
Submitted:
09 November 2024
Posted:
12 November 2024
You are already at the latest version
Abstract
Periodontal disease (PD) and myeloproliferative neoplasms (MPNs) are linked through common inflammatory mediators and immune dysregulation. Chronic inflammation plays a pivotal role in both conditions, with markers such as Chitinase-3-like protein 1, vascular endothelial growth factor, and interleukin-23 reflecting disease severity in PD and heightened inflammatory responses in MPNs. The NLRP3 and AIM2 inflammasomes are implicated in promoting excessive cytokine release, contributing to the pathogenesis of both diseases. This interplay underscores a bidirectional relationship between PD and MPNs, where pathogen-induced inflammation and immune dysregulation exacerbate disease progression. Understanding these mechanisms is crucial for developing targeted therapeutic strategies to manage inflammation and improve outcomes in affected individuals.
Keywords:
periodontal disease
; essential thrombocythemia
; immune dysregulation
; inflammation
; myelofibrosis
; myeloproliferative neoplasms
; polycythemia vera
; saliva pH
1. Introduction
Periodontal disease (PD) encompass a range of chronic inflammatory conditions affecting the gum, the supporting bone, and the connective tissue fibers that anchor the teeth to the alveolar bone. These diseases involve the soft tissue surrounding the teeth as well as the structures that provide support and stability [1]. Gingivitis is the most common and the incipient form of PD and is caused by the accumulation of bacteria and debris between the gumline and tooth (dental plaque) that consecutively leads to inflammation [2]. Progression of PD beyond the gingiva leads to Periodontitis a more destructive and permanent inflammatory state [3]. In the novel classification of Periodontal/Peri-implant Diseases and conditions, periodontitis can be subdivided in: Necrotizing periodontitis, Periodontitis and Periodontitis as a manifestation of systemic disease [4]. The same classification also includes systemic diseases associated with loss of dental supporting tissues like neoplastic conditions, and these can affect the periodontal apparatus non-related to biofilm induced periodontitis [5]. Tissue damage in PDs is mainly caused by the host's inflammatory response, with complement activation—via microbial components like LPS (lipopolysaccharide) playing a key role [6].
As PD progresses to periodontitis, anaerobic bacteria like Aggregatibacter actinomycetemcomitans and Porphyromonas gingivalis trigger a host inflammatory response, leading to the release of inflammatory markers such as C-reactive protein (CRP), tumor necrosis factor-alpha (TNF-α), matrix metalloproteinases (MMPs) and interleukins. Elevated CRP levels suggest a potential link between periodontitis and cardiovascular disease, while smoking exacerbates the disease by promoting pathogen growth [7,8].
Philadelphia negative-Chronic Myeloproliferative Neoplasms (MPNs) characterized by clonal expansion of hematopoietic cells. Among them they are three most common MPNs: Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF) [9]. Recent reports show that inflammasome NLPR3 genes and different cytokines are highly expressed in MPNs and they contribute to the progression of the disease, especially in patients who are positive for JAK2 V617F variant [10,11]. Moreover, allelic load of the JAK2 variant is correlated with plasma levels of inflammatory cytokines [12]. Inflammation in general is linked to atherosclerosis as study shows that LIGHT, a member of the TNF superfamily, decreases lipolytic gene expression and increases lipogenic gene expression in oxidized LDL-induced macrophages, promoting lipid accumulation through NF-κB activation and linking inflammation to hyperlipidemia. Additionally, the loss of Δ-5 fatty acid desaturase 1 has a proinflammatory effect in the liver, contributing to atherosclerosis and further reinforcing the connection between inflammation and atherogenesis [13,14,15]. These neoplasms are associated with higher cardiovascular risk and accelerated atherosclerosis mostly through dysregulated expression of cytokines like interleukin 1-β (IL1-β), Transforming growth factor -β (TGF-β), absent in melanoma 2 inflammasome (AIM2) and by disrupting the cholesterol efflux on foam cells [16,17].
In this review, we aim to comprehensively explore the potential relationships between chronic inflammation and the development of PD as a secondary condition in patients with MPNs. Our goal is to investigate both the direct and indirect mechanisms through which MPNs may contribute to periodontal pathology. Specifically, we will examine the role of biofilm-mediated pathways, where microbial colonization and dysbiosis may trigger inflammatory responses, as well as non-biofilm-mediated mechanisms, including systemic inflammatory mediators and immune dysregulation inherent to MPNs. By identifying the possible molecular and cellular pathways involved, we seek to provide a clearer understanding of how MPN-related inflammation influences PD progression and clinical implications.
2. Dysregulation of Inflammatory Cytokines in MPNs and Cytokine Pattern
In MPNs both healthy and mutated cells produce excessive amounts of inflammatory cytokines, with IL1-β, TNF-α, IL-6 and Colony stimulating factors (G-CSF) being the most important [18,19,20]. Chronic inflammation contributes to the initiation and expansion of leukocyte and platelet clones. These cells, in turn, release proinflammatory mediators, creating a self-sustaining cycle of inflammation [21]. While inflammation is a common feature across MPNs and more pronounced in JAK2-positive patients, each type of neoplasm exhibits a distinct cytokine profile, which influences clinical manifestations and prognosis in different ways [22]. Systemic consequences of the prolonged inflammation in MPNs have the most important impact on cardiovascular level with enhanced atherosclerosis, thrombotic events, myocardial fibrosis and pulmonary arterial hypertension (PAH) [23]. Additionally, these patients can present with constitutional symptoms osteoporotic features sharing similar traits with chronic inflammatory conditions like Lupus, rheumatoid arthritis and many more [22,24,25].
Major contributors to this inflammation are genetic driver mutations like the JAK2 46/1 haplotype which is associated with an abnormal myeloid response, promoting inflammation, myeloid neoplasms, and overexpression of genes like insulin-like 6 (INSL6) and insulin-like 4 (INSL4), which amplify pro-inflammatory cytokine production. Additionally, the telomerase reverse transcriptase (TERT) rs2736100 SNP, linked to increased enhancer activity and myeloproliferative neoplasm (MPN) susceptibility, may drive cytokine overproduction, particularly interleukin-6 (IL-6), contributing to MPN onset [26,27,28].
Overall, MPNs are characterized by the overexpression of a wide range of inflammatory mediators, including pro-inflammatory cytokines such as IL-1β, interferon alpha (IFN-α), interleukin-6 (IL-6), and interleukin-12 (IL-12), as well as anti-inflammatory cytokines like interleukin-4 (IL-4) and interleukin-10 (IL-10) [29,30,31]. Additionally, MPNs elevate chemokines like macrophage inflammatory protein 1 alpha and beta (MIP-1α/β), and interferon-inducible protein 9 (IP-9), growth factors such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and platelet-derived growth factor (PDGF), and pro-fibrotic cytokines, including macrophage inflammatory protein (MIP), platelet-derived growth factor (PDGF), and interleukin-8 (IL-8) [30,32,33,34,35].
Patients with ET are noted for elevated levels of circulating vascular endothelial growth factor (VEGF) and growth-related oncogene alpha (GRO-α) [12,36]. Although RANTES (regulated upon activation, normal T cell expressed and secreted) levels are also elevated in these patients, they do not exceed those observed in PMF [31]. Individuals with PV exhibit higher levels of interleukin-23 (IL-23) and, similar to ET patients, elevated levels of eotaxin [12,37]. PMF patients, however, display the highest cytokine burden, with increased levels of pro-inflammatory cytokines such as IL-2, IL-2R and TNF-α, along with chemokines like RANTES and MIP-1β. They also show elevated growth factors, including thrombopoietin (TPO), and pro-fibrotic molecules like fibroblast growth factor (FGF) and TGF-β [22,31,35,38].
3. Inflammatory Origin of the PD
There are three primary reasons why PD leads to chronic low-level systemic inflammation: oral inflammation, alterations in gut microbiota, and the activity of gingipains [39,40]. PD is caused by several hundred bacterial species known as periodontal pathogens. Among these, three key species—Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola—collectively referred to as the "red complex," have been strongly linked to the development of PD [41,42]. Studies suggest that oral bacteria can enter the bloodstream, particularly in individuals with gum inflammation or those undergoing multiple dental treatments. The inflammation driven by these bacterial infections is an independent risk factor for acute cardiovascular events. Improved oral hygiene, in turn, has been shown to reduce cardiovascular risk [43,44,45,46].
One key mechanism is oral inflammation from PD, which triggers the release of cytokines—crucial regulators of the immune response. In studies with mouse models and in vitro, cytokines have been shown to perform several functions, such as inducing neutrophil and lymphocyte activity and facilitating immune system communication. This cytokine release leads to chronic low-level systemic inflammation [47,48]. Furthermore, experiments on mice exposed to P. gingivalis have shown changes in gut microbiota composition, although P. gingivalis itself does not colonize the gut. Instead, it alters the microbial community, contributing to systemic inflammation [49].
Additionally, gingipains—proteases produced by periodontal pathogens—play a role in sustaining low-level systemic inflammation. These proteases include lysine-specific gingipain (Kgp) and arginine-specific gingipain (Rgp) [50]. Recent studies have indicated that periodontal pathogens also release outer membrane vesicles (OMVs), which prevent the resolution of inflammation caused by gingipains [51]. OMVs have been shown to elevate levels of cytokines such as interleukin-1 beta (IL-1β), interleukin-6 (IL-6), and interleukin-8 (IL-8), and inhibiting gingipains can reduce TNF-α levels, despite an initial increase in response to OMV stimulation [52].
4. Pathogen-Induced Inflammation Through JAK/STAT Signaling in PD
Reactive oxygen species (ROS) are reactive molecules formed by the partial reduction of oxygen and play a key role in diseases like rheumatoid arthritis, atherosclerosis, and periodontitis [53,54,55]. Besides their tissue-damaging effects, ROS act as signaling molecules in cell proliferation, immune responses, and inflammation [56]. P. gingivalis, triggers ROS production while evading immune defenses through antioxidant enzymes like superoxide dismutase and thiol peroxidase, and protective mechanisms like a hemin layer [57,58]. P. gingivalis can survive within epithelial cells by enhancing the antioxidant glutathione (GSH) response and inducing ROS [59]. This ROS production, driven by extracellular adenosine triphosphate (ATP), activates the JAK2 signaling pathway in both epithelial and myeloid cells, which regulates inflammatory cytokine production via Toll-like receptors (TLRs) and downstream pathways like signal transducer and activator of transcription (STAT) and mitogen-activated protein kinase (MAPK), including c-Jun amino-terminal kinase (JNK) [60,61]. Neutralizing ROS with N-acetyl-L-cysteine (NAC) blocks JAK2 activation and reduces P. gingivalis-induced production of IL-6 and IL-1β. JAK2 activation triggers the JNK/c-Jun pathway, and inhibiting JAK2 or JNK/c-Jun reduces cytokine levels. This shows that ROS-driven JAK2 activation is crucial for the inflammatory response to P. gingivalis (Figure 1) [62].
In a study, baricitinib treatment for rheumatoid arthritis (RA) showed significant improvement in both RA activity and periodontal inflammation, including reductions in gingival inflammation (GI), bleeding on probing (BOP), and probing depth (PD). However, changes in bacterial plaque were minor [63]. Another study indicates that baricitinib may help reverse periodontitis by reducing oxidative stress and inflammation while promoting the regeneration of periodontal tissues through enhanced osteogenic differentiation in a model of periodontal ligament stem cells (PDLSCs) by blocking the JAK/STAT signaling pathway, which helps restore cell viability, reduce pro-inflammatory cytokines like TNF-α, IL-1β, and IL-6, and support tissue regeneration [64].
Blockade of the Janus kinase (JAK) signaling pathway, particularly through JAK1-3 inhibitors, has shown promise in reducing inflammation and preventing bone resorption in periodontitis, suggesting its potential as a therapeutic approach. Studies indicate that JAK inhibition can improve periodontal clinical parameters and modulate the inflammatory response, warranting further investigation into its clinical efficacy, proving [65]. Interestingly, a study showed that patients with MPNs receiving ruxolitinib had higher incidence of tooth cracks and dental abscess [66]. However, JAK/STAT signaling pathway is not only an inflammatory pathway it is also a key signaling cascade for differentiation of neural crest stem cells [67]. Moreover, not all JAK/STAT pathways lead to inflammation, JAK3 plays a protective anti-inflammatory role in innate immune cells by inhibiting the ubiquitination of Wnt3a through the phospho-inactivation of Nedd4-2, thereby reducing pro-inflammatory cytokine production and attenuating inflammation in response to the periodontal pathogen P. gingivalis [68].
Inappropriate activation of the renin-angiotensin system (RAS) exacerbates atherosclerosis, endothelial damage, and insulin resistance, while promoting coronary heart disease (CHD) progression, potentially due to heightened JAK/STAT signaling, and angiotensin-converting enzyme (ACE) inhibitors have shown therapeutic benefits in conditions like PV and MF by optimizing hematocrit (HCT) levels and reducing fibrosis by interfering with JAK/STAT signaling [23,69,70]. A research showed that telmisartan, an angiotensin II receptor blocker, blocks LPS-induced nitric oxide (NO) and IL-1β production in macrophages, promotes M2 macrophage polarization, and downregulates JAK-STAT signaling by attenuating STAT proteins phosphorylation. As a consequence this may lead to reduction in inflammatory response in PD, highlighting the role of JAK/STAT signaling in bacterial-induced inflammation [71]. Another study revealed that JAK inhibitor ruxolitinib was shown to partially reduce necroptotic effects of INF-β, suggesting that JAK-STAT signaling plays a crucial role in regulating INF-β-induced cell death during bone formation [72].
5. Circulating Cells and Periodontal Disease
We have arguments to believe that circulating cells might have an impact on PD since a report has shown that CD34+ cells are positively correlated with the degree of PD [73].
MDSCs (myeloid-derived suppressor cells) expand in MPNs due to chronic inflammation and oxidative stress, driven in part by the JAK2 V617F mutation. This mutation promotes ROS production, which helps MDSCs suppress immune responses, contributing to disease progression in MPNs [74,75]. However, MDSC levels don't directly correlate with JAK2 allele burden, highlighting complex interactions in these diseases [76]. MDSCs are elevated in both the peripheral blood and bone marrow of patients with MPNs, including PMF, where they correlate with disease progression and fibrosis. These cells, influenced by chronic inflammation, exhibit immunosuppressive functions and may contribute to creating a fibrotic microenvironment, making them potential targets for therapy in MPNs [77,78]. MDSCs contribute to PD by suppressing T cell activity and promoting chronic inflammation in the periodontium. Key periodontal pathogens like P. gingivalis and Fusobacterium nucleatum activate MDSCs, leading to tissue damage through amino acid depletion and the production of reactive oxygen and nitrogen species [79]. PD associated with other inflammatory conditions as obesity, rheumatoid arthritis (RA) or cancer can induce systemic expansion of MDSC subpopulations or recruitment to tissues [80].
In patients with MPNs, an elevated neutrophil-to-lymphocyte ratio (NLR) and increased release of neutrophil extracellular traps (NETs) have been observed, potentially linked to heightened JAK2 kinase activity [81,82]. While NETs, composed of nucleic acids, proteins, and enzymes, are critical for pathogen defense and innate immunity, their excessive release in MPNs contributes to sterile inflammation, suggesting a role in the chronic inflammatory state associated with these conditions [83,84]. In murine models with PD, NETs promote macrophage polarization toward the pro-inflammatory M1 phenotype by inhibiting the JAK2/STAT3 pathway, which worsens inflammation, but this is rather the result of hyperglycemia, in contrast to MPNs where JAK2 variant induces a background sterile inflammation [85].
While NETs are essential for capturing and neutralizing pathogens, their excessive formation in periodontal tissues leads to heightened inflammation and immune dysregulation. This overaccumulation of NETs triggers the release of pro-inflammatory cytokines, drives the polarization of macrophages toward a destructive M1 phenotype, and exacerbates tissue damage, including alveolar bone resorption. Furthermore, gingival fibroblasts have been found to enhance NET production via the MIF-CD74/CXCR4 signaling pathway, further amplifying the inflammatory environment [86,87]. CXCR4 plays a critical role in immune regulation by modulating neutrophil dynamics, promoting their infiltration in periodontal tissues, and influencing inflammation through mechanisms such as the recruitment of proinflammatory monocytes/macrophages and the signaling of CXCL12, which enhances cytokine expression and chemotaxis [88].
A possible connection with platelets can be seen in the context of periodontitis, elevated levels of soluble P-selectin (sP-selectin), a marker of platelet activation, have been observed, and this activation is associated with the severity of the disease. The presence of higher numbers of platelets and activated platelets in periodontitis patients could explain the heightened risk of coronary heart disease and stroke seen in individuals with this condition, moreover mean platelet volume in these patients is also positively correlated with the degree of PD [89,90]. Interestingly, research on ET and PV show high levels of soluble P-selectin as a marker of thrombocyte activity when compared to healthy individuals [91].
6. Common Circulating Inflammatory Mediators in MPNs and Periodontal Disease
The new classification of periodontal diseases recognizes that neoplastic diseases can affect the periodontal tissues independently of plaque-induced periodontitis, and such cases should be classified based on the primary systemic disease, under the category of "Systemic Diseases or Conditions Affecting the Periodontal Supporting Tissues" [4]. Current evidence shows that conditions with systemic inflammation like auto-immune disease like LES and RA can manifest with PD [92,93].
Chitinase-3-like protein 1 (CHI3L1), also known as YKL-40, is an inflammatory protein elevated in chronic periodontitis (CP), showing a strong correlation with disease severity and periodontal pocket depth (PPD). This makes YKL-40 a promising biomarker for tracking PD progression [94]. Furthermore, YKL-40 levels increase progressively with the PD evolution, alongside other inflammatory cytokines like IL-6 [95]. YKL-40 is a promising biomarker for monitoring athero-protective therapy, as it is linked to inflammation and disease severity in cardio-metabolic disorders, potentially improving risk prediction for cardiovascular events [96]. Also, Anti-YKL-40 therapy has shown promising effects by inhibiting tumor migration and inducing apoptosis, proving YKL-40 pro-oncogenic effects [97]. Elevated circulating YKL-40 levels in Philadelphia-negative MPNs are associated with increased inflammation, higher C-reactive protein levels, poor performance status, and greater cardiovascular risk, with specific correlations showing that elevated YKL-40 predicts an increased risk of thrombosis in ET and PV patients and impaired survival in MF patients [98].
Inflammatory cytokines in gingival tissues around human gingival fibroblasts (HGFs) are vital for initiating inflammatory responses. Human gingival fibroblasts produce VEGF, which increases vascular permeability and contributes to the severity of periodontal disease [99,100]. Evidence so far indicate that VEGF production is generally upregulated in patients with periodontitis, suggesting it plays a crucial role in the etiology of gingivitis and its progression to periodontitis. VEGF enhances vascular permeability and angiogenesis, potentially linking it to periodontal disease [101]. In patients with ET, levels of VEGF are significantly elevated, particularly in untreated individuals [102]. This could explain partially the gums-bleeding tendency sometimes seen in these patients [103].
GRO-alpha is an important inflammatory mediator found in higher concentrations in gingival crevicular fluid from inflamed periodontal sites, where it stimulates neutrophil chemotaxis, and its secretion can be inhibited by compounds like A-type proanthocyanins AC-PACs, Epigallocatechin-3-gallate (EGCG) and LL-37, which prevent immune cell activation [104]. Even though in periodontal disease GRO-β is not a central element, in patients with ET, intracellular flow cytometry revealed that monocytes, particularly the proinflammatory CD56+/CD14+ subset, were the predominant producers of GRO-alpha, with significantly elevated levels correlating with an increased risk of disease transformation to MF, confirmed in an extended cohort [105,106,107].
IL-23 is crucial in the pathogenesis of periodontal disease, evidenced by its increase expression in gingival tissue samples from patients with chronic and aggressive periodontitis compared to healthy controls, correlating with inflammatory infiltrate intensity and clinical parameters such as probing depth and clinical attachment loss, highlighting its involvement in promoting Th17 cell responses that exacerbate tissue damage and inflammation during disease progression [108]. Moreover, IL-1β stimulates the production of IL-23 p19 in human periodontal ligament fibroblasts (hPDLFs) through NF-κB and MAPK-dependent pathways, suggesting that targeting this interaction could provide therapeutic benefits in managing Th17-driven inflammatory diseases such as periodontitis [109]. Plasma levels of IL-23 were significantly higher in patients with MPN compared to controls. Specifically, there was a significant difference in IL-23 levels between patients affected by PV and the control group [37]. Furthermore, the importance of this cytokine in periodontal disease is indicated by the fact that IL-23 not only enhances cell proliferation in oral cancer cells but also plays a critical role in modulating NF-kB activity, leading to tumor growth and survival in oral squamous cell carcinoma. However, there is no evidence that this could lead to a higher incidence of oral cancers in PV or other MPN [110].
Eotaxin-1 is categorized as a pro-inflammatory cytokine, which can contribute to the inflammatory microenvironment that often accompanies cancer, including OSCC, being involved in the recruitment of inflammatory cells, such as eosinophils, may play a role in both promoting and regulating tumor progression [111]. Eotaxin-1 is associated with prolonged inflammation in periodontitis, with levels that can be influenced by the A variant of interferon gamma inducible protein 16 (IFI16) [112]. Also they are elevated in PV and TE [12].
TGF-β plays a dual role in periodontitis, acting both as a pro-inflammatory and anti-inflammatory mediator. It induces immune cells to secrete pro-inflammatory cytokines like IL-1β and IL-6, contributing to inflammation, while also acting as a chemoattractant for neutrophils, monocytes, and lymphocytes. However, in inflammatory environments, TGF-β can lose its immunosuppressive properties, promoting tissue destruction instead of repair, as seen in PD [113,114]. Elevated TGF-β levels in MPNs are linked to arterial plaque growth and coronary injury, though its role remains unclear, making it a promising research focus. Additionally, TGF-β contributes to cell phenotype switching, which may play a role in oncogenesis, atherogenesis, and bone marrow fibrosis [115,116,117].
RANTES (Regulated upon Activation, Normal T Cell Expressed and Secreted) is a chemokine that significantly attracts immune cells like monocytes and T cells to inflamed tissues. In periodontal disease, elevated RANTES levels are linked with disease activity, particularly in aggressive periodontitis, where its high concentration correlates with severe tissue destruction. After periodontal therapy, RANTES levels decrease, reflecting a reduction in inflammation, but this alone is not enough to determine the need for further treatment or surgery [118,119,120,121]. In ET, platelets show increased secretion of the chemokine RANTES, particularly via TLR2 and TLR4 signaling, contributing to thromboinflammatory processes like monocyte recruitment and atherogenesis. However, despite elevated platelet-derived RANTES, its plasma levels remain low, suggesting that RANTES primarily mediates localized rather than systemic inflammation in ET [122,123,124,125]. Other reports have shown similar levels between bone marrow aspirate and peripheral blood of RANTES [126].
Hepatocyte growth factor (HGF) is a key angiogenic and regenerative factor with cytoprotective effects, playing a significant role in cardioprotection during coronary ischemia and infarction, and is linked to various chronic diseases, including coronary artery disease (CAD) [127]. In periodontal disease, HGF levels increase proportionally with disease progression, and its synthesis is stimulated by P. gingivalis, highlighting the relationship between oral health and systemic inflammation [128]. In MPNs, particularly those associated with the JAK2 V617F mutation, HGF is overproduced by both malignant myeloid progenitors and bone marrow stromal cells (BMSCs), independent of JAK2 signaling [129]. This deregulation of HGF not only enhances the proliferation and survival of hematopoietic progenitors but also contributes to an inflammatory microenvironment by promoting the production of various cytokines such as interleukin (IL)-6 and IL-11 [130,131]. While the production of HGF in MPNs occurs independently of JAK2, the mutation enhances the responsiveness of progenitor cells to various growth factors, including HGF [132]. The heightened responsiveness of progenitor cells to HGF due to JAK2 mutations may exacerbate inflammation and tissue remodeling in periodontal disease, potentially worsening tissue destruction and impairing immune responses against periodontal pathogens.
The NLRP3 inflammasome is a critical component of the host immune defense, activated by a wide range of structurally diverse stimuli such as extracellular ATP, pore-forming toxins, and RNA viruses. In myeloproliferative neoplasm (MPN) patients, NLRP3 inflammasome genes are upregulated in hematopoietic cells, contributing to the inflammatory state. When dysregulated, NLRP3 also plays a role in various inflammatory disorders, including Alzheimer's disease, diabetes, gout, and atherosclerosis, by triggering caspase-1 activation and the release of pro-inflammatory cytokines like IL-1β and IL-18 [10,133]. The dysregulation of the NLRP3 inflammasome in MPN may contribute to the pathogenesis of periodontitis by promoting chronic inflammation and excessive cytokine production, as a report shows that COVID-19 induced NLPR3 activation leads to periodontitis, supporting the hypothesis that biofilm-unrelated inflammatory response could lead to tissue damage by underlying inflammatory cytokine dysregulation [134]. Another inflammasome [112] AIM2 is activated in response to double-stranded DNA (dsDNA) fragments from pathogens. Upon activation, it associates with caspase-1, leading to the formation of the AIM2 inflammasome, which is crucial for the maturation of pro-inflammatory cytokines such as IL-1β, IL-18, and IL-33. This cytokine release contributes to the inflammatory processes associated with periodontitis [112]. In MPN AIM2 inflammasome hyperactivity is caused by JAK2 mutation leading to increased IL-1β signaling [135].
IL-1β and IL-6 are produced in response to infections in the root canal system, driven by the host immune response and epigenetic changes like DNA hypomethylation and pro-inflammatory signaling. Their increased levels lead to enhanced osteoclastic activity, resulting in bone resorption and tissue damage associated with apical periodontitis. Ultimately, the dysregulation of these cytokines exacerbates inflammation and contributes to the severity of the disease [136]. In MPN Il-1β and Il-6 are elevated in all subgroups (ET/PV/PMF) compared to healthy controls [22,29,30]. One could speculate that elevated pro-inflammatory signaling associated with NLPR3 and AIM2 inflammasome hyperactivity could lead to both biofilm dependent/independent inflammatory response and IL-1β signaling and contributing to periodontal disease.
7. The Bidirectional Impact of Periodontal Disease and Myeloproliferative Neoplasms
Recent studies have increasingly elucidated the relationship between periodontal disease (PD) and various forms of cancer, including lung, breast, prostate, pancreas and colorectal cancers [141,142,143,144,145]. Research, indicates that indicators of PD, like probing pocket depth and clinical attachment loss, are positively associated with an increased risk of lung cancer, particularly in smokers. Meta-analyses further confirm a significant link between PD and heightened cancer risks, with findings showing that individuals with PD may have up to a 51% higher likelihood of developing lung cancer and an increased risk of other cancers, such as breast and colorectal cancer, which can escalate with tooth loss [141,142,146,147]. New evidence suggest that the impact of PD could have implication even in hematological malignancies like Non-Hodgkin Lymphoma (NHL) [148]. Even though there are few researches on the impact of PD on MPN there are few links between cytokine expression and MPN occurrence. IL2Rα (IL-2 receptor alpha) gene variants may contribute to periodontal disease susceptibility in individuals with type 1 diabetes mellitus by altering T cell activation and immune responses, leading to increased inflammation and tissue damage in the periodontal region [137]. High IP-10 (interferon gamma-induced protein 10) levels are indicative of an ongoing inflammatory response, as they attract immune cells to the inflamed periodontal tissue, thus contributing to the disease's progression and severity [140]. There are at least two reports in IL2Rα and one for IP-10 that mention the risk associated of MPNs and the high circulating levels of both cytokines [138,139]. The potential bidirectional influence between PD and MPNs underscores a sophisticated interplay between chronic inflammation and hematological disorders (Figure 2) (Table 1).
This review highlights the intricate relationship between PD and MPNs, emphasizing the role of common inflammatory mediators. Elevated levels of cytokines such as YKL-40, VEGF, and IL-23 illustrate how chronic inflammation can impact both conditions. Genetic variants like IL2Rα and markers such as IP-10 further underscore the bidirectional influence of PD and MPNs, suggesting that managing systemic inflammation and oral health may be crucial for improving patient outcomes.
Author Contributions
R.G.M. and S.B.T.: conceptualization, methodology, investigation, writing—original draft. Both authors have read and agreed to the published version of the manuscript.
Acknowledgments
We thank the editors of the "Oral" magazine for the invitation to publish an article in this prestigious journal.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kinane, D.F.; Stathopoulou, P.G.; Papapanou, P.N. Periodontal diseases. Nat Rev Dis Primer 2017, 3, 17038. [Google Scholar] [CrossRef] [PubMed]
- Pihlstrom, B.L.; Michalowicz, B.S.; Johnson, N.W. Periodontal diseases. The Lancet 2005, 366, 1809–1820. [Google Scholar] [CrossRef]
- Highfield, J. Diagnosis and classification of periodontal disease. Aust Dent J 2009, 54. Available online: https://onlinelibrary.wiley.com/doi/10.1111/j.1834-7819.2009.01140.x (accessed on 7 October 2024). [CrossRef]
- Caton, J.G.; Armitage, G.; Berglundh, T.; Chapple, I.L.C.; Jepsen, S.; Kornman, K.S.; et al. A new classification scheme for periodontal and peri-implant diseases and conditions—Introduction and key changes from the 1999 classification. J Clin Periodontol 2018, 45. Available online: https://onlinelibrary.wiley.com/doi/10.1111/jcpe.12935 (accessed on 7 October 2024). [CrossRef] [PubMed]
- Jepsen, S.; Caton, J.G.; Albandar, J.M.; Bissada, N.F.; Bouchard, P.; Cortellini, P.; et al. Periodontal manifestations of systemic diseases and developmental and acquired conditions: Consensus report of workgroup 3 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J Clin Periodontol 2018, 45. Available online: https://onlinelibrary.wiley.com/doi/10.1111/jcpe.12951 (accessed on 7 October 2024). [CrossRef] [PubMed]
- Smalley, J.W. Pathogenic Mechanisms in Periodontal Disease. Adv Dent Res 1994, 8, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Zee, K. Smoking and periodontal disease. Aust Dent J 2009, 54. Available online: https://onlinelibrary.wiley.com/doi/10.1111/j.1834-7819.2009.01142.x (accessed on 7 October 2024). [CrossRef]
- Borgnakke, W.S. Does Treatment of Periodontal Disease Influence Systemic Disease? Dent Clin North Am 2015, 59, 885–917. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Zhou, Y.; Yan, S.; Liu, N.; He, N.; Zhang, A.; Meng, S.; et al. Genetic polymorphisms and expression of NLRP3 inflammasome-related genes are associated with Philadelphia chromosome-negative myeloproliferative neoplasms. Hum Immunol 2020, 81, 606–613. [Google Scholar] [CrossRef]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef]
- Øbro, N.F.; Grinfeld, J.; Belmonte, M.; Irvine, M.; Shepherd, M.S.; Rao, T.N.; et al. Longitudinal Cytokine Profiling Identifies GRO-α and EGF as Potential Biomarkers of Disease Progression in Essential Thrombocythemia. HemaSphere 2020, 4, e371. [Google Scholar] [CrossRef]
- Yuan, X.; Gu, Y.; Lai, X.; Gu, Q. LIGHT is increased in patients with coronary disease and regulates inflammatory response and lipid metabolism in oxLDL-induced THP-1 macrophages. Biochem Biophys Res Commun 2017, 490, 732–738. [Google Scholar] [CrossRef]
- Gromovsky, A.D.; Schugar, R.C.; Brown, A.L.; Helsley, R.N.; Burrows, A.C.; Ferguson, D.; et al. Δ-5 Fatty Acid Desaturase FADS1 Impacts Metabolic Disease by Balancing Proinflammatory and Proresolving Lipid Mediators. Arterioscler Thromb Vasc Biol 2018, 38, 218–231. [Google Scholar] [CrossRef]
- Mihăilă, R.G. Pragmatic Analysis of Dyslipidemia Involvement in Coronary Artery Disease: A Narrative Review. Curr Cardiol Rev 2020, 16, 36–47. [Google Scholar] [CrossRef]
- Todor, S.B.; Ichim, C.; Boicean, A.; Mihaila, R.G. Cardiovascular Risk in Philadelphia-Negative Myeloproliferative Neoplasms: Mechanisms and Implications—A Narrative Review. Curr Issues Mol Biol 2024, 46, 8407–8423. [Google Scholar] [CrossRef]
- Libby, P.; Molinaro, R.; Sellar, R.S.; Ebert, B.L. Jak-ing Up the Plaque’s Lipid Core…and Even More. Circ Res 2018, 123, 1180–1182. [Google Scholar] [CrossRef]
- Rai, S.; Zhang, Y.; Grockowiak, E.; Kimmerlin, Q.; Hansen, N.; Stoll, C.B.; et al. IL-1β promotes MPN disease initiation by favoring early clonal expansion of JAK2-mutant hematopoietic stem cells. Blood Adv 2024, 8, 1234–1249. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci 2009, 106, 17031–17036. [Google Scholar] [CrossRef] [PubMed]
- Arranz, L.; Arriero, M.D.M.; Villatoro, A. Interleukin-1β as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev 2017, 31, 306–317. [Google Scholar] [CrossRef]
- Landolfi, R.; Di Gennaro, L. Pathophysiology of thrombosis in myeloproliferative neoplasms. Haematologica 2011, 96, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Cacemiro, M.D.C.; Cominal, J.G.; Tognon, R.; Nunes, N.D.S.; Simões, B.P.; Figueiredo-Pontes, L.L.D.; et al. Philadelphia-negative myeloproliferative neoplasms as disorders marked by cytokine modulation. Hematol Transfus Cell Ther 2018, 40, 120–131. [Google Scholar] [CrossRef]
- Leiva, O.; Hobbs, G.; Ravid, K.; Libby, P. Cardiovascular Disease in Myeloproliferative Neoplasms. JACC CardioOncology 2022, 4, 166–182. [Google Scholar] [CrossRef]
- Farmer, S.; Ocias, L.F.; Vestergaard, H.; Broesby-Olsen, S.; Hermann, A.P.; Frederiksen, H. Bone morbidity in chronic myeloproliferative neoplasms. Expert Rev Hematol 2015, 8, 447–456. [Google Scholar] [CrossRef]
- Soyfer, E.M.; Fleischman, A.G. Myeloproliferative neoplasms—blurring the lines between cancer and chronic inflammatory disorder. Front Oncol 2023, 13, 1208089. [Google Scholar] [CrossRef]
- Hermouet, S.; Vilaine, M. The JAK2 46/1 haplotype: a marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica 2011, 96, 1575–1579. [Google Scholar] [CrossRef]
- Tapper, W.; Jones, A.V.; Kralovics, R.; Harutyunyan, A.S.; Zoi, K.; Leung, W.; et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat Commun 2015, 6, 6691. [Google Scholar] [CrossRef]
- Wang, F.; Fu, P.; Pang, Y.; Liu, C.; Shao, Z.; Zhu, J.; et al. TERT rs2736100T/G polymorphism upregulates interleukin 6 expression in non-small cell lung cancer especially in adenocarcinoma. Tumor Biol 2014, 35, 4667–4672. [Google Scholar] [CrossRef]
- Bourantas, K.L.; Hatzimichael, E.C.; Makis, A.C.; Chaidos, A.; Kapsali, E.D.; Tsiara, S.; Mavridis, A. Serum beta-2-microglobulin, TNF-α and interleukins in myeloproliferative disorders. Eur J Haematol 1999, 63, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independently Prognostic in Primary Myelofibrosis: A Comprehensive Cytokine Profiling Study. J Clin Oncol 2011, 29, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, R.; Gangat, N.; Jimma, T.; Finke, C.M.; Lasho, T.L.; Pardanani, A.; et al. Plasma cytokines in polycythemia vera: Phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am J Hematol 2012, 87, 1003–1005. [Google Scholar] [CrossRef]
- Hsu, H.C.; Tsai, W.H.; Jiang, M.L.; Ho, C.H.; Hsu, M.L.; Ho, C.K.; et al. Circulating levels of thrombopoietic and inflammatory cytokines in patients with clonal and reactive thrombocytosis. J Lab Clin Med 1999, 134, 392–397. [Google Scholar] [CrossRef]
- Ho, C.L.; Lasho, T.L.; Butterfield, J.H.; Tefferi, A. Global cytokine analysis in myeloproliferative disorders. Leuk Res 2007, 31, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Mambet, C.; Babosova, O.; Defour, J.P.; Leroy, E.; Necula, L.; Stanca, O.; et al. Cooccurring JAK2 V617F and R1063H mutations increase JAK2 signaling and neutrophilia in myeloproliferative neoplasms. Blood 2018, 132, 2695–2699. [Google Scholar] [CrossRef]
- Wong, W.J.; Baltay, M.; Getz, A.; Fuhrman, K.; Aster, J.C.; Hasserjian, R.P.; et al. Gene expression profiling distinguishes prefibrotic from overtly fibrotic myeloproliferative neoplasms and identifies disease subsets with distinct inflammatory signatures. PLOS ONE 2019, 14, e0216810. [Google Scholar] [CrossRef]
- Pourcelot, E.; Trocme, C.; Mondet, J.; Bailly, S.; Toussaint, B.; Mossuz, P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: Clinical implications. Exp Hematol 2014, 42, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, S.; Allegra, A.; Pace, E.; Alonci, A.; Ferraro, M.; Petrungaro, A.; et al. Evaluation of interleukin-23 plasma levels in patients with polycythemia vera and essential thrombocythemia. Cell Immunol 2012, 278, 91–94. [Google Scholar] [CrossRef]
- Panteli, K.E.; Hatzimichael, E.C.; Bouranta, P.K.; Katsaraki, A.; Seferiadis, K.; Stebbing, J.; et al. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br J Haematol 2005, 130, 709–715. [Google Scholar] [CrossRef]
- Martínez-García, M.; Hernández-Lemus, E. Periodontal Inflammation and Systemic Diseases: An Overview. Front Physiol 2021, 12, 709438. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Nakayama, K.; Okamoto, K.; Abe, N.; Baba, A.; Shi, Y.; et al. Porphyromonas gingivalis Proteinases as Virulence Determinants in Progression of Periodontal Diseases. J Biochem 2000, 128, 153–159. [Google Scholar] [CrossRef]
- Lopez, R.; Hujoel, P.; Belibasakis, G.N. On putative periodontal pathogens: an epidemiological perspective. Virulence 2015, 6, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L. Microbial complexes in subgingival plaque. J Clin Periodontol 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xuan, S.; Wang, Z. Oral microbiota: A new view of body health. Food Sci Hum Wellness 2019, 8, 8–15. [Google Scholar] [CrossRef]
- Emery, D.C.; Cerajewska, T.L.; Seong, J.; Davies, M.; Paterson, A.; Allen-Birt, S.J.; et al. Comparison of Blood Bacterial Communities in Periodontal Health and Periodontal Disease. Front Cell Infect Microbiol 2021, 10, 577485. [Google Scholar] [CrossRef]
- Aoki, S.; Hosomi, N.; Nishi, H.; Nakamori, M.; Nezu, T.; Shiga, Y.; et al. Serum IgG titers to periodontal pathogens predict 3-month outcome in ischemic stroke patients. PLOS ONE 2020, 15, e0237185. [Google Scholar] [CrossRef]
- Chang, Y.; Woo, H.G.; Park, J.; Lee, J.S.; Song, T.J. Improved oral hygiene care is associated with decreased risk of occurrence for atrial fibrillation and heart failure: A nationwide population-based cohort study. Eur J Prev Cardiol 2020, 27, 1835–1845. [Google Scholar] [CrossRef]
- Ling, M.R.; Chapple, I.L.; Matthews, J.B. Peripheral blood neutrophil cytokine hyper-reactivity in chronic periodontitis. Innate Immun 2015, 21, 714–725. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Farrar, W.L.; Yang, X. Transcriptional crosstalk between nuclear receptors and cytokine signal transduction pathways in immunity. Cell Mol Immunol 2004, 1, 416–424. [Google Scholar] [PubMed]
- Kato, T.; Yamazaki, K.; Nakajima, M.; Date, Y.; Kikuchi, J.; Hase, K.; et al. Oral Administration of Porphyromonas gingivalis Alters the Gut Microbiome and Serum Metabolome. mSphere 2018, 3, e00460-18. [Google Scholar] [CrossRef]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 2019, 5, eaau3333. [Google Scholar] [CrossRef]
- Seyama, M.; Yoshida, K.; Yoshida, K.; Fujiwara, N.; Ono, K.; Eguchi, T.; et al. Outer membrane vesicles of Porphyromonas gingivalis attenuate insulin sensitivity by delivering gingipains to the liver. Biochim Biophys Acta BBA - Mol Basis Dis 2020, 1866, 165731. [Google Scholar] [CrossRef] [PubMed]
- Castillo, Y.; Castellanos, J.; Lafaurie, G.; Castillo, D. Porphyromonas gingivalis outer membrane vesicles modulate cytokine and chemokine production by gingipain-dependent mechanisms in human macrophages. Arch Oral Biol 2022, 140, 105453. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol Rev 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Wang, H.; Li, L.; Feng, Y.; Xu, S.; Wang, Z. Trem2 mediated Syk-dependent ROS amplification is essential for osteoclastogenesis in periodontitis microenvironment. Redox Biol 2021, 40, 101849. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Kanai, T.; Okada, M. Rheumatoid Arthritis and Reactive Oxygen Species: A Review. Curr Issues Mol Biol 2023, 45, 3000–3015. [Google Scholar] [CrossRef]
- Padgett, L.E.; Broniowska, K.A.; Hansen, P.A.; Corbett, J.A.; Tse, H.M. The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci 2013, 1281, 16–35. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Ohara, N.; Sato, K.; Yoshimura, M.; Yukitake, H.; Sakai, E.; et al. Novel stationary-phase-upregulated protein of Porphyromonas gingivalis influences production of superoxide dismutase, thiol peroxidase and thioredoxin. Microbiology 2005, 151, 841–853. [Google Scholar] [CrossRef]
- Smalley, J.W.; Birss, A.J.; Silver, J. The periodontal pathogen Porphyromonas gingivalis harnesses the chemistry of the μ-oxo bishaem of iron protoporphyrin IX to protect against hydrogen peroxide. FEMS Microbiol Lett 2000, 183, 159–164. [Google Scholar] [CrossRef]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis -nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence: Porphyromonas inhibits ROS production. Cell Microbiol 2013, 15, 961–976. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol Rev 2009, 228, 273–287. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Faderl, S.; Ravandi, F.; Estrov, Z. The JAK-STAT Pathway: A Therapeutic Target in Hematological Malignancies. Curr Cancer Drug Targets 2006, 6, 671–679. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, H.; Duan, X.; Jotwani, R.; Vuddaraju, H.; Liang, S.; et al. Porphyromonas gingivalis-Induced Reactive Oxygen Species Activate JAK2 and Regulate Production of Inflammatory Cytokines through c-Jun. Infect Immun 2014, 82, 4118–4126. [Google Scholar] [CrossRef] [PubMed]
- Ancuța, C.; Pomîrleanu, C.; Mihailov, C.; Chirieac, R.; Ancuța, E.; Iordache, C.; et al. Efficacy of baricitinib on periodontal inflammation in patients with rheumatoid arthritis. Joint Bone Spine 2020, 87, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Shi, F.; Zhang, Y. Baricitinib alleviates lipopolysaccharide-induced human periodontal ligament stem cell injury and promotes osteogenic differentiation by inhibiting JAK/STAT signaling. Exp Ther Med 2022, 25, 3. [Google Scholar] [CrossRef] [PubMed]
- Godoi, M.A.; Camilli, A.C.; Gonzales, K.G.A.; Costa, V.B.; Papathanasiou, E.; Leite, F.R.M.; et al. JAK/STAT as a Potential Therapeutic Target for Osteolytic Diseases. Int J Mol Sci 2023, 24, 10290. [Google Scholar] [CrossRef]
- Orliaguet, M.; Boisramé, S.; Eveillard, J.; Pan-Petesch, B.; Couturier, M.; Rebière, V.; et al. Pegylated interferon 2a and ruxolitinib induce a high rate of oral complications among patients with myeloproliferative neoplasms. eJHaem 2020, 1, 350–352. [Google Scholar] [CrossRef]
- Tomokiyo, A.; Yoshida, S.; Hamano, S.; Hasegawa, D.; Sugii, H.; Maeda, H. Detection, Characterization, and Clinical Application of Mesenchymal Stem Cells in Periodontal Ligament Tissue. Stem Cells Int 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lü, L.; Yakoumatos, L.; Ren, J.; Duan, X.; Zhou, H.; Gu, Z.; et al. JAK3 restrains inflammatory responses and protects against periodontal disease through Wnt3a signaling. FASEB J 2020, 34, 9120–9140. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.K.; Kaur, G.; Buttar, H.S.; Bagabir, H.A.; Bagabir, R.A.; Bagabir, S.A.; et al. Role of the renin-angiotensin system in the pathophysiology of coronary heart disease and heart failure: Diagnostic biomarkers and therapy with drugs and natural products. Front Physiol 2023, 14, 1034170. [Google Scholar] [CrossRef]
- Corey, S.J.; Jha, J.; McCart, E.A.; Rittase, W.B.; George, J.; Mattapallil, J.J.; et al. Captopril mitigates splenomegaly and myelofibrosis in the Gata1 low murine model of myelofibrosis. J Cell Mol Med 2018, 22, 4274–4282. [Google Scholar] [CrossRef]
- Choe, S.H.; Choi, E.Y.; Hyeon, J.Y.; Keum, B.R.; Choi, I.S.; Kim, S.J. Telmisartan, an angiotensin II receptor blocker, attenuates Prevotella intermedia lipopolysaccharide-induced production of nitric oxide and interleukin-1β in murine macrophages. Int Immunopharmacol 2019, 75, 105750. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hayano, S.; Nagata, M.; Kosami, T.; Wang, Z.; Kamioka, H. Ruxolitinib altered IFN-β induced necroptosis of human dental pulp stem cells during osteoblast differentiation. Arch Oral Biol 2023, 155, 105797. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrowicz, P.; Kotuła, L.; Grabowska, K.; Krakowiak, R.; Kusa-Podkańska, M.; Agier, J.; et al. Evaluation of circulating CD34+ stem cells in peripheral venous blood in patients with varying degrees of periodontal disease. Ann Agric Environ Med 2021, 28, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Delano, M.J.; Scumpia, P.O.; Weinstein, J.S.; Coco, D.; Nagaraj, S.; Kelly-Scumpia, K.M.; et al. MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J Exp Med 2007, 204, 1463–1474. [Google Scholar] [CrossRef]
- Lu, M.; Xia, L.; Liu, Y.C.; Hochman, T.; Bizzari, L.; Aruch, D.; et al. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; et al. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef]
- Campanelli, R.; Carolei, A.; Catarsi, P.; Abbà, C.; Boveri, E.; Paulli, M.; et al. Circulating Polymorphonuclear Myeloid-Derived Suppressor Cells (PMN-MDSCs) Have a Biological Role in Patients with Primary Myelofibrosis. Cancers 2024, 16, 2556. [Google Scholar] [CrossRef]
- Wang, J.C.; Kundra, A.; Andrei, M.; Baptiste, S.; Chen, C.; Wong, C.; et al. Myeloid-derived suppressor cells in patients with myeloproliferative neoplasm. Leuk Res 2016, 43, 39–43. [Google Scholar] [CrossRef]
- Leija-Montoya, A.G.; González-Ramírez, J.; Serafín-Higuera, I.; Sandoval-Basilio, J.; Isiordia-Espinoza, M.; Serafín-Higuera, N. Emerging avenues linking myeloid-derived suppressor cells to periodontal disease. In International Review of Cell and Molecular Biology; Elsevier, 2023; pp. 165–189. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1937644822001459 (accessed on 22 October 2024).
- García-Arévalo, F.; Leija-Montoya, A.G.; González-Ramírez, J.; Isiordia-Espinoza, M.; Serafín-Higuera, I.; Fuchen-Ramos, D.M.; et al. Modulation of myeloid-derived suppressor cell functions by oral inflammatory diseases and important oral pathogens. Front Immunol 2024, 15, 1349067. [Google Scholar] [CrossRef]
- Kwon, S.S.; Yoon, S.Y.; Jeong, S.Y.; Lee, M.Y.; Kim, K.H.; Lee, N.; et al. Neutrophil–lymphocyte ratio and carotid plaque burden in patients with essential thrombocythemia and polycythemia vera. Nutr Metab Cardiovasc Dis 2022, 32, 1913–1916. [Google Scholar] [CrossRef]
- Wolach, O.; Sellar, R.S.; Martinod, K.; Cherpokova, D.; McConkey, M.; Chappell, R.J.; et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med 2018, 10, eaan8292. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, D.; Nagarkoti, S.; Kumar, A.; Dubey, M.; Singh, A.K.; Pathak, P.; et al. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Radic Biol Med 2016, 93, 190–203. [Google Scholar] [CrossRef]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front Immunol 2017, 8. Available online: http://journal.frontiersin.org/article/10.3389/fimmu.2017.00081/full (accessed on 25 June 2024). [CrossRef]
- Wen, J.; Li, J.; Wu, Z. Neutrophil extracellular traps induced by diabetes aggravate periodontitis by inhibiting janus kinase/signal transducers and activators of transcription signaling in macrophages. J Dent Sci 2024, S1991790224003179. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Ren, B.; Zou, L.; He, B.; Li, M. The Role of Neutrophil Extracellular Traps in Periodontitis. Front Cell Infect Microbiol 2021, 11, 639144. [Google Scholar] [CrossRef]
- Qiu, W.; Guo, R.; Yu, H.; Chen, X.; Chen, Z.; Ding, D.; et al. Single-cell atlas of human gingiva unveils a NETs-related neutrophil subpopulation regulating periodontal immunity. J Adv Res 2024, S2090123224003126. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, T.; Tang, J.; Wang, D.; Zhou, Y.; Gou, H.; et al. CXCR4-mediated neutrophil dynamics in periodontitis. Cell Signal 2024, 120, 111212. [Google Scholar] [CrossRef] [PubMed]
- Papapanagiotou, D.; Nicu, E.A.; Bizzarro, S.; Gerdes, V.E.A.; Meijers, J.C.; Nieuwland, R.; et al. Periodontitis is associated with platelet activation. Atherosclerosis 2009, 202, 605–611. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, Y.; Bai, J.; Luo, Y.; Song, J.; Feng, P. Mean platelet volume is associated with periodontitis: a cross-sectional study. BMC Oral Health 2024, 24, 461. [Google Scholar] [CrossRef]
- Musolino, C.; Alonci, A.; Bellomo, G.; Tringali, O.; Spatari, G.; Quartarone, C.; et al. Myeloproliferative Disease: Markers of Endothelial and Platelet Status in Patients with Essential Thrombocythemia and Polycythemia Vera. Hematol Amst Neth 2000, 4, 397–402. [Google Scholar]
- Kobayashi, T.; Bartold, P.M. Periodontitis and periodontopathic bacteria as risk factors for rheumatoid arthritis: A review of the last 10 years. Jpn Dent Sci Rev 2023, 59, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Sete, M.R.C.; Figueredo, C.M.D.S.; Sztajnbok, F. Periodontitis and systemic lupus erythematosus. Rev Bras Reumatol Engl Ed 2016, 56, 165–170. [Google Scholar] [CrossRef]
- Chavan, V.; Sabavath, S.; Babu, C.; Boyapati, L. Estimation of YKL-40 acute-phase protein in serum of patients with periodontal disease and healthy individuals: A clinical-biochemical study. Contemp Clin Dent 2019, 10, 249. [Google Scholar] [CrossRef]
- Keles, Z.P.; Keles, G.C.; Avci, B.; Cetinkaya, B.O.; Emingil, G. Analysis of YKL-40 Acute-Phase Protein and Interleukin-6 Levels in Periodontal Disease. J Periodontol 2014, 85, 1240–1246. [Google Scholar] [CrossRef]
- Deng, Y.; Li, G.; Chang, D.; Su, X. YKL-40 as a novel biomarker in cardio-metabolic disorders and inflammatory diseases. Clin Chim Acta 2020, 511, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, Z.; Zhu, B.; Jia, Z.; Luo, J.; Han, X.; et al. A humanized Anti-YKL-40 antibody inhibits tumor development. Biochem Pharmacol 2024, 225, 116335. [Google Scholar] [CrossRef] [PubMed]
- Krečak, I.; Gverić-Krečak, V.; Lapić, I.; Rončević, P.; Gulin, J.; Fumić, K.; et al. Circulating YKL-40 in Philadelphia-negative myeloproliferative neoplasms. Acta Clin Belg 2021, 76, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Afacan, B.; Öztürk, V.Ö.; Paşalı, Ç.; Bozkurt, E.; Köse, T.; Emingil, G. Gingival crevicular fluid and salivary HIF-1α, VEGF, and TNF-α levels in periodontal health and disease. J Periodontol 2019, 90, 788–797. [Google Scholar] [CrossRef]
- Dvorak, H.F. VPF/VEGF and the angiogenic response. Semin Perinatol 2000, 24, 75–78. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, C. Enhanced VEGF-A expression and mediated angiogenic differentiation in human gingival fibroblasts by stimulating with TNF-α in vitro. J Dent Sci 2022, 17, 876–881. [Google Scholar] [CrossRef]
- Treliński, J.; Wierzbowska, A.; Krawczyńska, A.; Sakowicz, A.; Pietrucha, T.; Smolewski, P.; et al. Plasma levels of angiogenic factors and circulating endothelial cells in essential thrombocythemia: correlation with cytoreductive therapy and JAK2–V617F mutational status. Leuk Lymphoma 2010, 51, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.R.; Anitha, J.R.; Pai, A.; Yaji, A.Y.; Jambunath, U.; Yadav, K. Oral manifestations of myeloid neoplasms and acute leukemia—a diagnostic perspective. Hematol Transfus Int J 2018, 6. Available online: https://medcraveonline.com/HTIJ/oral-manifestations-of-myeloid-neoplasms-and-acute-leukemia--a-diagnostic-perspective.html (accessed on 12 October 2024). [CrossRef]
- Lombardo Bedran, T.B.; Palomari Spolidorio, D.; Grenier, D. Green tea polyphenol epigallocatechin-3-gallate and cranberry proanthocyanidins act in synergy with cathelicidin (LL-37) to reduce the LPS-induced inflammatory response in a three-dimensional co-culture model of gingival epithelial cells and fibroblasts. Arch Oral Biol 2015, 60, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; MacLean, C.; Beer, P.A.; Buck, G.; Wheatley, K.; Kiladjian, J.J.; et al. Correlation of blood counts with vascular complications in essential thrombocythemia: analysis of the prospective PT1 cohort. Blood 2012, 120, 1409–1411. [Google Scholar] [CrossRef]
- Green, A.; Campbell, P.; Buck, G.; Wheatley, K.; East, C.; Bareford, D.; et al. The Medical Research Council PT1 Trial in Essential Thrombocythemia. Blood 2004, 104, 6–6. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Capsomidis, A.; Smits, E.L.; Van Tendeloo, V.F. CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front Immunol 2017, 8, 892. [Google Scholar] [CrossRef] [PubMed]
- De Sousa Lopes, M.L.D.; De Aguiar, J.N.M.; De Brito Monteiro, B.V.; De Oliveira Nóbrega, F.J.; Da Silveira, É.J.D.; Da Costa Miguel, M.C. PP-IMMUNOEXPRESSION OF IL-17, IL-23 AND RORγT IN THE PATHOGENESIS OF PERIODONTAL DISEASE. Oral Surg Oral Med Oral Pathol Oral Radiol 2017, 123, e52. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, Y.; Wei, H.; Yang, S.; Zhan, N.; Xing, X.; et al. Up-regulation of IL-23 p19 expression in human periodontal ligament fibroblasts by IL-1β via concurrent activation of the NF-κB and MAPKs/AP-1 pathways. Cytokine 2012, 60, 171–178. [Google Scholar] [CrossRef]
- Fukuda, M.; Ehara, M.; Suzuki, S.; Ohmori, Y.; Sakashita, H. IL-23 promotes growth and proliferation in human squamous cell carcinoma of the oral cavity. Int J Oncol 2010, 36. Available online: http://www.spandidos-publications.com/ijo/36/6/1355 (accessed on 13 October 2024). [CrossRef]
- Lee, L.T.; Wong, Y.K.; Hsiao, H.Y.; Wang, Y.W.; Chan, M.Y.; Chang, K.W. Evaluation of saliva and plasma cytokine biomarkers in patients with oral squamous cell carcinoma. Int J Oral Maxillofac Surg 2018, 47, 699–707. [Google Scholar] [CrossRef]
- Barrientos, M.O.; Cruz, Á.A.; Teixeira, H.M.P.; Silva, H.D.S.; Gomes-Filho, I.S.; Trindade, S.C.; et al. Variants in interferon gamma inducible protein 16 (IFI16) and absent in melanoma 2 (AIM2) genes that modulate inflammatory response are associated with periodontitis. Arch Oral Biol 2023, 147, 105640. [Google Scholar] [CrossRef] [PubMed]
- Balu, P.; Balakrishna Pillai, A.K.; Mariappan, V.; Ramalingam, S. Cytokine levels in gingival tissues as an indicator to understand periodontal disease severity. Curr Res Immunol 2024, 5, 100080. [Google Scholar] [CrossRef]
- Skalerič, U.; Kramar, B.; Petelin, M.; Pavllia, Z.; Wahl, S.M. Changes in TGF-β1 levels in gingiva, crevicular fluid and serum associated with periodontal inflammation in humans and dogs. Eur J Oral Sci 1997, 105, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; et al. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest 2015, 125, 4514–4528. [Google Scholar] [CrossRef]
- Janda, K.; Krzanowski, M.; Dumnicka, P.; Kusnierz-Cabala, B.; Krasniak, A.; Sulowicz, W. Transforming Growth Factor Beta 1 as a Risk Factor for Cardiovascular Diseases in End-Stage Renal Disease Patients Treated With Peritoneal Dialysis. Clin Lab 2014, 60. Available online: http://www.clin-lab-publications.com/article/1551 (accessed on 26 June 2024). [CrossRef] [PubMed]
- Wesseling, M.; Sakkers, T.R.; De Jager, S.C.A.; Pasterkamp, G.; Goumans, M.J. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vascul Pharmacol 2018, 106, 1–8. [Google Scholar] [CrossRef]
- Gamonal, J.; Bascones, A.; Jorge, O.; Silva, A. Chemokine RANTES in gingival crevicular fluid of adult patients with periodontitis. J Clin Periodontol 2000, 27, 675–681. [Google Scholar] [CrossRef]
- Gamonal, J.; Acevedo, A.; Bascones, A.; Jorge, O.; Silva, A. Characterization of cellular infiltrate, detection of chemokine receptor CCR5 and interleukin-8 and RANTES chemokines in adult periodontitis. J Periodontal Res 2001, 36, 194–203. [Google Scholar] [CrossRef]
- Emingil, G.; Atilla, G.; Hüseyinov, A. Gingival crevicular fluid monocyte chemoattractant protein-1 and RANTES levels in patients with generalized aggressive periodontitis. J Clin Periodontol 2004, 31, 829–834. [Google Scholar] [CrossRef]
- Sharma, E.; Sharma, A.; Nadela, M. RANTES comparison in patients with periodontal disease—A prospective clinical study. Future Dent J 2018, 4, 47–53. [Google Scholar] [CrossRef]
- Von Hundelshausen, P.; Weber, K.S.C.; Huo, Y.; Proudfoot, A.E.I.; Nelson, P.J.; Ley, K.; et al. RANTES Deposition by Platelets Triggers Monocyte Arrest on Inflamed and Atherosclerotic Endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap–mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Italiano, J.E., Jr.; Battinelli, E.M. Selective sorting of alpha-granule proteins. J Thromb Haemost 2009, 7, 173–176. [Google Scholar] [CrossRef]
- Marín Oyarzún, C.P.; Glembotsky, A.C.; Goette, N.P.; Lev, P.R.; De Luca, G.; Baroni Pietto, M.C.; et al. Platelet Toll-Like Receptors Mediate Thromboinflammatory Responses in Patients With Essential Thrombocythemia. Front Immunol 2020, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Wu, B.; Ji, L.; Zhan, Y.; Li, F.; Cheng, L.; et al. Cytokine Consistency Between Bone Marrow and Peripheral Blood in Patients With Philadelphia-Negative Myeloproliferative Neoplasms. Front Med 2021, 8, 598182. [Google Scholar] [CrossRef]
- Liu, Y.; Wilkinson, F.; Kirton, J.; Jeziorska, M.; Iizasa, H.; Sai, Y.; et al. Hepatocyte growth factor and c-Met expression in pericytes: implications for atherosclerotic plaque development. J Pathol 2007, 212, 12–19. [Google Scholar] [CrossRef]
- Lönn, J.; Starkhammar Johansson, C.; Kälvegren, H.; Brudin, L.; Skoglund, C.; Garvin, P.; et al. Hepatocyte growth factor in patients with coronary artery disease and its relation to periodontal condition. Results Immunol 2012, 2, 7–12. [Google Scholar] [CrossRef]
- Boissinot, M.; Cleyrat, C.; Vilaine, M.; Jacques, Y.; Corre, I.; Hermouet, S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in Polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene 2011, 30, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Coudriet, G.M.; He, J.; Trucco, M.; Mars, W.M.; Piganelli, J.D. Hepatocyte Growth Factor Modulates Interleukin-6 Production in Bone Marrow Derived Macrophages: Implications for Inflammatory Mediated Diseases. PLoS ONE 2010, 5, e15384. [Google Scholar] [CrossRef]
- Wang, H.; Yang, Y.F.; Zhao, L.; Xiao, F.J.; Zhang, Q.W.; Wen, M.L.; et al. Hepatocyte Growth Factor Gene-Modified Mesenchymal Stem Cells Reduce Radiation-Induced Lung Injury. Hum Gene Ther 2013, 24, 343–353. [Google Scholar] [CrossRef]
- Boissinot, M.; Vilaine, M.; Hermouet, S. The Hepatocyte Growth Factor (HGF)/Met Axis: A Neglected Target in the Treatment of Chronic Myeloproliferative Neoplasms? Cancers 2014, 6, 1631–1669. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci 2019, 20, 3328. [Google Scholar] [CrossRef]
- Şehirli, A.Ö.; Aksoy, U.; Koca-Ünsal, R.B.; Sayıner, S. Role of NLRP3 inflammasome in COVID-19 and periodontitis: Possible protective effect of melatonin. Med Hypotheses 2021, 151, 110588. [Google Scholar] [CrossRef]
- Fidler, T.P.; Xue, C.; Yalcinkaya, M.; Hardaway, B.; Abramowicz, S.; Xiao, T.; et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature 2021, 592, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Adeodato, C.S.R.; Soares-Lima, S.C.; Batista, P.V.; Fagundes, M.C.N.; Camuzi, D.; Tavares, S.J.O.; et al. Interleukin 6 and Interleukin 1β hypomethylation and overexpression are common features of apical periodontitis: A case-control study with gingival tissue as control. Arch Oral Biol 2023, 150, 105694. [Google Scholar] [CrossRef]
- D’Agostino, S.; Valentini, G.; Dolci, M. Exploring Interleukin Levels in Type 1 Diabetes and Periodontitis: A Review with a Focus on Childhood. Children 2024, 11, 238. [Google Scholar] [CrossRef]
- Li, Y.; Sun, T.; Chen, J.; Zhang, L. Identification of Novel Risk Variants of Inflammatory Factors Related to Myeloproliferative Neoplasm: A Bidirectional Mendelian Randomization Study. Glob Med Genet 2024, 11, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Zhang, H.; Bai, J.; Li, Y.; Li, L.; Zhang, L. Associations of the circulating levels of cytokines with the risk of myeloproliferative neoplasms: a bidirectional mendelian-randomization study. BMC Cancer 2024, 24, 531. [Google Scholar] [CrossRef]
- Ramadan, D.E.; Hariyani, N.; Indrawati, R.; Ridwan, R.D.; Diyatri, I. Cytokines and Chemokines in Periodontitis. Eur J Dent 2020, 14, 483–495. [Google Scholar] [CrossRef]
- Chrysanthakopoulos, N.A. Correlation between periodontal disease indices and lung cancer in Greek adults: a case-control study. Exp Oncol 2016, 38, 49–53. [Google Scholar] [CrossRef]
- Mai, X.; LaMonte, M.J.; Hovey, K.M.; Nwizu, N.; Freudenheim, J.L.; Tezal, M.; et al. History of periodontal disease diagnosis and lung cancer incidence in the Women’s Health Initiative Observational Study. Cancer Causes Control 2014, 25, 1045–1053. [Google Scholar] [CrossRef]
- Michaud, D.S.; Joshipura, K.; Giovannucci, E.; Fuchs, C.S. A Prospective Study of Periodontal Disease and Pancreatic Cancer in US Male Health Professionals. JNCI J Natl Cancer Inst 2007, 99, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Xuan, K.; Jha, A.R.; Zhao, T.; Uy, J.P.; Sun, C. Is periodontal disease associated with increased risk of colorectal cancer? A meta-analysis. Int J Dent Hyg 2021, 19, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Famili, P.; Cauley, J.A.; Greenspan, S.L. The Effect of Androgen Deprivation Therapy on Periodontal Disease in Men With Prostate Cancer. J Urol 2007, 177, 921–924. [Google Scholar] [CrossRef]
- Zeng, X.; Xia, L.; Zhang, Y.; Li, S.; Leng, W.; Kwong, J.S.W. Periodontal Disease and Incident Lung Cancer Risk: A Meta-Analysis of Cohort Studies. J Periodontol 2016, 87, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, X.; Zou, X.; Zhang, Y.; Wang, J.; Wang, Y. Relationship between periodontal disease and lung cancer: A systematic review and meta-analysis. J Periodontal Res 2020, 55, 581–593. [Google Scholar] [CrossRef]
- Dizdar, O.; Hayran, M.; Guven, D.C.; Yılmaz, T.B.; Taheri, S.; Akman, A.C.; et al. Increased cancer risk in patients with periodontitis. Curr Med Res Opin 2017, 33, 2195–2200. [Google Scholar] [CrossRef]
Figure 1.
Pathogen-induced inflammatory response in periodontal disease mediated through JAK2 signaling cascade; 1. ROS induced-JAk2 phosphorylation and activation as a result oxidative stress related to immune response to pathogens leading to JNK activation [60]; 2. Classical JAK2 signaling cascade with subsequent STAT3 phosphorylation [64]; 3. TLR-mediated JAK2 signaling through MYD88 association [62]; In this scenario, one can assume that a mutation with gain of function in JAK2 gene (like JAK2 V617F) could lead to pathogen dependent inflammatory response enhancement in periodontal disease, since JAk2 signaling is common to both epithelial cells and myeloid cells [60]. Abbreviations:IL-1β—Interleukin 1-beta; IL-6—Interleukin 6; JAK2—Janus kinase 2; JNK—c-Jun N-terminal kinase; MYD88—Myeloid differentiation primary response 88; MAPK—mitogen activated protein kinase; PAMPs—pathogen associated molecular patterns; P. gingivalis—Porphyromonas gingivalis; ROS—Reactive oxygen species; STAT—signal transducer and activator of transcription; Created in BioRender.com.
Figure 1.
Pathogen-induced inflammatory response in periodontal disease mediated through JAK2 signaling cascade; 1. ROS induced-JAk2 phosphorylation and activation as a result oxidative stress related to immune response to pathogens leading to JNK activation [60]; 2. Classical JAK2 signaling cascade with subsequent STAT3 phosphorylation [64]; 3. TLR-mediated JAK2 signaling through MYD88 association [62]; In this scenario, one can assume that a mutation with gain of function in JAK2 gene (like JAK2 V617F) could lead to pathogen dependent inflammatory response enhancement in periodontal disease, since JAk2 signaling is common to both epithelial cells and myeloid cells [60]. Abbreviations:IL-1β—Interleukin 1-beta; IL-6—Interleukin 6; JAK2—Janus kinase 2; JNK—c-Jun N-terminal kinase; MYD88—Myeloid differentiation primary response 88; MAPK—mitogen activated protein kinase; PAMPs—pathogen associated molecular patterns; P. gingivalis—Porphyromonas gingivalis; ROS—Reactive oxygen species; STAT—signal transducer and activator of transcription; Created in BioRender.com.
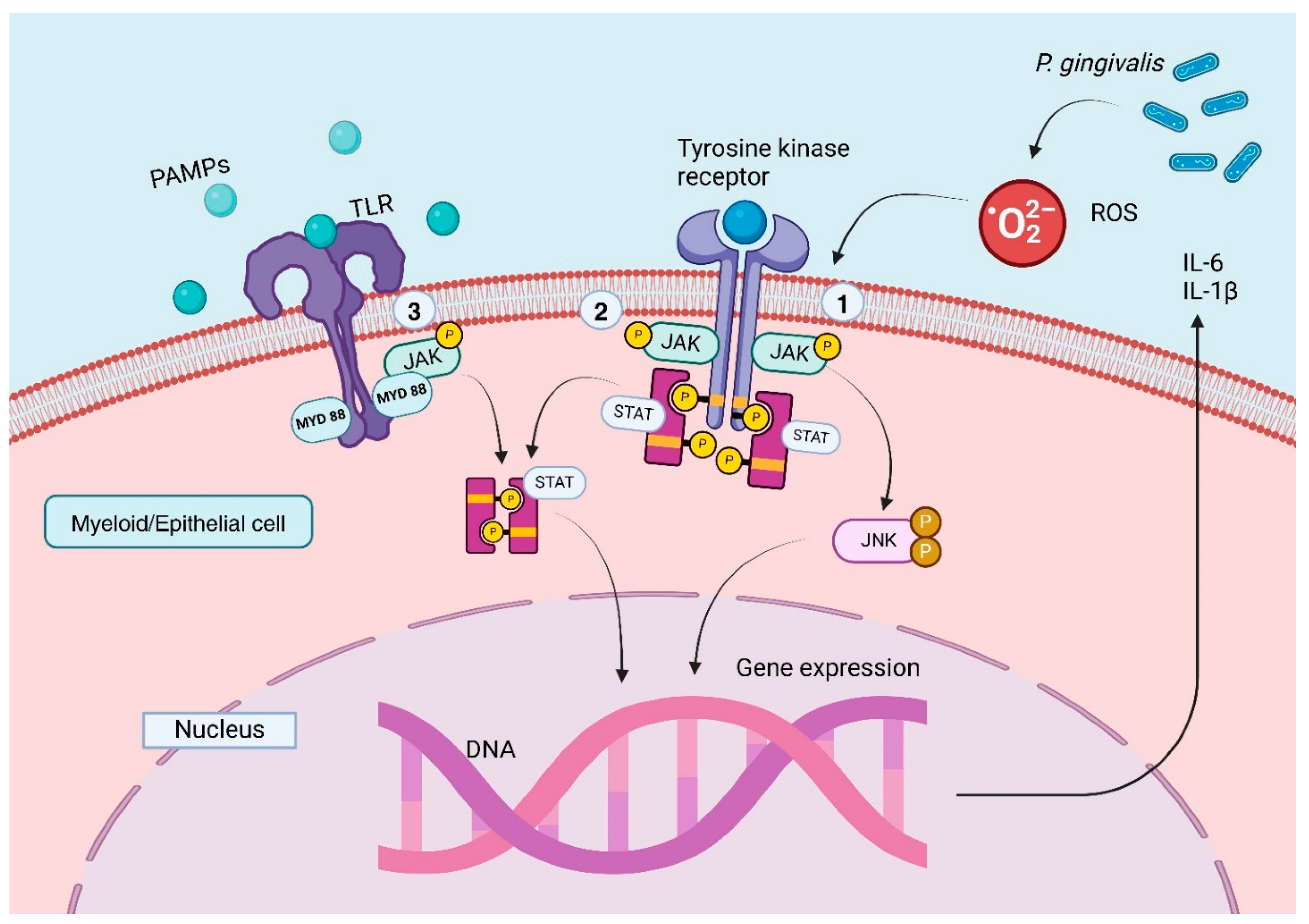
Figure 2.
Proposed Interrelationship Between Periodontal Disease Inflammation and Myeloproliferative Disorders; Created in BioRender.com.
Figure 2.
Proposed Interrelationship Between Periodontal Disease Inflammation and Myeloproliferative Disorders; Created in BioRender.com.
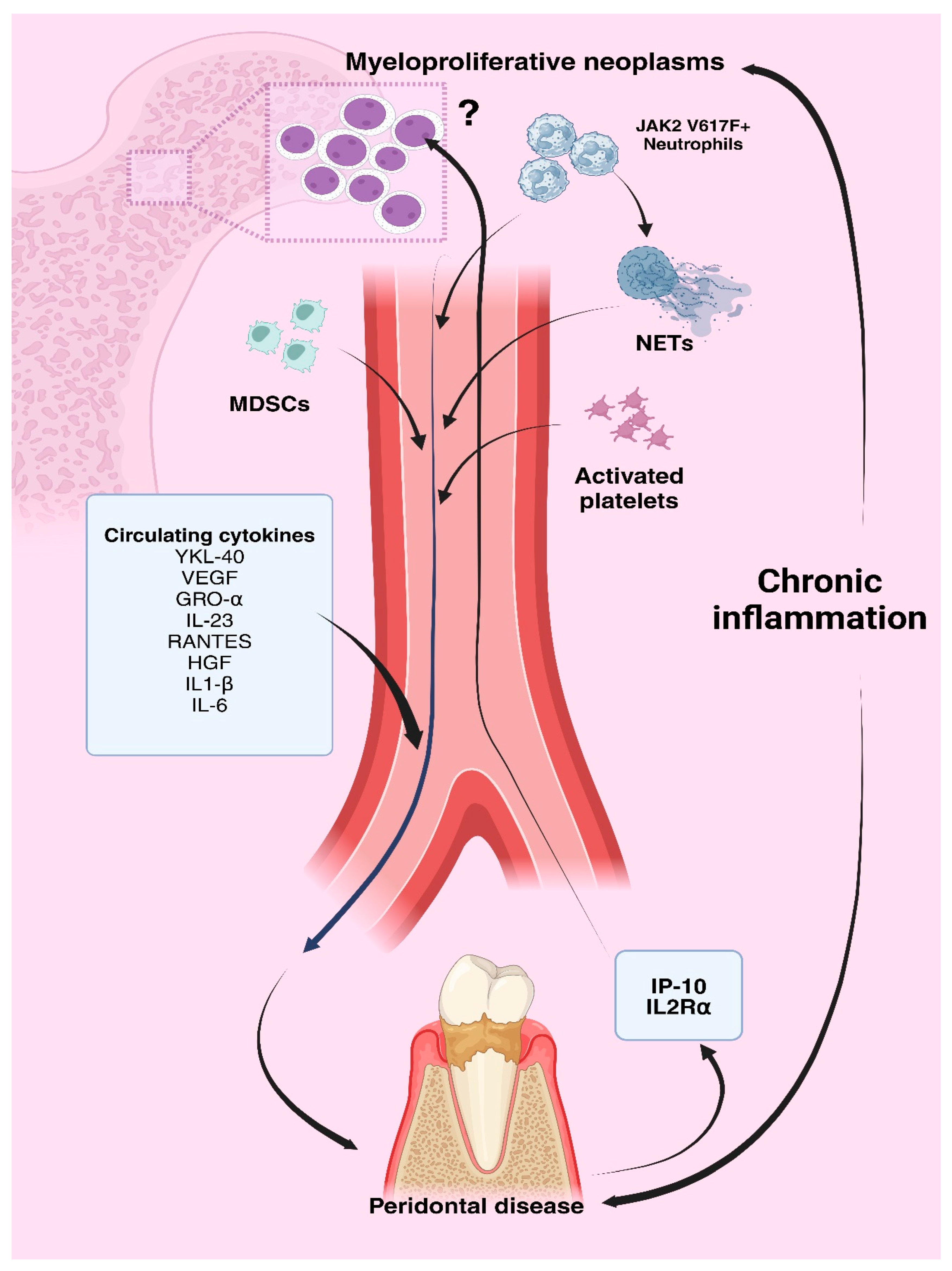

Table 1.
Key Circulating Inflammatory Mediators in PD and MPNs.
| Mediator | Role in PD | Role in MPNs | References |
|---|---|---|---|
| YKL-40 (CHI3L1) | Correlates with disease severity, elevated with IL-6 | Linked to increased inflammation, cardiovascular risk, thrombosis in ET/PV | [94,95,98] |
| VEGF | Increases vascular permeability, exacerbating inflammation | Elevated in ET, associated with bleeding tendencies | [99,100,102] |
| GRO-alpha | Promotes neutrophil chemotaxis in inflamed tissue | Elevated in ET, correlates with progression to MF | [104,105] |
| IL-23 | Enhances Th17 response and tissue damage | Elevated in PV, promotes cell proliferation and tumor growth | [37,109,110] |
| Eotaxin-1 | Recruits eosinophils, contributes to inflammation | Elevated in PV/ET, linked to chronic inflammation | [111,112] |
| TGF-β | Dual role in inflammation and tissue repair | Contributes to arterial plaque and coronary damage | [113,114,115,116,117] |
| RANTES | High levels in severe PD; attracts immune cells | Increased in ET, linked to localized thrombo-inflammatory processes | [118,119,124,125,126] |
| HGF | Increases with disease progression, stimulated by P. gingivalis | Overproduced in JAK2 V617F MPNs; promotes progenitor cell growth | [127,128,130,132] |
| NLRP3 inflammasome | Drives chronic inflammation, triggered by pathogens | Upregulated in MPNs, leading to excessive cytokine release | [10,133,134] |
| AIM2 inflammasome | Releases IL-1β, IL-18, and IL-33 in response to pathogens | Hyperactive in JAK2-mutant MPNs, driving IL-1β signaling | [112,135] |
| IL-1β and IL-6 | Induces bone resorption and tissue damage | Elevated in MPNs (ET/PV/PMF), leading to systemic inflammation | [22,29,30,33,36,136] |
| IL2Rα | Genetic variant linked to PD susceptibility in Type 1 Diabetes | High levels associated with MPN risk | [137,138,139] |
| IP-10 | Attracts immune cells to periodontal tissue, intensifying inflammation | High levels associated with MPN risk | [137,139,140] |
Legend: AIM2 = Interferon-Inducible Protein AIM2; GRO-alpha = Growth Regulated Oncogene Alpha; HGF = Hepatocyte Growth Factor; IP-10 = Interferon Gamma-Induced Protein 10; NLRP3 = NLR Family Pyrin Domain Containing 3; RANTES = Regulated on Activation, Normal T-Cell Expressed and Secreted; TGF-β = Transforming Growth Factor Beta; VEGF = Vascular Endothelial Growth Factor.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



