
Submitted:
19 November 2024
Posted:
20 November 2024
You are already at the latest version
Abstract
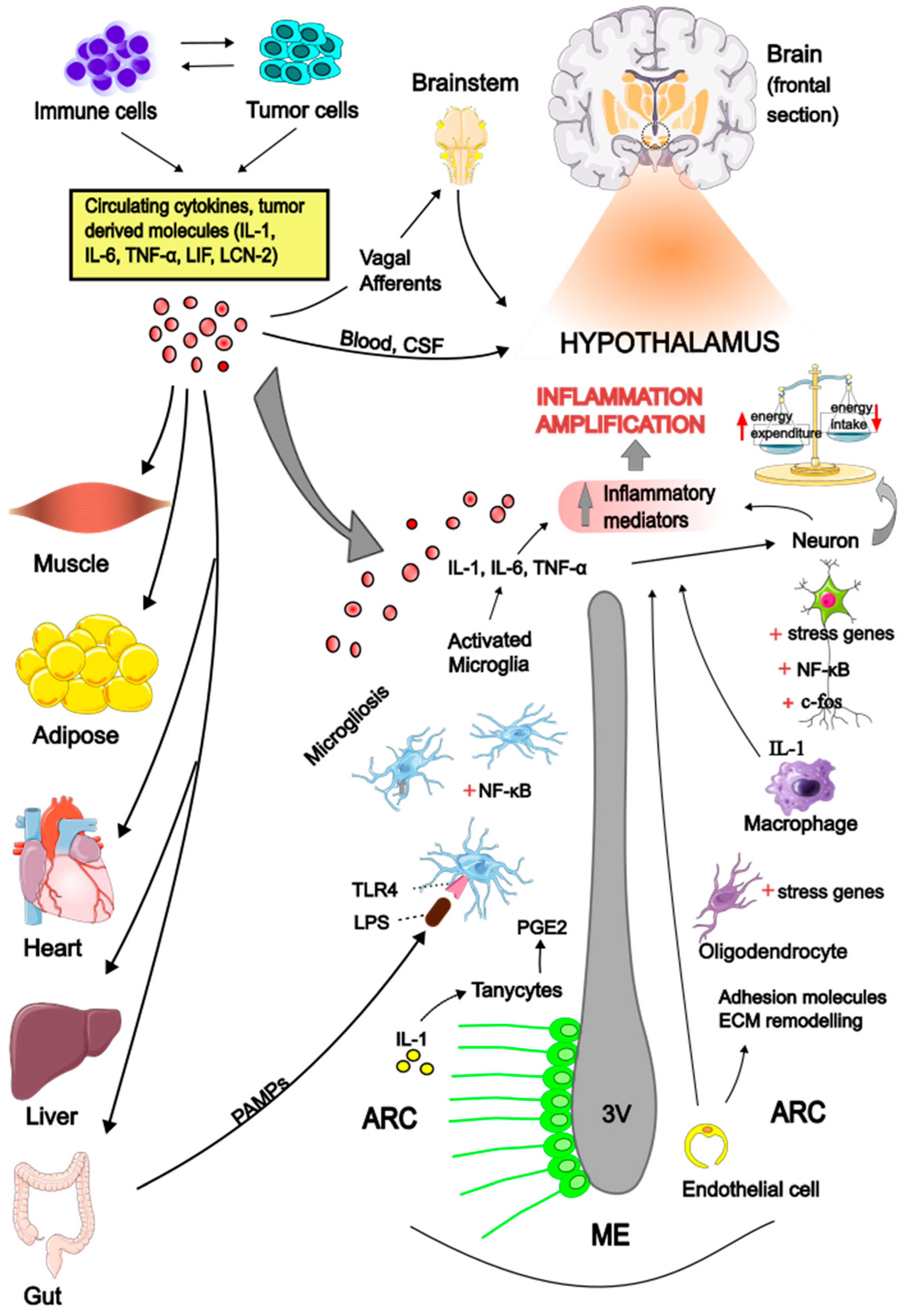
Cachexia is a complex multiorgan syndrome associated with various chronic diseases, characterized by anorexia and increased tissue wasting in the context of chronic inflammation. A specific form of this syndrome, known as cancer cachexia (CC), occurs alongside different types of tumors. The pathogenesis of CC is multifactorial, with inflammatory mediators and hormones released by either the tumor or the host identified as key drivers of the peripheral catabolic process through several direct mechanisms. Accumulating evidence indicates that the central nervous system (CNS) is also recognized as an integral component in the pathogenesis of CC. Hypothalamus has emerged as a critical brain area that senses and amplifies peripheral stimuli, generating an inappropriate neuronal signaling, leading to deregulation of feeding behavior and impaired control of energy homeostasis. Circulating cytokines may act in concert with hormones and neurotransmitters and perturbate the hypothalamic melanocortin system, shifting its activity towards the anorexigenic pathway and increase energy expenditure. The purpose of this review is to provide insights on the potential mechanisms mediating the hypothalamic inflammation in the context of anorexia and cachexia associated with cancer.
Keywords:
Introduction
Cytokines Signaling in the Brain
Hypothalamic Inflammation
Inflammatory Signals Across the Gut-Brain Axis
The Role of Hypothalamic Microglia
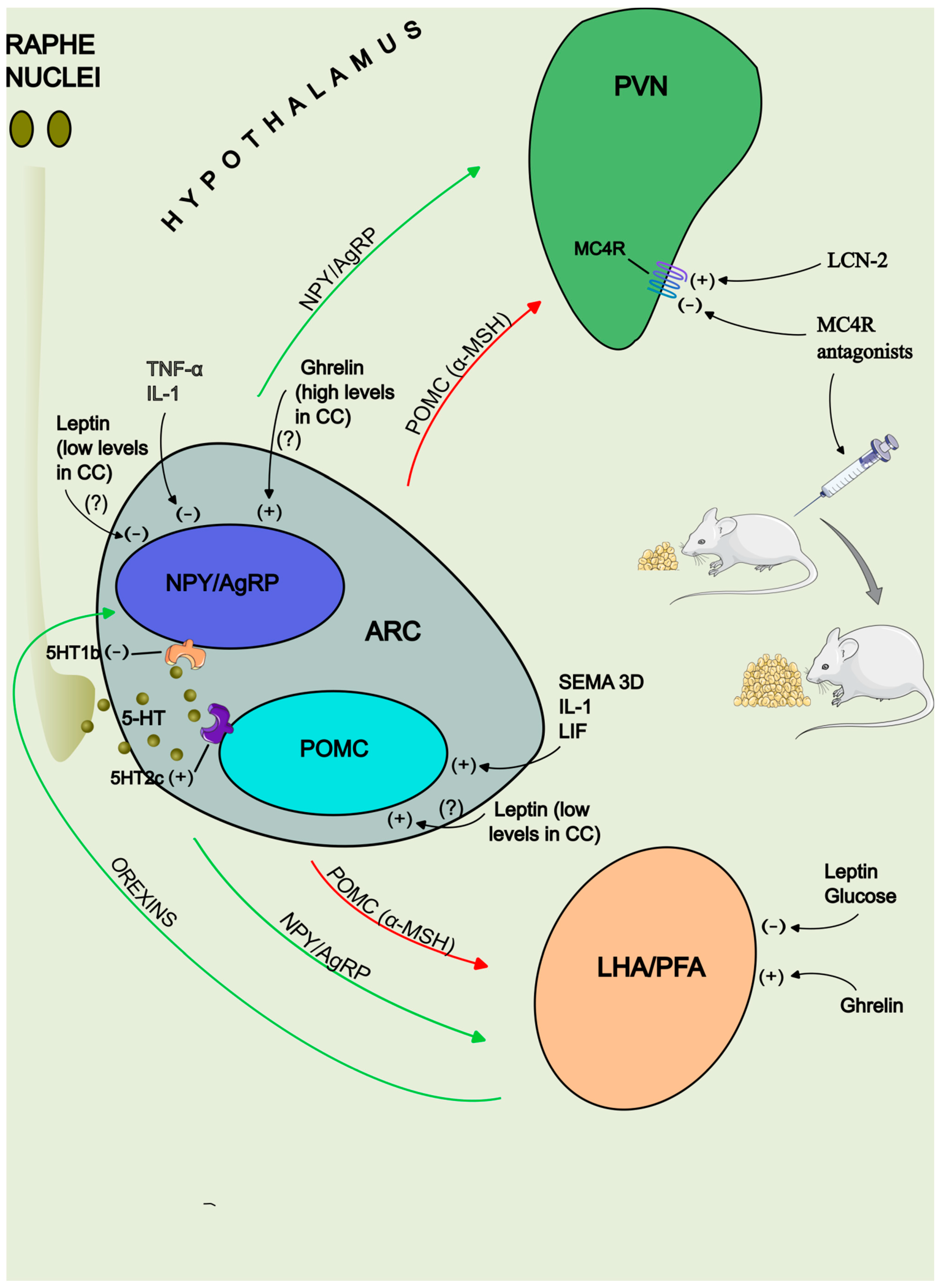
The Melanocortin System
Neurotransmitters Implicated in Energy Balance
1. NPY
2. 5-hydroxytryptamine
3. Orexins
4. Nesfatin-1
Peripheral Hormone Signals: Leptin and Ghrelin
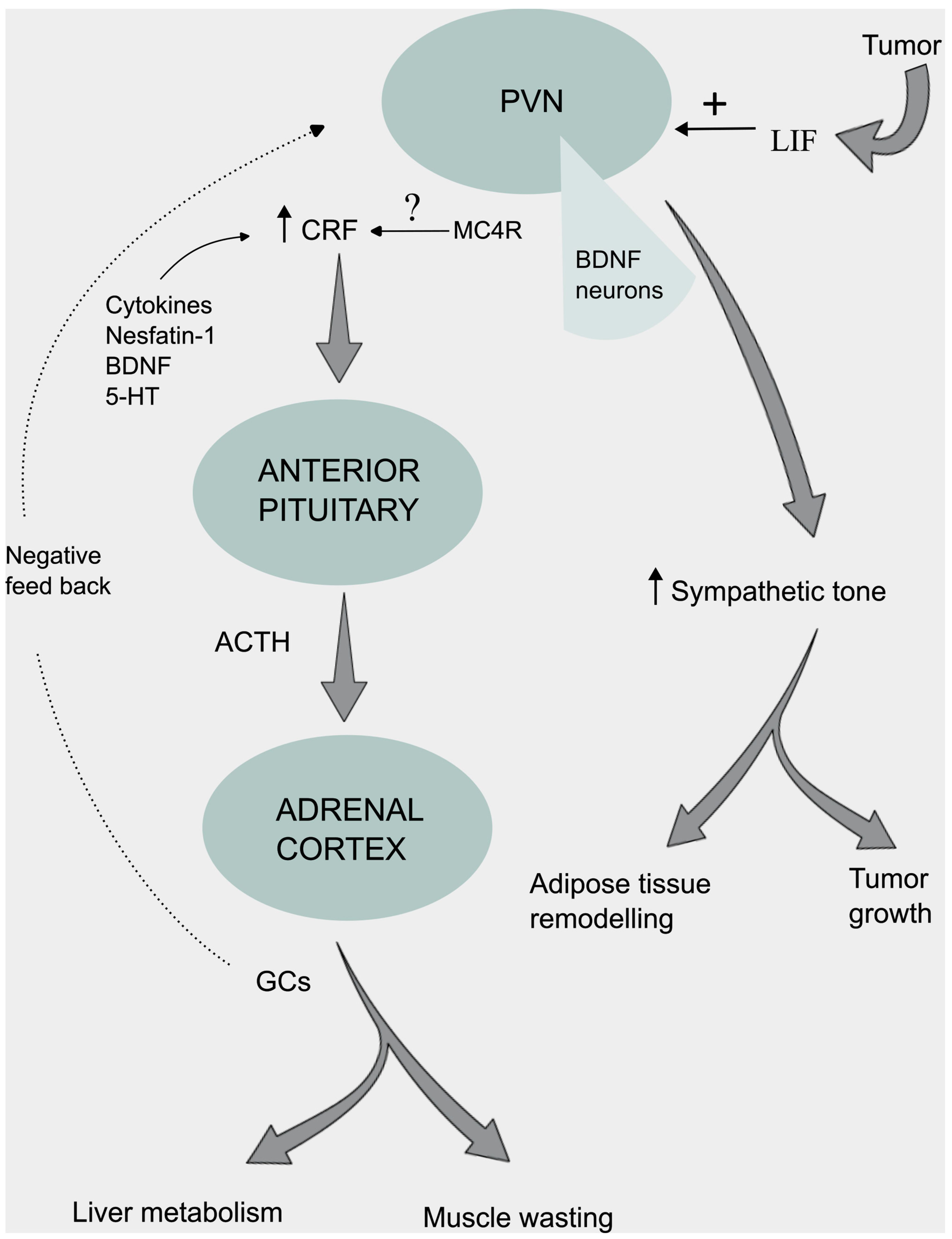
Neuroendocrine and Autonomic Regulation
Conclusions
Conflict of Interests
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
References
- von Haehling, S.; Anker, M.S.; Anker, S.D. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: facts and numbers update 2016. J. Cachex- Sarcopenia Muscle 2016, 7, 507–509. [Google Scholar] [CrossRef] [PubMed]
- M.J. Tisdale, Biology of cachexia. J. Natl. Cancer Inst. 1763; 89.
- Tisdale, M.J. Cachexia in cancer patients. Nat. Rev. Cancer 2002, 2, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Morley, J.E.; Argilés, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. 27. [CrossRef]
- R.D. Cone, M.A. R.D. Cone, M.A. Cowley, A.A. Butler, W. Fan, D.L. Marks, M.J. Low, The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int. J. 2001; 25. [Google Scholar]
- Cone, R.D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005, 8, 571–578. [Google Scholar] [CrossRef]
- Laviano, A.; Inui, A.; Marks, D.L.; Meguid, M.M.; Pichard, C.; Fanelli, F.R.; Seelaender, M. Neural control of the anorexia-cachexia syndrome. Am. J. Physiol. Metab. 2008, 295, E1000–E1008. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A Switch from White to Brown Fat Increases Energy Expenditure in Cancer-Associated Cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Suriben, R.; Chen, M.; Higbee, J.; Oeffinger, J.; Ventura, R.; Li, B.; Mondal, K.; Gao, Z.; Ayupova, D.; Taskar, P.; et al. Antibody-mediated inhibition of GDF15–GFRAL activity reverses cancer cachexia in mice. Nat. Med. 2020, 26, 1264–1270. [Google Scholar] [CrossRef]
- Fujitsuka, N.; Asakawa, A.; Uezono, Y.; Minami, K.; Yamaguchi, T.; Niijima, A.; Yada, T.; Maejima, Y.; Sedbazar, U.; Sakai, T.; et al. Potentiation of ghrelin signaling attenuates cancer anorexia–cachexia and prolongs survival. Transl. Psychiatry 2011, 1, e23–e23. [Google Scholar] [CrossRef]
- A. Martin, J. A. Martin, J. Castells, V. Allibert, A. Emerit, C. Zolotoff, V. Cardot-Ruffino, Y.S. Gallot, B. Vernus, V. Chauvet, L. Bartholin, L. Schaeffer, A.C. Durieux, C. Hourdé, F.B. Favier, L. Mazelin, D. Freyssenet, Hypothalamic-pituitary-adrenal axis activation and glucocorticoid-responsive gene expression in skeletal muscle and liver of Apc mice. J. 1686; 13. [Google Scholar]
- Suda, Y.; Nakamura, K.; Matsuyama, F.; Hamada, Y.; Makabe, H.; Narita, M.; Nagumo, Y.; Mori, T.; Kuzumaki, N.; Narita, M. Peripheral-central network analysis of cancer cachexia status accompanied by the polarization of hypothalamic microglia with low expression of inhibitory immune checkpoint receptors. Mol. Brain 2024, 17, 1–16. [Google Scholar] [CrossRef]
- Barrett-Jolley, R.; Nunn, N.; Womack, M.; Dart, C. Function and Pharmacology of Spinally-Projecting Sympathetic Pre-Autonomic Neurones in the Paraventricular Nucleus of the Hypothalamus. Curr. Neuropharmacol. 2011, 9, 262–277. [Google Scholar] [CrossRef]
- Watts, A.G.; Kanoski, S.E.; Sanchez-Watts, G.; Langhans, W. The physiological control of eating: signals, neurons, and networks. Physiol. Rev. 2022, 102, 689–813. [Google Scholar] [CrossRef]
- Mantovani, G.; Macciò, A.; Mura, L.; Massa, E.; Mudu, M.C.; Mulas, C.; Lusso, M.R.; Madeddu, C.; Dessì, A. Serum levels of leptin and proinflammatory cytokines in patients with advanced-stage cancer at different sites. J. Mol. Med. 2000, 78, 554–561. [Google Scholar] [CrossRef]
- Pfitzenmaier, J.; Vessella, R.; Higano, C.S.; Noteboom, J.L.; Wallace, D., Jr.; Corey, E. Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer 2003, 97, 1211–1216. [Google Scholar] [CrossRef]
- M. Krzystek-Korpacka, M. M. Krzystek-Korpacka, M. Matusiewicz, D. Diakowska, K. Grabowski, K. Blachut, I. Kustrzeba-Wojcicka, T. Banas, Impact of weight loss on circulating IL-1, IL-6, IL-8, TNF-alpha, VEGF-A, VEGF-C and midkine in gastroesophageal cancer patients. Clin. Biochem. 1353; 40. [Google Scholar]
- C. Scheede-Bergdahl, H.L. C. Scheede-Bergdahl, H.L. Watt, B. Trutschnigg, R.D. Kilgour, A. Haggarty, E. Lucar, A. Vigano, Is IL-6 the best pro-inflammatory biomarker of clinical outcomes of cancer cachexia? Clin. Nutr. 2012; 31. [Google Scholar]
- Lerner, L.; Hayes, T.G.; Tao, N.; Krieger, B.; Feng, B.; Wu, Z.; Nicoletti, R.; Chiu, M.I.; Gyuris, J.; Garcia, J.M. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J. Cachex- Sarcopenia Muscle 2015, 6, 317–324. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Wang, C.-J.; Chao, Y.-J.; Chen, H.-Y.; Wang, H.-C.; Tung, H.-L.; Lin, J.-T.; Shan, Y.-S. Elevated Serum Interleukin-8 Level Correlates with Cancer-Related Cachexia and Sarcopenia: An Indicator for Pancreatic Cancer Outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef]
- W.A. Banks, J.L. W.A. Banks, J.L. Lynch, O. Tulin O. Price, in The Neuroimmunological Basis of Behavior and Mental Disorder, ed. by A. Siegel, S.S. Zalcman (Springer New York, 2009), p.
- Johnston, G.R.; Webster, N.R. Cytokines and the immunomodulatory function of the vagus nerve. Br. J. Anaesth. 2009, 102, 453–462. [Google Scholar] [CrossRef]
- Plata-Salamán, C.R.; Ilyin, S.E.; Gayle, D. Brain cytokine mRNAs in anorectic rats bearing prostate adenocarcinoma tumor cells. Am. J. Physiol. Integr. Comp. Physiol. 1998, 275, R566–R573. [Google Scholar] [CrossRef]
- Ropelle, E.R.; Pauli, J.R.; Zecchin, K.G.; Ueno, M.; de Souza, C.T.; Morari, J.; Faria, M.C.; Velloso, L.A.; Saad, M.J.A.; Carvalheira, J.B.C. A Central Role for Neuronal Adenosine 5′-Monophosphate-Activated Protein Kinase in Cancer-Induced Anorexia. Endocrinology 2007, 148, 5220–5229. [Google Scholar] [CrossRef]
- Lira, F.S.; Yamashita, A.S.; Rosa, J.C.; Tavares, F.L.; Caperuto, E.; Carnevali, L.C.; Pimentel, G.D.; Santos, R.V.; Batista, M.L.; Laviano, A.; et al. Hypothalamic inflammation is reversed by endurance training in anorectic-cachectic rats. Nutr. Metab. 2011, 8, 60–60. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, K.A.; Zhu, X.; Burfeind, K.G.; Krasnow, S.M.; Levasseur, P.R.; Morgan, T.K.; Marks, D.L. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J. Cachex- Sarcopenia Muscle 2017, 8, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Burfeind, K.G.; Zhu, X.; Levasseur, P.R.; Michaelis, K.A.; Norgard, M.A.; Marks, D.L. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain, Behav. Immun. 2018, 73, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Burfeind, K.G.; Michaelis, K.A.; Braun, T.P.; Olson, B.; Pelz, K.R.; Morgan, T.K.; Marks, D.L. MyD88 signalling is critical in the development of pancreatic cancer cachexia. J. Cachex- Sarcopenia Muscle 2019, 10, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Norgard, M.A.; Levasseur, P.R.; Zhu, X.; Marks, D.L. Physiologic and molecular characterization of a novel murine model of metastatic head and neck cancer cachexia. J. Cachex- Sarcopenia Muscle 2021, 12, 1312–1332. [Google Scholar] [CrossRef] [PubMed]
- Cernackova, A.; Tillinger, A.; Bizik, J.; Mravec, B.; Horvathova, L. Dynamics of cachexia-associated inflammatory changes in the brain accompanying intra-abdominal fibrosarcoma growth in Wistar rats. J. Neuroimmunol. 2023, 376, 578033. [Google Scholar] [CrossRef] [PubMed]
- Layé, S.; Gheusi, G.; Cremona, S.; Combe, C.; Kelley, K.; Dantzer, R.; Parnet, P. Endogenous brain IL-1 mediates LPS-induced anorexia and hypothalamic cytokine expression. Am. J. Physiol. Integr. Comp. Physiol. 2000, 279, R93–R98. [Google Scholar] [CrossRef]
- Bluthe, R.-M.; Laye, S.; Michaud, B.; Combe, C.; Dantzer, R.; Parnet, P. Role of interleukin-1beta and tumour necrosis factor-alpha in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. Eur. J. Neurosci. 2000, 12, 4447–4456. [Google Scholar] [CrossRef] [PubMed]
- I Opara, E.; Laviano, A.; Meguid, M.M. Correlation between food intake and cerebrospinal fluid interleukin 1 alpha in anorectic tumor-bearing rats. . 1995, 11, 678–9. [Google Scholar]
- J.M. Scarlett, E.E. J.M. Scarlett, E.E. Jobst, P.J. Enriori, D.D. Bowe, A.K. Batra, W.F. Grant, M.A. Cowley, D.L. Marks, Regulation of central melanocortin signaling by interleukin-1 beta. 4217. [Google Scholar]
- Scarlett, J.M.; Zhu, X.; Enriori, P.J.; Bowe, D.D.; Batra, A.K.; Levasseur, P.R.; Grant, W.F.; Meguid, M.M.; Cowley, M.A.; Marks, D.L. Regulation of Agouti-Related Protein Messenger Ribonucleic Acid Transcription and Peptide Secretion by Acute and Chronic Inflammation. Endocrinology 2008, 149, 4837–4845. [Google Scholar] [CrossRef]
- Laviano, A.; Renvyle, T.; Meguid, M.M.; Yang, Z.J.; Cangiano, C.; Fanelli, F.R. Relationship between interleukin-1 and cancer anorexia. . 1995, 11, 680–3. [Google Scholar]
- Burfeind, K.G.; Michaelis, K.A.; Marks, D.L. The central role of hypothalamic inflammation in the acute illness response and cachexia. Semin. Cell Dev. Biol. 2015, 54, 42–52. [Google Scholar] [CrossRef]
- Arruda, A.P.; Milanski, M.; Romanatto, T.; Solon, C.; Coope, A.; Alberici, L.C.; Festuccia, W.T.; Hirabara, S.M.; Ropelle, E.; Curi, R.; et al. Hypothalamic Actions of Tumor Necrosis Factor α Provide the Thermogenic Core for the Wastage Syndrome in Cachexia. Endocrinology 2010, 151, 683–694. [Google Scholar] [CrossRef]
- G.F. Torelli, M.M. G.F. Torelli, M.M. Meguid, L.L. Moldawer, C.K. Edwards 3rd, H.J. Kim, J.L. Carter, A. Laviano, F. Rossi Fanelli, Use of recombinant human soluble TNF receptor in anorectic tumor-bearing rats. Am. J. Physiol. 1999. [Google Scholar]
- C.R. Plata-Salamán, Y. C.R. Plata-Salamán, Y. Oomura, Y. Kai, Tumor necrosis factor and interleukin-1 beta: suppression of food intake by direct action in the central nervous system. Brain Res. 1988. [Google Scholar]
- Chaves, F.M.; Mansano, N.S.; Frazão, R.; Donato, J. Tumor Necrosis Factor α and Interleukin-1β Acutely Inhibit AgRP Neurons in the Arcuate Nucleus of the Hypothalamus. Int. J. Mol. Sci. 2020, 21, 8928. [Google Scholar] [CrossRef] [PubMed]
- Arruda, A.P.; Milanski, M.; Romanatto, T.; Solon, C.; Coope, A.; Alberici, L.C.; Festuccia, W.T.; Hirabara, S.M.; Ropelle, E.; Curi, R.; et al. Hypothalamic Actions of Tumor Necrosis Factor α Provide the Thermogenic Core for the Wastage Syndrome in Cachexia. Endocrinology 2010, 151, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Müller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef] [PubMed]
- A.J. Grossberg, J.M. A.J. Grossberg, J.M. Scarlett, D.L. Marks, Hypothalamic mechanisms in cachexia Physiol. Behav. 2010. [Google Scholar]
- Burton, M.D.; Sparkman, N.L.; Johnson, R.W. Inhibition of interleukin-6 trans-signaling in the brain facilitates recovery from lipopolysaccharide-induced sickness behavior. J. Neuroinflammation 2011, 8, 54–54. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Denson, J.L.; Steculorum, S.M.; Heilinger, C.; Engström-Ruud, L.; Wunderlich, C.M.; Rose-John, S.; Wunderlich, F.T.; Brüning, J.C. IL-6 Improves Energy and Glucose Homeostasis in Obesity via Enhanced Central IL-6 trans-Signaling. Cell Rep. 2017, 19, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Bobbo, V.C.; Engel, D.F.; Jara, C.P.; Mendes, N.F.; Haddad-Tovolli, R.; Prado, T.P.; Sidarta-Oliveira, D.; Morari, J.; Velloso, L.A.; Araujo, E.P. Interleukin-6 actions in the hypothalamus protects against obesity and is involved in the regulation of neurogenesis. J. Neuroinflammation 2021, 18, 1–17. [Google Scholar] [CrossRef]
- Wallenius, K.; Wallenius, V.; Sunter, D.; Dickson, S.L.; Jansson, J.-O. Intracerebroventricular interleukin-6 treatment decreases body fat in rats. Biochem. Biophys. Res. Commun. 2002, 293, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Benrick, A.; Schéle, E.; Pinnock, S.B.; Wernstedt-Asterholm, I.; Dickson, S.L.; Karlsson-Lindahl, L.; Jansson, J. Interleukin-6 Gene Knockout Influences Energy Balance Regulating Peptides in the Hypothalamic Paraventricular and Supraoptic Nuclei. J. Neuroendocr. 2009, 21, 620–628. [Google Scholar] [CrossRef]
- Katashima, C.K.; Micheletti, T.d.O.; Braga, R.R.; Gaspar, R.S.; Goeminne, L.J.E.; Moura-Assis, A.; Crisol, B.M.; Brícola, R.S.; Silva, V.R.R.; Ramos, C.d.O.; et al. Evidence for a neuromuscular circuit involving hypothalamic interleukin-6 in the control of skeletal muscle metabolism. Sci. Adv. 2022, 8, eabm7355. [Google Scholar] [CrossRef]
- G. Arora, A. G. Arora, A. Gupta, T. Guo, A. Gandhi, A. Laine, D. Williams, C. Ahn, P. Iyengar, R. Infante, JAK Inhibitors Suppress Cancer Cachexia-Associated Anorexia and Adipose Wasting in Mice. JCSM Rapid Commun. 2020; 3. [Google Scholar]
- Xu, Q.; Cao, Y.; Kong, F.; Liu, J.; Chen, X.; Zhao, Y.; Lai, C.-H.; Zhou, X.; Hu, H.; Fu, W.; et al. Multiple cancer cell types release LIF and Gal3 to hijack neural signals. Cell Res. 2024, 34, 345–354. [Google Scholar] [CrossRef]
- Di Giorgio, C.; Marchianò, S.; Marino, E.; Biagioli, M.; Roselli, R.; Bordoni, M.; Bellini, R.; Urbani, G.; Zampella, A.; Distrutti, E.; et al. Next-Generation Sequencing Analysis of Gastric Cancer Identifies the Leukemia Inhibitory Factor Receptor as a Driving Factor in Gastric Cancer Progression and as a Predictor of Poor Prognosis. Front. Oncol. 2022, 12, 939969. [Google Scholar] [CrossRef]
- Zhang, F.; Yan, Y.; Cao, X.; Guo, C.; Wang, K.; Lv, S. TGF-β-driven LIF expression influences neutrophil extracellular traps (NETs) and contributes to peritoneal metastasis in gastric cancer. Cell Death Dis. 2024, 15, 1–13. [Google Scholar] [CrossRef]
- S.C. Kandarian, R.L. S.C. Kandarian, R.L. Nosacka, A.E. Delitto, A.R. Judge, S.M. Judge, J.D. Ganey, J.D. Moreira, R.W. Jackman, Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. 1109; 9. [Google Scholar]
- Yang, X.; Wang, J.; Chang, C.-Y.; Zhou, F.; Liu, J.; Xu, H.; Ibrahim, M.; Gomez, M.; Guo, G.L.; Liu, H.; et al. Leukemia inhibitory factor suppresses hepatic de novo lipogenesis and induces cachexia in mice. Nat. Commun. 2024, 15, 1–15. [Google Scholar] [CrossRef]
- Arora, G.K.; Gupta, A.; Narayanan, S.; Guo, T.; Iyengar, P.; Infante, R.E. Cachexia-associated adipose loss induced by tumor-secreted leukemia inhibitory factor is counterbalanced by decreased leptin. J. Clin. Investig. 2018, 3. [Google Scholar] [CrossRef]
- Pan, W.; Kastin, A.J.; Brennan, J. Saturable entry of leukemia inhibitory factor from blood to the central nervous system. J. Neuroimmunol. 2000, 106, 172–180. [Google Scholar] [CrossRef]
- Grossberg, A.J.; Scarlett, J.M.; Zhu, X.; Bowe, D.D.; Batra, A.K.; Braun, T.P.; Marks, D.L. Arcuate Nucleus Proopiomelanocortin Neurons Mediate the Acute Anorectic Actions of Leukemia Inhibitory Factor via gp130. Endocrinology 2010, 151, 606–616. [Google Scholar] [CrossRef]
- Terawaki, K.; Sawada, Y.; Kashiwase, Y.; Hashimoto, H.; Yoshimura, M.; Suzuki, M.; Miyano, K.; Sudo, Y.; Shiraishi, S.; Higami, Y.; et al. New cancer cachexia rat model generated by implantation of a peritoneal dissemination-derived human stomach cancer cell line. Am. J. Physiol. Metab. 2014, 306, E373–E387. [Google Scholar] [CrossRef]
- Elmquist, J.K.; Scammell, T.E.; Jacobson, C.D.; Saper, C.B. Distribution of fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. J. Comp. Neurol. 1996, 371, 85–103. [Google Scholar] [CrossRef]
- S. Tolchard, A.S. S. Tolchard, A.S. Hare, D.J. Nutt, G. Clarke, TNF alpha mimics the endocrine but not the thermoregulatory responses of bacterial lipopolysaccharide (LPS): correlation with 83. FOS-expression in the brain. 1996; 35. [Google Scholar]
- M. Herkenham, H.Y. M. Herkenham, H.Y. Lee, R.A. Baker, Temporal and spatial patterns of c-fos mRNA induced by intravenous interleukin-1: a cascade of non-neuronal cellular activation at the blood-brain barrier. J. Comp. Neurol. 1998. [Google Scholar]
- A. Ericsson, C. A. Ericsson, C. Liu, R.P. Hart, P.E. Sawchenko, Type 1 interleukin-1 receptor in the rat brain: distribution, regulation, and relationship to sites of IL-1-induced cellular activation. J. Comp. Neurol. 1995. [Google Scholar]
- Utsuyama, M.; Hirokawa, K. Differential expression of various cytokine receptors in the brain after stimulation with LPS in young and old mice. Exp. Gerontol. 2002, 37, 411–420. [Google Scholar] [CrossRef]
- H. Yamakuni, M. H. Yamakuni, M. Minami, M. Satoh, Localization of mRNA for leukemia inhibitory factor receptor in the adult rat brain. J. Neuroimmunol. 1996; 70. [Google Scholar]
- Patra, S.K.; Arora, S. Integrative role of neuropeptides and cytokines in cancer anorexia–cachexia syndrome. Clin. Chim. Acta 2012, 413, 1025–1034. [Google Scholar] [CrossRef]
- S.J. Hopkins, N.J. S.J. Hopkins, N.J. Rothwell, Cytokines and the nervous system I: expression and recognition. Trends Neurosci. 1995; 18. [Google Scholar]
- Reis, W.L.; Yi, C.-X.; Gao, Y.; Tschöp, M.H.; Stern, J.E. Brain Innate Immunity Regulates Hypothalamic Arcuate Neuronal Activity and Feeding Behavior. Endocrinology 2015, 156, 1303–1315. [Google Scholar] [CrossRef]
- P.G. Jang, C. P.G. Jang, C. Namkoong, G.M. Kang, M.W. Hur, S.W. Kim, G.H. Kim, Y. Kang, M.J. Jeon, E.H. Kim, M.S. Lee, M. Karin, J.H. Baik, J.Y. Park, K.U. Lee, Y.B Kim, M.S. Kim, NF-κB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. J. Biol. Chem. 9706. [Google Scholar]
- Valdearcos, M.; Douglass, J.D.; Robblee, M.M.; Dorfman, M.D.; Stifler, D.R.; Bennett, M.L.; Gerritse, I.; Fasnacht, R.; Barres, B.A.; Thaler, J.P.; et al. Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility. Cell Metab. 2017, 26, 185–197.e3. [Google Scholar] [CrossRef]
- Breder, C.D.; Dinarello, C.A.; Saper, C.B. Interleukin-1 Immunoreactive Innervation of the Human Hypothalamus. Science 1988, 240, 321–324. [Google Scholar] [CrossRef]
- Acarin, L.; González, B.; Castellano, B. Neuronal, astroglial and microglial cytokine expression after an excitotoxic lesion in the immature rat brain. Eur. J. Neurosci. 2000, 12, 3505–3520. [Google Scholar] [CrossRef]
- Dwarkasing, J.T.; Witkamp, R.F.; Boekschoten, M.V.; Ter Laak, M.C.; Heins, M.S.; van Norren, K. Increased hypothalamic serotonin turnover in inflammation-induced anorexia. BMC Neurosci. 2016, 17, 1–13. [Google Scholar] [CrossRef]
- Huisman, C.; Norgard, M.A.; Levasseur, P.R.; Krasnow, S.M.; van der Wijst, M.G.; Olson, B.; Marks, D.L. Critical changes in hypothalamic gene networks in response to pancreatic cancer as found by single-cell RNA sequencing. Mol. Metab. 2022, 58, 101441. [Google Scholar] [CrossRef]
- M. Böttcher, H. M. Böttcher, H. Müller-Fielitz, S.M. Sundaram, S. Gallet, V. Neve, K. Shionoya, A. Zager, N. Quan, X. Liu, R. Schmidt-Ullrich, R. Haenold, J. Wenzel, A. Blomqvist, D. Engblom, V. Prevot, M. Schwaninger, NF-κB signaling in tanycytes mediates inflammation-induced anorexia. Mol. Metab. 1010; 39. [Google Scholar]
- Costa, R.G.; Caro, P.L.; de Matos-Neto, E.M.; Lima, J.D.; Radloff, K.; Alves, M.J.; Camargo, R.G.; Pessoa, A.F.M.; Simoes, E.; Gama, P.; et al. Cancer cachexia induces morphological and inflammatory changes in the intestinal mucosa. J. Cachex- Sarcopenia Muscle 2019, 10, 1116–1127. [Google Scholar] [CrossRef]
- Bindels, L.B.; Neyrinck, A.M.; Loumaye, A.; Catry, E.; Walgrave, H.; Cherbuy, C.; Leclercq, S.; Van Hul, M.; Plovier, H.; Pachikian, B.; et al. Increased gut permeability in cancer cachexia: mechanisms and clinical relevance. Oncotarget 2018, 9, 18224–18238. [Google Scholar] [CrossRef]
- Liu, H.; Cheng, Y.; Qu, Y.; Wu, G. Unraveling the gut microbiota and short-chain fatty acids characteristics and associations in a cancer cachexia mouse model. Microb. Pathog. 2023, 183, 106332. [Google Scholar] [CrossRef]
- Li, X.; Holtrop, T.; Jansen, F.A.; Olson, B.; Levasseur, P.; Zhu, X.; Poland, M.; Schalwijk, W.; Witkamp, R.F.; Marks, D.L.; et al. Lipopolysaccharide-induced hypothalamic inflammation in cancer cachexia-anorexia is amplified by tumour-derived prostaglandin E2. J. Cachex- Sarcopenia Muscle 2022, 13, 3014–3027. [Google Scholar] [CrossRef]
- Carpentier, P.A.; Duncan, D.S.; Miller, S.D. Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain, Behav. Immun. 2007, 22, 140–147. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- T. Kawai, S. T. Kawai, S. Akira, Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007; 13. [Google Scholar]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Jin, S.; Kim, J.G.; Park, J.W.; Koch, M.; Horvath, T.L.; Lee, B.J. Hypothalamic TLR2 triggers sickness behavior via a microglia-neuronal axis. Sci. Rep. 2016, 6, 29424. [Google Scholar] [CrossRef]
- Kim, Y.T.; Park, B.S.; Yang, H.R.; Yi, S.O.; Nam-Goong, I.S.; Kim, J.G. Exploring the potential hypothalamic role in mediating cisplatin-induced negative energy balance. Chem. Interactions 2023, 385, 110733. [Google Scholar] [CrossRef]
- Kashihara, S.; Shinohara, K.; Ikeda, S.; Tsutsui, H. Microglia contribute to cancer cachexia through affecting PVN neurons and POMC neurons. FASEB J. 2020, 34, 1–1. [Google Scholar] [CrossRef]
- Burfeind, K.G.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Huisman, C.; Michaelis, K.A.; Olson, B.; Marks, D.L. Microglia in the hypothalamus respond to tumor-derived factors and are protective against cachexia during pancreatic cancer. Glia 2020, 68, 1479–1494. [Google Scholar] [CrossRef]
- Chang, C.-I.; Liao, J.C.; Kuo, L. Arginase modulates nitric oxide production in activated macrophages. Am. J. Physiol. Circ. Physiol. 1998, 274, H342–H348. [Google Scholar] [CrossRef]
- L. Ye, Y. L. Ye, Y. Huang, L. Zhao, Y. Li, L. Sun, Y. Zhou, G. Qian, J.C. Zheng, IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J. Neurochem. 2013. [Google Scholar]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Prinz, M. Microglia Heterogeneity in the Single-Cell Era. 30, 1271. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Mitchell, N.F.; Lin, S.; Macia, L.; Yulyaningsih, E.; Baldock, P.A.; Enriquez, R.F.; Zhang, L.; Shi, Y.-C.; Zolotukhin, S.; et al. Y1 and Y5 Receptors Are Both Required for the Regulation of Food Intake and Energy Homeostasis in Mice. PLoS ONE 2012, 7, e40191. [Google Scholar] [CrossRef]
- Wisse, B.E.; Frayo, R.S.; Schwartz, M.W.; Cummings, D.E. Reversal of Cancer Anorexia by Blockade of Central Melanocortin Receptors in Rats. Endocrinology 2001, 142, 3292–3301. [Google Scholar] [CrossRef]
- Marks, D.L.; Ling, N.; Cone, R.D. Role of the central melanocortin system in cachexia. . 2001, 61, 1432–8. [Google Scholar]
- Joppa, M.; Gogas, K.; Foster, A.; Markison, S. Central infusion of the melanocortin receptor antagonist agouti-related peptide (AgRP(83-132)) prevents cachexia-related symptoms induced by radiation and colon-26 tumors in mice. Peptides 2007, 28, 636–642. [Google Scholar] [CrossRef]
- Olson, B.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Butler, J.T.; Buenafe, A.; Burfeind, K.G.; Michaelis, K.A.; Pelz, K.R.; Mendez, H.; et al. Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Zhu, X.; Callahan, M.F.; Gruber, K.A.; Szumowski, M.; Marks, D.L. Melanocortin-4 receptor antagonist TCMCB07 ameliorates cancer- and chronic kidney disease–associated cachexia. J. Clin. Investig. 2020, 130, 4921–4934. [Google Scholar] [CrossRef]
- Axiak-Bechtel, S.M.; Leach, S.B.; Newton-Northup, J.R.; Milner, R.J.; Fox-Alvarez, S.A.; Fagman, L.I.; Young, K.A.; Tate, D.J.; Wright, Z.M.; Chretin, J.D.; et al. Safety of TCMCB07, a melanocortin-4 antagonist peptide, in dogs with naturally occurring cachexia. J. Veter- Intern. Med. 2023, 37, 2344–2355. [Google Scholar] [CrossRef]
- Bica, C.; Tirpe, A.; Nutu, A.; Ciocan, C.; Chira, S.; Gurzau, E.S.; Braicu, C.; Berindan-Neagoe, I. Emerging roles and mechanisms of semaphorins activity in cancer. Life Sci. 2023, 318, 121499. [Google Scholar] [CrossRef]
- Van Der Klaauw, A.; Croizier, S.; De Oliveira, E.M.; Stadler, L.; Park, S.; Kong, Y.; Banton, M.C.; Tandon, P.; E Hendricks, A.; Keogh, J.M.; et al. Human Semaphorin 3 Variants Link Melanocortin Circuit Development and Energy Balance. . 2019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xi, Q.; Zhong, M.; Jiang, Y.; Zhuang, Q.; Ding, Z.; Tan, S.; Wang, J.; Liu, H.; Zhang, Z.; et al. Tumor-derived semaphorin 3D promoting cancer cachexia via regulating hypothalamic pro-opiomelanocortin neurons. FASEB J. 2023, 37, e22980. [Google Scholar] [CrossRef]
- Wisse, B.E.; Schwartz, M.W.; Cummings, D.E. Melanocortin signaling and anorexia in chronic disease states. Ann. New York Acad. Sci. 2003, 994, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Dwarkasing, J.T.; van Dijk, M.; Dijk, F.J.; Boekschoten, M.V.; Faber, J.; Argilès, J.M.; Laviano, A.; Müller, M.; Witkamp, R.F.; van Norren, K. Hypothalamic food intake regulation in a cancer-cachectic mouse model. J. Cachex- Sarcopenia Muscle 2013, 5, 159–169. [Google Scholar] [CrossRef] [PubMed]
- H. Suzuki, H. H. Suzuki, H. Hashimoto, M. Kawasaki, M. Watanabe, H. Otsubo, T. Ishikura, H. Fujihara, H. Ohnishi, E. Onuma, H. Yamada-Okabe, Y. Takuwa, E. Ogata, T. Nakamura, Y. Ueta, Similar changes of hypothalamic feeding-regulating peptides mRNAs and plasma leptin levels in PTHrP-, LIF-secreting tumors-induced cachectic rats and adjuvant arthritic rats. Int. J. 2215. [Google Scholar]
- Suzuki, M.; Narita, M.; Ashikawa, M.; Furuta, S.; Matoba, M.; Sasaki, H.; Yanagihara, K.; Terawaki, K.; Suzuki, T.; Uezono, Y. Changes in the melanocortin receptors in the hypothalamus of a rat model of cancer cachexia. Synapse 2012, 66, 747–751. [Google Scholar] [CrossRef] [PubMed]
- Chance, W.T.; Sheriff, S.; Dayal, R.; Balasubramaniam, A. Refractory hypothalamic α-MSH satiety and AGRP feeding systems in rats bearing MCA sarcomas. Peptides 2003, 24, 1909–1919. [Google Scholar] [CrossRef] [PubMed]
- Sergeyev, V.; Broberger, C.; Hökfelt, T. Effect of LPS administration on the expression of POMC, NPY, galanin, CART and MCH mRNAs in the rat hypothalamus. Mol. Brain Res. 2001, 90, 93–100. [Google Scholar] [CrossRef]
- Shi, X.; Wang, X.; Li, Q.; Su, M.; Chew, E.; Wong, E.T.; Lacza, Z.; Radda, G.K.; Tergaonkar, V.; Han, W. Nuclear factor κB (NF-κB) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia 2013, 56, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Mosialou, I.; Shikhel, S.; Liu, J.-M.; Maurizi, A.; Luo, N.; He, Z.; Huang, Y.; Zong, H.; Friedman, R.A.; Barasch, J.; et al. Erratum: Corrigendum: MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 2017, 546, 440–440. [Google Scholar] [CrossRef]
- Dwarkasing, J.; Marks, D.; Witkamp, R.; van Norren, K. Hypothalamic inflammation and food intake regulation during chronic illness. Peptides 2016, 77, 60–66. [Google Scholar] [CrossRef] [PubMed]
- N. Nara-ashizawa, T. N. Nara-ashizawa, T. Tsukada, K. Maruyama, Y. Akiyama, N. Kajimura, K. Yamaguchi, Response of hypothalamic NPY mRNAs to a negative energy balance is less sensitive in cachectic mice bearing human tumor cells. Nutr. 2001; 41. [Google Scholar]
- W.T. Chance, A. W.T. Chance, A. Balasubramaniam, H. Thompson, B. Mohapatra, J. Ramo, J.E. Fischer, Assessment of feeding response of tumor-bearing rats to hypothalamic injection and infusion of neuropeptide Y. 1996; 17. [Google Scholar]
- Nara-Ashizawa, N.; Tsukada, T.; Maruyama, K.; Akiyama, Y.; Kajimura, N.; Nagasaki, K.; Iwanaga, T.; Yamaguchi, K. Hypothalamic appetite-regulating neuropeptide mRNA levels in cachectic nude mice bearing human tumor cells. Metabolism 2001, 50, 1213–1219. [Google Scholar] [CrossRef]
- Chance, W.T.; Xiao, C.; Dayal, R.; Sheriff, S. Alteration of NPY and Y1 receptor in dorsomedial and ventromedial areas of hypothalamus in anorectic tumor-bearing rats. Peptides 2007, 28, 295–301. [Google Scholar] [CrossRef]
- Dwarkasing, J.T.; Boekschoten, M.V.; Argilès, J.M.; van Dijk, M.; Busquets, S.; Penna, F.; Toledo, M.; Laviano, A.; Witkamp, R.F.; van Norren, K. Differences in food intake of tumour-bearing cachectic mice are associated with hypothalamic serotonin signalling. J. Cachex- Sarcopenia Muscle 2015, 6, 84–94. [Google Scholar] [CrossRef]
- Dhillon, S.S.; McFadden, S.A.; Chalmers, J.A.; Centeno, M.-L.; Kim, G.L.; Belsham, D.D. Cellular Leptin Resistance Impairs the Leptin-Mediated Suppression of Neuropeptide Y Secretion in Hypothalamic Neurons. Endocrinology 2011, 152, 4138–4147. [Google Scholar] [CrossRef]
- S.H. Cha, M.D. S.H. Cha, M.D. Lane, Central lactate metabolism suppresses food intake via the hypothalamic AMP kinase/malonyl-CoA signaling pathway. Biochem. Biophys Res Commun. 2009. [Google Scholar]
- Lockie, S.H.; Stark, R.; Mequinion, M.; Ch’ng, S.; Kong, D.; Spanswick, D.C.; Lawrence, A.J.; Andrews, Z.B. Glucose Availability Predicts the Feeding Response to Ghrelin in Male Mice, an Effect Dependent on AMPK in AgRP Neurons. Endocrinology 2018, 159, 3605–3614. [Google Scholar] [CrossRef]
- Shimizu, H.; Arima, H.; Watanabe, M.; Goto, M.; Banno, R.; Sato, I.; Ozaki, N.; Nagasaki, H.; Oiso, Y. Glucocorticoids Increase Neuropeptide Y and Agouti-Related Peptide Gene Expression via Adenosine Monophosphate-Activated Protein Kinase Signaling in the Arcuate Nucleus of Rats. Endocrinology 2008, 149, 4544–4553. [Google Scholar] [CrossRef]
- McCarthy, H.D.; McKibbin, P.E.; Perkins, A.V.; Linton, E.A.; Williams, G. Alterations in hypothalamic NPY and CRF in anorexic tumor-bearing rats. Am. J. Physiol. Metab. 1993, 264, E638–E643. [Google Scholar] [CrossRef] [PubMed]
- Meguid, M.M.; Ramos, E.J.; Laviano, A.; Varma, M.; Sato, T.; Chen, C.; Qi, Y.; Das, U.N. Tumor anorexia: effects on neuropeptide Y and monoamines in paraventricular nucleus. Peptides 2004, 25, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Voigt, J.-P.; Fink, H. Serotonin controlling feeding and satiety. Behav. Brain Res. 2015, 277, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Geyer, M.A.; Puerto, A.; Menkes, D.B.; Segal, D.S.; Mandell, A.J. Behavioral studies following lesions of the mesolimbic and mesostriatal serotonergic pathways. Brain Res. 1976, 106, 257–270. [Google Scholar] [CrossRef]
- Heisler, L.K.; Jobst, E.E.; Sutton, G.M.; Zhou, L.; Borok, E.; Thornton-Jones, Z.; Liu, H.Y.; Zigman, J.M.; Balthasar, N.; Kishi, T.; et al. Serotonin Reciprocally Regulates Melanocortin Neurons to Modulate Food Intake. Neuron 2006, 51, 239–249. [Google Scholar] [CrossRef]
- Xu, Y.; Jones, J.E.; Kohno, D.; Williams, K.W.; Lee, C.E.; Choi, M.J.; Anderson, J.G.; Heisler, L.K.; Zigman, J.M.; Lowell, B.B.; et al. 5-HT2CRs Expressed by Pro-Opiomelanocortin Neurons Regulate Energy Homeostasis. Neuron 2008, 60, 582–589. [Google Scholar] [CrossRef]
- Bláha, V.; Yang, Z.-J.; Meguid, M.M.; Chai, J.-K.; Oler, A.; Zadák, Z. Ventromedial Nucleus of Hypothalamus is Related to the Development of Cancer-Induced Anorexia: In Vivo Microdialysis Study. Acta Medica (Hradec Kralove, Czech Republic) 1998, 41, 3–11. [Google Scholar] [CrossRef]
- Ramos, E.J.; Suzuki, S.; Meguid, M.M.; Laviano, A.; Sato, T.; Chen, C.; Das, U. Changes in hypothalamic neuropeptide Y and monoaminergic system in tumor-bearing rats: Pre- and post-tumor resection and at death. Surgery 2004, 136, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, I.G.; Meguid, M.M.; Gatto, L.; Chen, C.; Ramos, E.J.; Goncalves, C.G.; Ugrumov, M.V. Normalization of hypothalamic serotonin (5-HT1B) receptor and NPY in cancer anorexia after tumor resection: An immunocytochemical study. Neurosci. Lett. 2005, 383, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Makarenko, I.G.; Meguid, M.M.; Gatto, L.; Goncalves, C.G.; Ramos, E.J.; Chen, C.; Ugrumov, M.V. Hypothalamic 5-HT1B-receptor changes in anorectic tumor bearing rats. Neurosci. Lett. 2005, 376, 71–75. [Google Scholar] [CrossRef]
- Laviano, A.; Gleason, J.R.; Meguid, M.M.; Yang, Z.J.; Cangiano, C.; Fanelli, F.R. Effects of intra-VMN mianserin and IL-1ra on meal number in anorectic tumor-bearing rats. . 2000, 48, 40–8. [Google Scholar]
- Sato, T.; Laviano, A.; Meguid, M.; Chen, C.; Rossi-Fanelli, F.; Hatakeyama, K. Involvement of plasma leptin, insulin and free tryptophan in cytokine-induced anorexia. Clin. Nutr. 2003, 22, 139–146. [Google Scholar] [CrossRef]
- Chance, W.T.; von Meyenfeldt, M.F.; Fischer, J.E. Changes in brain amines associated with cancer anorexia. Neurosci. Biobehav. Rev. 1984, 7, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Cangiano, C.; Cascino, A.; Ceci, F.; Laviano, A.; Mulieri, M.; Muscaritoli, M.; Rossi-Fanelli, F. Plasma and CSF tryptophan in cancer anorexia. J. Neural Transm. 1990, 81, 225–233. [Google Scholar] [CrossRef] [PubMed]
- J. Gatfield, C. J. Gatfield, C. Brisbare-Roch, F. Jenck, C. Boss, Orexin receptor antagonists: a new concept in CNS disorders? Chem. Med.Chem. 1197; 5. [Google Scholar]
- C.F. Elias, C.B. C.F. Elias, C.B. Saper, E. Maratos-Flier, N.A. Tritos, C. Lee, J. Kelly, J.B. Tatro, G.E. Hoffman, M.M. Ollmann, G.S. Barsh, T. Sakurai, M. Yanagisawa, J.K. Elmquist, Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J. Comp. Neurol. 1998. [Google Scholar]
- Sakurai, T.; Amemiya, A.; Ishii, M.; Matsuzaki, I.; Chemelli, R.M.; Tanaka, H.; Williams, S.C.; Richardson, J.A.; Kozlowski, G.P.; Wilson, S.; et al. Orexins and Orexin Receptors: A Family of Hypothalamic Neuropeptides and G Protein-Coupled Receptors that Regulate Feeding Behavior. Cell 1998, 92, 573–585. [Google Scholar] [CrossRef]
- Yamanaka, A.; Sakurai, T.; Katsumoto, T.; Yanagisawa, M.; Goto, K. Chronic intracerebroventricular administration of orexin-A to rats increases food intake in daytime, but has no effect on body weight. 849. [CrossRef]
- Cai, X.J.; Widdowson, P.S.; Harrold, J.; Wilson, S.; E Buckingham, R.; Arch, J.R.; Tadayyon, M.; Clapham, J.C.; Wilding, J.; Williams, G. Hypothalamic orexin expression: modulation by blood glucose and feeding. Diabetes 1999, 48, 2132–2137. [Google Scholar] [CrossRef]
- Grossberg, A.J.; Zhu, X.; Leinninger, G.M.; Levasseur, P.R.; Braun, T.P.; Myers, M.G.; Marks, D.L. Inflammation-Induced Lethargy Is Mediated by Suppression of Orexin Neuron Activity. J. Neurosci. 2011, 31, 11376–11386. [Google Scholar] [CrossRef]
- Guo, F.; Xu, L.; Gao, S.; Sun, X.; Zhang, N.; Gong, Y. Effect of orexin-A in the arcuate nucleus on cisplatin-induced gastric side effects in rats. Neurosci. Res. 2019, 143, 53–60. [Google Scholar] [CrossRef]
- Oh-I, S.; Shimizu, H.; Satoh, T.; Okada, S.; Adachi, S.; Inoue, K.; Eguchi, H.; Yamamoto, M.; Imaki, T.; Hashimoto, K.; et al. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature 2006, 443, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Rupp, S.K.; Stengel, A. Interactions between nesfatin-1 and the autonomic nervous system—An overview. Peptides 2022, 149, 170719. [Google Scholar] [CrossRef] [PubMed]
- D. Stephan, N. D. Stephan, N. Taege, R. Dore, J. Folberth, O. Jöhren, M. Schwaninger, H. Lehnert, C. Schulz, Knockdown of Endogenous Nucb2/Nesfatin-1 in the PVN Leads to Obese-Like Phenotype and Abolishes the Metformin- and Stress-Induced Thermogenic Response in Rats. Horm. Metab. Res. 2022; 54. [Google Scholar]
- L. Ren, D. L. Ren, D. Bao, L. Wang, Q. Xu, Y. Xu, Z. Shi, Nucleobindin-2/nesfatin-1 enhances the cell proliferation, migration, invasion and epithelial-mesenchymal transition in gastric carcinoma. J. Cell. Mol. Med. 4986; 26. [Google Scholar]
- S. Ning, C. S. Ning, C. Liu, K. Wang, Y. Cai, Z. Ning, M. Li, L. Zeng, NUCB2/Nesfatin-1 drives breast cancer metastasis through the up-regulation of cholesterol synthesis via the mTORC1 pathway. J. Transl. Med. 2023; 21. [Google Scholar]
- Burgos, J.R.; Iresjö, B.-M.; Smedh, U. MCG101-induced cancer anorexia-cachexia features altered expression of hypothalamic Nucb2 and Cartpt and increased plasma levels of cocaine- and amphetamine-regulated transcript peptides. Oncol. Rep. 2016, 35, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- G.L.C. Yosten, W.K. G.L.C. Yosten, W.K. Samson, Nesfatin-1 exerts cardiovascular effects in brain: possible interaction with the central melanocortin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009. [Google Scholar]
- Y. Maejima, U. Y. Maejima, U. Sedbazar, S. Suyama, D. Kohno, E. Takano, N. Yoshida, M. Koike, Y. Uchiyama, K. Fujiwara, T. Yashiro, T.L. Horvath, M.O. Dietrich, S. Tanaka, K. Dezaki, S. Oh -I, K. Hashimoto, H. Shimizu, M. Nakata, M. Mori, T. Yada, Nesfatin-1-regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin-independent melanocortin pathway. 2009; 10. [Google Scholar]
- Stengel, A.; Goebel, M.; Wang, L.; Rivier, J.; Kobelt, P.; Mönnikes, H.; Lambrecht, N.W.G.; Taché, Y. Central Nesfatin-1 Reduces Dark-Phase Food Intake and Gastric Emptying in Rats: Differential Role of Corticotropin-Releasing Factor2 Receptor. Endocrinology 2009, 150, 4911–4919. [Google Scholar] [CrossRef]
- Price, C.J.; Samson, W.K.; Ferguson, A.V. Nesfatin-1 inhibits NPY neurons in the arcuate nucleus. Brain Res. 2008, 1230, 99–106. [Google Scholar] [CrossRef]
- M. Tanida, H. M. Tanida, H. Gotoh, N. Yamamoto, M. Wang, Y. Kuda, Y. Kurata, M. Mori, T. 3725; 64. [Google Scholar]
- Liu, Y.; Chen, X.; Qu, Y.; Song, L.; Lin, Q.; Li, M.; Su, K.; Li, Y.; Dong, J. Central nesfatin-1 activates lipid mobilization in adipose tissue and fatty acid oxidation in muscle via the sympathetic nervous system. BioFactors 2020, 46, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M. Modern science versus the stigma of obesity. Nat. Med. 2004, 10, 563–569. [Google Scholar] [CrossRef]
- Baumann, H.; Morella, K.K.; White, D.W.; Dembski, M.; Bailon, P.S.; Kim, H.; Lai, C.F.; A Tartaglia, L. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc. Natl. Acad. Sci. 1996, 93, 8374–8378. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smart, J.L.; Rubinstein, M.; Cerdán, M.G.; Diano, S.; Horvath, T.L.; Cone, R.D.; Low, M.J. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 2001, 411, 480–484. [Google Scholar] [CrossRef]
- Ghamari-Langroudi, M.; Srisai, D.; Cone, R.D. Multinodal regulation of the arcuate/paraventricular nucleus circuit by leptin. Proc. Natl. Acad. Sci. 2010, 108, 355–360. [Google Scholar] [CrossRef]
- Simons, J.P.F.; Schols, A.M.; Campfield, L.A.; Wouters, E.F.; Saris, W.H. Plasma Concentration of Total Leptin and Human Lung-Cancer-Associated Cachexia. Clin. Sci. 1997, 93, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Smiechowska, J.; Utech, A.; Taffet, G.; Hayes, T.; Marcelli, M.; Garcia, J.M. Adipokines in Patients with Cancer Anorexia and Cachexia. J. Investig. Med. 2010, 58, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Bing, C.; Taylor, S.; Tisdale, M.J.; Williams, G. Cachexia in MAC16 adenocarcinoma: suppression of hunger despite normal regulation of leptin, insulin and hypothalamic neuropeptide Y. J. Neurochem. 2001, 79, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.D.; Anderson, K.D.; Sleeman, M.W.; Wong, V.; Tan, J.; Hijarunguru, A.; Corcoran, T.L.; Murray, J.D.; Thabet, K.E.; Yancopoulos, G.D.; et al. Ciliary neurotrophic factor activates leptin-like pathways and reduces body fat, without cachexia or rebound weight gain, even in leptin-resistant obesity. Proc. Natl. Acad. Sci. 2001, 98, 4652–4657. [Google Scholar] [CrossRef]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin Is a Growth-Hormone-Releasing Acylated Peptide from Stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Nakazato, M.; Murakami, N.; Date, Y.; Kojima, M.; Matsuo, H.; Kangawa, K.; Matsukura, S. A role for ghrelin in the central regulation of feeding. Nature 2001, 409, 194–198. [Google Scholar] [CrossRef]
- Willesen, M.G.; Kristensen, P.; Rømer, J. Co-Localization of Growth Hormone Secretagogue Receptor and NPY mRNA in the Arcuate Nucleus of the Rat. Neuroendocrinology 1999, 70, 306–316. [Google Scholar] [CrossRef]
- Kamegai, J.; Tamura, H.; Shimizu, T.; Ishii, S.; Sugihara, H.; Wakabayashi, I. Chronic Central Infusion of Ghrelin Increases Hypothalamic Neuropeptide Y and Agouti-Related Protein mRNA Levels and Body Weight in Rats. Diabetes 2001, 50, 2438–2443. [Google Scholar] [CrossRef]
- Cowley, M.A.; Smith, R.G.; Diano, S.; Tschöp, M.; Pronchuk, N.; Grove, K.L.; Strasburger, C.J.; Bidlingmaier, M.; Esterman, M.; Heiman, M.L.; et al. The Distribution and Mechanism of Action of Ghrelin in the CNS Demonstrates a Novel Hypothalamic Circuit Regulating Energy Homeostasis. Neuron 2003, 37, 649–661. [Google Scholar] [CrossRef]
- Chen, S.; Chen, H.; Zhou, J.; Pradhan, G.; Sun, Y.; Pan, H.; Li, D. Ghrelin receptors mediate ghrelin-induced excitation of agouti-related protein/neuropeptide Y but not pro-opiomelanocortin neurons. J. Neurochem. 2017, 142, 512–520. [Google Scholar] [CrossRef]
- Date, Y.; Murakami, N.; Toshinai, K.; Matsukura, S.; Niijima, A.; Matsuo, H.; Kangawa, K.; Nakazato, M. The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 2002, 123, 1120–1128. [Google Scholar] [CrossRef]
- Shimizu, Y.; Nagaya, N.; Isobe, T.; Imazu, M.; Okumura, H.; Hosoda, H.; Kojima, M.; Kangawa, K.; Kohno, N. Increased plasma ghrelin level in lung cancer cachexia. . 2003, 9, 774–8. [Google Scholar] [PubMed]
- Garcia, J.M.; Garcia-Touza, M.; Hijazi, R.A.; Taffet, G.; Epner, D.; Mann, D.; Smith, R.G.; Cunningham, G.R.; Marcelli, M. Active Ghrelin Levels and Active to Total Ghrelin Ratio in Cancer-Induced Cachexia. J. Clin. Endocrinol. Metab. 2005, 90, 2920–2926. [Google Scholar] [CrossRef] [PubMed]
- Murata, D.; Azuma, K.; Matsuo, N.; Murotani, K.; Matama, G.; Kawahara, A.; Sasada, T.; Tokito, T.; Hoshino, T. Survival and biomarkers for cachexia in non-small cell lung cancer receiving immune checkpoint inhibitors. Cancer Med. 2023, 12, 19471–19479. [Google Scholar] [CrossRef]
- Wang, W.; Andersson, M.; Iresjö, B.-M.; Lönnroth, C.; Lundholm, K. Effects of ghrelin on anorexia in tumor-bearing mice with eicosanoid-related cachexia. Int. J. Oncol. 2006, 28, 1393–1400. [Google Scholar] [CrossRef]
- K. Terawaki, Y. K. Terawaki, Y. Kashiwase, Y. Sawada, H. Hashimoto, M. Yoshimura, K. Ohbuchi, Y. Sudo, M. Suzuki, K. Miyano, S. Shiraishi, Y. Higami, K. Yanagihara, T. Hattori, Y Kase, Y. Ueta, Y Uezono, Development of ghrelin resistance in a cancer cachexia rat model using human gastric cancer-derived 85As2 cells and the palliative effects of the Kampo medicine rikkunshito on the model. 0173; 12. [Google Scholar]
- Yakabi, K.; Kurosawa, S.; Tamai, M.; Yuzurihara, M.; Nahata, M.; Ohno, S.; Ro, S.; Kato, S.; Aoyama, T.; Sakurada, T.; et al. Rikkunshito and 5-HT2C receptor antagonist improve cisplatin-induced anorexia via hypothalamic ghrelin interaction. Regul. Pept. 2010, 161, 97–105. [Google Scholar] [CrossRef]
- Tsubouchi, H.; Yanagi, S.; Miura, A.; Matsumoto, N.; Kangawa, K.; Nakazato, M. Ghrelin relieves cancer cachexia associated with the development of lung adenocarcinoma in mice. Eur. J. Pharmacol. 2014, 743, 1–10. [Google Scholar] [CrossRef]
- DeBoer, M.D.; Zhu, X.X.; Levasseur, P.; Meguid, M.M.; Suzuki, S.; Inui, A.; Taylor, J.E.; Halem, H.A.; Dong, J.Z.; Datta, R.; et al. Ghrelin Treatment Causes Increased Food Intake and Retention of Lean Body Mass in a Rat Model of Cancer Cachexia. Endocrinology 2007, 148, 3004–3012. [Google Scholar] [CrossRef]
- L.J. Rohrbasser, H. L.J. Rohrbasser, H. Alsaffar, J. Blair, in Principles of Endocrinology and Hormone Action, ed. by A. Belfiore, D. LeRoith (Springer, Cham, Switzerland, 2018) P.
- Goldberg, A.L. Protein turnover in skeletal muscle. II. Effects of denervation and cortisone on protein catabolism in skeletal muscle.. 1969, 244, 3223–9. [Google Scholar]
- Cala, M.P.; Agulló-Ortuño, M.T.; Prieto-García, E.; González-Riano, C.; Parrilla-Rubio, L.; Barbas, C.; Díaz-García, C.V.; García, A.; Pernaut, C.; Adeva, J.; et al. Multiplatform plasma fingerprinting in cancer cachexia: a pilot observational and translational study. J. Cachex- Sarcopenia Muscle 2018, 9, 348–357. [Google Scholar] [CrossRef]
- Russell, S.T.; Tisdale, M.J. The role of glucocorticoids in the induction of zinc-α2-glycoprotein expression in adipose tissue in cancer cachexia. Br. J. Cancer 2005, 92, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Costelli, P.; Carbó, N.; Tessitore, L.; Bagby, G.J.; Lopez-Soriano, F.J.; Argilés, J.M.; Baccino, F.M. Tumor necrosis factor-alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. J. Clin. Investig. 1993, 92, 2783–2789. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, K.; Kagaya, S.; Takayasu, S.; Hanada, K.; Iwasaki, Y.; Suda, T. Cytokines Induce NF-ĸB, Nurr1 and Corticotropin-Releasing Factor Gene Transcription in Hypothalamic 4B Cells. Neuroimmunomodulation 2010, 17, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Tasso, M.; Kageyama, K.; Iwasaki, Y.; Watanuki, Y.; Niioka, K.; Takayasu, S.; Daimon, M. Growth differentiation factor-15 stimulates the synthesis of corticotropin-releasing factor in hypothalamic 4B cells. Peptides 2023, 170, 171112. [Google Scholar] [CrossRef]
- Uehara, A.; Sekiya, C.; Takasugi, Y.; Namiki, M.; Arimura, A.; Grill, H.J.; Carmody, J.S.; Sadacca, L.A.; Williams, D.L.; Kaplan, J.M.; et al. Anorexia induced by interleukin 1: involvement of corticotropin-releasing factor. Am. J. Physiol. Integr. Comp. Physiol. 1989, 257, R613–R617. [Google Scholar] [CrossRef]
- Gotoh, K.; Masaki, T.; Chiba, S.; Ando, H.; Fujiwara, K.; Shimasaki, T.; Mitsutomi, K.; Katsuragi, I.; Kakuma, T.; Sakata, T.; et al. Brain-derived neurotrophic factor, corticotropin-releasing factor, and hypothalamic neuronal histamine interact to regulate feeding behavior. J. Neurochem. 2013, 125, 588–598. [Google Scholar] [CrossRef]
- Liposits, Z.; Phelix, C.; Paull, W.K. Synaptic interaction of serotonergic axons and corticotropin releasing factor (CRF) synthesizing neurons in the hypothalamic paraventricular nucleus of the rat. Histochem. 1987, 86, 541–549. [Google Scholar] [CrossRef]
- Van de Kar, L.D.; Javed, A.; Zhang, Y.; Serres, F.; Raap, D.K.; Gray, T.S. 5-HT2AReceptors Stimulate ACTH, Corticosterone, Oxytocin, Renin, and Prolactin Release and Activate Hypothalamic CRF and Oxytocin-Expressing Cells. J. Neurosci. 2001, 21, 3572–3579. [Google Scholar] [CrossRef]
- Lu, X.-Y.; Barsh, G.S.; Akil, H.; Watson, S.J. Interaction between α-Melanocyte-Stimulating Hormone and Corticotropin-Releasing Hormone in the Regulation of Feeding and Hypothalamo-Pituitary-Adrenal Responses. J. Neurosci. 2003, 23, 7863–7872. [Google Scholar] [CrossRef]
- A.V. Vergoni, A. A.V. Vergoni, A. Bertolini, J.E. Wikberg, H.B. Schioth Corticotropin-releasing factor (CRF) induced anorexia is not influenced by a melanocortin 4 receptor blockage. 1999; 20. [Google Scholar]
- S. Kawashima, S. S. Kawashima, S. Sakihara, K. Kageyama, T. Nigawara, T. Suda, Corti cotropin-releasing factor (CRF) is involved in the acute anorexic effect of α-melanocyte-stimulating hormone: A study using CRF-deficient mice. 2169; 29. [Google Scholar]
- Wang, J.; Sun, L.; You, J.; Peng, H.; Yan, H.; Wang, J.; Sun, F.; Cui, M.; Wang, S.; Zhang, Z.; et al. Role and mechanism of PVN–sympathetic–adipose circuit in depression and insulin resistance induced by chronic stress. Embo Rep. 2023, 24, e57176. [Google Scholar] [CrossRef]
- M. Erdem, D. M. Erdem, D. Möckel, S. Jumpertz, C. John, A. Fragoulis, I. Rudolph, J. Wulfmeier, J. Springer, H. Horn, M. Koch, G. Lurje, T. Lammers, S. Olde Damink, G. van der Kroft, F. Gremse, T. Cramer, Macrophages protect against loss of adipose tissue during cancer cachexia J. 1128; 10. [Google Scholar]
- Wang, P.; Loh, K.H.; Wu, M.; Morgan, D.A.; Schneeberger, M.; Yu, X.; Chi, J.; Kosse, C.; Kim, D.; Rahmouni, K.; et al. A leptin–BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 2020, 583, 839–844. [Google Scholar] [CrossRef] [PubMed]
- An, J.J.; Liao, G.-Y.; Kinney, C.E.; Sahibzada, N.; Xu, B. Discrete BDNF Neurons in the Paraventricular Hypothalamus Control Feeding and Energy Expenditure. Cell Metab. 2015, 22, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Lian, H.; Zhang, Y.; Tong, S.; Wang, Z. Dopamine receptor 2 downregulation and brain-derived neurotrophic factor upregulation in the paraventricular nucleus are correlated with brown adipose tissue thermogenesis in rats with bilateral substantia nigra lesions. J. Chem. Neuroanat. 2021, 117, 102016. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



