
Submitted:
12 November 2024
Posted:
13 November 2024
You are already at the latest version
Abstract
Background
Lesch-Nyhan syndrome (LNS) is a rare X-linked recessive metabolic disorder caused by mutations in the HPRT1 gene, resulting in hypoxanthine-guanine phosphoribosyltransferase (HPRT) deficiency. Early diagnosis is critical for optimizing management and improving outcomes. This study presents a case series of three Taiwanese patients diagnosed at a single medical center.
Methods
This case series involved three male patients with LNS. Exome sequencing and biochemical testing were used to confirm the diagnoses. Early clinical manifestations including hyperuricemia, hypotonia, and developmental delay were documented during the initial stages of the disease.
Results
All three patients had hyperuricemia, hypotonia, spasticity, and motor developmental delay. Pathogenic variants in the HPRT1 gene were identified in two patients, while the third was confirmed by biochemical testing. Two patients had orange-colored crystalline deposits in their diapers, indicative of hyperuricosuria. Self-injurious behavior had not yet developed in two patients due to their young age.
Conclusions
Early clinical features such as hyperuricemia, hypotonia, and motor delay may suggest LNS in infancy. Molecular genetic testing, particularly whole exome sequencing, can facilitate an early diagnosis before specific manifestations occur, enabling timely interventions and improving patient outcomes.
Keywords:
Lesch-Nyhan syndrome
; HPRT1 gene
; hyperuricemia
; developmental delay
; hypotonia
; self-mutilation
; hypoxanthine-guanine phosphoribosyltransferase
1. Introduction
Lesch-Nyhan syndrome (LNS) is a rare metabolic disorder first described by Lesch and Nyhan in 1964 [1]. The incidence of LNS is estimated to range between 1 in 235,000 and 1 in 380,000 live births [2,3]. In the UK, the incidence rate has been reported as 0.18 per 100,000 live births [4]. LNS is inherited in an X-linked recessive manner and results from pathogenic variants in the HPRT1 gene, which encodes the enzyme hypoxanthine-guanine phosphoribosyltransferase (HPRT), a key enzyme in the purine salvage pathway [5,6]. The HPRT1 gene is located on the X chromosome at Xq26.2-q26.3 and spans approximately 44 kb of DNA. It comprises 9 exons and 8 introns, and encodes a protein consisting of 218 amino acids with a molecular weight of 24.5 kDa [7,8]. Although a single gene is implicated in LNS, over 600 mutations have been identified, resulting in a spectrum of clinical severity due to varying levels of HPRT enzyme deficiency [7]. Males inheriting the defective X chromosome from carrier mothers manifest the disease, while females are typically carriers, although a few may exhibit symptoms if X-inactivation results in the expression of the defective X chromosome [9,10,11]. Genetic testing, specifically rapid exome sequencing, can facilitate an early diagnosis in infants, with detection possible as early as 3 weeks of age [12].
Patients with LNS typically present with hyperuricemia and neurodevelopmental abnormalities, including global developmental delay, involuntary movements, and characteristic behavioral abnormalities such as self-injurious behavior [11,13,14]. With effective clinical management, the life expectancy of patients with LNS can extend to 20–40 years [15]. Affected individuals may not exhibit apparent neurological dysfunction at birth, and developmental delay and neurological signs typically become evident after several months. By 4 months of age, hypotonia and recurrent vomiting are commonly observed. Poor head control is also considered one of the primary initial manifestations [16,17]. Extrapyramidal signs generally appear between 8 and 12 months [18]. While macrocytosis may be detectable as early as 6 months of age, it appears to progress in severity with advancing age [19]. Self-injurious behavior is rarely the initial presentation, but eventually develops in nearly all patients [14,20]. Cognitive impairment and behavioral disturbances typically emerge between 2 and 3 years of age [21].
Self-injurious behavior may manifest as early as 10 months of age, although in some patients it may not develop until adolescence [1,14,22]. Orange-colored crystalline deposits may occasionally be observed in diapers, caused by excess uric acid production [4,23,24]. This study aimed to describe Taiwanese patients with LNS from a single center, with a focus on enhancing understanding of early disease manifestations and providing a foundation for future management recommendations.
2. Clinical Reports
2.1. Case 1
A 1-year-old infant boy presented with hypotonia, intermittent spasticity, and poor head control. He was born at 39 weeks and 4 days with a birth weight of 3414 g. During a clinical evaluation, persistent hypotonia interspersed with episodes of spasticity, intermittent neck hyperextension, and poor head control were noted. In addition, he had mild hypotonia of the extremities, muscle strength graded at 4-5, brisk deep tendon reflexes (+++), and questionable bilateral ankle clonus. His mother noticed orange-colored crystalline sediment in his diaper when he was 4 months old (Figure 1). Developmentally, he showed significant motor delays, being unable to sit or stand independently by 1 year of age.
The family history was notable for a maternal uncle with suspected cerebral palsy who had similar neurological symptoms, including psychomotor delay, self-injurious behavior, and episodes of choking. Cardiac evaluation via echocardiography identified a patent foramen ovale measuring 0.187 cm. Brain magnetic resonance imaging (MRI) revealed mild enlargement of the subarachnoid spaces, particularly in the bilateral anterior frontal and temporal regions, as well as mild prominence of the lateral and third ventricles, suggestive of external hydrocephalus. Brain ultrasonography identified bilateral frontal horn cysts, a left subependymal cyst, and subdural fluid collection.
The patient had normal complete blood count, liver function, renal function, and electrolytes, and thyroid function tests were within normal limits. Venous blood gas analysis revealed a pH of 7.39 (reference range: 7.35–7.45), PCO2 of 42 mmHg (32–45), PO2 of 32 mmHg (20–49), HCO3- of 25 mmol/L (20–26), and a base excess of 0.3 mmol/L (-2–2). Further metabolic investigations demonstrated an elevated plasma total carnitine level of 81.9 μmol/L (reference range: 30.5–75.9), with a free carnitine level of 55.6 μmol/L (23.6–65.4) and bound carnitine level of 26.3 μmol/L (1.0–16.8). In addition, creatine kinase was elevated at 376 U/L (30–223), and he had hyperuricemia with a serum uric acid level of 10.2 mg/dL (reference range: 3.5–7.2). Based on these findings, advanced genetic counseling and further diagnostic evaluations were recommended.
Whole exome sequencing identified a splice site acceptor mutation at chromosome X position 134498607 (ChrX:134498607), specifically NM_000194.3
c.533-1G>C in the HPRT1 gene. Follow-up Sanger sequencing was conducted on the proband’s family members, and the results confirmed that both the mother and maternal grandmother were carriers of the HPRT1 c.533-1G>C variant in a heterozygous state. LNS was diagnosed at the age of 10 months, and he was initiated on pharmacological management including allopurinol, clonazepam, and carbidopa/levodopa. The family also considered hematopoietic stem cell transplantation as a potential therapeutic option.
2.2. Case 2
A male infant, aged 1 year and 2 months, born at a gestational age of 39 weeks and 3 days via Cesarean section due to placenta previa, presented with multiple medical complications. His birth weight was 3850 g, and his birth history was G1P1. Postnatally, he exhibited poor weight gain, prompting further evaluation. The nephrologist suspected renal tubular acidosis type I and nephrocalcinosis, leading to the initiation of sodium bicarbonate supplementation. The family reported observing occasional yellowish crystals in his urine, and also poor sleep quality with frequent startle reflexes.
At 4 months of age, he developed seizure activity, necessitating multiple hospital admissions. The seizures were managed with antiepileptic drugs, including levetiracetam and clonazepam. Associated symptoms included metabolic acidosis, poor appetite, and frequent regurgitation. Hyperuricemia was noted, raising the suspicion of LNS. A physical examination revealed hypotonia with intermittent spasticity and hypermobile joints. Brain ultrasonography and echocardiography were unremarkable; however, renal ultrasound at 1 year and 1 month revealed bilateral nephrocalcinosis and mild left hydronephrosis. Electroencephalography performed at 1 year and 2 months showed normal findings. Involuntary movements were noted during periods of excitement, and he began treatment with S-adenosylmethionine at 1 year and 3 months. Metabolic investigations during early infancy revealed abnormalities in renal function and metabolic acidosis. Further metabolic investigations demonstrated a reduced plasma total carnitine level of 39.6 μmol/L (reference range: 45.7–63.2), with a free carnitine level of 33.4 μmol/L (37.0–50.5) and bound carnitine level of 6.2 μmol/L (4.8–16.5). Urine organic acid analysis showed elevations in acetoacetic acid, vanillic acid, and sebacic acid. A urinary Benedict’s test was positive, and hyperuricemia was noted with a serum uric acid level of 17.3 mg/dL (reference range: 4.4–7.6). The urine urate-to-creatinine ratio was significantly elevated at 4.0 (111.2/27.6).
Genetic testing via Sanger sequencing of HPRT1 revealed a hemizygous guanine duplication at nucleotide position 212 (c.212dupG), resulting in a frameshift mutation and premature termination codon at amino acid position 71 (p.Gly71fs*3). This mutation produces a truncated and likely non-functional protein, consistent with the diagnosis of LNS. The diagnosis was confirmed at 1 year and 3 months of age. According to medical records, treatment included febuxostat, potassium citrate, S-adenosylmethionine, and diazepam. Unfortunately, he was lost to follow-up at 1 year and 6 months of age.
2.3. Case 3
This male patient presented to our outpatient department at the age of 4 years. He was born via Cesarean section at a gestational age of 39 weeks, with a birth weight of 2500 g and birth history of G1P1. Clinically, had had features reminiscent of cerebral palsy. A physical examination revealed dystonia, spasticity, bilateral ankle clonus, disuse muscle atrophy, and joint contractures involving both hands and ankles. A self-inflicted wound on the lower lip was observed. Hypotonia had been noted between the ages of 1 and 3 years. By the age of 5, he was able to ambulate with the assistance of a walker, but he developed hypertonia by 6 years of age. He had previously undergone a surgical intervention for C1-C2 subluxation. At the age of 10, due to respiratory failure, a tracheostomy was performed. Developmental milestones were delayed throughout infancy and early childhood. Brain MRI identified a focal bony fragment at the odontoid process with a well-defined border, suggestive of either os odontoideum or an old fracture of the odontoid process, with suspicion of focal narrowing of the upper cervical cord on the sagittal section. Additional findings included left-sided mastoiditis and bilateral sphenoid sinusitis, as well as a relatively brachycephalic skull configuration. Renal ultrasonography revealed bilateral nephrolithiasis with multiple stones in both kidneys, and a staghorn calculus was suspected in the right kidney. Non-contrast computed tomography of the abdomen confirmed the presence of bilateral renal stones, bladder stones, and a suspected stone in the left distal ureter.
Laboratory investigations showed an elevated serum uric acid level (12.1 mg/dL) and macrocytic anemia. Further testing at the Baylor College of Medicine in the USA revealed undetectable HPRT activity (0 nmol/min/mg protein; control range: 1.0–3.4 nmol/min/mg protein; affected control: 0.0 nmol/min/mg protein). Based on these findings, a diagnosis of LNS was confirmed when the patient was 12 years old. He was treated with allopurinol, folic acid, potassium citrate, S-adenosyl methionine, and piracetam during the therapeutic course. Unfortunately, the patient was lost to follow-up at the age of 24.
3. Discussion
Currently, the diagnosis of LNS mainly relies on molecular genetic testing and/or low HPRT enzyme activity. In most patients, the disease is caused by a hemizygous pathogenic variant in the HPRT1 gene in a male proband. Due to the potential lack of early clinical manifestations, hyperuricemia and hyperuricosuria may be among the first indicators of the disease. Some patients may present with normal serum uric acid levels but an elevated urine uric acid-to-creatinine ratio greater than 2 [25]. Due to the overproduction of uric acid, hyperuricemia or hyperuricosuria may manifest early in life. Orange-colored crystalline deposits in diapers and uric acid sediment in the kidneys can be observed as early as a few weeks of age [18]. In our series, all of the patients had hyperuricemia, with orange-colored crystalline deposits observed in the diapers of two patients during the first few months of life. In addition, hypotonia was observed in all patients as early as 4 to 6 months of age, accompanied by poor head control. Intermittent spasticity and hypotonia were frequently noted in subsequent evaluations. All three patients had developmental delay, with a particular emphasis on delayed motor function. None were able to sit independently, crawl, or walk without assistance. In comparison to previous studies, all three of our patients presented with hyperuricemia or hyperuricosuria, hypotonia, spasticity, and developmental delay [16]. These features may serve as early indicators of LNS during infancy. Self-injurious behavior typically emerges between 2 and 3 years of age; however, there have been reports of patients presenting as early as 10 months of age. Self-mutilation was not observed in two of our patients, possibly due to their young age. Although macrocytic erythrocytes are present in 81–92% of individuals with LNS or its neurological variants, macrocytic anemia was observed only in the older patient in this study, suggesting that this symptom may emerge in the later stages of disease progression [19]. While cognitive impairment may be a feature in all patients, it is challenging to accurately assess cognitive function during infancy. The clinical characteristics of our patients highlight the distinctive features of LNS (Table 1).
Molecular genetic testing, specifically sequencing of the HPRT1 gene or comprehensive genomic testing, offers a precise diagnostic tool for LNS. Biochemical testing, such as assessing HPRT enzyme activity, is an alternative method to confirm the diagnosis. However, an increasing number of patients are now being diagnosed through next-generation sequencing, which is becoming more prevalent than enzyme activity testing. One of our patients received whole exome sequencing when he was 9 months old, showing that a diagnosis can be made before all of the manifestations occur. Comprehensive evaluation and management led to improved outcomes and a slower progression of the disease in our patients (Table 2). In summary, hyperuricemia or hyperuricosuria, hypotonia, spasticity, and developmental delay may present in early infancy in patients with LNS. Our findings suggest that clinicians should consider LNS in patients presenting with these early manifestations.
Author Contributions
H.-H.F. was the primary author of the manuscript. S.-P.L., H.-Y.L., H.-J.C. and C.-L.L. contributed to patient follow-up and provided valuable input during the manuscript preparation. C.-K.C., Y.-R.T. and Y.-T.L. were responsible for conducting biochemical analyses and critically revising the manuscript. Y.-H.C. and H.-C.C. assisted with patient screening and provided crucial revisions to the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received grants from MacKay Memorial Hospital (MMH-MM-113-13, MMH E-113-13, MMH-MM-112-14, MMH-E-112-13, and MMH-E-111-13) and the Ministry of Science and Technology, Executive Yuan, Taiwan (NSTC-113-2314-B-195-003, NSTC-113-2314-B-195-004, NSTC-113-2314-B-715-002, NSTC-113-2314-B-195-021, NSTC-113-2811-B-195-001, NSTC-112-2314-B-195-014-MY3, NSTC-112-2811-B-195-001, NSTC-112-2314-B-195-003, NSTC-111-2314-B-195-017, NSTC-111-2811-B-195-002, NSTC-111-2811-B-195-001, NSTC-110-2314-B-195-014, NSTC-110-2314-B 195-010-MY3, and NSTC-110-2314-B-195-029). The authors confirm independence from the sponsors. The contents of the article, including the design of the study and collection, analysis, and interpretation of data, and writing of the manuscript, have not been influenced by the sponsors.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of MacKay Memorial Hospital (reference number: 21MMHIS109e, approval date: 1 October 2021).
Informed Consent Statement
Written informed consent has been obtained from the patients to publish this paper.
Data Availability Statement
The original data and findings obtained during this study are fully reported in the manuscript. The corresponding authors can be contacted for additional data or information inquiries.
Acknowledgments
We extend our heartfelt gratitude to the patients and families affected by LNS who participated in this study. We also sincerely appreciate the tireless efforts and dedication of the clinical staff and research laboratory personnel, without whom this study would not have been possible.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
LNS: Lesch-Nyhan syndrome
HPRT: hypoxanthine-guanine phosphoribosyltransferase
MRI: magnetic resonance imaging
References
- LESCH M, NYHAN WL. A FAMILIAL DISORDER OF URIC ACID METABOLISM AND CENTRAL NERVOUS SYSTEM FUNCTION. Am J Med. 1964 Apr;36:561-70. [CrossRef]
- Roche A, Pérez-Dueñas B, Camacho JA, Torres RJ, Puig JG, García-Cazorla A, Artuch R. Efficacy of rasburicase in hyperuricemia secondary to Lesch-Nyhan syndrome. Am J Kidney Dis. 2009 Apr;53(4):677-80. [CrossRef]
- Shoptaw JT, Reznik JI. Lesch-Nyhan Syndrome: report of three cases in one family. ASDC J Dent Child. 1978 Sep-Oct;45(5):403-7.
- McCarthy GT, Green EM, Ogunbona O, Simmonds HA, Fairbanks L, Pountney T, Bryant E. A population study of Lesch-Nyhan disease in the UK. Dev Med Child Neurol. 2011 Jan;53(1):34-9. [CrossRef]
- Jinnah HA, De Gregorio L, Harris JC, Nyhan WL, O’Neill JP. The spectrum of inherited mutations causing HPRT deficiency: 75 new cases and a review of 196 previously reported cases. Mutat Res. 2000 Oct;463(3):309-26. [CrossRef]
- Seegmiller JE, Rosenbloom FM, Kelley WN. Enzyme defect associated with a sex-linked human neurological disorder and excessive purine synthesis. Science. 1967 Mar 31;155(3770):1682-4. [CrossRef]
- Fu R, Ceballos-Picot I, Torres RJ, Larovere LE, Yamada Y, Nguyen KV, Hegde M, Visser JE, Schretlen DJ, Nyhan WL, Puig JG, O’Neill PJ, Jinnah HA; Lesch-Nyhan Disease International Study Group. Genotype-phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain. 2014 May;137(Pt 5):1282-303. [CrossRef]
- Torres RJ, Puig JG. Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J Rare Dis. 2007 Dec 8;2:48. [CrossRef]
- Nguyen KV, Silva S, Troncoso M, Naviaux RK, Nyhan WL. Lesch-Nyhan disease in two families from Chiloé Island with mutations in the HPRT1 gene. Nucleosides Nucleotides Nucleic Acids. 2017 Jul 3;36(7):452-462. [CrossRef]
- AlBakheet A, AlQudairy H, Alkhalifah J, Almoaily S, Kaya N, Rahbeeni Z. Detailed genetic and clinical analysis of a novel de novo variant in HPRT1: Case report of a female patient from Saudi Arabia with Lesch-Nyhan syndrome. Front Genet. 2023 Jan 26;13:1044936.
- Nanagiri A, & Shabbir N. (2024). Lesch-Nyhan Syndrome. In StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Nadeem Shabbir declares no relevant financial relationships with ineligible companies.
- Zhang T, Briere JM, Leeman KT, Wojcik MH, Agrawal PB. Ethical implications of early genetic diagnosis in an infant with Lesch-Nyhan syndrome. J Genet Couns. 2022 Dec;31(6):1434-1437. [CrossRef]
- Fu R, Sutcliffe D, Zhao H, Huang X, Schretlen DJ, Benkovic S, Jinnah HA. Clinical severity in Lesch-Nyhan disease: the role of residual enzyme and compensatory pathways. Mol Genet Metab. 2015 Jan;114(1):55-61. [CrossRef]
- Jathar P, Panse AM, Jathar M, Gawali PN. Lesch-Nyhan Syndrome: Disorder of Self-mutilating Behavior. Int J Clin Pediatr Dent. 2016 Apr-Jun;9(2):139-42. [CrossRef]
- Jinnah HA. (1993). HPRT1 Disorders. In M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, & A. Amemiya (Eds.), GeneReviews((R)). Seattle (WA).
- Li L, Qiao X, Liu F, Wang J, Shen H, Fu H, Mao JH. Description of the Molecular and Phenotypic Spectrum of Lesch-Nyhan Disease in Eight Chinese Patients. Front Genet. 2022 Apr 26;13:868942. [CrossRef]
- Yang L, Guo H. Case report: Early-onset renal failure as presenting sign of Lesch-Nyhan disease in infancy. Front Pediatr. 2022 Dec 19;10:1080486. [CrossRef]
- Nasser S, Nasser H, Michael J, N E, N B, Soboh S, Nasser W. Lesch-Nyhan syndrome. Case report and review of literature. Nephrol Renal Dis. 2020;5:1.
- Cakmakli HF, Torres RJ, Menendez A, Yalcin-Cakmakli G, Porter CC, Puig JG, Jinnah HA. Macrocytic anemia in Lesch-Nyhan disease and its variants. Genet Med. 2019 Feb;21(2):353-360. [CrossRef]
- Bozano A, Schiaffino A, Spessa A, Valeriani F, Mancinelli R, Micheli V, Dolcetta D. Description of the Lesch-Nyhan neurobehavioral disorder and its management through participant observation of three young individuals. JIMD Rep. 2020 Feb 18;52(1):63-71. [CrossRef]
- Anderson LT, Ernst M. Self-injury in Lesch-Nyhan disease. J Autism Dev Disord. 1994 Feb;24(1):67-81. [CrossRef]
- Jinnah HA, Ceballos-Picot I, Torres RJ, Visser JE, Schretlen DJ, Verdu A, Laróvere LE, Chen CJ, Cossu A, Wu CH, Sampat R, Chang SJ, de Kremer RD, Nyhan W, Harris JC, Reich SG, Puig JG; Lesch-Nyhan Disease International Study Group. Attenuated variants of Lesch-Nyhan disease. Brain. 2010 Mar;133(Pt 3):671-89.
- Gasperini S, Stagi S, Gasperini U, Guerrini R, la Marca G, Donati MA. Orange-colored diapers as first sign of Lesch-Nyhan disease in an asymptomatic infant. Pediatr Nephrol. 2010 Nov;25(11):2373-4. [CrossRef]
- Ambarsari CG, Cahyadi D, Sari L, Satria O, Sahli F, Darmadi TL, Kadaristiana A. Late diagnosis of Lesch-Nyhan disease complicated with end-stage renal disease and tophi burst: a case report. Ren Fail. 2020 Nov;42(1):113-121. [CrossRef]
- Tsai JD, Chen SM, Lin CH, Ku MS, Tsao TF, Sheu JN. Normal uricemia in Lesch-Nyhan syndrome and the association with pulmonary embolism in a young child-a case report and literature review. Pediatr Neonatol. 2014 Aug;55(4):312-5. [CrossRef]
Figure 1.
Orange-colored crystalline sediment in the diaper was noted when the baby (case 1) was 4 months old.
Figure 1.
Orange-colored crystalline sediment in the diaper was noted when the baby (case 1) was 4 months old.
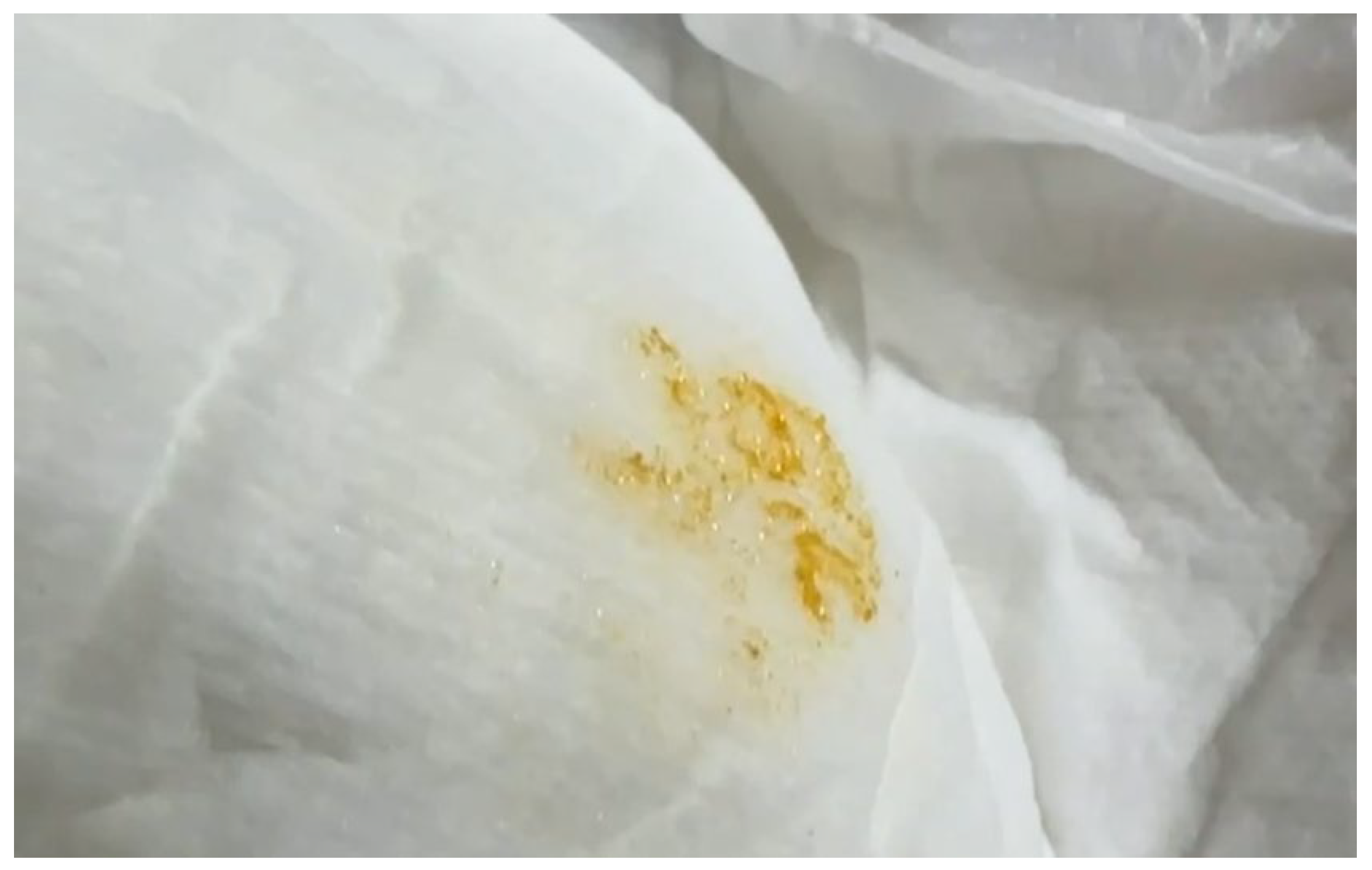

Table 1.
Clinical Characteristics of Three Patients with Lesch-Nyhan Syndrome at Our Hospital.
| Patients | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Sex | male | male | male |
| Year of birth | 2023 | 2013 | 1995 |
| Age at diagnosis | 10M | 1Y3M | 12Y |
| Diagnostic strategy | Whole exome sequencing: c.533-1G>C/Hemizygote |
HPRT1 gene sequencing: c.212dupG, p.Gly71fs*3/Hemizygote |
Hypoxanthine phosphoribosyl- transferase activity: 0 nmol/min/mg protein |
| Hyperuricemia | 10.2 mg/dl | 17.3 mg/dl | 12.1 mg/dl |
| Nephrolithiasis | NA | + | + |
| Hypotonia/ dystonia |
+ | + | + |
| Spasticity | + | + | + |
| Clonus | + | + | + |
| Cerebral palsy | - | - | + |
| Delayed motor skills | + | + | + |
| Intellectual disability | - | - | + |
| Seizure | - | + | + |
| Self-mutilation | - | - | + |
| Gouty arthritis | - | - | - |
| Macrocytic anemia | - | - | + |
| NA: no assessment, +: present, -: absent | |||
Table 2.
Clinical Evaluation and Management of Three Patients with Lesch-Nyhan Syndrome at Our Hospital.
Table 2.
Clinical Evaluation and Management of Three Patients with Lesch-Nyhan Syndrome at Our Hospital.
| Patients | Case 1 | Case 2 | Case 3 |
|---|---|---|---|
| Neuroimaging | Brain MRI: Mild enlargement of subarachnoid space most pronounced in bilateral anterior frontal and temporal convexities as well as mild prominence of lateral and third ventricles likely external hydrocephalus at this age.Brain ultrasonography:1. Frontal horn cyst, bilateral.2. Subependymal cyst, left3. Subdural fluid collection | Brain ultrasonography: Negative |
Brain MRI: Negative |
| Kidney ultrasonography | NA | Kidney ultrasonography: 1. Bilateral nephrocalcinosis2. Left mild hydronephrosis |
Kidney ultrasonography: 1. Right small kidney 2. Bilateral multiple stones in both kidneys (largest in right pelvis:19 mm, left pelvis: 19 mm) 3. Suspected staghorn stones, bilaterally |
| Treatment | Allopurinol,Clonazepam,Carbidopa/Levodopa | Febuxostat plus Potassium citrate, S-adenosyl methionine,Diazepam | Allopurinol,Folic acid,Potassium citrate,S-adenosyl methionine,Piracetam |
| NA: no assessment | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



