Submitted:
16 November 2024
Posted:
18 November 2024
You are already at the latest version
Abstract
Hemoglobin is a vital protein in red blood cells primarily responsible for transporting oxygen from the lungs to tissues. It binds to various ligands, including oxygen, carbon dioxide, carbon monoxide, and nitric oxide, influencing its function and regulation. Genetic mutations of Hb can lead to variants causing disorders like sickle cell disease and thalassemias, which present significant clinical challenges. Recent advances in therapies, such as allosteric modulators, pharmacological chaperones, and antioxidant treatments, have improved the management of hemoglobinopathies by enhancing hemoglobin stability and increasing oxygen affinity. According to the UniProt database (as of 7th August 2024), there are currently 819 variants of the -hemoglobin subunit and 771 variants of the -hemoglobin subunit. Over 116 hemoglobin subunit variants are classified as unstable, highlighting the need to further develop variant-specific hemoglobin-stabilizing molecules. In addition to several small-molecule binders, a potential in the engineering of proteins with hemoglobin-binding capabilities, e.g. falcilysin, lama-derived nanobodies, -hemoglobin-stabilizing proteins and others, offer promising options for further development. Continued research is essential for developing personalized treatments that consider the genetic diversity of hemoglobin variants, offering hope for more targeted therapies, improved management, and potential cures for hemoglobin-related disorders.
Keywords:
Hemoglobin (Hb)
; Oxygen affinity
; Allosteric regulation
; Bohr effect
; 2
; 3-Bisphosphoglycerate (2
; 3-BPG)
; Genetic variants
; Thalassemia
; Sickle cell disease (SCD)
; Oxygen-binding properties
; Protein engineering
1. Introduction
Hemoglobin (Hb) is a crucial protein for oxygen transport in mammals, with a complex structure and function. The cooperative binding of oxygen to Hb involves an equilibrium between two quaternary structures with different oxygen affinities [1,2]. This process is influenced by various factors, including pH (the Bohr effect), organic phosphates, and other allosteric effectors [3,4]. The two-state model proposed by Monod, Wyman, and Changeux has been widely used to explain Hb cooperativity, although some discrepancies have been observed [2].
Hb exhibits complex allosteric regulation, crucial for its oxygen-binding function. The interplay between tense and relaxed states governs Hb's behavior [5,6]. Allosteric effectors, both endogenous and synthetic, modulate Hb's oxygen affinity by binding to specific sites, altering its structure and function [7,8]. This allosteric control involves both homotropic and heterotropic effects, where ligand binding at one site influences binding at other sites [9]. Protein engineering strategies have explored mutations to optimize Hb's functional properties, revealing additive effects of interfacial and distal-heme pocket mutations on structure and function [10]. The evolution of Hb has resulted in diverse variants with altered oxygen-binding properties, while maintaining fundamental structural features [11]. Un[1,2derstanding these mechanisms is crucial for developing Hb-based oxygen carriers and treating blood-related disorders [12]. Hemoglobin oxygen affinity is influenced by several physiological ligands (Figure 1): (1) pH aka Bohr effect - a decrease in pH reduces hemoglobin’s oxygen affinity, promoting oxygen release in tissues [3]; (2) CO2 levels - increased CO2 levels form carbaminohemoglobin, stabilizing the T state and reducing oxygen affinity [13]; (3) NO, which can bind to Hb's heme groups, forming nitrosylhemoglobin, or to cysteine residues, creating S-nitrosohemoglobin (SNO-Hb) [14,15] and (4) 2,3-Bisphosphoglycerate (2,3-BPG) - this metabolite binds to deoxygenated hemoglobin, stabilizing the T state and promoting oxygen release in tissues. 2,3-Bisphosphoglycerate (2,3-BPG) is a crucial metabolite that binds to hemoglobin, regulating oxygen affinity and delivery to tissues. It binds more strongly to deoxyhemoglobin than oxyhemoglobin, with one binding site per tetramer [16] [17,18]. 2,3-BPG levels increase during hypoxia, enhancing oxygen delivery [19]. Its concentration is regulated by complex interactions between glycolysis, the 2,3-BPG shunt, and the pentose phosphate pathway [20]. Feedback inhibition of hexokinase and phosphofructokinase by 2,3-BPG, along with product inhibition of 2,3-BPG synthase, control its steady-state concentration. H+ and oxygen effectively regulate 2,3-BPG levels, primarily through hexokinase and phosphofructokinase. The 2,3-BPG shunt also plays a role in stabilizing ATP levels.
2. Hemoglobin Function and Clinical Variants
The α and β globin genes are located on different chromosomes, with α globin genes on chromosome 16 and β globin genes on chromosome 11. The arrangement and expression of these genes are tightly regulated to ensure proper hemoglobin formation and function. Two main natural variants exist: fetal hemoglobin (HbF, α2γ2) and adult hemoglobin (HbA, α2β2) [22]. HbF has higher oxygen affinity than HbA, facilitating oxygen transfer from mother to fetus [23]. During development, HbF levels decrease from 70% at birth to <1% in adults, while HbA becomes predominant [24,25]. HbA2 (α2δ2) comprises 1.5-3.5% of adult hemoglobin [26]. Genetic factors, including mutations and polymorphisms, can influence HbF production in adulthood [27]. Structural hemoglobinopathies and thalassemias result from mutations affecting globin genes, leading to abnormal or decreased hemoglobin production [28]. These variants can interfere with HbA1c measurements, impacting diabetes monitoring [29].
2.1. Diversity of Genetic Variants of Hemoglobin
Ca. 10 years ago, over 1000 naturally occurring hemoglobin variants have been identified; these variants show various effect hemoglobin structure and biochemical properties with varying physiological effects [30]. Since then, several new variants have been found and their sequences can be retrieved from UniProt database. UniProt entries for human hemoglobin α and β subunits are
In the UniProt database, there are 819 variants of the hemoglobin subunit α and 771 variants of the hemoglobin subunit β (as of 7th August 2024, Figure 2). Assuming homozygotes, there are 631,749 potential combinations of hemoglobin. However, the genetic diversity increases exponentially when considering heterozygotes, resulting in over 1011 possible combinations. This number surpasses the current human population on Earth, illustrating the astonishing potential variations in known combinations of hemoglobin variants. The actual diversity is likely much lower because Hb variants are often present in specific geographically constrained sub-populations. For example, HbS is prevalent in Africa and India, thalassemias are common in the Mediterranean, Middle East, and Southeast Asia, HbC is common in West Africa, HbD-Punjab has been found in the Indian subcontinent, HbE is highly prevalent in Southeast Asia, Hb Lepore is prevalent in Southern Italy and Mediterranean populations, Hb Constant Spring (Hb CS) is common in Southeast Asia, Hb Bart's (γ4) has been found in individuals with α-thalassemia, prevalent in Saudi Arabia and Thailand.
The classification of genetic variants, particularly in hemoglobinopathies, is crucial for accurate diagnosis and clinical management. The American College of Medical Genetics and Genomics (ACMG) guidelines provide a five-category system: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign [31,32]. However, this system has limitations in addressing the spectrum of variant effects. An expanded seven-category system, including "predisposing" and "likely predisposing," has been proposed to better classify variants in disease-causing genes [31,33]. Variant reclassification, driven by new evidence, can significantly impact patient care, with reclassification rates ranging from 3.6% to 58.8% [34]. For hemoglobinopathies, molecular genetic testing is essential for accurate diagnosis and genetic counseling [35,36]. The clinical implications of variant classification are profound, affecting organ surveillance, prophylactic surgery, and cascade testing [37].
Hemoglobinopathies are a group of disorders affecting the hemoglobin molecule. These disorders typically result from genetic mutations that lead to abnormal structure or production of hemoglobin, causing various health problems ranging from mild anemia to severe complications. For example, HbM variants lead to methemoglobinemia, where iron in the heme group is oxidized to the ferric form, impairing oxygen delivery. Methemoglobinemia is a blood disorder characterized by elevated levels of methemoglobin, which impairs oxygen delivery to tissues [38]. It can be inherited or acquired, with the latter being more common and often caused by medications or chemicals [39]. Symptoms include cyanosis unresponsive to oxygen therapy and chocolate-colored blood [40]. Diagnosis is confirmed through co-oximetry, arterial blood gas analysis, and serum methemoglobin levels [41]. Treatment involves removing the offending agent, administering oxygen, and using methylene blue as an antidote in severe cases [42]. Dapsone is a common cause of acquired methemoglobinemia, while benzocaine spray has been associated with severe cases [43]. In pediatric patients, underlying hematologic diseases and G6PD deficiency can predispose to methemoglobinemia [44]. Increased awareness and primary prevention efforts are crucial to reduce morbidity and mortality associated with this condition [43].
Sickle cell disease (SCD) is a genetic disorder caused by a single nucleotide mutation in the β-globin gene, resulting in the production of abnormal hemoglobin S (HbS) [45,46]. This E6V mutation leads to HbS polymerization under low oxygen conditions, causing red blood cell (RBC) sickling and hemolysis [45,47]. SCD is characterized by chronic inflammation, oxidative stress, and vascular dysfunction, contributing to various complications such as stroke, pulmonary hypertension, and renal disorders [46,48]. The disease affects millions worldwide, with higher prevalence in Africa and India [47]. Recent advancements in care, including newborn screening, penicillin prophylaxis, and hydroxyurea treatment, have improved patient outcomes [49,50]. Ongoing research explores gene editing techniques like CRISPR/Cas9 to target the β-globin gene in hematopoietic stem cells as a potential therapeutic approach [51]. Gene therapy approaches using βT87Q-globin have shown anti-sickling effects by reducing endogenous βS-globin expression [52]. These advancements in antisickling agents and therapies offer new possibilities for treating sickle cell disease.
Thalassemias are inherited disorders of hemoglobin synthesis, characterized by deficient production of globin chains [53,54]. They are prevalent in Mediterranean, Middle Eastern, and Southeast Asian regions [55,56]. Alpha and beta thalassemias are the most common types, resulting from mutations in α and β globin genes, respectively [57]. Symptoms range from asymptomatic in carriers to severe anemia requiring regular blood transfusions in major forms [58]. Complications include iron overload, cardiac issues, and endocrine disorders[59]. Treatment options include blood transfusions, iron chelation therapy, and bone marrow transplantation [55]. Gene therapy and editing strategies are emerging as potential curative treatments [53]. Prevention through premarital screening and prenatal diagnosis is crucial in reducing the prevalence of thalassemia [59]. Despite its significant impact, thalassemia is often underrecognized in global health assessments [54].
Hemoglobin C (HbC) is a common structural variant of normal hemoglobin, resulting from a mutation in the beta-globin gene [60]. HbC is less soluble than normal hemoglobin, leading to increased rigidity of red blood cells [61]. This can cause mild chronic hemolysis, splenomegaly, and jaundice in homozygous individuals [60]. HbC interacts more strongly with erythrocyte membranes than normal hemoglobin [62]. While HbC disease is generally mild, its inheritance with other hemoglobinopathies like HbS can have serious consequences [60]. Both HbC and HbS may affect the development of acquired immunity against malaria [63]. Efforts have been made to develop simple, inexpensive methods for detecting HbC in resource-limited settings, including microscopic visualization of HbC crystals [64,65]. These methods could potentially determine zygosity and aid in diagnosis in underdeveloped countries where HbC is prevalent.
Hemoglobin D-Punjab (HbD-Punjab) is a variant resulting from a mutation in codon 121 of the β-globin gene [66]. While heterozygous HbD-Punjab is generally asymptomatic, homozygous cases can cause mild hemolytic anemia and splenomegaly [66,67]. The clinical presentation of HbD-Punjab can vary, especially when coinherited with other hemoglobinopathies or thalassemias [68,69]. HbSD-Punjab, a compound heterozygous condition, can lead to severe symptoms resembling sickle cell disease [70]. HbD-Punjab/β-thalassemia combinations may result in moderate to severe anemia [67,71,72]. Diagnosis typically involves hemoglobin electrophoresis and molecular studies [68]. Treatment options include blood transfusions and hydroxyurea, which has shown promise in managing symptoms [70]. The prevalence of HbD-Punjab is relatively low, estimated at 0.06% in one study [66].
Hemoglobin E (HbE), a common β-globin variant in Southeast Asia, results from a Glu26Lys mutation [73,74]. This variant exhibits extended linkage disequilibrium and likely arose 1,240-4,440 years ago due to malarial selection [73]. HbE frequencies reach up to 0.433 in some Lao populations [74]. While HbE shows minimal allosteric changes, it demonstrates altered redox properties, including decreased nitrite reductase activity and accelerated reduction by cysteine [75]. HbE is more susceptible to oxidative damage, especially in the presence of free α subunits [76]. When co-inherited with β-thalassemia, HbE can cause severe clinical manifestations [77]. However, its association with protection against cerebral malaria remains inconclusive [78]. HbE disorders present a wide range of clinical severity, from asymptomatic carriers to severe anemia [79].
Hemoglobin [80]Bart's (γ4) is a homotetrameric hemoglobin found in individuals with α-thalassemia [81]. It lacks cooperativity and has a higher oxygen affinity than adult or fetal hemoglobin. Crystal structures reveal that Hb Bart's resembles the R state of adult hemoglobin [81,82]. Quantification of Hb Bart's in cord blood accurately indicates α-thalassemia status, with levels correlating to the number of deleted or inactivated α-globin genes [83,84]. The prevalence of Hb Bart's varies among populations, with high rates reported in Saudi Arabia and Thailand [85,86]. Spectroscopic studies show that Hb Bart's is structurally similar to Hb H (β4) [87]. In some populations, elevated Hb Bart's may result from developmental asynchrony rather than α-thalassemia [88].
Hemoglobin Chesapeake (α92 Arg→Leu) is a high-oxygen-affinity hemoglobin variant associated with erythrocytosis [89,90]. This mutation occurs at the α1β2 interface, similar to other high-affinity variants like Hb Wood and Hb Malmö [91]. The increased oxygen affinity results from an early conversion from the T to R state during ligand binding [90]. Hb Chesapeake causes mild erythrocytosis in carriers due to reduced oxygen delivery to tissues, necessitating increased red cell production [92]. This mechanism is also observed in other high-affinity variants like Hb Kempsey [93]. The fetal form of Hb Chesapeake (α2Chesγ2) also exhibits increased oxygen affinity, suggesting similar conformational changes in the γ chain [94]. Generally, high-affinity hemoglobin variants impair the formation of a stable T state or modify the heme environment, leading to compensatory erythrocytosis [95].
Hemoglobin Lepore is a structural variant formed by the fusion of δ and β globin genes, resulting from unequal crossover events [96,97]. It is associated with a β-thalassemia minor phenotype in carriers and can lead to severe anemia in homozygotes or compound heterozygotes with β-thalassemia [98,99]. Several variants exist, including Lepore-Boston, Lepore-Hollandia, and Lepore-Washington-Boston [100,101]. The condition is prevalent in Southern Italy and has spread globally due to migration [99]. Diagnosis requires hematological, biochemical, and molecular analyses [96]. Hb Lepore can interact with other hemoglobinopathies, resulting in various clinical phenotypes [101]. Recently, a novel variant, Hb Lepore-Hong Kong, was identified in a Chinese family [102]. Accurate diagnosis is crucial for genetic counseling and prenatal screening in affected populations.
Hemoglobin Constant Spring (Hb CS) is a variant caused by a mutation in the α2-globin gene termination codon, resulting in an elongated α-chain [103]. It is prevalent in Southeast Asian populations, with gene frequencies ranging from 0.008 in Thailand to 0.143 in Vietnam [104,105]. Hb CS can lead to thalassemia intermedia when combined with α-thalassemia [106]. Diagnosis is challenging due to low amounts of the mutant hemoglobin, but selective enzymatic amplification of α2-globin DNA allows for accurate detection [107]. Hb CS can interfere with glycated hemoglobin measurements using boronate affinity chromatography [108]. In rare cases, homozygous Hb CS can cause fetal anemia and hydrops [109]. Understanding Hb CS is crucial for genetic counseling and prenatal diagnosis in affected populations. Hb O-Arab (Glu121Lys) causes mild hemolytic anemia in populations from the Middle East and North Africa.
Hemoglobin O Arab is a rare abnormal hemoglobin variant characterized by a β121Glu → Lys mutation [110,111]. It can occur in homozygous form or in combination with other hemoglobinopathies. The homozygous form is generally well-tolerated [112,113], while compound heterozygous forms, particularly Hb S/O Arab, can result in severe clinical manifestations like sickle cell anemia [110,114]. Hb S/O Arab patients may experience acute chest syndrome, vasoocclusive events, and other complications [110]. Diagnosis requires specific electrophoresis techniques due to Hb O Arab's migration patterns [110]. The condition has been reported in various populations, including African Americans, West Africans, and Arabs [110,112,115]. Treatment may involve transfusions and splenectomy in cases of hypersplenism [113].
Hb Seal Rock, an extended α-chain variant, is associated with mild Hb H disease and α-thalassemia-2 trait [116]. Some α-chain variants can lead to chronic hemolytic anemia, with the mutation's location within the gene sequence playing a crucial role in determining its effect [117]. Hb Indianapolis, a rare and slightly unstable β-globin variant, was reported to cause moderate hemolytic anemia and renal damage in a Brazilian patient [118]. Routine DNA sequencing of α- and β-globin genes has revealed numerous novel mutations, including 11 new β-chain variants, 15 α-chain variants, 19 β-thalassemia mutations, and 15 α+-thalassemia mutations, highlighting the genetic diversity in hemoglobinopathies [119].
Several unstable hemoglobin variants have been identified that cause mild to moderate hemolytic anemia. Hb Madrid (β115Ala→Pro) was found in a Spanish boy and a Korean family, resulting in moderately severe hemolytic anemia [120,121]. Hb Showa-Yakushiji (β110Leu→Pro) was observed in four unrelated Indian patients [122]. Other variants include Hb Santander (β34Val→Asp) in a Spanish patient [123], Hb Yokohama (β31Leu→Pro) in a Japanese family [124], and Hb Seattle (β76Ala→Glu) in a mother and her two sons [125]. Hb Miami (β116His→Pro) and Hb Hershey (β70Ala→Gly) were found in association with beta-thalassemia mutations, while Hb Abington (β70Ala→Pro) was another unstable variant [126]. These variants demonstrate the range of clinical severity observed with unstable hemoglobin mutations, particularly when combined with thalassemic mutations.
2.2. Hemoglobin Instability and Heinz Bodies
Hemoglobin instability can lead to denaturation and precipitation, resulting in the formation of Heinz bodies, which are inclusions of denatured hemoglobin in red blood cells. This instability is often due to amino acid substitutions or deletions that disrupt the hemoglobin structure, enhancing oxidation to methemoglobin and subsequent conversion to hemichrome [127]. The loss of heme, particularly from beta chains, is a critical factor in Heinz body formation [128,129]. The presence of Heinz bodies can cause hemolytic anemia due to mechanical obstruction in microcirculation and oxidative damage to red cell membranes [80,130]. Additionally, factors such as temperature and oxidative stress can exacerbate the formation of Heinz bodies [131]. Oxidative stress occurs when there is an imbalance between reactive oxygen species (ROS) and the body's antioxidant defenses. Hemoglobin is particularly vulnerable to oxidative damage, which can lead to hemolysis. This oxidative damage is exacerbated in conditions such as thalassemia and sickle cell disease. Mechanisms leading to Heinz body formation include oxidative damage, genetic mutations, chemical exposure, and enzyme deficiencies. Reactive oxygen species (ROS) can oxidize hemoglobin, causing it to denature and form Heinz bodies. This oxidative stress is common in conditions with an elevated level of ROS, such as inflammation or chronic diseases.
As mentioned above genetic factors significantly influence the stability of hemoglobin. Mutations in the genes encoding hemoglobin can lead to structurally unstable hemoglobin variants, making them more susceptible to denaturation and resulting in disorders such as thalassemias and hemoglobinopathies. These genetic mutations can affect the function and stability of hemoglobin molecules, leading to various clinical manifestations. Out of the 819 known variants of the hemoglobin subunit α, 31 are classified as unstable. This represents approximately 3.8% of all α subunit variants. In contrast, 85 out of 771 known variants of the hemoglobin subunit β are considered unstable, accounting for about 11% of the β subunit variants. This comparison highlights a significant difference in the frequency of unstable variants between the two subunits. The frequency of unstable variants in the β subunit is nearly three times higher than that in the α subunit, suggesting that the β-globin gene may be more susceptible to mutations that result in instability or that the structural constraints of the β subunit allow for more potentially unstable configurations.
The human α-globin gene cluster, including HBA1 and HBA2, is located on chromosome 16 at position 16p13.2-pter [132,133]. This region is extremely gene-rich, containing 100 confirmed and 20 predicted genes [134]. The α-globin locus is highly polymorphic, with 15 dimorphic and 2 multiallelic genetic markers identified [135]. Upstream regulatory elements, particularly HS-40, play crucial roles in α-globin gene expression [136]. α-Thalassemia, a disorder of reduced α-chain synthesis, is often caused by large deletions within the gene cluster [137]. Interestingly, a 62 kb deletion upstream of the α-globin genes can also cause α-thalassemia without directly affecting the genes themselves [138]. The α-globin locus serves as an ideal genetic marker for constructing human linkage maps and studying molecular genetics of the cluster [135].
The β-globin gene cluster, located on chromosome 11 at position 11p15.5, includes the HBB gene, which encodes the β-globin chain of adult hemoglobin [139,140]. This cluster comprises several genes, including HBD, which encodes the δ-globin chain, and undergoes critical expression switches during development [141]. The precise localization of the β-globin gene has been confirmed through various studies, indicating its location within the 11p15.4 to 11p15.5 region [142,143,144]. The regulation of these genes is crucial for understanding hemoglobinopathies, with recent findings highlighting the role of factors like BCL11A in silencing fetal hemoglobin [145]. Overall, the β-globin gene cluster is essential for normal hemoglobin function and is implicated in disorders such as sickle cell anemia and β-thalassemia [140].
The observed difference in the frequency of unstable variants between the α and β subunits, 3.8% vs. 11%, may be due to several factors. One would expect that the α-globin gene cluster location on chromosome 16 and its gene duplication may confer additional stability, resulting in higher chance for accepting unstable variants compared to the β-globin gene cluster on chromosome 11. The presence of two nearly identical genes (HBA1 and HBA2) in the α-globin cluster means there is a redundancy that can potentially compensate for potential mutations. If one gene is mutated, the other can often continue to produce functional α-globin chains, reducing the impact of any single mutation and enhancing overall stability. This redundancy might act as a genetic buffer, making the α-globin production more resilient to genetic mutations and environmental pressures. However, as we found that less unstable variants of α-chain are nearly three-fold less indicating that other factors are responsible for higher number of unstable β-chain variants. Other factor would be structural differences between the α and β subunits might inherently affect their susceptibility to mutations that lead to instability. While both subunits share a high sequence identity of 43% and 50-60% sequence similarity, they may have distinct tolerance toward mutations constrained by their differences in function
The β-globin's versus α-globin's specific role and interactions in the hemoglobin tetramer may be reason for higher counts of unstable mutations. In fact, the β-globin plays a crucial role in hemoglobin structure and function. It forms strong interactions with α-globin to create stable α1β1 dimers, which then assemble into α2β2 tetramers [146]. The β-chains significantly influence oxygen binding characteristics, particularly through alterations in the T state properties [147]. The β112 Cys residue is critical for both homo- (β4) and hetero-tetramer (α2β2) formation, with mutations at this position affecting assembly and stability [148]. The β4 tetramer structure closely resembles the R state of liganded α2β2 hemoglobin, but with some unique interface interactions [149]. The β chains also interact with the cytoplasmic domain of band 3 in erythrocyte membranes, preferentially binding to deoxyhemoglobin [150]. On the other hand, α-globin plays a crucial role in hemoglobin formation and function. The α-Hemoglobin Stabilizing Protein (AHSP) is essential for maintaining α-globin stability and preventing its precipitation [151]. AHSP binds specifically to the G and H helices of α-globin, with the N-terminal part of the H helix being critical for this interaction[152,153]. AHSP acts as a molecular chaperone, facilitating α-globin folding, refolding after denaturation, and incorporation into hemoglobin A[154]. It also protects against oxidative damage and helps maintain the correct ratios of α and β globins during hemoglobin biosynthesis [155]. Mutations in α-globin that impair AHSP binding can lead to protein instability and anemia [156]. The complex interplay between α-globin expression, AHSP, and hemoglobin assembly is crucial for normal erythropoiesis [157]. Hence, we can speculate that these differences between globins may result in observed differences in frequencies of unstable variants that α-globin mutations produce a significant disadvantage, so they are much less tolerated than β-globin unstable variants. In addition, a possible correlation can be found between amino acid residues contacting heme and unstable variants (Fig. 2d).
2.3. Pharmacological Approaches to Hemoglobin Stabilization and Oxidative Stress in Hemolytic Disorders
Certain molecules and toxins can induce oxidative stress or directly interact with hemoglobin, leading to its denaturation and the formation of so-called Heinz bodies. Even below mentioned small molecules are not directly interacting with Hb, they can influence metabolism of erythrocytes, reduce oxidative stress, or indue other hemoglobin chain expression and by doing this they can provide stability for hemoglobin or prevent sickling.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency, affecting approximately 400 million people worldwide, is a crucial enzyme in protecting red blood cells from oxidative stress [158]. G6PD deficiency can lead to hemolytic anemia, neonatal jaundice, and increased susceptibility to oxidative damage [159]. The deficiency results in reduced NADPH production, compromising the cell's ability to maintain glutathione in its reduced state [160]. This leads to increased hemoglobin denaturation and ferriheme release, potentially causing hemolysis [161]. Contrary to previous beliefs, NADPH status, rather than glutathione levels, modulates oxidant sensitivity in G6PD-deficient erythrocytes [162]. G6PD deficiency also has broader implications, including increased risk of prenatal and postnatal death, infertility, and teratogenesis [163]. In transfusion medicine, G6PD-deficient blood may pose risks to certain patient populations [164].
Oxidative stress plays a significant role in hemolytic anemias, causing damage to red blood cells and exacerbating symptoms [165]. Antioxidants, such as N-acetylcysteine (NAC) and Vitamin E, have shown promise in reducing oxidative stress and improving hemoglobin levels, particularly in children with transfusion-dependent thalassemia [166]. These antioxidants can inhibit cation pathways responsible for red blood cell dehydration and reduce phosphatidylserine exposure, potentially improving cell rheology and reducing vascular adhesion [167]. Supplementation with vitamin C and NAC has been found to preserve glutathione homeostasis and reduce hemolysis in stored red blood cells [168]. However, high-dose vitamin C and E supplementation may worsen hemolysis in some cases [169]. Combining antioxidants with iron chelators may provide substantial improvements in the pathophysiology of hemolytic anemias, especially thalassemia [170].
Hemin is a pharmacological agent used in treating acute porphyrias, which are metabolic disorders characterized by heme biosynthesis defects. It functions primarily by inhibiting delta-aminolevulinic acid synthase (ALAS-1), the rate-limiting enzyme in heme production, thereby reducing the accumulation of toxic heme precursors like delta-aminolevulinic acid [171,172]. Hemin administration has been shown to alleviate symptoms during acute attacks by restoring heme levels and downregulating ALAS-1 activity [173,174]. Additionally, heme's feedback inhibition on ALAS-1 is mediated through various mechanisms, including proteolytic degradation of the enzyme [175]. The clinical efficacy of hemin underscores its role in managing acute porphyrias, as it directly addresses the biochemical disturbances underlying these conditions [176,177].
Hydroxyurea is a promising treatment for sickle cell disease, primarily due to its ability to induce fetal hemoglobin (HbF) production [178,179,180]. HbF inhibits the polymerization of sickle hemoglobin (HbS), reducing disease severity [179]. Hydroxyurea works by perturbing erythroid precursor maturation and potentially through nitric oxide-dependent mechanisms [181]. Clinical studies have shown that hydroxyurea treatment increases HbF levels, reduces hemolysis, and decreases the frequency of vaso-occlusive crises and hospitalizations [182,183]. The drug also improves red cell survival and reduces sickling at partial oxygen saturation [184]. While hydroxyurea's effects can be inconsistent, it has been shown to reduce morbidity and mortality in adults with sickle cell anemia [178]. Ongoing research is exploring its use in children and in combination with other HbF-inducing agents [179,185].
Mitapivat, an oral pyruvate kinase activator, has shown promising results in treating various hemoglobinopathies. In pyruvate kinase deficiency, it increased hemoglobin levels and reduced transfusion burden, with sustained long-term effects [186,187]. For non-transfusion-dependent α- and β-thalassemia, mitapivat improved hemoglobin levels, markers of erythropoiesis, and hemolysis [188,189,190]. In sickle cell disease, it demonstrated improvements in anemia, hemolysis, and hemoglobin S polymerization kinetics [191]. Mitapivat's mechanism of action involves increasing ATP production in red blood cells, which may help stabilize hemoglobin and improve cell integrity [192]. The drug has shown a favorable safety profile across studies, with common adverse events being generally mild and transient [187,188]. These findings suggest that mitapivat may represent a novel therapeutic approach for various hereditary anemias [193].
2.4. Small-Molecule Binders to Hemoglobin
Hemoglobin is also a direct drug target and hence several classes of molecules were found to bind and stabilize hemoglobin, which should improve oxygen delivery in patients with hemoglobinopathies such as sickle cell disease and β-thalassemia. Pharmacological chaperones are drugs that stabilize hemoglobin by promoting proper folding and preventing denaturation. These agents help maintain hemoglobin's functional state under stress conditions, reducing the likelihood of hemolysis (see above). Most of the development of drugs targeting hemoglobin focus on sickle cell diseases, which is caused by a single mutation that results in the substitution of valine for glutamic acid in the sixth position of the β-globin chain of hemoglobin, leading to the polymerization of hemoglobin S upon deoxygenation. Peptides and amino acids stabilizing hemoglobin have been summarized in detail in a recent review [194]. Recent advances in SCD treatment have expanded beyond the traditional therapies of hydroxyurea and L-glutamine. New approaches target various aspects of SCD pathophysiology, including HbS polymerization, cellular adhesion, inflammation, and oxidative stress [195]. The FDA has approved three new drugs: crizanlizumab, voxelotor, and L-glutamine, which have shown efficacy in reducing pain crises and improving hemoglobin levels [196]. Emerging therapies include peptide-based inhibitors of HbS polymerization, which represent a novel approach to SCD treatment [194]. Additionally, researchers are exploring combination therapies to address the complex nature of SCD manifestations [197,198]. While gene therapy holds promise for a cure, its widespread application remains limited due to technical and cost challenges [199]. These developments offer hope for improved quality of life and survival for SCD patients.
Compounds like TD7, VZHE039, and their nitric oxide-releasing derivatives have shown promising antisickling properties by increasing hemoglobin's oxygen affinity and disrupting HbS polymerization [200]. TD-1, a small molecule identified through screening, demonstrated dual allosteric and antioxidant effects by binding to βCys93 and βCys112, stabilizing hemoglobin's relaxed state and reducing oxidative changes [201,202]. Other compounds like TD-3 and hydroxyurea also target βCys93, providing both antioxidant and antisickling benefits [203]. These studies have elucidated the mechanisms of action for various antisickling agents, including their effects on oxygen affinity, HbS polymerization, and oxidative stress reduction. Further research on antisickling agents targeting hemoglobin's β-Cys93 has shown promising results for treating sickle cell disease. Other thiol reagents reacting with β-Cys93 have shown varying antisickling effects [204,205] Overall, increasing hemoglobin's oxygen affinity represents a promising therapeutic approach for sickle cell disease.
5-hydroxymethyl-2-furfural (5HMF) is a promising compound for treating sickle cell disease. It forms a Schiff-base adduct with hemoglobin, increasing oxygen affinity and inhibiting red blood cell sickling [206,207]. 5HMF demonstrates dose-dependent effects on hemoglobin oxygen affinity in phase 1 clinical trials [208]. It improves cardiac function during severe hypoxia [209] and reduces deoxygenation-induced dehydration of red blood cells by inhibiting cation conductance and Ca2+-activated K+ channels [210]. Researchers have developed ester and ether derivatives of 5HMF with enhanced antisickling properties [207] and explored nitric oxide-releasing prodrugs [211]. The mechanism of action involves stabilizing the R state of hemoglobin, which has higher oxygen affinity and slower polymerization kinetics [212,213]. These findings support 5HMF as a potential therapeutic agent for sickle cell disease.
Hb plays a crucial role in nitric oxide metabolism and oxygen transport. Hb can form various NO-related compounds, including nitrosylated Hb (HbNO) and S-nitrosohemoglobin (SNO-Hb) [214,215]. These compounds are involved in regulating vascular function and oxygen delivery [216,217]. The interaction between Hb and NO is complex, involving redox reactions and allosteric effects [214,218]. SNO-Hb has been proposed as a key mediator of NO transport and bioactivity, with its formation and release coupled to oxygen saturation [216,219]. However, the precise mechanisms of NO activity preservation and regulation by Hb remain debated [220,221]. Understanding these interactions is crucial for developing transfusion therapeutics and treating various cardiovascular diseases [217].
Recent research on SCD has also focused on developing aromatic aldehydes as potential treatments by increasing hemoglobin oxygen affinity and inhibiting red blood cell sickling. Novel compounds, including pyridyl derivatives of vanillin and azolylacryloyl derivatives, have shown improved potency, metabolic stability, and sustained antisickling effects in vitro [222,223]. Some compounds exhibit both oxygen-dependent and oxygen-independent antisickling mechanisms, potentially disrupting key intermolecular contacts necessary for hemoglobin S polymer formation. Crystal structure studies have provided insights into the binding modes and mechanisms of action of these compounds [223]. The development of hemoglobin modulators represents a promising approach for treating SCD, with ongoing research and growing intellectual property in this field [224]. These advancements complement other therapeutic strategies, including targeting cellular adhesion, inflammation, and oxidant injury.
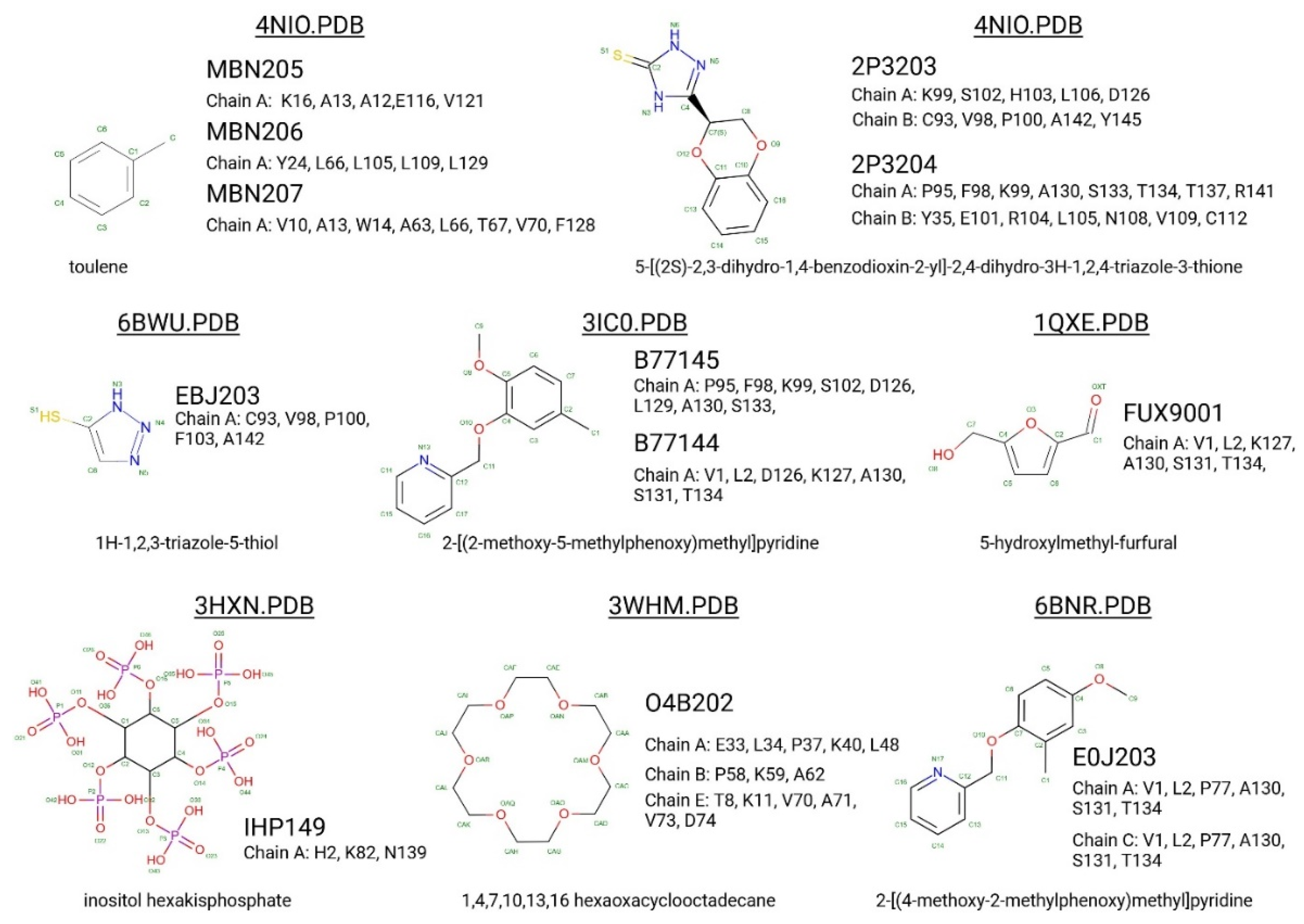
Figure 3.
Binding interactions of various small molecules with different Hb residues as visualized in their respective PDB structures. The figure shows the chemical structures of the ligands (such as toluene, 1H-1,2,3-triazole-5-thiol, inositol hexakisphosphate, and others) along with their binding pockets and the chains they interact with, as indicated by the PDB IDs (4NIO, 6BWU, 3HXN, etc.). The protein-ligand interactions are depicted through dashed lines, highlighting key residues involved in the binding. Residues interacting with hemoglobin chains (e.g. A, B, C) are listed. .
Figure 3.
Binding interactions of various small molecules with different Hb residues as visualized in their respective PDB structures. The figure shows the chemical structures of the ligands (such as toluene, 1H-1,2,3-triazole-5-thiol, inositol hexakisphosphate, and others) along with their binding pockets and the chains they interact with, as indicated by the PDB IDs (4NIO, 6BWU, 3HXN, etc.). The protein-ligand interactions are depicted through dashed lines, highlighting key residues involved in the binding. Residues interacting with hemoglobin chains (e.g. A, B, C) are listed. .
PF-07059013 (compound 23) is a noncovalent hemoglobin modulator that has advanced to phase 1 clinical trials for sickle cell disease (SCD) treatment. It demonstrated a 37.8% reduction in red blood cell sickling in a mouse model [225]. This compound represents a new approach to SCD treatment, targeting the root cause of the disease: hemoglobin S polymerization [226]. Unlike earlier covalent modifiers with potential safety concerns, PF-07059013 binds noncovalently to hemoglobin [227]. Similar approaches, such as GBT440, have shown promise in preclinical testing [228,229]. These developments are part of a broader effort to create new SCD therapies, including fetal hemoglobin inducers and agents targeting cellular adhesion, inflammation, and vascular tone [230]. Such advancements offer hope for improved SCD management beyond current treatments like hydroxyurea and blood transfusions [231].
Vanillin derivatives have shown promise as potential treatments for sickle cell disease and other conditions. Novel compounds like SAJ-009, SAJ-310, and SAJ-270 demonstrated improved binding and pharmacokinetic properties compared to vanillin, with enhanced allosteric and antisickling effects [223]. SAJ-310 exhibited sustained antisickling activity and potential metabolic stability [232]. Vanillin derivatives also showed antioxidant properties and inhibitory activity against acetylcholinesterase and β-amyloid aggregation, suggesting potential applications in Alzheimer's disease treatment [233]. Additionally, vanillin demonstrated analgesic effects on mechanical allodynia in a neuropathic pain model [234] and inhibitory activity against mushroom tyrosinase [235]. The compound's ability to react covalently with sickle hemoglobin and inhibit cell sickling supports its potential as a treatment for sickle cell anemia [236]. Vanillin's diverse pharmacological activities and safety profile make it a promising candidate for further research and development.
VZHE-039 is a novel antisickling agent that exhibits both oxygen-dependent and oxygen-independent mechanisms of action [237]. In this study, VZHE-039 demonstrates enhanced pharmacologic activities and metabolic stability. Its oxygen-independent activity is attributed to interactions with the αF-helix of hemoglobin, potentially destabilizing the sickle hemoglobin polymer [238]. Other antisickling agents, such as voxelotor and FT-4202, also show promise in treating sickle cell disease by increasing hemoglobin oxygen affinity.
Crown ethers, cyclic polyethers with oxygen atoms, have shown potential in modulating protein surface properties and interactions [239]. They can bind to metal ions and positively charged amino acid side chains, affecting protein behavior such as oligomerization and crystallization. In hemoglobin research, crown ethers and related compounds have been explored for their ability to modify hemoglobin's oxygen-binding properties. Acylation of hemoglobin with polyoxyethylene derivatives has been shown to alter its oxygen affinity [240]. Other synthetic allosteric effectors, such as RSR-4 and RSR-13, have demonstrated the ability to shift hemoglobin's allosteric equilibrium towards the low-affinity T-state [241]. These studies highlight the potential of crown ethers and related compounds in modulating hemoglobin function and developing novel therapeutic approaches.
Complex interactions between hemoglobin (Hb) can occur also with natural molecules such as including sphingosine 1-phosphate (S1P) and lipopolysaccharide (LPS). S1P is a bioactive lipid involved in immune responses, cell growth, and survival. It has been found to bind specifically to deoxygenated sickle hemoglobin (HbS), the form of hemoglobin that promotes the sickling of red blood cells in sickle cell disease. When S1P binds to HbS, it alters glucose metabolism and increases oxidative stress, both of which are significant contributors to the pathology of sickle cell disease. Altered glucose metabolism disrupts energy production in red blood cells, while increased oxidative stress leads to cellular damage and inflammation, further exacerbating the disease's symptoms [242]. Hb also interacts with lipopolysaccharide (LPS), a component of the outer membrane of Gram-negative bacteria, enhancing its biological activity and potentially contributing to the immune defence against bacterial infections. This interaction suggests that hemoglobin may play a role beyond oxygen transport, acting as a modulator of the immune response [243,244]. Multiple binding sites for LPS have been identified on both the α and β-subunits of hemoglobin, highlighting the molecule's multifunctional nature and its potential importance in pathogen defense [244,245]. Hemoglobin also binds to erythrocyte membranes, an interaction crucial for the stability and integrity of red blood cells. This binding is driven by electrostatic forces influenced by factors such as pH and ionic strength, which may vary under different physiological conditions, such as during oxygenation/deoxygenation cycles [246,247]. A critical protein involved in this interaction is band 3 protein, a major structural component of the erythrocyte membrane. The acidic N-terminal segment of the cytoplasmic domain of band 3 serves as a primary binding site for hemoglobin [150]. This interaction is essential for maintaining the structural integrity of red blood cells and regulating their lifespan. Disruptions in this binding can lead to membrane instability and hemolysis, a hallmark of hemolytic anemias such as sickle cell disease. In pathological conditions like sickle cell disease, these hemoglobin interactions are further altered, contributing to disease progression. The S1P-Hb interaction exacerbates oxidative stress, worsening the symptoms of sickle cell disease. The LPS-Hb interaction could play a dual role in immune defense and the potential triggering of exaggerated immune responses. Finally, hemoglobin's binding to the erythrocyte membrane via band 3 protein is critical for cell stability, and disruptions in this interaction could contribute to the shortened lifespan of red blood cells seen in sickle cell disease and other hemoglobinopathies. In summary, these complex interactions between hemoglobin, S1P, LPS, and the erythrocyte membrane are critical for both normal physiological processes and pathological conditions, especially sickle cell disease.
Voxelotor is a novel oral treatment for SCD that binds to hemoglobin, increasing its oxygen affinity and preventing polymerization of sickle hemoglobin (HbS) [248,249]. By forming a reversible covalent bond with hemoglobin, voxelotor reduces sickling of red blood cells and interrupts the molecular pathogenesis of SCD. Clinical trials have demonstrated that voxelotor increases hemoglobin concentration, reduces hemolysis, and improves hematological parameters in SCD patients [250,251]. The drug has shown a dose-dependent pharmacokinetic and pharmacodynamic response and is well-tolerated [252]. Studies in SCD mice have indicated that increasing hemoglobin oxygen affinity with voxelotor analogs improves brain oxygenation and hypoxia tolerance (Dufu et al., 2021). Voxelotor represents a new disease-modifying approach to SCD treatment, targeting the root cause of the disease [253].
3. Search for Potentially New Hb Stabilisers: Hemoglobin Binding Proteins
Innovative approaches targeting hemoglobin stability and function may enhance protein platforms for protein engineering by the use of protein chaperones, enzymes like falcilysin, and nanobodies.
Alpha-hemoglobin-stabilizing protein (AHSP) is an erythroid-specific molecular chaperone that binds and stabilizes free α-globin chains, preventing their precipitation and cytotoxic effects [254,255]. AHSP forms a 1:1 complex with α-globin, binding to its G and H helices in a manner similar to the α1-β1 interface in hemoglobin A [153,256]. This interaction facilitates the conversion of oxy-αHb to a non-reactive, oxidized form, limiting its ability to generate reactive oxygen species [257]. AHSP plays a crucial role in normal erythropoiesis and hemoglobin synthesis, as demonstrated by studies in AHSP-null mice [154,257]. It also acts as a folding chaperone for nascent α-globin, promoting its incorporation into hemoglobin A. The importance of AHSP in vivo is further supported by mutagenesis studies, which reveal its robust chaperone activity even when its biochemical interactions with α-globin are altered [258].
Falcilysin, a metalloprotease in the malaria parasite Plasmodium falciparum, plays a crucial role in hemoglobin degradation within the acidic food vacuole [259,260]. It operates downstream of aspartic and cysteine proteases, cleaving peptide fragments of hemoglobin rather than intact globin. Interestingly, falcilysin exhibits activity at both acidic and neutral pH, suggesting additional functions beyond hemoglobin catabolism, including potential roles in transit peptide degradation in the apicoplast [261,262]. The enzyme's essentiality for parasite development has led to the exploration of piperazine-based hydroxamic acid inhibitors, which effectively target falcilysin [263]. Overall, falcilysin's multifaceted roles highlight its importance in the parasite's survival and present it as a promising target for antimalarial drug development. Another attractive out-of-box option for falcilysin would be re-programming this protein to stabilise hemoglobin.
Recent research has explored the interactions between hemoglobin (Hb) and various molecules, including nanobodies and lipopolysaccharide (LPS). Nanobodies targeting BCL11A, a repressor of fetal hemoglobin, have been developed for potential therapeutic applications in sickle cell disease and β-thalassemia [264,265]. Studies have also identified LPS-binding sites on Hb, which may play a role in innate immune defense [266], but might be potentially used for drug development.
A recent study explored the use of llama-derived nanobodies, particularly NbE11, to develop sensitive diagnostic tools for detecting human hemoglobin [267]. The study determined the crystal structure of the NbE11-hemoglobin complex, revealing specific interactions primarily with the β-subunit of human hemoglobin. NbE11, a llama-derived nanobody, binds to human hemoglobin (Hb) with moderately high affinity (Kd = 147 nM) and the binding is predominantly driven by favorable enthalpy. These interactions are characterized by high specificity and moderate affinity, driven by favorable enthalpic changes. This specificity allows for the development of nanobody-based assays that can differentiate human hemoglobin from that of other species, reducing false positives in diagnostic applications. Structural differences between human Hb and Hb from other vertebrates at the NbE11 binding interface likely explain the lack of cross-reactivity observed in the NbE11-based sandwich ELISA assay. The incorporation of nanobodies in hemoglobin research highlights their potential as both diagnostic and therapeutic tools. The study showed that NbE11 binds specifically to human hemoglobin without cross-reacting with hemoglobin from other vertebrates, making it suitable for precise diagnostic applications.
The exploration of AHSP, falcilysin, and nanobodies as tools for modulating hemoglobin stability and function presents promising avenues for treating hemoglobin disorders and developing precise diagnostic tools. Additionally, molecularly imprinted polymeric nanoparticles have been developed for specific binding and purification of human Hb [268].
4. Conclusions
The wide range of hemoglobin variants presents challenges in drug development. Although most current drugs focus on SCD, there is potential to develop variant-specific therapies that address the molecular diversity of hemoglobinopathies. Personalized medicine approaches, which tailor treatments based on a patient's specific hemoglobin variant, may offer more precise and effective therapies in the future. Investigating the structural differences between hemoglobin variants and how these influence drug binding and efficacy is essential. Drugs that bind effectively to HbS may not work as well for other variants, such as those found in thalassemia or rare hemoglobinopathies like HbC or HbE. Advances in gene editing, such as CRISPR/Cas9, offer the potential for curative treatments by directly correcting the genetic mutations responsible for hemoglobin variants. However, widespread application remains limited due to technical challenges and high costs.
The relationship between drugs and hemoglobin variants is deeply intertwined with the molecular pathology of hemoglobin disorders. Small-molecule binders, pharmacological chaperones, antisickling agents, and novel therapies like nanobodies are all designed to stabilize hemoglobin or correct the aberrant behavior caused by specific variants. To advance research on hemoglobin variants, time-resolved X-ray powder diffraction, especially utilizing laser-driven X-ray sources, offers the potential to capture subtle dynamic differences at the atomistic level, providing deeper insights into the behavior of hemoglobin variants [269]. Complementing this, the development of 3D-printed in situ crystallization plates, which provide precise control over crystallization in an external electric field, represents a valuable advancement for studying challenging-to-crystallize proteins, including hemoglobin variants [270].
Based on understanding of molecular mechanism of actions, therapies target hemoglobin's functional aspects, such as oxygen affinity, polymerization, and oxidative stress, offering hope for patients with hemoglobinopathies like SCD and thalassemia. Continued research and development of both variant-specific treatments and broader hemoglobin modulators will be critical in improving patient outcomes and addressing the diverse challenges posed by hemoglobinopathies.
Author Contributions
Conceptualization, M.Z., G.Z. and K.K.; methodology, M.Z.; software, G.Z.; validation, M.N., M.Z., K.K. and Z.Z.; formal analysis, G.Z.; investigation, K.K., G.Z.; resources, G.Z., M.N.; data curation, G.Z.; writing—original draft preparation, G.Z.; writing—review and editing, G.Z., M.Z.; visualization, M.Z., G.Z. All authors have read and agreed to the published version of the manuscript.
Funding
The research was supported by the Slovak Research and Development Agency under the Contract no. APVV-23-0212 and Visegrad Grants from the International Visegrad Fund and Fellowship (No.#62410140). GZ is partly funded by the European Union’s NextGenerationEU through the Recovery and Resilience Plan for Slovakia under the project No. 09I03-03-V03-00008.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Hb | Hemoglobin |
| SNO | S-nitrosohemoglobin |
| BPG | Bisphosphoglycerate |
| HbF | Fetal Hemoglobin |
| HbA | Adult Hemoglobin |
| HbC | Hemoglobin C |
| HbD | Hemoglobin D |
| HbE | Hemoglobin E |
| Hb CS | Hb Constant Spring |
| ACMG | American College of Medical Genetics and Genomics |
| SCD | Sickle cell disease |
| RBC | Red Blood Cell |
| ROS | reactive oxygen species |
| AHSP | α-Hemoglobin Stabilizing Protein |
| G6PD | Glucose-6-phosphate dehydrogenase |
| NAC | N-acetylcysteine |
| ALAS-1 | aminolevulinic acid synthase |
| S1P | sphingosine 1-phosphate |
| LPS | lipopolysaccharide |
References
- Perutz, M.F. Stereochemical mechanism of oxygen transport by haemoglobin. Proc R Soc Lond B Biol Sci 1980, 208, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Bellelli, A. Hemoglobin and cooperativity: Experiments and theories. Curr Protein Pept Sci 2010, 11, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.F. The Bohr effect. Annu Rev Physiol 1988, 50, 181–204. [Google Scholar] [CrossRef] [PubMed]
- Tyuma, I.; Shimizu, K. Different response to organic phosphates of human fetal and adult hemo globins. Archives of Biochemistry and Biophysics 129, 404–405. [CrossRef] [PubMed]
- Safo, M.K.; Ahmed, M.H.; Ghatge, M.S.; Boyiri, T. Hemoglobin-ligand binding: understanding Hb function and allostery on atomic level. Biochimica et biophysica acta 2011, 1814, 797–809. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Ghatge, M.S.; Safo, M.K. Hemoglobin: Structure, Function and Allostery. Subcell Biochem 2020, 94, 345–382. [Google Scholar] [CrossRef] [PubMed]
- Randad, R.S.; Mahran, M.A.; Mehanna, A.S.; Abraham, D.J. Allosteric modifiers of hemoglobin. 1. Design, synthesis, testing, and structure-allosteric activity relationship of novel hemoglobin oxygen affinity decreasing agents. Journal of Medicinal Chemistry 34, 752–757. [CrossRef] [PubMed]
- Ronda, L.; Bruno, S.; Abbruzzetti, S.; Viappiani, C.; Bettati, S. Ligand reactivity and allosteric regulation of hemoglobin-based oxygen carriers. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 1784, 1365–1377. [CrossRef]
- Ciaccio, C.; Coletta, A.; De Sanctis, G.; Marini, S.; Coletta, M. Cooperativity and allostery in haemoglobin function. IUBMB Life 60, 112–123. [CrossRef] [PubMed]
- Maillett, D.H.; Simplaceanu, V.; Shen, T.-J.; Ho, N.T.; Olson, J.S.; Ho, C. Interfacial and Distal-Heme Pocket Mutations Exhibit Additive Effects on the Structure and Function of Hemoglobin. Biochemistry 47, 10551-10563. [CrossRef]
- Brunori, M.; Miele, A.E. Modulation of Allosteric Control and Evolution of Hemoglobin. Biomolecules 2023, 13, 572. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F. Hemoglobin structure and allosteric mechanism. In Hemoglobin, Oxford University Press: 2018, pp. 58-93. [CrossRef]
- Ferguson, J.K.; Roughton, F.J. The chemical relationships and physiological importance of carbamino compounds of CO(2) with haemoglobin. J Physiol 1934, 83, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Kavanaugh, J.; Rogers, P.; Arnone, A. Crystallographic analysis of the interaction of nitric oxide with quat ernary-T human hemoglobin. Biochemistry . [CrossRef]
- Allen, B.W.; Stamler, J.S.; Piantadosi, C.A. Hemoglobin, nitric oxide and molecular mechanisms of hypoxic vasodilat ion. Trends in Molecular Medicine 15, 452-460. [CrossRef]
- Hamasaki, N.; Rose, Z.B. The binding of phosphorylated red cell metabolites to human hemoglobin A. The Journal of biological chemistry 1974, 249, 7896–7901. [Google Scholar] [CrossRef] [PubMed]
- Garby, L.; De Verdier, C.H. Affinity of human hemoglobin A to 2,3--diphosphoglycerate. Effect of hemoglobin concentration and of pH. Scand J Clin Lab Invest 1971, 27, 345–350. [Google Scholar] [CrossRef]
- Beek, G.M.; Bruin, S.H.D. The pH Dependence of the Binding of d-Glycerate 2,3-Bisphosphate to De oxyhemoglobin and Oxyhemoglobin. [CrossRef]
- Isaacks, R.E. Can Metabolites Contribute in Regulating Blood Oxygen Affinity? In Advances in Experimental Medicine and Biology, Springer US: 1988; pp. 137-143. [CrossRef]
- Mulquiney, P.J.; Kuchel, P.W. Model of 2,3-bisphosphoglycerate metabolism in the human erythrocyte based on detailed enzyme kinetic equations: equations and parameter refinement. Biochemical Journal 1999, 342, 581–596. [Google Scholar] [CrossRef]
- Kayikci, M.; Venkatakrishnan, A.J.; Scott-Brown, J.; Ravarani, C.N.J.; Flock, T.; Babu, M.M. Visualization and analysis of non-covalent contacts using the Protein Contacts Atlas. Nature structural & molecular biology 2018, 25, 185–194. [Google Scholar] [CrossRef]
- Borrayo López, F.J.; Figarola Centurion, I.; Perea Díaz, F.J.; Millán Pacheco, C.; Ibarra Cortés, B.; Torres Mendoza, B.M.; Flores-Jimenez, J.A.; Rizo-De La Torre, L.D.C. Structural Effect of Gγ and Aγ Globin Chains in Fetal Hemoglobin Tetramer. Blood 2023, 142, 2291–2291. [Google Scholar] [CrossRef]
- Shiv k, S. Hematologic and Coagulation Disorders. Obstetric anesthesia: principles and practice 14.
- Manca, L.; Masala, B. Disorders of the synthesis of human fetal hemoglobin. IUBMB Life 2008, 60, 94–111. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Rockwood, A.L.; Agarwal, A.M.; Anderson, L.C.; Weisbrod, C.R.; Hendrickson, C.L.; Marshall, A.G. Diagnosis of Hemoglobinopathy and beta-Thalassemia by 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometry and Tandem Mass Spectrometry of Hemoglobin from Blood. Clin Chem 2019, 65, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, H.S. Diagnosis of hemoglobinopathy and beta-thalassemia by 21-Tesla Fourier transform ion cyclotron resonance mass spectrometry. Ann Transl Med 2019, 7, S239. [Google Scholar] [CrossRef]
- Carrocini, G.C.; Zamaro, P.J.; Bonini-Domingos, C.R. What influences Hb fetal production in adulthood? Rev Bras Hematol Hemoter 2011, 33, 231–236. [Google Scholar] [CrossRef]
- Hempe, J.M.; Craver, R.D. Laboratory Diagnosis of Structural Hemoglobinopathies and Thalassemias by Capillary Isoelectric Focusing. In Clinical Applications of Capillary Electrophoresis, Humana Press: pp. 81-98. [CrossRef]
- Little, R.R.; Roberts, W.L. A review of variant hemoglobins interfering with hemoglobin A1c measurement. J Diabetes Sci Technol 2009, 3, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Thom, C.S.; Dickson, C.F.; Gell, D.A.; Weiss, M.J. Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harb Perspect Med 2013, 3, a011858. [Google Scholar] [CrossRef] [PubMed]
- Masson, E.; Zou, W.B.; Genin, E.; Cooper, D.N.; Le Gac, G.; Fichou, Y.; Pu, N.; Rebours, V.; Ferec, C.; Liao, Z.; et al. Expanding ACMG variant classification guidelines into a general framework. Hum Genomics 2022, 16, 31. [Google Scholar] [CrossRef]
- Carss, K.; Goldstein, D.; Aggarwal, V.; Petrovski, S. Variant Interpretation and Genomic Medicine. Handbook of Statistical Genomics 2019, 25. [CrossRef]
- Chen, J.-M.; Masson, E.; Zou, W.-B.; Liao, Z.; Génin, E.; Cooper, D.N.; Férec, C. Validation of the ACMG/AMP guidelines-based seven-category variant cla ssification system. [CrossRef]
- Walsh, N.; Cooper, A.; Dockery, A.; O'Byrne, J.J. Variant reclassification and clinical implications. J Med Genet 2024, 61, 207–211. [Google Scholar] [CrossRef]
- Sabath, D.E. Molecular Diagnosis of Thalassemias and Hemoglobinopathies. American Journal of Clinical Pathology 148, 6-15. [CrossRef]
- Kountouris, P.; Stephanou, C.; Lederer, C.W.; Traeger-Synodinos, J.; Bento, C.; Harteveld, C.L.; Fylaktou, E.; Koopmann, T.T.; Halim-Fikri, H.; Michailidou, K.; et al. Adapting the ACMG/AMP variant classification framework: A perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Hum Mutat 2022, 43, 1089–1096. [Google Scholar] [CrossRef]
- Turner, S.A.; Rao, S.K.; Morgan, R.H.; Vnencak-Jones, C.L.; Wiesner, G.L. The impact of variant classification on the clinical management of hereditary cancer syndromes. Genet Med 2019, 21, 426–430. [Google Scholar] [CrossRef]
- David, S.; Nora Syahirah, S.; Muhammad Nur Salam Bin, H.; Rajan, R. The Blood Blues: A Review on Methemoglobinemia. Journal of Pharmacology and Pharmacotherapeutics 2018, 9, 5. [Google Scholar] [CrossRef]
- Boylston, M.; Beer, D. Methemoglobinemia: a case study. Crit Care Nurse 2002, 22, 50–55. [Google Scholar] [CrossRef]
- Ashurst, J.; Wasson, M. Methemoglobinemia: a systematic review of the pathophysiology, detecti on, and treatment. Delaware medical journal 2011, 83, 5. [Google Scholar]
- do Nascimento, T.S.; Pereira, R.O.; de Mello, H.L.; Costa, J. Methemoglobinemia: from diagnosis to treatment. Rev Bras Anestesiol 2008, 58, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Iolascon, A.; Bianchi, P.; Andolfo, I.; Russo, R.; Barcellini, W.; Fermo, E.; Toldi, G.; Ghirardello, S.; Rees, D.; Van Wijk, R.; et al. Recommendations for diagnosis and treatment of methemoglobinemia. Am J Hematol 2021, 96, 1666–1678. [Google Scholar] [CrossRef] [PubMed]
- Ash-Bernal, R.; Wise, R.; Wright, S.M. Acquired methemoglobinemia: a retrospective series of 138 cases at 2 teaching hospitals. Medicine (Baltimore) 2004, 83, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Friedman, N.; Scolnik, D.; McMurray, L.; Bryan, J. Acquired methemoglobinemia presenting to the pediatric emergency department: a clinical challenge. CJEM 2020, 22, 673–677. [Google Scholar] [CrossRef]
- Li, W. Biophysical Basis of Hb-S Polymerization in Red Blood Cell Sickling. bioRxiv 2019. [CrossRef]
- Karen, C. Sickle Cell Disease: A Genetic Disorder of Beta-Globin. Thalassemia and Other Hemolytic Anemias 2018. [CrossRef]
- Ellsworth, P.; Sparkenbaugh, E.M. Targeting the von Willebrand Factor-ADAMTS-13 axis in sickle cell disease. J Thromb Haemost 2023, 21, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Nader, E.; Romana, M.; Connes, P. The Red Blood Cell-Inflammation Vicious Circle in Sickle Cell Disease. Front Immunol 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Ashley-Koch, A.; Yang, Q.; Olney, R.S. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol 2000, 151, 839–845. [Google Scholar] [CrossRef]
- Al-Fatlawi, A.C.Y. A Review on Sickle Cells Disease. SJMR 2019, 03, 146–148. [Google Scholar] [CrossRef]
- Poli, M.C.; Orange, J. CRISPR/Cas9 β-globin Gene Targeting in Human Haematopoietic Stem Cells. Pediatrics 2017, 140, S226–S227. [Google Scholar] [CrossRef]
- Demirci, S.; Gudmundsdottir, B.; Li, Q.; Haro-Mora, J.J.; Nassehi, T.; Drysdale, C.; Yapundich, M.; Gamer, J.; Seifuddin, F.; Tisdale, J.F.; et al. βT87Q-Globin Gene Therapy Reduces Sickle Hemoglobin Production, Allowi ng for Ex Vivo Anti-sickling Activity in Human Erythroid Cells. Molecular Therapy - Methods & Clinical Development 17, 912-921. [CrossRef]
- De Simone, G.; Quattrocchi, A.; Mancini, B.; di Masi, A.; Nervi, C.; Ascenzi, P. Thalassemias: from gene to therapy. Mol Aspects Med 2022, 84, 101028. [Google Scholar] [CrossRef]
- Angastiniotis, M.; Lobitz, S. Thalassemias: An Overview. Int J Neonatal Screen 2019, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Weatherall, D.J. The thalassaemias. BMJ 1997, 314, 1675–1678. [Google Scholar] [CrossRef] [PubMed]
- Muncie, H.; James, C. Alpha and beta thalassemia. American Family Physician 2009, 80, 339–344. [Google Scholar] [PubMed]
- Vernimmen, D. Globins, from Genes to Physiology and Diseases. Blood Cells Mol Dis 2018, 70, 1. [Google Scholar] [CrossRef] [PubMed]
- Aksu, T.; Ş, Ü. Thalassemia. Trends in Pediatrics . 2021. [CrossRef]
- Ahmed Meri, M.; Hamid Al-Hakeem, A.; Saad Al-Abeadi, R. Overview on Thalassemia: A Review Article. Med.Sci.Jour.Adv.Res 2022, 3, 26–32. [Google Scholar] [CrossRef]
- Bibek, K.; Jha, S.; Eiman, A.A.Z. Hemoglobin C Disease. Definitions 2023. [Google Scholar]
- Charache, S.; Conley, C.L.; Waugh, D.F.; Ugoretz, R.J.; Spurrell, J.R. Pathogenesis of hemolytic anemia in homozygous hemoglobin C disease. J Clin Invest 1967, 46, 1795–1811. [Google Scholar] [CrossRef]
- Reiss, G.H.; Ranney, H.M.; Shaklai, N. Association of hemoglobin C with erythrocyte ghosts. J Clin Invest 1982, 70, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Verra, F.; Simpore, J.; Warimwe, G.M.; Tetteh, K.K.; Howard, T.; Osier, F.H.; Bancone, G.; Avellino, P.; Blot, I.; Fegan, G.; et al. Haemoglobin C and S role in acquired immunity against Plasmodium falciparum malaria. PLoS One 2007, 2, e978. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.L.; Randolph, T.R. Development of a Microscopic Method to Identify Hemoglobin C Conditions for Use in Developing Countries. Clin Lab Sci 2023, 32, 61–66. [Google Scholar] [CrossRef]
- Ohiri, C.D.; Randolph, T.R. Development of an easy, inexpensive, and precise method to identify Hemoglobin C for use in underdeveloped countries. The FASEB Journal 2016, 30. [Google Scholar] [CrossRef]
- Ghosh, A.; Basak, J.; Mukhopadhyay, A. Coexistence of rare variant HbD Punjab [alpha2beta2(121(Glu-->Gln))] and alpha 3.7 kb deletion in a young boy of Hindu family in West Bengal, India. Cell Mol Biol Lett 2015, 20, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Seth, T.; Tyagi, S. Review of Clinical and Hematological Profile of Hemoglobin D Cases in a Single Centre. Journal of Marine Medical Society 2023, 25, S74–S79. [Google Scholar] [CrossRef]
- Pandey, S.; Mishra, R.M.; Pandey, S.; Shah, V.; Saxena, R. Molecular characterization of hemoglobin D Punjab traits and clinical-hematological profile of the patients. Sao Paulo Med J 2012, 130, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Italia, K.; Upadhye, D.; Dabke, P.; Kangane, H.; Colaco, S.; Sawant, P.; Nadkarni, A.; Gorakshakar, A.; Jain, D.; Italia, Y.; et al. Clinical and hematological presentation among Indian patients with common hemoglobin variants. Clin Chim Acta 2014, 431, 46–51. [Google Scholar] [CrossRef]
- Oberoi, S.; Das, R.; Trehan, A.; Ahluwalia, J.; Bansal, D.; Malhotra, P.; Marwaha, R.K. HbSD-Punjab: clinical and hematological profile of a rare hemoglobinopathy. J Pediatr Hematol Oncol 2014, 36, e140–144. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.F. Globin chain synthesis in HbD (Punjab)-beta-thalassemia. Blood 47, 113-120. [CrossRef]
- Petrenko, A.A.; Pivnik, A.V.; Kim, P.P.; Demidova, E.Y.; Surin, V.L.; Abdullaev, A.O.; Sudarikov, A.B.; Petrova, N.A.; Maryina, S.A. Coinheritance of HbD-Punjab/beta+-thalassemia (IVSI+5 G-C) in patient with Gilbert's syndrome. Ter Arkh 2018, 90, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, J.; Naka, I.; Patarapotikul, J.; Hananantachai, H.; Brittenham, G.; Looareesuwan, S.; Clark, A.G.; Tokunaga, K. Extended linkage disequilibrium surrounding the hemoglobin E variant due to malarial selection. Am J Hum Genet 2004, 74, 1198–1208. [Google Scholar] [CrossRef]
- Flatz, G.; Sanguansermsri, T.; Sengchanh, S.; Horst, D.; Horst, J. The ‘Hot Spot’ of Hb E [β26(B8)Glu→Lys] in Southeast Asia: β-Globin An omalies in the Lao Theung Population of Southern Laos. Hemoglobin 28, 197-204. [CrossRef]
- Camille, J.R.; Malashkevich, V.; Tatiana, C.B.; Dantsker, D.; Qiu-Mao, C.; Juan, C.M.; Almo, S.; Friedman, J.; Hirsch, R.E. Structural and Functional Studies Indicating Altered Redox Properties of Hemoglobin E. Journal of Biological Chemistry . [CrossRef]
- Strader, M.B.; Kassa, T.; Meng, F.; Wood, F.B.; Hirsch, R.E.; Friedman, J.M.; Alayash, A.I. Oxidative instability of hemoglobin E (beta26 Glu-->Lys) is increased in the presence of free alpha subunits and reversed by alpha-hemoglobin stabilizing protein (AHSP): Relevance to HbE/beta-thalassemia. Redox Biol 2016, 8, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Jamsai, D.; Zaibak, F.; Vadolas, J.; Voullaire, L.; Fowler, K.J.; Gazeas, S.; Peters, H.; Fucharoen, S.; Williamson, R.; Ioannou, P.A. A humanized BAC transgenic/knockout mouse model for HbE/beta-thalassemia. Genomics 2006, 88, 309–315. [Google Scholar] [CrossRef]
- Naka, I.; Ohashi, J.; Nuchnoi, P.; Hananantachai, H.; Looareesuwan, S.; Tokunaga, K.; Patarapotikul, J. Lack of association of the HbE variant with protection from cerebral malaria in Thailand. Biochem Genet 2008, 46, 708–711. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhyay, M.; Jha, A.K.; Karmakar, S.; Sengupta, M.; Basu, K.; Das, M.; Mukherjee, S. HbE Variants: An Experience from Tertiary Care Centre of Eastern India. Annals of Pathology and Laboratory Medicine 2020, 7, A570–575. [Google Scholar] [CrossRef]
- Winterbourn, C. Oxidative denaturation in congenital hemolytic anemias: the unstable hemoglobins. Proceedings of Seminars in hematology; p. 41.
- Kidd, R.D.; Baker, H.M.; Mathews, A.J.; Brittain, T.; Baker, E.N. Oligomerization and ligand binding in a homotetrameric hemoglobin: Two high-resolution crystal structures of hemoglobin Bart's (γ4), a marker for α-thalassemia. Protein Science 10, 1739-1749. [CrossRef]
- Czerwinski, E.W.; Risk, M.; Matustik, M.C. Crystallization and preliminary X-ray diffraction studies of methemogl obin Bart's. Journal of Biological Chemistry 256, 13128-13129. [CrossRef]
- Rugless, M.J.; Fisher, C.A.; Stephens, A.D.; Amos, R.J.; Mohammed, T.; Old, J.M. Hb Bart's in Cord Blood: An Accurate Indicator of α-Thalassemia. Hemoglobin 30, 57-62. [CrossRef]
- Papassotiriou, I.; Traeger-Synodinos, J.; Vlachou, C.; Karagiorga, M.; Metaxotou, A.; Kanavakis, E.; Stamoulakatou, A. Rapid and accurate quantitation of Hb Bart's and Hb H using weak cation exchange high performance liquid chromatography: correlation with the alpha-thalassemia genotype. Hemoglobin 1999, 23, 203–211. [Google Scholar] [CrossRef]
- Pootrakul, S.; Wasi, P.; Na-Nakorn, S. Haemoglobin Bart's hydrops foetalis in Thailand. Ann Hum Genet 1967, 30, 293–311. [Google Scholar] [CrossRef] [PubMed]
- Pembrey, M.E.; Weatherall, D.J.; Clegg, J.B.; Bunch, C.; Perrine, R.P. Haemoglobin Bart's in Saudi Arabia. British Journal of Haematology 29, 221-234. [CrossRef]
- Sasazuki, T.; Isomoto, A.; Nakajima, H. Circular dichroism and absorption spectra of haemoglobin Bart's. Journal of molecular biology 65, 365-369. [CrossRef]
- Folayan Esan, G.J. Haemoglobin Bart's in Newborn Nigerians. British Journal of Haematology 22, 73-86. [CrossRef]
- Jones, C.M.; Charache, S.; Hathaway, P.J. The effect of hemoglobin F-Chesapeake (alpha 2 92 Arg. leads to Leu gamma 2) on fetal oxygen affinity and erythropoiesis. Pediatr Res 1979, 13, 851–853. [Google Scholar] [CrossRef]
- Gibson, Q.H.; Nagel, R.L. Allosteric Transition and Ligand Binding in Hemoglobin Chesapeake. Journal of Biological Chemistry 249, 7255-7259. [CrossRef]
- Taketa, F.; Huang, Y.; Libnoch, J.; Dessel, B. Hemoglobin Wood beta97(FG4) His replaced by Leu. A new high-oxygen-aff inity hemoglobin associated with familial erythrocytosis. Biochimica et biophysica acta . [CrossRef]
- Charache, S. A manifestation of abnormal hemoglobins of man: altered oxygen affinity. Hemoglobin Chesapeake: from the clinic to the laboratory, and back again. Ann N Y Acad Sci 1974, 241, 449–455. [Google Scholar] [CrossRef]
- Reed, C.S.; Hampson, R.; Gordon, S.; Jones, R.T.; Novy, M.J.; Brimhall, B.; Edwards, M.J.; Koler, R.D. Erythrocytosis Secondary to Increased Oxygen Affinity of a Mutant Hemo globin, Hemoglobin Kempsey. Blood 31, 623-632. [CrossRef]
- Jones, C.M.; Charache, S.; Hathaway, P. The effect of hemoglobin F-Chesapeake (alpha 2 92 Arg. leads to Leu ga mma 2) on fetal oxygen affinity and erythropoiesis. Pediatric Research 1979, 851-853.
- Wajcman, H.; Galactéros, F. Hemoglobins With High Oxygen Affinity Leading to Erythrocytosis. New V ariants and New Concepts. Hemoglobin 29, 91-106. [CrossRef]
- Pirastru, M.; Manca, L.; Trova, S.; Mereu, P. Biochemical and Molecular Analysis of the Hb Lepore Boston Washington in a Syrian Homozygous Child. Biomed Res Int 2017, 2017, 1261972. [Google Scholar] [CrossRef] [PubMed]
- Harteveld, C.L.; Wijermans, P.W.; Arkesteijn, S.G.; Van Delft, P.; Kerkhoffs, J.L.; Giordano, P.C. Hb Lepore-Leiden: a new delta/beta rearrangement associated with a beta-thalassemia minor phenotype. Hemoglobin 2008, 32, 446–453. [Google Scholar] [CrossRef]
- Seward, D.P.; Ware, R.E.; Kinney, T.R. Hemoglobin sickle-Lepore: report of two siblings and review of the literature. Am J Hematol 1993, 44, 192–195. [Google Scholar] [CrossRef]
- Mirabile, E.; Testa, R.; Consalvo, C.; Dickerhoff, R.; Schilirò, G. Association of Hb S/Hb lepore and δβ-thalassemia/Hb lepore in Sicilian patients: Review of the presence of Hb lepore in Sicily. European Journal of Haematology 55, 126-130. [CrossRef]
- Efremov, G.D. Hemoglobins Lepore and anti-Lepore. Hemoglobin 1978, 2, 197–233. [Google Scholar] [CrossRef] [PubMed]
- Chaibunruang, A.; Srivorakun, H.; Fucharoen, S.; Fucharoen, G.; Sae-Ung, N.; Sanchaisuriya, K. Interactions of hemoglobin Lepore (deltabeta hybrid hemoglobin) with v arious hemoglobinopathies: A molecular and hematological characteristi cs and differential diagnosis. Blood Cells, Molecules & Diseases . [CrossRef]
- Jiang, F.; Tang, X.W.; Li, J.; Zhou, J.Y.; Zuo, L.D.; Li, D.Z. Hb Lepore-Hong Kong: First Report of a Novel delta/beta-Globin Gene Fusion in a Chinese Family. Hemoglobin 2021, 45, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.M.; Higgs, D.R.; Winichagoon, P.; Clegg, J.B.; Weatherall, D.J. Haemoglobin Constant Spring has an unstable α chain messenger RNA. British Journal of Haematology 51, 405-413. [CrossRef]
- Thonglairoam, V.; Winichagoon, P.; Fucharoen, S.; Tanphaichitr, V.S.; Pung-amritt, P.; Embury, S.H.; Wasi, P. Hemoglobin constant spring in Bangkok: molecular screening by selective enzymatic amplification of the alpha 2-globin gene. Am J Hematol 1991, 38, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.; Sanchaisuriya, K.; Wongprachum, K.; Nguyen, M.D.; Phan, T.T.; Vo, V.T.; Sanchaisuriya, P.; Fucharoen, S.; Schelp, F.P. Hemoglobin Constant Spring is markedly high in women of an ethnic minority group in Vietnam: a community-based survey and hematologic features. Blood Cells Mol Dis 2014, 52, 161–165. [Google Scholar] [CrossRef]
- Jomoui, W.; Fucharoen, G.; Sanchaisuriya, K.; Nguyen, V.H.; Fucharoen, S. Hemoglobin Constant Spring among Southeast Asian Populations: Haplotypic Heterogeneities and Phylogenetic Analysis. PLoS One 2015, 10, e0145230. [Google Scholar] [CrossRef] [PubMed]
- Kropp, G.L.; Fucharoen, S.; Embury, S.H. Selective enzymatic amplification of alpha 2-globin DNA for detection of the hemoglobin Constant Spring mutation. Blood 1989, 73, 1987–1992. [Google Scholar] [CrossRef]
- Roberts, W.L. Hemoglobin constant spring can interfere with glycated hemoglobin measurements by boronate affinity chromatography. Clin Chem 2007, 53, 142–143. [Google Scholar] [CrossRef] [PubMed]
- Charoenkwan, P.; Sirichotiyakul, S.; Chanprapaph, P.; Tongprasert, F.; Taweephol, R.; Sae-Tung, R.; Sanguansermsri, T. Anemia and hydrops in a fetus with homozygous hemoglobin constant spring. J Pediatr Hematol Oncol 2006, 28, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.A.; O'Branski, E.E.; Rosse, W.F.; Ware, R.E. Hemoglobin S/O(Arab): thirteen new cases and review of the literature. Am J Hematol 1999, 60, 279–284. [Google Scholar] [CrossRef]
- Dror, S. Clinical and hematological features of homozygous hemoglobin O-Arab [b eta 121 Glu → Lys]. Pediatric Blood & Cancer 60, 506-507. [CrossRef]
- Sangaré, A.; Sanogo, I.; Meité, M.; Ambofo, Y.; Abesopie, V.; Ségbéna, A.; Tolo, A. [Hemoglobin O Arab in Ivory Coast and western Africa]. Medecine tropicale : revue du Corps de sante colonial 52, 5.
- Hafsia, R.; Gouider, E.; Ben Moussa, S.; Ben Salah, N.; Elborji, W.; Hafsia, A. [Hemoglobin O Arab: about 20 cases]. Tunis Med 2007, 85, 637–640. [Google Scholar]
- Milner, P.F.; Miller, C.; Grey, R.; Seakins, M.; DeJong, W.W.; Went, L.N. Hemoglobin O Arab in Four Negro Families and Its Interaction with Hemo globin S and Hemoglobin C. N Engl J Med 283, 1417-1425. [CrossRef]
- El-Hazmi, M.A.F.; Lehmann, H. Human Haemoglobins and Haemoglobinopathies in Arabia: Hb O Arab in Sau di Arabia. Acta Haematol 63, 268-273. [CrossRef]
- Merritt, D.; Jones, R.T.; Head, C.; Thibodeau, S.N.; Fairbanks, V.F.; Steinberg, M.H.; Coleman, M.B.; Rodgers, G.P. Hb Seal Rock [(alpha 2)142 term-->Glu, codon 142 TAA-->GAA]: an extended alpha chain variant associated with anemia, microcytosis, and alpha-thalassemia-2 (-3.7 Kb). Hemoglobin 1997, 21, 331–344. [Google Scholar] [CrossRef]
- Prehu, C.; Moradkhani, K.; Riou, J.; Bahuau, M.; Launay, P.; Martin, N.; Wajcman, H.; Goossens, M.; Galacteros, F. Chronic hemolytic anemia due to novel alpha-globin chain variants: critical location of the mutation within the gene sequence for a dominant effect. Haematologica 2009, 94, 1624–1625. [Google Scholar] [CrossRef] [PubMed]
- Fattori, A.; Kimura, E.M.; Albuquerque, D.M.; Oliveira, D.M.; Costa, F.F.; Sonati, M.F. Hb Indianapolis [beta112 (G14) Cys-->Arg] as the probable cause of moderate hemolytic anemia and renal damage in a Brazilian patient. Am J Hematol 2007, 82, 672–675. [Google Scholar] [CrossRef]
- Henderson, S.J.; Timbs, A.T.; McCarthy, J.; Gallienne, A.E.; Proven, M.; Rugless, M.J.; Lopez, H.; Eglinton, J.; Dziedzic, D.; Beardsall, M.; et al. Ten Years of Routine alpha- and beta-Globin Gene Sequencing in UK Hemoglobinopathy Referrals Reveals 60 Novel Mutations. Hemoglobin 2016, 40, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Outeirino, J.; Casey, R.; White, J.M.; Lehmann, H. Haemoglobin Madrid β 115 (G17) Alanine→Proline: an U nstable Variant Associated with Haemolytic Anaemia. Acta Haematol 52, 53-60. [CrossRef]
- Kim, J.Y.; Park, S.S.; Jung, H.L.; Keum, D.H.; Park, H.; Chang, Y.H.; Lee, Y.J.; Cho, H.I. Hb Madrid [beta115(G17)Ala-->Pro] in a Korean family with chronic hemolytic anemia. Hemoglobin 2000, 24, 133–138. [Google Scholar] [CrossRef]
- Edison, E.S.; Shaji, R.V.; Devi, S.G.; Kumar, S.S.; Srivastava, A.; Chandy, M. Hb Showa-Yakushiji [beta110(G12)Leu-->Pro] in four unrelated patients from west Bengal. Hemoglobin 2005, 29, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Villegas, A.; Ropero, P.; Nogales, A.; González, F.A.; Mateo, M.; Mazo, E.; Rodrigo, E.; Arias, M. Hb Santander [β34(B16)Val→Asp (GTC→GAC)]: A New Unstable Variant Found as a De Novo Mutation in a Spanish Patient. Hemoglobin 27, 31-35. [CrossRef]
- Nakatsuji, T.; Miwa, S.; Ohba, Y.; Hattori, Y.; Miyaji, T.; Hino, S.; Matsumoto, N. A new unstable hemoglobin, Hb Yokohama beta 31 (B13)Leu substituting for Pro, causing hemolytic anemia. Hemoglobin 1981, 5, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Huehns, E.R.; Hecht, F.; Yoshida, A.; Stamatoyannopoulos, G.; Hartman, J.; Motulsky, A.G. Hemoglobin-Seattle (α2Aβ276 Glu): An Unstable Hemoglobin Causing Chron ic Hemolytic Anemia. Blood 36, 209-218. [CrossRef]
- Hoyer, J.D.; Baxter, J.K.; Moran, A.M.; Kubic, K.S.; Ehmann, W.C. Two unstable beta chain variants associated with beta-thalassemia: Hb Miami [beta116(G18)his-->Pro], and Hb Hershey [beta70(E14)Ala-->Gly], and a second unstable Hb variant at 170: Hb Abington [beta70(E14)Ala-->Pro]. Hemoglobin 2005, 29, 241–248. [Google Scholar] [CrossRef]
- Ideguchi, H. [Effects of abnormal Hb on red cell membranes]. Rinsho Byori 1999, 47, 232–237. [Google Scholar] [PubMed]
- Jacob, H.S.; Winterhalter, K.H. The role of hemoglobin heme loss in Heinz body formation: studies with a partially heme-deficient hemoglobin and with genetically unstable h emoglobins. Journal of Clinical Investigation 49, 2008-2016. [CrossRef]
- Jacob, H.; Winterhalter, K. Unstable Hemoglobins: The Role of Heme Loss in Heinz Body Formation. Proc. Natl. Acad. Sci. U.S.A. 65, 697-701. [CrossRef]
- Winterbourn, C.C.; Carrell, R.W. Studies of hemoglobin denaturation and Heinz body formation in the unstable hemoglobins. J Clin Invest 1974, 54, 678–689. [Google Scholar] [CrossRef]
- Sugawara, Y.; Hayashi, Y.; Shigemasa, Y.; Abe, Y.; Ohgushi, I.; Ueno, E.; Shimamoto, F. Molecular biosensing mechanisms in the spleen for the removal of aged and damaged red cells from the blood circulation. Sensors (Basel) 2010, 10, 7099–7121. [Google Scholar] [CrossRef] [PubMed]
- Simmers, R.N.; Mulley, J.C.; Hyland, V.J.; Callen, D.F.; Sutherland, G.R. Mapping the human alpha globin gene complex to 16p13.2----pter. J Med Genet 1987, 24, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, R.D.; Jonasson, J.A.; McGee, J.O.; Patil, S.; Ionasescu, V.V.; Weatherall, D.J.; Higgs, D.R. High resolution gene mapping of the human alpha globin locus. J Med Genet 1987, 24, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.J.; Peden, J.F.; Lloyd, C.; Horsley, S.W.; Clark, K.; Tufarelli, C.; Kearney, L.; Buckle, V.J.; Doggett, N.A.; Flint, J.; et al. Sequence, structure and pathology of the fully annotated terminal 2 Mb of the short arm of human chromosome 16. Hum Mol Genet 2001, 10, 339–352. [Google Scholar] [CrossRef]
- Higgs, D.R.; Wainscoat, J.S.; Flint, J.; Hill, A.V.; Thein, S.L.; Nicholls, R.D.; Teal, H.; Ayyub, H.; Peto, T.E.; Falusi, A.G.; et al. Analysis of the human alpha-globin gene cluster reveals a highly informative genetic locus. Proc Natl Acad Sci U S A 1986, 83, 5165–5169. [Google Scholar] [CrossRef] [PubMed]
- Coelho, A.; Picanco, I.; Seuanes, F.; Seixas, M.T.; Faustino, P. Novel large deletions in the human alpha-globin gene cluster: Clarifying the HS-40 long-range regulatory role in the native chromosome environment. Blood Cells Mol Dis 2010, 45, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Higgs, D.R.; Hill, A.V.; Nicholls, R.; Goodbourn, S.E.; Ayyub, H.; Teal, H.; Clegg, J.B.; Weatherall, D.J. Molecular rearrangements of the human alpha-globin gene cluster. Annals of the New York Academy of Sciences 1985, 445, 45–56. [Google Scholar] [CrossRef]
- Hatton, C.S.; Wilkie, A.O.; Drysdale, H.C.; Wood, W.G.; Vickers, M.A.; Sharpe, J.; Ayyub, H.; Pretorius, I.M.; Buckle, V.J.; Higgs, D.R. Alpha-thalassemia caused by a large (62 kb) deletion upstream of the human alpha globin gene cluster. Blood 1990, 76, 221–227. [Google Scholar] [CrossRef]
- Steinberg, M.H. A New Trans-Acting Modulator of Fetal Hemoglobin? Acta Haematol 2018, 140, 112–113. [Google Scholar] [CrossRef]
- Cao, A.; Galanello, R. Beta-thalassemia. Genetics in Medicine 12, 61-76. [CrossRef]
- Ana, M.; Seixas, S.; Lopes, A.; Bento, C.; Prata, M.; Amorim, A. Evolutionary Constraints in the β-Globin Cluster: The Signature of Pur ifying Selection at the δ-Globin (HBD) Locus and Its Role in Developme ntal Gene Regulation. Genome Biology and Evolution . [CrossRef]
- Lin, C.C.; Draper, P.N.; De Braekeleer, M. High-resolution chromosomal localization of the β-gene of the human β- globin gene complex by in situ hybridization. Cytogenet Genome Res 39, 269-274. [CrossRef]
- Gusella, J.; Varsanyi-Breiner, A.; Kao, F.T.; Jones, C.; Puck, T.T.; Keys, C.; Orkin, S.; Housman, D. Precise localization of human beta-globin gene complex on chromosome 11. Proc Natl Acad Sci U S A 1979, 76, 5239–5242. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, A.; Nienhuis, A.; Lawrence, J.; Giles, R.; Turner, P.; Ruddle, F.H. Chromosomal localization of human beta globin gene on human chromosome 11 in somatic cell hybrids. Proc Natl Acad Sci U S A 1978, 75, 1456–1460. [Google Scholar] [CrossRef]
- Bauer, D.E.; Orkin, S.H. Update on fetal hemoglobin gene regulation in hemoglobinopathies. Curr Opin Pediatr 2011, 23, 1–8. [Google Scholar] [CrossRef]
- Manning, J.M.; Dumoulin, A.; Li, X.; Manning, L.R. Normal and abnormal protein subunit interactions in hemoglobins. J Biol Chem 1998, 273, 19359–19362. [Google Scholar] [CrossRef] [PubMed]
- Kidd, R.D.; Russell, J.E.; Watmough, N.J.; Baker, E.N.; Brittain, T. The role of beta chains in the control of the hemoglobin oxygen binding function: chimeric human/mouse proteins, structure, and function. Biochemistry 2001, 40, 15669–15675. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Pang, J.; Reddy, K.S.; Surrey, S.; Adachi, K. Role of beta112 Cys (G14) in homo- (beta4) and hetero- (alpha2 beta2) tetramer hemoglobin formation. J Biol Chem 1998, 273, 14179–14185. [Google Scholar] [CrossRef] [PubMed]
- Borgstahl, G.; Rogers, P.; Arnone, A. The 1.8 A structure of carbonmonoxy-beta 4 hemoglobin. Analysis of a h omotetramer with the R quaternary structure of liganded alpha 2 beta 2 hemoglobin. Journal of molecular biology . [CrossRef]
- Walder, J.A.; Chatterjee, R.; Steck, T.L.; Low, P.S.; Musso, G.F.; Kaiser, E.T.; Rogers, P.H.; Arnone, A. The interaction of hemoglobin with the cytoplasmic domain of band 3 of the human erythrocyte membrane. The Journal of biological chemistry 1984, 259, 10238–10246. [Google Scholar] [CrossRef] [PubMed]
- Gell, D.; Kong, Y.; Eaton, S.A.; Weiss, M.J.; Mackay, J.P. Biophysical Characterization of the α-Globin Binding Protein α-Hemoglo bin Stabilizing Protein. Journal of Biological Chemistry 277, 40602-40609. [CrossRef]
- Domingues-Hamdi, E.; Vasseur, C.; Fournier, J.B.; Marden, M.C.; Wajcman, H.; Baudin-Creuza, V. Role of alpha-globin H helix in the building of tetrameric human hemoglobin: interaction with alpha-hemoglobin stabilizing protein (AHSP) and heme molecule. PLoS One 2014, 9, e111395. [Google Scholar] [CrossRef]
- Feng, L.; Gell, D.A.; Zhou, S.; Gu, L.; Kong, Y.; Li, J.; Hu, M.; Yan, N.; Lee, C.; Rich, A.M.; et al. Molecular mechanism of AHSP-mediated stabilization of alpha-hemoglobin. Cell 2004, 119, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Kong, Y.; Dore, L.C.; Abdulmalik, O.; Katein, A.M.; Zhou, S.; Choi, J.K.; Gell, D.; Mackay, J.P.; Gow, A.J.; et al. An erythroid chaperone that facilitates folding of alpha-globin subunits for hemoglobin synthesis. J Clin Invest 2007, 117, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Mollan, T.L.; Yu, X.; Weiss, M.J.; Olson, J.S. The Role of Alpha-Hemoglobin Stabilizing Protein in Redox Chemistry, D enaturation, and Hemoglobin Assembly. Antioxidants & Redox Signaling 12, 219-231. [CrossRef]
- Yu, X.; Mollan, T.L.; Butler, A.; Gow, A.J.; Olson, J.S.; Weiss, M.J. Analysis of human alpha globin gene mutations that impair binding to the alpha hemoglobin stabilizing protein. Blood 2009, 113, 5961–5969. [Google Scholar] [CrossRef]
- Voon, H.P.; Vadolas, J. Controlling alpha-globin: a review of alpha-globin expression and its impact on beta-thalassemia. Haematologica 2008, 93, 1868–1876. [Google Scholar] [CrossRef]
- Ronquist, G.; Theodorsson, E. Inherited, non-spherocytic haemolysis due to deficiency of glucose-6-phosphate dehydrogenase. Scand J Clin Lab Invest 2007, 67, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.Y.; Cheng, M.L.; Chiu, D.T. Glucose-6-phosphate dehydrogenase--from oxidative stress to cellular functions and degenerative diseases. Redox Rep 2007, 12, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Arese, P.; Gallo, V.; Pantaleo, A.; Turrini, F. Life and Death of Glucose-6-Phosphate Dehydrogenase (G6PD) Deficient E rythrocytes – Role of Redox Stress and Band 3 Modifications. Transfusion Medicine and Hemotherapy . [CrossRef]
- Janney, S.K.; Joist, J.J.; Fitch, C.D. Excess release of ferriheme in G6PD-deficient erythrocytes: possible cause of hemolysis and resistance to malaria. Blood 1986, 67, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.D.; Zuo, L.; Lubin, B.H.; Chiu, D.T. NADPH, not glutathione, status modulates oxidant sensitivity in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Blood 1991, 77, 2059–2064. [Google Scholar] [CrossRef]
- Nicol, C.J.; Zielenski, J.; Tsui, L.C.; Wells, P.G. An embryoprotective role for glucose-6-phosphate dehydrogenase in deve lopmental oxidative stress and chemical teratogenesis. The FASEB Journal 14, 111-127. [CrossRef]
- Francis, R.O.; Jhang, J.S.; Pham, H.P.; Hod, E.A.; Zimring, J.C.; Spitalnik, S.L. Glucose-6-phosphate dehydrogenase deficiency in transfusion medicine: the unknown risks. Vox Sang 2013, 105, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Fibach, E.; Rachmilewitz, E. The role of oxidative stress in hemolytic anemia. Curr Mol Med 2008, 8, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Haghpanah, S.; Hosseini-Bensenjan, M.; Zekavat, O.R.; Bordbar, M.; Karimi, M.; Ramzi, M.; Asmarian, N. The Effect of N-Acetyl Cysteine and Vitamin E on Oxidative Status and Hemoglobin Level in Transfusion-Dependent Thalassemia Patients: A Syst ematic Review and Meta-Analysis. 2021. [CrossRef]
- Halima, W.M.A.B.; Hannemann, A.; Rees, D.; Brewin, J.; Gibson, J. The Effect of Antioxidants on the Properties of Red Blood Cells From P atients With Sickle Cell Anemia. Frontiers in Physiology.
- Pallotta, V.; Gevi, F.; D'Alessandro, A.; Zolla, L. Storing red blood cells with vitamin C and N-acetylcysteine prevents oxidative stress-related lesions: a metabolomics overview. Blood Transfus 2014, 12, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Delesderrier, E.; Curioni, C.; Omena, J.; Macedo, C.R.; Cople-Rodrigues, C.; Citelli, M. Antioxidant nutrients and hemolysis in sickle cell disease. Clin Chim Acta 2020, 510, 381–390. [Google Scholar] [CrossRef]
- Fibach, E.; Rachmilewitz, E.A. The role of antioxidants and iron chelators in the treatment of oxidative stress in thalassemia. Annals of the New York Academy of Sciences 2010, 1202, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Bonkovsky, H.L.; Guo, J.T.; Hou, W.; Li, T.; Narang, T.; Thapar, M. Porphyrin and heme metabolism and the porphyrias. Compr Physiol 2013, 3, 365–401. [Google Scholar] [CrossRef]
- Bissell, D.M.; Lai, J.C.; Meister, R.K.; Blanc, P.D. Role of delta-aminolevulinic acid in the symptoms of acute porphyria. Am J Med 2015, 128, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.J. Editorial: Hematin and porphyria. N Engl J Med 1975, 293, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Mustajoki, P. Acute Porphyria: Treatment with Heme. Semin Liver Dis 18, 53-55. [CrossRef]
- Tian, Q.; Li, T.; Hou, W.; Zheng, J.; Schrum, L.W.; Bonkovsky, H.L. Lon peptidase 1 (LONP1)-dependent breakdown of mitochondrial 5-aminolevulinic acid synthase protein by heme in human liver cells. The Journal of biological chemistry 2011, 286, 26424–26430. [Google Scholar] [CrossRef] [PubMed]
- Hift, R.J.; Thunell, S.; Brun, A. Drugs in porphyria: From observation to a modern algorithm-based system for the prediction of porphyrogenicity. Pharmacol Ther 2011, 132, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Gardner, L.C.; Smith, S.J.; Cox, T.M. Biosynthesis of delta-aminolevulinic acid and the regulation of heme formation by immature erythroid cells in man. Journal of Biological Chemistry 1991, 266, 22010–22018. [Google Scholar] [CrossRef]
- Steinberg, M.H. Hydroxyurea Treatment for Sickle Cell Disease. The Scientific World JOURNAL 2, 1706-1728. [CrossRef]
- Steinberg, M.H. Therapies to increase fetal hemoglobin in sickle cell disease. Curr Hematol Rep 2003, 2, 95–101. [Google Scholar] [PubMed]
- Fathallah, H.; Atweh, G.F. Induction of fetal hemoglobin in the treatment of sickle cell disease. Hematology Am Soc Hematol Educ Program 2006, 2006, 58–62. [Google Scholar] [CrossRef]
- Cokic, V.P.; Smith, R.D.; Beleslin-Cokic, B.B.; Njoroge, J.M.; Miller, J.L.; Gladwin, M.T.; Schechter, A.N. Hydroxyurea induces fetal hemoglobin by the nitric oxide–dependent activation of soluble guanylyl cyclase. Journal of Clinical Investigation 2003, 111, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, G.P.; Dover, G.J.; Noguchi, C.T.; Schechter, A.N.; Nienhuis, A.W. Hematologic Responses of Patients with Sickle Cell Disease to Treatmen t with Hydroxyurea. N Engl J Med 322, 1037-1045. [CrossRef]
- Anderson, N. Hydroxyurea therapy: improving the lives of patients with sickle cell disease. Pediatr Nurs 2006, 32, 541–543. [Google Scholar] [PubMed]
- Goldberg, M.A.; Brugnara, C.; Dover, G.J.; Schapira, L.; Charache, S.; Bunn, H.F. Treatment of Sickle Cell Anemia with Hydroxyurea and Erythropoietin. N Engl J Med 323, 366-372. [CrossRef]
- Dover, G.; Samuel, C. Hydroxyurea induction of fetal hemoglobin synthesis in sickle-cell dis ease. Seminars in Oncology 1992, 19, 5. [Google Scholar]
- Grace, R.F.; Glenthøj, A.; Barcellini, W.; Verhovsek, M.; Rothman, J.A.; Morado, M.; Layton, D.M.; Andres, O.; Galactéros, F.; van Beers, E.J.; et al. Long-Term Hemoglobin Response and Reduction in Transfusion Burden Are Maintained in Patients with Pyruvate Kinase Deficiency Treated with Mitapivat. Blood 2022, 140, 5313–5315. [Google Scholar] [CrossRef]
- Grace, R.F.; Rose, C.; Layton, D.M.; Galacteros, F.; Barcellini, W.; Morton, D.H.; van Beers, E.J.; Yaish, H.; Ravindranath, Y.; Kuo, K.H.M.; et al. Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. N Engl J Med 2019, 381, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.H.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E.P. Results from a Phase 2 Study of Mitapivat in Adults with Non–Transfusion-Dependent Alpha- or Beta-Thalassemia. Hematology, Transfusion and Cell Therapy 2021, 43, S28. [Google Scholar] [CrossRef]
- Kuo, K.H.M.; Layton, D.M.; Lal, A.; Al-Samkari, H.; Kosinski, P.A.; Tong, B.; Estepp, J.H.; Uhlig, K.; Vichinsky, E.P. Mitapivat Improves Markers of Erythropoietic Activity in Long-Term Study of Adults with Alpha- or Beta-Non-Transfusion-Dependent Thalassemia. Blood 2022, 140, 2479–2480. [Google Scholar] [CrossRef]
- Kuo, K.; Layton, D.; Lal, A.; Al-Samkari, H.; Bhatia, J.; Kosinski, P.; Tong, B.; Lynch, M.; Uhlig, K.; Vichinsky, E. S116: Long-Term Efficacy and Safety of the Oral Pyruvate Kinase Activator Mitapivat in Adults with Non—Transfusion-Dependent Alpha- or Beta-Thalassemia. HemaSphere 2022, 6, 8–9. [Google Scholar] [CrossRef]
- Xu, J.Z.; Conrey, A.; Frey, I.; Gwaabe, E.; Menapace, L.A.; Tumburu, L.; Lundt, M.; Li, Q.; Glass, K.; Iyer, V.; et al. Mitapivat (AG-348) Demonstrates Safety, Tolerability, and Improvements in Anemia, Hemolysis, Oxygen Affinity, and Hemoglobin S Polymerization Kinetics in Adults with Sickle Cell Disease: A Phase 1 Dose Escalation Study. Blood 2021, 138, 10–10. [Google Scholar] [CrossRef]
- Pilo, F.; Angelucci, E. Mitapivat for sickle cell disease and thalassemia. Drugs Today (Barc) 2023, 59, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Cappellini, M.D. Right in time: Mitapivat for the treatment of anemia in alpha- and beta-thalassemia. Cell Rep Med 2022, 3, 100790. [Google Scholar] [CrossRef] [PubMed]
- Olubiyi, O.O.; Olagunju, M.O.; Strodel, B. Rational Drug Design of Peptide-Based Therapies for Sickle Cell Disease. Molecules 2019, 24. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Ogu, U.O. Sickle cell disease in the new era: advances in drug treatment. Transfus Apher Sci 2022, 61, 103555. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Ahmad, A.; Chaudry, H.; Aiman, W.; Aamir, S.; Anwar, M.Y.; Khan, A. Efficacy and safety of recently approved drugs for sickle cell disease: a review of clinical trials. Exp Hematol 2020, 92, 11–18. [Google Scholar] [CrossRef]
- Ataga, K.I.; Desai, P.C. Advances in new drug therapies for the management of sickle cell disease. Expert Opin Orphan Drugs 2018, 6, 329–343. [Google Scholar] [CrossRef]
- Torres, L.; Conran, N. Emerging pharmacotherapeutic approaches for the management of sickle cell disease. Expert Opin Pharmacother 2019, 20, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Carden, M.A.; Little, J. Emerging disease-modifying therapies for sickle cell disease. Haematologica 2019, 104, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Ghatge, M.S.; Donkor, A.K.; Musayev, F.N.; Deshpande, T.M.; Al-Awadh, M.; Alhashimi, R.T.; Zhu, H.; Omar, A.M.; Telen, M.J.; et al. Design, Synthesis, and Investigation of Novel Nitric Oxide (NO)-Releasing Aromatic Aldehydes as Drug Candidates for the Treatment of Sickle Cell Disease. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Nakagawa, A.; Lui, F.E.; Wassaf, D.; Yefidoff-Freedman, R.; Casalena, D.; Palmer, M.A.; Meadows, J.; Mozzarelli, A.; Ronda, L.; Abdulmalik, O.; et al. Identification of a small molecule that increases hemoglobin oxygen affinity and reduces SS erythrocyte sickling. ACS chemical biology 2014, 9, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Kassa, T.; Strader, M.B.; Nakagawa, A.; Zapol, W.M.; Alayash, A.I. Targeting betaCys93 in hemoglobin S with an antisickling agent possessing dual allosteric and antioxidant effects. Metallomics 2017, 9, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Kassa, T.; Wood, F.; Strader, M.B.; Alayash, A.I. Antisickling Drugs Targeting betaCys93 Reduce Iron Oxidation and Oxidative Changes in Sickle Cell Hemoglobin. Front Physiol 2019, 10, 931. [Google Scholar] [CrossRef]
- Garel, M.C.; Domenget, C.; Caburi-Martin, J.; Prehu, C.; Galacteros, F.; Beuzard, Y. Covalent binding of glutathione to hemoglobin. I. Inhibition of hemogl obin S polymerization. Journal of Biological Chemistry 261, 14704-14709. [CrossRef]
- Garel, M.C.; Domenget, C.; Galacteros, F.; Martin-Caburi, J.; Beuzard, Y. Inhibition of erythrocyte sickling by thiol reagents. Molecular pharmacology 1984, 26, 559–565. [Google Scholar] [PubMed]
- Abdulmalik, O.; Safo, M.; Qiukan, C.; Jisheng, Y.; Brugnara, C.; Ohene-Frempong, K.; Abraham, D.; Asakura, T. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin a nd inhibits sickling of red blood cells †,‡. British Journal of Haematology . [CrossRef]
- Xu, G.G.; Pagare, P.P.; Ghatge, M.S.; Safo, R.P.; Gazi, A.; Chen, Q.; David, T.; Alabbas, A.B.; Musayev, F.N.; Venitz, J.; et al. Design, Synthesis, and Biological Evaluation of Ester and Ether Derivatives of Antisickling Agent 5-HMF for the Treatment of Sickle Cell Disease. Mol Pharm 2017, 14, 3499–3511. [Google Scholar] [CrossRef] [PubMed]
- Stern, W.; Mathews, D.; McKew, J.; Shen, X.; Kato, G.J. A Phase 1, First-in-Man, Dose-Response Study of Aes-103 (5-HMF), an Anti-Sickling, Allosteric Modifier of Hemoglobin Oxygen Affinity in Healthy Norman Volunteers. Blood 2012, 120, 3210–3210. [Google Scholar] [CrossRef]
- Lucas, A.; Ao-Ieong, E.S.Y.; Williams, A.T.; Jani, V.P.; Muller, C.R.; Yalcin, O.; Cabrales, P. Increased Hemoglobin Oxygen Affinity With 5-Hydroxymethylfurfural Supports Cardiac Function During Severe Hypoxia. Front Physiol 2019, 10, 1350. [Google Scholar] [CrossRef]
- Hannemann, A.; Cytlak, U.M.; Rees, D.C.; Tewari, S.; Gibson, J.S. Effects of 5-hydroxymethyl-2-furfural on the volume and membrane permeability of red blood cells from patients with sickle cell disease. J Physiol 2014, 592, 4039–4049. [Google Scholar] [CrossRef]
- Alhashimi, R.T.; Ghatge, M.S.; Donkor, A.K.; Deshpande, T.M.; Anabaraonye, N.; Alramadhani, D.; Danso-Danquah, R.; Huang, B.; Zhang, Y.; Musayev, F.N.; et al. Design, Synthesis, and Antisickling Investigation of a Nitric Oxide-Releasing Prodrug of 5HMF for the Treatment of Sickle Cell Disease. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Safo, M.; Abdulmalik, O.; Danso-Danquah, R.; Burnett, J.; Samuel, N.; Joshi, G.; Musayev, F.; Asakura, T.; Abraham, D. Structural basis for the potent antisickling effect of a novel class o f five-membered heterocyclic aldehydic compounds. Journal of Medicinal Chemistry 2004, 47, 4665–4676. [Google Scholar] [CrossRef] [PubMed]
- Safo, M.K.; Kato, G.J. Therapeutic strategies to alter the oxygen affinity of sickle hemoglobin. Hematol Oncol Clin North Am 2014, 28, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, C.; Fago, A.; Henkens, R.; Crumbliss, A.L. Critical Redox and Allosteric Aspects of Nitric Oxide Interactions wit h Hemoglobin. Antioxidants & Redox Signaling 6, 979-991. [CrossRef]
- Gladwin, M.T.; Ognibene, F.P.; Pannell, L.K.; Nichols, J.S.; Pease-Fye, M.E.; Shelhamer, J.H.; Schechter, A.N. Relative role of heme nitrosylation and beta-cysteine 93 nitrosation in the transport and metabolism of nitric oxide by hemoglobin in the human circulation. Proc Natl Acad Sci U S A 2000, 97, 9943–9948. [Google Scholar] [CrossRef]
- Allen, B.; Stamler, J.; Piantadosi, C. Hemoglobin, nitric oxide and molecular mechanisms of hypoxic vasodilat ion. Trends in Molecular Medicine . [CrossRef]
- Sonveaux, P.; Lobysheva, I.I.; Feron, O.; McMahon, T.J. Transport and peripheral bioactivities of nitrogen oxides carried by red blood cell hemoglobin: role in oxygen delivery. Physiology (Bethesda) 2007, 22, 97–112. [Google Scholar] [CrossRef]
- Stamler, J.S.; Jia, L.; Eu, J.P.; McMahon, T.J.; Demchenko, I.T.; Bonaventura, J.; Gernert, K.; Piantadosi, C.A. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science 1997, 276, 2034–2037. [Google Scholar] [CrossRef] [PubMed]
- Frehm, E.; Bonaventura, J.; Gow, A. Serial Review: Biomedical Implications for Hemoglobin Interactions wit h Nitric Oxide Serial Review Editors: Mark T. Gladwin and Rakesh Patel S-NITROSOHEMOGLOBIN: AN ALLOSTERIC MEDIATOR OF NO GROUP FUNCTION IN M AMMALIAN VASCULATURE. Free Radical Biology & Medicine 2004, 37, 11. [Google Scholar]
- Su, H.; Liu, X.; Du, J.; Deng, X.; Fan, Y. The role of hemoglobin in nitric oxide transport in vascular system. Medicine in Novel Technology and Devices 2020, 5. [Google Scholar] [CrossRef]
- Lancaster, J.; Hutchings, A.; Kerby, J.D.; Patel, R.P. The hemoglobin-nitric oxide axis: implications for transfusion therapeutics. Transfusion Alternatives in Transfusion Medicine 2008, 9, 273–280. [Google Scholar] [CrossRef]
- Omar, A.M.; Abdulmalik, O.; Ghatge, M.S.; Muhammad, Y.A.; Paredes, S.D.; El-Araby, M.E.; Safo, M.K. An Investigation of Structure-Activity Relationships of Azolylacryloyl Derivatives Yielded Potent and Long-Acting Hemoglobin Modulators for Reversing Erythrocyte Sickling. Biomolecules 10, 1508. [CrossRef]
- Pagare, P.P.; Ghatge, M.S.; Musayev, F.N.; Deshpande, T.M.; Chen, Q.; Braxton, C.; Kim, S.; Venitz, J.; Zhang, Y.; Abdulmalik, O.; et al. Rational design of pyridyl derivatives of vanillin for the treatment o f sickle cell disease. Bioorganic & Medicinal Chemistry 26, 2530-2538. [CrossRef]
- Pagare, P.P.; Rastegar, A.; Abdulmalik, O.; Omar, A.M.; Zhang, Y.; Fleischman, A.; Safo, M.K. Modulating hemoglobin allostery for treatment of sickle cell disease: current progress and intellectual property. Expert Opinion on Therapeutic Patents 32, 115-130. [CrossRef]
- Gopalsamy, A.; Aulabaugh, A.E.; Barakat, A.; Beaumont, K.C.; Cabral, S.; Canterbury, D.P.; Casimiro-Garcia, A.; Chang, J.S.; Chen, M.Z.; Choi, C.; et al. PF-07059013: A Noncovalent Modulator of Hemoglobin for Treatment of Si ckle Cell Disease. Journal of Medicinal Chemistry 64, 326-342. [CrossRef]
- Saunthararajah, Y. Targeting sickle cell disease root-cause pathophysiology with small molecules. Haematologica 2019, 104, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Janz, J.M.; Piotrowski, D.W.; Beaumont, K.; Chang, J.S.; Jasti, J.; Narula, J.; Sahasrabudhe, P.; Wenzel, Z.; Rao, D.; Barakat, A.; et al. A Novel Non-Covalent Modulator of Hemoglobin Improves Anemia and Reduces Sickling in a Mouse Model of Sickle Cell Disease. Blood 2019, 134, 207–207. [Google Scholar] [CrossRef]
- Oder, E.; Safo, M.K.; Abdulmalik, O.; Kato, G.J. New developments in anti-sickling agents: can drugs directly prevent the polymerization of sickle haemoglobin in vivo? Br J Haematol 2016, 175, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, B.; Chuang, C.; Dufu, K.; Patel, M.P.; Silva-Garcia, A.; Johnson, C.; Lu, Q.; Partridge, J.R.; Patskovska, L.; Patskovsky, Y.; et al. Discovery of GBT440, an Orally Bioavailable R-State Stabilizer of Sick le Cell Hemoglobin. ACS Medicinal Chemistry Letters 8, 321-326. [CrossRef]
- Kapoor, S.; Little, J.A.; Pecker, L.H. Advances in the Treatment of Sickle Cell Disease. Mayo Clin Proc 2018, 93, 1810–1824. [Google Scholar] [CrossRef]
- Archer, N.; Galacteros, F.; Brugnara, C. 2015 Clinical trials update in sickle cell anemia. American J Hematol 90, 934-950. [CrossRef]
- Deshpande, T.M.; Pagare, P.P.; Ghatge, M.S.; Chen, Q.; Musayev, F.N.; Venitz, J.; Zhang, Y.; Abdulmalik, O.; Safo, M.K. Rational modification of vanillin derivatives to stereospecifically de stabilize sickle hemoglobin polymer formation. Acta Crystallogr D Struct Biol 74, 956-964. [CrossRef]
- Scipioni, M.; Kay, G.; Megson, I.L.; Kong Thoo Lin, P. Synthesis of novel vanillin derivatives: novel multi-targeted scaffold ligands against Alzheimer's disease. Med. Chem. Commun. 10, 764-777. [CrossRef]
- Beaudry, F.; Ross, A.; Lema, P.P.; Vachon, P. Pharmacokinetics of vanillin and its effects on mechanical hypersensit ivity in a rat model of neuropathic pain. Phytotherapy Research 24, 525-530. [CrossRef]
- Ashraf, Z.; Rafiq, M.; Seo, S.-Y.; Babar, M.M.; Zaidi, N.-u.-S.S. Synthesis, kinetic mechanism and docking studies of vanillin derivativ es as inhibitors of mushroom tyrosinase. Bioorganic & Medicinal Chemistry 23, 5870-5880. [CrossRef]
- Abraham, D.J.; Mehanna, A.S.; Wireko, F.C.; Whitney, J.; Thomas, R.P.; Orringer, E.P. Vanillin, a potential agent for the treatment of sickle cell anemia. Blood 1991, 77, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Abdulmalik, O.; Pagare, P.P.; Huang, B.; Xu, G.G.; Ghatge, M.S.; Xu, X.; Chen, Q.; Anabaraonye, N.; Musayev, F.N.; Omar, A.M.; et al. VZHE-039, a novel antisickling agent that prevents erythrocyte sickling under both hypoxic and anoxic conditions. Sci Rep 2020, 10, 20277. [Google Scholar] [CrossRef] [PubMed]
- Pagare, P.P.; Ghatge, M.S.; Chen, Q.; Musayev, F.N.; Venitz, J.; Abdulmalik, O.; Zhang, Y.; Safo, M.K. Exploration of Structure-Activity Relationship of Aromatic Aldehydes Bearing Pyridinylmethoxy-Methyl Esters as Novel Antisickling Agents. J Med Chem 2020, 63, 14724–14739. [Google Scholar] [CrossRef]
- Lee, C.C.; Maestre-Reyna, M.; Hsu, K.C.; Wang, H.C.; Liu, C.I.; Jeng, W.Y.; Lin, L.L.; Wood, R.; Chou, C.C.; Yang, J.M.; et al. Crowning proteins: modulating the protein surface properties using crown ethers. Angew Chem Int Ed Engl 2014, 53, 13054–13058. [Google Scholar] [CrossRef]
- Leonard, M.; Dellacherie, E. Acylation of human hemoglobin with polyoxyethylene derivatives. Biochimica et biophysica acta 1984, 791, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Abraham, D.J.; Wireko, F.C.; Randad, R.S.; Poyart, C.; Kister, J.; Bohn, B.; Liard, J.F.; Kunert, M.P. Allosteric modifiers of hemoglobin: 2-[4-[[(3,5-disubstituted anilino)carbonyl]methyl]phenoxy]-2-methylpropionic acid derivatives that lower the oxygen affinity of hemoglobin in red cell suspensions, in whole blood, and in vivo in rats. Biochemistry 1992, 31, 9141–9149. [Google Scholar] [CrossRef]
- Sun, K.; D’Alessandro, A.; Ahmed, M.H.; Zhang, Y.; Song, A.; Ko, T.-P.; Nemkov, T.; Reisz, J.A.; Wu, H.; Adebiyi, M.; et al. Structural and Functional Insight of Sphingosine 1-Phosphate-Mediated Pathogenic Metabolic Reprogramming in Sickle Cell Disease. Scientific reports 7. [CrossRef]
- Wiesław, K.; Roth, R.; Levin, J. Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protei n that enhances LPS biological activity. Journal of Biological Chemistry . [CrossRef]
- Bahl, N.; Du, R.; Winarsih, I.; Ho, B.; Tucker-Kellogg, L.; Tidor, B.; Ding, J.L. Delineation of lipopolysaccharide (LPS)-binding sites on hemoglobin: from in silico predictions to biophysical characterization. J Biol Chem 2011, 286, 37793–37803. [Google Scholar] [CrossRef] [PubMed]
- Lechuga, G.C.; Souza-Silva, F.; Sacramento, C.Q.; Trugilho, M.R.O.; Valente, R.H.; Napoleao-Pego, P.; Dias, S.S.G.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Carels, N.; et al. SARS-CoV-2 Proteins Bind to Hemoglobin and Its Metabolites. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Nagel, R.L.; Bookchin, R.M.; Roth, E.F., Jr.; Tellez-Nagel, I. The binding of hemoglobin to membranes of normal and sickle erythrocytes. Biochimica et biophysica acta 1975, 375, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Shaklai, N.; Yguerabide, J.; Ranney, H.M. Interaction of hemoglobin with red blood cell membranes as shown by a fluorescent chromophore. Biochemistry 1977, 16, 5585–5592. [Google Scholar] [CrossRef] [PubMed]
- Yenamandra, A.; Marjoncu, D. Voxelotor: A Hemoglobin S Polymerization Inhibitor for the Treatment of Sickle Cell Disease. J Adv Pract Oncol 2020, 11, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Maggo, S.; Sadananden, U.K. Voxelotor: novel drug for sickle cell disease. International Journal of Basic & Clinical Pharmacology 2020, 9, 513. [Google Scholar] [CrossRef]
- Han, J.; Saraf, S.L.; Gordeuk, V.R. Systematic Review of Voxelotor: A First-in-Class Sickle Hemoglobin Polymerization Inhibitor for Management of Sickle Cell Disease. Pharmacotherapy 2020, 40, 525–534. [Google Scholar] [CrossRef]
- Howard, J.; Hemmaway, C.J.; Telfer, P.; Layton, D.M.; Porter, J.; Awogbade, M.; Mant, T.; Gretler, D.D.; Dufu, K.; Hutchaleelaha, A.; et al. A phase 1/2 ascending dose study and open-label extension study of voxelotor in patients with sickle cell disease. Blood 2019, 133, 1865–1875. [Google Scholar] [CrossRef]
- Hutchaleelaha, A.; Patel, M.; Washington, C.; Siu, V.; Allen, E.; Oksenberg, D.; Gretler, D.D.; Mant, T.; Lehrer-Graiwer, J. Pharmacokinetics and pharmacodynamics of voxelotor (GBT440) in healthy adults and patients with sickle cell disease. Br J Clin Pharmacol 2019, 85, 1290–1302. [Google Scholar] [CrossRef]
- Vissa, M.; Vichinsky, E. Voxelotor for the treatment of sickle cell disease. Expert Rev Hematol 2021, 14, 253–262. [Google Scholar] [CrossRef]
- Vasseur, C.; Baudin-Creuza, V. [Role of alpha-hemoglobin molecular chaperone in the hemoglobin formation and clinical expression of some hemoglobinopathies]. Transfus Clin Biol 2015, 22, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.E.; Costa, F.F. Alpha-hemoglobin-stabilizing protein: an erythroid molecular chaperone. Biochem Res Int 2011, 2011, 373859. [Google Scholar] [CrossRef]
- Gell, D.; Kong, Y.; Eaton, S.A.; Weiss, M.J.; Mackay, J.P. Biophysical characterization of the alpha-globin binding protein alpha-hemoglobin stabilizing protein. J Biol Chem 2002, 277, 40602–40609. [Google Scholar] [CrossRef]
- Weiss, M.J.; Zhou, S.; Feng, L.; Gell, D.A.; Mackay, J.P.; Shi, Y.; Gow, A.J. Role of alpha-hemoglobin-stabilizing protein in normal erythropoiesis and beta-thalassemia. Annals of the New York Academy of Sciences 2005, 1054, 103–117. [Google Scholar] [CrossRef]
- Khandros, E.; Mollan, T.L.; Yu, X.; Wang, X.; Yao, Y.; D'Souza, J.; Gell, D.A.; Olson, J.S.; Weiss, M.J. Insights into hemoglobin assembly through in vivo mutagenesis of alpha-hemoglobin stabilizing protein. J Biol Chem 2012, 287, 11325–11337. [Google Scholar] [CrossRef]
- Eggleson, K.K.; Duffin, K.L.; Goldberg, D.E. Identification and characterization of falcilysin, a metallopeptidase involved in hemoglobin catabolism within the malaria parasite Plasmodium falciparum. J Biol Chem 1999, 274, 32411–32417. [Google Scholar] [CrossRef] [PubMed]
- Murata, C.E.; Goldberg, D.E. Plasmodium falciparum falcilysin: a metalloprotease with dual specificity. The Journal of biological chemistry 2003, 278, 38022–38028. [Google Scholar] [CrossRef]
- Ralph, S.A. Subcellular multitasking - multiple destinations and roles for the Plasmodium falcilysin protease. Mol Microbiol 2007, 63, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Klemba, M.; Park, M.; Gluzman, I.Y.; Lamppa, G.K.; Goldberg, D.E. A role for falcilysin in transit peptide degradation in the Plasmodium falciparum apicoplast. Mol Microbiol 2007, 63, 314–334. [Google Scholar] [CrossRef] [PubMed]
- Chance, J.P.; Fejzic, H.; Hernandez, O.; Istvan, E.S.; Andaya, A.; Maslov, N.; Aispuro, R.; Crisanto, T.; Nguyen, H.; Vidal, B.; et al. Development of piperazine-based hydroxamic acid inhibitors against falcilysin, an essential malarial protease. Bioorg Med Chem Lett 2018, 28, 1846–1848. [Google Scholar] [CrossRef]
- Shen, F.; Zheng, G.; Setegne, M.; Tenglin, K.; Izada, M.; Xie, H.; Zhai, L.; Orkin, S.H.; Dassama, L.M.K. A cell-permeant nanobody-based degrader that induces fetal hemoglobin. [CrossRef]
- Yin, M.; Izadi, M.; Tenglin, K.; Viennet, T.; Zhai, L.; Zheng, G.; Arthanari, H.; Dassama, L.M.K.; Orkin, S.H. Evolution of nanobodies specific for BCL11A. Proc Natl Acad Sci U S A 2023, 120, e2218959120. [Google Scholar] [CrossRef] [PubMed]
- Bahl, N.; Du, R.; Winarsih, I.; Ho, B.; Tucker-Kellogg, L.; Tidor, B.; Ding, J.L. Delineation of Lipopolysaccharide (LPS)-binding Sites on Hemoglobin. Journal of Biological Chemistry 286, 37793-37803. [CrossRef]
- Fox, D.R.; Samuels, I.; Binks, S.; Grinter, R. The structure of a haemoglobin-nanobody complex reveals human beta-subunit-specific interactions. Febs Lett 2024. [CrossRef]
- Aylaz, G.; Andac, M.; Denizli, A.; Duman, M. Recognition of human hemoglobin with macromolecularly imprinted polymeric nanoparticles using non-covalent interactions. J Mol Recognit 2021, 34, e2935. [Google Scholar] [CrossRef]
- Khakurel, K.P.; Zoldak, G.; Angelov, B.; Andreasson, J. On the feasibility of time-resolved X-ray powder diffraction of macromolecules using laser-driven ultrafast X-ray sources. Journal of Applied Crystallography 2024, 57, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Khakurel, K.P.; Nemergut, M.; Dzupponova, V.; Kropielnicki, K.; Savko, M.; Zoldak, G.; Andreasson, J. Design and fabrication of 3D-printed in situ crystallization plates for probing microcrystals in an external electric field. J Appl Crystallogr 2024, 57, 842–847. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) Structure of hemoglobin (α2β2) showing its interaction with natural ligands such as oxygen, carbon dioxide, carbon monoxide, and nitric oxide, highlighted in a close-up view of the heme-binding site (b) Heme-protein interactions in hemoglobin, illustrating the key residues in the first layer (L83, L86, A65, V62, K61, H58, F46, H45, F43, Y42, T39, L136, M32, V132, L101, F98, N97, L91, H87, V93) involved in stabilizing the heme group within the protein. Asteroid plot was calculated using web server Protein Contacts Atlas (https://pca.mbgroup.bio/index.html) [21]. Created with BioRender.com.
Figure 1.
(a) Structure of hemoglobin (α2β2) showing its interaction with natural ligands such as oxygen, carbon dioxide, carbon monoxide, and nitric oxide, highlighted in a close-up view of the heme-binding site (b) Heme-protein interactions in hemoglobin, illustrating the key residues in the first layer (L83, L86, A65, V62, K61, H58, F46, H45, F43, Y42, T39, L136, M32, V132, L101, F98, N97, L91, H87, V93) involved in stabilizing the heme group within the protein. Asteroid plot was calculated using web server Protein Contacts Atlas (https://pca.mbgroup.bio/index.html) [21]. Created with BioRender.com.
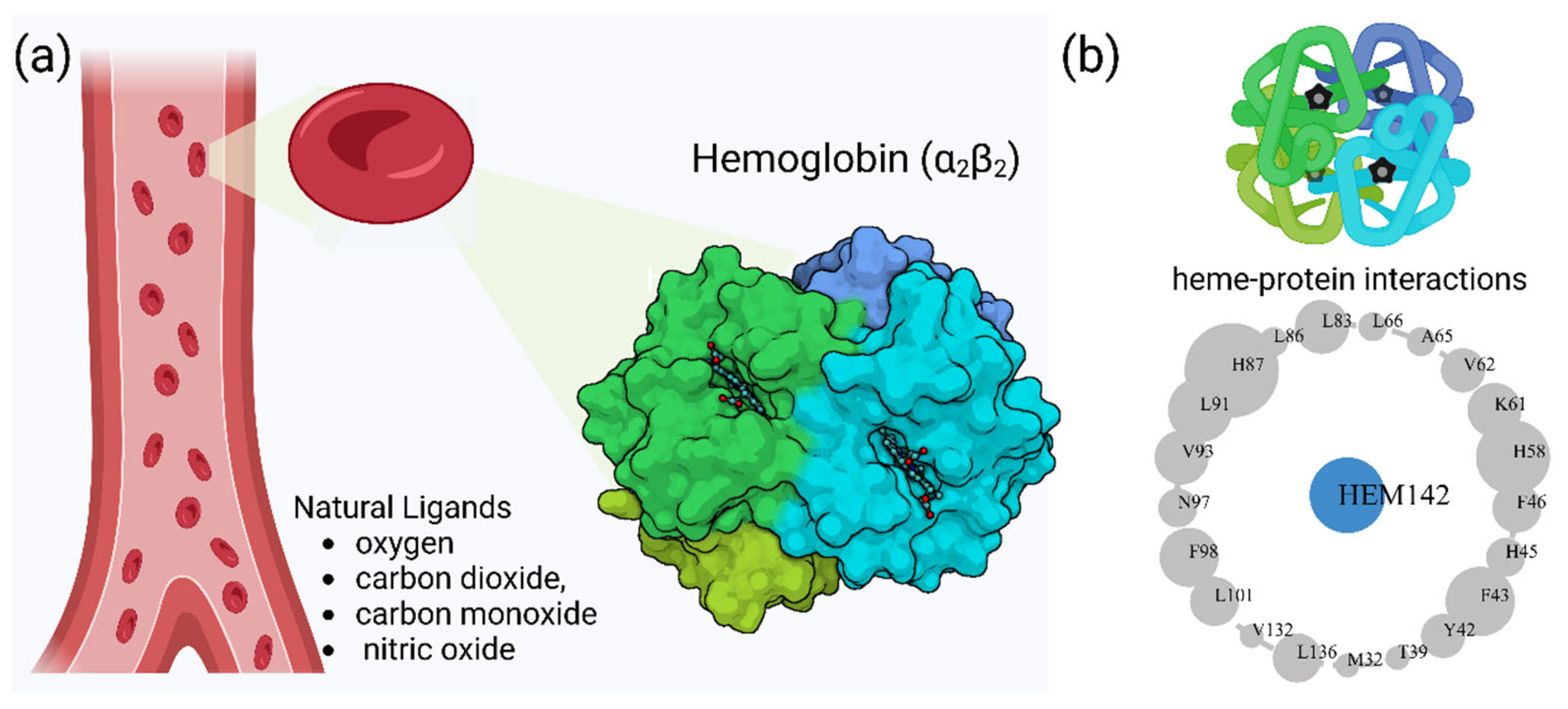
Figure 2.
(a) Depiction of hemoglobin (Hb) instability leading to the formation of Heinz bodies. (b) Distribution of 819 unstable α-globin variants, with 3.8% classified as unstable, mapped on chromosome 16 (16p13.3) and divided into clinical significance categories: benign, pathogenic, and uncertain significance. (c) Distribution of 771 unstable β-globin variants, with 11% classified as unstable, mapped on chromosome 11 (11p15.5), similarly categorized. (d) Graph showing the number of unstable variants along the amino acid sequence for both α- and β-globin chains, highlighting regions where residues contact the heme group (grey-filled circles). Created with BioRender.com.
Figure 2.
(a) Depiction of hemoglobin (Hb) instability leading to the formation of Heinz bodies. (b) Distribution of 819 unstable α-globin variants, with 3.8% classified as unstable, mapped on chromosome 16 (16p13.3) and divided into clinical significance categories: benign, pathogenic, and uncertain significance. (c) Distribution of 771 unstable β-globin variants, with 11% classified as unstable, mapped on chromosome 11 (11p15.5), similarly categorized. (d) Graph showing the number of unstable variants along the amino acid sequence for both α- and β-globin chains, highlighting regions where residues contact the heme group (grey-filled circles). Created with BioRender.com.
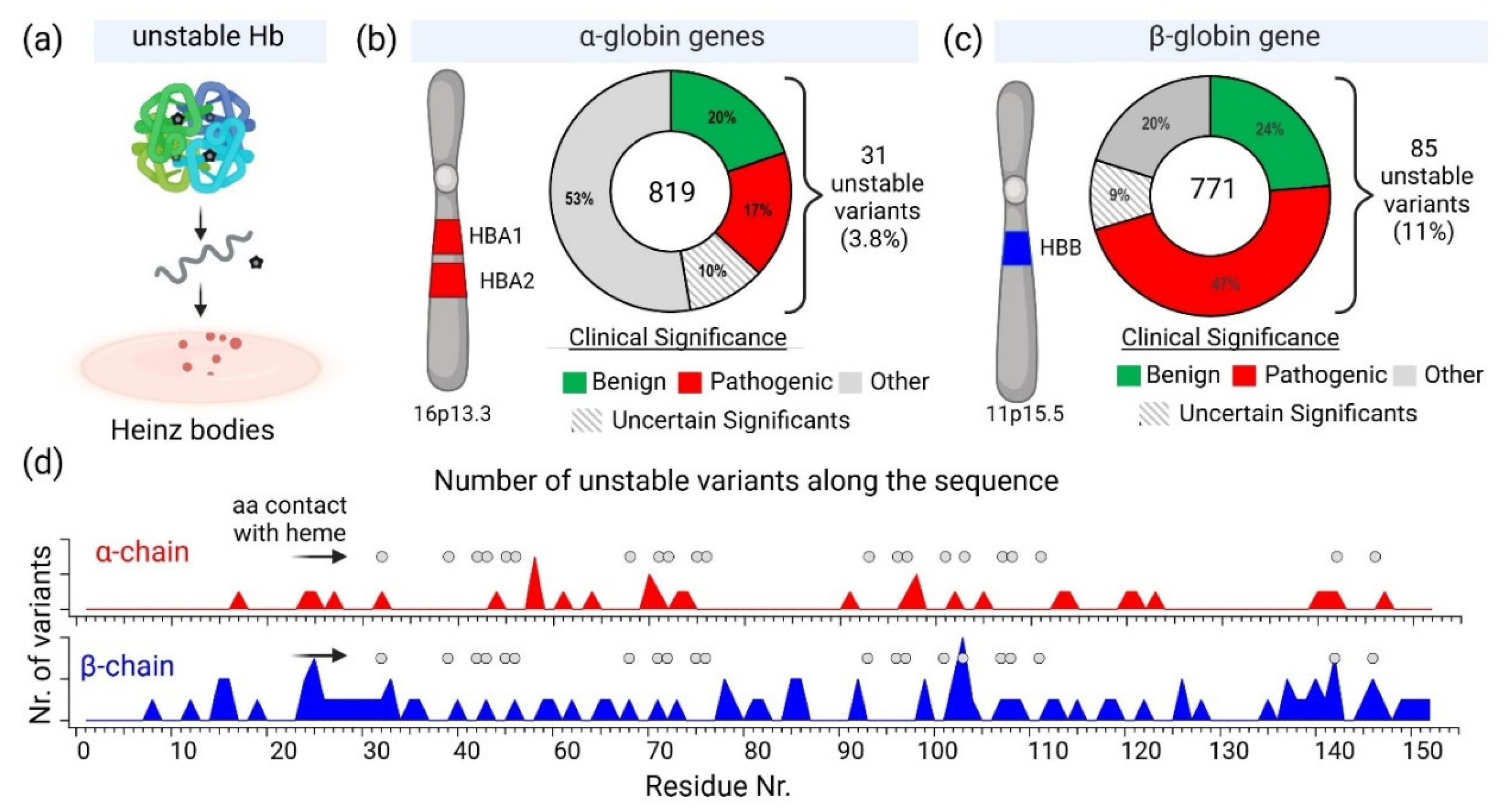
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



