Submitted:
23 December 2024
Posted:
24 December 2024
You are already at the latest version
Abstract
In contrast to the classical paradigm of treating enzymes as passive structural scaffolds that bring together and properly orient other participants in the catalyzed reactions, increasing evidences suggest that the internal motions and conformational alterations of intrinsically flexible enzymes play a vital role throughout catalysis. Such a biophysical view, based on a comprehensive survey of the literature, indicates that a number of enzymes contain distinct regions of conserved residues that are in relation to the active site and presumably serve as pathways of energy transfer for the thermodynamic coupling of the surrounding solvent with enzyme catalysis. It means that some particular conformational fluctuations within enzyme structure essentially encode dynamics and the catalytic rate acceleration, that is, enzymatic features that promote function. Certain computational methods are shown to be capable of establishing the underlying hierarchy of motions that drive the conformational and energetic coupling between enzyme and solvent through internal regions of different protein folds. Theoretical studies that deal with the dynamics of enzyme structure are based on molecular mechanics simulations associated with affordable computational costs. This advantage is limited by the fact that enzyme electronic structure is treated as being unaffected throughout the entire simulation. The impossibility of observing the formation or break up of intra- and intermolecular interactions in order to grasp charge transfer means that it is necessary to exploit quantum-chemical methods that allow the change of the electronic structure in the underlying regions of enzyme function. Recent applications of quantum-chemical wave function-based calculations to this problem, being well calibrated relative to experimental observations, are reviewed. Accordingly, new insights into the function of several enzymes, including viral enzymes and those involved in drug metabolism, intracellular signal transduction pathways and cell cycle control, are summarized. Possible trends in further research are discussed.
Keywords:
catalysis
; conformational fluctuation
; enzyme
; function
; molecular dynamics
; quantum mechanics
; structure
1. Introduction
Enzymes are biological catalysts that play a pivotal role in the essential chemical reactions taking place in living organisms and in technological applications. For example, they are needed for providing cellular energy, repairing tissues, organs and cells, stimulating brain functions, digesting food etc. Research interest in the mechanisms of enzyme-catalyzed reactions and their kinetics dates from the middle of the 18th century on, particularly in terms of the relation between enzyme structure and the determinants of the specificity and efficiency of catalysis (Fersht, 1985). Experimental and biomolecular modeling studies, based on the lock-and-key, induced-fit and transition-state stabilization hypotheses, enabled to conceive enzymes as structural scaffolds that selectively recognize and bind substrates and cofactors (Fischer, 1894; Koshland, 1958; Robertus et al., 1972) in a reaction environment that is different from the bulk solvent (Bruice & Benkovic, 2000). They are able to speed up reactions by about 1017 folds or more (Richard, 2013). Active site residues that are in direct contact with substrates and cofactors are critical for orienting other reactants and keeping them in fixed positions properly (Nagel & Klinman, 2009; Nagel et al., 2013), indicating that mutations of these specific residues allow for the change of the rate of reactions in a desired way (Nagel et al., 2013). Active sites presumably have shapes that are complementary to the transition states of catalyzed reactions, and thus provide complementary structural and electrostatic environments for the preferential binding and/or stabilization of the transition states (Warshel et al., 2006). Electrostatic effects are one of the key factors for understanding the mechanism of enzyme catalysis. In contrast to the classical view that considers enzymes as rigid receptors responsible for interacting with properly-oriented ligands (Figure 1, left), growing evidences suggest essential impact of the bulk and hydration-shell solvent (Figure 1, right) on enzymatic structure-function relationships (Agarwal, 2005; Fenimore et al., 2004). The question of how other factors than direct structural interactions, such as long-range effects (electrostatic effects, external and internal solvent molecules) and the energy released from the binding of substrate and/or cofactor, contribute to catalysis is a research challenge. Noteworthy is that bio-organic and protein molecules, designed to mimic enzyme active site, are mainly associated with lower catalytic efficiencies than their natural counterparts (Benkovic, 1992; Siegel et al., 2010). In other words, a particular mechanism by which each factor contributes to overcoming the activation energy barrier depends on enzyme system and type of chemical catalysis.
Understanding enzymes as intrinsically flexible macromolecules implicated substantial interest in studying the impact of internal motions, driven by environment and temperature, on their function (Antoniou & Schwartz, 2011; Arora & Brooks, 2009; Aviram et al., 2018; Boehr et al., 2018; Choy et al., 2017; Clouthier et al., 2012; Garcia-Viloca et al., 2004; Henzler-Wildman et al., 2007; Hirschi et al., 2010; Holliday et al., 2017; Kim et al., 2017; Klinman, 2013; Klinman et al., 2017; Kohen, 2015; Kondo et al., 2017; Lisi & Loria, 2016; Machleder et al., 2010; Micheletti, 2013; Onions et al., 2016; Otten et al., 2018; Quaytman & Schwartz, 2007; Schramm et al., 2008). The role of internal motions is a matter of intensive debate in the literature with several contradictory views (Benkovic et al., 2008; Boehr et al., 2006; Kamerlin & Warshel, 2010; Kohen, 2015; Nagel & Klinman, 2009; Ramanathan & Agarwal, 2011) due to various definitions of protein motions (Kohen, 2015). It means that some particular conformational fluctuations may be associated with encoding dynamics and the catalytic rate acceleration being enzyme’s features that promote function (Agarwal, 2019). The origin and hierarchy of these internal motions in enzyme structure are discussed hereafter. Also, recent applications of hybrid quantum mechanics/molecular mechanics (QM/MM) calculations in understanding enzyme function, which are well calibrated relative to experimental observations by allowing the change of electronic structure in the functionally critical regions of enzyme structure, are reviewed.
2. Reaction-promoting conformational fluctuations in the enzyme structure
The term enzyme dynamics often has a misleading meaning, considering a wide range of motions associated with. A physically meaningful distinction of the internal motions is based upon their timescales (Agarwal, 2019). The first type of motions includes those at the sub-picosecond timescales, involving a small number of atoms or a few residues, sampling the same area of conformational landscape, averaging around the same mean structure over time, and being driven by temperature (Figure 2a). The second type of motions includes those at the nanosecond, microsecond and higher timescales, involving fluctuations within protein domains (loops, secondary structure elements) or entire protein, sampling remote areas of conformational landscape, and being driven by coupling global and local vibrational modes in dynamic allostery of proteins (Figure 2b). The third type of motions includes temporary (or even permanent) change in protein structure, shifting the thermodynamic equilibrium through changes in the energetic landscape due to the binding (or release) of other molecules (Figure 2c), and leading to a change in average structure (Agarwal, 2019; Hawkins & McLeish, 2006).
A variety of experimental methods, including nuclear magnetic resonance (NMR) (Boehr et al., 2006; Holliday et al., 2017; Palmer, 2015), single molecule techniques (Zhang et al., 2004), hydrogen/deuterium exchange (Zavodszky et al., 1998) and neutron scattering (Tehei et al., 2007), have been combined with computational methods (Garcia-Viloca et al., 2004; Holliday et al., 2017) in order to identify motions that are coupled to enzyme reaction mechanisms (Boehr et al., 2006; Klinman et al., 2017). It was shown that a number of enzymes, including cyclophilin A (Agarwal et al., 2004; Camilloni et al., 2014; Howard et al., 2003), dihydrofolate reductase (DHFR) (Agarwal et al., 2002a; Boehr et al., 2013; Sawaya & Kraut, 1997; Wang et al., 2006), liver alcohol dehydrogenase (Agarwal et al., 2000; Bandaria et al., 2008; Billeter et al., 2001; Nagel et al., 2013), ribonuclease A (Veeraraghavan et al., 2010), lipase B (Layfield & Hammes-Schiffer, 2013) and so on, contain functionally relevant residues being distant from the active site and associated with a hierarchy of conformational fluctuations that mediate the thermodynamic coupling of the surrounding solvent with enzyme catalysis (Agarwal, 2019). These particular residues are arranged in networks (Figure 3) linking loop regions on the enzyme surface to the active site (Agarwal, 2004; Agarwal et al., 2002b; Agarwal et al., 2004; Agarwal et al., 2012; Ramanathan & Agarwal, 2011) through internal motions having the rates that are measurable and reproducible in both apo enzyme and enzyme complexes (Boehr et al., 2006; Eisenmesser et al., 2002; Fraser et al., 2009). Thus, the nature of the protein flexibility is not random, but intrinsic to the topology of enzyme structure (Agarwal, 2019). Functionally important conformational substates can be leveraged to overcome the activation barrier through selectively directed energy transfer into the active site, while dynamic regions of the enzyme structure involved in slower conformational shifts are thought to be evolutionary conserved (Agarwal, 2019; Ramanathan & Agarwal, 2011). This biophysical standpoint stimulates the development of hyper-catalytic enzymes by identifying and modulating reaction-promoting conformational fluctuations (Agarwal et al., 2012).
Although allosteric modulation is a common mechanism for regulation of biochemical processes (Monod et al., 1963), an insufficiently clarified aspect of the control of enzyme activity in the presence of long-range effects was related to electrostatics and correlated structural interactions (Baldwin & Chothia, 1979) in terms of energy flow in the enzyme structure (Frauenfelder et al., 2001; Lee et al., 2008; Leitner, 2008; Reynolds et al., 2011). In addition to enzyme involvement in lowering the effective transition state barrier by raising the ground state and/or lowering the transition state through stabilization, additional barriers that need to be overcome by enzyme-catalyzed reactions were energetically estimated to cost circa 5-10 kcal mol-1 (Agarwal, 2019). The random thermal fluctuations in enzyme structure provide about 1 kcal mol-1 at room temperature. Thus, the remaining amount of energy of 4-9 kcal mol-1 was hypothesized to come from the thermodynamic energy of the solvent on the protein surface (Agarwal, 2005; Agarwal, 2006; Ramanathan & Agarwal, 2011). It indicates that the pathways of connected residues, which span from the surface regions to the active site (Figure 3), serve as energetic channels for coupling the surrounding solvent to the enzyme reaction through intrinsic long-range effects (Fenimore et al., 2004). The thermodynamic coupling with solvent is a consequence of the energy produced by the fast motions of the surface loop residues with long side chains providing their high flexibility (Bellissent-Funel et al., 2016; Borreguero et al., 2011; Klinman et al., 2017; Ramanathan & Agarwal, 2009). This energy is transferred to the intermediate motions and eventually to the slow protein motions in the active site through preserved hydrogen bonds (Agarwal, 2005). This energy flow is not dissipative due to random collisions (Leitner, 2008; Moritsugu & Kidera, 2004), but likely directed flow of coupled conformational motions, mainly transfer of vibrational energy from a specific normal mode to several other resonant modes (Moritsugu & Kidera, 2004), as observed between α-helices (Leitner, 2008; Yu & Leitner, 2005). This aspect may be quantitatively rationalized using the configuartional entropy, being the entropy of the solute without translational and rotational contributions (Andricioaei & Karplus, 2001; Schlitter, 1993). The separation of translational entropy of the solute is generally acceptable without any approximation, while the removal of rotational entropy is associated with the assumption of negligible correlation between the internal degrees of freedom and the global rotation of the solute. The configurational entropy, estimated by the quasiharmonic approximation that treats a system of atoms with many degrees of freedom as a system of uncoupled harmonic oscillators and assumes a multivariate normal distribution of atomic fluctuations, gives the upper bound of the true entropy contribution to the free energy of non-covalent binding (Andricioaei & Karplus, 2001). The configurational entropy change can pragmatically be considered as the thermodynamic consequence of the change in receptor flexibility when binding ligands (Mitrasinovic, 2015, 2018, 2019). Understanding that slower global motions are consequences of faster local motions in proteins speaks in favor of the hypothesized model of enzyme activity (Figure 1, right) that was supported by a few experimental studies (Boehr et al., 2013; Chi et al., 2015; Eisenmesser et al., 2005; Gagne et al., 2012, 2015; Holliday et al., 2017; Wang et al., 2006).
Protein motions that occur at short timescales allow conformational sampling within the well (Figure 2a), while those at longer timescales stretch over multiple minima and allow interconversion between different conformational substates (Figure 2b). The rate of conformational transitions in proteins was related to slower anharmonic energy relaxation rate at the nanosecond timescale with the assistance of multilevel vibrational excitations (Xie et al., 2002). It was reported that functionally relevant conformational substates, characterized by small populations, contain structural and dynamic features that promote catalytic reactions (Ramanathan et al., 2014). The observed anharmonic nature of protein motions at long timescales (Borreguero et al., 2011; Ichiye & Karplus, 1988; Pontiggia et al., 2007) led to the development of a new computational method, termed “quasi anharmonic analysis” (QAA), for the identification of functionally relevant conformational substates by use of higher order statistics (Ramanathan et al., 2011). Conformational transitions obtained by QAA (Ramanathan et al., 2011) were in agreement with results of the NMR spin relaxation studies (Eisenmesser et al., 2002; Lange et al., 2008), showing that long-range fluctuations primarily refer to both loop areas on protein surface and loops located in the vicinity of the active site and represent rare, short-lived enzyme conformations that facilitate the acquisition of the transition states through their geometrical features (Agarwal, 2019). The impact of the hidden conformational substates on enzyme catalysis was highlighted experimentally (Fraser et al., 2009).
Although enzymes undergo a range of motions, the lack of clear definition of protein dynamics has been making a number of computational and experimental studies unsuccessful in providing consistent explanations on which particular protein motions promote catalysis (Agarwal, 2019). The development of models, based on the QAA methodology and followed by experimental validation, is a promising approach to getting mechanistic insights into the identification of functionally relevant conformational substates. For example, the study of enzyme activity in miscible organic solvents has shown that not all types of protein dynamics alter the catalytic rate, but a particular subset of conformational transitions into functionally relevant substates (Duff et al., 2018).
3. On the relevant structural basis of enzymatic function
Essential conformational fluctuations that subtly drive receptor structure into functionally relevant substates are reflected through the formation or break up of intra- and intermolecular interactions in the functionally critical regions of enzyme structure on the nanosecond timescale (Pavlov & Mitrasinovic, 2010). The advent of computers has enabled the development and application of theoretical models that aid in decoding ligand/enzyme binding as a major topic in medicine, chemistry and biology. All the main computational methods have some pros and cons, so that any particular choice should be carefully made after considering the chemical nature of the system under investigation, the available structural and experimental data, and the ultimate goal of the research (Limongelli, 2020).
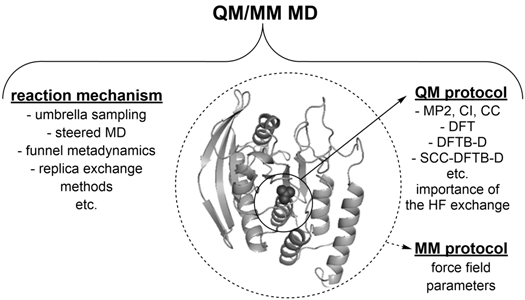
The possibility of observing various conformations in static crystal structures, as a consequence of different crystallization conditions or procedures, implies that the dynamics of ligand/enzyme binding is a functionally remarkable standpoint of consideration. Theoretical studies that deal with the molecular dynamics (MD) of enzyme structure are based on molecular mechanics (MM) simulations associated with acceptable computational costs (Limongelli, 2020). This advantage is, however, balanced with the fact that enzyme electronic structure is treated as being unchanged throughout the entire simulation. Thus, it is hard to grasp the underlying charge transfer of intra- and intermolecular interactions by applying only MD methods (Pavlov & Mitrasinovic, 2010). Instead, it is necessary to exploit quantum mechanical (QM) methods that model the distributions of electrons in molecules explicitly. The geometries, electronic structure and reactions of small molecules are commonly studied by this approach. As an enzyme system typically consists of thousands of atoms means that the modeling of an entire enzyme structure using QM methods is a computationally challenging task. Furthermore, the need to account for solvent effects using adequate descriptions of the non-bonded interactions between solvent molecules, and between the solvent and solute, additionally makes required quantum chemical wave functions a lot more sophisticated. Therefore, fully realistic models of the solvation of macromolecular complexes are currently out of reach by calculation. In need of the strategies capable of treating efficiently charge-related effects in the functionally relevant regions of the large biomolecular structures, hybrid - QM/MM approach was developed with the ultimate goal to yield subtle conformational insights into the recognition of biological macromolecules. The general idea was to approximately treat electron correlation by QM in the key functional region of biomolecular structure, while having the rest of the system treated by MM (Lonsdale et al., 2012).
QM methods mutually differ depending on the approximations made to the Schrodinger equation that cannot be solved exactly for molecular systems containing more than one electron. Three main types of QM methods are semi-empirical approaches, ab initio and density functional theory (DFT). Semi-empirical molecular orbital methods, such as AM1 and PM3, are based on the Modified Neglect of Diatomic Differential Overlap (MNDO) approximation, the least computationally demanding and the least accurate of the QM methods. In some cases a MM treatment may be better than a low level semi-empirical one. Semi-empirical methods can be applied to larger systems than ab initio or DFT methods, typically to those of hundreds of atoms. The foundation of the most straightforward ab initio method is based on Hartree-Fock (HF) theory, which makes the very fundamental approximation that each electron's spatial distribution is independent of the other electrons’ instantaneous motion. By ignoring electron correlation or the tendency of electrons to avoid each other, HF calculations on chemical reactions are often misleading and impractical. Other ab initio methods that include electron correlation, such as those based on Moller-Plesset perturbation theory (e.g., MP2), configuration interaction (CI), or coupled cluster theory (CC), exploit HF wavefunctions as a starting point and provide a significant improvement in accuracy over HF calculations (Figure 4), but with much higher computational costs that make their application to systems with many tens of atoms difficult (Lonsdale et al., 2012). DFT provides a theoretical framework for determining the ground-state energy of a molecule from knowledge of the electron density distribution. The electron density is a function of only three variables, and thus much less complex for calculation than the ab intio wave function being a function of 3N variables, where N denotes the number of electrons. As an exact form of the functional for connecting the density to the energy is unknown, many approximate functionals have been empirically developed (Ahmed et al., 2012; Mitrasinovic, 2001, 2002, 2003a, 2003b, 2004, 2005, 2010a, 2012, 2013, 2014a, 2014b); e.g., B3LYP – a combination of the Becke’s three-parameter hybrid exchange functional (Becke, 1993) and the Lee-Yang-Parr correlation functional (Lee et al., 1988). DFT methods are capable of approaching accuracy of the correlated ab initio methods at much more reasonable computational expense (Lonsdale et al., 2012; Pavlov & Mitrasinovic, 2010), but often incapable of describing dispersive forces adequately (Grimme et al., 2007), particularly within unconventional systems (Hobza et al., 1995; Kristyan & Pulay, 1994). By including a dispersion correction (D) into DFT, DFT-D has become capable of performing the energy minimizations and vibrational analyses of extended molecular systems containing hundreds of atoms (Hachim et al., 2020; Jurecka et al., 2007; Mohammadkhani et al., 2020; Wang & Zhu, 2020). The second order Taylor expansion of the DFT total energy around a reference density has been a starting point for further approximations leading to the the Self-Consistent-Charge Density-Functional Tight-Binding (SCC-DFTB) model that contains additional induced-charges term for a proper description of charge-transfer phenomena (Elstner et al., 1998, 2001). SCC-DFTB with an empirical correction for dispersion energy (SCC-DFTB-D) (Grimme et al., 2007) has contributed to a more balanced description of the interactions inside the systems (Kubar et al., 2007). By being several orders of magnitude faster than DFT-D (Pavlov & Mitrasinovic, 2010) and providing geometries and relative energies comparable to DFT and ab initio calculations (Elstner et al., 2003), SCC-DFTB-D is an optimal, quantum chemical choice for modeling the structural parts of enzyme systems that are mainly responsible for their functions (Figure 4).
In order to provide insights into enzyme function by biomolecular modeling, a high-resolution (less than 2 Å) X-ray crystallographic structure of an enzyme complex is usually a starting point that tends to accurately represent the reacting system. A crystal structure is a result of averaging over all the conformations in the crystal and over the entire time of data acquisition. It means that the alternative conformations may be observed for some structural elements of the system, particularly for those that are very mobile and have no well-defined single conformation or position such as amino acid side chains, surface loops, and terminal regions. It is difficult to structurally isolate the active enzyme-substrate complexes as their reactions proceed too fast. The crystal structures of less efficient mutants or substrates can sometimes be resolved by varying redox conditions, if the reactions are slow enough to allow the observation of the complexes. It is widely accepted that the crystallographic structure of an enzyme-inhibitor complex, recognized as a closed or “receptor inaccessible” conformation, is an appropriate choice, where the inhibitor mimics the substrate, product, and transition state (or an intermediate) in its bound conformation (Lonsdale et al., 2012). Therefore, an apo (ligand-free) crystal structure of an enzyme, denoted by an open or “receptor accessible” conformation, may be seen as one of little use, by being incapable of predicting conformational changes upon binding. However, it has been shown that the open conformations in the structures of several enzymes, such as influenza A/H5N1 virus neuraminidase (Mihajlovic & Mitrasinovic, 2008, 2009a, 2009b; Mitrasinovic, 2009, 2010b; Wang et al., 2010), HIV-1 protease (Yu et al., 2017), Yersinia protein tyrosine phosphatase (YopH) (Mitrasinovic, 2023) and others, should be considered as conformations of some relevance for exploring the dynamics of their recognition by ligands. In all those cases, a common point is that an open state is defined by the conformation of a loop that is located nearby the active site and affects ligand binding through the conformational flexibility of some of its residues (Figure 2c). For example, the apo HIV-1 protease shows a variety of flap conformations, ranging from closed (Pillai et al., 2001; Robbins et al., 2010) to semi open (Heaslet et al., 2007; Lapatto et al., 1989; Liu et al., 2006; Spinelli et al., 1991) and to wide open (Coman et al., 2008; Heaslet et al., 2007; Martin et al., 2005). It means that the wild-type (wt) receptor may initially approach to the ligand in an open conformation and, then, likely adopts the higher energy state or closed conformation of the particular loop via a subtle conformational change. A more realistic interpretation, based on the thermodynamics of protein-ligand binding, is that a semi-open intermediate reduces the free energy barrier between the closed and open states (Gur et al., 2020), despite the fact that it is difficult to capture a semi-open state by currently available experimental techniques (Lapatto et al., 1989; Spinelli et al., 1991; Navia et al., 1989) due to its short transient lifetime. In the case of YopH, this intermediate has been observed by means of computational simulations, which demonstrate that the closure of the so-called “WPD” loop occurs in a half-closed state in the apo, native protein structure (Hu & Erec Stebbins, 2006). The possibility of having the conformational flexibility of the WPD loop correlated with the conformational behavior of another loop as the second binding site in the YopH structure (Hu & Erec Stebbins, 2006) has been elaborated in more detail (Mitrasinovic, 2024a, 2024b), thereby indicating that the potential allosteric binding sites offer new opportunities for the design of novel, selective YopH inhibitors (Mitrasinovic, 2023, 2024a, 2024b) as antibacterial agents (Kuban-Jankowska et al., 2016). It has also been reported that the catalytic process during glycolysis is associated with the synergistic effect of conformational conversions through a semi-open state in phosphoglycerate kinase 1 (PGK1) due to the releasing/binding of products/substrates, as well as with post-translational modifications, thereby enabling to better understand the PGK1 substrates/products conversion process and the development of small drug molecules targeting PGK1 (Liu et al., 2022). A similar approach would apply to the structural understanding of the inhibition mechanism of a lipid peroxidase pathway associated with therapy-resistant cancer cells in various mesenchymal cell state contexts (Viswanathan et al., 2017). It has even been suggested that forming the semi-open or intermediate state in a protein structure is a prerequisite to the recognition of host cells (Gur et al., 2020). In some cases, specific single point mutations might affect the attachment of proteins to cell surface receptors by improving the stability of proteins in their open and closed conformations and driving the thermodynamic preferences toward the open mutant form (Verkhivker et al., 2022).
Cytochrome P450 enzymes (P450s or CYPs) are mainly involved in Phase I drug metabolism, which includes the introduction of polar functional groups into non-polar molecules and can lead to inactivation or activation of drugs. Therefore, modeling P450-catalyzed reactions is important for drug discovery (Lonsdale et al., 2012). QM calculations using small cluster models improved the understanding of the mechanisms through which these enzymes stimulate oxidative metabolism (Bathelt et al., 2003, 2004, 2008; Guallar et al., 2003; Shaik et al., 2005, 2010; Zurek et al., 2006). The modeling of such systems, based on the B3LYP density functional, is in good agreement with the available experimental data (Bathelt et al., 2008; Harvey et al., 2006; Lonsdale et al., 2010, 2011; Shaik et al., 2010; Zurek et al., 2006). The determination of the crystal structures of P450 enzymes, combined with the development of QM (DFT)/MM methods (Figure 4), made it possible to model the mechanisms of oxidation in the enzymatic environment. The QM/MM (SCC-DFTB-D/Amber-FF99SB) methodology, being well calibrated against relevant experimental data, was an inevitable choice between alternatives to address several vital issues by studying the ligand/enzyme binding interactions and evaluating the competitive inhibition parameters. Both quantitative and qualitative insights into the functional ability of structurally diversified flavonoids to inhibit P4503A4, the most versatile enzyme involved in drug metabolism, were reported (Mitrasinovic, 2022). The same approach was employed to rationalize the affinity and specificity of peptide inhibitors to caspase-2, one of the most evolutionarily conserved caspases that is involved in cell-cycle regulation and DNA repair as well as in mediating neurodegenerative disorders and non-alcoholic steatohepatitis pathogenesis (Mitrasinovic, 2020). In a similar manner, the active/inactive functional behavior of YopH, an extraordinarily active protein tyrosine phosphatase involved in breaking down signal transduction mechanisms in immune cells and inhibiting the immune response, was investigated by observing its modes of recognition by carboxylic acid derivatives (Mitrasinovic, 2023) and identifying a pharmacophore for designing novel inhibitors capable of achieving specificity against phosphatases (Mitrasinovic, 2024a, 2024b). In all the QM/MM models (Mitrasinovic, 2020, 2022, 2023), the QM region was defined to comprise not only active site residues and the ligand, but also loops proximal to the active site and flexible surface loop regions previously shown by using experimental means to promote function through their conformational fluctuations within the enzymatic structure of P4503A4 (Sevrioukova & Poulos, 2015), caspase-2 (Tang et al., 2011), and YopH (Schubert et al., 1995), respectively. Thus, SCC-DFTB-D essentially reconciled an implicit description of charge-transfer phenomena with a balanced description of the interactions inside the systems, enabling to quantitatively express the impact of the underlying conformational substates on the enzyme’s performances. It was for this reason necessary to avoid unrealistic effects by choosing the adequate positions of atoms on the QM/MM frontier (Figure 4), so that they do not cut across any polar bond (Mitrasinovic, 2020, 2022, 2023). A pseudo-QM atom can be added to create an appropriate bond, e.g., modeling edge atoms as hydrogen atoms and neglecting some of their interactions with MM atoms. The charges of MM groups next to QM groups are commonly set to zero in order to escape unbalanced charge interactions across the boundary, even though more sophisticated electrostatic settings can be applied too (Lonsdale et al., 2012). Finally, in order to reasonably describe conformational fluctuations within an enzyme structure as pathways of energy transfer that encode dynamics, an evaluation of chosen protein force field for MM part is needed in terms of how accurately it predicts the structure of multiple flexible loop domains present in proteins (Love et al., 2022).
To sum up, it is pragmatic to note two sides - the mechanistic and electronic structure aspects of modeling enzyme-catalyzed reactions. The mechanistic aspect of this topic can be explored using the QM/MM strategy, the empirical valence bond (EVB) methodology and the cluster approach. Although useful, the cluster approach is out of the scope of this review, because it includes only a small portion of the enzyme structure in the models for exploring chemical properties and reaction mechanisms (Himo, 2017). An insufficient amount of available experimental data about the catalytic efficiencies of enzyme substrates often means that a more complete understanding of the true substrate preferences is hardly possible. The above-mentioned QM/MM results (Mitrasinovic, 2020, 2022, 2023) illustrate the way in which additional experiments can be stimulated by biomolecular modeling, in order to better understand the kinetics of ligand/enzyme recognition (e.g., in terms of specificity (Mitrasinovic, 2024a, 2024b)) for further research or therapeutic product development (Mitrasinovic, 2020). We advocate that both mechanistic and electronic structure standpoints can be reconciled by applying a more advanced strategy based on molecular dynamics simulations with the QM/MM potentials (Figure 4). For example, in the case of hydrolysis by glutamate carboxypeptidase, it was possible to construct reaction energy profiles with realistic barrier heights on the reaction pathways for the wt and mutated enzymes (Krivitskaya et al., 2021). The QM/MM potentials were evaluated by means of atom-centered basis sets and various hybrid DFT functionals in QM parts, as well as commonly accepted force field parameters in MM parts (Krivitskaya et al., 2021). To generate MD trajectories on 10 picosecond timescale (Krivitskaya et al., 2021), the umbrella sampling (US) scan of the Gibbs free energy landscape was exploited with the subsequent statistical analysis (Limongelli, 2020), using the umbrella integration methods to quantify the elementary steps of chemical reactions in the enzyme active sites (Krivitskaya et al., 2021). Anyway, the conformational flexibility of molecules (large proteins or small ligands) is an important feature. It means that conformational sampling that generates an ensemble of biologically meaningful conformations is a precondition for computer-aided drug design. It was observed that funnel metadynamics (FM) at the nanosecond timescales leads to an efficient characterization of the binding free energy surface and an accurate calculation of the absolute ligand/protein (or enzyme) binding free energy compared to an experimental value (Limongelli et al., 2013). FM also yields important insights into the binding process, such as the presence of alternative binding modes and the role of water, at an affordable computational cost (Limongelli, 2020; Limongelli et al., 2013). Some of the generally applicable correction terms can further improve the accuracy of physical pathways methods for calculating the free energy, such as Markov state modeling, FM, US and steered MD (Hall et al., 2020). Continued advances in increasing computational power are expected to make QM/MM MD calculations (Figure 4) routine in the timescales involving conformational fluctuations within protein domains.
4. Future outlook
The origin of the enormous catalytic power of enzymes is one of the key questions in biology today. To address this issue, besides electrostatic effects that stabilize the transition states of enzymatic reactions, it is necessary to investigate the impact of other possible factors (Pisliakov et al., 2009). An interesting possibility is that the dynamics of conformational transitions makes a remarkable contribution to enzyme catalysis through a hierarchy of internal motions in intrinsically flexible enzyme structures (Agarwal, 2019). A multiscale approach has been developed to explore the dynamic nature of enzyme catalysis in the millisecond timescale, indicating that there exists basically no coupling between chemistry and protein structural fluctuations as long as the chemical barrier is greater than a few kcal/mol (which is the case in enzymatic reactions) (Pisliakov et al., 2009). In spite of that, the nature of this coupling is a question of great importance that deserves continued research interest. This view may be relevant for the development of hyper-catalytic enzymes by identifying and modulating reaction-favoring conformational fluctuations (Agarwal, 2019; Agarwal et al., 2012).
Advanced QM/MM MD calculations are getting feasible due to advances in computer power that enables to reconcile mechanistic and electronic structure aspects (two sides of the same enzyme-catalyzed reaction) in the nanosecond, microsecond and higher timescales (Figure 4). In the context of ligand/protein (or enzyme) recognition, advanced MD techniques, such as replica exchange methods (Yang et al., 2015; Yang & MacKerell, 2015), can enhance the sampling efficiency of the conformational landscape at the microsecond scales and reveal new modes of interaction (Mitrasinovic, 2019). The integration of more rigorous physics-based methods, such as emerging methods for ensemble based virtual screening, enables the use of multiple enzyme conformations, reveals new modes of activation/inhibition, provides a better understanding of the structural dynamics of disease-related target enzymes, and supports the development of novel patient-specific therapeutics that are less prone to drug resistance (Amaro & Li, 2010). Thus, computational protocols, such as SILCS (site identification by ligand competitive saturation) for structure-based drug design and CSP (conformationally sampled pharmacophore) for ligand-based drug design, are more advantageous in practice than CADD (computer-aided drug design) methods being based on single crystal structure or a limited number of ligand conformations (Yu & MacKerell, 2017).
Acknowledgment
Prof. David A. Case of Rutgers University is acknowledged for granting the author an academic license for using the molecular dynamics software package Amber 11 in combination with Amber Tools 1.5.
Disclosure statement
No potential conflict of interest was reported by the author.
References
- Agarwal, P. K. (2004). Cis/trans isomerization in HIV-1 capsid protein catalyzed by cyclophilin A: insights from computational and theoretical studies. Proteins, 56(3), 449-463. [CrossRef]
- Agarwal, P. K. (2005). Role of protein dynamics in reaction rate enhancement by enzymes. Journal of the American Chemical Society, 127(43), 15248-15256. [CrossRef]
- Agarwal, P. K. (2006). Enzymes: an integrated view of structure, dynamics and function. Microbial Cell Factories, 5, 2. [CrossRef]
- Agarwal, P. K. (2019). A biophysical perspective on enzyme catalysis. Biochemistry, 58(6), 438-449. [CrossRef]
- Agarwal, P. K., Billeter, S. R., & Hammes-Schiffer, S. (2002a). Nuclear quantum effects and enzyme dynamics in dihydrofolate reductase catalysis. The Journal of Physical Chemistry B, 106(12), 3283-3293. [CrossRef]
- Agarwal, P. K., Billeter, S. R., Rajagopalan, P. T. R., Benkovic, S. J., & Hammes-Schiffer, S. (2002b). Network of coupled promoting motions in enzyme catalysis. Proceedings of the National Academy of Sciences of the United States of America, 99(5), 2794-2799. [CrossRef]
- Agarwal, P. K., Geist, A., & Gorin, A. (2004). Protein dynamics and enzymatic catalysis: investigating the peptidyl-prolyl cis-trans isomerization activity of cyclophilin A. Biochemistry, 43(33), 10605-10618. [CrossRef]
- Agarwal, P. K., Schultz, C., Kalivretenos, A., Ghosh, B., & Broedel, S. E. (2012). Engineering a hyper-catalytic enzyme by photoactivated conformation modulation. The Journal of Physical Chemistry Letters, 3(9), 1142-1146. [CrossRef]
- Agarwal, P. K., Webb, S. P., & Hammes-Schiffer, S. (2000). Computational studies of the mechanism for proton and hydride transfer in liver alcohol dehydrogenase. Journal of the American Chemical Society, 122(19), 4803-4812. [CrossRef]
- Ahmed, N., Babu, B. V., Singh, S., & Mitrasinovic, P. M. (2012). An efficient one-pot three-component synthesis of highly functionalized coumarin fused indenodihydropyridine and chromeno[4,3-b]quinoline derivatives. Heterocycles, 85(7), 1629-1653. [CrossRef]
- Amaro R. E., & Li, W. W. (2010). Emerging methods for ensemble based virtual screening. Current Topics in Medicinal Chemistry, 10(1), 3-13. [CrossRef]
- Andricioaei, I., & Karplus, M. (2001). On the calculation of entropy from covariance matrices of the atomic fluctuations. The Journal of Chemical Physics, 115(14), 6289-6292. [CrossRef]
- Antoniou, D., & Schwartz, S. D. (2011). Protein dynamics and enzymatic chemical barrier passage. The Journal of Physical Chemistry B, 115(51), 15147-15158. [CrossRef]
- Arora, K., & Brooks, C. L. III (2009). Functionally important conformations of the Met20 loop in dihydrofolate reductase are populated by rapid thermal fluctuations. Journal of the American Chemical Society, 131(15), 5642-5647. [CrossRef]
- Aviram, H. Y., Pirchi, M., Mazal, H., Barak, Y., Riven, I., & Haran, G. (2018). Direct observation of ultrafast large-scale dynamics of an enzyme under turnover conditions. Proceedings of the National Academy of Sciences of the United States of America, 115(13), 3243-3248. [CrossRef]
- Baldwin, J., & Chothia, C. (1979). Hemoglobin: the structural changes related to ligand binding and its allosteric mechanism. Journal of Molecular Biology, 129(2), 175-200. [CrossRef]
- Bandaria, J. N., Dutta, S., Hill, S. E., Kohen, A., & Cheatum, C. M. (2008). Fast enzyme dynamics at the active site of formate dehydrogenase. Journal of the American Chemical Society, 130(1), 22-23. [CrossRef]
- Bathelt, C. M., Ridder, L., Mulholland, A. J., & Harvey, J. N. (2003). Aromatic hydroxylation by cytochrome P450: model calculations of mechanism and substituent effects. Journal of the American Chemical Society, 125(49), 15004-15005. [CrossRef]
- Bathelt, C. M., Ridder, L., Mulholland, A. J., & Harvey, J. N. (2004). Mechanism and structure-reactivity relationships for aromatic hydroxylation by cytochrome P450. Organic & Biomolecular Chemistry, 2(20), 2998-3005. [CrossRef]
- Bathelt, C. M., Mulholland, A. J., & Harvey, J. N. (2008). QM/MM modeling of benzene hydroxylation in human cytochrome P450 2C9. The Journal of Physical Chemistry A, 112(50), 13149-13156. [CrossRef]
- Becke, A. D. (1993). A new mixing of Hartree-Fock and local density-functional theories. The Journal of Chemical Physics, 98(2), 1372-1377. [CrossRef]
- Bellissent-Funel, M. C., Hassanali, A., Havenith, M., Henchman, R., Pohl, P., Sterpone, F., van der Spoel, D., Xu, Y., & Garcia, A. E. (2016). Water determines the structure and dynamics of proteins. Chemical Reviews, 116(13), 7673-7697. [CrossRef]
- Benkovic, S. J. (1992). Catalytic antibodies. Annual Review of Biochemistry, 61, 29-54. [CrossRef]
- Benkovic, S. J., Hammes, G. G., & Hammes-Schiffer, S. (2008). Free-energy landscape of enzyme catalysis. Biochemistry, 47(11), 3317-3321. [CrossRef]
- Billeter, S. R., Webb, S. P., Agarwal, P. K., Iordanov, T., & Hammes-Schiffer, S. (2001). Hydride transfer in liver alcohol dehydrogenase: quantum dynamics, kinetic isotope effects, and role of enzyme motion. Journal of the American Chemical Society, 123(45), 11262-11272. [CrossRef]
- Boehr, D. D., D’Amico, R. N., & O’Rourke, K. F. (2018). Engineered control of enzyme structural dynamics and function. Protein Science, 27(4), 825-838. [CrossRef]
- Boehr, D. D., Dyson, H. J., & Wright, P. E. (2006). An NMR perspective on enzyme dynamics. Chemical Reviews, 106(8), 3055-3079. [CrossRef]
- Boehr, D. D., Schnell, J. R., McElheny, D., Bae, S. H., Duggan, B. M., Benkovic, S. J., Dyson, H. J., & Wright, P. E. (2013). A distal mutation perturbs dynamic amino acid networks in dihydrofolate reductase. Biochemistry, 52(27), 4605-4619. [CrossRef]
- Borreguero, J. M., He, J. H., Meilleur, F., Weiss, K. L., Brown, C. M., Myles, D. A., Herwig, K. W., & Agarwal, P. K. (2011). Redox-promoting protein motions in rubredoxin. The Journal of Physical Chemistry B, 115(28), 8925-8936. [CrossRef]
- Bruice, T. C., & Benkovic, S. J. (2000). Chemical basis for enzyme catalysis. Biochemistry, 39(21), 6267-6274. [CrossRef]
- Camilloni, C., Sahakyan, A. B., Holliday, M. J., Isern, N. G., Zhang, F. L., Eisenmesser, E. Z., & Vendruscolo, M. (2014). Cyclophilin A catalyzes proline isomerization by an electrostatic handle mechanism. Proceedings of the National Academy of Sciences of the United States of America, 111(28), 10203-10208. [CrossRef]
- Chi, C. N., Vogeli, B., Bibow, S., Strotz, D., Orts, J., Guntert, P., & Riek, R. (2015). A structural ensemble for the enzyme cyclophilin reveals an orchestrated mode of action at atomic resolution. Angewandte Chemie International Edition, 54(40), 11657-11661. [CrossRef]
- Choy, M. S., Li, Y., Machado, L., Kunze, M. B. A., Connors, C. R., Wei, X., Lindorff-Larsen, K., Page, R., & Peti, W. (2017). Conformational rigidity and protein dynamics at distinct timescales regulate PTP1B activity and allostery. Molecular cell, 65(4), 644-658.e5. [CrossRef]
- Clouthier, C. M., Morin, S., Gobeil, S. M. C., Doucet, N., Blanchet, J., Nguyen, E., Gagne, S. M., & Pelletier, J. N. (2012). Chimeric beta-lactamases: global conservation of parental function and fast time-scale dynamics with increased slow motions. PLoS One, 7(12): e52283. [CrossRef]
- Coman, R. M., Robbins, A. H., Goodenow, M. M., Dunn, B. M., & McKenna, R. (2008). High-resolution structure of unbound human immunodeficiency virus 1 subtype C protease: implications of flap dynamics and drug resistance. Acta Crystallographica Section D: Biological Crystallography, D64(Pt7), 754-763. [CrossRef]
- Duff, M. R., Jr., Borreguero, J. M., Cuneo, M. J., Ramanathan, A., He, J., Kamath, G., Chennubhotla, S. C., Meilleur, F., Howell, E. E., Herwig, K. W., Myles, D. A. A., & Agarwal, P. K. (2018). Modulating enzyme activity by altering protein dynamics with solvent. Biochemistry, 57(29), 4263-4275. [CrossRef]
- Eisenmesser, E. Z., Bosco, D. A., Akke, M., & Kern, D. (2002). Enzyme dynamics during catalysis. Science, 295(5559), 1520-1523. [CrossRef]
- Eisenmesser, E. Z., Millet, O., Labeikovsky, W., Korzhnev, D. M., Wolf-Watz, M., Bosco, D. A., Skalicky, J. J., Kay, L. E., & Kern, D. (2005). Intrinsic dynamics of an enzyme underlies catalysis. Nature, 438(7064), 117-121. [CrossRef]
- Elstner, M., Frauenheim, T., & Suhai, S. (2003). An approximate DFT method for QM/MM simulations of biological structures and processes. Journal of Molecular Structrure: THEOCHEM, 632(1-3), 29-41. [CrossRef]
- Elstner, M., Hobza, P., Frauenheim, T., Suhai, S., & Kaxiras, E. (2001). Hydrogen bonding and stacking interactions of nucleic acid base pairs: a density-functional-theory based treatment. The Journal of Chemical Physics, 114(12), 5149-5155. [CrossRef]
- Elstner, M., Porezag, D., Jungnickel, G., Elsner, J., Haugk, M., Frauenheim, T., Suhai, S., & Seifert, G. (1998). Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Physical Review B, 58(11), 7260-7268. [CrossRef]
- Fenimore, P. W., Frauenfelder, H., McMahon, B. H., & Young, R. D. (2004). Bulk-solvent and hydration-shell fluctuations, similar to alpha- and beta-fluctuations in glasses, control protein motions and functions. Proceedings of the National Academy of Sciences of the United States of America, 101(40), 14408-14413. [CrossRef]
- Fersht Alan (1985). Enzyme structure and mechanism (2nd edition). New York, NY: W. H. Freeman and Company.
- Fischer, E. (1894). Einfluss der configuration auf die wirkung den enzyme. Berichte der Deutschen Chemischen Gesellschaft, 27(3), 2985-2993. [CrossRef]
- Fraser, J. S., Clarkson, M. W., Degnan, S. C., Erion, R., Kern, D., & Alber, T. (2009). Hidden alternative structures of proline isomerase essential for catalysis. Nature, 462(7273), 669-673. [CrossRef]
- Frauenfelder, H., McMahon, B. H., Austin, R. H., Chu, K., & Groves, J. T. (2001). The role of structure, energy landscape, dynamics, and allostery in the enzymatic function of myoglobin. Proceedings of the National Academy of Sciences of the United States of America, 98(5), 2370-2374. [CrossRef]
- Gagne, D., Charest, L. A., Morin, S., Kovrigin, E. L., & Doucet, N. (2012). Conservation of flexible residue clusters among structural and functional enzyme homologues. Journal of Biological Chemistry, 287(53), 44289-44300. [CrossRef]
- Gagne, D., Narayanan, C., & Doucet, N. (2015). Network of long-range concerted chemical shift displacements upon ligand binding to human angiogenin. Protein Science, 24(4), 525-533. [CrossRef]
- Garcia-Viloca, M., Gao, J., Karplus, M., & Truhlar, D. G. (2004). How enzymes work: analysis by modern rate theory and computer simulations. Science, 303(5655), 186-195. [CrossRef]
- Grimme, S., Antony, J., Schwabe, T., & Muck-Lichtenfeld, C. (2007). Density functional theory with dispersion corrections for supramolecular structures, aggregates, and complexes of (bio)organic molecules. Organic and Biomolecular Chemistry, 5, 741-758. [CrossRef]
- Guallar, V., Baik, M.-H., Lippard, S. J., & Friesner, R. A. (2003). Peripheral heme substituents control the hydrogen-atom abstraction chemistry in cytochromes P450. Proceedings of the National Academy of Sciences of the United States of America, 100(12), 6998-7002. [CrossRef]
- Gur, M., Taka, E., Yilmaz, S. Z., Kilinc, C., Aktas, U., & Golcuk, M. (2020). Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states. The Journal of Chemical Physics, 153(7), 075101. [CrossRef]
- Hachim, M. E., Sadik, K., Byadi, S., & Aboulmouhajir, A. (2020). Electronic investigation and spectroscopic analysis using DFT with the long-range dispersion correction on the six lowest conformers of 2.2. 3-trimethyl pentane. Journal of Molecular Modeling, 26(7), 168. [CrossRef]
- Hall, R., Dixon, T., & Dickson, A. (2020). On calculating free energy differences using ensembles of transition paths. Frontiers in Molecular Biosciences, 7, 106. [CrossRef]
- Harvey, J. N., Bathelt, C. M., & Mulholland, A. J. (2006). QM/MM modeling of compound I active species in cytochrome P450, cytochrome C peroxidase, and ascorbate peroxidase. Journal of Computational Chemistry, 27(12), 1352-1362. [CrossRef]
- Hawkins, R. J., & McLeish, T. C. B. (2006). Coupling of global and local vibrational modes in dynamic allostery of proteins. Biophysical Journal, 91(6), 2055-2062. [CrossRef]
- Heaslet, H., Rosenfeld, R., Giffin, M., Lin, Y. C., Tam, K., Torbett, B. E., Elder, J. H., McRee, D. E., & Stout, C. D. (2007). Acta Crystallographica Section D: Biological Crystallography, 63(Pt8), 866-875. [CrossRef]
- Henzler-Wildman, K. A., Lei, M., Thai, V., Kerns, S. J., Karplus, M., & Kern, D. (2007). A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature, 450(7171), 913-916. [CrossRef]
- Himo, F. (2017). Recent trends in quantum chemical modeling of enzymatic reactions. Journal of the American Chemical Society, 139(20), 6780-6786. [CrossRef]
- Hirschi, J. S., Arora, K., Brooks, C. L., & Schramm, V. L. (2010). Conformational dynamics in human purine nucleoside phosphorylase with reactants and transition-state analogues. The Journal of Physical Chemistry B, 114(49), 16263-16272. [CrossRef]
- Hobza, P., Sponer, J., & Reschel, T. (1995). Density functional theory and molecular clusters. Journal of Computational Chemistry, 16(11), 1315-1325. [CrossRef]
- Holliday, M. J., Camilloni, C., Armstrong, G. S., Vendruscolo, M., & Eisenmesser, E. Z. (2017). Networks of dynamic allostery regulate enzyme function. Structure, 25(2), 276-286. [CrossRef]
- Howard, B. R., Vajdos, F. F., Li, S., Sundquist, W. I., & Hill, C. P. (2003). Structural insights into the catalytic mechanism of cyclophilin A. Nature Structural & Molecular Biology, 10(6), 475-481. [CrossRef]
- Hu, X., & Erec Stebbins, C. (2006). Dynamics of the WPD loop of the Yersinia protein tyrosine phosphatase. Biophysical Journal, 91(3), 948-956. [CrossRef]
- Ichiye, T., & Karplus, M. (1988). Anisotropy and anharmonicity of atomic fluctuations in proteins - implications for X-ray-analysis. Biochemistry, 27(9), 3487-3497. [CrossRef]
- Jurecka, P., Cerny, J., Hobza, P., & Salahub, D. R. (2007). Density functional theory augmented with an empirical dispersion term. Interaction energies and geometries of 80 noncovalent complexes compared with ab initio quantum mechanics calculations. Journal of Computational Chemistry, 28(2), 555-569. [CrossRef]
- Kamerlin, S. C. L., & Warshel, A. (2010). At the dawn of the 21st century: is dynamics the missing link for understanding enzyme catalysis? Proteins, 78(6), 1339-1375. [CrossRef]
- Kim, T. H., Mehrabi, P., Ren, Z., Sljoka, A., Ing, C., Bezginov, A., Ye, L., Pomes, R., Prosser, R. S., & Pai, E. F. (2017). The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science, 355(6322), eaag2355. [CrossRef]
- Klinman, J. P. (2013). Importance of protein dynamics during enzymatic C-H bond cleavage catalysis. Biochemistry, 52(12), 2068-2077. [CrossRef]
- Klinman, J. P., Offenbacher, A. R., & Hu, S. (2017). Origins of enzyme catalysis: experimental findings for C-H activation, new models, and their relevance to prevailing theoretical constructs. Journal of the American Chemical Society, 139(51), 18409-18427. [CrossRef]
- Kohen, A. (2015). Role of dynamics in enzyme catalysis: substantial versus semantic controversies. Accounts of Chemical Research, 48(2), 466-473. [CrossRef]
- Kondo, T., Pinnola, A., Chen, W. J., Dall’Osto, L., Bassi, R., & Schlau-Cohen, G. S. (2017). Single-molecule spectroscopy of LHCSR1 protein dynamics identifies two distinct states responsible for multi-timescale photosynthetic photoprotection. Nature Chemistry, 9(8), 772-778. [CrossRef]
- Koshland, D. E. (1958). Application of a theory of enzyme specificity to protein synthesis. Proceedings of the National Academy of Sciences of the United States of America, 44(2), 98-104. [CrossRef]
- Kristyan, S., & Pulay, P. (1994). Can (semi)local density functional theory account for the London dispersion forces? Chemical Physics Letters, 229(3), 175-180. [CrossRef]
- Krivitskaya, A. V., Khrenova, M. G., & Nemukhin, A. V. (2021). Two sides of quantum-based modeling of enzyme-catalyzed reactions: mechanistic and electronic structure aspects of the hydrolysis by glutamate carboxypeptidase. Molecules, 26(20), 6280. [CrossRef]
- Kuban-Jankowska, A., Sahu, K. K., Gorska, M., Niedzialkowski, P., Tuszynski, J. A., Ossowski, T., & Wozniak, M. (2016). Aurintricarboxylic acid structure modifications lead to reduction of inhibitory properties against virulence factor YopH and higher cytotoxicity. World Journal of Microbiology and Biotechnology, 32, 163. [CrossRef]
- Kubar, T., Jurecka, P., Cerny, J., Rezac, J., Otyepka, M., Valdes, H., & Hobza, P. (2007). Density-functional, density-functional tight-binding, and wave function calculations on biomolecular systems. The Journal of Physical Chemistry A, 11(26), 5642-5647. [CrossRef]
- Lange, O. F., Lakomek, N. A., Fares, C., Schroder, G. F., Walter, K. F. A., Becker, S., Meiler, J., Grubmuller, H., Griesinger, C., & de Groot, B. L. (2008). Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science, 320(5882), 1471-1475. [CrossRef]
- Lapatto, R., Blundell, T., Hemmings, A., Overington, J., Wilderspin, A., Wood, S., Merson, J. R., Whittle, P. J., Danley, D. E., Geoghegan, K. F., Hawrylik, S. J., Lee, S. E., Scheld K. G., & Hobart, P. M. (1989). X-ray analysis of HIV-1 proteinase at 2.7 Å resolution confirms structural homology among retroviral enzymes. Nature, 342(6247), 299-302. [CrossRef]
- Layfield, J. P., & Hammes-Schiffer, S. (2013). Calculation of vibrational shifts of nitrile probes in the active site of ketosteroid isomerase upon ligand binding. Journal of the American Chemical Society, 135(2), 717-725. [CrossRef]
- Lee, J., Natarajan, M., Nashine, V. C., Socolich, M., Vo, T., Russ, W. P., Benkovic, S. J., & Ranganathan, R. (2008). Surface sites for engineering allosteric control in proteins. Science, 322(5900), 438-442. [CrossRef]
- Lee, C. T., Yang, W. T., & Parr, R. G. (1988). Development of the Cole-Salvetti correlation energy formula into a functional of the electron density. Physical Review B Condens Matter, 37(2), 785-789. [CrossRef]
- Leitner, D. M. (2008). Energy flow in proteins. Annual Review of Physical Chemistry, 59, 233-259. [CrossRef]
- Limongelli, V. (2020). Ligand binding free energy and kinetics calculation in 2020. WIREs Computational Molecular Science, 10(4), e1455. [CrossRef]
- Limongelli, V., Bonomi, M., & Parrinello, M. (2013). Funnel metadynamics as accurate binding free-energy method. Proceedings of the National Academy of Sciences of the United States of America, 110(16), 6358-6363. [CrossRef]
- Lisi, G. P., & Loria, J. P. (2016). Solution NMR spectroscopy for the study of enzyme allostery. Chemical Reviews, 116(11), 6323-6369. [CrossRef]
- Liu, F., Kovalevsky, A. Y., Louis, J. M., Boross, P. I., Wang, Y. F., Harrison, R. W., & Weber, I. T. (2006). Mechanism of drug resistance revealed by the crystal structure of the unliganded HIV-1 protease with F53L mutation. Journal of Molecular Biology, 358(5), 1191-1199. [CrossRef]
- Liu, Y., Li, Y., Wu, S., Li, G., & Chu, H. (2022). Synergistic effect of conformational changes in phosphoglycerate kinase 1 product release. Journal of Biomolecular Structure and Dynamics, 41(19), 10059-10069. [CrossRef]
- Lonsdale, R., Harvey, J. N., & Mulholland, A. J. (2010). Compound I reactivity defines alkene oxidation selectivity in cytochrome P450cam. The Journal of Physical Chemistry B, 114(2), 1156-1162. [CrossRef]
- Lonsdale, R., Olah, J., Mulholland, A. J., & Harvey, J. N. (2011). Does compound I vary significantly between isoforms of cytochrome P450? Journal of the American Chemical Society, 133(39), 15464-15474. [CrossRef]
- Lonsdale, R., Harvey, J. N., & Mulholland, A. J. (2012). A practical guide to modelling enzyme-catalysed reactions. Chemical Society Reviews, 41(8), 3025-3038. [CrossRef]
- Love, O., Pacheco Lima, M. C., Clark, C., Cornillie, S., Roalstad, S., & Cheatham III, T. E. (2022). Evaluating the accuracy of the AMBER protein force fields in modeling dihydrofolate reductase structures: misbalance in the conformational arrangements of the flexible loop domains. Journal of Biomolecular Structure and Dynamics, 41(13), 5946-5960. [CrossRef]
- Machleder, S. Q., Pineda, J. R. E. T., & Schwartz, S. D. (2010). On the origin of the chemical barrier and tunneling in enzymes. Journal of Physical Organic Chemistry, 23(7), 690-695. [CrossRef]
- Martin, P., Vickrey, J. F., Proteasa, G., Jimenez, Y. L., Wawrzak, Z., Winters, M. A., Merigan, T. C., & Kovari, L. C. (2005). “Wide-open” 1.3 Å structure of a multidrug-resistant HIV-1 protease as a drug target. Structure, 13(12), 1887-1895. [CrossRef]
- Micheletti, C. (2013). Comparing proteins by their internal dynamics: exploring structure-function relationships beyond static structural alignments. Physics of Life Reviews, 10(1), 1-26. [CrossRef]
- Mihajlovic, M. L., & Mitrasinovic, P. M. (2008). Another look at the molecular mechanism of the resistance of H5N1 influenza A virus neuraminidase (NA) to oseltamivir (OTV). Biophysical Chemistry, 136(2-3), 152-158. [CrossRef]
- Mihajlovic, M. L., & Mitrasinovic, P. M. (2009a). Applications of the ArgusLab4/AScore protocol in the structure-based binding affinity prediction of various inhibitors of group-1 and group-2 influenza virus neuraminidases (NAs). Molecular Simulation, 35(4), 311-324. [CrossRef]
- Mihajlovic, M. L., & Mitrasinovic, P. M. (2009b). Some novel insights into the binding of oseltamivir and zanamivir to H5N1 and N9 influenza virus neuraminidases: a homology modeling and flexible docking study. Journal of the Serbian Chemical Society, 74(1), 1-13. [CrossRef]
- Mitrasinovic, P. M. (2001). Quantitative characterization of the P-C bonds in ylides of phosphorus. Journal of Computational Chemistry, 22(13), 1387-1395. [CrossRef]
- Mitrasinovic, P. M. (2002). Qualitative characterization of the P-C bonds in ylides of phosphorus. The Journal of Physical Chemistry A, 106(30), 7026-7033. [CrossRef]
- Mitrasinovic, P. M. (2003a). Acrylonitrile (AN)/Cu9(100) interfaces: electron distribution and nature of bonded interactions. Canadian Journal of Chemistry, 81(6), 542-554. [CrossRef]
- Mitrasinovic, P. M. (2003b). Sharing analysis of the behavior of electrons in some simple ylides. Chemical Physics, 286(1), 1-13. [CrossRef]
- Mitrasinovic, P. M. (2004). Invariant description of the behavior of electrons in donor–acceptor molecules. Chemical Physics Letters, 392(4-6), 419-427. [CrossRef]
- Mitrasinovic, P. M. (2005). Cross-linking between thymine and indolyl radical: Possible mechanisms for cross-linking of DNA and tryptophan-containing peptides. Bioconjugate Chemistry, 16(3), 588-597. [CrossRef]
- Mitrasinovic, P. M. (2009). On the structure-based design of novel inhibitors of H5N1 influenza A virus neuraminidase (NA). Biophysical Chemistry, 140(1-3), 35-38. [CrossRef]
- Mitrasinovic, P. M. (2010a). Electronic processes at organic/metal interfaces: recent progress and pitfalls. Current Organic Chemistry, 14(2), 198-211. [CrossRef]
- Mitrasinovic, P. M. (2010b). Advances in the structure-based design of the influenza A neuraminidase inhibitors. Current Drug Targets, 11(3), 315-326. [CrossRef]
- Mitrasinovic, P. M. (2012). Advances in boron-based supramolecular architecture. Current Organic Synthesis, 9(2), 233-246. [CrossRef]
- Mitrasinovic, P. M. (2013). Progress in structure-based design of EGFR inhibitors. Current Drug Targets, 14(7), 817-829. [CrossRef]
- Mitrasinovic, P. M. (2014a). Structural elucidation of unique inhibitory activities of two thiazolo[4,5-d]pyrimidines against epidermal growth factor receptor (EGFR): implications for successful drug design. Medicinal Chemistry, 10(1), 46-58. [CrossRef]
- Mitrasinovic, P. M. (2014b). Inhibitory activity against epidermal growth factor receptor (EGFR) based on single point mutations of active site residues. Medicinal Chemistry, 10(3), 252-270. [CrossRef]
- Mitrasinovic, P. M. (2015). Sequence-dependent binding of flavonoids to duplex DNA. Journal of Chemical Information and Modeling, 55(2), 421-433. [CrossRef]
- Mitrasinovic, P. M. (2018). Structural insights into the binding of small ligand molecules to a G-quadruplex DNA located in the HIV-1 promoter. Journal of Biomolecular Structure and Dynamics, 36(9), 2292-2302. [CrossRef]
- Mitrasinovic, P. M. (2019). Small-molecule interaction with G-quadruplex DNA: context of anti-cancer drug design. Croatica Chemica Acta, 92(1), 43-57. [CrossRef]
- Mitrasinovic, P. M. (2020). Quantum mechanics/molecular mechanics study on caspase-2 recognition by peptide inhibitors. Acta Chimica Slovenica, 67(3), 876-884. [CrossRef]
- Mitrasinovic, P. M. (2022). On the inhibition of cytochrome P450 3A4 by structurally diversified flavonoids. Journal of Biomolecular Structure and Dynamics, 40(20), 9713-9723. [CrossRef]
- Mitrasinovic, P. M. (2023). On the recognition of Yersinia protein tyrosine phosphatase by carboxylic acid derivatives. Journal of Biomolecular Structure and Dynamics, 41(5), 1879-1894. [CrossRef]
- Mitrasinovic, P. M. (2024a). On the specificity of aurintricarboxylic acid toward some phosphatases. ChemRxiv. [CrossRef]
- Mitrasinovic, P. M. (2024b). On the potential pharmacophore for the structure-based inhibitor design against phosphatases, ChemRxiv. [CrossRef]
- Mohammadkhani, R., Ramezanzadeh, M., Akbarzadeh, S., Bahlakeh, G., & Ramezanzadeh, B. (2020). Graphene oxide nanoplatforms reduction by green plant-sourced organic compounds for construction of an active anti-corrosion coating; experimental/electronic-scale DFT-D modeling studies. Chemical Engineering Journal, 397, 125433. [CrossRef]
- Monod, J., Changeux, J. P., & Jacob, F. (1963). Allosteric proteins and cellular control systems. Journal of Molecular Biology, 6(4), 306-329. [CrossRef]
- Moritsugu, K., & Kidera, A. (2004). Protein motions represented in moving normal mode coordinates. The Journal of Physical Chemistry B, 108(12), 3890-3898. [CrossRef]
- Nagel, Z. D., & Klinman, J. P. (2009). A 21st century revisionist’s view at a turning point in enzymology. Nature Chemical Biology, 5(8), 543-550. [CrossRef]
- Nagel, Z. D., Cun, S. J., & Klinman, J. P. (2013). Identification of a long-range protein network that modulates active site dynamics in extremophilic alcohol dehydrogenases. Journal of Biological Chemistry, 288(20), 14087-14097. [CrossRef]
- Navia, M. A., Fitzgerald, P. M., McKeever, B. M., Leu, C. T., Heimbach, J. C., Herber, W. K., Sigal, I. S., Darke, P. L., & Springer, J. P. (1989). Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature, 337(6208), 615-620. [CrossRef]
- Onions, S. T., Ito, K., Charron, C. E., Brown, R. J., Colucci, M., Frickel, F., Hardy, G., Joly, K., King-Underwood, J., Kizawa, Y., Knowles, I., Murray, P. J., Novak, A., Rani, A., Rapeport, G., Smith, A., Strong, P., Taddei, D. M., & Williams, J. G. (2016). The discovery of narrow spectrum kinase inhibitors: new therapeutic agents for the treatment of COPD and steroid-resistant asthma. Journal of Medicinal Chemistry, 59(5), 1727-1746. [CrossRef]
- Otten, R., Liu, L., Kenner, L. R., Clarkson, M. W., Mavor, D., Tawfik, D. S., Kern, D., & Fraser, J. S. (2018). Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nature Communications, 9, 1314. [CrossRef]
- Palmer, A. G. III (2015). Enzyme dynamics from NMR spectroscopy. Accounts of Chemical Research, 48(2), 457-465. [CrossRef]
- Pavlov, A., & Mitrasinovic, P. M. (2010). Theoretical insights into dispersion and hydrogen-bonding interactions in biomolecular systems. Current Organic Chemistry, 14(2), 129-137. [CrossRef]
- Pillai, B., Kannan, K. K., & Hosur, M. V. (2001). 1.9 Å x-ray study shows closed flap conformation in crystals of tethered HIV-1 PR. Proteins, 43(1), 57-64. [CrossRef]
- Pisliakov, A. V., Cao, J., Kamerlin, S. C. L., & Warshel, A. (2009). Enzyme millisecond conformational dynamics do not catalyze the chemical step. Proceedings of the National Academy of Sciences of the United States of America, 106(41), 17359-17364. [CrossRef]
- Pontiggia, F., Colombo, G., Micheletti, C., & Orland, H. (2007). Anharmonicity and self-similarity of the free energy landscape of protein G. Physical Review Letters, 98(4), 048102. [CrossRef]
- Quaytman, S. L., & Schwartz, S. D. (2007). Reaction coordinate of an enzymatic reaction revealed by transition path sampling. Proceedings of the National Academy of Sciences of the United States of America, 104(30), 12253-12258. [CrossRef]
- Ramanathan, A., & Agarwal, P. K. (2009). Computational identification of slow conformational fluctuations in proteins. The Journal of Physical Chemistry B, 113(52), 16669-16680. [CrossRef]
- Ramanathan, A., & Agarwal, P. K. (2011). Evolutionarily conserved linkage between enzyme fold, flexibility, and catalysis. PLoS Biology, 9(11), e1001193. [CrossRef]
- Ramanathan, A., Savol, A., Burger, V., Chennubhotla, C., & Agarwal, P. K. (2014). Protein conformational populations and functionally relevant sub-states. Accounts of Chemical Research, 47(1), 149-156. [CrossRef]
- Ramanathan, A., Savol, A. J., Langmead, C. J., Agarwal, P. K., & Chennubhotla, C. S. (2011). Discovering conformational sub-states relevant to protein function. PLoS One, 6(1), e15827. [CrossRef]
- Reynolds, K. A., McLaughlin, R. N., & Ranganathan, R. (2011). Hot spots for allosteric regulation on protein surfaces. Cell, 147(7), 1564-1575. [CrossRef]
- Richard, J. P. (2013). Enzymatic rate enhancements: a review and perspective. Biochemistry, 52(12), 2009-2011. [CrossRef]
- Robbins, A. H., Coman, R. M., Bracho-Sanchez, E., Fernandez, M. A., Gilliland, C. T., Li, M., Agbandje-McKenna, M., Wlodawer, A., Dunn, B. M., & McKenna R. (2010). Structure of the unbound form of HIV-1 subtype A protease: comparison with unbound forms of proteases from other HIV subtypes. Acta Crystallographica Section D: Biological Crystallography, 66(Pt3), 233-242. [CrossRef]
- Robertus, J. D., Kraut, J., Alden, R. A., & Birktoft, J. J. (1972). Subtilisin. Stereochemical mechanism involving transition-state stabilization. Biochemistry, 11(23), 4293-4303. [CrossRef]
- Sawaya, M. R., & Kraut, J. (1997). Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: crystallographic evidence. Biochemistry, 36(3), 586-603. [CrossRef]
- Schlitter, J. (1993). Estimation of absolute and relative entropies of macromolecules using the covariance matrix. Chemical Physics Letters, 215(6), 617-621.
- Schramm, A. M., Mehra-Chaudhary, R., Furdui, C. M., & Beamer, L. J. (2008). Backbone flexibility, conformational change, and catalysis in a phosphohexomutase from Pseudomonas aeruginosa. Biochemistry, 47(35), 9154-9162. [CrossRef]
- Schubert, H. L., Fauman, E. B., Stuckey, J. A., Dixon, J. E., & Saper, M. A. (1995). A ligand-induced conformational change in the Yersinia protein tyrosine phosphatase. Protein Science, 4(9), 1904-1913. [CrossRef]
- Sevrioukova, I. F., & Poulos, T. L. (2015). Anion-dependent stimulation of CYP3A4 monooxygenase. Biochemistry, 54, 4083-4096. [CrossRef]
- Shaik, S,, Kumar, D., de Visser, S. P., Altun, A., & Thiel, W. (2005). Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chemical Reviews, 105(6), 2279-2328 . [CrossRef]
- Shaik, S., Cohen, S., Wang, Y., Chen, H., Kumar, D., & Thiel, W. (2010). P450 enzymes: their structure, reactivity, and selectivity-modeled by QM/MM calculations. Chemical Reviews, 110(2), 949-1017. [CrossRef]
- Siegel, J. B., Zanghellini, A., Lovick, H. M., Kiss, G., Lambert, A. R., St Clair, J. L., Gallaher, J. L., Hilvert, D., Gelb, M. H., Stoddard, B. L., Houk, K. N., Michael, F. E., & Baker, D. (2010). Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science, 329(5989), 309-313. [CrossRef]
- Spinelli, S., Liu, Q. Z., Alzari, P. M., Hirel, P. H., & Poljak, R. J. (1991). The three-dimensional structure of the aspartyl protease from the HIV-1 isolate BRU. Biochimie, 73(11), 1391-1396. [CrossRef]
- Tang, Y., Wells, J. A., & Arkin, M. R. (2011). Structural and enzymatic insights into caspase-2 protein substrate recognition and catalysis. Journal of Biological Chemistry, 286(39), 34147-34154. [CrossRef]
- Tehei, M., Franzetti, B., Wood, K., Gabel, F., Fabiani, E., Jasnin, M., Zamponi, M., Oesterhelt, D., Zaccai, G., Ginzburg, M., & Ginzburg, B. Z. (2007). Neutron scattering reveals extremely slow cell water in a Dead Sea organism. Proceedings of the National Academy of Sciences of the United States of America, 104(3), 766-771. [CrossRef]
- Veeraraghavan, N., Bevilacqua, P. C., & Hammes-Schiffer, S. (2010). Long-distance communication in the HDV ribozyme: insights from molecular dynamics and experiments. Journal of Molecular Biology, 402(1), 278-291. [CrossRef]
- Verkhivker G. M., Agajanian, S., Oztas, D., & Gupta, G. (2022). Computational analysis of protein stability and allosteric interaction networks in distinct conformational forms of the SARS-CoV-2 spike D614G mutant: reconciling functional mechanisms through allosteric model of spike regulation. Journal of Biomolecular Structure and Dynamics, 40(20), 9724-9741. [CrossRef]
- Viswanathan, V. S., Ryan, M. J., Dhruv, H. D., Gill, S., Eichhoff, O. M., Seashore-Ludlow, B., Kaffenberger, S. D., Eaton, J. K., Shimada, K., Aguirre, A. J., Viswanathan, S. R., Chattopadhyay, S., Tamayo, P., Yang, W. S., Rees, M. G., Chen, S., Boskovic, Z. V., Javaid, S., Huang, C., Wu, X., Tseng, Y. Y., Roider, E. M., Gao, D., Cleary, J. M., Wolpin, B. M., Mesirov, J. P., Haber, D. A., Engelman, J. A., Boehm, J. S., Kotz, J. D., Hon, C. S., Chen, Y., Hahn, W. C., Levesque, M. P., Doench, J. G., Berens, M. E., Shamji, A. F., Clemons, P. A., Stockwell, B. R., & Schreiber, S. L. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature, 547(7664), 453-457. [CrossRef]
- Wang, S.-Q., Cheng, X.-C., Dong, W.-L., Wang, R.-L., & Chou, K.-C. (2010). Three new powerful oseltamivir derivatives for inhibiting the neuraminidase of influenza virus. Biochemical and Biophysical Research Communications, 401(2), 188-191. [CrossRef]
- Wang, L., Goodey, N. M., Benkovic, S. J., & Kohen, A. (2006). Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proceedings of the National Academy of Sciences of the United States of America, 103(43), 15753-15758. [CrossRef]
- Wang, K., & Zhu, W. (2020). Insight into the roles of small molecules in CL-20 based host-guest crystals: a comparative DFT-D study. CrystEngComm, 22(37), 6228-6238. [CrossRef]
- Warshel, A., Sharma, P. K., Kato, M., Xiang, Y., Liu, H. B., & Olsson, M. H. M. (2006). Electrostatic basis for enzyme catalysis. Chemical Reviews, 106(8), 3210-3235. [CrossRef]
- Xie, A. H., van der Meer, L., & Austin, R. H. (2002). Excited-state lifetimes of far-infrared collective modes in proteins. Journal of Biological Physics, 28(2), 147-154. [CrossRef]
- Yang, M., Huang, J., & MacKerell, A. D. Jr. (2015). Enhanced conformational sampling using replica exchange with concurrent solute scaling and hamiltonian biasing realized in one dimension. Journal of Chemical Theory and Computation, 11(6), 2855-2867. [CrossRef]
- Yang, M., & MacKerell, A. D. Jr. (2015). Conformational sampling of oligosaccharides using Hamiltonian replica exchange with two-dimensional dihedral biasing potentials and the weighted histogram analysis method (WHAM). Journal of Chemical Theory and Computation, 11(2), 788-799. [CrossRef]
- Yu, X., & Leitner, D. M. (2005). Heat flow in proteins: computation of thermal transport coefficients. The Journal of Chemical Physics, 122(5), 54902. [CrossRef]
- Yu, W., & MacKerell, A. D. Jr. (2017). Computer-aided drug design methods. Methods in Molecular Biology, 1520, 85-106. [CrossRef]
- Yu, Y., Wang, J., Chen, Z., Wang, G., Shao, Q., Shi, J., & Zhu, W. (2017). Structural insights into HIV-1 protease flap opening processes and key intermediates. RSC Advances, 7(71), 45121-45128. [CrossRef]
- Zavodszky, P., Kardos, J., Svingor, A., & Petsko, G. A. (1998). Adjustment of conformational flexibility is a key event in the thermal adaptation of proteins. Proceedings of the National Academy of Sciences of the United States of America, 95(13), 7406-7411. [CrossRef]
- Zhang, Z. Q., Rajagopalan, P. T. R., Selzer, T., Benkovic, S. J., & Hammes, G. G. (2004). Single-molecule and transient kinetics investigation of the interaction of dihydrofolate reductase with NADPH and dihydrofolate. Proceedings of the National Academy of Sciences of the United States of America, 101(9), 2764-2769. [CrossRef]
- Zurek, J., Foloppe, N., Harvey, J. N., & Mulholland, A. J. (2006). Mechanisms of reaction in cytochrome P450: hydroxylation of camphor in P450cam. Organic & Biomolecular Chemistry, 4(21), 3931-3937. [CrossRef]
Figure 1.
(left) The traditional (lock-and-key) hypothesis of enzyme-substrate interaction. (right) An emerging biophysical model indicates that enzymes as intrinsically flexible molecules play an active role in the enzyme-substrate recognition (Agarwal, 2019).
Figure 1.
(left) The traditional (lock-and-key) hypothesis of enzyme-substrate interaction. (right) An emerging biophysical model indicates that enzymes as intrinsically flexible molecules play an active role in the enzyme-substrate recognition (Agarwal, 2019).
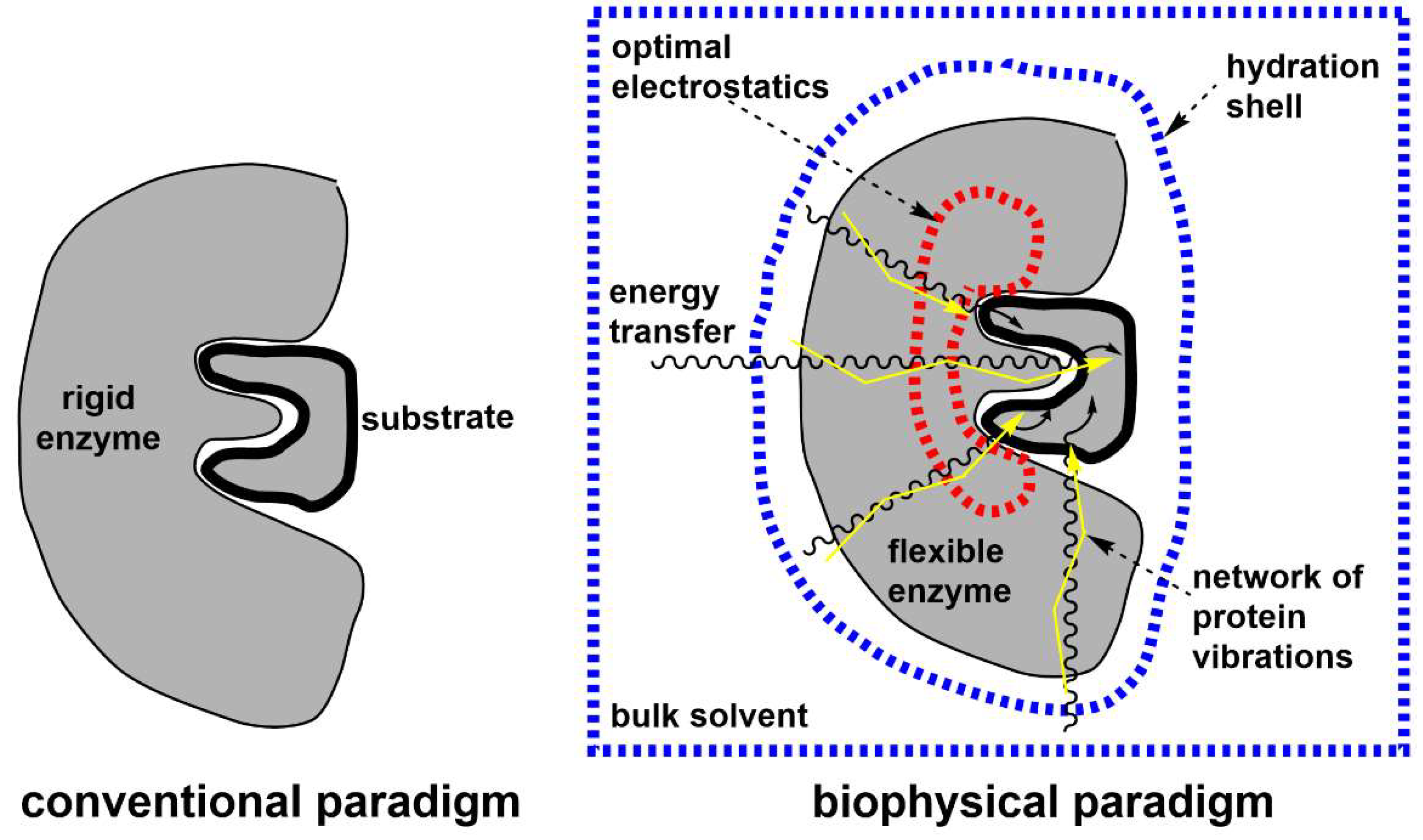
Figure 2.
Different types of internal motions in protein structure (Agarwal, 2019). (a) Fast motions involve a small number of atoms or a few residues at the sub-picosecond timescales. Every unique conformation is denoted by a black circle. (b) Conformational fluctuations within protein domains (loops, secondary structure elements) or entire protein at the nanosecond, microsecond and higher timescales. (c) Temporary (or even permanent) structural change induced by binding (or releasing) other molecules. Shown is the tertiary structure of Yersinia protein tyrosine phosphatase (YopH) in the apo form (left, PDB ID: 1YPT) and in the closed form with bound nitrate (right, PDB ID: 1YTN) that is denoted by black spheres in the active site (Mitrasinovic, 2023, 2024a, 2024b).
Figure 2.
Different types of internal motions in protein structure (Agarwal, 2019). (a) Fast motions involve a small number of atoms or a few residues at the sub-picosecond timescales. Every unique conformation is denoted by a black circle. (b) Conformational fluctuations within protein domains (loops, secondary structure elements) or entire protein at the nanosecond, microsecond and higher timescales. (c) Temporary (or even permanent) structural change induced by binding (or releasing) other molecules. Shown is the tertiary structure of Yersinia protein tyrosine phosphatase (YopH) in the apo form (left, PDB ID: 1YPT) and in the closed form with bound nitrate (right, PDB ID: 1YTN) that is denoted by black spheres in the active site (Mitrasinovic, 2023, 2024a, 2024b).
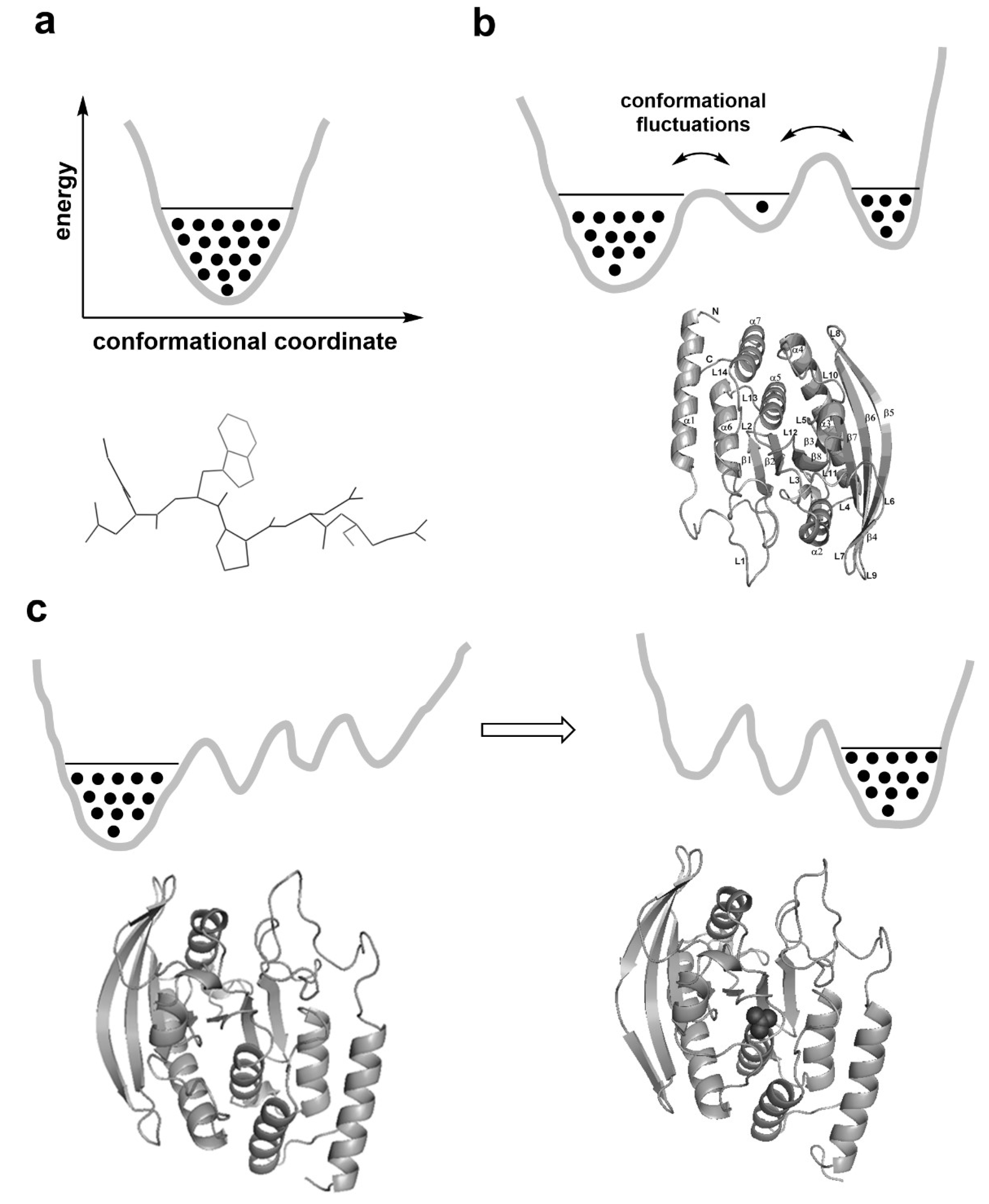
Figure 3.
A biophysical standpoint that hypothesizes the transfer of thermodynamic energy (from enzyme surface regions to the active site) through a hierarchy of conformational motions in order to drive the catalytic reaction (Agarwal, 2019).
Figure 3.
A biophysical standpoint that hypothesizes the transfer of thermodynamic energy (from enzyme surface regions to the active site) through a hierarchy of conformational motions in order to drive the catalytic reaction (Agarwal, 2019).
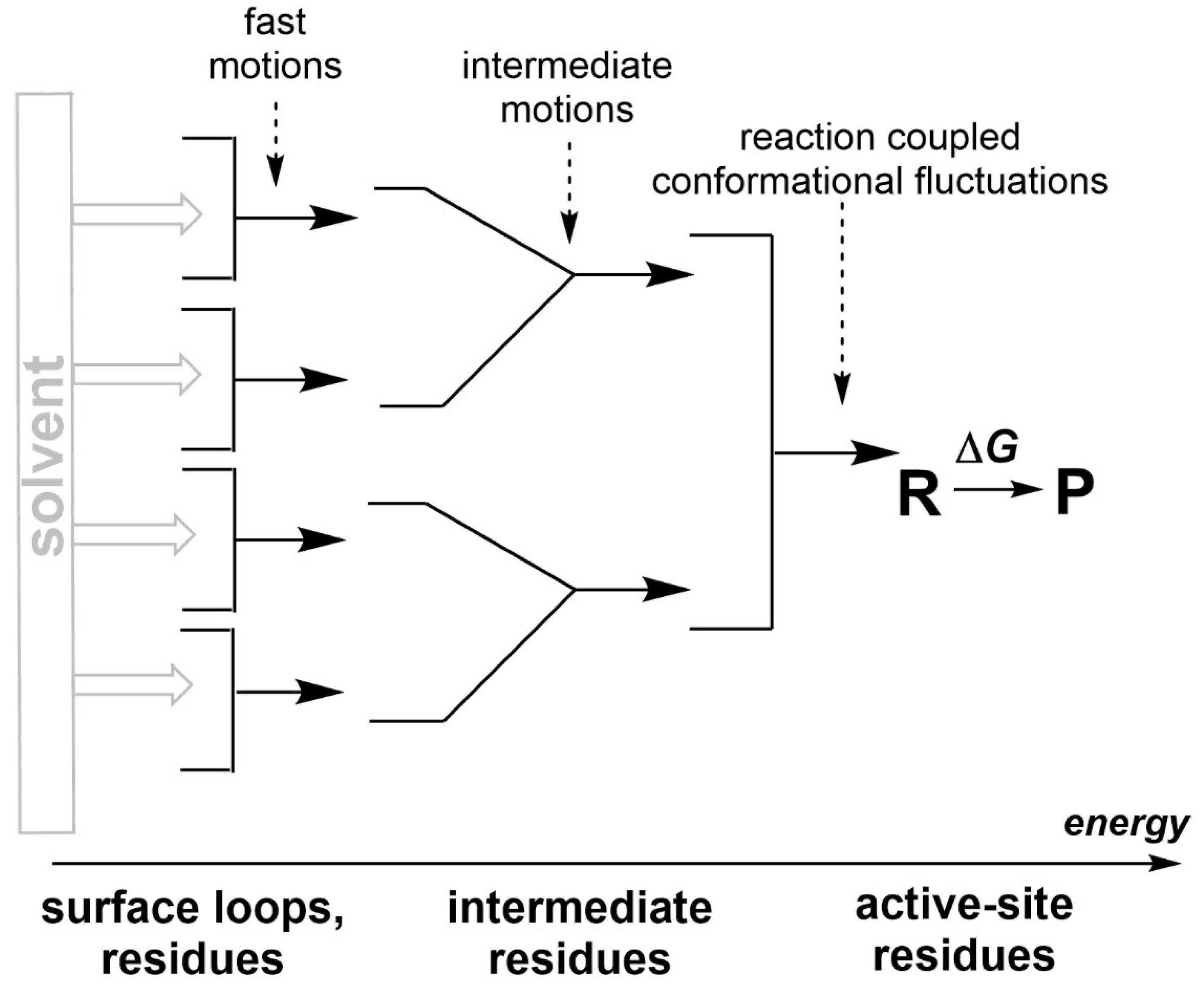
Figure 4.
The conformational flexibility of molecules (proteins or ligands) is an important feature, meaning that mechanistic and electronic structure aspects of an enzyme-catalyzed reaction can presumably be accounted for by using molecular dynamics (MD) simulations with the quantum mechanics/molecular mechanics (QM/MM) potentials in the enzymatic environment (Krivitskaya et al., 2021). E.g., funnel metadynamics yields an efficient characterization of the binding free energy (FE) surface, an accurate calculation of the absolute binding FE, and insights into both the presence of alternative binding modes and the role of water at the nanosecond scales (Limongelli, 2020; Limongelli et al., 2013). Replica exchange methods (Yang et al., 2015; Yang & MacKerell, 2015) are able to enhance the sampling efficiency of the conformational landscape and reveal new modes of interaction at the microsecond scales (Mitrasinovic, 2019).
Figure 4.
The conformational flexibility of molecules (proteins or ligands) is an important feature, meaning that mechanistic and electronic structure aspects of an enzyme-catalyzed reaction can presumably be accounted for by using molecular dynamics (MD) simulations with the quantum mechanics/molecular mechanics (QM/MM) potentials in the enzymatic environment (Krivitskaya et al., 2021). E.g., funnel metadynamics yields an efficient characterization of the binding free energy (FE) surface, an accurate calculation of the absolute binding FE, and insights into both the presence of alternative binding modes and the role of water at the nanosecond scales (Limongelli, 2020; Limongelli et al., 2013). Replica exchange methods (Yang et al., 2015; Yang & MacKerell, 2015) are able to enhance the sampling efficiency of the conformational landscape and reveal new modes of interaction at the microsecond scales (Mitrasinovic, 2019).
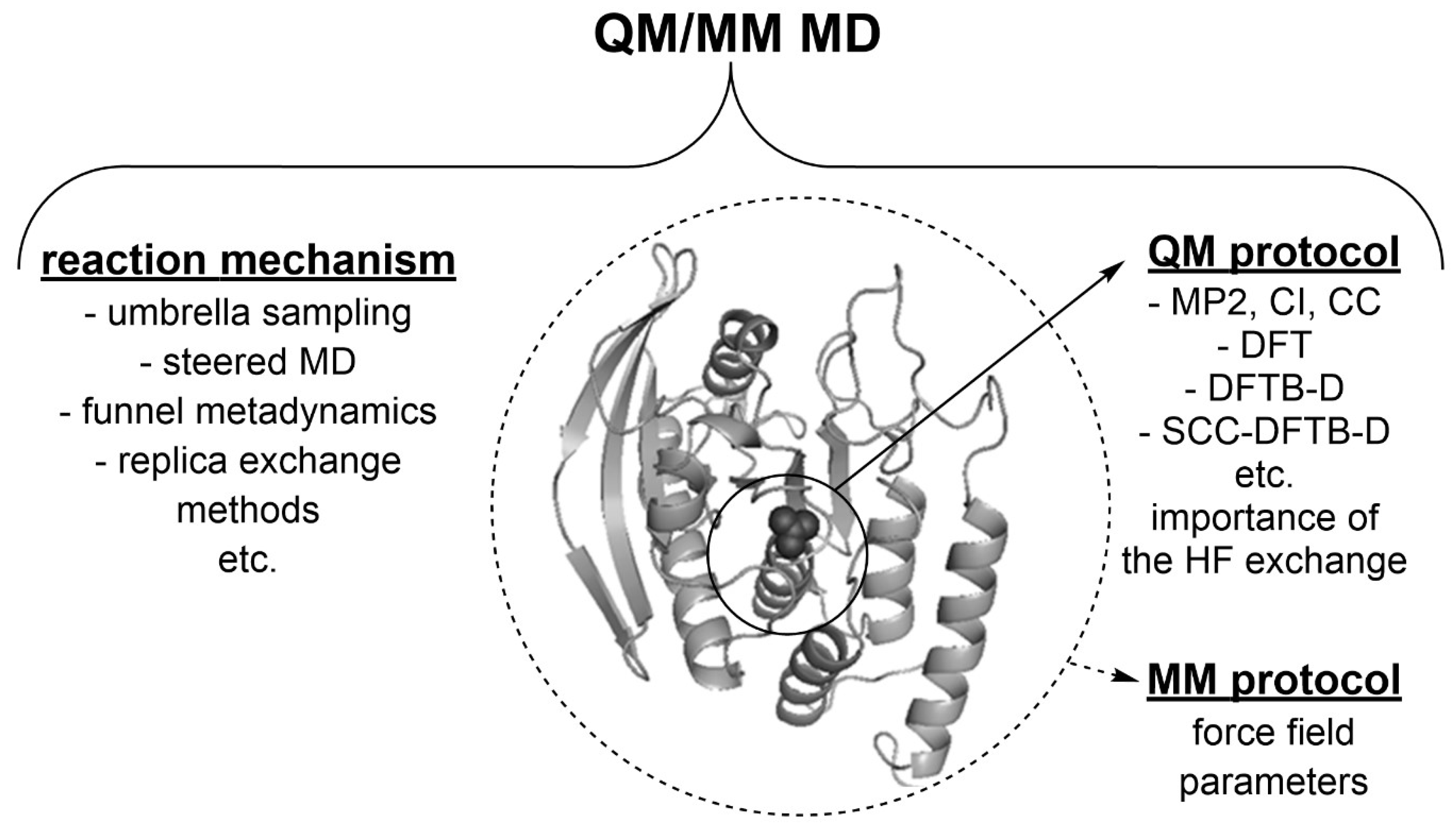
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



