
Submitted:
09 November 2024
Posted:
12 November 2024
You are already at the latest version
Abstract
Background/Objectives: Transcriptomic studies of diffuse large cell lymphoma (DLCL) have made it possible to distinguish several profiles, including Germinal Centres (GC) and Activated B-Cells. These different types present a different pathophysiology and evolution, which may lead to different treatments. However, these profiles were determined in patients not infected with the human immunodeficiency virus (HIV), whereas lymphomas in the immunocompromised present certain specific characteristics. We therefore set out to determine the transcriptomic profile of DLCL occurring in HIV patients, in order to more precisely determine the pathophysiology of the different subtypes and to identify deregulated molecular pathways that could have a theragnostic value.
Methods: We analysed 12 paraffin-embeddd samples of DLCL linked to HIV infection (two replicates per biological samples) using the Agilent's SurePrint G3 Human GE 8x60K v2 transccriptome chip. Unsupervised hierarchical clustering was performed on the normalized data (quantile normalization) to define the groups that separate the samples according to their gene expression profile.
Results: From this sample of 12 DLCLs, we were able to clearly define two transcriptomic subgroups. Among the differences in gene expression, TP53 and BCL7A were overexpressed in cluster I, and BCL2 in cluster II. In terms of signalling pathways, the ‘immune system development’ pathway was under-expressed in cluster I and over-expressed in cluster II.
Conclusions: Our results strongly suggest the existence of two different transcriptomic signatures which certainly underlie significant differences in pathophysiology. These differences may be due to the specific tumour environment associated with HIV infection. Our very preliminary study will need to be continued with a larger number of patients, and integrate proteomic, metabolomic and clinical data in order to define a theragnostic approach.
Keywords:
lymphoma
; human immunodeficiency virus type 1
; transcriptome
1. Introduction
Diffuse large B-cell lymphomas (DLBCL) represent 30-40% of lymphomas and are treated with a combination of chemotherapy and immunotherapy drugs. The International Prognostic Index (IPI), used to classify and treat patients, is calculated on the basis of clinical parameters: age, LDH level, anatomical stage, extra-nodal localizations, and performance status [1]. On this basis, four prognostic groups are defined with a 5-year survival rate varying from 26% for IPI 4-5 to 70% for IPI 0-1. Modifications of the IPI have been proposed, that have only marginal consequences on the prognosis in the era of anti-CD20 associated chemotherapy regimens. Nonetheless in a seminal paper, Alizadeh et al. [2] used microarrays analysis of tumor biopsies from patients with DLBCLs to propose the distinction of two different subtypes. The GC (germinal center) subtype is defined by the overexpression of CD10, CD38, A-myb, OGG1, BCL-6, BCL-7A, and LMO2, while the ABC (activated B cell) subtype is characterized by the overexpression of IRF4, FLIP, and BCL-2, with its proliferation seemingly relying on the NF-κB pathway. The GC lymphomas are considered to have a better prognosis than the ABC type. Based on these molecular profiles, attempts have been made to specifically target the metabolisms pathways used in each subtypes [3]. Moreover, attempts to use a more simple technique for its use in routine practice have been proposed, such as the immunochemistry algorithm of Hans et al. [4] or Muris et al. [5], which strict correlation with transcriptomic data is not observed. Since ABC-DLCL proliferation and survival rely on BcR-dependent NF-kB signaling, inhibitors of Bruton tyrosine kinase (BTK) could be useful, although more trials are required before the validation for routine treatment [6,7]. Additional subgroups have further been identified, since a 17 gene model allowed to divide DLCL in quartiles with five-year survival ranging from 15% to 73% [8]. These profiles correspond to different prognostic groups, at least in relapsed/refractory patients [9] although this issue is debated [10] and probably depends on the technical approach used, transcriptomic profiling versus various algorithms [11].
Notably, these studies were performed in HIV-negative patients, although non-Hodgkin's lymphomas in HIV-infected patients include specific pathological forms (serous lymphomas, multicentric Castleman's disease), in addition to histological forms found in non-immunocompromised patients, including Burkitt's lymphoma and DLBCL [12,13]. Although this recent WHO classification is no more exclusively based on the lymphoma disease background (HIV-related, post-transplantation, primary immunodeficiency, iatrogenic immunodeficiencies), HIV infection has some specificity [14]. The presence of HIV induces a particular microenvironment in the lymph node that induces, among other effects, activation and proliferation of B cells in the absence of the immune response. This proliferation increases the deregulation of genes such as p53 and the activation of proto-oncogenes such as c-myc and BCL-6 in addition to the absence of immune surveillance. Moreover, the immunosuppression linked to CD4 T cell depletion allows the proliferation of EBV and of KSHV/HHV8 viruses which encoded proteins stimulate B cell proliferation and thus lymphomagenesis [15]. Nonetheless, HIV itself could have a direct impact on lymphomagenesis, more particularly via the expression of HIV p17 protein variants that accumulate in lymph node (even in the absence of HIV detection) and are able to activate the I3K/Akt signaling pathway [16]. These HIV p17 protein variants share insertions in their C-terminal region that modify their biologic properties Although recent advances in anti-retroviral treatment allow most patients with controlled HIV infection to be treated like the general population [17], due to these particularities of HIV-related lymphomas, the existence of the two GEP signatures GC vs. ABC in HIV-related lymphomas was not evident. Thapa et al. [18] have focused their study on the expression of microRNAs, since these molecules have been shown to play a significant role more particularly in EBV-related tumorigenesis [19]. In this study they have shown that the mi-17-92 paralog clusters were upregulated in B cells more particularly at the GC stage, in the eight analyzed DLCL but also in three other subtypes of HIV-related lymphomas (Burkitt, central nervous system, primary effusion lymphoma) [18]. The study of Ramos et al. [20] analyzed the expression of NF-kB target genes in HIV-related lymphomas. They observed the expression of tissue origin-specific markers in PEL (CD69, CSF-1, CIQBP), of IL1beta, cyclin D3 and CD48 in KS, and identified CCR5 as a key marker in Burkitt lymphoma [20]. To differentiate HIV-related from the other DLBCL, Capello et al./Rinaldi et al. performed a genome-wide DNA profiling [21,22]. They concluded that HIV-related had specific genetic lesions since fragile sites-associated genes were more frequently inactivated, more particularly FHIT(FRA3B), WWOX (FRA16D), DCC(FRA18B) and PARK2(FRA6E), in comparison with non-HIV related DLBCL. In the study of Chapman et al. [23] thirty HIV-related DLBCL were analyzed. Interestingly the HIV-related lymphomas had more frequent MYC rearrangements or mutations than non-HIV related lymphoma, and in contrast had rarely BCL2 rearrangements. The authors used the Hans algorithm [4] to classify these HIV-related lymphoma, and concluded that half lymphoma was of GC type, but the other half could not be classified as ABC. These results were confirmed by cell of origin (COO) LymphGen tool [24]. Of note, the percentage of lymphoma classified as other than GC but not ABC (50%) is quite high in comparison with the results usually obtained in non-HIV related lymphoma, i.e ≈ 50% of ABC types 2. The very interesting study of Madan et al. [25] analyzed by immunohistochemical staining of tissue microarrays the expression of GC markers (BCL6, CD10, CyclinH) vs ABC markers (MUM1, CD138, PAK1, CD44, BCL2) in 12 HIV-related and 27 non-HIV related DLBCL. The immunostaining, as expected, clearly identified two distinct clusters of GC and ABC types in non-HIV related DLBCL, while in the case of HIV-related lymphomas only a single aggregate was identified, that moreover expressed an intermediate GG/ABC phenotype [25]. Interestingly, the study of Patrone et al. [26] allows some different conclusions in comparison with the previous studies. They analyzed by suppression subtractive hybridization (SSH) to isolate differentially expressed genes from HIV-related and non-related DLBCL. They progressively restricted the study from 1800 to 18 candidate genes. Unfortunately, there was no preferential expression of these genes in HIV-related vs non-HIV-related lymphomas. This study had nonetheless significant limitations, i.e the number of analyzed samples and the restriction of analyzed genes to those already annotated in data banks [26].
Finally, the validation of the GC/ABC subtypes in HIV-related lymphoma has to be confirmed. For these reasons, the objective of our study was to analyze the gene expression profile of DLBCL in order to verify the possible existence of subgroups described in immunocompetent patients and/or to define new gene profiles specific to HIV-infected NHL. Our goal was also to define the pathophysiology of the different subtypes and to identify deregulated molecular pathways that may have prognostic value.
2. Materials and Methods
2.1. Biological Samples
The tumor library of the ANRS (Agence Nationale de la Recherche sur le Sida et les hépatites virales) provided us with seven paraffin-embedded samples as well as clinical and biological data (ANRS CO16 Lympho-VIR cohort). Five other samples were provided by Pr. H Lepidi (CHU de la Timone, Marseille). We used two technical replicates per biological sample. All samples were obtained from patients who gave their informed consent for the use of their samples for research.
2.2. RNA Extraction, Quantification, and Quality Control
The RNeasy FFPE kit from Qiagen was used for the purification of total RNA from paraffin-embedded fixed organ microdissection. This protocol includes a deparaffinization and rehydration step of the tissue, a lysis step with proteinase K, followed by a heat treatment step. RNA was then extracted on-column with DNase treatment, successive washes, and elution with RNase-free water. RNA quantification and quality control were performed using the Agilent NanoDrop and Bioanalyzer.
2.3. Transcriptomic Study
For transcriptomic analysis, Agilent's SurePrint G3 Human GE 8x60K v2 chip was used. The chip provides high gene and transcript coverage with high sensitivity. The chip has eight arrays, each with 62,976 probes. Several probes of the same transcript are synthesized on the chip that target different locations of the gene, and these have a size of 60 nucleotides. Agilent's Gene Expression FFPE Workflow protocol was used for sample labeling and hybridization. The protocol includes a repair, amplification, purification, and labeling step of the library, followed by a purification step of the labeled complementary DNA and hybridization on genome-wide arrays.
2.4. Statistical Analysis
Unsupervised hierarchical clustering was performed on the normalized data (quantile normalization) to define the groups that can separate the samples according to their gene expression profile. A SAM analysis was then performed, taking into account the groups found after hierarchical clustering. Ten thousand permutations and an FDR of 1% were used with a fold change of 2.
2.5. Gene Enrichment Analysis
Gene enrichment analysis was performed using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost : g:Profiler version e111_eg58_p18_f463989d, database updated on 25/01/2024) to identify enriched biological pathways, molecular functions, and cellular components based on the list of differentially expressed genes. The input gene set was derived from microarray analysis, filtered based on an FDR of 1% and fold change of 2. The analysis was employed with default settings, using the Ensembl genome database as the reference background, and accounting for multiple testing correction with the g:profiler algorithm to reduce false positives. Specific attention was given to Gene Ontology (GO) terms, with significant enrichment set at an adjusted p-value threshold of 0.05.
3. Results
After hierarchical clustering of the normalized data of the 24 samples (2 replicates per sample), two groups were highlighted according to their gene expression profile. Based on this, a SAM analysis was performed on the normalized data using Pearson correlation and 10,000 permutations, with the FDR set to 1% and a fold change of 2. The two groups identified by the initial unsupervised hierarchical clustering were confirmed, characterized by the differential expression of 4045 genes (Figure 1). These two clearly defined subgroups did not correspond to the GC versus non-GC transcriptomic subgroups, except for the expression of the BCL2 and BCL7a genes. Among the most significant differences, the BCL2 gene was over-expressed in patients’ group II, while BCL7a was over-expressed in patients’ group I. In contrast with the transcriptomic classification of non-HIV related DLBCL, we failed to identify differential functional annotations specific to one subgroup. We were not able to define differential prognosis between the two groups due to the small number of samples analyzed. Functional annotation has enabled us to identify very general signaling pathways that are not directly related to pathology. The several signaling pathways identified are summarized in Table 1 and Table 2. Our findings indicate that HIV-related DLBCLs exhibit a distinct transcriptomic profile compared to HIV-negative DLBCLs. This distinct profile could be mainly attributed to the different microenvironment, cytokine profiles, and immune responses in these different lymphoma populations. The overexpression of BCL2 and BCL7a aligns with the notion that these genes play a critical role in the pathogenesis of HIV-related lymphomas. The lack of clear GC versus ABC subtype differentiation suggests that the underlying biology of HIV-associated DLBCL may involve unique pathogenic mechanisms not present in immunocompetent individuals.
For example, the signaling pathway “immune system development“ identified in the cluster I functional annotation is under-expressed in patients’ group I and over- represented in patients’ group II. This pathway includes significative genes such as STAT1, BATF, IRF7, HLA-E and IL4-R, thus differentially expressed between the patients’ groups. STAT1 is a key transcription factor involved in interferon signaling pathways, crucial for antiviral responses and tumor surveillance. Dysregulation of STAT1 has been associated with immune evasion mechanisms in various lymphomas, including diffuse large B-cell lymphoma [27]. Other pivotal genes of the immune responses have also been identified. BATF is an important transcription factor that drives the differentiation of T cells and B cells. It is essential for the development of follicular helper T cells, which support antibody responses. BATF mutations or dysregulation can affect immune cell function and are implicated in lymphomagenesis [28]. IRF7 is a transcription factor that regulates the production of type I interferons, critical for antiviral immunity and immune surveillance against tumors. It has been linked to immune evasion in lymphomas due to its role in regulating innate immunity [29]. HLA-E is involved in immune recognition and modulation, particularly through interactions with NK cells. Overexpression of HLA-E contributes to immune escape in lymphomas by inhibiting NK cell-mediated cytotoxicity [30]. IRF8 is another transcription factor implicated in the differentiation of myeloid cells and B cells. Its dysregulation has been associated with certain subtypes of lymphoma, particularly those affecting B cell maturation [31]. IL4R encodes the receptor for interleukin-4, a cytokine that promotes B cell proliferation and survival. Aberrant signaling through IL4R has been linked to the development of B cell lymphomas [32].
4. Discussion
Diffuse large B-cell lymphoma represents a heterogeneous group of aggressive lymphoid malignancies characterized by diverse genetic alterations and distinct clinical behaviors. Among the differentially expressed genes, we identified TP53, BCL2 and BCL7A. These pivotal genes implicated in the pathogenesis of DLBCL stand out for their significant roles in tumor development and progression.
The TP53 gene is over-expressed in patients’ group I, this gene is a crucial tumor suppressor gene known for its role in maintaining genomic stability by regulating the cell cycle and initiating apoptosis in response to DNA damage [33]. Mutations in TP53 are frequently observed in DLBCL, leading to a loss of function that allows neoplastic cells to escape apoptosis. This dysfunction contributes not only to the survival of genetically unstable cells but also to the accumulation of further genetic aberrations, which can drive tumorigenesis The correlation between TP53 mutations and poor prognosis in DLBCL emphasizes the need for targeted therapies aimed at restoring p53 function or mimicking its pro-apoptotic effects [34].
The BCL2 gene, a master regulator of apoptosis, is often found to be overexpressed in DLBCL and is more particularly over-expressed in patients’ group II. The dysregulation of BCL2 expression enables cancer cells to evade programmed cell death, a hallmark of many malignancies. The overexpression of BCL2 can occur through various mechanisms, including chromosomal translocations, such as t(14;18), which juxtaposes the BCL2 gene to the immunoglobulin locus. This aberrant expression is associated with a poor prognosis in DLBCL patients, as it contributes to the aggressive nature of the disease. Therapeutic strategies targeting BCL2, such as BH3 mimetics, have shown promise in preclinical and clinical settings, suggesting that modulation of apoptosis pathways could be a viable approach for treating DLBCL despite various escape mechanisms [35]. Previous studies have demonstrated the pivotal role of the immune microenvironment in influencing lymphoma behavior [36]. The presence of HIV alters the lymph node microenvironment significantly, leading to chronic B-cell activation and an increased risk of lymphomagenesis [37]. The high degree of gene expression dysregulation observed in our study is consistent with the known impact of HIV on B-cell proliferation and oncogene activation [38].
The BCL7A gene is over-expressed in patients’ group I. This gene was initially identified as part of the t(8;14) translocation in Burkitt lymphoma and plays a significant role in the pathogenesis of DLBCL. Its product is involved in chromatin remodeling, influencing transcription regulation, which is critical for maintaining normal B-cell function. Mutations or dysregulation of BCL7A have been associated with alterations in the B-cell receptor (BCR) signaling pathway, a key driver in the oncogenesis of DLBCL [39]. In the context of HIV-associated DLBCL, BCL7A may have a more prominent role. HIV infection leads to chronic immune activation and an increased likelihood of genetic instability in B-cells. This environment may promote mutations in BCL7A, exacerbating its oncogenic potential. HIV-positive patients with DLBCL often present with more aggressive disease, and the dysregulation of genes involved in chromatin remodeling, such as BCL7A, could contribute to this phenotype [40]. Studies have shown that BCL7A mutations are more frequent in HIV-positive DLBCL compared to non-HIV-associated cases, suggesting that this gene may act as a crucial link between the immunodeficiency state and lymphomagenesis.
5. Conclusions
Preliminary results may indicate that HIV-related DLBCLs have a clearly distinct transcriptomic profile compared to HIV-negative lymphomas. This difference could be mainly related to the distinct microenvironment, cytokine profiles, and immune responses in these populations. Although we were unable to define differential prognostic groups, the identification of unique gene expression patterns in HIV-related DLBCL provides a foundation for future research into targeted therapies and personalized medicine. Further studies are warranted to explore the potential of transcriptionally guided therapeutic approaches in improving outcomes for patients with HIV-associated lymphomas. Future research should aim to expand the sample size and include comprehensive clinical data to validate the prognostic significance of the identified subgroups. Additionally, integrating multi-omics approaches, including proteomics and metabolomics, could offer deeper insights into the molecular underpinnings of HIV-related DLBCL. Investigating the role of the tumor microenvironment and immune landscape in modulating gene expression and lymphoma progression will also be crucial. Ultimately, our goal is to translate these findings into clinical practice, developing personalized treatment strategies that leverage the unique molecular characteristics of HIV-associated lymphomas.
Funding
This work was supported by grants from the ANRS (Agence Nationale de la Recherche sur le Sida et les hépatites virales), Aix-Marseille Université and Advanced BioDesign. The TGML platform is supported by the France Génomique.
Acknowledgments
The authors thank INSERM, Aix-Marseille Université, and Advanced BioDesign (Lyon) for grants; the patients who gave their informed consent for the use of their samples for research; the France Génomique National infrastructure for the IBiSA Transcriptomics and Genomics Marseille–Luminy (TGML) platform. The autors thank also Nicolas Mounier for providing the samples for tests and Christophe Avignon for the preparation of the paraffin sections.
References
- International Non-Hodgkin's Lymphoma Prognostic Factors P. A predictive model for aggressive non-Hodgkin's lymphoma. N Engl J Med. Sep 30 1993:329(14):987-94. [CrossRef]
- Alizadeh AA, Eisen MB, Davis RE, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. Feb 3 2000;403(6769):503-11. [CrossRef]
- Karmali R, Gordon LI. Molecular Subtyping in Diffuse Large B Cell Lymphoma: Closer to an Approach of Precision Therapy. Curr Treat Options Oncol. Feb 2017;18(2):11. [CrossRef]
- Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. Jan 1 2004;103(1):275-82. [CrossRef]
- Muris JJ, Meijer CJ, Vos W, et al. Immunohistochemical profiling based on Bcl-2, CD10 and MUM1 expression improves risk stratification in patients with primary nodal diffuse large B cell lymphoma. J Pathol. Apr 2006;208(5):714-23. [CrossRef]
- Younes A, Sehn LH, Johnson P, et al. Randomized Phase III Trial of Ibrutinib and Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Non-Germinal Center B-Cell Diffuse Large B-Cell Lymphoma. J Clin Oncol. May 20 2019;37(15):1285-1295. [CrossRef]
- Wilson WH, Wright GW, Huang DW, et al. Effect of ibrutinib with R-CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell. Dec 13 2021;39(12):1643-1653 e3. [CrossRef]
- Rosenwald A, Staudt LM. Gene expression profiling of diffuse large B-cell lymphoma. Leuk Lymphoma. 2003;44 Suppl 3:S41-7. [CrossRef]
- Thieblemont C, Briere J, Mounier N, et al. The germinal center/activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J Clin Oncol. Nov 1 2011;29(31):4079-87. [CrossRef]
- Rutherford SC, Leonard JP. DLBCL Cell of Origin: What Role Should It Play in Care Today? Oncology (Williston Park). Sep 15 2018;32(9):445-9.
- Boltezar L, Prevodnik VK, Perme MP, Gasljevic G, Novakovic BJ. Comparison of the algorithms classifying the ABC and GCB subtypes in diffuse large B-cell lymphoma. Oncol Lett. May 2018;15(5):6903-6912. [CrossRef]
- Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia. Jul 2022;36(7):1720-1748. [CrossRef]
- Alaggio R, Amador C, Anagnostopoulos I, et al. Correction: "The 5th edition of The World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms" Leukemia. 2022 Jul;36(7):1720-1748. Leukemia. Sep 2023;37(9):1944-1951. [CrossRef]
- Carbone A, Chadburn A, Gloghini A, Vaccher E, Bower M. Immune deficiency/dysregulation -associated lymphoproliferative disorders. Revised classification and management. Blood Rev. Mar 2024;64:101167. [CrossRef]
- Lurain K, Ramaswami R, Yarchoan R. The role of viruses in HIV-associated lymphomas. Semin Hematol. Oct 2022;59(4):183-191. [CrossRef]
- Dolcetti R, Gloghini A, Caruso A, Carbone A. A lymphomagenic role for HIV beyond immune suppression? Blood. Mar 17 2016;127(11):1403-9. [CrossRef]
- Vaccher E, Gloghini A, Carbone A. HIV-related lymphomas. Curr Opin Oncol. Sep 1 2022;34(5):439-445. [CrossRef]
- Thapa DR, Li X, Jamieson BD, Martinez-Maza O. Overexpression of microRNAs from the miR-17-92 paralog clusters in AIDS-related non-Hodgkin's lymphomas. PLoS One. 2011;6(6):e20781. [CrossRef]
- Torne AS, Robertson ES. Epigenetic Mechanisms in Latent Epstein-Barr Virus Infection and Associated Cancers. Cancers (Basel). Feb 29 2024;16(5). [CrossRef]
- Ramos JC, Sin SH, Staudt MR, et al. Nuclear factor kappa B pathway associated biomarkers in AIDS defining malignancies. Int J Cancer. Jun 1 2012;130(11):2728-33. [CrossRef]
- Capello D, Scandurra M, Poretti G, et al. Genome wide DNA-profiling of HIV-related B-cell lymphomas. Br J Haematol. Jan 2010;148(2):245-55. [CrossRef]
- Rinaldi A, Mian M, Chigrinova E, et al. Genome-wide DNA profiling of marginal zone lymphomas identifies subtype-specific lesions with an impact on the clinical outcome. Blood. Feb 3 2011;117(5):1595-604. [CrossRef]
- Chapman JR, Bouska AC, Zhang W, et al. EBV-positive HIV-associated diffuse large B cell lymphomas are characterized by JAK/STAT (STAT3) pathway mutations and unique clinicopathologic features. Br J Haematol. Sep 2021;194(5):870-878. [CrossRef]
- Scott DW, Wright GW, Williams PM, et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood. Feb 20 2014;123(8):1214-7. [CrossRef]
- Madan R, Gormley R, Dulau A, et al. AIDS and non-AIDS diffuse large B-cell lymphomas express different antigen profiles. Mod Pathol. Mar 2006;19(3):438-46. [CrossRef]
- Patrone L, Henson SE, Teodorovic J, et al. Gene expression patterns in AIDS versus non-AIDS-related diffuse large B-cell lymphoma. Exp Mol Pathol. Apr 2003;74(2):129-39. [CrossRef]
- Wang W, Lopez McDonald MC, Kim C, et al. The complementary roles of STAT3 and STAT1 in cancer biology: insights into tumor pathogenesis and therapeutic strategies. Front Immunol. 2023;14:1265818. [CrossRef]
- Trujillo-Ochoa JL, Kazemian M, Afzali B. The role of transcription factors in shaping regulatory T cell identity. Nat Rev Immunol. Dec 2023;23(12):842-856. [CrossRef]
- Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. Sep 2011;12(6):399-414. [CrossRef]
- Liu X, Song J, Zhang H, et al. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell. Feb 13 2023;41(2):272-287 e9. [CrossRef]
- Cai M, Chen N. The Roles of IRF-8 in Regulating IL-9-Mediated Immunologic Mechanisms in the Development of DLBCL: A State-of-the-Art Literature Review. Front Oncol. 2022;12:817069. [CrossRef]
- Natoli A, Lupertz R, Merz C, et al. Targeting the IL-4/IL-13 signaling pathway sensitizes Hodgkin lymphoma cells to chemotherapeutic drugs. Int J Cancer. Oct 15 2013;133(8):1945-54. [CrossRef]
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. Jan 2010;2(1):a001008. [CrossRef]
- Duffy MJ, O'Donovan N, McDermott E, Crown J. Validated biomarkers: The key to precision treatment in patients with breast cancer. Breast. Oct 2016;29:192-201. [CrossRef]
- Adams CM, McBride A, Michener P, et al. Identifying Targetable Vulnerabilities to Circumvent or Overcome Venetoclax Resistance in Diffuse Large B-Cell Lymphoma. Cancers (Basel). Jun 3 2024;16(11). [CrossRef]
- Takahara T, Nakamura S, Tsuzuki T, Satou A. The Immunology of DLBCL. Cancers (Basel). Jan 29 2023;15(3). [CrossRef]
- Mu W, Patankar V, Kitchen S, Zhen A. Examining Chronic Inflammation, Immune Metabolism, and T Cell Dysfunction in HIV Infection. Viruses. Jan 31 2024;16(2). [CrossRef]
- Isaguliants M, Bayurova E, Avdoshina D, Kondrashova A, Chiodi F, Palefsky JM. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers (Basel). Jan 15 2021;13(2). [CrossRef]
- Ramos-Medina R, Montes-Moreno S, Maestre L, et al. BCL7A protein expression in normal and malignant lymphoid tissues. Br J Haematol. Jan 2013;160(1):106-9. [CrossRef]
- Scott DW, Gascoyne RD. The tumour microenvironment in B cell lymphomas. Nat Rev Cancer. Aug 2014;14(8):517-34. [CrossRef]
Figure 1.
Expression profile of diffuse large-cell B lymphomas associated with HIV infection. Under-expressed genes are represented in green, over-expressed genes are in red. On the abscissa samples are separated into two groups. On the ordinate are the probes that represent the differentially expressed genes, these are separated into two clusters, i.e the Cluster I and Cluster II.
Figure 1.
Expression profile of diffuse large-cell B lymphomas associated with HIV infection. Under-expressed genes are represented in green, over-expressed genes are in red. On the abscissa samples are separated into two groups. On the ordinate are the probes that represent the differentially expressed genes, these are separated into two clusters, i.e the Cluster I and Cluster II.
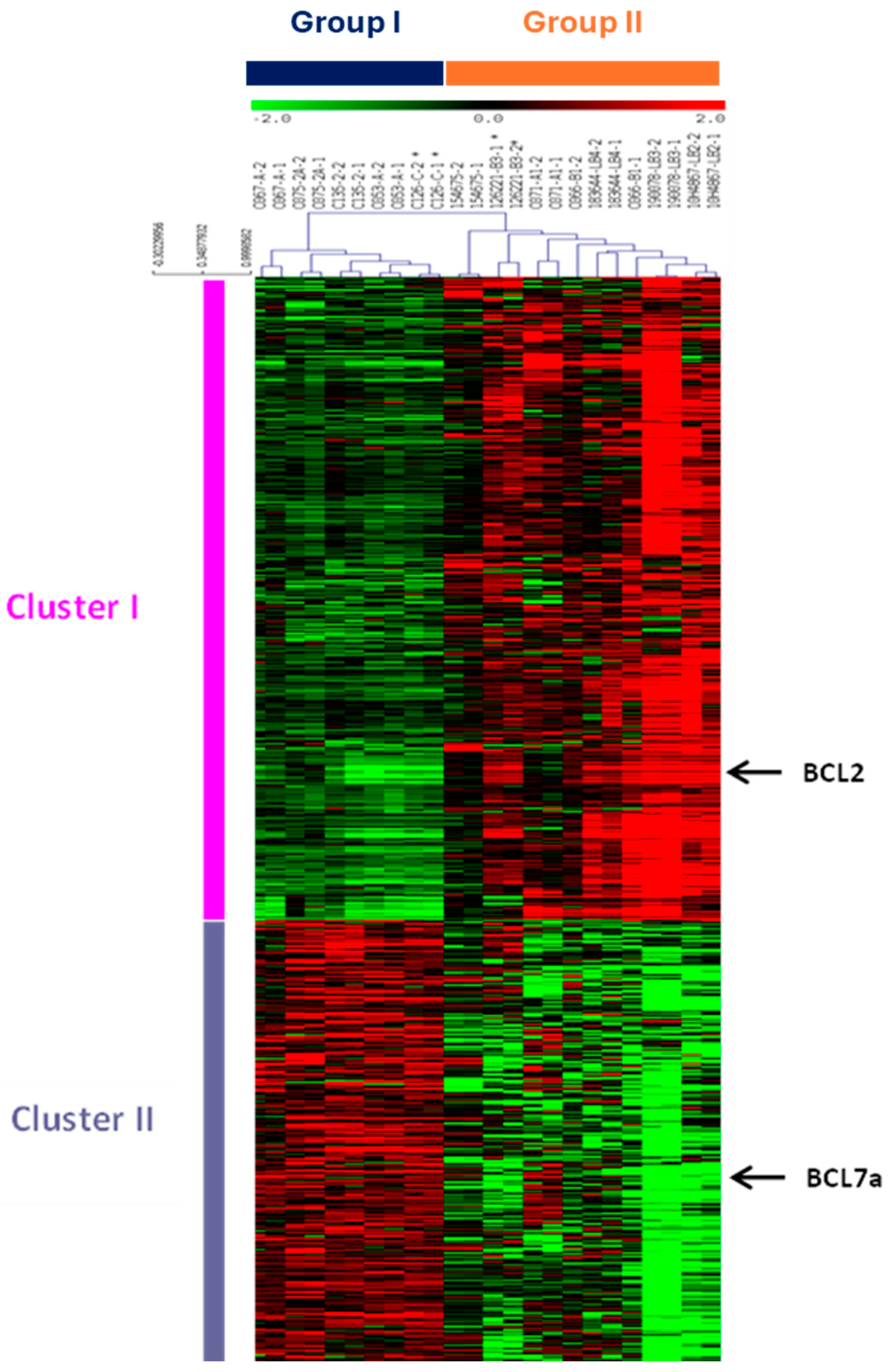

Table 1.
Cluster I functional annotation. Cluster I genes are under-expressed in patients’ group I and over-expressed in patients’ group II. (MF: Molecular functions, BP: Biological Process).
Table 1.
Cluster I functional annotation. Cluster I genes are under-expressed in patients’ group I and over-expressed in patients’ group II. (MF: Molecular functions, BP: Biological Process).
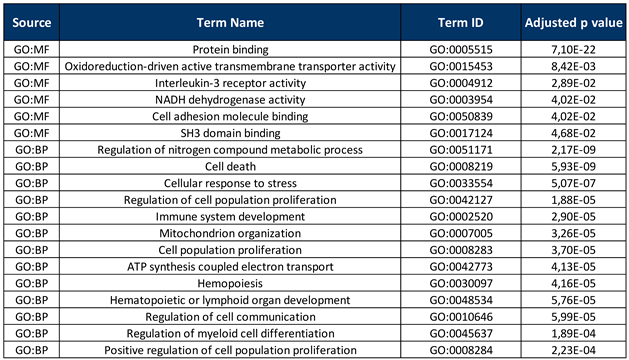 |
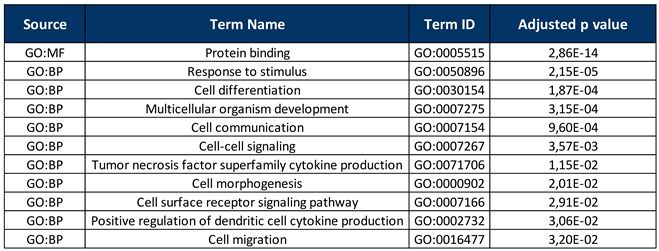
Table 2.
Cluster II functional annotation. The cluster II genes are over-expressed genes in patients’ group I and under-expressed in patients’ group II. (MF: Molecular functions, BP: Biological Process).
Table 2.
Cluster II functional annotation. The cluster II genes are over-expressed genes in patients’ group I and under-expressed in patients’ group II. (MF: Molecular functions, BP: Biological Process).
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



