Submitted:
16 June 2025
Posted:
17 June 2025
Read the latest preprint version here
Abstract
The increasing complexity of cancer treatments and the emergence of new therapeutic targets have made it more challenging to identify the most effective evidence-based therapies for patients at any given time. This review revisits the core principles of cancer therapy to propose flexible clinical trial frameworks that can expedite the identification of optimal treatments for locally advanced, recurrent, and metastatic cancers. It emphasizes four key abnormal pathways in cancer progression: abnormal blood vessel formation, metabolic alterations, immune system dysfunction, and phenotypic transitions, all significantly influenced by hypoxia-driven Hypoxia-Inducible Factor-1 alpha (HIF-1α). Beyond standard chemotherapy and immunotherapy, the current therapeutic arsenal comprises nanoparticles, chimeric antigen receptor (CAR) cells, cellular and antibody therapies, vaccines, labeled radionuclides, phagocytic agents, and intralesional therapies. This article examines how combining these modalities can synergistically enhance the currently modest benefits of immunotherapy. By prioritizing immunotherapy, the review outlines strategies to “unmask” cancer cells and modify the tumor and its microenvironment to maximize the immunotherapeutic response. It advocates for improving the delivery of anticancer agents into the resistant cancer cell microenvironment (CCME) within the complex, layered barriers of the tumor microenvironment (TME). The authors propose an approach focused on the Combinations, Timing, and Sequencing (CTS) of therapies for the early determination of optimal treatment schedules in preclinical and clinical studies. The integration of pulsed stereotactic radiotherapy is also emphasized as a crucial part of this strategy.
Keywords:
Hypoxia
; HIF-1 α
; vascular-normalization
; clinical trials
; Antiangiogenics
; Chemotherapy
; RT-SBRT
; Immunotherapy
; Nanoparticles
; Vaccines
; CAR-T-Therapy
; CCME
; CTS Therapy Strategy
Introduction
The evolution of cancer therapy has always centered around combinations, timing, and sequencing. A network of potential combinations exists for effective treatments, but determining the optimal combination through clinical trials is time-consuming. New targets and effective drugs are continuously being added, complicating the establishment of up-to-date, evidence-based therapy at any given moment. Recently, an understanding of hypoxia-induced interactive and interdependent vascular, metabolic, immune, and phenotypic cascades has directed clinical trials toward targeting multiple pathways using bispecific and multispecific approaches for both cancer and its tumor microenvironment (TME). [1][Passariello, M]. Clinical trial outcomes are being established for multitargeted triplet therapies. [2] [Pascual J]. This strategy also addresses multidrug resistance. (MDR). [3] [Zhi Li]. However, these studies may still lack a comprehensive approach regarding the resources and time needed to address the complex evolution of cancer and its TME for the foreseeable future.
a). Importance of Immunotherapy: In recent years, immunotherapy has become the leading approach in cancer therapy. However, the overall benefit is, on average, 12% across all tumor types. [4] [Appleton]. Reported long-term survival rates in nonsmall cell lung cancer have increased to over 15%, compared to just 5% during the earlier immunotherapy era. [5][Putzu C]. Secondly, despite the initial strategic targeting and good response, developing secondary resistance after a variable period has motivated efforts to optimize these promising approaches with enhanced adjuvanticity and antigenicity. [4] [Appleton E],
b). Highlighting Reversion, Differentiation, Regression, and Reprogramming as Therapeutic Endpoints: Evidence of tumor reversion dates back to Askanazy’’s’ 1907 documentation of teratoma regression and Braun’’s’ work on plant tumor cells in the 1950s. Recent interest in regression has reemerged due to advances in cell reprogramming and epigenetic mechanisms. [6] [Pensotti A]. The term “reversion” specifically refers to the loss of malignant features and the reintegration with normal tissues. Acute promyelocytic leukemia (APL) is the only clinically proven application of differentiation/reversion. Spontaneous regression has been observed in neuroblastoma, breast cancer, melanoma, renal carcinoma, and lung metastases. [6] [Pensotti A]. Phenotypic reversion in cancer cell lines was shown in a study to involve approximately 300 genes that redirect cellular pathways away from malignancy. At the same time, other mechanisms involve integrins and cell adhesion pathways that can restore normal phenotypes despite ongoing genetic mutations. The embryonic microenvironment can also encourage reversion because of its similarities in processes. [6] [Pensotti A].
Reversion, differentiation, regression, and reprogramming refer to processes of ’t’umor reversal.’ One definition describes reversion as “the process by which cancer cells lose their malignant phenotype... involving a cellular reprogramming mechanism that overrides genetic changes by triggering alternative pathways leading to suppression of tumorigenicity.” (Tuynder et al., 2002). [7] [Tuynder M]. While accumulated genetic mutations are generally irreversible, evidence suggests the phenomenon of tumor reversion warrants attention. The loss of a malignant phenotype and the conversion of cells to benign forms, getting closer to the stable original state, do not necessarily indicate a return to the original state where the malignant process began. This makes the term “reversion” (to a benign state) more suitable than the term “reversal” for this process. Reverted cancer cells can start proliferating again if treatment is stopped early, making long-term consolidation therapy necessary, even after reversion, to prevent recurrence. [8] [Shin D] [6] [Pensotti A].
DNA methyltransferase or histone inhibitors can reverse epigenetic changes. miRNA-targeting drugs (miR-125/20/24) can reverse multidrug resistance at the gene level. [9] [Garg, P] [10] [Lei ZN]. The mesenchymal-to-epithelial transition (MET) represents the reversal of Epithelial–Mesenchymal Transition (EMT) and is facilitated by specific miRNAs. In various cancers, silencing the Translationally Controlled Tumor Protein (TCTP) is critical for MET reversion. Normalizing the tumor microenvironment (TME) is a key strategy for cancer reversion, as it restores the functional relationships with normal cells. As recently reported, although poorly understood, senescent cells, which are considered irreversibly phenotypic, can be reactivated to re-enter the cell cycle by promigratory cytokines. [11] [Škarková A].
The most fundamental approach to cancer reversion involves normalizing the tumor microenvironment (TME). It consists of restoring the proper acidic potential of hydrogen (pH) levels, oxygen, nutrients, and immune cell–cytokine cross-talk, which can re-educate cancer cells toward less malignant phenotypes. Evidence suggests that cancer may result from progressive extracellular matrix (ECM) deregulation and could be reversed by mimicking embryonic microenvironments. [12] [Ingber DE].
c). Tumor Heterogeneity: As a significant barrier to successful therapy, intratumoral cells exhibit biologically diverse behavior, dynamically evolving subclonal variability in both space and time. These cells develop with varied metabolic functions through linear or branching mechanisms and are significantly influenced by hypoxia. Increasing precision in evaluating functional heterogeneity in tumor cell populations, demonstrated by single-cell RNA sequencing (scRNAseq), has made identifying heterogeneity at the single-cell level feasible. In practical terms, most druggable targets are not homogeneously expressed within a tumor. Additionally, drug sequestration by endosomes/exosomes inhibited drug transport inside cancer cells due to pH gradients, complicating drug delivery to cancer cells. [13] [Jacquemin, V].
d). Unmasking the tumor for immunotherapy: The “unmasking” strategy involves redirecting multicomponent TME modulation away from the immunosuppressive elements and triggering immunologic rejection of cancer cells to unlock the true benefits of immunotherapy. [14] [Newton JM]. Immunoediting theory describes a three-step process of immunosurveillance, equilibrium, and escape for cancer initiation to progress. [15] [Dunn GP]. Unmasking of cancer is a strategy by which cancer cells become recognizable to the body’’s’ immune surveillance and elimination. Advanced and recurrent malignancies have adopted the usual milieu to their advantage by developing multiple immune escape mechanisms and deceptive barriers. Hence, upfront use of immunotherapy, especially in cold tumors, may not yield the expected results. [4] [Appleton]. A few baseline conditions must be restored to enhance the effectiveness of immunotherapy. One condition entails generating tumor-specific antigens linked to non-synonymous mutations that prime T cells, activating CTLs to target cancer cells. Progress is being made in antigen profiling to identify targets in cancer cells and in developing in vitro cancer vaccines for personalized treatment. [16] [Battaglia S]. Some chemotherapy agents and radiotherapy, particularly SBRT, induce immunological cell death, resulting in significant cell death among tumor cells. This process leads to an increase in tumor cell-specific neoantigens, which subsequently generate tumor-specific CTLs. [14] [Newton JM]. Another requirement is to optimize the immune-ECM cross-talk. [17] [Zhao].
Objectives of the Present Review: This review article centers on immunotherapy, focusing on cancer reversion and reprogramming as key endpoints, supported by a foundational analysis of cancer genesis to progression. It proposes a Combination, timing, and sequencing (CTS) strategy in actionable phases to “unmask” and prepare the tumor for effective immunotherapy. The review offers a classification system and framework for a phase-wise CTS strategy to develop the necessary algorithm. The functional heterogeneity of individual cells, as demonstrated by scRNAseq, can now be assessed. [13] [Jacquemin, V]. Cancers typically consist of various coexisting subclonal mutations, with even a single cell exhibiting diverse mutations. [18] [Schmitt MW]. Therefore, the secondary objective is to focus on strategies for targeting and delivering anticancer agents in the cancer cell microenvironment (CCME) within the tumor microenvironment (TME).
1. Foundational Analysis
Need for Foundational Analysis: The present review article is structured to discuss the ecology of cancer and to understand the critical points of targeting (foundational analysis) to prevent the evolution of resistant clones (actionable analysis). The predator and prey game will be initiated the instant anticancer therapy begins, and surviving cells (even if few) begin to evolve a cascade of ecological and evolutionary resistance strategies. Cancer cells have a unique advantage (unlike infectious conditions) in accessing the vast information encoded in the human genome. Intervention by one mechanism encourages the emergence of an alternative mechanism due to various resistant phenotypic pathways. [19] [Gatenby RA]. This foundational analysis section examines how complex interactions among six foundational factors (some aspects may be relevant to the future) influence ecological evolutionary cascades to develop strategies that prevent the initiation of resistant pathways at the outset of anticancer therapy. [19] [Gatenby RA]. and establish conditions for the cancer “reversion” process.
1.1. Foundational Factor 1: Hypoxia, HIF1-α, and the Domino Effects
1.1.1. 100-micron Factor—The Genesis
Hypoxia can manifest when the intercapillary distance exceeds 100 microns. Hypoxia occurs in the arterial ends of microvessels when the intercapillary distance in a breast xenograft exceeds 140 microns. Hypoxia is further aggravated by decreased blood flow caused by vessel tortuosity, arteriovenous shunts, abnormal blood cell circulation, and the elongation of distorted tumor microvessels resulting from rapidly proliferating cancer cells. [20] [Groebe K et al.]. The average size of tumor cells can range from about 20 to 30 microns, depending on their site of origin [21] [MOORE GE et al.]. Therefore, hypoxia sets in when the tumor size approaches 4 to 7 cell clumps (around 101 cells), far ahead of the imaging detection levels. These changes align with the phases of equilibrium and escape in cancer evolution’’s’ immuno-editing theory [15] [Dunn GP et al.] and added immune exhaustion (Figure 1A).
1.1.2. HIF-1α Cascading Effects
Figure 1B illustrates the vascular, metabolic, immunosuppressive, and genomic-phenotypic changes HIF1-α orchestrates following hypoxia in cancer cells. Theoretically, HIF1-α could be the primary target to block subsequent downstream developments. However, HIF1-α has an extensive list of target genes, and a specific subset of HIF-1α target genes varies by cancer types and subtypes, which adds to the complexity of its regulation in cancer. Another limiting factor is the significant toxic effects associated with targeting HIF-1α. [22] [Jun JC, ] [23] [Yong L].
1.2. Foundational Factor 2: Vascular Changes
1.2.1. Angiogenesis in Healing vs. Tumor Angiogenesis (Neoangiogenesis)
Healing angiogenesis is typically a well-orchestrated series of events that supplies additional oxygen and nutrients to the proliferating healing cells. It is a well-balanced process involving growth factors and inhibitors. The progression of angiogenesis involves the migration, proliferation, and differentiation of endothelial cells (Ecs), which form lumen-bearing cords with stable branching structures, led by “tip” cells and covered by appropriately formed encasing cells—the pericytes. [24] [Al-Ostoot FH].
The fundamental distinction between normal tissue healing, angiogenesis, and tumor angiogenesis induced by hypoxia (referred to as neoangiogenesis) lies in the presence of malformed, leaky, tortuous, dilated, saccular, and shunted blood vessels in the latter, known as aberrant angiogenesis. [25] [Multhoff] (Figure 2).
Tumor cell hypoxia is a crucial driver of neoangiogenesis, primarily by stabilizing HIF-1α when cancer cells encounter low oxygen levels. [26] [Wegiel B]. HIF-1α subsequently transactivates various pro-angiogenic genes, including growth factors, that promote tumor angiogenesis. [27] [Montemagno C]. Therefore, the upregulation of the hypoxia-induced HIF-1α transcription factor is central to the cellular response, leading to multiple downstream effects through Vascular Endothelial Growth Factor (VEGF) and Vascular Endothelial Growth Factor Receptors (VEGFRs). Additional downstream factors include Placental Growth Factor (PlGF), Angiopoietin-1 (Ang-1)/Angiopoietin-2 (Ang-2), Platelet-Derived Growth Factor (PDGF), Transforming Growth Factor-β (TGF-β), among others. Collectively, these factors promote cell survival, endothelial migration, anaerobic metabolism, and metastasis. The PDGF pathway influences pericyte recruitment, fibroblast growth factor (FGF), and various inflammatory mediators. [28] [Clarke J M,].
The VEGF family includes VEGF A, B, C, D, and PlGF. VEGF operates through receptors VEGFR 1, 2, and 3, activating phosphatidylinositol 3-kinase (PI3K) and protein kinase C (PKC). Alternative pathways involve the inhibition of cyclooxygenase-2 (COX-2) by microRNAs (miRNAs) that compete with messenger RNA (mRNA) targets and suppress matrix metalloproteinases (MMPs). [29] [Caporarello N]. Additionally, 18 glycoproteins from the FGF family interact with four transmembrane receptors (FGFR1–4) to effectively modulate tumor angiogenesis independently of VEGF signaling. [30] [Zahra, F.T et al.]. Stromal pathways include bone marrow-derived cells (BMDCs) acting as endothelial and pericyte progenitors, as well as pro-angiogenic tumor-infiltrating lymphocytes (TILs). Ang-2 increases the pro-angiogenic receptor tyrosine kinase (Tie2+), which is associated with monocytes/macrophages linked to blood vessels. [28] [Clarke J M,].
1.2.2. Process and Types of Tumor Angiogenesis
Endothelial cells communicate with pericytes via N-cadherin, relying on PDGF-PDGFR signaling to sustain normal microcirculation. Disruption of this interaction results in dysregulation of tumor angiogenesis, characterized by heterogeneous endothelial cells. [27] [Montemagno C] [31] [Jiang Z]. This altered angiogenesis primarily results from the accumulation of Hypoxia-Inducible Factor 1-alpha (HIF-1α) due to the excessive production of Reactive Oxygen Species (ROS). The tumor vasculature is marked by instability and chaos, characterized by tortuous, leaky vessels that arise from weak endothelial junctions, a thickened basement membrane, and insufficient pericyte coverage. This “aberrant” vasculature, in turn, stimulates HIF-1α, which increases resistance to flow, reduces perfusion, and creates a vicious cycle of tumor progression and hypoxia. [25] [Multhoff, G]. The tumor and aberrant angiogenesis take several forms, and some of them are reasons for developing resistance to therapies.
- Sprouting and Splitting Angiogenesis: In sprouting angiogenesis, the formation of “tip cells” is supported by the growth of “stalk cells,” which help elongate from the side of the blood vessel through the activation of endothelial cells. Intussusceptive angiogenesis (or splitting angiogenesis) involves dividing an existing blood vessel into two. Splitting/intussusceptive angiogenesis, which does not require endothelial proliferation, has a lower metabolic demand. Therefore, relapse after post-tyrosine kinase inhibitor (TKI) therapy may occur due to extensive splitting angiogenesis. [32] [Ribatti D et al.]
- Vasculogenesis: This is the type of angiogenesis through which precursor cells differentiate into endothelial cells. The term neo-vascularization encompasses both neoangiogenesis and vasculogenesis. [24] [Al-Ostoot FH et al.]. Contribution to vasculogenesis can arise from differentiating hematopoietic (stem) cells or cancer stem cells through direct endothelial differentiation. This vasculogenesis operates as a VEGF-independent mechanism and remains unaffected by anti-VEGF bevacizumab. However, it could be inhibited by the tyrosine kinase inhibitor sunitinib or the anti-VEGF-receptor-2 neutralizing antibody. [33] [Brossa A]
- Vascular cooption: The immediate adoption of preexisting vasculature is known as vascular cooption. This process increases metastatic potential and the Ang2-mediated apoptotic cascade, which worsens neoangiogenesis. It is resistant to AAGs because it is independent of angiogenic switches. [29] [Caporarello N]. [34] [Haibe Y et al.]. [32] [Ribatti D et al.].
- Vascular mimicry: This occurs when cancer cells directly form vascular-like structures. Research has shown that this process is upregulated after treatment with bevacizumab or during the induction of hypoxia in the resistance phase. [34] [Haibe Y]. It is proposed that vasculogenic mimicry might depend on cancer stem cells, as classical angiogenics do not play a role. [32] [Ribatti D].
1.2.3. Neoangiogenesis, Hypoxia, and Treatment Resistance
The ability of normal and cancer cell biological systems to reroute through existing local bypasses, activate latent pathways, or adapt to genetic or environmental perturbations is a poorly understood aspect of adaptive evolution at the molecular level. [36] [Fong SS] [19] [Gatenby]. The development of resistance to AAGs can primarily be classified into VEGF (sprouting and splitting angiogenesis) and non-VEGF-dependent (vasculogenesis, co-option, mimicry) pathways. [28] [Clarke J M]). Continued long-term administration of VGFR blockade leads to Ang-2 accumulation, excessive vascular pruning, destabilizes the vessels, and worsens hypoxia. However, evidence suggests that, even with ongoing vascular pruning, long-term AAG use provides a small survival benefit beyond disease progression [32] [Ribatti D]. EMT in the TME drives aggressive tumor phenotypes through upregulated angiogenic factors, HIF-1a, vessel over-pruning, myeloid cell recruitment, CAFs, increased co-option/mimicry, and AAG delivery impediments, promoting aberrant angiogenesis, tumor growth, EMT, and metastasis. Continued use causes AAG-induced hypoxia, which stimulates β1 integrin, decreases adherens junction proteins, and impacts pericyte coverage, promoting invasiveness. [34] [Haibe Y] [37] [Ayoub NM]. This tumor hypoxia induces angiogenic dormancy, autophagy, lactate production, stem cell proliferation, and lymphangiogenesis, contributing to AAG therapy resistance [27] [Montemagno C].
1.2.4. The Changing Paradigm of the Antiangiogenesis Approach (From Vascular Disruption to Normalization)
Vascular Disruption Approach
Introducing the concept of disrupting tumor vasculature and starving cancer cells of oxygen and nutrients is logical, highly innovative, and promising. Initial work on this concept in preclinical studies raised immense expectations; however, it fell short in clinical settings, yielding only modest gains in clinical benefit. Following the initial response, vascular disruption led to evasive or acquired resistance over time due to alternative progression routes, dramatically increasing hypoxic cells [38] [Zahra]. [29] [Caporarello N] [39] [Winkler F]. Additionally, disruption-induced hypoxia caused mutational alterations or modifications that influenced drug uptake and efflux, affecting concurrent therapies [40] [Chae SS]. The effect was more pronounced in total “vascular disruption” trials, where rebound growth was observed at the viable edge of the cancer mass [41] [Thomas Nielsen]. In other words, the therapeutic approach of cutting off the vasculature to the tumor, intending to eliminate cancer cells, may initiate additional resistance pathways after short-term control. Vascular disruption with SBRT dose per fraction (> 20 Gy) can also convert a radical intent into a palliative one, ultimately resulting in short-term tumor control. [42] [Moding EJ].
Vascular Normalization Approach
- Vascular Normalization, Normalization Window Approach (first switch): An alternative concept emerged that favors vascular normalization over shutting it off [43] [Jain RK]. RK Jain et al. (2005) showed specific antiangiogenics transiently “normalized” tumor vasculature, alleviating hypoxia and improving oxygen/drug delivery, thereby enhancing conventional therapies [43] [Jain RK]. Cediranib resulted in a consistent and dramatic reduction in tumor enhancement within 24 hours of therapy and a decrease in vascular permeability, indicating the onset of normalization. This aligns with the onset of normalization following blockade of the VEGF/VEGFR2 pathway in preclinical studies. Upregulation of tumor Ang-1 gene expression, typically produced by perivascular cells (PVCs), activates the Tie-2 receptor on ECs. The reorganization of the basement membrane of vessels, which is usually haphazard and thick in cancer, is restored to a thinner and more closely associated structure, beginning in 48–72 hours [35] [Goel S]. Tumor vascularization and oxygenation typically improve from day 1 of AAG administration. However, by Day 28, continued AAG administration leads to excessive pruning/regression of blood vessels due to anti-VEGF overaction, and the features of the aberrant vasculature, anoxia, and reproliferation of cancer cells return. This interval between the onset of normalization (around Day 2) and the excessive pruning of vessels with continued AAGs (around Day 28) is defined as a normalization time window. The time window opens up a period of enhanced delivery of cytotoxic drugs into the CCME. Additionally, during this normalization window, the effectiveness of combinatorial radiation therapy and chemotherapy/immunotherapy is enhanced due to improved drug delivery and oxygenation. [43] [RK Jain ] [35] [Goel S].
- Reversibility of Vascular Pruning/regression During the Therapy Gap of AAGs (second switch): This is another critical finding after a long course of AAGs, when the hypoxia resets due to excessive pruning of the vasculature at the end of the normalization window (around day 28). This was initially demonstrated in patients who required “drug holidays” due to toxicity, with the reversion to the normalization phenotype observed on MRa (second “normalization window”). Stopping AAGs at this stage (Day 28) reverses the excessive regression of long-term AAGs and restores vascular normalization. [35] [Goel S].
- Restarting AAGs and Extending the Normalization Window (third switch): Later, when aberrant angiogenesis sets in again (at the end of the drug holiday gap), restarting the AAGs restores normalization once more. [35] [Goel S].
- Stages of Normalization and Cyclical AAG Therapy Opportunity: Studying the initiation process of the second and potentially additional normalization windows may be foundational to adopting the “cyclical” AAG treatment concept. Theoretically, extending the normalization window reasonably indefinitely while dynamically mapping tumor vasculature during therapy is feasible and can guide the on-and-off scheduling of AAGs. [32] [Ribatti D].
- Other Methods of Extending the Normalization Window: Hypoxia-driven fibroblast growth factor (bFGF) may be responsible for escaping the normalization window, and bFGF levels increased alongside microvessel density and tumor cell proliferation after 7 weeks of ongoing VEGF blockade. [38] [Zahra].. Temporal changes in circulating fibroblast growth factor 2 (FGF2) levels due to the inhibition of the VEGF axis are associated with the disease progression observed in glioblastoma. [28] [Clarke J M].
Targeting excessive pericytes pharmacologically or genetically can prolong the effects of anti-VEGF therapy. Imatinib, a potent inhibitor of PDGFR-β, can downregulate pericyte coverage. A preclinical study suggested a 4-day imatinib regimen for excessive pericyte coverage to normalize vessels and HIF1-α, as monitored by mpMRI. [44] [ Hu, X].
Angiopoietins (Ang-1, Ang-2) and the tyrosine kinase receptor (Tie-2) axis collaborate with VEGF in neoangiogenesis. During normal healing, newly formed vessels are “matured” and “sealed” by Ang-1, while Ang-2’’s’ action is restricted during maturation. In cancer, hypoxia-induced Ang-2 overwhelms Ang-1, inhibiting the maturation process and resulting in thin-walled vessels that lead to myeloid cell infiltration, polarizing them into Tumor-Associated Macrophages (TAMs). Soluble Tie-2, Ang-2 neutralization antibodies, and small peptides have demonstrated antitumor effectiveness. [45] [Xiaolan Yu et al.]. A mouse model study showed that dual inhibition of VEGFRs and Ang-2 was more effective than single inhibition because of a longer normalization period. [46] [Li S]. The simultaneous activation of Tie-2 and the blockade of Ang-2 in mice with certain cancers led to more effective normalization. This approach improved progression-free survival (PFS). Still, it did not enhance overall survival (OS) in a phase III trial of recurrent ovarian cancer, indicating that a comprehensive blockade of the Ang-1/2 and receptor tyrosine kinase (Tie2) axis did not benefit survival. [47] [Ramjiawan RR].
Mapping the Tumor Angiogenesis During Anticancer Therapy—Vascular Guided Therapy
Dynamic tracking of the “window for vascular normalization” will optimize drug dosages, combinations, and cycles. [48] [Yang T] Indirectly, the vasculature can be evaluated using dynamic contrast-enhanced perfusion MRI, perfusion computed tomography (CT), positron emission tomography (PET), or a combination of these methods to assess the duration of therapy and drug holidays. [32] [Ribatti D]. Dynamic contrast-enhanced CT (DCE CT) can evaluate intratumoral vascular physiological states such as perfusion, permeability surface area, and interstitial space. Additionally, paramagnetic nanoparticles targeting αvβ3 integrin in positron emission tomography or ultrasound can also directly track angiogenesis. [32] [Ribatti D]. The serum level of soluble truncated VEGFR (soluble fms-like tyrosine kinase-1 1-sFlt1), produced by endothelial cells (ECs), as well as the Ang 1/Ang 2 ratio, hypoxia-regulated Apelin and its mRNA, circulating type IV collagen, and hypoxia-correlating thrombospondin-1 (TSP-1), can also track neoangiogenesis. [48] [Yang T].
1.3. Foundational Factor 3: Metabolic Aspects—Hypoxia, Acidic pH, Differential glycolysis TILs and Tumor Cells
Cancer cells have “preferential dependence on glycolysis,” even though this process is less efficient than normal oxidative phosphorylation regarding net ATP yield. This mathematical disadvantage leads to increased glucose uptake and a further rise in glycolysis, resulting in elevated intracellular ATP (which contributes to chemoresistance) despite a reduced nutrient availability supply. [49] [Ganapathy-Kanniappan, S]. These changes improve cancer cell survival and lead to therapy resistance. [35] [Goel S].
The heterodimer of HIF-1α and HIF-1β (Figure 1B) promotes the expression of a series of target genes, including glucose transporters (GLUTs) and the glycolysis enzyme lactate dehydrogenase A (LDHA) upon formation under hypoxia, initiating anaerobic glycolysis—the Warburg effect. LDHA accelerates the conversion of pyruvate to lactate, facilitating the proliferation of cancer cells. [26] [Wegiel B]. Lactate, a byproduct of glycolysis, seeps into the tumor microenvironment (TME), resulting in an acidic pH. This process leads to a significant and diverse accumulation of immunosuppressive cells, which fosters genomic mutations and further fuels tumor growth, as well as the development of resistant phenotypic sub-clones (Figure 1B). [50] [Weihua Wu] [51] [Liberti MV].
The critical aspect of TILs regarding immunotherapy is that the nutrient-deprived TME limits the effector function of the former, resulting in immunotherapy resistance. T-cells acquire a highly glycolytic phenotype when exposed to an antigen and undergo a metabolic switch. Conversely, T cell-targeted immunotherapies in preclinical trials suggest that low-glycolysis tumor cells respond more effectively. [52] [Schreier A]. Elevated serum LDH, which indicates glycolysis, has been found to correlate with the poorest response to melanoma anti-PD1 treatment. In a mouse model of triple-negative breast cancer, LDHA knockdown was directly associated with reduced tumor growth and glycolytic activity. Neoadjuvant CTLA-4 blockade demonstrated increased T cell infiltration in glycolysis-low tumors, and Tregs are reprogrammed into effector T cells with higher extracellular glucose that produce IFN-γ and TNF-α. These findings emphasize the paradox for nutrition-deprived tumor cells in a nutrition-rich TME. Moreover, there are mechanistic interactions between hypoxia and glycolysis, each promoting the other. [52] [Schreier A].
1.4. Foundational Factor 4: Genomic–Phenotypic Alterations and Cancer Cell Heterogeneity
The cancer mass transforms into various sub-clonal cells, ranging from hypoxic-dormant cancer stem cells to resistant-proliferating types, evolving with each therapy. Treatment responses can be misleading when only the susceptible sub-clones respond. Meanwhile, resistant cells escape into a “temporal” web of mutations (Figure 1B) based on complex ecological evolutionary dynamics. [19] [Gatenby RA]. Ralston Paterson noted the recurrence of dormant cells in the necrotic wall as early as the 1960s, following a therapeutic intervention with a supralethal (ablative) radiation dose. This concept remains valid today, adding another dimension to the heterogeneity in pretreated patients. [53] [James Ralston Kennedy Paterson].
1.4.1. Clonal–Sub-Clonal Evolution
Clonal and subclonal expansion leads to widespread intratumor heterogeneity. The complexity of the deterministic process, compared to the stochastic process, results in three fundamental changes: clonal selection, drift, and mutation. These changes influence cancer evolution and contribute to infiltration, progression, and metastasis. The alterations are also reflected in dormancy, recurrence, and driver landscapes. The diverse, multidimensional changes hinder the predictability of the overall cancer mutational path, limiting strategies for the preemptive targeting of expected mutations [54] [Lipinski KA]. The inappropriate and suboptimal combination of anticancer agents lacking favorable synergism, along with hypoxia, promotes the temporal emergence of resistant phenotypes, with each line and pressure of treatment leading to cancer relapse [55] [Brady SW] [19] [Gatenby] (Figure 3).
In the history of anticancer therapies, the initial excitement over targeted therapy results has been tempered by an increasing understanding of significant intratumor genetic heterogeneity. Even a few cells within pre-existing sub-clones resistant to the drug can lead to recurrence. The complete clonal composition cannot be measured accurately. [18] [Schmitt MW] [19] [Gatenby].
1.5. Foundational Factor 5: Changes in CCME Within TME and Optimizing Immunological Cross-Talk
Figure 1B illustrates the domino effects of HIF-1α immunosuppression on TME immune cells, including the associated cytokines and chemokines.
1.5.1. Normal Vascular-Immune Crosstalk
Innate immune cells, such as mature dendritic cells (mDCs) and M1-type tumor-associated macrophages (TAMs-M1), produce various immune-activating cytokines. These cytokines include interferon-alpha (IFN-α), interleukin-12 (IL-12), interleukin-18 (IL-18), and tumor necrosis factor (TNF). Additionally, C-X-C motif chemokine ligands (CXCL), including CXCL9, CXCL10, and C-C motif chemokine ligand 21 (CCL21), affect the phenotypic and functional characteristics of tumor vasculature, leading to normalization. Adaptive immune cells secrete the cytokine IFN-γ, which possesses anticancer effects and contributes to normalizing the tumor microenvironment (TME) vasculature. Furthermore, mDCs, Cluster-Differentiation 8 (CD8), and enhanced T helper type 1 (Th1) immune cells promote the polarization of macrophages from the M2 to the M1 phenotype. [56] [Lee W S].
1.5.2. Endothelial Cell–Immune Crosstalk During Vascular Normalization
The role of high-plump endothelial venule cells (HEVs) in cross-talk: The vascular phenotype characterized by high-plump endothelial venule cells (HEVs) is functionally specialized in lymphocyte extravasation by producing IFN-γ in the TME. The lymphotoxin-β receptor (LTβR) signaling pathway generates HEVs in tumor vasculature through a combination of anti-VEGFR2 and anti-programmed death-ligand 1 (anti-PD-L1) blockade. Stimulator of Interferon Genes (STING), a stimulator of IFN genes, has shown synergism with anti-VEGFR2 and immune checkpoint inhibitors (either anti-PD-1 or anti-cytotoxic T lymphocyte-associated protein 4 (anti-CTLA-4)). This synergism occurs through the activation of type I IFN signaling and the upregulation of genes, which promotes endothelial–CD8+ CTL interaction and complete regression of tumors resistant to either monotherapy. [56] [Lee W S].
In vitro immunomodulation occurs in mouse ECs induced by interferon-γ (IFN-γ) and TNF during antigen uptake, processing, and presentation. Additionally, IFN-γ triggers antigen degradation and loading via the ‘immunoproteasome’. [57] [Amersfoort J].
1.5.3. Pericyte–Immune Crosstalk
Depleting CD4+ TH1 cells reduces pericyte coverage and increases vascular distortion. IFN-γ signaling downregulates VEGF-A while simultaneously upregulating CXCL9, CXCL10, and CXCL11. These regulatory changes facilitate pericyte recruitment along the endothelial cells (ECs), leading to vascular maturation. [56] [Lee W S].
1.6. Foundational Factor 6: Multi-Dimensional, Multi-Layered Cancer Cell Protective Shields and Cancer Cell Sequestration Within CCME
1.6.1. TME/CCME Mechanobiological Forces
The tumor mass comprises a heterogeneous group of unrestrained, proliferating cancer cells. Other components include vasculature, lymphatics, and stromal fibroblasts, which later transform into cancer-associated fibroblasts (CAFs) and immune-modifying cell infiltrates, all embedded in fibrocollagenous tissue collectively known as ECM. [58] [Finger AM]. The mechanical shear stress exerted by flowing or stagnant blood further assists cancer cells and immunosuppressive host cells. [59] [Jain RK]. The increasingly acidic interstitial fluid serves as another immune-suppressive variable in the TME. As the cell mass and vascular leakiness increase, interstitial pressure rises. Generally, cancer cells modify the TME through hypoxia and sequestration to ensure their survival and progression. The TME primarily acts as an “immunosuppressive sink,” where all the CTLs/TILs are exhausted and transformed to protect cancer cells. [60] [Karin E].
Vascular and lymphatic compression forces worsen the onset of hypoxia, and increased blood viscosity creates a vicious cycle for rising interstitial pressure (ISP). Consequently, the concentration of systemically administered therapeutic agents near the cancer cells is compromised, diminishing the effectiveness of radiotherapy as well. Targeting the abnormalities in mechanosensing, mechanobiological coupling, and signal transduction between cancer cells and the surrounding extracellular matrix (ECM) architecture, which exacerbate hypoxia, improves conventional tumor cell targeting. Tissue mechanics drive tumor progression, promote the expansion of cancer stem cells, increase tumor mutational burden, and hinder antitumor immunity, ultimately broadening the tumor’s’ heterogeneity. [61] Zhou H; [58] [Finger AM]. These barriers also facilitate the transition of macrophages from M1 to M2, promote immunosuppressive crosstalk, and divert tumor-infiltrating lymphocytes (TILs) from directly interacting with cancer cells. Inflammatory products and tumoral edema elevate ISP, compress lymphatics, and create a vicious cycle of increasing edema that further obstructs the entry of drugs and TILs into the TME [62] [Chung, S.W] [61] [Zhou H et al.]. Additionally, CAFs provide another protective layer for cancer cells. Even adoptively transferred cell therapies can be neutralized. [63] [Kim G B].
1.6.2. Acidic pH—Metabolic Shield
According to Huber V (2017), acidic pH in the TME represents the ultimate immune escape frontier, acting as a “global protection shield” that forms an “apoptosis sink” for CTLs and NK cells. Furthermore, immune-suppressive cells such as MDSCs and Tregs impede immune surveillance and promote tumor growth. Additionally, low pH significantly impacts the local and systemic bioactivities of therapeutic antibodies and checkpoint inhibitors. [64] [Huber V].
Radiation therapy can significantly improve the immunotherapy response by reducing the lactate-driven acidic microenvironment [65] [Liu, S.]. While chemotherapy is expected to yield similar results following cancer cell depletion, the advantages of alkalinization during chemotherapy and using anticancer drug conjugates with “pH-responsible” nanoparticles remain unclear. [66] [Bogdanov].
Taxol exhibited decreased intracellular pH in cultured cells and tumors. [67] [Druzhkova I]. Studies have reported similar results of increased intracellular pH with cisplatin, causing cytoplasm acidification early in therapy, favoring the metabolic reorganization of cells with a shift to oxidative metabolism. [68] [Shirmanova MV]. This could be one of the mechanisms of TME immune modulation with cisplatin, serving as the backbone in chemo-immuno combinations. [69] [[Matthew D].
1.6.3. Fibroblasts and Rigid ECM as Physical Shields
Fibroblasts, the engineers of the ECM, are responsible for tissue repair and homeostasis, but they transform into CAFs. The CAFs cross-link with the ECM, increasing its stiffness and contractility. Consequently, they induce hypoxia, promote aberrant angiogenesis, trap or shunt immune cells, and set the stage for cancer progression and metastasis. [70] [Piersma B] (Figure 4A and B).
Radiotherapy is an established anticancer treatment that fully penetrates the tumor, disrupting and normalizing the extracellular matrix (ECM). However, dosing schedules are crucial for inducing apoptotic and immunogenic cell death while preserving ECM flexibility. [71] [Martinez-Zubiaurre I]. An endothelial non-disruptive stereotactic radiotherapy dose of 6 Gy to 10 Gy per fraction enhances cancer cell killing and is recognized as an immunogenic dose. [72] [Dewan MZ]. In stereotactic body radiotherapy (SBRT), a single dose of 12 Gy prompted the release of excessive TGF-β; however, this was not observed with a single fraction of 6 Gy. Since elevated TGF-β plays a vital role in both short- and long-term radiation-induced fibrosis, limiting the dose per fraction to below 10 Gy can help prevent SBRT-induced fibrosis. TGF-β blockers/normalizers can further reduce this to maintain an immune-stimulatory and flexible ECM. An 8 Gy dose per fraction also inflicts minimal damage to microvessels and endothelial cell linings. A previous study indicated that 10 Gy per fraction is the threshold dose, above which endothelial apoptosis occurs, leading to the hypoxia pathway and excessive ECM fibrosis. [71] [Martinez-Zubiaurre I].
1.6.4. Cancer Cell Lysis Products and ISP Shield
Solid tumors pose challenges due to poor penetration and uneven distribution of therapeutic agents (chemotherapy, immunotherapy, Chimeric Antigen Receptor T (CAR-T) cells, macrophages, natural killer (NK) cells, or vaccines). Contributing factors include dysfunctional vasculature, ECM barriers, and increased interstitial pressure. [73] [Nguyen DT]. Chimeric antigen receptor macrophage (CAR-M)-tagged nanoparticle therapies achieve better penetration but still fall short. Consequently, effective drug concentration near individual cancer cells is limited because of inadequate capillary wall openings, insufficient transport, heterogeneous diffusion, and extravasation compounded by ISP. [74] [Miao L].
Radiotherapy and chemotherapy cause significant cell lysis, creating debris that elevates interstitial pressure. Professional phagocytosis by macrophages, neutrophils, and immature dendritic cells is essential for clearing this debris. Macrophages (“big eaters”) enhance anticancer treatment efficacy when activated, despite cancer cells’ “don’t’ eat me” signals. Chimeric antigen receptor macrophages (CAR-Ms) can enhance phagocytosis and reduce interstitial pressure. [75] [Li SY].
Vascular normalization reduces interstitial pressure, improves oxygen perfusion, overcomes physical barriers to drug transport, and enhances TIL infiltration, effects that can be amplified by CAR-M therapy [75] [Li SY]. The CD47, CD24, major histocompatibility class I (MHC-I), PD-L1, stanniocalcin-1 (STC-1), and disialoganglioside-2 (GD-2) have emerged as phagocytosis checkpoints, with CD47 especially linking innate and adaptive immunity in cancer immunotherapy [76] [Liu, Y].
2. Actionable Analytics
Actionable Elements of Foundational Analysis: The remission outcome appears promising during and after anticancer therapy. However, resistance may have developed much earlier, and the predator-prey dynamic might already favor the “prey” (cancer). Consequently, the evolution of resistance begins long before its progression is documented. [19] [Gatenby RA] This highlights the importance of preventing the initiation of resistant pathways and maintaining the treatment strategy until cancer “reversion” occurs, rather than merely accepting the end point of disappearance of cancer in imaging or biomarkers. Following the advocacy of evolution-based therapy of Darwinian Dynamics [19] [Gatenby RA], this section emphasizes the need to prevent the development of resistance. Therefore, the present article underscores the significance of selecting the appropriate combination with different mechanisms of action, optimizing timing through five phases for enhanced effectiveness, and sequencing for maximum probability of cancer cell elimination. This approach aims to disrupt the ecological evolution of resistance throughout all therapeutic interventions and beyond.
2.1. “Unmasking” the Tumor and Preparing CCME for Enhanced Immunotherapy Response
Despite impressive clinical efficacy, immune checkpoint inhibitors for solid tumors do not optimally achieve long-term remissions. To unlock the therapeutic benefits of immunotherapies, tumor unmasking is required through TME modulation combination therapies [14] [Newton JM].
For advanced malignancies, CT is typically combined with surgery and radiotherapy. Since the emergence of chemoradiotherapy for Ewing’s’ sarcoma in the 1970s, progress in other solid malignancies has largely been empirically driven due to the unclear understanding of how each treatment enhances the others. Further developments occurred with chemotherapy applied before (neoadjuvant), during (concurrent), or after (adjuvant) surgery and radiotherapy, leading to an empirical clinical approach that considered the sequencing and spatial cooperation of timing. [77] [Brunner TB].
2.1.1. Rationale and Importance of Developing a Planned, Verifiable Combination, Timing, and Sequencing (CTS) Strategy
The summation of all the changes discussed in the six foundational factors above leads to one significant resistance factor: tumor heterogeneity. Tumor heterogeneity is the primary reason for treatment failure and becomes a key condition for combination therapies. As recent single-cell analysis highlights, each cell is functionally different from the neighboring cell. [78] [Kang, D.H] [79] [S.Sordo-Bahamonde] [13] [Jacquemin, V].
The first justification for planned CTS is that tumor heterogeneity encompasses varying vascular perfusion (oxic/hypoxic/anoxic/necrotic) and diverse metabolic, immune-suppressive environments. [13][Jacquemin, V]. The vascular heterogeneity within the tumor also happens due to a variation in the imbalance of pro- and anti-angiogenic factors in the different regions of TME. [35] [Goel S]. Vascular normalization by creating a better TME supports immunotherapy. [80] [Sun, X.X] Vascular enhancement/ promotion over a period of time is part of radiotherapy and chemotherapy primarily due to cancer cell lysis, phagocytosis, and decreased ISP. [81] [Ferretti S] [82] [Hoffman KE] [83] [Taghian AG].
The second aspect of CTS is decreasing the ISP. Tumor hypoxia closely relates to interstitial pressure in the TME, which has traditionally required combinations of conventional chemotherapy and radiotherapy. Most anticancer therapeutics reduce ISP sooner (2 to 3 days) or later (6 to 7 days). [81] [Ferretti S]. Hoffman KE et al. noted as early as 2007 the impact of sequencing chemotherapy drugs on reducing ISP, improving tumor oxygenation, providing better responses, and promoting long-term local control/survival in the neoadjuvant therapy of breast carcinoma. [82] [K.E. Hoffman]. Large randomized clinical trials demonstrated significant improvements in survival with the combination of chemotherapy and antiangiogenic bevacizumab compared to either therapy alone. [35] [Goel S]. Fundamentally, cancer cells killed by the initial cycles of chemotherapy, initial fractions of radiotherapy, and the activation of professional phagocytosis can reduce ISP. Enhanced oxygenation and drug delivery from subsequent doses improve susceptibility to immunological sensitization. [83] [Taghian AG]. Both conventional radiotherapy and SBRT regimens reduce ISP and increase the delivery and retention of intratumorally administered drugs. [84] [Barsoumian HB]. Even with SBRT, the oxygen utilization rate increased to 87% after six to eight fractions, demonstrating an enhanced response in a three-sittings-per-week schedule. [85] [Shibamoto Y].
The third support for CTS is that cancer cells display diverse genomic variations, transdifferentiation, phenotype alterations, and distinct changing genomic-epigenetic patterns, prioritizing targeting each cancer cell. [86] [Sophie A]. Additionally, various types of stem and progenitor cells and evolving receptors contribute to evading treatment responses through resistance pathways. Trimodal therapy, which includes chemotherapy, radiotherapy/SBRT, and immunotherapy, is the foundational approach currently used to address tumor heterogeneity and stimulate an immune response through all three modalities. Many studies have confirmed the efficacy of this combination approach. The remaining aspects are dose, timing, and sequencing, requiring trials with a thorough understanding of underlying mechanisms. [87] [Li Q] [88] [ [Ling, S.P].
The fourth essential aspect for CTS, dealing with metabolic heterogeneity, arises from the need to reduce cancer glycolysis through nutritional deprivation while simultaneously facilitating nutrition-rich TILs. [52] [Schreier A].
The fifth CTS need arises from CAFs stiffening the ECM, which activates cellular mechanotransduction, driving tumor aggression and increasing the genomic mutational burden and evolution. A stiffened ECM serves as a reservoir for immune-suppressive cytokines and growth factors, drives genetic instability, and promotes EMT. Additionally, CAF-ECM stiffness creates cancer stem cell niches, inhibits the expression of tumor suppressors, hinders the delivery of anticancer oxygen and drugs, and prevents the infiltration of CTLs to CCME. Therefore, disrupting and normalizing the ECM is crucial for better long-term outcomes. [58] [Finger AM].
Therefore, the complex genomic, phenotypic, metabolic, and functional variability of cancer cells requires a systematic strategy. Given this complexity, a single or limited targeting strategy is unlikely to be effective; therefore, a combination of antitumor and immune-suppressive cell targeting is necessary. Moreover, the dosage, timing, sequence of administration, and drug delivery strategies must be emphasized. [89] [Tie, Y.]
2.1.2. Classification of Treatments, Combinations, and Slotting (BOX 1)
Given the wide range of therapeutic options available and the increasing identification of specific targets expected in the future, achieving transmissible consensus is expected. Figure 5 illustrates the network of major therapeutic options. Each option has been found to play an independent role in the evolution of cancer management.
These interconnected and complex combinations propose the following strategy for practical utilization. The first level involves classifying these (and future methods) to fit into a “slot” within the primary mechanisms (BOX 1) of vascular normalization/promotion, excision/cell kill, immunogenicity (antigenicity—adjuvanticity), phagocytosis, and ECM suppleness as described in the literature. [4] [Appleton E] [58] [Finger AM]. The second level prioritizes existing options; for example, four universally reliable approaches in metastatic cancers are chemotherapy, SBRT, immunotherapy, and nanotherapies, which can ultimately achieve complete cell kill and immunorestoration due to their ability to modulate all steps of the immunity cycle. [90] [Liu, N]. The third level designs the CTS strategy with a phase-wise approach using the “Fit a slot in the template” technique, which employs available therapeutic options (and any future ones) with different mechanisms of action that synergize spatial precision and temporal sensitization. [91] [Mortaja M]. This strategy aims to maximize the anticancer effect by debulking the tumor, reducing immune suppressive factors, and enhancing long-term anticancer immunity while minimizing side effects. [79] [S.Sordo-Bahamonde]. [Box 1 and Figure 6]. The proposed strategy is discussed below in five phases.
Box 1. Classification of Treatment Options Based on Primary Mechanisms. Newer and other treatment methods (not included in the box) can be organized into slots and combinations based on their primary mechanisms of action.
Classification Treatment Options Based on Primary Mechanisms
Phase Slot Mechanism
- Vascular Normalization: Antiangiogenics, genetic modulation, newer approaches.
- Cancer Cell Kill / Vascular Enhancement or Promotion: Radiotherapy/SBRT/Chemotherapy/Targeted therapies/Radionuclide therapies
- Immune Therapy / Immune Promotion: Immunotherapies/Cell-CAR & Antibody Therapies
- Consolidation therapies
- Cancer Reversion: Maintenance/metronomic therapies, ROS targeting, repurposing drugs, senolytics, Lifestyle modifications
Others – Non-specific to the Phases – Used as adjuvants to the above
- *
- ECM Normalization agents, eg, TGF-β blockers: Applicable from Phase I to Phase V
- *
- Phagocytic Agents – Applicable from Phase II to Phase V
- *
- Immune Adjuvants - Applicable from Phase II to Phase V
- *
- Nanotherapies - Applicable from Phase I to Phase V
2.1.3. Mitigating Toxicities Through CTS Strategy
With the possibility of long-term survival and an increasing number of patients coming within the purview of cure in the coming years, any strategy will be redundant if toxicity affecting the quality of life is not taken into account. The implied undertone of the CTS strategy is to fulfill the criteria of long-term mitigation of toxicities. This happens by a) selecting a combination with nonoverlapping toxicities [4] [Appleton]. ; b) Use of available methods by sequencing treatment during expected optimum time of action [14] [Newton] eg. initiating Immunotherapy after prepriming and priming therapy enumerated in the present article [16] [Battaglia S]; c) Titrating the intensity of individual therapy in different phases—eg. To limit the dose of SBRT to <10 Gy. [71] [51] [Martinez-Zubiaurre I]. and limit to less that four metastatic lesions [92] [Chmura S] and strategise SBRT as pulsed/(boost) therapy [115] [He K], judicious use of metronomic maintanence chemotherapy [67] [67] [Pasquier], d) keeping the option open for future lessor toxic targeted approaches [93] [Wang, M] ; e) Repurposing the well established relatively non-toxic drugs. [94] [68] [Xia ]
2.1.4. Fundamental Conditions for Effective Combinations, Timing, and Sequencing (CTS Strategy)
According to the principles of vascular normalization, therapy in any combination should induce cancer cell lysis without compromising endothelial integrity and ECM rigidity in the tumor/tumor bed. [59] [Jain RK]. In SBRT, interfering with endothelial cells does not eliminate the primary sarcoma model, and doses exceeding 20 Gy per fraction shift irradiated sarcoma from expected tumor eradication to short-term control. In a study in a mouse model, extensive endothelial cell death with a dose of 50 Gy did not prevent the recurrence. Therefore, according to the study, the critical target for SBRT is tumor cells and not endothelial cells for sarcoma eradication. [42] [Moding EJ]. Generally, ablative therapies leave the tumor bed as a “scar/nidus” for late recurrences with disturbed, rigid ECMs, hindering drug delivery and immunological cross-talk. Dormant cancer cells may reactivate later due to inadequate immunological cross-talk. [17] [Zhao Y].
2.2. CTS Therapy Strategy Phases
2.2.1. CTS Therapy Phase I: Normalization of vasculature and Targeting Hypoxia
Box 2. Begin anticancer therapy to stop the hypoxic resistance cascade and improve the response to later stages of interventions.
Therapy Phase I (The prerequisite for other phases)
- Strategy:
- Prepriming with AAGs to initiate vascular normalization & hypoxia targeting.
- Time of Start:
- Day 0, Day 2: Improves the vasculature.
- Objective 1:
- Normalization of Vasculature for effective subsequent chemo-immunotherapy delivery and improves sensitivity to RT/SBRT.
- Objective 2:
- Improve by adding newer strategies/drugs for vascular normalization.
Supporting Literature:
- Targeting HIF-1α, the root: Efforts have been made to develop HIF inhibitors, such as dissociating HIF-1α and HIF-1β dimers, which inhibit the transcriptional activation of HIF-2. This treatment has been tested in patients with multi-treated renal cell carcinoma and can be combined with classical antiangiogenic drugs or immunotherapies. [27] [Montemagno C]. Targeting HIF-1α is impractical due to a wide range of gene mutations and its normal presence in the homeostatic environment, which contributes to toxicity [22] [Jun JC, ] [23] [Yong L]. Therefore, hypothetically, the optimal approach to targeting HIF-1α involves normalizing oxygenation, restoring normal homeostasis, and reducing cancer-induced inflammation to decrease ROS. For this reason, vascular normalization is essential for restoring normal homeostasis and “reversion/reprogramming” of the cancer process. [95] [Swamy K].
- Vascular normalization and its window: The most accessible vascular normalization agents include antiangiogenics and several small-molecule TKIs, which enhance the effects of subsequent RT/SBRT/CT and immunotherapy during their window period. [35] [Goel S] (Figure 6). Initially, VEGF was the preferred target for normalization, as it was deemed the primary driver. Soon, it became clear that the initial response was followed by re-growth due to the emergence of multiple complementary VEGF and non-VEGF angiogenic pathways [28] [Clarke J M]. Consequently, to inhibit the parallel vertical and horizontal paths, combinations of anti-angiogenics were introduced. However, toxicity and the subsequent development of resistance emerged as significant issues [28] [Clarke J M]. In clinical settings (unlike animal studies), bevacizumab improved survival only when administered with chemotherapy. [32] [Ribatti D].
- Extending the Normalization Window—Temporal Kinetics—Cyclic antiangiogenic schedules: Stopping AAGs during the excessive regression of vessels (around day 28) reverses normalization phenotype, reducing the vascular obstruction and reestablishes normalization due to the withdrawal effect of anti-VEGF. This was observed clinically (MRI) during toxicity-mandated drug holidays. After this drug holiday, restarting AAGs reinitiates the normalization process. [35] (Figure 2) [Goel S]. This finding theoretically enables continuous vascular normalization through cyclical three-to-four-week on/off AAG administration. Several other approaches have been tried to extend the normalization window by targeting bFGF [38] [Zahra]; PDGFR-β [44] [ Hu, X]; soluble Tie-2, Ang-2 neutralization antibodies [45] [Xiaolan Yu et al.]; combined VEGFRs and ANG-2 dual blockade [46] [Li S]; as discussed previously.
- Monitoring normalization window: The normalization window has been identified using functional magnetic resonance imaging (fMRI) 2–4 days after starting sunitinib, demonstrating improved perfusion, reduced hypoxia, and metabolic shifts. FDG-PET measurements of reduced glucose uptake indicate vessel normalization. [76] [Liu, Y]. Therefore, the cyclical AAGs or other approaches discussed above can be personalized for individual patients in clinical trials (“Vascular-guided therapy”) using modern imaging techniques and specific systemic biomarkers like VEGF and miRNA. [37] [Ayoub NM].
- Timing Ang2 & Ang-1: An alternative strategy is to target Ang-1 and Ang-2. A sufficient presence of Ang-1 (with restricted Ang-2) at the maturation stage results in vessels with robust “tip cells” and a thinner vasculature penetrating deeper into the tumor’s’ hypovascular/avascular part. Theoretically, alongside the restriction of Ang-2 at the maturation stage of the vasculature, Ang-1 would induce normalization, overcoming the “trimming effect” of long-term AAGs. [96] [Biel NM].
- Moderate/Lower-Dose Antiangiogenic Therapy: Using low to moderate doses of AAGs effectively normalizes aberrant tumor microvessels and optimizes blood perfusion. [97] [Chatterjee S]. The normalization window is dose- and time-dependent, with higher doses causing excessive vascular pruning earlier. [98] [Huang Y]. High-dose AAG leads to exaggerated hypoxia, shortens the normalization window, and promotes post-window immunosuppressive cell infiltration. [46] [Li S]. Studies showed that short-term high-dose sunitinib (120 mg/kg/day) increased tumor growth, while low doses (30–60 mg/kg/day) did not stimulate metastasis in mice. [34] [Haibe Y]. Low-dose bevacizumab yielded better results in glioblastoma. [46] [Li S]. VEGFR-2 blockade doses increased perfusion and promoted M1-type TAMs and CD8+ T-cell infiltration, while higher doses expanded immunosuppressive Tregs in breast cancer. [47] [Ramjiawan RR]. While optimal normalization dosing remains unclear, lower bevacizumab doses (5 mg/kg for colorectal cancer, 15 mg/kg for lung cancer) warrant further investigation. [56] [Lee W S].
2.2.2. CTS Therapy Phase II: Normalization of Cancer Cell Sequestration, Cancer Cell Lysis, Vascular Enhancement, and Immune-Suppressive Cells Depletion—Making the TME Immunotherapy Ready
Box 3. Focusing on maximum cell kill and enhancement of the wheel of immunity cycle.
Therapy Phase II
Strategy: Priming therapy to “unmask” the cancer (for4 the subsequent immunotherapy)
Time of Start: Start CT/RT (SBRT) in the 1st week of AAGs (> Day 2)
- Objective 1:
- Cancer cell lysis (preferably by ICD); waves of neoantigen generation; Immune suppressive/exhausted cell depletion in TME; decreased ISP; enhanced vascularity (vascular promotion), improved lymphatic drainage for neoantigen presentation in the lymph nodes, and reinvigorating the Immunity cycle; enhanced fresh TME CTLs infiltration.
- Objective 2:
- Endothelial integrity eg. Gene editing (moding ref); improving lymphatic functions, eg, TGF-β blockers; Improving antigenicity and adjuvanticity by immune adjuvants/ nanoedjuvants [4] [Appleton E], professional phagocytic agents [75] [Li SY]; metabolic & pH modulators; Surgery/SBRT for residual resistant mass.
Supporting Literature:
- Effective Cancer Cell-Killing/Lysis and Immune-Suppressive Cell Population Depletion: Routinely used radiotherapy, SBRT, and chemotherapy enhance the normalization window discussed in Phase 1, [35] [Goel S] [76] [Liu, Y.] for subsequent immunotherapy. This ensures more efficient drug distribution due to the space created by cancer cell lysis, normalization of ECM, depletion of immune-suppressive (and immune-exhausted) cells, and reduction of CAF populations. Depleting cancer cells decreases ISP, leading to lower oxygen demand and increased vascularization. Professional phagocytic agents can further aid in decreasing ISP. [75] [Li SY]. TGF-β blockers can also mitigate ISP by reducing inflammation in the ECM and improving the function of uncollapsed lymphatics for presenting neoantigens by APCs at lymph nodes. [99] [Ariffin AB]. An activated immunity cycle and reduced ISP promote the fresh infiltration of antigen-specific CTLs/TILs [100] [Mellman I] [48] [Yang T]. SBRT, at a dose range of 6 Gy to 10 Gy per fraction, induces ICD and vascularization without disrupting endothelial function or stimulating immunosuppressive pathways, thereby preserving the flexible ECM. [71] [Martinez-Zubiaurre I]. Theoretically, SBRT within a non-disruptive vascular range (6 to 10 Gy per fraction) can be scheduled during the extended normalization window of AAG administration as a booster dose against residual disease in advanced malignancies and oligometastases. [101] [Swamy K].
- Fortification of the Vascular Normalization Window—Beyond Antiangiogenics: Several targets can be utilized to normalize the vasculature. Angiostatic factors like TNFα, TSP-1, and endostatin enhance vascular perfusion. Injection of TILs and TNFα intratumorally improves in vivo vaccination effects. The use of the Herpes virus entry mediator Ligand (HVEM-L) repairs abnormal tumor vasculature by activating various intermediaries. Incorporating a modified type 1 repeat peptide of thrombospondin (ABT-510), a TSP-1 mimic that promotes normalization and immune modulation without reducing vascular density. [48] [Yang T]. miRNAs can restore vascular integrity, as with miR-20b inhibiting the nuclear aggregation of HIF-1α and Signal Transducer and Activator of Transcription 3 (STAT3) activation. [102] [Cascio S]. Research has evaluated miRNA-153 suppression of HIF-1α and Ang-1 targeting in breast cancer. [37] [Ayoub NM]. Studies show that miRNA-140-5p silences VEGF-A; miRNA-29b inhibits angiogenesis by downregulating VEGF. Cellular myelocytomatosis oncogene (c-Myc) and miRNA-497 overexpression decrease VEGF and HIF-1α. [48] [Yang T].
- Exploiting Ang2–Ang1 interplay: Combining AAGs with properly timed Ang-1 can produce more viable vasculature during the normalization window period. Even with prolonged treatment of pancreatic tumors in transgenic mouse models using anti-VEGFR2 antibody, a delay in growth and modest survival benefit was noted due to increased expression of the pro-angiogenic Ang-1 and associated growth factors. The Ang–1–Tie pathway leads to the maturation or stabilization of blood vessels when accumulated Ang-2 is blocked. [34] [Haibe Y].
- Genetic modification: Targeting G-protein signaling 5 (Rgs5) can enhance TILs in the tumor parenchyma and the survival of tumor-bearing mice through an unknown mechanism. [46] [Li S]. Another approach is to utilize Dual Recombinase Technology to preferentially radiosensitize tumor cells while protecting the endothelium, particularly during SBRT. [42] [Moding EJ].
- Enhancing primary therapies (chemo-radio-immunotherapy)—antigenicity and adjuvanticity strategies: Localized/intralesional therapies, including oncolytic viruses, oncolytic peptides, STING, and Toll-like receptors (TLR) agonists, can activate immunogenic cold tumors through adjuvanticity and antigenicity [4] [Appleton]. Immunoadjuvants, hyperthermia, and novel nanotechnology-based targeting of the cancer cell cycle enhance the efficacy of chemo-radiotherapy. [103] [Vlastou, E]. The vascular non-disruptive, immunogenic pulsed-SBRT schedule generates an in situ/in vivo therapeutic vaccine effect. This strategy amplifies immune activation and drug penetration within the CCME [81] [Ferretti S] [104] [Liu Z] [105] [Magnussen AL]. Facilitate the neoantigen–APC flow to the lymph nodes by restoring lymphatic functions after opening the lymphatic lumens with reduced interstitial pressure [106] [Padera TP] [107] [Liao S] [108] [Avraham T]. Nanodynamic therapies are open to exploration and can be incorporated for their ability to modify pH and improve oxygen diffusion [109] [Zhang, B].
2.2.3. CTS Therapy Phase III: Initiation of Immunotherapy, Immune Promotion/Enhancement, and Memory Cell Pool Expansion
Box 4. Cancer Cells Sensitized and TME modulated for accelerated immune response.
Therapy Phase III
Strategy: Cancer and TME Immunotherapy Ready Phase and Immune Promotion
Time of Start: 2 to 3 months after neoadjuvant CT/ RT (SBRT)
- Objective 1:
- Optimize the immunotherapy schedule by starting immunotherapy when cancer cells are unmasked and CCME modulated for maximum response & least toxicity.
- Objective 2:
- Integrated Boost/Pulsed SBRT for dynamic generation of contemporary neoantigen for in-situ vaccination effect and to improve memory cell pool.
The literature review in foundational analysis discussed above indicates that approximately eight major conditions must be met before the cancer can be identified as ready for an optimized immunotherapy response. (Figure 7A and 7B).
Supporting Literature:
- Basis for unmasking for immunotherapy as a priming strategy: Vascular normalization and modulation are gaining traction, enhancing the efficacy of immunotherapies such as PD-1/PD-L1 and CTLA-4 antibodies, CAR T cells, and cancer vaccines. Before initiating the immunotherapy strategy of establishing vascular normalization, promotion, or modulation, increases in cancer cell sensitivity, oxygen diffusion, effective drug delivery, and TILs. Chemoradiotherapy depletes TME immune-suppressive elements, particularly Tregs, sensitizing cancer cells and priming the immunity cycle [17] [Zhao Y] [110] [Newport EL]. Infiltrating immune-suppressive cells, stromal cells, and abnormal vascular and lymphatic vessels—key components of the TME—are initially immunologically compromised due to hypoxia, characterized by high interstitial pressure and low pH [46] [Li S]. By the end of Phase II, following chemotherapy or radiotherapy (SBRT) or both, vascular promotion and remodeling facilitate optimal delivery of immunotherapies, immunoadjuvants, newer nanotechnological targets, and oxygen perfusion to the CCME. [17] [Zhao Y] [85] [Shibamoto Y] [35] [Goel S] [111] [Potiron V]. This vascular reset and remodeling, alongside normalized angiogenesis after RT/SBRT, form the foundation for subsequent treatment interventions of other types. [105] [Magnussen AL]. Cancer cell lysis by CTLs initiates the immunity cycle, leading to antigen uptake and presentation by dendritic cells for subsequent iterations of the immunity cycle, adapting to tumor evolution. This creates a virtuous cycle that sustains active immunity. A better understanding of the checkpoint mechanism and the maintenance of anti-tumor immunity by dendritic cells becomes relevant. Tumors exhibit three classical “immunotypes” (immunological phenotypes): immune inflamed (hot), immune excluded (cold), and immune desert. This simplification of tumors’ immunological classification should be considered in the context of the heterogeneity of all three within each particular immunotype. The proposed CTS strategy in the present article is relevant because even next-generation checkpoint inhibitors are unlikely to overcome immune-excluded and immune-desert barriers. Overall, 60%–70% of all cancers exhibit features demonstrating an immune-restrictive phenotype, with shared immune escape essentials refractory to immunotherapy. Amid all the complexities, the goal of a therapeutic strategy should be to ensure the wheel of the cancer immunity cycle keeps turning continuously. [100] [Mellman I] [48] [Yang T].
- Synergism of Immunotherapy with AAGs: Immunotherapy agents also enhance vascular enhancement by improving the immune-vascular and ECM-immune cross-talk. [17] [Zhao Y]. The vascular promotion/ remodelling induced by immunotherapy forms an “enhanced loop” by improving TILs, which improves the response to further immunotherapy doses. [48] [Yang T]. Additionally, resistant phenotypes become sensitive as “immunotypes.” [100] [Mellman I]. For example, PDL-1 expression was upregulated in tumor endothelial cells after AAG treatment, with increased infiltration of CD4+ and CD8+ T cells resulting in improved tumor control following immunotherapy. Also, VEGFA and Ang-2 blockade with the added bispecific antibody Ang-2-VEGF-A CrossMab (A2 V) resulted in enhanced infiltration of CD8+ T cells (leading to increased tumor antigen presentation) and promoted perivascular T cell accumulation. [46] [Li S]. Combining AAG and immunotherapy agents such as bevacizumab, atezolizumab, and apatinib with an anti-PD-L1 antibody could normalize the TME. [38] [Zahra, F.T] [48] [Yang T]. IFN-ϒ secreted by Th1 cells is positively associated with vessel normalization [76] [Liu, Y]. When PD-1/PD-L1 antibody is combined with an anti-VEGF, it has positive results in several phase III studies. This combination overcomes the inhibition of dendritic cell (DC) maturation, which facilitates the recruitment of T cells to the TME with improved perfusion [112] [Hack SP].
- Pulsed Immunogenic Dose SBRT and In situ Vaccination Effects Complementing Immunotherapy in Phase III: This technique has dual advantages. One is preparing the CCME for better penetration of TILs and immunotherapy drugs due to normalization of vessels and perfusion. [85] [Shibamoto Y]. Second, He K et al. (2021), based on indirect clinical evidence and preclinical studies, propose that repeated “pulsed-RT” resulted in the release of tumor antigens, expanded the tumor-specific T-cell receptor repertoire, high-affinity antibodies against the tumor, and memory cells with an in situ vaccination effect. [113] [Sezen D] [114] [Moore C] [115] [He K]. ECM suppleness is paramount for improving immune cells’ memory pool and cross-talk, highlighting all combination therapies’ appropriate dose, timing, and sequence. [17] [Zhao Y].
- Antigenicity and Adjuvanticity acceleration: In vitro-designed vaccines and cell therapies will be adjuncts at the immune-promotion level. Immune adjuvants enhance immune promotion via immune antigenicity–adjuvanticity actions, especially in cancer vaccines. [116] [Baxevanis, C.N]. Intralesional therapies add value to immunological promotion methods through adjuvanticity and antigenicity [117] [DePalo][4] [Appleton E]. Surgical excision after neoadjuvant therapy to downstage the tumor is prevalent in resectable/unresectable locally advanced head and neck cancer. [118] [Chen Y]. Additionally, in neoadjuvant therapy, an optimum immunological cascade may set in before surgery in the presence of intact drainage lymph nodes. [119] [Baniel CC]. The infrequent possibility of progression during neoadjuvant therapy can be minimized by evaluating for response to treatment often [120] [Caudle AS], and the results of several ongoing clinical trials are pending [121] [Rao, Y.J].
2.2.4. CTS Therapy Phase IV: Therapy Consolidation and Cancer Reversion/Reprogramming
Box 5. Beginning of Cancer reversion/ reprogramming to normalcy.
Therapy Phase: IV
- Strategy:
- Consolidation of Immunotherapy Effects, Normalization of CCME, Enhanced Long-term Control and Cancer reversion/reprogramming
Time of Start: After completion of the immunotherapy course.
- Objective 1:
- Design the maintenance therapy with the least long-term side effects.
- Objective 2:
- Develop anticancer drugs suitable for long-term medications to prevent the recurrence/ eliminate dormant cells, like any other chronic disease.
Supporting Literature:
- The maintenance treatments for various malignancies and their duration are still in the early stages of development. A critical issue is creating methods to monitor the quantification of microscopic, quiescent, or dormant cancer cells. However, this represents a crucial step toward evolving therapies for “reversion” and a cure. The presence of dying or apoptotic cells also contributes to the measured cfDNA levels, limiting their usefulness as indirect information about living cancer cells. [122] [Heitzer E]. The marker for living cancer cell activity, which is reasonably stable, is microRNA (miRNA). [123] [Ruksha TG] Even though specific cirRNA has not made its foray into monitoring dormancy, due to its abundance, high stability, functional diversity, ability to act as a protein sponge, and evolutionary conservation, cirRNA is the dormancy biomarker of the future. [124] [Bach DH]. CirRNA may finally turn out to be the choice for cancer elimination monitoring. Hence, it is worth exploring how to extend the cancer therapy intervention period beyond the current strategy of merely achieving remission and “leaving it” halfway, much before the first step in cancer elimination is completed.
- <105 Cells and Dormancy Factor: The stage is now set for “reversion/reprogramming” of cancer by reestablishing normalized phenotypes and the TME, potentially overriding persistent genetic mutations. The adoption of the embryonic microenvironment within the TME can further support this. [6] [Pensotti A].
- Consolidation therapy / Cancer Cell “Reversion” monitoring: Using liquid biopsy to detect minimal residual disease (MRD) by adopting circulating tumor cells (CTC) and cell-free DNA (cfDNA) for establishing the presence of residual cells is gaining acceptance over imaging in follow-up for solid tumors. This is particularly useful for guiding maintenance treatment. [125] [Ma Y] (Figure 1B).
2.2.5. CTS Therapy Phase V: Prevention of late recurrences and Cancer Reversion/Reprogramming
Box 6. The crucial phase of returning to a “benign state” to prevent late recurrences.
Therapy Phase: V
- Strategy:
- To prevent late recurrences
Time of Start: Starts from Phase I
- Objective 1:
- To keep the ECM supple. Secondly, to target HIF1-α and ROS to flip towards normalization of CCME and elimination of dormant cells.
- Objective 2:
- Reducing inflammation by maintenance therapy, senolytics, and lifestyle modifications or combinations thereof.
Supporting Literature:
- Steps to Prevent Late Recurrences: The treatment mandate at this stage is to preserve a supple ECM following all combination therapies to prevent late recurrences while minimizing long-term side effects. [126] [Abyaneh HS]. Therefore, planning for this step begins with Actionable Phase I. Two factors meet this criterion: ensuring vascular endothelial integrity (especially of the endothelial stem cells) [42] [Moding EJ] and preventing fibrosis in the ECM. [17] [Zhao Y]. When SBRT is part of treatment, the dose per fraction (total dose in regular radiotherapy) and the complex role of TGF-β on ECM are crucial for ECM suppleness. TGF-β plays a significant role in normal homeostasis and can contribute to post-therapy tissue inflammation and fibrosis. TGF-β blockers help keep the ECM supple, maintaining immunological cross-talk and preventing breakthroughs in cancer cells dormancy [71] [ Martinez-Zubiaurre I ] [17] [Zhao Y]. The elements of the consolidation phase include metronomic chemotherapy. [71] [Martinez-Zubiaurre I]. repurposing drugs [94] [Xia, Y], senolytics [127] [Wyld, L] [128] [Kirkland, J.L], lifestyle modifications [129] [Berrino F], or a combination of these for the cancer reversion/reprogramming process. The future should focus on developing targets that emphasize the fundamental processes involved in the initiation and progression of cancer. This includes targeting increased ROS, persistent HIF-1α in the cytosol, and the HIF-1α-HIF-1β dimer within the cell nucleus. [130] [Ziello JE].
- Immune Protection—Expanding the Memory Cell Pool “Repertoire”: The background objective of Phases I through IV of the CTS strategy is to establish a memory cell pool that provides long-term immunological protection while targeting cancer cells. Although CT has traditionally been viewed as harmful to the immune system, a growing body of evidence suggests its capacity to engage innate and adaptive immunity. Studies have demonstrated a CD44hi memory T cell response associated with specific CT low-dose schedules and an enhanced peptide-specific CD8+ effector T cell response to DTIC. [131] [Emens LA]. After CT, resilient effector memory CD8+ T cells, which inherently possess strong resistance, are the primary contributors to the re-expansion of the memory pool. Reforming the T cell memory subpopulation is supported by an in vitro study showing strong proliferation of these subsets after IL-7/IL-15 supplementation. Eventually, there will be a shift from terminal effector phenotype cells to effector memory cells following the recovery period, alongside reduced MDSCs and Tregs. Importantly, subsequent adoptive cell therapy regimens may lead to immune memory in solid tumors. [132] [Truong, N]. Pretreatment with CT can activate virtual memory (VM) CD8+ T cells in an antigen-independent manner, mediating cancer cell cytotoxicity. [133] [Schmiechen ZC].
Reports indicate that RT is not contributing to the pool of memory cells in the mouse model. [134][Huang, J]. Radiotherapy is a double-edged sword, with a “dark” and “bright” side that does not always synergize with immunotherapy. Synergism depends on dose per fraction, timing, and sequencing. The combination of RT and immunotherapy should have an RT immunogenic endothelial non-disruptive dose per sitting of 6 to 10 Gy, within 2 to 7 days (timing), and should follow RT (sequencing). Studies have shown drug accumulation of 1.2 to 3.3 fold enhancement intratumorally after RT and DC recruitment into tumors between days 5 and 10. [71] [Martinez-Zubiaurre I]. Initiating Immunotherapy within one week of SBRT in treatment naïve patients can be a potential standard practice until further evidence emerges [135] [Breen WG] . The adjuvanticity effect of immunogenic cell death by SBRT can induce in situ vaccine effects due to neoantigen generation, which would activate the immunity cycle, mediated by damage-associated molecular patterns (DAMPs), high mobility group box 1 (HMGB1), calreticulin, and heat shock proteins (HSPs). [136] [N.E. Donlon]. Timed pulsed-SBRT (or SBRT boosts) can replenish and expand this pool by delivering waves of neoantigens with each therapy pulse, in line with the mutational changes in the cancer cells’ subclones. (Figure 6 & Figure 8). Additionally, combining pulsed XRT with anti-CTLA-4 resulted in delayed growth at sites beyond indexed lesions and improved survival rates. More importantly, compared to single-cycle RT, pulsed RT expanded the CD4+ effector memory repertoire, similar to a booster concept with conventional vaccines, generating long-term memory. [113] [Sezen D] [114] [Moore C] [115] [He K]. Transcriptomic analysis shows that tissue-resident memory T cells are more resistant to chemotherapy and RT than measurements usually done with naïve T cells in circulation. The other critical finding in a study is that newly infiltrating T cells can be reprogrammed into tissue resident memory cells more effectively when combined with PD1 blockade. [137] [Zhai D].
2.3. Methodology for Adopting the CTS Strategy
- Regardless of how innovative the treatment may be, a singular or limited approach cannot sufficiently address the complex and evolved architecture of cancer lesions in today’s context. Even in lung cancer, where immunotherapy, targeted drug therapy, and conventional chemotherapy have advanced significantly over the decades, drug resistance remains a major challenge. Tumor heterogeneity, physical barriers, adaptive dynamic mutations, and dormancy contribute to treatment failures. Understanding and targeting multiple pathways is essential for progressing toward more reliable therapies. [78] [Kang, D.H].
- The CTS strategy outlined above could be crucial for addressing the complexities of cancer. Efforts to explore the right combination of radiotherapy, chemotherapy, and immunotherapy have demonstrated benefits in only a few trials. Insights from spatial transcriptomics and single-cell sequencing may aid individual CCME in determining optimal scheduling. [79] [S.Sordo-Bahamonde].
- During trials, an adaptive multi-arm, multi-stage (MAMS) methodology is used by adding or removing therapies from a combination. This can be achieved through either the drop-the-losers design or a Bayesian design by selecting all promising treatments in the order of their treatment effects based on their mechanisms of action, as proposed in the classification above. Designing the testing models with artificial intelligence and machine learning can be accomplished using the analytical approach discussed. [138] [Wason J] [139] [Serra A].
- An earlier version of this work is available on Preprint.org [140] [Swamy, K.]. A detailed exploration of Pulsed SBRT as a practical, precisely timed in situ/in vivo vaccine modulator is discussed elsewhere. These findings highlight the potential of incorporating advanced radiotherapy techniques like SBRT into evolving therapeutic strategies. [141] [Swamy, K].
Conclusions
Immunotherapy has fundamentally transformed the landscape of cancer treatment, offering new hope to patients across all stages of the disease. While its average benefit remains modest in solid tumors, integrating immunotherapy with other modalities, such as chemotherapy, targeted agents, antiangiogenics, and radiotherapy, has significantly improved many patients’ survival and quality of life. The Combination, Timing, and Sequencing (CTS) strategy outlined in this review provides a logical, adaptable framework for maximizing the effectiveness of immunotherapy by “unmasking” cancer cells and optimizing the tumor microenvironment.
Foundational factors such as hypoxia lead to core phenotypes of aberrant vasculature, metabolic reprogramming, cancer stem cell trait acquisition, and immune dysregulation, which play critical roles in cancer progression, EMT/metastases, and resistance. [142] [Bae T]. Addressing these factors with tailored, step-by-step interventions can enhance immune recognition and response without creating opportunities for additional resistance to develop. ECM normalization enhances the likelihood of the “reversion” of cancer. The authors propose focusing on the cancer cell microenvironment (CCME), which aligns with recent developments in transcriptomics, amid the various barriers in the tumor microenvironment (TME). Ensuring the effective delivery of diverse anticancer agents, including nanoparticles, chimeric antigen receptor (CAR) cells, antibodies, vaccines, radionuclides, phagocytic agents, intralesional drugs, and TILs to the CCME is critical. Pulsed SBRT presents a promising opportunity to enhance in situ vaccination effects and optimize short- and long-term immune cycle activation.
In summary, despite present-day advances, challenges remain. The CTS approach represents a promising paradigm for harnessing the full potential of immunotherapy. It involves strategically combining and sequencing available therapies and personalizing interventions based on tumor evolutionary biology and patient tolerance. Integrating dynamic biomarkers (e.g., soluble VEGFR, circulating tumor DNA) into the Combination, Timing, and Sequencing (CTS) framework offers a powerful approach to optimizing real-time therapy and individualized patient care.
Author Contributions
Equal—Concepts and review
Funding
None.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declared no conflicts of interest.
Grammar Check
https://www.grammarly.com/ and MDPI English Language Editing Services.
References
- Passariello, M., Manna, L., Rapuano Lembo, R. et al. Tri-specific tribodies targeting 5T4, CD3, and immune checkpoint drive stronger functional T-cell responses than combinations of antibody therapeutics. Cell Death Discov. 11, 58 (2025). [CrossRef]
- Pascual J, Lim JSJ, Macpherson IR, et al., Triplet Therapy with Palbociclib, Taselisib, and Fulvestrant in PIK3CA-Mutant Breast Cancer and Doublet Palbociclib and Taselisib in Pathway-Mutant Solid Cancers. Cancer Discov. 2021 Jan;11(1):92-107. [CrossRef]
- Zhi Li, Peihao Yin, Tumor microenvironment diversity and plasticity in cancer multidrug resistance, Biochimica et Biophysica Acta (BBA)—Reviews on Cancer, Volume 1878, Issue 6, 2023, 188997. [CrossRef]
- Appleton E, Hassan J, Chan Wah Hak C, Sivamanoharan N, Wilkins A, Samson A, Ono M, Harrington KJ, Melcher A, Wennerberg E. Kickstarting Immunity in Cold Tumours: Localised Tumour Therapy Combinations With Immune Checkpoint Blockade. Front Immunol. 2021 Oct 18;12:754436. [CrossRef]
- Putzu C, Canova S, Paliogiannis P, Lobrano R, Sala L, Cortinovis DL, Colonese F. Duration of Immunotherapy in Non-Small Cell Lung Cancer Survivors: A Lifelong Commitment? Cancers (Basel). 2023 Jan 22;15(3):689. [CrossRef]
- Pensotti A, Bizzarri M, Bertolaso M. The phenotypic reversion of cancer: Experimental evidence on cancer reversibility through epigenetic mechanisms (Review). Oncol Rep. 2024 Mar;51(3):48 . https://pmc.ncbi.nlm.nih.gov/articles/PMC10835663. [CrossRef]
- Tuynder M, Susini L, Prieur S, Besse S, Fiucci G, Amson R, Telerman A. Biological models and genes of tumor reversion: cellular reprogramming through tpt1/TCTP and SIAH-1. Proc Natl Acad Sci U S A. 2002 Nov 12;99(23):14976-81. [CrossRef]
- Shin D, Cho KH. Critical transition and reversion of tumorigenesis. Exp Mol Med. 2023 Apr;55(4):692-705. [CrossRef]
- Garg, P.; Malhotra, J.; Kulkarni, P.; Horne, D.; Salgia, R.; Singhal, S.S. Emerging Therapeutic Strategies to Overcome Drug Resistance in Cancer Cells. Cancers 2024, 16, 2478. [CrossRef]
- Lei ZN, Tian Q, Teng QX, Wurpel JND, Zeng L, Pan Y, Chen ZS. Understanding and targeting resistance mechanisms in cancer. MedComm (2020). 2023 May 22;4(3):e265. [CrossRef]
- Škarková A, Bizzarri M, Janoštiak R, Mašek J, Rosel D, Brábek J. Educate, not kill: treating cancer without triggering its defenses. Trends Mol Med. 2024 Jul;30(7):673-685. [CrossRef]
- Ingber DE. Can cancer be reversed by engineering the tumor microenvironment? Semin Cancer Biol. 2008 Oct;18(5):356-64. [CrossRef]
- Jacquemin, V.; Antoine, M.; Dom, G.; Detours, V.; Maenhaut, C.; Dumont, J.E. Dynamic Cancer Cell Heterogeneity: Diagnostic and Therapeutic Implications. Cancers 2022, 14, 280. [CrossRef]
- Newton JM, Hanoteau A, Liu HC, Gaspero A, Parikh F, Gartrell-Corrado RD, Hart TD, Laoui D, Van Ginderachter JA, Dharmaraj N, Spanos WC, Saenger Y, Young S, Sikora AG. Immune microenvironment modulation unmasks therapeutic benefit of radiotherapy and checkpoint inhibition. J Immunother Cancer. 2019 Aug 13;7(1):216. [CrossRef]
- Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004 Aug;21(2):137-48. PMID: 15308095. [CrossRef]
- Battaglia S, Muhitch JB. Unmasking targets of antitumor immunity via high-throughput antigen profiling. Curr Opin Biotechnol. 2016 Dec;42:92-97. [CrossRef]
- Zhao Y, Yu X, Li J. Manipulation of immune‒vascular cross-talk: new strategies towards cancer treatment. Acta Pharm Sin B. 2020 Nov;10(11):2018-2036. [CrossRef]
- Schmitt MW, Loeb LA, Salk JJ. The influence of subclonal resistance mutations on targeted cancer therapy. Nat Rev Clin Oncol. 2016 Jun;13(6):335-47. [CrossRef]
- Gatenby RA, Brown JS. The Evolution and Ecology of Resistance in Cancer Therapy. Cold Spring Harb Perspect Med. 2020 Nov 2;10(11):a040972. https://pmc.ncbi.nlm.nih.gov/articles/PMC7605238/. [CrossRef]
- Groebe K, Vaupel P. Evaluation of oxygen diffusion distances in human breast cancer xenografts using tumour-specific in vivo data: role of various mechanisms in the development of tumour hypoxia. Int J Radiat Oncol Biol Phys. 1988 Sep;15(3):691-7. [CrossRef]
- MOORE GE, SANDBERG AA, WATNE AL. The comparative size and structure of tumour cells and clumps in the blood, bone marrow, and tumour imprints. Cancer. 1960 Jan-Feb;13:111-7. [CrossRef]
- Jun JC, Rathore A, Younas H, Gilkes D, Polotsky VY. Hypoxia-Inducible Factors and Cancer. Curr Sleep Med Rep. 2017 Mar;3(1):1-10. [CrossRef]
- Yong L, Tang S, Yu H, Zhang H, Zhang Y, Wan Y, Cai F. The role of hypoxia-inducible factor-1 alpha in multidrug-resistant breast cancer. [CrossRef]
- Al-Ostoot FH, Salah S, Khamees HA, Khanum SA. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat Res Commun. 2021;28:100422. [CrossRef]
- Multhoff, G., Radons, J., & Vaupel, P. (2014). Critical Role of Aberrant Angiogenesis in Developing Tumor Hypoxia and Associated Radioresistance. Cancers, 2014, 6(2), 813-828. [CrossRef]
- Wegiel B, Vuerich M, Daneshmandi Sand Seth P (2018) Metabolic Switch in the Tumor Microenvironment Determi[nes Immune Responses to Anticancer Therapy. Front. Oncol. (2018) 8:284. [CrossRef]
- Montemagno C and Pagès G (2020) Resistance to Antiangiogenic Therapies: A Mechanism Depending on the Time of Exposure to the Drugs. Front. Cell Dev. Biol. 8:584. 2020. [CrossRef]
- Clarke J M, Hurwitz H I. Understanding and targeting resistance to antiangiogenic therapies. J Gastrointest Oncol 2013;4(3):253-263. [CrossRef]
- Caporarello N, Lupo G, Olivieri M et al. Classical VEGF, Notch, and Ang signalling in cancer angiogenesis, alternative approaches and future directions (Review). Mol Med Rep 16: 4393-4402, 2017. PMCID: https://pubmed.ncbi.nlm.nih.gov/28791360/. [CrossRef]
- Zahra, F.T.; Sajib, M. S.; Mikelis, C.M. Role of bFGF in Acquired Resistance upon Anti-VEGF Therapy in Cancer. Cancers 2021, 13, 1422. [CrossRef]
- Jiang Z, Zhou J, Li L, Liao S, He J, Zhou S, Zhou Y. Pericytes in the tumor microenvironment. Cancer Lett. 2023 Mar 1;556:216074. [CrossRef]
- Ribatti D. Tumor refractoriness to anti-VEGF therapy. Oncotarget. 2016 Jul 19;7(29):46668-46677. [CrossRef]
- Brossa A, Grange C, Mancuso L, Annaratone L, Satolli MA, Mazzone M, Camussi G, Bussolati B. Sunitinib but not VEGF blockade inhibits cancer stem cell endothelial differentiation. Oncotarget. 2015 May 10;6(13):11295-309. [CrossRef]
- Haibe Y, Kreidieh M, El Hajj H et al. Resistance Mechanisms to Antiangiogenic Therapies in Cancer. Front. Oncol. (2020) 10:221. [CrossRef]
- Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalisation of the vasculature is needed to treat cancer and other diseases. Physiol Rev. 2011 Jul;91(3):1071-121. [CrossRef]
- Fong SS, Nanchen A, Palsson BO, Sauer U. Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J Biol Chem. 2006 Mar 24;281(12):8024-33. [CrossRef]
- Ayoub NM, Jaradat SK, Al-Shami KM, Alkhalifa AE. Targeting Angiogenesis in Breast Cancer: Current Evidence and Future Perspectives of Novel Antiangiogenic Approaches. Front Pharmacol. 2022 Feb 25;13:838133. [CrossRef]
- Zahra, F.T.; Sajib, M. S.; Mikelis, C.M. Role of bFGF in Acquired Resistance upon Anti-VEGF Therapy in Cancer. Cancers 2021, 13, 1422. [CrossRef]
- Winkler F, Kozin SV, Tong RT, Chae SS, Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E, Munn LL, Jain RK. Kinetics of vascular normalisation by VEGFR2 blockade governs brain tumour response to radiation: role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell. 2004 Dec;6(6):553-63. [CrossRef]
- Chae SS, Kamoun WS, Farrar CT, Kirkpatrick ND, Niemeyer E, de Graaf AM, Sorensen AG, Munn LL, Jain RK, Fukumura D. Angiopoietin-2 interferes with anti-VEGFR2-induced vessel normalisation and survival benefit in mice bearing gliomas. Clin Cancer Res. 2010 Jul 15;16(14):3618-27. [CrossRef]
- Thomas Nielsen; Lise Bentzen; Michael Pedersen; Trine Tramm; Paul F.J.W. Rijken; Johan Bussink; Michael R. Horsman; Leif Østergaard. Combretastatin A-4 Phosphate Affects Tumor Vessel Volume and Size Distribution as Assessed Using MRI-Based Vessel Size Imaging. Clin Cancer Res (2012) 18 (23): 6469–6477. [CrossRef]
- Moding EJ, Castle KD, Perez BA, Oh P, Min HD, Norris H, et al. Tumor cells, but not endothelial cells, mediate the eradication of primary sarcomas by stereotactic body radiation therapy. Sci Trans Med (2015) 7(278):278ra34– 278ra34. [CrossRef]
- Jain RK. Normalisation of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005 Jan 7;307(5706):58-62. [CrossRef]
- Hu, X., Ye, K., Bo, S. et al. Monitoring imatinib decreasing pericyte coverage and HIF-1α level in a colorectal cancer model by an ultrahigh-field multiparametric MRI approach. J Transl Med 22, 712 (2024). [CrossRef]
- Xiaolan Yu and Fengchun Ye. Role of Angiopoietins in Development of Cancer and Neoplasia Associated with Viral Infection. Cells 2020, 9, 457. [CrossRef]
- Li S, Zhang Q, Hong Y. Tumor Vessel Normalization: A Window to Enhancing Cancer Immunotherapy. Technol Cancer Res Treat. 2020 Jan-Dec;19:1533033820980116. [CrossRef]
- Ramjiawan RR, Griffioen AW, Duda DG. Antiangiogenesis for Cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis. 2017 May;20(2):185-204. [CrossRef]
- Yang T, Xiao H, Liu X, Wang Z, Zhang Q, Wei N, Guo X. Vascular Normalization: A New Window Opened for Cancer Therapies. Front Oncol. 2021 Aug 12;11:719836. [CrossRef]
- Ganapathy-Kanniappan, S., Geschwind, JF.H. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer 12, 152 (2013). [CrossRef]
- Weihua Wu, Shimin Zhao, Metabolic changes in Cancer: beyond the Warburg effect, Acta Biochimica et Biophysica Sinica, Volume 45, Issue 1, January 2013, Pages 18–26. [CrossRef]
- Liberti MV, Locasale JW. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci. 2016 Mar;41(3):211-218. Erratum in: Trends Biochem Sci. 2016 Mar;41(3):287. Erratum in: Trends Biochem Sci. 2016 Mar;41(3):287. https://doi.org/10.1016/j.tibs.2016.01.004. [CrossRef]
- Schreier A, Zappasodi R, Serganova I, Brown KA, Demaria S and Andreopoulou E (2023) Facts and Perspectives: Implications of tumor glycolysis on immunotherapy response in triple negative breast cancer. Front. Oncol. 12:1061789. [CrossRef]
- James Ralston Kennedy Paterson. The Treatment of Malignant Disease by Radiotherapy. Williams & Wilkins Company, 1963. Page 45.
- Lipinski KA, Barber LJ, Davies MN, Ashenden M, Sottoriva A, Gerlinger M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer. 2016 Jan;2(1):49-63. [CrossRef]
- Brady SW, McQuerry JA, Qiao Y, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun. 2017 Nov 1;8(1):1231. Erratum in: Nat Commun. 2018 Feb 5;9(1):572. [CrossRef]
- Lee W S, Yang H, Chon H J and Kim C. Combination of antiangiogenic therapy and immune checkpoint blockade normalise vascular immune cross-talk to potentiate cancer immunity. Experimental & Molecular Medicine (2020) 52:1475–1485. [CrossRef]
- Amersfoort J, Eelen G, and Carmeliet P. Immunomodulation by endothelial cells — partnering up with the immune system? PERSPECTIVES Nature Reviews. Nat Rev Immunol 2022 Mar 14;1-13. [CrossRef]
- Finger AM, Hendley AM, Figueroa D, Gonzalez H, Weaver VM. Tissue mechanics in tumor heterogeneity and aggression. Trends Cancer. 2025 Apr 29:S2405-8033(25)00096-2. [CrossRef]
- Jain RK, Martin JD, Stylianopoulos T. The role of mechanical forces in tumour growth and therapy. Annu Rev Biomed Eng. 2014 Jul 11;16:321-46. [CrossRef]
- Karin E. de Visser, Johanna A. Joyce, The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth, Cancer Cell, Volume 41, Issue 3, 2023, Pages 374-403. [CrossRef]
- Zhou H, Wang M, Zhang Y, Su Q, Xie Z, Chen X, Yan R, Li P, Li T, Qin X, Yang H, Wu C, You F, Li S, Liu Y. Functions and Clinical significance of mechanical tumour microenvironment: cancer cell sensing, mechanobiology and metastasis. Cancer Commun (Lond). 2022 May;42(5):374-400. [CrossRef]
- Chung, S.W., Xie, Y. & Suk, J.S. Overcoming physical stromal barriers to cancer immunotherapy. Drug Deliv. and Transl. Res. 11, 2430–2447 (2021). [CrossRef]
- Kim G B, Riley, J L, Levine L B. Engineering T cells to survive and thrive in the hostile tumor microenvironment, Current Opinion in Biomedical Engineering, Volume 21, 2022, 100360. [CrossRef]
- Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T, Tuccitto A, et al. Cancer acidity: An ultimate frontier of tumour immune escape and a novel target of immunomodulation. Semin Cancer Biol. 2017 Apr;43:74-89. [CrossRef]
- Liu, S., Wang, W., Hu, S. et al. Radiotherapy remodels the tumour microenvironment to enhance immunotherapeutic sensitivity. Cell Death Dis 14, 679 (2023). [CrossRef]
- Bogdanov A, Bogdanov A, Chubenko V, Volkov N, Moiseenko F, Moiseyenko V. Tumor acidity: From hallmark of cancer to target of treatment. Front Oncol. 2022 Aug 29;12:979154. [CrossRef]
- Druzhkova I, Lukina M, Dudenkova V, Ignatova N, Snopova L, Gavrina A, Shimolina L, Belousov V, Zagaynova E, Shirmanova M. Tracing of intracellular pH in cancer cells in response to Taxol treatment. Cell Cycle. 2021 Aug;20(16):1540-1551. [CrossRef]
- Shirmanova MV, Druzhkova IN, Lukina MM, Dudenkova VV, Ignatova NI, Snopova LB, Shcheslavskiy VI, Belousov VV, Zagaynova EV. Chemotherapy with cisplatin: insights into intracellular pH and metabolic landscape of cancer cells in vitro and in vivo. Sci Rep. 2017 Aug 21;7(1):8911. [CrossRef]
- Matthew D. Galsky, Xiangnan Guan, et al. Immunomodulatory effects and improved outcomes with cisplatin- versus carboplatin-based chemotherapy plus atezolizumab in urothelial cancer, Cell Reports Medicine, Volume 5, Issue 2, 2024, 101393. [CrossRef]
- Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev Cancer. 2020 Apr;1873(2):188356. [CrossRef]
- Martinez-Zubiaurre I, Chalmers AJ, Hellevik T. Radiation-induced transformation of immunoregulatory networks in the tumour stroma. Front Immunol (2018) 9:167. [CrossRef]
- Dewan MZ, Galloway AE, Kawashima N, et al. Fractionated But Not Single-Dose Radiotherapy Induces an ImmuneMediated Abscopal Effect When Combined With Anti-CTLA–4 Antibody. Clin Cancer Res (2009) 15(17):5379–88. [CrossRef]
- Nguyen DT, Ogando-Rivas E, Liu R, Wang T, Rubin J, Jin L, Tao H, Sawyer WW, Mendez-Gomez HR, Cascio M, Mitchell DA, Huang J, Sawyer WG, Sayour EJ, Castillo P. CAR T Cell Locomotion in Solid Tumor Microenvironment. Cells. 2022 Jun 20;11(12):1974. [CrossRef]
- Miao L, Huang L. Exploring the tumour microenvironment with nanoparticles. Cancer Treat Res. 2015;166:193-226. PMID: 25895870; PMCID: PMC5010228. [CrossRef]
- Li SY, Guo YL, Tian JW, Zhang HJ, Li RF, Gong P, Yu ZL. Antitumor Strategies by Harnessing the Phagocytosis of Macrophages. Cancers (Basel). 2023 May 11;15(10):2717. [CrossRef]
- Liu, Y., Wang, Y., Yang, Y. et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Sig Transduct Target Ther 8, 104 (2023). [CrossRef]
- Brunner TB. The rationale of combined radiotherapy and chemotherapy—Joint action of Castor and Pollux. Best Pract Res Clin Gastroenterol. 2016 Aug;30(4):515-28. [CrossRef]
- Kang, D.H.; Lee, J.; Im, S.; Chung, C. Navigating the Complexity of Resistance in Lung Cancer Therapy: Mechanisms, Organoid Models, and Strategies for Overcoming Treatment Failure. Cancers 2024, 16, 3996. [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; Gonzalez-Rodriguez, A.P.; Martínez-Pérez, A.; Rodrigo, J.P.; García-Pedrero, J.M.; Gonzalez, S. Chemo-Immunotherapy: A New Trend in Cancer Treatment. Cancers 2023, 15, 2912. [CrossRef]
- Sun, X.X.; Nosrati, Z.; Ko, J.; Lee, C.-M.; Bennewith, K.L.; Bally, M.B. Induced Vascular Normalization—Can One Force Tumors to Surrender to a Better Microenvironment? Pharmaceutics 2023, 15, 2022. [CrossRef]
- Ferretti S, Allegrini PR, Becquet MM, McSheehy PM. Tumor interstitial fluid pressure as an early-response marker for anticancer therapeutics. Neoplasia. 2009 Sep;11(9):874-81. [CrossRef]
- K.E. Hoffman ∙ R. Abi-Raad ∙ M. Ancukiewicz ∙ E. Yeh ∙ P. Ryan ∙ L. Schapira ∙ J. Younger ∙ B.L. Smith ∙ I. Kuter ∙ A.G. Taghian. Impact of Interstitial Fluid Pressure, Tumor Oxygenation, and Chemotherapy Drug Sequencing on Response to Neoadjuvant Chemotherapy and Long-Term Local Control in Women Treated for Locally Advanced Breast Cancer. International Journal of Radiation Oncology, Biology, Physics, Volume 69, Issue 3, Supplement S61-S62November 01, 2007.
- 83. [CrossRef]
- Taghian AG, Abi-Raad R, Assaad SI, Casty A, Ancukiewicz M, Yeh E, Molokhia P, Attia K, Sullivan T, Kuter I, Boucher Y, Powell SN. Paclitaxel decreases the interstitial fluid pressure and improves oxygenation in breast cancers in patients treated with neoadjuvant chemotherapy: clinical implications. J Clin Oncol. 2005 Mar 20;23(9):1951-61. [CrossRef]
- Barsoumian HB, Sheth RA, Ramapriyan R, Hsu E, Gagea M, Crowley K, Sezen D, Williams M, Welsh JW. Radiation Therapy Modulates Tumor Physical Characteristics to Reduce Intratumoral Pressure and Enhance Intratumoral Drug Delivery and Retention. Adv Radiat Oncol. 2022 Dec 5;8(2):101137. [CrossRef]
- Shibamoto Y, Miyakawa A, Otsuka S, Iwata H. Radiobiology of hypofractionated stereotactic radiotherapy: what are the optimal fractionation schedules? J Radiat Res. 2016 Aug;57 Suppl 1(Suppl 1):i76-i82. [CrossRef]
- Sophie A. Lelièvre, Kurt B. Hodges, Pierre-Alexandre Vidi. Chapter 26—Application of Theranostics to Measure and Treat Cell Heterogeneity in Cancer. Editor(s): Xiaoyuan Chen, Stephen Wong, Cancer Theranostics, Academic Press, 2014, Pages 493-516. [CrossRef]
- Li Q, Lei X, Zhu J, Zhong Y, Yang J, Wang J, Tan H. Radiotherapy/Chemotherapy-Immunotherapy for Cancer Management: From Mechanisms to Clinical Implications. Oxid Med Cell Longev. 2023 Feb 2;2023:7530794. [CrossRef]
- Ling, S.P.; Ming, L.C.; Dhaliwal, J.S.; Gupta, M.; Ardianto, C.; Goh, K.W.; Hussain, Z.; Shafqat, N. Role of Immunotherapy in the Treatment of Cancer: A Systematic Review. Cancers 2022, 14, 5205. [CrossRef]
- Tie, Y., Tang, F., Wei, Yq. et al. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol 15, 61 (2022). [CrossRef]
- Liu, N., Wang, X., Wang, Z. et al. Nanomaterials-driven in situ vaccination: a novel frontier in tumor immunotherapy. J Hematol Oncol 18, 45 (2025). [CrossRef]
- Mortaja M, Adams SR, McKay RR, Gutkind JS, Advani SJ. Spatially precise chemo-radio-immunotherapy by antibody drug conjugate directed tumor radiosensitization to potentiate immunotherapies. NPJ Precis Oncol. 2025 Apr 4;9(1):97. [CrossRef]
- Chmura S, Winter KA, Robinson Cet al., Evaluation of Safety of Stereotactic Body Radiotherapy for the Treatment of Patients With Multiple Metastases: Findings From the NRG-BR001 Phase 1 Trial. JAMA Oncol. 2021 Jun 1;7(6):845-852. [CrossRef]
- Wang, M., Yu, F. & Zhang, Y. Present and future of cancer nano-immunotherapy: opportunities, obstacles and challenges. Mol Cancer 24, 26 (2025). [CrossRef]
- Xia, Y., Sun, M., Huang, H. et al. Drug repurposing for cancer therapy. Sig Transduct Target Ther 9, 92 (2024). [CrossRef]
- Swamy K. Vascular-Immuno-Phenotypic (VIP) Model for Locally Advanced and Oligo-Metastatic Cancer: A Hypothesis. Med Hypotheses (2021) 152:110618. [CrossRef]
- Biel NM, Siemann DW. Targeting the Angiopoietin-2/Tie-2 axis in conjunction with VEGF signal interference. Cancer Lett. 2016 Oct 1;380(2):525-533. [CrossRef]
- Chatterjee S, Wieczorek C, Schöttle J, Siobal M, Hinze Y, Franz T, Florin A, Adamczak J, Heukamp LC, Neumaier B, Ullrich RT. Transient antiangiogenic treatment improves the delivery of cytotoxic compounds and therapeutic outcomes in lung cancer. Cancer Res. 2014 May 15;74(10):2816-24. [CrossRef]
- Huang Y, Stylianopoulos T, Duda DG, Fukumura D, Jain RK. The benefits of vascular normalisation are dose and time-dependent--letter. Cancer Res. 2013 Dec 1;73(23):7144-6. [CrossRef]
- Ariffin AB, Forde PF, Jahangeer S, Soden DM, Hinchion J. Releasing pressure in tumours: what do we know so far and where do we go from here? A review. Cancer Res. 2014 May 15;74(10):2655-62. [CrossRef]
- Mellman I, Chen DS, Powles T, Turley SJ. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity. 2023 Oct 10;56(10):2188-2205. [CrossRef]
- Swamy K. Stereotactic Body Radiotherapy Immunological Planning-A Review With a Proposed Theoretical Model. Front Oncol. 2022 Jan 26;12:729250. [CrossRef]
- Cascio S, D’Andrea A, Ferla R, Surmacz E, Gulotta E, Amodeo V, Bazan V, Gebbia N, Russo A. miR-20b modulates VEGF expression by targeting HIF-1 alpha and STAT3 in MCF-7 breast cancer cells. J Cell Physiol. 2010 Jul;224(1):242-9. [CrossRef]
- Vlastou, E.; Kougioumtzopoulou, A.; Platoni, K.; Georgakopoulos, I.; Lagopati, N.; Kouloulias, V.; Zygogianni, A. The Emerging Role of Nanoparticles Combined with Either Radiotherapy or Hyperthermia in Head and Neck Cancer: A Current Review. Cancers 2025, 17, 899. [CrossRef]
- Liu Z, Zhao Q, Zheng Z, Liu S, Meng L, Dong L, Jiang X. Vascular normalisation in immunotherapy: A promising mechanisms combined with radiotherapy. Biomed Pharmacother. 2021 Jul;139:111607. [CrossRef]
- Magnussen AL, Mills IG. Vascular normalisation as the stepping stone into tumour microenvironment transformation. Br J Cancer. 2021 Aug;125(3):324-336. [CrossRef]
- Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumor vessels. Nature. 2004 Feb 19;427(6976):695. [CrossRef]
- Liao S, Liu J, Lin P, Shi T, Jain RK, Xu L. TGF-beta blockade controls ascites by preventing abnormalization of lymphatic vessels in orthotopic human ovarian carcinoma models. Clin Cancer Res. 2011 Mar 15;17(6):1415-24. [CrossRef]
- Avraham T, Daluvoy S, Zampell J, Yan A, Haviv YS, Rockson SG, Mehrara BJ. Blockade of transforming growth factor-beta1 accelerates lymphatic regeneration during wound repair. Am J Pathol. 2010 Dec;177(6):3202-14. [CrossRef]
- Zhang, B.; Huang, Y.; Huang, Y. Advances in Nanodynamic Therapy for Cancer Treatment. Nanomaterials 2024, 14, 648. [CrossRef]
- Newport EL, Pedrosa AR, Njegic A, Hodivala-Dilke KM, Muñoz-Félix JM. Improved Immunotherapy Efficacy by Vascular Modulation. Cancers (Basel). 2021 Oct 17;13(20):5207. [CrossRef]
- Potiron V, Clément-Colmou K, Jouglar E, Pietri M, Chiavassa S, Delpon G, Paris F, Supiot S. Tumor vasculature remodeling by radiation therapy increases doxorubicin distribution and efficacy. Cancer Lett. 2019 Aug 10;457:1-9. [CrossRef]
- Hack SP, Zhu AX and Wang Y (2020) Augmenting Anticancer Immunity Through Combined Targeting of Angiogenic and PD-1/PD-L1 Pathways: Challenges and Opportunities. Front. Immunol. 11:598877. [CrossRef]
- Sezen D, Barsoumian HB, He K, Y, Wang Q, Abana CO, Puebla-Osorio N, Hsu EY, high tumour burden and generate immune memory. Front Immunol. 2022 Oct 6;13:984318. [CrossRef]
- Moore C, Hsu CC, Chen WM, Chen BPC, Han C, Story M, Aguilera T, Pop LM, Hannan R, Fu YX, Saha D, Timmerman R. Personalized Ultrafractionated Stereotactic Adaptive Radiotherapy (PULSAR) in Preclinical Models Enhances Single-Agent Immune Checkpoint Blockade. Int J Radiat Oncol Biol Phys. 2021 Aug 1;110(5):1306-1316. [CrossRef]
- He K, Barsoumian HB, Sezen D, Puebla-Osorio N, Hsu EY, Verma V, Abana CO, Chen D, Patel RR, Gu M, Cortez MA, Welsh JW. Pulsed Radiation Therapy to Improve Systemic Control of Metastatic Cancer. Front Oncol. 2021 Aug 23;11:737425. [CrossRef]
- Baxevanis, C.N.; Tsitsilonis, O.E.; Goulielmaki, M.; Tsakirakis, N.; Gritzapis, A.D. The Role of Therapeutic Vaccines in Cancer Immunotherapy. Onco 2025, 5, 11. [CrossRef]
- DePalo, D.K.; Zager, J.S. Advances in Intralesional Therapy for Locoregionally Advanced and Metastatic Melanoma: Five Years of Progress. Cancers 2023, 15, 1404. [CrossRef]
- Chen Y, Zhong NN, Cao LM, Liu B, Bu LL. Surgical margins in head and neck squamous cell carcinoma: A narrative review. Int J Surg. 2024 Jun 1;110(6):3680-3700. [CrossRef]
- Baniel CC, Heinze CM, Hoefges A, Sumiec EG, Hank JA, Carlson PM, Jin WJ, Patel RB, Sriramaneni RN, Gillies SD, Erbe AK, Schwarz CN, Pieper AA, Rakhmilevich AL, Sondel PM, Morris ZS. In situ, Vaccine Plus Checkpoint Blockade Induces Memory Humoral Response. Front Immunol. 2020 Jul 24;11:1610. [CrossRef]
- Caudle AS, Gonzalez-Angulo AM, Hunt KK, Pusztai L, Kuerer HM, Mittendorf EA, Hortobagyi GN, Meric-Bernstam F. Impact of progression during neoadjuvant chemotherapy on surgical management of breast cancer. Ann Surg Oncol. 2011 Apr;18(4):932-8. [CrossRef]
- Rao, Y.J.; Goodman, J.F.; Haroun, F.; Bauman, J.E. Integrating Immunotherapy into Multimodal Treatment of Head and Neck Cancer. Cancers 2023, 15, 672. [CrossRef]
- Ellen Heitzer, Lisa Auinger, Michael R. Speicher, Cell-Free DNA and Apoptosis: How Dead Cells Inform About the Living, Trends in Molecular Medicine, Volume 26, Issue 5, 2020, Pages 519-528. [CrossRef]
- Ruksha TG. MicroRNAs’ control of cancer cell dormancy. Cell Div. 2019 Oct 10;14:11. [CrossRef]
- Bach DH, Lee SK, Sood AK. Circular RNAs in Cancer. Mol Ther Nucleic Acids. 2019 Jun 7;16:118-129. [CrossRef]
- Ma Y, Gan J, Bai Y, Cao D, Jiao Y. Minimal residual disease in solid tumors: an overview. Front Med. 2023 Aug;17(4):649-674. [CrossRef]
- Abyaneh HS, Regenold M, McKee TD, Allen C, Gauthier MA. Towards extracellular matrix nor-malization for improved treatment of solid tumors. Theranostics. 2020 Jan 12;10(4):1960-1980. [CrossRef]
- Wyld, L., Bellantuono, I., Tchkonia, T., Morgan, J., Turner, O., Foss, F., George, J., Danson, S., & Kirkland, J. L. (2020). Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers, 12(8), 2134. [CrossRef]
- Kirkland, J.L. Tumor dormancy and disease recurrence. Cancer Metastasis Rev 42, 9–12 (2023). [CrossRef]
- Berrino F. Lifestyle prevention of cancer recurrence: the yin and the yang. Cancer Treat Res. 2014;159:341-51. [CrossRef]
- Ziello JE, Jovin IS, Huang Y. Hypoxia-Inducible Factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med. 2007 Jun;80(2):51- 60. PMID: 18160990; PMCID: PMC2140184.
- Emens LA. Chemoimmunotherapy. Cancer J. 2010 Jul-Aug;16(4):295-303. [CrossRef]
- Truong, N.T.H.; Gargett, T.; Brown, M.P.; Ebert, L.M. Effects of Chemotherapy Agents on Circulating Leukocyte Populations: Potential Implications for the Success of CAR-T Cell Therapies. Cancers 2021, 13, 2225. [CrossRef]
- Schmiechen ZC, Burrack AL, Stromnes IM. Chemotherapy brings virtual memory T cells into reality for cancer therapy. Cell Mol Immunol. 2021 May;18(5):1339-1340. [CrossRef]
- Huang, J., Michaud, E., Shinde-Jadhav, S. et al. (2024). Effects of combined radiotherapy with immune checkpoint blockade on immunological memory in luminal-like subtype murine bladder cancer model. Cancer Biology & Therapy, 25(1). [CrossRef]
- Breen WG, Leventakos K, Dong H, Merrell KW. Radiation and immunotherapy: emerging mechanisms of synergy. J Thorac Dis. 2020 Nov;12(11):7011-7023. [CrossRef]
- N.E. Donlon, R. Power, C. Hayes, J.V. Reynolds, J. Lysaght. Radiotherapy, immunotherapy, and the tumour microenvironment: Turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Letters, Volume 502, 2021, Pages 84-96. [CrossRef]
- Zhai D, An D, Wan C, Yang K. Radiotherapy: Brightness and darkness in the era of immunotherapy. Transl Oncol. 2022 May;19:101366. [CrossRef]
- Wason J, Stallard N, Bowden J, Jennison C. A multi-stage drop-the-losers design for multi-arm clinical trials. Stat Methods Med Res. 2017 Feb;26(1):508-524. [CrossRef]
- Serra A, Mozgunov P, Jaki T. A Bayesian multi-arm multi-stage clinical trial design incorporating information about treatment order. Stat Med. 2023 Jul 20;42(16):2841-2854. [CrossRef]
- Swamy, K.; Basavalingaiah S, A. Bridging the Foundational Versus Actionable Cycle in Cancer: Hypoxia Domino Effect and Cancer Cell Microenvironment (CCME) “Drug” Delivery. Preprints 2024, 2024111830. [CrossRef]
- Swamy, K. (2023). Therapeutic In Situ Cancer Vaccine Using Pulsed Stereotactic Body Radiotherapy—A Translational Model. Vaccines, 12(1), 7. [CrossRef]
- Bae, T., Hallis, S.P. & Kwak, MK. Hypoxia, oxidative stress, and the interplay of HIFs and NRF2 signaling in cancer. Exp Mol Med 56, 501–514 (2024). [CrossRef]
Figure 1.
A: Three critical oxygenation-linked transformational phases in cancer progression: (1) The first phase of normoxia, where the multiplication and elimination of cancer cells are balanced. (2) The immune escape phase, during which growth exceeds the oxygen supply when some cells are more than 100 µm away from the vasculature, becoming hypoxic. In this phase, the diffusion and penetration of tumor-infiltrating lymphocytes (TILs), oxygen, and anticancer drugs are impeded. (3) Persisting hypoxia results in excessive Reactive Oxygen Species, preventing the anticipated degradation of hypoxia-inducing factor (HIF)-1α, which stabilizes the latter. This stabilization of HIF-1α in the cytosol causes it to re-enter the cell’’s’ nucleus, forming a dimer with HIF-1β, triggering a cascade that ultimately results in immune exhaustion (Figure 1A & 1B). Created with BioRender.com. B: Hypoxia–HIF-1α domino effects—why is hypoxia a primary foundational factor in cancer initiation and progression? A. The genesis: Persistently increased Reactive Oxygen Species (ROS) (A1) stabilizes hypoxia-inducible factor 1 subunit alpha (HIF-1α) and accumulates in the setting of hypoxia (A2). HIF-1α, re-enters the nucleus (A3), forms a transcription complex with the stable hypoxia-inducible factor 1 beta subunit (HIF-1β) to create a heterodimer, which binds to DNA motifs (A4), influencing numerous homeostatic genes and leading to cascading effects in cancer progression. (1) Shift to anaerobic metabolism—Warburg effect: lactate production alters pH levels. (2) Vascular Endothelial Growth Factor (VEGF) transcription influences angiogenesis, resulting in abnormal neoangiogenesis, worsening hypoxia, and creating a vicious cycle. (3) Activation of genomic mutations that lead to cancer progression and the formation of clones and subclones with diverse phenotypically resistant populations of cancer cells. (4) Accumulation of various types of immunosuppressive cells, cancer-associated fibroblasts (CAFs), and cytokines in the tumor microenvironment (TME). B. With successful treatment, ROS and HIF-1α normalization ensues with the possibility of a cure. X, Y, Z (Bidirectional arrows) represent the interactive and interdependent vascular–metabolic–immune–phenotypic (VMIP) cancer model of the Combination, Timing, and Sequencing (CTS) strategy. Created with BioRender.com.
Figure 1.
A: Three critical oxygenation-linked transformational phases in cancer progression: (1) The first phase of normoxia, where the multiplication and elimination of cancer cells are balanced. (2) The immune escape phase, during which growth exceeds the oxygen supply when some cells are more than 100 µm away from the vasculature, becoming hypoxic. In this phase, the diffusion and penetration of tumor-infiltrating lymphocytes (TILs), oxygen, and anticancer drugs are impeded. (3) Persisting hypoxia results in excessive Reactive Oxygen Species, preventing the anticipated degradation of hypoxia-inducing factor (HIF)-1α, which stabilizes the latter. This stabilization of HIF-1α in the cytosol causes it to re-enter the cell’’s’ nucleus, forming a dimer with HIF-1β, triggering a cascade that ultimately results in immune exhaustion (Figure 1A & 1B). Created with BioRender.com. B: Hypoxia–HIF-1α domino effects—why is hypoxia a primary foundational factor in cancer initiation and progression? A. The genesis: Persistently increased Reactive Oxygen Species (ROS) (A1) stabilizes hypoxia-inducible factor 1 subunit alpha (HIF-1α) and accumulates in the setting of hypoxia (A2). HIF-1α, re-enters the nucleus (A3), forms a transcription complex with the stable hypoxia-inducible factor 1 beta subunit (HIF-1β) to create a heterodimer, which binds to DNA motifs (A4), influencing numerous homeostatic genes and leading to cascading effects in cancer progression. (1) Shift to anaerobic metabolism—Warburg effect: lactate production alters pH levels. (2) Vascular Endothelial Growth Factor (VEGF) transcription influences angiogenesis, resulting in abnormal neoangiogenesis, worsening hypoxia, and creating a vicious cycle. (3) Activation of genomic mutations that lead to cancer progression and the formation of clones and subclones with diverse phenotypically resistant populations of cancer cells. (4) Accumulation of various types of immunosuppressive cells, cancer-associated fibroblasts (CAFs), and cytokines in the tumor microenvironment (TME). B. With successful treatment, ROS and HIF-1α normalization ensues with the possibility of a cure. X, Y, Z (Bidirectional arrows) represent the interactive and interdependent vascular–metabolic–immune–phenotypic (VMIP) cancer model of the Combination, Timing, and Sequencing (CTS) strategy. Created with BioRender.com.
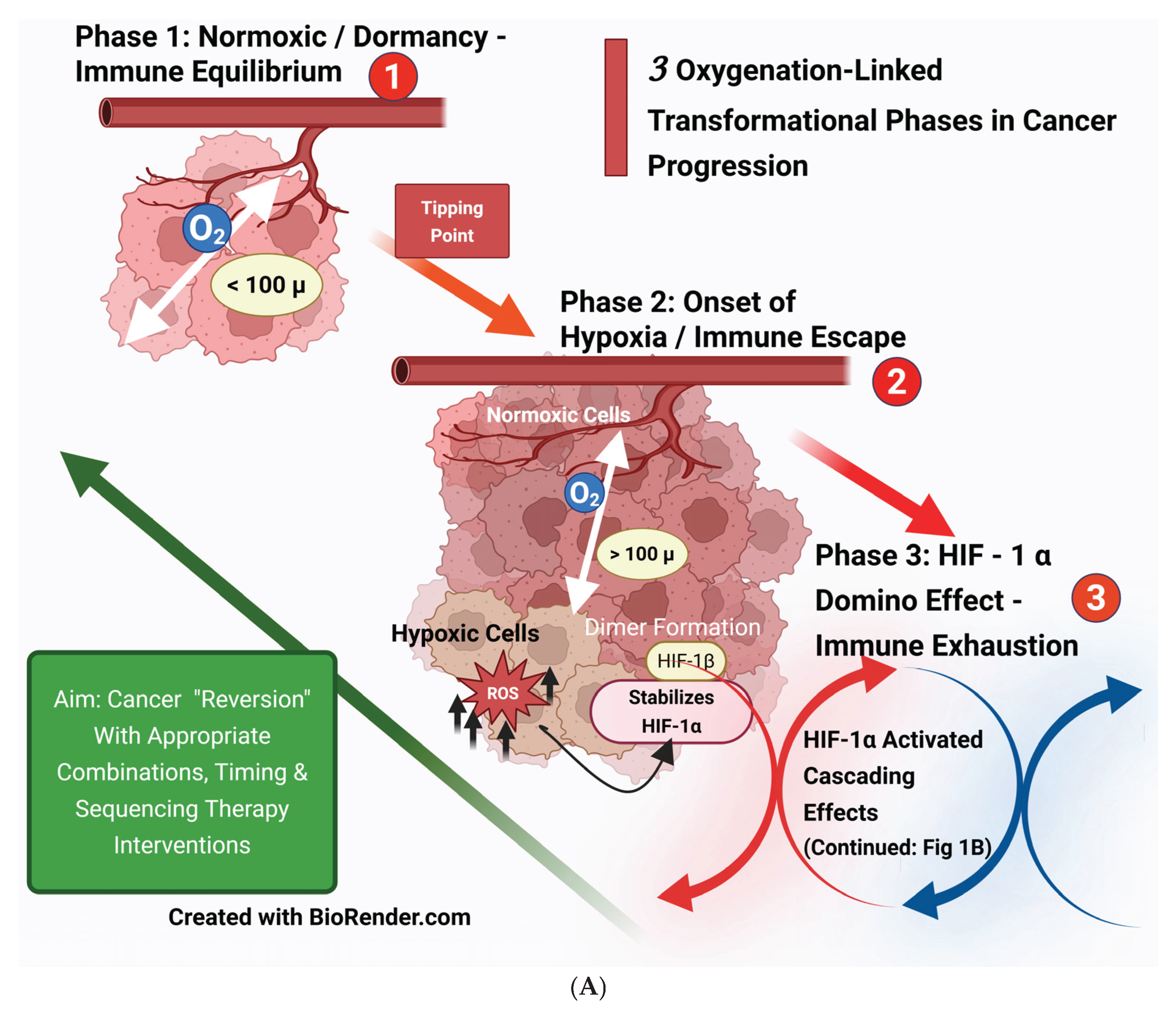
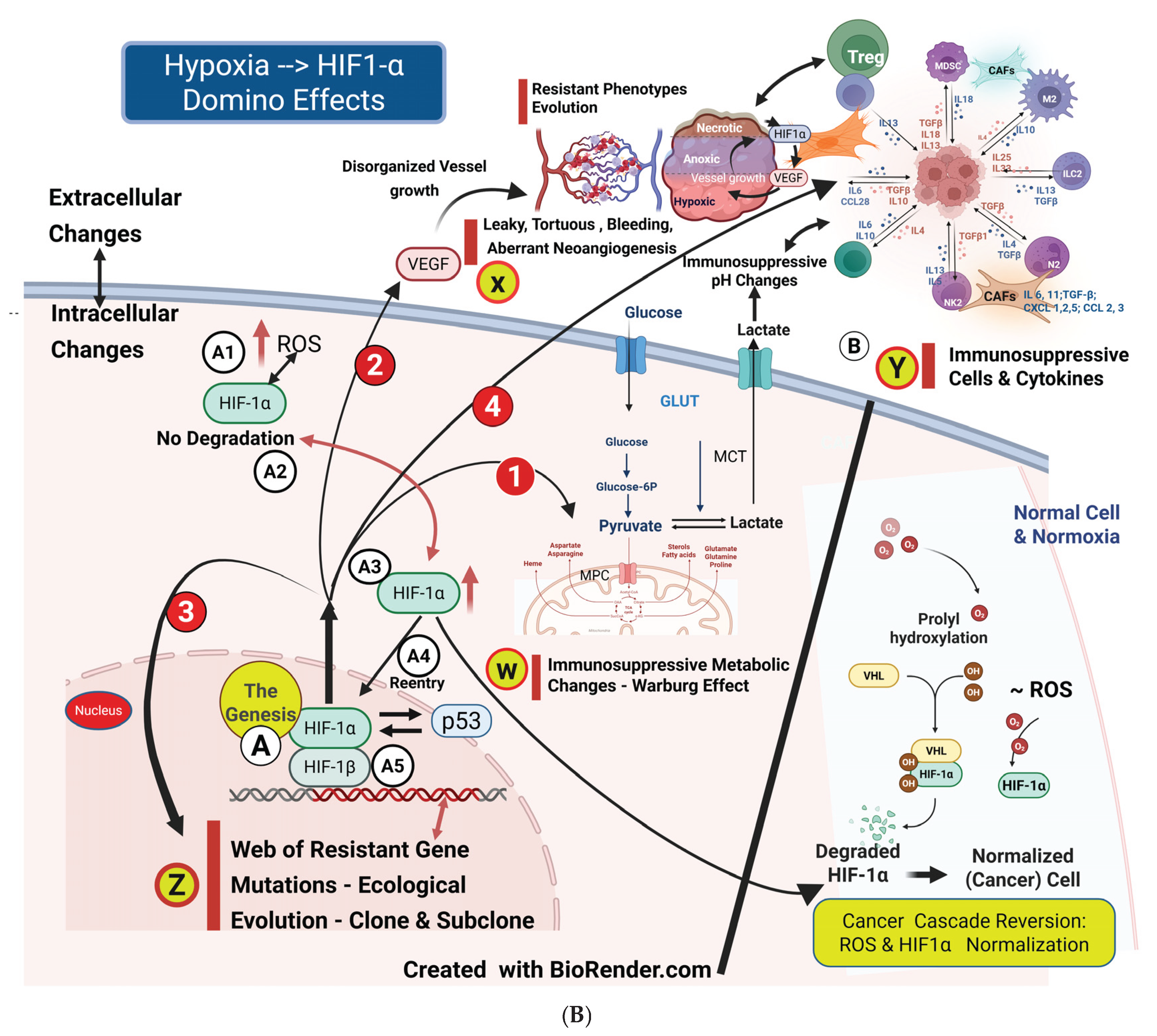
Figure 2.
Pathophysiological features of cancer vasculature: A) The typical inner diameter of a cancer cell is about 20–30 µm, depending on its site of origin. B) Oxygen and nutrient diffusion (as well as drugs) ceases beyond 100 µm (approximately 5 to 3 cancer cell layers). Cancer cells become overtly hypoxic and anoxic resistant/dormant cell populations, with some eventually undergoing necrosis. C) Leaky and abnormal angiogenesis, along with the extravasation of blood, results in uneven drug delivery and immune cell infiltration in CCME. Evolution of Angiogenesis, Tumor (Neo) Angiogenesis, Vascular Normalization, and Extended Vascular Normalization with a Cyclicity Strategy: (1) Normal angiogenesis results in well-formed, tube-like blood vessels featuring stalk and tip cells that are closely surrounded by pericytes and in contact with the vessel, responding to physiological signals for wound healing. (2) In contrast, tumor/neoangiogenesis is characterized by elongation and distortion, with weak, displaced pericytes and weak or absent tip cells, leading to dysfunctional, leaky, and aberrant angiogenesis. (3) Within hours of AAG (Day 0) commencing, the normalization process of tumor angiogenesis begins both physically and functionally. This leads to the restoration of endothelial junctions, enhanced pericyte coverage, improved oxygenation, reduced pH, increased drug delivery, immune cell infiltration/activation, and heightened tumor sensitivity to therapies. (4) These vascular normalization features persist from Day 0 to Day 28. (5) Around Day 28, the excessive vascular trimming (pruning) phase begins, leading to hypoxia re-emerging. (6) Stopping AAGs at this stage (Day 28) reverses the excessive pruning process, and the normalization phase of the vasculature begins again, lasting about four weeks. (7) At this stage, restarting antiangiogenics repeats the normalization process (like on Day 0), lasting until Day 28. [35] [Goel]. (8) By using the cyclic administration of antiangiogenics for about 4 weeks on and 4 weeks off (with vascular guidance methods), it is possible to achieve an extended normalization period. Created with BioRender.com.
Figure 2.
Pathophysiological features of cancer vasculature: A) The typical inner diameter of a cancer cell is about 20–30 µm, depending on its site of origin. B) Oxygen and nutrient diffusion (as well as drugs) ceases beyond 100 µm (approximately 5 to 3 cancer cell layers). Cancer cells become overtly hypoxic and anoxic resistant/dormant cell populations, with some eventually undergoing necrosis. C) Leaky and abnormal angiogenesis, along with the extravasation of blood, results in uneven drug delivery and immune cell infiltration in CCME. Evolution of Angiogenesis, Tumor (Neo) Angiogenesis, Vascular Normalization, and Extended Vascular Normalization with a Cyclicity Strategy: (1) Normal angiogenesis results in well-formed, tube-like blood vessels featuring stalk and tip cells that are closely surrounded by pericytes and in contact with the vessel, responding to physiological signals for wound healing. (2) In contrast, tumor/neoangiogenesis is characterized by elongation and distortion, with weak, displaced pericytes and weak or absent tip cells, leading to dysfunctional, leaky, and aberrant angiogenesis. (3) Within hours of AAG (Day 0) commencing, the normalization process of tumor angiogenesis begins both physically and functionally. This leads to the restoration of endothelial junctions, enhanced pericyte coverage, improved oxygenation, reduced pH, increased drug delivery, immune cell infiltration/activation, and heightened tumor sensitivity to therapies. (4) These vascular normalization features persist from Day 0 to Day 28. (5) Around Day 28, the excessive vascular trimming (pruning) phase begins, leading to hypoxia re-emerging. (6) Stopping AAGs at this stage (Day 28) reverses the excessive pruning process, and the normalization phase of the vasculature begins again, lasting about four weeks. (7) At this stage, restarting antiangiogenics repeats the normalization process (like on Day 0), lasting until Day 28. [35] [Goel]. (8) By using the cyclic administration of antiangiogenics for about 4 weeks on and 4 weeks off (with vascular guidance methods), it is possible to achieve an extended normalization period. Created with BioRender.com.
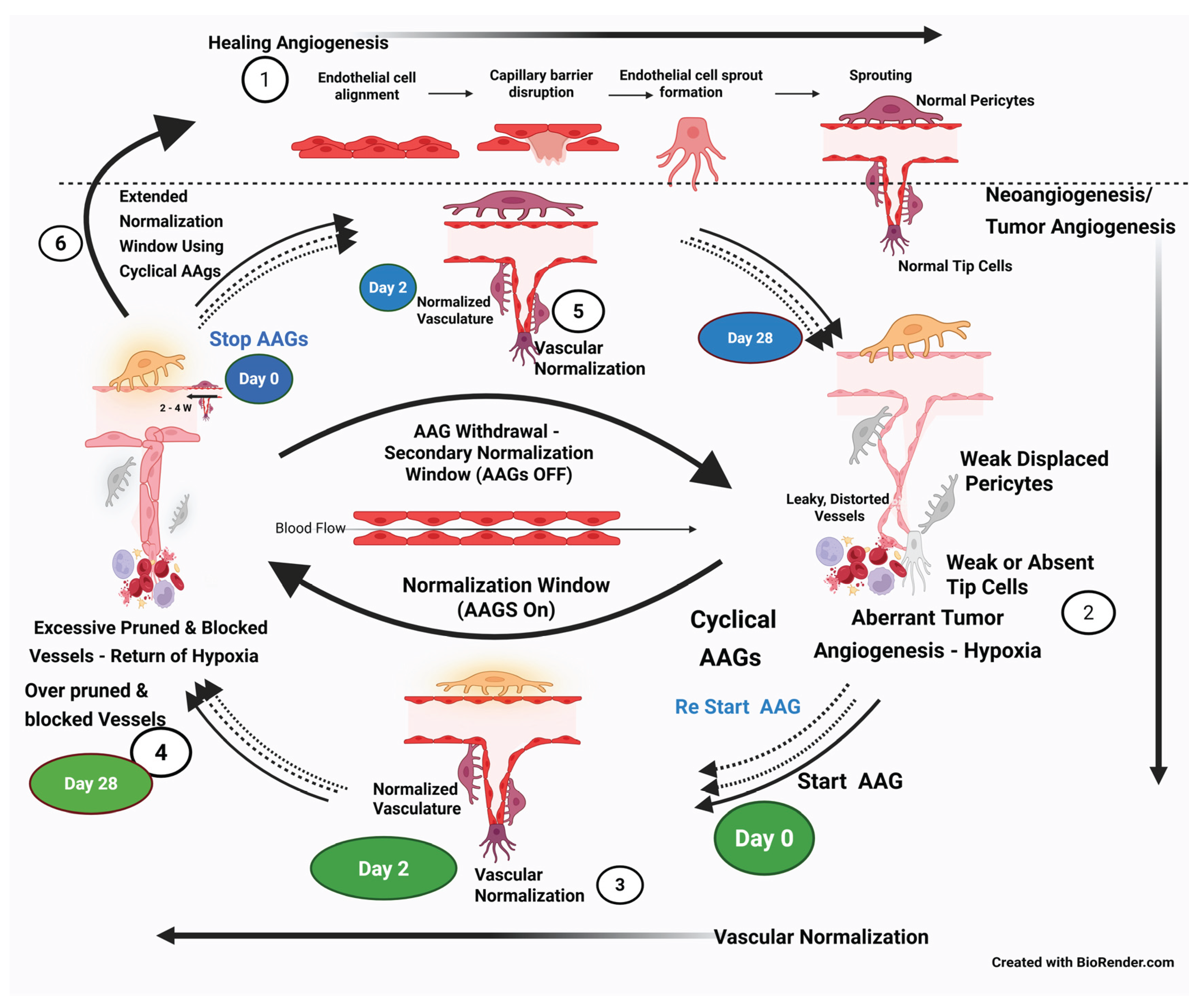
Figure 3.
Genomic–phenotypic evolution. A & B). Hypoxia is the “spider” that weaves a web of mutational cell types. Clonogenic cancer stem cells can proliferate and evolve into different sub-clones, contributing to cancer heterogeneity with varied resistant phenotypes that confer primary resistance to therapies. Consequently, the cancer tissue mass displays diverse spatial heterogeneity, necessitating combinations of curative methods with distinct mechanisms of action. C). The two major types of cells are those with targetable mutations/receptors and those without. D). Meanwhile, metastatic driver mutations arise, leading to distant metastases whose responses may differ from the primary lesions. E). Temporal mutations emerge, with or without therapeutic interventions, increasing complexity and leading to resistant phenotypes (adaptive/acquired resistance). F). (Circle) Mutations show increasing intricacy in response to failed first-line therapies. Created with BioRender.com.
Figure 3.
Genomic–phenotypic evolution. A & B). Hypoxia is the “spider” that weaves a web of mutational cell types. Clonogenic cancer stem cells can proliferate and evolve into different sub-clones, contributing to cancer heterogeneity with varied resistant phenotypes that confer primary resistance to therapies. Consequently, the cancer tissue mass displays diverse spatial heterogeneity, necessitating combinations of curative methods with distinct mechanisms of action. C). The two major types of cells are those with targetable mutations/receptors and those without. D). Meanwhile, metastatic driver mutations arise, leading to distant metastases whose responses may differ from the primary lesions. E). Temporal mutations emerge, with or without therapeutic interventions, increasing complexity and leading to resistant phenotypes (adaptive/acquired resistance). F). (Circle) Mutations show increasing intricacy in response to failed first-line therapies. Created with BioRender.com.
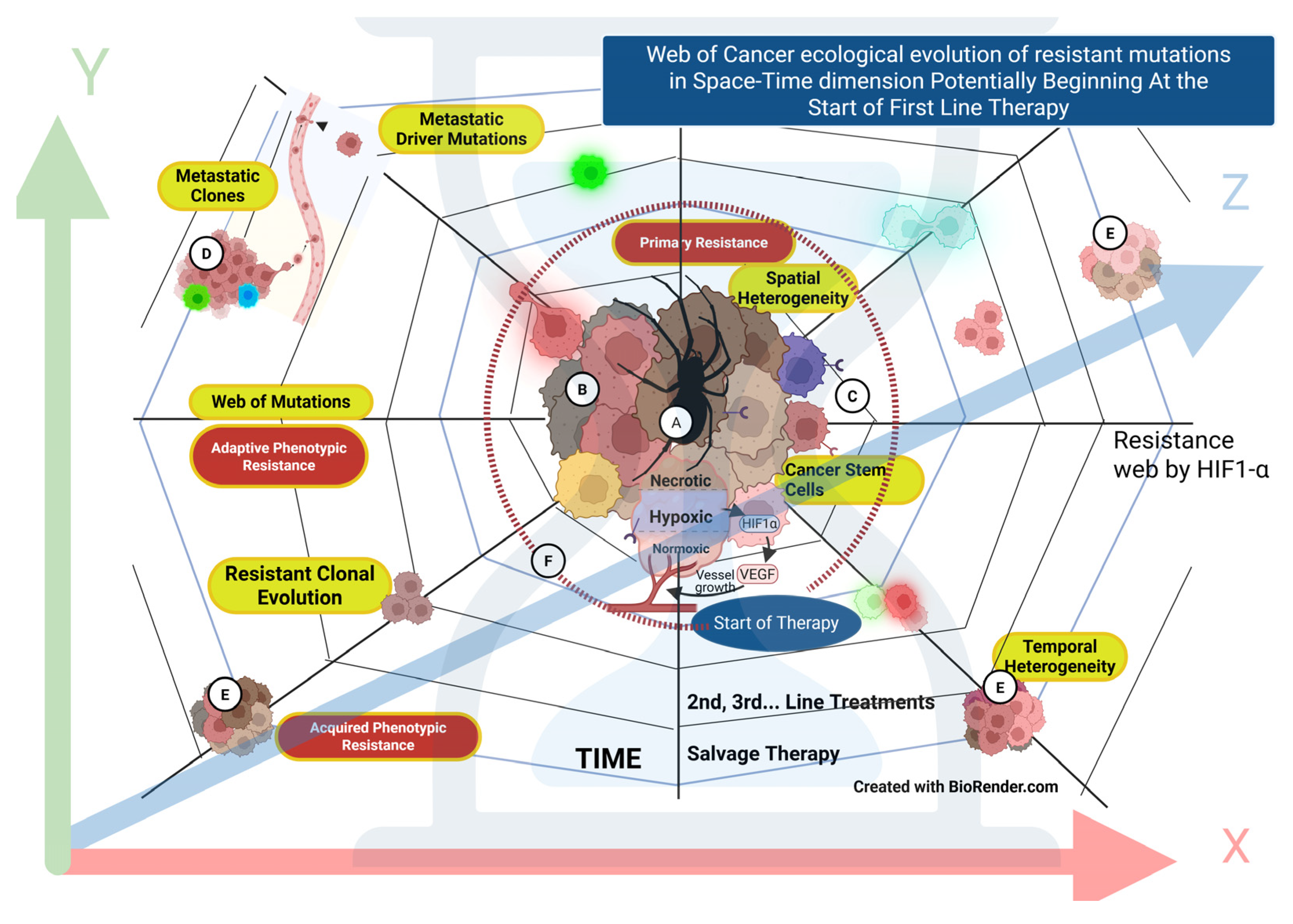
Figure 4.
A: TME physical barriers for “drug” delivery and reasons for treatment failure. While radiation can penetrate these barriers, its effectiveness in killing cancer cells may be limited by hypoxic resistance, as radiation requires oxygen to minimize DNA damage repair. 1): Drug diffusion barrier. A major reason for CAR-T cell therapy’s’ failure in solid tumors is its difficulty in reaching the CCME. Even nanoparticles (and tagged drugs) with exceptional diffusion capabilities cannot fully access the CCME due to the combined effects of abnormal angiogenesis and physical barriers. 2): Other consequences of these barriers include hypoxia, reduced nutrient supply, increased metabolic stress, and drug efflux, leading to depleted concentrations of the drugs [55] [Brady SW]. 3 & 4): The trafficking of anticancer immune cells into the CCME is hindered by immune cell trapping and immune exhaustion. 5): The walls of the necrotic areas within the cancer mass serve as a sanctuary for dormant or the most resistant clonogenic cancer cells. [53] [James Paterson]. Created with BioRender.com. B: The resistant clonal evolution of cancer cells results from insufficient drug concentrations in the CCME, which arise from several foundational changes: 1) Inadequate delivery to the CCME caused by aberrant angiogenesis. 2) A complex extracellular matrix and immunosuppressive cells transformed or differentiated from T-infiltrating lymphocytes (TILs). 3) Drugs, vaccines, and cell therapies, including oxygen, are either diverted by physical barriers or rendered ineffective. 4) Clonal evolution of resistant cancer cells. 5) A metabolic barrier: acidic pH in the CCME within the TME reduces the effectiveness of drugs and immune cells. Created with BioRender.com.
Figure 4.
A: TME physical barriers for “drug” delivery and reasons for treatment failure. While radiation can penetrate these barriers, its effectiveness in killing cancer cells may be limited by hypoxic resistance, as radiation requires oxygen to minimize DNA damage repair. 1): Drug diffusion barrier. A major reason for CAR-T cell therapy’s’ failure in solid tumors is its difficulty in reaching the CCME. Even nanoparticles (and tagged drugs) with exceptional diffusion capabilities cannot fully access the CCME due to the combined effects of abnormal angiogenesis and physical barriers. 2): Other consequences of these barriers include hypoxia, reduced nutrient supply, increased metabolic stress, and drug efflux, leading to depleted concentrations of the drugs [55] [Brady SW]. 3 & 4): The trafficking of anticancer immune cells into the CCME is hindered by immune cell trapping and immune exhaustion. 5): The walls of the necrotic areas within the cancer mass serve as a sanctuary for dormant or the most resistant clonogenic cancer cells. [53] [James Paterson]. Created with BioRender.com. B: The resistant clonal evolution of cancer cells results from insufficient drug concentrations in the CCME, which arise from several foundational changes: 1) Inadequate delivery to the CCME caused by aberrant angiogenesis. 2) A complex extracellular matrix and immunosuppressive cells transformed or differentiated from T-infiltrating lymphocytes (TILs). 3) Drugs, vaccines, and cell therapies, including oxygen, are either diverted by physical barriers or rendered ineffective. 4) Clonal evolution of resistant cancer cells. 5) A metabolic barrier: acidic pH in the CCME within the TME reduces the effectiveness of drugs and immune cells. Created with BioRender.com.
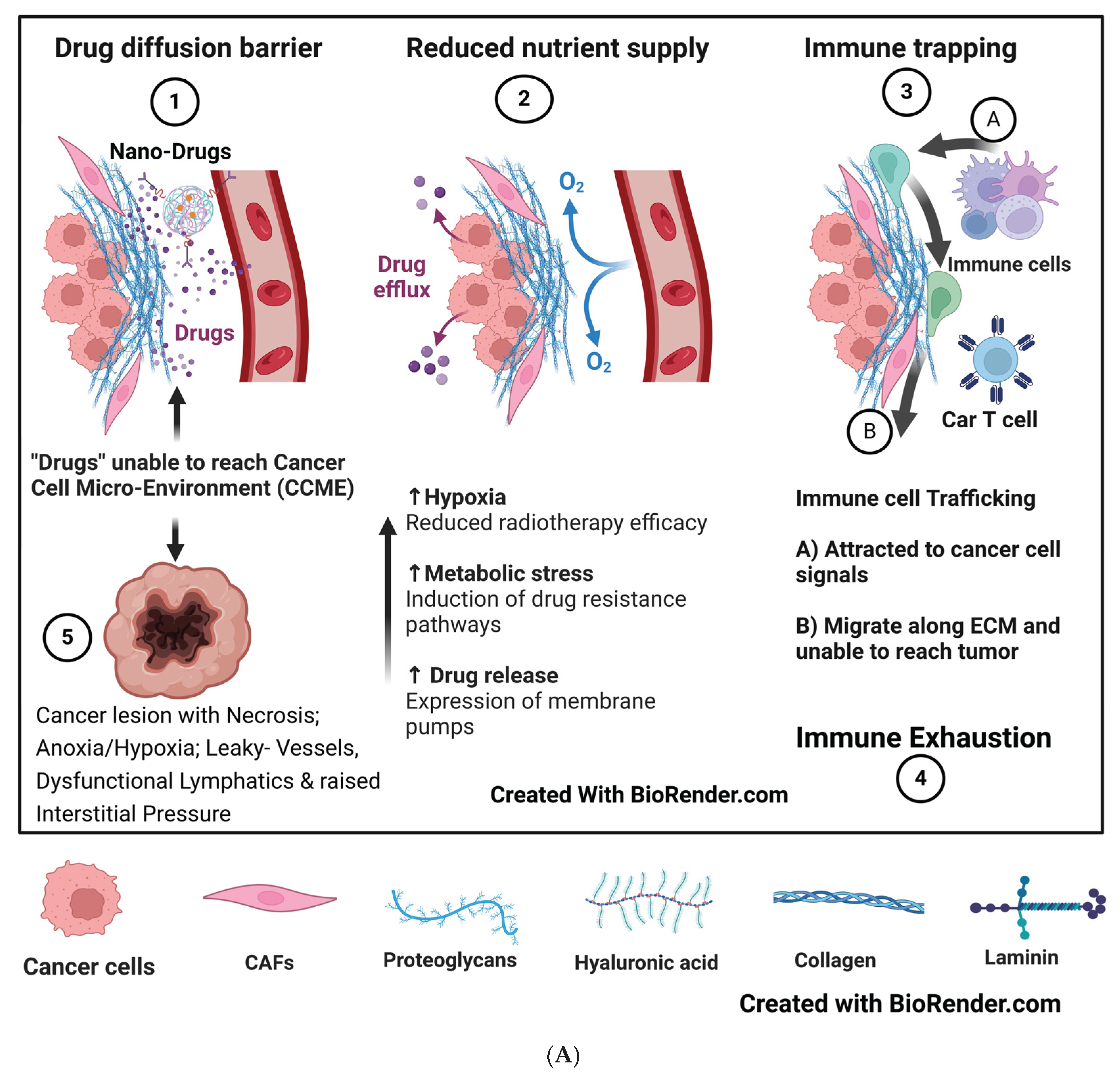
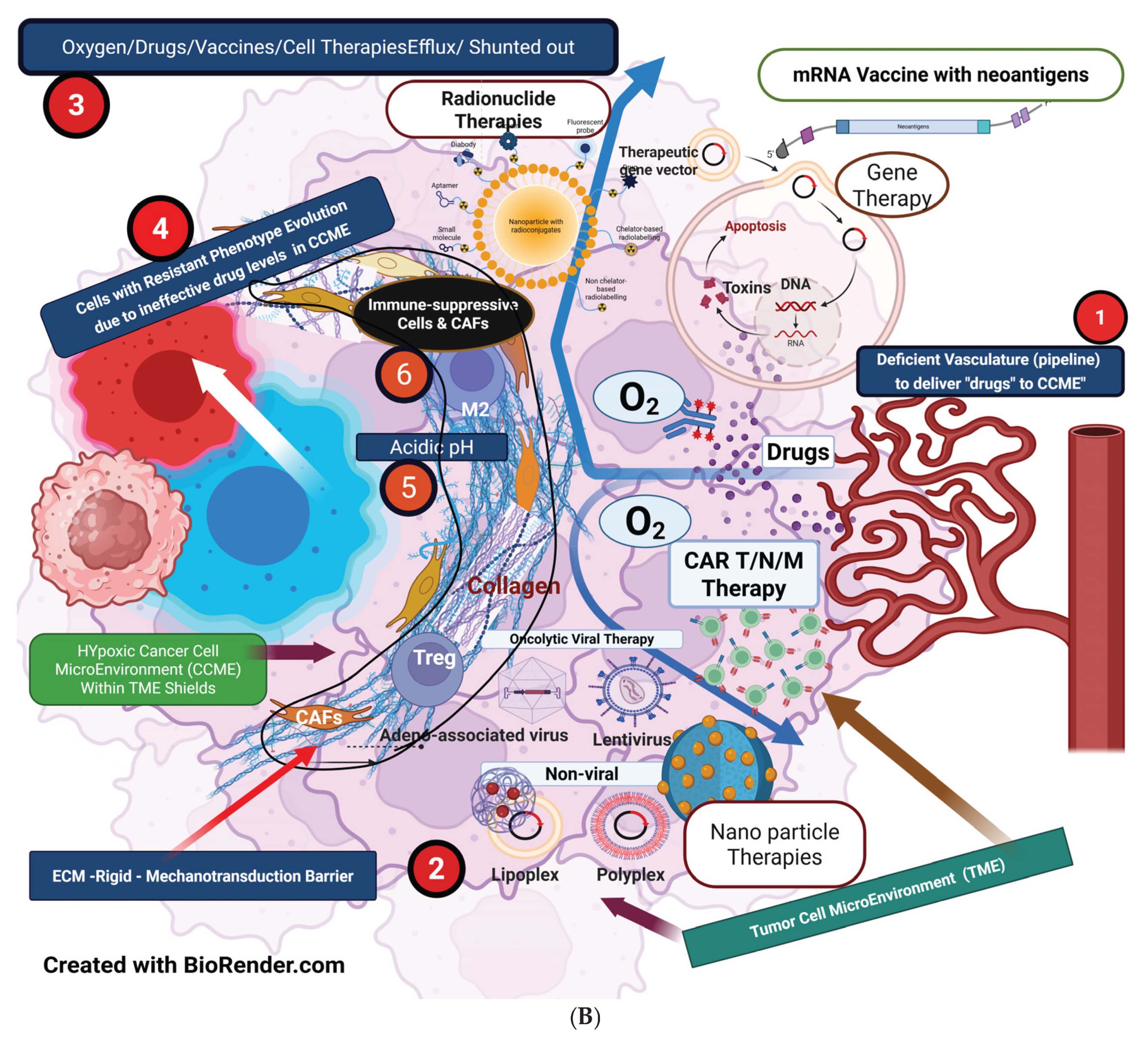
Figure 5.
Network of therapy options: The evolution of cancer treatments has depended on therapies that operate independently and, in summary, on various combinations, timing, and sequencing [77] [Brunner TB]. Based on the potentially effective limitless combinations charted here, it is necessary to classify and design their applicability in clinical practice appropriately.
Figure 5.
Network of therapy options: The evolution of cancer treatments has depended on therapies that operate independently and, in summary, on various combinations, timing, and sequencing [77] [Brunner TB]. Based on the potentially effective limitless combinations charted here, it is necessary to classify and design their applicability in clinical practice appropriately.
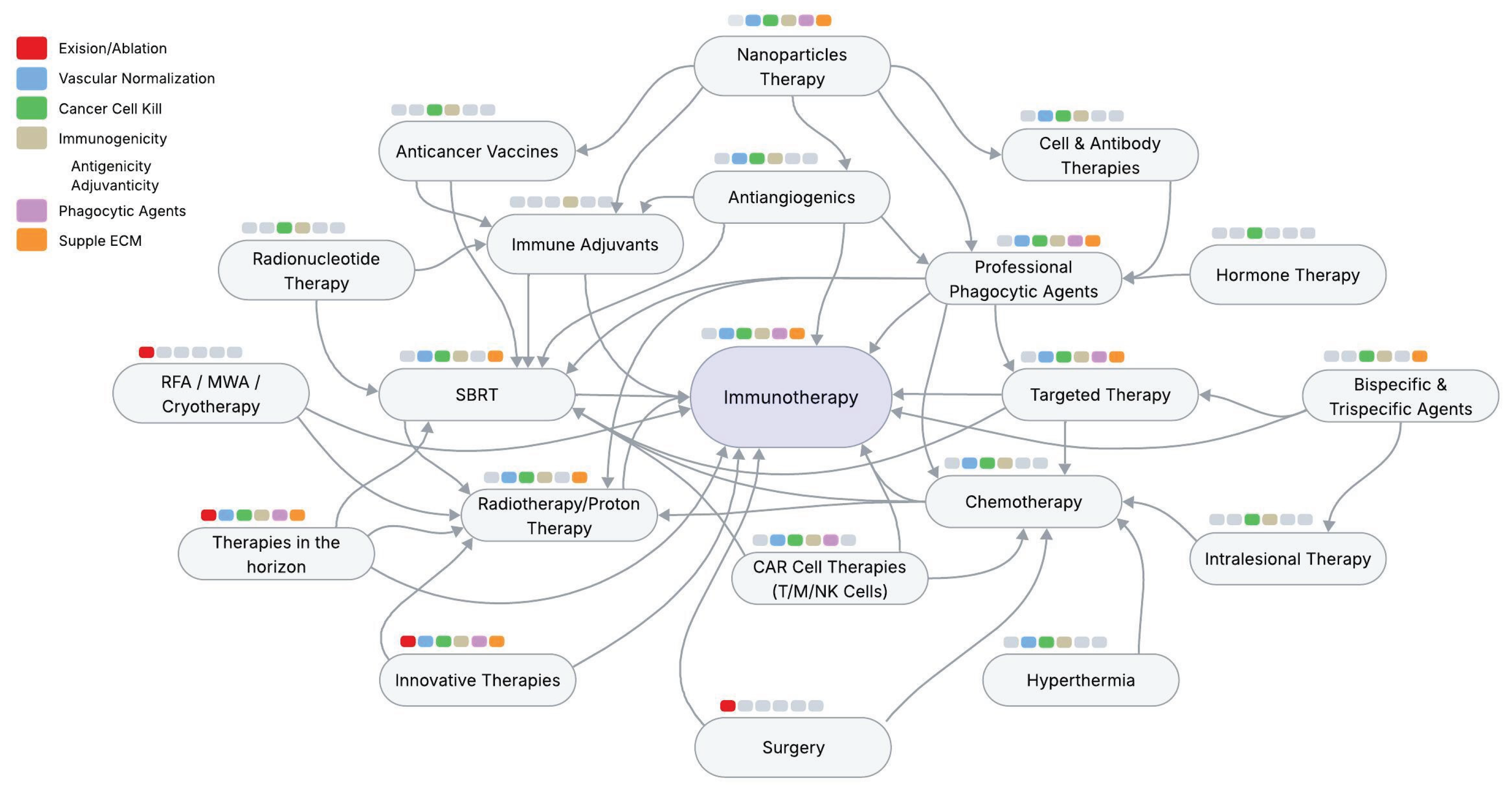
Figure 6.
Unmasking cancer cells and preparing the CCME for immunotherapy and beyond. Phase 1: vascular normalization; Phase 2: Cell Kill/phagocytosis/ vascular promotion; Phase 3: Immunotherapy/ immune promotion; Phase 4: Consolidation; Phase 5: Cancer “Reversion”. CT/RT = chemotherapy/Radiotherapy; SBRT = stereotactic radiotherapy. Created with BioRender.com.
Figure 6.
Unmasking cancer cells and preparing the CCME for immunotherapy and beyond. Phase 1: vascular normalization; Phase 2: Cell Kill/phagocytosis/ vascular promotion; Phase 3: Immunotherapy/ immune promotion; Phase 4: Consolidation; Phase 5: Cancer “Reversion”. CT/RT = chemotherapy/Radiotherapy; SBRT = stereotactic radiotherapy. Created with BioRender.com.
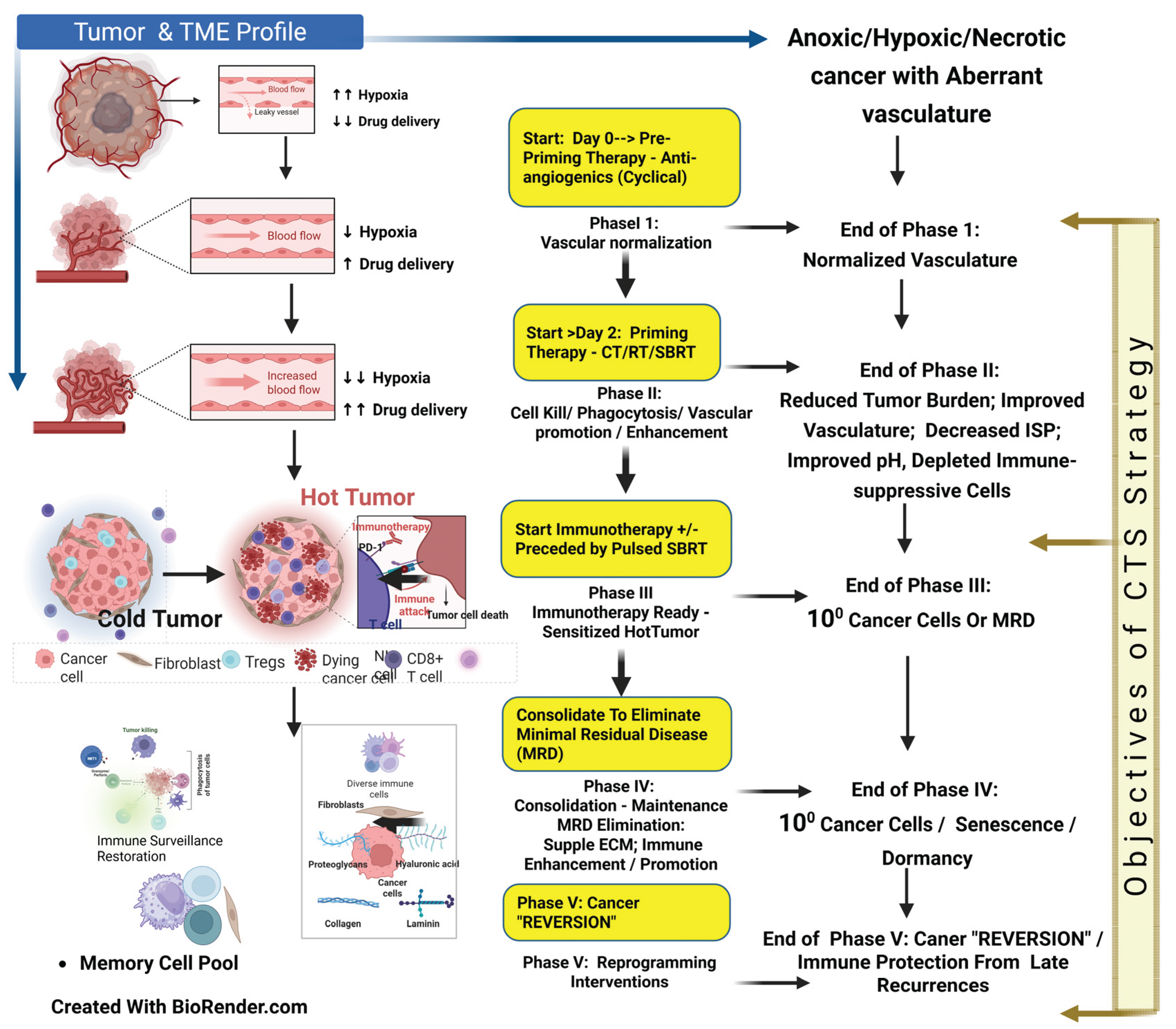
Figure 7.
A: CTS strategy in Unmasking of cancers, making it ready for optimized immunotherapy response. TME = Tumor microenvironment; ECM = Extracellular matrix; ISP = Interstitial pressure; CTLs = Cytotoxic lymphocytes; TILS = Tumor infiltrating lymphocytes. ECM = Extracellular matrix; CCME = Cancer Cell Microenvironment; TME = Tumor MicroEnvironment; TILs = Tumor Infiltrating Lymphocytes; CTLs = Cytotoxic Lymphocytes; CAFs = Cancer Associated Fibroblasts. B: Unmasking cancer making Immunotherapy ready—Beyond Hot and Cold Tumor: The figure indicates the conditions to be fulfilled for optimum immunotherapy results. The cancer cell shrouded within the barriers and protection (left side) will be open for immune attack, as enumerated (left side). ECM = Extracellular matrix; CCME = Cancer Cell Microenvironment; TME = Tumor MicroEnvironment; RT = Radiotherapy; SBRT = Stereotactic Body Radiotherapy; CT = Chemotherapy; AAGS = Antianiongenics; TILs = Tumor Infiltrating Lymphocytes; CTLs = Cytotoxic Lymphocytes; CAFs = Cancer Associated Fibroblasts.
Figure 7.
A: CTS strategy in Unmasking of cancers, making it ready for optimized immunotherapy response. TME = Tumor microenvironment; ECM = Extracellular matrix; ISP = Interstitial pressure; CTLs = Cytotoxic lymphocytes; TILS = Tumor infiltrating lymphocytes. ECM = Extracellular matrix; CCME = Cancer Cell Microenvironment; TME = Tumor MicroEnvironment; TILs = Tumor Infiltrating Lymphocytes; CTLs = Cytotoxic Lymphocytes; CAFs = Cancer Associated Fibroblasts. B: Unmasking cancer making Immunotherapy ready—Beyond Hot and Cold Tumor: The figure indicates the conditions to be fulfilled for optimum immunotherapy results. The cancer cell shrouded within the barriers and protection (left side) will be open for immune attack, as enumerated (left side). ECM = Extracellular matrix; CCME = Cancer Cell Microenvironment; TME = Tumor MicroEnvironment; RT = Radiotherapy; SBRT = Stereotactic Body Radiotherapy; CT = Chemotherapy; AAGS = Antianiongenics; TILs = Tumor Infiltrating Lymphocytes; CTLs = Cytotoxic Lymphocytes; CAFs = Cancer Associated Fibroblasts.
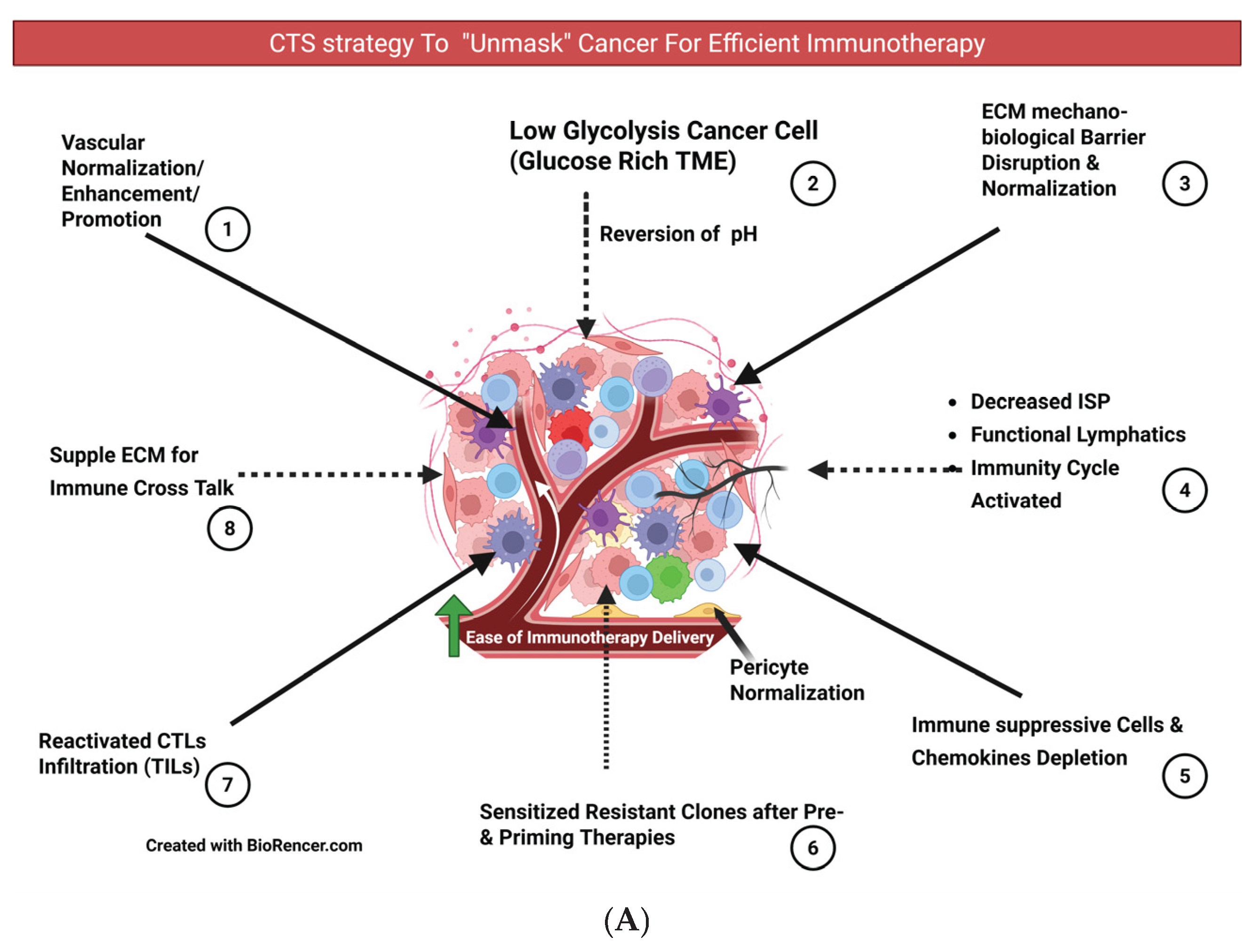
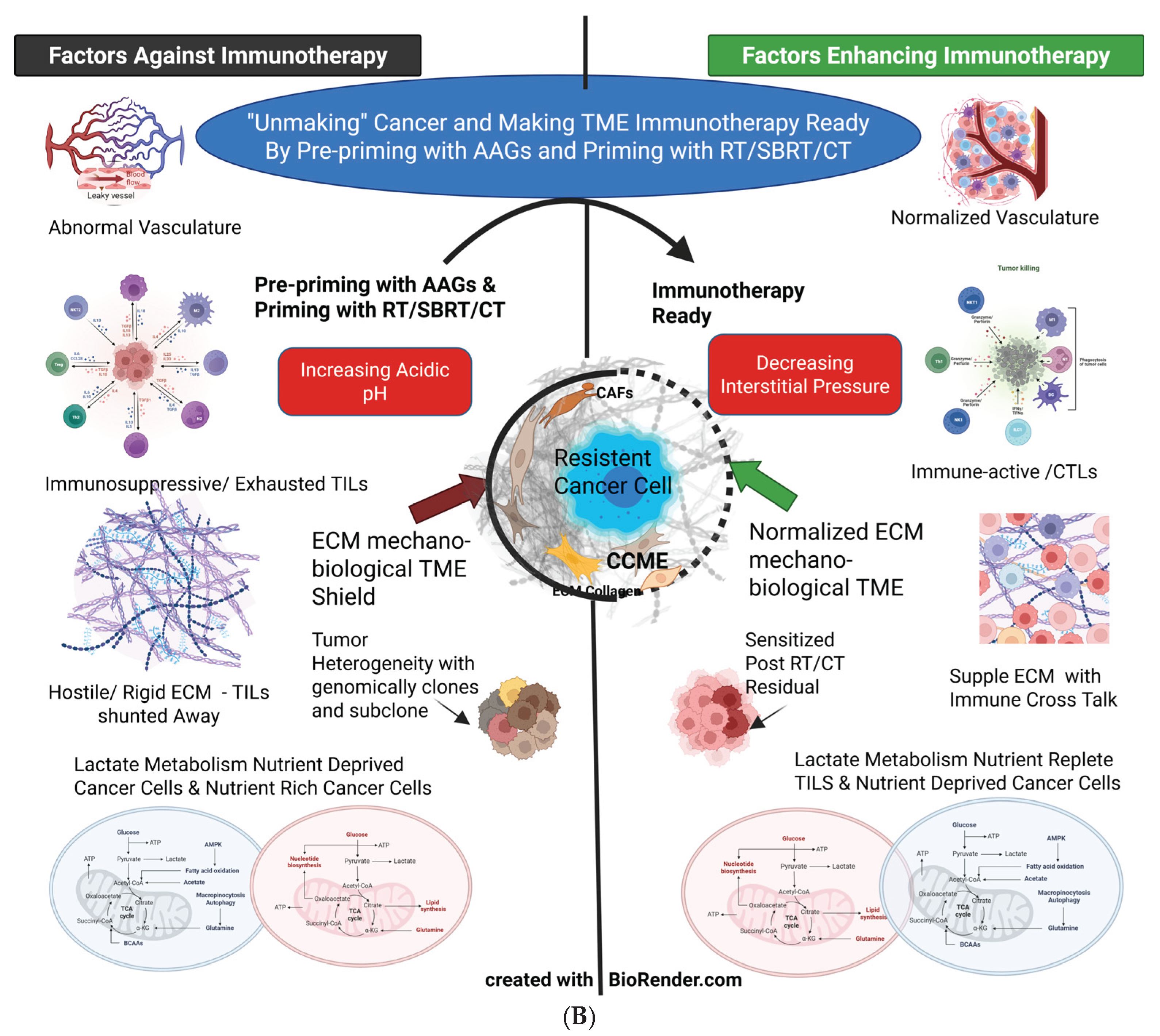
Figure 8.
In situ vaccination using pulsed immunogenic dose SBRT: Synchronized pulsed SBRT and immunotherapy optimize the immunity cycles. 1. An initial hypoxic or anoxic lesion during the initial pulse of SBRT generates the first wave of neoantigens. 2. A better-oxygenated subclonal tumor mass during the second pulse, following the response to the first dose, produces a second set of altered neoantigens. 3. Normoxic residual tumors contain potentially resistant clones that persist after the first and second pulses during the third pulse of SBRT, generating neoantigens from those resistant clones. These changes result in increasing radiation sensitivity (following normalization of the vasculature, improved oxygenation, dormant cells recruited into susceptible phases of the cell cycle, decreased interstitial pressure, etc.), expanding the tumor-specific T-cell receptor repertoire [115] [He K], and broadening of the spectrum of the memory cell pool. Created with BioRender.com.
Figure 8.
In situ vaccination using pulsed immunogenic dose SBRT: Synchronized pulsed SBRT and immunotherapy optimize the immunity cycles. 1. An initial hypoxic or anoxic lesion during the initial pulse of SBRT generates the first wave of neoantigens. 2. A better-oxygenated subclonal tumor mass during the second pulse, following the response to the first dose, produces a second set of altered neoantigens. 3. Normoxic residual tumors contain potentially resistant clones that persist after the first and second pulses during the third pulse of SBRT, generating neoantigens from those resistant clones. These changes result in increasing radiation sensitivity (following normalization of the vasculature, improved oxygenation, dormant cells recruited into susceptible phases of the cell cycle, decreased interstitial pressure, etc.), expanding the tumor-specific T-cell receptor repertoire [115] [He K], and broadening of the spectrum of the memory cell pool. Created with BioRender.com.
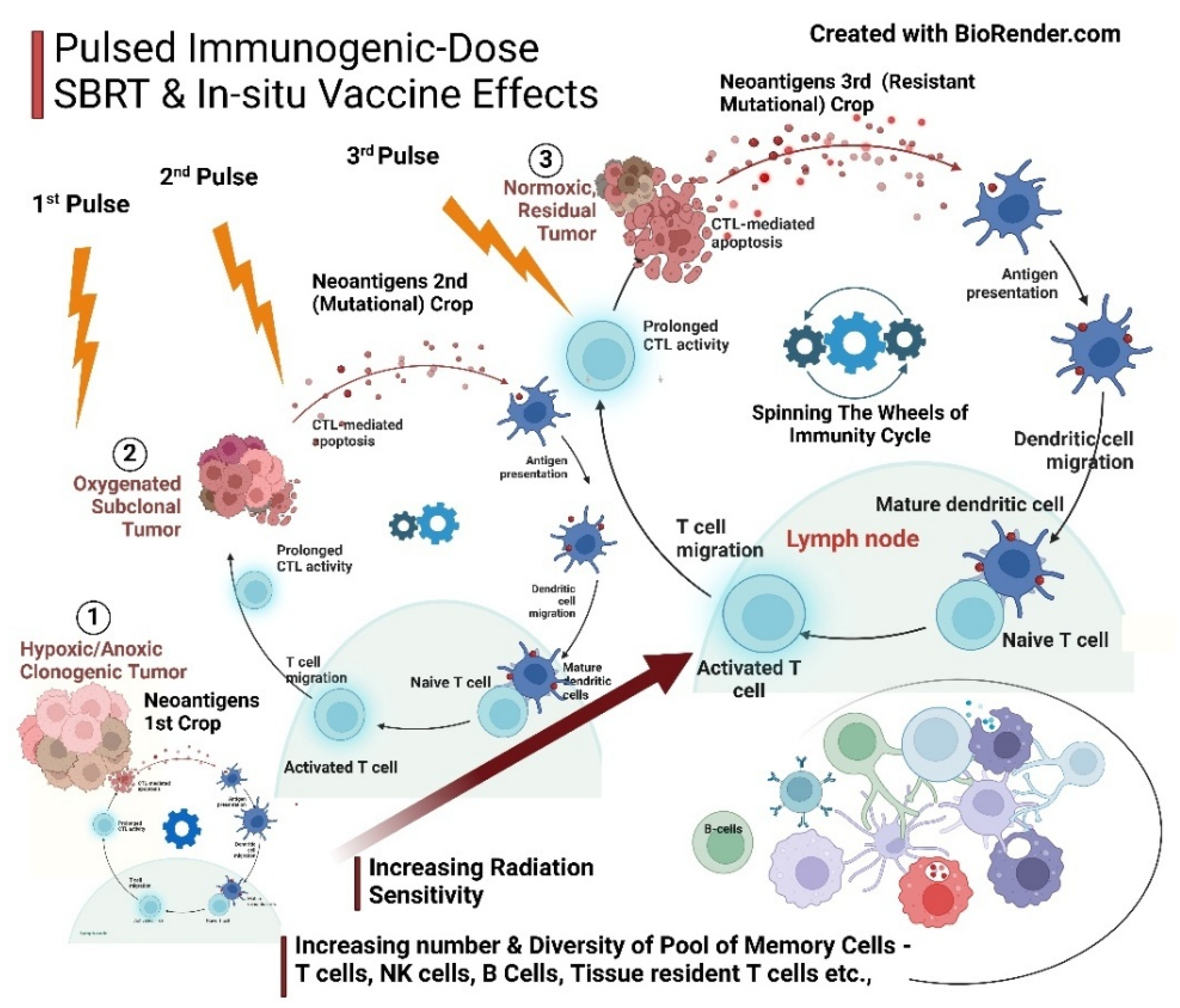
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



