Submitted:
24 December 2024
Posted:
25 December 2024
You are already at the latest version
Abstract
The ability to recognize hidden symmetry in a highly asymmetric world is a key factor in how we view and understand the world around us. Despite the fact it is an intrinsic property of the natural world, we have an innate ability to find hidden symmetry in asymmetric objects. The inherent asymmetry of the natural world is a fundamental property built into its chemical building blocks (e.g., proteins, carbohydrates, etc.). This review highlights the role of asymmetry in the structure of the carbohydrates and how these stereochemical complexities present synthetic challenges. This survey starts with an overview of the role synthetic chemistry plays in the discovery of carbohydrates and their 3D structure. The review then introduces various de novo asymmetric synthetic approaches that have been developed for the synthesis of carbohydrates and in particular oligosaccharides. The two most successful strategies for oligosaccharide synthesis rely on diastereoselective palladium-catalyzed glycosylation. The first uses an Achmatowicz reaction to asymmetrically prepare the pyranose building blocks along with a substrate-controlled Pd-glycosylation. The other strategy couples a ligand-controlled Pd-glycosylation with a ring-closing metathesis for oligosaccharide assembly.
Keywords:
Carbohydrates
; hexoses
; pyranoses
; oligosaccharides
; de novo asymmetric synthesis
1. Introduction
There appears to be an innate attraction to symmetry. This attraction is evident in how we explain and apply mathematical concepts, especially in geometry, where seeing potential symmetry helps simplify and better understand concepts. In fact, an appreciation for symmetry likely underlies aphorisms such as that “symmetrical faces are more beautiful”.[1] The natural tendency to notice hidden symmetry in the physical objects we see and the mathematical formulas we derive is probably best viewed as a generalization of how we view and understand nature rather than a perfect model of nature.[2] This simplification is necessary as a hallmark of all living organisms is an inherent asymmetry. In fact, it has been suggested that overuse of symmetry is behind our ability to recognize very lifelike robot faces as being artificial (aka, the uncanny valley).[3] In contrast to humans’ preference for seeing symmetry, asymmetry is a hallmark to the natural world.
Young artists often encounter nature's asymmetry when taught to break the symmetry in a drawing.[4] For example, when they lower a branch on one side of a tree branch to create a more natural appearance. This is a process called desymmetrization. When viewed in this way, inherent asymmetry in natural systems allows access to more states or the more efficient filling of space. This phenomenon of asymmetrically filling three-dimensional space can also be seen in organic chemistry. Asymmetry arises when four different atoms are attached to a single carbon atom, forming what is called a chiral carbon. The term 'chirality' comes from the Greek word for 'hand,' referring to two mirror-image molecules called enantiomers. In chemistry, enantiomers of chiral molecules are distinguished from one another with (+)/ (–), D-/L- or R-/S-labeling systems.[5]
There is a fundamental preference of one enantiomer over another in the biomolecules of nature (e.g., D-sugars and L-amino acids) that results from various diastereomeric interactions (i.e., right/right-handed vs right/left-handed interactions) in biosynthesis. This enantiomeric preference was amplified when nature’s chiral building blocks oligomerized into the biopolymers. Proteins which are made of many amino acids, have more chiral centers which leads them to exhibit significantly greater asymmetry. This increase in stereochemical complexity can also be seen in the structures of DNA (from nucleosides) and oligosaccharides (from sugars). In this context, stereochemical complexity can be defined in terms of both the number of stereocenters (n) and the possible stereoisomers (2n). Correspondingly, nature’s stereochemical bias is evident by the fact that only one of the possible isomers is produced. One can view nature’s predetermined stereochemical bias (e.g., D-sugars, L-amino acids) as resulting from the downstream effect of the diastereoselectivities inherent in the replication chemistry of life. An interesting result of the chiral bias in biosynthesis is that there are a greater number of molecular structures and three-dimensional spaces that are not used in the biomolecules of nature (Scheme 1).
Of the building blocks that make the biopolymers, carbohydrates possess the greatest degree of stereochemical complexity. For example, the hexose, D-glucose 1, the most common six-carbon carbohydrate, has four chiral centers in its acyclic form and five in its cyclic forms. In their simplest forms, the carbohydrates are compositionally defined by the molecular formula (CH2O)n, where n is the number of carbon atoms in the chain. When the carbon count reaches three (triose), the simplest chiral carbohydrate, glyceraldehyde 2, is formed, with a single chiral center at the C-2 position. Glyceraldehyde can exist in a D- or L-configuration, where the D-glyceraldehyde has an R-configured alcohol at C-2 and the L-glyceraldehyde is S-configured at C-2. This simple choice of D- vs L-sugars fundamentally established the chiral bias of the carbohydrates in nature. In four-carbon sugars (tetroses), a second stereocenter arises, resulting in four possible acyclic stereoisomers (2²) and eight cyclic, five-membered ring (furanose) isomers (2³). Five-carbon sugars (pentoses), having three chiral centers, yield eight acyclic stereoisomers (2³). Additionally, they form two sets of sixteen cyclic isomers (2⁴), consisting of furanoses and pyranoses (six-membered rings). Finally, hexose sugars, with four stereocenters, yield sixteen acyclic stereoisomers (2⁴) and three sets of thirty-two cyclic isomers (2⁵), including furanose, pyranose, and septanose (seven-membered ring) forms (Scheme 2).
Among the eight possible diastereomers of acyclic D-hexoses, nature primarily uses three in its biopolymers: glucose, mannose (C-2 epi-glucose) and galactose (C-4 epi-glucose), with glucose representing the majority of the monosaccharides used. In fact, glucose monomers make up the most prevalent oligosaccharides in nature: cellulose and amylose (aka, starch). When glucose is in an aqueous solution, it exists primarily in its cyclic form (~99%) with only a negligible amount in its acyclic form (< 1%). Of the possible cyclic forms, glucose exists almost exclusively in the pyranose form with minimal amounts (< 1%) of the furanose form and undetectable amounts of the septanose forms.
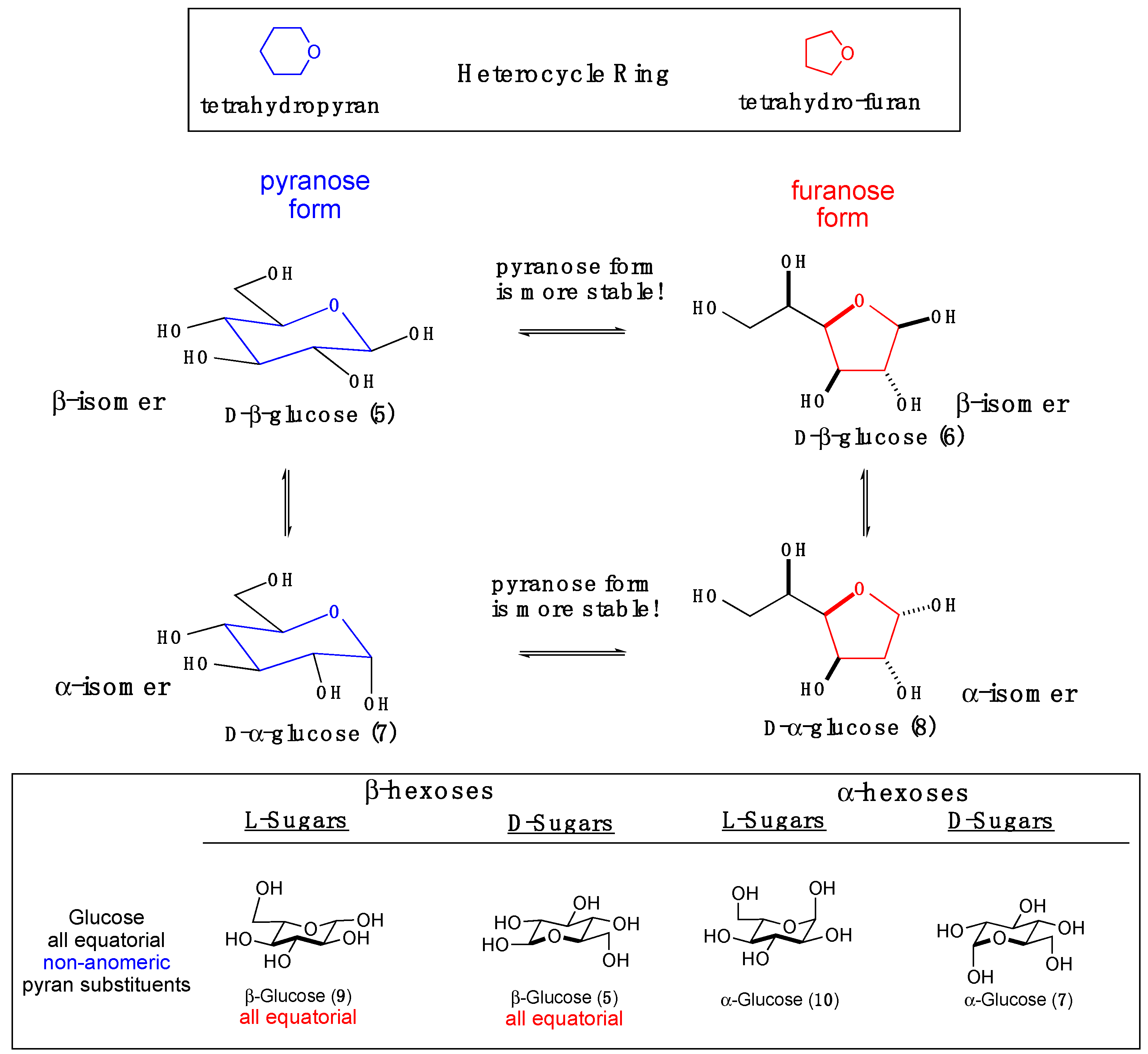
The hydroxy-aldehyde functionality of the hexoses can reversibly form cyclic structures, primarily five- and six-membered rings with a hemiacetal functionality at the C-1 position (Scheme 2). In the case of the furanose and pyranose rings, the equilibrium constant lies heavily in favor of the cyclic structures. In the cyclic form, the C-1 group is called the anomeric position, which is the point of attachment for the glycosidic bond. When in a chair conformation, the newly formed hemiacetal introduces a hydroxyl group and a new chiral center at the anomeric position. When the hydrogen of the anomeric hydroxyl group (OH) is replaced with an alkyl-group (OR), the newly formed acetal product is called an alkyl-glycoside. The anomeric group can have a configuration that is either trans (alpha (α)-stereochemistry) or cis (beta (β)-stereochemistry) relative to the C-6 substituent. There is a balance between countervailing steric and electronic effects that favor the α- and β-stereoisomers. While the equatorial β-isomer is sterically preferred, the axial α-isomer is electronically preferred. This electronic preference, called anomeric effect, arises from the antiperiplanar arrangement, where hyperconjugation provides electronic stabilization to the α-isomer.
A final element of structural complexity can be introduced when a sugar is connected via the anomeric position to another sugar (i.e., disaccharide). In the disaccharide case, when both hexoses are in their pyranose forms, there are ten possible isomers. This results from a combination of the five possible connections (regioisomers) with two possible stereochemistries (α/β). The structural complexity increases quickly when progressing from disaccharides and trisaccharides to oligosaccharides. In mammalian cells, the most common oligosaccharide connections are limited, due to enzymatic specificity and biosynthetic constraints. For example, cellulose is usually a 1,4-linked oligomer of glucose with the anomeric position in the β-configuration; whereas, amylose (starch) contains 1,4- and 1,6-linked oligomers of glucose with the anomeric position in the α-configuration. A greater degree of structural complexity in terms of sugars and connectivities exists in the various mammalian glycoprotein and glycolipid structures. This complexity is used by cells and proteins to communicate identity via the immune system. The situation becomes even more complex in the non-mammalian systems, as a significantly larger range of carbohydrate structures and oligosaccharide connectivity can be found therein.
In mammalian cells there are at least nine common monosaccharides, as defined by sugars that function as enzyme substrates. In addition to the three main monosaccharides in mammalian cells (glucose, mannose, and galactose), there are six other monosaccharides. These include four hexoses (N-acetylglucosamine, N-acetylgalactosamine, glucuronic acid, and fucose), a pentose (xylose) and, a nonose (sialic acid). Of these mammalian sugars the hexose fucose is unique in that it is an L-sugar (6-deoxy-L-galactose). Other non-mammalian rare sugars that are commonly contained in natural products are the six-deoxy sugars, D-quinovose (6-deoxy-D-glucose) and L-rhamnose (6-deoxy-L-mannose), the 2,6-dideoxy sugars D-olivose (2,6-dideoxy-D-glucose) and D-digitoxose (2,6-dideoxy-D-allose), and the 2,3,6-trideoxy sugars, L-amicetose (2,3,6-trideoxy-L-glucose) and rhodinose (2,3,6-trideoxy-L-galactose).
Historically, organic synthesis has played a critical role in the development of carbohydrate chemistry. This began in the 1800s with the formose synthesis of sugars as part of an effort to prove their molecular formulae (CH2O)n.[6] Then in the 1890s, carbohydrate synthesis evolved into the use of stereodivergent synthesis to assign the stereochemistry of the hexoses. This was followed by the development of glycosylation reaction methods for forming the glycosidic bond stereoselectively. This glycosylation chemistry was developed in combination with protecting group strategies for the regioselective construction of oligosaccharides. As the need for greater structural diversity and complexity grew, the synthetic chemistry evolved from diastereoselective synthesis to enantioselective chemistry. This growth in synthetic capability ultimately led to the use of asymmetric catalysis for the synthesis of the hexoses, which is called “de novo asymmetric synthesis.”[7],[8],[9],[10] Herein, this review will explore the historical development of these asymmetric synthetic methods.
2. Formose Synthesis
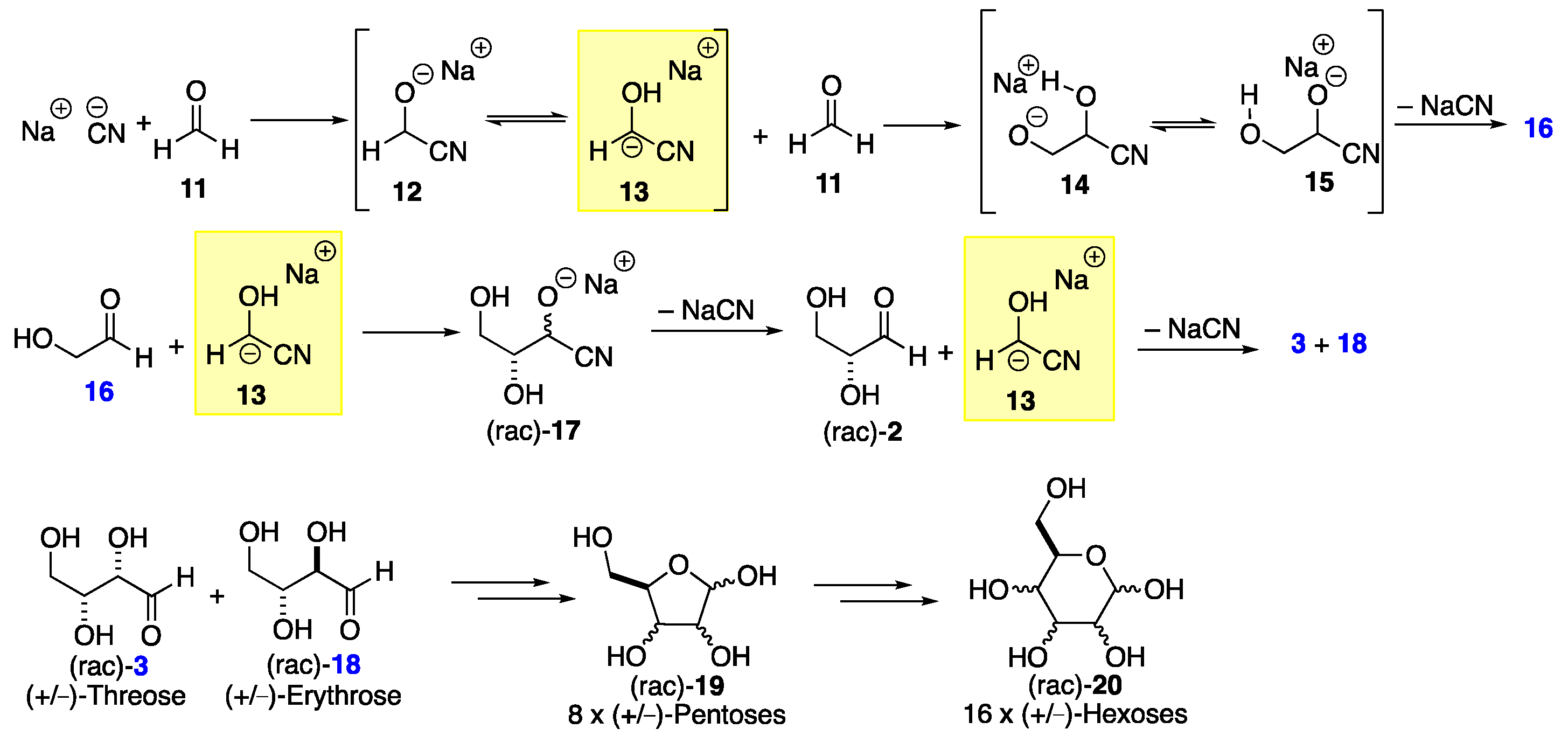
The structure and synthesis of sugars was an early topic of discovery during the emergence of organic chemistry in the 19th century. This research effort began with the realization that simple sugars shared the empirical formula CH2O with formaldehyde 11, one of the simplest organic molecules (O=CH2). Thus, the hexose glucose (CH2O)6 and the related shorter sugars (CH2O)n can be viewed as oligomers of formaldehyde (CH2O)1. Importantly, this oligomerization mechanism for the formose synthesis is significantly different than for the oligomerization process that converts formaldehyde to paraformaldehyde (O–CH2)n (Scheme 3). The initial attempts to oligomerize formaldehyde into sugars using Ca(OH)2 bases was met with limited success. Better results were found when the base catalyst was switched to cyanide salts. The use of NaCN enabled an iterative benzoin-type condensation for carbohydrate chain growth (e.g., 11 to 16 to 3 + 18).[11] Further improvements were achieved when the cyanide was replaced with N-heterocyclic carbene (NHC) catalysts like thiazolium salts.[12] However, it should be noted that these processes lack the ability to control stereochemistry or the number of carbons in the sugar products. When aqueous formaldehyde 11 is treated with NaCN as a catalyst, stereochemically complex mixtures of the two-carbon sugar (glycolaldehyde 16), trioses (racemic glyceraldehyde 2), and tetroses (racemic threose 3 and erythrose 18) are formed. Once sugars capable of forming stable hemiacetals are produced, the oligomerization process begins to slow down, as this hemiacetal formation removes a significant amount of the reactive aldehydes. However, longer chain sugars like the pentoses (rac-19) and the hexoses (rac-20) can also be detected.
The mechanism of chain propagation in the formose synthesis begins with the cyanide attack on formaldehyde 11 to form oxyanion 12, which equilibrates to carbanion 13. The insight gained the from understanding of the mechanism underlying this equilibrium process was fundamental in the development of many related Stetter-type processes and our understanding of thiamine biochemistry.[13],[14],[15] The nucleophilic carbanion 13 can be added to another molecule of formaldehyde 11 to form alkoxide 14, which can equilibrate further to alkoxide 15. Because of the proximity of the alkoxide anion in 15 to the cyano group, 15 can eliminate an equivalent of sodium cyanide, forming the simplest of the sugars, glycolaldehyde 16. Under the same reaction conditions, the aldehyde in 16 can further react with 13 via an analogous addition and proton migration to form 17, which can similarly eliminate cyanide to form glyceraldehyde 2 as a racemic mixture. After another round of addition/isomerization/elimination between rac-glyceraldehyde 2 and 13, a mixture of the racemic cis-triol threose 3 and trans-triol erythrose 18 is produced. Further extension by one or two carbons (via carbanion 13) leads to eight possible pentose diastereomers 19 or 16 possible hexose diastereomers 20, both as racemates. A full appreciation of the extent of the product mixture formed, including stereochemical diversity and chain lengths, only became possible with the development of modern analytical methods, although it should be noted how this work inspired the synthetically practical stepwise approach of Dondoni.[16]
3. Fischer Synthesis of the Hexoses
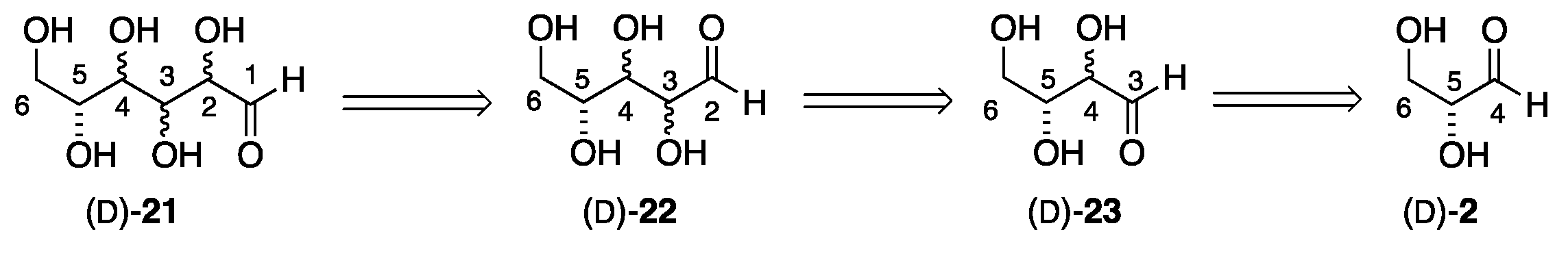
The full stereochemical complexity of hexoses was not completely understood until the late 19th century. This insight was gained from the synthetic and structural chemistry studies by Emil Fischer. The Fischer synthesis and structural proof of the hexoses were accomplished through a combination of a stepwise one-carbon homologation (chain growth) and a stepwise one-carbon degradative (chain reduction) reaction sequence. The homologation chemistry involved cyanohydrin formation (HCN addition to the aldehyde carbon of the sugar), followed by reduction and hydrolysis to form the aldehyde carbon of the new sugar. This process builds a two-carbon sugar into the trioses and onward (e.g., glyceraldehyde 2, to erythrose/threose 23, etc.).
In contrast to the stereo-randomness of the formose process, the Fischer approach is stepwise and as a result, introduces only one stereocenter at a time (Scheme 4). This allows for the separation of the diastereomers, which can then be subjected again to the cyanohydrin formation, reduction, and hydrolysis to exclusively prepare two new homologated stereoisomers (Scheme 5). Thus, glyceraldehyde 2 is converted into a mixture of tetrose sugars 23, which can be separated into erythrose 8 and threose 3. In turn, the mixture of tetroses can be converted into a mixture of pentoses 22, and finally to a mixture of hexoses 21 (Scheme 4). Alternatively, the purified erythrose 18 can be selectively converted into arabinose 27 and ribose 26. Similarly, threose 3 can be selectively converted into xylose 4 and lyxose 31(Scheme 6).
The basic three-step process developed by Fischer is outlined in Scheme 5. This process involved cyanohydrin formation, reduction/hydrolysis, and separation. The process began with the addition of HCN to D- or L-glyceraldehyde 2 with the addition to the D-enantiomer shown. The nucleophilic addition of cyanide to either face of the aldehyde in glyceraldehyde (D)-2 (via 24) affords triol 25 as a diastereomeric mixture. The carbon/nitrogen triple bond in nitrile 25 was reduced to form a diastereomeric mixture of aldehydes. At this point, the two aldehydes, (D)-3 and (D)-18, can be separated into optically pure forms. Importantly, the optical purity of (D)-3 and (D)-18 is determined by the configuration of the starting D-glyceraldehyde (D)-2. Thus, if L-glyceraldehyde (L)-2 was used as the starting material, the products would be L-erythrose (L)-18 and L-threose (L)-3. Conversely, using racemic glyceraldehyde (rac)-2 would yield racemic erythrose (rac)-18 and threose (rac)-3.
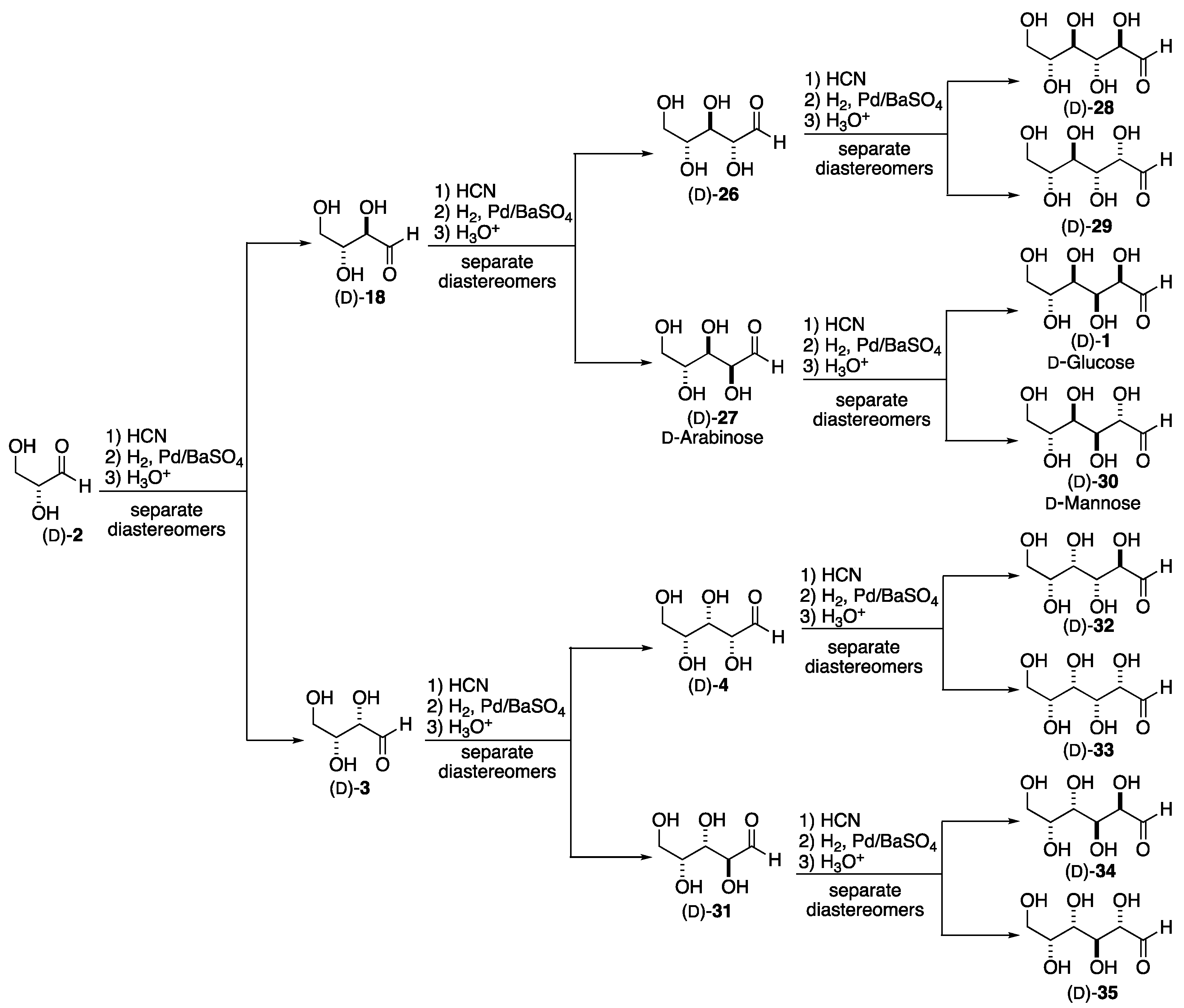
This same synthetic sequence of one-carbon chain extension and diastereomer separation can be further utilized to construct all the hexoses. Repeating the cyanohydrin formation, reduction/hydrolysis, and separation on the tetrose aldehydes, threose 3 yields the five-carbon sugars xylose 4 and lyxose 31, featuring C-2/3 syn-stereochemistry derived from threose 3. A similar three-step transformation was used to convert erythrose 18 into arabinose 27 and ribose 26. In an analogous fashion, the four pentose sugars ribose 26, arabinose 27, xylose 4, and lyxose 31—can be converted into the eight hexose sugars: allose 28, altrose 29, glucose 1, mannose 30, gulose 32, idose 33, galactose 34, and talose 35, respectively.
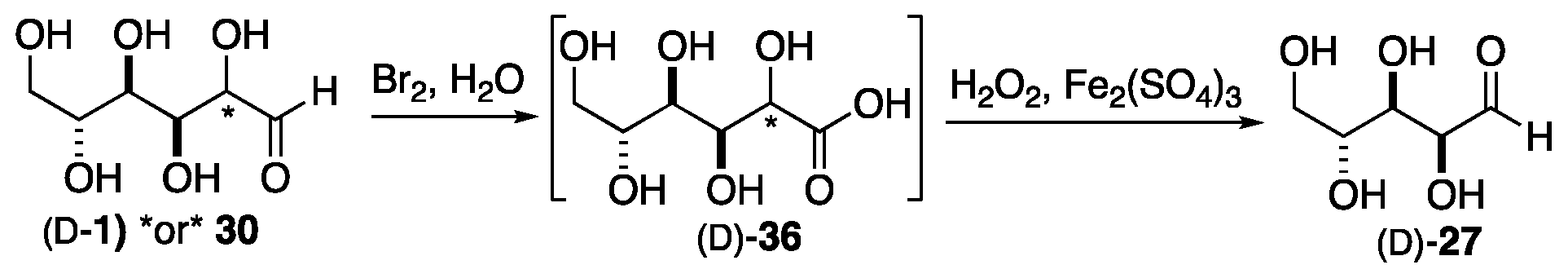
In practice, the essentially optically pure form of D- (or L-) glyceraldehyde 2 was obtained from readily available naturally sourced hexoses via a retro version of the Fischer methodology. This was accomplished using the Wohl degradation reaction (Scheme 7) to dismantle the carbohydrate one carbon at a time, from the reducing end of the sugar. The two-step oxidative degradation begins with an oxidation of the aldehyde to a carboxylic acid, followed by an oxidative decarboxylation of the hydroxy acid to yield a shortened sugar with a C-1 (formerly C-2) aldehyde. Thus, the Wohl degradation can produce pentose sugars from a hexose. For example, either glucose 1 or mannose 30 could be converted into arabinose 27 through the intermediate carboxylic acid 36. Two further rounds of this chain length reduction would produce D-glyceraldehyde (D-2) from any D-hexose, or L-glyceraldehyde (L-2) from any L-hexose. Importantly, the resulting pentose sugar is produced as a single diastereomer, where the C-3 to C-5 stereochemistry of the hexose starting material matches the C-2 to C-4 stereochemistry of the pentose product. The high stereospecificity of both the cyanohydrin formation and the Wohl degradation were key factors in enabling Fischer to assign the stereochemistry of all the hexoses.
4. Sharpless De Novo Asymmetric Synthesis of the Hexoses
Since the seminal work of Fischer, synthetic chemists have used the structures of carbohydrates as inspiration for the development of new asymmetric methods for the synthesis carbohydrate motifs. Of particular interest are those routes to sugars that start from achiral starting materials, where asymmetric catalysis is used to install the initial asymmetry (i.e., D- or L-sugar). These asymmetric routes, starting from achiral materials, are referred to as 'de novo' or 'de novo asymmetric' routes. It should be noted that in the carbohydrate synthesis community the term de novo has taken on very different meanings. The de novo asymmetric synthesis of the hexoses has been an important and ongoing interest to the synthetic community. Of these approaches, only the iterative epoxidation strategy of Masamune and Sharpless provides access to all eight hexoses. Their work utilized iterative asymmetric epoxidation of allylic alcohols to obtain the eight possible L-hexoses.[17],[18]
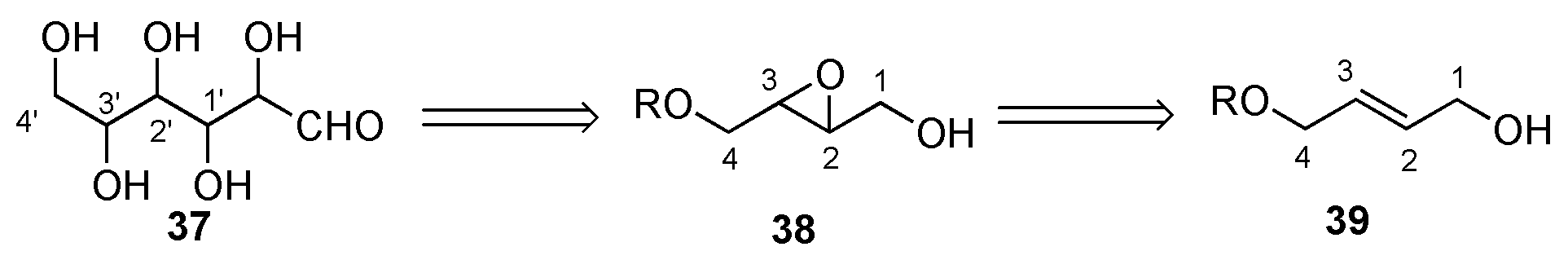
Retrosynthetically, the Masamune-Sharpless approach derives the asymmetry of the hexoses from a single trans-epoxide 38, which can be prepared in either enantiomeric form by means of Sharpless Asymmetric epoxidation of achiral allylic alcohol 39 (Scheme 8). Like the Fischer synthesis of the hexoses, the route developed by Masamune and Sharpless is stereodivergent. That is to say, the route systematically builds stereochemical complexity in a synthetic method that branches such as to allow for the construction of all the possible stereoisomers. The Sharpless epoxidation is accomplished by the use of an alkyl peroxide (e.g., cumene hydroperoxide) as the stoichiometric reagent and a catalytic amount of the complex that from the admixture of Ti(Oi-Pr)4 and diisopropyl tartrate. As the process is catalytic in its use of chiral reagent, the asymmetric epoxidation can be viewed as a transformation that amplifies the net asymmetry. Thus, a single molecule of the Sharpless reagent can be used to generate a mole of enantiomerically pure chiral epoxide. Importantly, tartaric acid is readily available in both its D- and L-enantiomeric forms.
The Masamune-Sharpless approach involves the ability to add to hydroxyl groups across the trans-alkene of an allylic alcohol from either face in a net syn- or net anti-selective manner. In addition to the Sharpless epoxidation (40 to 41), the route developed by Masamune and Sharpless inventively involves the use of a Payne rearrangement (41 to 43), Pummerer reaction (42 to 45 and acetonide equilibration (46 to 47). The use of these reactions is outlined in Scheme 9 and Scheme 10.
This Masamune-Sharpless synthesis of the hexoses begins with the asymmetric epoxidation of (E)-4-diphenylmethoxy-2-en-1-ol 40 using the (+)-DIPT ligand system to give the epoxide 41 in 92% yield and excellent enantioselectivity. Next, a base-mediated Payne rearrangement of internal epoxide 41 to intermediate terminal epoxide 43 was performed, preceding a regioselective epoxide opening using thiophenol to form a diol which was acetonide protected as acetonide 42. Next, thioether 42 was oxidized using mCPBA to give sulfoxide 44. A subsequent Pummerer rearrangement of 44 using Ac2O/NaOAc afforded acetate ester 45 as a diastereomeric mixture at the thio-substituted carbon (Scheme 9).
With two stereocenters established in 45, stereochemical divergence was possible based on the different hydrolysis conditions used. When DIBAL-H was used to hydrolyze 45, it produced aldehyde 46 in 91% yield without epimerization. Alternatively, when 45 was treated with K2CO3/MeOH, hydrolysis occurred in tandem with the desired epimerization at the adjacent stereocenter to form 47. A Wittig reaction was applied to both 46 and 47 to extend each molecule by two carbons, followed by NaBH4 reduction to afford terminal alcohols 48 and 53, respectively. Asymmetric epoxidation using either the (+)-DIPT or (–)-DIPT was applied to 48 and 53 to generate four new diastereomeric epoxides: 49, 50, 54, and 55 in 76%, 84%, 71%, and 73%, respectively. These epoxides were subjected to the NaSPH/Payne rearrangement, once again, to induce regioselective epoxide opening and hydrolysis. After acetonide formation, the protected tetraols 51, 52, 56, and 57 were produced in 77%, 63%, 79%, and 86% yields, respectively (Scheme 10).
Each of the four acetonides (51, 52, 56 and 57) could be converted into two L-sugars through three steps: oxidation, Pummerer rearrangement, and hydrolysis (with or without epimerization). For example, 51 was oxidized and rearranged to afford 58 in 90% yield. 58 was then hydrolyzed, with or without epimerization, to afford aldehydes 60 and 59, respectively. Finally, these two protected L-sugars were acetonide deprotected, followed by hydrogenation to liberate the terminal alcohol, to afford L-allose (L)-28 and L-altrose (L)-29 in good yields. Correspondingly, L-mannose (L)-30 and L-glucose (L)-1 were produced from 52 in 77% and 22% yields, respectively. L-Gulose (L)-32 and L-idose (L)-33 were produced from 56 in 59% and 49% yields, respectively. Lastly, L-talose (L)-35 and L-galactose (L)-34 were produced from 57 in 33% and 8% yields, respectively (Scheme 11).
5. Other De Novo Asymmetric Carbohydrate Syntheses
In terms of synthetic efficiency, the Masamune-Sharpless approach to the hexoses is particularly impressive in how few total steps are required to prepare all eight hexoses. However, it should be noted that in terms of an asymmetric synthesis of a specific hexose, the Masamune-Sharpless route is step-intensive (~15 steps). In the subsequent years, significantly shorter and more efficient de novo asymmetric approaches have been developed to specific sugars (Scheme 12). Like the Masamune-Sharpless approach, these routes use highly enantio- and diastereo-selective reactions to install the two initial stereocenters, followed by diastereoselective transformations to establish the remaining sugar stereochemistry. A notable example of this can be seen in the use of the Jacobsen variant of the Danishefsky hetero-Diels-Alder reaction (63+ 64) for the synthesis of pyran 62, which was used in the synthesis of fucose sugar 61.[19],[20] An asymmetric approach to an allose sugar 65 was developed by MacMillan from chiral fragment 66, which was prepared by the dimerization of 67 via a proline-catalyzed aldol reaction.[21],[22] Hudlicky developed an enantioselective synthesis of protected mannose derivative 68 from chiral diol 69, which was prepared by the enzymatic oxidation of chlorobenzene 70.[23],[24] A related biocatalytic approach was developed by Johnson that uses the enzymatic desymmetrization of triol 72 in the synthesis of dideoxyglucose 71.[25],[26] There is also a mixed asymmetric dihydroxylation and enzymatic aldol approach by Wong and Sharpless.[27] Finally, another Sharpless asymmetric oxidation[28],[29] was applied in the iterative dihydroxylation of dienoates, such as 76,[30] yielding galacto-lactones through the intermediate diol 75.[31],[32],[33],[34],[35] At the end of the 20th century, the O’Doherty group developed a more systematic de novo asymmetric approach to the hexoses, based upon the Achmatowicz oxidation of furan alcohols to pyran ketones (pyranones) in combination with a Pd-glycosylation reaction (vide infra).[36],[37],[38],[39] The route has also been applied to several higher order sugars.[40],[41],[42],[43],[44]
6. De Novo Asymmetric Achmatowicz Approach to Carbohydrates
The O’Doherty approach to de novo synthesis of sugars is based on the idea of simplifying all the pyranoses into just four pyranones (Scheme 13). These four pyranones each contain two chiral centers, at C-1 (setting α/β configuration) and C-5 (setting D-/L-configuration). In terms of absolute stereochemistry, the enantiocontrol is established in a furan alcohol by means of an asymmetric reduction of an acylfuran or oxidation of a vinylfuran.[45],[46] An Achmatowicz rearrangement can convert the furan alcohol into a pyranone ring where the stereochemistry at C-5 is derived from the furan alcohol A diastereoselective acylation or carbonate formation can then be used to control the C-1 stereochemistry (α or β). A stereochemically retentive Pd-allylation is then used transfer the α- or β-stereochemistry to the pyranone product, which can then be converted into various pyranoses (vide infra).[47],[48],[49]
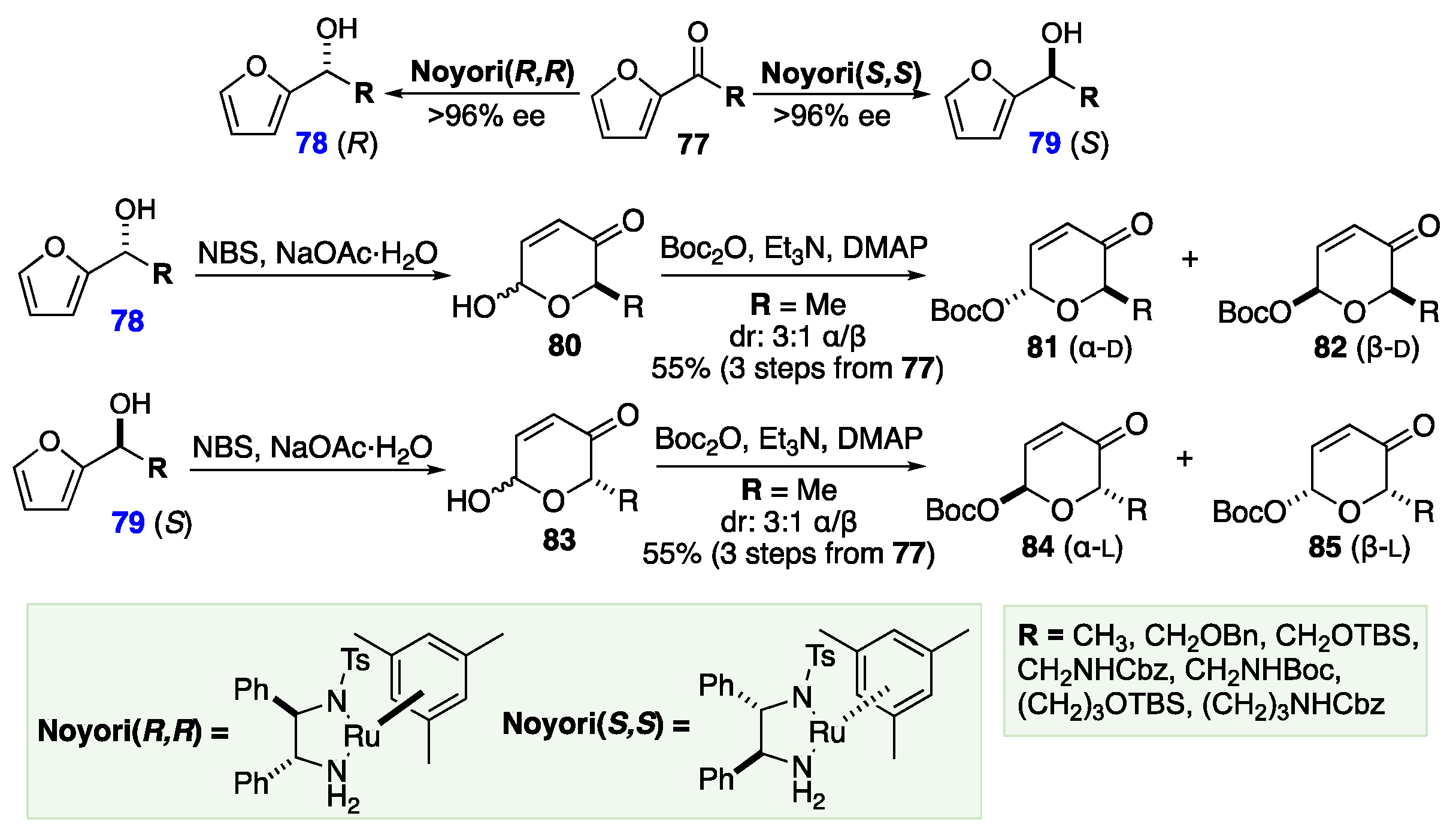
In practice, the process begins with achiral furyl ketone 77, which can be functionalized at the α-methyl position (C-6) with a variety of useful substituents. 77 can be reduced to the R- or S-chiral alcohol (78 or 79 respectively), with excellent enantioselectivity, via Noyori asymmetric hydrogenation using a chiral ruthenium catalyst.[50],[51],[52],[53] Next, these alcohols are subjected to Achmatowicz rearrangement to pyran alcohols 80 and 83, from furan alcohols 78 and 79, respectively; the C-5 stereochemistry being set by the previous asymmetric reduction. Next, the equilibrating mixture of anomers is OH-protected, locking the pyranone into the α-configuration (81 and 84), or the β-configuration (82 or 85). The α/β ratio can be controlled by altering the protecting groups, or reaction conditions (Scheme 13). When the anomeric protecting group is an ester or carbonate group, it also serves as a Pd-π-allyl-leaving group for a Pd-catalyzed allylation reaction for coupling carbon, nitrogen, and oxygen nucleophiles. [54],[55],[56]
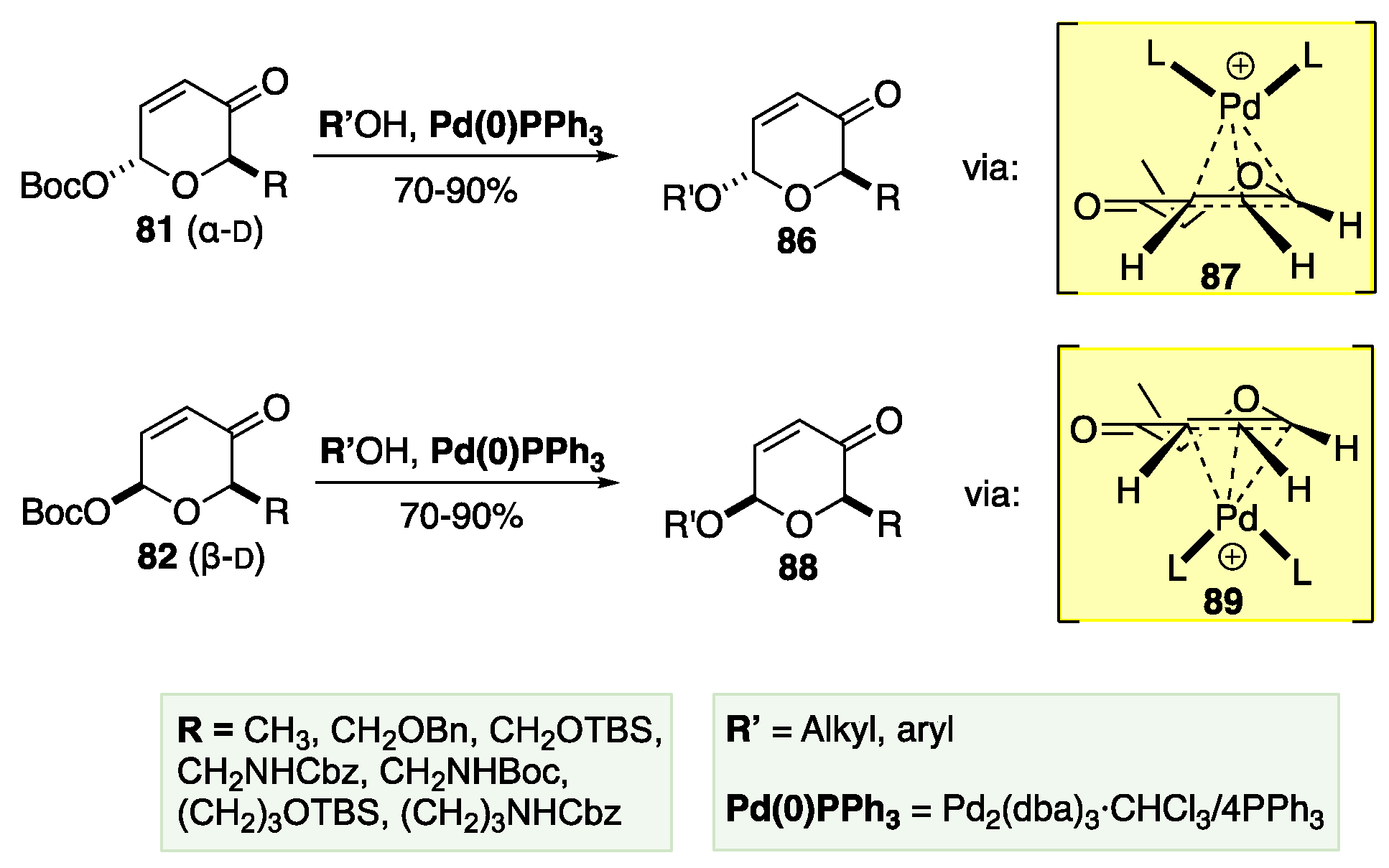
The next critical part of O’Doherty methodology is the utilization of a highly diastereoselective Pd-catalyzed Tsuji-Trost glycosylation (Scheme 14).[57],[58],[59],[60] This allows the anomeric position to be functionalized with a variety of different nucleophiles with complete stereochemical retention of the established C-1 stereochemistry. The diastereoselectivity of these transformations stems from the stereochemistry of the intermediate Pd complex. For example, α-pyranone 81 is converted into α-pyranone 86 via the β-intermediate 87 in 70-90% yield. The Pd catalyst coordinates to form the π-allyl on the opposite face of the C-1 leaving group. Therefore, when the alcohol attacks the π-allyl, it must approach from the opposite face as the Pd-group, which leads to the overall stereochemical retention. The same principle guides the transformation of β-82 to β-88 through the α-transition state 89 with a similarly strong yield. The route has been successfully applied to several oligosaccharide targets,[61],[62],[63],[64],[65],[66] with no or minimal use of protecting groups.[67]
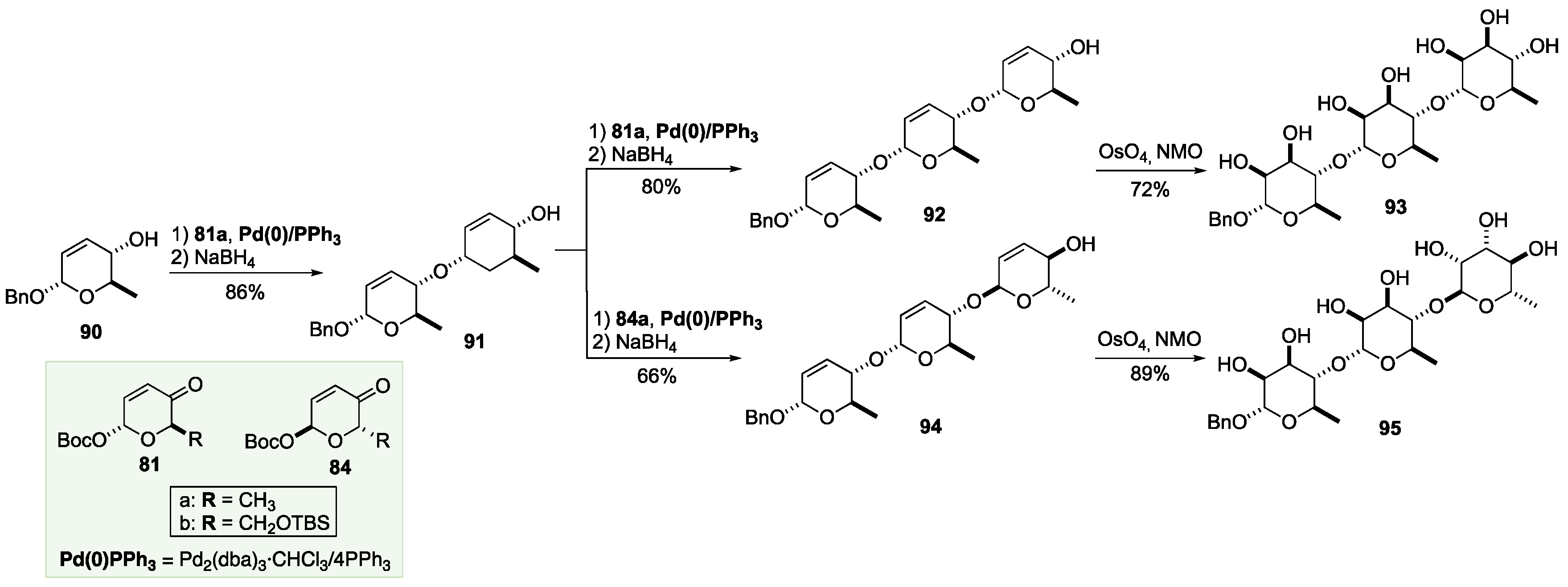
Where the Achmatowicz approach really distinguishes itself from the other de novo asymmetric approaches to carbohydrates, is in its application to oligosaccharides, where the C-2/C-3 alkene can function as an atomless-protecting group (Scheme 15 and Scheme 16). By carrying the alkene functionality to the end of the oligosaccharide, these routes have excellent step economy when applied to both linear and branched oligosaccharides.[68],[69] The route to the linear sugars started with allylic alcohol 90, which was prepared from ketone 81a in two steps (Scheme 15). A Pd-catalyzed glycosylation was accomplished using pyranone 81a, followed by a reduction to afford disaccharide precursor 91. By repeating this diastereoselective elongation of 91 (glycosylation/reduction) with two possible D/L-diastereomeric α-pyranones, trisaccharide precursors 92 and 94 were synthesized; 92 was by glycosylation and reduction using pyranone 81a, and 94 through a glycosylation and reduction using pyranone 84a. These two diastereomeric routes occurred with equivalent excellent overall efficiency and stereochemical control. A similar diastereoselective osmium-catalyzed dihydroxylation of the trienes 92 and 94 afforded trisaccharides 93 and 95, respectively (Scheme 15).
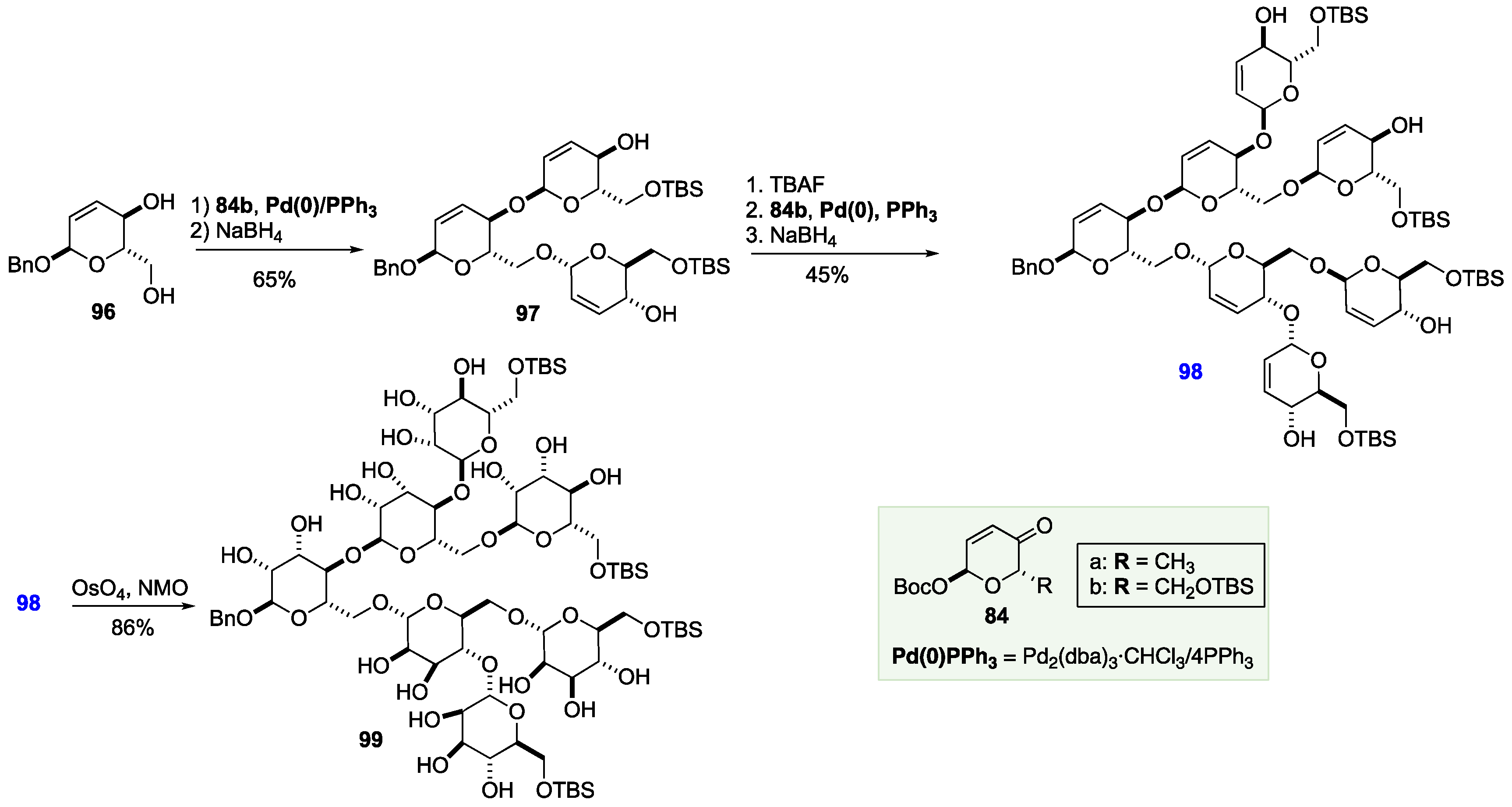
The power of the de novo Achmatowicz approach in terms of step economy is particularly preannounced in the application to the branched oligosaccharides. This route began with a Pd-catalyzed bis-glycosylation of two equivalents of pyranone 84b with diol 96, which was synthesized from 84b in three steps. Following reduction with NaBH4, allylic alcohol 97 was obtained in 65% yield. Following TBAF-mediated removal of the two TBS groups, the resulting tetraol was again subjected to the two-step protocol of glycosylation (with four equivalents of 84b) and reduction to yield heptasaccharide precursor 98 in 45% yield over three steps. Finally, dihydroxylation was performed using the Upjohn conditions of OsO4/NMO to give the manno-hepta-saccharide 99 in 86% yield. When viewed from the perspective its high degree of stereochemical complexity, it is worth noting the heptasaccharide 99, with its 35 stereocenters, was prepared in only 11-steps from an achiral furan starting material (Scheme 16).
6. Rhee Synthesis of Carbohydrates
More recently, Rhee and co-workers developed an alternative de novo approach to hexose sugars,[70],[71] which is particularly compatible with oligosaccharide synthesis. Like the Achmatowicz approach, the Rhee approach relies on the use of a Pd-allylation reaction. This alternative approach builds upon the use of asymmetric Pd-allylation of alcohols with alkoxy-allenes that was developed by Trost and Lee.[72],[73] In the Rhee application, the chiral ligands on the palladium catalyst (108(R,R) or 108(S,S)) are used to control the sugar acetal stereochemistry in the asymmetric allylation (107 to 109 or 110). After the allylation, a ring-closing metathesis (RCM) reaction is used to close the pyran ring (109 to 111 and 110 to 113 via 112). The approach is compatible with all the D-/L- and α-/β-sugar stereochemical combinations (Scheme 17).
En lieu of asymmetric catalysis, Rhee’s synthesis derives its asymmetry from lactic acid, which is readily available in either D- or L-enantiomeric form. Therefore, the Rhee de novo carbohydrate synthesis is technically not a de novo asymmetric approach (Scheme 17). Nevertheless, the Rhee synthesis begins with the conversion of either D- or L-lactic acid into the four possible ene-diols 102-105 that contain the C-3 to C-6 sugar fragment. Thus, in three steps, (i.e., protection, DibalH reduction and vinyl-Grignard addition) D-lactic acid was converted into a separable mixture of a variably protected syn- and anti-diols (102 (D, syn) or 103(D, anti)). Similarly, L-lactic acid can be converted into syn- and anti-diols (105 (L, syn) or 105 (L, anti)). These chiral fragments can be converted into the desired pyran by a six-step protocol. The approach begins with a four-step introduction of an allenyl ether at the C-4 position, which functions as a Pd-glycosylation precursor. The key to the approach is a two-step allenylation propargylation (NaH/BrCH2CCH) and isomerization (KOt-Bu) to an allene (106 to 107).
Using a Pd(0) complex with the Trost C-2 symmetric chiral ligand Pd(0) (108(R,R)), Rhee showed that allenyl ethers like 107 (D, anti) could be converted into diastereomeric acetals 109 or 110. After Grubbs II catalyzed ring-closing metathesis reaction diene 107 can be converted into pyran 109 (β-D). Thus, the 2-step Pd-allylation/RCM reaction results in a net β-glycosylation, where the anomeric stereochemistry is controlled by the chiral ligand on the Pd catalyst. Similarly, the α-isomer 112 can be prepared by Grubbs II-catalyzed RCM on 110. A subsequent osmium-catalyzed dihydroxylation converted 112 into the α-rhamno-sugar 113 (α-D). To prove the utility of his approach, Rhee applied this approach to the synthesis of numerous oligosaccharide motifs, such as, cervimycin K[74], saccharomicin[75], and landomycin (Schemes 20 and 21).[76]
7. The De Novo Asymmetric Synthesis of the Landomycins
The landomycins (Scheme 18) are a family of glycosylated angucycline anticancer antibiotics with an unusual structural motif that were first isolated in the 1990s. Of the landomycins, landomycin A 114 has received the most attention from the synthetic community, as it is reported to have a novel mechanism of anticancer activity. Landomycin A consists of a novel deoxygenated hexasaccharide with two repeating trisaccharides (α-L-rhodinose-(1→3)-β-D-olivose-(1→4)-β-D-olivose). More recently, the landomycin family has grown with the discovery of the 11-Deoxylandomycins, which has an alternative dehydrated aglycon motif and the same basic oligosaccharide repeat unit. Of the 11-Deoxylandomycins, the largest member of this family of angucycline natural products is landomycin Y 116 with the same landomycin A hexasaccharide. Most synthetic approaches to the landomycin oligosaccharides have focused on using traditional carbohydrate approaches to assemble the deoxy-sugar oligomer.[77],[78],[79],[80],[81],[82],[83],[84],[85],[86],[87],[88],[89] In contrast to these more conventional approaches, there have been two de novo approaches to the landomycin carbohydrates. The first de novo approach was by O’Doherty and used the asymmetric Achmatowicz chemistry, and the other by Rhee used a mix traditional carbohydrate/de novo approach. This approach (Pd-allylation/RCM) also heavily relies on the 2-deoxy-sugar glycosylation chemistry developed by Bennett.[90],[91],[92]
The landomycin oligosaccharides offer an excellent opportunity to apply the de novo Achmatowicz approach to a sugar oligomer that repeats the sugar subunit. For example, the trisaccharide repeat unit of the hexasaccharide landomycin A, has two rare β-D-olivose sugars and one α-L-rhoodinose. This asymmetry of the oligomer chain is another form of structural complexity, which adds steps when compared to the oligosaccharide syntheses of Scheme 15 and Scheme 16. Note that these de novo approaches, compared to traditional ones, save steps in the preparation of the monosaccharide building blocks.
The application of the de novo Achmatowicz approach to the landomycins, begins with β-pyranone 82, which can be prepared in three steps from commercially available 2-acetylfuran. Pyranone 82 was subjected to a palladium-catalyzed glycosylation with BnOH followed by a Luche reduction of the keto group to yield allylic alcohol 118. This alcohol was then subjected to a Myers’ reductive 1,3-allylic transposition followed by a stereoselective Upjohn dihydroxylation to afford diol 119 in 56% overall yield over four steps. Mitsunobu reaction conditions were then utilized to selectively invert the axial C-3 alcohol, converting 119 into a protected β-D-olivose 120 in 71% yield.[93],[94] Next, 120 was used to glycosylate pyranone 82 using the typical palladium-catalyzed glycosylation conditions to form the C-4 glycosylated disaccharide 121 in 85% yield. A Luche reduction of the keto group in 121 yielded a diastereomeric mixture of allylic alcohols. However, when this mixture was exposed to the Myers’ reductive 1,3-allylic transposition conditions, the desired olefin 122 was produced in good yield. Next, the p-nitrobenzoate group of 122 was hydrolyzed and the resulting free alcohol protected with TBSCl to give silyl ether 123 in 71% yield. Following subjection of 123 to OsO4-catalyzed dihydroxylation, the resulting diol was applied to a regioselective Mitsunobu reaction to produce benzoate 124. The remaining free alcohol was then protected with a TBS group, after which the PNB-protected alcohol was liberated via DIBAL-H reduction to afford the desired glycosyl acceptor 125. Next, pyranone 84a was glycosylated with 125, producing allylic alcohol 126 after Luche reduction, providing an intermediate trisaccharide precursor. The allylic alcohol 126 was inverted using the Mitsunobu reaction followed by the removal of the resulting benzoate, affording axial alcohol 127 in 95% yield. Lastly, olefin 127 was reduced to provide the protected trisaccharide, which was liberated by TBAF-mediated protection with TBSCl to afford benzyl glycoside 128 in 83% yield over the final two steps (Scheme 19).
The Rhee groups’ synthesis of landomycin Y began with naphthyl-protection of rhamnal 129 to form 130 in nearly quantitative yield. Next, glycal 130 was converted to a 2-deoxy sugar 131, as an equilibrating mixture of unprotected anomers. 131 was then selectively converted to α-tosylate intermediate 132, followed by glycosylation with C-4 unprotected sugar 133 to afford protected disaccharide 134. Next, the anomeric PMP group was selectively removed via ceric ammonium nitrate (CAN) oxidation to afford acetal 135. Next, the aglycone component was coupled at the liberated anomeric position to afford 136 with the desired β-configuration, followed by protection of the phenol in the aglycone as n-hexyl ester 137. 137 was then sequentially deprotected: first by oxidative removal of the naphthyl groups with DDQ, followed by hydrogenation of the benzyl group, affording the first key coupling partner 139 (Scheme 20).
The synthesis of the second coupling partner also began with 129, which was iteratively protected as a C-3 acetate and C-4 TBS ether 140. Next, mono-protected diol 102b was coupled using Pd(II) chemistry to afford 141 with the desired β-linkage. Next, the PMB-group was removed, followed by propargyl addition to the resulting alcohol, and then TBS-deprotection yielding allylic alcohol 142. This alcohol was coupled with 129 to give the β-linked trisaccharide precursor 143. This was coupled with allene 144 using the appropriate chiral Pd catalyst to yield C-3 α-linked tetra saccharide precursor 145. Finally, terminal alkyne 145 was isomerized to allene 146 under standard isomerization conditions (Scheme 21).
Allene 146 and alcohol 139 were coupled together with both the (R,R) and (S,S) Pd catalysts to install the α- or β-linkage, respectively. This was followed by ring-closing metathesis using the Grubbs catalyst to cyclize the terminal sugar residue, giving hexasaccharide precursors 147 and 148, respectively. Finally, the benzyl groups were removed by hydrogenation, and the n-hexyl esters of the aglycones were subjected to base hydrolysis to afford landomycin Y 116 in two steps from 147, and epi-landomycin Y 149 from 148. The use of 108(R,R) versus 108(S,S) to produce either 116 or 149 demonstrates the stereochemical versatility of this reagent-controlled approach to anomeric stereochemical control (Scheme 22).
8. Conclusions
In summary, this review introduces the concept of de novo asymmetric synthesis in the context of carbohydrate synthesis, particularly for the synthesis of oligosaccharides. This was accomplished by introducing the role asymmetry plays in nature and natural substances and describing how asymmetry is particularly prevalent in the chemistry of biomolecules, especially carbohydrates with their many chiral carbon centers. As asymmetry plays an important role in the structure of carbohydrates, it also plays a similarly critical role in their syntheses. As our ability to synthesize carbohydrate motifs advances, the complexity of the targets that can be accessed synthetically and has also progressed. This can be seen in the progression of the early diastereoselective syntheses to enantioselective syntheses, and finally, to de novo asymmetric syntheses. This advancement culminated in the development of two de novo asymmetric synthetic methods for oligosaccharides, with application to oligosaccharide medicinal chemistry.[95],[96],[97],[98],[99],[100],[101],[102],[103],[104] The first approach was the Achmatowicz approach, which relies on the asymmetric synthesis of furan alcohols, a Pd-glycosylation reaction, and post-glycosylation reactions. The Achmatowicz approach synthesizes stereochemically simplified D- and L-pyranones with α- and β-stereochemistry. The Achmatowicz approach reduces the 32 possible hexose monosaccharides to just 4 stereoisomers. The second approach developed by the Rhee group, led to an alternative method for pyran construction with similar applications to oligosaccharides like the landomycins.
Author Contributions
All authors contributed to the writing of the review. The order of the first three Co-first authors (IH, SK, AS) is alphabetical.
Funding
Financial support of this research program was provided by NSF (CHE-2102649 and CHE-2400110 to GAO) and NIH (AI146485 and AI154860 to GAO).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- “When Did We Become So Obsessed With Being Symmetrical,” Rhonda Garelick, Published NY Times, 23 August 2022.
- Leigh, W. Simmons, Gillian Rhodes, Marianne Peters, Nicole Koehler, Are human preferences for facial symmetry focused on signals of developmental instability? Behavioral Ecology, Volume 15, Issue 5, 04, Pages 864–871. 20 September. [CrossRef]
- Misselhorn, C. "Empathy with inanimate objects and the uncanny valley". Minds and Machines. 2009, 19, 345–359. [Google Scholar] [CrossRef]
- McManus, I.C. "Symmetry and asymmetry in aesthetics and the arts". Eur. Rev. 2005, 13, 157–180. [Google Scholar] [CrossRef]
- Eliel, E.; Willen, S.H.; Mander, L.N. "Stereochemistry of Organic Compounds". John Wiley & Sons, Inc., New York, NY. 1994. 0167. [Google Scholar]
- Mizuno, T.; Weiss, A.H. Synthesis and utilization of formose sugars. Adv. Carbohydr. Chem. Biochem. 1974, 29, 173–227. [Google Scholar]
- Aljahdali, A.Z.; Shi, P.; Zhong, Y.; O’Doherty, G.A. , De Novo Asymmetric Synthesis of the Pyranoses: From Monosaccharides To Oligosaccharides. Advances in Carbohydrate Chemistry and Biochemistry, 2013; 69. [Google Scholar]
- Kim, S.; Oiler, J.; Xing, Y.; O’Doherty, G.A. Asymmetric Achmatowicz Approach to Oligosaccharides. Chem. Commun. 2022, 58, 12913–12926. [Google Scholar] [CrossRef]
- Kim, S.; O’Doherty, G.A. De novo asymmetric synthesis of the pyranoses—from monosaccharides to oligosaccharides: An update. Advances in Carbohydrate Chemistry and Biochemistry David Baker Ed. 2024; 85. [Google Scholar]
- Zheng, J.; O’Doherty, G.A. De Novo Synthesis of Oligosaccharides Via Metal Catalysis in Comprehensive Glycoscience, 2nd Edition, Joseph Barchi Ed., Elsevier: Oxford 2021; Volume 2., pp. 435-463.
- Adams, R.; C. S. Marvel. Benzoin. Org. Synth. 1921, 1, 33. [Google Scholar]
- Matsumoto, T.; Yamamoto, H.; Inoue, S. Selective Formation of Triose from Formaldehyde Catalyzed by Thiazolium Salt. J. Am. Chem. Soc. 1984, 106, 4829–4832. [Google Scholar] [CrossRef]
- Stetter, H. Catalyzed addition of aldehydes to activated double bonds-a new synthetic approach. Angew. Chem. Int. Ed. 1976, 15, 639. [Google Scholar] [CrossRef]
- de Alaniz, J.R.; Kerr, M.S.; Moore, J.L.; Rovis, T. Scope of the asymmetric intramolecular Stetter reaction catalyzed by chiral nucleophilic triazolinylidene carbenes. J. Org. Chem. 2008, 73, 2033. [Google Scholar] [CrossRef]
- Breslow, R. On the Mechanism of Thiamine Action. IV. Evidence from Studies on Model Systems. J. Am. Chem. Soc. 1958, 80, 3719. [Google Scholar] [CrossRef]
- Dondoni, A.; Perrone, D. Thiazole-based routes to amino hydroxyl aldehydes, and their use of the synthesis of biologically active compounds. Aldrichimica Acta 1997, 30, 35–46. [Google Scholar]
- Ko, S.Y.; Lee, A.W. M.; Masamune, S.; Reed III, L.A.; Sharpless, K.B.; Walker, F.J. Total Synthesis of the L-Hexoses. Science 1983, 220, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.Y.; Lee, A.W. M.; Masamune, S.; Reed III, L.A.; Sharpless, K.B.; Walker, F.J. Total synthesis of the L-hexoses. Tetrahedron 1990, 46, 245–264. [Google Scholar] [CrossRef]
- Danishefsky, S.J. Cycloaddition and cyclocondensation reactions of highly functionalized dienes: Applications to organic synthesis. Chemtracts 1989, 273–297. [Google Scholar] [CrossRef]
- Schaus, S.E.; Branalt, J.; Jacobsen, E.N. Asymmetric Hetero-Diels-Alder Reactions Catalyzed by Chiral (Salen)Chromium(III) Complexes. J. Org. Chem. 1998, 63, 403–405. [Google Scholar] [CrossRef]
- Northrup, B.; Macmillan, D.W.C. Two-step synthesis of carbohydrates by selective aldol reactions, Science 2004, 305 1752--1755.
- B. Northhrup, I.K. Mangion, F. Hettche, and D. W. C MacMillan, Enantioselective organocatalytic direct aldol reactions of α-oxyaldehydes: Step one in a two-Step synthesis of carbohydrates, Angew. Chem., Int. Ed. 2004, 43, 2152–2154.
- Hudlicky, T.; Pitzer, K.K.; Stabile, M.R.; Thorpe, A.J.; Whited, G.M. , Biocatalytic Syntheses of Protected D-Mannose-d(5), D-Mannose-d(7), D-Mannitol-2,3,4,5,6-d(5), and D-Mannitol-1,1,2,3,4,5,6,6-d(8) J. Org. Chem. 1996, 61, 4151–4153. [Google Scholar] [CrossRef]
- Gibson, D.T.; Koch, J.R.; Kallio, R.E. Oxidative degradation of aromatic hydrocarbons by microorganisms. I. Enzymatic formation of catechol from benzene. Biochemistry 1968, 7, 2653-–2662. [Google Scholar] [CrossRef]
- Johnson, C.R.; Golebiowski, A.; Steensma, D.H.; Scialdone, M.A. Enantio- and diastereoselective transformations of cycloheptatriene to sugars and related products. J. Org. Chem. 1993, 58, 7185–7194. [Google Scholar] [CrossRef]
- JBackvall, K.; Bystrom, S.E.; Nordberg, R.E. Stereo- and regioselective palladium-catalyzed 1,4-diacetoxylation of 1,3-dienes. J.Org. Chem. 1984, 49, 4619–4631. [Google Scholar]
- Henderson, I.; Sharpless, K.B.; Wong, C.-H. Synthesis of Carbohydrates via Tandem Use of the Osmium-Catalyzed Asymmetric Dihydroxylation and Enzyme-Catalyzed Aldol Addition Reactions. J. Am. Chem. Soc. 1994, 116, 558–561. [Google Scholar] [CrossRef]
- Kolb, H.C.; Van Nieuwenhze, M.S.; Sharpless, K.B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Kolb, H.C.; Anderson, P.G.; Sharpless, K.B. Toward an Understanding of the High Enantioselectivity in the Osmium-Catalyzed Asymmetric Dihydroxylation (AD). 1. Kinetics. J. Am. Chem. Soc. 1994, 116, 1278. [Google Scholar] [CrossRef]
- Zhang, Y.; O’Doherty, G.A. Remote steric effect on the regioselectivity of Sharpless asymmetric dihydroxylation, Tetrahedron 2005, 61 6337-6351. 61.
- Ahmed, M.M.; O'Doherty, G.A. De Novo Asymmetric Syntheses of D- and L-Talose via an Iterative Dihydroxylation of Dienoates. J. Org. Chem. 2005, 70, 10576–10578. [Google Scholar] [CrossRef]
- Ahmed, M.M.; O'Doherty, G.A. De novo synthesis of a galacto-papulacandin moiety via an iterative dihydroxylation strategy. Tetrahedron Lett. 2005, 46, 4151–4155. [Google Scholar] [CrossRef]
- Gao, D.; O'Doherty, G.A. Enantioselective Synthesis of 10-epi-Anamarine via an Iterative Dihydroxylation Sequence. Org. Lett. 2005, 7, 1069–1072. [Google Scholar] [CrossRef]
- Ahmed, M.M.; O'Doherty, G.A. De novo synthesis of galacto-sugar δ-lactones via a catalytic osmium/palladium/osmium reaction sequence. Tetrahedron Lett. 3019; 46, 3015–3019. [Google Scholar]
- Ahmed, M.M.; Berry, B.P.; Hunter, T.J.; Tomcik, D.J.; O'Doherty, G.A. De Novo Enantioselective Syntheses of Galacto-Sugars and Deoxy Sugars via the Iterative Dihydroxylation of Dienoate. Org. Lett. 2005, 7, 745–748. [Google Scholar] [CrossRef]
- Harris, J.M.; Keranen, M.D.; O’Doherty, G.A. Syntheses of d- and l-mannose, gulose, and talose via diastereoselective and enantioselective dihydroxylation reactions, J. Org. Chem. 1999, 64, 2982-–2983. [Google Scholar] [CrossRef]
- Harris, J.M.; Keranen, M.D.; Nguyen, H.; Young, V.G.; O’Doherty, G.A. Syntheses of four d- and l-hexoses via diastereoselective and enantioselective dihydroxylation reactions, Carbohydr. Res. 2000, 328, 17–36. [Google Scholar]
- Bushey, M.L.; Haukaas, M.H.; O’Doherty, G.A. Asymmetric aminohydroxylation of vinylfuran, J. Org. Chem. 1999, 64, 2984–2985. [Google Scholar] [CrossRef]
- Cuccarese, M.F.; Li, J.J.; O’Doherty, G.A. De Novo Approaches to Monosaccharides and Complex Glycans in Modern Synthetic Methods in Carbohydrate Chemistry, Sebastien Vidal and Daniel B. Werz Eds., Wiley-VCH Verlag GmbH & Co. KG, Weinheim 2014, pp. 1-28.
- Ashmus, R.; Jayasuriya, A.; Lim, Y.-J.; O’Doherty, G.A.; Lowary, T.L. De novo asymmetric synthesis of a 6-O-methyl-D-glycero-L-gluco-heptopyranose-derived thioglycoside for the preparation of Campylobacter jejuni NCTC11168 capsular polysaccharide fragments. J. Org. Chem. 2016, 81, 3058–3063. [Google Scholar] [CrossRef]
- Cunha, V.L.S.; O'Doherty, G.A.; Lowary, T.L. Exploring a de novo route to bradyrhizose and its diastereomers: synthesis and isomeric equilibrium of reducing bicyclic carbohydrates. Chem. Eur. J. 2024, 30. [Google Scholar] [CrossRef] [PubMed]
- Balachari, D.; O’Doherty, G.A. Sharpless Asymmetric Dihydroxylation of 5-Aryl-2-vinylfurans: Application to the Synthesis of the Spiroketal Moiety of Papulacandin D. Org. Lett. 2000, 2, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Balachari, D.; O’Doherty, G.A. Enantioselective Synthesis of the Papulacandin Ring System: Conversion of the Mannose Diastereoisomer into a Glucose Stereoisomer. Org. Lett. 2000, 2, 4033–4036. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M.; O'Doherty, G.A. De Novo Asymmetric Synthesis of a Galacto-Papulacandin Moiety Via an Iterative Dihydroxylation Strategy. Tetrahedron Lett. 4155; 46, 4151–4155. [Google Scholar]
- Haukaas, M.H.; O’Doherty, G.A. Enantioselective synthesis of N-Cbz-protected 6-amino-6-deoxy-mannose, gulose and talose. Org. Lett. 3899; 3, 3899–3992. [Google Scholar]
- Haukaas, M.H.; O’Doherty, G.A. Enantioselective Synthesis of 2-Deoxy and 2,3-Dideoxy-hexoses. Org. Lett. 2002, 4, 1771–1774. [Google Scholar] [CrossRef]
- Guo, H.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Daumone via a Palladium Catalyzed Glycosylation Org. Lett. 2005, 7, 3921–3924. [Google Scholar]
- Xing, Y. , O’Doherty, G. A. De Novo Asymmetric Approach to Aspergillide-C. ChemistrySelect 2022, 7, e202200266. [Google Scholar]
- Guppi, S.R.; Zhou, M.; O’Doherty, G.A. De Novo Asymmetric Synthesis of Homo-Adenosine via a Palladium Catalyzed N-Glycosylation Org. Lett. 2006, 8, 293–296. [Google Scholar]
- Noyri, R.; Ohkuma, T. Asymmetric catalysis by architectural and functional molecular engineering: practical chemo-and stereoselective hydrogenation of ketones. Angew. Chem. Int. Ed. 2001, 40, 40. [Google Scholar] [CrossRef]
- Noyori, R.; Yamakawa, M.; Hashiguchi, S. Metal- ligand bifunctional catalysis: a nonclassical mechanism for asymmetric hydrogen transfer between alcohols and carbonyl compounds. J. Org. Chem. 2001, 66. [Google Scholar] [CrossRef]
- Li, M.; O’Doherty, G.A. An enantioselective synthesis of phomopsolide D. Tetrahedron Lett. 2004, 45, 6407–6411. [Google Scholar] [CrossRef]
- Li, M.; Scott, J.G.; O’Doherty, G.A. Synthesis of 7-oxa-phomopsolide E and its C-4 epimer. Tetrahedron Lett. 2004, 45, 1005–1009. [Google Scholar] [CrossRef]
- Babu, R.S.; O’Doherty, G.A. A Palladium-Catalyzed Glycosylation Reaction: The De Novo Synthesis of Natural and Unnatural Glycosides. J. Am. Chem. Soc. 2003, 125, 12406–12407. [Google Scholar] [CrossRef] [PubMed]
- Babu, R.S.; O’Doherty, G.A. Palladium Catalyzed Glycosylation Reaction: De-Novo Synthesis of Trehalose Analogues. J. Carb. Chem. 2005, 24, 169–177. [Google Scholar] [CrossRef]
- Sharif, E.U.; Wang, H.-Y. L.; Akhmedov, N.G.; O’Doherty G., A. Merremoside D: De novo synthesis of its purported structure, NMR analysis and comparison of spectral data. Org. Lett. 2014, 16, 492–495. [Google Scholar] [CrossRef]
- Babu, R.S.; Guppi, S.R.; O’Doherty, G.A. Synthetic Studies Towards Mannopeptimycin-E: Synthesis of a O-Linked Tyrosine 1,4-,-Manno,Manno-Pyanosyl-Pyranoside. Org. Lett. 2006, 8, 1605–1608. [Google Scholar] [CrossRef]
- Feringa, L.; Comely, A.C.; Eelkema, R.; Minnaard, A.J.; Feringa, B.L. De novo asymmetric bio- and chemocatalytic synthesis of saccharides - stereoselective formal O-glycoside bond formation using palladium catalysis. J. Am. Chem. Soc. 2003, 125, 8714–8715. [Google Scholar]
- Kim, H.; Men, H.; Lee, C. Stereoselective palladium-catalyzed O-glycosylation using glycals. J. Am. Chem. Soc. 2004, 126, 1336–1337. [Google Scholar] [CrossRef]
- Kim, H.; Lee, C. A mild and efficient method for the stereoselective formation of C-O bonds: palladium-catalyzed allylic etherification using zinc(II) alkoxides. Org. Lett. 2002, 4, 4369–4372. [Google Scholar] [CrossRef]
- Wu, B.; Li, M.; O’Doherty, G.A. Synthesis of several cleistrioside and cleistetroside natural products via a divergent de novo asymmetric approach, Org. Lett. 2010, 12, 5466–5469. [Google Scholar]
- Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of the anthrax tetrasaccharide by a palladium-catalyzed glycosylation reaction. Angew. Chem. Int. Ed. 2007, 46, 5206–5208. [Google Scholar] [CrossRef]
- Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of anthrax tetrasaccharide and related tetrasaccharide, J. Org. Chem. 2008, 73, 5211–5220. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y. L.; Guo, H.; O’Doherty, G.A. De novo asymmetric synthesis of rhamno di- and tri-saccharides related to the anthrax tetrasaccharide, Tetrahedron 2013, 69 3432-3436. 69.
- Bajaj, S.O.; Sharif, E.U.; Akhmedov, N.G.; O’Doherty, G.A. De novo asymmetric synthesis of the mezzettiaside family of natural products via the iterative use of a dual B-/Pd-catalyzed glycosylation. Chem. Sci. 2014, 5, 2230–2234. [Google Scholar] [CrossRef] [PubMed]
- Sharif, E.U.; Wang, H.-Y. L.; Akhmedov, N.G.; O’Doherty G., A. Merremoside D: De novo synthesis of its purported structure, NMR analysis and comparison of spectral data. Org. Lett. 2014, 16, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Ray, D. and O’Doherty, G.A. De Novo Asymmetric Synthesis of Oligosaccharides Using Atom-Less Protecting Group in Protecting Groups: Strategies and Applications in Carbohydrate Chemistry, Sebastien Vidal Ed., Wiley-VCH Verlag GmbH Co. KG, Weinheim, 2019, pp. 327-351Weinheim, 2019, pp. 327-351.
- Babu, R.S.; Zhou, M.; O’Doherty, G.A. De-Novo Synthesis of Oligosaccharides Using a Palladium Catalyzed Glycosylation Reaction. J. Am. Chem. Soc. 2004, 126, 3428–3429. [Google Scholar] [CrossRef]
- Babu, R.S.; Chen, Q.; Kang, S.-W.; Zhou, M.; O’Doherty, G.A. De Novo Synthesis of Oligosaccharides Using Green Chemistry Principles. J. Am. Chem. Soc. 2012, 134, 11952–11955. [Google Scholar] [CrossRef]
- Jang, S.H.; Kim, H.W.; Jeong, W.; Moon, D.; Rhee, Y.H. Palladium-catalyzed asymmetric nitrogen-selective addition reaction of indoles to alkoxyallenes. Org. Lett. 2018, 20, 1248–1251. [Google Scholar] [CrossRef]
- Lim, W.; Kim, J.; Rhee, Y.H. Pd-catalyzed asymmetric intermolecular hydroalkoxylation of allene: an entry to cyclic acetals with activating group-free and flexible anomeric control. J. Am. Chem. Soc. 2014, 136, 13618–13621. [Google Scholar] [CrossRef]
- Trost, M.; Fandrick, D.R.; Dinh, D.C. . Dynamic kinetic asymmetric allylic alkylations of allenes. J. Am. Chem. Soc. 2005, 127, 14186–14187. [Google Scholar] [CrossRef]
- Trost, M.; Lee, C.B. . Geminal dicarboxylates as carbonyl surrogates for asymmetric synthesis. Part I. Asymmetric addition of malonate nucleophiles. J. Am. Chem. Soc. 2001, 123, 3671–3686. [Google Scholar] [CrossRef]
- Kang, J.; Rhee, Y.H. . Convergent Synthesis of Tetrasaccharide Fragment of Cervimycin K. Org. Lett. 2021, 23, 4468–4472. [Google Scholar] [CrossRef]
- Barpuzary, B.; Kim, M.; Rhee, Y.H. . Synthetic Study toward Saccharomicin Based upon Asymmetric Metal Catalysis. Org. Lett. 2021, 23, 5969–5972. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kang, J.; Lee, S.; Rhee, Y.H. . Flexible Total Synthesis of 11-Deoxylandomycins and Their Non-Natural Analogues by Way of Asymmetric Metal Catalysis. Angew. Chem. 2020, 132, 2369–2373. [Google Scholar]
- Yang, X.; Fu, B.; Yu, B. Total Synthesis of Landomycin A, a Potent Antitumor Angucycline Antibiotic. J. Am. Chem. Soc. 2011, 133, 12433–12435. [Google Scholar] [CrossRef]
- Yang, X.; Wang, P.; Yu, B. Tackling the Challenges in the Total Synthesis of Landomycin A. Chem. Rec. 2013, 13, 70–84. [Google Scholar] [CrossRef]
- Lai, Y-H. ; Mondal, S.; Su, H-T.; Huang, S-C.; Wu, M-H.; Huang, I-W.; Lauderdale, T-S. Y.; Song, J-S.; Shia, K-S.; Mong, K-K. T. Total synthesis of landomycins Q and R and related core structures for exploration of the cytotoxicity and antibacterial properties. RSC Adv. 2021, 11, 9426–9432. [Google Scholar] [CrossRef]
- Lee, J.; Kang, S.; Kim, J.; Moon, D.; Rhee, Y.H. A Convergent Synthetic Strategy towards Oligosaccharides containing 2,3,6-Trideoxypyranoglycosides. Angew. Chem. Int. Ed. 2018, 58, 628–631. [Google Scholar] [CrossRef]
- Yalamanchili, S.; Lloyd, D.; Bennet, C.S. Synthesis of the Hexasaccharide Fragment of Landomycin A Using a Mild, Reagent-Controlled Approach. Org. Lett. 2019, 21, 3684–3677. [Google Scholar] [CrossRef]
- Guo, Y.; Sulikowski, G.A. Synthesis of the Hexasaccharide Fragment of Landomycin A: Application of Glycosyl Tetrazoles and Phosphites in the Synthesis of Deoxyoligosaccharide. J. Am. Chem. Soc. 1998, 120, 1392–1397. [Google Scholar] [CrossRef]
- Roush, W.R.; Bennett, C.E. A Highly Stereoselective Synthesis of the Landomycin A Hexasaccharide Unit. J. Am. Chem. Soc. 2000, 122, 6124–6125. [Google Scholar] [CrossRef]
- Yu, B.; Wang, P. Efficient Synthesis of the Hexasaccharide Fragment of Landomycin A: Using Phenyl 2,3-O-Thionocarbonyl-1-thioglycosides as 2-Deoxy-β-glycoside Precursors. Org. Lett. 2002; 4, 1919–1922. [Google Scholar]
- Tanaka, H.; Yamaguchi, S.; Yoshizawa, A.; Takagi, M.; Shin-ya, K.; Takahashi, T. Combinatorial Synthesis of Deoxyhexasaccharides Related to the Landomycin A Sugar Moiety, Based on an Orthogonal Deprotection Strategy. Chem. Asian. J. 2010, 5, 1407–1424. [Google Scholar] [CrossRef]
- Zhou, M.; O’Doherty, G.A. De Novo Synthesis of the Trisaccharide Subunit of Landomycins A and E. Org. Lett. 2008, 10, 2283–2286. [Google Scholar] [CrossRef] [PubMed]
- Ruei, J-H. ; Venukumar, P.; Ingle, A.B.; Mong, K-K. T. C6 picoloyl protection: a remote stereodirecting group for 2-deoxy-β-glycoside formation. Chem. Commun. 2015, 51, 5394–5397. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Baryal, K.N.; Adhikari, S.; Zhu, J. Direct Synthesis of 2-Deoxy-β-glycosides via Anomeric O-Alkylation with Secondary Electrophiles. J. Am. Chem. Soc. 2014, 136, 3172–3175. [Google Scholar] [CrossRef] [PubMed]
- McDonald, F.E.; Reddy, K.S. Discovery of the tungsten carbonyl-catalyaed endo-selective alkynyl alcohol cylcoisomerization reaction: applications to stereoselective syntheses of D-olivose, D-olivose disaccharide substructures of landomycin and mithramycin. J. Organomet. Chem. 2001, 617, 444–452. [Google Scholar] [CrossRef]
- Issa, J.P.; Bennett, C.S. A Reagent-Controlled SN2-Glycosylation for the Direct Synthesis of β-Linked 2-Deoxy-Sugars. J. Am. Chem. Soc. 2014, 136, 5740–5744. [Google Scholar] [CrossRef]
- Lloyd, D.; Bylsma, M.; Bright, D.K.; Chen, X.; Bennett, C.S. Mild method for 2-naphthylmethyl ether protecting group removal using a combination of 2, 3-dichloro-5, 6-dicyano-1, 4-benzoquinone (DDQ) and β-pinene. J. Org. Chem. 2017, 82, 3926–3934. [Google Scholar] [CrossRef]
- Lloyd, D.; Bennett, C.S. An Improved Approach to the Direct Construction of 2-Deoxy-β-Linked Sugars: Applications to Oligosaccharide Synthesis. Chem. Eur. J. 2018, 24, 7610–7614. [Google Scholar] [CrossRef]
- Yu, X.; O’Doherty, G.A. De Novo Asymmetric Synthesis and Biological Evaluation of the Trisaccharide Portion of PI-080 and Vineomycin B2. Org. Lett. 2008, 10, 4529–4532. [Google Scholar] [CrossRef]
- Yu, X.; Li, M.; O’Doherty, G.A. De Novo Asymmetric Synthesis of the D-/L-Disaccharide Portion of Sch 47555. Heterocycles 2011, 82, 1577–1584. [Google Scholar]
- Shi, P.; Silva, M.; Wu, B. ; Wang, H-Y. L.; Akhmedov, N.G.; Li, M.; Beuning, P.; O’Doherty, G.A. Structure activity relationship study of the cleistrioside/cleistetroside natural products for antibacterial/anticancer activity, ACS Med. Chem. Lett. 2012, 3, 1086–1090. [Google Scholar]
- Bajaj, S.O.; Shi, P.; Beuning, P.J.; O’Doherty, G.A. Structure activity relationship study of Mezzettiasides natural products and its four new disaccharide analogues for anticancer/antibacterial activity. ChemMedComm. 2014, 5, 1138–1142. [Google Scholar]
- Zhou, M.; O’Doherty, G.A. De novo approach to 2-deoxy-O-glycosides: asymmetric syntheses of digioxose and digitoxin. J. Org. Chem. 2007, 72, 2485–2493. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; O’Doherty, G.A. De novo asymmetric synthesis of digitoxin via a palladium catalyzed glycosylation reaction. Org. Lett. 2006, 8, 4339–4342. [Google Scholar] [CrossRef]
- Iyer, A.; Zhou, M.; Azad, N.; Elbaz, H.; Wang, L.; Rogalsky, D.K.; Rojanasakul, Y.; O’Doherty, G.A.; Langenhan, J.M. A direct comparison of the anticancer activities of digitoxin MeON-neoglycosides and O-glycosides: oligosaccharide chain length-dependant induction of caspase-9-mediated apoptosis. ACS Med. Chem. Lett. 2010, 1, 326–330. [Google Scholar] [CrossRef]
- Zhou, M.; O’Doherty, G.A. The de novo synthesis of oligosaccharides: application to the medicinal chemical study of digitoxin. Curr. Top. Med. Chem 2008, 8, 114–125. [Google Scholar]
- Hinds, J.W.; McKenna, S.B.; Sharif, E.U.; Wang, H-Y. L.; Akhmedov N. G.; O’Doherty, G.A. C3'/C4'-stereochemical effects of digitoxigenin -L-/-D-glycoside in cancer cytotoxicity. ChemMedChem. 2013, 8, 63–69. [Google Scholar] [CrossRef]
- Wang, H.-Y. L.; Xin, W.; Zhou, M.; Stueckle, T.A.; Rojanasakul, Y.; O’Doherty, G.A. Stereochemical survey of digitoxin monosaccharides, ACS Med. Chem. Lett. 2011, 2, 73–78. [Google Scholar]
- Wang, H.-Y.L.; Wu, B.; Zhang, Q.; Kang, S.-W.; Rojanasakul, Y.; O’Doherty, G.A. C5’-Alkyl Substitution Effects on Digitoxigenin -L-Glycoside Epithelial Human Lung Cancer Cells Cytotoxicity, ACS Med. Chem. Lett. 2011, 2, 259–263. [Google Scholar]
- Wang, H.-Y.L.; Rojanasakul, Y.; O’Doherty, G.A. Synthesis and Evaluation of the α-D-/α-L-Rhamnosyl and Amicetosyl Digitoxigenin Oligomers as Anti-tumor Agents, ACS Med. Chem. Lett. 2011, 2, 264–259. [Google Scholar]
Scheme 1.
The monosaccharides and their stereochemical complexity from C-3 to C-6.
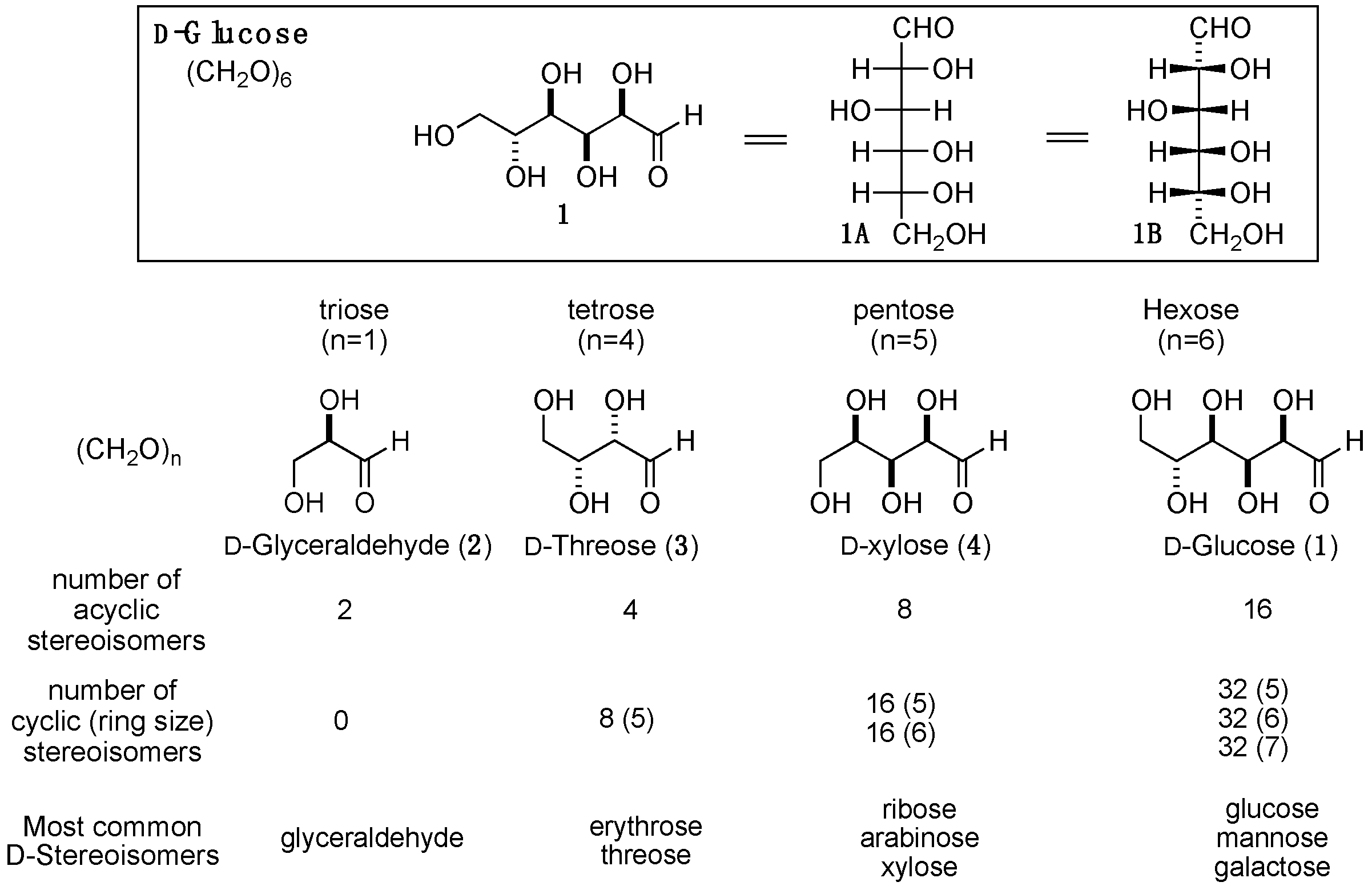
Scheme 2.
The cyclic hexoses in their cyclic furanose and pyranose forms.
Scheme 3.
The formose synthesis of sugars and mechanism.
Scheme 4.
Retrosynthesis for the Fischer stereodivergent synthesis of the hexoses.
Scheme 5.
The Fischer stereodivergent cyanohydrin formation/reduction and separation (chain homologation).
Scheme 5.
The Fischer stereodivergent cyanohydrin formation/reduction and separation (chain homologation).
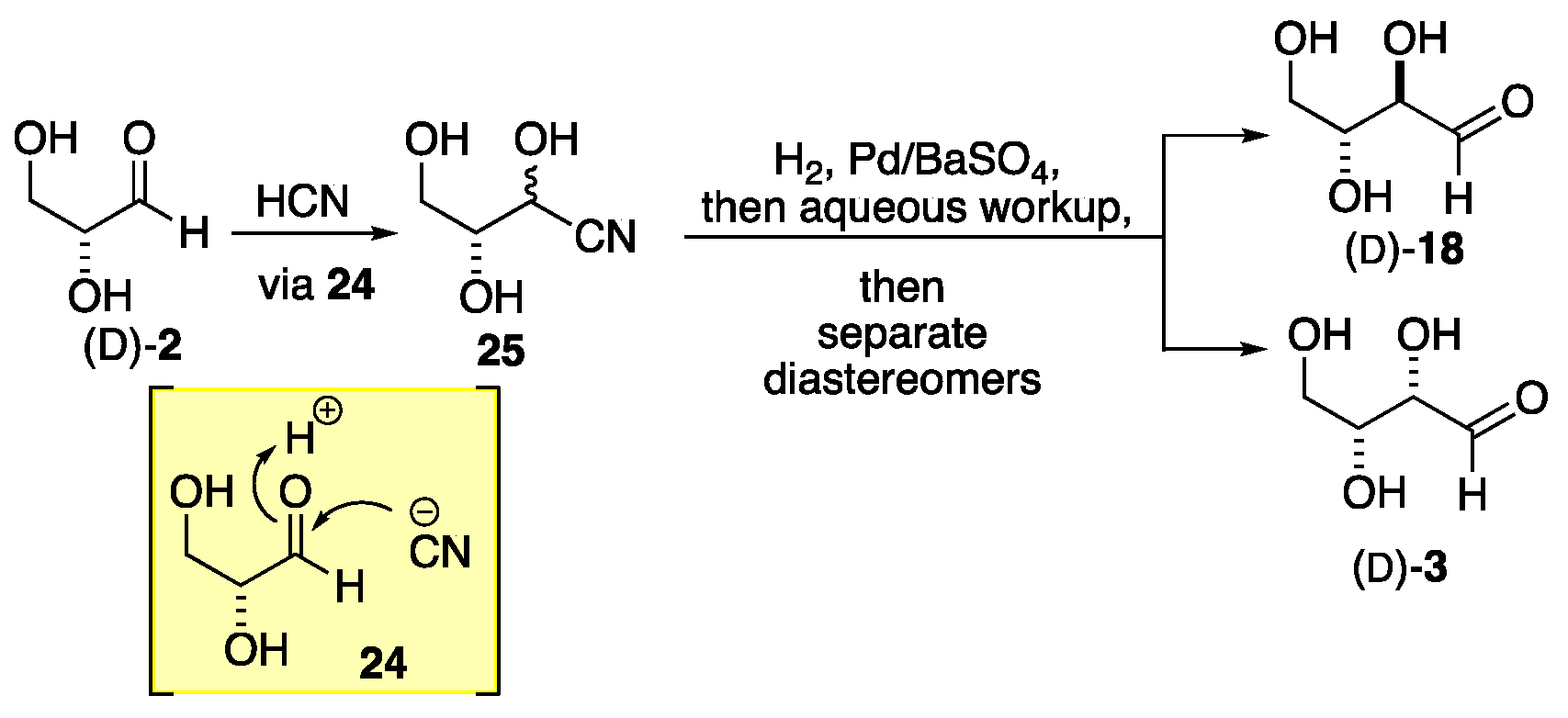
Scheme 6.
The Fischer synthesis of all the D-hexoses.
Scheme 7.
The conversion of glucose/mannose to arabinose via the Wohl degradation.
Scheme 8.
Retrosynthesis for the Sharpless de novo asymmetric synthesis of the L-hexoses.
Scheme 9.
Sharpless epoxidation/Payne rearrangement for the synthesis of cis-acetonide.
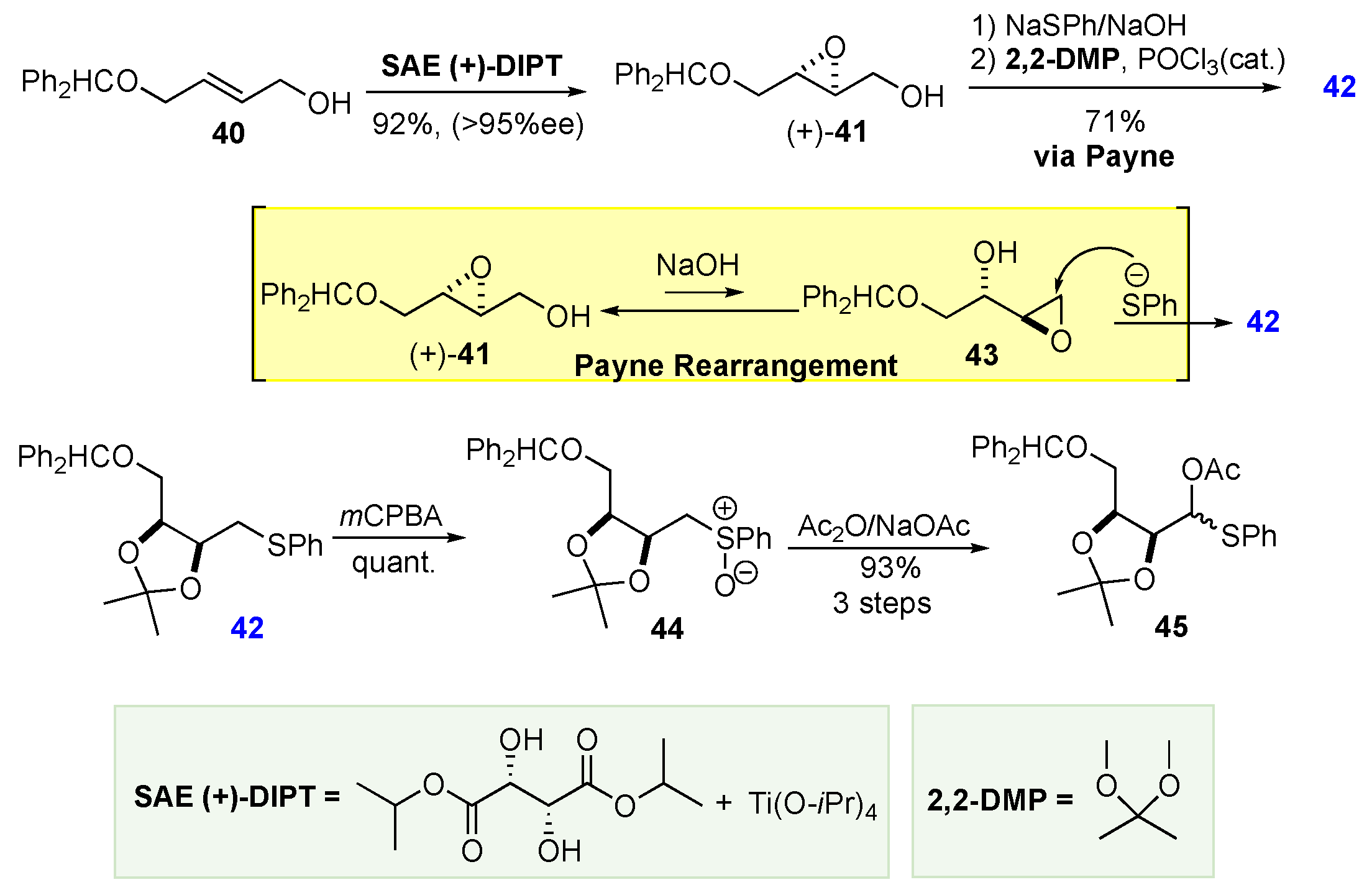
Scheme 10.
Sharpless synthesis of C-6 sugars via cis-/trans-acetonide isomerization.
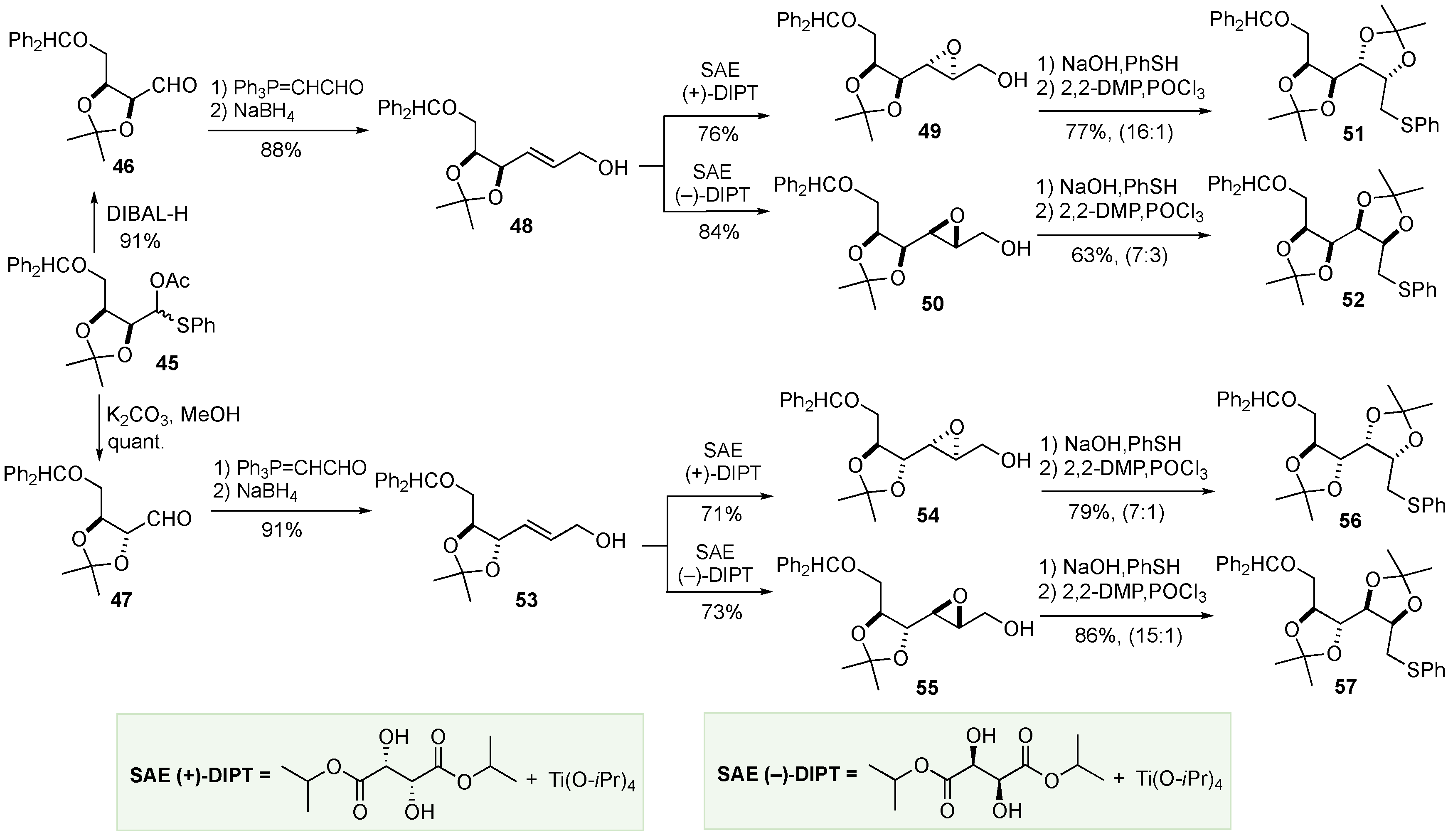
Scheme 11.
End game for the Sharpless de novo asymmetric synthesis of the L-hexoses.
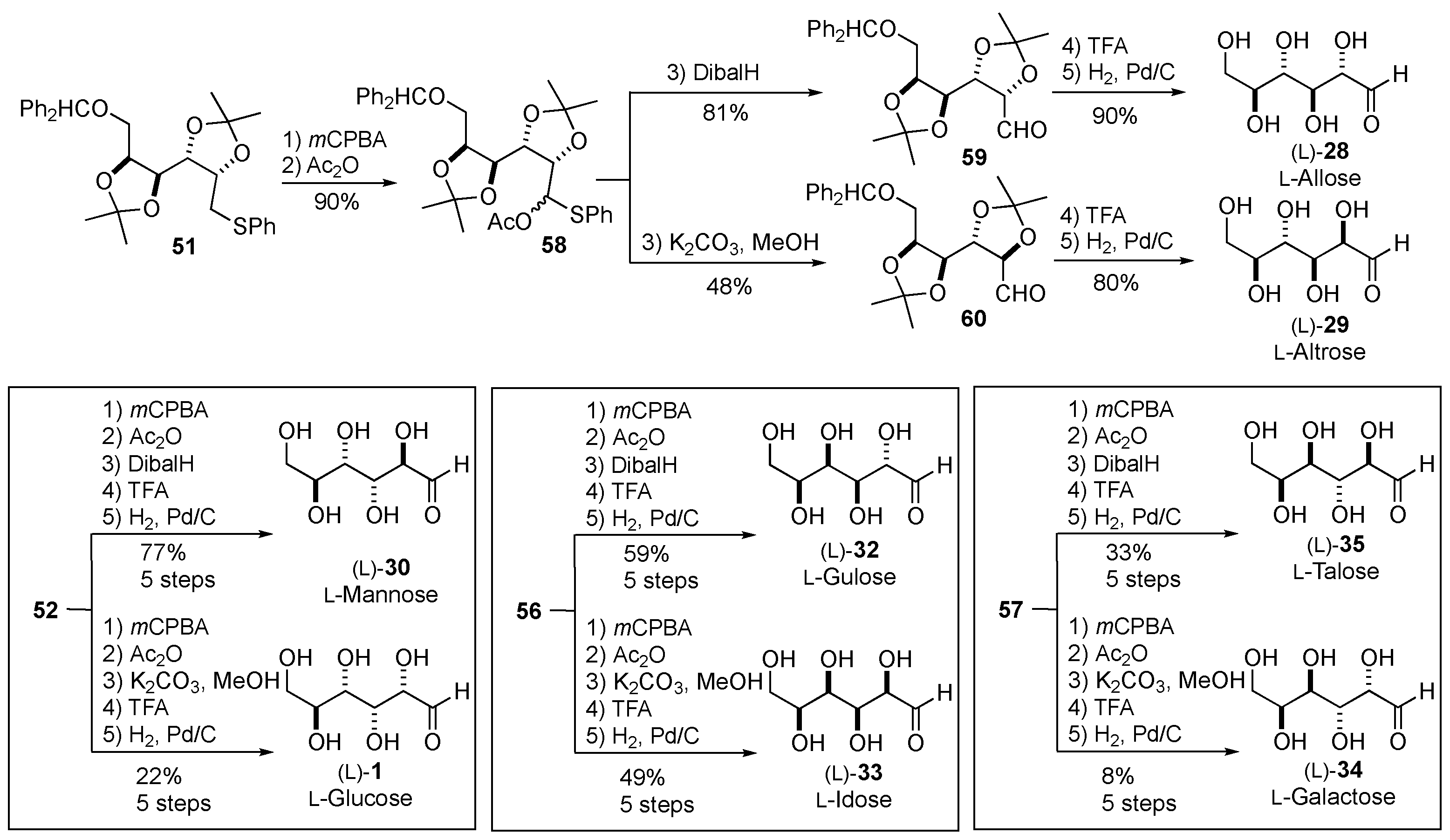
Scheme 12.
Other de novo asymmetric synthesis of hexoses.
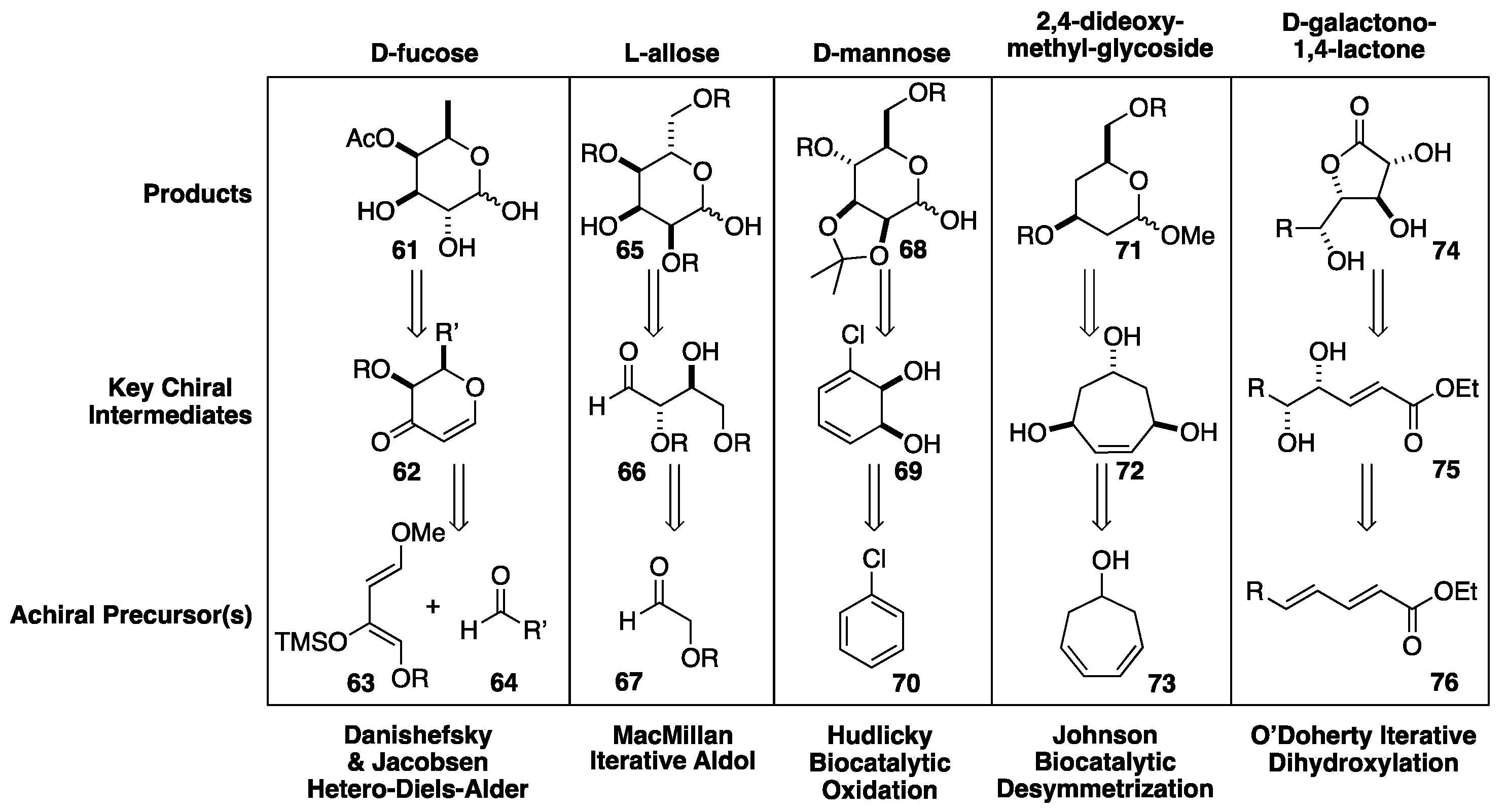
Scheme 13.
O’Doherty de novo asymmetric synthesis of pyranone based Pd-glycosylation donors.
Scheme 14.
Diastereoselective Pd-glycosylation.
Scheme 15.
O’Doherty de novo asymmetric synthesis of 1,4-linked oligosaccharides.
Scheme 16.
O’Doherty de novo asymmetric synthesis of branched 1,4-/1,6-linked oligosaccharides.
Scheme 17.
Rhee asymmetric synthesis of hexoses via a Pd-glycosylation (Pd-allylation/RCM).
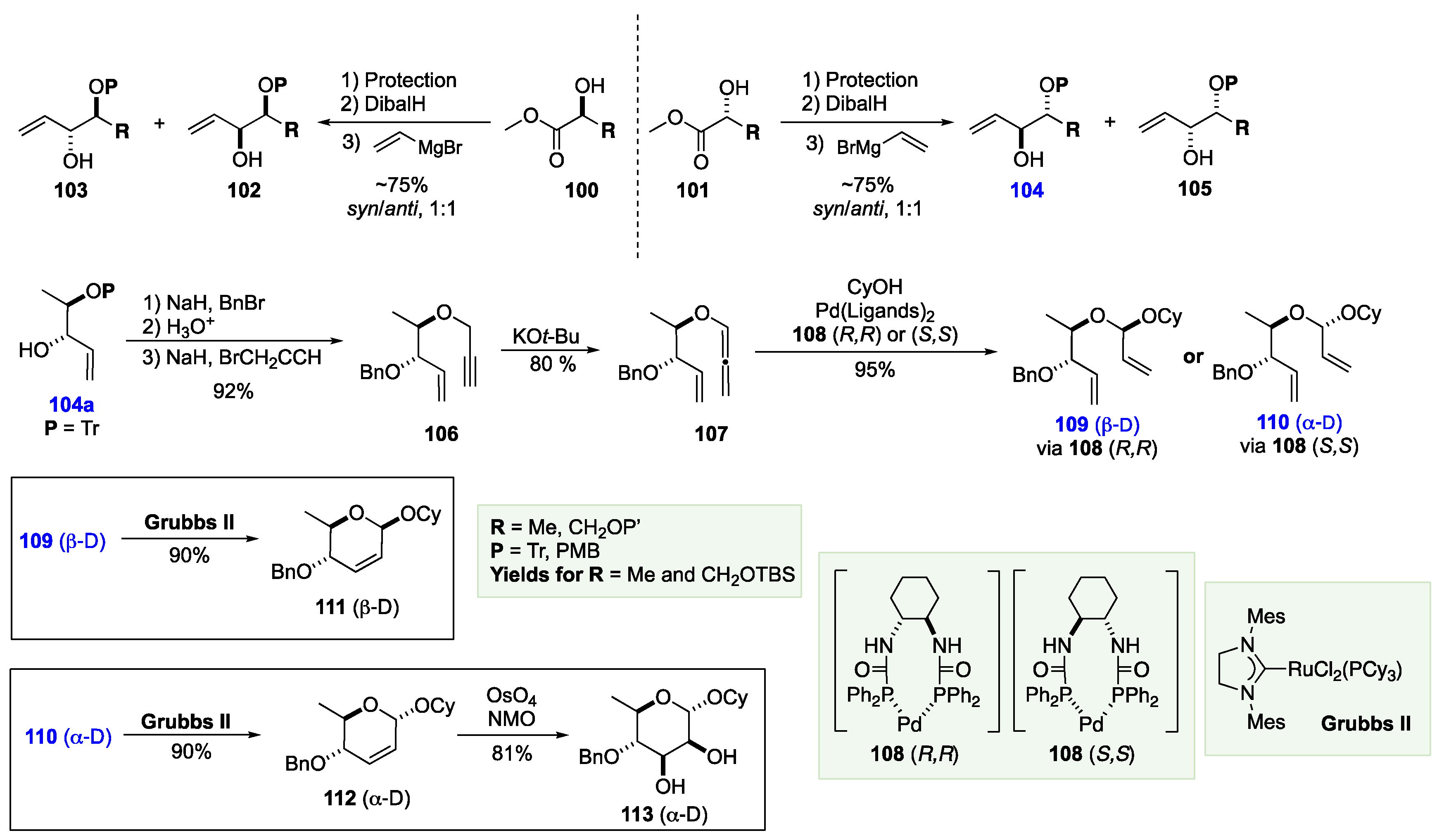
Scheme 18.
The landomycin family of oligosaccharides.
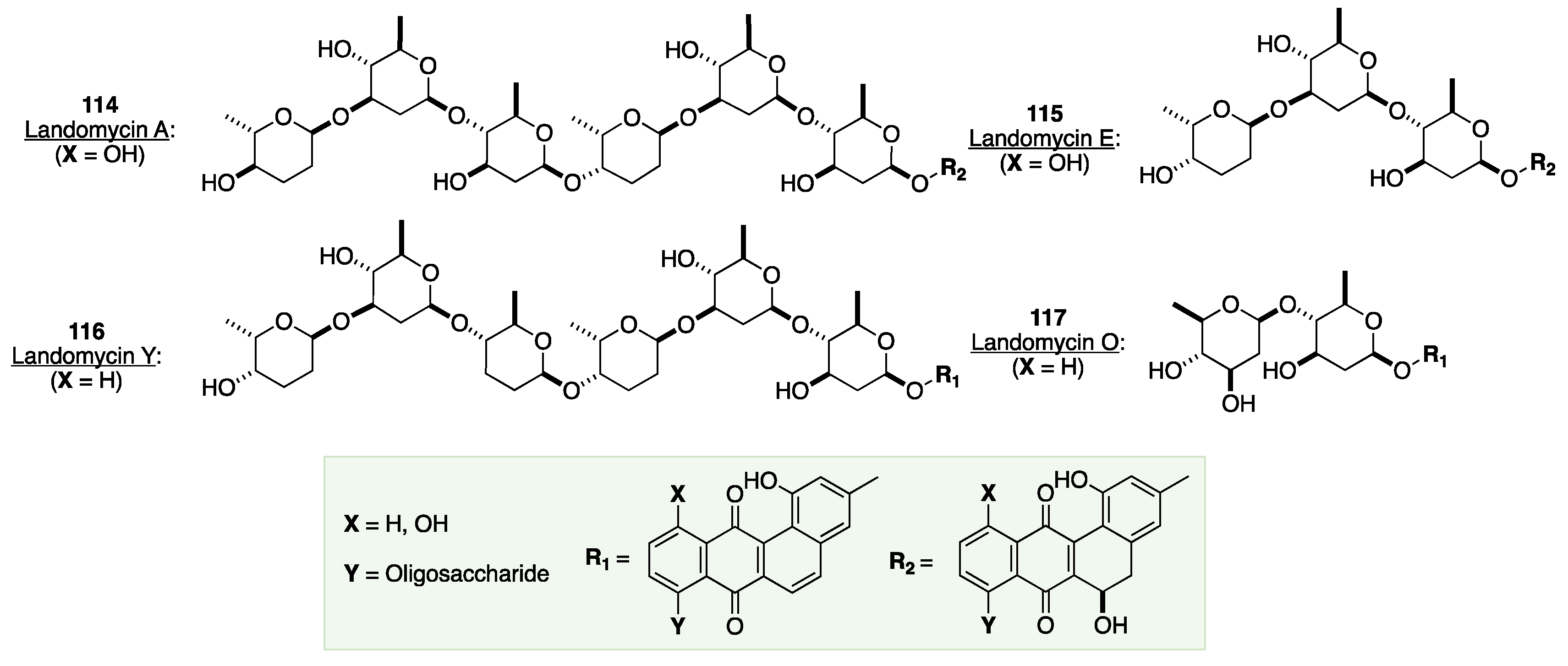
Scheme 19.
O’Doherty de novo asymmetric synthesis of the trisaccharide repeat of landomycin A.
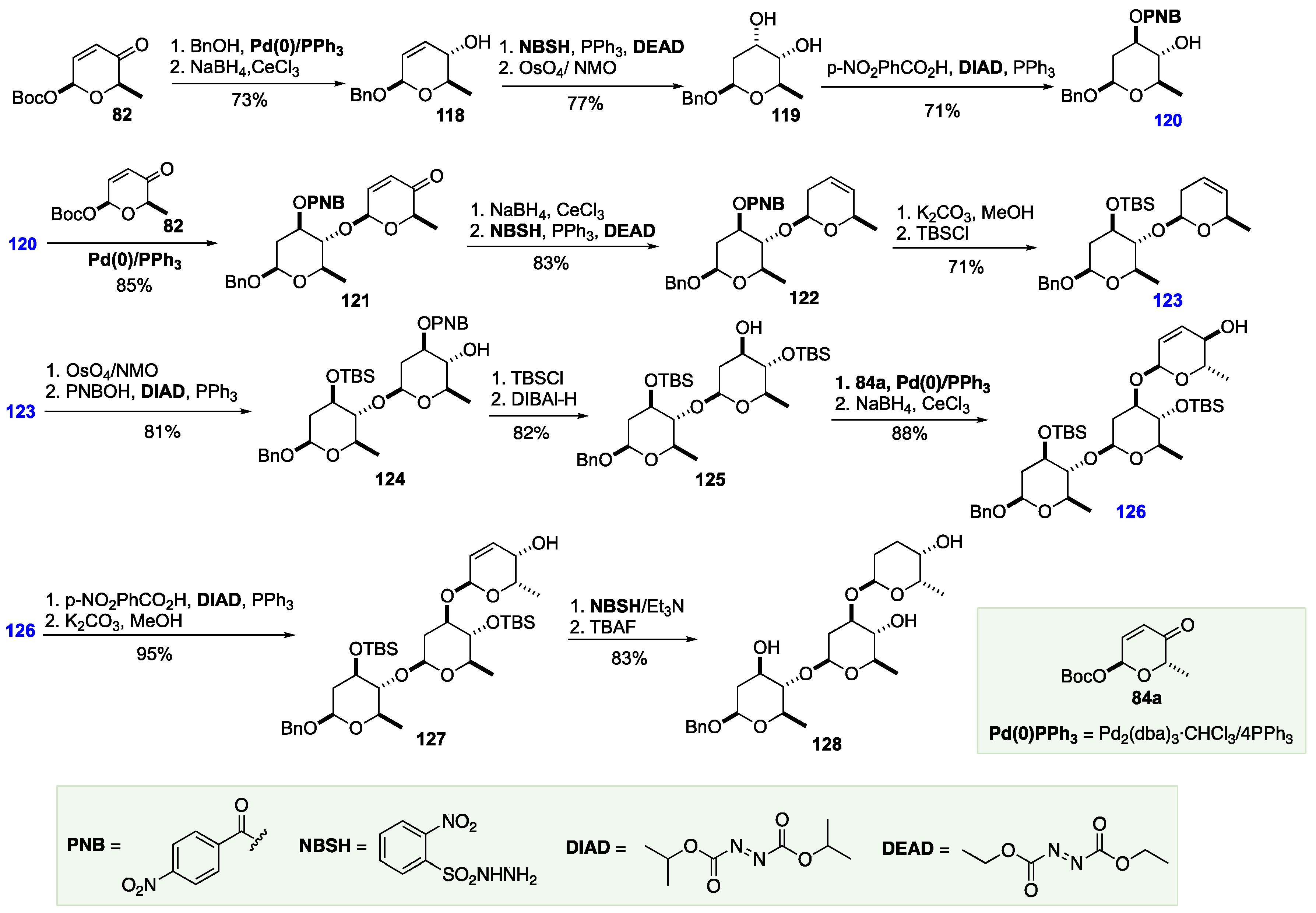
Scheme 20.
Rhee asymmetric synthesis of the landomycin disaccharide.
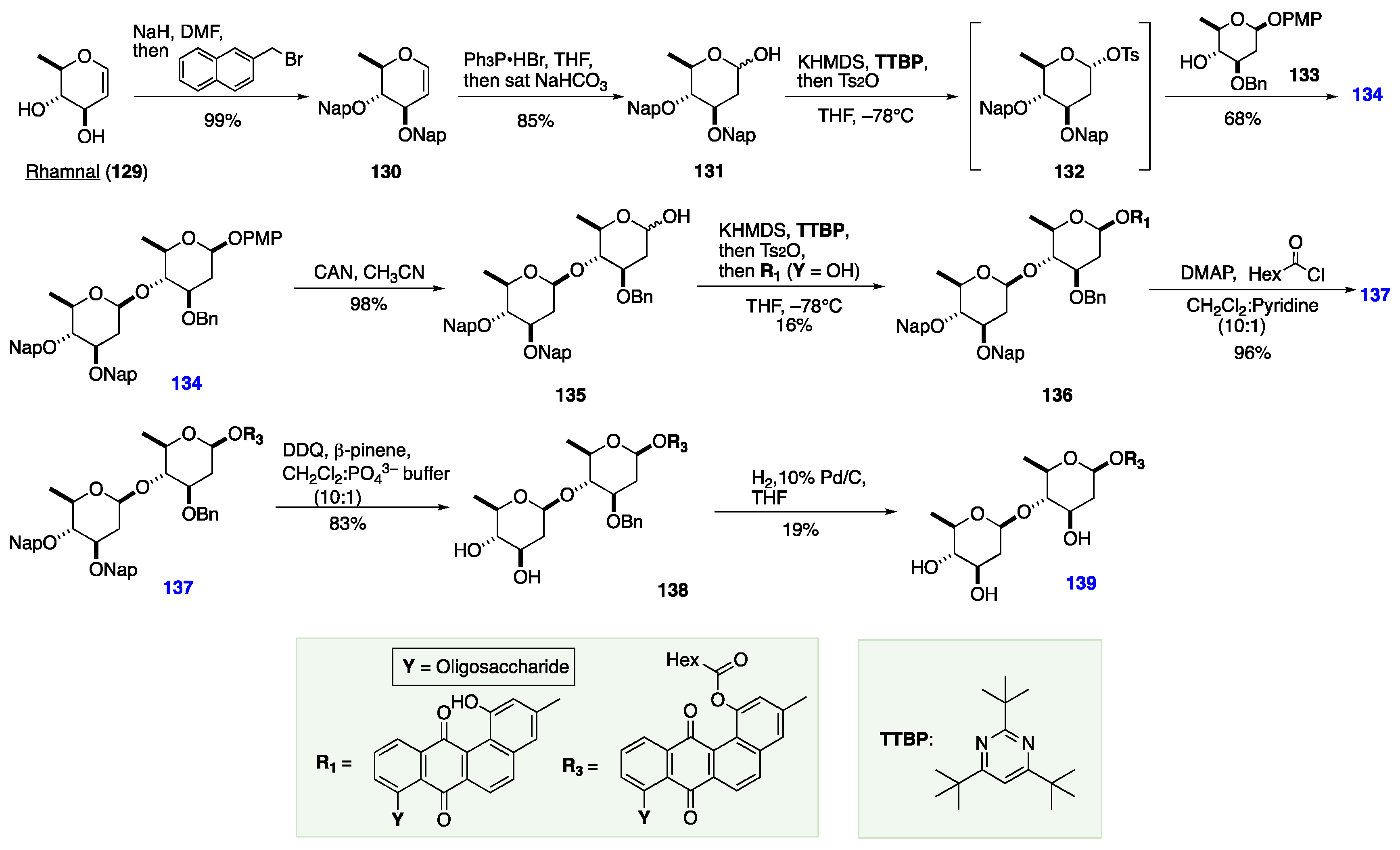
Scheme 21.
Rhee Pd-allylation approach to a landomycin tetrasaccharide precursor.
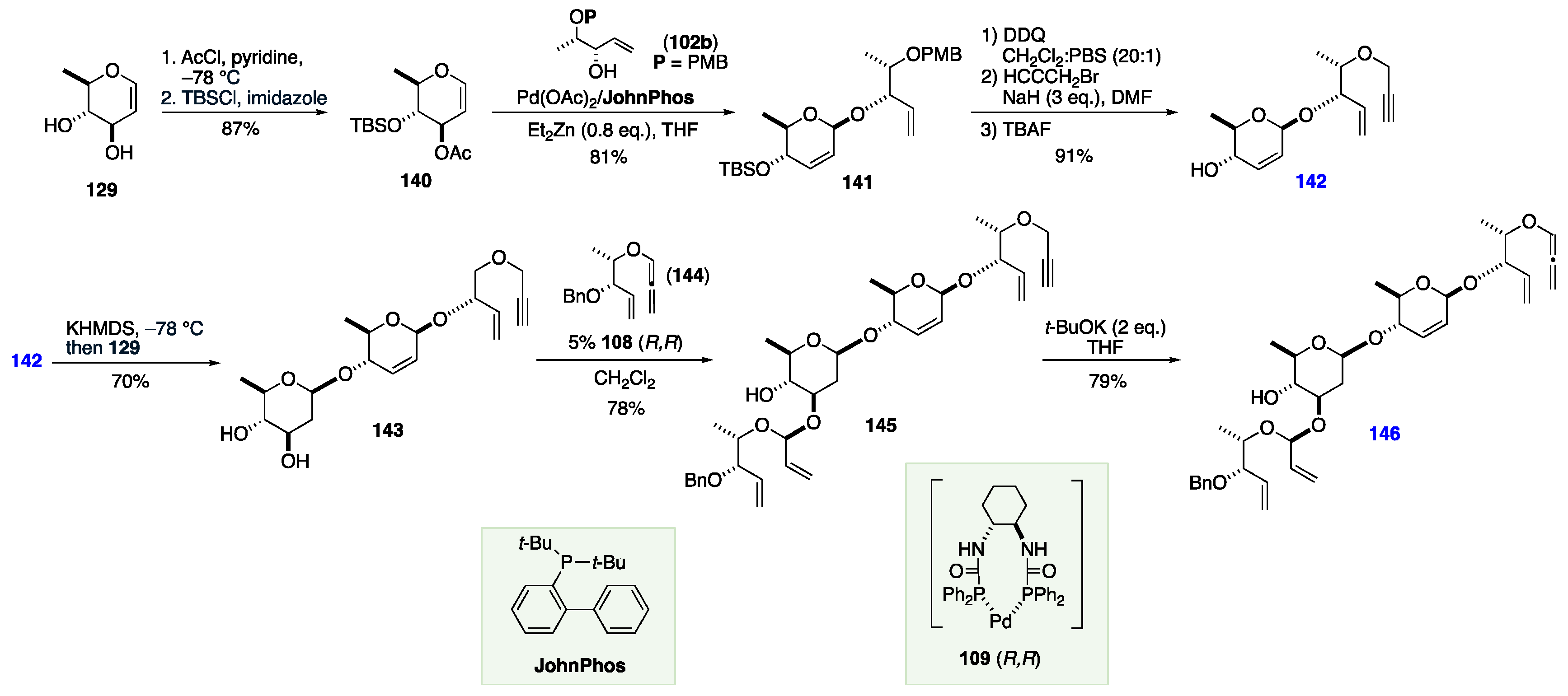
Scheme 22.
Rhee Pd-allylation and RCM approach to a landomycin Y.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



