstrating the connection between acetaminophen and ASD. These factors, in turn, make it more difficult for parents and parents-to-be to receive and act upon information regarding the role of acetaminophen in the etiology of ASD (Figure 2). As a result, exposure of susceptible babies and children to acetaminophen persists, increasing the conflicts and emotional compromises already imposed on the medical and scientific communities.
Clinical Implications
At the present time, no evidence supports the idea that the benefits of acetaminophen use outweigh the risks of neurodevelopmental injury. The drug has never been shown to be lifesaving or to provide any long-term benefits in any study. Fears over fevers tend to be unfounded, with no evidence supporting the view that acetaminophen can block adverse fever-associated events with long-term negative consequences. This topic has been reviewed in detail recently [3]. At the same time, acetaminophen exposure to babies and children seems to be performed with a cavalier attitude, likely due to the erroneous assumption that it is extremely safe because hepatoxicity is not generally induced, even at doses exceeding the recommended dose [45]. Hospital pharmacy recommendations, for example, generally involve doses of acetaminophen up to 45 mg/kg when the drug is administered via the rectum [46,47,48], three times more than the recommended oral dose. The recommended dose for rectal administration is greater than the recommended dose for oral administration because, on average, the rectally administered drug has less bioavailability than the orally administered drug. However, the bioavailability of the drug administered via the rectal route is variable among children and especially neonates [47,48,49,50], which could result in some babies and children receiving considerably more acetaminophen than is possible via the oral route. Further, it seems unlikely that acetaminophen use with circumcision in the first hours of life is always considered with the possibility that some acetaminophen might remain in the neonate’s body as a result of the mother receiving the drug during labor and delivery. In addition, reports abound describing the common occurrence of acetaminophen administration more frequently than recommended, at doses higher than recommended, and for reasons that are not recommended (for review, see Patel et al. [2]), supporting the view that a cavalier attitude exists toward the drug among medical health professionals and possibly other caregivers.
Acetaminophen use during sensitive periods of neurodevelopment continues to be recommended by medical authorities without apparent awareness of the risks. An excellent example of such a policy is the recommendation of three doses of acetaminophen concurrent with the meningococcal group B (MenB) vaccine at 2, 4, and sometimes 12 months of life [51], when acetaminophen-induced induction of ASD is likely [3]. Children older than 1 year of age may also receive the MenB vaccine if they are not already vaccinated. Israel, the UK, Australia, New Zealand and Canada are among the countries that have implemented such recommendations [51,52,53,54,55] for the MenB vaccine, which became available in 2014 [56]. Although measures of the prevalence of ASD tend to take several years to compile, some health services have provided relatively current estimates that include individuals receiving the MenB vaccine early in life. For example, the prevalence of ASD in Australia in 2022, eight years after the MenB vaccine recommendation was put into place, was 4.3 % of the total population aged 5-14 years, a 34 % increase in four years (up from 3.2 % of 5-14 year olds with ASD in 2018) [57]. Unfortunately, 73 % of those affected were found to have severe and profound disabilities [57]. In another example, the prevalence of ASD was found to be rising dramatically and rapidly in Israel in 2021, with two independent data sources showing that the prevalence of ASD in 1–17 year-old children had almost doubled within 4 years [58]. The most rapid increases were seen in children ages 2-3 years old, some of whom could have received three doses of acetaminophen with the MenB vaccine more than once during the first four months of life, when ASD might be more readily induced than in older children [3].
The rising prevalence of ASD has been a fact of life in high-income countries for more than 40 years, and can be affected by several factors other than actual changes in the occurrence of ASD. Thus, it remains unknown whether dramatic increases in the prevalence of ASD in some Westernized countries observed at the present time are attributable in part to acetaminophen exposure at the time of the MenB vaccine. However, available evidence (Table 1) makes it abundantly clear that acetaminophen use with any vaccine is exceedingly risky for the long-term well-being of the child. Fortunately, Australian and Canadian authorities have determined that the MenB vaccine can be given separate from other vaccines and without acetaminophen [52,54]. It is this approach, in addition to avoidance of other acetaminophen exposures from the start of labor and delivery until after age 5, that should be strongly recommended by medical authorities. Finally, the risk of acetaminophen exposure to the fetus during pregnancy is presently uncertain, and parents-to-be should be made aware of this fact prior to pregnancy in order to make informed decisions and have plans in place to treat fever and pain during pregnancy.
Conclusions
The call for “more research (and funds)” rather than calls for action is encouraged by perverse incentives within in the practice of science today [59]. However, the time for superficial analyses of the acetaminophen/ASD connection examining only a few lines of evidence with substantial limitations is in the past. Although no single line of evidence is conclusive, the weight of total evidence (Table 1) is overwhelming, demonstrating that acetaminophen is, in fact, a developmental neurotoxin.
The conclusions regarding the role of acetaminophen in the induction of ASD and the recommendations for pediatric practice expressed in this manuscript do not constitute an “opinion” in the classical sense of the word. The authors have reached the conclusion, without reasonable doubt, that many if not most cases of ASD are induced by exposure of susceptible individuals to acetaminophen. We have also concluded that the best explanation for all known observations is that the vast majority of ASD is induced by exposure of susceptible individuals to acetaminophen. Given these conclusions, and given the fact that acetaminophen use in the pediatric population has never been shown to have long-term benefits, the cost/benefit ratio of the drug in the pediatric population is insufficient to merit its continued use in that population. The axiom in medicine, “do no harm”, which is more reasonably stated as “do not knowingly do more harm than good”, is not a matter of question. Rather, the axiom is accepted as unquestionable and foundational, to the point of being included in an oath taken by all physicians. Thus, discontinuation of acetaminophen use from labor and delivery until the age of 5 years should be considered as a matter of course, not a matter of opinion. In contrast, use of acetaminophen during pregnancy merits additional study, and parents-to-be should be educated regarding the potential benefits and risks of acetaminophen exposure to their fetus.
Acknowledgments
The authors wish to thank Kathryn J. Reissner, R. Randy Bollinger, Doron Goldberg, Paul Corrigan, and John Poulton for invaluable insight and discussion.
References
- Zhao L, Jones J, Anderson L, Konsoula Z, Nevison C, Reissner K, et al. Acetaminophen causes neurodevelopmental injury in susceptible babies and children: no valid rationale for controversy. Clinical and experimental pediatrics. 2023. [CrossRef] [PubMed]
- Patel E, Jones Iii JP, 3rd, Bono-Lunn D, Kuchibhatla M, Palkar A, Cendejas Hernandez J, et al. The safety of pediatric use of paracetamol (acetaminophen): a narrative review of direct and indirect evidence. Minerva pediatrics. 2022, 74, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Parker W, Anderson LG, Jones JP, Anderson R, Williamson L, Bono-Lunn D, et al. The Dangers of Acetaminophen for Neurodevelopment Outweigh Scant Evidence for Long-Term Benefits. Children. 2024, 11, 44. [Google Scholar] [CrossRef]
- Jones JP, 3rd, Williamson L, Konsoula Z, Anderson R, Reissner KJ, Parker W. Evaluating the Role of Susceptibility Inducing Cofactors and of Acetaminophen in the Etiology of Autism Spectrum Disorder. Life (Basel, Switzerland). 2024, 14. [CrossRef] [PubMed]
- Graeca M, Kulesza R. Impaired brainstem auditory evoked potentials after in utero exposure to high dose paracetamol exposure. Hear Res. 2024, 454, 109149. [Google Scholar] [CrossRef] [PubMed]
- Hinz B, Cheremina O, Brune K. Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J. 2008, 22, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Hogestatt ED, Jonsson BA, Ermund A, Andersson DA, Bjork H, Alexander JP, et al. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005, 280, 31405–31412. [Google Scholar] [CrossRef] [PubMed]
- Hirai T, Umeda N, Harada T, Okumura A, Nakayasu C, Ohto-Nakanishi T, et al. Arachidonic acid-derived dihydroxy fatty acids in neonatal cord blood relate symptoms of autism spectrum disorders and social adaptive functioning: Hamamatsu Birth Cohort for Mothers and Children (HBC Study). Psychiatry and clinical neurosciences. 2024, 78, 546–557. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schultz ST, Klonoff-Cohen HS, Wingard DL, Akshoomoff NA, Macera CA, Ji M. Acetaminophen (paracetamol) use, measles-mumps-rubella vaccination, and autistic disorder. The results of a parent survey. Autism. 2008, 12, 293–307. [Google Scholar] [CrossRef]
- Cendejas-Hernandez J, Sarafian J, Lawton V, Palkar A, Anderson L, Lariviere V, et al. Paracetamol (Acetaminophen) Use in Infants and Children was Never Shown to be Safe for Neurodevelopment: A Systematic Review with Citation Tracking. . Eur J Pediatr. 2022, 181, 1835–1857. [Google Scholar] [CrossRef]
- Ertmann RK, Møller JJ, Waldorff FB, Siersma V, Reventlow S, Söderström M. The majority of sick children receive paracetamol during the winter. Danish medical journal. 2012, 59, A4555. [Google Scholar] [PubMed]
- Bornehag CG, Reichenberg A, Hallerback MU, Wikstrom S, Koch HM, Jonsson BA, et al. Prenatal exposure to acetaminophen and children's language development at 30 months. Eur Psychiatry. 2018, 51, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Parker W, Hornik CD, Bilbo S, Holzknecht ZE, Gentry L, Rao R, et al. The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. J Int Med Res. 2017, 45, 407–438. [Google Scholar] [CrossRef] [PubMed]
- Ji Y, Azuine RE, Zhang Y, Hou W, Hong X, Wang G, et al. Association of Cord Plasma Biomarkers of In Utero Acetaminophen Exposure With Risk of Attention-Deficit/Hyperactivity Disorder and Autism Spectrum Disorder in Childhood. JAMA Psychiatry. 2020, 77, 180–189. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Baker BH, Lugo-Candelas C, Wu H, Laue HE, Boivin A, Gillet V, et al. Association of Prenatal Acetaminophen Exposure Measured in Meconium With Risk of Attention-Deficit/Hyperactivity Disorder Mediated by Frontoparietal Network Brain Connectivity. JAMA pediatrics. 2020, 174, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahlqvist VH, Sjöqvist H, Dalman C, Karlsson H, Stephansson O, Johansson S, et al. Acetaminophen Use During Pregnancy and Children's Risk of Autism, ADHD, and Intellectual Disability. Jama. 2024, 331, 1205–1214. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lexchin J, Bero LA, Djulbegovic B, Clark O. Pharmaceutical industry sponsorship and research outcome and quality: systematic review. BMJ. 2003, 326, 1167–1170. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lundh A, Lexchin J, Mintzes B, Schroll JB, Bero L. Industry sponsorship and research outcome. Cochrane Database Syst Rev. 2017, 2, Mr000033 Epub 2017/02/17. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bero L, Oostvogel F, Bacchetti P, Lee K. Factors associated with findings of published trials of drug-drug comparisons: why some statins appear more efficacious than others. PLoS Med. 2007, 4, e184. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bero, LA. Tobacco industry manipulation of research. Public health reports (Washington, DC : 1974). 2005, 120, 200–208. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fabbri A, Holland TJ, Bero LA. Food industry sponsorship of academic research: investigating commercial bias in the research agenda. Public health nutrition. 2018, 21, 3422–3430. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Whitstock, M. Manufacturing the truth: From designing clinical trials to publishing trial data. Indian journal of medical ethics. 2018, 3, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Gupta R, Chernesky J, Lembke A, Michaels D, Tomori C, Greene JA, et al. The opioid industry's use of scientific evidence to advance claims about prescription opioid safety and effectiveness. Health affairs scholar. 2024, 2, qxae119 Epub 2024/10/25. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Legg T, Hatchard J, Gilmore AB. The Science for Profit Model-How and why corporations influence science and the use of science in policy and practice. PLoS One. 2021, 16, e0253272. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tovo-Rodrigues L, Schneider BC, Martins-Silva T, Del-Ponte B, Loret de Mola C, Schuler-Faccini L, et al. Is intrauterine exposure to acetaminophen associated with emotional and hyperactivity problems during childhood? Findings from the 2004 Pelotas birth cohort. BMC Psychiatry. 2018, 18, 368. [Google Scholar] [CrossRef]
- Vlenterie R, Wood ME, Brandlistuen RE, Roeleveld N, van Gelder MM, Nordeng H. Neurodevelopmental problems at 18 months among children exposed to paracetamol in utero: a propensity score matched cohort study. Int J Epidemiol. 2016, 45, 1998–2008. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liew Z, Ritz B, Virk J, Arah OA, Olsen J. Prenatal Use of Acetaminophen and Child IQ: A Danish Cohort Study. Epidemiology. 2016, 27, 912–918. [Google Scholar] [CrossRef] [PubMed]
- Liew Z, Bach CC, Asarnow RF, Ritz B, Olsen J. Paracetamol use during pregnancy and attention and executive function in offspring at age 5 years. Int J Epidemiol. 2016, 45, 2009–2017. [Google Scholar] [CrossRef] [PubMed]
- Avella-Garcia CB, Julvez J, Fortuny J, Rebordosa C, Garcia-Esteban R, Galan IR, et al. Acetaminophen use in pregnancy and neurodevelopment: attention function and autism spectrum symptoms. Int J Epidemiol. 2016, 45, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Alemany S, Avella-García C, Liew Z, García-Esteban R, Inoue K, Cadman T, et al. Prenatal and postnatal exposure to acetaminophen in relation to autism spectrum and attention-deficit and hyperactivity symptoms in childhood: Meta-analysis in six European population-based cohorts. Eur J Epidemiol. 2021, 36, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Skovlund E, Handal M, Selmer R, Brandlistuen RE, Skurtveit S. Language competence and communication skills in 3-year-old children after prenatal exposure to analgesic opioids. Pharmacoepidemiol Drug Saf. 2017, 26, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Liew Z, Ritz B, Rebordosa C, Lee PC, Olsen J. Acetaminophen use during pregnancy, behavioral problems, and hyperkinetic disorders. JAMA pediatrics. 2014, 168, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Liew Z, Ritz B, Virk J, Olsen J. Maternal use of acetaminophen during pregnancy and risk of autism spectrum disorders in childhood: A Danish national birth cohort study. Autism research : official journal of the International Society for Autism Research. 2016, 9, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Ystrom E, Gustavson K, Brandlistuen RE, Knudsen GP, Magnus P, Susser E, et al. Prenatal Exposure to Acetaminophen and Risk of ADHD. Pediatrics. 2017. [CrossRef] [PubMed]
- Thompson JM, Waldie KE, Wall CR, Murphy R, Mitchell EA. Associations between acetaminophen use during pregnancy and ADHD symptoms measured at ages 7 and 11 years. PLoS One. 2014, 9, e108210. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stergiakouli E, Thapar A, Davey Smith G. Association of Acetaminophen Use During Pregnancy With Behavioral Problems in Childhood: Evidence Against Confounding. JAMA pediatrics. 2016, 170, 964–970. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brandlistuen RE, Ystrom E, Nulman I, Koren G, Nordeng H. Prenatal paracetamol exposure and child neurodevelopment: a sibling-controlled cohort study. Int J Epidemiol. 2013, 42, 1702–1713. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Masarwa R, Levine H, Gorelik E, Reif S, Perlman A, Matok I. Prenatal Exposure to Acetaminophen and Risk for Attention Deficit Hyperactivity Disorder and Autistic Spectrum Disorder: A Systematic Review, Meta-Analysis, and Meta-Regression Analysis of Cohort Studies. Am J Epidemiol. 2018, 187, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Khan FY, Kabiraj G, Ahmed MA, Adam M, Mannuru SP, Ramesh V, et al. A Systematic Review of the Link Between Autism Spectrum Disorder and Acetaminophen: A Mystery to Resolve. Cureus. 2022, 14, e26995. [Google Scholar] [CrossRef]
- Viberg H, Eriksson P, Gordh T, Fredriksson A. Paracetamol (Acetaminophen) Administration During Neonatal Brain Development Affects Cognitive Function and Alters Its Analgesic and Anxiolytic Response in Adult Male Mice. Toxicol Sci. 2013, 138, 139–147. [Google Scholar] [CrossRef]
- Frisch M, Simonsen J. Ritual circumcision and risk of autism spectrum disorder in 0- to 9-year-old boys: national cohort study in Denmark. J R Soc Med. 2015, 108, 266–279. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bauer A, Kriebel D. Prenatal and perinatal analgesic exposure and autism: an ecological link. Environmental Health. 2013, 12, 41. [Google Scholar] [CrossRef]
- Morris BJ, Krieger JN, Klausner JD. CDC's Male Circumcision Recommendations Represent a Key Public Health Measure. Global health, science and practice. 2017, 5, 15–27. [CrossRef] [PubMed] [PubMed Central]
- Sneppen I, Thorup J. Foreskin Morbidity in Uncircumcised Males. Pediatrics. 2016; 137. [CrossRef] [PubMed]
- Rumack BH. Aspirin versus acetaminophen: a comparative view. Pediatrics. 1978, 62(5 Pt 2 Suppl):943-6. [PubMed]
- Chen L, Zhang M, Yung J, Chen J, McNair C, Lee KS. Safety of Rectal Administration of Acetaminophen in Neonates. The Canadian journal of hospital pharmacy. 2018, 71, 364–369. [Google Scholar] [PubMed] [PubMed Central]
- Anderson BJ, van Lingen RA, Hansen TG, Lin YC, Holford NH. Acetaminophen developmental pharmacokinetics in premature neonates and infants: a pooled population analysis. Anesthesiology. 2002, 96, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Birmingham PK, Tobin MJ, Fisher DM, Henthorn TK, Hall SC, Coté CJ. Initial and subsequent dosing of rectal acetaminophen in children: a 24-hour pharmacokinetic study of new dose recommendations. Anesthesiology. 2001, 94, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Montgomery CJ, McCormack JP, Reichert CC, Marsland CP. Plasma concentrations after high-dose (45 mg.kg-1) rectal acetaminophen in children. Canadian journal of anaesthesia = Journal canadien d'anesthesie. 1995, 42, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Coulthard KP, Nielson HW, Schroder M, Covino A, Matthews NT, Murray RS, et al. Relative bioavailability and plasma paracetamol profiles of Panadol suppositories in children. J Paediatr Child Health. 1998, 34, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Paracetamol use with Bexsero New Zealand: Immunisation Advisory Centre; 2018 [updated 2023, cited 2024 December 21]. Available from: https://www.immune.org.nz/factsheets/paracetamol-use-with-bexsero-r.
- Meningococcal disease. In: Care HaA, editor.: Australian Government; 2024.
- Using paracetamol to prevent and treat fever after MenB vaccination. In: Agency UHS, editor. UK: National Health Service; 2022.
- Meningococcal vaccines: Canadian Immunization Guide. In: Canada PHAo, editor.: Government of Canada; 2024.
- Vaccines for Children Israel: Ministry of Health; 2021 [cited 2024 21 December]. Available from: https://www.gov.il/en/pages/vaccines-for-children-pamphlet.
- Significant events in meningococcal vaccination practice in Australia Australia: National Centre for Immunisation Research and Surveillance; 2020 [cited 2024 21 December]. Available from: https://www.ncirs.org.au/sites/default/files/2020-07/Meningococcal-history-July%202020.pdf.
- Autism in Australia, 2022.: Australian Bureau of Statistics; 2024. Available from: https://www.abs.gov.au/articles/autism-australia-2022.
- Dinstein I, Solomon S, Zats M, Shusel R, Lottner R, Gershon BB, et al. Large increase in ASD prevalence in Israel between 2017 and 2021. Autism research : official journal of the International Society for Autism Research. 2024, 17, 410–418. [Google Scholar] [CrossRef] [PubMed]
- O'Ryan ML, Saxena S, Baum F. Time for a revolution in academic medicine? BMJ. 2024, 387, q2508. [CrossRef] [PubMed]
- Alberti A, Pirrone P, Elia M, Waring RH, Romano C. Sulphation deficit in "low-functioning" autistic children: a pilot study. Biol Psychiatry. 1999, 46, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Geier DA, Kern JK, Garver CR, Adams JB, Audhya T, Geier MR. A prospective study of transsulfuration biomarkers in autistic disorders. Neurochem Res. 2009, 34, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Pagan C, Benabou M, Leblond C, Cliquet F, Mathieu A, Lemière N, et al. Decreased phenol sulfotransferase activities associated with hyperserotonemia in autism spectrum disorders. Translational Psychiatry. 2021, 11, 23. [Google Scholar] [CrossRef]
- Brzezinski MR, Boutelet-Bochan H, Person RE, Fantel AG, Juchau MR. Catalytic activity and quantitation of cytochrome P-450 2E1 in prenatal human brain. J Pharmacol Exp Ther. 1999, 289, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Santos JX, Rasga C, Marques AR, Martiniano H, Asif M, Vilela J, et al. A Role for Gene-Environment Interactions in Autism Spectrum Disorder Is Supported by Variants in Genes Regulating the Effects of Exposure to Xenobiotics. Frontiers in neuroscience. 2022, 16, 862315. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Braam W, Keijzer H, Struijker Boudier H, Didden R, Smits M, Curfs L. CYP1A2 polymorphisms in slow melatonin metabolisers: a possible relationship with autism spectrum disorder? J Intellect Disabil Res. 2013, 57, 993–1000. [CrossRef] [PubMed]
- Kinoshita M, Stempel KS, Borges do Nascimento IJ, Bruschettini M. Systemic opioids versus other analgesics and sedatives for postoperative pain in neonates. Cochrane Database Syst Rev. 2023, 3, Cd014876. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Suda N, Hernandez JC, Poulton J, Jones JP, Konsoula Z, Smith C, et al. Therapeutic doses of paracetamol with co-administration of cysteine and mannitol during early development result in long term behavioral changes in laboratory rats. PLoS One. 2020, 16, e0253543. [Google Scholar] [CrossRef]
- Philippot G, Gordh T, Fredriksson A, Viberg H. Adult neurobehavioral alterations in male and female mice following developmental exposure to paracetamol (acetaminophen): characterization of a critical period. J Appl Toxicol. 2017, 37, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Dean SL, Knutson JF, Krebs-Kraft DL, McCarthy MM. Prostaglandin E2 is an endogenous modulator of cerebellar development and complex behavior during a sensitive postnatal period. Eur J Neurosci. 2012, 35, 1218–1229. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Philippot G, Hosseini K, Yakub A, Mhajar Y, Hamid M, Buratovic S, et al. Paracetamol (Acetaminophen) and its Effect on the Developing Mouse Brain. Frontiers in toxicology. 2022, 4, 867748. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Klein RM, Rigobello C, Vidigal CB, Moura KF, Barbosa DS, Gerardin DCC, et al. Gestational exposure to paracetamol in rats induces neurofunctional alterations in the progeny. Neurotoxicol Teratol. 2020, 77, 106838. [Google Scholar] [CrossRef] [PubMed]
- Baker BH, Rafikian EE, Hamblin PB, Strait MD, Yang M, Pearson BL. Sex-specific neurobehavioral and prefrontal cortex gene expression alterations following developmental acetaminophen exposure in mice. Neurobiol Dis. 2023, 177, 105970. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blecharz-Klin K, Wawer A, Jawna-Zboińska K, Pyrzanowska J, Piechal A, Mirowska-Guzel D, et al. Early paracetamol exposure decreases brain-derived neurotrophic factor (BDNF) in striatum and affects social behaviour and exploration in rats. Pharmacol Biochem Behav. 2018, 168, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kanno SI, Tomizawa A, Yomogida S, Hara A. Glutathione peroxidase 3 is a protective factor against acetaminophen-induced hepatotoxicity in vivo and in vitro. Int J Mol Med. 2017, 40, 748–754. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Posadas I, Santos P, Blanco A, Muñoz-Fernández M, Ceña V. Acetaminophen induces apoptosis in rat cortical neurons. PLoS One. 2010, 5, e15360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Donovan AP, Basson MA. The neuroanatomy of autism - a developmental perspective. J Anat. 2017, 230, 4–15. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Casanova MF, Sokhadze EM, Casanova EL, Opris I, Abujadi C, Marcolin MA, et al. Translational Neuroscience in Autism: From Neuropathology to Transcranial Magnetic Stimulation Therapies. The Psychiatric clinics of North America. 2020, 43, 229–248. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dong D, Zielke HR, Yeh D, Yang P. Cellular stress and apoptosis contribute to the pathogenesis of autism spectrum disorder. Autism research : official journal of the International Society for Autism Research. 2018, 11, 1076–1090. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lv MN, Zhang H, Shu Y, Chen S, Hu YY, Zhou M. The neonatal levels of TSB, NSE and CK-BB in autism spectrum disorder from Southern China. Translational neuroscience. 2016, 7, 6–11. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stancioiu F, Bogdan R, Dumitrescu R. Neuron-Specific Enolase (NSE) as a Biomarker for Autistic Spectrum Disease (ASD). Life (Basel, Switzerland). 2023, 13. [CrossRef] [PubMed]
- Ham A, Chang AY, Li H, Bain JM, Goldman JE, Sulzer D, et al. Impaired macroautophagy confers substantial risk for intellectual disability in children with autism spectrum disorders. Mol Psychiatry. 2024. [CrossRef] [PubMed]
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Du K, Farhood A, Jaeschke H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch Toxicol. 2017, 91, 761–773. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Frye RE, Sequeira JM, Quadros EV, James SJ, Rossignol DA. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol Psychiatry. 2013, 18, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Hutabarat RM, Unadkat JD, Kushmerick P, Aitken ML, Slattery JT, Smith AL. Disposition of drugs in cystic fibrosis. III. Acetaminophen. Clin Pharmacol Ther. 1991, 50, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Kearns, GL. Hepatic drug metabolism in cystic fibrosis: recent developments and future directions. Ann Pharmacother. 1993, 27, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Roberts ID, Krajbich I, Way BM. Acetaminophen influences social and economic trust. Scientific Reports. 2019, 9, 4060. [Google Scholar] [CrossRef] [PubMed]
- Dewall CN, Macdonald G, Webster GD, Masten CL, Baumeister RF, Powell C, et al. Acetaminophen reduces social pain: behavioral and neural evidence. Psychol Sci. 2010, 21, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Durso GRO, Luttrell A, Way BM. Over-the-Counter Relief From Pains and Pleasures Alike: Acetaminophen Blunts Evaluation Sensitivity to Both Negative and Positive Stimuli. Psychol Sci. 2015, 26, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Randles D, Kam JWY, Heine SJ, Inzlicht M, Handy TC. Acetaminophen attenuates error evaluation in cortex. Social Cognitive and Affective Neuroscience. 2016, 11, 899–906. [Google Scholar] [CrossRef]
- Donohue, J. A history of drug advertising: the evolving roles of consumers and consumer protection. Milbank Q. 2006, 84, 659–699. [Google Scholar] [CrossRef] [PubMed]
- Rimland, B. The autism increase: research needed on the vaccine connection. Autism Research Review International. 2000.
- Howard CR, Howard FM, Weitzman ML. Acetaminophen analgesia in neonatal circumcision: the effect on pain. Pediatrics. 1994, 93, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Kim YS, Leventhal BL, Koh YJ, Fombonne E, Laska E, Lim EC, et al. Prevalence of autism spectrum disorders in a total population sample. Am J Psychiatry. 2011, 168, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Baird, G. 2.64% of South Korean children aged 7 to 12 have autism spectrum disorders. Evidence Based Mental Health. 2012, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Hall C, Smith M. Increased cGMP enforcement has gone international: South Korean action against Johnson & Johnson serves as warning. White Collar Watch. 2013.
- Green MD, Shires TK, Fischer LJ. Hepatotoxicity of acetaminophen in neonatal and young rats. I. Age-related changes in susceptibility. Toxicol Appl Pharmacol. 1984, 74, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Raz R, Weisskopf MG, Davidovitch M, Pinto O, Levine H. Differences in autism spectrum disorders incidence by sub-populations in Israel 1992-2009: a total population study. J Autism Dev Disord. 2015, 45, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Kacker S, Tobian AA. Male circumcision: integrating tradition and medical evidence. The Israel Medical Association journal : IMAJ. 2013, 15, 37–38. [Google Scholar] [PubMed] [PubMed Central]
- Anvik, JO. Acetaminophen toxicosis in a cat. The Canadian veterinary journal = La revue veterinaire canadienne. 1984, 25, 445–447. [Google Scholar] [PubMed] [PubMed Central]
- Savides MC, Oehme FW, Nash SL, Leipold HW. The toxicity and biotransformation of single doses of acetaminophen in dogs and cats. Toxicol Appl Pharmacol. 1984, 74, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Court, MH. Feline drug metabolism and disposition: pharmacokinetic evidence for species differences and molecular mechanisms. The Veterinary clinics of North America Small animal practice. 2013, 43, 1039–1054. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lautz LS, Jeddi MZ, Girolami F, Nebbia C, Dorne J. Metabolism and pharmacokinetics of pharmaceuticals in cats (Felix sylvestris catus) and implications for the risk assessment of feed additives and contaminants. Toxicology letters. 2021, 338, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Miller RP, Roberts RJ, Fischer LJ. Acetaminophen elimination kinetics in neonates, children, and adults. Clin Pharmacol Ther. 1976, 19, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Cook SF, Stockmann C, Samiee-Zafarghandy S, King AD, Deutsch N, Williams EF, et al. Neonatal Maturation of Paracetamol (Acetaminophen) Glucuronidation, Sulfation, and Oxidation Based on a Parent-Metabolite Population Pharmacokinetic Model. Clin Pharmacokinet. 2016, 55, 1395–1411. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Freed GL, Clark SJ, Butchart AT, Singer DC, Davis MM. Parental vaccine safety concerns in 2009. Pediatrics. 2010, 125, 654–659. [CrossRef] [PubMed]
- Bazzano A, Zeldin A, Schuster E, Barrett C, Lehrer D. Vaccine-related beliefs and practices of parents of children with autism spectrum disorders. American journal on intellectual and developmental disabilities. 2012, 117, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Wakefield AJ, Murch SH, Anthony A, Linnell J, Casson DM, Malik M, et al. Ileal-lymphoid-nodular hyperplasia, non-specific colitis, and pervasive developmental disorder in children. Lancet. 1998, 351, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S. Understanding Autism: My Quest for Nathan: Schultz Publishing LLC; 2013. 92 p.
- Aguiar AG, Mainegra FD, García RO, Hernandez FY. Diagnosis in children with autism spectrum disorders in their development in textual comprehension. Rev Medical Sciences. 2016, 20, 729–737. [Google Scholar]
- Kosteva, D. A Peek at Pharmacy Practice in Cuba: Pharmacy Times; 2016. Available from: https://www.pharmacytimes.com/view/a-peek-at-pharmacy-practice-in-cuba.
- Yeldham C. Going to Cuba? Here’s What Else To Pack: STARTUP CUBA.TV; 2021 [cited 2024 December 18]. Available from: https://startupcuba.tv/2021/11/10/going-to-cuba-heres-what-else-to-pack/.
- Sainsbury B. 20 things to know before visiting Cuba: Lonely Planet; 2024 [cited 2024 December 18]. Available from: https://www.lonelyplanet.com/articles/things-to-know-before-traveling-to-cuba.
- Preparing for Cuba: The Ultimate Packing List For Health & Safety: Simply Cuba Tours; 2023 [cited 2024 December 18]. Available from: https://simplycubatours.com/preparing-for-cuba-the-ultimate-packing-list-for-health-safety/.
- How to Support Local Cubans When You Travel to Cuba: Locally Sourced Cuba Tours; [cited 2024 December 18]. Available from: https://locallysourcedcuba.com/how-to-support-locals-in-cuba/.
- Maher, B. Personal genomes: The case of the missing heritability. Nature. 2008, 456, 18–21. [Google Scholar] [CrossRef]
- Roberts AL, Lyall K, Rich-Edwards JW, Ascherio A, Weisskopf MG. Maternal exposure to childhood abuse is associated with elevated risk of autism. JAMA psychiatry. 2013, 70, 508–515. [Google Scholar] [CrossRef]
Figure 1.
Hazards ratios calculated by Ahlqvist et al [16] depending on the amount of acetaminophen (APAP) exposure and the number of factors adjusted for in the analysis. Model 1 adjusted only for time of birth and for sex. Model 2 adjusted for the same factors as Model 1, plus 7 factors associated with inflammation and oxidative stress. Model 3 adjusted for the same factors as Model 2, plus 19 additional factors, including at least a dozen factors associated with inflammation and oxidative stress. Model 4 adjusted for the same factors as Model 3, plus “unobserved genetic and environmental confounders shared by full siblings”, many of which may be related to inflammation and oxidative stress. These results demonstrate that acetaminophen is not sufficient to induce ASD by itself, a fact that has been known for some years [13]. The results are consistent with the possibility that acetaminophen interacts with a variety of inflammatory-related factors to induce ASD, a possibility confirmed by a variety of other observations (Table 1). As demonstrated previously [4], the conclusion reached by the original study team, that there is no real association between acetaminophen and ASD [16], cannot be drawn from these results. Error bars indicate 95 % confidence intervals reported by Ahlqvist and colleagues [16].
Figure 1.
Hazards ratios calculated by Ahlqvist et al [16] depending on the amount of acetaminophen (APAP) exposure and the number of factors adjusted for in the analysis. Model 1 adjusted only for time of birth and for sex. Model 2 adjusted for the same factors as Model 1, plus 7 factors associated with inflammation and oxidative stress. Model 3 adjusted for the same factors as Model 2, plus 19 additional factors, including at least a dozen factors associated with inflammation and oxidative stress. Model 4 adjusted for the same factors as Model 3, plus “unobserved genetic and environmental confounders shared by full siblings”, many of which may be related to inflammation and oxidative stress. These results demonstrate that acetaminophen is not sufficient to induce ASD by itself, a fact that has been known for some years [13]. The results are consistent with the possibility that acetaminophen interacts with a variety of inflammatory-related factors to induce ASD, a possibility confirmed by a variety of other observations (Table 1). As demonstrated previously [4], the conclusion reached by the original study team, that there is no real association between acetaminophen and ASD [16], cannot be drawn from these results. Error bars indicate 95 % confidence intervals reported by Ahlqvist and colleagues [16].
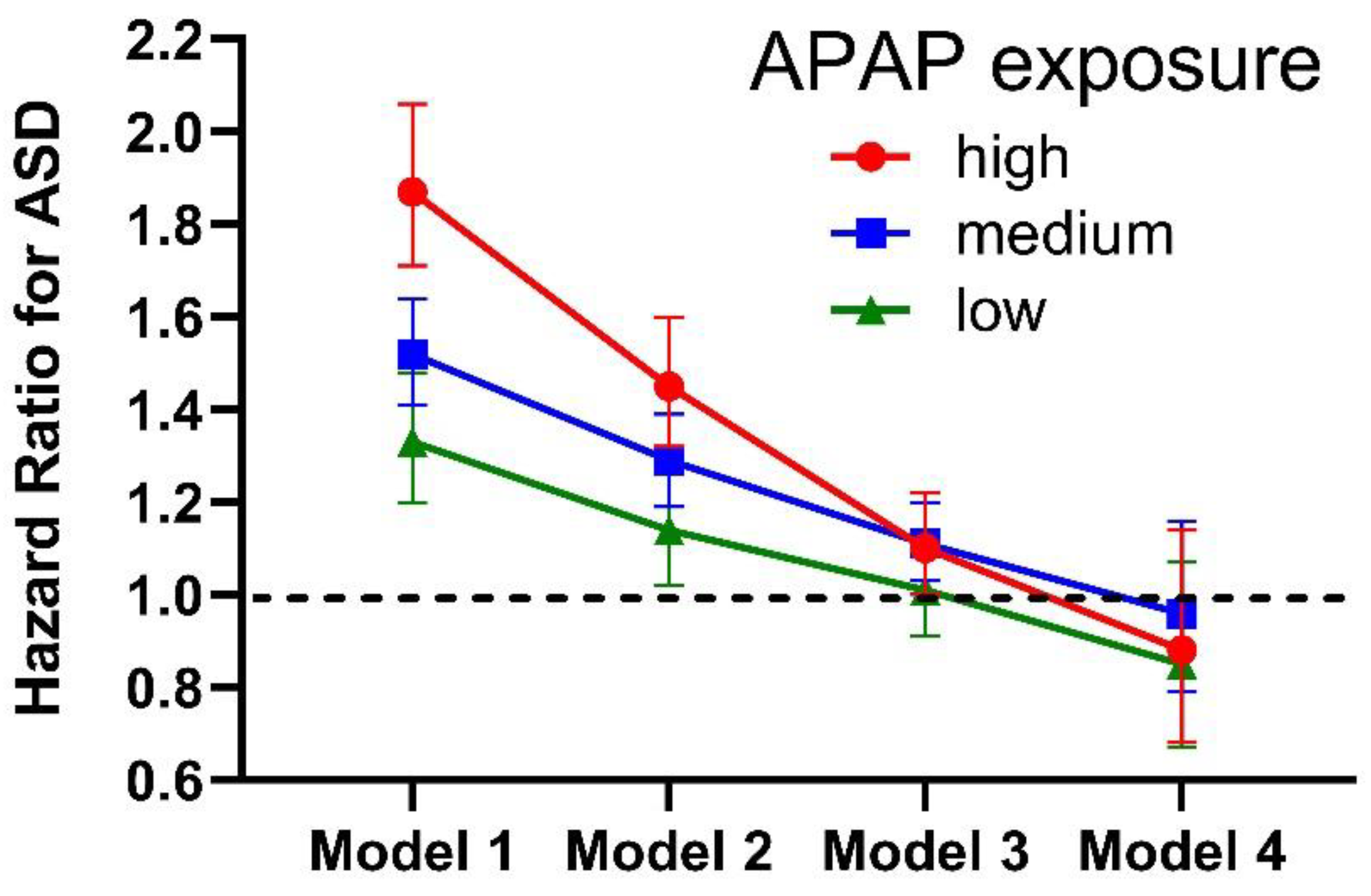
Figure 2.
Potential factors affecting continued use of acetaminophen before the age of 6 years despite overwhelming evidence that the drug is a developmental neurotoxin involved in the etiology of both infantile and regressive ASD. The list is not exhaustive. None of the factors affecting continued use of acetaminophen are evidence-based, but rather are aspects of human nature or results of aspects of human nature manifested as a result of the current environment.
Figure 2.
Potential factors affecting continued use of acetaminophen before the age of 6 years despite overwhelming evidence that the drug is a developmental neurotoxin involved in the etiology of both infantile and regressive ASD. The list is not exhaustive. None of the factors affecting continued use of acetaminophen are evidence-based, but rather are aspects of human nature or results of aspects of human nature manifested as a result of the current environment.
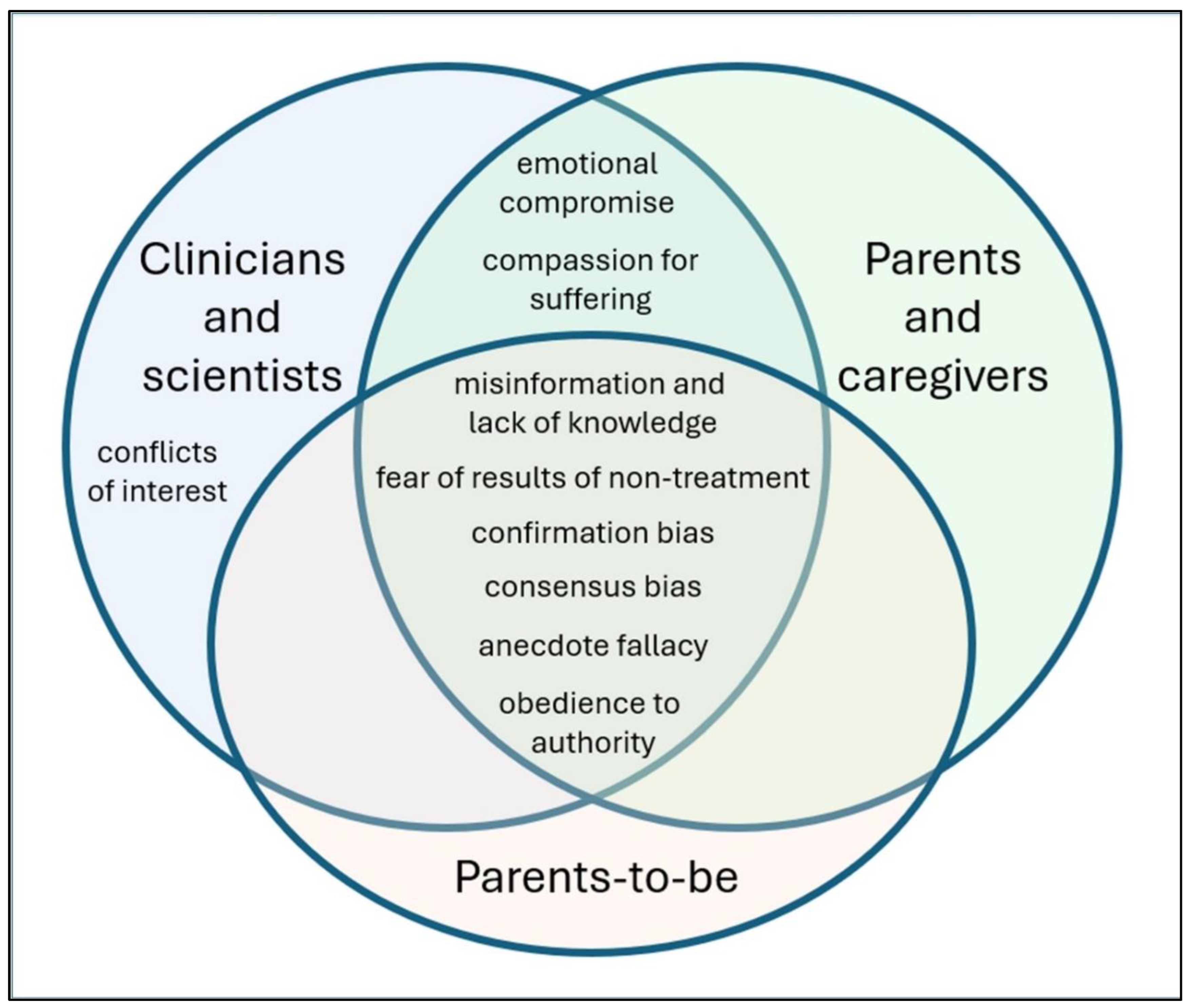

Table 1.
Lines of evidence leading to the conclusion, without reasonable doubt, that exposure of susceptible babies and children to acetaminophen (N-acetyl-p-aminophenol; APAP) leads to many if not most cases of ASD. Lines of evidence are independent, except for two lines of evidence (#2 and #3), which are dependent on the same study. Division of evidence into discrete “lines”, particularly the evidence from laboratory animal studies, is somewhat subjective. For example, studies examining the effect of APAP on learning in mice are lumped together with studies of the effect of APAP on social behavior in rats (line of evidence #5), and the numerous similarities between fetal alcohol spectrum disorder and ASD are lumped together as one line of evidence (#27).
Table 1.
Lines of evidence leading to the conclusion, without reasonable doubt, that exposure of susceptible babies and children to acetaminophen (N-acetyl-p-aminophenol; APAP) leads to many if not most cases of ASD. Lines of evidence are independent, except for two lines of evidence (#2 and #3), which are dependent on the same study. Division of evidence into discrete “lines”, particularly the evidence from laboratory animal studies, is somewhat subjective. For example, studies examining the effect of APAP on learning in mice are lumped together with studies of the effect of APAP on social behavior in rats (line of evidence #5), and the numerous similarities between fetal alcohol spectrum disorder and ASD are lumped together as one line of evidence (#27).
| Evidence/References | Background/Additional Information |
|---|---|
| 1. Mechanisms of APAP-mediated injury are plausible. For review, see Jones et al. [4] | The first study showing that children with ASD are deficient in a metabolic pathway necessary to safely detoxify APAP in babies (sulfation) is now more than a quarter of a century old [60], and was subsequently corroborated [61,62]. One enzyme (CyP450 2E1) which produces the toxic metabolite of APAP (NAPQI) is expressed in the human brain from before birth [63], and polymorphisms in another enzyme (CyP450 1A2) that produces the same toxic metabolite of APAP is associated with ASD [64,65]. |
| 2. APAP use during early childhood is associated with a 20-fold greater risk of regressive ASD [9]. | This case-controlled study, now more than 16 years old, has been widely criticized, but careful analysis does not reveal any valid objections [1]. |
| 3. APAP use with an adverse reaction to a vaccine, but not an adverse reaction to a vaccine alone, is associated with ASD [9]. | This study, the same as in line of evidence #2, was the first study to separate the impact of vaccines from APAP on neurodevelopment, and the first to directly connect APAP with ASD. |
| 4. APAP was never demonstrated to be safe for neurodevelopment [10]. Over two thousand papers in the medical literature claim that APAP is safe for babies and/or children when used as directed, but all studies were based on the false assumption that adverse reactions in babies would be the same as in adults [10]. | Like APAP, opioids have also never been shown to be safe for neurodevelopment [66]. However, unlike APAP, opioids are not generally assumed to be safe for neurodevelopment when used as directed. Further, one study probing the safety of in utero opioid exposure found reductions in communication skills in children associated with in utero APAP exposure, but not with in utero opioid exposure [31]. |
| 5. Numerous studies in laboratory animals from multiple laboratories indicate that early life exposure to APAP causes long term changes in brain function [40,67,68,69,70,71,72,73]. | After adjusting for weight, the amount of APAP that causes profound changes in laboratory animals in some studies is very close to [40] or even less than [67] the amount administered to human babies and children. Thus, APAP could never be used in babies or children if current guidelines for drug safety were applied. |
| 6. Early life exposure to APAP has a greater long-term impact on male laboratory animals than female laboratory animals [69,72,74]. ASD is more common in males than in females. | The reason or reasons why males are more susceptible to APAP-mediated injury has been considered in some detail [69,74], but the answer is not entirely known. |
| 7. In utero exposure of laboratory rats to APAP causes problems with the processing of sound [5]. Some degree of auditory dysfunction is seen in the majority of individuals with ASD. Reviewed by Graeca and Kulesza [5]. | The investigators found developmental delays with ear opening and, essentially, difficulty with hearing later in life after exposure to APAP as a fetus. It is unknown whether these affects in laboratory animals are related to impairments in some individuals with ASD. |
| 8. APAP causes apoptosis-mediated death of cortical neurons in laboratory rats [75], and cortical neurons may be involved in the pathology of ASD [76,77]. | Increased levels of biomarkers for neuronal apoptosis [78,79,80] and impaired autophagy [81] are associated with ASD. Autophagy is necessary to clearing damaged organelles such as mitochondria [82], which are created by aberrant metabolism of APAP [83]. |
| 9. Genetic, epigenetic, and environmental factors associated with an increased risk of ASD have an adverse effect on the body’s ability to safely metabolize APAP [13,60,84]. | The wide array of factors associated with ASD have led to the hypothesis that many things can come together to cause ASD, but ASD is characterized by impairment of social function and other particular phenotypes, suggesting specificity in the etiology of the condition. |
| 10. Cystic fibrosis is associated with unusually efficient (effective) metabolism of APAP [85,86], and the prevalence of ASD is apparently very low in patients with cystic fibrosis [13]. | The mental health of patients with cystic fibrosis has been characterized extensively, but no association between ASD and cystic fibrosis has been reported. |
| 11. APAP temporarily blunts social trust [87] and awareness [88], emotional responses to external stimuli [89], and the ability to identify errors [90] in adults. | Although the mechanisms are unknown, these studies show that APAP affects aspects of mental function that are impaired in individuals with ASD. |
| 12. Higher levels of APAP in cord blood are associated with ASD [14]. | For the analysis, the authors divided the women into three groups based on cord blood APAP levels. The third with the highest levels had 3.6 times more likelihood of having a child with ASD that the third with the lowest levels of APAP. |
| 13. Use of APAP during pregnancy has been associated with adverse long-term effects on the mental function of offspring in numerous studies [14,25,26,27,28,29,30,31,32,33,34,35,36,37]. | This line of evidence has received more attention than any other line of evidence, to the point of being the only line of evidence considered by many investigators. However, the numerous studies underpinning this line of evidence are hampered by several factors which can cause underestimation or overestimation of the association between APAP and ASD [4]. A recent study found a dramatic association (OR for ASD with APAP use = 1.8) [16], but incorrectly and completely cancelled out that association using an error in the assumptions underlying the statistical analysis [2,4]. |
| 14. Analysis of the Danish National Birth Cohort (DNBC) revealed an odds ratio (OR) of 1.3 (CI 1.02–1.66) for ASD associated with postnatal APAP exposure [30], despite the fact that the use of APAP appears to be dramatically underreported in the DNBC [1]. | The study authors averaged the results from the DNBC with assessments of autism-like symptoms (not ASD) from smaller data sets, and reported no association between APAP use and those symptoms (not ASD) in the abstract of the paper. This issue has been addressed in detail by us in the literature [1,2], but unfortunately may still result in confusion [5]. In addition, the study employed invalid statistical adjustments expected to underestimate the association between APAP and ASD [2,4]. See text for additional discussion. |
| 15. The incidence of ASD began to increase in the early 1980s, coinciding with the increase in APAP use after aspirin was associated with Reye’s syndrome [13] | Temporal associations do not prove causality, but are a necessary prerequisite for causality to exist. Alternative explanations for the rise in prevalence of ASD face several insurmountable problems, previously reviewed [1,4]. |
| 16. The incidence of ASD has steadily increased [13] as direct-to-consumer advertising [91] and perhaps other factors such as mandated use of APAP with the MenB vaccine (see discussion) have led to increased APAP exposure early in life. | Temporal associations do not prove causality, but are a necessary prerequisite for causality to exist. Alternative explanations for the rise in prevalence of ASD face several insurmountable problems, previously reviewed [1,4]. A likely explanation for the persistence of such alternative explanations is that most investigators are unaware of a satisfactory explanation consistent with available evidence. |
| 17. The ratio of regressive to infantile ASD rose at the same time as pediatric APAP use rose [92] after aspirin was associated with Reye’s syndrome [13]. | This observation, made in 2000, would suggest that something was introduced into the environment that could induce ASD after months or even years of neurodevelopment. This factor was tragically and incorrectly suspected to be a vaccine at that time, an issue that was decisively addressed by Stephen Schultz eight years later (see line of evidence #3). |
| 18. Circumcision of males is associated with a 2-fold increase in the risk for early-onset (infantile) ASD [41]. | Circumcision is often performed using APAP as an analgesic despite the fact that such use is of highly questionable effectiveness [93]. |
| 19. The popularity of APAP use and the prevalence of ASD was substantially higher in Denmark than in Finland in the mid-2000s [3]. | Geographic-dependent associations do not prove causality, but do provide evidence to be considered. Particularly in the absence of alternative explanations, these associations can be compelling. |
| 20. An exceptionally high prevalence of ASD was identified in South Korea [94,95] following repeated findings of levels of APAP exceeding the package label of children’s products [96]. | Repeating mistakes made when the initial determination of APAP safety for pediatric use was determined (See line of evidence #4), public health authorities assessed the prevalence of reports of liver failure in the pediatric population, and determined that no harm was caused by the excess active ingredient (APAP) in the formulation. Liver failure is the primary adverse event from APAP overdose in adults. However, a study in laboratory animals in the 1980s demonstrated that the liver is not susceptible to APAP-mediated injury in very young animals, even with lethal doses of APAP [97]. |
| 21. Ultra-Orthodox Jews [98] in Israel have a reported prevalence of ASD less than half of that of reform Jews. Traditional circumcision practices employed by Ultra-Orthodox Jews do not utilize APAP. | Circumcision is often performed using APAP as an analgesic despite the fact that such use is of highly questionable effectiveness [93]. Almost all Israeli Jews are circumcised [99]. |
| 22. APAP is not used in domestic cats because they lack of a robust glucuronidation-dependent capacity for metabolism [100,101,102,103], making them susceptible to APAP-mediated injury. Human neonates also lack a robust glucuronidation-dependent pathway [104,105]. | Based on liver function in human babies and children, APAP was incorrectly determined to be safe for pediatric use in the 1960s and 1970s (see line of evidence # 4), before this evidence from veterinary science became available in the 1980s. One study in laboratory animals in the 1980s showed that even lethal doses of APAP do not cause liver failure in neonates [97], but the first study showing APAP-mediated neurodevelopmental brain injury in laboratory animals was not published until 2013 [40]. |
| 23. Surveys show that up to 50% of parents who have a child with ASD believe that their children’s ASD was induced by a vaccine [106,107]. | Although this belief has been widely attributed to a 1998 report describing 12 patients [108], the title of that report is not intelligible to individuals outside of the medical profession, and medical papers have seldom affected public opinion. A more likely explanation involves the induction of ASD by APAP use concurrent with vaccination, as suggested by Schultz [9,109]. |
| 24. Studies in several countries with chronic shortages of medication found dramatically lower-than-expected levels of ASD relative to other developmental issues, including Down syndrome. Reviewed by Jones et al. [4] | Not included in the previous review of this issue [4] is the apparently low levels of ASD in Cuba, where 241 cases of ASD in the entire nation (1 in 25,000 children) have been identified based on a 2016 report [110]. APAP is available in Cuba by prescription only [111], and multiple travel advisors cite APAP in particular as being in short supply in Cuba [112,113,114,115]. |
| 25. APAP binds directly to arachidonic acid [7] and affects arachidonic acid metabolism [6]. Alterations of arachidonic acid are associated with ASD [8]. | It is unknown what role arachidonic acid plays in ASD, but arachidonic acid plays a role in both the analgesic and antipyretic properties of APAP, and its metabolism is associated with ASD. |
| 26. The “missing heritability” paradox of ASD suggests that epigenetic factors or very early exposure to environmental factors might influence the onset of ASD [116]. | The role of APAP in the induction of ASD nicely resolves the missing heritability paradox connected with ASD, in which sibling studies indicate a high contribution of genetics, but genome wide studies fail to identify the genes involved [116]. The observation that abuse of a mother when she was a child is associated with ASD in the offspring [117] is one example of evidence that supports this view. |
| 27. ASD and fetal alcohol spectrum disorder (FASD) are similar in many regards. Reviewed by Jones et al. [4] | This observation demonstrates that a complex spectrum disorder (FASD) sharing many similarities with ASD can (a) be induced by a single chemical and (b) be influenced by a variety of genetic and environmental factors. |
Table 2.
Consequences of assumptions underlying multivariate analysis of observational data in a virtual computer construct. Acetaminophen (APAP) is “shown” to be protective against ASD (HR < 1.0), even though it induced 50 % of all cases of ASD in the virtual construct. The virtual data set was constructed and analyzed as previously described [4] using a Cox regression analysis, with 240,000 individuals, 60 % exposure to APAP and one in 36 individuals with ASD. In this virtual data set, 50 % of ASD was induced by exposure to acetaminophen combined with the sum of 10 cofactors, modeling the contribution of oxidative stress (OS) related factors in the induction of ASD. The other 50 % of ASD cases were randomly assigned. The propensity for acetaminophen exposure was dependent on levels of oxidative stress, as previously described [4]. n = 96,000 virtual individuals without APAP use, and n = 144,000 virtual individuals with APAP use. CI = confidence interval. NA = not applicable.
Table 2.
Consequences of assumptions underlying multivariate analysis of observational data in a virtual computer construct. Acetaminophen (APAP) is “shown” to be protective against ASD (HR < 1.0), even though it induced 50 % of all cases of ASD in the virtual construct. The virtual data set was constructed and analyzed as previously described [4] using a Cox regression analysis, with 240,000 individuals, 60 % exposure to APAP and one in 36 individuals with ASD. In this virtual data set, 50 % of ASD was induced by exposure to acetaminophen combined with the sum of 10 cofactors, modeling the contribution of oxidative stress (OS) related factors in the induction of ASD. The other 50 % of ASD cases were randomly assigned. The propensity for acetaminophen exposure was dependent on levels of oxidative stress, as previously described [4]. n = 96,000 virtual individuals without APAP use, and n = 144,000 virtual individuals with APAP use. CI = confidence interval. NA = not applicable.
| Variable | HR (CI) | p |
|---|---|---|
| APAP, actual risk built into virtual construct | 2.667 (NA) | NA |
| APAP, result of regression analysis | 2.55 (2.41-2.71) | 2 x 10-16 |
| APAP, adjusted for all contributing cofactors | 0.85 (0.80-0.90) | 2 x 10-7 |
| OS factor 1, all individuals | 1.24 (1.23-1.26) | 2 x 10-16 |
| OS factor 1, virtual individuals with APAP use only | 1.32 (1.30-1.34) | 2 x 10-16 |
| OS factor 1, virtual individuals with no APAP use only | 1.00 (0.97-1.03) | 0.90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



