
Submitted:
09 December 2024
Posted:
11 December 2024
You are already at the latest version
Abstract
Congenital mesoblastic nephroma represents 3 - 10 % of all pediatric renal tumors. With the advancement of ultrasound diagnostics and magnetic resonance imaging, the diagnosis of this renal neoplasm is increasingly being established prenatally and at birth. It usually presents as a benign tumor, but it can severely affect pregnancy outcomes, contributing to serious complications, perinatal morbidity and mortality. Lissencephaly belongs to a rare category of neurodevelopmental disorders marked by the absence of substantial reduction of the typical folds and grooves in the cerebral cortex. The prognosis for patients with lissencephaly is extremely poor, carryng with it a high mortality rate. The aim of this study is to point out the characteristics and unique correlation between CMN and lissencephaly, and to illustrate the histopathological features of CMN and lissencephaly through an educational example derived from our presented index case. To the best to our knowledge, the association of CMN with lissencephaly has not been decribed in the literature so far. Outlining the prenatal progression of CMN and the outcome of pregnancies involving fetal CMN and lissencephaly this case underscores the importance of comprenhesive ultrasound examionations, including central nervous system evaluation, to identify potenial coexisting anomalies and refine prenatal diagnostic practices.
Keywords:
congenital mesoblastic nephroma
; lissencephaly
; pachygyria
; prenatal course
1. Introduction
Renal tumors present at birth are uncommon in general. Among them, the most common are congenital mesoblastic nephroma (CMN), nephroblastoma (Wilms tumor), rhabdoid tumor, and clear cell sarcoma. CMN represents 3 – 10% of all pediatric renal tumors and is mostly diagnosed within the first three months of life [1,2,3]. However, with the advancement of ultrasound diagnostics and magnetic resonance imaging, the diagnosis of this renal neoplasm is increasingly being established prenatally and at birth [1,4].
Ultrasound is the primary tool for the prenatal diagnosis of CMN and magnetic resonance imaging (MRI) can aid in assessing the origin and morphological characteristics of a fetal abdominal mass [5,6]. Diagnosing CMN before the third trimester of pregnancy is exceptionally challenging, and it is most commonly established in the third or late second trimester. Polyhydramnios emerges as the primary clinical indication in the majority of cases, prompting a comprehensive ultrasound examination during the later part of pregnancy [6,7].
Histopathologically, CMN is classified into three types: classic, cellular, and mixed. It typically exhibits characteristics of a benign tumor and is most effectively managed through surgical removal in neonates [1,2,4]. However, fetal CMN significantly impacts pregnancy outcomes and contributes to perinatal morbidity and mortality, carrying potentially serious complications such as polyhydramnios, premature rupture of membranes and preterm labor [2,4,7]. Early diagnosis during pregnancy is crucial, as it plays a vital role in shaping the antenatal plan and can influence the management strategy.
Lissencephaly belongs to a rare category of neurodevelopmental disorders marked by the absence or substantial reduction of the typical folds and grooves in the cerebral cortex, leading to an unusually smooth surface [8]. It results from a defective neuronal migration happening around 12 – 20 weeks of gestation [9]. The prenatal diagnosis of lissencephaly can be achieved through ultrasound and MRI in the early second trimester [10]. The outlook for patients diagnosed with lissencephaly is quite poor, as a significant proportion do not survive long after birth, or experience postnatal difficulties in daily activities and failure to thrive [9].
We present a case of congenital mesoblastic nephroma (CMN) diagnosed with polyhydramnios at 28 weeks of gestation, which led to preterm delivery at 29 weeks and a fatal outcome for the newborn. Histopathological examination confirmed the diagnosis of CMN along with fetal pachygyria/lissencephaly. To our knowledge, the association of CMN with lissencephaly has not been previously described in the literature. The aim of this study is to point out the distinctive and singular correlation between CMN and lissencephaly, illustrating the histopathological features of CMN and lissencephaly.
2. Case Report
2.1. Clinical Presentation
A 22-year-old female patient was admitted as an emergency case to the Clinic of Gynecology and Obstetrics at the University Clinical Center of Serbia in the 28th week of her first pregnancy, which was spontaneously conceived, due to a significantly increased amount of amniotic fluid. Initial ultrasound examination confirmed fetal vitality and appropriate fetal biometrics for gestational age, as well as an increased amount of amniotic fluid (Amniotic Fluid Index 170 mm). A tumor-like hyperechoic formation in the fetal abdomen, measuring 58 x 50 mm, was noted but could not be reliably identified (Figure 1).
No fetal breathing or limb movements were detected and end-diastolic block of the umbilical artery was observed. Amnioreduction was performed, evacuating 2000 mL of amniotic fluid. Microbiological and biochemical analysis of the amniotic fluid was normal. During the course of hospitalization, at 29 weeks of gestation, premature rupture of the fetal membranes and spontaneous initiation of labor occurred, resulting in the birth of a live male newborn, weighing 870 g, with an Apgar score of 3 at 5 minutes. Histopathological examination of the placenta revealed chronic multifocal villitis, acute purulent chorioamnionitis, and subchorionic thrombosis.
After admission to the Neonatal Intensive Care Unit (NICU), the newborn was transferred to the University Children's Clinic (UCC) for further diagnosis and treatment. The abdomen was markedly protruding above the chest level with a palpable firm swelling in the central and right parts of the abdomen. An ultrasound examination revealed a large intraabdominal tumor mass in the right renal fossa, measuring approximately 63 x 50 x 47 mm. The mass was well-delineated from the liver, highly heterogeneous, well-vascularized, with a striated zone of calcifications, and it compressed the inferior vena cava (IVC). The renal parenchyma and the right adrenal gland showed no differentiation. Other organs appeared normal. Although neurological findings were consistent with the gestational age, ultrasound examination of the central nervous system (CNS) revealed symmetric cerebral hemispheres with a completely agyric cortex and no dilation of the ventricular system. A subsequent ultrasound examination of the CNS indicated a normal gyral pattern with the presence of ventriculomegaly. From the third day of hospitalization, despite the administered therapy, the newborn's condition worsened, leading to pulmonary hemorrhage. Due to the patient's severe overall condition, only basic diagnostics of the renal tumor were performed, and surgical treatment was not an option. Despite all implemented measures, the patient remained hemodynamically unstable, with severe metabolic and respiratory acidosis, as well as bradycardia. Cardiopulmonary resuscitation efforts were unsuccessful, and the patient's death was confirmed on the fifth day of life.
2.2. Autopsy Findings
Through clinical autopsy, the pathoanatomical diagnosis is established: the right kidney is almost entirely altered, enlarged, replaced by tumor tissue that does not breach the renal capsule and does not infiltrate the adrenal gland. On cross-section, it has a yellowish color, interspersed with darker brownish areas (Figure 2).
The tumor of the right kidney was composed of uniform spindle cells arranged in clusters and fascicles, with indistinct intercellular boundaries. The tumor cells exibited oval vesicular nuclei and scant eosinophilic cytoplasm. The tumor almost completely replaced the normal renal parenchyma, trapping small islands of non-neoplastic renal tissue (Figure 3).
Immunohistochemically, the tumor cells exhibit diffuse vimentin and INI1 positivity along with focal positivity for WT1, and cyclin D1. The tumor cells were negative for SMA, CD34, bcl-2, ALK, and BCOR. The differential diagnosis considered several spindle cell tumors of the kidney, each with distinct morphological and immunohistochemical features. These morphological and immunohistochemical findings were consistent with a diagnosis of congenital mesoblastic nephroma. However, the differential diagnosis included several spindle cell tumors of the kidney that were carefully evaluated based on their distinct features. Metanephric stromal tumor (MST) was considered due to its spindle cell morphology, but it typically shows alternating nodular cellularity, angiodysplasia, and "onion skinning" around vessels, none of which were present. Additionally, MST is characteristically CD34-positive and may harbor a BRAF V600E mutation, whereas this tumor was CD34-negative. Stromal-type Wilms tumor was another consideration, as it can present as a spindle cell lesion, often containing rhabdomyoblasts or adipose tissue, and is associated with nephrogenic rests. However, these features were not observed in the current tumor, and BCL2, which is often expressed in Wilms tumor, was negative. Moreover, there was no evidence of epithelial or blastemal components upon detailed examination. Clear cell sarcoma of the kidney was excluded due to the absence of its characteristic arborizing vascular pattern and the lack of BCOR expression. Rhabdoid tumor of the kidney, a high-grade spindle cell tumor, was also considered, however, these tumors typically exhibit INI1 loss, whereas the current tumor demonstrated retained INI1 expression. Inflammatory myofibroblastic tumor, another potential differential diagnosis, was ruled out as it is typically ALK-positive, and the current tumor was ALK-negative. The lack of features characteristic of the other entities considered supports the final diagnosis (Figure 3).
Furthermore, the gross morphological findings suggested an anomaly of cerebral cortical folding, predominantly of the lissencephalic type (absence of gyri in the large brain), as illustrated on Figure 4, although the possibility of pachygyria (presence of a small number of wide gyri) could not be completely ruled out with certainty.
Moreover, intraventricular bleeding originating from the periventricular germinal matrix was also observed on gross (Figure 4), as well as on the microscopic slides analysis (Figure 5).
Sections taken from the cortex of the large brain did not reveal gyri (the cortex of the large brain was almost completely flat, with only occasional rudimentary sulci). The periventricular germinal matrix was well-developed, containing numerous merged fresh hemorrhages. Other organs showed appropriate morphology. The histopathological finding in the lungs indicated diffuse alveolar damage (Figure 5).
2.3. Genetic Testing
During the pregnancy, molecular genetic testing (QF-PCR) did not reveal any fetal numerical chromosomal aberrations. Exome sequencing of the DNA from the deceased newborn did not detect any gene mutations potentially causative for the condition, although this cannot be completely ruled out. Three partially relevant mutations were identified, classified as variants of unknown significance. These include heterozygous variants in the autosomal-dominant genes ARID2 and HIVEP2, as well as a hemizygous variant in the OFD1 gene. Although genetic indications suggested targeted testing of the mother for the OFD1 gene variant, given the potential correlation between the genotype and phenotype of the deceased newborn, the parents chose not to undergo genetic testing.
3. Discussion
Congenital mesoblastic nephroma was first described by Bolande and colleagues in 1967. as the most common renal neoplasm in newborns and early childhood [11]. Histopathologically, it is classified into three types: classic, cellular, and mixed types. The classic form closely resembles leiomyoma, primarily characterized as a solid, firm mass with a yellowish hue, lacking a capsule, featuring indistinct margins and comprising spindle cells in a fusiform arrangement with occasional mitoses. In contrast, the cellular variant, histologically identical to infantile fibrosarcoma, exhibits elevated cell density, increased mitotic activity, necrosis and hemorrhage. It consists of solid ovoid or fusiform spindle cells with reduced cytoplasm [1,4]. Classic type is generally considered a benign tumor. The potential to invade neighboring structures, increased aggressiveness, recurrence, metastasis and poorer prognosis are associated with the cellular and mixed types, with a survival rate of 85% when compared to 100% for the classic variant [12].
Ultrasound is the primary tool for prenatal diagnosis of CMN. The tumors exhibit solid, unilateral, varied to uniform echogenicity, well-defined boundaries, significant vascularity [5]. Color Doppler flow can be used to highlight a concentric hyperechoic and hypoechoic ring pattern, known as the vascular ‘ring’ sign, running along the tumor border [6,13,14]. It can displace the abdominal aorta and also move the bowel loops to the right [15]. The cellular type of CMN can be identified in ultrasound and CT scans by its features of cystic areas, hemorrhage, necrosis, and calcification. In contrast, the classic CMN typically presented as a predominantly solid mass [1]. The most prevalent ultrasound finding is polyhydramnios. Specifically, polyhydramnios can be diagnosed several days and even weeks before the detection of the fetal renal mass, making it a potential first clinical manifestation, prompting a comprehensive ultrasound examination during the latter part of pregnancy (6,14,15). Polyhydramnios arises from polyuria resulting from elevated renal perfusion and hypercalcemia influenced by the prostaglandins of the kidney tumor. Additionally, it is associated with bowel obstruction induced by the pressure exerted by the renal mass and almost occupying the abdominal cavity [6]. Managing polyhydramnios is crucial to prevent premature rupture of the membranes [7]. There have been documented occurrences where CMN appears alongside a typical level of amniotic fluid [1,16]. However, particular attention is warranted in cases of CMN associated with oligohydramnios [17]. CMN may exhibit significant vascularity and be connected to high-output cardiac failure, resulting in hydrops fetalis, serious indication of fetal mortality outcome [6].
Characteristic of CMN and simultaneously a challenge for clinicians in prenatal diagnosis, is the fact that it is rarely detected during routine second-trimester anomaly screening. Cases have been described where the ultrasound findings were normal from the 20th to the 32nd week of gestation, with CMN being diagnosed later in the course of pregnancy [6,13,14,17]. The diagnosis is typically established in the third trimester, with an average gestational age of 32 weeks. However, it should be noted that the diagnosis of CMN is most commonly not made prenatally but rather during infancy, with a median age at diagnosis ranging from 0 days (newborn) to 2.9 months [2]. The primary symptoms observed at the time of diagnosis include the emergence of an abdominal mass, along with hematuria, hypertension, polyuria and hypercalcemia [1,2,18,19]. In support of how challenging it can be to establish an accurate diagnosis is the fact that even postnatally, based on ultrasound examination, CMN can be misdiagnosed as a hematoma [20]. In contrast to ultrasound, fetal MRI offers superior soft tissue contrast and various imaging planes, exceling in detecting fetal anomalies, delineating the tumor's relationship with adjacent structures and identifying the tumor's origin, size and characteristics. Consequently, MRI serves as a valuable adjunct to ultrasound in the prenatal diagnosis of CMN and confirmes the ultrasound findings without altering the management plan [6,13,14,15]. Computed Tomography (CT) serves as the primary diagnostic method for identifying kidney tumors and assessing possible metastases.
Utilizing CT during pregnancy poses the risk of fetal exposure to radiation, making it an ideal strategy for postnatal tumor evaluation [1,21,22,23,24].
The most commonly reported genetic aberrations in CMN are trisomy 11 and t(12;15)(p13;q25), leading to a fusion of the genes ETV6 and NTRK3, exclusively observed in mixed and cellular type CMN and never in the classic form [2,25]. ETV6 is identified as an oncogene in various leukemias and myeloproliferative syndromes, functioning as a transcription factor. In contrast, NTRK3 belongs to the neurotrophic tyrosine kinase receptor (NTRK) family [26]. The presence of the ETV6-NTRK3 chimeric oncoprotein has demonstrated oncogenicity in various tumor types, including mammary analog secretory carcinoma, secretory breast carcinoma and papillary thyroid carcinoma [27,28,29]. There is optimism regarding the potential utility of targeted therapy for NTRK-positive congenital mesoblastic nephroma in the neoadjuvant treatment of recurrent or metastatic congenital mesoblastic nephroma [30]. Other recurrently reported genetic aberrations in mixed and cellular type CMN include trisomies 8, 17, 20, 7, 18, and 9 [2].
Congenital mesoblastic nephroma is considered tumor with a favorable prognosis by nature. However, serious complications are frequently described with CMN, including polyhydramnios, hydrops fetalis and spontaneous premature rupture of membranes resulting in preterm birth. These complications are strongly associated with adverse outcomes in newborns, such as respiratory distress syndrome, necrotizing enterocolitis, intraventricular hemorrhage, neonatal hypertension and the development of metastases [4,6,24]. Surgery constitutes the primary therapeutic intervention and neonates with live birth demonstrate the capability to lead a healthy life without necessitating adjuvant therapy [1,2,4,6,7].
When it comes to lissencephaly the term is derived from the Greek words "lissos," meaning smooth, and "enkephalos," meaning brain. It comes with an estimated prevalence of 1 in 100,000 births. Embryologically it results from a defective neuronal migration along with more complex and subtle anomalies affecting cell proliferation and differentiation, neurite outgrowth, axonal pathfinding transport, connectivity and myelination [8,9]. It has been divided into two categories: classic lissencephaly (type 1 lissencephaly or agyria-pachygyria) and cobblestone complex (type 2 lissencephaly). So far, over 19 genes associated with lissencephaly have been identified. The most common of them are LIS1, DCX, TUBA1A, RELN, VLDLR, ARX, WDR62 and the PAFAH1B1 gene [8,9]. The functions of certain lissencephaly genes, including LIS1, DCX, and TUBA1A, are closely associated with microtubules which, as integral components of the cytoskeleton, play crucial roles in cellular processes such as mitosis and cytokinesis. The prenatal diagnosis of lissencephaly can be achieved through ultrasound and magnetic resonance imaging in the early second trimester, revealing abnormal development of sulci and gyri [10]. Prenatal ultrasonography features commonly involve commissural anomalies, ventriculomegaly, hydrocephalus, asymmetric ventricles, cerebellar vermian or hemispheric anomalies, abnormal head circumference [10]. MRI can detect fetal lissencephaly between 20 and 24 weeks of gestation. However, false positives are possible, especially during the second trimester [31]. The prognosis for individuals with lissencephaly is notably grim, as a significant proportion do not survive long after birth, or experience postnatal failure to thrive. Upon searching the literature, we did not encounter a case similar to ours, neither when considering a potential genetic factor nor an association with a similar condition. However, in a 2019. study a case of CMN was described where signs of microcephaly, hypotonia and developmental delay were noted at 6 months of age. An MRI of the brain performed at this age revealed cortical and subcortical atrophy, indicative of prolonged hypoxia. By the age of 1, the clinical condition was characterized by severe microcephaly, right-sided tetraparesis, global hypotonia and developmental delay [4].
4. Conclusions
Congenital mesoblastic nephroma (CMN) often manifests in late pregnancy, characterized by rapid tumor growth, as observed in our case. The substantial tumor size at diagnosis may result from its echogenic similarity to normal renal pyramids, complicating differentiation during prenatal ultrasound. The tumor's proximity to normal parenchyma, absence of a distinct capsule in smaller masses, and blending with surrounding kidney tissue further obscure detection. Early-stage tumors are often undetectable due to their small size, but abnormal renal enlargement or asymmetry warrants careful examination of the kidneys, adrenal glands, and contralateral kidney. While detailed ultrasound remains the gold standard for CMN diagnosis, MRI can provide additional clarity in cases of diagnostic uncertainty. Prenatal detection of CMN is crucial for optimal management, improved outcomes and informed parental counseling. Emphasis should be placed on thorough evaluation of the fetal urinary tract, especially in polyhydramnios cases requiring close monitoring to prevent preterm birth. Although genetic testing in our case did not confirm a specific genetic condition, targeted testing of the OFD1 gene in the mother is warranted due to potential genotype-phenotype correlations with the deceased newborn. Our investigation did not identify a genetic link between CMN and lissencephaly, a previously unreported association. This case underscores the importance of comprehensive ultrasound examinations, including central nervous system evaluation, to identify potential coexisting anomalies and refine prenatal diagnostic practices.
Author Contributions
Conceptualization, H.Z., O.K.V., J.S.; and M.Ž; Writing—Original Draft Preparation, H.Z., M.Ž., J.S.; Writing—Review and Editing, H.Z., O.K.V., J.S.; J.J., M.P.M., M.Ž.; Visualization, H.Z., O.K.V., J.S.; J.J., M.P.M., M.Ž.; supervision, J.S.J. and M.Ž. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Ministry of Science, Technological Development and Innovation (MSTDI) of the Republic of Serbia (Grant No. 451-03-66/2024-03/200110) and the Science Fund of the Republic of Serbia (SFRS), as the funding sources, had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Institutional Review Board Statement
The patient consented to the publication of this anonymized case report, and her consent has been formally documented. As per our institutional guidance, there is no requirement to submit this for ethical review.
Informed Consent Statement
Written informed consent has been obtained from the patient to publish this paper.
Data Availability Statement
The original contributions presented in this report are included in the article. Further enquiries can be directed to the corresponding authors.
Acknowledgments
This research was supported by the Ministry of Science, Technological Development and Innovation (MSTDI) of the Republic of Serbia (Grant No. 451-03-66/2024-03/200110) and the Science Fund of the Republic of Serbia (SFRS), as the funding sources, had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, Z.P.; LI, K.; Dong, K.R.; Xiao, X.M.; Zheng, S. Congenital Mesoblastic Nephroma: Clinical Analysis of Eight Cases and a Review of the Literature. Oncol Lett 2014, 8, 2007–2011. [Google Scholar] [CrossRef]
- Gooskens, S.L.; Houwing, M.E.; Vujanic, G.M.; Dome, J.S.; Diertens, T.; Coulomb-l’Herminé, A.; Godzinski, J.; Pritchard-Jones, K.; Graf, N.; van den Heuvel-Eibrink, M.M. Congenital Mesoblastic Nephroma 50 Years after Its Recognition: A Narrative Review. Pediatr Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Whittle, S.; Gosain, A.; Scott Brown, P.Y.; Debelenko, L.; Raimondi, S.; Wilimas, J.A.; Jenkins, J.J.; Davidoff, A.M. Regression of a Congenital Mesoblastic Nephroma. Pediatr Blood Cancer 2010, 55, 364–368. [Google Scholar] [CrossRef]
- Soyaltın, E.; Alaygut, D.; Alparslan, C.; Özdemir, T.; Çamlar, S.A.-; Mutlubaş, F.; Kasap-Demir, B.; Yavaşcan, Ö. A Rare Cause of Neonatal Hypertension: Congenital Mesoblastic Nephroma. Turk J Pediatr 2018, 60, 198–200. [Google Scholar] [CrossRef]
- Won, H.S.; Jung, E.; Lee, P.R.; Lee, I.S.; Kim, A.; Kim, J.K.; Cho, K.S.; Nam, J.H. Prenatal Detection of Mesoblastic Nephroma by Sonography and Magnetic Resonance Imaging. Ultrasound Obstet Gynecol 2002, 19, 197–199. [Google Scholar] [CrossRef]
- Tongsong, T.; Palangmonthip, W.; Chankhunaphas, W.; Luewan, S. Prenatal Course and Sonographic Features of Congenital Mesoblastic Nephroma. Diagnostics (Basel) 2022, 12, 1951. [Google Scholar] [CrossRef]
- Goldstein, I.; Shoshani, G.; Ben-Harus, E.; Sujov, P. Prenatal Diagnosis of Congenital Mesoblastic Nephroma. Ultrasound Obstet Gynecol 2002, 19, 209–211. [Google Scholar] [CrossRef]
- Spalice, A.; Parisi, P.; Nicita, F.; Pizzardi, G.; Del Balzo, F.; Iannetti, P. Neuronal Migration Disorders: Clinical, Neuroradiologic and Genetics Aspects. Acta Paediatr 2009, 98, 421–433. [Google Scholar] [CrossRef]
- Friocourt, G.; Marcorelles, P.; Saugier-Veber, P.; Quille, M.-L.; Marret, S.; Laquerrière, A. Role of Cytoskeletal Abnormalities in the Neuropathology and Pathophysiology of Type I Lissencephaly. Acta Neuropathol 2011, 121, 149–170. [Google Scholar] [CrossRef]
- Tonni, G.; Pattacini, P.; Bonasoni, M.P.; Araujo Júnior, E. Prenatal Diagnosis of Lissencephaly Type 2 Using Three-Dimensional Ultrasound and Fetal MRI: Case Report and Review of the Literature. Rev Bras Ginecol Obstet 2016, 38, 201–206. [Google Scholar] [CrossRef]
- Bolande, R.P.; Brough, A.J.; Izant, R.J. Congenital Mesoblastic Nephroma of Infancy. A Report of Eight Cases and the Relationship to Wilms’ Tumor. Pediatrics 1967, 40, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Furtwaengler, R.; Reinhard, H.; Leuschner, I.; Schenk, J.P.; Goebel, U.; Claviez, A.; Kulozik, A.; Zoubek, A.; von Schweinitz, D.; Graf, N.; et al. Mesoblastic Nephroma--a Report from the Gesellschaft Fur Pädiatrische Onkologie Und Hämatologie (GPOH). Cancer 2006, 106, 2275–2283. [Google Scholar] [CrossRef] [PubMed]
- Mata, R.P.; Alves, T.; Figueiredo, A.; Santos, A. Prenatal Diagnosis of Congenital Mesoblastic Nephroma: A Case with Poor Prognosis. BMJ Case Rep 2019, 12, e230297. [Google Scholar] [CrossRef]
- Che, M.; Yang, F.; Huang, H.; Zhang, H.; Han, C.; Sun, N. Prenatal Diagnosis of Fetal Congenital Mesoblastic Nephroma by Ultrasonography Combined with MR Imaging. Medicine (Baltimore) 2021, 100, e24034. [Google Scholar] [CrossRef]
- Ko, S.; Kim, M.-J.; Im, Y.-J.; Park, K.-I.; Lee, M.-J. Cellular Mesoblastic Nephroma with Liver Metastasis in a Neonate: Prenatal and Postnatal Diffusion-Weighted MR Imaging. Korean J Radiol 2013, 14, 361–365. [Google Scholar] [CrossRef]
- Schild, R.L.; Plath, H.; Hofstaetter, C.; Hansmann, M. Diagnosis of a Fetal Mesoblastic Nephroma by 3D-Ultrasound. Ultrasound Obstet Gynecol 2000, 15, 533–536. [Google Scholar] [CrossRef]
- Kim, C.H.; Kim, Y.H.; Cho, M.K.; Kim, K.M.; Ha, J.A.; Joo, E.H.; Kim, S.M.; Song, T.-B. A Case of Fetal Congenital Mesoblastic Nephroma with Oligohydramnios. J Korean Med Sci 2007, 22, 357–361. [Google Scholar] [CrossRef]
- Santos, L.G. dos; Carvalho, J. de S.R. de; Reis, M.A.; Sales, R.L.J.B. Cellular Congenital Mesoblastic Nephroma: Case Report. J Bras Nefrol 2011, 33, 109–112. [Google Scholar] [CrossRef]
- Robertson-Bell, T.; Newberry, D.M.; Jnah, A.J.; DeMeo, S.D. Congenital Mesoblastic Nephroma Presenting With Refractory Hypertension in a Premature Neonate: A Case Study. Neonatal Netw 2017, 36, 32–39. [Google Scholar] [CrossRef]
- Anakievski, D.; Ivanova, P.; Kitanova, M. Laparoscopic Management of Congenital Mesoblastic Nephroma- Case Report. Urol Case Rep 2019, 26, 100979. [Google Scholar] [CrossRef]
- Do, A.Y.; Kim, J.-S.; Choi, S.-J.; Oh, S.-Y.; Roh, C.-R.; Kim, J.-H. Prenatal Diagnosis of Congenital Mesoblastic Nephroma. Obstet Gynecol Sci 2015, 58, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Simkhada, A.; Paudel, R.; Sharma, N. Congenital Mesoblastic Nephroma: A Case Report. JNMA J Nepal Med Assoc 2023, 61, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Al-Kzayer, L.F.Y.; Sarsam, S.N.; Chen, L.; Saeed, R.M.; Ali, K.H.; Nakazawa, Y. Cellular Congenital Mesoblastic Nephroma Detected by Prenatal MRI: A Case Report and Literature Review. Transl Pediatr 2022, 11, 163–173. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Wang, S.; Gao, Y.; Liang, L.; Yang, H. Prenatal Diagnosis and Postnatal Management of Congenital Mesoblastic Nephroma: A Case Report and Literature Review. Front Pediatr 2022, 10, 1040304. [Google Scholar] [CrossRef]
- Lei L, Stohr BA, Berry S, Lockwood CM, Davis JL, Rudzinski ER, Kunder CA. Recurrent EGFR alterations in NTRK3 fusion negative congenital mesoblastic nephroma. Pract Lab Med. 2020 May 16;21:e00164. [CrossRef] [PubMed] [PubMed Central]
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.G.; Tosi, F.; Siena, S. Tropomyosin Receptor Kinase (TRK) Biology and the Role of NTRK Gene Fusions in Cancer. Ann Oncol 2019, 30, viii5–viii15. [Google Scholar] [CrossRef]
- Tognon, C.; Knezevich, S.R.; Huntsman, D.; Roskelley, C.D.; Melnyk, N.; Mathers, J.A.; Becker, L.; Carneiro, F.; MacPherson, N.; Horsman, D.; et al. Expression of the ETV6-NTRK3 Gene Fusion as a Primary Event in Human Secretory Breast Carcinoma. Cancer Cell 2002, 2, 367–376. [Google Scholar] [CrossRef]
- Majewska, H.; Skálová, A.; Stodulski, D.; Klimková, A.; Steiner, P.; Stankiewicz, C.; Biernat, W. Mammary Analogue Secretory Carcinoma of Salivary Glands: A New Entity Associated with ETV6 Gene Rearrangement. Virchows Arch 2015, 466, 245–254. [Google Scholar] [CrossRef]
- Leeman-Neill, R.J.; Kelly, L.M.; Liu, P.; Brenner, A.V.; Little, M.P.; Bogdanova, T.I.; Evdokimova, V.N.; Hatch, M.; Zurnadzy, L.Y.; Nikiforova, M.N.; et al. ETV6-NTRK3 Is a Common Chromosomal Rearrangement in Radiation-Associated Thyroid Cancer. Cancer 2014, 120, 799–807. [Google Scholar] [CrossRef]
- Pachl, M.; Arul, G.S.; Jester, I.; Bowen, C.; Hobin, D.; Morland, B. Congenital Mesoblastic Nephroma: A Single-Centre Series. Ann R Coll Surg Engl 2020, 102, 67–70. [Google Scholar] [CrossRef]
- Williams, F.; Griffiths, P.D. In Utero MR Imaging in Fetuses at High Risk of Lissencephaly. Br J Radiol 2017, 90, 20160902. [Google Scholar] [CrossRef]
Figure 1.
Hyperechoic tumor formation in the abdomen of the fetus, size 50 x 50 mm.
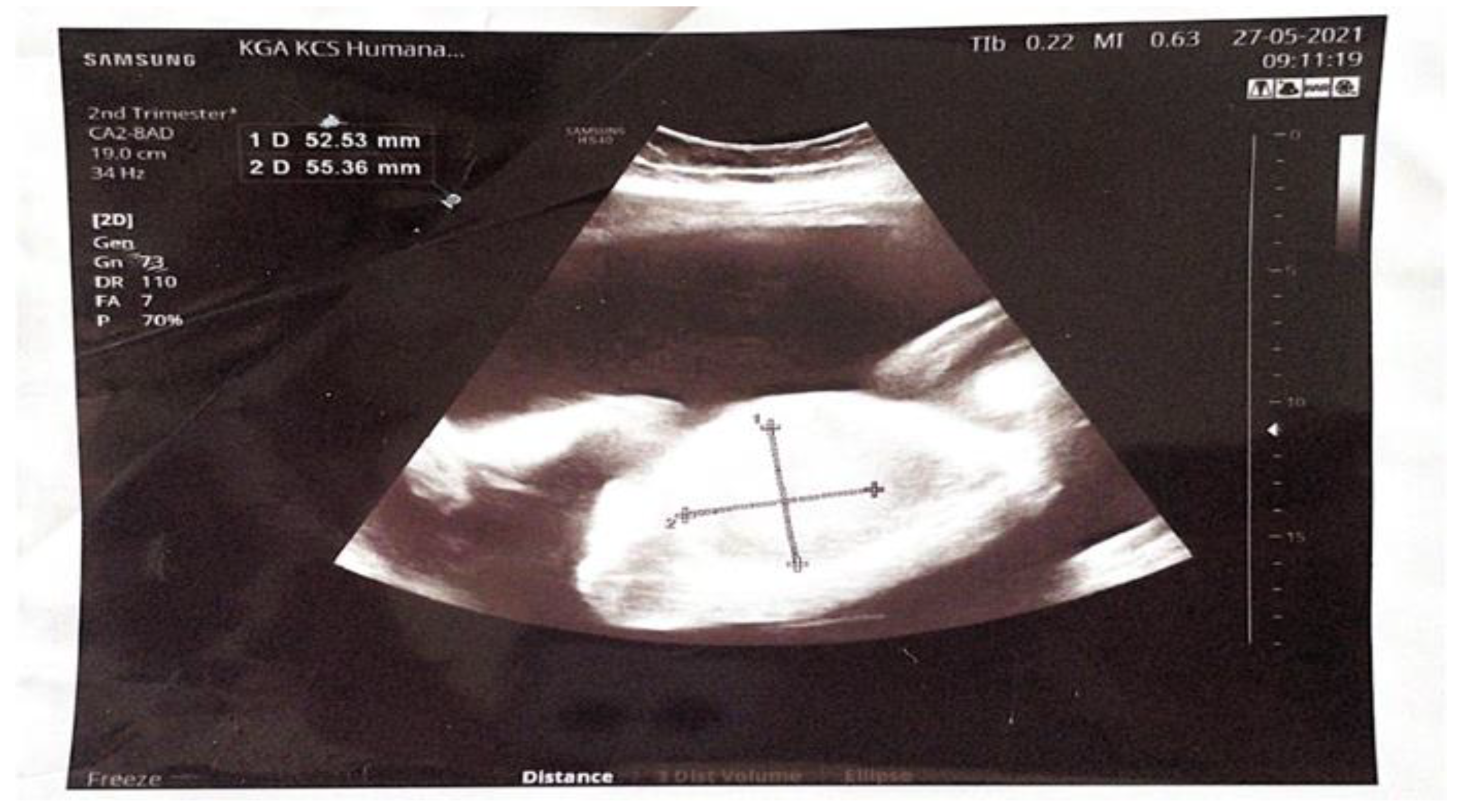
Figure 2.
Gross autopsy findings of the abdominal cavity and the right kidney. A) Enlarged right kidney compresses abdominal organs (liver and intestinal tract); B) Cross section of the right kidney illustrates tumor mass which completely replaced the normal kidney parenchyma.
Figure 2.
Gross autopsy findings of the abdominal cavity and the right kidney. A) Enlarged right kidney compresses abdominal organs (liver and intestinal tract); B) Cross section of the right kidney illustrates tumor mass which completely replaced the normal kidney parenchyma.
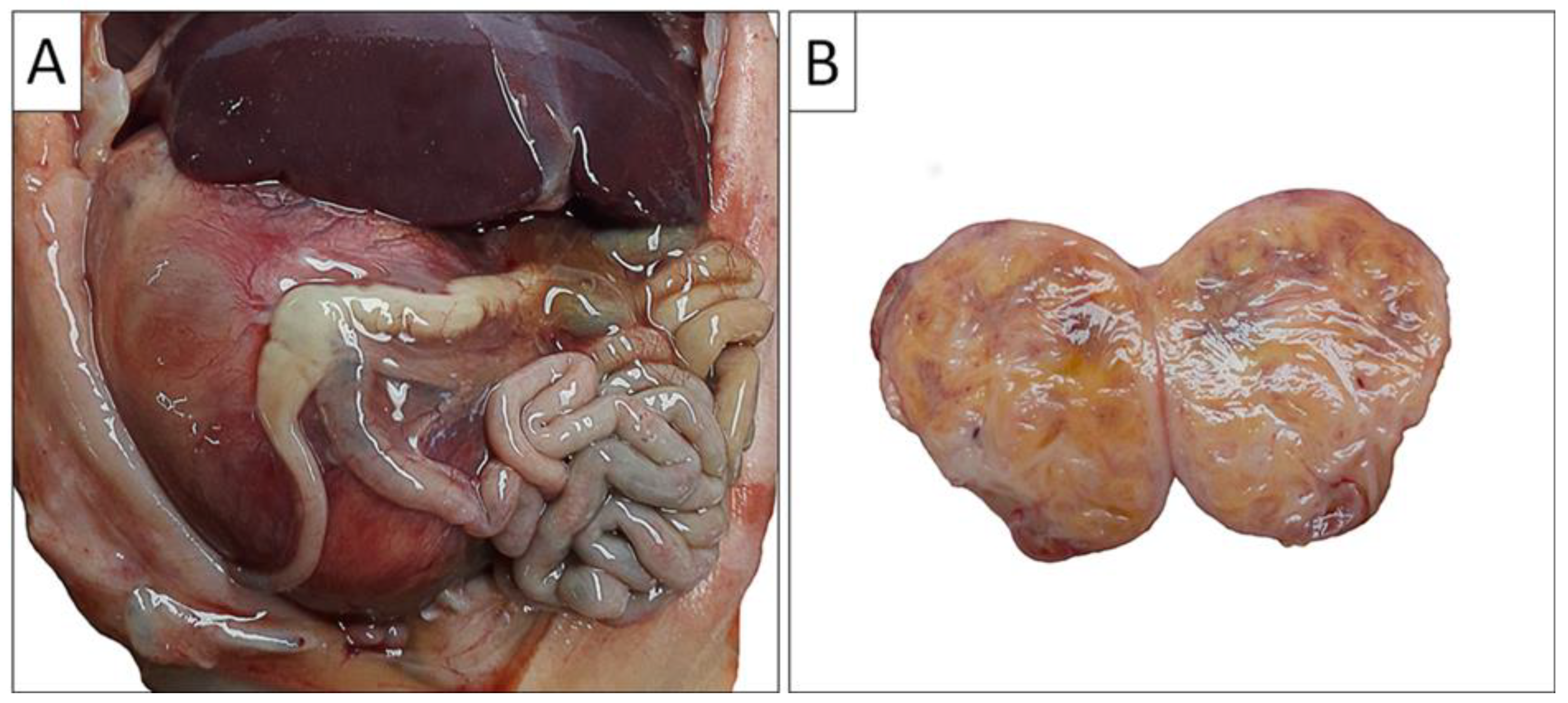
Figure 3.
Pathohistology of congenital mesoblastic nephroma. A) and B) The tumor was composed of uniform spindle cells arranged in clusters and fascicles, with indistinct intercellular boundaries, trapping small islands of non-neoplastic renal tissue (HE stain); Immunohistochemically, the tumor cells exhibited diffuse C) vimentin and F) INI1 positivity along with focal positivity for D) WT1, and E) cyclin D1. The tumor cells were negative for G) SMA (internal positive control are blood vessels), H) CD34 (internal positive control are blood vessels), I) bcl-2, J) ALK, and K) BCOR.
Figure 3.
Pathohistology of congenital mesoblastic nephroma. A) and B) The tumor was composed of uniform spindle cells arranged in clusters and fascicles, with indistinct intercellular boundaries, trapping small islands of non-neoplastic renal tissue (HE stain); Immunohistochemically, the tumor cells exhibited diffuse C) vimentin and F) INI1 positivity along with focal positivity for D) WT1, and E) cyclin D1. The tumor cells were negative for G) SMA (internal positive control are blood vessels), H) CD34 (internal positive control are blood vessels), I) bcl-2, J) ALK, and K) BCOR.
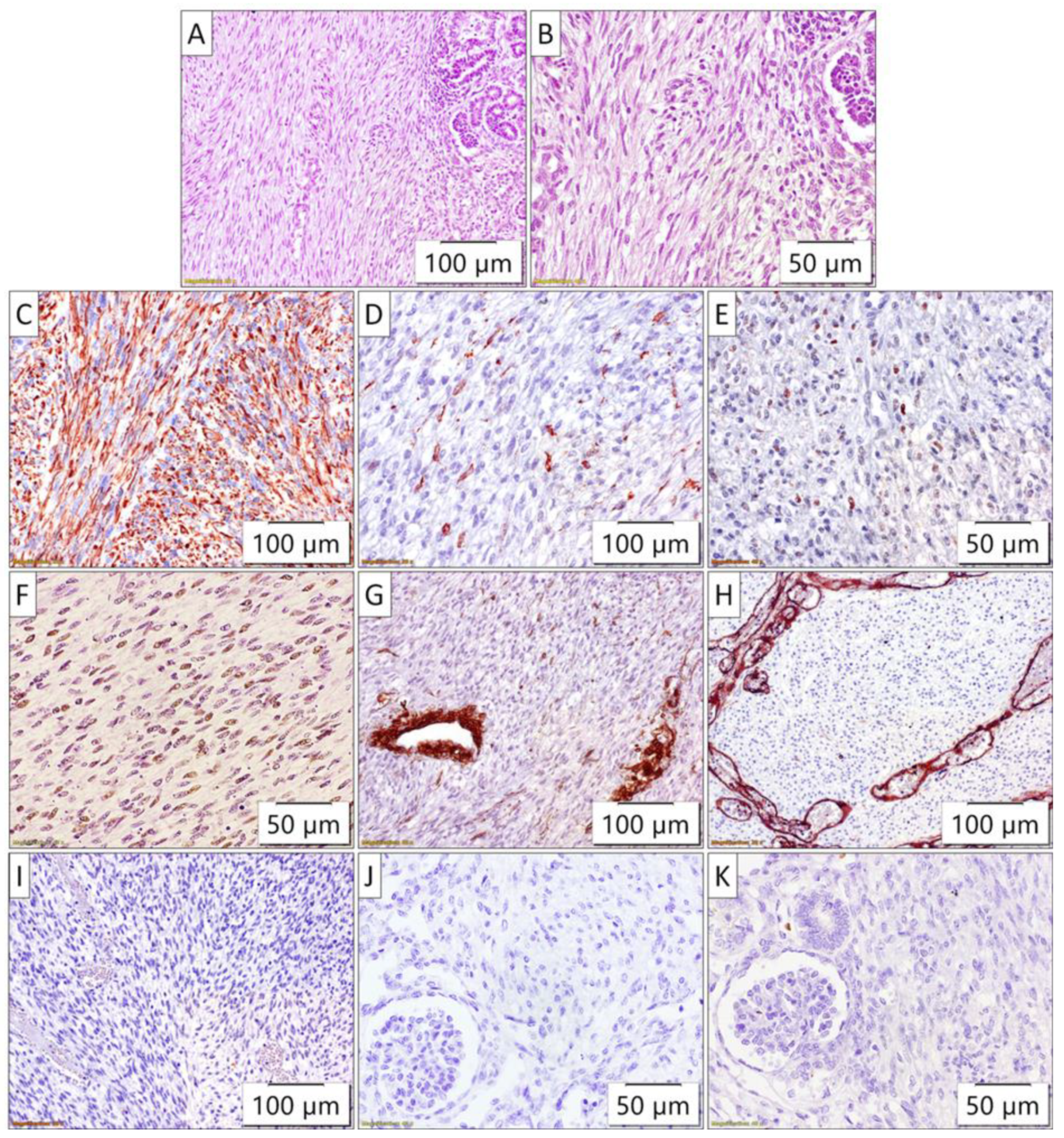
Figure 4.
Gross autopsy findings of the brain. A) Antero-posterior view of left and right cerebral hemispheres revealed abnormal cortical folding, predominantly of the lissencephalic type; B) Temporal-parietal view of the right cerebral hemisphere revealed abnormal cortical folding, predominantly of the lissencephalic type, although the possibility of pachygyria cannot be completely ruled out with certainty; C) Coronal planes of brain sections revealed massive intraventricular bleeding.
Figure 4.
Gross autopsy findings of the brain. A) Antero-posterior view of left and right cerebral hemispheres revealed abnormal cortical folding, predominantly of the lissencephalic type; B) Temporal-parietal view of the right cerebral hemisphere revealed abnormal cortical folding, predominantly of the lissencephalic type, although the possibility of pachygyria cannot be completely ruled out with certainty; C) Coronal planes of brain sections revealed massive intraventricular bleeding.
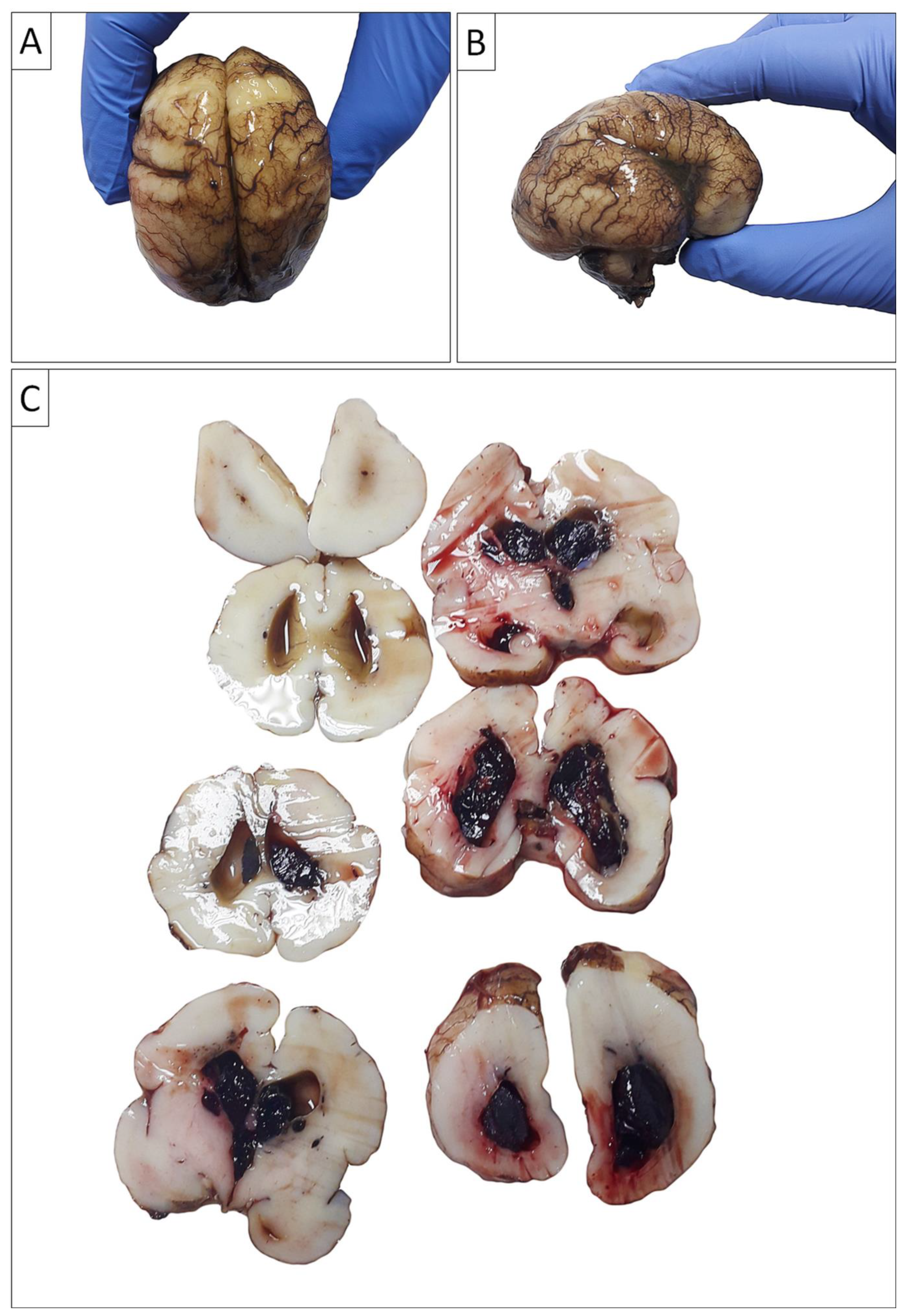
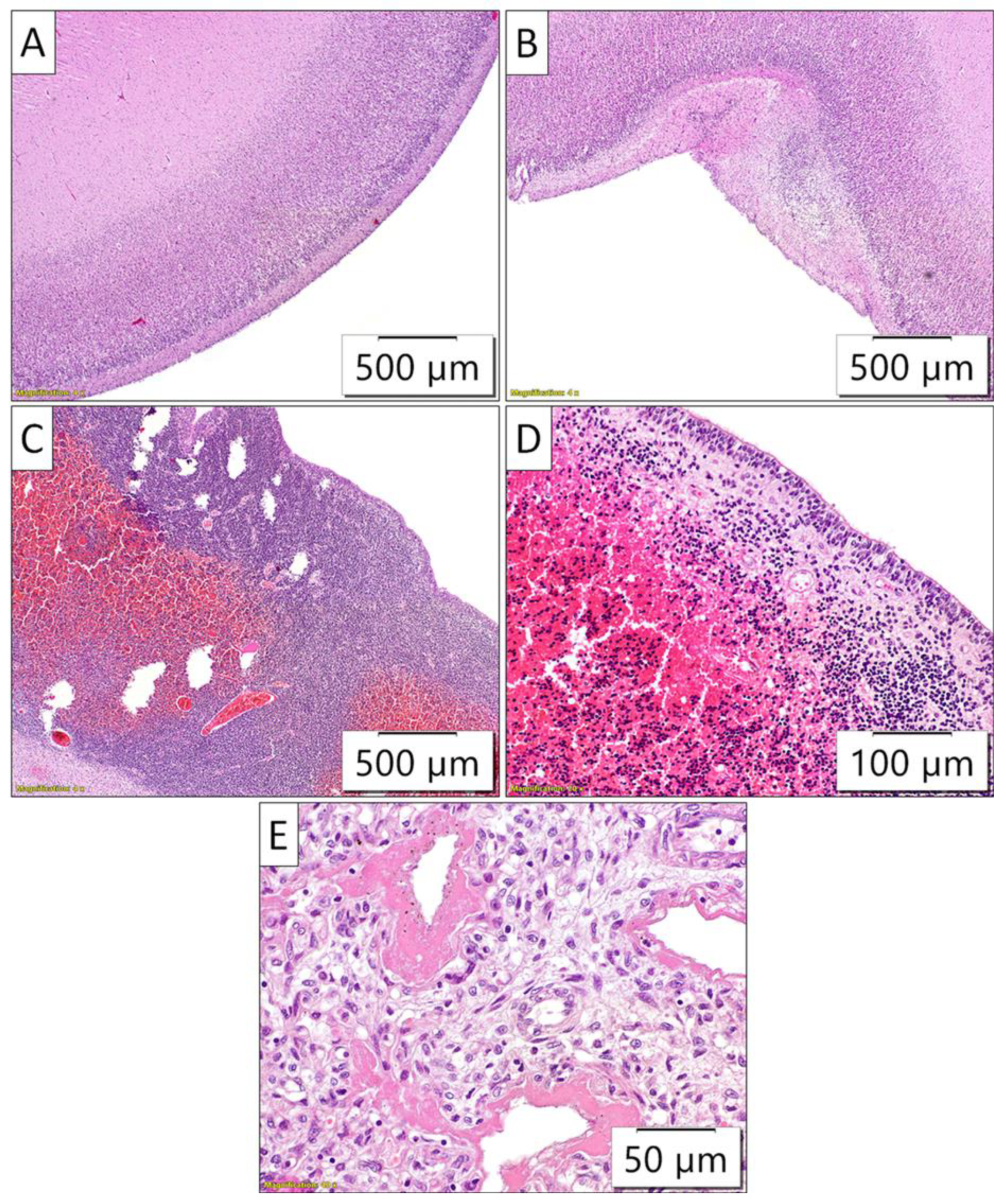
Figure 5.
Pathohistology of the brain and lung sections. A) The absence of large brain gyri, thus, the brain surface was almost completely flat; B) Occasional rudimentary sulci of the large brain cortex. C) and D) Well-developed periventricular germinal matrix with pronounced fresh hemorrhages; E) Diffuse and thick intraalveolar hyaline membranes illustrated the morphology of severe diffuse alveolar damage.
Figure 5.
Pathohistology of the brain and lung sections. A) The absence of large brain gyri, thus, the brain surface was almost completely flat; B) Occasional rudimentary sulci of the large brain cortex. C) and D) Well-developed periventricular germinal matrix with pronounced fresh hemorrhages; E) Diffuse and thick intraalveolar hyaline membranes illustrated the morphology of severe diffuse alveolar damage.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



