
Submitted:
02 December 2024
Posted:
03 December 2024
You are already at the latest version
Abstract
Well-defined amorphous/semi-crystalline statistical copolymers of n-dodecyl isocyanate, DDIC, and allyl isocyanate, ALIC, were synthesized via coordination polymerization, using the chiral half-titanocene complex CpTiCl2(O-(S)-2-Bu) as initiator. In the frame of the terminal model the monomer reactivity ratios of the statistical copolymers were calculated using both well-known linear graphical methods and the computer program COPOINT. The molecular and structural characteristics of the copolymers were also calculated. The thermal properties of these samples were studied by Differential Scanning Calorimetry, DSC, measurements. The kinetics of the thermal decomposition of the statistical copolymers was studied by Thermogravimetric Analysis, TGA, and Differential Thermogravimetry, DTG, and the activation energy of this process was calculated employing several theoretical models. Moreover, block copolymers with the structure P[DDIC-b-(DDIC-co-ALIC)] were synthesized by sequential addition of monomers and coordination polymerization methodologies. The samples were characterized by nuclear magnetic resonance, NMR, spectroscopy, size exclusion chromatography, SEC, and DSC. The thermal stability of the blocks was also studied by TGA and DTG and compared to the corresponding statistical copolymers, showing that the macromolecular architecture greatly affects the properties of the copolymers. Thiol-ene click post-polymerization reaction was performed to introduce aromatic groups along the polyisocyanate chain in order to improve the thermal stability of the parent polymers.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Materials
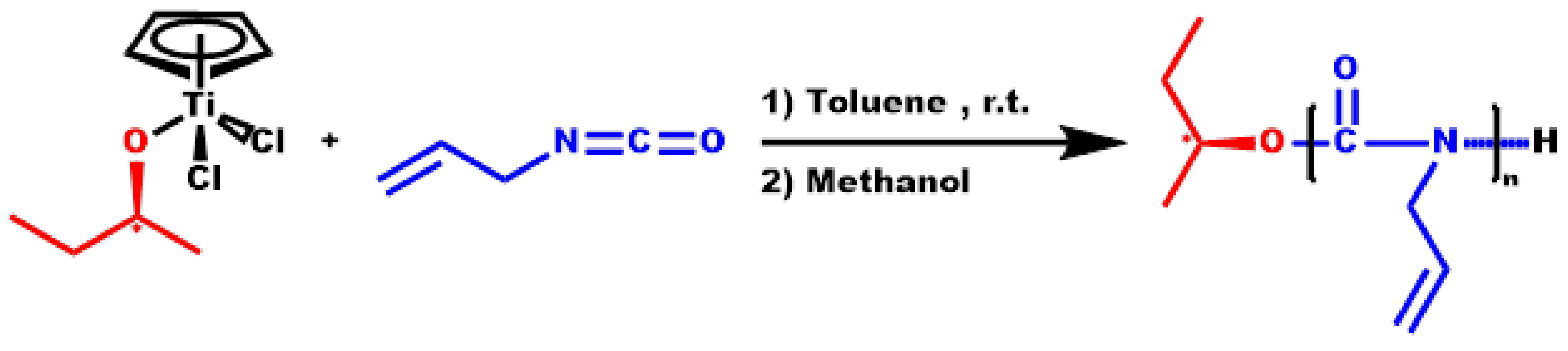
2.2. Synthesis of Homopolymer PDDIC
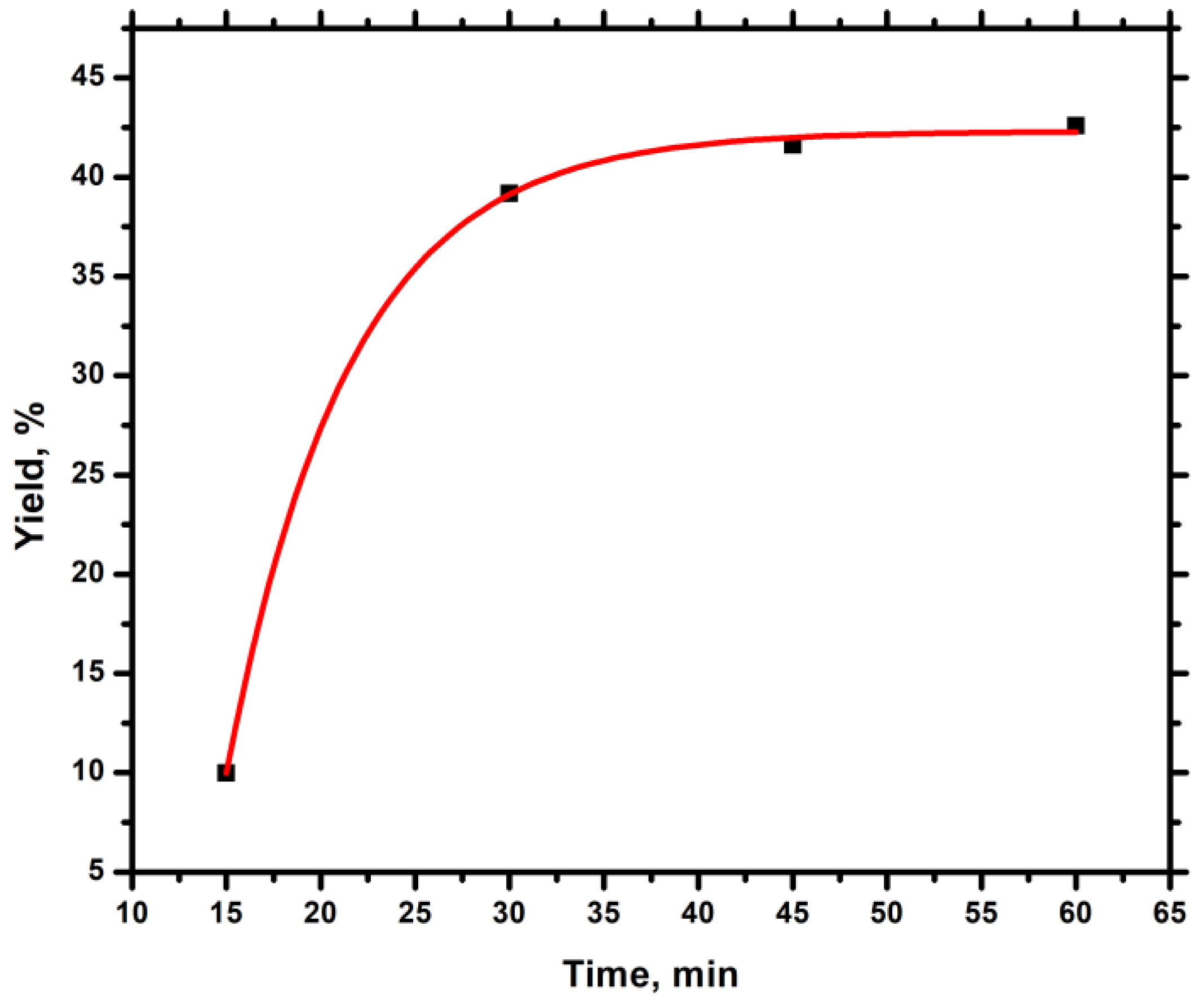
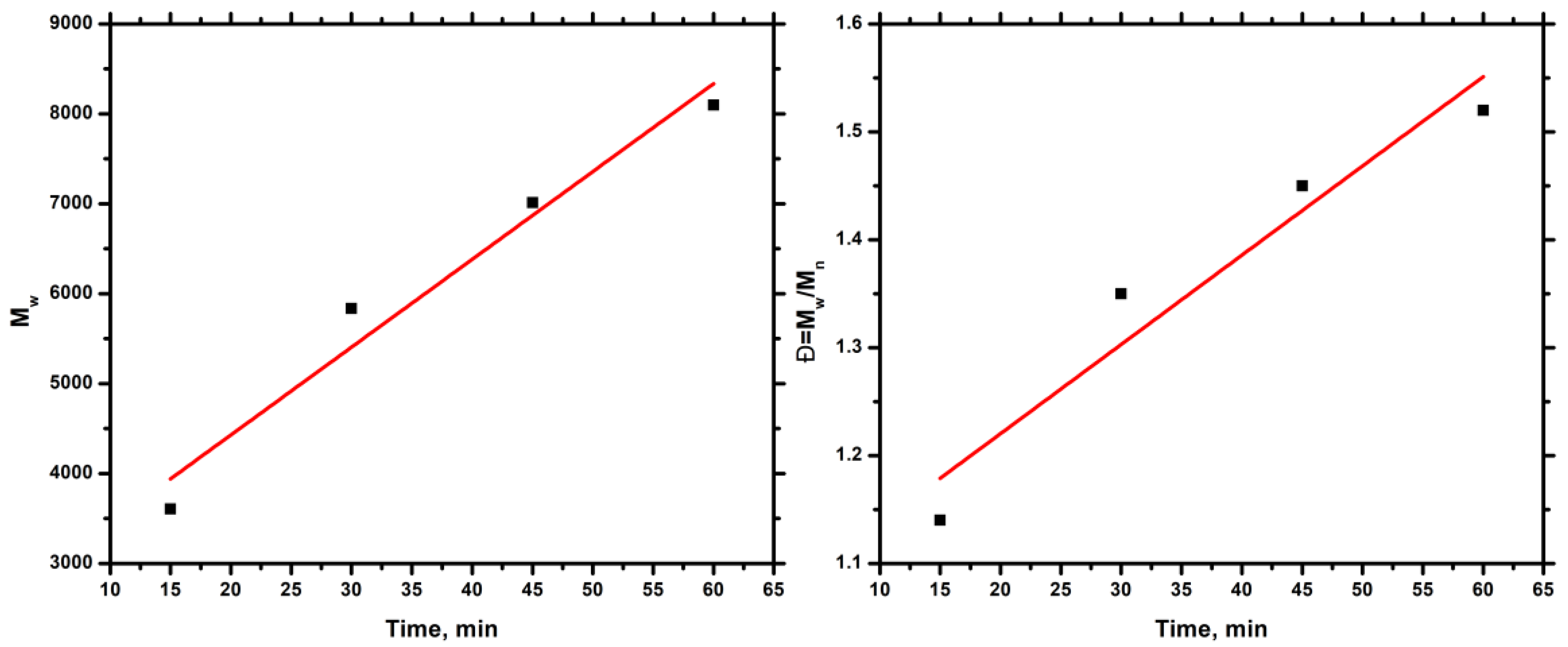
2.3. Synthesis of Homopolymer PALIC & Kinetic Study
2.4. Synthesis of Statistical Copolymers, P(ALIC-co-DDIC)
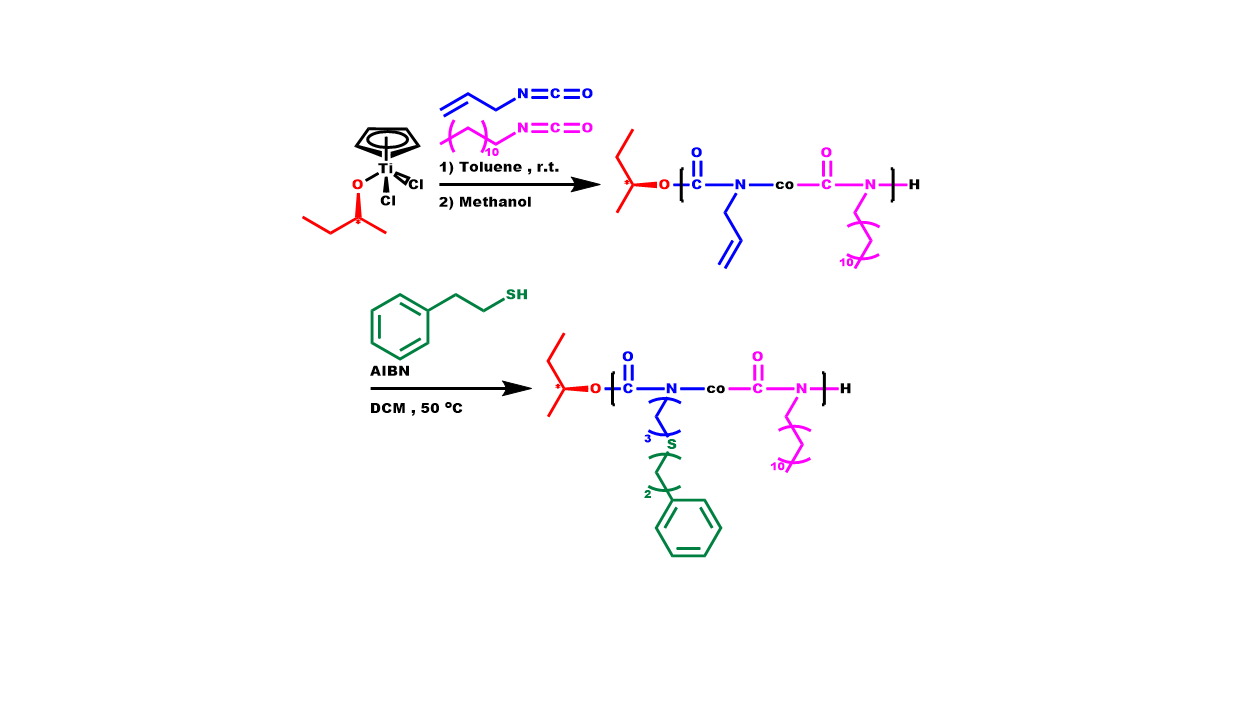
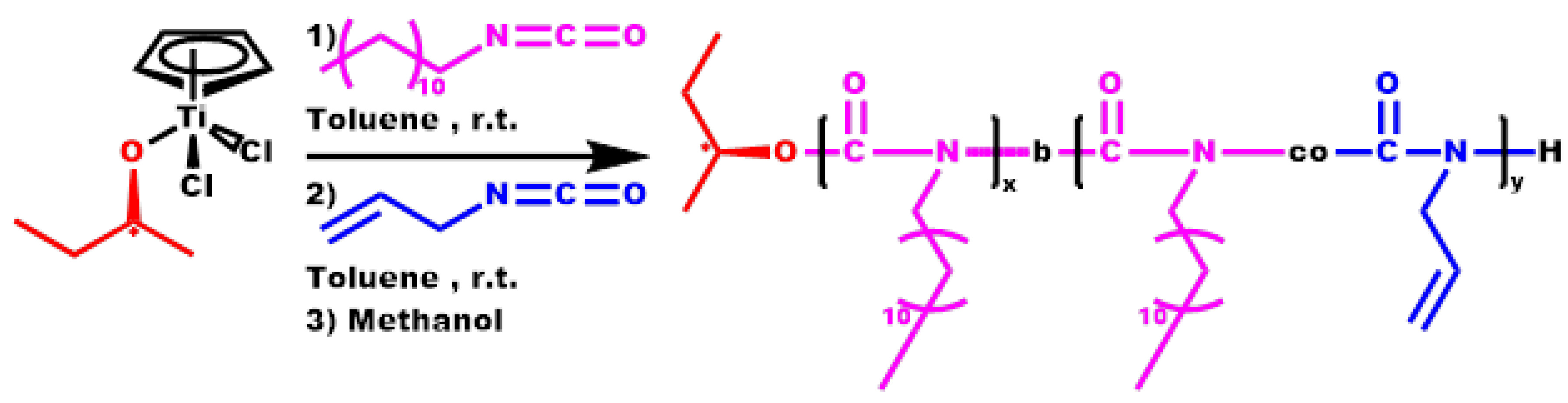
2.5. Synthesis of Block Copolymers, P[DDIC-b-(DDIC-co-ALIC)]
2.6. Synthesis of P3PETPIC & P(3PETPIC-co-DDIC)
2.7. Characterization
3. Results and Discussion
3.1. Homopolymerization of ALIC & Kinetic Study

| Time (min) | Mn a | Mw a | Đ a | Yield (%) |
|---|---|---|---|---|
| 15 | 3169 | 3607 | 1.14 | 10.0 |
| 30 | 4328 | 5836 | 1.35 | 39.2 |
| 45 | 4830 | 7012 | 1.45 | 41.6 |
| 60 | 5336 | 8096 | 1.52 | 42.6 |
3.2. Homopolymerization of DDIC
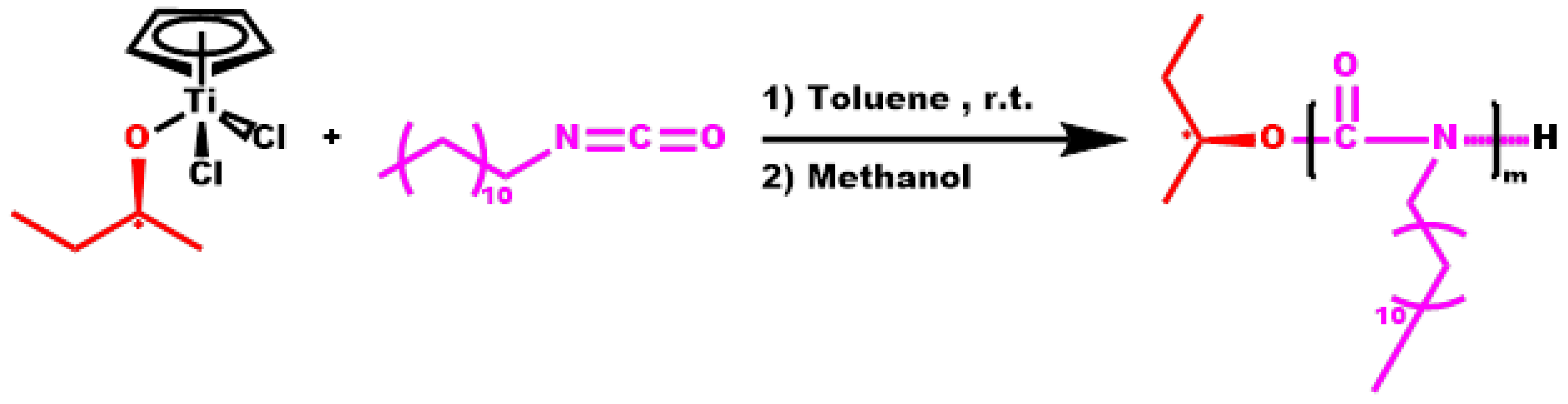
3.3. Statistical Copolymers, P(ALIC-co-DDIC)
| Feed molar ratio ALIC/DDIC | Yield (%) | Molar ratio ALIC/DDIC a | Mw b | Đ b |
|---|---|---|---|---|
| 80/20 | 34.0 | 92/8 | 19800 | 1.06 |
| 60/40 | 35.3 | 73/27 | 17600 | 1.07 |
| 50/50 | 30.5 | 63/37 | 16800 | 1.06 |
| 40/60 | 38.5 | 51/49 | 21600 | 1.09 |
| 20/80 | 42.7 | 35/65 | 26100 | 1.08 |
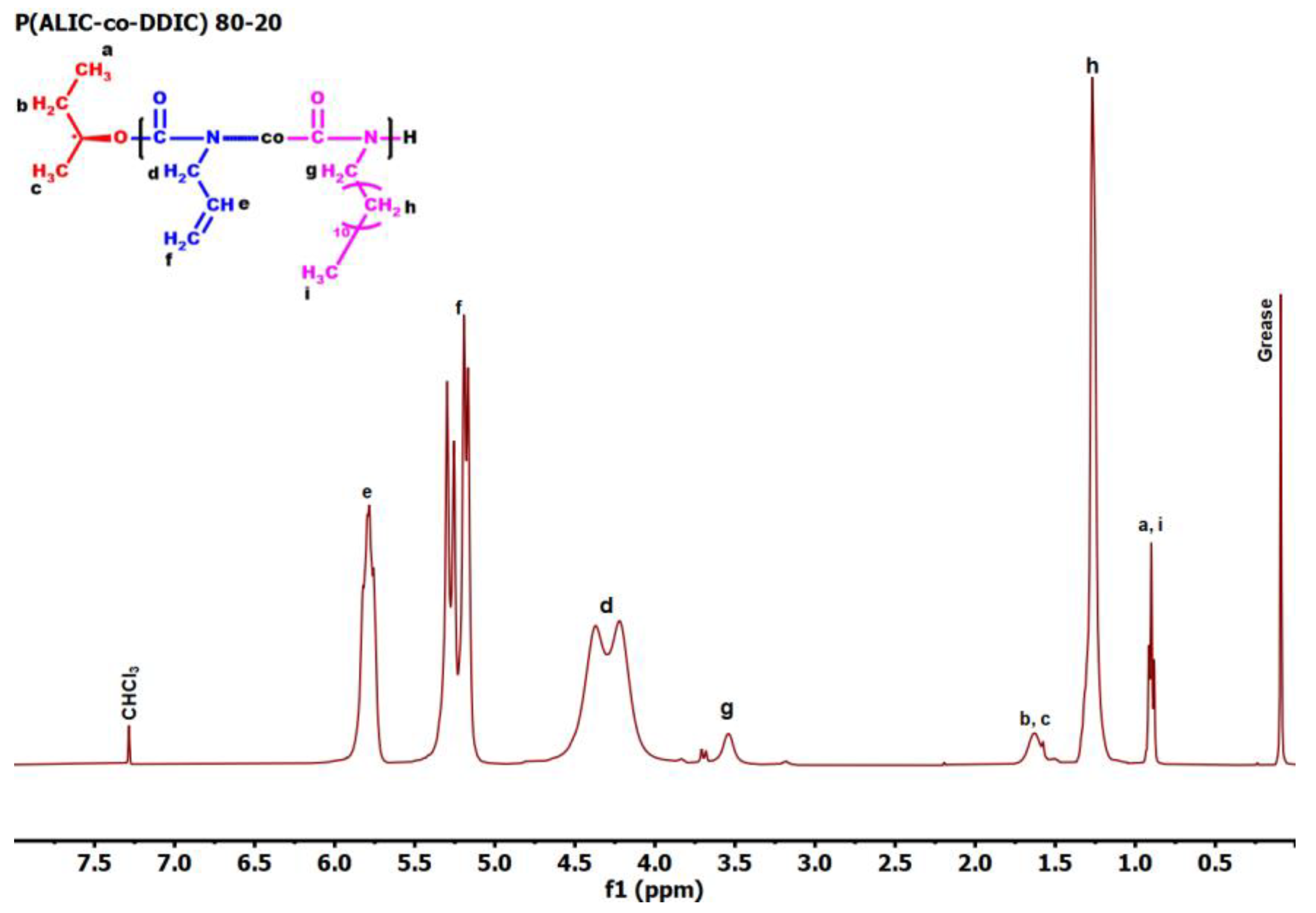
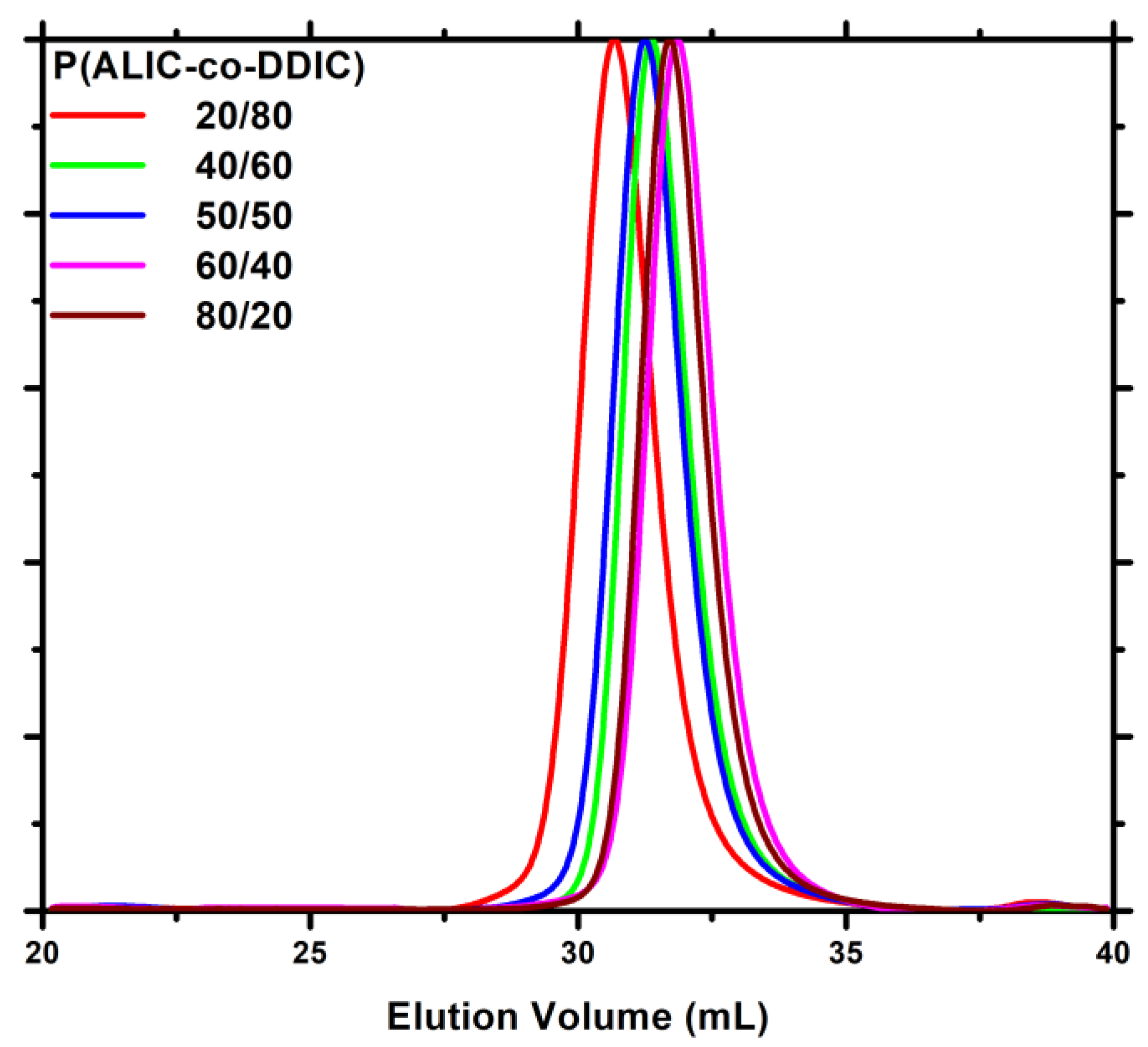
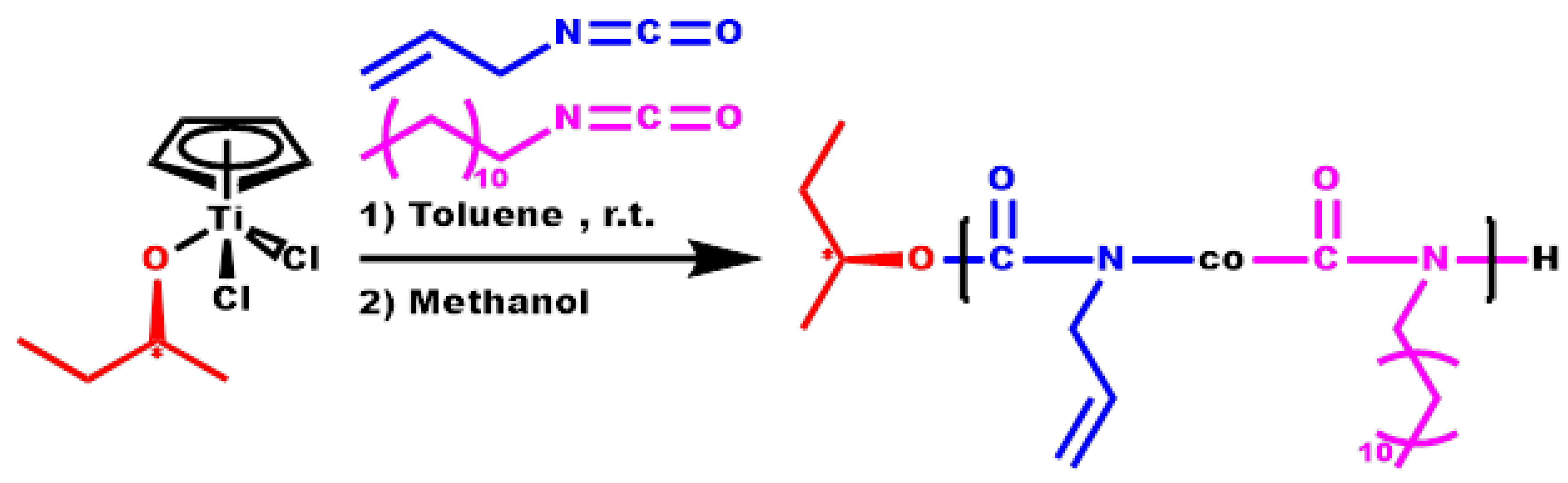
3.4. Monomer Reactivity Ratios and Statistical Analysis of the Copolymers
| Method | r ALIC | rDDIC |
|---|---|---|
| F-R | 2.63 | 1.14 |
| IF-R | 2.60 | 1.13 |
| K-T | 2.60 | 1.12 |
| ext K-T | 3.04 | 1.08 |
| COPOINT | 2.64 | 1.14 |
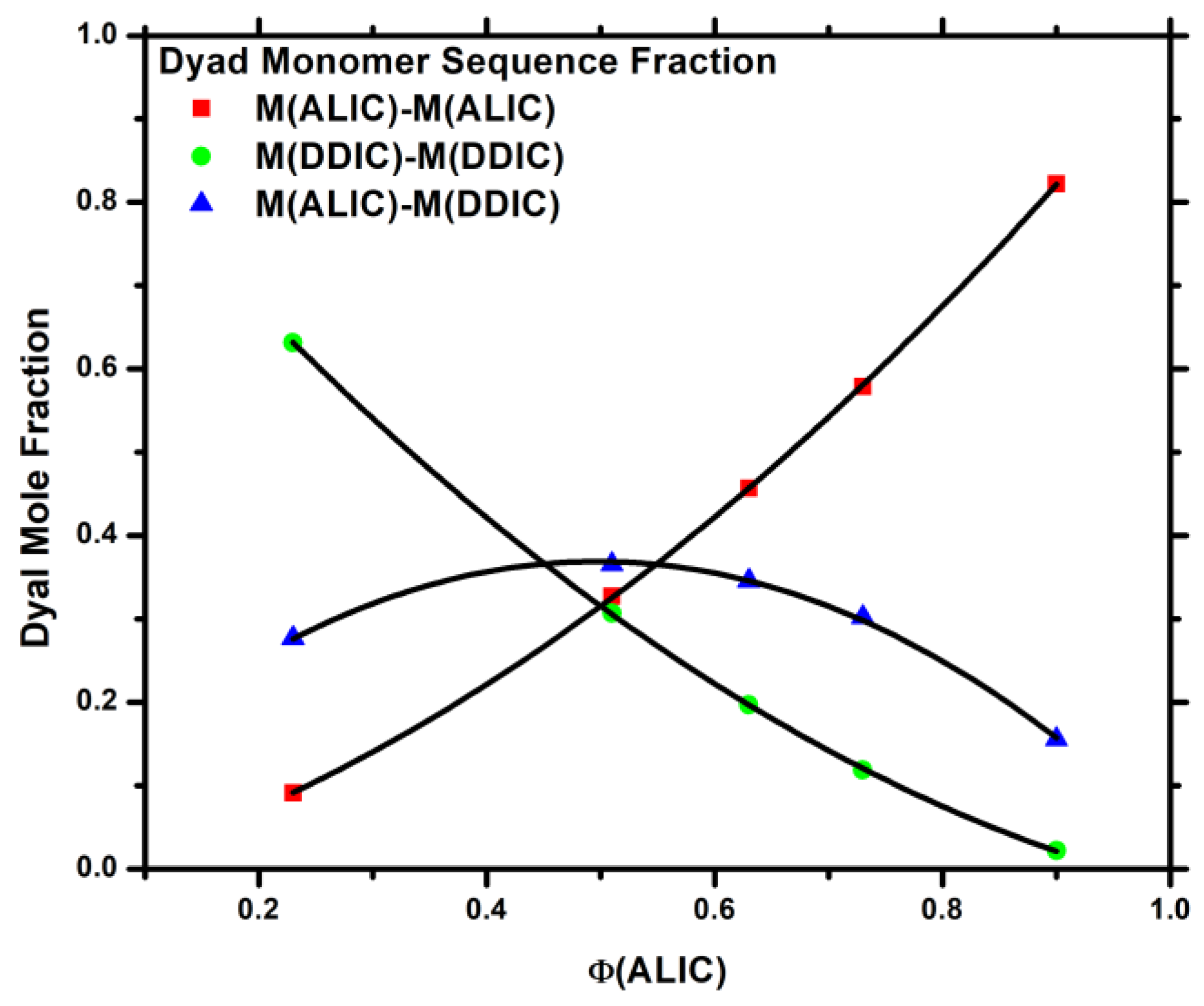
3.5. Block Copolymers, P[DDIC-b-(DDIC-co-ALIC)]
| Feed molar ratio ALIC/DDIC | Molar ratio ALIC/DDIC a | Yield (%) | Mw b | Đ b |
|---|---|---|---|---|
| 80/20 | 76/24 | 42.8 | 9200 | 1.11 |
| 50/50 | 44/56 | 38.0 | 11100 | 1.17 |
| 20/80 | 8/91 | 41.7 | 8300 | 1.13 |
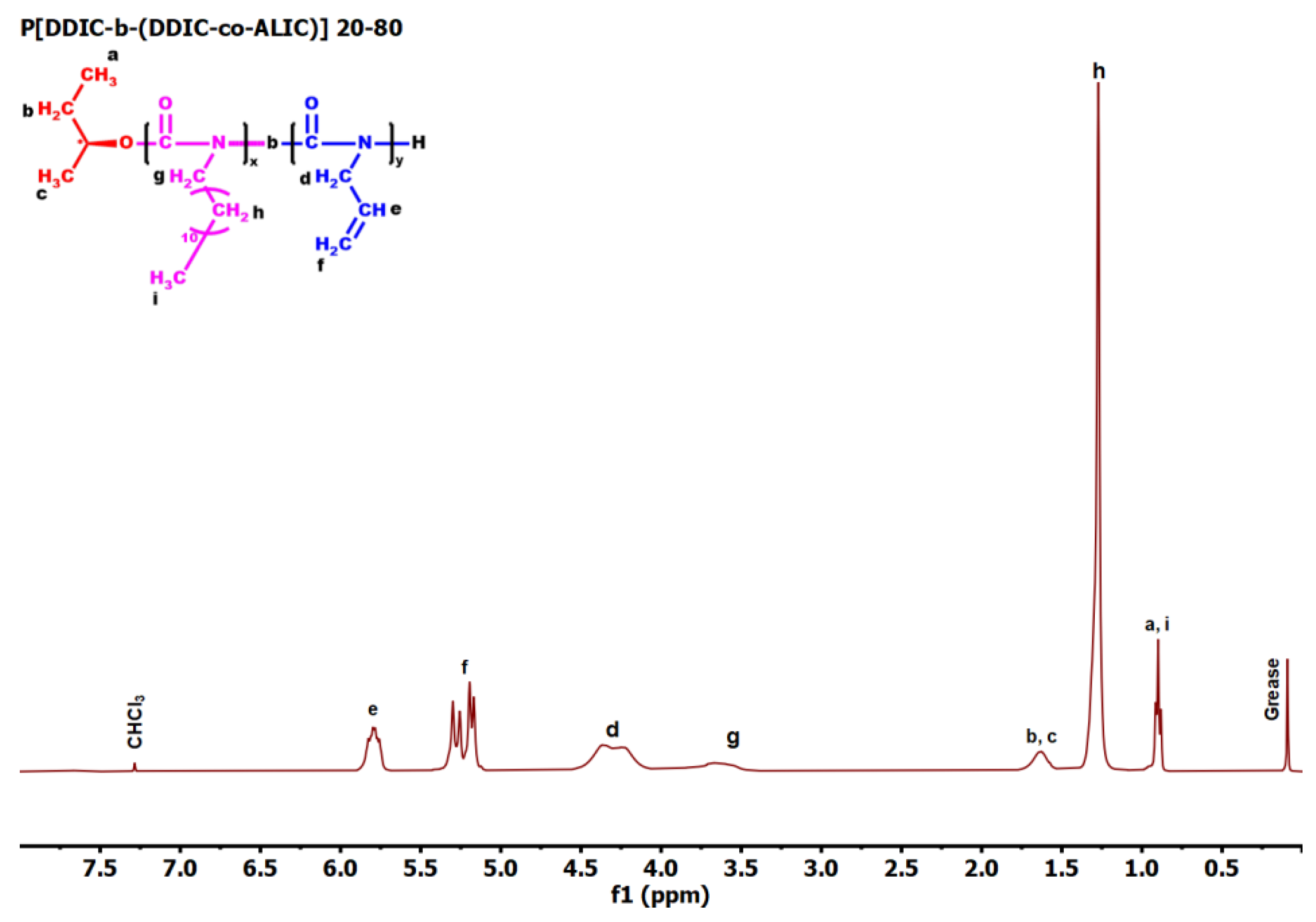
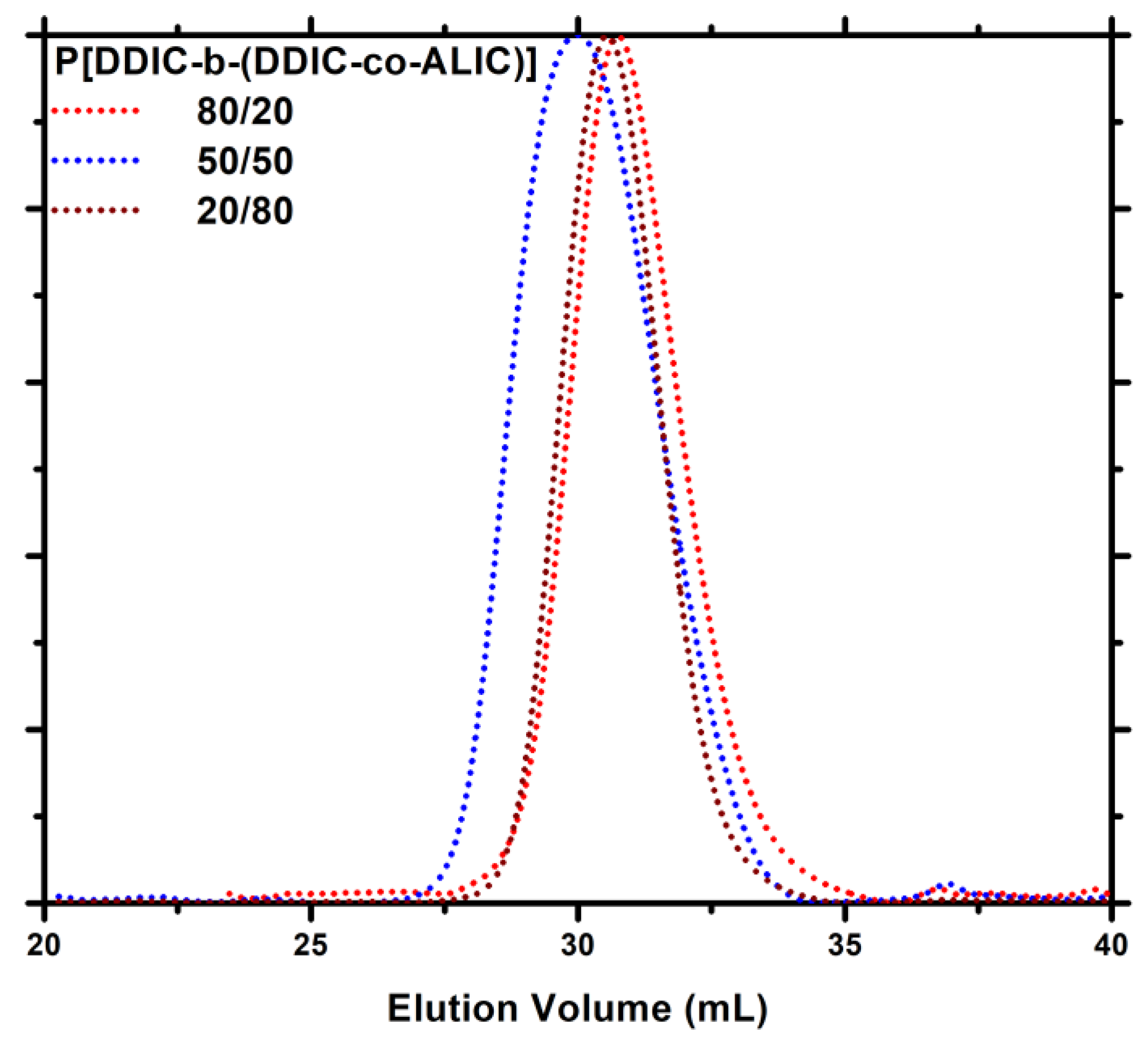
3.6. Synthesis of P3PETPIC & P(3PETPIC-co-DDIC) via Thiol-Ene Click Reaction
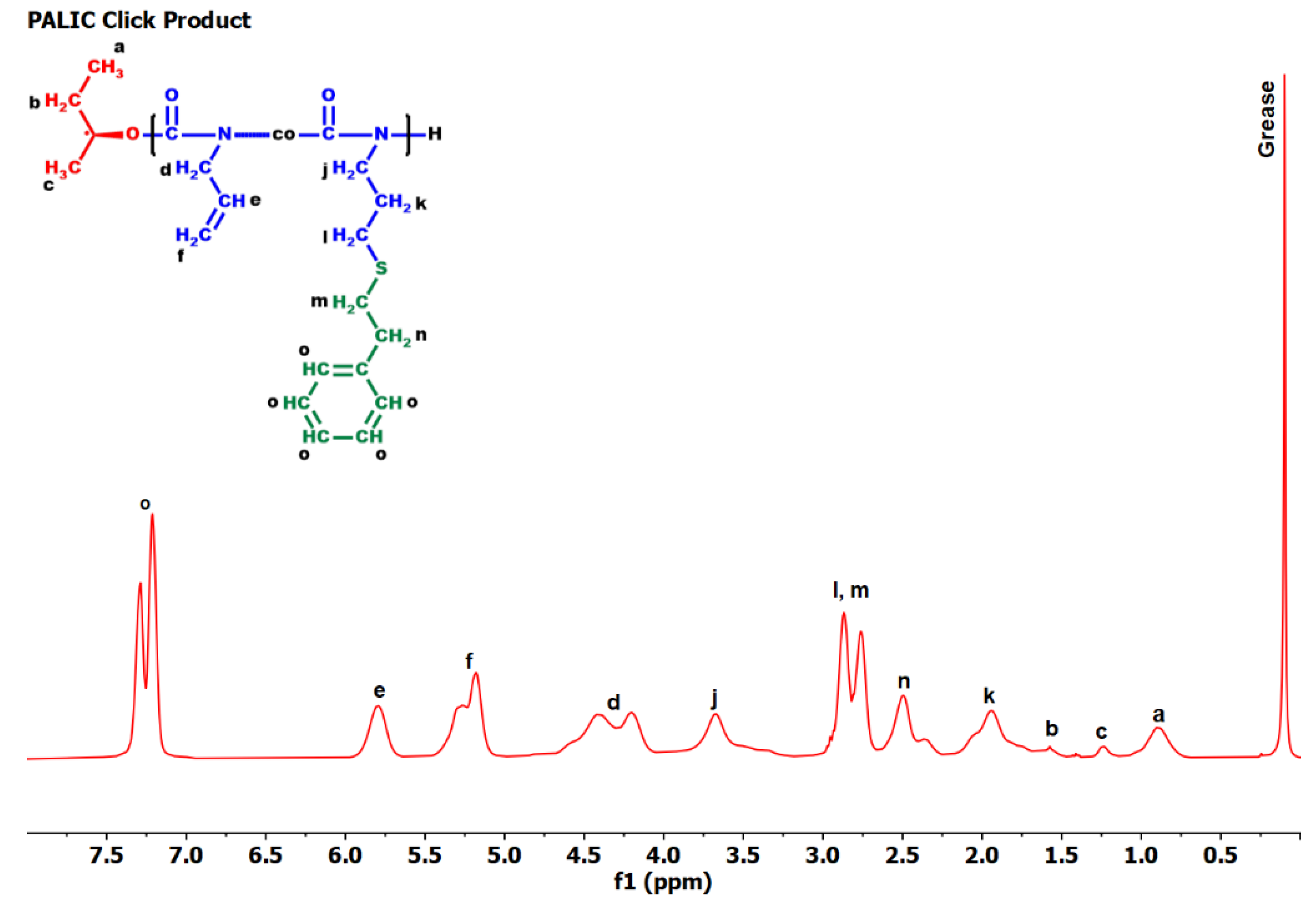
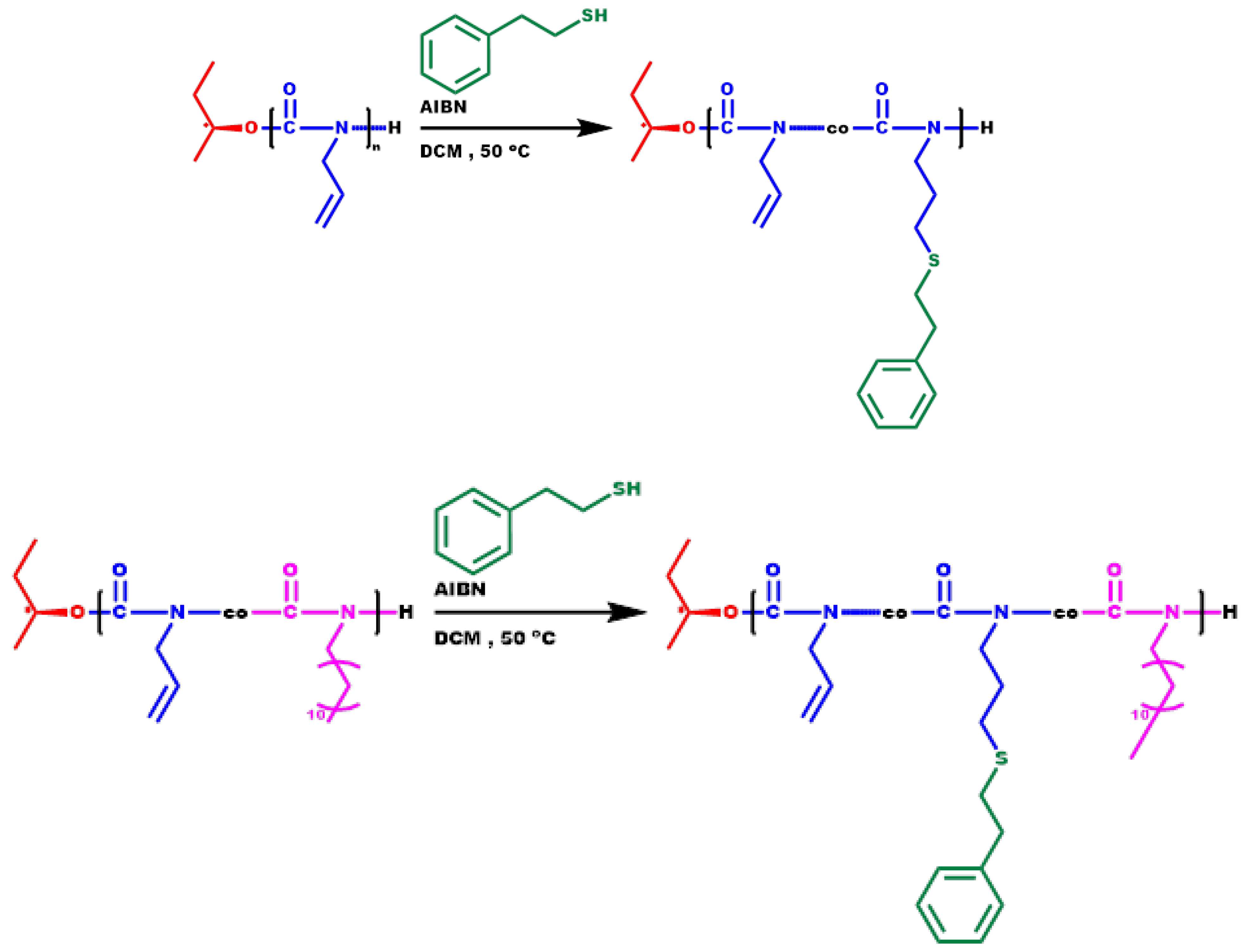
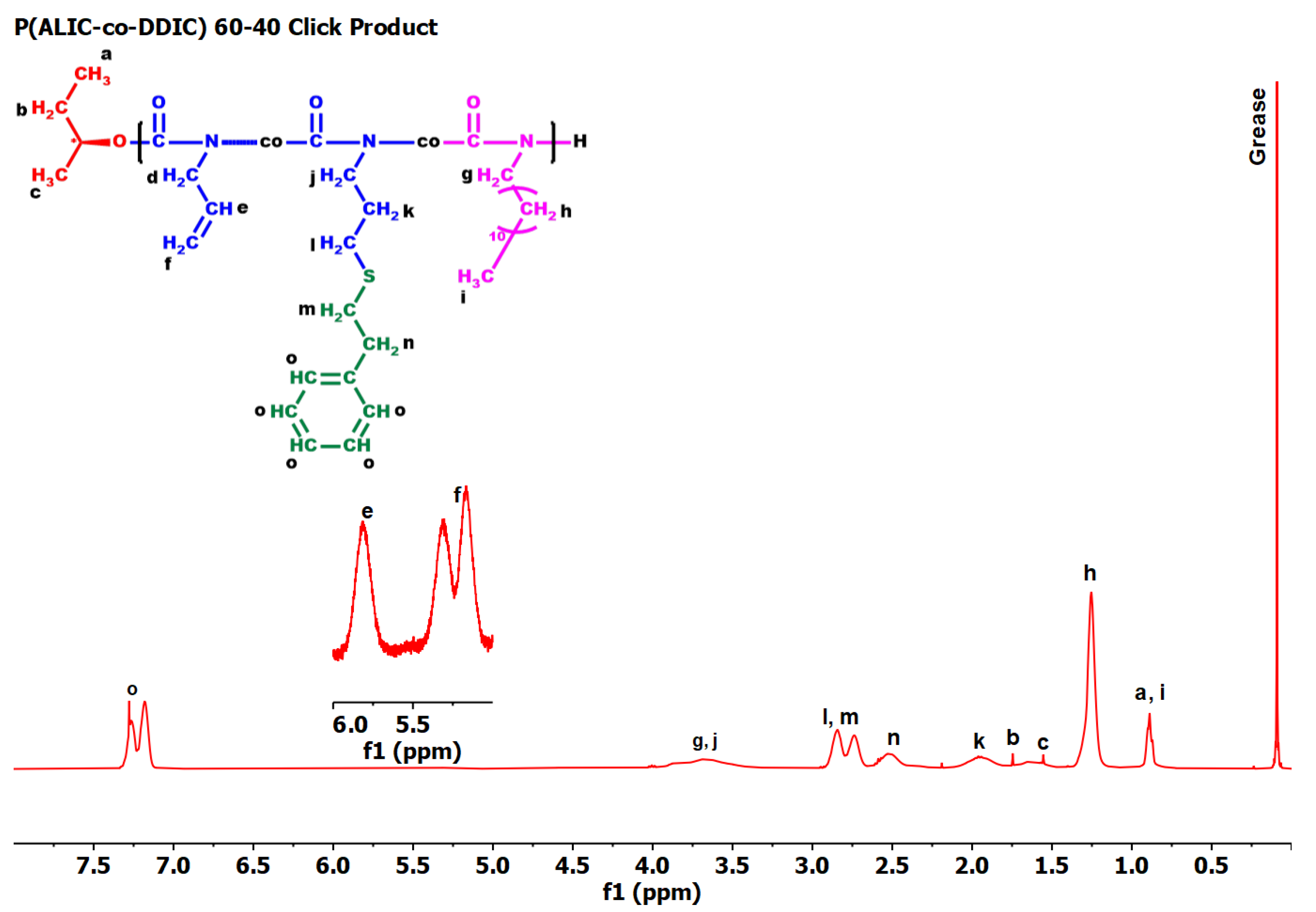
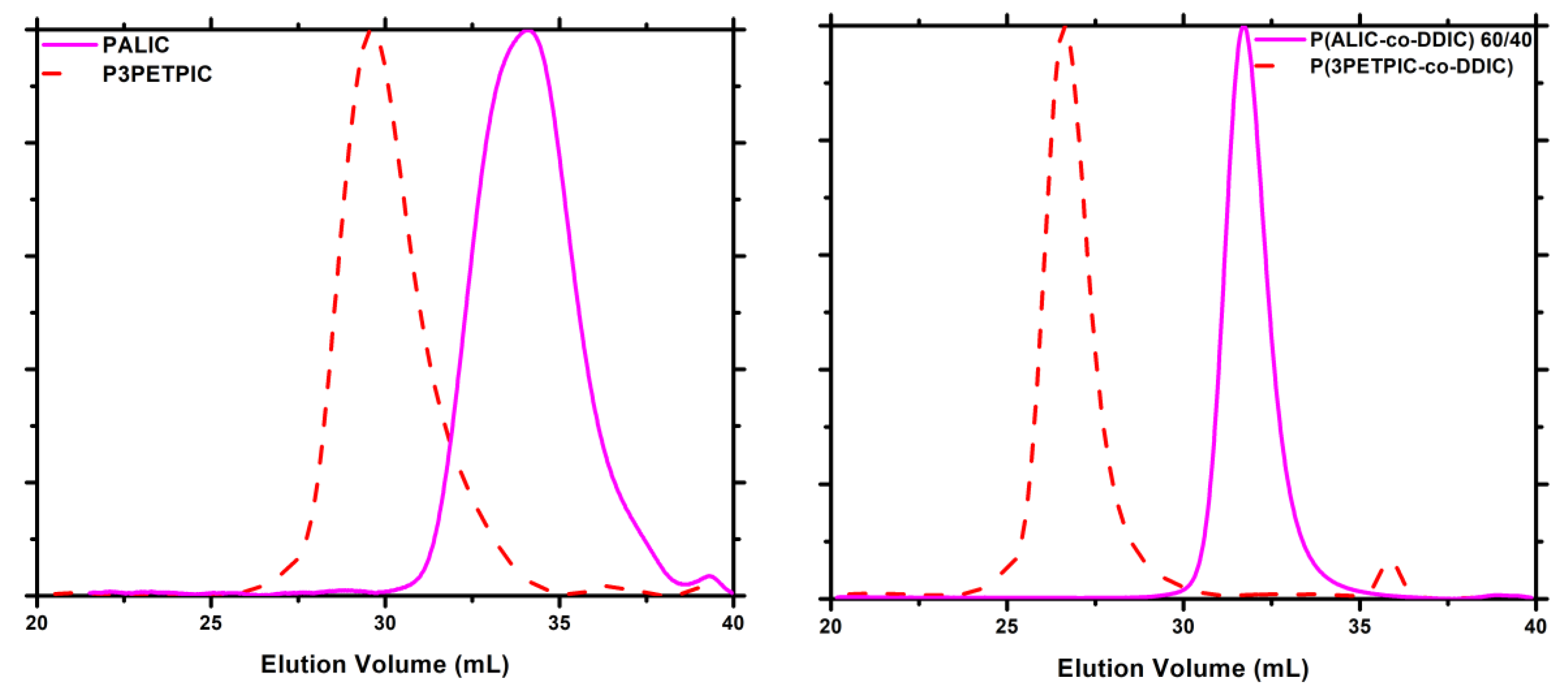
3.7. Thermal Analysis
3.7.1. DSC Analysis
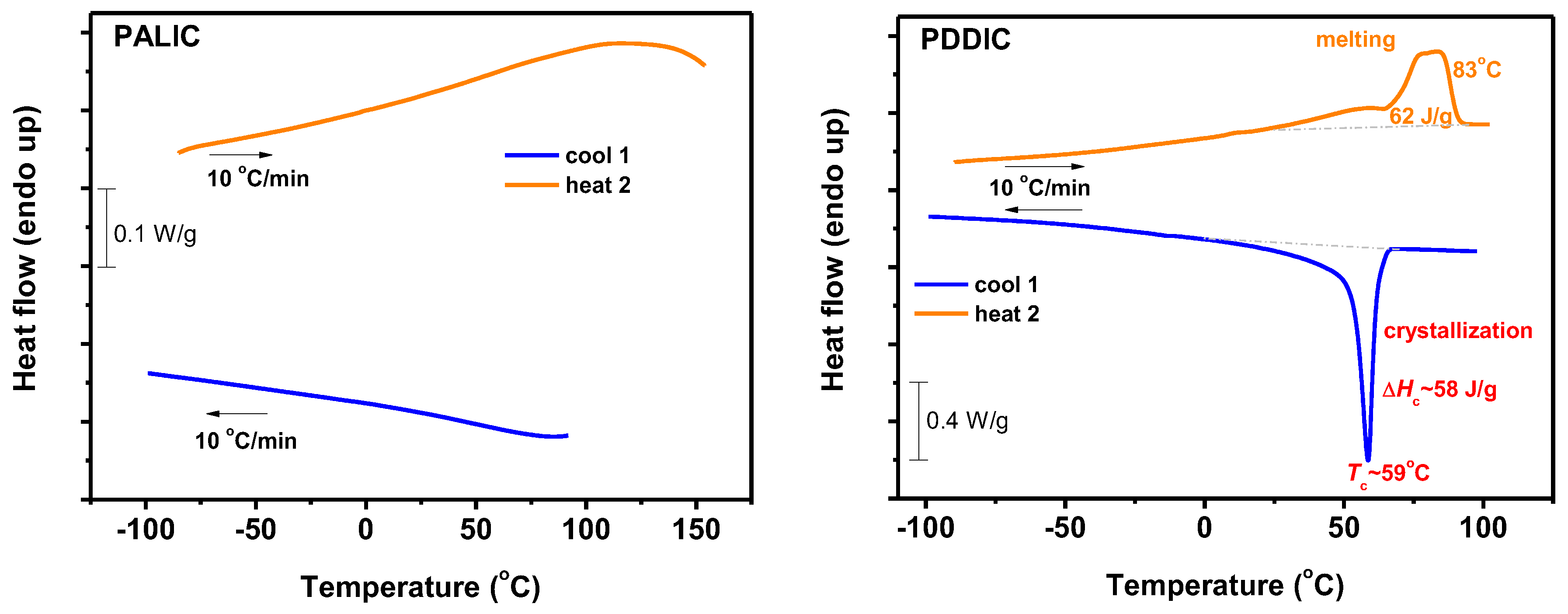
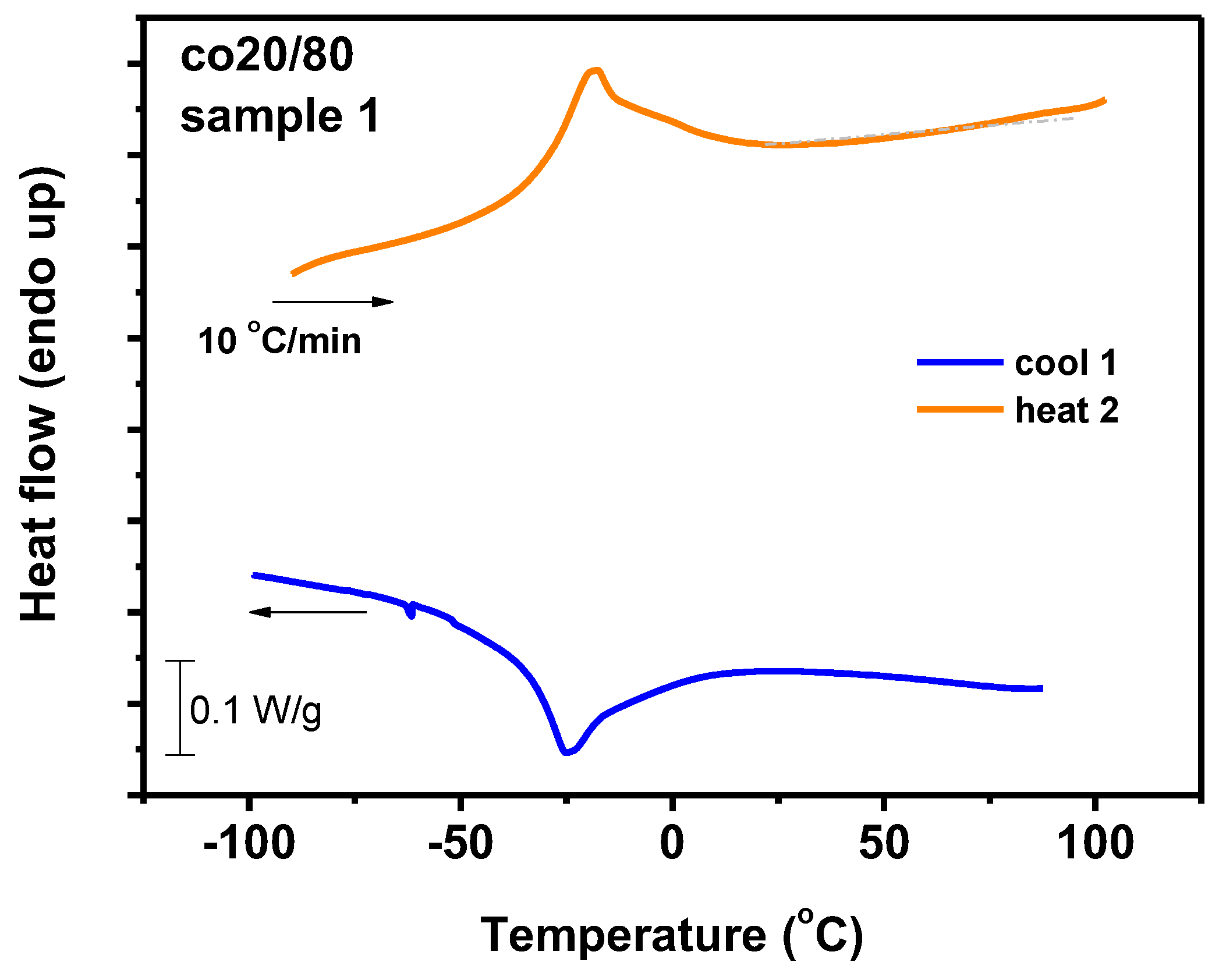
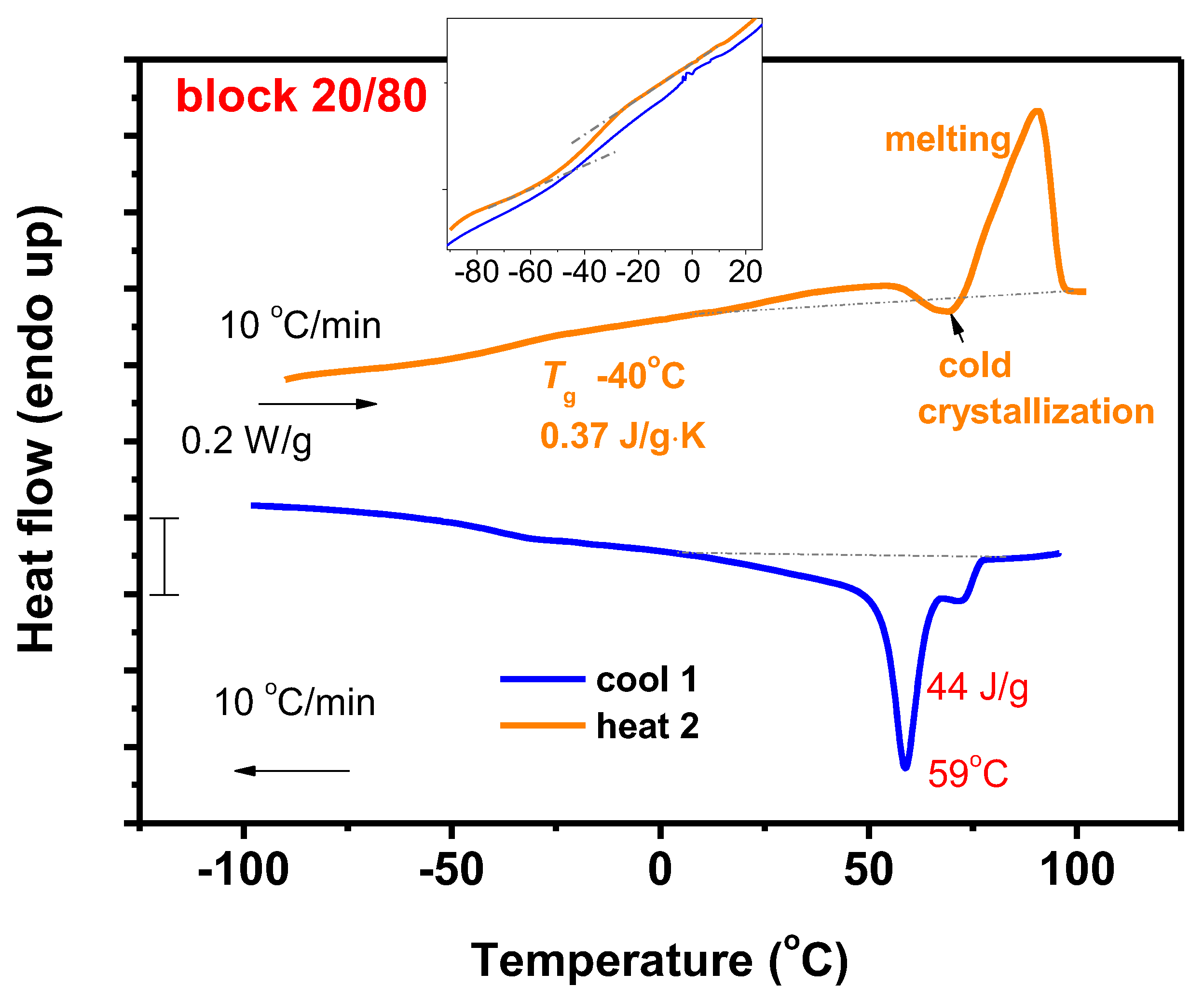
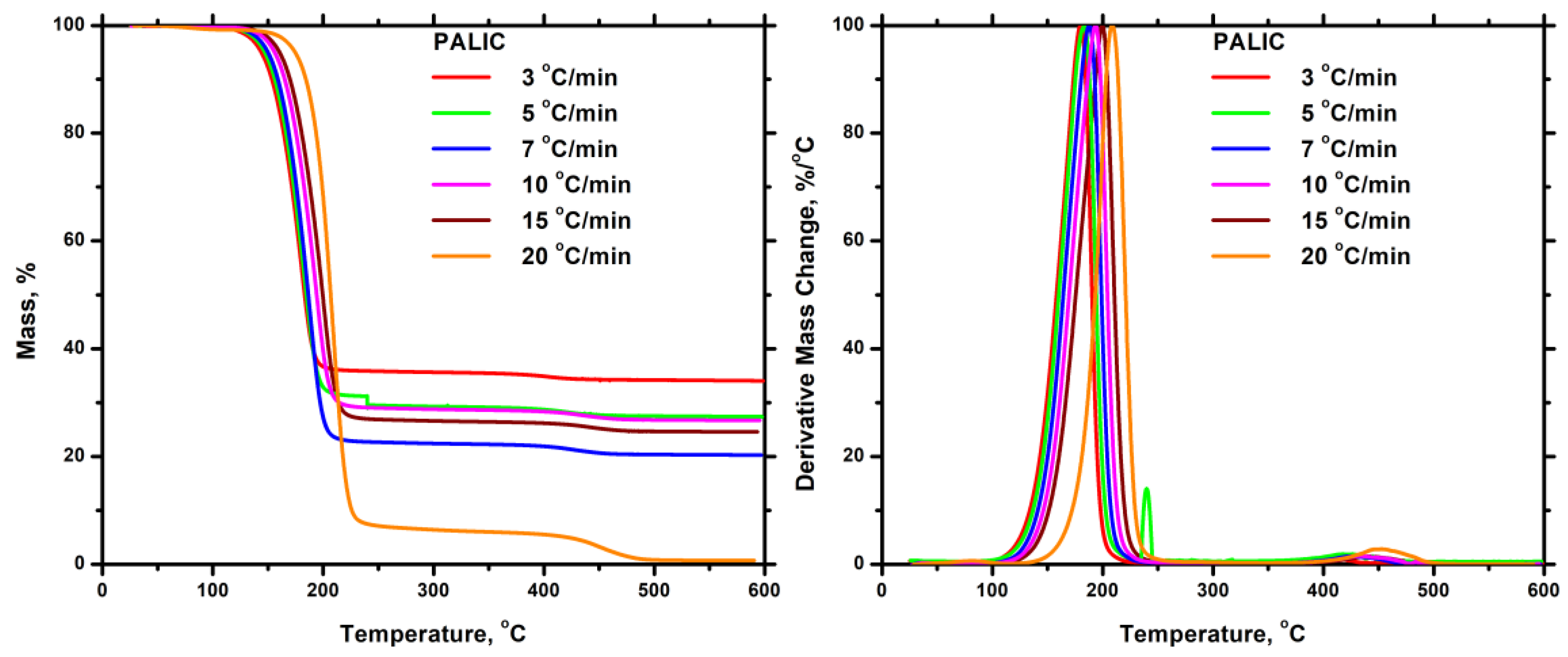
3.7.2. Thermal Decomposition
3.7.2.1. Homopolymers
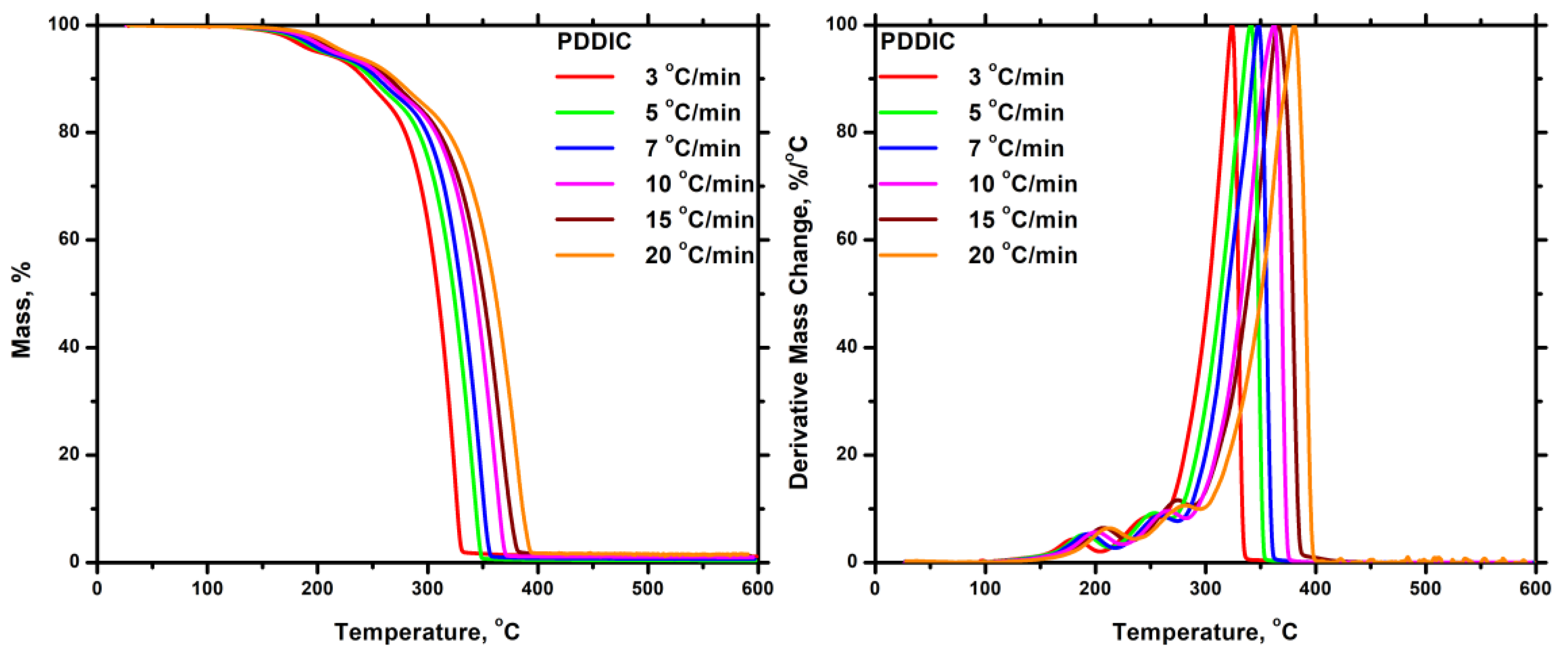
3.7.2.2. Statistical Copolymers
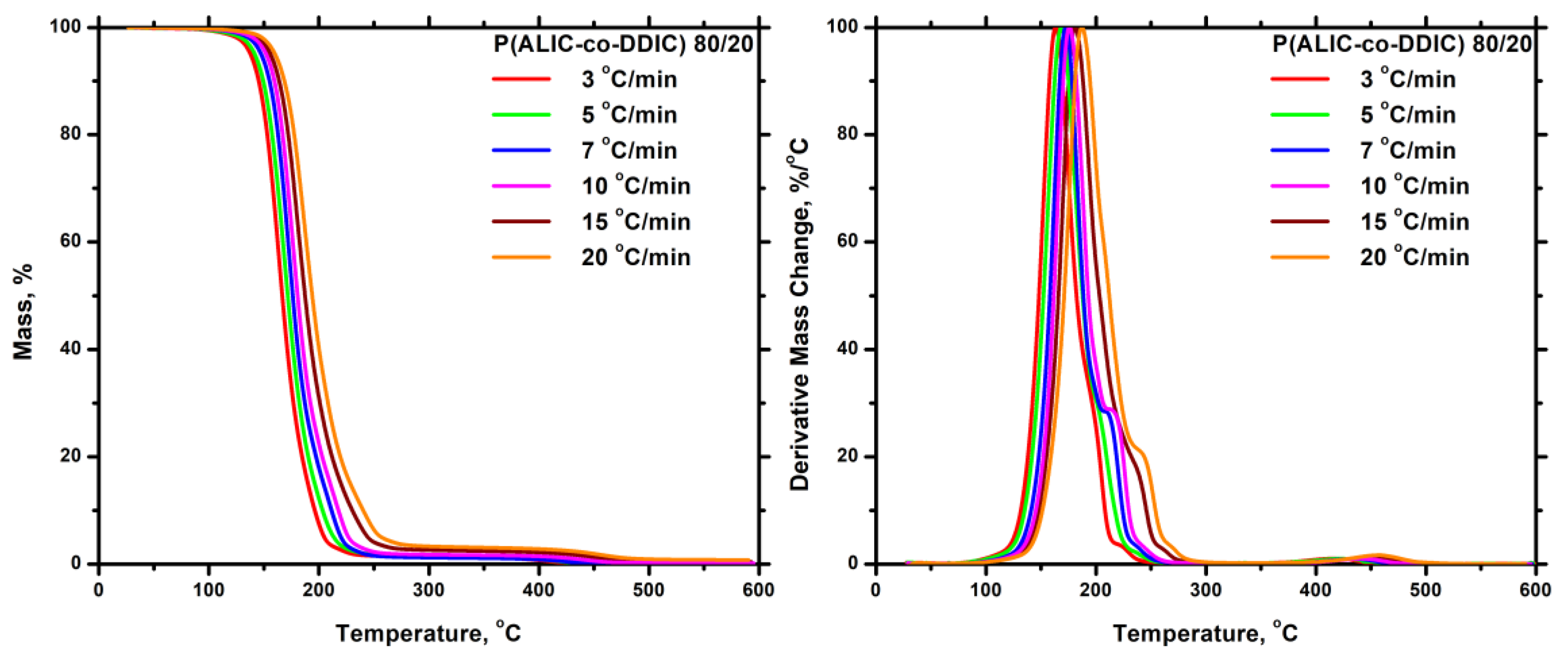
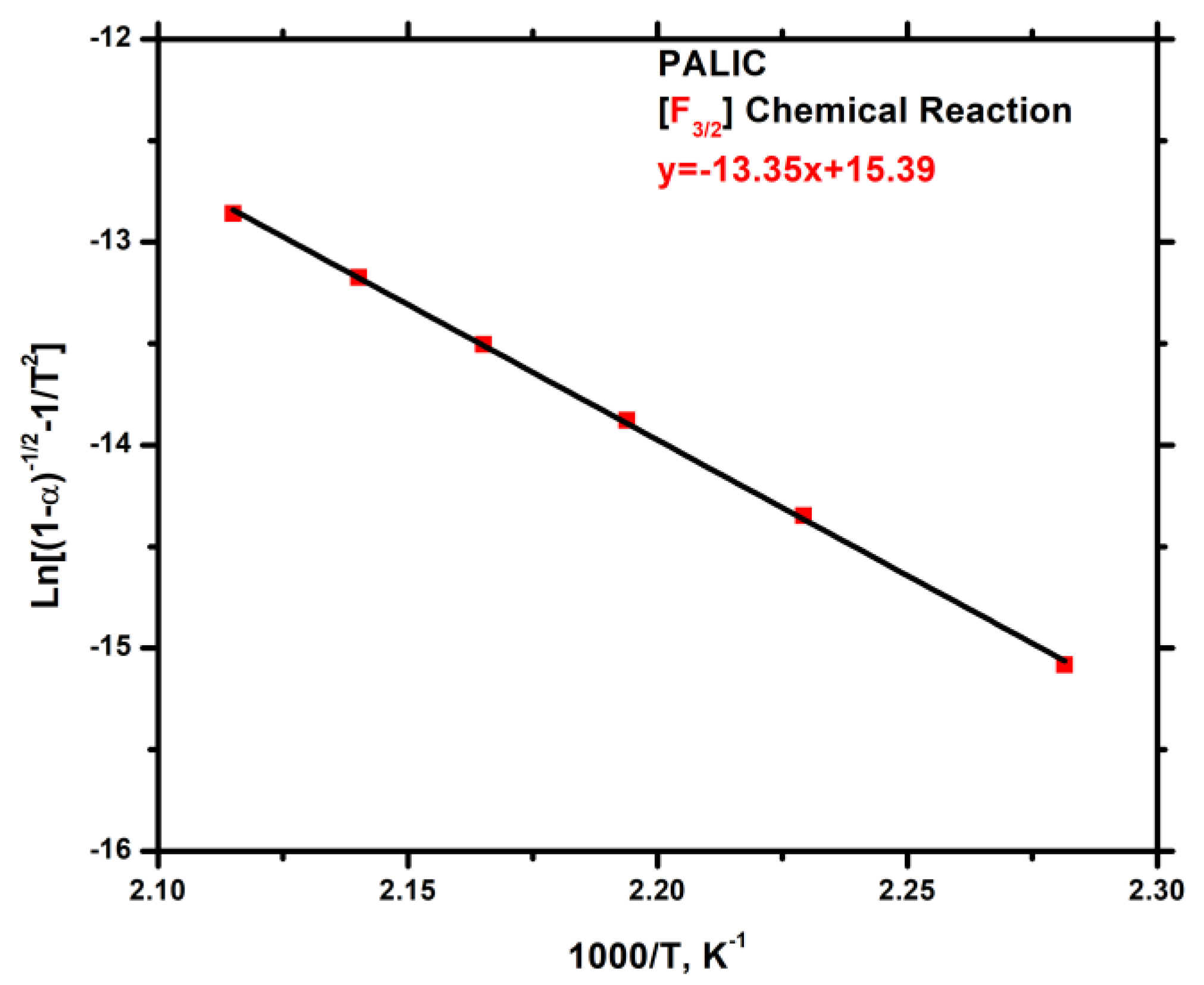
3.7.3. Kinetics of Thermal Degradation of Homopolymers and Statistical Copolymers
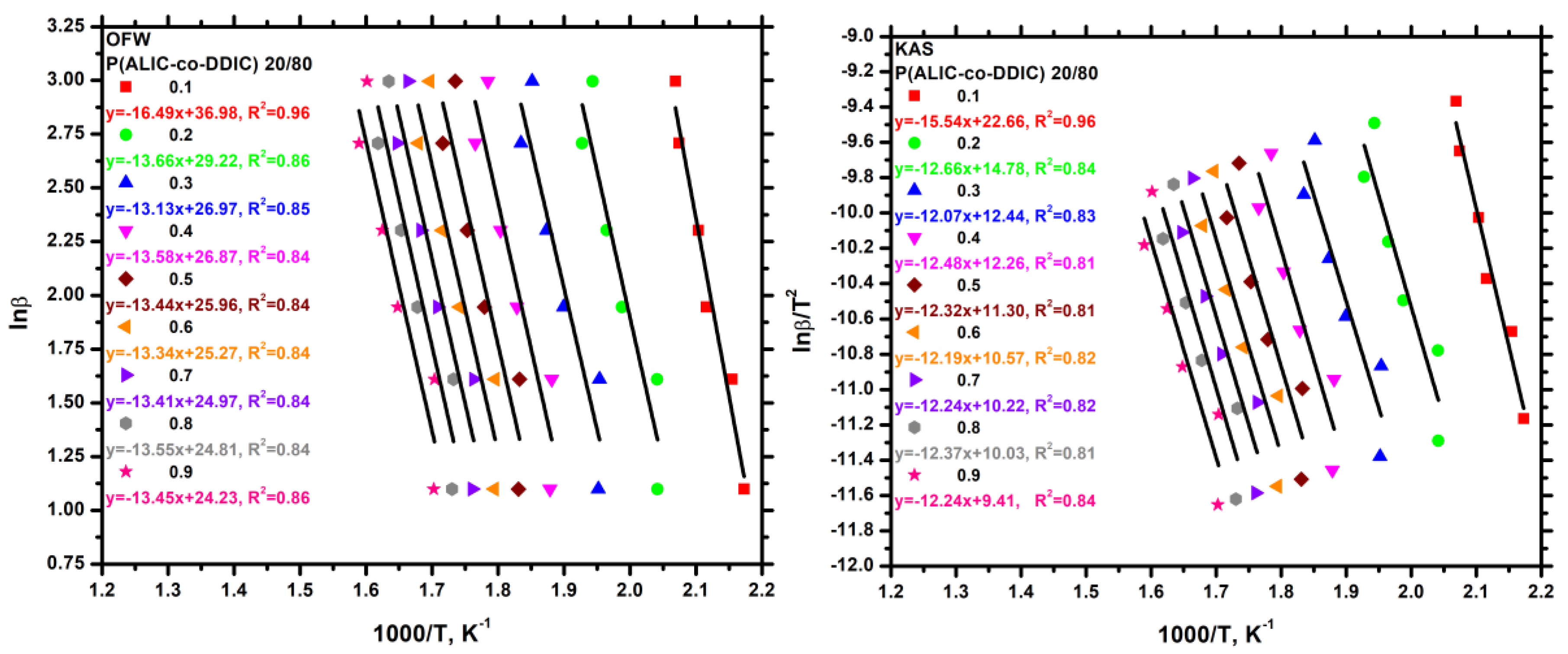
| Weight Loss (a) | PALIC | PDDIC | 20/80 | 40/60 | 50/50 | 60/40 | 80/20 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | OFW | KAS | |
| 0.1 | 94.56 | 87.26 | 162.04 | 153.24 | 136.95 | 129.14 | 117.67 | 110.11 | 111.94 | 104.46 | 143.26 | 135.95 | 122.32 | 115.18 |
| 0.2 | 102.13 | 94.65 | 135.12 | 125.65 | 113.51 | 105.12 | 111.02 | 103.13 | 107.78 | 100.05 | 128.22 | 120.66 | 122.49 | 115.26 |
| 0.3 | 110.94 | 103.29 | 121.49 | 111.77 | 109.03 | 100.30 | 96.15 | 87.84 | 100.63 | 95.65 | 124.15 | 116.42 | 122.66 | 115.26 |
| 0.4 | 120.49 | 112.77 | 116.42 | 106.45 | 112.85 | 103.71 | 93.16 | 84.60 | 88.34 | 80.12 | 119.33 | 111.44 | 121.24 | 113.85 |
| 0.5 | 130.55 | 122.82 | 113.76 | 103.71 | 111.69 | 102.30 | 96.89 | 88.00 | 84.76 | 76.26 | 104.71 | 96.64 | 118.33 | 110.77 |
| 0.6 | 145.43 | 137.53 | 112.27 | 102.05 | 110.77 | 101.22 | 99.97 | 90.83 | 88.50 | 79.77 | 96.31 | 88.09 | 113.60 | 106.04 |
| 0.7 | 110.86 | 100.55 | 111.44 | 101.71 | 99.72 | 90.41 | 92.99 | 83.931 | 95.07 | 86.59 | 107.03 | 99.30 | ||
| 0.8 | 109.28 | 98.89 | 112.68 | 102.71 | 100.14 | 90.58 | 92.90 | 83.60 | 99.39 | 90.66 | 100.14 | 92.24 | ||
| 0.9 | 106.20 | 95.81 | 111.69 | 101.63 | 101.88 | 92.07 | 95.81 | 86.09 | 105.54 | 96.40 | 84.60 | 76.44 | ||
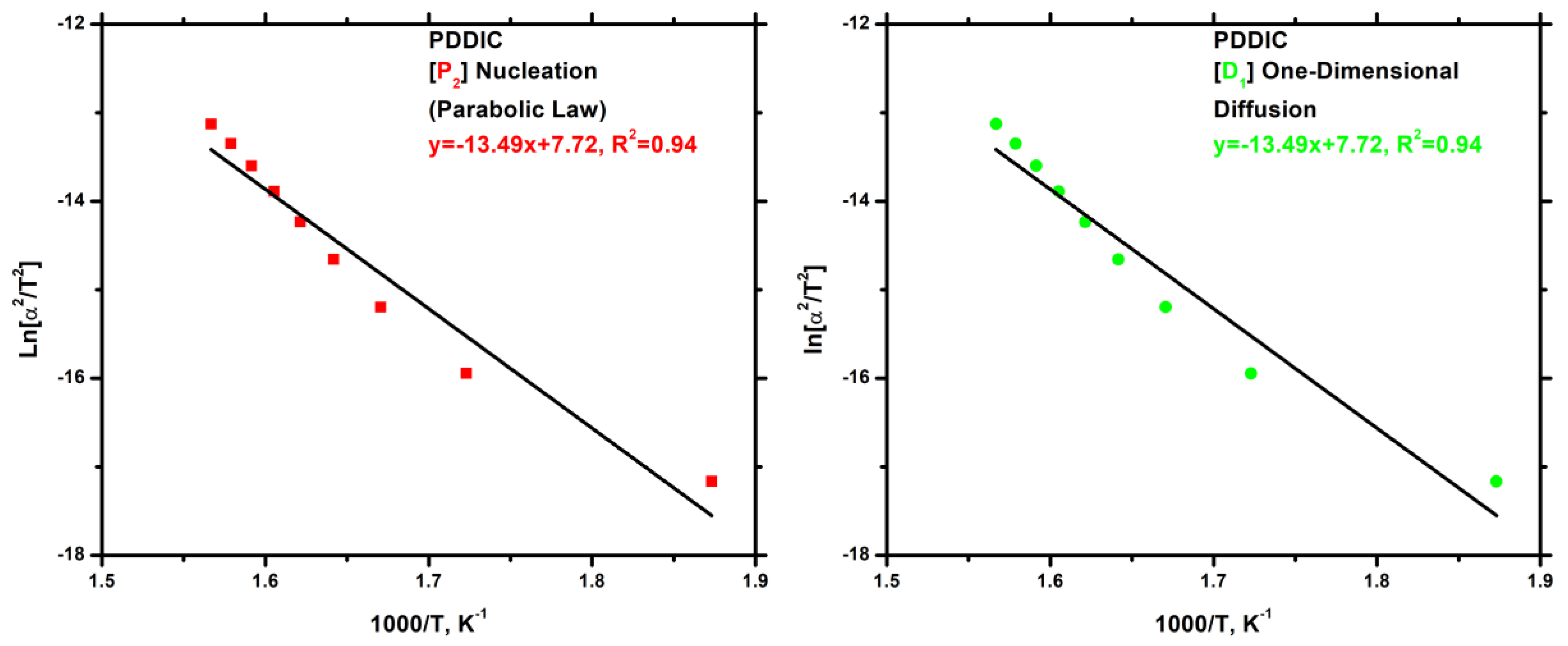
3.7.4. Block Copolymers
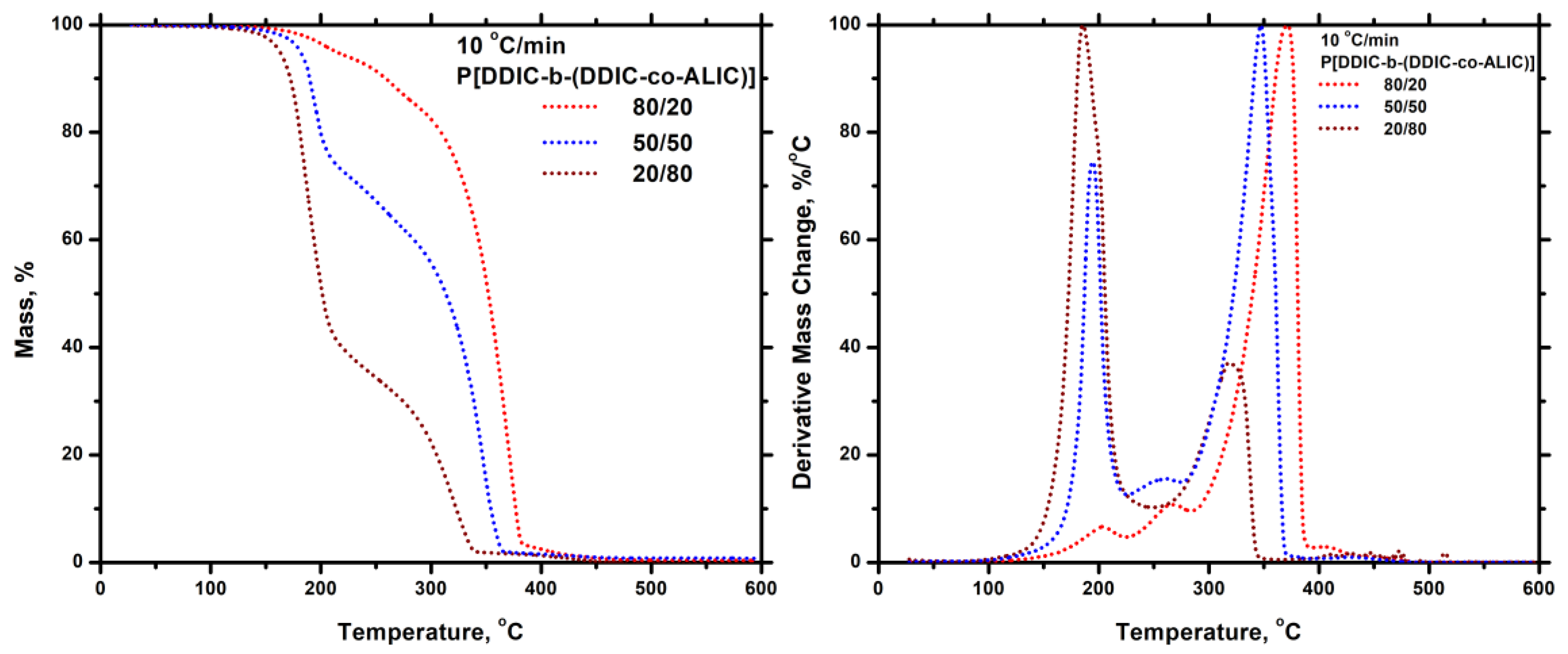
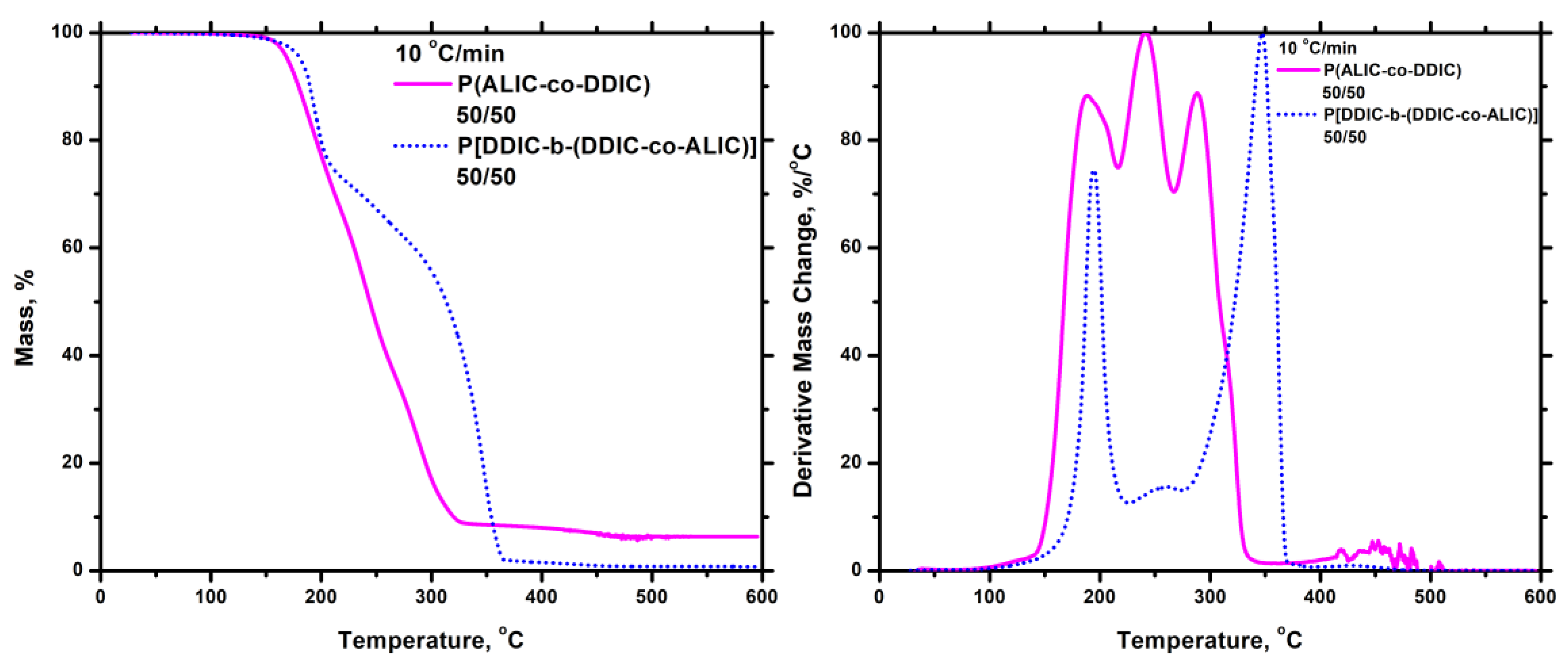
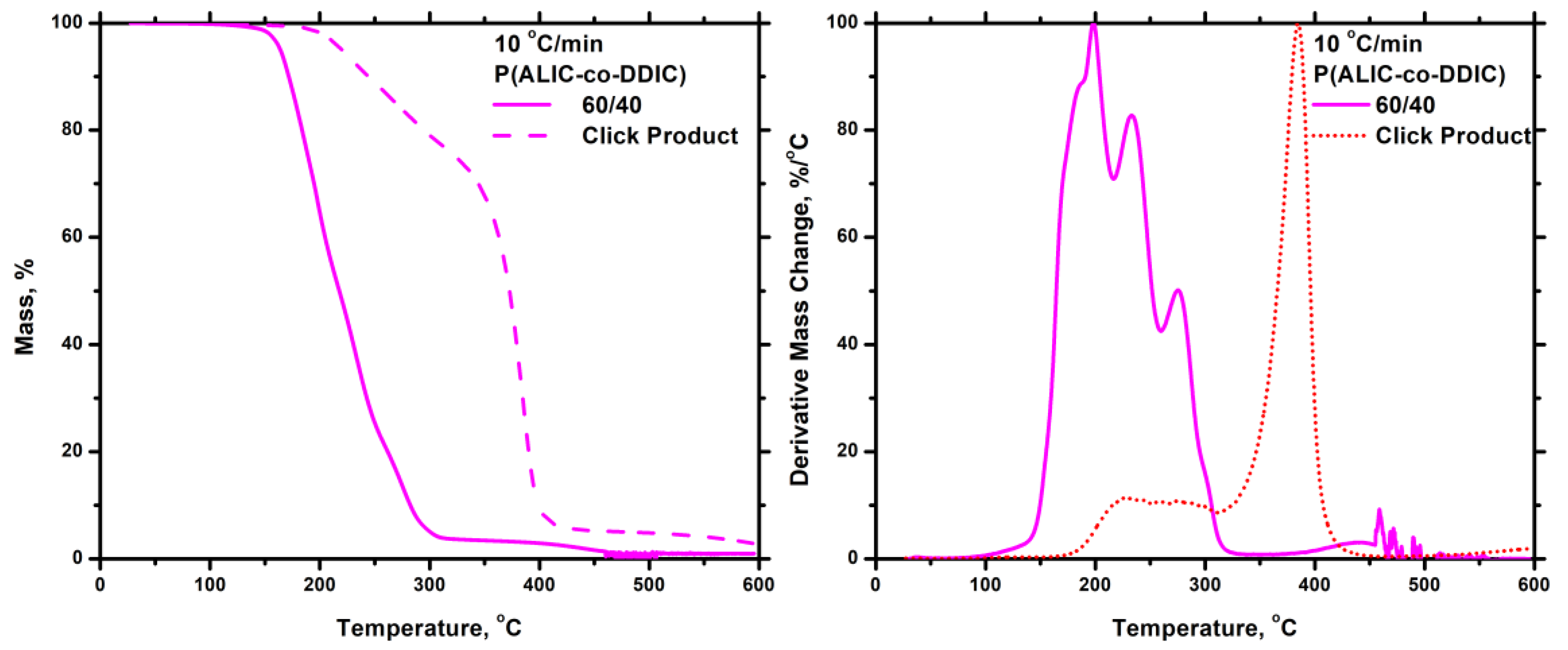
3.7.5. Thiol-Ene Products
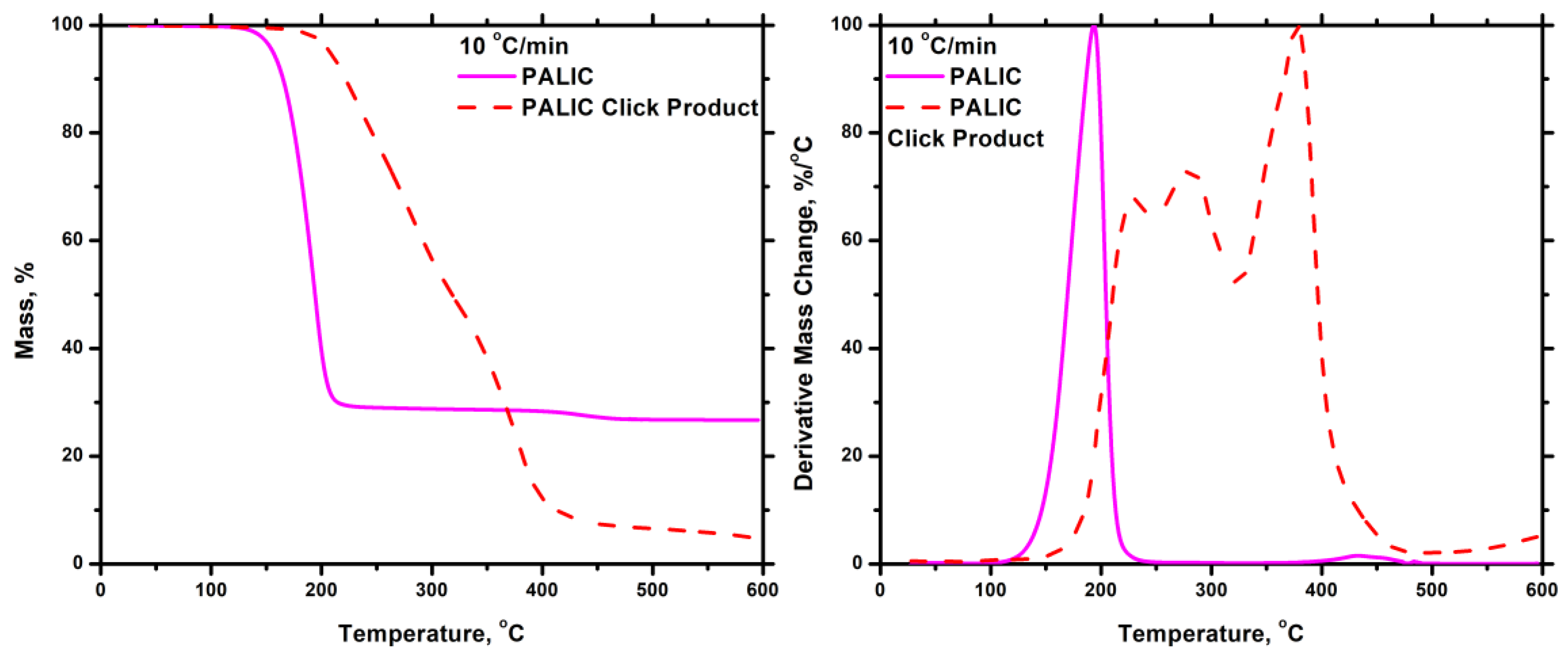
4. Conclusions
Supplementary Materials
Author Contributions
Data Availability Statement
Conflicts of Interest
References
- Kuran, W. Principles of coordination polymerization. J. Wiley & Sons LTD: Chichester, England, 2001. [Google Scholar]
- Novokshonova, L.A.; Zakharov, V.A. Kinetics of Olefin Polymerization and Active Sites of Heterogeneous Ziegler-Natta Catalysts. Adv. Polym. Sci. 2013, 257, 99–134. [Google Scholar] [CrossRef]
- Soga, K.; Shiono, T. Ziegler-Natta catalysts for olefin polymerizations. Progr. Polym. Sci. 1997, 22, 1503–1546. [Google Scholar] [CrossRef]
- Baugh, L.S.; Canich, J.A.M. (Eds.) Stereospecific polymerization with single site catalysts. CRC Press, Taylor and Francis Group: Boca Raton, USA, 2008. [Google Scholar]
- Brintzinger, H.H.; Fischer, D. Development of ansa-Metallocene Catalysts for Isotactic Olefin Polymerization. Adv. Polym. Sci. 2013, 258, 29–42. [Google Scholar] [CrossRef]
- Karanikolopoulos, G.; Batis, C.; Pitsikalis, M.; Hadjichristidis, N. The influence of the nature of the catalytic systems on the zirconocenes catalyzed polymerization of alkyl methacrylates. Macromol. Chem. Phys. 2003, 204, 831–840. [Google Scholar] [CrossRef]
- Chen, E.Y.X. Coordination Polymerization of Polar Vinyl Monomers by Single-Site Metal Catalysts. Chem. Rev. 2009, 109, 5157–5214. [Google Scholar] [CrossRef] [PubMed]
- Kourti, M.E.; Fotinogiannopoulou, G.; Fega, E.; Pitsikalis, M. Statistical Copolymers of Methyl and Phenyl Oxazoline by Metallocene Mediated Cationic Polymerization. Reactivity Ratios and Kinetics of Thermal Decomposition. J. Macromol. Sci. Part A: Pure Appl. Chem. 2015, 52, 630–641. [Google Scholar] [CrossRef]
- Batagianni, E.; Marathianos, A.; Koraki, A.; Maroudas, A.-P.; Pitsikalis, M. Metallocene-mediated cationic polymerization of vinyl ethers: Kinetics of polymerization and synthesis and characterization of statistical copolymers. J. Macromol. Sci. Part A: Pure Appl. Chem. 2016, 53, 140–151. [Google Scholar] [CrossRef]
- Zouganelis, S.; Choinopoulos, I.; Goulas, I.; Pitsikalis, M. Statistical copolymers of n-butyl vinyl ether and 2-chloroethyl vinyl ether via metallocene-mediated cationic polymerization. A scaffold for the synthesis of graft copolymers. Polymers 2019, 11, 1510. [Google Scholar] [CrossRef] [PubMed]
- Kostakis, K.; Mourmouris, S.; Karanikolopoulos, G.; Pitsikalis, M.; Hadjichristidis, N. Ring-opening polymerization of lactones using zirconocene catalytic systems. Block copolymerization with methyl methacrylate. J. Polym. Sci. Polym. Chem. Ed. 2007, 45, 3524–3537. [Google Scholar] [CrossRef]
- Khosravi, E.; Szymanska-Buzar, T. (Eds.) Ring opening metathesis polymerization and realated chemistry NATO Science Series. Springer-Science + Business Media: B.V. Dordrecht, 2002; Volume 56. [Google Scholar]
- Grubbs, R.H. (Ed.) Handbook of metathesis. Wiley-VCH: Weinheim, 2003. [Google Scholar]
- Giambastiani, G.; Cámpora, J. (Eds.) Olefin Upgrading catalysis by nitrogen-based metal complexes I. State of the art and perspectives. In Catalysis by metal complexes; Springer, 2011; Volume 35. [Google Scholar]
- Osakada, K. Olefin Polymerization with Non-metallocene Catalysts (Early Transition Metals). Organometallic Reactions and Polymerization 2014, 85, 89–117. [Google Scholar] [CrossRef]
- Takeuchi, D. Recent progress in olefin polymerization catalyzed by transition metal complexes: new catalysts and new reactions. Dalton Trans. 2010, 39, 311–328. [Google Scholar] [CrossRef]
- Wu, X.; Tamm, M. Transition metal complexes supported by highly basic imidazolin-2-iminato and imidazolin-2-imine N-donor ligands. Coord. Chem. Rev. 2014, 260, 116–138. [Google Scholar] [CrossRef]
- Batis, C.; Karanikolopoulos, G.; Pitsikalis, M.; Hadjichristidis, N. Complex macromolecular architectures utilizing metallocene catalysts. Macromolecules 2003, 36, 9763–9774. [Google Scholar] [CrossRef]
- Wang, Y.; Lai, J.; Gou, Q.Q.; Gao, R.; Zheng, G.; Zhang, R.D.; Song, Z.H.; Yue, Q.; Guo, Z.F. Development of well-defined olefin block (co)polymers achieved by late transition metal catalysts: Catalyst, synthesis and characterization. Coord. Chem. Rev. 2024, 522, 216195. [Google Scholar] [CrossRef]
- Βur, A.J.; Fetters, L.J. The Chain Structure, Polymerization and Conformation of Polyisocyanates. Chem. Rev. 1976, 76, 727–745. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. Organotitanium(IV) compounds as catalysts for the polymerization of isocyanates: the polymerization of isocyanates with functionalized side chains. Macromolecules 1993, 26, 436–439. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B. "Living" titanium(IV) catalyzed coordination polymerizations of isocyanates. J. Am. Chem. Soc. 1991, 113, 5065–5066. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. Living Organotitanium(IV)-Catalyzed Polymerizations of Isocyanates. J. Am. Chem. Soc. 1996, 118, 1906–1916. [Google Scholar] [CrossRef]
- Chae, C.-G.; Seo, H.-B.; Lee, J.-S. Living anionic polymerization of isocyanates in Anionic Polymerization. Priciples, practice, strength, consequences and applications. Hadjichristidis, N., Hirao, A., Eds.; Springer: Japan, 2015; pp. 339–386. [Google Scholar] [CrossRef]
- Shashoua, V.E.; Sweeny, W.E.; Tiet, R.F. The Homopolymerization of Monoisocyanates. J. Am. Chem. Soc. 1960, 82, 866–873. [Google Scholar] [CrossRef]
- Shashoua, V.E. The Homopolymerization of Monoisocyanates. J. Am. Chem. Soc 1959, 81, 3156. [Google Scholar] [CrossRef]
- Shin, Y.D.; Ahn, J.H.; Lee, J. Anionic polymerization of isocyanates with optical functionalities. Polymer 2001, 42, 7979–7985. [Google Scholar] [CrossRef]
- Shin, Y.D.; Kim, S.Y.; Ahn, J.H.; Lee, J.S. Synthesis of Poly(n-hexyl isocyanate) by Controlled Anionic Polymerization in the Presence of NaBPh4. Macromolecules 2001, 34, 2408–2410. [Google Scholar] [CrossRef]
- Ahn, J.H.; Shin, Y.D.; Kim, S.Y.; Lee, J.S. Synthesis of well-defined block copolymers of n-hexyl isocyanate with isoprene by living anionic polymerization. Polymer 2003, 44, 3847–3854. [Google Scholar] [CrossRef]
- Lee, J.-S.; Ryu, S.-W. Anionic living polymerization of 3-(triethoxysilyl)propyl isicyanate. Macromolecules 1999, 32, 2085–2087. [Google Scholar] [CrossRef]
- Kang, N.-G.; Kang, B.-G.; Koh, H.-D.; Changez, M.; Lee, J.-S. Block copolymers containing pyridine moieties: Precise synthesis and applications. React. Funct. Polym. 2009, 69, 470–479. [Google Scholar] [CrossRef]
- Vazaios, A.; Pitsikalis, M.; Hadjichristidis, N. Triblock copolymers and penatblock terpolymers of n-hexyl isocyanate with styrene and isoprene: synthesis, characterization and thermal properties. J. Polym. Sci. Polym. Chem. Ed. 2003, 41, 3094–3102. [Google Scholar] [CrossRef]
- Zorba, G.; Vazaios, A.; Pitsikalis, M.; Hadjichristidis, N. Anionic polymerization of n-hexyl isocyanate with monofunctional initiators. Application in the synthesis of diblock copolymers with styrene and isoprene. J. Polym. Sci. Polym. Chem. Ed. 2005, 43, 3533–3542. [Google Scholar] [CrossRef]
- Min, J.; Yoo, H.-S.; Shah, P.N.; Chae, C.-G.; Lee, J.-S. Enolate anionic initiator, sodium deoxybenzoin, for leading living natures by formation of aggregates at the growth chain ends. J. Polym. Sci. Polym. Chem. Ed. 2013, 51, 1742–1748. [Google Scholar] [CrossRef]
- Ann, J.-H.; Shin, Y.-D.; Nath, G.Y.; Park, S.-Y.; Rahman, M.S.; Samal, S.; Lee, J.-S. Unprecedented Control over Polymerization of n-Hexyl Isocyanate using an Anionic Initiator Having Synchronized Function of Chain-End Protection. J. Am. Chem. Soc. 2005, 127, 4132–4133. [Google Scholar] [CrossRef]
- Rahman, M.S.; Yoo, H.-S.; Changez, M.; Lee, J.-S. Living anionic polymerization of isocyanate containing a reactive carbamate group. Macromolecules 2009, 42, 3927–3932. [Google Scholar] [CrossRef]
- Zorba, G.; Pitsikalis, M.; Hadjichristidis, N. Novel well-defined star homopolymers and star-block copolymers of poly(n-hexyl isocyanate) by anionic polymerization. J. Polym. Sci. Polym. Chem. Ed. 2007, 45, 2387–2399. [Google Scholar] [CrossRef]
- Wu, J.; Pearce, E.M.; Kwei, T.K. A Novel Rod−Coil Block Copolymer and Its Compatible Blends. Macromolecules 2001, 34, 1828–1836. [Google Scholar] [CrossRef]
- Liu, H.; Deng, J.; Wu, Y.; Zhang, L. Amphiphilic triblock terpolymers of poly(n-hexyl isocyanate) and poly(ethylene glycol): Preparation and characterization. Polymer 2012, 53, 5717–5722. [Google Scholar] [CrossRef]
- Hoff, S.M.; Novak, B.M. Complex architectures through living polymerizations. The synthesis of "once-broken worms" and triblock copolymers using bimetallic initiators. Macromolecules 1993, 26, 4067–4069. [Google Scholar] [CrossRef]
- Touris, A.; Kostakis, K.; Mourmouris, S.; Kotzabasakis, V.; Pitsikalis, M.; Hadjichristidis, N. Complex Macromolecular Architectures Based on n-Hexyl Isocyanate and ϵ-Caprolactone Using Titanium-Mediated Coordination Polymerization. Macromolecules 2008, 41, 2426–2438. [Google Scholar] [CrossRef]
- Hoff, S.M.; Novak, B.M. Synthesis and Characterization of Wormlike Three-Arm Poly(n-hexyl isocyanate) Star Polymers. Macromolecules 2001, 34, 3849–3855. [Google Scholar] [CrossRef]
- Choinopoulos, I.; Patias, G.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of chiral poly(l-lactide-b-hexyl isocyanate) macromonomers with norbornenyl end groups and their homopolymerization through ring opening metathesis polymerization to afford polymer brushes. J. Polym. Sci. Part A: Polym. Chem. 2017, 55, 1102–1112. [Google Scholar] [CrossRef]
- Choinopoulos, I.; Patias, G.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of brush diblock and triblock copolymers bearing polynorbornene backbone and poly(l-lactide) and/or poly(hexyl isocyanate) side chains by a combination of coordination and ring opening metathesis polymerization. J. Polym. Sci. Part A: Polym. Chem. 2017, 55, 3455–3465. [Google Scholar] [CrossRef]
- Bhatt, M.P.; Du, J.; Rainbolt, E.A.; Pathiranage, T.M.S.K.; Huang, P.; Reuther, J.F.; Novak, B.M.; Biewer, M.C.; Stefan, M.C. A semiconducting liquid crystalline regioregular poly(3-hexylthiophene) and nematic poly(n-hexyl isocyanate) and its application in bulk heterojunction solar cells. J. Mater. Chem. 2014, 2, 16148–16156. [Google Scholar] [CrossRef]
- Miyake, G.M.; Weitekamp, R.A.; Piunova, V.A.; Grubbs, R.H. Synthesis of isocyanate-based brush block copolymers and their rapid self-assembly to infrared-reflecting photonic crystals. J. Am. Chem. Soc. 2012, 134, 14249–14254. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Nishikawa, N.; Kawato, D.; Suemasa, D.; Jung, S.; Kim, Y.Y.; Ree, M.; Kakuchi, T. Precise synthesis of a rod-coil miktoarm star copolymer containing poly(n-hexyl isocyanate) and aliphatic ester. Polym. Chem. 2014, 5, 588–599. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Mihara, T.; Kikuchi, M.; Lien, L.T.N.; Nagai, K. Synthesis of Methacrylate-Ended Poly(n-hexyl isocyanate) Rodlike Macromonomers and Their Radical Copolymerization Behavior. Macromolecules 2007, 40, 950–958. [Google Scholar] [CrossRef]
- Deike, S.; Binder, W.H. Induction of Chirality in β-Turn Mimetic Polymer Conjugates via Postpolymerization “Click” Coupling. Macromolecules 2017, 50, 2637–2644. [Google Scholar] [CrossRef]
- Schneider, N.S.; Furusaki, S. Chain stiffness in polyisocyanates. J. Polym. Sci. Part A 1965, 3, 933–948. [Google Scholar] [CrossRef]
- Zhao, W.; Kloczkowski, A.; Mark, J.E. Main-Chain Lyotropic Liquid-Crystalline Elastomers. 1. Syntheses of Cross-Linked Polyisocyanate Gels Acquiring Liquid-Crystalline Behavior in the Swollen State. Macromolecules 1996, 26, 2796–2804. [Google Scholar] [CrossRef]
- Gu, H.; Sato, T.; Teramoto, A.; Varichon, L.; Green, M.M. Molecular Mechanisms for the Optical Activities of Polyisocyanates Induced by Intramolecular Chiral Perturbations. Polym. J. 1997, 29, 77–84. [Google Scholar] [CrossRef]
- Tonelli, A. Conformational Characteristics of the Poly(n-alkyl isocyanates). Macromolecules 1974, 7, 628–631. [Google Scholar] [CrossRef]
- Lecomte, L.; Desreux, V. Dielectric properties of poly(4-methylphenylisocyanate) and poly(4-methoxyphenylisocyanate) in solution. Eur. Polym. J. 1976, 12, 741–747. [Google Scholar] [CrossRef]
- Chen, J.T.; Thomas, E.L.; Ober, C.K.; Mao, G.P. Self-Assembled Smectic Phases in Rod-Coil Block Copolymers. Science 1996, 273, 343–346. [Google Scholar] [CrossRef]
- Chen, J.T.; Thomas, E.L.; Ober, C.K.; Hwang, S.S. Zigzag morphology of a poly(styrene-b-hexyl isocyanate) rod-coil block copolymer. Macromolecules 1995, 28, 1688–1697. [Google Scholar] [CrossRef]
- Vazaios, A.; Touris, A.; Echeverria, M.; Zorba, G.; Pitsikalis, M. Micellization behaviour of linear and nonlinear block copolymers based on poly(n-hexyl isocyanate) in selective solvents. Polymers 2020, 12, 1678. [Google Scholar] [CrossRef] [PubMed]
- Green, M.M.; Peterson, N.C.; Sato, T.; Teramoto, A.; Cook, R. A Helical Polymer with a Cooperative Response to Chiral Information. Science 1995, 268, 1860–1866. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Matsuda, M.; Nakano, T.; Yashima, E. Asymmetric Polymerization of Isocyanates with Optically Active Anionic Initiators. Polym. J. 1993, 4, 391–396. [Google Scholar] [CrossRef]
- Μayer, S.; Zentel, R. A new chiral polyisocyanate: an optical switch triggered by a small amount of photochromic side groups. Macromol. Chem. Phys. 1998, 199, 1675–1682. [Google Scholar] [CrossRef]
- Baudis, S.; Ligon, S.C.; Seidler, K.; Weigel, G.; Grasl, C.; Bergmeister, H.; Schima, H.; Liska, R. Hard-Block Degradable Thermoplastic Urethane-Elastomers for Electrospun Vascular Prostheses. J. Polym. Sci. Part A: Polym. Chem. 2012, 50, 1272–1280. [Google Scholar] [CrossRef]
- Godfrey, R.A.; Miller, G.W. Block polymers of isocyanates and vinyl monomers by homogeneous anionic polymerization. J. Polym. Sci. 1969, 7, 2387–2404. [Google Scholar] [CrossRef]
- Chae, C.-G.; Shah, P.N.; Min, J.; Seo, H.-B.; Lee, J.-S. Synthesis of novel amphiphilic polyisocyanate block copolymer with hydroxyl side group. Macromolecules 2014, 47, 1561–1569. [Google Scholar] [CrossRef]
- Patten, T.E.; Novak, B.M. Well-defined polyisocyanates via organotitanium(IV) catalyzed living polymerization of isocyanates. Makromol. Chem. Macromol. Symp. 1993, 67, 203–211. [Google Scholar] [CrossRef]
- Zhao, W.; Kloczkowski, A.; Mark, J.E.; Erman, B.; Bahar, I. Main-chain lyotropic liquid-crystalline elastomers. 1. Syntheses of cross-linked polyisocyanate gels acquiring liquid-crystalline behavior in the swollen state. Macromolecules 1996, 29, 2796–2804. [Google Scholar] [CrossRef]
- Ratkanthwar, K.; Zhao, J.; Zhang, H.; Hadjichristidis, N.; Mays, J.W. Schlenk techniques for anioinic polymerization. In Anionic Polymerization: Principles, practice, strength, consequences and applications; Hadjichristidis, N., Hirao, A., Eds.; Springer: Japan, 2015. [Google Scholar] [CrossRef]
- Hadjichristidis, N.; Iatrou, H.; Pispas, S.; Pitsikalis, M. Anionic polymerization: High vacuum techniques. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3211–3234. [Google Scholar] [CrossRef]
- Hagiopol, C. Copolymerization: Toward a Systematic Approach. Springer Science & Business Media: Berlin, Germany, 2012. [Google Scholar]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Macromol. Sci. Part: A Chem. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Kelen, T.; Tüdos, F. Analysis of the Linear Methods for Determining Copolymerization Reactivity Ratios. I. A New Improved Linear Graphic Method. J. Macromol. Sci. Part: A Chem. 1975, 9, 1–27. [Google Scholar] [CrossRef]
- Beginn, U. COPOINT—A simple computer program to determine copolymerization parameters by numerical integration. E-Polymers 2005, 5, 759–773. [Google Scholar] [CrossRef]
- Odian, G. Principles of Polymerization. John Wiley & Sons: Hoboken, NJ, USA, 2004. [Google Scholar]
- Igarashi, S. Representation of composition and blockiness of the copolymer by a triangular coordinate system. J. Polym. Sci. Part: B Polym. Lett. 1963, 1, 359–363. [Google Scholar] [CrossRef]
- Elias, H.G. Synthesis, Materials, and Technology. Springer: New York, 1984; Volume 2. [Google Scholar]
- Mantzara, D.; Katara, A.; Panteli, M.; Choinopoulos, I.; Pitsikalis, M. Synthesis and characterization of statistical and block copolymers of n-hexyl isocyanate and 2-chloroethyl isocyanate via coordination polymerization. J. Polym. Sci. 2024, 62, 2484–2502. [Google Scholar] [CrossRef]
- Katara, A.; Mantzara, D.; Panteli, M.; Choinopoulos, I.; Pitsikalis, M. Statistical and Block Copolymers of n-Hexyl Isocyanate and 2-Phenylethyl Isocyanate via Coordination Polymerization. Synthesis, Characterization and Thermal Properties. Eur. Polym. J. 2023, 199, 112441. [Google Scholar] [CrossRef]
- Panteli, M.; Mantzara, D.; Katara, A.; Choinopoulos, I.; Pitsikalis, M. Synthesis and characterization of Statistical and Block Copolymers of n-Hexyl Isocyanate and 3-(triethoxysilyl) propyl Isocyanate via Coordination Polymerization. Polymers 2023, 15, 4113. [Google Scholar] [CrossRef]
- Lowe, A.B. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polymer Chemistry 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Jordan, E.F., Jr. Side-chain crystallinity. III. Influence of side-chain crystallinity on the glass transition temperatures of selected copolymers incorporating n-octadecyl acrylate or vinyl stearate. J. Polym. Sci. Part A-1 1971, 9, 3367–3378. [Google Scholar] [CrossRef]
- Alig, I.; Jarek, M.; Hellmann, G.P. Restricted segmental mobility in side-chain crystalline comblike polymers, studied by dielectric relaxation measurements. Macromolecules 1998, 31, 2245–2251. [Google Scholar] [CrossRef]
- Ozawa, T. A new method of analyzing thermogravimetric data. Bull. Chem. Soc. Jpn. 1965, 38, 1881–1886. [Google Scholar] [CrossRef]
- Flynn, J.H.; Wall, L.A. A quick, direct method for the determination of activation energy from thermogravimetric data. J. Polym. Sci. Polym. Lett. Ed. 1966, 4, 323–328. [Google Scholar] [CrossRef]
- Ozawa, T. Kinetic analysis of derivative curves in thermal analysis. J. Therm. Anal. Calorim. 1970, 2, 301–324. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction Kinetics in Differential Thermal Analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Vyazovkin, S.; Sbirrazzuoli, N. Isoconversional Kinetic Analysis of Thermally Stimulated Processes in Polymers. Macromol. Rapid Commun. 2006, 27, 1515–1532. [Google Scholar] [CrossRef]
- Doyle, C.D. Kinetic analysis of thermogravimetric data. J. Appl. Polym. Sci. 1961, 5, 285–292. [Google Scholar] [CrossRef]
- Coats, A.W.; Redfern, J. Kinetic parameters from thermogravimetric data. Nature 1964, 201, 68–69. [Google Scholar] [CrossRef]
- Trache, D.; Abdelaziz, A.; Siouani, B. A simple and linear isoconversional method to determine the pre-exponential factors and the mathematical reaction mechanism functions. J. Therm. Anal. Calorim. 2017, 128, 335–348. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



