
Submitted:
26 November 2024
Posted:
27 November 2024
You are already at the latest version
Abstract
Glioblastomas are the most aggressive tumors of the nervous system, whose treatment success is associated with many problems, including resistance to therapy and the permeability of the blood-brain barrier (BBB). The blood-brain barrier is equipped with ATP-binding cassette (ABC) transporters that play an important function by excreting toxic compounds. Two transporters in particular, ABCB1 (P-glycoprotein) and ABCG2 (BCRP), play an important role both at the BBB and in the development of drug resistance, leading to low intracellular drug concentrations. We investigated the antiproliferative activity of fifteen hybrids comprising harmine and quino-line-based antimalarials with triazole- and ureido-type linkers on neuroblastoma and glioblastoma cell lines. We also investigated their inhibitory effect on the ABCB1 and ABCG2 transporters using cell-based functional assays. We demonstrated that all hybrids showed significantly stronger anti-proliferative effect on glioblastoma cells than the parent compounds and also than the reference drug temozolomide. In addition, a number of derivatives showed remarkable inhibitory effects on ABC transporters, with ureido-type derivatives, harmiprime PU-9 and harmiquine MU-9 exerting the most pronounced dual inhibitory effect on both transporters. These results indicate that hy-brids of harmine and quinoline scaffolds have potential for glioblastoma drug discovery and for the discovery of new dual (ABCB1/ABCG2) inhibitors.
Keywords:
hybrid compounds
; harmine
; chloroquine
; mefloquine
; primaquine
; ATP-binding cassette (ABC) transporters
; dual inhibitors
; multi-drug resistance
; antitumor
; glioblastoma
1. Introduction
The concept of molecular hybridization is a rational strategy for the development of new and efficient biological agents. It involves the synthesis of a new hybrid compound by combining two or more different and independently acting molecules or their pharmacophores [1,2]. This strategy is a sophisticated form of combination therapy that can be particularly valuable in the treatment of complex and multifactorial diseases such as cancer, infectious diseases and neurological disorders, where conventional targeted therapies often fail. One of the most important reasons for the low clinical efficacy of therapies, especially cancer chemotherapies, is multidrug resistance (MDR). Therefore, the use of properly designed hybrids can ensure that different features of cancer cells are affected simultaneously to circumvent the pre-existing resistance or avoid the emergence of resistance mechanisms [3].
These challenging conditions require further continuous efforts in the development of new treatment strategies that are not only more efficient but also economically acceptable. The slow pace and increasing failure rate of drug discovery has led to the exploration of complementary strategies based on drug repurposing. This is a low-cost, low-risk and efficient approach for identifying new uses for approved or investigational drugs that are outside the scope of the original medical indication [4]. Similarly, hybrid drug design by integrating different pharmacophores from known or failed drugs and/or natural products is an alternative valuable strategy to combat cancer [5].
A number of antimalarial drugs have been developed directly or indirectly from the naturally occurring substances quinine (e.g. mefloquine, chloroquine and primaquine) and artemisinin (e.g. artesunate). In addition to their antiparasitic effect, these drugs have a variety of biological activities, including antibiotic, antitumor and anti-inflammatory effects [5,6,7,8,9]. An intensive area of research is therefore the repurposing and derivatization of antimalarials, particularly as anticancer drugs, as several classes of antimalarials have either direct or adjuvant anticancer effects, inhibit the development of drug resistance or have synergistic effects with known anticancer drugs. In addition, many of them are currently being investigated in clinical trials, alone or in combination with conventional anticancer drugs [5,8]. At the same time, a number of anticancer agents have shown remarkable potential to fight malaria and are now being repurposed against malaria [10,2].
Our recent research has been focused on the hybridization of quinoline-based antimalarials with harmine and related β-carboline alkaloids. Harmine, a prominent member of the β-carboline family, exhibits diverse biological activities, including anti-inflammatory, neuroprotective, antidiabetic, and antitumor effects, along with insecticidal, antiviral, and antibacterial properties [11,12]. It shows antimalarial activity by selective inhibition of Plasmodium falciparum heat shock protein 90 (PfHsp90) and enhances the effects of chloroquine and artemisinin in vitro and in a Plasmodium berghei mouse model [13]. Additionally, harmine’s antitumor effects involve cell cycle arrest, apoptosis, and reversal of drug resistance, making it a promising candidate for the treatment of both cancer and malaria [12,2,14].
To this end, we prepared two related series of compounds, harmiquins (hybrids of β -carbolines and chloroquine (CQ) or mefloquine (MQ)) and harmiprims (hybrids of b-carbolines and primaquine (PQ)), by varying the position of the substitution at the β -carboline ring (1, 3, 6, 7 and 9) and type of the linker between two parent moities. In both cases, urea or triazole were used as linkers due to their advantageous features (stability, ability to form hydrogen bonds, bioisosteric and favorable pharmacokinetic properties), resulting in urea- and triazole-type hybrids. We were able to show that these compounds exhibit improved activity and selectivity against Plasmodium and/or cancer cells, establishing them as valuable hits for further optimization [15,16,2].
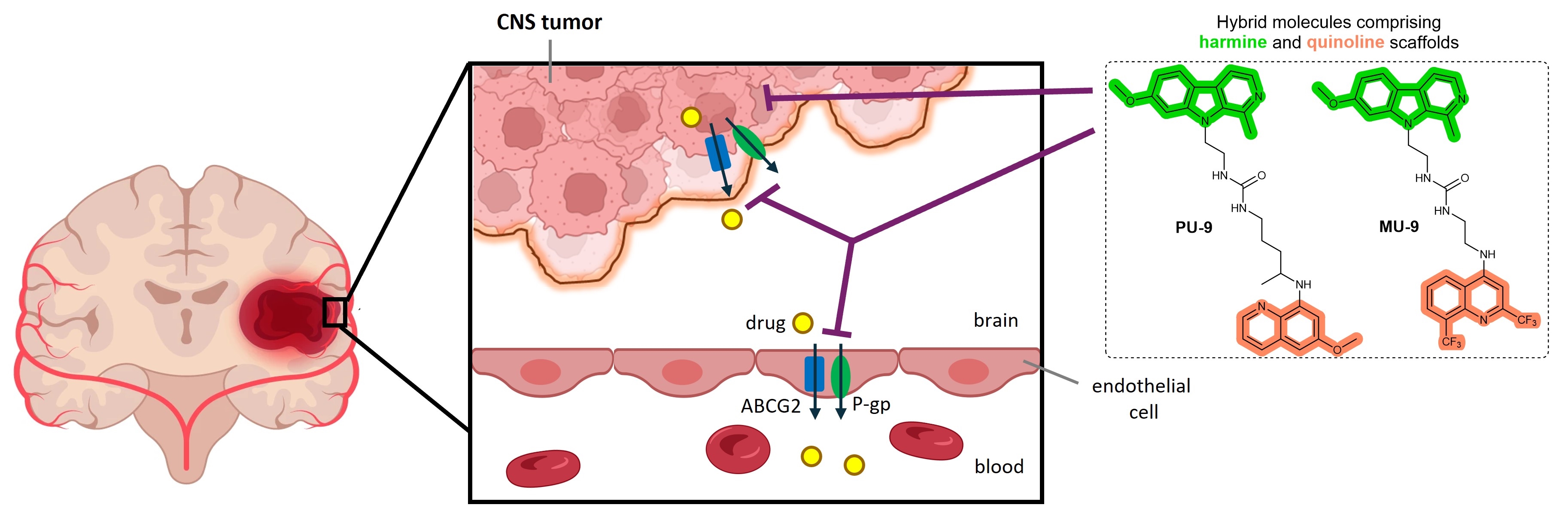
In this study, we investigated the antiproliferative activity of fifteen ureido- and triazole-type harmiquins and harmiprims (Figure 1) on neuroblastoma and glioblastoma cell lines. We also evaluated their potential inhibitory effect on ATP-binding cassette transporters (ABC transporters) – P-glycoprotein (ABCB1) and BCRP (ABCG2), as their expression hinders the penetration of chemotherapeutics into the brain and thus impedes the successful therapy of brain tumors. We demonstrated that all tested hybrids showed significantly stronger antiproliferative effects on glioblastoma cells compared to the parent compounds, i.e. harmine and the antimalarials PQ or CQ, as well as the reference drug temozolomide (TZM). In addition, a number of derivatives showed remarkable inhibitory effects on both ABC transporters, with ureido-type harmiprime PU-9 and harmiquine MU-9 exerting the most pronounced activity on both transporters, comparable to the inhibitory reference compounds. These results show that hybrids of harmine and quinoline-based antimalarials could be promising hits for further development as anti-glioblastoma agents.
2. Results
2.1. Synthesis of hybrid compounds
The title hybrids, harmiprims and harmiquins, comprising tethered β-carboline and a quinoline moiety derived from antimalarials (CQ, MQ, or PQ) were prepared using previously established procedures [17,16,2]. Based on the linker type, the compounds are classified as triazole- or ureido-type hybrids. Triazole-type harmiquins (CT-6, CT-7, and CT-9) [17] and harmiprims (PT-6, PT-7, and PT-9)[2] were obtained by Cu(I)-catalyzed azide-alkyne cycloaddition between harmine/β-carboline-based alkynes and quinoline-based azides. Ureido-type harmiquins (CU-6, CU-7, CU-9, MU-6, MU-7, and MU-9) were synthesized by the 1,1′-carbonyldiimidazole-mediated coupling of harmine/β-carboline-based amines with quinoline-based amines[16]. Meanwhile, ureido-type harmiprims (PU-6, PU-7, and PU-9) were prepared through an addition–elimination reaction involving PQ-benzotriazolide and harmine-based amines under microwave irradiation [2]. Preparation of the amine, azide, and alkyne intermediates followed previously reported procedures [15,17,18,19,20,21].
2.2. In vitro antiproliferative activity
We previously prepared ureido-type and triazole-type harmiprims [2] as well as ureido-type [16] and triazole-type harmiquins [17] and reported their antitumor activity on a panel of human tumor cell lines: HepG2 (hepatocellular carcinoma), SW620 and HCT116 (colorectal adenocarcinoma), and MCF-7 (breast adenocarcinoma), along with a non-tumor cell line Hek293T (embryonic kidney). We also analyzed the influence of the linker between two structural motifs and the position of the β-carboline substitution. In general, the ureido-type harmiprims were slightly more active than their triazole-type analogs and less selective towards the Hek293T cell line. Significant differences in the activity of the compounds prepared at different β-carboline positions were also found.
Based on these results, in the present study, we chose ureido- and triazole-type harmiquins and harmiprims based on CQ or MQ scaffolds, prepared at the positions 6, 7, and 9 of the β-carboline alkaloid harmine and tested their antiproliferative activity against glioblastoma and neuroblastoma cell lines – U251 and SH-SY5Y. The parent compound harmine as well as CQ, PQ, and the reference drug TMZ were used as controls. We also compared their activities to those against non-tumor cell line Hek293T. The results obtained are shown in Table 1 and Supporting Figure S1. All compounds tested showed remarkable antiproliferative activity in a glioma cell line, while the activity in neuroblastoma cell line SH-SY5Y is less prominent but still significant. The ureido-type MQ derivatives were the most active and least selective, followed by ureido-type PQ hybrids, while ureido-type CQ derivatives were more selective. On the other hand, triazole-type hybrids were less active and more selective, with PT-6, PT-9, CT-6, and CT-9 being the most selective in regard to non-tumor cells. These results are in accordance with previously published results on other cell lines [16,2].
2.3. Effects of hybrids on ABCG2-mediated fluorescent substrate accumulation in model cells
To test the inhibitory potential of the hybrids against the ABCG2 transporter, we performed a cell-based functional assay. For the cell-based assay, we used PLB/ABCG2 and its parental line without significant ABC transporter expression (PLB-985) using the Hoechst 33342 (H 33342) uptake assay (Figure 2). The assay is based on the incubation of cells with a fluorescent ABCG2 substrate (Hoechst 33342) in the presence or absence of the tested compounds (we used a concentration of 10 µM for all compounds). The selected concentration allowed testing without toxic effects on the cells. Ko143, a high-affinity specific inhibitor of ABCG2, was used as a reference inhibitor at a concentration of 1 µM, resulting in complete inhibition of ABCG2 activity. In the case of a potent inhibitor, increased fluorescence within the cell can be measured as a result of ABCG2 inhibition.
The results clearly show that at the concentration tested (10 µM) a number of hybrids reduce the efflux of H 3342 from PLB/ABCG2 cells in a similar way or even more than the reference compound Ko143. The most effective hybrids were MU-9 and CU-9, while triazole-type harmiquins were inactive together with PT-9, CU-6, CU-7 and TMZ.
To test the potency of ABCG2 inhibition and to compare the strength of inhibition between the selected compounds, we performed the Hoechst 33342 uptake assay at a range of concentrations for each compound and calculated their IC50 concentrations (Figure 3). Our results show that the most potent hybrid is MU-9 (IC50 = 0.6 µM), followed by PU-9 (IC50 = 1.5 µM), and CU-9 (IC50 = 5 µM). The maximum inhibition was 100 % for all three compounds and was achieved at a concentration of 5 µM for MU-9 and 10 µM for PU-9 and CU-9.
2.4. Sensitization of ABCG2-expressing tumor cells to mitoxantrone by hybrid compounds
Based on the results obtained, we decided to test whether the hybrids could sensitize cells resistant to mitoxantrone (MX), a known substrate of ABCG2. Therefore, we used acute myeloid leukemia cells PLB-985, which lack significant expression of the ABC transporter, and their MX-resistant counterpart (PLB/ABCG2), a stable cell line overexpressing the ABCG2 transporter, as a model to study MDR in the context of ABCG2 overexpression. We treated them with non-toxic concentrations of the most active compounds PU-9, MU-9 and CU-9 (1 µM) (Figure 4). Interestingly, PU-9 and MU-9 lead to a pronounced sensitization of PLB/ABCG2 cells to MX, comparable to Ko143 (Table 2 and Figure 4A). On the other hand, harmiquine CU-9 at the concentration tested did not sensitize the cells to MX, which is consistent with the efficacy of ABCG2 inhibition in the previous experiment and shows that CU-9 was not active at the concentration used in the sensitization assay (1 µM). We also examined the toxicity of hybrids and MX in the parental cell line (Table 2 and Figure 4B). The compounds had no significant effect on the toxicity of MX in the parental PLB-985 cell line, which is expected since the parental cells do not express the ABCG2 transporter.
2.5. Effects of hybrids on P-gp-mediated fluorescent substrate accumulation in model cells
To test the ability of the hybrid derivatives to inhibit another important transporter involved in the MDR phenotype – P-gp – we used a different cell model: the ovarian carcinoma cell line A2780 and its adriamycin (doxorubicin)-resistant counterpart A2780/Adr, which overexpress P-gp (A2780/Adr) [23] and performed the calcein uptake assay. Calcein is a substrate for P-gp and therefore the presence of P-gp in the cells leads to an efflux of calcein, thereby reducing cellular fluorescence. The assay is similar to the H 33342 accumulation assay, in which the efflux of calcein is reduced in the presence of the inhibitory substance, as evidenced by increased fluorescence within the cells. We used verapamil, a known P-gp inhibitor, as a reference compound. The results show that the parent compounds primaquine and chloroquine as well as TMZ have no activity at the tested concentration of 10 µM (Figure 5). Harmiquins, with the exception of MU-6 and MU-9, also showed no significant inhibition of P-gp activity. On the other hand, all tested harmiprims together with MU-6 and MU-9 showed a pronounced inhibitory effect (Figure 5). In summary, these results indicate that some of the hybrid compounds may have dual inhibitory potential, i.e. they may be able to inhibit both ABCG2 and P-gp transporters.
We selected the most active hybrids in both experimental setups (ABCG2 and P-gp inhibition): PU-9 and MU-9, and measured their inhibitory effect, i.e. we tested their inhibitory effect at a range of concentrations (Figure 6). We also tested the harmiprime with the most pronounced activity towards P-gp, PT-9. Our results show that all compounds tested have similar potency, with the most potent hybrid being PU-9 (IC50 = 0.57 µM), followed by MU-9 (IC50 = 0.6 µM) and PT-9 (IC50 = 0.8 µM). The maximum inhibition of 100 % was achieved at a concentration of 5 µM (for PU-9 and MU-9) and 10 µM (for PT-9).
2.6. Sensitization of P-gp-expressing tumor cells to doxorubicine by hybrid compounds
MDR-reversing activity associated with P-gp overexpression was also investigated for hybrid compounds that showed promising inhibitory potential against P-gp (PT-9, PU-9, MU-9). Their ability to restore doxorubicin (DOXO) sensitivity of cells overexpressing P-gp was tested. We treated both parental and P-gp-overexpressing cell lines with a previously determined non-toxic concentration of 1 µM of the compounds in combination with DOXO at five serial tenfold dilutions and assessed cell viability by MTT assay. The compounds had no significant effect on the toxicity of DOXO in the parental A2780 cell line (Table 3, Figure 7). On the other hand, the combination treatment of the A2780/Adr cell line with PU-9 resulted in a clear sensitization of the A2780/Adr cell line to DOXO, followed by MU-9 and PT-9. However, none of the compounds had the same inhibitory effect as the reference inhibitor verapamil. To summarise, the hybrid compounds PU-9 and MU-9 exhibit dual inhibitory activity – for both the ABCG2 and P-gp transporters, while PT-9 exhibits selective inhibition of P-gp.
3. Discussion
Glioblastomas are the most common tumors of the nervous system. Their treatment is associated with a number of problems, including systemic toxicity, resistance to therapy, poor targeting and problems crossing the blood-brain barrier (BBB) [24]. Glioblastoma multiforme (GBM) is one of the most aggressive brain tumors, but treatment success is inadequate and the prognosis is still very poor. The standard treatment of glioblastoma is very limited and is based on chemotherapy with TMZ as the only drug with proven efficacy against high-grade gliomas [25]. The main reason for this is the cytological heterogeneity of GBM and drug resistance. Considerable efforts have been made in the field of drug discovery against GBM in the last decade, but success has been mainly limited to the preclinical level [24]. Therefore, new small molecule inhibitors with the ability to penetrate the CNS and new targets and modalities for treatment are needed.
In this study, we show that ureido- and triazole-type harmiquins and harmiprims exhibit significant antiproliferative activity on the glioblastoma cell line U251, while their effect on neuroblastoma cells SH-SY5Y is somewhat less potent. Most of the tested hybrids showed a significantly stronger antiproliferative effect on glioblastoma cells than the parent compounds, i.e. harmine and the antimalarials PQ or CQ. In addition, all compounds were significantly more effective compared to TMZ, demonstrating their potential for glioblastoma drug development.
We opted for hybrid molecules consisting of two active parts, both of which have previously shown potential antitumor activity against glioblastoma cells. Recently, harmine was shown to suppress the proliferation and migration of U251-MG and U373-MG cells by inhibiting the FAK/AKT signaling pathway, making it a promising drug for glioblastoma therapy [26]. In addition, antimalarials have already been repurposed as potential antitumor agents, with chloroquine showing a particularly interesting effect on glioma cell [27].
Another important obstacle to the successful treatment of tumors of the nervous system is caused by the aforementioned BBB [28]. The BBB is the inherent biological barrier that supplies nutrients to the central nervous system (CNS) through selective uptake of small molecules, but also protects the CNS from harmful endogenous and xenobiotic molecules [25]. In addition to more or less passive restrictions, the BBB is also equipped with efflux transporters, which belong to the superfamily of ATP-binding cassette transporters (ABC transporters) and exert an important barrier function on the endothelial cells (ECs) of the brain by effluxing toxic compounds. Consequently, they limit the penetration of almost all classical chemotherapeutics and new targeted cancer drugs into the brain [29,25,30].
Two transporters in particular, ABCB1 (also known as P-glycoprotein or multidrug resistance protein 1 (MDR1)) and ABCG2 (breast cancer resistance protein (BCRP)), play an important role at the BBB, as they are highly expressed in the ECs of the brain. In addition, their expression on tumor cells has long been recognized as a major cause of MDR of cancer cells and cancer stem-like cells (CSCs) [31,32,33]. Consequently, the active efflux of drugs such as temozolomide leads to low intracellular drug concentrations and thus to an insufficient antitumor effect [34].
It was previously shown that co-treatment with mefloquine and primaquine sensitizes drug-resistant cancer cells by increasing P-gp inhibition[35]. In addition, we and colleagues have previously investigated the influence of a hybrid molecule, sahaquine on glioblastoma cells [36]. Sahaquine consists of a hydroxamic acid and a primaquine component. Hydroxamic acid is the active part of suberoylanilide hydroxamic acid (SAHA, vorinostat), which has been tested in clinical trials for GBM as monotherapy and in combination with radiotherapy. The primaquine portion of sahaquine has been shown to reduce the activity of the P-glycoprotein, which contributes to resistance to glioblastoma multiforme.
Furthermore, harmine`s β-carboline core resembles the tetrahydro- β-carboline moiety found in the structure of known ABCG2 inhibitors Ko143 and FTC [37] and was indeed identified as an ABCG2 reversal agent; it inhibited ABCG2-mediated drug efflux and increased the cytotoxicity of MX (1 µM), while it did not inhibit ABCB1 (P-gp)-mediated drug efflux [14,38].
As our study focused in particular on the potential inhibitory effect of hybrids against ABCG2 and/or P-gp transporters, we hypothesized that the combination of two components with inhibitory effects on ABC transporters could lead to even more active compounds with potential dual inhibitory properties. Namely, several studies show that inhibition of ABCB1 and/or ABCG2 and thus enhancement of intracellular exposure to potent chemotherapeutic agents can improve the efficacy of TMZ therapy alone or in combination with other drugs in glioblastoma patients [28]. For example, using genetically modified mice, de Gooijer et al. demonstrated that the combined deletion of P-gp and ABCG2 significantly increased the brain penetration of temozolomide without altering systemic drug exposure. Furthermore, the same increase was achieved when temozolomide was administered in combination with the dual P-gp/BCRP inhibitor elacridar (GF120918) [25].
Our results have clearly shown that two ureido-type hybrid molecules – harmiprime PU-9 and harmiquine MU-9 – have a strong inhibitory effect on the transporters ABCG2 and P-gp. In addition, they successfully sensitized both cells resistant to mitoxantrone (an ABCG2 substrate) and cells resistant to doxorubicin (a canonical P-gp substrate) at low concentration (1 µM). Although several other hybrids also showed the ability to inhibit one or both transporters at higher concentrations (i.e. 10 µM), they were not effective at lower, non-toxic concentrations. This suggests that the substitution at position 9 of harmine, and urea linker, are the most favorable structural features for the inhibitory effect on the ABC transporter. These results encourage further testing in combination with temozolomide to see if they could improve the brain penetration of temozolomide. This offers the prospect of further improving the anti-tumor efficacy of this already active agent or new agents. Besides, GBM tumor cells themselves can also express these transporters, further contributing to the multidrug-resistant phenotype of GBM [30,34]. Many other tumor types, primarily cancer stem cells, achieve MDR phenotype by expression of multiple ABC transporters. Consequently, simultaneous inhibition of P-gp and BCRP may also increase the sensitivity of these cells to chemotherapy and may be used to prevent cancer recurrence.
In summary, this work demonstrates that novel hybrid compounds comprising β-carbolines and CQ- or MQ-based 4-aminoquinoline motifs – harmiquins – and PQ-based 8-aminoquinolines – harmiprims, with triazole-type and ureido-type linkers, exert prominent antiproliferative activity towards glioblastoma cells. In addition, ureido-type harmiprime PU-9 and harmiquine MU-9 demonstrate strong dual inhibitory effect against ABCG2 and P-pg transporter. These results form the basis for the discovery of novel dual (P-gp/ABCG2) inhibitors and for improving the anti-tumor efficacy of TMZ and/or other chemotherapeutics.
4. Materials and Methods
4.1. Materials
Hybrid compounds constituted of β-carbolines and CQ- or MQ-based 4-aminoquinoline motifs (harmiquins) and PQ-based 8-aminoquinolines (harmiprims) with triazole-type or ureido-type linker (Figure 1) were prepared as described in [17,16,2].
Chemotherapeutic agents mitoxantrone (MX) and adriamycin (doxorubicine – DOXO) as well as conventional P-gp and ABCG2 inhibitors verapamil and Ko143 were purchased from Sigma-Aldrich. Fluorescent dye Hoechst 33342 was purchased from Sigma-Aldrich and calcein-AM from Thermo Fisher.
4.2. Cell lines
The human glioblastoma cell line U251 (originally obtained from the National Cancer Institute) and the neuroblastoma cell line SH-SY5Y (CRL-2266) (originally obtained from the American Type Culture Collection, ATCC, USA) were utilized for cell proliferation assays. To investigate transporter inhibition, the human acute myeloid leukemia cell line PLB/ABCG2, with overexpressed ABCG2 (achieved through retroviral transduction and selection using the ABCG2 substrate MX, as described by Ujhelly et al., [39], was used along with its parental PLB-985 counterpart. These cell lines were generously provided by Dr. Balazs Sarkadi from the Institute of Enzymology at the Centre for Natural Sciences in Budapest, Hungary. In addition, an ovarian carcinoma cell line with P-gp transporter overexpression A2780/Adr (ECACC cat. no. 93112520) and its parental counterpart A2780 (ECACC cat. no. 93112519), both obtained from The European Collection of Authenticated Cell Cultures, were included in the transporter study.
The cell lines were cultured either in suspension (PLB-985) or as monolayers (U251, SH-SY5Y, A2780 and A2780/Adr), using RPMI-1640 or DMEM medium, respectively. Both mediums were supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
4.3. Cell proliferation assay
Cell proliferation and cytotoxicity assays were carried out following the method described previously [23,32]. Cells were plated in 96-well plates at different concentrations: 3×10⁴ cells per well for PLB-985 and PLB/ABCG2, and 3×10³ cells per well for U251, SH-SY5Y, A2780, and A2780/Adr. The following day, the test compounds were added in serial dilutions (ranging from 10⁻⁸ to 10⁻⁴ M), either alone or at a fixed concentration of 1 µM in combination with ABCG2 substrate (MX, 10⁻⁹–10⁻⁵ M) or P-gp transporter substrate (DOXO, 10⁻⁹–10⁻⁵ M). After 72 hours of incubation, cell viability was assessed using the MTT assay. The concentration of each compound required to inhibit 50% of cell growth (IC50) was determined from dose-response curves using linear regression analysis.
4.4. Calcein-AM accumulation assay
The Calcein-AM accumulation assay was conducted as described by Guberović et al. [23,32]. Cells A2780 and A2780/Adr were seeded in 96-well plates 24 hours prior to the experiment at a concentration of 3 × 10⁴ cells per well. On the day of the assay, the cells were washed with PBS, and the test compounds or verapamil were added at a final concentration of 10 µM in RPMI medium without FBS. Calcein-AM (Thermo Fisher) was then added at a final concentration of 0.25 µM, and the cells were incubated for 60 minutes at 37 °C in the dark. After incubation, the cells were washed with PBS, and calcein fluorescence was measured using a Tecan Genios microplate reader (Tecan, Männedorf, Switzerland) with excitation and emission wavelengths of 485 nm and 530 nm, respectively.
4.5. Hoechst 33342 uptake assay
The Hoechst 33342 uptake assay was carried out following the protocol described by Mioč et al. [32]. On the day of the experiment, PLB-985 and PLB/ABCG2 suspension cells were diluted in 96-well plates at a concentration of 3 × 10⁴ cells per well. The cells were washed with PBS, and the test compounds were added at final concentrations of 10 µM and 1 µM (for Ko143) in RPMI medium without FBS. For the ABCG2 transporter inhibition assay, the test compounds were added at concentrations of 0.1, 0.5, 5, and 10 µM. Hoechst 33342 fluorescent dye (Sigma Aldrich) was then added at a final concentration of 0.5 µM, and the mixture was incubated for 60 minutes at 37 °C in the dark. After incubation, the cells were washed with PBS, and Hoechst 33342 fluorescence was measured using a Tecan Genios microplate reader (Tecan, Männedorf, Switzerland) at excitation/emission wavelengths of 360 nm/465 nm. The inhibition of transporter activity was calculated as described by Telbisz et al. [40], by comparing the fluorescence intensity in the presence of the test compounds with the fluorescence intensity observed with the reference inhibitor Ko143, which induces maximal transporter inhibition.
4.6. Statistics
Data are presented as the mean ± standard deviation (SD). Statistical significance was evaluated using one-way ANOVA followed by Dunnett’s post-hoc test. All statistical analyses were performed using GraphPad Prism 5, with significance levels indicated as follows: NS (not significant), *p < 0.05, **p < 0.01, and ***p < 0.001.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Antiproliferative activity of hybrids of harmine and quinoline-based antimalarials in U251 and SH-SY5Y cell lines.
Author Contributions
Conceptualization, M.K.; methodology, M.M.,K.P., G.P.; writing—original draft preparation, M.K., M.M. G.P., Z.R.; writing—review and editing, M.M. G.P., Z.R., M.K.; funding acquisition, M.K. and Z.R. All authors have read and agreed to the published version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
We greatly appreciate the financial support of the Croatian Science Foundation under the projects IP-2013-5660 and UIP-2017-05-5160, as well as the FarmInova project (KK.01.1.1.02.0021) funded by the European Regional Development Fund..
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable
Data Availability Statement
The data presented in this study are available in this article and the Supplementary Materials.
Acknowledgments
We would like to thank Dr. Agnes Telbisz and Dr. Balazs Sarkadi from the ELKH Research Center for Natural Sciences, Budapest, Hungary, for the donation of the PLB/ABCG2 cell line.
Conflicts of Interest
The authors declare no conflicts of interest. “The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results”.
References
- Kucuksayan, E.; Ozben, T. Hybrid Compounds as Multitarget Directed Anticancer Agents. Curr. Top. Med. Chem. 2017, 17, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Pavić, K.; Poje, G.; Pessanha de Carvalho, L.; Tandarić, T.; Marinović, M.; Fontinha, D.; Held, J.; Prudêncio, M.; Piantanida, I.; Vianello, R.; et al. Discovery of Harmiprims, Harmine-Primaquine Hybrids, as Potent and Selective Anticancer and Antimalarial Compounds. Bioorg. Med. Chem. 2024, 105. [Google Scholar] [CrossRef] [PubMed]
- Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid Drugs—A Strategy for Overcoming Anticancer Drug Resistance? Mol. 2021, Vol. 26, Page 2601 2021, 26, 2601. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Zorc, B.; Perković, I.; Pavić, K.; Rajić, Z.; Beus, M. Primaquine Derivatives: Modifications of the Terminal Amino Group. Eur. J. Med. Chem. 2019, 182. [Google Scholar] [CrossRef]
- Hu, Y.Q.; Gao, C.; Zhang, S.; Xu, L.; Xu, Z.; Feng, L.S.; Wu, X.; Zhao, F. Quinoline Hybrids and Their Antiplasmodial and Antimalarial Activities. Eur. J. Med. Chem. 2017, 139, 22–47. [Google Scholar] [CrossRef]
- Kucharski, D.J.; Jaszczak, M.K.; Boratyński, P.J. A Review of Modifications of Quinoline Antimalarials: Mefloquine and (Hydroxy)Chloroquine. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Ellis, T.; Eze, E.; Raimi-Abraham, B.T. Malaria and Cancer: A Critical Review on the Established Associations and New Perspectives. Infect. Agent. Cancer 2021, 16, 1–14. [Google Scholar] [CrossRef]
- Ma, Z.; Woon, C.Y.N.; Liu, C.G.; Cheng, J.T.; You, M.; Sethi, G.; Wong, A.L.A.; Ho, P.C.L.; Zhang, D.; Ong, P.; et al. Repurposing Artemisinin and Its Derivatives as Anticancer Drugs: A Chance or Challenge? Front. Pharmacol. 2021, 12, 828856. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, H.; Yang, Y.; Chen, Z.S.; Zou, C.; Zhang, J. Chloroquine against Malaria, Cancers and Viral Diseases. Drug Discov. Today 2020, 25, 2012–2022. [Google Scholar] [CrossRef]
- Luo, B.; Song, X. A Comprehensive Overview of β-Carbolines and Its Derivatives as Anticancer Agents. Eur. J. Med. Chem. 2021, 224, 113688. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, D.; Yu, S. Pharmacological Effects of Harmine and Its Derivatives: A Review. Arch. Pharmacal Res. 2020 4312 2020, 43, 1259–1275. [Google Scholar] [CrossRef] [PubMed]
- Shahinas, D.; MacMullin, G.; Benedict, C.; Crandall, I.; Pillaid, D.R. Harmine Is a Potent Antimalarial Targeting Hsp90 and Synergizes with Chloroquine and Artemisinin. Antimicrob. Agents Chemother. 2012, 56, 4207. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wink, M. The Beta-Carboline Alkaloid Harmine Inhibits BCRP and Can Reverse Resistance to the Anticancer Drugs Mitoxantrone and Camptothecin in Breast Cancer Cells. Phyther. Res. 2010, 24, 146–149. [Google Scholar] [CrossRef]
- Poje, G.; Marinović, M.; Pavić, K.; Mioč, M.; Kralj, M.; de Carvalho, L.P.; Held, J.; Perković, I.; Rajić, Z. Harmicens, Novel Harmine and Ferrocene Hybrids: Design, Synthesis and Biological Activity. Int. J. Mol. Sci. 2022, 23, 9315. [Google Scholar] [CrossRef]
- Pavić, K.; Poje, G.; de Carvalho, L.P.; Held, J.; Rajić, Z. Synthesis, Antiproliferative and Antiplasmodial Evaluation of New Chloroquine and Mefloquine-Based Harmiquins. Acta Pharm. 2023, 73, 537–558. [Google Scholar] [CrossRef]
- Poje, G.; Pessanha de Carvalho, L.; Held, J.; Moita, D.; Prudêncio, M.; Perković, I.; Tandarić, T.; Vianello, R.; Rajić, Z. Design and Synthesis of Harmiquins, Harmine and Chloroquine Hybrids as Potent Antiplasmodial Agents. Eur. J. Med. Chem. 2022, 238, 114408. [Google Scholar] [CrossRef]
- Džimbeg, G.; Zorc, B.; Kralj, M.; Ester, K.; Pavelić, K.; Andrei, G.; Snoeck, R.; Balzarini, J.; De Clercq, E.; Mintas, M. The Novel Primaquine Derivatives of N-Alkyl, Cycloalkyl or Aryl Urea: Synthesis, Cytostatic and Antiviral Activity Evaluations. Eur. J. Med. Chem. 2008, 43. [Google Scholar] [CrossRef]
- Perković, I.; Raić-Malić, S.; Fontinha, D.; Prudêncio, M.; Pessanha de Carvalho, L.; Held, J.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Harmicines − Harmine and Cinnamic Acid Hybrids as Novel Antiplasmodial Hits. Eur. J. Med. Chem. 2020, 187, 111927. [Google Scholar] [CrossRef]
- Marinović, M.; Perković, I.; Fontinha, D.; Prudêncio, M.; Held, J.; de Carvalho, L.P.; Tandarić, T.; Vianello, R.; Zorc, B.; Rajić, Z. Novel Harmicines with Improved Potency against Plasmodium. Mol. 2020, Vol. 25, Page 4376 2020, 25, 4376. [Google Scholar] [CrossRef]
- Rajić, K.P.Z.; Mlinarić, Z.; Uzelac, L.; Kralj, M.; Zorc, B. Chloroquine Urea Derivatives: Synthesis and Antitumor Activity in Vitro. Acta Pharm. 2018, 68. [Google Scholar] [CrossRef]
- Poje, G.; Šakić, D.; Marinović, M.; You, J.; Tarpley, M.; Williams, K. P.; Golub, N.; Dernovšek, J.; Tomašić, T.; Bešić, E.; Rajić, Z. Unveiling the Antiglioblastoma Potential of Harmicens, Harmine and Ferrocene Hybrids. Acta Pharm. 2024, 74, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Guberović, I.; Marjanović, M.; Mioč, M.; Ester, K.; Martin-Kleiner, I.; Šumanovac Ramljak, T.; Mlinarić-Majerski, K.; Kralj, M. Crown Ethers Reverse P-Glycoprotein-Mediated Multidrug Resistance in Cancer Cells. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.; Faujdar, C.; Sharma, R.; Sharma, S.; Malik, B.; Nepali, K.; Liou, J.P. Glioblastoma: Current Status, Emerging Targets, and Recent Advances. J. Med. Chem. 2022, 65, 8596–8685. [Google Scholar] [CrossRef] [PubMed]
- de Gooijer, M.C.; de Vries, N.A.; Buckle, T.; Buil, L.C.M.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Improved Brain Penetration and Antitumor Efficacy of Temozolomide by Inhibition of ABCB1 and ABCG2. Neoplasia 2018, 20, 710. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Lv, Y.X.; Guo, C.Y.; Xiao, Z.M.; Jiang, Q.G.; Kuang, H.; Zhang, W.H.; Hu, P. Harmine Inhibits the Proliferation and Migration of Glioblastoma Cells via the FAK/AKT Pathway. Life Sci. 2021, 270, 119112. [Google Scholar] [CrossRef]
- Weyerhäuser, P.; Kantelhardt, S.R.; Kim, E.L. Re-Purposing Chloroquine for Glioblastoma: Potential Merits and Confounding Variables. Front. Oncol. 2018, 8, 335. [Google Scholar] [CrossRef]
- Lin, F.; De Gooijer, M.C.; Roig, E.M.; Buil, L.C.M.; Christner, S.M.; Beumer, J.H.; WEurdinger, T.; Beijnen, J.H.; Van Tellingen, O. ABCB1, ABCG2, and PTEN Determine the Response of Glioblastoma to Temozolomide and ABT-888 Therapy. Clin. Cancer Res. 2014, 20, 2703–2713. [Google Scholar] [CrossRef]
- Wijaya, J.; Fukuda, Y.; Schuetz, J.D. Obstacles to Brain Tumor Therapy: Key ABC Transporters. Int. J. Mol. Sci. 2017, Vol. 18, Page 2544 2017, 18, 2544. [Google Scholar] [CrossRef]
- Dréan, A.; Rosenberg, S.; Lejeune, F.X.; Goli, L.; Nadaradjane, A.A.; Guehennec, J.; Schmitt, C.; Verreault, M.; Bielle, F.; Mokhtari, K.; et al. ATP Binding Cassette (ABC) Transporters: Expression and Clinical Value in Glioblastoma. J. Neurooncol. 2018, 138, 479–486. [Google Scholar] [CrossRef]
- Sajid, A.; Rahman, H.; Ambudkar, S. V. Advances in the Structure, Mechanism and Targeting of Chemoresistance-Linked ABC Transporters. Nat. Rev. Cancer 2023, 23, 762–779. [Google Scholar] [CrossRef] [PubMed]
- Mioč, M.; Telbisz, Á.; Radman, K.; Bertoša, B.; Šumanovac, T.; Sarkadi, B.; Kralj, M. Interaction of Crown Ethers with the ABCG2 Transporter and Their Implication for Multidrug Resistance Reversal. Histochem. Cell Biol. 2022. [CrossRef] [PubMed]
- Begicevic, R.R.; Falasca, M. ABC Transporters in Cancer Stem Cells: Beyond Chemoresistance. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Radtke, L.; Majchrzak-Celińska, A.; Awortwe, C.; Vater, I.; Nagel, I.; Sebens, S.; Cascorbi, I.; Kaehler, M. CRISPR/Cas9-Induced Knockout Reveals the Role of ABCB1 in the Response to Temozolomide, Carmustine and Lomustine in Glioblastoma Multiforme. Pharmacol. Res. 2022, 185, 106510. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, A.R.; Kim, Y.K.; Yoon, S. Co-Treatment with the Anti-Malarial Drugs Mefloquine and Primaquine Highly Sensitizes Drug-Resistant Cancer Cells by Increasing P-Gp Inhibition. Biochem. Biophys. Res. Commun. 2013, 441, 655–660. [Google Scholar] [CrossRef]
- Zhang, I.; Beus, M.; Stochaj, U.; Le, P.U.; Zorc, B.; Rajić, Z.; Petrecca, K.; Maysinger, D. Inhibition of Glioblastoma Cell Proliferation, Invasion, and Mechanism of Action of a Novel Hydroxamic Acid Hybrid Molecule. Cell Death Discov. 2018 41 2018, 4, 1–14. [Google Scholar] [CrossRef]
- Spindler, A.; Stefan, K.; Wiese, M. Synthesis and Investigation of Tetrahydro-β-Carboline Derivatives as Inhibitors of the Breast Cancer Resistance Protein (ABCG2). J. Med. Chem. 2016, 59, 6121–6135. [Google Scholar] [CrossRef]
- Peña-Solórzano, D.; Stark, S.A.; König, B.; Sierra, C.A.; Ochoa-Puentes, C. ABCG2/BCRP: Specific AndNonspecific Modulators. Med. Res. Rev. 2017, 37, 987–1050. [Google Scholar] [CrossRef]
- Ujhelly, O.; Özvegy, C.; Várady, G.; Cervenak, J.; Homolya, L.; Grez, M.; Scheffer, G.; Roos, D.; Bates, S.E.; Váradi, A.; et al. Application of a Human Multidrug Transporter (ABCG2) Variant as Selectable Marker in Gene Transfer to Progenitor Cells. Hum. Gene Ther. 2003, 14, 403–412. [Google Scholar] [CrossRef]
- Telbisz, Á.; Ambrus, C.; Mózner, O.; Szabó, E.; Várady, G.; Bakos, É.; Sarkadi, B.; Özvegy-Laczka, C. Interactions of Potential Anti-COVID-19 Compounds with Multispecific ABC and OATP Drug Transporters. Pharmaceutics 2021, 13, 1–18. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of harmine-based hybrids (harmine/ β-carboline is marked in green, linker (L) in grey, pharmacophore 2 (P2) in red, triazole in blue, urea in yellow, C atom of pharmacophore directly attached to linker in violet).
Figure 1.
Chemical structures of harmine-based hybrids (harmine/ β-carboline is marked in green, linker (L) in grey, pharmacophore 2 (P2) in red, triazole in blue, urea in yellow, C atom of pharmacophore directly attached to linker in violet).

Figure 2.
Inhibition of ABCG2-mediated transport by hybrids, primaquine (PQ), chloroquine (CQ), and temozolomide (TMZ). The inhibitory potential of the compounds was assessed using the Hoechst 33342 accumulation assay in the PLB/ABCG2 cell line. The mean fluorescence intensity (MFI) of Hoechst 33342 was measured after incubation with substrates and treatment with 10 µM compounds. Uptake of Hoechst 33342 is shown relative to 100% uptake at maximum inhibition of ABCG2 by Ko143 (1 µM). All data are expressed as mean ± SD of 3 individual experiments. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 2.
Inhibition of ABCG2-mediated transport by hybrids, primaquine (PQ), chloroquine (CQ), and temozolomide (TMZ). The inhibitory potential of the compounds was assessed using the Hoechst 33342 accumulation assay in the PLB/ABCG2 cell line. The mean fluorescence intensity (MFI) of Hoechst 33342 was measured after incubation with substrates and treatment with 10 µM compounds. Uptake of Hoechst 33342 is shown relative to 100% uptake at maximum inhibition of ABCG2 by Ko143 (1 µM). All data are expressed as mean ± SD of 3 individual experiments. One-way ANOVA with Dunnett’s post hoc test was used for statistical analysis *p < 0.05; **p < 0.01; ***p < 0.001.
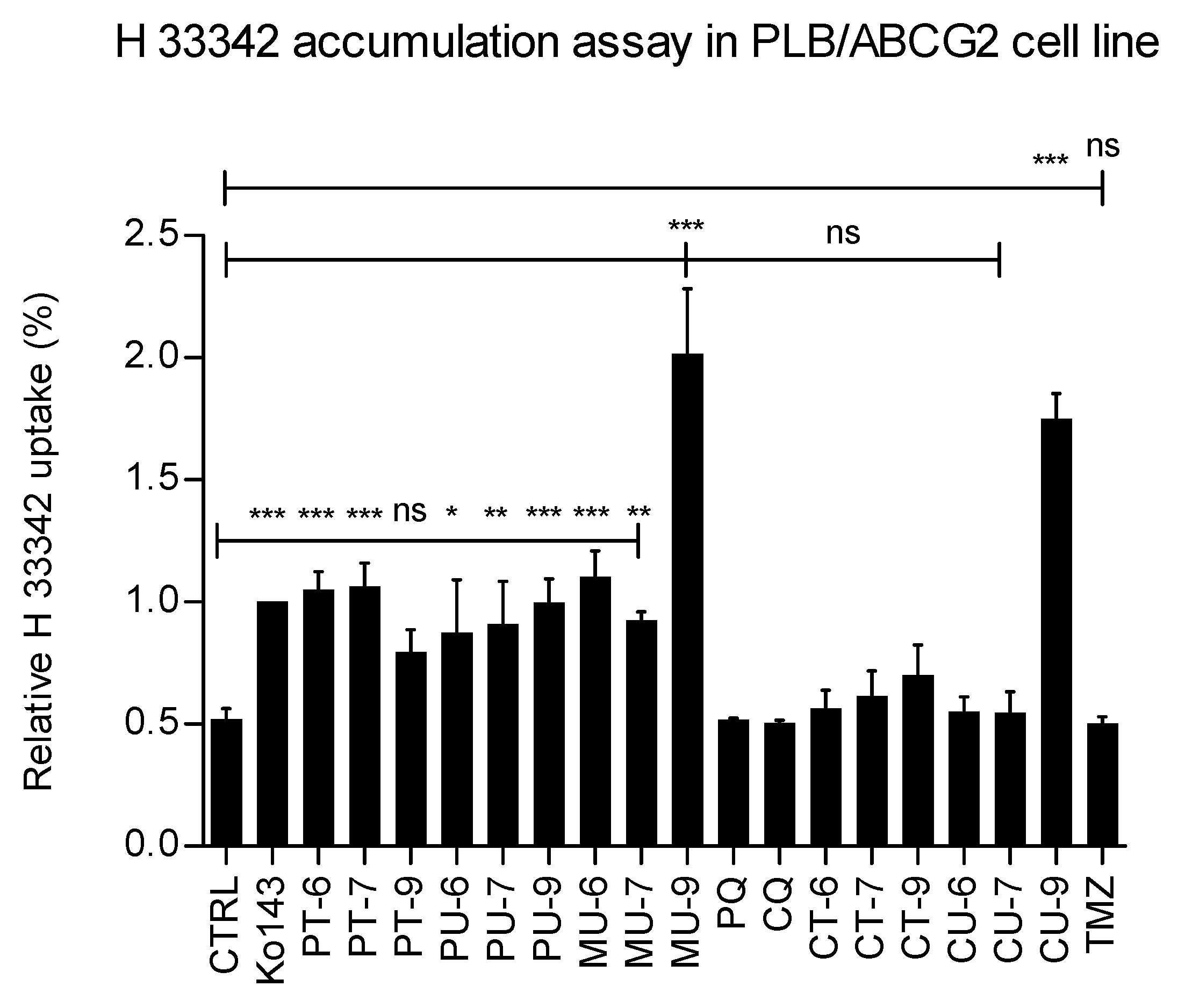
Figure 3.
The potency of the hybrid compounds PU-9, MU-9, and CU-9 to inhibit ABCG2 as measured by the Hoechst 33342 transport assay in the PLB/ABCG2 cell line. Inhibition of transporter activity was calculated by comparing fluorescence intensity in the presence of the tested compound with that in the presence of Ko143 (maximal inhibition). Data were plotted in triplicate ± SD from three independent experiments. The IC50 of each compound was calculated using a non-linear regression curve three independent experiments in triplicate.
Figure 3.
The potency of the hybrid compounds PU-9, MU-9, and CU-9 to inhibit ABCG2 as measured by the Hoechst 33342 transport assay in the PLB/ABCG2 cell line. Inhibition of transporter activity was calculated by comparing fluorescence intensity in the presence of the tested compound with that in the presence of Ko143 (maximal inhibition). Data were plotted in triplicate ± SD from three independent experiments. The IC50 of each compound was calculated using a non-linear regression curve three independent experiments in triplicate.
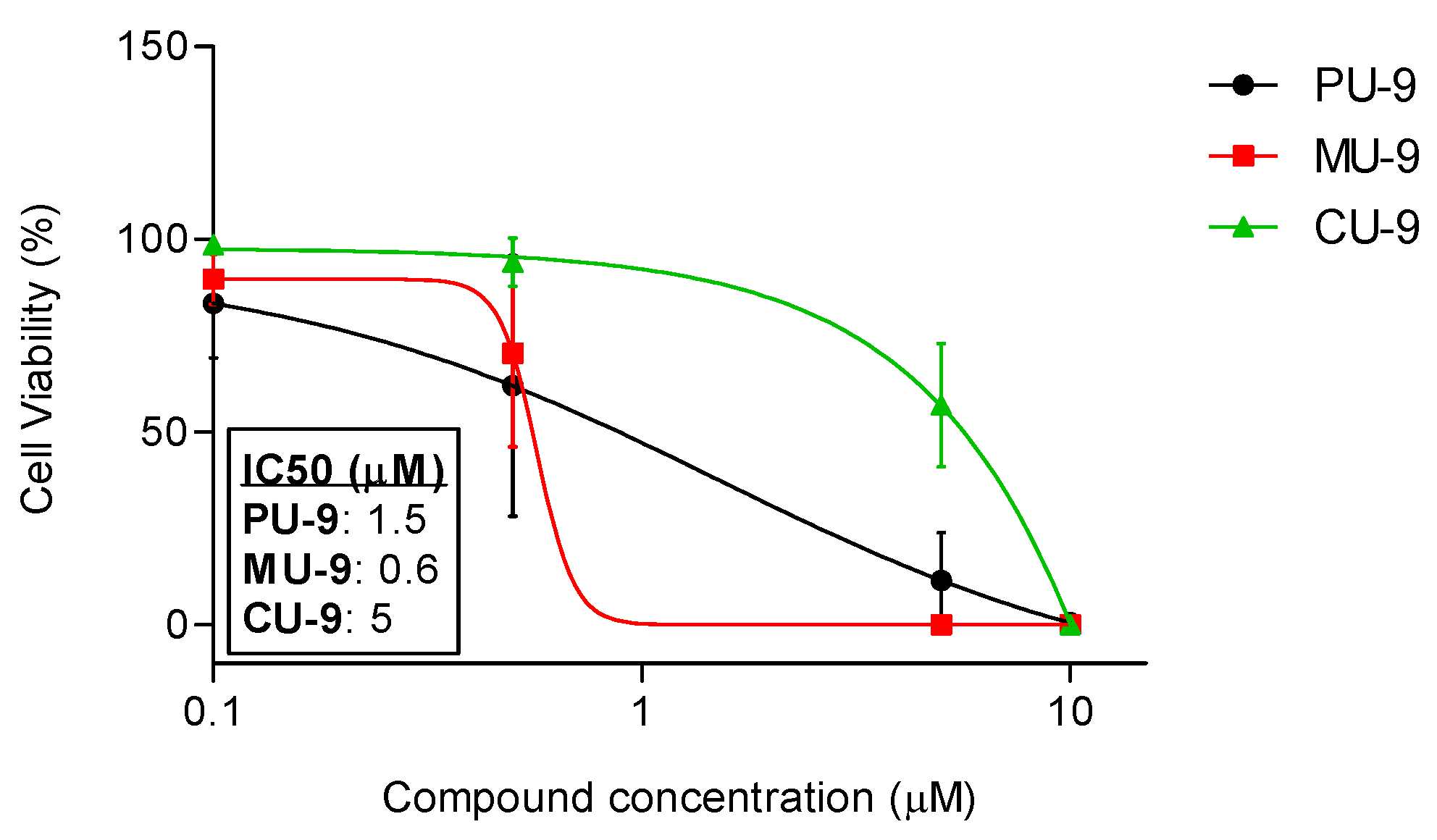
Figure 4.
Harmiquins sensitize resistant cells to mitoxantrone (MX). The PLB/ABCG2 (A) and PLB-985 (B) cell lines were treated with increasing concentrations of MX alone (MX) or in combination with Ko143 or other tested agents as indicated at a concentration of 1 µM. Cell viability was determined after 72 hours of incubation using the MTT assay, and the percentage of viable cells was calculated. Each bar represents a mean ± SD of at least three individual experiments performed in quadruplicate.
Figure 4.
Harmiquins sensitize resistant cells to mitoxantrone (MX). The PLB/ABCG2 (A) and PLB-985 (B) cell lines were treated with increasing concentrations of MX alone (MX) or in combination with Ko143 or other tested agents as indicated at a concentration of 1 µM. Cell viability was determined after 72 hours of incubation using the MTT assay, and the percentage of viable cells was calculated. Each bar represents a mean ± SD of at least three individual experiments performed in quadruplicate.
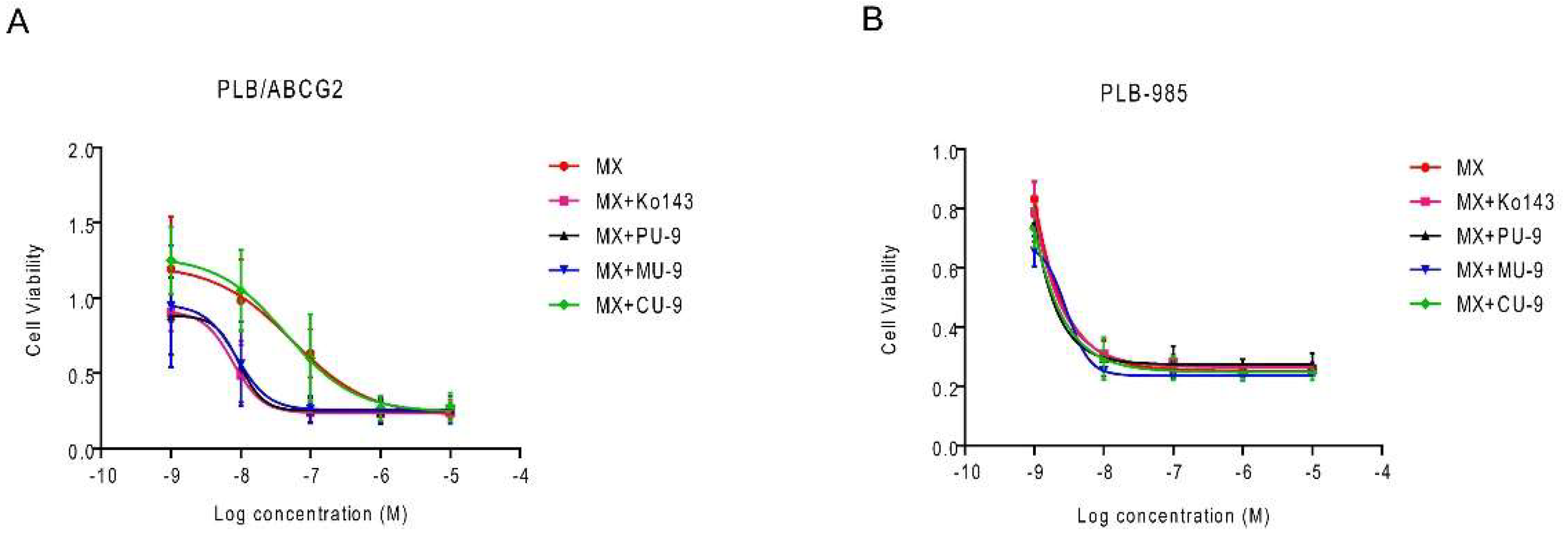
Figure 5.
Inhibition of P-gp-mediated transport by hybrids, primaquine (PQ), chloroquine (CQ) and temozolomide (TMZ). Functional activity of P-gp was evaluated by calcein-AM accumulation assay measured in A2780/Adr cell line. The mean fluorescence intensity (MFI) of calcein was measured in A2780/Adr cells after incubation with 0.25 μM calcein-AM (CTRL) and treatment with verapamil and tested compounds at 10 μM concentration. Uptake of calcein-AM is shown relative to 100 % uptake at maximum inhibition of P-gp by verapamil (10 µM). All data are shown as means ± SD of 3 individual experiments. One-way ANOVA with Dunnett’s post-hoc test was used for statistical analysis *p < 0.05; **p < 0.01; ***p < 0.001.
Figure 5.
Inhibition of P-gp-mediated transport by hybrids, primaquine (PQ), chloroquine (CQ) and temozolomide (TMZ). Functional activity of P-gp was evaluated by calcein-AM accumulation assay measured in A2780/Adr cell line. The mean fluorescence intensity (MFI) of calcein was measured in A2780/Adr cells after incubation with 0.25 μM calcein-AM (CTRL) and treatment with verapamil and tested compounds at 10 μM concentration. Uptake of calcein-AM is shown relative to 100 % uptake at maximum inhibition of P-gp by verapamil (10 µM). All data are shown as means ± SD of 3 individual experiments. One-way ANOVA with Dunnett’s post-hoc test was used for statistical analysis *p < 0.05; **p < 0.01; ***p < 0.001.
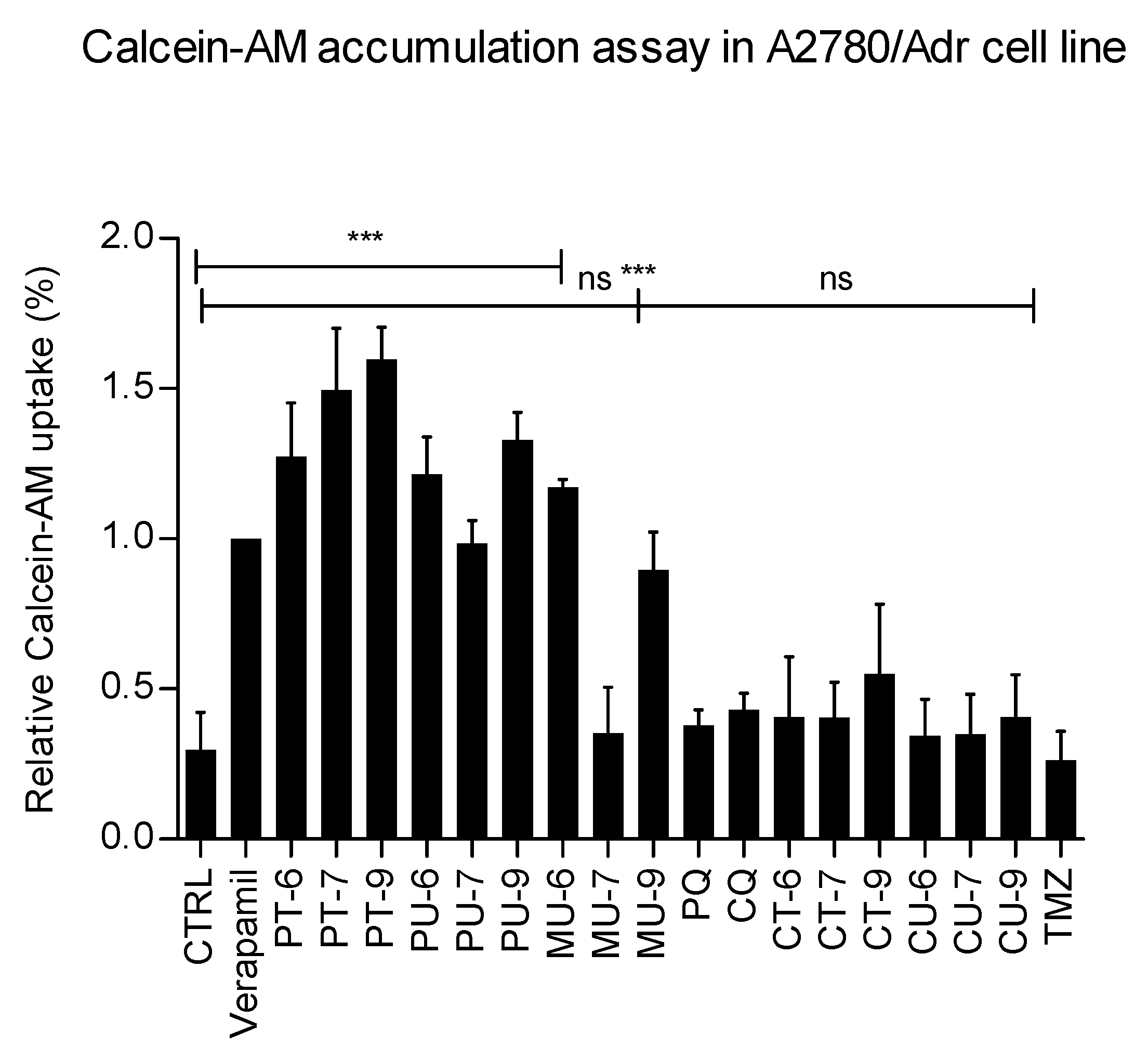
Figure 6.
Potency of the hybrid compounds PU-9, MU-9, and PT-9 to inhibit P-gp as measured by the calcein-AM transport assay in the A2780/Adr cell line. Inhibition of transporter activity was calculated by comparing the fluorescence intensity in the presence of the tested compound with that in the presence of verapamil (maximal inhibition). Data were plotted in triplicate ± SD from three independent experiments. The IC50 of each compound was calculated using a non-linear regression curve from three independent experiments performed in triplicate.
Figure 6.
Potency of the hybrid compounds PU-9, MU-9, and PT-9 to inhibit P-gp as measured by the calcein-AM transport assay in the A2780/Adr cell line. Inhibition of transporter activity was calculated by comparing the fluorescence intensity in the presence of the tested compound with that in the presence of verapamil (maximal inhibition). Data were plotted in triplicate ± SD from three independent experiments. The IC50 of each compound was calculated using a non-linear regression curve from three independent experiments performed in triplicate.
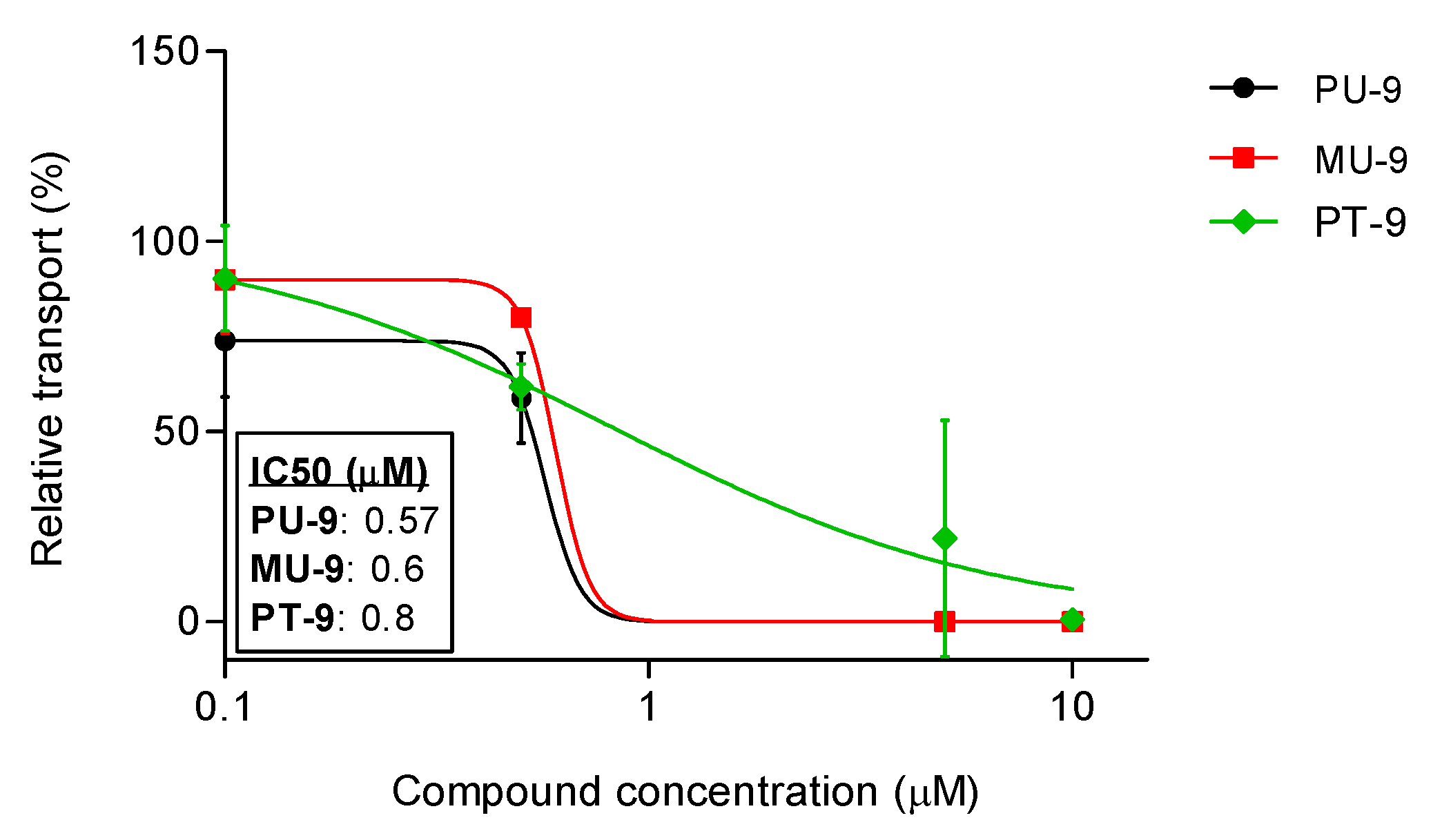
Figure 7.
Harmiprims PU-9 and PT-9 and harmiquine MU-9 sensitize resistant cells to doxorubicin (DOXO). The P-gp overexpressing cells A2780/ADR (A) and the parental A2780 (B) cell lines were treated with increasing concentrations of DOXO alone (DOXO) or in combination with verapamil or the tested compounds as indicated. Cell viability was determined after 72 hours of incubation using the MTT assay, and the percentage of viable cells was calculated. Each bar represents a mean ± SD of at least three individual experiments performed in quadruplicate.
Figure 7.
Harmiprims PU-9 and PT-9 and harmiquine MU-9 sensitize resistant cells to doxorubicin (DOXO). The P-gp overexpressing cells A2780/ADR (A) and the parental A2780 (B) cell lines were treated with increasing concentrations of DOXO alone (DOXO) or in combination with verapamil or the tested compounds as indicated. Cell viability was determined after 72 hours of incubation using the MTT assay, and the percentage of viable cells was calculated. Each bar represents a mean ± SD of at least three individual experiments performed in quadruplicate.
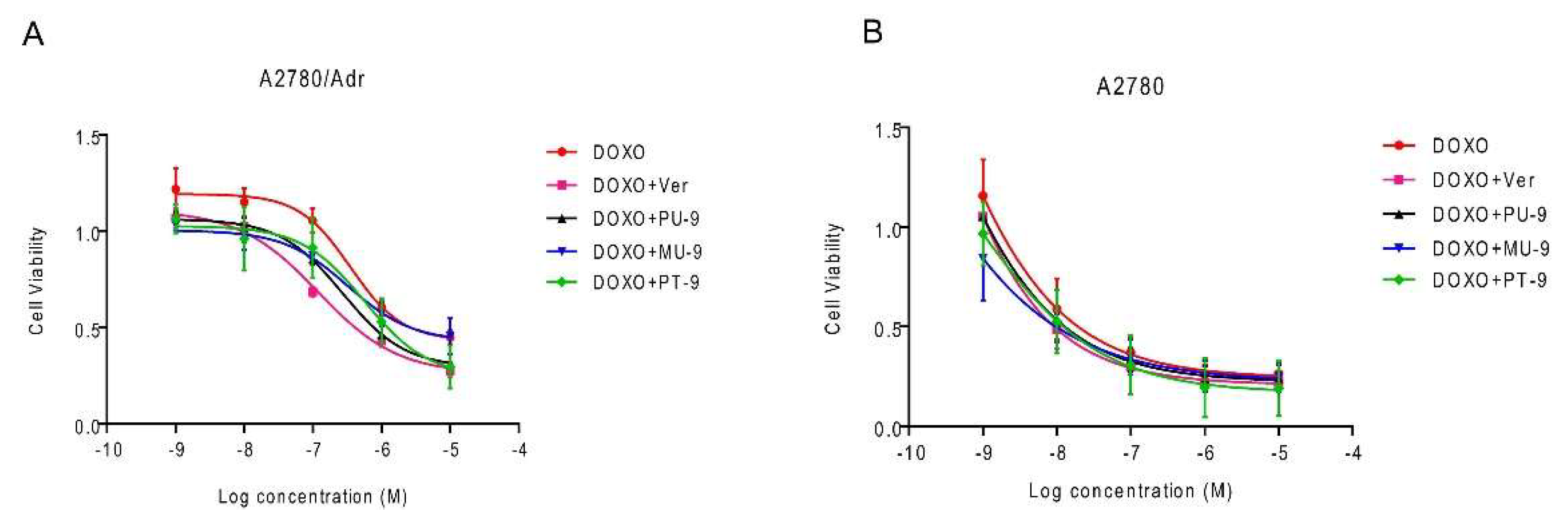

Table 1.
Antiproliferative activity in vitro of hybrids of harmine and quinolone antimalarials. Data represent the mean ± SD of three independent experiments performed in quadruplicate.
Table 1.
Antiproliferative activity in vitro of hybrids of harmine and quinolone antimalarials. Data represent the mean ± SD of three independent experiments performed in quadruplicate.
| IC50/µM | ||||
|---|---|---|---|---|
| Cpd. | Cell line | |||
| Structure | U251 | SH-SY5Y | Hek293T | |
| PT-6 | 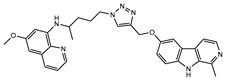 |
4.4±0.3 | >100 | 25.2±3.6a |
| PT-7 | 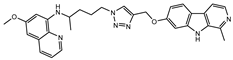 |
2.9±1.7 | >100 | 7.6±0.01a |
| PT-9 | 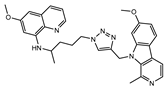 |
33.5±3 | >100 | >50a |
| PU-6 | 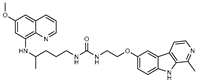 |
4±0.4 | 8.2±0.9 | 12±0.9a |
| PU-7 | 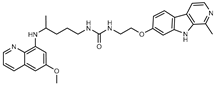 |
4±0.5 | 7.4±0.3 | 7.6±0.007a |
| PU-9 | 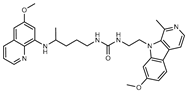 |
4±0.6 | 7.5±0.7 | 13±0.06a |
| CT-6 | 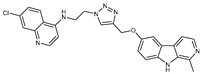 |
5±0.4 | 72±28 | >50 |
| CT-7 | 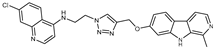 |
3.6±0.4 | 8.6±0.5 | 5.7±0.13 |
| CT-9 | 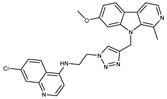 |
4.5±0.09 | 8.8±1 | 26.1±0.4 |
| CU-6 | 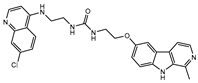 |
35.7±25.3 | >100 | >50b |
| CU-7 | 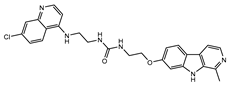 |
6.3±0.8 | >100 | 38.8±.5b |
| CU-9 | 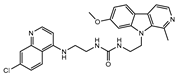 |
4.6±0.4 | >100 | 5.5±0.3b |
| MU-6 | 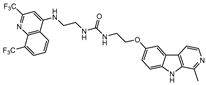 |
1.8±0.3 | 6.2±0.04 | 3.1±0.03b |
| MU-7 | 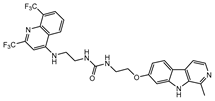 |
0.6±0.2 | 7.5±4.5 | 16.8±0.2b |
| MU-9 | 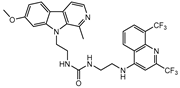 |
4±0.3 | 7.9±1.6 | 7.8±0.1b |
| PQc | 44.3±3 | >100 | 37.6±0.6a | |
| CQd | 24±0.95 | 77.8±18 | n.te | |
| HARf | 20 ± 1.7g | n.te | 21±3.04a | |
| TMZh | >100 | >100 | >100 | |
Table 2.
Growth inhibition of PLB-985 and PLB/ABCG2 cell lines by mitoxantrone (MX) alone or in combination with tested hybrids or Ko143. IC50 values were calculated from cell viability curves plotted from the corresponding cell viability curves, from 3 independent experiments performed in quadruplicate (mean ± SD). All concentrations are given in µM (IC50 - the concentration that causes 50 % of cell viability).
Table 2.
Growth inhibition of PLB-985 and PLB/ABCG2 cell lines by mitoxantrone (MX) alone or in combination with tested hybrids or Ko143. IC50 values were calculated from cell viability curves plotted from the corresponding cell viability curves, from 3 independent experiments performed in quadruplicate (mean ± SD). All concentrations are given in µM (IC50 - the concentration that causes 50 % of cell viability).
| IC50 (µM) | ||
| treatment | PLB/ABCG2 | PLB-985 |
| MX | 0.20±0.17 | 0.004±0.001 |
| MX + Ko143 | 0.006±0.002 | 0.004±0.001 |
| MX + PU-9 | 0.006±0.003 | 0.005±0.003 |
| MX + MU-9 | 0.005±0,003 | 0.002±0.0004 |
| MX + CU-9 | 0.14±0.1 | 0.003±0.0001 |
Table 3.
Growth inhibition of A2780/Adr and A2780 cell lines by doxorubicine (DOXO) alone or in combination with tested hybrids or verapamil. IC50 values were calculated from cell viability curves plotted from the corresponding cell viability curves, from 3 independent experiments performed in quadruplicate (mean ± SD). All concentrations are given in µM (IC50 – the concentration that causes 50 % of cell viability).
Table 3.
Growth inhibition of A2780/Adr and A2780 cell lines by doxorubicine (DOXO) alone or in combination with tested hybrids or verapamil. IC50 values were calculated from cell viability curves plotted from the corresponding cell viability curves, from 3 independent experiments performed in quadruplicate (mean ± SD). All concentrations are given in µM (IC50 – the concentration that causes 50 % of cell viability).
| IC50 (µM) | ||
| treatment | A2780/Adr | A2780 |
| DOXO | 4.57±3.0 | 0.01±0.003 |
| DOXO+Ver | 0.58±0.1 | 0.01±0.007 |
| DOXO + PU-9 | 0.95±0.3 | 0.01±0.003 |
| DOXO + MU-9 | 1.56±0.4 | 0.02±0.01 |
| DOXO + PT-9 | 1.9±1 | 0.02±0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



