
Submitted:
31 December 2024
Posted:
03 January 2025
You are already at the latest version
Abstract
Rare diseases impose a significant burden on affected individuals, caregivers, and healthcare systems worldwide. Developing effective therapeutics for these small patient populations presents substantial challenges. Antisense oligonucleotides (ASOs) have emerged as a promising therapeutic approach that targets the underlying genetic cause of disease at the RNA level. Several ASOs have gained FDA approval for the treatment of genetic conditions, including use in personalized N-of-1 trials. However, despite their potential, ASOs often exhibit limited clinical efficacy, and optimizing their design is a complex process influenced by numerous factors. Machine learning-based platforms, including eSkip-Finder and ASOp-timizer, have been developed to address these challenges by predicting optimal ASO sequences and chemical modifications to enhance efficacy. eSkip-Finder focuses on exon-skipping applications, while ASOptimizer aims to optimize ASOs for RNA degradation. Preliminary in vitro results have demon-strated the promising predictive power of these platforms. However, limitations remain, including their generalizability to alternative targets and gaps in their consideration of all factors influencing ASO efficacy and safety. Continued advancements in machine learning models, alongside efforts to incorporate ad-ditional features affecting ASO efficacy and safety, hold significant promise for the field. These platforms have the potential to streamline ASO development, reduce associated costs, and improve clinical out-comes, positioning machine learning as a key tool in the future of ASO therapeutics.
Keywords:
Antisense oligonucleotides (ASOs)
; machine learning
; design
; eSkip-Finder
; ASOptimizer
; rare disease
1. Introduction
Rare diseases, commonly referred to as “orphan diseases,” are defined by a prevalence of fewer than 1 in 2,000 individuals [1]. Although each disease affects only a small number of people, there are approximately 8,000 identified rare diseases, collectively impacting an estimated 3.5-5.9% of the global population [2,3]. Around 80% of these diseases are attributed to genetic causes, many of which manifest in early stages of life and can be associated with reduced survival [4,5,6]. In the United States alone, the economic burden of rare diseases, encompassing both direct medical expenses and indirect costs, is estimated to approach $1 trillion annually [7,8]. The limited patient populations for rare diseases pose significant challenges in developing treatments and conducting meaningful clinical trials [9]. Pharmaceutical companies are often reluctant to invest in these treatments due to the low expected revenues from such a small patient group. Although legislative initiatives and government funding have enhanced efforts toward rare disease therapeutics [9,10], additional challenges remain. These include the use of suboptimal endpoints, lack of appropriate controls, and difficulties in recruiting patients for clinical trials [11,12]. Consequently, only 5% of patients with rare diseases currently have access to potentially disease-modifying treatments [13]. There is a critical need to improve processes for researching and developing therapeutics for rare diseases, accelerating their time to market while reducing associated costs.
Antisense oligonucleotides (ASOs) have emerged as promising therapeutic strategies for rare diseases due to their versatility [14]. By modulating the splicing or degradation of target RNA with high specificity, ASOs hold the potential to address a broad range of rare diseases [15]. Several ASOs have already received FDA approval for treating rare genetic conditions [16,17,18,19]. Furthermore, collaborative efforts with the FDA have enabled multiple patients to access personalized N-of-1 ASOs, which has subsequently led to the establishment of guidelines for N-of-1 ASO development [20]. Despite these advances, there is still significant room to improve the clinical efficacy of ASOs. Designing optimal ASOs is intrinsically complex, requiring careful consideration of a vast array of potential sequences and numerous factors that impact their safety and effectiveness [21]. Historically, identifying the most effective sequence and chemistry relied on labor-intensive and time-consuming experimental trials [22,23,24,25]. However, the importance of optimizing ASO design has become increasingly evident, as ASOs with optimized target sequences have demonstrated greater efficacy than many currently FDA-approved counterparts [25,26,27].
To leverage the extensive experimental data available on ASO efficacy, encompassing parameters such as target sequence and chemical modifications, machine learning-based platforms have been developed [21,28]. These platforms hold significant potential to predict the efficacy of novel ASOs, paving the way for the identification of more effective therapies [29]. This review provides a comprehensive overview of the challenges associated with ASO design and current machine learning-based platforms that have been developed in an attempt to improve ASO design.
2. Antisense Oligonucleotides (ASOs)
Initially discovered in 1978 [30], ASOs are single-stranded, RNA or DNA-like molecules that are capable of regulating gene expression through a range of mechanisms [31]. They are typically composed of between 12 to 25 nucleotides that are complimentary to a specific region of the target mRNA and/or pre-mRNA. Through Watson-Crick pairing, ASOs bind to their targets, where, with high specificity, they can regulate RNA processing and translation [15].
2.1. Chemical Modifications
Although these versatile molecules are highly specific, they face significant challenges penetrating biological membranes to reach target cells in vivo when they are unmodified [32]. In addition, the presence of the phosphodiester bond on unmodified ASOs causes it to be relatively unstable due to degradation by nucleases [33]. To improve their stability, safety, and overall efficacy, numerous chemical modifications have been identified that can be applied to some or all nucleotides, depending on the desired mechanism of the ASO. The first chemical modification utilized to improve ASO stability, known as phosphorothioates (PS) ASOs, was made by replacing the non-bridging oxygen of the phosphodiester bond with a sulfur group [34]. PS ASOs have a significantly longer half-life than unmodified ASOs due to their ability to evade degradation by nucleases [35,36]. However, the PS modifications reduce an ASOs binding affinity for its target, which can be improved by combining PS modifications with modifications to the sugar moiety [37]. ASOs commonly used possessing chemical modifications to their sugar component include locked nucleic acids (LNAs), 2’-O-methyl (2’-OME), 2’-O-methoxyethyl (2’-MOE), and 2’fluoro ASOs. LNA modifications are composed of a methylene link that creates a bridge between the 2’ oxygen and 4’ carbon of the sugar moiety, which locks the nucleotide in a conformation that substantially increases the ASOs affinity for its target RNA [38]. Alternatively, 2’-OME, 2’-MOE, and 2’fluoro ASOs are created through modifications to the 2’ position of the ribose ring [39]. These 2’ substituents promote the molecule’s stability by protecting it from nuclease degradation, as well as improving its target affinity [40,41]. 2’-OME and 2’-MOE modifications are also associated with improved safety due to limited immune activation [15]. Additional chemical modifications, including phosphorodiamidate morpholino oligonucleotides (PMOs) and peptide nucleic acids (PNAs), result in a more substantial change to the overall nucleotide chemistry. Specifically, PMOs are charge-neutral ASOs that utilize a six-membered morpholine ring, in place of the natural five-membered ribose ring, linked through phosphorodiamidate bonds [42]. PNAs are composed of an N-2-aminoethyl backbone [43]. Both PMOs and PNAs are resistant to nuclease degradation and have improved target binding affinity [44,45]. The neutral charge uniquely associated with these ASOs improves their safety, by reducing immune stimulation, and allows them to be conjugated to cell-penetrating-peptides for the potential to improve delivery [37,46].
2.2. Mechanisms of Action
ASOs are highly versatile therapeutic tools capable of employing various mechanisms to mitigate the effects of a wide range of pathogenic mutations. These mechanisms can be broadly classified into two primary categories: RNA degradation (RNase H-dependent) and steric blockage (RNase H-independent) [15]. Depending on the desired mechanism of action, ASOs can be chemically modified and designed to target specific RNA regions to achieve their intended therapeutic effects [47,48].
RNase H-dependent RNA degradation is particularly effective for addressing diseases caused by gain-of-function mutations, where reducing or eliminating the production of the mutated protein can improve the disease phenotype [49]. RNase H is an endogenous enzyme that plays an important role in cleaving RNA when it becomes hybridized to DNA [50]. ASOs, due to their DNA-like qualities, exploit this natural mechanism to target and degrade disease-causing mRNA (Figure 1). To enable RNase H-mediated degradation, ASOs must retain some unmodified regions, as full chemical modification prevents recognition by RNase H [51,52]. However, PS modifications can be applied to all nucleotides in an ASO without compromising RNase H recognition [53]. To balance the advantages of additional chemical modifications with the need for RNase H activity, a specialized ASO design known as a gapmer is used. In this configuration, the central region of the ASO remains unmodified to allow RNase H recognition, while the flanking regions incorporate alternative chemical modifications for enhanced stability and efficacy [54,55].
In cases where a disease arises from a loss-of-function mutation that disrupts the production of functional proteins, ASOs can regulate pre-mRNA splicing to compensate for these pathogenic mutations [56]. These ASOs are designed to bind to specific regions of pre-mRNA typically recognized by splicing factors. By sterically blocking the binding of splicing machinery, ASOs can modulate the inclusion or exclusion of specific exons to achieve the desired therapeutic effect [57]. ASO-mediated exon-skipping has received significant interest due to its ability to exclude cryptic exons, exons containing pathogenic mutations, exons adjacent to large out-of-frame deletions, or poison exons from the final mRNA transcript, offering a powerful approach to treating genetic diseases [58,59,60]. Additionally, ASOs can promote exon inclusion, although this strategy presents greater technical challenges, as identifying effective target regions to promote exon inclusion is more complex [61]. To avoid RNase H-mediated degradation of RNA, ASOs designed for steric blockage are typically fully chemically modified to not be detected by RNase H [62,63]. These fully modified ASOs have also proven effective in inhibiting translation by sterically blocking the interaction of translation machinery with the bound RNA, further broadening their therapeutic potential [64,65].
2.3. FDA-Approved ASOs
There are currently many ASOs that have received FDA approval, utilizing different chemical modifications and mechanisms of action to treat genetic diseases (Table 1). Fomiversen provided the first clinical proof-of-concept of the effectiveness of ASOs in patients and was approved for the treatment of cytomegalovirus (CMV) retinitis, particularly for patients with acquired immune deficiency syndrome (AIDS) [16]. It is composed of 21 nucleotides with PS backbone modifications, targeting the major immediate-early (MIE) gene of the virus to prevent the translation of critical proteins for its replication [66,67]. Through intravitreal injections, Fomiversen was found to effectively prevent the progression of CMV retinitis in affected patients, with a tolerable safety profile [68,69]. Despite its promising results in early clinical studies, the approval of highly active antiretroviral therapy (HAART) led to its FDA approval being withdrawn in 2001, as HAART was effective in curing or preventing CMVR in patients with AIDS [70,71].
In recent years, additional ASOs utilizing RNase H-dependent RNA degradation have received FDA approval for the treatment of disease. Mipomersen, approved in January 2013, is designed for homozygous familial hypercholesterolemia (HoFH), a genetic condition characterized by an impaired ability to clear low-density lipoproteins (LDL), placing patients at high risk for heart disease [17]. Mipomersen is a PS gapmer, with 2’-MOE-modified ends, targeting ApoB-100 mRNA for RNase H-mediated degradation [72]. By reducing ApoB translation, Mipomersen is effective in reducing very-low-density lipoprotein, and subsequently lowering the levels of circulating LDLs [73]. Similarly, Inotersen, also a PS gapmer with 2’-MOE-modified ends, targets transthyretin (TTR) mRNA to prevent the production of dysfunctional TTR proteins, which accumulate as deposits causing sensorimotor and autonomic neuropathy in hereditary transthyretin amyloidosis (hATTR) [74,75]. Inotersen received FDA approval in October 2018 [74]. Most recently, Tofersen, employing the same chemical modifications as Mipomersen and Inotersen, was approved in April 2023 for amyotrophic lateral sclerosis (ALS) associated with SOD1 gene mutations [76].
Alternatively, several ASOs designed to regulate RNA splicing have been FDA-approved for treating rare diseases. Among these, four ASOs with PMO chemical modifications, designed to enhance stability and safety, have been approved for Duchenne muscular dystrophy (DMD) [18]. DMD typically arises from mutations in the DMD gene that disrupt the reading frame, preventing the production of functional dystrophin [77,78]. Patients with in-frame mutations, including large deletions spanning multiple exons, often present with the milder Becker muscular dystrophy phenotype [79]. ASOs can restore the reading frame by promoting the exclusion of exons flanking the disrupted region, allowing for the production of truncated but partially functional dystrophin [80]. Since these DMD-causing frame-shift mutations occur throughout the DMD gene [81,82], each exon-skipping approach targets specific subsets of DMD patients [83]. Eteplirsen, targeting exon 51, became the first ASO approved for DMD in September 2016 [19]. This was followed by the approvals of Golodirsen and Viltolarsen, both targeting exon 53, and Casimersen, targeting exon 45 [84].
Splice-switching ASOs have also been approved for spinal muscular atrophy (SMA), a severe rare disease caused by pathogenic mutations in the SMN1 gene [85]. Unlike ASOs for DMD, which target the mutated gene, these ASOs modulate the splicing of SMN2, a pseudogene that differs from SMN1 by a single C-to-T transition in exon 7, leading to its exclusion in most SMN2 transcripts [86]. Nusinersen, approved in December 2016, promotes the inclusion of exon 7 in SMN2 transcripts, effectively compensating for the loss of SMN1 function [42]. As this therapeutic targets SMN2 pre-mRNA, its applicability is independent of the specific mutations in the SMN1 gene.
2.4. ASOs as N-of-1 Therapies
Recent advances in N-of-1 therapeutics, designed to target disease-causing mutations unique to a single patient or very small groups, have provided hope for individuals with ultra-rare genetic conditions. ASOs have emerged as a promising approach for such cases due to their high specificity, ability to address the root cause of disease, and demonstrated safety profile [15]. With their growing popularity and recent FDA approvals, the processes for preclinical ASO testing and evaluating relevant potential adverse effects are now well established [87]. As the focus on personalize medicine grows and regulations evolve to enable single-patient clinical trials, ASOs have started to become accessible to patients through N-of-1 trials (Table 2).
Milasen represents a landmark in N-of-1 therapeutics as the first ASO designed to treat a single patient [88]. Mila, a 6-year-old girl at the time of the study in 2017, was experiencing rapid health decline, including sudden blindness, ataxia, seizures, and developmental delay. Genetic testing revealed that she carried two distinct mutations in each allele of the CLN7 gene, associated with Batten disease [89]. Batten disease is associated with the accumulation of lipofuscin in the nervous system, leading to severe neurological symptoms and premature death [90]. One mutation Mila possessed had been previously reported. The other mutation was a unique SINE-VNTR-Alu (SVA) insertion in intron 6, which caused the inclusion of a cryptic intronic sequence in the final mRNA product [91], resulting in a dysfunctional protein. Milasen, a 22-nucleotide 2’-MOE-modified ASO, was designed to target the intron 6 cryptic splice-acceptor site. Preclinical in vitro and in vivo studies demonstrated that Milasen improved the ratio of wild-type CLN7 mRNA [88]. With Mila’s health beginning to deteriorate, the N-of-1 trial was approved by the FDA, following a dosing regimen like Nusinersen due to similarities in chemistry, target tissue, and mechanism of action. While Milasen significantly reduced the frequency of Mila’s seizures without adverse effects, she ultimately succumbed to Batten disease three years after treatment began. Despite this outcome, the rapid development of Milasen, completed within a year of identifying Mila’s unique variant, set a powerful precedent for the use of ASOs in personalized medicine for rare diseases [88,92].
Following the success of Milasen, the same team developed Atipeksen, another N-of-1 ASO, for a 1-year-old girl named Ipek diagnosed with severe ataxia-telangiectasia (A-T) [93]. A-T, also known as Louis Bar syndrome, is a rare autosomal recessive disease, caused by mutations in the ATM gene [94], leading to progressive neurodegeneration, immune dysfunction, and significantly shortened life expectancy [95]. Ipek was found to have compound heterozygous ATM mutations, one of which created a splice donor site in exon 53 that prevents the entire exon 53 from being included in the final transcript, resulting in a frameshift [93]. To restore normal splicing, 32 novel 2’-MOE-modified ASOs were evaluated targeting either the aberrant splice donor site or an adjacent splice silencer site. After further functional testing, a single ASO, named Atipeksen, was identified as the most effective candidate. Following animal safety studies, Atipeksen was approved for the clinical trial in Ipek and was administered intracerebroventricularly every three months for three years. By age six, Ipek exhibited only mild symptoms of A-T, with no reported adverse effects from the treatment [92].
To date, Milasen and Atipeksen are the only N-of-1 ASOs that have received official or academic publication [92]. However, reports from patient-run sites and news articles suggest additional N-of-1 ASOs are being developed to address unique mutations [92]. Since the promising preliminary findings from these N-of-1 trials, the FDA has introduced specific guidelines for N-of-1 ASO development to streamline the process for patients without existing treatment options [20]. These guidelines include requirements for a non-clinical report involving the results of short-term animal studies, a chemistry, manufacturing, control report outlining the drug’s known structure and properties, as well as a clinical considerations report [92]. By addressing the regulatory challenges inherent in personalized drug development, the FDA’s efforts are expected to pave the way for more N-of-1 ASOs in the future.
2.5. Challenges Associated with ASO Design
Despite the recent successful transitions of many ASOs from the bench to the bedside, there remains considerable room for improvement in their clinical efficacy. For instance, Eteplirsen’s effectiveness in treating DMD remains a subject of ongoing debate [19]. Patients receiving weekly 30 mg/kg doses of Eteplirsen demonstrated functional benefits, such as improved performance on a 6-minute walk test and a reduced incidence of ambulation loss [96]. However, the small sample size and reliance on historical control data significantly limit the reliability of these findings. Additionally, treated patients achieved an average of up to only 1% functional dystrophin protein restoration in muscle tissues and showed no improvement in dystrophin expression in cardiac tissues after at least 48 weeks of therapy [97,98]. Whether Eteplirsen can prevent long-term disease progression at this dose remains uncertain.
It has been well established that various factors in ASO design profoundly impact their safety and ability to achieve the intended RNA-targeting effects. These factors include RNA secondary structure, ASO binding energy, chemical modifications, conjugation to alternative molecules, and the specific tissues being targeted [99,100,101,102]. Designing effective ASOs is inherently challenging due to the vast number of potential RNA sequences that could theoretically serve the same purpose [21]. This complexity is further compounded by the integration of diverse chemical modifications, which introduce additional hurdles in the design process. Optimizing their design is further complicated when considering the incorporation of different combinations of chemical modifications, which bring additional challenges to the design process [21].
Improving ASO design remains a critical barrier to achieving more clinically effective therapies. Studies have demonstrated that optimized ASO sequences can be significantly more effective than current FDA-approved options [26,27,103]. For instance, in silico predictive screening tools have been utilized to refine ASO sequences targeting exon 51 of DMD pre-mRNA for exclusion [104]. These tools successfully identified a sequence that increased exon 51 skipping by 12-fold compared to Eteplirsen analogs in vitro. However, while numerous in silico tools have been developed to improve ASO sequence design [105,106,107], they currently lack the ability to account for the impact of chemical modifications and other critical factors on ASO efficacy [28].
3. Machine Learning-Based Platforms to Improve Antisense Oligonucleotide Design
To address the complexities and challenges of designing ASOs, machine learning-based platforms have been developed to optimize ASO design by leveraging insights from previous studies. ASOptimizer was developed for designing ASOs utilizing RNase-H dependent RNA degradation, while eSkip-Finder focuses on optimizing the design of splice-switching ASOs [21,28]. As ASOs gain popularity as a therapeutic strategy, extensive testing has been conducted on ASOs with various RNA targets, with diverse lengths, sequences, and chemical modifications, generating a wealth of experimental data [15,48]. These machine learning-based platforms capitalize on this vast and multifaceted dataset, integrating information about key features, such as sequence composition, chemical modifications, and experimental design, into comprehensive databases. By training predictive models on these data, they can either predict the efficacy of an individual ASO or rank multiple ASOs based on their predictions (Figure 2). This application of machine learning holds immense promise for accelerating ASO development while enhancing their precision and effectiveness, ultimately facilitating their transition from experimental stages to clinically viable therapies [29].
3.1. eSkip-Finder
eSkip-Finder (https://eskip-finder.org) is a web-based resource that employs machine learning algorithms to predict the efficacy of ASOs specifically designed for exon-skipping [28]. Its database was curated through the manual collection of all available data on exon-skipping ASOs from patents and published literature. To develop a comprehensive predictive model, the database includes information on targeted genes, ASO sequences, chemical modifications, experimental protocols, and observed exon-skipping efficacy. A total of 32 distinct features related to ASOs or experimental design were initially assessed for their impact on predictive accuracy. Features with the highest importance, determined through permutation importance methods [108], were incorporated into the final model [28]. Given the unique effects of different chemical modifications on ASO behavior, separate predictive models were developed for PMOs and 2’MOE ASOs.
The eSkip-Finder platform, involving a support vector regressor predictive model, was trained using data from 566 exon-skipping trials involving 298 novel ASOs targeting DMD pre-mRNA. The features selected for the final PMO model included ASO concentration, GC content of exon/intron when blocked by the ASO, predicted binding energy, and predicted accessibility scores of the 3’ end of the target [28]. Similarly, for 2’-MOE ASOs, features such as ASO concentration, ASO GC content, distance from splice acceptor site, remaining GC content of exon when blocked by the ASO, cumulative NI score [109], and predicted target accessibility were incorporated into the model. To evaluate the model’s predictive accuracy, 10% of the DMD exon-skipping values were reserved for validation. The test sets for PMO and 2’MOE ASOs yielded R2 value of 0.6 and 0.7, respectively [28]. To further assess the platform’s generalizability, eSkip-Finder predictions for three unique PMOs targeting exon 72 of COL7A1 were compared with their experimental outcomes [110]. The strong correlation between the predictions and experimental results highlight the potential of eSkip-Finder for broader applications beyond DMD exon-skipping [28].
3.2. ASOptimizer
Alternatively, ASOptimizer was developed to enhance the efficacy of ASOs designed to treat diseases via RNase H-dependent RNA degradation [21]. Similar to eSkip-Finder, ASOptimizer relies on an extensive database compiled from patents and scientific literature. This database encompasses 187,090 entries detailing ASOs targeting 67 unique mRNA targets for degradation. Recognizing the critical importance of both sequence design and chemical modifications, the ASOptimizer platform employs a two-stage optimization approach. The first stage, a sequence optimizer, utilizes a linear factor model to evaluate candidate ASOs. This model analyzes key variables influencing sequence potency, such as GC content, length, the secondary structure of target RNA, and potential off-target effects, to identify the most effective sequences. In the second stage, chemical modifications, including changes to nucleotide structure and inter-nucleotide bonds, are optimized using an edge-augmented graph transformer. This deep graph neural network identifies patterns and relationships within previously reported combinations of chemical modifications, enabling informed decisions about the optimal modifications for ASOs [111].
To determine its effectiveness in improving ASO design, ASOptimizer was applied to optimize ASOs targeting indoleamine 2,3-dioxygenase 1 (IDO1) mRNA. The IDO1 gene encodes an enzyme involved in the kynurenine pathway, which is often upregulated in cancers, enabling the tumor to evade immune detection [112]. Initially, ASOptimizer’s predictive performance was compared to Sfold, a widely used in silico ASO design tool employing statistical sampling [105]. Both tools were trained using a subset of 155 ASOs targeting IDO1 mRNA and tested on the remaining values. ASOptimizer demonstrated superior predictive accuracy, achieving a Pearson correlation of 0.66, compared to Sfold’s 0.5 [21]. Next, ASOptimizer was used to identify six optimized 19-nucleotide sequence candidates for improved IDO1 inhibition. Through in vitro testing, all 6 ASO sequences, developed with only PS backbone modifications, were successful in reducing IDO1 expression. Subsequently, the chemical modifications for these six ASOs were optimized in the second stage of the ASOptimizer. Current literature on ASOs targeting IDO1 predominantly employs LNA modifications with a PS backbone. As such, ASOptimizer introduced diverse combinations of LNA modifications at the ends of the ASO to form LNA-PS gapmers. Interestingly, several candidates lacked the typical gapmer structure, where flanked outer regions are fully modified, and instead featured a combination of modified and unmodified nucleotides at the end. When compared to their unoptimized PS-ASO counterparts, the optimized LNA-PS gapmers demonstrated improved efficacy and reduced cytotoxicity as measured by lactate dehydrogenase levels [21]. These findings validated the effectiveness of ASOptimizer’s chemical engineering stage in enhancing both potency and cellular safety profile of ASOs.
3.3. Limitations and Future Directions of Current Machine Learning-Based Platforms
Despite the promising potential of these machine learning-based platforms, they are not without limitations. Both eSkip-Finder and ASOptimizer have been validated using data from only one or two ASO targets, raising questions about the generalizability to alternative RNA targets [28,29]. Additionally, while the Pearson correlation and R2 values achieved in their test sets indicate their utility in optimizing ASO design [113], there remains considerable room for improvement. Updating and expanding the databases used to train these models could improve their predictive accuracy and robustness [28]. Additionally, more extensive validation of these platforms’ generalizability to alternative targets, as well as their efficacy in vivo, is essential to bolster confidence in their practical utility.
Significant gaps persist in the current landscape of machine learning platforms for ASO design. For instance, no platform has been developed specifically for optimizing ASOs intended for exon inclusion, likely because this mechanism has narrower applicability compared to exon-skipping and RNA degradation [114]. However, the increased complexity of exon-inclusion design highlights the potential value of such a platform to enhance sequence optimization [61]. Furthermore, while ASOptimizer effectively integrates a sequence engineering model, it does not address critical factors such as ASO delivery to target tissues in vivo [29]. As several delivery systems and conjugation with alternative molecules have resulted in promising pre-clinical results [115,116,117], these factors may become an important feature in future platforms for ASO design. Similarly, despite promising in vitro cytotoxicity data, neither platform accounts for in vivo safety, which must still be rigorously evaluated for ASOs designed using these tools [29].
Nevertheless, these platforms hold immense promise for advancing ASO design and development. Notably, eSkip-Finder has undergone enhancements to its predicative algorithm since its release. A three-way voting system combining random forest, gradient boosting, and XGBoost was shown to significantly reduce computing costs compared to the original support vector regressor model, while also improving R2 values in test sets analyzed [118]. By further expanding the underlying databases and refining their predictive algorithms, these platforms could revolutionize ASO development. They enable the selection of highly effective ASOs through computational modeling, reducing the need for costly and time-consuming in vitro and in vivo screening experiments. These advancements are anticipated to substantially improve the clinical efficacy of ASOs, particularly in time-sensitive cases such as N-of-1 trials, where rapid development is crucial [92,93]. The ability of these platforms to accelerate the design of therapeutically effective ASOs has the potential to modify disease progression more efficiently and enhance patient outcomes.
4. Conclusions
Machine learning holds immense promise in addressing the complexities of designing effective ASOs for rare diseases [29]. Existing platforms have demonstrated success in optimizing ASO sequences and chemical modifications by accounting for a wide range of critical factors [21,28]. However, significant limitations remain, including the need for further validation of their generalizability and addressing gaps in ASO design features that are not yet incorporated into these models [29]. Advancing these machine learning-based platforms to predict the most effective sequences and chemical structures will not only reduce the time and costs associated with early preclinical trials but also enhance our ability to deliver highly efficient therapies to patients more rapidly.
Author Contributions
J.L.; writing—original draft preparation, T.Y.; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. T.Y. is supported by Muscular Dystrophy Canada, the Friends of Garrett Cumming Research Fund, the HM Toupin Neurological Science Research Fund, the Canadian Institute of Health Research (CIHR), Alberta Innovates: Health Solutions (AIHS), Jesse’s Journey, the Women and Children’s Health Research Institute (WCHRI), the Heart and Stroke Foundation Canada, and the US Department of Defense. J.L. is supported by scholarships from the University of Alberta Faculty of Medicine and Dentistry, and the CIHR Strategic Master’s Award.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We thank the University of Alberta Faculty of Medicine and Dentistry, the Canadian Institute of Health Research, the Friends of Garrett Cumming Research Funds, HM Toupin Neurological Science Research Funds, Muscular Dystrophy Canada, and the Women and Children’s Health Research Institute for their support.
Conflicts of Interest
T.Y. is a co-founder and shareholder of OligomicsTx Inc., which aims to commercialize antisense technology. Both authors declare that the research was conducted in absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
References
- Aronson, J. Rare Diseases, Orphan Drugs, and Orphan Diseases. BMJ 2006, 333, 127.
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating Cumulative Point Prevalence of Rare Diseases: Analysis of the Orphanet Database. Eur. J. Hum. Genet. EJHG 2020, 28, 165–173. [CrossRef]
- Haendel, M.; Vasilevsky, N.; Unni, D.; Bologa, C.; Harris, N.; Rehm, H.; Hamosh, A.; Baynam, G.; Groza, T.; McMurry, J.; et al. How Many Rare Diseases Are There? Nat. Rev. Drug Discov. 2020, 19, 77–78. [CrossRef]
- Health, T.L.G. The Landscape for Rare Diseases in 2024. Lancet Glob. Health 2024, 12, e341. [CrossRef]
- Lee, C.E.; Singleton, K.S.; Wallin, M.; Faundez, V. Rare Genetic Diseases: Nature’s Experiments on Human Development. iScience 2020, 23, 101123. [CrossRef]
- Mazzucato, M.; Visonà Dalla Pozza, L.; Minichiello, C.; Toto, E.; Vianello, A.; Facchin, P. Estimating Mortality in Rare Diseases Using a Population-Based Registry, 2002 through 2019. Orphanet J. Rare Dis. 2023, 18, 362. [CrossRef]
- Yang, G.; Cintina, I.; Pariser, A.; Oehrlein, E.; Sullivan, J.; Kennedy, A. The National Economic Burden of Rare Disease in the United States in 2019. Orphanet J. Rare Dis. 2022, 17, 163. [CrossRef]
- Tisdale, A.; Cutillo, C.M.; Nathan, R.; Russo, P.; Laraway, B.; Haendel, M.; Nowak, D.; Hasche, C.; Chan, C.-H.; Griese, E.; et al. The IDeaS Initiative: Pilot Study to Assess the Impact of Rare Diseases on Patients and Healthcare Systems. Orphanet J. Rare Dis. 2021, 16, 429. [CrossRef]
- Fehr, A.; Prütz, F. Rare Diseases: A Challenge for Medicine and Public Health. J. Health Monit. 2023, 8, 3–6. [CrossRef]
- Ng, Q.X.; Ong, C.; Chan, K.E.; Ong, T.S.K.; Lim, I.J.X.; Tang, A.S.P.; Chan, H.W.; Koh, G.C.H. Comparative Policy Analysis of National Rare Disease Funding Policies in Australia, Singapore, South Korea, the United Kingdom and the United States: A Scoping Review. Health Econ. Rev. 2024, 14, 42. [CrossRef]
- Augustine, E.F.; Adams, H.R.; Mink, J.W. Clinical Trials in Rare Disease: Challenges and Opportunities. J. Child Neurol. 2013, 28, 1142–1150. [CrossRef]
- Griggs, R.C.; Batshaw, M.; Dunkle, M.; Gopal-Srivastava, R.; Kaye, E.; Krischer, J.; Nguyen, T.; Paulus, K.; Merkel, P.A.; Rare Diseases Clinical Research Network Clinical Research for Rare Disease: Opportunities, Challenges, and Solutions. Mol. Genet. Metab. 2009, 96, 20–26. [CrossRef]
- Fermaglich, L.J.; Miller, K.L. A Comprehensive Study of the Rare Diseases and Conditions Targeted by Orphan Drug Designations and Approvals over the Forty Years of the Orphan Drug Act. Orphanet J. Rare Dis. 2023, 18, 163. [CrossRef]
- Lauffer, M.C.; van Roon-Mom, W.; Aartsma-Rus, A. Possibilities and Limitations of Antisense Oligonucleotide Therapies for the Treatment of Monogenic Disorders. Commun. Med. 2024, 4, 1–11. [CrossRef]
- Dhuri, K.; Bechtold, C.; Quijano, E.; Pham, H.; Gupta, A.; Vikram, A.; Bahal, R. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J. Clin. Med. 2020, 9, 2004. [CrossRef]
- Perry, C.M.; Balfour, J.A. Fomivirsen. Drugs 1999, 57, 375–380; discussion 381. [CrossRef]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen Sodium: First Global Approval. Drugs 2013, 73, 487–493. [CrossRef]
- Gupta, S.; Sharma, S.N.; Kundu, J.; Pattanayak, S.; Sinha, S. Morpholino Oligonucleotide-Mediated Exon Skipping for DMD Treatment: Past Insights, Present Challenges and Future Perspectives. J. Biosci. 2023, 48, 38.
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [CrossRef]
- Research, C. for D.E. and IND Submissions for Individualized Antisense Oligonucleotide Drug Products: Administrative and Procedural Recommendations Guidance for Sponsor-Investigators Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/ind-submissions-individualized-antisense-oligonucleotide-drug-products-administrative-and-procedural (accessed on 28 December 2024).
- Hwang, G.; Kwon, M.; Seo, D.; Kim, D.H.; Lee, D.; Lee, K.; Kim, E.; Kang, M.; Ryu, J.-H. ASOptimizer: Optimizing Antisense Oligonucleotides through Deep Learning for IDO1 Gene Regulation. Mol. Ther. Nucleic Acids 2024, 35, 102186. [CrossRef]
- Yanagihara, N.; Tadakuma, H.; Ishihama, Y.; Okabe, K.; Funatsu, T. Determination of Potent Antisense Oligonucleotides In Vitro by Semiempirical Rules. J. Biosci. Bioeng. 2007, 103, 270–277. [CrossRef]
- Aupy, P.; Echevarría, L.; Relizani, K.; Zarrouki, F.; Haeberli, A.; Komisarski, M.; Tensorer, T.; Jouvion, G.; Svinartchouk, F.; Garcia, L.; et al. Identifying and Avoiding tcDNA-ASO Sequence-Specific Toxicity for the Development of DMD Exon 51 Skipping Therapy. Mol. Ther. Nucleic Acids 2020, 19, 371–383. [CrossRef]
- Jiang, Q.-S.; Wang, S.-Q. Design and Screening of Antisense Oligodeoxynucleotides against PAI-1 mRNA in Endothelial Cells in Vitro. Acta Pharmacol. Sin. 2006, 27, 1018–1023. [CrossRef]
- Kamola, P.J.; Kitson, J.D.A.; Turner, G.; Maratou, K.; Eriksson, S.; Panjwani, A.; Warnock, L.C.; Douillard Guilloux, G.A.; Moores, K.; Koppe, E.L.; et al. In Silico and in Vitro Evaluation of Exonic and Intronic Off-Target Effects Form a Critical Element of Therapeutic ASO Gapmer Optimization. Nucleic Acids Res. 2015, 43, 8638–8650. [CrossRef]
- Lee, J.J.A.; Maruyama, R.; Duddy, W.; Sakurai, H.; Yokota, T. Identification of Novel Antisense-Mediated Exon Skipping Targets in DYSF for Therapeutic Treatment of Dysferlinopathy. Mol. Ther. Nucleic Acids 2018, 13, 596–604. [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Trieu, N.; Bao, B.; Miskew Nichols, B.; Vila, M.C.; Novak, J.S.; Hara, Y.; Lee, J.; Touznik, A.; et al. Quantitative Antisense Screening and Optimization for Exon 51 Skipping in Duchenne Muscular Dystrophy. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2561–2572. [CrossRef]
- Chiba, S.; Lim, K.R.Q.; Sheri, N.; Anwar, S.; Erkut, E.; Shah, M.N.A.; Aslesh, T.; Woo, S.; Sheikh, O.; Maruyama, R.; et al. eSkip-Finder: A Machine Learning-Based Web Application and Database to Identify the Optimal Sequences of Antisense Oligonucleotides for Exon Skipping. Nucleic Acids Res. 2021, 49, W193–W198. [CrossRef]
- Lin, S.; Hong, L.; Wei, D.-Q.; Xiong, Y. Deep Learning Facilitates Efficient Optimization of Antisense Oligonucleotide Drugs. Mol. Ther. Nucleic Acids 2024, 35, 102208. [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous Sarcoma Virus Replication and Cell Transformation by a Specific Oligodeoxynucleotide. Proc. Natl. Acad. Sci. U. S. A. 1978, 75, 280–284. [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [CrossRef]
- Dowdy, S.F. Overcoming Cellular Barriers for RNA Therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [CrossRef]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.-D. The Powerful World of Antisense Oligonucleotides: From Bench to Bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [CrossRef]
- Eckstein, F. Phosphorothioates, Essential Components of Therapeutic Oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [CrossRef]
- Geary, R.S.; Henry, S.P.; Grillone, L.R. Fomivirsen: Clinical Pharmacology and Potential Drug Interactions. Clin. Pharmacokinet. 2002, 41, 255–260. [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [CrossRef]
- Qassem, S.; Breier, D.; Naidu, G.S.; Hazan-Halevy, I.; Peer, D. Unlocking the Therapeutic Potential of Locked Nucleic Acids through Lipid Nanoparticle Delivery. Mol. Ther. Nucleic Acids 2024, 35, 102224. [CrossRef]
- Juliano, R.L. The Delivery of Therapeutic Oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [CrossRef]
- Manoharan, M. 2’-Carbohydrate Modifications in Antisense Oligonucleotide Therapy: Importance of Conformation, Configuration and Conjugation. Biochim. Biophys. Acta 1999, 1489, 117–130. [CrossRef]
- Hebb, M.O.; Robertson, H.A. End-Capped Antisense Oligodeoxynucleotides Effectively Inhibit Gene Expression in Vivo and Offer a Low-Toxicity Alternative to Fully Modified Phosphorothioate Oligodeoxynucleotides. Brain Res. Mol. Brain Res. 1997, 47, 223–228. [CrossRef]
- Summerton, J.; Weller, D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1497–1500. [CrossRef]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskii, M.D.; Egholm, M.; Buchard, O.; Sönnichsen, S.H.; Nielsen, P.E. Stability of Peptide Nucleic Acids in Human Serum and Cellular Extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [CrossRef]
- Hudziak, R.M.; Barofsky, E.; Barofsky, D.F.; Weller, D.L.; Huang, S.B.; Weller, D.D. Resistance of Morpholino Phosphorodiamidate Oligomers to Enzymatic Degradation. Antisense Nucleic Acid Drug Dev. 1996, 6, 267–272. [CrossRef]
- Lee, J.J.A.; Yokota, T. Antisense Therapy in Neurology. J. Pers. Med. 2013, 3, 144–176. [CrossRef]
- Thakur, S.; Sinhari, A.; Jain, P.; Jadhav, H.R. A Perspective on Oligonucleotide Therapy: Approaches to Patient Customization. Front. Pharmacol. 2022, 13. [CrossRef]
- Egli, M.; Manoharan, M. Chemistry, Structure and Function of Approved Oligonucleotide Therapeutics. Nucleic Acids Res. 2023, 51, 2529–2573. [CrossRef]
- Sumner, C.J.; Miller, T.M. The Expanding Application of Antisense Oligonucleotides to Neurodegenerative Diseases. J. Clin. Invest. 2024, 134, e186116. [CrossRef]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and Mechanism. DNA Repair 2019, 84, 102672. [CrossRef]
- Wu, H.; Lima, W.F.; Zhang, H.; Fan, A.; Sun, H.; Crooke, S.T. Determination of the Role of the Human RNase H1 in the Pharmacology of DNA-like Antisense Drugs. J. Biol. Chem. 2004, 279, 17181–17189. [CrossRef]
- ten Asbroek, A.L.M.A.; van Groenigen, M.; Nooij, M.; Baas, F. The Involvement of Human Ribonucleases H1 and H2 in the Variation of Response of Cells to Antisense Phosphorothioate Oligonucleotides. Eur. J. Biochem. 2002, 269, 583–592. [CrossRef]
- Khvorova, A.; Watts, J.K. The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol. 2017, 35, 238–248. [CrossRef]
- Kuespert, S.; Heydn, R.; Peters, S.; Wirkert, E.; Meyer, A.-L.; Siebörger, M.; Johannesen, S.; Aigner, L.; Bogdahn, U.; Bruun, T.-H. Antisense Oligonucleotide in LNA-Gapmer Design Targeting TGFBR2—A Key Single Gene Target for Safe and Effective Inhibition of TGFβ Signaling. Int. J. Mol. Sci. 2020, 21, 1952. [CrossRef]
- Kiełpiński, Ł.J.; Hagedorn, P.H.; Lindow, M.; Vinther, J. RNase H Sequence Preferences Influence Antisense Oligonucleotide Efficiency. Nucleic Acids Res. 2017, 45, 12932–12944. [CrossRef]
- Bauman, J.; Jearawiriyapaisarn, N.; Kole, R. Therapeutic Potential of Splice-Switching Oligonucleotides. Oligonucleotides 2009, 19, 1–13. [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-Switching Antisense Oligonucleotides as Therapeutic Drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [CrossRef]
- Tomkiewicz, T.Z.; Suárez-Herrera, N.; Cremers, F.P.M.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4. Int. J. Mol. Sci. 2021, 22, 4621. [CrossRef]
- Evers, M.M.; Tran, H.-D.; Zalachoras, I.; Pepers, B.A.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.-J.B.; Aartsma-Rus, A.; van Roon-Mom, W.M.C. Ataxin-3 Protein Modification as a Treatment Strategy for Spinocerebellar Ataxia Type 3: Removal of the CAG Containing Exon. Neurobiol. Dis. 2013, 58, 49–56. [CrossRef]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Meena, null; Aznarez, I.; et al. Antisense Oligonucleotides Increase Scn1a Expression and Reduce Seizures and SUDEP Incidence in a Mouse Model of Dravet Syndrome. Sci. Transl. Med. 2020, 12, eaaz6100. [CrossRef]
- Singh, N.N.; Howell, M.D.; Androphy, E.J.; Singh, R.N. How the Discovery of ISS-N1 Led to the First Medical Therapy for Spinal Muscular Atrophy. Gene Ther. 2017, 24, 520–526. [CrossRef]
- Rigo, F.; Seth, P.P.; Bennett, C.F. Antisense Oligonucleotide-Based Therapies for Diseases Caused by Pre-mRNA Processing Defects. Adv. Exp. Med. Biol. 2014, 825, 303–352. [CrossRef]
- Summerton, J. Morpholino Antisense Oligomers: The Case for an RNase H-Independent Structural Type. Biochim. Biophys. Acta 1999, 1489, 141–158. [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA Targeting Therapeutics: Molecular Mechanisms of Antisense Oligonucleotides as a Therapeutic Platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [CrossRef]
- Dias, N.; Dheur, S.; Nielsen, P.E.; Gryaznov, S.; Van Aerschot, A.; Herdewijn, P.; Hélène, C.; Saison-Behmoaras, T.E. Antisense PNA Tridecamers Targeted to the Coding Region of Ha-Ras mRNA Arrest Polypeptide Chain Elongation. J. Mol. Biol. 1999, 294, 403–416. [CrossRef]
- Anderson, K.P.; Fox, M.C.; Brown-Driver, V.; Martin, M.J.; Azad, R.F. Inhibition of Human Cytomegalovirus Immediate-Early Gene Expression by an Antisense Oligonucleotide Complementary to Immediate-Early RNA. Antimicrob. Agents Chemother. 1996, 40, 2004–2011. [CrossRef]
- Arend, K.C.; Ziehr, B.; Vincent, H.A.; Moorman, N.J. Multiple Transcripts Encode Full-Length Human Cytomegalovirus IE1 and IE2 Proteins during Lytic Infection. J. Virol. 2016, 90, 8855–8865. [CrossRef]
- Vitravene Study Group A Randomized Controlled Clinical Trial of Intravitreous Fomivirsen for Treatment of Newly Diagnosed Peripheral Cytomegalovirus Retinitis in Patients with AIDS. Am. J. Ophthalmol. 2002, 133, 467–474. [CrossRef]
- Vitravene Study Group. Safety of Intravitreous Fomivirsen for Treatment of Cytomegalovirus Retinitis in Patients with AIDS. Am. J. Ophthalmol. 2002, 133, 484–498. [CrossRef]
- Piper, H.; Ciulla, T.A.; Danis, R.P.; Pratt, L.M. Changing Therapeutic Paradigms in CMV Retinitis in AIDS. Expert Opin. Pharmacother. 2000, 1, 1343–1352. [CrossRef]
- Alhamadani, F.; Zhang, K.; Parikh, R.; Wu, H.; Rasmussen, T.P.; Bahal, R.; Zhong, X.-B.; Manautou, J.E. Adverse Drug Reactions and Toxicity of the Food and Drug Administration-Approved Antisense Oligonucleotide Drugs. Drug Metab. Dispos. Biol. Fate Chem. 2022, 50, 879–887. [CrossRef]
- Crooke, S.T.; Geary, R.S. Clinical Pharmacological Properties of Mipomersen (Kynamro), a Second Generation Antisense Inhibitor of Apolipoprotein B. Br. J. Clin. Pharmacol. 2013, 76, 269–276. [CrossRef]
- Santos, R.D.; Duell, P.B.; East, C.; Guyton, J.R.; Moriarty, P.M.; Chin, W.; Mittleman, R.S. Long-Term Efficacy and Safety of Mipomersen in Patients with Familial Hypercholesterolaemia: 2-Year Interim Results of an Open-Label Extension. Eur. Heart J. 2015, 36, 566–575. [CrossRef]
- Gales, L. Tegsedi (Inotersen): An Antisense Oligonucleotide Approved for the Treatment of Adult Patients with Hereditary Transthyretin Amyloidosis. Pharm. Basel Switz. 2019, 12, 78. [CrossRef]
- Sekijima, Y.; Nakamura, K. Hereditary Transthyretin Amyloidosis. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle (WA), 1993.
- Blair, H.A. Tofersen: First Approval. Drugs 2023, 83, 1039–1043. [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne Muscular Dystrophy. Nat. Rev. Dis. Primer 2021, 7, 13. [CrossRef]
- Vengalil, S.; Preethish-Kumar, V.; Polavarapu, K.; Mahadevappa, M.; Sekar, D.; Purushottam, M.; Thomas, P.T.; Nashi, S.; Nalini, A. Duchenne Muscular Dystrophy and Becker Muscular Dystrophy Confirmed by Multiplex Ligation-Dependent Probe Amplification: Genotype-Phenotype Correlation in a Large Cohort. J. Clin. Neurol. Seoul Korea 2017, 13, 91–97. [CrossRef]
- Takeshima, Y.; Yagi, M.; Okizuka, Y.; Awano, H.; Zhang, Z.; Yamauchi, Y.; Nishio, H.; Matsuo, M. Mutation Spectrum of the Dystrophin Gene in 442 Duchenne/Becker Muscular Dystrophy Cases from One Japanese Referral Center. J. Hum. Genet. 2010, 55, 379–388. [CrossRef]
- Aartsma-Rus, A.; Janson, A.A.M.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.-J.B.; van Deutekom, J.C.T. Therapeutic Antisense-Induced Exon Skipping in Cultured Muscle Cells from Six Different DMD Patients. Hum. Mol. Genet. 2003, 12, 907–914. [CrossRef]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. Genotype-Phenotype Correlations in Duchenne and Becker Muscular Dystrophy Patients from the Canadian Neuromuscular Disease Registry. J. Pers. Med. 2020, 10, 241. [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7,000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [CrossRef]
- Matsuo, M. Antisense Oligonucleotide-Mediated Exon-Skipping Therapies: Precision Medicine Spreading from Duchenne Muscular Dystrophy. JMA J. 2021, 4, 232–240. [CrossRef]
- Sabrina Haque, U.; Kohut, M.; Yokota, T. Comprehensive Review of Adverse Reactions and Toxicology in ASO-Based Therapies for Duchenne Muscular Dystrophy: From FDA-Approved Drugs to Peptide-Conjugated ASO. Curr. Res. Toxicol. 2024, 7, 100182. [CrossRef]
- Mercuri, E.; Sumner, C.J.; Muntoni, F.; Darras, B.T.; Finkel, R.S. Spinal Muscular Atrophy. Nat. Rev. Dis. Primer 2022, 8, 52. [CrossRef]
- Ruhno, C.; McGovern, V.L.; Avenarius, M.R.; Snyder, P.J.; Prior, T.W.; Nery, F.C.; Muhtaseb, A.; Roggenbuck, J.S.; Kissel, J.T.; Sansone, V.A.; et al. Complete Sequencing of the SMN2 Gene in SMA Patients Detects SMN Gene Deletion Junctions and Variants in SMN2 That Modify the SMA Phenotype. Hum. Genet. 2019, 138, 241–256. [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of Antisense Oligonucleotide and siRNA-Based Therapeutics. Drug Discov. Today 2017, 22, 823–833. [CrossRef]
- Kim, J.; Hu, C.; Achkar, C.M.E.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [CrossRef]
- McBride, J.L.; Neuringer, M.; Ferguson, B.; Kohama, S.G.; Tagge, I.J.; Zweig, R.C.; Renner, L.M.; McGill, T.J.; Stoddard, J.; Peterson, S.; et al. Discovery of a CLN7 Model of Batten Disease in Non-Human Primates. Neurobiol. Dis. 2018, 119, 65–78. [CrossRef]
- Kousi, M.; Lehesjoki, A.-E.; Mole, S.E. Update of the Mutation Spectrum and Clinical Correlations of over 360 Mutations in Eight Genes That Underlie the Neuronal Ceroid Lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [CrossRef]
- Ray, D.A.; Batzer, M.A. Reading TE Leaves: New Approaches to the Identification of Transposable Element Insertions. Genome Res. 2011, 21, 813–820. [CrossRef]
- Wilton-Clark, H.; Yan, E.; Yokota, T. Preparing for Patient-Customized N-of-1 Antisense Oligonucleotide Therapy to Treat Rare Diseases. Genes 2024, 15, 821. [CrossRef]
- Kim, J.; Woo, S.; de Gusmao, C.M.; Zhao, B.; Chin, D.H.; DiDonato, R.L.; Nguyen, M.A.; Nakayama, T.; Hu, C.A.; Soucy, A.; et al. A Framework for Individualized Splice-Switching Oligonucleotide Therapy. Nature 2023, 619, 828–836. [CrossRef]
- Shiloh, Y. ATM and Related Protein Kinases: Safeguarding Genome Integrity. Nat. Rev. Cancer 2003, 3, 155–168. [CrossRef]
- Verhagen, M.M.M.; Abdo, W.F.; Willemsen, M. a. a. P.; Hogervorst, F.B.L.; Smeets, D.F.C.M.; Hiel, J. a. P.; Brunt, E.R.; van Rijn, M.A.; Majoor Krakauer, D.; Oldenburg, R.A.; et al. Clinical Spectrum of Ataxia-Telangiectasia in Adulthood. Neurology 2009, 73, 430–437. [CrossRef]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Eteplirsen Study Group and Telethon Foundation DMD Italian Network Longitudinal Effect of Eteplirsen versus Historical Control on Ambulation in Duchenne Muscular Dystrophy. Ann. Neurol. 2016, 79, 257–271. [CrossRef]
- Sheikh, O.; Yokota, T. Pharmacology and Toxicology of Eteplirsen and SRP-5051 for DMD Exon 51 Skipping: An Update. Arch. Toxicol. 2022, 96, 1–9. [CrossRef]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 2017, 27, 1–3. [CrossRef]
- Chan, J.H.P.; Lim, S.; Wong, W.S.F. Antisense Oligonucleotides: From Design to Therapeutic Application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [CrossRef]
- Gagliardi, M.; Ashizawa, A.T. The Challenges and Strategies of Antisense Oligonucleotide Drug Delivery. Biomedicines 2021, 9, 433. [CrossRef]
- Vázquez-Domínguez, I.; Anido, A.A.; Duijkers, L.; Hoppenbrouwers, T.; Hoogendoorn, A.D.M.; Koster, C.; Collin, R.W.J.; Garanto, A. Efficacy, Biodistribution and Safety Comparison of Chemically Modified Antisense Oligonucleotides in the Retina. Nucleic Acids Res. 2024, 52, 10447–10463. [CrossRef]
- Seth, P.P.; Tanowitz, M.; Bennett, C.F. Selective Tissue Targeting of Synthetic Nucleic Acid Drugs. J. Clin. Invest. 129, 915–925. [CrossRef]
- Echigoya, Y.; Mouly, V.; Garcia, L.; Yokota, T.; Duddy, W. In Silico Screening Based on Predictive Algorithms as a Design Tool for Exon Skipping Oligonucleotides in Duchenne Muscular Dystrophy. PloS One 2015, 10, e0120058. [CrossRef]
- Echigoya, Y.; Lim, K.R.Q.; Trieu, N.; Bao, B.; Miskew Nichols, B.; Vila, M.C.; Novak, J.S.; Hara, Y.; Lee, J.; Touznik, A.; et al. Quantitative Antisense Screening and Optimization for Exon 51 Skipping in Duchenne Muscular Dystrophy. Mol. Ther. 2017, 25, 2561–2572. [CrossRef]
- Ding, Y.; Chan, C.Y.; Lawrence, C.E. Sfold Web Server for Statistical Folding and Rational Design of Nucleic Acids. Nucleic Acids Res. 2004, 32, W135-141. [CrossRef]
- Shao, Y.; Wu, Y.; Chan, C.Y.; McDonough, K.; Ding, Y. Rational Design and Rapid Screening of Antisense Oligonucleotides for Prokaryotic Gene Modulation. Nucleic Acids Res. 2006, 34, 5660–5669. [CrossRef]
- Sciabola, S.; Xi, H.; Cruz, D.; Cao, Q.; Lawrence, C.; Zhang, T.; Rotstein, S.; Hughes, J.D.; Caffrey, D.R.; Stanton, R.V. PFRED: A Computational Platform for siRNA and Antisense Oligonucleotides Design. PLoS ONE 2021, 16, e0238753. [CrossRef]
- Altmann, A.; Toloşi, L.; Sander, O.; Lengauer, T. Permutation Importance: A Corrected Feature Importance Measure. Bioinforma. Oxf. Engl. 2010, 26, 1340–1347. [CrossRef]
- Stadler, M.B.; Shomron, N.; Yeo, G.W.; Schneider, A.; Xiao, X.; Burge, C.B. Inference of Splicing Regulatory Activities by Sequence Neighborhood Analysis. PLoS Genet. 2006, 2, e191. [CrossRef]
- Ham, K.A.; Aung-Htut, M.T.; Fletcher, S.; Wilton, S.D. Nonsequential Splicing Events Alter Antisense-Mediated Exon Skipping Outcome in COL7A1. Int. J. Mol. Sci. 2020, 21, 7705. [CrossRef]
- Reiser, P.; Neubert, M.; Eberhard, A.; Torresi, L.; Zhou, C.; Shao, C.; Metni, H.; van Hoesel, C.; Schopmans, H.; Sommer, T.; et al. Graph Neural Networks for Materials Science and Chemistry. Commun. Mater. 2022, 3, 1–18. [CrossRef]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in Cancer: A Gemini of Immune Checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [CrossRef]
- Akoglu, H. User’s Guide to Correlation Coefficients. Turk. J. Emerg. Med. 2018, 18, 91–93. [CrossRef]
- McNally, E.M.; Wyatt, E.J. Welcome to the Splice Age: Antisense Oligonucleotide–Mediated Exon Skipping Gains Wider Applicability. J. Clin. Invest. 126, 1236–1238. [CrossRef]
- McClorey, G.; Banerjee, S. Cell-Penetrating Peptides to Enhance Delivery of Oligonucleotide-Based Therapeutics. Biomedicines 2018, 6, 51. [CrossRef]
- Yang, L.; Ma, F.; Liu, F.; Chen, J.; Zhao, X.; Xu, Q. Efficient Delivery of Antisense Oligonucleotides Using Bioreducible Lipid Nanoparticles In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2020, 19, 1357–1367. [CrossRef]
- Huang, S.; Hao, X.-Y.; Li, Y.-J.; Wu, J.; Xiang, D.-X.; Luo, S. Nonviral Delivery Systems for Antisense Oligonucleotide Therapeutics. Biomater. Res. 2022, 26, 49. [CrossRef]
- Zhu, A.; Chiba, S.; Shimizu, Y.; Kunitake, K.; Okuno, Y.; Aoki, Y.; Yokota, T. Ensemble-Learning and Feature Selection Techniques for Enhanced Antisense Oligonucleotide Efficacy Prediction in Exon Skipping. Pharmaceutics 2023, 15, 1808. [CrossRef]
Figure 1.
Mechanisms by which ASOs can be utilized to treat genetic diseases. A) standard gapmers and unmodified ASOs bind to target RNA, recruiting RNase H to induce RNA degradation. B) Fully chemically modified ASOs can bind to target RNA to block splicing or translation machinery, facilitating exon-skipping or inclusion and translation inhibition, respectively. Chemically modified nucleotides are depicted in dark blue while unmodified nucleotides are shown in light blue. .
Figure 1.
Mechanisms by which ASOs can be utilized to treat genetic diseases. A) standard gapmers and unmodified ASOs bind to target RNA, recruiting RNase H to induce RNA degradation. B) Fully chemically modified ASOs can bind to target RNA to block splicing or translation machinery, facilitating exon-skipping or inclusion and translation inhibition, respectively. Chemically modified nucleotides are depicted in dark blue while unmodified nucleotides are shown in light blue. .
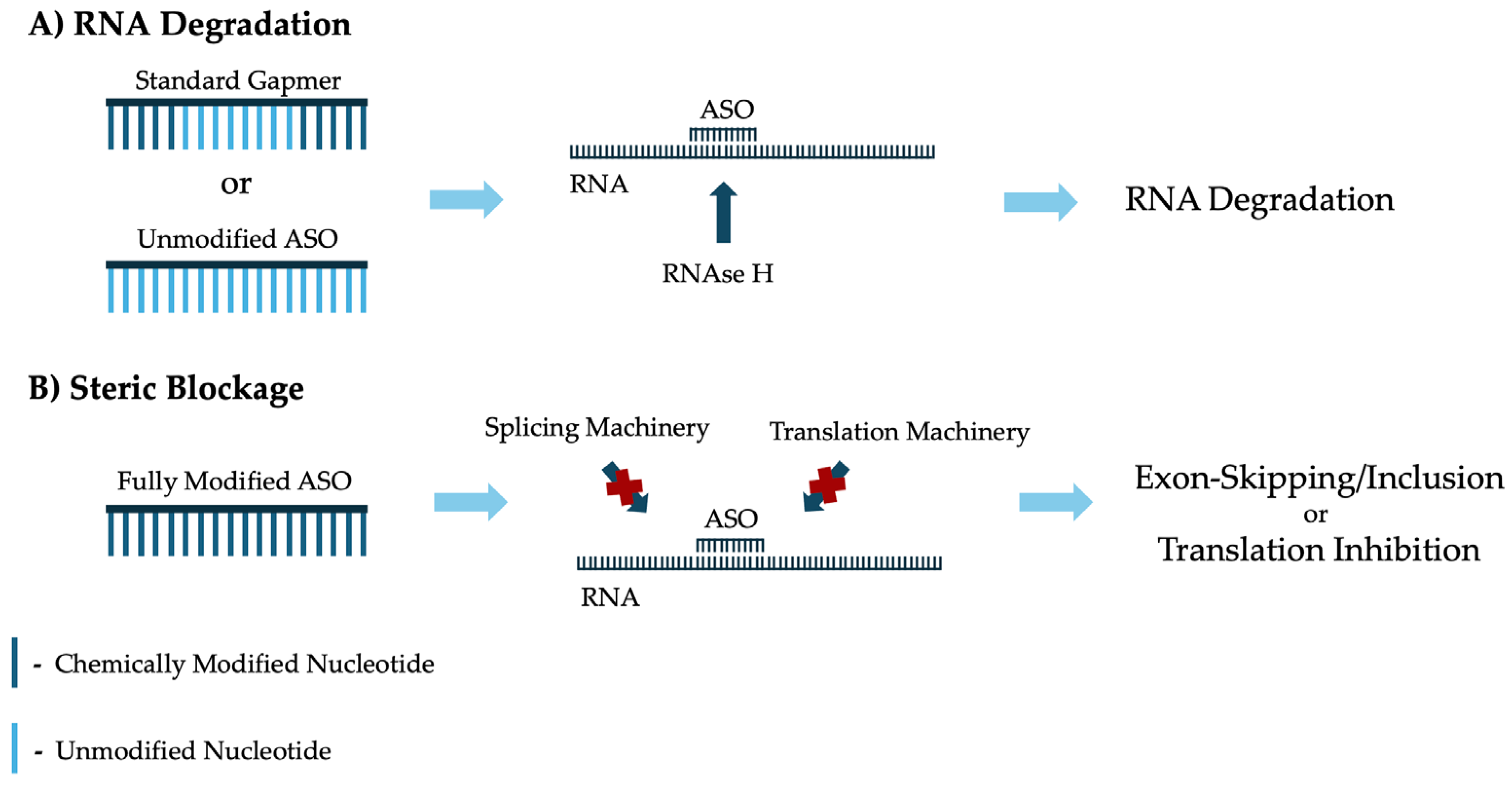
Figure 2.
Overview of machine learning-based platforms designed to enhance ASO efficacy through sequence optimization and optimized chemical engineering. A) A comprehensive database is built using data extracted from relevant literature and patents. B) A predictive model is trained on the curated database, leveraging key features associated with ASO performance. C) The trained model predicts the efficacy of individual ASO candidates or ranks multiple ASOs based on their predicted therapeutic potential.
Figure 2.
Overview of machine learning-based platforms designed to enhance ASO efficacy through sequence optimization and optimized chemical engineering. A) A comprehensive database is built using data extracted from relevant literature and patents. B) A predictive model is trained on the curated database, leveraging key features associated with ASO performance. C) The trained model predicts the efficacy of individual ASO candidates or ranks multiple ASOs based on their predicted therapeutic potential.
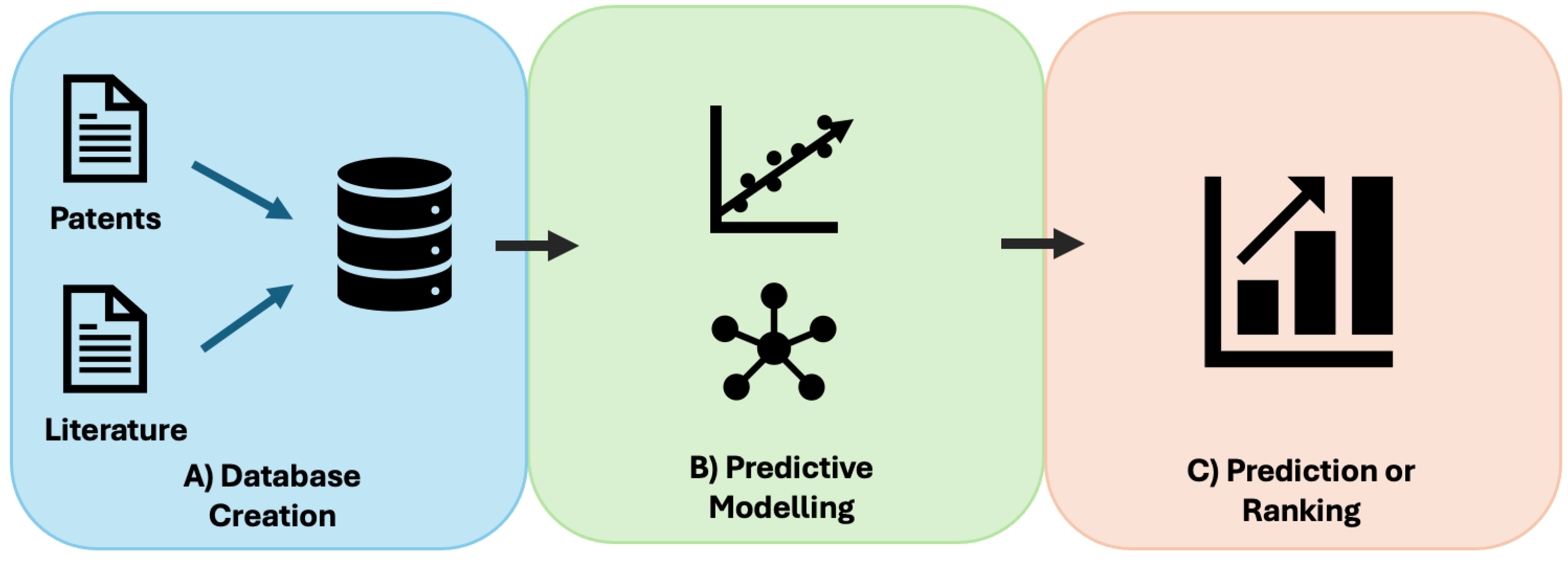

Table 1.
Overview of FDA-approved ASOs, highlighting the diseases they treat, their RNA targets, chemical modifications, and mechanisms of action used to alleviate symptoms.
Table 1.
Overview of FDA-approved ASOs, highlighting the diseases they treat, their RNA targets, chemical modifications, and mechanisms of action used to alleviate symptoms.
| Therapy Name | Disease | Target RNA | Mechanism of Action | Chemical Modifications |
|---|---|---|---|---|
| Fomiversen | CMV retinitis | MIE | RNA degradation | PS |
| Mipomersen | HoFH | APOB | RNA degradation | PS and 2’-MOE |
| Eteplirsen | DMD | DMD (exon 51) | Splice switching | PMO |
| Nusinersen | SMA | SMN2 (exon 7) | Splice switching | PS and 2’-MOE |
| Inotersen | hATTR | TTR | RNA degradation | PS and 2’-MOE |
| Golodirsen | DMD | DMD (exon 53) | Splice switching | PMO |
| Viltolersen | DMD | DMD (exon 53) | Splice switching | PMO |
| Casimersen | DMD | DMD (exon 45) | Splice switching | PMO |
| Tofersen | ALS | SOD1 | RNA degradation | PS and 2’-MOE |
Table 2.
Overview of ASOs evaluated through N-of-1 clinical trials with published data reporting their results highlighting the diseases they treat, their RNA targets, chemical modifications, and mechanisms of action used to alleviate symptoms.
Table 2.
Overview of ASOs evaluated through N-of-1 clinical trials with published data reporting their results highlighting the diseases they treat, their RNA targets, chemical modifications, and mechanisms of action used to alleviate symptoms.
| Therapy Name | Disease | Target | Mechanism of Action | Chemical Modifications |
|---|---|---|---|---|
| Milasen | Batten Disease | CLN7 (intron 6) | Splice Switching | PS and 2’-MOE |
| Atipeksen | HoFH | ATM (exon 53) | Splice Switching | PS and 2’-MOE |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



