
Submitted:
10 December 2024
Posted:
11 December 2024
You are already at the latest version
Abstract
Alzheimer's disease (AD) is a common form of dementia and a neurodegenerative disorder that affects the nervous system and patients’ cognitive abilities. Current evidence implies that iron dysregulation and bacterial inflammatory factors may be associated with the oral and gut microbiome as critical factors in developing AD. Iron homeostasis disruption may lead to excessive iron accumulation within the cell, forming aggressive reactive oxygen species (ROS). Inflammatory agents produced by bacteria can infiltrate the body via two distinct routes: either through the gut or through a two-step process, starting with the oral cavity and proceeding through the bloodstream to the brain, disrupting cell equilibrium within the body's circulation and tissues, leading to neuroinflammation. Personalized treatment plans targeted at the core attributes of AD should be implemented to prevent disease development and progression.
Keywords:
Alzheimer’s disease
; iron dysregulation
; bacterial inflammatory factors
; oral microbiota
; dysbiosis
; neuroinflammation
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease and the most common form of dementia, with an increasing number of patients worldwide [1]. Currently, there are approximately 55 million dementia patients worldwide, and due to a rapidly aging global population, this number is expected to triple by 2050, leading to increased disability, disease burden, and healthcare expenditures [2].
AD development remains challenging due to its etiology and pathogenesis [3]. Although dementia is mainly diagnosed based on cognitive impairment during a patient’s life, an accurate and definite AD diagnosis can only be established post-mortem by analyzing specific brain lesions (including extracellular senile plaques and intracellular neurofibrillary tangles with synaptic and neuronal loss) [4]. One of the leading causes of AD is senile plaque formation, which results from amyloid beta precursor protein fission (AβPP) [5]. AβPP develops cell differentiation functions and likely shapes synapses; however, it is also expressed in neurons in response to cell injury [6,7]. Neurofibrillary tangles consist of tau protein, paramount in healthy neurons [8]. Under normal conditions, tau is an essential microtubule component (the internal support structure responsible for transporting critical components such as nutrients, vesicles, mitochondria, and chromosomes within the neuron, both towards the axon terminals and back to the cell body) [9]. However, AD tau is hyperphosphorylated, leading to tau protein aggregation and twisted filament formation [10,11]. Interestingly, this pathological process can be reversed by iron chelation [12].
Recently, Wang and Masaldan have emphasized the correlation between heightened copper and iron levels in the body and the brain [13,14,15,16]. Furthermore, they have investigated the role of the microbiome and how imbalances in the inflammatory biomarker circulation, including those produced by bacteria, may interact with AD pathology [17,18].
This article analyzes the current literature and presents AD development interrelated with a complex series of co-factors affecting the condition. We will present a collection of scientific publications that indicate that AD is both driven by and associated with impaired inflammatory factors in the blood, which lead to cellular inflammation, swelling, and cytokine release. Additionally, there is compelling evidence that inflammatory factors generated by bacteria contribute to systemic inflammation and may be a critical factor in AD development [19,20].
Figure 1.
AD Pathomechanisms. Description: This figure illustrates the fundamental factors contributing to Alzheimer’s disease (AD) pathomechanisms. Recent studies have highlighted the impact of lifestyle/environment, iron dysregulation, and oral/intestinal dysbiosis on immune system activation during AD onset. These three factors directly influence the AD-characteristic brain neurodegeneration process.
Figure 1.
AD Pathomechanisms. Description: This figure illustrates the fundamental factors contributing to Alzheimer’s disease (AD) pathomechanisms. Recent studies have highlighted the impact of lifestyle/environment, iron dysregulation, and oral/intestinal dysbiosis on immune system activation during AD onset. These three factors directly influence the AD-characteristic brain neurodegeneration process.
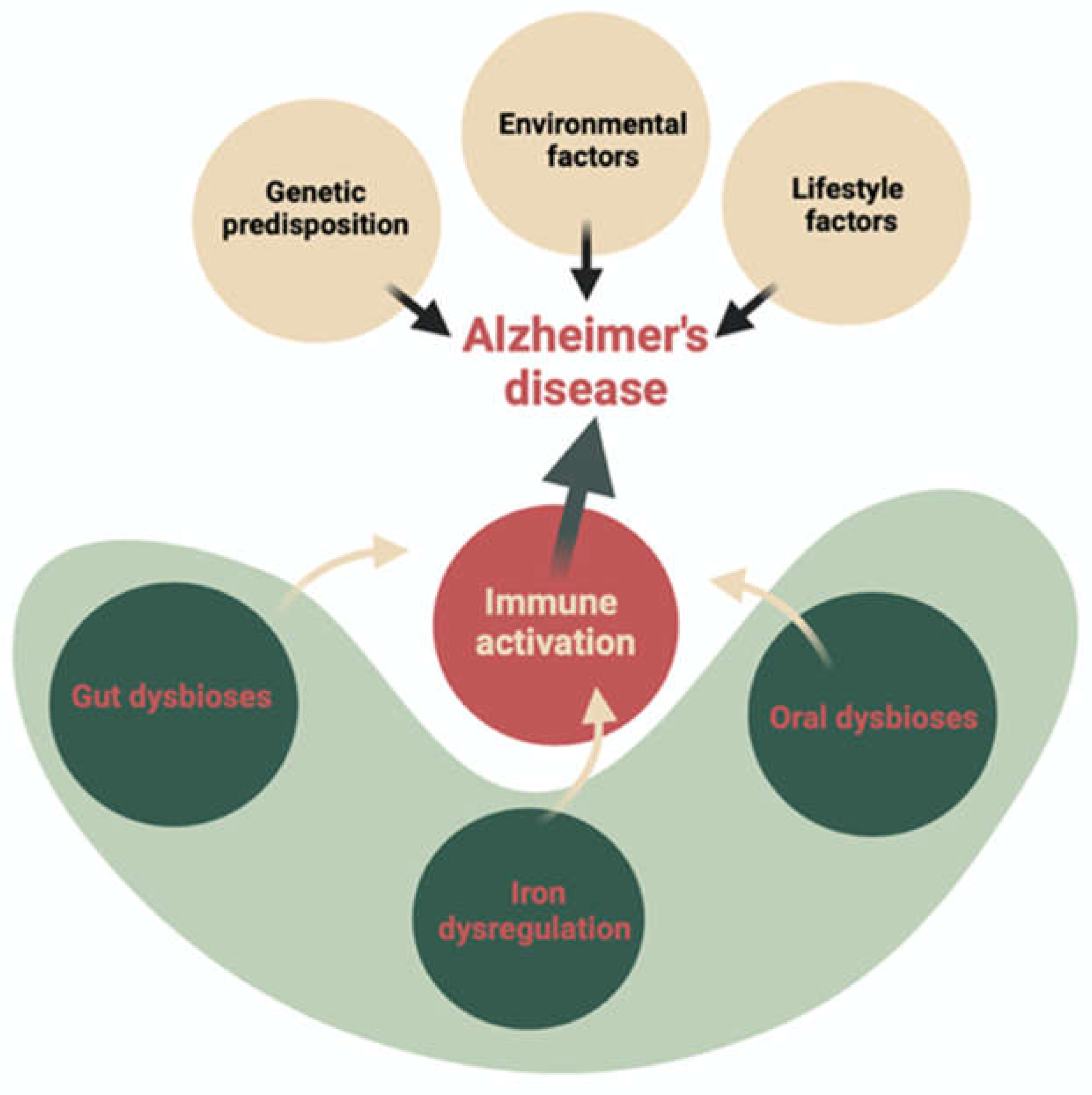
Numerous basic and clinical studies have recently demonstrated the special role of intestinal microbiota in both the metabolism of iron ions and the breakdown of ROS (reactive oxygen species) through the extracellular production of bacterial antioxidant enzymes (catalase, pseudocatalase, dismutase). However, the understanding of the complex iron- microbiome-cognition crosstalk remains elusive [21,22,23,24].
2. The Role of Iron, Oxidative Stress Ferroptosis, and Systemic Inflammation in AD Pathomechanism
2.1. Role of Iron
Iron is an essential trace element in various physiological and biological processes [25]. It is the second most abundant metal in the Earth’s crust. Iron is essential for brain functions such as neuronal development, myelination, neurotransmitter synthesis and breakdown, electron transport, and respiration [24]. Iron readily donates and accepts electrons to participate in energy production and enzyme function (including coordination compound formation) [26,27]. The efficiency of Fe2+ ions as electron donors and Fe3+ ions as electron acceptors is fundamental to many biochemical reactions and makes iron indispensable to life. On the other hand, the same properties that make iron useful also make it toxic and dangerous. In fact, iron is a strong promoter of reactive oxygen species, which can promote protein oxidation, lipid peroxidation, and nucleic acid modification [24,28]. Moreover, iron is indispensable for replicating and growing almost all bacterial species [29]. Approximately 5–20% of ingested iron is absorbed in the duodenum, with the gut microbiota utilizing about 80% of ingested iron, primarily in the colon [29,30]. Iron serves as a cofactor in proteins critical for the metabolic pathways essential for bacterial survival, such as short-chain fatty acid production, DNA synthesis, redox reactions, and the electron transport chain [30]. Iron dysregulation refers to its storage, uptake, and release to the body [31].
2.2. Oxidative Stress and Ferroptosis Promoting Inflammation in Neurodegeneration?
Iron homeostasis disruption can lead to excessive iron accumulation within the cell, forming one of the most aggressive forms of ROS, the hydroxyl radical [32]. The involvement of non-ligand iron and the accompanying oxidative damage strongly correlate to AD nervous system inflammation [27]. Iron dysregulation is any deviation from the typical regulation of iron metabolism at the homeostasis; this is most evident in the regulation of iron transport [33]. Abnormal iron levels are observed in brain and peripheral tissues in AD patients [34]. Iron dysregulation is one of the primary causes of dopaminergic neuronal dysfunction and death [35]. Major causes of iron imbalances are external triggers of stress [36,37]. This form of iron dysregulation can be initiated by multiple factors that contribute to or lead to cell death, including mechanical trauma, nutritional stress, and, of course, oxidative stress [38,39,40,41].
Another source to obtaining free iron is through the heme degradation pathway, as a result of the function of heme oxygenase-1 (HO-1), which catalyzes the destruction of heme [42]. Since the activity of HO-1 is increased in disorders of inflammation that involve the destruction of erythrocytes, it may also serve as an important indicator of inflammation and iron misbalance [43]. Intestinal inflammation caused by dysbiosis in the gut can adversely affect iron regulation in the GI tract [44]. In some chronic diseases (e.g., Crohn’s disease), bleeding into the gastrointestinal tract may result in excessive accumulation of free Fe ions, which consequently leads to an increase in the number, mainly of Gram-negative bacilli and their metabolites. This directly stimulates the activity of immune system cells and intensifies strong oxidative stress, leading to necrosis of intestinal epithelial cells. Therefore, too high levels of Fe ions remaining in our body may directly affect the phenomenon of necrosis of eukaryotic host cells but may also indirectly regulate the number of certain bacterial species (and their metabolites), which in the long run may result in the occurrence of many chronic diseases, including those related to dysfunction nervous system [45]. However, the association between these findings and iron regulation in the serum has not been established yet. While iron misbalance within the GI tract and dysbiosis in the gut could theoretically increase each other, it is reported that the luminal heme found in dietary components or bleeding from the gastrointestinal tract is more likely to cause dysbiosis of the gut microbiome in mice than the reverse. However, the consumption of non-heme iron in food has been linked to a 30% increase in the risk of Parkinson’s disease (p = 0.02)[46]. In the same research, authors also observed that iron supplements were inversely proportional to the risk of Parkinson’s Disease in men [46]. However, the most common cause of iron dysregulation in the form of increased serum ferritin levels is cell death [47].
Research on ferroptosis has grown significantly in recent years since the term was introduced in 2012 [48]. This distinctive form of cell death, characterized by iron-dependent peroxidation of phospholipids, is subject to several cellular metabolic processes. These include redox homeostasis, iron regulation, mitochondrial function, amino acid, lipid, and glucose metabolism, and various disease-related signaling pathways [49]. More recently, ferroptosis has been related to neurodegenerative diseases, including AD [50,51]. Ferroptosis occurs due to cell membrane unsaturated fatty acid depletion and lipid iron-induced reactive oxygen species accumulation, which leads to lethal protein, nucleic acid, and cell lipid damage [52]. Serum ferritin (SF) levels are increased in AD and other inflammatory conditions [53]. Cohort studies have shown that elevated SF levels in cerebrospinal fluid are negatively associated with cognitive performance [54]. Therefore, systemic SF levels have clinical relevance as indicators of cognitive function [54]. Increased iron levels can lead to oxidative stress. Thus, abnormal redox activity is one of the early pathological changes in AD [55]. Oxidative stress contributes to increased lipid peroxidation of DNA and protein oxidation products in AD-affected brains [56].
Oxidative stress promotes amyloid beta (Aβ) deposition, hyperphosphorylation of tau protein, and loss of synapses and neurons. A mimicry is observed in AD; Aβ behaves similarly to the prion protein in prion diseases [57]. Aβ can become a prooxidant, and when combined with iron, it can form hydrogen peroxide [58]. The link between oxidative stress and AD suggests that it is an essential part of the pathological process, which indicates that weakly liganded iron can participate in the Fenton reaction (Fe 2 + + H 2 O 2 ⟶ Fe 3 + + OH • + OH -) leading to the reactive hydroxyl radical (OH-) formation [59]. Ligand-free iron is particularly toxic, confirming the Haber-Weiss reaction indicating that iron is more catalytic than stoichiometric (Fe3 + + O2 -• ⟶ Fe2 + + O2 converts Fe3 + to Fe 2 +) [15]. Krewulak and Vogel emphasize the meaningful role of iron in regulating most bacteria populations, including gram-negative bacilli [60]. Pathogens’ growth is restricted when free iron is uniableilable, suggesting that the presence of free iron facilitates their multiplication and potential spread [61]. Specifically, the invasion of microorganisms causing cytotoxicity leads to iron release, which, to a certain extent, allows them to reproduce and release additional inflammatory bacterial products [55].
3. The Microbiota and Microbiome as a Potential Source of Excess Iron and Inflammatory Biomarkers in the Context of Alzheimer’s Disease
Nothing has attracted as much scientific and public interest as the microbiome and microbiota since the publication of the Human Genome Project (HGP) results [62]. The microbiota is the collection of all microorganisms, including bacteria, viruses, fungi, and other microscopic life forms that inhabit a particular area in living organisms or the environment. The microbiota is found in the skin, intestines, oral cavity, nasal passages, and more [63]. However, the microbiome is a more encompassing concept. It refers to microorganisms inhabiting a specific environment, along with their genomes, metabolic functions, and interactions with the living organism or its environment [64]. The microbiome considers the genetic and functional aspects of microorganisms, their impact on the health of the host or environment, and their distribution and diversity [65]. To identify the source and initiating factors of AD pathology, researchers have begun to focus on the systemic features that characterize patients with the disease [66,67].
3.1. Bi-Directional Communication Between Gut Microbiome and Brain
Available data suggest a bidirectional communication between the gut microbiome and the central nervous system (CNS), referred to as the ‘gut-brain microbiota axis’, is responsible for dysregulation of the gut microbiota among patients with neurodegenerative diseases [68]. Although the gut and brain are anatomically separated, several pathways by which the gut microbiota communicates with the CNS have been proposed. These include modulation of the immune system, the vagus nerve, enteric nervous system (ENS), neuroendocrine system, and circulatory system via the production of neuroactive substances, metabolites, and hormones demonstrated that the gut microbiota is capable of producing or stimulating the production of neurotransmitters, including serotonin, dopamine, and γ-aminobutyric acid (GABA) [69,70,71,72,73].
The altered gut microbiome may induce neuroinflammation through its effects on microglial function and activation [74,75]. Therefore, a possible origin of disturbed inflammatory biomarkers circulating in the blood are bacteria and fungi and inflammatory products, which enter the body thought an imbalanced gut microbiome and translocate to other niches [76].
The interaction between microglia and the gut microbiota begins during the earliest stages of life, and throughout the lifespan of the host [73,77,78]. The gut microbiome provides essential signaling to microglia in both physiological and pathological conditions. Of the neuronal and glial cells, microglia are the most vulnerable to alterations in the gut microbiome [18,55]. Recent studies have demonstrated that the gut microbiota plays a pivotal role in iron metabolism, which is involved in the production of myelin and neurotransmitter synthesis in the central nervous system [79,80]. Gut bacteria influence cognition via the gut-brain axis. Inflammation from gut bacteria can compromise the blood-brain barrier, leading to neuroinflammation and degeneration, worsened by neurotransmitter reduction and oxidative stress from aging and poor diet [81]. Differences in microbiota diversity and composition correlate with Alzheimer’s disease (AD) severity, with AD patients having less diversity and specific bacterial changes compared to healthy individuals [82,83].
3.2. Iron Homeostasis Is Associated with the Microbiome
It has been shown that alterations in host iron homeostasis can affect the luminal iron content of the gut and thus the composition of the gut microbiota [16]. Analysis of mouse feces showed that iron regulatory protein 2 (Irp2) and the protein from mutated genes found in diseases such as hereditary haemochromatosis (Hfe) are highly involved in iron regulation [84,85,86]. The results of the test showed that the composition of the microbiota was significantly altered in mutant mice (Irp2-/- or Hfe-/-) compared to wild-type control mice [87]. Therefore, it can be concluded that iron metabolism in the host has a significant effect on the type of host gut microbiota [88]. Furthermore, the pH of the colon contents is a factor that dictates iron absorption. For instance, microorganisms can ferment galacto-oligosaccharides, resulting in a reduction in the pH of the intestine, thereby facilitating iron absorption at this level. Consequently, the acetic acid-containing products produced by probiotics can be incorporated into the diet, which may enhance iron absorption [23,89]. The experiment conducted by Das et al. demonstrated that gut microbiota regulate systemic iron homeostasis in the host in two ways: by repressing intestinal iron-absorptive pathways via the inhibition of basal HIF-2α function, and by promoting cellular iron storage via the induction of FTN expression [22]. Their results indicate that the host iron-sensing mechanism is intimately connected to and regulated by the gut microbiome [22]. The presence of intestinal iron deficiency results in the positive selection of a specific group of bacteria that produce metabolites that suppress HIF-2α and induce FTN. Two gut microbial metabolites, reuterin and 1,3-diaminopropane (DAP), were identified as effective suppressors of intestinal HIF-2α activity, both in vitro and in vivo [22]. Both of these metabolites prevent the dimerization of HIF-2α with the aryl hydrocarbon receptor nuclear translocator (ARNT), thereby preventing the accumulation of iron in tissues in a mouse model of iron overload. In some cases, these microbes possess cell-wall surface receptors that enable them to intercept host iron sources. In other instances, they deploy high-affinity siderophores that effectively strip iron from host transferrin or lactoferrin [22]. Bacterial iron metabolism has been described in detail in several articles and discussed many times [88,90]. Here we summarize the most commonly described processes.
Bacteria use three main strategies to acquire iron: through the production and use of siderophores (iron-specific chelators), through the selective reduction of iron (III) followed by uptake of iron (II) and using host iron compounds such as heme and transferrin [91]. Siderophores are small ferric iron chelating molecules [92]. They are produced and secreted by many microorganisms in response to iron limitation caused by environmental iron deficiency. Hundreds of siderophores have been characterized and can be divided into four groups according to their function: catecholate, hydroxamate, phenolate and carboxylate. Siderophores form a ferric siderophore complex which is then internalized into the bacterial cell. In Gram-negative bacteria, ferric siderophore complexes are internalized via specific outer membrane (OM) receptors, a periplasmic binding protein (PBP) and an ATP-binding cassette (ABC) transporter in the inner membrane [93].
The OM siderophore receptors are induced by iron deficiency and, as a consequence, are not present under iron-sufficient conditions. The ligand binding sites of the receptors are specific for each siderophore. However, bacteria have multiple OM receptors, allowing them to use siderophores that they are unable to synthesise. Gram-negative outer membranes do not have an established ion gradient or ATP to provide the energy for transport. This energy requirement is met by coupling the proton motive force of the cytoplasmic membrane to the outer membrane via three proteins, TonB, ExbB and ExbD. Periplasmic binding proteins shuttle ferric siderophores from OM receptors to CM ATP-binding cassette (ABC) transporters, which in turn deliver the ferric siderophores to the cytosol, where the complexes are likely to be dissociated by reduction [88].
Conversely, Gram-positive bacteria lack an outer membrane, a cell wall composed of murein, polysaccharides and teichoic acids, and only cell wall proteins separate the bacterial cytoplasm from its environment. Iron is taken up by membrane-anchored binding proteins that direct the iron to a permease and ABC transporter system [94]. Bacteria are also able to transport ferrous iron, which is the most abundant form of iron under anaerobic conditions or at low pH. Ferric iron can also be reduced to the ferrous form by extracellular reductases and is then transported by completely different routes than ferric iron [95].
Feo is the widespread dedicated ferric iron transport system in bacteria. The feo system was first identified in E. coli K12 and was found to be encoded by the feoABC geneb [96]. FeoB is an integral IM protein, whereas FeoA and FeoC are thought to be cytoplasmic. Feo systems play an important role in bacterial virulence in low oxygen environments [94]. This has been demonstrated by deleting the feo genes in many pathogenic/non-pathogenic Gram-negative and Gram-positive bacteria. For example, it has been shown that when the feo genes are deleted, strains of Escherichia coli, Helicobacter pylori and Campylobacter jeuni are unable to take up ferrous iron or colonize the intestines of mice. In contrast, other pathogenic bacteria such as Escherichia coli 08 strain x7122 and Shigella flexneri were shown to survive despite the deletion of the gene, strongly suggesting that a different mode of iron transport is involved.
Other ferrous iron transporters, including zinc-regulated transporters (ZIP-like transporters), natural resistance-associated macrophage protein (Nramp) transporters, EfeUOB systems and P19 iron transporters, have also been described [88].
Bacteria also have receptors for host transferrin and lactoferrin and can therefore take them up directly. These receptors are located in the outer membrane and are induced by iron starvation. In this case, iron is removed from transferrin and lactoferrin at the bacterial cell surface and the iron-free proteins are released extracellularly. The transport of iron released from transferrin and lactoferrin across the periplasmic and cytosolic membranes depends on a periplasmic binding protein ABC permease system [94].
Some pathogenic bacteria also have haem acquisition systems; haem is released from red blood cells by haemolysins and protease. Once released, heme can be bound by host proteins (haemopexin, albumin); however, it can also be taken up directly by bacteria. Extracellular iron is not the only source of iron in bacteria, as they can also use intracellular iron, especially under iron-restricted conditions. Three types of iron storage proteins have been identified in bacteria: ferritin, haem-containing bacterioferritins, and the smaller iron detoxification proteins, which protect the chromosome from iron-induced free radical damage [95].
4. Correlation Between Intestinal and Oral Dysbiosis and the Number of Free Bacterial Antigens
The human gut is the most diverse regarding bacteria species and genetics (occupying an area of 200-300 m2 of the mucosal layer—often called a ‘secret garden’. It is common knowledge that, in the human body bacteria outnumber human cells by a ratio of approximately 1:1. Some studies imply that the gut may contain more than 5000 bacterial taxa, mainly belonging to Bacteroidetes, Firmicutes, Proteobacterium, and Actinobacterium types. In a healthy digestive tract, Bacteroidetes and Firmicutes represent about 90% of the bacteria compared to the remainder [97,98,99].
4.1. Gut-Microbiota
However, microbiota composition changes with age, antibiotic and anti-cancer drug administration, dietary habits, and stress, ultimately leading to imbalances between different microorganisms [100]. Estimates suggest that the microbiome is ten times more numerous than all the somatic and germ cells in the human body. Thousands of microbial species benefit from their favorable, nutrient-rich intestinal environment and perform protective, metabolic, and structural functions that influence the physiology and maintenance of a host’s well-being. Microorganisms regulate the gastrointestinal tract’s pH in a healthy organism, forming a protective barrier against infectious agents [101]. Intestinal colonization in infants begins soon after birth and depends on the delivery method. Babies born vaginally are colonized mainly by Lactobacillus and Bifidobacterium species (their mother’s vaginal microflora [102,103]. However, babies born by cesarean section acquire skin microflora species, such as Propionibacterium, Staphylococcus, and Corynebacterium [81]. It is estimated that in approximately 72% of newborns born vaginally, the intestinal microflora is comparable to their mothers’ microbiota. In contrast, for babies born by cesarean section, this percentage drops to only 41% [104]. With aging, this microbiome balance is often disrupted, resulting in a higher prevalence of Gram-negative bacteria, increased intestinal permeability, and reduced tight junction protein production [105]. The intestinal microflora lives and replicates on the gut’s surface, creating a stable system to prevent invasion by pathogenic microorganisms. The gut microflora remains relatively steady throughout adulthood, ensuring the unchanging operation of the host organism [83]. The intestinal microflora can synthesize a variety of metabolic substances—products that can have positive or negative effects on human health through interaction with the host [106]. For instance, some bacteria in the microbiome that are accountable for vitamin production (such as B12 and K2) directly affect nervous system function [107]. Due to vitamin B12’s anti-oxidative nature, its deficiency may result in the oxidation of lipids, nucleic acids, and proteins, which may contribute to the onset of age-related illnesses [108]. Another study indicates that vitamin K2 enhances neuron health through various pathways, such as limiting oxidative stress, reducing apoptosis caused by β-amyloid (Aβ), reversing microglial activation, suppressing neuroinflammation, and enhancing vascular health [109].
4.2. Oral Microbiota
The oral microbiota plays a vital role in the human microbiome, constituting the second largest microbiota after the gut microbiota [110]. The oral cavity is a complex ecosystem with habitats such as the lips, tongue, tonsils, palate, gingiva, and subgingival spaces [111]. Various microorganisms live under favorable conditions in these areas due to food residues, exfoliated epithelial cells, secretions, and a controlled temperature with high humidity [112].
The oral cavity is characterized by its dynamic nature, as the oral microbiota experiences constant environmental fluctuations. These fluctuations are due to daily physicochemical disturbances caused by ingesting foods and dietary components, antimicrobial agents, smoking, specific hygiene practices, and pH alterations [111]. The oral environment shifts throughout life due to physiological, hormonal, and behavioral modifications, affecting the oral microbial population. Many oral microorganisms acclimate to specific oral environments that are not readily replicable in vitro. The survival of many microorganisms relies on factors such as specific nutrients, temperature, pH, and other microorganisms. Coaggregation and metabolic cooperation are indispensable for bacteria to survive in the oral cavity. Since bacterial microorganisms dominate the oral cavity, most studies on the oral microbiome concentrate primarily on bacteria, with more infrequent reports on the fungal microbiome, referred to as the mycobiome. A number of species of microorganisms have been identified in the oral microbiota ecosystem. Bacteria such as Streptococcus, Neisseria, Veillonella and Actinomyces are among the most prevalent [113]. The oral microbiota exists as biofilms throughout the oral cavity, forming an ecosystem that maintains homeostasis and health [114]. Therefore, oral dysbiosis may alter the function of the bacterial community and have a significant impact on health [113,115].Two of the major dental diseases caused by a dysbiosis of oral microbiota are dental caries and periodontal disease (PD) [116] The latter is an inflammatory disease characterized by a progressive destruction of the tissues of the periodontal complex, mediated by a bacterial dysbiosis predominated by gram-negative anaerobic bacteria such as Porphyromonas gingivalis, Actinobacillus actinmycetemcomitans and Tannerella forsythia [117,118]. Periodontitis and gingivitis are associated with AD [119]. Periodontitis is a potential source facilitating bacterial translocation. Franciotti et al. suggest that patients with AD may develop periodontal disease as a result of dental negligence [120]. However, a retrospective cohort study by Chen and colleagues showed an increased risk of developing AD in people with chronic periodontitis [121]. Kim and the teams suggest that reducing the severity of chronic periodontitis may aid in diminishing dementia risks. Dental care in early dementia prevention programs, especially for men under 70, is recommended to prevent periodontitis progression from mild to severe [122]. Porphyromonas gingivalis (P. gingivalis), a class of Gram-negative bacteria with pro-inflammatory qualities, is widely recognized as a primary culprit behind periodontal and gingival infections (periodontitis and gingivitis) [123,124]. Although primarily found in the oral cavity, animal studies have exhibited that P. gingivalis can cause intestinal dysbiosis, intestinal barrier dysfunction, and systemic inflammation [125]. After examining the metabolome serum of mice infected with P. gingivalis, Verma and Sansores-España observed elevated levels of amino acids like alanine, glutamine, histidine, tyrosine, and phenylalanine, which suggests that bacteria producing these metabolites may have increased [126,127]. Since metabolic and periodontal diseases are strictly correlated, P. gingivalis may impact the metabolites generated in the gut, suggesting a spike in bacteria producing these metabolites [128].
Figure 2.
Iron Dysregulation and Inflammatory Factors. Description: This figure shows the relationship between iron dysregulation and inflammatory factors associated with oral and gut microbiota, which are implicated in the development of Alzheimer’s disease.
Figure 2.
Iron Dysregulation and Inflammatory Factors. Description: This figure shows the relationship between iron dysregulation and inflammatory factors associated with oral and gut microbiota, which are implicated in the development of Alzheimer’s disease.
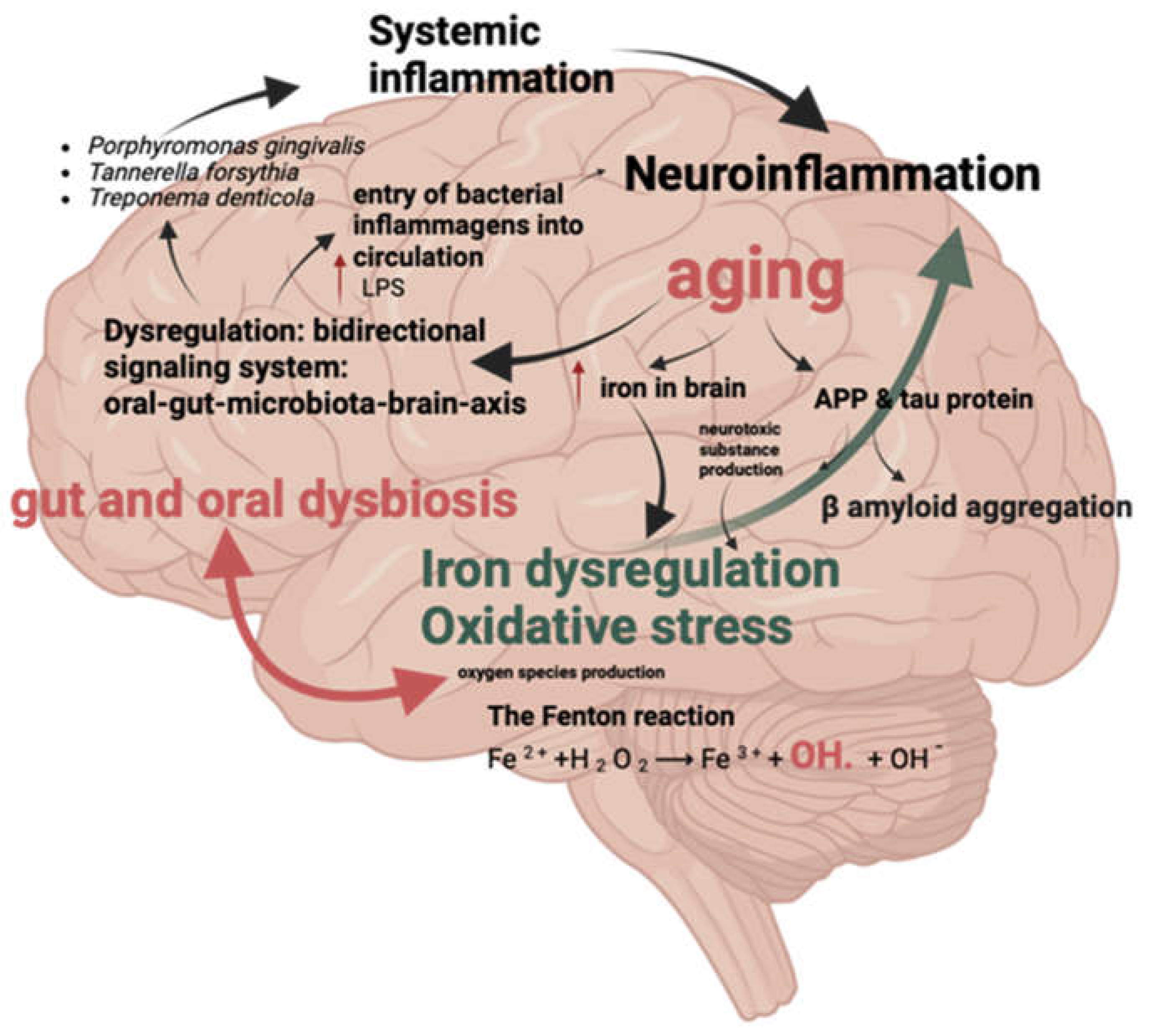
4.3. Oral and Gut Dysbiosis Factors Promoting Neuroinflammation
In physiological conditions, the oral and gut microbiome is thought to have a symbiotic character [78,129]. However, various environmental and immunological factors can lead to a loss of microbial homeostasis, which is associated with an increase in disease-promoting, proinflammatory microorganisms and impaired immunological tolerance [19,78]. This could result in tissue destruction with a systemic inflammatory response [130]. The inflammatory response in the central nervous system (CNS) may originate from inflammasomes, which are multi-protein complexes that mediate innate immunity. In the CNS, the inflammasome is predominantly found in the cytoplasm of immune cells, neuronal cells, astrocytes and microglia, where it recognises pathogen-associated molecular patterns (PAMPs) or host-derived danger-associated molecular patterns (DAMPs). Based on the receptor structure, sensors can be classified into two types: nucleotide-binding oligomerization domain-like receptors (NLRs) and absent in melanoma 2 receptors (ALRs) [19].
4.4. Bacterial Antigens
Bacterial strains impact the system (CNS) by synthesizing neurotransmitters like catecholamines, gamma-aminobutyric acid (GABA), glutamate, norepinephrine, dopamine, acetylcholine, histamine, and other neuromodulatory substances(Narengaowa et al., 2021; Timofeeva et al., 2022). These include short-chain fatty acids (SCFAs), long-chain fatty acids (LCFAs), propionate, and linoleic acid, which are linked with bacterial metabolites that affect host physiology. These microbial influences extend to the behavior of glial cells in both the CNS and the enteric nervous system (ENS). Consequently, the gut microbiota has emerged as a significant environmental factor capable of modulating both the CNS and ENS. This microbial involvement is particularly notable in the pathogenesis of neurodegenerative disorders such as AD, a leading cause of dementia and a major public health concern. One of the key players in this context is the nucleotide-binding domain, leucine-rich repeat protein-3 (NLRP3) inflammasome, a component of the innate immune system. This inflammasome, composed of sensor protein NLRP3, adaptor protein apoptosis-associated speck-like protein containing caspase activation and recruitment domain (ASC), and effector protein pro-caspase-1, triggers the production of proinflammatory cytokines IL-18 and IL-1β upon activation. Notably, NLRP3 inflammasome activation is closely associated with AD pathogenesis [131].
Bacteria produce specific inflammatory agents that can contribute to inflammation [42,103]. Agents that contribute to the formation of biofilms on cell surfaces and intercellular interactions include such proteolytic enzymes as carbonic anhydrases, gingipains, and peptidyl deiminases, as well as bacterial surface components like fimbriae and Curli filaments [132,133] Pathogenic bacteria produce amyloid-like ‘curli’ proteins that form biofilms and have functional similarities to human amyloid-β (Aβ). Ganesh et al. found fundamental evidence for vagus nerve activation in response to bacterial curli. They used a three-dimensional human mini-epithelial monolayer system in an in vitro model and demonstrated increased TLR2 levels after stimulation with purified bacterial curli fibers [134]. The data presented here are associated with an increased colonization of Gram-positive bacteria in the ileum of mice with AD symptoms [134].
Moreover, dysbiosis in oral microbiota is suggested to directly contribute to the production of beta-amyloid peptides, a hallmark of AD, through the trigeminal nervous system and systemic circulation [115]. Furthermore, brain-nose proximity (the nose home to a separate microbiome) indicates the probability of interactions between olfactory receptors, microorganisms, and bacterial metabolites [68]. In addition, oral bacteria (especially Porphyromonas gingivalis, Treponema spp.—including T. denticola, and Tannerella forsythia species) [110,112,115].
4.5. Lipopolysaccharide—Endotoxin
LPS and other toxic products cause neuroinflammation and contribute to Aβ plaque accumulation and tau hyperphosphorylation in the brain [135]. Additional contributing factors include LPSs and lipoteichoic acid (LTA) [136]. In some cases, certain bacteria can produce functional amyloid fibers on the cell surface, and these fibers may also play a role in forming biofilms and intercellular interactions [137,138].
Friedland et al. suggest bacterial amyloids may impact immune system activation and neuronal amyloid production, potentially contributing to brain disorders [5] This process involves using TLR receptors on the epithelial surface and neural connections of enteroendocrine and other epithelial cells. Exposure to bacterial amyloids intensifies the buildup of neuronal amyloids, resulting in protein misfolding in the brain [99]. Furthermore, bacterial inflammatory agents can indirectly contribute to both the onset and progression of AD by triggering peripheral immune cells such as astrocytes, microglia, monocytes, and macrophages [5]. These cells can cross the blood-brain barrier and promote inflammation within the nervous system. In addition, they can initiate structural changes in proteins and encourage the transition to β-sheet-rich amyloid fibers, which directly affects AD pathology. Senile plaque formation, an indicator of AD, is caused by the accumulation of amyloid proteins within the brain [139,140].
Additionally, the presence of amyloid fibrin(ogen) in the bloodstream causes hypercoagulation, a newly identified co-occurring pathology [137]. The relationship between proteopathy, neurological inflammation, and gut microbiota may hold considerable potential for further exploration, particularly regarding therapeutic interventions [136].
One fascinating bacterial inflammatory factor is LPSs from the bacterial cell membrane. LPSs are large molecules composed of a hydrophobic lipid A domain, a repeating oligosaccharide ‘core,’ and a polysaccharide chain known as the O antigen, defining the serotype [141,142]. The lipid A domain is usually considered to be the most inflammatory region of this molecule [143]. There are differences between commensal gut bacteria LPSs, which are less immunogenic, and LPSs exhibiting solid pro-inflammatory properties. Microglia activation is another characteristic histopathological feature in AD [144,145]. Research indicates that LPSs may be responsible for microglia activation, which plays a significant part in nervous system inflammation. In addition, LPSs induce changes in microglia function, suggesting that it may affect blood-brain barrier (BBB) dysfunction through the generation of ROS via nicotinamide adenine dinucleotide oxidase (NADPH) [105]. When activated by LPSs, microglia can release cytokines like IL-1β, IL-6, and TNFα, which leads to an upsurge in inducible nitric oxide synthase (iNOS) expression and ROS production [146]. It is also riveting to note that in a rat model, substantia nigra had the highest density of microglia, making it particularly susceptible to LPS-induced damage [105]. LPSs can also induce inflammation and BBB damage in recipients, allowing peripheral cytokines to infiltrate the brain. Therefore, LPSs in the circulation, both directly and indirectly, lead to neurodegeneration by inducing a robust inflammatory response, leading to BBB damage, inflammation, and oxidative stress in the central nervous system, as well as stimulating abnormal folding and aggregation of amyloid beta [105].
Studies have ascertained a relationship between this pathogen and host matrix metalloproteinases (MMPs) in periodontal disease pathogenesis [147]. MMPs are essential in tissue destruction associated with periodontal disease pathogenesis [147]. P. gingivalis also promotes tissue destruction [148]. The activation of MMP genes by P. gingivalis is accompanied by the activation of the TIMP-2 gene, which regulates MMP activity [148]. Furthermore, P. gingivalis causes an up-regulation of MMP-2 and MMP-9 mRNA expression in oral epithelial cells. The interaction between P. gingivalis and host MMPs is complex and critical in periodontal disease pathogenesis [148]. Chronic inflammation caused by the oral microbiota leads to immune reactions, free radical production, apoptosis, and Aβ deposition [18,110,115]. Oral microbiota can enter the brain via the bloodstream through tooth brushing, flossing, chewing, or using a toothpick, especially in people with periodontal disease (which may result in bacteremia) [119].
Peptidoglycan (PGN) is a dominant component of the Gram-negative cell wall that is recognized by specific pattern-recognition receptors (PRRs) of the innate immune system [149]. Gut microbiota-derived PGN could potentially traverse the blood-brain barrier (BBB) and affect gene transcription communication is possible through the Nod2 receptor, which is present in both the gut and the brain [150]. The Nod2 receptor plays a role in immune regulation [151]. The activation of the Nod2 receptor by bacterial peptidoglycan can initiate signaling pathways that affect brain function and communication [76].
Tryptophan plays a pivotal role in the metabolism of two major pathways, kynurenine and serotonin, by acting as a precursor for their activity [152]. Metabolism of tryptophan through the kynurenine pathway is upregulated during the pathophysiological processes that precede the development of AD [153]. This upregulation appears to play a role in the oxidative and neuroinflammatory mechanisms that precede the onset of AD symptoms. However, the kynurenine metabolites can exert either neurotoxic or neuroprotective effects [154]. Indeed, excitotoxicity may be related to the increase of 3-hydroxykynurenine (3-HK) and quinolinic acid (QUIN) metabolites. While the inhibition of 3-HK formation through the blockade of kynurenine monooxygenase (KMO) has been demonstrated to have beneficial effects against neuronal and synaptic loss, as well as memory deficits, in mouse models of AD [154]
The synthesis and availability of serotonin is facilitated by tryptophan intake, with improved memory acquisition in rodents and decreased intraneuronal Aβ accumulation in the brain of 3xTg-AD animals [155]. Both serotonin selective re-uptake inhibitors (SSRIs) and increased dietary tryptophan intake reduce amyloid plaques and induce antidepressant effects in humans [154].
5. Therapeutic Possibilities and Pharmaceutical Interventions
The dominant pathophysiological model for AD is the amyloid cascade hypothesis, first proposed in 1992 [156]. Numerous anti-amyloid therapies, such as β-secretase converting enzyme inhibitors and anti-amyloid-β monoclonal antibodies, reduce amyloid in the brain and cerebrospinal fluid, but none of these drugs has been shown to delay disease progression [157,158]. Some argue that the use of anti-amyloid-β therapeutics in the symptomatic stage of AD may be too late [159]. This hypothesis is being tested in ongoing trials in presymptomatic amyloid-positive individuals at risk of sporadic Alzheimer’s disease [160]. Despite considerable investment and effort, numerous clinical trials have so far failed to produce clinically meaningful results [161].
5.1. Iron Chelation
In the search for new directions in the application of novel AD therapies and given the availability of compounds used against other disease entities, it is reasonable to attempt the use of these compounds given the abundant evidence for a link between iron dyshomeostasis and many aspects of neurodegenerative disease pathophysiology [45]. Iron chelators are currently used clinically in transfusion-dependent patients with iron overload, beta thalassemia, sickle cell anaemia, myelodysplasia and aplastic anaemia [162]. There are three iron chelates in common clinical use: deferoxamine, deferasirox deferiprone [45]. Iron chelation therapy, aiming to sequester unliganded iron, holds promise for mitigating iron-induced damage in AD [163]. Compounds like VK-28, with potent iron-chelating and MAO inhibitory properties, demonstrate potential therapeutic benefits [45,164]. Clinical trials, notably with deferiprone, show some efficacy in reducing iron levels in brain regions affected by AD. However, their overall impact on disease progression remains uncertain due to PD’s multifaceted nature [163]. Multifunctional iron chelators, such as DHC12 and CT51, are under experimental investigation for their potential to target multiple pathological mechanisms simultaneously [164]
On the other hand, iron sequestration drugs used to treat another neurodegenerative disease: Parkinson’s disease (PD) may influence gut microbiome composition by eliminating iron-dependent bacteria as demonstrated very recently in vitro in a very recent paper However, postulated by the article authors oral iron supplementation in AD patients treated with iron—depleting drugs may cause even more dysregulation of their gut microbiome in favor of the potentially pathogenic and drug-resistant bacteria [165].
5.2. Antibiotics and Probiotics
Antibiotics and probiotics are being explored as potential treatments for PD, primarily targeting gut-related issues. Probiotics show promise in altering disease progression and alleviating gastrointestinal symptoms [164,166].
Probiotics have different mechanisms of action, although the exact way in which they exert their effects is not yet fully understood [166]. These range from bacteriocin, production of short-chain fatty acids and competition for nutrients to stimulation of the role of the gut-brain axis and immunomodulation [167]. SCFAs are produced in the gut depending on the fibre content of the diet. Metabolites such as acetate, butyrate and propionate are produced by fermentation mediated by Bacteriodes, Clostridium, Lactobacillus, Bifidobacterium and Eubacterium species [166,168]. SCFAs may affect brain function through three main pathways: immune modulation, endocrine pathways and neuronal factors [166]. Through immune modulation, SCFAs improve barrier integrity and maintain mucus production, which may influence gut mucosal immunity and barrier function. Systemically, SCFAs mediate immunomodulation through the secretion of cytokines that affect the proliferation and differentiation of immune cells. This interaction generates an anti-inflammatory response while suppressing pro-inflammatory cytokines (such as IL-1β, IL-6, TNF-α) [169]. In addition, SCFAs can cross the BBB via monocarboxylate transporters and affect BBB integrity by upregulating the expression of tight junction proteins. In the CNS, SCFAs influence neuroinflammation by affecting microglial cell morphology and function, thereby preventing neuronal cell death [170].
Antibiotics like rifaximin and minocycline demonstrate neuroprotective effects, possibly through modulating gut microbiota composition and reducing inflammatory responses [171]. Rifampicin, with its multifaceted neuroprotective functions, presents another potential avenue for PD treatment, although certain antibiotics may carry risks of exacerbating the condition [172].
5.3. Faecal Microbiota Transplantation
Fecal microbiota transplantation (FMT) is a widely accepted and safely used treatment for recurrent Clostridium difficile infections as well as other metabolic diseases such as diabetes mellitus [173]. FMT has also been shown to be a potential treatment for AD [174]. While initial studies suggest potential benefits in reducing neuroinflammation and motor dysfunction, controlled clinical trials are essential to validate its efficacy and safety [175]. According to Elangovan et al., older Tg-FY mice showed better overall cognition after FMT treatment, while older Tg-FO mice also showed some cognitive improvement. The increasing trend of cognitive improvement observed in the older groups correlated with decreasing Aβ load [173]. In other research, FMT treatment has been demonstrated to exert beneficial effects against neuropsychiatric disorders by modulating the gut microbiota. However, its effect on AD remains unclear. This study demonstrated the neuroprotective effects of FMT against AD in APPswe/PS1dE9 transgenic mice [174]. These results showed that FMT treatment significantly ameliorated cognitive deficits, Aβ accumulation, synaptic dysfunction and neuroinflammation. The observed protective effects of FMT may be related to the reversal of changes in the gut microbiota and its metabolites SCFAs [174]. However, there is currently a lack of evidence to support the use of faecal transplantation in AD.
5.4. Additional Therapeutic Options
Therapeutic strategies targeting both LPS and gingipains, such as small molecule inhibitors or IgY antibody-containing lozenges against gingipains, represent additional potential treatment avenues [176]. These options, though intriguing, require further research and clinical validation for their effectiveness in AD management [177].
6. Conclusions
This review analyzes the evidence that AD and its development depend on certain interrelated factors. These factors include iron dysregulation, intestinal and oral dysbiosis (which causes local inflammation leading to translocation of bacterial metabolites and inflammation in the nervous system), inflammation at a systemic level, cellular deterioration (resulting from the beta-amyloid accumulation and neurofibrillary tangles), abnormalities in blood vessel function and iron regulation, as well as digestive disorders and disruptions in oral microbiota. The evidence published supports the notion that AD is linked to and brought about by dysregulated inflammatory factors circulating in the body. Iron is continuously deposited in the brain with aging, leading to iron dysregulation and potential oxidative stress. Iron homeostasis dysregulation produces neurotoxic substances and reactive oxygen species, causing iron-induced oxidative stress. Bacteria-produced inflammatory agents can infiltrate the body via two distinct routes: either through the gut or through a two-step process, starting with the oral cavity and proceeding through the bloodstream to the brain, leading to neuroinflammation. At this disease stage, pathological changes that initially affect the olfactory system progress to the temporal lobe and ultimately regress to the brainstem.
These processes are potential targets for therapeutic intervention. Several treatments have demonstrated potential for treating AD (including prebiotics, probiotics, antibiotics, and fecal transplant treatments). At present, the primary obstacle in drug development aimed at treating intricate cognitive illnesses, including AD, is the pursuit of multimodal drugs that can alter the course of the disease. The pursuit of identifying the root causes of AD, such as gut dysbiosis and iron toxicity, has resulted in the creation of diverse and innovative treatments. These treatments not only have the potential to alleviate the loss of motor control seen in AD but also can significantly decelerate the disease’s advancement.
To prevent AD and its progression, we propose pinpointing the origins of the illness and any accompanying health issues and then implementing a tailored treatment plan that emphasizes its core attributes. This personalized treatment plan should encompass a thorough investigation of all factors involved. To achieve this goal, it is essential to commence extensive longitudinal studies with sizable groups of individuals. Prospective tactics could center on biomarker utilization to predict AD advancement before blatant cognitive impairment emergence. This guideline aims to provide medication that can modify the trajectory of a disease in its early stages, typically before any symptoms manifest themselves. The intended outcome is to hinder or postpone disease onset. Ultimately, attempts to restore the gut flora in AD patients may significantly delay neurodegeneration by reducing inflammatory responses and amyloidogenesis.
Author Contributions
Conceptualization, MS. and P.H.; software, A.K.; resources, A.K.; writing—original draft preparation, A.K.; writing—review and editing, M.S, A.P. and P.H..; supervision, M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Data Availability Statement
Not applicable.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. The Lancet 2021;397:1577–90. [CrossRef]
- Nichols E, Steinmetz JD, Vollset SE, Fukutaki K, Chalek J, Abd-Allah F, et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the Global Burden of Disease Study 2019. Lancet Public Health 2022;7:e105–25. [CrossRef]
- Chandra S, Sisodia SS, Vassar RJ. The gut microbiome in Alzheimer’s disease: what we know and what remains to be explored. Mol Neurodegener 2023;18. [CrossRef]
- Gaweł M, Potulska-Chromik A. Choroby neurodegeneracyjne: choroba Alzheimera i Parkinsona. 2015.
- Friedland RP, McMillan JD, Kurlawala Z. What are the molecular mechanisms by which functional bacterial amyloids influence amyloid beta deposition and neuroinflammation in neurodegenerative disorders? Int J Mol Sci 2020;21. [CrossRef]
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 2016;8:595–608. [CrossRef]
- Shcherbinin S, Evans CD, Lu M, Andersen SW, Pontecorvo MJ, Willis BA, et al. Association of Amyloid Reduction after Donanemab Treatment with Tau Pathology and Clinical Outcomes: The TRAILBLAZER-ALZ Randomized Clinical Trial. JAMA Neurol 2022;79:1015–24. [CrossRef]
- Frisoni GB, Altomare D, Thal DR, Ribaldi F, van der Kant R, Ossenkoppele R, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci 2022;23:53–66. [CrossRef]
- Gulisano W, Maugeri D, Baltrons MA, Fà M, Amato A, Palmeri A, et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. Journal of Alzheimer’s Disease 2018;64:S611–31. [CrossRef]
- Gulisano W, Maugeri D, Baltrons MA, Fà M, Amato A, Palmeri A, et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. Journal of Alzheimer’s Disease 2018;64:S611–31. [CrossRef]
- Ashrafian H, Zadeh EH, Khan RH. Review on Alzheimer’s disease: Inhibition of amyloid beta and tau tangle formation. Int J Biol Macromol 2021;167:382–94. [CrossRef]
- Kim AC, Lim S, Kim YK. Metal ion effects on Aβ and tau aggregation. Int J Mol Sci 2018;19. [CrossRef]
- Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med 2019;133:221–33. [CrossRef]
- Ndayisaba A, Kaindlstorfer C, Wenning GK. Iron in neurodegeneration—Cause or consequence? Front Neurosci 2019;13. [CrossRef]
- Wang C, Wang Z, Zeng B, Zheng M, Xiao N, Zhao Z. Fenton-like reaction of the iron(ii)-histidine complex generates hydroxyl radicals: Implications for oxidative stress and Alzheimer’s disease. Chemical Communications 2021;57:12293–6. [CrossRef]
- Zeidan RS, Han SM, Leeuwenburgh C, Xiao R. Iron homeostasis and organismal aging. Ageing Res Rev 2021;72. [CrossRef]
- Stadlbauer V, Engertsberger L, Komarova I, Feldbacher N, Leber B, Pichler G, et al. Dysbiosis, gut barrier dysfunction and inflammation in dementia: A pilot study. BMC Geriatr 2020;20. [CrossRef]
- Zenobia C, Darveau RP. Does Oral Endotoxin Contribute to Systemic Inflammation? Frontiers in Oral Health 2022;3. [CrossRef]
- Lukiw WJ. The microbiome, microbial-generated proinflammatory neurotoxins, and Alzheimer’s disease. J Sport Health Sci 2016;5:393–6. [CrossRef]
- Quigley, Eamonn M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr Neurol Neurosci Rep 2017;17. [CrossRef]
- Rusu IG, Suharoschi R, Vodnar DC, Pop CR, Socaci SA, Vulturar R, et al. Iron supplementation influence on the gut microbiota and probiotic intake effect in iron deficiency—A literature-based review. Nutrients 2020;12:1–17. [CrossRef]
- Das NK, Schwartz AJ, Barthel G, Inohara N, Liu Q, Sankar A, et al. Microbial Metabolite Signaling Is Required for Systemic Iron Homeostasis. Cell Metab 2020;31:115-130.e6. [CrossRef]
- Holbein BE, Lehmann C. Dysregulated Iron Homeostasis as Common Disease Etiology and Promising Therapeutic Target. Antioxidants 2023;12. [CrossRef]
- Levi S, Ripamonti M, Moro AS, Cozzi A. Iron imbalance in neurodegeneration. Mol Psychiatry 2024. [CrossRef]
- Ni S, Yuan Y, Kuang Y, Li X. Iron Metabolism and Immune Regulation. Front Immunol 2022;13. [CrossRef]
- Elstrott B, Khan L, Olson S, Raghunathan V, DeLoughery T, Shatzel JJ. The role of iron repletion in adult iron deficiency anemia and other diseases. Eur J Haematol 2020;104:153–61. [CrossRef]
- Peng Y, Chang X, Lang M. Iron homeostasis disorder and alzheimer’s disease. Int J Mol Sci 2021;22. [CrossRef]
- Gutteridge JMC, Halliwell B. Mini-Review: Oxidative stress, redox stress or redox success? Biochem Biophys Res Commun 2018;502:183–6. [CrossRef]
- Mayneris-Perxachs J, Moreno-Navarrete JM, Fernández-Real JM. The role of iron in host–microbiota crosstalk and its effects on systemic glucose metabolism. Nat Rev Endocrinol 2022;18:683–98. [CrossRef]
- Rosell-Díaz M, Santos-González E, Motger-Albertí A, Ramió-Torrentà L, Garre-Olmo J, Pérez-Brocal V, et al. Gut microbiota links to serum ferritin and cognition. Gut Microbes 2023;15. [CrossRef]
- Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem Sci 2016;41:274–86. [CrossRef]
- Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim Biophys Acta Mol Cell Res 2019;1866. [CrossRef]
- Pretorius L, Kell DB, Pretorius E. Iron dysregulation and dormant microbes as causative agents for impaired blood rheology and pathological clotting in Alzheimer’s type dementia. Front Neurosci 2018;12. [CrossRef]
- Wang F, Wang J, Shen Y, Li H, Rausch WD, Huang X. Iron Dyshomeostasis and Ferroptosis: A New Alzheimer’s Disease Hypothesis? Front Aging Neurosci 2022;14. [CrossRef]
- Yan N, Zhang J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. n.d.
- Apple CG, Miller ES, Kannan KB, Thompson C, Darden DB, Efron PA, et al. Prolonged Chronic Stress and Persistent Iron Dysregulation Prevent Anemia Recovery Following Trauma. Journal of Surgical Research 2021;267:320–7. [CrossRef]
- Reid BM, Georgieff MK. The Interaction between Psychological Stress and Iron Status on Early-Life Neurodevelopmental Outcomes. Nutrients 2023;15. [CrossRef]
- Zhang L, Hu R, Li M, Li F, Meng H, Zhu G, et al. Deferoxamine attenuates iron-induced long-term neurotoxicity in rats with traumatic brain injury. Neurological Sciences 2013;34:639–45. [CrossRef]
- Charlebois E, Pantopoulos K. Nutritional Aspects of Iron in Health and Disease. Nutrients 2023;15. [CrossRef]
- Zeidan RS, Martenson M, Tamargo JA, McLaren C, Ezzati A, Lin Y, et al. Iron homeostasis in older adults: balancing nutritional requirements and health risks. Journal of Nutrition, Health and Aging 2024;28. [CrossRef]
- Loveikyte R, Bourgonje AR, van Goor H, Dijkstra G, van der Meulen—de Jong AE. The effect of iron therapy on oxidative stress and intestinal microbiota in inflammatory bowel diseases: A review on the conundrum. Redox Biol 2023;68. [CrossRef]
- Haines DD, Tosaki A. Heme degradation in pathophysiology of and countermeasures to inflammation-associated disease. Int J Mol Sci 2020;21:1–25. [CrossRef]
- Mendoza E, Duque X, Moran S, Martínez-Andrade G, Reyes-Maldonado E, Flores-Huerta S, et al. Hepcidin and other indicators of iron status, by alpha-1 acid glycoprotein levels, in a cohort of Mexican infants Content courtesy of Springer Nature, terms of use apply. Rights reserved n.d. [CrossRef]
- Malesza IJ, Bartkowiak-Wieczorek J, Winkler-Galicki J, Nowicka A, Dzięciołowska D, Błaszczyk M, et al. The Dark Side of Iron: The Relationship between Iron, Inflammation and Gut Microbiota in Selected Diseases Associated with Iron Deficiency Anaemia—A Narrative Review. Nutrients 2022;14. [CrossRef]
- Gleason A, Bush AI. Iron and Ferroptosis as Therapeutic Targets in Alzheimer’s Disease. Neurotherapeutics 2021;18:252–64. [CrossRef]
- Logroscino G, Gao X, Chen H, Wing A, Ascherio A. Dietary iron intake and risk of Parkinson’s disease. Am J Epidemiol 2008;168:1381–8. [CrossRef]
- Kernan KF, Carcillo JA. Hyperferritinemia and inflammation. Int Immunol 2017;29:401–9. [CrossRef]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–72. [CrossRef]
- Li J, Cao F, Yin H liang, Huang Z jian, Lin Z tao, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis 2020;11. [CrossRef]
- Sfera A, Bullock K, Price A, Inderias L, Osorio C. Ferrosenescence: The iron age of neurodegeneration? Mech Ageing Dev 2018;174:63–75. [CrossRef]
- Ma H, Dong Y, Chu Y, Guo Y, Li L. The mechanisms of ferroptosis and its role in alzheimer’s disease. Front Mol Biosci 2022;9. [CrossRef]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017;171:273–85. [CrossRef]
- Smith M. Increased Iron and Free Radical Generation in Preclinical Alzheimer Disease and Mild Cogni 2010.
- Ayton S, Faux NG, Bush AI, Weiner MW, Aisen P, Petersen R, et al. Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat Commun 2015;6. [CrossRef]
- Seyoum Y, Baye K, Humblot C. Iron homeostasis in host and gut bacteria–a complex interrelationship. Gut Microbes 2021;13:1–19. [CrossRef]
- Ashraf A, Jeandriens J, Parkes HG, So PW. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol 2020;32. [CrossRef]
- Goedert M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science (1979) 2015;349. [CrossRef]
- The prion-like propagation hypothesis in Alzheimerʼs and Parkinsonʼs disease. Current Opin n.d.
- Formanowicz D, Radom M, Rybarczyk A, Formanowicz P. The role of Fenton reaction in ROS-induced toxicity underlying atherosclerosis—modeled and analyzed using a Petri net-based approach. BioSystems 2018;165:71–87. [CrossRef]
- Krewulak KD, Vogel HJ. Structural biology of bacterial iron uptake. Biochim Biophys Acta Biomembr 2008;1778:1781–804. [CrossRef]
- Schaible UE, Kaufmann SHE. Iron and microbial infection. Nat Rev Microbiol 2004;2:946–53. [CrossRef]
- Lyte M, Cryan JF. Advances in Experimental Medicine and Biology 817 Microbial Endocrinology Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease. n.d.
- Marchesi JR, Ravel J. The vocabulary of microbiome research: a proposal. Microbiome 2015;3. [CrossRef]
- Berg G, Rybakova D, Fischer D, Cernava T, Vergès MCC, Charles T, et al. Microbiome definition re-visited: old concepts and new challenges. Microbiome 2020;8. [CrossRef]
- Shanahan F, Ghosh TS, O’Toole PW. The Healthy Microbiome—What Is the Definition of a Healthy Gut Microbiome? Gastroenterology 2021;160:483–94. [CrossRef]
- Bhattacharjee S, Lukiw WJ. Alzheimer’s disease and the microbiome. Front Cell Neurosci 2013. [CrossRef]
- Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep 2017;7. [CrossRef]
- Rawls M, Ellis AK. The microbiome of the nose. Annals of Allergy, Asthma and Immunology 2019;122:17–24. [CrossRef]
- Strandwitz P, Kim KH, Terekhova D, Liu JK, Sharma A, Levering J, et al. GABA-modulating bacteria of the human gut microbiota. Nat Microbiol 2019;4:396–403. [CrossRef]
- Stasi C, Sadalla S, Milani S. The Relationship Between the Serotonin Metabolism, Gut-Microbiota and the Gut-Brain Axis. Curr Drug Metab 2019;20:646–55. [CrossRef]
- Wang Y, Tong Q, Ma SR, Zhao ZX, Pan L Bin, Cong L, et al. Oral berberine improves brain dopa/dopamine levels to ameliorate Parkinson’s disease by regulating gut microbiota. Signal Transduct Target Ther 2021;6. [CrossRef]
- Braga JD, Thongngam M, Kumrungsee T. Gamma-aminobutyric acid as a potential postbiotic mediator in the gut–brain axis. NPJ Sci Food 2024;8. [CrossRef]
- Loh JS, Mak WQ, Tan LKS, Ng CX, Chan HH, Yeow SH, et al. Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct Target Ther 2024;9. [CrossRef]
- Abdel Haq R, Schlachetzki JCM, Glass CK, Mazmanian SK. Microbiome–microglia connections via the gut–brain axis. Journal of Experimental Medicine 2019;216:41–59. [CrossRef]
- Chandra S, Di Meco A, Dodiya HB, Popovic J, Cuddy LK, Weigle IQ, et al. The gut microbiome regulates astrocyte reaction to Aβ amyloidosis through microglial dependent and independent mechanisms. Mol Neurodegener 2023;18. [CrossRef]
- Arabi TZ, Alabdulqader AA, Sabbah BN, Ouban A. Brain-inhabiting bacteria and neurodegenerative diseases: the “brain microbiome” theory. Front Aging Neurosci 2023;15. [CrossRef]
- Mossad O, Erny D. The microbiota–microglia axis in central nervous system disorders. Brain Pathology 2020;30:1159–77. [CrossRef]
- Lynch CMK, Cowan CSM, Bastiaanssen TFS, Moloney GM, Theune N, van de Wouw M, et al. Critical windows of early-life microbiota disruption on behaviour, neuroimmune function, and neurodevelopment. Brain Behav Immun 2023;108:309–27. [CrossRef]
- Arnoriaga-Rodríguez M, Mayneris-Perxachs J, Burokas A, Contreras-Rodríguez O, Blasco G, Coll C, et al. Obesity Impairs Short-Term and Working Memory through Gut Microbial Metabolism of Aromatic Amino Acids. Cell Metab 2020;32:548-560.e7. [CrossRef]
- Yang Y, Xu Z, Guo J, Xiong Z, Hu B. Exploring the gut microbiome-Postoperative Cognitive Dysfunction connection: Mechanisms, clinical implications, and future directions. Brain Behav Immun Health 2024;38. [CrossRef]
- Martin CR, Osadchiy V, Kalani A, Mayer EA. The Brain-Gut-Microbiome Axis. CMGH 2018;6:133–48. [CrossRef]
- Clemmensen C, Müller TD, Woods SC, Berthoud HR, Seeley RJ, Tschöp MH. Gut-Brain Cross-Talk in Metabolic Control. Cell 2017;168:758–74. [CrossRef]
- Kesika P, Suganthy N, Sivamaruthi BS, Chaiyasut C. Role of gut-brain axis, gut microbial composition, and probiotic intervention in Alzheimer’s disease. Life Sci 2021;264. [CrossRef]
- Saitoh O SKMR et al. The forms and the levels of fecal PMN-elastase in patients with colorectal diseases. American Journal of Gastroenterology 1995;103:162–9. [CrossRef]
- Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, et al. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol 2016;22:501–18. [CrossRef]
- Ng O. Iron, microbiota and colorectal cancer. Wiener Medizinische Wochenschrift 2016;166:431–6. [CrossRef]
- Buhnik-Rosenblau K, Moshe-Belizowski S, Danin-Poleg Y, Meyron-Holtz EG. Genetic modification of iron metabolism in mice affects the gut microbiota. BioMetals 2012;25:883–92. [CrossRef]
- Andrews SC, Robinson AK, Rodríguez-Quiñones F. Bacterial iron homeostasis. FEMS Microbiol Rev 2003;27:215–37. [CrossRef]
- Pérez-Conesa D, López G, Ros G. Effect of probiotic, prebiotic and synbiotic follow-up infant formulas on iron bioavailability in rats. Food Science and Technology International 2007;13:69–77. [CrossRef]
- Cornelis P, Wei Q, Andrews SC, Vinckx T. Iron homeostasis and management of oxidative stress response in bacteria. Metallomics 2011;3:540–9. [CrossRef]
- Timofeeva AM, Galyamova MR, Sedykh SE. Bacterial Siderophores: Classification, Biosynthesis, Perspectives of Use in Agriculture. Plants 2022;11. [CrossRef]
- Timofeeva AM, Galyamova MR, Sedykh SE. Bacterial Siderophores: Classification, Biosynthesis, Perspectives of Use in Agriculture. Plants 2022;11. [CrossRef]
- Řezanka T, Palyzová A, Faltýsková H, Sigler K. Siderophores: Amazing Metabolites of Microorganisms. Studies in Natural Products Chemistry, vol. 60, Elsevier B.V.; 2018, p. 157–88. [CrossRef]
- Lau CKY, Krewulak KD, Vogel HJ. Bacterial ferrous iron transport: The Feo system. FEMS Microbiol Rev 2016;40:273–98. [CrossRef]
- Seyoum Y, Baye K, Humblot C. Iron homeostasis in host and gut bacteria–a complex interrelationship. Gut Microbes 2021;13:1–19. [CrossRef]
- Çelen E, Kiliç A, Moore GR. The role of Escherichia coli bacterroferrrttn n stress nduced conddttons. vol. 74. 2015.
- Sender R, Fuchs S, Milo R. Are We Really Vastly Outnumbered? Revisiting the Ratio of Bacterial to Host Cells in Humans. Cell 2016;164:337–40. [CrossRef]
- Das B, Ghosh TS, Kedia S, Rampal R, Saxena S, Bag S, et al. Analysis of the gut microbiome of rural and urban healthy indians living in sea level and high altitude areas. Sci Rep 2018;8. [CrossRef]
- Kitamura K, Sasaki M, Matsumoto M, Shionoya H, Iida K. Protective effect of Bacteroides fragilis LPS on Escherichia coli LPS-induced inflammatory changes in human monocytic cells and in a rheumatoid arthritis mouse model. Immunol Lett 2021;233:48–56. [CrossRef]
- Chang C, Lin H. Dysbiosis in gastrointestinal disorders. Best Pract Res Clin Gastroenterol 2016;30:3–15. [CrossRef]
- Angelucci F, Cechova K, Amlerova J, Hort J. Antibiotics, gut microbiota, and Alzheimer’s disease. J Neuroinflammation 2019;16:108. [CrossRef]
- Wang B, Yao M, Lv L, Ling Z, Li L. The Human Microbiota in Health and Disease. Engineering 2017;3:71–82. [CrossRef]
- Sochocka M, Donskow-Łysoniewska K, Diniz BS, Kurpas D, Brzozowska E, Leszek J. The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease—a Critical Review. Mol Neurobiol 2019;56:1841–51. [CrossRef]
- Pluta R, Ułamek-Kozioł M, Januszewski S, Czuczwar SJ. Gut microbiota and pro/prebiotics in Alzheimer’s disease. Aging 2020;12:5539–50. [CrossRef]
- Kim H soo, Kim S, Shin SJ, Park YH, Nam Y, Kim C won, et al. Gram-negative bacteria and their lipopolysaccharides in Alzheimer’s disease: pathologic roles and therapeutic implications. Transl Neurodegener 2021;10. [CrossRef]
- Cortés-Albornoz MC, García-Guáqueta DP, Velez-Van-meerbeke A, Talero-Gutiérrez C. Maternal nutrition and neurodevelopment: A scoping review. Nutrients 2021;13. [CrossRef]
- Baltrusch S. The Role of Neurotropic B Vitamins in Nerve Regeneration. Biomed Res Int 2021;2021. [CrossRef]
- Lauer AA, Grimm HS, Apel B, Golobrodska N, Kruse L, Ratanski E, et al. Mechanistic Link between Vitamin B12 and Alzheimer’s Disease. Biomolecules 2022;12. [CrossRef]
- Popescu A, German M. Vitamin K2 holds promise for Alzheimer’s prevention and treatment. Nutrients 2021;13. [CrossRef]
- Zhang Y, Wang X, Li H, Ni C, Du Z, Yan F. Human oral microbiota and its modulation for oral health. Biomedicine and Pharmacotherapy 2018;99:883–93. [CrossRef]
- DIbello V, Lozupone M, Manfredini D, DIbello A, Zupo R, Sardone R, et al. Oral frailty and neurodegeneration in Alzheimer’s disease. Neural Regen Res 2021;16:2149–53. [CrossRef]
- Sureda A, Daglia M, Argüelles Castilla S, Sanadgol N, Fazel Nabavi S, Khan H, et al. Oral microbiota and Alzheimer’s disease: Do all roads lead to Rome? Pharmacol Res 2020;151. [CrossRef]
- Bello-Corral L, Alves-Gomes L, Fernández-Fernández JA, Fernández-García D, Casado-Verdejo I, Sánchez-Valdeón L. Implications of gut and oral microbiota in neuroinflammatory responses in Alzheimer’s disease. Life Sci 2023;333. [CrossRef]
- Narengaowa, Kong W, Lan F, Awan UF, Qing H, Ni J. The Oral-Gut-Brain AXIS: The Influence of Microbes in Alzheimer’s Disease. Front Cell Neurosci 2021;15. [CrossRef]
- Orr ME, Reveles KR, Yeh CK, Young EH, Han X. Can oral health and oral-derived biospecimens predict progression of dementia? Oral Dis 2020;26:249–58. [CrossRef]
- Dioguardi M, Crincoli V, Laino L, Alovisi M, Sovereto D, Mastrangelo F, et al. The role of periodontitis and periodontal bacteria in the onset and progression of alzheimerʹs disease: A systematic review. J Clin Med 2020;9. [CrossRef]
- Bui FQ, Almeida-da-Silva CLC, Huynh B, Trinh A, Liu J, Woodward J, et al. Association between periodontal pathogens and systemic disease. Biomed J 2019;42:27–35. [CrossRef]
- Sedghi LM, Bacino M, Kapila YL. Periodontal Disease: The Good, The Bad, and The Unknown. Front Cell Infect Microbiol 2021;11. [CrossRef]
- Mysak J, Podzimek S, Duskova J. Porphyromonas gingivalis: Major Periodontopathic Pathogen Overview. 2014.
- Franciotti R, Pignatelli P, Carrarini C, Romei FM, Mastrippolito M, Gentile A, et al. Exploring the connection between porphyromonas gingivalis and neurodegenerative diseases: A pilot quantitative study on the bacterium abundance in oral cavity and the amount of antibodies in serum. Biomolecules 2021;11. [CrossRef]
- Chen CK, Wu YT, Chang YC. Association between chronic periodontitis and the risk of Alzheimer’s disease: A retrospective, population-based, matched-cohort study. Alzheimers Res Ther 2017;9. [CrossRef]
- Kim SR, Son M, Kim YR, Kang HK. Risk of dementia according to the severity of chronic periodontitis in Korea: a nationwide retrospective cohort study. Epidemiol Health 2022;44. [CrossRef]
- Ryder MI. Porphyromonas gingivalis and Alzheimer disease: Recent findings and potential therapies. J Periodontol 2020;91:S45–9. [CrossRef]
- Zhang Z, Liu D, Pan Y. The Role of Porphyromonas gingivalis Outer Membrane Vesicles in Periodontal Disease and Related Systemic Diseases. n.d.
- Narengaowa, Kong W, Lan F, Awan UF, Qing H, Ni J. The Oral-Gut-Brain AXIS: The Influence of Microbes in Alzheimer’s Disease. Front Cell Neurosci 2021;15. [CrossRef]
- Sansores-España LD, Melgar-Rodríguez S, Olivares-Sagredo K, Cafferata EA, Martínez-Aguilar VM, Vernal R, et al. Oral-Gut-Brain Axis in Experimental Models of Periodontitis: Associating Gut Dysbiosis With Neurodegenerative Diseases. Frontiers in Aging 2021;2. [CrossRef]
- Verma A, Azhar G, Zhang X, Patyal P, Kc G, Sharma S, et al. P. gingivalis-LPS Induces Mitochondrial Dysfunction Mediated by Neuroinflammation through Oxidative Stress. Int J Mol Sci 2023;24:950. [CrossRef]
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. 2019.
- Cox LM, Weiner HL. Microbiota Signaling Pathways that Influence Neurologic Disease. Neurotherapeutics 2018;15:135–45. [CrossRef]
- Weber C, Dilthey A, Finzer P. The role of microbiome-host interactions in the development of Alzheimer’s disease. Front Cell Infect Microbiol 2023;13. [CrossRef]
- Cook J, Prinz M. Regulation of microglial physiology by the microbiota. Gut Microbes 2022;14. [CrossRef]
- Cherny I, Rockah L, Levy-Nissenbaum O, Gophna U, Ron EZ, Gazit E. The formation of Escherichia coli curli amyloid fibrils is mediated by prion-like peptide repeats. J Mol Biol 2005;352:245–52. [CrossRef]
- Smith DR, Price JE, Burby PE, Blanco LP, Chamberlain J, Chapman MR. The production of curli amyloid fibers is deeply integrated into the biology of escherichia coli. Biomolecules 2017;7. [CrossRef]
- Das TK, Blasco-Conesa MP, Korf J, Honarpisheh P, Chapman MR, Ganesh BP. Bacterial Amyloid Curli Associated Gut Epithelial Neuroendocrine Activation Predominantly Observed in Alzheimer’s Disease Mice with Central Amyloid-β Pathology. Journal of Alzheimer’s Disease 2022;88:191–205. [CrossRef]
- Dua P ZY. Microbial Sources of Amyloid and Relevance to Amyloidogenesis and AlzheimerÂ’s Disease (AD). J Alzheimers Dis Parkinsonism 2015;05. [CrossRef]
- Zhao Y, Jaber V, Lukiw WJ. Secretory products of the human GI tract microbiome and their potential impact on Alzheimer’s disease (AD): Detection of lipopolysaccharide (LPS) in AD hippocampus. Front Cell Infect Microbiol 2017;7. [CrossRef]
- Zhou Y, Smith D, Leong BJ, Brännström K, Almqvist F, Chapman MR. Promiscuous cross-seeding between bacterial amyloids promotes interspecies biofilms. Journal of Biological Chemistry 2012;287:35092–103. [CrossRef]
- R. Deane. RAGE mediates amyloid-β peptide transport across theblood-brain barrier and accumulation in brain 2003.
- Vargas-Caraveo A, Sayd A, Maus SR, Caso JR, Madrigal JLM, García-Bueno B, et al. Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci Rep 2017;7. [CrossRef]
- Kim W-G, Mohney RP, Wilson B, Jeohn G-H, Liu B, Hong J-S. Regional Difference in Susceptibility to Lipopolysaccharide-Induced Neurotoxicity in the Rat Brain: Role of Microglia. 2000.
- Zhao Y, Cong L, Jaber V, Lukiw WJ. Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front Immunol 2017;8. [CrossRef]
- Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep 2019;9. [CrossRef]
- Zhao Y, Jaber VR, Pogue AI, Sharfman NM, Taylor C, Lukiw WJ. Lipopolysaccharides (LPSs) as Potent Neurotoxic Glycolipids in Alzheimer’s Disease (AD). Int J Mol Sci 2022;23. [CrossRef]
- Kantarci A, Tognoni CM, Yaghmoor W, Marghalani A, Stephens D, Ahn JY, et al. Microglial response to experimental periodontitis in a murine model of Alzheimer’s disease. Sci Rep 2020;10. [CrossRef]
- Fakhoury M. Microglia and astrocytes in Alzheimer’s disease: implications for therapy. Curr Neuropharmacol 2017;15. [CrossRef]
- di Penta A, Moreno B, Reix S, Fernandez-Diez B, Villanueva M, Errea O, et al. Oxidative Stress and Proinflammatory Cytokines Contribute to Demyelination and Axonal Damage in a Cerebellar Culture Model of Neuroinflammation. PLoS One 2013;8. [CrossRef]
- Zhou J, Windsor LJ. Porphyromonas gingivalis affects host collagen degradation by affecting expression, activation, and inhibition of matrix metalloproteinases. J Periodontal Res 2006;41:47–54. [CrossRef]
- Andrian E, Mostefaoui Y, Rouabhia M, Grenier D. Regulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases by porphyromonas gingivalis in an engineered human oral mucosa model. J Cell Physiol 2007;211:56–62. [CrossRef]
- Wolf AJ, Underhill DM. Peptidoglycan recognition by the innate immune system. Nat Rev Immunol 2018;18:243–54. [CrossRef]
- Arentsen T, Qian Y, Gkotzis S, Femenia T, Wang T, Udekwu K, et al. The bacterial peptidoglycan-sensing molecule Pglyrp2 modulates brain development and behavior. Mol Psychiatry 2017;22:257–66. [CrossRef]
- Gabanyi I, Lepousez G, Wheeler R, Vieites-Prado A, Nissant A, Wagner S, et al. Bacterial sensing via neuronal Nod2 regulates appetite and body temperature n.d. [CrossRef]
- Halliwell B. Oxidative stress and neurodegeneration: Where are we now? J Neurochem 2006;97:1634–58. [CrossRef]
- Gupta SK, Vyavahare S, Duchesne Blanes IL, Berger F, Isales C, Fulzele S. Microbiota-derived tryptophan metabolism: Impacts on health, aging, and disease. Exp Gerontol 2023;183. [CrossRef]
- Maitre M, Klein C, Patte-Mensah C, Mensah-Nyagan AG. Tryptophan metabolites modify brain Aβ peptide degradation: A role in Alzheimer’s disease? Prog Neurobiol 2020;190. [CrossRef]
- Sun DS, Gao LF, Jin L, Wu H, Wang Q, Zhou Y, et al. Fluoxetine administration during adolescence attenuates cognitive and synaptic deficits in adult 3×TgAD mice. Neuropharmacology 2017;126:200–12. [CrossRef]
- John A. Hardy. Alzheimer’s Disease: The Amyloid Cascade Hypothesis | Science 1992.
- Takasugi N, Komai M, Kaneshiro N, Ikeda A, Kamikubo Y, Uehara T. The Pursuit of the “Inside” of the Amyloid Hypothesis—Is C99 a Promising Therapeutic Target for Alzheimer’s Disease? Cells 2023;12. [CrossRef]
- Rudge JDA. A New Hypothesis for Alzheimer’s Disease: The Lipid Invasion Model. J Alzheimers Dis Rep 2022;6:129–61. [CrossRef]
- Sperling RA, Rentz DM, Johnson KA, Karlawish J, Donohue M, Salmon DP, et al. The A4 study: Stopping AD before symptoms begin? Sci Transl Med 2014;6. [CrossRef]
- Javitt DC, Martinez A, Sehatpour P, Beloborodova A, Habeck C, Gazes Y, et al. Disruption of early visual processing in amyloid-positive healthy individuals and mild cognitive impairment. Alzheimers Res Ther 2023;15. [CrossRef]
- O’Connor A, Weston PSJ, Pavisic IM, Ryan NS, Collins JD, Lu K, et al. Quantitative detection and staging of presymptomatic cognitive decline in familial Alzheimer’s disease: A retrospective cohort analysis. Alzheimers Res Ther 2020;12. [CrossRef]
- Elliott P Vichinsky. Iron chelators: Choice of agent, dosing, and adverse effects. 2024.
- Schreiner OD, Schreiner TG. Iron chelators as a therapeutic option for Alzheimer’s disease—A mini-review. Frontiers in Aging 2023;4. [CrossRef]
- Nuñez MT, Chana-Cuevas P. New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 2018;11. [CrossRef]
- Pereira FC, Ge X, Kristensen JM, Kirkegaard RH, Maritsch K, Szamosvári D, et al. The Parkinson’s disease drug entacapone disrupts gut microbiome homeostasis via iron sequestration. Nat Microbiol 2024;9:3165–83. [CrossRef]
- Naomi R, Embong H, Othman F, Ghazi HF, Maruthey N, Bahari H. Probiotics for alzheimer’s disease: A systematic review. Nutrients 2022;14. [CrossRef]
- Cheng Y, Liu J, Ling Z. Short-chain fatty acids-producing probiotics: A novel source of psychobiotics. Crit Rev Food Sci Nutr 2022;62:7929–59. [CrossRef]
- Plaza-Diaz J, Ruiz-Ojeda FJ, Gil-Campos M, Gil A. Mechanisms of Action of Probiotics. Advances in Nutrition, vol. 10, Oxford University Press; 2019, p. S49–66. [CrossRef]
- Cristofori F, Dargenio VN, Dargenio C, Miniello VL, Barone M, Francavilla R. Anti-Inflammatory and Immunomodulatory Effects of Probiotics in Gut Inflammation: A Door to the Body. Front Immunol 2021;12. [CrossRef]
- Nagpal R, Wang S, Ahmadi S, Hayes J, Gagliano J, Subashchandrabose S, et al. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci Rep 2018;8. [CrossRef]
- Angelucci F, Cechova K, Amlerova J, Hort J. Antibiotics, gut microbiota, and Alzheimer’s disease. J Neuroinflammation 2019;16. [CrossRef]
- Angelucci F, Cechova K, Amlerova J, Hort J. Antibiotics, gut microbiota, and Alzheimer’s disease. J Neuroinflammation 2019;16. [CrossRef]
- Elangovan S, Borody TJ, Holsinger RMD. Fecal Microbiota Transplantation Reduces Pathology and Improves Cognition in a Mouse Model of Alzheimer’s Disease. Cells 2023;12. [CrossRef]
- Sun J, Xu J, Ling Y, Wang F, Gong T, Yang C, et al. Fecal microbiota transplantation alleviated Alzheimer’s disease-like pathogenesis in APP/PS1 transgenic mice. Transl Psychiatry 2019;9. [CrossRef]
- Nassar ST, Tasha T, Desai A, Bajgain A, Ali A, Dutta C, et al. Fecal Microbiota Transplantation Role in the Treatment of Alzheimer’s Disease: A Systematic Review. Cureus 2022. [CrossRef]
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 2019;5. [CrossRef]
- Nguyen S V., Nguyen MTH, Tran BC, Ho MTQ, Umeda K, Rahman S. Evaluation of lozenges containing egg yolk antibody against porphyromonas gingivalis gingipains as an adjunct to conventional non-surgical therapy in periodontitis patients: A randomized controlled clinical trial. J Periodontol 2018;89:1334–9. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



