
Submitted:
30 December 2024
Posted:
31 December 2024
You are already at the latest version
Abstract
Background: Diffuse intrinsic pontine glioma (DIPG) is a devastating childhood brainstem tumor. The median survival of DIPG is 16–24 months independently of the treatment received. Therefore, new therapeutic strategies against DIPG are urgently needed. Substance P (SP) peptide, through the neurokinin Neurokin-1 receptor (NK-1R) is involved in glioma progression, induces glioma cells proliferation by activating, MAPKs (p38 MAPK, ERK1/2 and JNK), c-Myc, AP-1, NF-κB, and induces antiapoptotic by PI3K/Akt/mTOR in glioma cells. SP favors glycogen breakdown that is essential for glycolysis. The SP/NK-1R system regulates also the migration and invasion of glioma cells, stimulates angiogenesis and triggering inflammation which contributes to glioma progression. Moreover, all glioma cells express NK-1R and NK-1R is essential for the viability of glioma cells and non for normal cells. In contrast, in glioma, NK-1R antagonists, such as aprepitant drug penetrate brain and reach therapeutic concentrations: inhibit mitogenesis, induces apoptosis and inhibit breakdown of glycogen in glioma cells. In addition, they inhibit angiogenesis and exert antimetastatic and anti-inflammatory effects. The combination of radiotherapy with NK-1R antagonists produces radiosensitization, radioneuroprotection, reduces both peritumoral and radiation-induced inflammation, and is also antinausea and antivomiting. Objective: This review updates the involvement of the SP/NK-1R system in glioma promotion and progression and the potential clinical application of NK-1R antagonist drugs in DIPG therapy. Conclusion: NK-1R plays a crucial role in glioma progression and NK-1R antagonists such as aprepitant could be used in combination with radiotherapy as a potent therapeutic strategy for the treatment of patients with DIPG.
Keywords:
DIPG
; glioma
; substance P
; Neurokinin-1 receptor
; aprepitant
; antitumor
; apoptosis
; angiogenesis
; metastases
1. Introduction
Glioblastoma or glioma is an astrocytoma WHO grade IV. Diffuse intrinsic pontine glioma (DIPG) has been classified as a new pathologic entity called diffuse midline glioma, H3K27M-altered by the World Health Organization [1]. DIPG is a childhood malignancy of the brainstem with a dismal prognosis, and the median survival of DIPG is 16–24 months independently of the treatment received [2]. The combination of multiple cranial neuropathies, long tract and cerebellar signs is known as the “classic triad” of DIPG and only a small percentage of pediatric patients (<10%) develop symptoms of hydrocephalus [2]. Recent studies analyzing the molecular profiles of DIPG found that most harbor a mutation in histone H3 (H3K27M-altered) and that they are more aggressive and have worse outcomes compared to their non-mutant counterparts, regardless of histology. Despite recent advances in the understanding of the molecular basis of the tumor in the last decade, the prognosis of DIPG has remained unchanged [2]. DIPG is resistant to conventional chemotherapeutic agents used for brain tumors, such as temozolomide. Radiotherapy (RT) is the standard treatment of DIPG; however, it is only palliative. RT is expected to increase survival for patients about 3 months on average. DIPG remains universally fatal [3]. Unfortunately, new drugs only slightly improve overall survival in DIPG. Recently has been published a systematic review about clinical trial on DIPG involving radiation therapy in combination with chemotherapeutic and/or included immunotherapeutic options even CAR T cells therapy and oncolytic virus that showed a lacks response to both traditional and highly cytotoxic chemotherapeutic options [4]. Therefore, it is necessary and urgent to investigate new and different molecular bases of DIPG tumor development and to search for new therapeutic targets, as well as new effective and safe drugs that can improve the ominous prognosis of DIPG.
In the last twenty years, considerable evidence has been published that substance P (SP) peptide and its receptor, Neurokinin-1 receptor (NK-1R), are involved in glioma promotion and progression. NK-1Rs are overexpressed in glioma tumors [5,6,7] and SP are overexpressed in glioma tumors [7]. SP in a concentration dependent manner after binding to NK-1R of the glioma cells increases tumor cell proliferation (mitogenesis), has antiapoptotic effects, is involved in the Warburg effect. SP after binding to NK-1R increases endothelial cells proliferation and induces angiogenesis, and produces inflammation, as well SP after binding to NK-1R of glioma cells induces migration and invasion (invasion and metastases) of glioma cells [8,9]. Moreover, NK-1R is essential for the viability of glioma cells and non for normal cells [10]. In contrast, NK-1R antagonists in a concentration dependent manner counteract all the pathophysiological effects of SP in glioma: inhibit glioma cells proliferation (antimitogenic effect), induces programmed cell death in glioma cells (apoptosis), counteract the Warburg effect in glioma cells, decrease both endothelial cells proliferation and angiogenesis, it has anti-inflammatory effects, prevent invasion and migration (infiltration and metastases) of glioma cells [8,9,11]. Therefore, NK-1Rs could be considered a new therapeutic target in gliomas and NK-1R antagonists such as aprepitant drug could be new selective, effective and safe drugs in the treatment of gliomas such as DIPG.
2. SP and NK-1R in Glioma
SP and hemokinin-1 (HK-1) area member of the tachykinin peptide family, such as neurokinin A (NKA) and NKB. Tachykinins, via the NK-1R, NK-2R and NK-3R, exert many biological actions. SP/HK-1 is known to have a preferential affinity for NK-1R, they are the natural ligands of NK-1R. The TAC1 gene encodes SP and TAC4 encodes HK-1 [12]. SP and HK-1 are two undecapeptides. The C-terminus sequence of SP 6-11 (Gln-Phe-Phe-Gly-Leu-Met) [13] is essential for affinity for the NK-1R and HK-1 has a similar affinity for the NK-1R because the C-terminus 6-11 is the same [14]. This suggests that NK-1R is very important for cells because it has two natural ligands SP and HK-1. Glioma cells expresses both SP and HK-1 [15], however SP is not essential for the viability of glioma cells [10], probably the absence of SP is compensated by HK-1 in glioma cells. Furthermore, SP is ubiquitous throughout the organism, tissues, cells and body fluids (blood, cerebrospinal fluid, saliva, etc.) [16]. However, SP is overexpressed in cancer tissues and cancer cells [17] and cancer patients have higher serum SP levels than normal subjects [18]. SP is located in cytoplasm and in the nucleus of glioma cells [10]. In addition, SP is well known to be a universal mitogen in cancer (including glioma) cells [9,11].
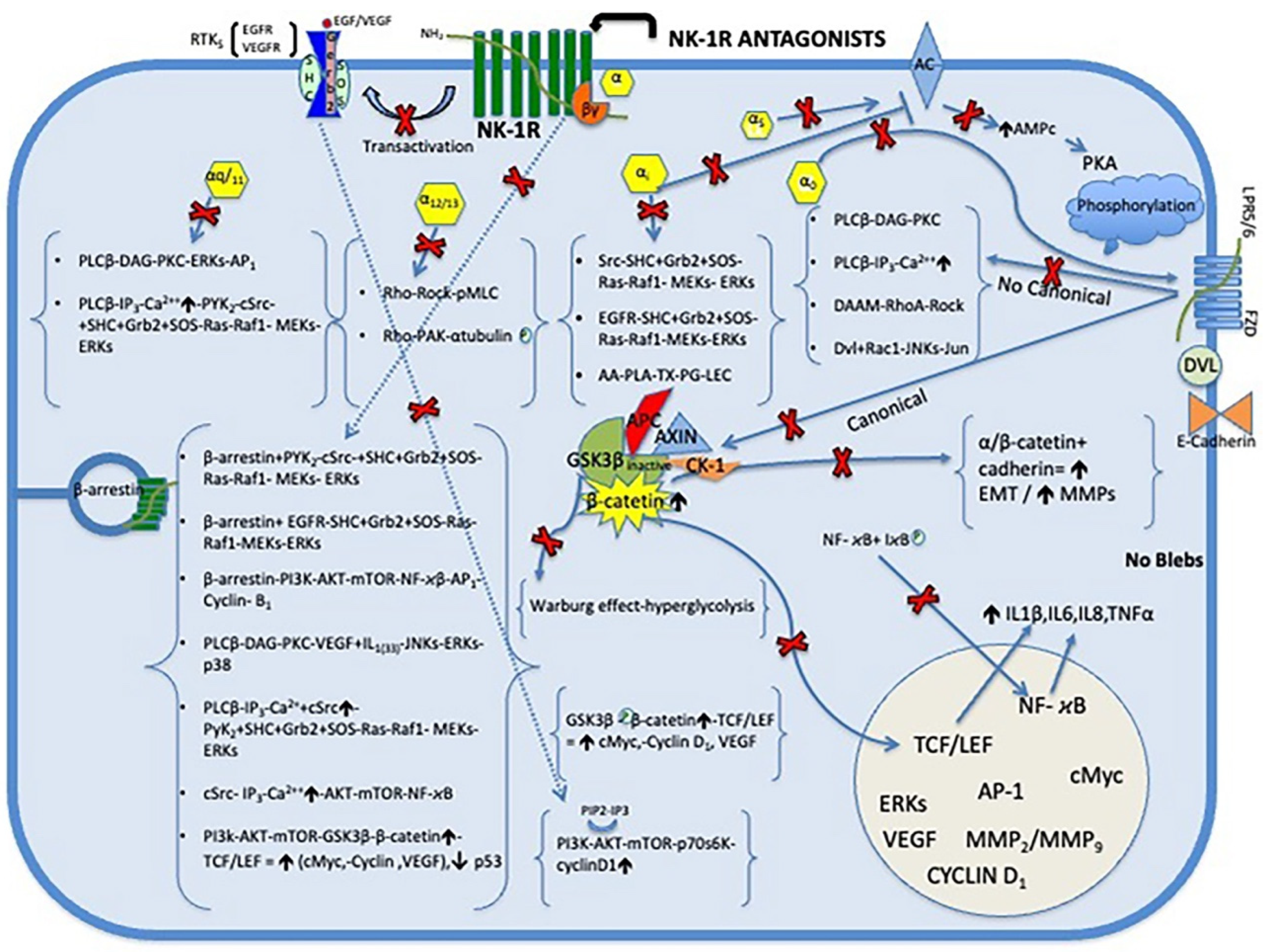
The NK-1R belongs to the class of seven transmembrane domain (7-TM) receptors is functionally coupled with G protein-coupled receptors (GPCRs) and mediates the biological activities of SP or HK-1, which are the most potent agonist for the NK-1R compared to neurokinin A or neurokinin B [19]. This G complex consists of three different subunits: Gα induces a exchange GTP for GDP and Gβ and Gγ subunits that form the complex Gβγ. The Gαs subunit produces the activation of the second messenger adenylate cyclase (AC) which catalyzes the conversion of ATP into cyclic adenosine monophosphate cytoplasmic (cAMP) by increasing the level of cAMP the activation of protein kinase A (PKA), while the Gαi subunit stimulates the release of [3 H] araquidonic acid (AA) and induces epidermal growth factor receptor (EGFR) transactivation and subsequent DNA synthesis, also inhibits AC. Other effector of the Gαq/11 pathway is the phospholipase C-β (PLCβ), which stimulates the hydrolysis of phosphatidylinositol (PI) and the formation of IP3 (IP3) and diacylglycerol (DAG) eliciting the release of Ca2+, whereas DAG diffuses along the plasma membrane where it may activate a ser/thr kinase called protein kinase C (PKC) which are also activated by an increase in the intracellular level of Ca2+. Moreover, Gαo is too crucial for the activation of Wnt-β-catenin signaling pathway and Gα12/13 regulates changes in cyto-skeletal pathway Rho [Encyclopedia of Signaling Molecules Second Edition]. For the other hand, the Gβγ. subunit activates effectors such as PLCβ, AC, PI3K, K++ ion channels, and Src [20]. The NK-1R activates members of the mitogen-activated protein kinase (MAPK) cascade, including extracellular signal-regulated kinases 1 and 2 (ERK1/2), also called (p42/44 MAPK) or by various signaling pathways, one pathway depends on β-arrestin mediated receptor endocytosis, and other pathway depended on tyrosine kinases or c-Src and transactivation of receptor tyrosine kinases, suggesting that this complex (β-arrestin and c-Src) might serve to link the receptor to the ras-dependent pathway of ERK1/2 activation mediated through Gβγ subunit and independent of protein kinase C through Gαq/11 between other [21] (Figure 1).
The NK-1R has two isoforms: a full-length 407 amino acid isoform NK-1R-Fl and a truncated isoform NK-1R-Tr (311 amino acids; because 96 residues are lost at the C-terminus) [22]. Due to the different structure of both isoforms, they have a different functional significance, differing in cell signaling capability [23]. The NK-1R-Fl exhibits a 10-fold higher binding affinity for SP than the NK-1R-Tr isoform [24]. It has been reported that all glioma cell lines LN71, LN229, LN319, LN405 expressed NK-1R, however, the two NK-1R isoforms NK-1R-Fl and NK-1R-Tr are expressed in different concentrations. Only the LN319 glioma cell line presents the highest concentration of the NK1R-Fl isoform. This could have therapeutic implications [25]. In contrast, it has been reported that β-Arrestin deficiency increases the sensitivity of glioma cells to the treatment with NK-1R antagonists and besides is essential for NK1-mediated glioblastoma cell proliferation [26]. Moreover, it has been shown that the U-373 MG, U87MG and GAMG glioma cell lines express both the NK-1R-Fl and NK-1R-Tr isoforms [10,27] and that regardless of the amount of expression of the two isoforms, NK-1R antagonists for instance L-733,060, L-732,138 and aprepitant drug, in a concentration-dependent manner have the same therapeutic effect on GAMG, U-373 MG and U87 MG glioma cells, inhibit mitogenesis (at certain concentrations they inhibit 100%) and induce apoptosis of glioma cells [28,29,30,31]. Therefore, from a therapeutic point of view, the different expressions of the NK-1R-Fl and NK-1R-Tr isoforms in gliomas have no therapeutic implications. Indeed, the NK-1R is essential for glioma viability of GAMG glioma cells [10]. In glioma samples also called glioblastoma (grade 4 astrocytoma) all samples studied, (10 of 10) [5] and (17 of 17) [6], express NK-1Rs and in the case of astrocytoma samples, in 9 of 12; furthermore, NK-1R density was more marked and more extensive in gliomas than in astrocytomas [5,7]. Glioma cell lines express NK-1Rs (e.g., LN71, LN229, LN319, LN405, U-373 MG, U251 MG, DBTRG-05 MG, UC-11 MG, SNB-19, GAMG, U-87 MG) [10,15,25,27,29,32,33,34,35,36]. Moreover, it has been reported that glioma cell expresses 40,000-60,000 NK-1Rs per cells in UC-11 MG glioma [36]. It known that the number of NK-1Rs was lower in cultures of glioma cell lines than in glioma tumors [5] and that poor prognosis and advanced tumor stages were related with the most malignant phenotypes overexpress NK-1Rs [37]. It has been suggested that the degree of malignancy of a tumor is correlated with the number of NK-1Rs: greater number, higher malignancy [17]. The overexpression of NK-1Rs in glioma is very useful for therapeutic intervention using NK-1R antagonists. In glioma samples, it has been also reported that the number of these receptors was higher in tumors than in normal surrounding brain and that also NK-1Rs were found in blood vessels located within the tumor [5,7].
3. SP Is Involved in Many Types of Signaling in Glioma
It is known that SP is located in the cytoplasm and the nucleus of glioma cells [10], indicating potential autocrine, paracrine, intracrine, and/or endocrine actions [38]. Autocrine function: cancer cells synthesize and release SP into the extracellular space by binding to the tumor cell's own NK-1Rs, producing both glioma mitogenesis and antiapoptotic effects. Paracrine function: glioma cells synthesize and release SP into the extracellular space by binding to NK-1Rs from other glioma cells, endothelial cells and inflammatory cells. As a result, it produces some types of actions; an increased mitogenesis of glioma cells and endothelial cells and an antiapoptotic effect on glioma cells and it stimulates the secretion of cytokines such as IL-1β, IL-6, IL-8, TNF-α, granulocyte macrophage colony-stimulating factor (GM-CSF) and leukemia inhibitory factor (LIF) in inflammatory and gliomas cells [32,39,40]. Intracrine function: glioma cells synthesize SP in the cytoplasm, which in turn can migrate to the nucleus, playing a nuclear regulatory role in the glioma cells. It is known that the TATA box binding protein (TPB), a transcription factor in the minor groove of DNA, leads to DNA deformation and that DNA unfolding is facilitated by the projection of two phenylalanine residues from TPB [38,41]. SP contains two phenylalanine residues at its C-terminus [13] being able to produce large-scale deformations in the DNA and initiating a transcription similar to that produced by TBP, so SP can be considered a transcription factor. Endocrine function: glioma tumor is composed of glioma cells which synthesize SP in the cytoplasm and release SP that can be released into the bloodstream and bind to NK-1Rs that express different cells throughout the body. Moreover, SP plasma concentrations were higher in cancer patients than in healthy subjects [18].
4. Role of SP and NK-1R in Mitogenesis and Antiapoptotic Effect in Glioma Cells (Figure 1)
One of the hallmarks of cancer is the uncontrolled increase in mitogenesis in tumor cells. Furthermore, it is well known that SP is a universal mitogen in tumor cells [9,11,42,43]. There are numerous studies showing that SP after binding to NK-1R of glioma cells induces mitogenesis of glioma cells [28,44,45,46,47]. Moreover, it has been reported that SP binds to NK-1R of U-373 MG glioma cells producing: a rapid increase in the synthesis of IP3, an increase in the concentration of cytosolic Ca2+ and the induction of immediate early transcription of the expression of the c-Fos and c-Jun (Activator protein 1 (AP-1) a transcription factor) genes and an increase in DNA synthesis [44]. SP activates NK-1R and induces the incorporation of [3H] thymidine into DNA and SP potently induces c-Myc mRNA and c-Myc protein expression. These genes are required for cell progression from the G1 phase to the S phase of the cell cycle in U-373 MG glioma cells. SP also stimulates NK-1R and activates tyrosine phosphorylation and enzymatic activity of MAPKs ERK1/2 [45]. SP activates phosphorylation of PHAS-I protein (also known as 4E-BP1) and p70S6 kinase (p70(S6K)), which is inhibited by rapamycin. In contrast, rapamycin does not suppress SP-induced activation of MAPK, c-Fos, and DNA synthesis in U-373 MG astrocytoma cells [48]. p70S6K is member of the serine/threonine protein kinase family, it is one of the downstream effectors of the PI3K/Akt/mTOR signal transduction pathway. It phosphorylates S6 protein of 40S ribosomal subunit and thus functions in protein synthesis and cell growth. It has been suggested that inhibition of the PI3K/Akt/mTOR pathway is an effective therapeutic strategy in tumors overexpressing phospho-p70S6K [49]. Similarly, also it has been suggested the PI3K/Akt/mTOR pathway is an effective therapeutic strategy in glioma [50]. Moreover, in U-373 MG glioma cells SP induced tyrosine phosphorylation of several proteins including EGFR and activates EGFR complex containing the adapter proteins, Sos, Shc and Grb2, but not c-Src. SP activates the MAPK pathway increasing ERK2 kinase activity and DNA synthesis in glioma cells and EGFR is an essential regulator in SP/NK-1R-induced activation of the MAPK pathway and cell proliferation in U-373 MG glioma cells, and these events are mediated by subunit Gαi protein [46]. However, it was later reported that blockade of the EGFR, which is transactivated by NK-1R, has minimal effect on NK-1R-mediated ERK1/2 phosphorylation. NK-1R-mediated ERK1/2 phosphorylation is significantly reduced by the c-Src kinase inhibitor PP2. Interestingly, ERK1/2 in U-373 MG glioma cells is also activated by several other mitogenic GPCRs, including alpha(1B)-adrenergic, M(3)-muscarinic, and H(1)-histaminergic in a c-Src-dependent manner. Thus, c-Src is a mediator of SP-stimulated ERK1/2 phosphorylation in human U-373 MG glioma cells [47]. In addition, EGFR transactivation by the NK-1R is only marginally involved in SP-dependent ERK1/2 phosphorylation because Src is a major component of NK-1R signaling, and the NK-1R also stimulates PKC δ phosphorylation [51]. Furthermore, β-arrestin-mediated signaling pathway is essential for NK-1R-mediated glioma cell proliferation. β-arrestin knockdown inhibits NK-1R-mediates glioma cell proliferation and induces G2/M phase cell cycle arrest. β-arrestin knockdown cells showed remarkable down-regulation of CDC25C/CDK1/cyclin B1 activity. β-arrestin-mediates ERK1/2 and Akt phosphorylation regulates the transcriptional activity of both NF-κB and AP-1, which were involved in cyclin B1 expression [26]. CDC25C/CDK1/cyclin B1 is a cyclin-dependent protein kinase that controls the cell cycle entry from G2 to M phase. It has been suggested that inhibition the G1 and G2 phases of the cell cycle progression is an effective therapeutic strategy in tumors overexpressing CDC25/CDK1 induced by NK-1R activation [49]. On the other hand, Akt, or Protein Kinase B (PKB), a serine-threonine protein kinase which becomes activates via phosphatidyl-3-kinase (PI3K) enzyme and suppresses apoptosis. In addition, SP binds to NK-1R-mediated Akt phosphorylation and the activation occurs through Src and PI3K, but only partially through the EGFR kinase. NK-1R activates Akt through both EGFR-dependent and EGFR-independent pathways. This action of SP is counteracted by both NK-1R antagonists L-733,060 and L-732,138 [29]. Another effect of Akt is the inactivation of the tumor suppressor gene, p53 preventing its activity as a transcription factor of proapoptotic genes, decreasing its levels [52]. Once activated, Akt translocate to the various subcellular compartments where it phosphorylates several targets, including GSK-3β and the phosphorylation leads to the inactivation of GSK-3β, it creates a complex intracellular network [53]. In addition, it has been reported that the majority of DIPG tumors were positive for EGFR and p53 suggesting that deregulation of EGFR and p53 may play an important role in the development of DIPG and therefore therapies targeting these proteins might be beneficial in the treatment of DIPG [54].
5. Warburg Effect
Tumor cells are characterized by the production of hyperglycolysis followed by lactic fermentation, a phenomenon known as the Warburg effect [55]. Therefore, cancer cells need to obtain a large amount of glucose for hyperglycolysis. It is known that SP promotes the degradation of glycogen into glucose and increases the intracellular concentration of Ca2+ in glioma cells overexpressing NK-1R [56]. In contrast, these effects are completely blocked by the NK-1R antagonist CP-96,345 in a concentration-dependent manner [56]; Therefore, without glucose there can be no hyperglycolysis and glioma cells can starve. Moreover, it has suggested that such effects are mediated by the NK-1R and that the glycolytic function is directly related to the number of NK-1R receptors expressed in each cell, with tumor cells exhibiting higher expression levels and increased glycolytic rates [43]. Moreover, SP induces phosphorylation of GSK-3α/β (renders it inactive) in glioma cells [29]. In contrast, both NK-1R antagonists L-733,060 and L-732,138 counteract phosphorylation of GSK-3α/β in glioma cells [29]. Indeed, GSK-3 inhibitors reduce glucose production and increase glycogen synthesis from L-glucose. Therefore, GSK-3 inhibitors may reduce glucose levels [57]. The anti-Warburg effect of NK-1R antagonists is particularly important, as currently only this type of drug specifically counteracts the Warburg effect by preventing glucose formation in cancer therapy [43]. GSK-3 is activated by the canonical Wnt/β-catenin pathway and inhibits autophagy and the production of the autophagic adaptor p62. However, nutrient deprivation (such as glucose starvation) promotes the destruction of β-catenin through the inactivation of the Wnt pathway and Dvl is destroyed [50]. In summary, SP binds to the NK-1R of glioma cells and induces GSK-3α/β phosphorylation, glycogen degradation and glucose formation. In contrast, NK-1R antagonists decrease both GSK-3α/β phosphorylation and glucose formation, counteracting the Warburg effect and inducing autophagy in glioma cells.
6. SP/HK-1 and NK-1R in Glioma Angiogenesis
Angiogenesis is a sequential process, in which there is early proliferation of endothelial cells and subsequent formation of new blood vessels, resulting in increased blood flow, accompanied by maturation of endogenous neurovascular regulatory systems, which occurs late in this process in inflamed tissues [11]. Moreover, angiogenesis is a hallmark of tumor development, associated with both increased tissue innervation and NK-1R expression [43]. Furthermore, NK-1Rs are found in intra- and peritumoral blood vessels. SP, through the NK-1R receptor, which is found in high density in vessels, can strongly influence vascular structure and function in and around tumors by increasing tumor blood flow, which promotes, for example, stromal development and facilitates metastatic spread [5]. SP is expressed in brain capillary endothelial cells and is secreted by these cells in response to treatment with high doses of cytokines such as IL-1β and TNF-α. [42,58]. In addition, NK-1R agonists, such as SP, can directly stimulate the process of neovascularization through induction of endothelial cell proliferation [59], and SP-enhanced angiogenesis results from a direct action on microvascular NK-1Rs. Subsequently it has been shown that endothelial cells express NK-1R and NK-2R [60]. In addition, vascular endothelial growth factor (VEGF) is a growth factor that promotes angiogenesis, has a mitogenic and antiapoptotic effect on endothelial cells, increases vascular permeability, and promotes cell migration [61]. Overexpression of VEGF in tumors is associated with increased angiogenesis, proliferation, and metastasis [62]. Furthermore, HK-1 have been reported to be key endogenous regulators of angiogenesis. HK-1 after binding to NK-1R of human umbilical vein endothelial cells (HUVECs) concentration-dependently stimulated proliferation, migration, adhesion, and tube formation and also had angiogenic effects in vivo. In contrast, the angiogenic effects of HK-1 were inhibited by the selective NK-1R antagonist. In addition, HKs activated ERK1/2 phosphorylation, stimulated nitric oxide production, and increased the expression of endothelial nitric oxide synthase (eNOS) and VEGF in HUVECs [63]. VEGF overexpression plays an essential role in glioma progression and indicates poor glioma outcome, so VEGF may be an important prognostic factor in the overall survival of glioma patients [64]. In summary, glioma cells express SP and HK-1 [10,15], upon release, SP and HK-1 bind to the NK-1R of glioma vessel endothelial cells, inducing endothelial cell proliferation, migration and adhesion and tube formation and activates ERK1/2 phosphorylation, stimulates nitric oxide production and increases eNOS and VEGF expression which are involved in glioma angiogenesis.
7. SP and NK-1R in Inflammation and Microenvironment of Glioma (Figure 1)
In 1863, Rudolf Virchow observed the presence of leukocytes in neoplastic tissues and established a connection between inflammation and cancer. Accordingly, it has been suggested that inflammatory cells and cytokines found and released in tumors contribute more to tumor growth, progression and immunosuppression than to generating an effective antitumor response from the host. Therefore, the use of cytokine and chemokine blockers and nonsteroidal anti-inflammatory drugs could be useful in the chemoprevention and treatment of malignant diseases [65]. SP is a main mediator of neuroimmunomodulatory activities and neurogenic inflammation within the central and peripheral nervous system. Neurogenic inflammation, which is characterized by vasodilation, increased permeability of postcapillary venules, plasma leakage, leukocyte infiltration and adherence of neutrophils to blood vessels [40] and the release of neuropeptides including SP and calcitonin gene related peptide (CGRP). Inflammatory and glioma cells express and can release SP that regulates neurogenic inflammation mediated by the release of IL-1β, IL-6, IL-8, TNF-α, GM-CSF, and LIF [32,39,40]. It is known that human astrocytic and microglial cells constitutively overexpress the NK-1R-Fl isoform but not NK-1R-Tr. Furthermore, SP can enhance inflammatory and/or neurotoxic immune responses of glial cells to different inflammatory processes. So, SP by binding to NK-1R of glia cells elicits nuclear translocation of NF-κB [66]. Furthermore, microglia are known to secrete numerous inflammatory mediators, such as tumor necrosis factor-α (TNF-α,) IL-1β, IL-6, and monocyte chemoattractant protein 1 (MCP-1) [67]. SP activates NF-κB, a transcription factor involved in the control of cytokine expression and apoptosis. SP increased the expression and secretion of interleukin-8 (IL-8), a target gene controlled by NF-κB, this effect is specific, since an NK-1R antagonist completely prevented NF-κB activation in response to SP, but not to IL-1β in U-373 MG glioma cells [68]. SP and histamine also induce interleukin-6 expression by a mechanism involving PKC and nuclear factor-IL-6 in U-373 MG glioma cells [69]. Furthermore, SP also induces phosphorylation of p38 MAPK which mediates SP-induced IL-6 expression in U-373 MG glioma cells by mechanism independent of ERK1/2 (p42/44 MAPK), PKC and NF-κB activation [70]. However, ketamine a parenteral an anesthetic drug, acts as a competitive antagonist of the excitatory neurotransmitter N-methyl-D-aspartate receptor and antagonizes also SP functions by binding to the NK-1R of glioma cells by inhibiting the synthesis of IL-6 and IL-8 in U-373 MG glioma cells through the suppression of the phosphorylation of several signaling molecules such as NF-κB, p38MAPK and p42/44 MAPK [71]. SP is involved in inflammatory conditions, in fact the human plasma levels of SP are increased under certain pathologic conditions, including HIV infection [72] and cancer [18]. Furthermore, elevated circulating SP levels are associated with decreased numbers of natural killer (NK) cells and impaired function. SP inhibits the cytotoxic capacity and reduces NK degranulation. NK cells express NK-1R and this inhibitory effect of SP on cytotoxicity was partially prevented by the NK-1R antagonist CP-96,345. Thus, SP regulates NK cell functions and acts downstream of NK-1Rs to modulate NK cell activation signaling [73]. This mechanism may contribute to the impaired NK cell function in cancer patients in whom circulating SP is increased [18]. In contrast, using selective NK-1R antagonist can increases NK functions in cancer patients [73]. In addition, NK cells have been reported to lyse all DIPG cell cultures, suggesting that NK cells may represent a promising avenue for targeting DIPG [74]. Macrophages are component of tumor microenvironment. Macrophages can differentiate along a spectrum of phenotypes, M1 and M2 are the extremes of the same. M1 macrophages are considered antitumor and M2 macrophages are predominantly expressed within the tumor microenvironment and are considered protumoral. SP can induce M2 polarization of inflammatory macrophages. SP induces differentiation of GM-CSF-differentiated proinflammatory macrophages into M2-type alternatively activated phagocytic macrophages (M2SP) by direct activation of the PI3K/Akt/mTOR/S6kinase pathway and induction of arginase-1, CD163 and CD206, whereas treatment with the NK-1R antagonist RP67580 prevents all these processes [75]. However, it has been reported that DIPG tumors do not have a highly immunosuppressive or inflammatory microenvironment. Therefore, potential immunotherapeutic should target the recruitment, activation and retention of tumor-specific immune effector cells, such as NK cells [74]. Mast cells are another component of tumor microenvironment. SP at nanomolar concentrations induces gene expression and secretion of VEGF in human mast cells and cultured mast cells derived from human umbilical cord blood (hCBMC). This effect is enhanced by co-administration of IL-33 in both cell types. In contrast, this effect of SP on VEGF release is inhibited by treatment with the NK-1R antagonist L-733,060. Although PKC pathway is important for induction of VEGF by SP this is not obligatory for its maximum induction. Also, SP stimulates phosphorylation of the MAPKs: ERK and JNK, which can be activated by PKC-dependent and PKC-independent mechanisms. Activation of these MAPKs leads to activation of the transcription factor AP-1, a heterodimer of c-Fos and c-Jun. The VEGF promoter has several AP-1 binding sites that increase transcription, which may explain the increased abundance of VEGF mRNA in SP-stimulated cells. These results imply that functional interactions between SP, IL-33, and mast cells leading to VEGF release contribute to inflammatory conditions [76]. In addition, it has been reported that the inhibition of NK-1R expression can decrease the phosphorylation levels of MAPKs (ERK1/2, JNK, and p38 MAPK) in RBL-2H3 mast cells [77]. Since SP is a mediator of neurogenic inflammation, SP induces prostaglandin (PG) production in various cell types, and these eicosanoids are responsible for numerous inflammatory and vascular effects. Cyclooxygenase (COX) are needed to convert arachidonic acid to PGs. COX-2 protein expression was upregulated by SP and exist a correlation between COX-2 expression and PGI (2) and PGE (2) release. Dexamethasone (DEX) inhibited SP-mediated COX-2 expression. MAPK p38 and ERKs (p42/44 MAPK) were activated by SP. NK-1R and NK-2R but not NK-3R receptors are present on HUVEC. Selective NK-1R and NK-2R agonists, upregulated COX-2 protein expression and PG production. The NK-1R antagonist L-703,606 and the NK-2R antagonist SR 48,968 competitively antagonized SP-induced effects [60]. Furthermore, early angiogenesis is a key step in the transition from acute to persistent inflammation. SP can initiate angiogenesis during acute neurogenic inflammation by increasing the proliferation rate of endothelial cells. In contrast, administration of an NK-1R antagonist, SR140333 (nolpitantium), prevents this [78]. In addition, the growth of new vessels from pre-existing vasculature, is a common feature of chronic inflammation, and early angiogenesis is a key step in the transition from acute to persistent inflammation [43]. In summary, the tumor inflammatory microenvironment is composed of glioma cells, the glioma tumor, inflammatory cells and endothelial cells, all express NK-1R, and SP, so they all release SP that when binding to the NK-1R of the cells of the tumor microenvironment release different inflammatory interleukins, producing two negative effects, one stimulates tumor growth and the other depresses the patient's immunity. Therefore, the use of the NK-1R antagonist would counteract the pathological effects mediated by SP, decreasing inflammation and tumor growth and improving the patient's immunity.
8. SP/HK-1 and NK-1R in Migration and Invasion of Glioma Cells (Figure 1)
The migration of tumor cells is the previous step for the invasion and dissemination of primary solid tumors, causing infiltration and secondary metastases in distant organs. It is known that tumor cell migration is induced by classical neurotransmitters (dopamine, noradrenaline) and peptides such as SP [79].
8.1. Epithelial-Mesenchymal Transition (EMT)
EMT is a biological process that allows a polarized epithelial cell to undergo multiple biochemical changes that allow it to assume a mesenchymal cell phenotype, which includes greater migratory capacity, invasiveness, high resistance to apoptosis and greatly increased production of ECM components [80]. In general, in tumors, EMT contributes to initiation migration, by loss of cell–cell adhesion, invasion by acquisition of migratory and metastasis to distant sites by loss of cell polarity, and resistance to chemotherapy and/or radiotherapy. In adults, intrinsic chemoresistance to glioma has been linked to EMT. Additionally, pediatric high-grade glioma (HGG) and DIPG are extremely chemoresistant, partially due to the blood–brain barrier (BBB) and partially due to intrinsic factors [Meel 2018]. EMT has also been suggested to play an important, yet poorly understood, role in DIPG biology and therapy resistance [81]. EMT is induced by external factors, such as cytokines, growth factors and hypoxia. The activation of EMT transcription factors is produced by different signaling pathways, most importantly SNAI1 (SNAIL), SNAI2 (SLUG), ZEB1, ZEB2 and TWIST. These transcription factors inhibit the expression of genes that promote cell–cell contact, most importantly E-cadherin, and induce genes that promote migration and stemness [81]. The cadherin is a transmembrane glycoprotein involved in intercellular adhesions and signaling, the cadherin switch is the result of activation of the EMT-related signaling pathways. E-cadherin (CDH1) is expressed in epithelial tissues and N-cadherin (CDH2) is found in neural tissue, muscle and fibroblast. EMT transcription factors after binding to cadherins causes the loss of expression of E-cadherin, and induction of expression of N-cadherin triggering a series of events that allows cancer cells to become less dependent of their micro-environment, thereby promoting cell migration, invasion and metastasis in epithelial tumor cells [82]. This possibility of a switch from E- to N-cadherin and its relationship to EMT is unsettled due to it has been report a double labeling for E-and N-cadherin in MPAP (invasive micropapillary carcinomas of breast cancer) tumors revealed that within individual tumors, N- and E-cadherin expression can be heterogeneous, suggesting the possibility that the tumor cells first down-regulate E-cadherin and invade and then can re-express E-cadherin as the tumor develops [83]. Several studies have also shown that changes in N-cadherin levels occur in malignant gliomas [84]. Interestingly, it is known that SP inducing EMT, and matrix metalloproteases that promote tumor invasion in head and neck cancers (HNC) cells [85]. In contrast, NK-1R antagonist L-703,606 counteracts several of these upregulated EMT-associated genes, producing an antimetastatic effects [85]. In addition, it has been reported the NK-1R antagonist aprepitant, counteracts the progression of EMT in a Balb/C mouse model of endometriosis [86]. Thus, the use of NK-1R antagonists such as the drug aprepitant may counteract the progression of EMT which may play an important, yet poorly understood, role in DIPG biology and therapy resistance.
Activation of EMT by Wnt/ β-catetin pathway allows cancer cells to survive individually, invade surrounding tissues, and metastasize [50]. It is known that self-renewal, proliferation and differentiation of neural progenitor cells (NPCs) in the brain during different stages of the central nervous system (CNS) is regulated by Wnt signaling. [50]. Wnt signaling can be subdivided in canonical and non-canonical (β-catenin independent). In glioma and other cancers have been linked to abnormal Wnt pathway activity and aberrant activation of Wnt/β-catenin signaling may play a crucial role in tumorigenesis. Importantly, active Wnt/β-catenin signaling has been associated with decreased glioma patient survival [87]. In the absence of Wnt, cytoplasmic β-catenin is constantly degraded by the Axin complex, composed of the Axin protein, the adenomatous polyposis coli (APC) tumor suppressor gene, casein kinase 1 (CK1), and GSK-3.Whereas Wnt ligands bind to the transmembrane receptor frizzled (Frz) and the co-receptor lipoprotein-related proteins 5 and 6 (LRP-5/6) inactivates GSK-3β and prevents it from phosphorylating β-catenin, thus stabilizing β-catenin in the cytoplasm as β-catenin accumulates, it translocates into the nucleus where it binds to TCF/LEF and increases their transcriptional activity. Genes up-regulated by TCF/LEF include proto-oncogenes, such as c-Myc and cyclin-D1, and genes regulating cell invasion/migration, such as MMP-7 [88]. Wnt proteins are also able to regulate the β-catenin- independent pathways, so-called noncanonical Wnt pathways that activate RhoA-Rock, Rac-1-JNKs, calcium/calmodulin-dependent protein kinase II, PKC and phospholipase C, for cell motility and polarity [89]. It has been reported that SP increases the expression of the genes and proteins associated with activation of the Wnt/ β-catenin signalling pathway of bone marrow stromal stem cells (BMSC) so SP increases the proliferation of stem cells [90]. Moreover, Wnt signaling is involved in the SP inhibition of apoptosis. SP exerts a protective effect, reduces the apoptotic rate: reduces nuclear condensation and inhibits the activation of caspase-3 and caspase-9 and reduced the percentage of cleaved caspase-3 positive cells. SP promotes the mRNA and protein expression of Wnt signaling molecules such as β-catenin, GSK-3β, c-Myc and cyclin D1 in bone marrow stromal stem cells [91]. Furthermore, the increased transcriptional activity of β-catenin results from the activation of EGFR through ERK that directly interacts with casein kinase 2 (CK2) without altering its stability and the level of phosphorylation by GSK-3 β in human glioblastoma cells U87E, U373 and LN229 and correlate with levels of ERK1/2 activity with grades of glioma malignancy. [92].
8.2. Blebbing on Glioma Cell Membranes
Blebbing on tumor cell membranes is important in the spread and invasion of cancer cells [93]. SP, after binding to NK-1Rs, induces changes in cellular cancer shape, including blebbing, which is crucial in cell movement/spreading and in cancer cell infiltration [94]. Moreover, it has been reported that Rho-associated protein kinase (Rock) is also involved in these changes, and in glioma cells SP induced the phosphorylation of p21-activated kinase (PAK) and enhanced phosphorylation of myosin regulatory light chain kinase (MLCK) resulting in blebs formation; however, this phenomenon was not observed in human normal cells [95]. In non-canonical Wnt/PCP signaling, the interaction of Wnt/Fzd leads to the recruitment of Dvl, which utilizes its domains (PDZ and DIX) to produce a complex with DAAM and then stimulates RhoA which can activate Rock [50]. Therefore, SP produces blebbing via non-canonical Wnt/PCP signaling mediated by subunit Gα0 and via subunit Gα12/13 [20].
8.3. The Extracelular Matrix (ECM)
ECM is the non-cellular component of tissues, which has been compared to the "glue" that holds cells together in connective tissues, where it is an important component of the tissue [96]. Matrix metalloproteinases (MMPs) are a group of active protein hydrolases found in the extracellular matrix. MMPs can degrade the ECM leading to tumor invasion and metastasis. The overexpression and secretion of MMP-2 and MMP-9 is increased in several types of human cancers and is associated with poor prognosis. Increased MMP-2 and MMP-9 expression has also been reported to correlate with cancer invasion [97]. MMP-2 and MMP-9 have also been closely associated with glioma progression and malignancy [98]. In glioma, it is known that HK-1, through NK-1R, dose-dependently promoted the migration of U-251 and U-87 glioma cells. Furthermore, HK-1 increased MMP-2 activity, as well as the expression of MMP-2 and MT1-MMP (makes MMP-2 active), which regulate cell migration. In U-25 glioma cells, HK-1 by NK-1R induces an increase in calcium release. However, inhibiting PLC reduces calcium release, MMP-2 and MT1-MMP activation, and HK-1-induced cell migration, suggesting that the NK-1R-mediated migration effect is by the Gαq-PLC pathway. HK-1 also increases phosphorylation of ERK, JNK and Akt and inhibition of ERK and Akt decreases the induction of MMP-2 and MT1-MMP by HK-1. Furthermore, HK-1 phosphorylates p65 and c-Jun and increases both AP-1 and NF-kB activity in U-251 glioma cells [30].
In summary, SP/HK-1 by NK-1R causes strong regulation and progression of EMT-associated genes in tumor cells, SP also induces blebbing of glioma cell membranes and increases the expression of MMP-2 and MT1-MMP in glioma cells, resulting in migration and invasion of human glioma cells.
9. NK-1R Antagonists
It is known that NK-1R receptor antagonists after binding to the NK-1R and in a concentration-dependent manner counteract all SP-mediated pathophysiological actions. NK-1Rs constitute a heterogeneous group of compounds. There are two types of NK-1R antagonists: non-peptide antagonists and peptide antagonists.
Peptide NK-1R antagonists (in the SP molecule, L-amino acids are changed by D-amino acids) are also known as SP analogue antagonists, synthetic analogues of SP or broad-spectrum peptide antagonists. Peptide antagonists of NK-1R show lower affinity for NK-1R than natural peptide agonists; they are metabolically unstable because they are hydrolyzed by peptidases; they are not lipid-soluble so they do not penetrate the brain; and some of them have toxic effects; therefore, they cannot be used for the treatment of glioma [8,43].
Non-peptide NK-1R antagonists include compounds that exhibit similar sterochemical characteristics and different chemical compositions; The therapeutic activity is linked to the affinity of these compounds for the NK-1R and the concentration [J Bioscience and Glioma Muñoz]. Non-peptide antagonists of the NK-1R are not degraded by peptidases and are lipid soluble/brain penetrants [8,43]. Currently there are the following NK-1R antagonist drugs: aprepitant, fosaprepitant, netupitant, fosnetupitant, rolapitant, tradipitant [99].
10. NK-1R Antagonists in Glioma Therapy (Figure 2)
10.1. Brain Penetrant
BBB prevents the use of most anti-tumor drugs, as they cannot cross the BBB. Therefore, it is essential that the drugs used can cross the BBB in the treatment of glioma. It is well known that NK-1R antagonist aprepitant is brain penetrant. In fact, aprepitant is used in chemotherapy induces nausea and vomiting (CINV)
Figure 2.
NK-1R antagonists, via the NK-1R, block these pathways and inhibit the effects mediated by SP on tumor. Abbreviations see Figure 1.
Figure 2.
NK-1R antagonists, via the NK-1R, block these pathways and inhibit the effects mediated by SP on tumor. Abbreviations see Figure 1.
10.2. Antitumor Action of NK-1R Antagonist in Glioma
NK-1R antagonists CP-96,345, L-733,060, L-732,138, MEN-11,467, MEN-11,149 the drug aprepitant and cyclosporine A (CsA) are known to have antitumor activity in glioma. All of them produce inhibition of proliferation and death by apoptosis in glioma cells. In addition, it is known that inducing apoptosis in tumor cells is a key anti-cancer treatment strategy [100].
10.3. NK-1R Antagonists Inhibit Mitogenesis and Induce Apoptosis in Glioma Cells
SP via NK-1Rs of U-373 MG glioma cells induces mitogenesis and induces the synthesis of c-Myc mRNA and protein, and that SP activates the MAPKs: ERK1/2 and p38MAPK. In contrast, NK-1R antagonist CP-96,345 in a concentration dependent manner suppresses DNA synthesis and activation of the MAPK pathway [45]. Furthermore, in a human glioma xenograft model, NK-1R antagonists (MEN-11,467 and MEN-11,149) have been shown to decrease tumor growth and exhibit antitumor activity [101]. In addition, three different NK-1R antagonists (L-733,060, aprepitant, L-732,138), at micromolar concentrations, inhibit the proliferation of GAMG glioma cells [28,31]. The antiproliferative action of the three NK-1R antagonists mentioned above is related to the affinity for NK-1R: the most potent antagonist against GAMG glioma cells was L-733,060 followed by aprepitant and L-732,138 [28,31]. The immunosuppressive drug CsA has also been shown to exert an antitumor action against GAMG glioma cells [102]. However, in clinical practice, the use of CsA is not possible, since the required antitumor dose induces nephrotoxicity. On the other hand, β-Arrestin deficiency increases the sensitivity of U251 MG and U87 MG glioma cells to treatment with NK-1R antagonists L-732,138 and [D-Arg1, DTrp5,7,9, Leu11] SP antagonist by increasing cell apoptosis and G2/M phase arrest [26].
GSK-3, a serine/threonine kinase, is involved in diverse cellular processes such as nutrient and energy homeostasis to glioma cell proliferation and apoptosis. Inhibition of GSK-3 activity induces phosphorylation of c-Myc site S62 and results in increased expression of apoptosis related molecules Bax, Bim, DR4/DR5 and TRAIL expression and subsequent cytotoxicity and apoptosis of glioma cell. Thus, GSK-3 may therefore be an important therapeutic target for glioma treatment [103]. In addition, NK-1R antagonists L-733,060 and L-732,138 in a concentration dependent manner counteract SP-induced GSK-3 phosphorylation [29]. Moreover, SP phosphorylates Akt and exerts an antiapoptotic effect in glioma cells [29]. In contrast, NK-1R antagonist aprepitant drug have been shown to induces apoptosis in GAMG glioma cells [31]. Furthermore, the NK-1R antagonist L-733,060 via NK-1R inhibits the basal kinase activity of Akt; it increases apoptosis and results in caspase-3 cleavage and poly (ADP-ribose) polymerase proteolysis in U-373 MG glioma cells [29]. Moreover, basal Akt activity has been linked to poor prognosis in glioma [29]. It should be noted that NK-1R is essential for glioma cell viability [10]. Furthermore, it is known that all glioma cells overexpress NK-1R. That means that H3K27M-mutated diffuse midline glioma necessarily overexpresses the NK-1Rs. Thus, the use of NK-1R antagonists as aprepitant drug may be an effective therapy in the treatment of this type of glioma, producing inhibition of mitogenesis and death by apoptosis. [29,31].
10.4. NK-1R Antagonists Antiangiogenic Effects
SP is a main mediator of neurogenic inflammation. SP and a selective peptide NK-IR agonist [β-Ala4, Sar9, Met(O2)11]-SP (4-11) induces a marked neovascularization. Moreover, SP through NK-1R of endothelial cells increases their proliferation in a concentration-dependent manner. Endothelial cell proliferation was followed by blood vessel formation and increased blood flow. Thus, SP can directedly stimulate the process of angiogenesis and neovascularization through induction of endothelial cells. In contrast, two NK-1R antagonists [D-Pro2, D-Trp7,9]-SP and [D-Pro4, D-Trp7,9, Ph11]-SP (4-11) counteract SP-mediates angiogenesis and proliferation endothelial cells in a concentration-dependent manner [59]. NK-1R antagonists also inhibit the angiogenic effects of HK-1, such as nitric oxide production, eNOS and VEGF expression in endothelial cells [63]. Furthermore, non-peptide NK-1 receptor antagonists, including the drug aprepitant, have been shown in xenograft models to block both angiogenesis and tumor growth [104,105]. NK-1R antagonist aprepitant also reduces the expression of VEGF and VEGFR in tumor cells [106,107,108].
10.5. NK-1R Antagonists Antimetastatic Effects in Glioma
Because metastasis causes 90% of cancer deaths [109]. Thus, one of the main goals of cancer treatment is to prevent this process. For cancer progression is necessary the migration, invasion and metastasis of cancer cells. In this sense, it has been reported that NK-1R antagonist L-733,060 inhibit SP-mediated tumor cell migration in MDA-MB-468 breast and PC-3 prostate carcinoma cells [79]. Regarding the glioma cells can invade and spread to the brain parenchyma over long distances. This is not a metastatic process, but rather an active dissemination, either along myelinated axons or perivascular spaces, or through the cerebrospinal fluid. Therefore, the therapeutic goal of glioma invasion is to prevent the detachment of cancer cells from the initial tumor mass to limit its dissemination [84]. Furthermore, it has been reported that downregulation of N-cadherin in glioma cells results in cell polarization defects leading to abnormal motility behavior with increased cell velocity and decreased persistence of directionality. In contrast, re-expression of N-cadherin in glioma cells restores cell polarity and limits glioma cell migration, providing a potential therapeutic tool for diffuse glioma [84]. In addition, it has been also reported that targeted inhibition of ZEB2 induced upregulation of E-cadherin expression and downregulation of vimentin expression. Furthermore, it reduced invasion and migration of pediatric glioma and adult glioma cells [110]. The cell adhesion molecule E-cadherin acts as an indispensable suppressor of cancer metastasis, and furthermore, increased E-cadherin can decrease β-catenin transcription, thereby decreasing Wnt/β-catenin signaling activity [Niu]. In colon cancer HCT116 cells, NK-1R antagonist NKP-608 reduces expressions of Wnt-3a, β-catenin, Cyclin D1, and VEGF while induces expression of E-cadherin. NK-1R antagonist NKP-608 also triggering apoptosis and induces apoptosis-related proteins reduces Bcl-2 and increases Bax and active Caspase-3 [106]. Therefore, treatment with the NK-1 receptor antagonist aprepitant could increase N-cadherin in glioma cells, restore cell polarity, and limit glioma cell migration, similar to E-cadherin in colon cancer cells. Moreover, NK-1R antagonist aprepitant inhibits the expression of MMP-2, MMP-9, VEGF-A, and VEGF receptor1 (VEGFR1) in ovarian carcinoma cells and in osteosarcoma cells [107,108]. NK-1R antagonist aprepitant also inhibits the SP-induced proliferation, cell migration and invasion and induced apoptosis of human squamous cells carcinoma (ESCC) cells, by downregulating the PI3K/AKT/mTOR signaling pathways [111].
In summary, therapeutic use of NK-1R antagonists such as aprepitant or similar drugs in DIPG patients can be useful because are brain penetrant and reach therapeutic concentration in the central nervous system (CNS). The antitumor action of NK-1R receptor antagonist drugs consists of inhibiting mitogenesis and promoting apoptosis of glioma cells, inhibiting angiogenesis by inhibiting both endothelial cell proliferation and VEGF, decreased migration of tumor cells, counteracting blebbing formation, and decreasing the expression of MMP-2 and MT1-MMP, and increases E-cadherin in glioma cells, resulting in decreased migration and invasion (metastasis).
10.6. NK-1R Antagonists Anti-Inflammatory
It has been reported in a model of inflammatory pain, NK-1R antagonist aprepitant suppresses microglia activation, JNK and p38 MAPK phosphorylation, and mRNA and protein expressions of TNF-α, IL-6, IL-1β and MCP-1, in vitro and in vivo [67].
Moreover, In an experimental brain tumor model, it has been reported that untreated animals showed increased albumin, SP, and NK-1R receptors in the peritumoral area, as well as increased perivascular staining in the surrounding brain tissue. Brain water content and blood-brain barrier permeability were significantly increased in tumor-inoculated animals compared with controls. However, treatment with the NK-1R antagonist aprepitant reduced BBB dysfunction and edema formation. This suggests that SP plays a role in the genesis of peritumoral edema and, therefore, could be a possible anti-edema treatment in brain tumors [112].
10.7. Combination of RT Plus NK-1R Antagonist
The current standard of treatment for DIPG consists of external beam RT at a dose of 54–60 Gy with conventional fractionation [113]. Radiotherapy produces a temporary improvement in neurological function, although the overall prognosis remains poor. Median overall survival is approximately 12 months after conventionally fractionated radiotherapy [114]. Unfortunately, it is well known that radiotherapy induces adverse effects on the surrounding normal brain, mainly producing inflammation and radionecrosis in the CNS [115]. The development of effective methods to enhance tumor radiosensitivity is crucial to improve the therapeutic efficacy of RT. Thus, it is a need to find drugs that increase the antitumor activity of RT (radiosensitization) and reduce serious side effects such as inflammation and radionecrosis in the treatment of patients with DIPG. It has been reported that the combination of radiotherapy and NK-1R antagonist aprepitant exerted a synergistic antitumor activity and decreased the side effects promoted by radiotherapy [116]. In a lung cancer patient was treated with palliative radiotherapy plus compassionate use of NK-1R antagonist aprepitant. After 45 days of treatment the lung tumor mass disappears and the side effects of radiotherapy were not observed [116]. In addition, it is known that radiotherapy elicits neurogenic inflammation through processes involving the NK-1R [117]. Radiotherapy-induced neurogenic inflammation in rats. In contrast, pretreatment with NK-1R antagonists GR203040 or GR205171 (at low dose 0,3/mg/kg) or dexamethasone inhibits radiotherapy-induces plasma protein extravasation in the duodenum and jejunum; this inhibition was increased when the combination of an NK-1R antagonist plus dexamethasone was used [117]. Interestingly, NK-1R antagonists GR203040 or GR205171 penetrate the brain [118]. Therefore, NK-1R antagonist decrease neurogenic inflammation induces by radiotherapy [117]. In addition, it has shown the radioprotective potential of N-acetyl-L-tryptophan (L-NAT) a NK-1R antagonist in neuronal cells [119]. Pretreatment with the NK-1R antagonist L-NAT resulted in DNA protection. NK-1R antagonist L-NAT pretreatment of irradiated Neuro2a cells neutralizes oxidative stress and reduce mitochondrial dysfunction. The expression of caspase-3 and γ-H2aX proteins was decreased, while the expression of p-ERK1/2 and p53 was increased in irradiated cells pretreated with L-NAT compared with irradiated cells. Therefore, NK-1R antagonist L-NAT could be a potential radioprotective that can inhibit oxidative stress and DNA damage and maintain mitochondrial health and Ca2+ levels by activating the expression of p-ERK1/2 and p53 in neuronal cell [119]. In addition, recently it has been reported that combination therapy of chemotherapy or radiotherapy and NK1R antagonist as a new strategy in cancer therapy [120]. In summary, combination of radiotherapy plus NK-1R antagonist aprepitant or similar drugs produces both radiosensitization of the tumor and can decrease the severe side effects of radiotherapy. Thus, this combination therapy could be useful in DIPG treatment.
10.10. Clinical Experience with the Drug Aprepitant, an NK-1R Antagonist
The drug aprepitant was approved the clinical use of aprepitant for chemotherapy induces nausea and vomiting (CINV) twenty years ago. Therefore, it is a well-known drug in oncology and there is a lot of experience with its use. The side-effects of aprepitant and fosaprepitant (a prodrug of aprepitant for intravenous use) are minimal, the most common (incidence higher than 10%) of which are constipation, fatigue, headache, anorexia, hiccups, and diarrhea [121]. Chemotherapy has an extremely low therapeutic index (at therapeutic doses, chemotherapeutics is also cytotoxic to normal tissues). In contrast, the therapeutic index of aprepitant is high. It has been reported adverse events associated with aprepitant in pediatric bone cancer patients includes febrile neutropenia in frequencies over 43.8 per 100 patients in one study [122]. In contrast, in a meta-analysis of 23 randomized controlled trials with 7956 patients, aprepitant was shown to be safe with no statistically significant differences in the incidence of febrile neutropenia [123]. Moreover, the cytotoxicity effect of aprepitant has been shown to be eight-fold higher in cancer cells than in normal cells (lymphocytes) [124]. As bone marrow toxicity is a requirement for neutropenia and aprepitant does not produce bone marrow toxicity, there should be no reason why aprepitant would cause febrile neutropenia in the setting of cancer patients. Furthermore, it is well known that aprepitant at 300 mg/day was safe, well-tolerated and showed side-effects similar to placebo [125]. Moreover, it has been reported, the use of a combination of palliative radiotherapy plus compassionate use of aprepitant in a lung cancer patient has generated very good clinical outcomes. The patient was treated for 45 days with radiotherapy plus aprepitant (1,140 mg/day for 45 days). Radiotherapy was administered to the right lung and mediastinum, reaching 50.4 Gy and then overprinted to 65 Gy. In each successive control, the tumor volume decreased and, after 6 months of treatment, the chest computed tomography scan showed that the tumor mass had disappeared. No side effects were observed during the combination therapy; and rather, the patient was in very good general health and weight gain and showed no biochemical analytical alteration [116]. Moreover, aprepitant may be useful in metastasis. It has been reported that a patient with brain metastasis from breast refractory to standard antiemetic therapies, was treated with aprepitant 80 mg/day and subsequently 125 mg/3 days for six months, improved clinically and the tumor marker CA153 decreased from 187 to 122, achieving control of nausea and vomiting without side effects [126]. Administration of aprepitant (375 mg/day/2 weeks) to HIV patients was shown to be safe and well tolerated and resulted in a decrease in the number of CD4+ PD-1 positive cells, reduction of both soluble CD163 and plasma SP levels [127]. Furthermore, overexpression of CD163 in glioma samples correlates with poor patient prognosis. CD163 expression is increased in glioma cells, especially in primary glioma cells. Loss of CD163 expression inhibits both cell cycle progression and proliferation of glioma cell lines and primary glioma cells. CD163 directly interacts with casein kinase 2 (CK2), and silencing of CD163 reduced the activity of the AKT/GSK-3β/β-catenin/cyclin D1 pathway through CK2. Thus, CD163 contributes to gliomagenesis through CK2 and suggests that the CD163 pathway could serve as a therapeutic target for glioma [128].
The different doses of aprepitant are different therapeutic effects: aprepitant doses (first day 125 mg; second day 80 mg; third day 80 mg) low doses for CINV. Aprepitant 300 md/day as antidepressant [125]. Aprepitant 375 mg/day as anti-inflammatory effects in HIV patients [127], both are moderate doses. The suggested dose for the treatment of glioma is 20–40 mg/kg/day (based on extrapolation from the concentration used in preclinical studies) are high doses [9,31]. Interestingly a high dose all the therapeutic effects are added. In 1907 (more than 100 years ago), German Nobel Prize winner Paul Ehrlich developed a revolutionary scientific concept in pharmacology, the magic bullet, which would be specific against tumor cells but not against the patient's normal cells. NK-1R antagonist drugs are new and promising anticancer drugs, and could be considered as a new magic bullet called an "intelligent bullet" [31]. The concept goes further because, in addition to their multiple anti-tumor actions, they have other therapeutic effects on patients such as CINV, antidepressant, anti-inflammatory and immunoprotective, and they are also safe.
11. Conclusions
Radiotherapy is current and generally nonspecific antitumor treatments that present two major problems, the resistance of glioma cells and serious side effects. For this reason, new antitumor procedures targeting glioma cells must be urgently developed; they must be effective drugs without serious side effects. Many preclinical studies have shown that the SP/NK-1R system is a good antitumor target because it is overexpressed in glioma cells and NK-1R are essential for the viability of glioma cells and non for normal cells. The SP/NK-1R system is involved in the proliferation of glioma cells, has antiapoptotic effects on glioma cells, induces the Warburg effect, triggering angiogenesis, produces inflammation, as well as the progression of EMT increasing the migration and invasion of glioma cells (infiltration and metastasis) and resistance to chemotherapy and/or radiotherapy. In contrast, NK-1R antagonists such as aprepitant or similar drugs penetrate the brain and in a concentration-dependent manner (high doses) have many therapeutic effects on glioma such as: inhibiting mitogenesis and inducing apoptosis in glioma cells, counteracting the Warburg effect in glioma cells, inhibiting angiogenesis, having anti-inflammatory effects, counteracting the progression of EMT by decreasing glioma cell migration and invasion (invasion and metastasis) as well as resistance to chemotherapy and/or radiotherapy. Furthermore, the combination of radiotherapy plus aprepitant may increase the antitumor activity of radiotherapy (radiosensitization of glioma). In addition, it has an anti-inflammatory effect both on peritumoral inflammation of the glioma tumor and on inflammation caused by radiotherapy and reduces the serious side effects of radiotherapy, it is neuroradioprotective by preventing radiotoxicity and radionecrosis. Moreover, aprepitant or similar drugs present other therapeutic effects in glioma patients such as antinausea and vomiting. Therefore, NK-1R antagonist drugs can be considered as a new magic bullet in the treatment of glioma, perhaps more accurately called a “intelligent bullet”, and we suggest the use of aprepitant to treat DIPG in combination with radiotherapy. In summary, the SP/NK-1R system provides a promising therapeutic target (NK-1R) in glioma and novel antitumor drugs (NK-1R antagonists) in glioma therapy. Therefore, aprepitant or similar drugs should be repurposed using NK-1R antagonist drugs in combination with radiotherapy in patients with DIPG. Furthermore, we suggest that phase I clinical trials are needed to evaluate the safety and phase II trials of aprepitant in combination with radiotherapy to evaluate the efficacy in patients with DIPG.
Author Contributions
“Conceptualization, M.M.; writing: original draft preparation, M.M. and M.R.; design of the Figure: M.R.; writing: review and editing, M.M. and M.R.; All authors have read and accepted the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Acknowledgments
RCP in memoriam.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, SM.; Reifenberger, G.; Soffietti, R.; von Deimling, A.; Ellison, D.W. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021, 23, 1231-12. doi: 10.1093/neuonc/noab106. [CrossRef]
- Johung, T.B.; Monje, M. Diffuse intrinsic pontine glioma: new pathophysiological insights and emerging therapeutic targets. Curr. Neuropharmacol. 2017, 15, 88-97.doi: 10.2174/1570159x14666160509123229. [CrossRef]
- Damodharan, S.; Lara-Velazquez, M.; Williamsen, B.C.; Helgager, J.; Dey, M. Diffuse intrinsic pontine glioma: molecular landscape, evolving treatment strategies and emerging clinical trials. J. Pers. Med. 2022,12,840. doi: 10.3390/jpm12050840. [CrossRef]
- Davidson, C.; Woodford, S.; Valle, D. Diffuse intrinsic pontine gliomas in pediatric patients: management updates. Egypt J Neurosurg. 2023, 38, 64. doi: 10.3390/jpm12050840. [CrossRef]
- Hennig, IM.; Laissue, JA.; Horisberger, U.; Reubi, JC. Substance-P receptors in human primary neoplasms: tumoral and vascular localization. Int. J. Cancer. 1995, 61, 786-92.doi: 10.1002/ijc.2910610608. [CrossRef]
- Cordier, D.; Forrer, F.; Kneifel, S.; Sailer, M.; Mariani, L.; Mäcke, H.; Müller-Brand, J.; Merlo, A. Neoadjuvant targeting of glioblastoma multiforme with radiolabeled DOTAGA-substance P results from a phase I study. J. Neurooncol. 2010, 100, 129-36. doi: 10.1007/s11060-010-0153-5. [CrossRef]
- Mehboob, R.; Kurdi, M.; Baeesa, S.; Fawzy Halawa, T.; Tanvir, I.; Maghrabi, Y.; Hakamy, S.; Saeedi, R.; Moshref, R.; Nasief, H.; Hassan, A.; Waseem, H.; Rasool, S.; Bahakeem, B.; Faizo, E.; Katib, Y.; Bamaga, A.; Aldardeir, N. Immunolocalization of neurokinin 1 receptor in WHO grade 4 astrocytomas, oral squamous cell and urothelial carcinoma. Folia Neuropathol. 2022, 60, 165-176. doi: 10.5114/fn.2022.116469. [CrossRef]
- Muñoz, M.; Coveñas, R. Glioma and Neurokinin-1 receptor antagonists: a new approach in glioma therapy. Anticancer Agents Med. Chem. 2019a, 19, 92-100. doi: 10.2174/1871520618666180420165401. [CrossRef]
- Muñoz, M.; Coveñas, R. The neurokinin-1 receptor antagonist aprepitant: an intelligent bullet against cancer?. Cancers. 2020, 12, 2682. doi: 10.3390/cancers12092682. [CrossRef]
- Muñoz, M.F.; Argüelles, S.; Rosso, M.; Medina, R.; Coveñas, R.; Ayala, A.; Muñoz, M. The Neurokinin-1 Receptor Is Essential for the Viability of Human Glioma Cells: A Possible Target for Treating Glioblastoma. Biomed. Res. Int. 2022, 6291504.doi: 10.1155/2022/6291504. [CrossRef]
- Muñoz, M.; Rosso M.; Coveñas, R. A new frontier in the treatment of cancer: NK-1 receptor antagonists. Curr. Med. Chem. 2010, 17, 504-516.doi: 10.2174/092986710790416308. [CrossRef]
- Pennefather, J.N.; Lecci, A.; Candenas, M.L.; Patak, E.; Pinto, F.M.; Maggi, C.A. Tachykinins and tachykinin receptors: a growing family. Life Sci. 2004, 74, 1445-63. doi: 10.1016/j.lfs.2003.09.039. [CrossRef]
- Chang, M.M.; Leeman, S.E.; Niall, H.D. Amino-acid sequence of substance P. Nat. New. Biol. 1971, 232, 86-7.doi: 10.1038/newbio232086a0. [CrossRef]
- Muñoz, M.; Coveñas, R.; Kramer, M. The involvement of the substance P/neurokinin 1 receptor system in viral infection: focus on the gp120 fusion protein and homologous dipeptide domains. Acta Virol. 2019b, 63, 253-260. doi: 10.1038/newbio232086a0. [CrossRef]
- Berger, A.; Paige, C.J. Hemokinin-1 has Substance P-like function in U-251 MG astrocytoma cells: a pharmacological and functional study. J. Neuroimmunol. 2005, 164, 48-56. doi: 10.1016/j.jneuroim.2005.03.016. [CrossRef]
- Muñoz, M.; Coveñas, R. Involvement of substance P and NK-1 receptor in human pathology. Amino Acids. 2014; 46, 1727–50. doi: 10.1007/s00726-014-1736-9. [CrossRef]
- Castro, T.A.; Cohen, M.C.; Rameshwar, P. The expression of neurokinin-1 and preprotachykinin-1 in breast cancer cells depends on the relative degree of invasive and metastatic potential. Clin. Exp. Metastasis. 2005, 22, 621-8. doi: 10.1007/s10585-006-9001-6. [CrossRef]
- Davoodian, M.; Boroumand, N.; Mehrabi, Bahar, M.; Jafarian, A.H.; Asadi, M.; Hashemy S.I. Evaluation of serum level of substance P and tissue distribution of NK-1 receptor in breast cancer. Mol. Biol. Rep. 2019, 46, 1285-1293. doi: 10.1007/s11033-019-04599-9. [CrossRef]
- Quartara, L.; Maggi, C.A.; The tachykinin receptor. Part II: distribution and pathophysiological roles. Neuropeptides. 1998, 32, 1-49. doi: 10.1016/s0143-4179(98)90015-4. [CrossRef]
- Muñoz, M.; Rosso, M.; Coveñas, R. Neurokinin-1 Receptor. In Encyclopedia of Signaling Molecules, 2nd ed.; Sangdun Choi Eds.; Publisher: Springer Nature Suwon, Korea, 2018; pp 3437-3445. doi.org/10.1007/978-3-319-67199-4. [CrossRef]
- DeFea, K.A.; Vaughn, Z.D.; O'Bryan, E.M. Nishijima, D.; Déry, O.; Bunnett, N.W. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta-arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 11086-91doi: 10.1073/pnas.190276697. [CrossRef]
- Fong, T.M.; Anderson, S.A.; Yu, H.; Huang, R.R.; Strader, C.D. Differential activation of intracellular effector by two isoforms of human neurokinin-1 receptor. Mol. Pharmacol. 1992, 41, 24–30.
- Douglas, S.D.; Leeman, S.E. Neurokinin-1 receptor: functional significance in the immune system in reference to selected infections and inflammation. Ann. N. Y. Acad. Sci. 2010, 1217, 83–95. doi: 10.1111/j.1749-6632.2010.05826.x. [CrossRef]
- Li, H.; Leeman, S.E.; Slack, B.E.; Hauser, G.; Saltsman, W.S.; Krause, J.E.; Blusztajn, J.K.; Boyd, N.D. A substance P (neurokinin-1) receptor mutant carboxyl-terminally truncated to resemble a naturally occurring receptor isoform displays enhanced responsiveness and resistance to desensitization. Proc. Natl. Acad. Sci. U.S. A. 1997, 94, 9475-80. doi: 10.1073/pnas.94.17.9475. [CrossRef]
- Cordier, D.; Gerber, A.; Kluba, C.; Bauman, A.; Hutter, G.; Mindt, T.L.; Mariani, L. Expression of different neurokinin-1 receptor (NK1R) isoforms in glioblastoma multiforme: potential implications for targeted therapy. Cancer Biother. Radiopharm. 2014, 29, 221-6. doi: 10.1089/cbr.2013.1588. [CrossRef]
- Zhang, Y.X.; Li, X.F.; Yuan, G.Q.; Hu, H. Song, X.Y.; Li, J.Y.; Miao, X.K.; Zhou, T.X.; Yang, W.L.; Zhang, X.W.; Mou, L.Y.; Wang, R. β-Arrestin 1 has an essential role in neurokinin-1 receptor-mediated glioblastoma cell proliferation and G2/M phase transition. J. Biol. Chem. 2017, 292, 8933-8947. doi: 10.1074/jbc.M116.770420. [CrossRef]
- Ogo, H.; Kuroyanagi, N.; Inoue, A.; Nishio, H.; Hirai, Y.; Akiyama, M.; DiMaggio, D.A.; Krause, J.E.; Nakata, Y. Human astrocytoma cells (U-87 MG) exhibit a specific substance P-binding site with the characteristics of an NK-1 receptor. J. Neurochem. 1996, 67, 1813-1820. doi: 10.1046/j.1471-4159.1996.67051813. x. [CrossRef]
- Muñoz, M.; Rosso, M.; Perez, A.; Coveñas, R.; Rosso, R.; Zamarriego, C.; Piruat, J.I. The NK1 receptor is involved in the antitumoral action of L-733,060 and the mitogenic action of substance P on neuroblastoma and glioma cell lines. Neuropeptides. 2005, 39, 427-32. doi: 10.1016/j.npep.2005.03.004. [CrossRef]
- Akazawa, T.; Kwatra, S.G.; Goldsmith, L.E.; Richardson, M.D.; Cox, E.A.; Sampson, J.H.; Kwatra, M.M. A constitutively active form of neurokinin 1 receptor and neurokinin 1 receptor-mediated apoptosis in glioblastomas. J. Neurochem. 2009, 109, 1079-86.doi: 10.1111/j.1471-4159.2009.06032.x. [CrossRef]
- Mou, L.; Kang, Y.; Zhou, Y.; Zeng, Q.; Song, H.; Wang, R. Neurokinin-1 receptor directly mediates glioma cell migration by up-regulation of matrix metalloproteinase-2 (MMP-2) and membrane type 1-matrix metalloproteinase (MT1-MMP). J. Biol. Chem. 2013, 288, 306-18. doi: 10.1074/jbc.M112.389783. [CrossRef]
- Muñoz, M.; Rosso, M. The NK-1 receptor antagonist aprepitant as a broad-spectrum antitumor drug. Invest. New. Drugs. 2010, 28, 187-193. doi: 10.1007/s10637-009-9218-8. [CrossRef]
- Palma, C.; Manzini, S. Substance P induces secretion of immunomodulatory cytokines by human astrocytoma cells. J. Neuroimmunol. 1998, 81, 127-37. doi: 10.1016/s0165-5728(97)00167-7. [CrossRef]
- Palma, C.; Nardelli, F.; Manzini, S.; Maggi, C.A. Substance P activates responses correlated with tumour growth in human glioma cell lines bearing tachykinin NK1 receptors. Br. J. Cancer. 1999, 79, 236-43. doi: 10.1038/sj.bjc.6690039. [CrossRef]
- Gitter, B.D.; Bruns, R.F.; Howbert, J.J.; Waters, D.C.; Threlkeld, P.G.; Cox, L.M.; Nixon, J.A.; Lobb, K.L.; Mason, N.R.; Stengel, P.W. Pharmacological characterization of LY303870: a novel, potent and selective nonpeptide substance P (neurokinin-1) receptor antagonist. J. Pharmacol. Exp. Ther. 1995, 275, 737-44.
- Johnson, C.L.; Johnson, C.G. Characterization of receptors for substance P in human astrocytoma cells: radioligand binding and inositol phosphate formation. J. Neurochem. 1992, 58, 471-7. doi: 10.1111/j.1471-4159.1992.tb09745.x. [CrossRef]
- Fowler, C.J.; Brännström, G. Substance P enhances forskolin-stimulated cyclic AMP production in human UC11MG astrocytoma cells. Methods Find. Exp. Clin. Pharmacol. 1994, 16, 21-8.
- Friess, H.; Zhu, Z.; Liard, V.; Shi, X.; Shrikhande, S.V.; Wang, L.; Lieb, K.; Korc, M.; Palma, C.; Zimmermann, A.; Reubi, J.C.; Büchler, M.W. Neurokinin-1 receptor expression and its potential effects on tumor growth in human pancreatic cancer. Lab. Invest. 2003, 83, 731-42.doi: 10.1097/01.lab.0000067499. 57309.f6. [CrossRef]
- Isorna, I.; González-Moles, M.A.; Muñoz, M.; Esteban, F. Substance P and Neurokinin-1 Receptor System in Thyroid Cancer: Potential Targets for New Molecular Therapies. J. Clin. Med. 2023, 12, 6409. doi: 10.3390/jcm12196409. [CrossRef]
- Lotz, M.; Vaughan, J.H.; Carson, D.A. Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science. 1988, 241, 1218-21. doi: 10.1126/science.2457950. [CrossRef]
- O'Connor, T.M.; O'Connell, J.; O'Brien, D.I.; Goode, T.; Bredin, C.P.; Shanahan, F. The role of substance P in inflammatory disease. J. Cell. Physiol. 2004, 201, 167. doi: 10.1002/jcp.20061. PMID: 15334652. [CrossRef]
- Mondal, M.; Mukherjee, S.; Bhattacharyya, D. Contribution of phenylalanine side chain intercalation to the TATA-box binding protein-DNA interaction: molecular dynamics and dispersion-corrected density functional theory studies. J. Mol. Model. 2014, 20, 2499. doi: 10.1007/s00894-014-2499-7. [CrossRef]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides. 2013, 48, 1-9. doi: 10.1016/j.peptides.2013.07.024. [CrossRef]
- Muñoz, M.; Coveñas, R.; Esteban, F.; Redondo, M. The substance P/NK-1 receptor system: NK-1 receptor antagonists as anti-cancer drugs. J. Biosciences. 2015 ,40, 441-463. doi: 10.1007/s12038-015-9530-8. [CrossRef]
- Eistetter, H.R.; Mills, A.; Brewster, R.; Alouani, S.; Rambosson, C.; Kawashima, E. Functional characterization of neurokinin-1 receptors on human U373MG astrocytoma cells. Glia. 1992, 6, 89-95. doi: 10.1002/glia.440060203. [CrossRef]
- Luo, W.; Sharif, T.R.; Sharif, M. Substance P-induced mitogenesis in human astrocytoma cells correlates with activation of the mitogen-activated protein kinase signaling pathway. Cancer Res. 1996, 56, 4983-91.
- Castagliuolo, I.; Valenick, L.; Liu, J.; Pothoulakis, C. Epidermal growth factor receptor transactivation mediates substance P-induced mitogenic responses in U-373 MG cells. J. Biol. Chem. 2000, 275, 26545-50. doi: 10.1074/jbc.M003990200. [CrossRef]
- Yamaguchi, K.; Kugimiya, T.; Miyazaki, T. Substance P receptor in U373 MG human astrocytoma cells activates mitogen-activated protein kinases ERK1/2 through Src. Brain Tumor Pathol. 2005 a, 22, 1-8.doi: 10.1007/s10014-005-0178-1. [CrossRef]
- Sharif, M.; Sharif, T.; Dilling, M.; Hosoi, H.; Lawrence, J.; Houghton, P. Rapamycin inhibits substance P-induced protein synthesis and phosphorylation of PHAS-I (4E-BP1) and p70 S6 kinase (p70(S6K)) in human astrocytoma cells. Int. J. Oncol. 1997, 11, 797-805. doi: 10.3892/ijo.11.4.797. [CrossRef]
- Zhao, X.F.; Gartenhaus, R.B. Phospho-p70S6K and cdc2/cdk1 as therapeutic targets for diffuse large B-cell lymphoma. Expert. Opin. Ther. Targets. 2009, 13, 1085-93. doi: 10.1517/14728220903103833. [CrossRef]
- Barzegar Behrooz, A.; Talaie, Z.; Jusheghani, F.; Łos, M.J.; Klonisch, T.; Ghavami, S. Wnt and PI3K/Akt/mTOR Survival Pathways as Therapeutic Targets in Glioblastoma. Int. J. Mol. Sci. 2022, 23, 1353. doi: 10.3390/ijms23031353. [CrossRef]
- Yamaguchi, K.; Richardson, M.D.; Bigner, D.D.; Kwatra, M.M. Signal transduction through substance P receptor in human glioblastoma cells: roles for Src and PKCdelta. Cancer Chemother. Pharmacol. 2005b, 56, 585-93. doi: 10.1007/s00280-005-1030-3. [CrossRef]
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000,10,94-9.
- Majewska, E.; Szeliga, M. AKT/GSK3β Signaling in Glioblastoma. Neurochem. Res. 2017; 42, 918-924. doi: 10.1007/s11064-016-2044-4. [CrossRef]
- Ballester, L.Y.; Wang, Z.; Shandilya, S.; Miettinen, M.; Burger, P.C.; Eberhart, C.G.; Rodriguez, F.J.; Raabe, E.; Nazarian, J.; Warren, K.; Quezado, M.M. Morphologic characteristics and immunohistochemical profile of diffuse intrinsic pontine gliomas. Am. J. Surg. Pathol. 2013, 37, 1357-64. doi: 10.1097/PAS.0b013e318294e817. [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science. 1956, 123, 309-314.doi: 10.1126/science.123.3191.309. [CrossRef]
- Medrano, S.; Gruenstein, E.; Dimlich, R.V. Substance P receptors on human astrocytoma cells are linked to glycogen breakdown. Neurosci. Lett. 1994, 167, 14-8. doi: 10.1016/0304-3940(94)91017-0. [CrossRef]
- Lochhead, P.A.; Coghlan, M.; Rice, S.Q.; Sutherland, C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes. 2001, 50, 937-46. doi: 10.2337/diabetes.50.5.937. [CrossRef]
- Harford-Wright, E.; Lewis, K.M.; Vink, R.; Ghabriel, M.N. Evaluating the role of substance P in the growth of brain tumors. Neurosci. 2014a, 261, 85-94. doi: 10.1016/j.neuroscience. [CrossRef]
- Ziche, M.; Morbidelli, L.; Pacini, M.; Geppetti, P.; Alessandri, G.; Maggi, CA. Substance P stimulates neovascularization in vivo and proliferation of cultured endothelial cells. Microvasc Res. 1990, 40, 264-78. doi: 10.1016/0026-2862(90)90024-l. [CrossRef]
- Gallicchio, M.; Rosa, A.C.; Benetti, E.; Collino, M.; Dianzani, C.; Fantozzi, R. Substance P-induced cyclooxygenase-2 expression in human umbilical vein endothelial cells. Br. J. Pharmacol. 2006, 147, 681-9. doi: 10.1038/sj.bjp.0706660. [CrossRef]
- Melincovici CS, Boşca AB, Şuşman S, Mărginean M, Mihu C, Istrate M, Moldovan IM, Roman AL, Mihu CM. Vascular endothelial growth factor (VEGF) - key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455-467.
- Takahashi, Y.; Kitadai, Y.; Bucana, C.D.; Cleary, K.R.; Ellis, L.M. Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res. 1995, 55, 3964-396.
- Song, H.; Yin, W.; Zeng, Q.; Jia, H.; Lin, L.; Liu, X.; Mu, L.; Wang, R. Hemokinins modulate endothelium function and promote angiogenesis through neurokinin-1 receptor. Int. J. Biochem. Cell Biol. 2012, 44, 1410-142. doi: 10.1016/j.biocel.2012.04.014. [CrossRef]
- Chen, W.; He, D.; Li, Z.; Zhang, X.; Pan, D.; Chen, G. Overexpression of vascular endothelial growth factor indicates poor outcomes of glioma: a systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 8709-19.
- Balkwill, F.; Mantovani, A. Inflammation and cancer: back to Virchow? Lancet. 2001, 357, 539-45. doi: 10.1016/S0140-6736(00)04046-0. [CrossRef]
- Burmeister, A.R.; Johnson, M.B.; Chauhan, V.S.; Moerdyk-Schauwecker, M.J.; Young, A.D.; Cooley, I.D.; Martinez, AN.; Ramesh. G.; Philipp, M.T.; Marriott, I.: Human microglia and astrocytes constitutively express the neurokinin-1 receptor and functionally respond to substance P. J. Neuroinflammation. 2017, 14, 245. doi: 10.1186/s12974-017-1012-5. [CrossRef]
- Yang, Y.; Zhou, W.; Xu, X.; Ge, X.; Wang, F.; Zhang, G.Q.; Miao, L.; Deng, X.;Aprepitant Inhibits JNK and p38/MAPK to Attenuate Inflammation and Suppresses Inflammatory Pain. Front. Pharmacol. 2022, 12,811584. doi: 10.3389/fphar.2021.811584. [CrossRef]
- Lieb, K.; Fiebich, B.L.; Berger, M.; Bauer, J.; Schulze-Osthoff, K. The neuropeptide substance P activates transcription factor NF-kappa B and kappa B-dependent gene expression in human astrocytoma cells. J. Immunol. 1997, 159, 4952-8.
- Lieb, K.; Schaller, H.; Bauer, J.; Berger, M.; Schulze-Osthoff, K.; Fiebich, B.L. Substance P and histamine induce interleukin-6 expression in human astrocytoma cells by a mechanism involving protein kinase C and nuclear factor-IL-6. J. Neurochem. 1998, 70, 1577-1583. doi: 10.1046/j.1471-4159.1998.70041577. x. [CrossRef]
- Fiebich, B.L.; Schleicher, S.; Butcher, R.D.; Craig, A.; Lieb, K. The neuropeptide substance P activates p38 mitogen-activated protein kinase resulting in IL-6 expression independently from NF-kappa B. J. Immunol. 2000, 165, 5606-11. doi: 10.4049/jimmunol.165.10.5606. [CrossRef]
- Yamaguchi, K.; Kumakura, S.; Murakami, T.; Someya, A.; Inada, E.; Nagaoka, I. Ketamine suppresses the substance P-induced production of IL-6 and IL-8 by human U373MG glioblastoma/astrocytoma cells. Int. J. Mol. Med. 2017, 39, 687–692. doi: 10.3892/ijmm.2017.2875. [CrossRef]
- Douglas, S.D.; Ho, W.Z.; Gettes, D.R.; Cnaan, A.; Zhao, H.; Leserman, J.; Petitto, J.M.; Golden, R.N.; Evans, D.L. Elevated substance P levels in HIV-infected men. A.I.D.S. 2001, 15, 2043-5. doi: 10.1097/00002030-200110190-00019. [CrossRef]
- Monaco-Shawver, L.; Schwartz, L.; Tuluc, F.; Guo, C.J.; Lai, J.P.; Gunnam, S.M.; Kilpatrick, L.E.; Banerjee, P.P.; Douglas, S.D.; Orange, J.S. Substance P inhibits natural killer cell cytotoxicity through the neurokinin-1 receptor. J. Leukoc. Biol. 2011, 89, 113-25. doi: 10.1189/jlb.0410200. [CrossRef]
- Lieberman, N.A.P.; DeGolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; Leary, S.E.S.; Crane, C.A. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: implications for development of immunotherapy. Neuro. Oncol. 2019, 21, 83-94.doi: 10.1093/neuonc/noy145.
- Lim, J.E.; Chung, E.; Son, Y. A neuropeptide, Substance-P, directly induces tissue-repairing M2 like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNγ. Sci. Rep. 2017, 7, 9417. doi: 10.1038/s41598-017-09639-7. [CrossRef]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; Peterson, E.; Leeman, S.; Conti, P. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4448-53. doi: 10.1073/pnas.1000803107. [CrossRef]
- Fang, X.; Hu, H.; Xie, J.; Zhu, H.; Zhang, D.; Mo, W.; Zhang, R.; Yu, M. An involvement of neurokinin-1 receptor in FcεRΙ-mediated RBL-2H3 mast cell activation. Inflamm. Res. 2012, 61,1257-63. doi: 10.1007/s00011-012-0523-x. [CrossRef]
- Seegers, H.C.; Hood, V.C.; Kidd, B.L.; Cruwys, S.C.; Walsh, D.A. Enhancement of angiogenesis by endogenous substance P release and neurokinin-1 receptors during neurogenic inflammation. J. Pharmacol. Exp. Ther. 2003, 306, 8-12. doi: 10.1124/jpet.103.050013. [CrossRef]
- Lang, K.; Drell, T.L. 4th; Lindecke, A.; Niggemann, B.; Kaltschmidt, C.; Zaenker, K.S.; Entschladen, F. Induction of a metastatogenic tumor cell type by neurotransmitters and its pharmacological inhibition by established drugs. Int. J. Cancer. 2004, 112, 231-8. doi: 10.1002/ijc.20410. [CrossRef]
- Kalluri, R.; Neilson, E.G. Epithelial- mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003, 112, 1776–1784. doi: 10.1124/jpet.103.050013. [CrossRef]
- Meel, M.H.; Schaper, S.A.; Kaspers, G.J.L.; Hulleman, E. Signaling pathways and mesenchymal transition in pediatric high-grade glioma. Cell Mol. Life Sci. 2018, 75, 871-887. doi: 10.1007/s00018-017-2714-7. [CrossRef]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727-35. doi: 10.1242/jcs.000455. [CrossRef]
- Agiostratidou, G.; Hulit, J.; Phillips, G.R.; Hazan, R.B. Differential cadherin expression: potential markers for epithelial to mesenchymal transformation during tumor progression. J. Mammary Gland Biol. Neoplasia. 2007, 12, 127-33. doi: 10.1007/s10911-007-9044-6. [CrossRef]
- Péglion, F.; Etienne-Manneville, S. N-cadherin expression level as a critical indicator of invasion in non-epithelial tumors. Cell Adh. Migr. 2012, 6, 327-32. doi: 10.4161/cam.20855. [CrossRef]
- Singh, S.; Kumaravel, S.; Dhole, S.; Roy, S.; Pavan, V.; Chakraborty, S. Neuropeptide Substance P Enhances Inflammation-Mediated Tumor Signaling Pathways and Migration and Proliferation of Head and Neck Cancers. Indian J. Surg. Oncol. 2021, 12, 93-102. doi: 10.1007/s13193-020-01210-7. [CrossRef]
- Liu, X.; Yan, D.; Guo, S.W. Sensory nerve-derived neuropeptides accelerate the development and fibrogenesis of endometriosis. Hum. Reprod. 2019, 34, 452-468. doi: 10.1093/humrep/dey392. [CrossRef]
- Liu, C.; Tu, Y.; Sun, X.; Jiang, J.; Jin, X.; Bo, X.; Li, Z.; Bian, A.; Wang, X.; Liu, D.; Wang, Z.; Ding, L. Wnt/beta-Catenin pathway in human glioma: Expression pattern and clinical/prognostic correlations. Clin. Exp. Med. 2010, 11, 105–112. doi: 10.1007/s10238-010-0110-9. [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell. 2009, 17, 9-26. doi: 10.1016/j.devcel.2009.06.016. [CrossRef]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell. 2003, 5, 367-77. doi: 10.1016/s1534-5807(03)00266-1. [CrossRef]
- Mei, G.; Xia, L.; Zhou, J.; Zhang, Y.; Tuo, Y.; Fu, S.; Zou, Z.; Wang, Z.; Jin, D. Neuropeptide SP activates the WNT signal transduction pathway and enhances the proliferation of bone marrow stromal stem cells. Cell Biol. Int. 2013, 37, 1225-32. doi: 10.1002/cbin.10158. [CrossRef]
- Fu, S.; Jin, D.; Liu, S.; Wang, L.; Wang, Z.; Mei, G.; Zou, Z.L.; Wu, J.Q.; Xu, Z.Y. Protective effect of neuropeptide substance P on bone marrow mesenchymal stem cells against apoptosis induced by serum deprivation. Stem Cells Int. 2015, 270328. doi: 10.1155/2015/270328. [CrossRef]
- Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q, Fang B, Fang X, Fang D, Litchfield DW, Aldape K, Lu Z. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol. Cell. 2009, 36, 547-59. doi: 10.1016/j.molcel.2009.09.034. [CrossRef]
- Fackler, O.T.; Grosse, R. Cell motility through plasma membrane blebbing. J. Cell. Biol. 2008, 181, 879-84. doi: 10.1083/jcb.200802081. [CrossRef]
- Meshki, J.; Douglas, S.D.; Lai, J.P.; Schwartz, L.; Kilpatrick, L.E.; Tuluc, F. Neurokinin 1 receptor mediates membrane blebbing in HEK293 cells through a Rho/Rho-associated coiled-coil kinase-dependent mechanism. J. Biol. Chem. 2009, 284, 9280-9. doi: 10.1074/jbc.M808825200. [CrossRef]
- Meshki, J.; Douglas, S.D.; Hu, M.; Leeman, S.E.; Tuluc, F. Substance P induces rapid and transient membrane blebbing in U373MG cells in a p21-activated kinase-dependent manner. PLoS One. 2011, 6, e25332. doi: 10.1371/journal.pone.0025332. [CrossRef]
- Rolfe, K.J.; Grobbelaar, A.O. A review of fetal scarless healing. I.S.R.N. Dermatol. 2012, 698034. doi: 10.5402/2012/698034. [CrossRef]
- Roomi, M.W.; Monterrey, J.C.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Patterns of MMP-2 and MMP-9 expression in human cancer cell lines. Oncol. Rep. 2009, 5, 1323-33. doi: 10.3892/or_00000358. [CrossRef]
- VanMeter, T.E.; Rooprai, H.K.; Kibble, M.M.; Fillmore, H.L.; Broaddus, W.C.; Pilkington, G.J. The role of matrix metalloproteinase genes in glioma invasion: co-dependent and interactive proteolysis. J. Neurooncol. 2000, 53, 213-35. doi: 10.1023/a:1012280925031. [CrossRef]
- Rodríguez, F.D.; Coveñas, R. Substance P receptor antagonists. In Molecular Mediators in Health and Disease: How Cells Communicate, Substance P; Vink, R. Eds.; Academic Press, Elsevier Inc, 2025; pp. 95-117. doi.org/10.1016/B978-0-443-22194-1.00010-0. [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: a link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. doi: 10.1016/s0092-8674(02)00625-6. [CrossRef]
- Palma, C.; Bigioni, M.; Irrissuto, C.; Nardelli, F.; Maggi, C.A.; Manzini, S. Anti-tumour activity of tachykinin NK1 receptor antagonists on human glioma U373 MG xenograft. Br. J. Cancer 2000, 82, 480-7. doi: 10.1054/bjoc.1999.0946. [CrossRef]
- Muñoz, M.; Rosso, M.; González, A.; Saenz, J.; Coveñas, R. The broad-spectrum antitumor action of cyclosporin A is due to its tachykinin receptor antagonist pharmacological profile. Peptides. 2010, 31, 1643-1648. 0.1016/j.peptides.2010.06.002. [CrossRef]
- Kotliarova, S.; Pastorino, S.; Kovell, L.C.; Kotliarov, Y.; Song, H.; Zhang, W.; Bailey, R.; Maric, D.; Zenklusen, J.C.; Lee, J.; Fine, H.A. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 2008, 68, 6643-51. doi: 10.1158/0008-5472.CAN-08-0850. [CrossRef]
- Guha, S.; Eibl, G.; Kisfalvi, K., Fan, R. S.; Burdick, M.; Reber, H., ... & Rozengurt, E. Broad-spectrum G protein–coupled receptor antagonist, [D-Arg1, D-Trp5, 7, 9, Leu11] SP: a dual inhibitor of growth and angiogenesis in pancreatic cancer. Cancer research. 2005, 65, 2738-2745.
- Berger, M.; Neth, O.; Ilmer, M.; Garnier, A.; Salinas-Martín, M.V.; de Agustín Asencio, J.C.; von Schweinitz, D., Kappler, R.; Muñoz, M. Hepatoblastoma cells express truncated neurokinin-1 receptor and can be growth inhibited by aprepitant in vitro and in vivo. J. Hepatol. 2014, 60, 985-94. doi: 10.1016/j.jhep.2013.12.024. [CrossRef]
- Niu, X.L.; Hou, J.F.; Li, J.X. The NK1 receptor antagonist NKP608 inhibits proliferation of human colorectal cancer cells via Wnt signaling pathway. Biol. Res. 2018, 51, 14. doi: 10.1186/s40659-018-0163-x. [CrossRef]
- Momen Razmgah, M.; Ghahremanloo, A.; Javid, H.; AlAlikhan, A.; Afshari, A.R.; Hashemy, S.I. The effect of substance P and its specific antagonist (aprepitant) on the expression of MMP-2, MMP-9, VEGF, and VEGFR in ovarian cancer cells. Mol. Biol. Rep. 2022, 49, 9307-9314. doi: 10.1007/s11033-022-07771-w. [CrossRef]
- Alsaeed, M.A.; Ebrahimi, S.; Alalikhan, A.; Hashemi, S.F.; Hashemy, S.I. The Potential In Vitro Inhibitory Effects of Neurokinin-1 Receptor (NK-1R) Antagonist, Aprepitant, in Osteosarcoma Cell Migration and Metastasis. Biomed. Res. Int. 2022, 8082608. doi: 10.1155/2022/8082608. [CrossRef]
- Sporn, M.B. The war on cancer. Lancet. 1996, 347, 1377-81. doi: 10.1016/s0140-6736(96)91015-6. [CrossRef]
- Myung, J.K.; Ah Choi, S.; Kim, S.K.; Ik Kim S, Park JW, Park SH. The role of ZEB2 expression in pediatric and adult glioblastomas. Anticancer Res. 2021, 41,175-185. doi: 10.21873/anticanres.14763. [CrossRef]
- Zheng, Y.; Sang, M.; Liu, F.; Gu, L.; Li, J.; Wu, Y.; Shan, B. Aprepitant inhibits the progression of esophageal squamous cancer by blocking the truncated neurokinin-1 receptor. Oncol. Rep. 2023, 50, 131. doi: 10.3892/or.2023.8568. [CrossRef]
- Harford-Wright E, Lewis KM, Ghabriel MN, Vink R. Treatment with the NK1 antagonist emend reduces blood brain barrier dysfunction and edema formation in an experimental model of brain tumors. PLoS One. 2014b,9, doi: 10.1371/journal.pone.0097002. [CrossRef]
- Hargrave, D.; Bartels, U.; Bouffet, E. Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol. 2006, 3, 241-8. doi: 10.1016/S1470-2045(06)70615-5. [CrossRef]
- Kim, H.J.; Suh, C.O. Radiotherapy for Diffuse Intrinsic Pontine Glioma: Insufficient but Indispensable. Brain Tumor Res. Treat. 2023, 11, 79-85. doi: 10.14791/btrt.2022.0041. [CrossRef]
- Limon, D.; McSherry, F.; Herndon, J.; Sampson, J.; Fecci, P.; Adamson, J.; Wang, Z.; Yin, F.F.; Floyd, S.; Kirkpatrick, J.; Kim, G.J. Single fraction stereotactic radiosurgery for multiple brain metastases. Adv. Radiat. Oncol. 2017, 2, 555-563. doi: 10.1016/j.adro.2017.09.002. [CrossRef]
- Muñoz, M.; Crespo, J.C.; Crespo, J.P.; Coveñas, R. Neurokinin 1 receptor antagonist aprepitant and radiotherapy, a successful combination therapy in a patient with lung cancer: A case report. Mol. Clin. Oncol. 2019, 11, 50-54. doi: 10.3892/mco.2019.1857. [CrossRef]
- Alfieri, A.B.; Cubeddu, LX. Efectos de los antagonistas de los receptores NK1 y de la dexametasona sobre la inflamación neurogénica inducida por ciclofosfamida y por radiación X, en la rata. A.V.F.T. 2004, 23, 61-66.
- Bergström, M.; Fasth, K.J.; Kilpatrick, G.; Ward, P.; Cable, K.M.; Wipperman, M.D.; Sutherland, D.R.; Långström, B. Brain uptake and receptor binding of two [11C]labelled selective high affinity NK1-antagonists, GR203040 and GR205171--PET studies in rhesus monkey. Neuropharmacology. 2000, 39, 664-70. doi: 10.1016/s0028-3908(99)00182-3. [CrossRef]
- Kumar, R.; Kumari, P.; Pandey, S.; Singh, S.K.; Kumar, R. Amelioration of Radiation-Induced Cell Death in Neuro2a Cells by Neutralizing Oxidative Stress and Reducing Mitochondrial Dysfunction Using N-Acetyl-L-Tryptophan. Oxid. Med. Cell Longev. 2022, 9124365. doi: 10.1155/2022/9124365. [CrossRef]
- Robinson, P.; Coveñas, R.; Muñoz. M. Combination Therapy of Chemotherapy or Radiotherapy and the Neurokinin-1 Receptor Antagonist Aprepitant: A New Antitumor Strategy?. Curr. Med. Chem. 2023, 30,1798-1812. doi: 10.2174/0929867329666220811152602. [CrossRef]
- Muñoz, M.; Coveñas, R. Safety of neurokinin-1 receptor antagonists. Expert. Opin. Drug Saf. 2013b, 12, 673-85. doi: 10.1517/14740338.2013.804059. [CrossRef]
- Okumura, L.M.; da Silva Ries, S.A.; Meneses, C.F.; Michalowski, M.B.; Ferreira, M.A.P.; Moreira, L.B. Adverse events associated with aprepitant pediatric bone cancer patients. J. Oncol. Pharm. Pract. 2019, 25, 735-738. doi: 10.1177/1078155218755547. [CrossRef]
- Zhang, M.; Guo, Q.L.; Zhang, T.T.; Fu, M.; Bi, H.T.; Zhang, J.Y.; Zou, K.L. Efficacy and safety of Aprepitant-containing triple therapy for the prevention and treatment of chemotherapy-induced nausea and vomiting: A meta-analysis. Medicine (Baltimore). 2023, 102, e35952. doi: 10.1097/MD.0000000000035952. [CrossRef]
- Molinos-Quintana, A.; Trujillo-Hacha, P.; Piruat, J.I.; Bejarano-García, J.A.; García-Guerrero, E.; Pérez-Simón, J.A.; Muñoz, M. Human acute myeloid leukemia cells express neurokinin-1 receptor, which is involved in the antileukemic effect of neurokinin-1 receptor antagonists. Invest. New Drugs. 2019, 37, 17–26. doi: 10.1007/s10637-018-0607-8. [CrossRef]
- Kramer MS, Cutler N, Feighner J, Shrivastava R, Carman J, Sramek JJ, Reines SA, Liu G, Snavely D, Wyatt-Knowles E, Hale JJ, Mills SG, MacCoss M, Swain CJ, Harrison T, Hill RG, Hefti F, Scolnick EM, Cascieri MA, Chicchi GG, Sadowski S, Williams AR, Hewson L, Smith D, Carlson EJ, Hargreaves RJ, Rupniak NM. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science. 1998, 281, 1640-5. doi: 10.1126/science.281.5383.1640. [CrossRef]
- Lee, M.; McCloskey, M.; Staples, S. Prolonged use of aprepitant in metastatic breast cancer and a reduction in CA153 tumour marker levels. Int. J. Cancer Clin. Res. 2016, 3, 071. doi: 10.23937/2378-3419/3/6/1071. [CrossRef]
- Tebas, P.; Spitsin, S.; Barrett, J.S.; Tuluc, F.; Elci, O.; Korelitz, J.J.; Wagner, W.; Winters, A.; Kim, D.; Catalano, R.; Evans, D.L.; Douglas, S.D. Reduction of soluble CD163, substance P, programmed death 1 and inflammatory markers: phase 1B trial of aprepitant in HIV-1-infected adults. A.I.D.S. 2015, 29, 931-9. doi: 10.1097/QAD.0000000000000638. [CrossRef]
- Chen, T.; Chen, J.; Zhu, Y.; Li, Y.; Wang, Y.; Chen, H.; Wang, J.; Li, X.; Liu, Y.; Li, B.; Sun, X.; Ke, Y. CD163, a novel therapeutic target, regulates the proliferation and stemness of glioma cells via casein kinase 2. Oncogene. 2019, 38, 1183-1199. doi: 10.1038/s41388-018-0515-6. [CrossRef]
Figure 1.
The substance P/neurokinin-1 receptor (SP/NK-1R) system regulates multiple cells signaling pathways involved in cancer progression: cell proliferation and migration, angiogenesis, inflammation, antiapoptotic mechanisms, Warburg effect and in the suppression of the immune system. Abbreviations: AA, Arachidonic acid ; AC, Adenylate cyclase ;AKT or PKB, Protein kinase B; APC, adenomatous polyposis coli; CK-1, Casein kinase 1;DAAM, disheveled-associated activator of morphogenesis; DAG, diacylglycerol; Dvl; disheveled; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; EMT, Epithelial-Mesenchymal Transition; ERK1/2, extracellular signal-regulated protein kinase; Fzd, frizzled; GSK3β, glycogen synthase kinase 3β; HK-1, hemokinin-1; IP3, inositol trisphosphate; LEC, leukotrienes; LRP5/6, low-density lipoprotein receptor-related protein 5/6; MAPKs, mitogen-activated protein kinases; MEKs: mitogen-activated protein kinase kinases; MMPs, Matrix metalloproteinases; NF-κB, nuclear factor kappa B; PKC, protein kinase C; PG, Prostaglandin; pMLC, Myosin light-chain kinase; PI3K, Phosphoinositide 3-kinase; PLA, Phospholipase A; PLC β, 1-Phosphatidylinositol-4,5-bisphosphate phospholipase beta; ROCK, Rho-associated kinase; RTK, Receptor tyrosine kinase; TCF/LEF, T-cell factor/lymphoid enhancer-binding factor; TNFα, Tumor necrosis factor alpha; TX, Thromboxane; VEGF, Vascular endothelial growth factor.
Figure 1.
The substance P/neurokinin-1 receptor (SP/NK-1R) system regulates multiple cells signaling pathways involved in cancer progression: cell proliferation and migration, angiogenesis, inflammation, antiapoptotic mechanisms, Warburg effect and in the suppression of the immune system. Abbreviations: AA, Arachidonic acid ; AC, Adenylate cyclase ;AKT or PKB, Protein kinase B; APC, adenomatous polyposis coli; CK-1, Casein kinase 1;DAAM, disheveled-associated activator of morphogenesis; DAG, diacylglycerol; Dvl; disheveled; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; EMT, Epithelial-Mesenchymal Transition; ERK1/2, extracellular signal-regulated protein kinase; Fzd, frizzled; GSK3β, glycogen synthase kinase 3β; HK-1, hemokinin-1; IP3, inositol trisphosphate; LEC, leukotrienes; LRP5/6, low-density lipoprotein receptor-related protein 5/6; MAPKs, mitogen-activated protein kinases; MEKs: mitogen-activated protein kinase kinases; MMPs, Matrix metalloproteinases; NF-κB, nuclear factor kappa B; PKC, protein kinase C; PG, Prostaglandin; pMLC, Myosin light-chain kinase; PI3K, Phosphoinositide 3-kinase; PLA, Phospholipase A; PLC β, 1-Phosphatidylinositol-4,5-bisphosphate phospholipase beta; ROCK, Rho-associated kinase; RTK, Receptor tyrosine kinase; TCF/LEF, T-cell factor/lymphoid enhancer-binding factor; TNFα, Tumor necrosis factor alpha; TX, Thromboxane; VEGF, Vascular endothelial growth factor.
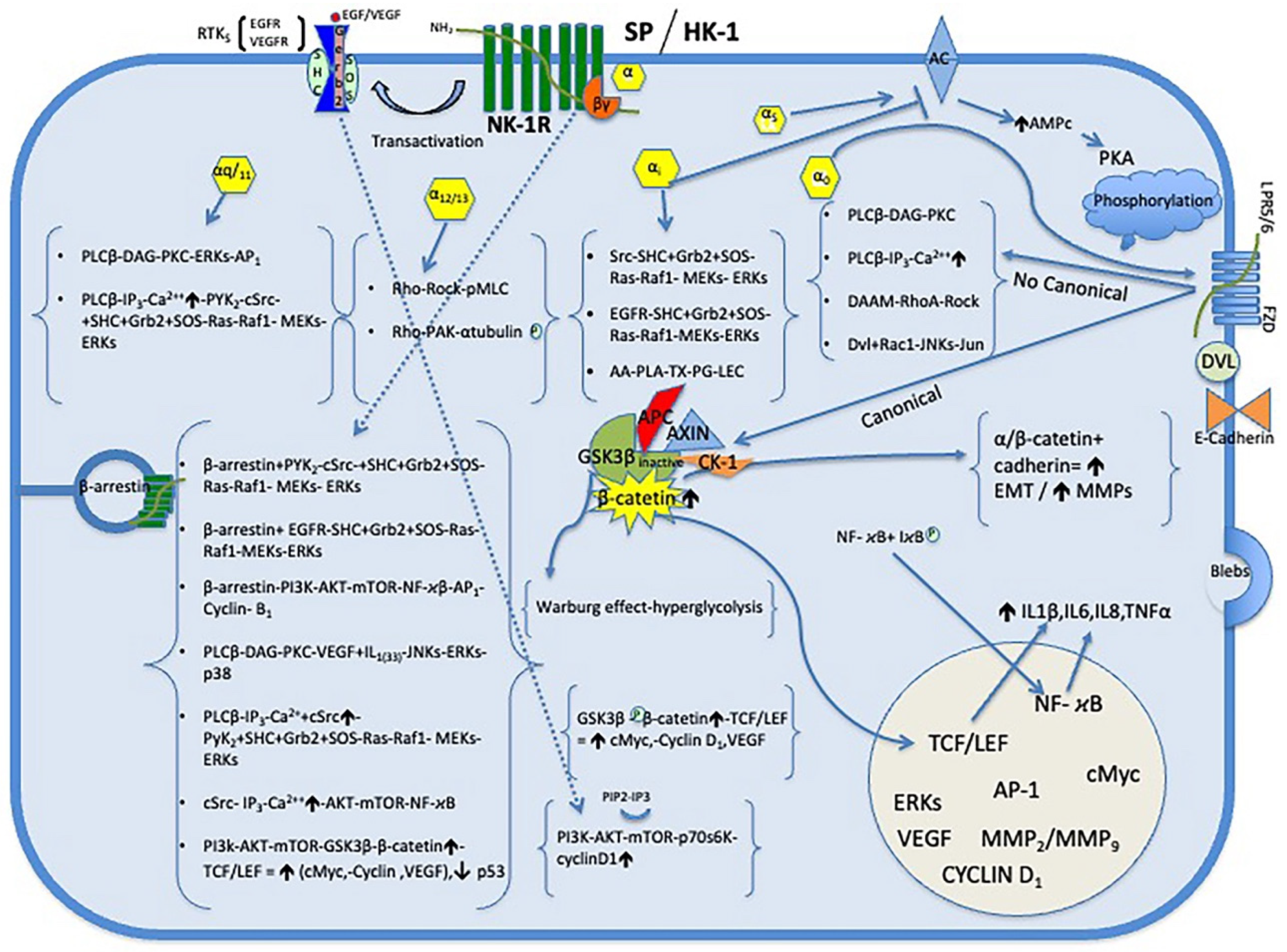
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



